
PLUVICTO injektio-/infuusioneste, liuos 1000 MBq/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Potilasopas / Patientguide - PLUVICTO
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi millilitra liuosta sisältää 1 000 MBq lutetium(177Lu)vipivotiditetraksetaania kalibrointiajankohtana.
Yhden kerta-annosinjektiopullon kokonaisradioaktiivisuus on 7 400 MBq ± 10 % valmisteen antohetkellä. Koska valmisteen kiinteä volumetrinen aktiivisuus on 1 000 MBq/ml kalibrointihetkellä, liuoksen tilavuutta injektiopullossa voidaan muuttaa 7,5 ml ja 12,5 ml välillä tarvittavan radioaktiivisuusmäärän tuottamiseksi valmisteen antohetkellä.
Fysikaaliset ominaisuudet
Lutetium-177:n fysikaalinen puoliintumisaika on 6,647 vrk. Se hajoaa stabiiliksi hafnium-177:ksi säteilemällä β−-säteilyä maksimienergialla 0,498 MeV (79 %) ja fotonisäteilyä (γ) maksimienergialla 0,208 MeV (11 %) ja 0,113 MeV (6,4 %).
Apuaine, jonka vaikutus tunnetaan
Yksi millilitra liuosta sisältää enintään 0,312 mmol (7,1 mg) natriumia. Yksi injektiopullo sisältää enintään 88,75 mg natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektio-/infuusioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
Pluvicto yhdessä androgeenideprivaatiohoidon kanssa tai androgeenideprivaatiohoidon ja androgeenireseptorireitin estäjähoidon kanssa on tarkoitettu sellaisten aikuispotilaiden hoitoon, joilla on edennyt PSMA (prostataspesifinen membraaniantigeeni) -positiivinen etäpesäkkeinen kastraatioresistentti eturauhassyöpä (mCRPC) ja jotka ovat saaneet androgeenireseptorireitin estäjähoitoa ja taksaanipohjaista solunsalpaajahoitoa (ks. kohta Farmakodynamiikka).
Ehto
Valmistetta saavat antaa vain radioaktiivisten aineiden käsittelyyn valtuutetut henkilöt tähän tarkoitukseen varatuissa tiloissa sen jälkeen, kun pätevä lääkäri on tutkinut potilaan.
Annostus ja antotapa
Tärkeät turvallisuusohjeet
Pluvicto-valmistetta saavat antaa vain radiofarmaseuttisten valmisteiden käsittelyyn valtuutetut henkilöt siihen tarkoitetussa hoitolaitoksessa (ks. kohta Käyttö- ja käsittelyohjeet) ja sen jälkeen, kun asiaan perehtynyt lääkäri on arvioinut potilaan.
Radiofarmaseuttisten valmisteiden (mukaan lukien Pluvicto) anto ja annon valvonta ovat sellaisten terveydenhuollon ammattilaisten vastuulla, joilla on radiofarmaseuttisten valmisteiden turvalliseen käyttöön ja käsittelyyn tarvittava koulutus ja kokemus ja joiden kokemuksen ja koulutuksen on hyväksynyt asianmukainen, radiofarmaseuttisten valmisteiden antolupia myöntävä valtion virasto.
Potilasvalinta
Hoitoon soveltuvat potilaat on tunnistettava PSMA:n kuvantamisella.
Annostus
Suositellussa Pluvicto hoito-ohjelmassa annetaan 7 400 MBq:n annos laskimoon 6 viikon välein (± 1 viikko) yhteensä enintään 6 kertaa tai vähemmän, jos sairaus etenee tai esiintyy sietämätöntä toksisuutta.
Lääkkeellistä kastraatiota on jatkettava gonadotropiinia vapauttavan hormonin analogilla niillä potilailla, joita ei ole kirurgisesti kastroitu.
Hoidon seuranta
Ennen Pluvicto-hoidon aloitusta ja hoidon aikana on tehtävä laboratoriotutkimuksia. Annostusta voi olla tarpeen muuttaa tutkimustulosten perusteella (ks. taulukko 1).
