
NPLATE injektiokuiva-aine ja liuotin, liuosta varten 250 mikrog, 500 mikrog
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Ohjelehtiö: Nplate-kotipistoshoidon kuvalliset ohjeet
NPLATE
Malli suurempaan kokoon painetusta pistospakkauksen käyttöön tarkoitetusta työskentelyalustasta.
Terveydenhuollon ammattilainen
Havainnekuva annosteluoppaasta (annoksen laskeminen ja säätäminen), jonka voi tilata osoitteesta medinfo.finland@amgen.com
Kotipistoshoidon tarkistuslista
Opas kotipistoshoitoon soveltuvien potilaiden valintaan ja koulutukseen
Vaikuttavat aineet ja niiden määrät
Nplate 250 mikrogrammaa injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää 250 mikrog romiplostiimia. Liuottamisen jälkeen käytettävissä oleva 0,5 ml liuosta sisältää 250 mikrog romiplostiimia (500 mikrog/ml). Jokaisessa injektiopullossa on ylitäyttöä, joka varmistaa, että pullosta saadaan 250 mikrog romiplostiimia.
Nplate 500 mikrogrammaa injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää 500 mikrog romiplostiimia. Liuottamisen jälkeen käytettävissä oleva 1 ml liuosta sisältää 500 mikrog romiplostiimia (500 mikrog/ml). Jokaisessa injektiopullossa on ylitäyttöä, joka varmistaa, että pullosta saadaan 500 mikrog romiplostiimia.
Romiplostiimi valmistetaan yhdistelmä‑DNA‑tekniikalla Escherichia coli (E. coli) ‑bakteerissa.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten (injektiokuiva-aine).
Kliiniset tiedot
Käyttöaiheet
Nplate on tarkoitettu primaarin immunotrombosytopenian (ITP) hoitoon aikuispotilaille, kun muut hoidot (esim. kortikosteroidit, immunoglobuliinit) eivät tehoa (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Ehto
Hoito on toteutettava verisairauksien hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito on toteutettava verisairauksien hoitoon perehtyneen lääkärin valvonnassa.
Annostus
Nplate annetaan kerran viikossa injektiona ihon alle.
Aloitusannos
Romiplostiimin aloitusannos on 1 mikrog/kg painon mukaan.
Annoksen laskeminen
Aloitusannos tai myöhemmät viikoittaiset annokset: | Paino* kg x annos mikrog/kg = potilaan yksilöllinen annos mikrogrammoina (mikrog) |
Annettava määrä: | Annos mikrog x (1 ml/500 mikrog) = Injisoitava määrä millilitroina (ml) |
Esimerkki: | 75 kg painavan potilaan aloitusannos on 1 mikrog/kg romiplostiimia. Potilaan yksilöllinen annos = 75 kg x 1 mikrog/kg = 75 mikrog Vastaava injisoitava määrä Nplate‑liuosta = 75 mikrog x (1 ml/500 mikrog) = 0,15 ml |
| * Romiplostiimiannoksen laskemiseen käytetään aina potilaan painoa hoidon alkaessa. Myöhemmin annosta säädetään pelkästään trombosyyttiarvon muutosten perusteella 1 mikrog/kg kerrallaan (ks. taulukko alla). | |
Annoksen säätäminen
Annoksen laskemiseen käytetään potilaan painoa hoidon alkaessa. Kerran viikossa annettavaa romiplostiimiannosta suurennetaan 1 mikrog/kg kerrallaan, kunnes potilaan trombosyyttiarvo on ≥ 50 x 109/l. Trombosyyttiarvo määritetään viikon välein, kunnes se pysyy vakaana (≥ 50 x 109/l vähintään 4 viikon ajan ilman annosmuutoksia). Tämän jälkeen trombosyyttimääritys tehdään kuukauden välein. Enimmäisannosta, 10 mikrog/kg kerran viikossa, ei saa ylittää.
Annosta säädetään seuraavasti:
Trombosyyttiarvo (x 109/l) | Toimenpide |
< 50 | Kerran viikossa annettavaa annosta suurennetaan 1 mikrog/kg. |
> 150 kahden peräkkäisen viikon aikana | Kerran viikossa annettavaa annosta pienennetään 1 mikrog/kg. |
> 250 | Annosta ei anneta. Trombosyyttiarvoa seurataan edelleen viikon välein. Kun trombosyyttiarvo on laskenut tasolle < 150 x 109/l, hoito aloitetaan uudelleen 1 mikrog/kg pienemmällä viikkoannoksella. |
Trombosyyttivasteen yksilöllisten vaihteluiden vuoksi joidenkin potilaiden trombosyyttiarvo saattaa laskea äkillisesti tason 50 x 109/l alapuolelle annoksen pienentämisen tai hoidon lopettamisen jälkeen. Näissä tapauksissa voidaan harkita korkeampia trombosyyttipitoisuuden raja‑arvoja annoksen pienentämiselle (200 x 109/l) ja hoidon keskeyttämiselle (400 x 109/l) lääkärin arvion mukaan, mikäli se on kliinisesti perusteltua.
Jos hoitovaste häviää tai trombosyyttivastetta ei onnistuta säilyttämään suositelluilla romiplostiimiannoksilla, vasteen puuttumisen syyt tulee selvittää (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet, romiplostiimivasteen häviäminen).
Hoidon keskeyttäminen
Kun romiplostiimihoitoa on jatkettu neljä viikkoa suurimmalla sallitulla viikkoannoksella (10 mikrog/kg), hoito lopetetaan, ellei trombosyyttimäärä nouse tasolle, joka riittää estämään kliinisesti merkittävät verenvuodot.
Potilaiden tila arvioidaan kliinisesti säännöllisin välein, ja hoitava lääkäri päättää kunkin potilaan hoidon jatkamisesta yksilöllisesti. Jos potilaan perna on tallella, on arvioitava myös splenektomian tarve. Trombosytopenian uusiutuminen on todennäköistä hoidon lopettamisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Iäkkäät (≥ 65‑vuotiaat) potilaat
Hoidon turvallisuudessa tai tehossa ei ole havaittu yleisiä eroja 65 vuotta täyttäneiden ja nuorempien potilaiden välillä (ks. kohta Farmakodynamiikka). Näiden tietojen perusteella annostusta ei tarvitse muuttaa iäkkäitä potilaita hoidettaessa, mutta varovaisuutta on kuitenkin syytä noudattaa, sillä toistaiseksi kliinisissä tutkimuksissa on ollut vasta vähän iäkkäitä potilaita.
Pediatriset potilaat
Romiplostiimin 250/500 mikrog injektiokuiva-aine ja liuotin, liuosta varten turvallisuutta ja tehoa alle 18‑vuotiaiden potilaiden hoidossa ei ole vielä varmistettu. Tätä lääkemuotoa käyttävät myös aikuispotilaat, jotka ovat saaneet luvan pistää annoksensa itse. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset ja Farmakodynamiikka, ei voida antaa suosituksia annostuksesta.
Lapsipotilaat eivät saa pistää romiplostiimiannoksia itse. Tietoja ei ole saatavilla.
Muut lääkemuodot/vahvuudet voivat soveltua paremmin tälle potilasryhmälle.
Potilaat, joilla on maksan vajaatoiminta
Romiplostiimihoitoa ei pidä antaa potilaille, joilla on kohtalainen tai vaikea maksan vajaatoiminta (Child‑Pugh‑pistearvo ≥ 7), paitsi jos hoidon odotettu hyöty on suurempi kuin todettu porttilaskimotromboosin riski potilailla, jotka ovat saaneet trombopoietiinireseptorin (TPO‑reseptorin) agonisteja maksan vajaatoimintaan liittyvän trombosytopenian hoitoon (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jos romiplostiimihoito katsotaan välttämättömäksi, trombosyyttiarvoja on seurattava tarkoin tromboembolisten komplikaatioiden riskin pienentämiseksi.
Potilaat, joilla on munuaisten vajaatoiminta
Näistä potilasryhmistä ei ole tehty varsinaisia kliinisiä tutkimuksia. Nplaten käytössä on noudatettava varovaisuutta näitä potilasryhmiä hoidettaessa.
Antotapa
Ihon alle.
Kuiva-aineen liuottamisen jälkeen Nplate‑injektioliuos annetaan ihon alle. Injisoitava määrä voi olla hyvin pieni. Nplate‑injektioliuosta valmistettaessa on noudatettava huolellisuutta annoksen laskemisessa ja kuiva-aineen liuottamisessa oikeaan määrään steriiliä injektionesteisiin käytettävää vettä. Erityisen huolellisesti on varmistettava, että injektiopullosta vedetään oikea määrä Nplate‑injektioliuosta ihonalaista injektiota varten. Tähän on käytettävä ruiskua, jonka asteikon tarkkuus on 0,01 ml.
Potilaat, joilla on vakaa trombosyyttiarvo ≥ 50 x 109/l vähintään 4 viikon ajan ilman annosmuutoksia, voivat pistää Nplate‑injektioliuoksen itse hoitoa valvovan lääkärin harkinnan mukaan. Jos potilaalle tarjotaan mahdollisuutta pistää Nplate‑annokset itse, hänelle on opetettava siihen kuuluvat toimenpiteet.
Kun potilas on ensimmäiset neljä viikkoa pistänyt Nplate‑annokset itse, hänen tekniikkansa on tarkistettava uudelleen, kun hän valmistaa injektioliuoksen ja pistää Nplate‑annoksensa. Potilas voi jatkaa Nplate‑annosten pistämistä itse vain, jos hän pystyy osoittamaan, että hän osaa valmistaa Nplate‑injektioliuoksen ja pistää annoksen.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja pistämisestä.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille tai E. coli ‑bakteerissa tuotetuille proteiineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Trombosytopenian uusiutuminen ja verenvuoto hoidon lopettamisen jälkeen
Trombosytopenia ilmaantuu todennäköisesti uudelleen romiplostiimihoidon lopettamisen jälkeen. Verenvuotoriski on suurentunut, jos romiplostiimihoito keskeytetään antikoagulanttihoidon tai trombosyyttien toimintaa estävän lääkityksen aikana. Romiplostiimihoidon lopettamisen yhteydessä potilaiden tilaa seurataan tarkoin trombosyyttiarvon laskun havaitsemiseksi ja annetaan asianmukaista hoitoa verenvuotojen välttämiseksi. Jos romiplostiimihoito lopetetaan, ITP:n hoito tulisi aloittaa uudelleen voimassa olevien hoitosuositusten mukaisesti. Muuhun lääketieteelliseen hoitoon voi kuulua antikoagulanttihoidon ja/tai trombosyyttien toimintaa estävän lääkityksen lopettaminen, antikoagulaation kumoaminen tai trombosyyttituki.
Lisääntynyt luuytimen retikuliini
Luuytimen retikuliinin lisääntymisen arvellaan johtuvan TPO‑reseptorin stimulaatiosta, joka johtaa megakaryosyyttien lisääntymiseen luuytimessä, ja tämä voi puolestaan johtaa sytokiinien vapautumiseen. Morfologiset muutokset perifeerisissä verisoluissa voivat olla merkkejä retikuliinin lisääntymisestä, joka voidaan todeta luuydinbiopsian avulla. Solujen morfologiset poikkeavuudet tulisi tutkia perifeerisen veren sivelyvalmisteesta ja tehdä täydellisen verenkuvan määritykset ennen romiplostiimihoitoa ja hoidon aikana. Kohdassa Haittavaikutukset on lisätietoa romiplostiimin kliinisissä tutkimuksissa havaitusta retikuliinin lisääntymisestä.
Jos hoitoteho häviää ja perifeerisen veren sivelyvalmisteessa havaitaan poikkeavuuksia, romiplostiimihoito on keskeytettävä, tehtävä fysikaalinen tutkimus ja harkittava luuydinbiopsiaa, josta tehdään asianmukainen retikuliinivärjäys. Jos aikaisempi luuydinnäyte on käytettävissä, tehdään vertailu aikaisempaan biopsianäytteeseen. Jos hoitoteho säilyy mutta perifeerisen veren sivelyvalmisteessa havaitaan poikkeavuuksia, lääkärin tulee käyttää asianmukaista kliinistä harkintaa ja harkita myös luuydinbiopsian ottamista, ja romiplostiimihoidon ja vaihtoehtoisten ITP:n hoitomuotojen riski‑hyötysuhde on arvioitava uudelleen.
Tromboottiset/tromboemboliset komplikaatiot
Tromboottisia/tromboembolisia tapahtumia, kuten syvä laskimotromboosi, keuhkoembolia ja sydäninfarkti, on havaittu käytettäessä romiplostiimia ITP‑potilaiden hoidossa. Näitä tapahtumia esiintyi trombosyyttiarvoista riippumatta (ks. kohta Haittavaikutukset). Kliinisissä tutkimuksissa tromboottisten/tromboembolisten tapahtumien ilmaantuvuus oli romiplostiimia saaneilla potilailla 6,0 % ja lumehoitoa saaneilla 3,6 %. Romiplostiimihoidossa on noudatettava varovaisuutta, jos potilaalla on tunnettuja tromboembolian riskitekijöitä, joita voivat olla muun muassa perinnölliset (esim. Tekijä V:n Leiden‑mutaatio) tai hankinnaiset (esim. ATIII:n puute, fosfolipidivasta‑aineoireyhtymä) riskitekijät, korkea ikä, pitkään jatkunut immobilisaatio, pahanlaatuiset sairaudet, ehkäisyvalmisteiden käyttö ja hormonikorvaushoito, leikkaus/vamma, lihavuus ja tupakointi. Potilaita on syytä tarkkailla tromboottisten/tromboembolisten tapahtumien oireiden ja löydösten varalta ja hoitaa ne viipymättä hoitoyksikön ohjeiden ja vakiintuneiden hoitokäytäntöjen mukaisesti.
Tromboembolisia tapahtumia, myös porttilaskimotromboosia, on raportoitu romiplostiimia saaneilla potilailla, joilla on krooninen maksasairaus. Romiplostiimin käytössä on noudatettava varovaisuutta näitä potilasryhmiä hoidettaessa. Annoksen säätämistä koskevia ohjeita on noudatettava (ks. kohta Annostus ja antotapa).
Lääkitysvirheet
Nplate‑hoidon yhteydessä on raportoitu lääkitysvirheitä, kuten yli- ja aliannostelua. Annoksen laskemista ja säätämistä koskevia ohjeita on noudatettava (ks. kohta Annostus ja antotapa).
Yliannostus voi johtaa liian suureen trombosyyttiarvon nousuun ja tromboottisiin/tromboembolisiin komplikaatioihin. Jos trombosyyttiarvo on kohonnut huomattavasti, Nplate‑hoito lopetetaan ja trombosyyttiarvoja seurataan. Nplate‑hoito aloitetaan uudelleen annostusta ja antotapaa koskevia suosituksia noudattaen. Aliannostelutapauksissa trombosyyttiarvo voi jäädä odotettua pienemmäksi, mikä voi altistaa verenvuodoille. Potilaiden trombosyyttiarvoja on seurattava Nplate‑hoidon aikana (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Yliannostus).
Jo olemassa olevien myelodysplastisten oireyhtymien (MDS) eteneminen
Romiplostiimin positiivinen hyöty-riskisuhde on osoitettu vain ITP:hen liittyvän trombosytopenian hoidossa (ks. kohta Käyttöaiheet), eikä romiplostiimia saa käyttää muissa kliinisissä tiloissa, joihin liittyy trombosytopeniaa.
Aikuisten ja iäkkäiden potilaiden ITP‑diagnoosi on vahvistettava sulkemalla pois muut kliiniset tilat, joihin liittyy trombosytopeniaa, varsinkin myelodysplastisen oireyhtymän (MDS) diagnoosi on suljettava pois. Luuydinaspiraatio ja ‑biopsia on tehtävä normaalisti sairauden ja hoidon aikana, jos potilaalla on systeemisiä oireita tai poikkeavia löydöksiä, kuten perifeeristen blastisolujen lisääntymistä.
Kliinisissä tutkimuksissa, joissa romiplostiimia annettiin MDS‑potilaille, havaittiin blastisolujen ohimenevää lisääntymistä joissakin tapauksissa ja raportoitiin tapauksia, joissa MDS eteni akuutiksi myelooiseksi leukemiaksi (AML). MDS‑potilaiden satunnaistetussa lumevertailututkimuksessa romiplostiimihoito lopetettiin ennenaikaisesti, koska romiplostiimiryhmässä oli enemmän potilaita, joiden tauti eteni AML:ksi, ja potilaita, joiden blastisolujen määrä veressä lisääntyi > 10 %. Havaituissa tapauksissa MDS:n eteneminen AML:ksi oli todennäköisempää potilailla, joilla oli RAEB‑1‑luokan MDS lähtötilanteessa, kuin niillä, joilla oli pienemmän riskin tauti.
Romiplostiimia ei saa käyttää myelodysplastisesta oireyhtymästä johtuvan trombosytopenian eikä minkään muun trombosytopenian aiheuttajan kuin ITP:n hoitoon muutoin kuin kliinisissä tutkimuksissa.
Romiplostiimivasteen häviäminen
Jos hoitovaste häviää tai trombosyyttivaste ei säily suositeltua romiplostiimiannosta käytettäessä, tulisi pyrkiä selvittämään taustalla olevat syyt, joita voivat olla esimerkiksi immunogeenisuus (ks. kohta Haittavaikutukset) ja lisääntynyt luuytimen retikuliini (ks. edellä).
Romiplostiimin vaikutukset puna- ja valkosoluihin
Veren punasolu- ja valkosoluarvojen muutoksia (punasolujen vähenemistä ja valkosolujen lisääntymistä) on havaittu eläinkokeissa (rotilla ja apinoilla) ja myös ITP‑potilailla. Anemiaa ja leukosytoosia voi esiintyä samanaikaisesti (4 viikon aikaikkunan sisällä) riippumatta siitä, onko perna poistettu vai ei, mutta tämä on ollut yleisempää potilailla, joille on aikaisemmin tehty pernan poisto. Potilaiden puna- ja valkosoluarvojen seuraamista tulisi harkita romiplostiimihoidon aikana.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. Mahdollisia yhteisvaikutuksia, jotka johtuvat sitoutumisesta plasman proteiineihin, ei tunneta romiplostiimin ja muiden samanaikaisesti käytettyjen lääkkeiden välillä.
Kliinisissä tutkimuksissa romiplostiimia on annettu yhdessä seuraavien ITP:n hoidossa käytettävien lääkkeiden kanssa: kortikosteroidit, danatsoli ja/tai atsatiopriini, laskimoon annettava immunoglobuliinihoito (IVIG) ja anti‑D‑immunoglobuliini. Trombosyyttiarvoja on seurattava, kun romiplostiimia annetaan yhdessä muiden ITP‑lääkkeiden kanssa, jotta vältettäisiin suositellun raja-alueen ulkopuoliset trombosyyttiarvot (ks. kohta Annostus ja antotapa).
Kortikosteroidien, danatsolin ja atsatiopriinin käyttöä voidaan vähentää tai se voidaan keskeyttää, kun romiplostiimi lisätään hoitoon (ks. kohta Farmakodynamiikka). Trombosyyttiarvoja on seurattava, kun muiden ITP‑lääkkeiden käyttöä vähennetään tai niiden käyttö lopetetaan, jotta vältettäisiin suositellun raja-alueen alittavat trombosyyttiarvot (ks. kohta Annostus ja antotapa).
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja romiplostiimin käytöstä raskaana oleville naisille.
Eläinkokeet ovat osoittaneet, että romiplostiimi läpäisee istukan ja suurentaa sikiön trombosyyttiarvoja. Eläinkokeissa havaittiin myös implantaation jälkeisiä keskenmenoja ja poikasten perinataalikuolleisuuden vähäistä lisääntymistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Romiplostiimin käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyvätkö romiplostiimi/metaboliitit äidinmaitoon. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Päätettäessä lopetetaanko imetys vai romiplostiimihoito, on otettava huomioon toisaalta imetyksen hyöty lapselle ja toisaalta hoidosta koituva hyöty äidille.
Hedelmällisyys
Valmisteen vaikutuksista hedelmällisyyteen ei ole tutkimustietoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Nplatella on kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Kliinisissä tutkimuksissa joillakin potilailla on esiintynyt lieviä tai kohtalaisia ohimeneviä huimauskohtauksia.
Haittavaikutukset
Tiivistelmä turvallisuustiedoista
Analyysi kaikista aikuisista ITP‑potilaista, jotka saivat romiplostiimia neljässä kliinisessä vertailututkimuksessa ja viidessä ei-vertailevassa kliinisessä tutkimuksessa, osoitti, että 91,5 prosentille (248/271) romiplostiimihoitoa saaneista tutkittavista ilmaantui jokin haittavaikutus. Romiplostiimialtistuksen kesto oli tässä tutkimusjoukossa keskimäärin 50 viikkoa.
Vakavimpia haittavaikutuksia, joita voi esiintyä Nplate‑hoidon aikana, ovat trombosytopenian uusiutuminen ja verenvuoto hoidon lopettamisen jälkeen, lisääntynyt luuytimen retikuliini, tromboottiset/tromboemboliset komplikaatiot, lääkitysvirheet ja jo olemassa olevan MDS:n eteneminen AML:ksi. Yleisimpiä havaittuja haittavaikutuksia ovat yliherkkyysreaktiot (joihin kuuluvat ihottuma-, nokkosihottuma- ja angioedeematapaukset) ja päänsärky.
Haittavaikutustaulukko
Yleisyysluokat on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin MedDRA‑elinjärjestelmäluokassa ja kussakin yleisyysluokassa haittavaikutuksen ilmaantuvuuden mukaan alenevassa järjestyksessä.
| Elinjärjestelmä (MedDRA) | Hyvin yleinen | Yleinen | Melko harvinainen |
| Infektiot | Ylähengitystieinfektio Nuha*** | Maha-suolitulehdus Nielutulehdus*** Sidekalvotulehdus*** Korvatulehdus*** Sivuontelotulehdus***/**** Keuhkoputkitulehdus**** | Influenssa Paikallinen infektio Nenän ja nielun tulehdus |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Multippeli myelooma Myelofibroosi | ||
| Veri ja imukudos | Luuytimen toimintahäiriö* Trombosytopenia* Anemia | Aplastinen anemia Luuytimen vajaatoiminta Leukosytoosi Splenomegalia Trombosytemia Kohonnut trombosyyttiarvo Poikkeava trombosyyttiarvo | |
| Immuunijärjestelmä | Yliherkkyys** | Angioedeema | |
| Aineenvaihdunta ja ravitsemus | Alkoholi-intoleranssi Ruokahaluttomuus Heikentynyt ruokahalu Kuivuminen Kihti | ||
| Psyykkiset häiriöt | Unettomuus | Masennus Poikkeavat unet | |
| Hermosto | Päänsärky | Huimaus Migreeni Parestesiat | Klonus Makuhäiriöt Heikentynyt tuntoaisti Heikentynyt makuaisti Perifeerinen neuropatia Sinus transversuksen tromboosi |
| Silmät | Sidekalvon verenvuoto Akkommodaatiohäiriö Sokeutuminen Silmäsairaus Silmien kutina Lisääntynyt kyynelvuoto Näköhermon nystyn turvotus Näköhäiriöt | ||
| Kuulo ja tasapainoelin | Pyörrytys | ||
| Sydän | Sydämentykytys | Sydäninfarkti Nopeutunut syke | |
| Verisuonisto | Kasvojen ja kaulan punoitus Syvä laskimotromboosi | Hypotensio Perifeerinen embolia Perifeerinen iskemia Laskimotulehdus Pinnallinen tromboflebiitti Verisuonitukos Erytromelalgia | |
| Hengityselimet, rintakehä ja välikarsina | Suunielun kipu*** | Keuhkoembolia* | Yskä Vetinen nuha Kurkun kuivuminen Hengenahdistus Nenän tukkoisuus Hengityskipu |
| Ruoansulatuselimistö | Ylävatsakipu*** | Pahoinvointi Ripuli Vatsakipu Ummetus Dyspepsia | Oksentelu Peräsuolen verenvuoto Pahanhajuinen hengitys Nielemisvaikeus Refluksitauti Veriulosteet Suun verenvuoto Vatsavaivat Suutulehdus Hampaiden värjäytyminen |
| Maksa ja sappi | Porttilaskimotromboosi Kohonneet aminotransferaasiarvot | ||
| Iho ja ihonalainen kudos | Kutina Mustelmat Ihottuma | Hiustenlähtö Valoyliherkkyysreaktio Akne Kosketusihottuma Ihon kuivuminen Ekseema Punoitus Kesivä ihottuma Poikkeava karvoituksen kasvu Prurigo Purppura Näppyläinen ihottuma Kutiava ihottuma Ihon kyhmyt Ihon poikkeava haju Nokkosihottuma | |
| Luusto, lihakset ja sidekudos | Nivelkipu Lihaskipu Lihaskouristukset Raajakipu Selkäkipu Luukipu | Lihaskireys Lihasheikkous Hartiakipu Lihasten nykiminen | |
| Munuaiset ja virtsatiet | Proteinuria | ||
| Sukupuolielimet ja rinnat | Verenvuoto emättimestä | ||
| Yleisoireet ja antopaikassa todettavat haitat | Väsymys Perifeerinen edeema Influenssan kaltainen sairaus Kipu Voimattomuus Kuume Vilunväristykset Injektiokohdan reaktiot Perifeerinen turvotus*** | Injektiokohdan verenvuoto Rintakipu Ärtyisyys Yleinen huonovointisuus Kasvojen turvotus Kuumotus Hermostuneisuuden tunne | |
| Tutkimukset | Kohonnut verenpaine Kohonnut veren laktaattidehydrogenaasiarvo Lämmönnousu Painon lasku Painon nousu | ||
| Vammat, myrkytykset ja hoitokomplikaatiot | Ruhje | ||
* ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ** Yliherkkyysreaktiot, joihin kuuluvat ihottuma-, nokkosihottuma- ja angioedeematapaukset *** Pediatrisissa tutkimuksissa havaittuja muita haittavaikutuksia **** Enintään 12 kuukautta ITP:tä sairastaneilla aikuispotilailla havaittuja muita haittavaikutuksia | |||
Aikuispotilaat, joilla ITP on kestänyt enintään 12 kuukautta
Romiplostiimin turvallisuusprofiili oli samanlainen kaikilla aikuispotilailla riippumatta ITP:n kestosta. Integroidussa 9 ITP‑tutkimuksen analyysissä oli mukana ≤ 12 kuukautta ITP:tä sairastaneita aikuispotilaita (n = 311), joista 277 oli saanut vähintään yhden annoksen romiplostiimia (ks. myös kohta Farmakodynamiikka). Tässä integroidussa analyysissa romiplostiimia saaneilla potilailla, joilla ITP oli kestänyt enintään 12 kuukautta, havaittiin seuraavia haittavaikutuksia (ilmaantuvuus vähintään 5 % ja ilmaantuvuus vähintään 5 % suurempi Nplate‑hoitoa saaneilla potilailla kuin lumevalmistetta tai tavanomaista hoitoa saaneilla potilailla), joita ei havaittu aikuispotilailla, joilla ITP oli kestänyt > 12 kuukautta: keuhkoputkitulehdus ja sivuontelotulehdus (raportoitu yleisesti (≥ 1/100, < 1/10)).
Pediatriset potilaat
Pediatrisissa tutkimuksissa 282 pediatriselle ITP‑potilaalle annettiin romiplostiimia kahdessa kliinisessä vertailututkimuksessa ja kolmessa ei-vertailevassa kliinisessä tutkimuksessa. Altistuksen keston mediaani oli 65,4 viikkoa. Yleinen turvallisuusprofiili oli samanlainen kuin aikuisilla.
Pediatrisilla potilailla esiintyneet haittavaikutukset ovat peräisin kummankin pediatrisen ITP‑tutkimuksen satunnaistetusta turvallisuusaineistosta (2 kliinistä vertailututkimusta) ja pediatristen ITP‑tutkimusten turvallisuusaineistosta (2 vertailevaa ja 3 ei-vertailevaa tutkimusta), joissa haittavaikutusten ilmaantuvuus oli vähintään 5 % suurempi romiplostiimihaarassa kuin lumevalmistetta saaneessa haarassa ja ilmaantuvuus oli vähintään 5 % romiplostiimihoitoa saaneilla potilailla.
Yleisimmät haittavaikutukset 1‑vuotiailla ja sitä vanhemmilla pediatrisilla ITP‑potilailla olivat ylähengitystieinfektio, nuha, yskä, suunielun kipu, ylävatsakipu, ripuli, ihottuma, kuume, ruhje (raportoitu hyvin yleisesti (≥ 1/10)) sekä nielutulehdus, sidekalvotulehdus, korvatulehdus, maha-suolitulehdus, sivuontelotulehdus, purppura, nokkosihottuma ja perifeerinen turvotus (raportoitu yleisesti (≥ 1/100, < 1/10)).
Suunielun kipu, ylävatsakipu, nuha, nielutulehdus, sidekalvotulehdus, korvatulehdus, sivuontelotulehdus ja perifeerinen turvotus olivat haittavaikutuksia, joita esiintyi pediatristen potilaiden tutkimuksissa mutta ei aikuisilla tehdyissä tutkimuksissa.
Joitakin aikuisilla havaittuja haittavaikutuksia esiintyi yleisemmin pediatrisilla potilailla. Tällaisia olivat esimerkiksi yskä, ripuli, ihottuma, kuume ja ruhje, joita raportoitiin pediatrisilla potilailla hyvin yleisesti (≥ 1/10), sekä purppura ja nokkosihottuma, joita raportoitiin pediatrisilla potilailla yleisesti (≥ 1/100, < 1/10).