-
Hematologia (hemoglobiini, valkosoluarvo, absoluuttinen neutrofiiliarvo, verihiutalearvo)
-
Munuaisten toiminta (seerumin kreatiniini, laskennallinen kreatiniinipuhdistuma)
-
Maksan toiminta (alaniiniaminotransferaasi, aspartaattiaminotransferaasi, alkalinen fosfataasi, seerumin albumiini, kokonaisbilirubiini).
Annosmuutokset haittavaikutusten yhteydessä
Haittavaikutusten yhteydessä suositellut Pluvicto-annosmuutokset esitetään taulukossa 1. Vaikeiden tai sietämättömien haittavaikutusten hoito voi edellyttää Pluvicto-hoidon tauottamista, annoksen pienentämistä tai hoidon pysyvää lopettamista. Jos Pluvicto-hoito viivästyy haittavaikutuksen takia yli 4 viikkoa, hoito on lopetettava. Pluvicto-annosta voidaan kerran pienentää 20 % eli annokseen 5 900 MBq; tämän jälkeen annosta ei saa enää suurentaa. Jos potilaalla esiintyy muita haittavaikutuksia, jotka vaatisivat annoksen pienentämistä uudelleen, Pluvicto-hoito on lopetettava.
Taulukko 1 Suositellut Pluvicto-annosmuutokset haittavaikutusten yhteydessä
Haittavaikutus | Vaikeusastea | Annosmuutokset |
Suun kuivuus | Aste 3 | Pluvicto-annosta pienennetään 20 % eli annokseen 5 900 MBq. |
Ruoansulatuskanavaan kohdistuva toksisuus | Aste ≥ 3 (ei vastetta lääketieteelliseen hoitoon) | Pluvicto tauotetaan, kunnes haitta lievittyy asteeseen 2 tai korjautuu lähtötasolle. Pluvicto-annosta pienennetään 20 % eli annokseen 5 900 MBq. |
Myelosuppressio (anemia, trombosytopenia, leukopenia, neutropenia, pansytopenia) | Aste 2 | Pluvicto tauotetaan, kunnes haitta lievittyy asteeseen 1 tai korjautuu lähtötasolle. Asianmukainen hoito tarpeen mukaan. Kasvutekijöiden käyttö on sallittua mutta lopetettava, kun haitta lievittyy asteeseen 1 tai korjautuu lähtötasolle. Tutkitaan rauta-, B12- ja folaattiarvot ja määrätään ravintolisiä. Verivalmisteita voidaan antaa kliinisen tarpeen mukaan. |
Aste ≥ 3 | Pluvicto tauotetaan, kunnes haitta lievittyy asteeseen 1 tai korjautuu lähtötasolle. Pluvicto-annosta pienennetään 20 % eli annokseen 5 900 MBq. | |
Munuaistoksisuus | Määritelmä:
| Pluvicto tauotetaan haitan lievittymiseen asti. |
Määritelmä:
ja
| Pluvicto tauotetaan, kunnes haitta lievittyy tai korjautuu lähtötasolle. Pluvicto-annosta pienennetään 20 % eli annokseen 5 900 MBq. | |
Uusiutunut munuaistoksisuus (aste ≥ 3) | Pluvicto lopetetaan pysyvästi. | |
Selkäydinkompressio | Kaikki | Pluvicto tauotetaan, kunnes kompressio on hoidettu riittävästi ja neurologiset jälkitilat ja ECOG-toimintakykyluokka ovat stabiloituneet. |
Kantavan luun murtuma | Kaikki | Pluvicto tauotetaan, kunnes murtuma on stabiloitunut/hoidettu riittävästi ja ECOG-toimintakykyluokka on stabiloitunut. |
Uupumus | Aste ≥ 3 | Pluvicto tauotetaan, kunnes haitta lievittyy asteeseen 2 tai lähtötasolle. |
Elektrolyyttien tai aineenvaihdunnan poikkeavuudet | Aste ≥ 2 | Pluvicto tauotetaan, kunnes haitta lievittyy asteeseen 1 tai lähtötasolle. |
Ei-hematologinen toksisuus (kliinisesti merkittävä, ei toisin mainittu) | Aste ≥ 2 | Pluvicto tauotetaan, kunnes haitta lievittyy asteeseen 1 tai lähtötasolle. |