Tärkeimpien haittavaikutusten kuvaus
Myös seuraavien vaikutusten on katsottu liittyneen romiplostiimihoitoon.
Verenvuototapahtumat
Aikuisten ITP‑potilaiden kliinisessä tutkimusohjelmassa verenvuototapahtumien määrä oli kaikissa tutkimuksissa kääntäen verrannollinen trombosyyttiarvoihin. Kaikki kliinisesti merkittävät (≥ 3. asteen) verenvuototapahtumat tapahtuivat, kun trombosyyttiarvo oli < 30 x 109/l. Kaikki ≥ 2. asteen verenvuototapahtumat tapahtuivat, kun trombosyyttiarvo oli < 50 x 109/l. Verenvuototapahtumien kokonaismäärässä ei ollut tilastollisesti merkitsevää eroa Nplate‑hoitoa ja lumevalmistetta saaneiden potilaiden välillä.
Kahdessa aikuispotilaiden lumevertailututkimuksessa vakavaksi luokiteltu verenvuototapahtuma todettiin 9 potilaalla (viidellä (6,0 %) romiplostiimiryhmässä ja neljällä (9,8 %) lumeryhmässä, ristitulosuhde (odds ratio, OR) (romiplostiimi/lume) = 0,59; 95 % CI = (0,15–2,31)). Vähintään 2. asteen verenvuototapahtumia raportoitiin 15 prosentilla romiplostiimia saaneista ja 34 prosentilla lumevalmistetta saaneista potilaista (OR (romiplostiimi/lume) = 0,35; 95 % CI = (0,14–0,85)).
Vaiheen 3 pediatrisessa tutkimuksessa yhdistettyjen verenvuotoepisodien (ks. kohta Farmakodynamiikka) määrä (keskiarvo (SD)) oli romiplostiimihaarassa 1,9 (4,2) ja lumevalmistetta saaneessa haarassa 4,0 (6,9).
Trombosytoosi
Analyysi kaikista aikuisista ITP‑potilaista, jotka saivat romiplostiimia neljässä kliinisessä vertailututkimuksessa ja viidessä ei-vertailevassa kliinisessä tutkimuksessa, osoitti, että tutkimuksessa raportoitiin kolme trombosytoositapausta, n = 271. Kohonneisiin trombosyyttiarvoihin liittyviä kliinisiä jälkiseurauksia ei raportoitu yhdelläkään näistä kolmesta potilaasta.
Trombosytoosia esiintyi pediatrisilla potilailla melko harvoin (≥ 1/1 000, < 1/100), ja sen ilmaantuvuus oli 1 (0,4 %). Joko ≥ 3. asteen tai vakavan trombosytoosin ilmaantuvuus oli 1 (0,4 %).
Trombosytopenia hoidon päättymisen jälkeen
Analyysi kaikista aikuisista ITP‑potilaista, jotka saivat romiplostiimia neljässä kliinisessä vertailututkimuksessa ja viidessä ei-vertailevassa kliinisessä tutkimuksessa, osoitti, että hoidon päättymisen jälkeen raportoitiin neljä trombosytopeniatapausta, n = 271 (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jo olemassa olevien myelodysplastisten oireyhtymien (MDS) eteneminen
MDS‑potilaiden satunnaistetussa kliinisessä lumevertailututkimuksessa romiplostiimihoito lopetettiin ennenaikaisesti, koska MDS:n etenemistä akuutiksi myelooiseksi leukemiaksi (AML) ja blastisolujen ohimenevää lisääntymistä esiintyi enemmän romiplostiimiryhmässä kuin lumeryhmässä. Havaituissa tapauksissa MDS:n eteneminen AML:ksi oli todennäköisempää potilailla, joilla oli RAEB‑1‑luokan MDS lähtötilanteessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kokonaiselinaika oli samanlainen kuin lumeryhmässä.
Lisääntynyt luuytimen retikuliini
Kliinisissä tutkimuksissa 271 potilaasta neljällä romiplostiimihoito keskeytettiin luuytimen retikuliinikertymän vuoksi. Lisäksi kuudella potilaalla todettiin retikuliinia luuydinbiopsiassa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatristen potilaiden kliinisessä tutkimuksessa (ks. kohta Farmakodynamiikka) niistä potilaista, joilta saatiin arvioitavissa oleva luuydinnäyte tutkimuksen aikana, viidellä 27 potilaasta (18,5 %) kohortissa 1 havaittiin retikuliinin lisääntymistä 1 vuoden kohdalla romiplostiimihoidon alusta ja seitsemällätoista 36 potilaasta (47,2 %) kohortissa 2 havaittiin retikuliinin lisääntymistä 2 vuoden kohdalla romiplostiimihoidon alusta. Yhdelläkään potilaalla ei kuitenkaan todettu lähtötilanteessa eikä hoidon aikana sellaisia luuydinmuutoksia, jotka eivät olisi sopineet ITP:n perusdiagnoosiin.
Immunogeenisuus
Immunogeenisuuden mahdollisuus on olemassa, kuten kaikkia proteiinilääkkeitä käytettäessä. Aikuisilla ITP‑potilailla tehdyissä kliinisissä tutkimuksissa määritettiin romiplostiimin ja trombopoietiinin vasta-aineet. Potilaista 5,7 prosentille (60/1 046) kehittyi sitoutuvia romiplostiimin vasta-aineita ja 3,2 prosentille (33/1 046) sitoutuvia trombopoietiinin vasta-aineita, mutta vain neljällä potilaalla todettiin neutraloivia romiplostiimin vasta-aineita, jotka eivät kuitenkaan reagoineet ristiin endogeenisen trombopoietiinin kanssa. Näistä neljästä potilaasta kahdella ei tavattu neutraloivia romiplostiimin vasta-aineita potilaan viimeisessä aikapisteessä (ohimenevä positiivinen) ja potilaista kahdella todettiin neutraloivia romiplostiimin vasta-aineita vielä potilaan viimeisessä aikapisteessä (pysyvät vasta-aineet). Ennen hoidon alkua romiplostiimin vasta‑aineita todettiin 3,3 prosentilla (35/1 046) ja trombopoietiinin vasta-aineita 3,0 prosentilla (31/1 046) potilaista.
Pediatrisissa tutkimuksissa sitoutuvien vasta-aineiden ilmaantuvuus ajankohdasta riippumatta oli 9,6 % (27/282). Näistä 27 potilaasta kahdella oli sitoutuvia ei‑neutraloivia romiplostiimin vasta‑aineita jo lähtötilanteessa. Lisäksi 2,8 prosentille (8/282) kehittyi neutraloivia romiplostiimin vasta-aineita. Yhteensä 3,9 prosentilla (11/282) potilaista oli sitoutuvia trombopoietiinin vasta-aineita ajankohdasta riippumatta romiplostiimihoidon aikana. Näistä 11 potilaasta kahdella oli jo aikaisemmin todettu sitoutuvia ei‑neutraloivia trombopoietiinin vasta‑aineita. Yhdellä potilaalla (0,35 %) lähtötilanteen jälkeinen neutraloivia trombopoietiinin vasta-aineita mittaava tulos oli heikosti positiivinen tutkimuksen aikana (missään vaiheessa ei todettu romiplostiimin vasta-aineita), kun tulos lähtötilanteessa oli negatiivinen. Potilaalla todettiin ohimenevä trombopoietiinia neutraloivien vasta-aineiden vaste testauksen tuloksen ollessa negatiivinen potilaan viimeisessä aikapisteessä tutkimusjakson aikana.
Markkinoille tulon jälkeen tehdyssä rekisteritutkimuksessa oli mukana 19 iältään pediatriseksi varmistettua potilasta. Sitoutuvien romiplostiimin vasta-aineiden ilmaantuvuus hoidon jälkeen oli 16 % (3/19), ja näistä potilaista 5,3 prosentilla (1/19) todettiin neutraloivia romiplostiimin vasta-aineita. Trombopoietiinin vasta-aineita ei todettu. Tässä tutkimuksessa oli mukana yhteensä 184 iältään aikuiseksi varmistettua potilasta. Näillä potilailla sitoutuvien romiplostiimin vasta-aineiden ilmaantuvuus hoidon jälkeen oli 3,8 % (7/184), ja 0,5 prosentilla (1/184) todettiin neutraloivia romiplostiimin vasta-aineita. Aikuispotilaista 2,2 prosentille (4/184) kehittyi sitoutuvia, ei-neutraloivia trombopoietiinin vasta-aineita.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta: www.fimea.fi.
Yliannostus
Haittavaikutuksia ei esiintynyt, kun romiplostiimia annettiin rotille kerta-annoksena 1 000 mikrog/kg (100‑kertainen annos verrattuna kliiniseen enimmäisannokseen, 10 mikrog/kg) ja apinoille toistuvina annoksina 500 mikrog/kg (50‑kertainen annos kliiniseen enimmäisannokseen verrattuna).
Yliannostustapauksissa trombosyyttiarvo saattaa nousta huomattavasti, mikä voi johtaa tromboottisiin/tromboembolisiin komplikaatioihin. Jos trombosyyttiarvo on kohonnut huomattavasti, Nplate‑hoito lopetetaan ja trombosyyttiarvoja seurataan. Nplate‑hoito aloitetaan uudelleen annostusta ja antotapaa koskevia suosituksia noudattaen (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: hemostaatit, muut systeemisesti käytettävät hemostaatit, ATC‑koodi: B02BX04
Vaikutusmekanismi
Romiplostiimi on peptidi‑Fc‑fuusioproteiini (peptibody), joka TPO‑reseptorin (tunnetaan myös nimellä cMpl) välittämänä aktivoi solunsisäisiä transkriptioon vaikuttavia signaalireittejä trombosyyttien tuotannon lisäämiseksi. Tämä peptibody‑molekyyli koostuu ihmisen immunoglobuliini IgG1:n Fc‑domeenista, jonka kumpikin yksiketjuinen alayksikkö on C‑terminaalisesta päästään kovalenttisesti kiinnittyneenä kaksi TPO‑reseptoria sitovaa domeenia sisältävään peptidiketjuun.
Romiplostiimin aminohappojärjestys ei ole homologinen endogeenisen trombopoietiinin aminohappojärjestyksen kanssa. Prekliinisissä ja kliinisissä tutkimuksissa yksikään romiplostiimin vasta‑aine ei reagoinut ristiin endogeenisen trombopoietiinin kanssa.
Kliininen teho ja turvallisuus
Romiplostiimin turvallisuutta ja tehoa on seurattu enintään 3 vuotta kestäneen jatkuvan hoidon aikana. Kliinisissä tutkimuksissa romiplostiimihoito lisäsi trombosyyttien määrää annoksesta riippuvasti. Vaikutus trombosyyttimäärään on voimakkaimmillaan noin 10–14 vuorokauden kuluttua. Tämä maksimivaikutuksen saavuttamiseen kuluva aika on annoksesta riippumaton. Kun ITP‑potilaille annettiin romiplostiimia 1–10 mikrog/kg kerta-annoksena ihon alle, trombosyyttien huippuarvo, joka saavutettiin 2–3 viikon kuluessa, oli 1,3–14,9 kertaa suurempi kuin lähtöarvo, ja vaste vaihteli potilaiden välillä. Kun romiplostiimia annettiin ITP‑potilaille 1 tai 3 mikrog/kg viikossa 6 viikon ajan, useimpien potilaiden trombosyyttiarvo oli 50–450 x 109/l. Kliinisissä tutkimuksissa romiplostiimia saaneista 271:stä ITP‑potilaasta 55 (20 %) oli yli 65‑vuotiaita ja 27 (10 %) yli 75‑vuotiaita. Lumevertailututkimuksissa ei havaittu yleisiä eroja hoidon turvallisuudessa tai tehossa iäkkäiden ja nuorempien potilaiden välillä.
Keskeisten lumevertailututkimusten tulokset
Kahdessa kaksoissokkoutetussa lumevertailututkimuksessa arvioitiin romiplostiimin turvallisuutta ja tehoa aikuisilla ITP‑potilailla, jotka olivat saaneet vähintään yhden hoidon ennen tutkimukseen osallistumista ja jotka edustavat tällaisten ITP‑potilaiden koko kirjoa.
Tutkimukseen S1 (20030212) osallistui potilaita, joiden perna oli tallella ja joille aikaisemmat hoidot eivät olleet tuoneet riittävää hoitovastetta tai he eivät olleet sietäneet niitä. Potilaiden aloittaessa tutkimuksessa mediaaniaika ITP‑diagnoosista oli 2,1 vuotta (vaihteluväli 0,1–31,6 vuotta). Potilaat olivat saaneet kolmea (mediaani, vaihteluväli 1–7) eri ITP‑hoitoa ennen tutkimukseen osallistumista. Aikaisempia hoitoja olivat kortikosteroidit (90 % kaikista potilaista), immunoglobuliinit (76 %), rituksimabi (29 %), solunsalpaajat (21 %), danatsoli (11 %) ja atsatiopriini (5 %). Potilaiden trombosyyttiarvojen mediaani tutkimukseen tullessa oli 19 x 109/l.
Tutkimukseen S2 (20030105) osallistui potilaita, joiden perna oli poistettu mutta joilla oli edelleen trombosytopenia. Potilaiden aloittaessa tutkimuksessa mediaaniaika ITP‑diagnoosista oli 8 vuotta (vaihteluväli 0,6–44,8 vuotta). Pernan poiston lisäksi potilaat olivat saaneet kuutta (mediaani, vaihteluväli 3–10) eri ITP‑hoitoa ennen tutkimukseen osallistumista. Aikaisempia hoitoja olivat kortikosteroidit (98 % kaikista potilaista), immunoglobuliinit (97 %), rituksimabi (71 %), danatsoli (37 %), solunsalpaajat (68 %) ja atsatiopriini (24 %). Potilaiden trombosyyttiarvojen mediaani tutkimukseen tullessa oli 14 x 109/l.
Tutkimusasetelma oli molemmissa tutkimuksissa sama. Potilaat (ikä ≥ 18 vuotta) satunnaistettiin suhteessa 2:1 romiplostiimiryhmään, joka sai aloitusannosta 1 mikrog/kg, ja lumeryhmään. Potilaille annettiin yksi injektio ihon alle kerran viikossa 24 viikon ajan. Annoksia säädettiin trombosyyttimäärän (50–200 x 109/l) ylläpitämiseksi. Molemmissa tutkimuksissa tehon kriteerinä oli kestävän trombosyyttivasteen saavuttaneiden potilaiden suhteellisen osuuden suureneminen. Viikkoannoksen mediaani oli 3 mikrog/kg potilailla, joiden perna oli poistettu, ja 2 mikrog/kg potilailla, joiden perna oli tallella.
Kestävän trombosyyttivasteen saavuttaneiden potilaiden osuus oli molemmissa tutkimuksissa merkitsevästi suurempi romiplostiimia saaneessa ryhmässä kuin lumeryhmässä. Lumevertailututkimuksissa neljän ensimmäisen tutkimusviikon jälkeen trombosyyttimäärä pysyi tasolla ≥ 50 x 109/l kuuden kuukauden hoitojakson loppuun asti 50–70 prosentilla romiplostiimia saaneista potilaista. Lumeryhmässä 0–7 % potilaista saavutti trombosyyttivasteen kuuden kuukauden hoitojakson aikana. Alla on yhteenveto tärkeimmistä tehoa mittaavista päätetapahtumista.
Yhteenveto lumevertailututkimusten tärkeimmistä tehoa mittaavista tuloksista
Tutkimus 1 potilaat, joilla perna tallella | Tutkimus 2 potilaat, joilta perna poistettu | Yhdistetyt tulokset tutkimukset 1 & 2 | ||||
romiplostiimi (n = 41) | lume (n = 21) | romiplostiimi (n = 42) | lume (n = 21) | romiplostiimi (n = 83) | lume (n = 42) | |
Kestävä trombosyyttivaste, potilaiden lkm (%)a | 25 (61 %) | 1 (5 %) | 16 (38 %) | 0 (0 %) | 41 (50 %) | 1 (2 %) |
(95 %:n luottamusväli (CI)) | (45 %, 76 %) | (0 %, 24 %) | (24 %, 54 %) | (0 %, 16 %) | (38 %, 61 %) | (0 %, 13 %) |
p‑arvo | < 0,0001 | 0,0013 | < 0,0001 | |||
Trombosyyttivaste, potilaiden lkm (%)b | 36 (88 %) | 3 (14 %) | 33 (79 %) | 0 (0 %) | 69 (83 %) | 3 (7 %) |
(95 % CI) | (74 %, 96 %) | (3 %, 36 %) | (63 %, 90 %) | (0 %, 16 %) | (73 %, 91 %) | (2 %, 20 %) |
p‑arvo | < 0,0001 | < 0,0001 | < 0,0001 | |||
Trombosyyttivasteen kesto, viikkoa (keskiarvo)c | 15 | 1 | 12 | 0 | 14 | 1 |
(SD) | 3,5 | 7,5 | 7,9 | 0,5 | 7,8 | 2,5 |
p‑arvo | < 0,0001 | < 0,0001 | < 0,0001 | |||
Varahoitoa tarvinneiden potilaiden lkm (%)d | 8 (20 %) | 13 (62 %) | 11 (26 %) | 12 (57 %) | 19 (23 %) | 25 (60 %) |
(95 % CI) | (9 %, 35 %) | (38 %, 82 %) | (14 %, 42 %) | (34 %, 78 %) | (14 %, 33 %) | (43 %, 74 %) |
p‑arvo | 0,001 | 0,0175 | < 0,0001 | |||
Kestävä trombosyyttivaste annoksen pysyessä vakaana, potilaiden lkm (%)e | 21 (51 %) | 0 (0 %) | 13 (31 %) | 0 (0 %) | 34 (41 %) | 0 (0 %) |
(95 % CI) | (35 %, 67 %) | (0 %, 16 %) | (18 %, 47 %) | (0 %, 16 %) | (30 %, 52 %) | (0 %, 8 %) |
p‑arvo | 0,0001 | 0,0046 | < 0,0001 | |||
a Kestävän trombosyyttivasteen kriteerit olivat: viikoittainen trombosyyttiarvo ≥ 50 x 109/l vähintään 6 kertaa tutkimusviikoilla 18–25 eikä varahoitoja missään vaiheessa koko hoitojakson aikana. b Trombosyyttivasteen kriteeri on kestävän tai tilapäisen trombosyyttivasteen saavuttaminen. Tilapäisen trombosyyttivasteen kriteerit olivat: viikoittainen trombosyyttiarvo ≥ 50 x 109/l vähintään 4 kertaa tutkimusviikoilla 2–25 mutta ei kestävää trombosyyttivastetta. Jos potilas sai jotakin varahoitoa, häntä ei voitu määritellä viikoittaisen vasteen saavuttaneeksi 8 seuraavan viikon aikana. c Trombosyyttivasteen kesto viikkoina on niiden viikkojen lukumäärä, joiden aikana trombosyyttiarvo oli ≥ 50 x 109/l, tutkimusviikoilla 2–25. Jos potilas sai jotakin varahoitoa, häntä ei voitu määritellä viikoittaisen vasteen saavuttaneeksi 8 seuraavan viikon aikana. d Varahoidolla tarkoitetaan mitä tahansa hoitoa, jolla pyritään suurentamaan trombosyyttimäärää. Varahoitoa tarvinneiden potilaiden ei katsottu saavuttaneen kestävää trombosyyttivastetta. Tutkimuksessa sallittuja varahoitoja olivat laskimoon annettava immunoglobuliini (IVIG), trombosyyttisiirrot, anti‑D‑immunoglobuliini ja kortikosteroidit. e Vakaa annos tarkoittaa, että annos on muuttunut enintään ± 1 mikrog/kg edellisten 8 hoitoviikon aikana. | ||||||
Tutkimustulokset aikuispotilailla, joilla on äskettäin diagnosoitu tai persistoiva ITP
Tutkimus S3 (20080435) oli yhden hoitohaaran avoin tutkimus aikuispotilailla, joiden vaste ensilinjan hoitoon oli jäänyt riittämättömäksi (trombosyyttimäärä ≤ 30 x 109/l). Tutkimukseen osallistui 75 potilasta, joiden mediaani-ikä oli 39 vuotta (vaihteluväli 19–85 vuotta) ja joista 59 % oli naisia.
Mediaaniaika ITP‑diagnoosista tutkimukseen osallistumiseen oli 2,2 kuukautta (vaihteluväli 0,1–6,6 kuukautta). Kuudellakymmenellä prosentilla potilaista (n = 45) ITP oli kestänyt < 3 kuukautta ja neljälläkymmenellä prosentilla (n = 30) ≥ 3 kuukautta. Trombosyyttiarvojen mediaani seulonnan aikaan oli 20 x 109/l. Aikaisempia ITP‑hoitoja olivat kortikosteroidit, immunoglobuliinit ja anti‑D‑immunoglobuliinit. Muita ITP‑lääkkeitä käyttäneet potilaat, joiden annostusohjelma oli vakaa, saivat jatkaa samaa lääkitystä koko tutkimuksen ajan. Tutkimuksessa sallittiin varahoidot, kuten kortikosteroidit, laskimoon annettava immunoglobuliini (IVIG), trombosyyttisiirrot, anti‑D‑immunoglobuliini, dapsoni, danatsoli ja atsatiopriini.
Potilaat saivat romiplostiimia injektiona ihon alle kerran viikossa 12 kuukauden ajan. Annosta säädettiin yksilöllisesti siten, että trombosyyttimäärä pysyi tasolla 50–200 x 109/l. Romiplostiimin viikoittaisen annoksen mediaani oli tutkimuksen aikana 3 mikrog/kg (25.–75. persentiili: 2–4 mikrog/kg).
Tutkimukseen 20080435 osallistuneista 75 potilaasta 70 (93 %) saavutti 12 kuukauden hoitojakson aikana trombosyyttivasteen ≥ 50 x 109/l. Trombosyyttivasteen keston keskiarvo 12 kuukauden hoitojakson aikana oli 9,2 kuukautta (95 % CI: 8,3–10,1 kuukautta) ja mediaani 11 kuukautta (95 % CI: 10–11 kuukautta). Kaplan-Meierin estimaatti ensimmäisen trombosyyttivasteen saavuttamiseen kuluvan ajan mediaanille oli 2,1 viikkoa (95 % CI: 1,1–3,0 viikkoa). Kaksikymmentäneljä potilasta (32 %) saavutti ilman hoitoa kestävän remission, jonka määritelmänä oli kaikkien mitattujen trombosyyttimäärien pysyminen tasolla ≥ 50 x 109/l vähintään 6 kuukauden ajan ilman romiplostiimia tai muuta samanaikaista ITP‑lääkehoitoa tai varahoitoa. Mediaaniaika siihen, että kaikki mitatut trombosyyttimäärät pysyivät tasolla ≥ 50 x 109/l vähintään 6 kuukautta, oli 27 viikkoa (vaihteluväli 6–57 viikkoa).
Integroitu 9 ITP‑tutkimuksen (mukaan lukien tutkimus S3) tehoanalyysi kattoi 277 aikuispotilasta, joilla ITP oli kestänyt ≤ 12 kuukautta ja jotka saivat vähintään yhden annoksen romiplostiimia. Kaikkiaan 277:stä romiplostiimihoitoa saaneesta potilaasta 140:llä oli äskettäin diagnosoitu ITP (ITP kestänyt < 3 kuukautta) ja 137:llä oli persistoiva ITP (ITP kestänyt ≥ 3 kuukautta, mutta ≤ 12 kuukautta). Kestävän trombosyyttivasteen, jonka määritelmänä oli viikoittainen trombosyyttiarvo ≥ 50 x 109/l vähintään 6 kertaa hoitoviikoilla 18–25, saavutti 50 % (95 % CI: 41,4–58,6 %) niistä 140 potilaasta, joilla oli äskettäin diagnosoitu ITP ja 55 % (95 % CI: 46,7–64,0 %) niistä 137 potilaasta, joilla oli persistoiva ITP. Trombosyyttivasteen ≥ 50 x 109/l keston prosenttiosuuden mediaani (Q1, Q3) oli 100,0 % (70,3 %, 100,0 %) potilailla, joilla oli äskettäin diagnosoitu ITP ja 93,5 % (72,2 %, 100,0 %) potilailla, joilla oli persistoiva ITP. Tämän lisäksi varahoitoa tarvitsi 47,4 % potilaista, joilla oli äskettäin diagnosoitu ITP ja 44,9 % potilaista, joilla oli persistoiva ITP.
Tutkimustulokset verrattuna tavanomaiseen hoitoon potilailla, joiden perna on tallella
Tutkimus S4 (20060131) oli avoin satunnaistettu 52 viikon tutkimus, jossa potilaat saivat romiplostiimia tai tavanomaisen hoitokäytännön mukaista lääkehoitoa. Potilaiden aloittaessa tutkimuksessa mediaaniaika ITP‑diagnoosista oli 2 vuotta (vaihteluväli 0,01–44,2 vuotta). Tutkimuksessa oli mukana ITP‑potilaita, joiden perna oli tallella ja joiden trombosyyttiarvo oli < 50 x 109/l. Romiplostiimia annettiin 157 potilaalle injektiona ihon alle kerran viikossa. Aloitusannos oli 3 mikrog/kg, ja annosta sovitettiin koko tutkimuksen ajan alueella 1–10 mikrog/kg siten, että trombosyyttipitoisuus pysyi välillä 50–200 x 109/l. Tavanomaista lääkehoitoa annettiin 77 potilaalle laitoksen omaa hoitokäytäntöä tai yleisiä hoitosuosituksia noudattaen.
Pernan poisto tehtiin kaiken kaikkiaan 8,9 prosentille (14 potilaalle 157:stä) romiplostiimiryhmän potilaista ja 36,4 prosentille (28/77) tavanomaista hoitoa saaneista potilaista, ja ristitulosuhde (odds ratio, OR) (romiplostiimi / tavanomainen hoito) oli 0,17 (95 % CI: 0,08–0,35).
Hoito osoittautui tehottomaksi kaiken kaikkiaan 11,5 prosentilla potilaista (18/157) romiplostiimiryhmässä ja 29,9 prosentilla (23/77) tavanomaista hoitoa saaneessa ryhmässä, ja ristitulosuhde (romiplostiimi / tavanomainen hoito) oli 0,31 (95 % CI: 0,15–0,61).
Romiplostiimiryhmään satunnaistetuista 157 potilaasta kolme ei saanut romiplostiimia. Romiplostiimia saaneiden 154 potilaan ryhmässä romiplostiimialtistuksen kesto oli keskimäärin 52,0 viikkoa (mediaani) ja vaihteluväli oli 2–53 viikkoa. Yleisimmin käytetty viikoittainen annos oli 3–5 mikrog/kg (25.–75. persentiili, mediaani 3 mikrog/kg).
Tavanomaista hoitoa saaneeseen ryhmään satunnaistetuista 77 potilaasta kaksi ei saanut mitään tavanomaista hoitoa. Niiden 75 potilaan ryhmässä, jotka saivat vähintään yhden annoksen tavanomaista hoitoa, hoidon kesto oli keskimäärin 51 viikkoa (mediaani) ja vaihteluväli oli 0,4–52 viikkoa.
Sallitun muun samanaikaisen ITP‑lääkityksen väheneminen
Molemmissa kaksoissokkoutetuissa lumevertailututkimuksissa muita ITP‑lääkkeitä käyttäneet potilaat, joiden annostusohjelma oli vakaa, saivat jatkaa samaa lääkitystä koko tutkimuksen ajan (kortikosteroideja, danatsolia ja/tai atsatiopriinia). Yhteensä 21 potilasta, joiden perna oli tallella, ja 18 potilasta, joilta perna oli poistettu, sai tutkimuksessa käytettäviä ITP‑lääkkeitä (pääasiassa kortikosteroideja) tutkimuksen alkaessa. Niistä potilaista, joiden perna oli poistettu, kaikkien romiplostiimia saaneiden potilaiden (100 %) muun samanaikaisen ITP‑lääkityksen annosta voitiin pienentää yli 25 % tai se voitiin lopettaa kokonaan hoitojakson loppuun mennessä. Lumeryhmässä tämä onnistui 17 prosentilla potilaista. Niistä potilaista, joiden perna oli tallella, 73 prosentilla romiplostiimia saaneista potilaista muun samanaikaisen ITP‑lääkityksen annosta voitiin pienentää yli 25 % tai se voitiin lopettaa kokonaan tutkimuksen loppuun mennessä. Lumeryhmässä tämä onnistui 50 prosentilla potilaista (ks. kohta Yhteisvaikutukset).
Verenvuototapahtumat
ITP:n kliinisessä tutkimusohjelmassa verenvuototapahtumien määrä oli kaikissa tutkimuksissa kääntäen verrannollinen trombosyyttiarvoihin. Kaikki kliinisesti merkittävät (≥ 3. asteen) verenvuototapahtumat tapahtuivat, kun trombosyyttiarvo oli < 30 x 109/l. Kaikki ≥ 2. asteen verenvuototapahtumat tapahtuivat, kun trombosyyttiarvo oli < 50 x 109/l. Verenvuototapahtumien kokonaismäärässä ei ollut tilastollisesti merkitsevää eroa romiplostiimihoitoa ja lumevalmistetta saaneiden potilaiden välillä.