ASAT- tai ALAT-arvon suureneminen | ASAT tai ALAT > 5 x ULN ilman maksametastaaseja | Pluvicto lopetetaan pysyvästi. |
Lyhenteet: ECOG, Eastern Cooperative Oncology Group; ASAT, aspartaattiaminotransferaasi; ALAT, alaniiniaminotransferaasi; ULN, viitealueen yläraja. Vaikeusasteet perustuvat uusimpiin CTCAE-kriteereihin (Common Terminology Criteria for Adverse Events). a Samat kynnysarvot koskevat myös Pluvicto-hoidon aloituksen yhteydessä mitattavia lähtöarvoja. | ||
Erityisryhmät
Iäkkäät
Annosta ei tarvitse muuttaa 65 vuotta täyttäneillä potilailla.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta (lähtötilanteen kreatiniinipuhdistuma ≥ 50 ml/min Cockcroft–Gaultin kaavalla). Pluvicto-hoitoa ei suositella, jos potilaalla on keskivaikea tai vaikea munuaisten vajaatoiminta (lähtötilanteen kreatiniinipuhdistuma < 50 ml/min) tai loppuvaiheen munuaissairaus, sillä Pluvicto-valmisteen turvallisuutta ja farmakokineettistä profiilia ei ole tutkittu näillä potilailla (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on maksan vajaatoiminta. Pluvicto-valmistetta ei ole tutkittu keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastavilla potilailla (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Ei ole asianmukaista käyttää Pluvicto-valmistetta pediatrisille potilaille PSMA:ta ilmentävän eturauhassyövän hoitoon.
Antotapa
Pluvicto on käyttövalmis injektio-/infuusioneste (liuos) vain yhtä käyttökertaa varten.
Anto-ohjeet
Suositeltu Pluvicto-annos voidaan antaa injektiona laskimoon ruiskumenetelmällä, infuusiona painovoimamenetelmällä tai infuusiona peristalttisen pumpun menetelmällä.
Jos käytetään painovoimamenetelmää tai peristalttisen pumpun menetelmää, Pluvicto infusoidaan suoraan alkuperäispakkauksestaan.
Kun Pluvicto-annosta on muutettu haittavaikutuksen vuoksi, tulee antoon käyttää ruiskumenetelmää tai peristalttisen pumpun menetelmää. Jos pienennetyn Pluvicto-annoksen antoon käytetään painovoimamenetelmää, on annos säädettävä oikeaksi ennen antoa, jotta vältetään väärän suuruisen Pluvicto-tilavuuden saaminen.
Yksinomaan Pluvicto-valmisteen antoon käytettävä laskimokanyyli on huuhdeltava ennen valmisteen antoa vähintään 10 ml:lla steriiliä 9 mg/ml (0,9 %) NaCl-injektionestettä, jotta varmistetaan kanyylin aukiolo ja minimoidaan ekstravasaatioriski. Ekstravasaatiotapaukset on hoidettava hoitolaitoksen ohjeiden mukaisesti. Potilasta on ohjeistettava huolehtimaan hyvästä nesteytyksestä ja virtsaamaan tiheästi ennen Pluvicto-valmisteen antoa ja annon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ks. kohdasta 12 ohjeet valmisteen saattamisesta käyttökuntoon ja antotavoista laskimoon.
Potilaan valmistelu, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Yksilöllinen hyöty-riskiarvio
Kunkin potilaan kohdalla säteilyaltistus on oltava perusteltavissa todennäköisellä hyödyllä. Annettavan aktiivisuuden on oltava aina niin vähäistä kuin kohtuullisesti on mahdollista tavoitellun hoitovaikutuksen saavuttamiseksi.