Kahdessa lumevertailututkimuksessa vakavaksi luokiteltu verenvuototapahtuma todettiin 9 potilaalla (viidellä (6,0 %) romiplostiimiryhmässä ja neljällä (9,8 %) lumeryhmässä, ristitulosuhde (odds ratio, OR) (romiplostiimi/lume) = 0,59; 95 % CI = (0,15–2,31)). Vähintään 2. asteen verenvuototapahtumia raportoitiin 15 prosentilla romiplostiimia saaneista ja 34 prosentilla lumevalmistetta saaneista potilaista (OR (romiplostiimi/lume) = 0,35; 95 % CI = (0,14–0,85)).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset käytöstä alle 1‑vuotiaiden lasten hoidossa.
Romiplostiimin turvallisuutta ja tehoa arvioitiin kahdessa kaksoissokkoutetussa lumevertailututkimuksessa. Tutkimus S5 (20080279) oli vaiheen 3 tutkimus, jossa romiplostiimia annettiin 24 viikon ajan, ja tutkimus S6 (20060195) oli vaiheen 1/2 tutkimus, jossa romiplostiimia annettiin 12 viikon ajan (enintään 16 viikon ajan niille hoitovasteen saavuttaneille potilaille, jotka valittiin 4 viikkoa kestävälle farmakokineettiselle arviointijaksolle).
Molempiin tutkimuksiin otettiin pediatrisia potilaita (ikäjakauma ≥ 1 ‑ < 18 vuotta), joilla oli trombosytopenia (kriteerinä molemmissa tutkimuksissa oli kahden trombosyyttiarvon keskiarvo ≤ 30 x 109/l, eikä kumpikaan lukema saanut olla > 35 x 109/l) ja siihen liittyvä ITP, riippumatta siitä, oliko perna poistettu vai ei.
S5‑tutkimuksessa 62 potilasta satunnaistettiin suhteessa 2:1 romiplostiimia (n = 42) tai lumevalmistetta (n = 20) saavaan ryhmään ja ositettiin johonkin kolmesta ikäkohortista. Romiplostiimin aloitusannos oli 1 mikrog/kg, ja annokset säädettiin siten, että trombosyyttiarvo pysyi vakaana (50–200 x 109/l). Yleisin käytetty viikkoannos oli 3–10 mikrog/kg, ja suurin sallittu annos tutkimuksessa oli 10 mikrog/kg. Potilaille annettiin yksi injektio ihon alle kerran viikossa 24 viikon ajan. Tutkimuksen 62 potilaasta 48:lla ITP oli kestänyt yli 12 kuukautta (32 potilasta sai romiplostiimia ja 16 potilasta sai lumevalmistetta).
Ensisijainen päätetapahtuma oli kestävä hoitovaste, jonka määritelmänä oli viikoittainen trombosyyttiarvo ≥ 50 x 109/l vähintään 6 kertaa hoitoviikoilla 18–25. Ensisijaisen päätetapahtuman saavutti merkitsevästi suurempi osa potilaista romiplostiimia kuin lumevalmistetta saaneessa haarassa (p = 0,0018). Romiplostiimia saaneessa haarassa yhteensä 22 potilaalla (52 %) oli kestävä trombosyyttivaste ja lumevalmistetta saaneessa haarassa 2 potilaalla (10 %): vastaavat luvut olivat ≥ 1 ‑ < 6‑vuotiaiden ikäryhmässä 38 % ja 25 %, ≥ 6 ‑ < 12‑vuotiaiden ikäryhmässä 56 % ja 11 %, ≥ 12 ‑ < 18‑vuotiaiden ikäryhmässä 56 % ja 0 %.
Niiden potilaiden alaryhmässä, joilla ITP oli kestänyt yli 12 kuukautta, kestävän vasteen saavutti myös merkitsevästi suurempi osa romiplostiimihaaran kuin lumehaaran potilaista (p = 0,0022). Romiplostiimia saaneessa haarassa yhteensä 17 potilaalla (53,1 %) oli kestävä trombosyyttivaste ja lumevalmistetta saaneessa haarassa 1 potilaalla (6,3 %): vastaavat luvut olivat ≥ 1 ‑ < 6‑vuotiaiden ikäryhmässä 28,6 % ja 25 %, ≥ 6 ‑ < 12‑vuotiaiden ikäryhmässä 63,6 % ja 0 %, ≥ 12 ‑ < 18‑vuotiaiden ikäryhmässä 57,1 % ja 0 %.
Yhdistettyjen verenvuotoepisodien määritelmä oli kliinisesti merkittäviä verenvuototapahtumia tai varahoidon käyttö kliinisesti merkittävän verenvuototapahtuman ehkäisemiseksi hoitojakson viikoilla 2–25. Kliinisesti merkittäväksi verenvuototapahtumaksi katsottiin ≥ 2. asteen verenvuototapahtuma CTCAE‑kriteeristön (Common Terminology Criteria for Adverse Events, versio 3.0) mukaan. Yhdistettyjen verenvuotoepisodien määrä (keskiarvo (SD)) oli romiplostiimihaarassa 1,9 (4,2) ja lumehaarassa 4,0 (6,9), ja verenvuototapahtumien lukumäärän mediaani (Q1, Q3) oli romiplostiimihaarassa 0,0 (0, 2) ja lumehaarassa 0,5 (0, 4,5). Niiden potilaiden alaryhmässä, joilla ITP oli kestänyt yli 12 kuukautta, yhdistettyjen verenvuotoepisodien määrä (keskiarvo (SD)) oli romiplostiimihaarassa 2,1 (4,7) ja lumehaarassa 4,2 (7,5), ja verenvuototapahtumien lukumäärän mediaani (Q1, Q3) oli romiplostiimihaarassa 0,0 (0, 2) ja lumehaarassa 0,0 (0, 4). Koska tilastollinen testaus varahoidon käytön yleisyydestä ei ollut merkitsevä, yhdistettyjen verenvuotoepisodien määrän päätetapahtumasta ei tehty tilastollista testiä.
S6‑tutkimuksessa 22 potilasta satunnaistettiin suhteessa 3:1 romiplostiimia (n = 17) tai lumevalmistetta (n = 5) saavaan ryhmään. Annoksia suurennettiin 2 mikrog/kg kerrallaan kahden viikon välein, ja trombosyyttipitoisuuden tavoitearvo oli ≥ 50 x 109/l. Trombosyyttivasteen ilmaantuvuus oli romiplostiimiryhmässä tilastollisesti merkitsevästi suurempi kuin lumeryhmässä (p = 0,0008). Tutkimuksen 22 potilaasta 17:llä ITP oli kestänyt yli 12 kuukautta (14 potilasta sai romiplostiimia ja 3 potilasta sai lumevalmistetta). Trombosyyttivasteen ilmaantuvuus oli romiplostiimiryhmässä tilastollisesti merkitsevästi suurempi kuin lumeryhmässä (p = 0,0147).
Pediatriset potilaat, jotka olivat aiemmin olleet mukana romiplostiimitutkimuksessa (S5‑tutkimus mukaan lukien) tutkimuksen loppuun asti, voivat osallistua S7 (20090340) -tutkimukseen. Tässä avoimessa jatkotutkimuksessa arvioitiin pitkäaikaisen romiplostiimihoidon turvallisuutta ja tehoa pediatristen ITP‑potilaiden trombosytopenian hoidossa.
Tähän tutkimukseen osallistui yhteensä 66 potilasta, joista 54 (82 %) oli ollut mukana S5‑tutkimuksessa sen loppuun asti. Näistä 66 potilaasta 65 (98,5 %) sai vähintään yhden annoksen romiplostiimia. Hoidon keston mediaani (Q1, Q3) oli 135,0 viikkoa (95,0 viikkoa, 184,0 viikkoa). Keskimääräisen viikkoannoksen mediaani (Q1, Q3) oli 4,82 mikrog/kg (1,88 mikrog/kg, 8,79 mikrog/kg). Hoitojakson aikana potilaille yleisimmin annetun annoksen mediaani (Q1, Q3) oli 5,0 mikrog/kg (1,0 mikrog/kg, 10,0 mikrog/kg). Tutkimukseen tulleista 66 potilaasta 63:lla ITP oli kestänyt > 12 kuukautta. Kaikki nämä 63 potilasta saivat vähintään yhden annoksen romiplostiimia. Hoidon keston mediaani (Q1, Q3) oli 138,0 viikkoa (91,1 viikkoa, 186,0 viikkoa). Keskimääräisen viikkoannoksen mediaani (Q1, Q3) oli 4,82 mikrog/kg (1,88 mikrog/kg, 8,79 mikrog/kg). Hoitojakson aikana potilaille yleisimmin annetun annoksen mediaani (Q1, Q3) oli 5,0 mikrog/kg (1,0 mikrog/kg, 10,0 mikrog/kg).
Koko tutkimuksessa trombosyyttivasteen (vähintään yksi trombosyyttiarvo ≥ 50 x 109/l ilman varahoitoa) kokonaisilmaantuvuus oli 93,8 % (n = 61), ja se oli samanlainen kaikissa ikäryhmissä. Koko potilasjoukossa trombosyyttivasteen keston mediaani (Q1, Q3) kuukausina oli 30,0 kuukautta (13,0 kuukautta, 43,0 kuukautta) ja tutkimukseen osallistumisajan mediaani (Q1, Q3) oli 34,0 kuukautta (24,0 kuukautta, 46,0 kuukautta). Koko potilasjoukossa niiden kuukausien prosentuaalisen osuuden mediaani (Q1, Q3), joina potilailla todettiin trombosyyttivaste, oli 93,33 % (67,57 %, 100,00 %), ja se oli samanlainen kaikissa ikäryhmissä.
Niiden potilaiden alaryhmässä, joilla ITP oli kestänyt > 12 kuukautta, trombosyyttivasteen kokonaisilmaantuvuus oli 93,7 % (n = 59), ja se oli samanlainen kaikissa ikäryhmissä. Koko potilasjoukossa trombosyyttivasteen keston mediaani (Q1, Q3) kuukausina oli 30,0 kuukautta (13,0 kuukautta, 43,0 kuukautta) ja tutkimukseen osallistumisajan mediaani (Q1, Q3) oli 35,0 kuukautta (23,0 kuukautta, 47,0 kuukautta). Koko potilasjoukossa niiden kuukausien prosentuaalisen osuuden mediaani (Q1, Q3), joina potilailla todettiin trombosyyttivaste, oli 93,33 % (67,57 %, 100,00 %), ja se oli samanlainen kaikissa ikäryhmissä.
Yhteensä 31 potilasta (47,7 %) sai samanaikaisesti muuta ITP‑hoitoa tutkimuksen aikana. Näihin kuului 23 potilasta (35,4 %), jotka saivat varahoitoa, ja 5 potilasta (7,7 %), jotka käyttivät muuta samanaikaista ITP‑lääkitystä lähtötilanteessa. Muuta samanaikaista ITP‑lääkitystä käyttäneiden potilaiden osuudessa oli havaittavissa vähenemistä tutkimuksen aikana: 30,8 prosentista (viikoilla 1–12) < 20,0 prosenttiin (viikoilla 13–240), ja sen jälkeen 0 % viikosta 240 tutkimuksen loppuun.
Niiden potilaiden alaryhmässä, joilla ITP oli kestänyt > 12 kuukautta, 29 potilasta (46,0 %) sai samanaikaisesti muuta ITP‑hoitoa tutkimuksen aikana. Näihin kuului 21 potilasta (33,3 %), jotka saivat varahoitoa, ja 5 potilasta (7,9 %), jotka käyttivät muuta samanaikaista ITP‑lääkitystä lähtötilanteessa. Muuta samanaikaista ITP‑lääkitystä käyttäneiden potilaiden osuudessa oli havaittavissa vähenemistä tutkimuksen aikana: 31,7 prosentista (viikoilla 1–12) < 20,0 prosenttiin (viikoilla 13–240), ja sen jälkeen 0 % viikosta 240 tutkimuksen loppuun.
Varahoitoa saaneiden potilaiden osuudessa oli havaittavissa vähenemistä tutkimuksen aikana: 24,6 prosentista (viikoilla 1–12) < 13,0 prosenttiin (viikoilla 13–216), ja sen jälkeen 0 % viikosta 216 tutkimuksen loppuun. Vastaava väheneminen varahoitoa saaneiden potilaiden osuudessa oli havaittavissa tutkimuksen aikana niiden potilaiden alaryhmässä, joilla ITP oli kestänyt > 12 kuukautta: 25,4 prosentista (viikoilla 1–12) ≤ 13,1 prosenttiin (viikoilla 13–216), ja sen jälkeen 0 % viikosta 216 tutkimuksen loppuun.
Tutkimus S8 (20101221) oli vaiheen 3 pitkäkestoinen yhden hoitohaaran avoin monikeskustutkimus, johon osallistui 203 pediatrista potilasta, joiden ITP‑diagnoosista oli vähintään 6 kuukautta ja jotka olivat saaneet aikaisemmin vähintään yhtä ITP‑hoitoa (mutta ei romiplostiimia) tai joille muut ITP‑hoidot eivät sopineet. Romiplostiimia annettiin injektiona ihon alle kerran viikossa. Aloitusannos oli 1 mikrog/kg, ja sitä nostettiin viikoittain enintään annokseen 10 mikrog/kg, jotta saavutettaisiin trombosyyttimäärän tavoitetaso 50–200 x 109/l. Potilaiden mediaani-ikä oli 10 vuotta (vaihteluväli 1–17 vuotta), ja hoidon keston mediaani oli 155,9 viikkoa (vaihteluväli 8,0–163,0 viikkoa).
Sen ajan prosentuaalisen osuuden keskiarvo (SD), jona potilailla todettiin trombosyyttivaste (trombosyyttimäärä ≥ 50 x 109/l) ensimmäisten 6 kuukauden aikana romiplostiimihoidon aloittamisesta ilman varahoidon käyttöä edellisen 4 viikon aikana, oli 50,57 % (37,01) ja mediaani 50,0 %. Kaikkiaan 60 potilasta (29,6 %) sai varahoitoa. Tutkimuksessa sallittiin varahoidot, kuten kortikosteroidit, trombosyyttisiirrot, laskimoon annettava immunoglobuliini (IVIG), anti‑D‑immunoglobuliini ja danatsoli.
Lisäksi S8‑tutkimuksessa arvioitiin romiplostiimihoitoa saavien pediatristen ITP‑potilaiden luuydinnäytteistä retikuliinin ja kollageenin muodostumista ja muita poikkeavuuksia. Retikuliini- ja kollageeniarvioihin käytettiin muokattua Bauermeisterin arviointiasteikkoa, kun taas luuydinmuutosten arviointiin käytettiin sytogeneettisiä menetelmiä ja fluoresenssi in situ ‑hybridisaatiota (FISH). Potilailta arvioitiin retikuliinin ja kollageenin määrä luuytimessä lähtötilanteeseen verrattuna 1 vuoden (kohortti 1) ja 2 vuoden kuluttua (kohortti 2) tutkimuksen alussa tehdyn kohortteihin jaon mukaisesti. Näissä kahdessa kohortissa oli kaikkiaan 79 potilasta. Kohortin 1 30 potilaasta 27:ltä (90 %:lta) ja kohortin 2 49 potilaasta 36:lta (73,5 %:lta) saatiin tutkimuksen aikana arvioitavissa oleva luuydinnäyte. Retikuliinisäikeiden muodostuksen lisääntymistä raportoitiin 18,5 prosentilla kohortin 1 potilaista (viidellä 27 potilaasta) ja 47,2 prosentilla kohortin 2 potilaista (seitsemällätoista 36 potilaasta). Yhdellekään kummankaan kohortin potilaista ei kehittynyt kollageenifibroosia tai ITP:n perusdiagnoosiin sopimattomia luuydinmuutoksia.
Farmakokinetiikka
Romiplostiimin farmakokinetiikkaan kuului kohdemekanismin kautta välittyvä ("target‑mediated") poistuma, joka välittyy todennäköisesti trombosyyttien ja muiden trombopoieettisen linjan solujen, kuten megakaryosyyttien, TPO‑reseptorien kautta.
Imeytyminen
Kun romiplostiimia annettiin ITP‑potilaille 3–15 mikrog/kg ihonalaisena injektiona, romiplostiimin maksimipitoisuus seerumissa saavutettiin 7–50 tunnin kuluttua (mediaani 14 tuntia). Pitoisuudet seerumissa vaihtelivat potilaiden välillä eivätkä korreloineet annettuun annokseen. Romiplostiimin pitoisuus seerumissa näyttää olevan kääntäen verrannollinen trombosyyttimääriin.
Jakautuminen
Laskimoon annettujen annosten jälkeen romiplostiimin jakautumistilavuus pieneni terveillä tutkittavilla epälineaarisesti tasolta 122 ml/kg, kun annos oli 0,3 mikrog/kg, tasolle 78,8 ml/kg, kun annos oli 1,0 mikrog/kg, ja tasolle 48,2 ml/kg, kun annos oli 10 mikrog/kg. Tämä jakautumistilavuuden epälineaarinen pieneneminen sopii (megakaryosyyttien ja trombosyyttien) kohdereseptorien kautta välittyvään romiplostiimin sitoutumiseen, joka saattaa kyllästyä suurempia annoksia annettaessa.
Eliminaatio
Romiplostiimin eliminaation puoliintumisaika oli ITP‑potilailla 1–34 vuorokautta (mediaani 3,5 vrk).
Seerumin romiplostiimin eliminaatio on osittain riippuvainen trombosyyttien TPO‑reseptorista. Tämän seurauksena tietyn annoksen aikaansaama pitoisuus seerumissa on pieni, jos potilaan trombosyyttimäärä on suuri, ja päinvastoin. Toisessa kliinisessä ITP‑tutkimuksessa seerumin lääkeainepitoisuuksissa ei havaittu kumuloitumista kuuden viikoittaisen romiplostiimiannoksen (3 mikrog/kg) jälkeen.
Erityisryhmät
Romiplostiimin farmakokinetiikkaa ei ole tutkittu munuaisten eikä maksan vajaatoiminnan yhteydessä. Potilaan ikä, paino ja sukupuoli eivät näytä vaikuttavan merkittävästi romiplostiimin farmakokinetiikkaan.
Prekliiniset tiedot turvallisuudesta
Toistuvilla annoksilla tehdyissä toksisuustutkimuksissa romiplostiimia annettiin rotille 4 viikon ja apinoille enintään 6 kuukauden ajan. Näissä tutkimuksissa esiin tulleet vaikutukset liittyivät yleisesti romiplostiimin trombopoieettiseen vaikutukseen, ja ne olivat samanlaisia tutkimuksen kestosta riippumatta. Injektiokohdan reaktiot liittyivät myös romiplostiimin antoon. Rottien luuytimessä on todettu myelofibroosia kaikilla testatuilla annostasoilla. Näissä tutkimuksissa eläimillä ei havaittu myelofibroosia enää 4 viikon toipumisen jälkeen hoidon päätyttyä, mikä osoittaa, että muutos on palautuva.
Kuukauden kestäneissä toksisuustutkimuksissa rotilla ja apinoilla havaittiin lievää punasolujen vähenemistä ja hematokriitti‑ ja hemoglobiiniarvon laskua. Myös valkosolujen tuotantoa stimuloiva vaikutus havaittiin, sillä perifeerisen veren neutrofiili-, lymfosyytti-, monosyytti- ja eosinofiilimäärä oli suurentunut hiukan. Pitempään kestäneessä tutkimuksessa apinoilla ei havaittu punasolu- eikä valkosolulinjaan kohdistuneita vaikutuksia, kun romiplostiimia annettiin 6 kuukauden ajan ja annostelua harvennettiin kolmesta kerrasta yhteen kertaan viikossa. Myöskään keskeisissä vaiheen 3 tutkimuksissa romiplostiimilla ei ollut punasolu- eikä valkosolulinjaan kohdistuvia vaikutuksia lumevalmistetta saaneisiin potilaisiin verrattuna.
Neutraloivien vasta-aineiden muodostumisen vuoksi romiplostiimin farmakodynaamiset vaikutukset heikkenivät usein pitkään jatkuneen käytön aikana rotilla. Toksikokineettisissä tutkimuksissa vasta-aineilla ei havaittu yhteisvaikutuksia mitatuissa pitoisuuksissa. Eläinkokeissa testatuista suurista annoksista huolimatta turvallisuusmarginaaleja ei voida luotettavasti arvioida, koska herkkyydet romiplostiimin farmakodynaamiselle vaikutukselle ja neutraloivien vasta-aineiden vaikutukselle ovat erilaiset laboratorioeläimillä ja ihmisellä.
Karsinogeneesi
Romiplostiimin karsinogeenisuutta ei ole arvioitu. Siksi romiplostiimin mahdollisen karsinogeenisuuden riskiä ihmisille ei tunneta.
Lisääntymistoksisuus
Kaikissa yksilönkehitystä koskevissa tutkimuksissa muodostui neutraloivia vasta-aineita, jotka ovat saattaneet estää romiplostiimin vaikutuksia. Hiirien ja rottien alkion‑ ja sikiönkehitystä selvittäneissä tutkimuksissa emojen painon laskua todettiin vain hiirillä. Hiirillä havaittiin viitteitä implantaation jälkeisten keskenmenojen lisääntymisestä. Rottien pre‑ ja postnataalista kehitystä koskevassa tutkimuksessa esiintyi tiineysajan pitenemistä ja poikasten perinataalikuolleisuuden vähäistä lisääntymistä. Romiplostiimin tiedetään läpäisevän rottien istukan, joten se saattaa siirtyä äidistä kehittyvään sikiöön ja stimuloida sikiön trombosyyttituotantoa. Romiplostiimilla ei ollut havaittavaa vaikutusta rottien hedelmällisyyteen.
Farmaseuttiset tiedot
Apuaineet
Mannitoli (E421), sakkaroosi, l‑histidiini, kloorivetyhappo (pH:n säätöön), polysorbaatti 20. Liuotin: Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
3 vuotta.
Liuottamisen jälkeen: Kemiallisen ja fysikaalisen käytönaikaisen säilyvyyden on osoitettu olevan 24 tuntia 25 °C:ssa ja 24 tuntia 2–8 °C:ssa säilytettäessä valolta suojattuna alkuperäisessä injektiopullossa.
Mikrobiologisista syistä lääkevalmiste on käytettävä heti. Ellei sitä käytetä heti, käytönaikaiset säilytysajat ja käyttöä edeltävät olosuhteet ovat käyttäjän vastuulla, ja ne saavat normaalisti olla enintään 24 tuntia 25 °C:ssa tai 24 tuntia jääkaapissa (2–8 °C), valolta suojattuna.
Säilytys
Säilytä jääkaapissa (2°C ‑ 8°C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Voidaan ottaa jääkaapista huoneenlämpöön (enintään 25 °C) ja säilyttää huoneenlämmössä 30 vuorokautta alkuperäispakkauksessa.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
NPLATE injektiokuiva-aine ja liuotin, liuosta varten
250 mikrog (L:ei) 1 pakkaus (562,88 €)
500 mikrog (L:ei) 1 pakkaus (1089,70 €)
PF-selosteen tieto
Kuiva-aine:
5 ml:n kertakäyttöinen injektiopullo (tyypin 1 kirkasta lasia), jossa on tulppa (klooributyylikumia), suljin (alumiinia) ja suojakansi (polypropyleeniä).
Liuotin:
Nplate 250 mikrogrammaa injektiokuiva-aine ja liuotin, liuosta varten: Esitäytetty ruisku (tyypin 1 kirkasta lasia, mäntä bromobutyylikumia), joka sisältää 0,72 ml injektionesteisiin käytettävää vettä liuottamista varten.
Nplate 500 mikrogrammaa injektiokuiva-aine ja liuotin, liuosta varten: Esitäytetty ruisku (tyypin 1 kirkasta lasia, mäntä bromobutyylikumia), joka sisältää 1,2 ml injektionesteisiin käytettävää vettä liuottamista varten.
Pakkauskoko:
Nplate 250 mikrogrammaa injektiokuiva‑aine ja liuotin, liuosta varten:
Nplate toimitetaan 1 kpl:n pakkauksessa. Yhden pakkauksen sisältö:
1 injektiopullo, jossa 250 mikrogrammaa romiplostiimia.
1 esitäytetty ruisku, jossa 0,72 ml injektionesteisiin käytettävää vettä liuottamista varten.
1 männän varsi esitäytettyyn ruiskuun.
1 steriili injektiopullon liitin.
1 steriili 1 ml:n Luer lock ‑ruisku.
1 steriili turvaneula.
4 desinfiointipyyhettä.
Nplate 500 mikrogrammaa injektiokuiva-aine ja liuotin, liuosta varten:
Nplate toimitetaan 1 kpl:n pakkauksessa. Yhden pakkauksen sisältö:
1 injektiopullo, jossa 500 mikrogrammaa romiplostiimia.
1 esitäytetty ruisku, jossa 1,2 ml injektionesteisiin käytettävää vettä liuottamista varten.
1 männän varsi esitäytettyyn ruiskuun.
1 steriili injektiopullon liitin.
1 steriili 1 ml:n Luer lock ‑ruisku.
1 steriili turvaneula.
4 desinfiointipyyhettä.
Valmisteen kuvaus:
Jauhe on valkoista.
Liuotin on kirkasta väritöntä nestettä.
Käyttö- ja käsittelyohjeet
Nplate on steriili lääkevalmiste, joka ei sisällä säilytysaineita, ja kukin injektiopullo on tarkoitettu vain yhtä käyttökertaa varten. Nplate saatetaan käyttökuntoon hyvää aseptista menettelytapaa noudattaen.
Nplate 250 mikrogrammaa injektiokuiva-aine ja liuotin, liuosta varten
Nplate 250 mikrogrammaa injektiokuiva-aine liuotetaan 0,72 ml:aan steriiliä injektionesteisiin käytettävää vettä, ja näin saatava käytettävissä oleva määrä on 0,5 ml. Jokaisessa injektiopullossa on ylitäyttöä, joka varmistaa, että pullosta saadaan 250 mikrog romiplostiimia (ks. injektiopullon sisältö alla olevasta taulukosta).
Nplate 500 mikrogrammaa injektiokuiva-aine ja liuotin, liuosta varten
Nplate 500 mikrogrammaa injektiokuiva-aine liuotetaan 1,2 ml:aan steriiliä injektionesteisiin käytettävää vettä, ja näin saatava käytettävissä oleva määrä on 1 ml. Jokaisessa injektiopullossa on ylitäyttöä, joka varmistaa, että pullosta saadaan 500 mikrog romiplostiimia (ks. injektiopullon sisältö alla olevasta taulukosta).
Injektiopullon sisältö:
Kertakäyttöinen Nplate‑injektiopullo | Romiplostiimin kokonaismäärä injektiopullossa | Steriilin injektionesteisiin käytettävän veden määrä | Käytettävissä oleva annos ja liuoksen määrä | Lopullinen pitoisuus | ||
250 mikrog | 375 mikrog | + | 0,72 ml | = | 250 mikrog / 0,50 ml | 500 mikrog/ml |
500 mikrog | 625 mikrog | + | 1,20 ml | = | 500 mikrog / 1,00 ml | 500 mikrog/ml |
Mikrobiologisista syistä valmiste tulisi käyttää heti. Ellei sitä käytetä heti, käytönaikaiset säilytysajat ja käyttöä edeltävät olosuhteet ovat käyttäjän vastuulla, ja ne saavat normaalisti olla enintään 24 tuntia 25 °C:ssa tai 24 tuntia jääkaapissa (2–8 °C), valolta suojattuna.