Säteilyaltistukseen liittyvä riski
Pluvicto vaikuttaa osaltaan potilaan pitkän aikavälin kumulatiiviseen kokonaissäteilyaltistukseen, joka on yhteydessä suurentuneeseen syöpäriskiin.
Potilaaseen, hoitohenkilökuntaan ja toisiin henkilöihin kohdistuva säteilyaltistus on minimoitava Pluvicto-hoidon aikana ja hoidon jälkeen noudattamalla hoitolaitoksen asianmukaisia säteilyturvakäytäntöjä, hoitokäytäntöjä ja ohjeistusta, joka annetaan potilaalle kotona noudatettavien säteilysuojaustoimien osalta.
Potilaan valmistelu
Potilasta on muistutettava juomaan tavallista enemmän ja virtsaamaan mahdollisimman usein, jotta virtsarakkoon kohdistuva säteilymäärä vähenisi etenkin suuren radioaktiivisuuden hoitotoimien jälkeen (esim. radionuklidihoito).
Toimenpiteen jälkeen
Ennen potilaan kotiuttamista isotooppilääkärin tai muun terveydenhuollon ammattilaisen on selitettävä potilaalle, mitä säteilysuojaustoimia hänen on noudatettava muihin henkilöihin kohdistuvan säteilyaltistuksen minimoimiseksi.
Jokaisen Pluvicto-hoitokerran jälkeen voidaan kansallisten, paikallisten ja toimipaikkakohtaisten menettelyjen ja säädösten ohella huomioida seuraavat yleiset suositukset:
- Rajoita alle metrin etäisyydellä tapahtuvaa kanssakäymistä toisten kanssa 2 vrk ajan ja lasten ja raskaana olevien naisten kanssa 7 vrk ajan.
- Pidättäydy seksistä 7 vrk ajan.
- Nuku erillään (eri huoneessa) toisista 3 vrk ajan, lapsista 7 vrk ajan ja raskaana olevista 15 vrk ajan.
Myelosuppressio
VISION-tutkimuksessa myelosuppressiota, mukaan lukien kuolemaan johtaneita tapauksia, esiintyi useammin Pluvicto-hoitoa ja parasta standardihoitoa (best standard of care, BSoC) saaneilla kuin pelkkää BSoC-hoitoa saaneilla (ks. kohta Haittavaikutukset).
Hematologisia laboratoriotutkimuksia, mukaan lukien hemoglobiini, valkosoluarvo, absoluuttinen neutrofiiliarvo ja verihiutalearvo, on tehtävä ennen Pluvicto-hoitoa ja sen aikana. Myelosuppression vaikeusasteen perusteella Pluvicto-annosta on pienennettävä tai hoito on tauotettava tai lopetettava pysyvästi ja potilasta on hoidettava kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa).
Munuaistoksisuus
VISION-tutkimuksessa munuaistoksisuutta esiintyi useammin Pluvicto- ja BSoC-hoitoa saaneilla kuin pelkkää BSoC-hoitoa saaneilla (ks. kohta Haittavaikutukset).
Ennen Pluvicto-hoitoa ja hoidon jälkeen potilasta on muistutettava juomaan tavallista enemmän ja virtsaamaan mahdollisimman usein etenkin suuren radioaktiivisuuden hoitotoimien jälkeen (esim. radionuklidihoito). Munuaisten toimintaa tutkivia laboratoriokokeita, mukaan lukien seerumin kreatiniini ja laskennallinen kreatiniinipuhdistuma, on tehtävä ennen Pluvicto-hoitoa ja hoidon aikana. Munuaistoksisuuden vaikeusasteen perusteella Pluvicto-annosta on pienennettävä tai hoito on tauotettava tai lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Munuaisten/maksan vajaatoiminta
Valmisteen hyöty-riskisuhde on arvioitava tarkoin näillä potilailla, sillä säteilyaltistus saattaa olla tavanomaista suurempi.
Lutetium(177Lu)vipivotiditetraksetaanialtistuksen (AUC) odotetaan suurenevan munuaisten vajaatoiminnan vaikeusasteen mukaan (ks. kohta Farmakokinetiikka). Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla haittojen riski voi olla suurentunut. Munuaisten toimintaa ja haittavaikutuksia on seurattava tiheään, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta (ks. kohta Annostus ja antotapa). Pluvicto-hoitoa ei suositella, jos potilaalla on keskivaikea tai vaikea munuaisten vajaatoiminta (lähtötilanteen kreatiniinipuhdistuma < 50 ml/min) tai loppuvaiheen munuaissairaus.
Hedelmällisyys
Lutetium(177Lu)vipivotiditetraksetaanin säteily saattaa aiheuttaa miesten sukupuolirauhasiin ja spermatogeneesiin liittyviä haittoja. Suositeltu kumulatiivinen Pluvicto-annos 44 400 MBq aiheuttaa säteilyn absorboitumisen kiveksiin annosarvoilla, joilla hedelmättömyyttä voi esiintyä. Perinnöllisyysneuvontaan hakeutuminen on suositeltavaa, jos potilas toivoo saavansa lapsia hoidon jälkeen. Miespotilaiden kanssa voidaan keskustella hoitoa edeltävästä sperman pakastuksesta (ks. kohta Raskaus ja imetys).
Ehkäisy miehillä
Miespotilaita ohjeistetaan olemaan siittämättä lasta ja käyttämään yhdynnässä kondomia Pluvicto-hoidon aikana ja vielä 14 viikon ajan viimeisen annoksen jälkeen (ks. kohta Raskaus ja imetys).
Erityisvaroitukset
Natriumpitoisuus
Tämä lääkevalmiste sisältää enintään 3,9 mmol (88,75 mg) natriumia per injektiopullo, joka vastaa 4,4 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Ympäristöriskejä koskevat varotoimet, ks. kohta Käyttö- ja käsittelyohjeet.
Yhteisvaikutukset
Kliinisiä yhteisvaikutustutkimuksia ei ole tehty.
Raskaus ja imetys
Ehkäisy miehillä
Lutetium(177Lu)vipivotiditetraksetaanin säteily saattaa aiheuttaa spermatogeneesiin liittyviä haittoja, joten miespotilaita ohjeistetaan olemaan siittämättä lasta ja käyttämään yhdynnässä kondomia Pluvicto-hoidon aikana ja vielä 14 viikon ajan viimeisen annoksen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus
Pluvicto ei ole tarkoitettu käytettäväksi naisille. Eläimillä ei ole tehty tutkimuksia lutetium(177Lu)vipivotiditetraksetaanin vaikutuksista naaraan lisääntymiskykyyn tai alkion- ja sikiönkehitykseen. Kaikenlainen radioaktiivinen säteily, myös Pluvicton aiheuttama, voi kuitenkin vahingoittaa sikiötä, jos valmistetta annetaan raskaana olevalle naiselle.
Imetys
Pluvicto ei ole tarkoitettu käytettäväksi naisille. Ei ole olemassa tietoja lutetium(177Lu)vipivotiditetraksetaanin erittymisestä ihmisen rintamaitoon tai vaikutuksista imetettävään vastasyntyneeseen/imeväiseen tai maidoneritykseen.
Hedelmällisyys
Lutetium(177Lu)vipivotiditetraksetaanin vaikutuksista hedelmällisyyteen ei ole tehty tutkimuksia. Lutetium(177Lu)vipivotiditetraksetaanin säteily saattaa aiheuttaa miesten sukupuolirauhasiin ja spermatogeneesiin liittyviä haittoja. Suositeltu kumulatiivinen Pluvicto-annos 44 400 MBq aiheuttaa säteilyn absorboitumisen kiveksiin annosarvoilla, joilla hedelmättömyyttä voi esiintyä. Perinnöllisyysneuvontaan hakeutuminen on suositeltavaa, jos potilas toivoo saavansa lapsia hoidon jälkeen. Miespotilaiden kanssa voidaan keskustella hoitoa edeltävästä sperman pakastuksesta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Pluvicto-valmisteella saattaa olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Ellei toisin mainita, lueteltujen haittavaikutusten esiintymistiheydet perustuvat VISION-tutkimukseen, jossa 529 potilasta sai vähintään yhden 7 400 MBq:n annoksen valmistetta (annosten mediaanimäärä oli viisi).