1. Poista injektiokuiva-ainetta sisältävän Nplate‑injektiopullon muovinen suojakansi ja puhdista kumitulppa pakkauksessa mukana olevalla desinfiointipyyhkeellä. | ||
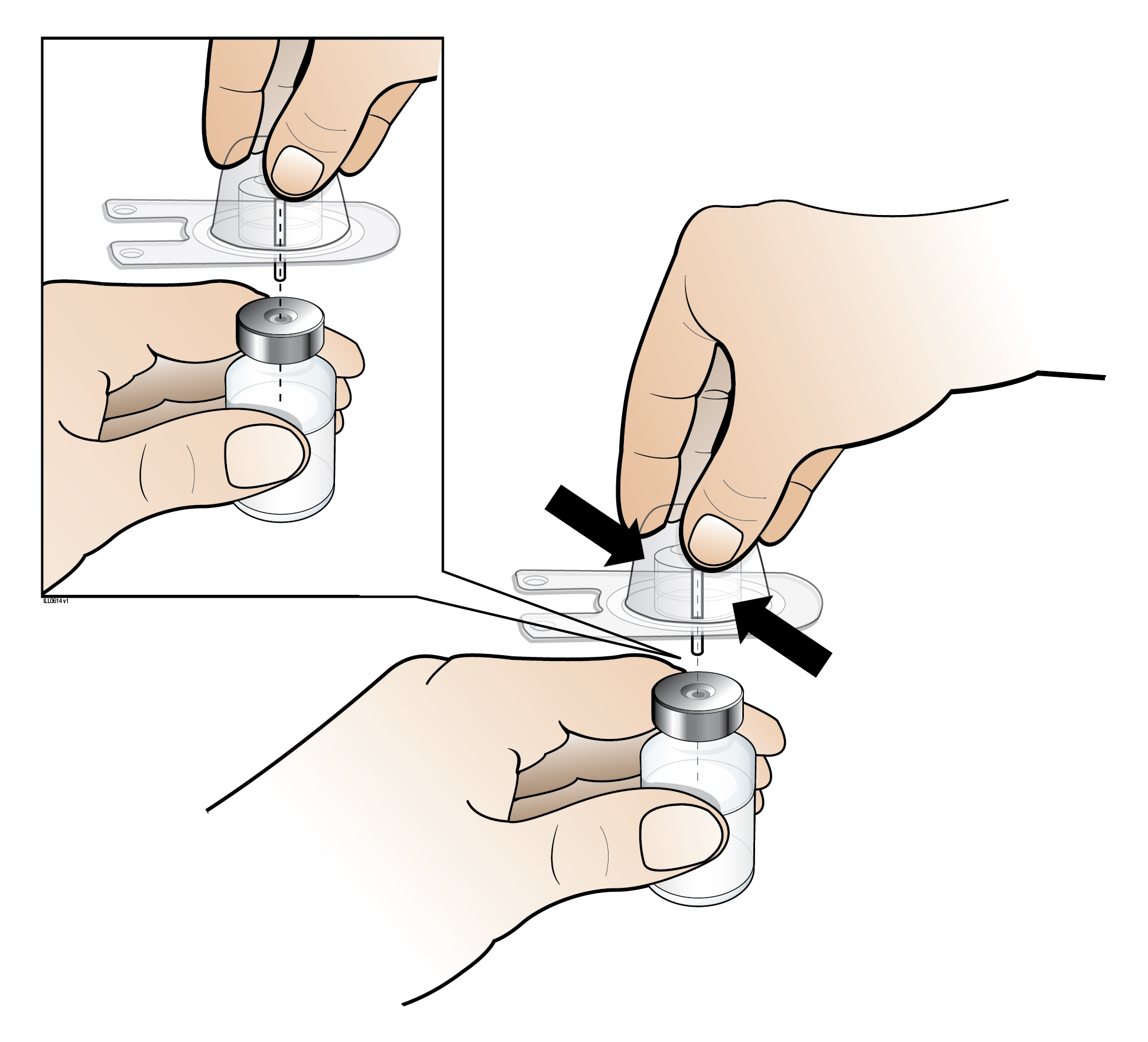
2. Poista injektiopullon liittimen paperisuojus niin, että liitin pysyy koko ajan pakkauksen sisällä, ja yhdistä liitin Nplate‑injektiopulloon. Pidä injektiopullo pöydällä ja paina liitin aivan tulpan keskeltä injektiopulloon, kunnes se on tukevasti paikoillaan. Huom: Älä koske injektiopullon liittimen kärkeen äläkä Luer lock ‑liitoskohtaan, jotta valmisteeseen ei pääse epäpuhtauksia. |
| |
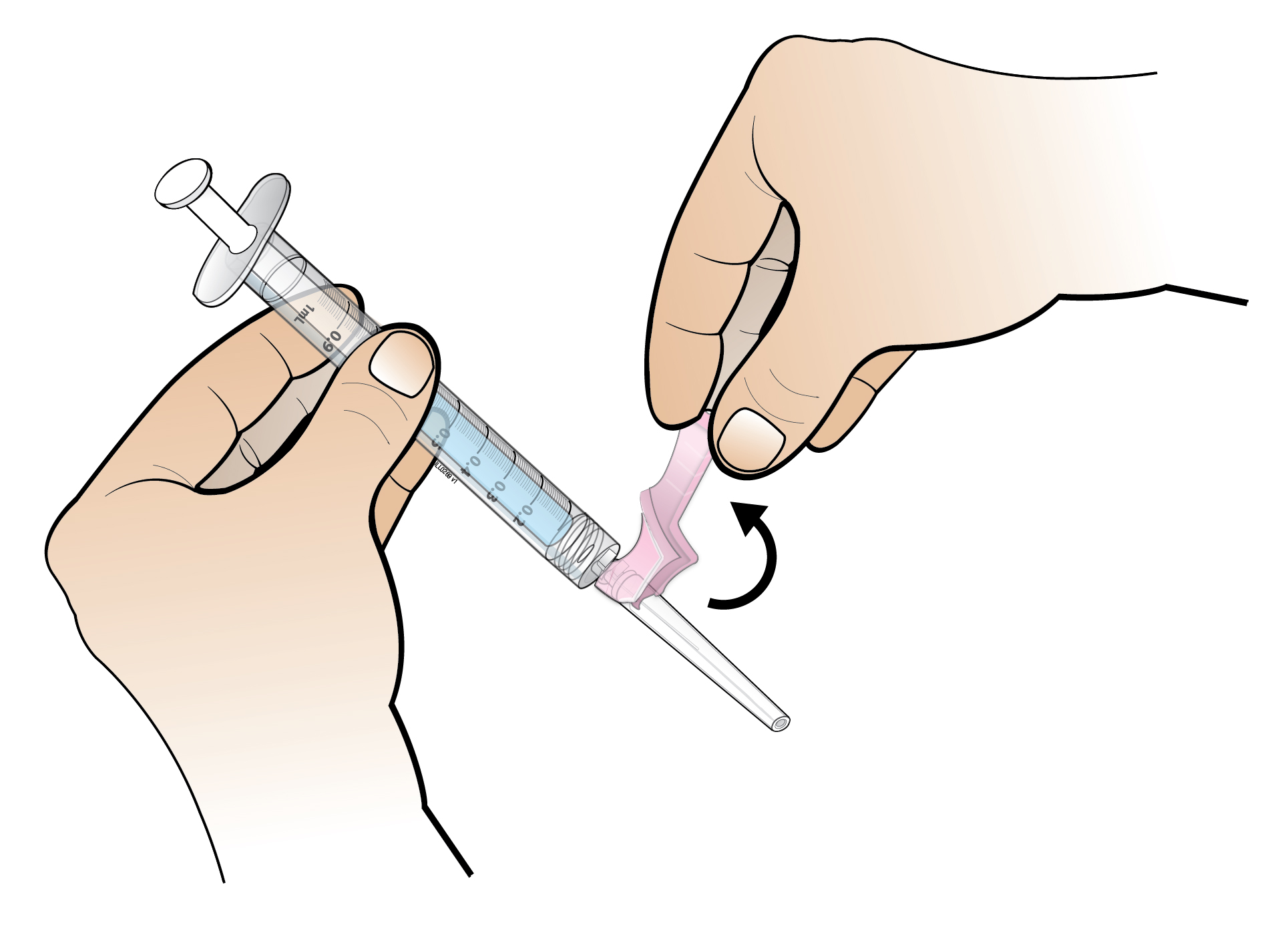
3. Poista liittimen pakkaus ja hävitä se. | ||
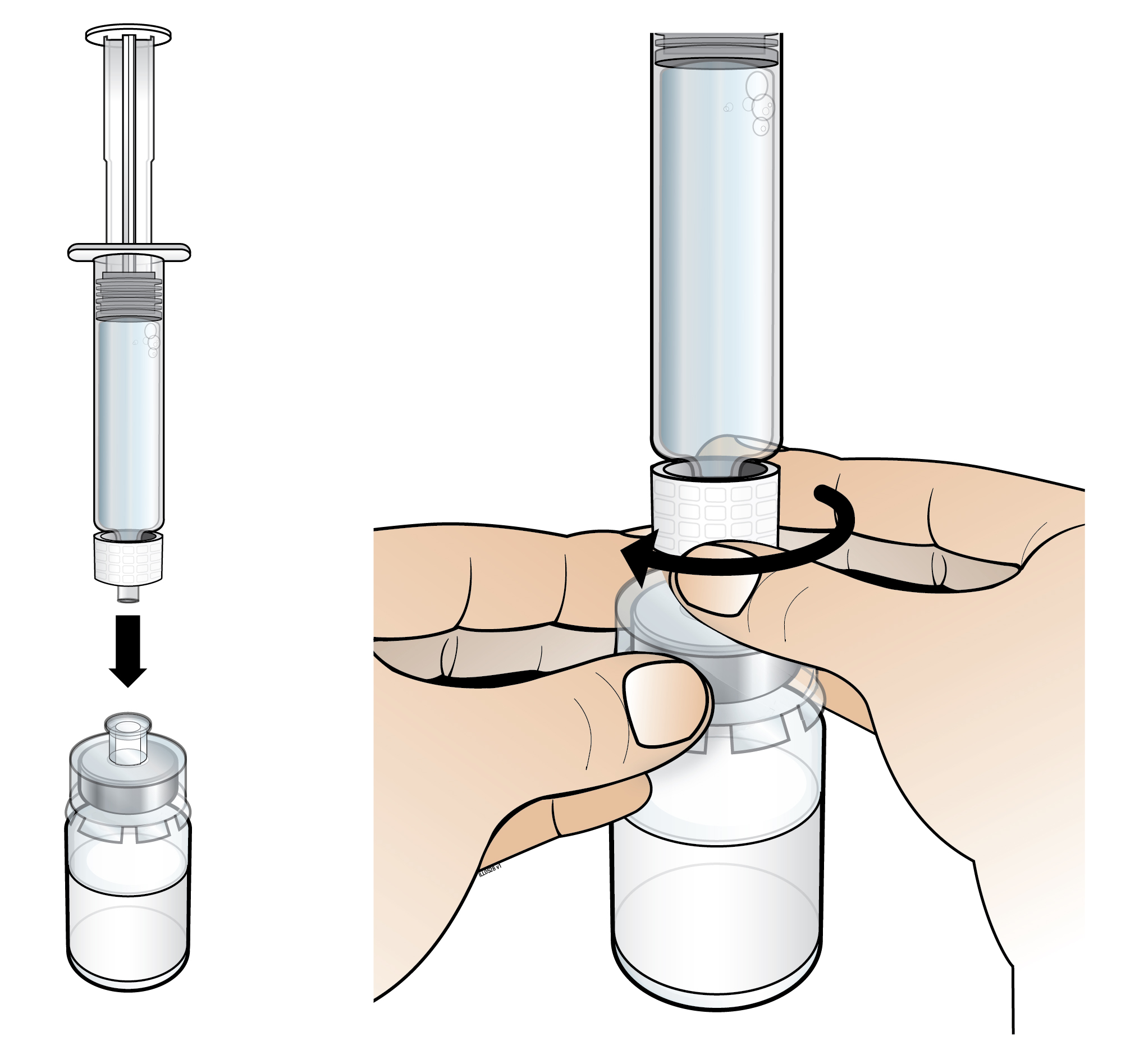
| 4. Kiinnitä männän varsi esitäytettyyn ruiskuun, jossa on injektionesteisiin käytettävää vettä: kierrä varsi myötäpäivään ruiskun mäntään, kunnes tunnet kevyen vastuksen | ||
| 5. Pidä injektionesteisiin käytettävää vettä sisältävää esitäytettyä ruiskua toisessa kädessä ja käännä toisella kädellä valkoisen muovisuojuksen kärkeä alaspäin. Tämä rikkoo valkoisen muovisuojuksen sinetöinnin. Kun sinetöinti on rikkoutunut, vedä suojus pois, jotta harmaa kumitulppa irtoaa ruiskun kirkkaasta muovikärjestä. |
| |
| 6. Pidä injektiopullo pöydällä ja kiinnitä injektionesteisiin käytettävää vettä sisältävä esitäytetty ruisku injektiopullon liittimeen: pidä toisella kädellä kiinni injektiopullon liittimen ulkoreunasta ja kierrä toisella kädellä myötäpäivään ruiskun kärki liittimeen, kunnes tunnet kevyen vastuksen. |
| |
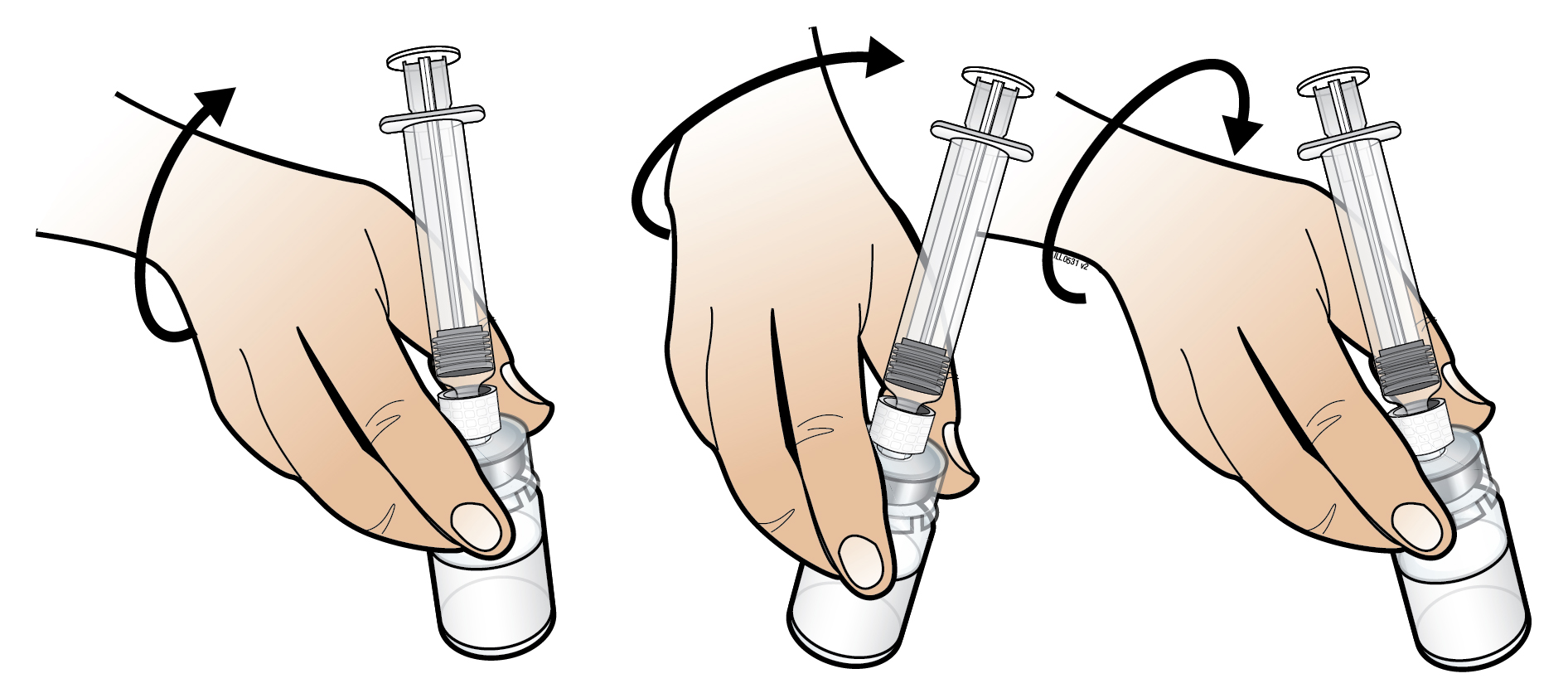
7. Ruiskuta hyvin hitaasti ja varovasti kaikki vesi kuiva-ainetta sisältävään injektiopulloon. Veden pitää valua hitaasti jauheen päälle. Pyörittele VAROVASTI injektiopulloa, kunnes kaikki jauhe on liuennut ja injektiopullossa oleva neste on kirkasta ja väritöntä. Älä ravista injektiopulloa. Huom: Mikrobiologisista syistä valmiste on käytettävä heti liuottamisen jälkeen. Ellei valmista liuosta käytetä heti, ruiskua ei pidä irrottaa injektiopullon liittimestä, jotta valmisteen mikrobiologinen puhtaus säilyy. |
Huom: Saattaa kestää jopa 2 minuuttia ennen kuin jauhe on kokonaan liuennut. | |
Ennen kuin jatkat: Tarkista silmämääräisesti, ettei valmiissa liuoksessa ole hiukkasia eikä sen väri ole muuttunut. Valmiin liuoksen tulee olla kirkasta ja väritöntä, eikä liuosta saa antaa, jos siinä havaitaan hiukkasia ja/tai värimuutoksia. Varmista, että liuos on täysin liuennut ennen kuin irrotat ruiskun. | ||
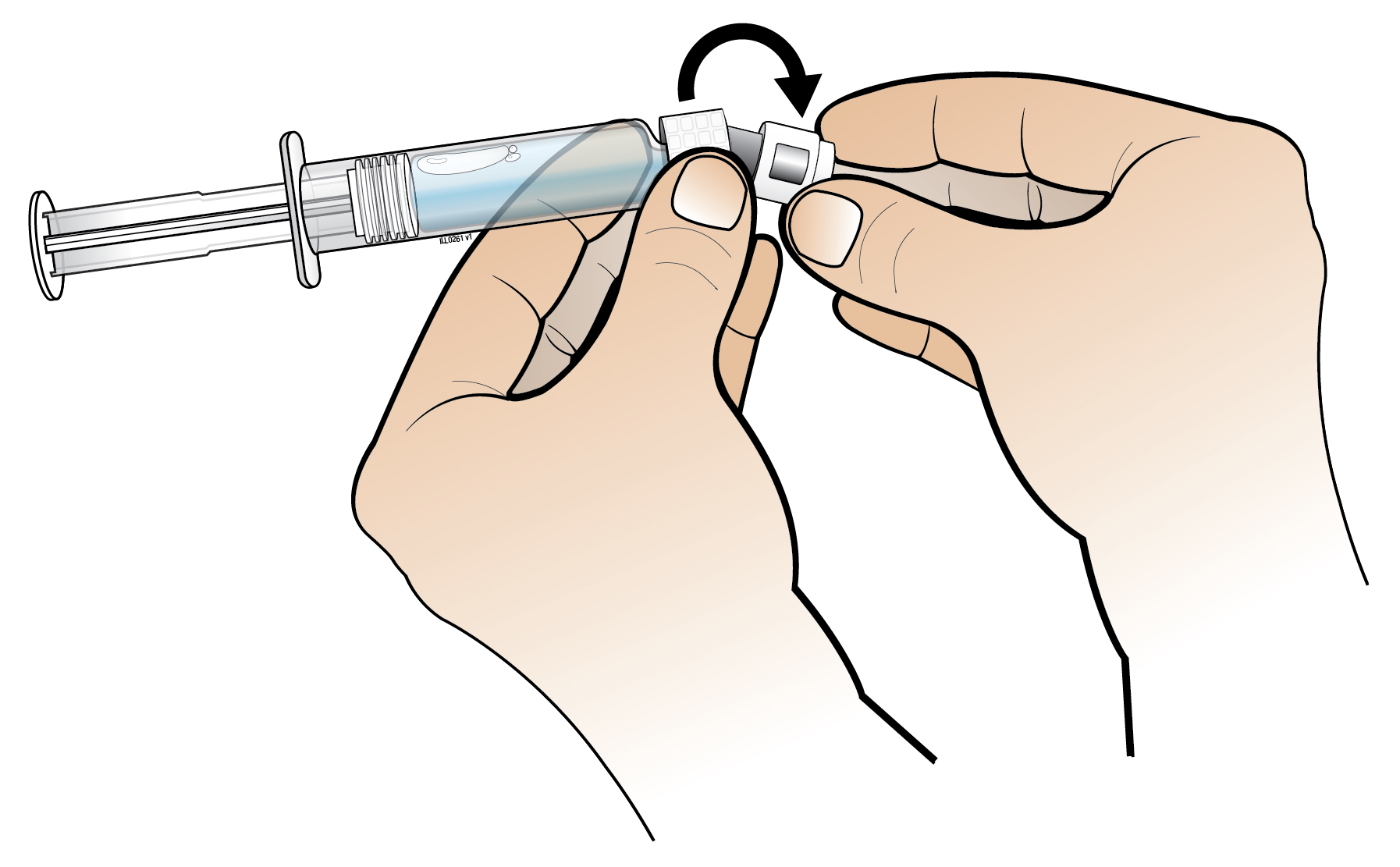
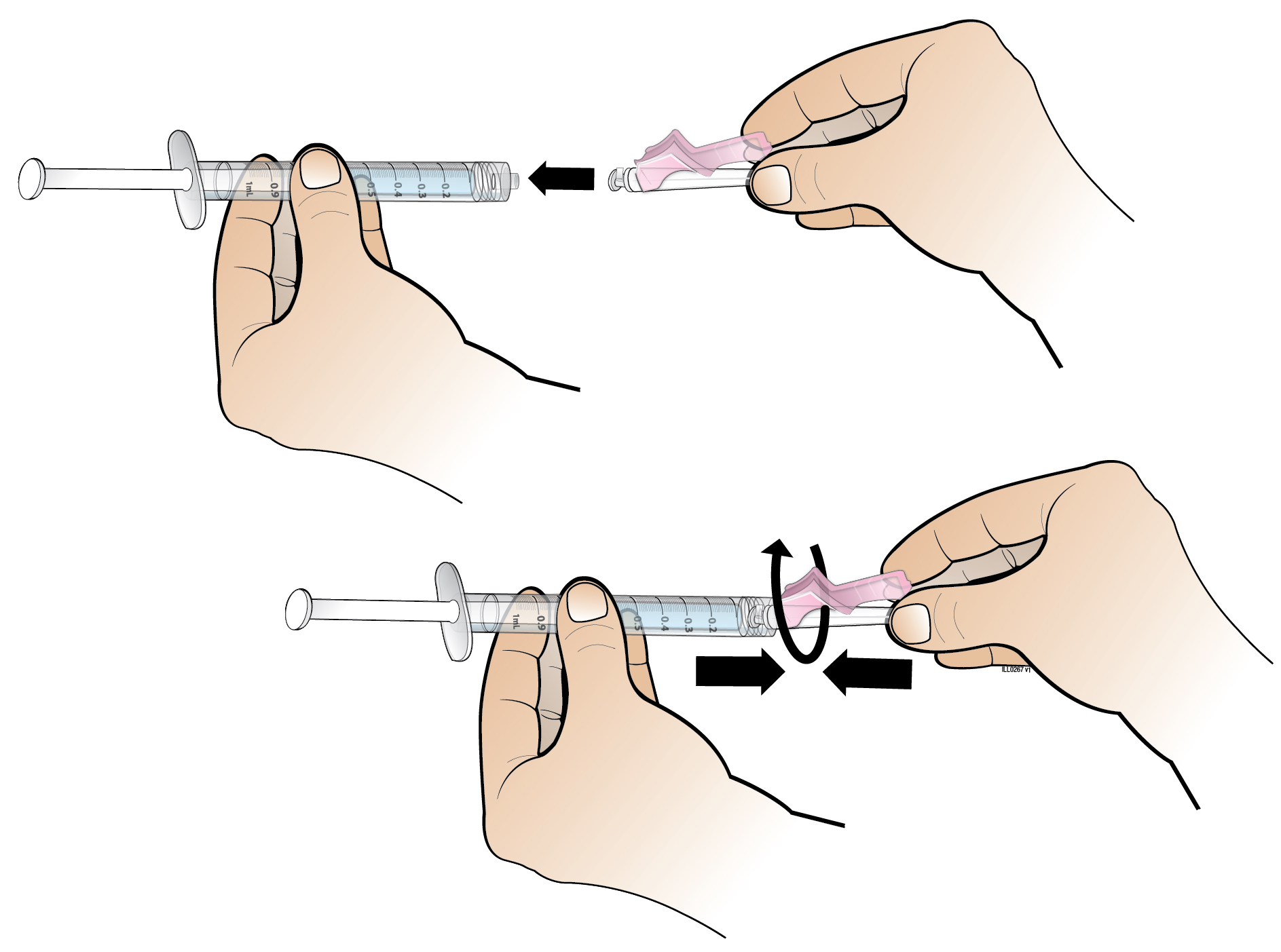
| 8. Irrota tyhjä esitäytetty ruisku injektiopullon liittimestä. | ||
| 9. Ota 1 ml:n injektioruisku pois pakkauksestaan. Kiinnitä 1 ml:n ruiskukäyttövalmista liuosta sisältävän injektiopullon liittimeen kiertämällä ruiskun kärki liittimeen, kunnes tunnet kevyen vastuksen. | ||
10. Käännä ruiskuun kiinnitetty injektiopullo ylösalaisin siten, että käyttövalmista liuosta sisältävä injektiopullo on ruiskun yläpuolella. Vedä kaikki lääkeliuos injektioruiskuun. Varmista, että mäntä pysyy ruiskun sisällä. |
| |
11. Varmista, että injektioruiskussa on potilaan annokseen tarvittava oikea määrä liuosta, ja ruiskuta tarvittaessa ylimääräinen liuos takaisin injektiopulloon. Huom: Poista kaikki ilmakuplat ruiskusta, jotta voit varmistaa, että ruiskussa on täsmälleen oikea määrä liuosta. | | |
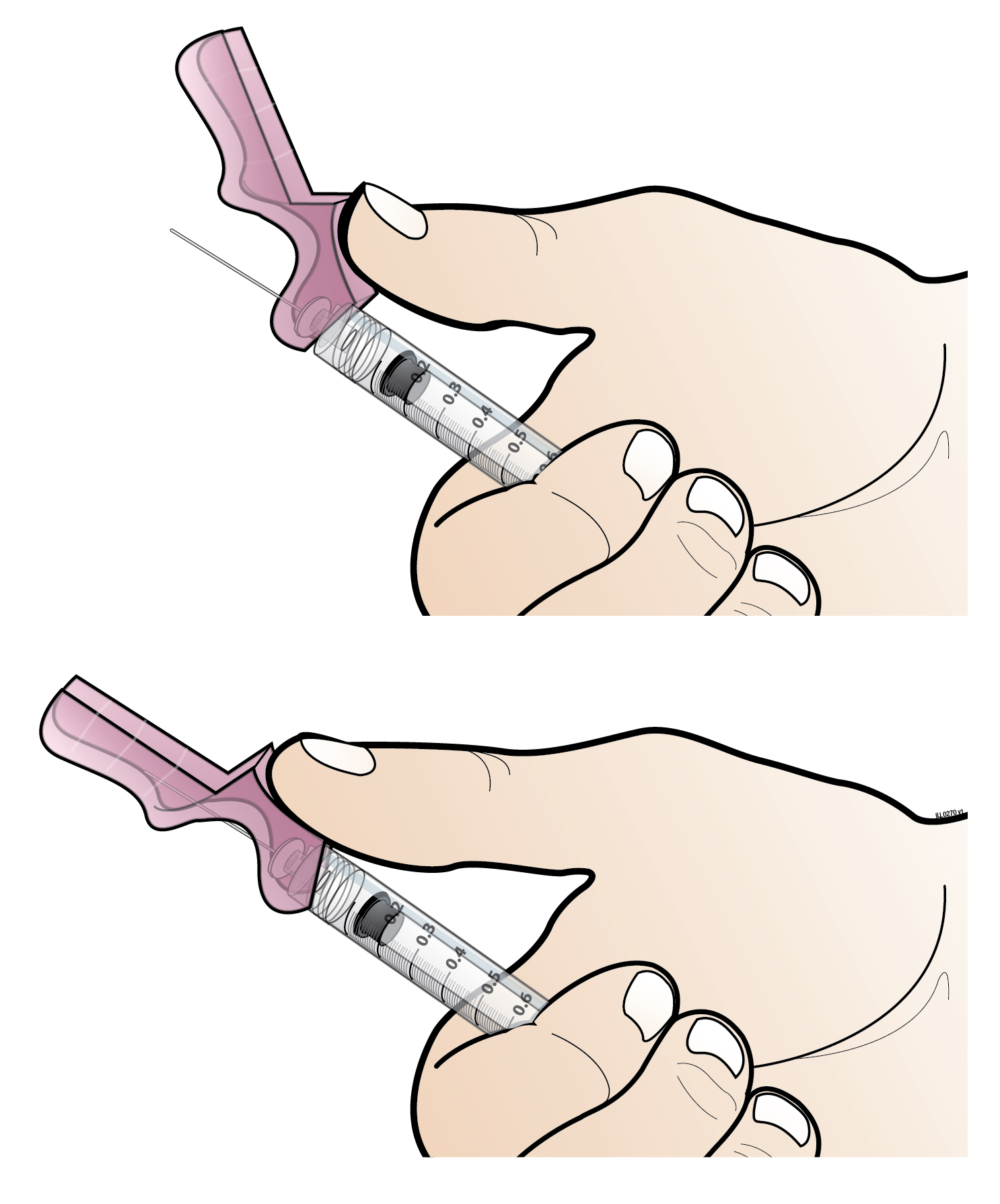
12. Kierrä injektioruisku irti injektiopullon liittimestä. Kiinnitä turvaneula täytettyyn injektioruiskuun kiertämällä neula myötäpäivään ruiskun Luer lock ‑kärkeen. |
| |
13. Puhdista pistoskohta uudella desinfiointipyyhkeellä. Vedä vaaleanpunainen turvasuojus taakse ruiskua kohti ja poispäin neulasta. Ota väritön neulansuojus pois ruiskuun kiinnitetystä neulasta siten, että pidät ruiskua toisessa kädessä ja vedät toisella kädellä suojuksen suoraan pois. |
| |
| 14. Pistä ruiske ihon alle paikallista käytäntöä ja hyvää aseptista menettelytapaa noudattaen. | ||
| 15. Kun ruiske on pistetty, aktivoi vaaleanpunainen turvasuojus työntämällä sitä eteenpäin samalla kädellä, kunnes kuulet ja/tai tunnet sen naksahtavan/lukkiutuvan. |
| |
16. Hävitä ruisku ja neula heti asianmukaiseen keräysastiaan. | ||
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
NPLATE injektiokuiva-aine ja liuotin, liuosta varten
250 mikrog 1 pakkaus
500 mikrog 1 pakkaus
- Ylempi erityiskorvaus (100 %). Romiplostiimi: Aikuispotilaiden kroonisen immunologisen (idiopaattisen) trombosytopeenisen purppuran hoito erityisin edellytyksin (1516).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Romiplostiimi: Aikuispotilaiden kroonisen immunologisen (idiopaattisen) trombosytopeenisen purppuran (ITP) hoito erityisin edellytyksin (3038).
ATC-koodi
B02BX04
Valmisteyhteenvedon muuttamispäivämäärä
04.09.2025
Yhteystiedot
Keilaranta 10, PL 86
02101 Espoo
09 5490 0500