Yleisimmät haittavaikutukset olivat uupumus (48,0 %), suun kuivuus (39,3 %), pahoinvointi (35,7 %), anemia (31,9 %), ruokahalun heikkeneminen (21,4 %) ja ummetus (20,2 %). Yleisimmät asteen 3–4 haittavaikutukset olivat anemia (12,9 %), trombosytopenia (7,9 %), lymfopenia (7,8 %) ja uupumus (6,6 %).
VISION-tutkimuksen lopullisen analyysin ajankohtana, kun seuranta-ajan mediaani oli 14,2 kuukautta (vaihteluväli: 0,6–60,9 kuukautta), yleinen turvallisuusprofiili pysyi yhdenmukaisena aiemmin raportoidun kanssa.
Haittavaikutustaulukko
Haittavaikutukset (taulukko 2) esitetään MedDRA-elinjärjestelmäluokittain. Kunkin elinjärjestelmäluokan haittavaikutukset esitetään yleisyysjärjestyksessä yleisimmästä alkaen. Lisäksi kunkin haittavaikutuksen kohdalla mainittava yleisyysluokka perustuu seuraavaan jaotteluun (CIOMS III): hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000).
Taulukko 2 Haittavaikutukset, joiden ilmaantuvuus oli VISION-tutkimuksessa suurempi Pluvicto- ja BSoC-hoitoa saaneilla kuin pelkkää BSoC-hoitoa saaneillaa
Elinjärjestelmäluokka Haittavaikutus | Yleisyysluokka | Kaikki asteet n (%) | Asteet 3–4b n (%) |
| Infektiot | |||
| Suun sieni-infektioc | Yleinen | 13 (2,5) | 0 (0,0) |
| Veri ja imukudos | |||
| Anemia | Hyvin yleinen | 169 (31,9) | 68 (12,9) |
| Trombosytopenia | Hyvin yleinen | 91 (17,2) | 42 (7,9) |
| Leukopeniad | Hyvin yleinen | 83 (15,7) | 22 (4,2) |
| Lymfopenia | Hyvin yleinen | 75 (14,2) | 41 (7,8) |
| Pansytopeniae | Yleinen | 9 (1,7) | 7 (1,3)b |
| Luuytimen vajaatoiminta | Melko harvinainen | 1 (0,2) | 1 (0,2)b |
| Hermosto | |||
| Huimaus | Yleinen | 44 (8,3) | 5 (0,9) |
| Päänsärky | Yleinen | 37 (7,0) | 4 (0,8) |
| Dysgeusiaf | Yleinen | 37 (7,0) | 0 (0,0) |
| Silmät | |||
| Silmien kuivuus | Yleinen | 16 (3,0) | 0 (0,0) |
| Kuulo ja tasapainoelin | |||
| Kiertohuimaus | Yleinen | 11 (2,1) | 0 (0,0) |
| Ruoansulatuselimistö | |||
| Suun kuivuusg | Hyvin yleinen | 208 (39,3) | 0 (0,0) |
| Pahoinvointi | Hyvin yleinen | 189 (35,7) | 7 (1,3) |
| Ummetus | Hyvin yleinen | 107 (20,2) | 6 (1,1) |
| Oksenteluh | Hyvin yleinen | 101 (19,1) | 5 (0,9) |
| Ripuli | Hyvin yleinen | 101 (19,1) | 4 (0,8) |
| Vatsakipui | Hyvin yleinen | 61 (11,5) | 7 (1,3) |
| Ruokatorven häiriöj | Yleinen | 18 (3,4) | 1 (0,2) |
| Suutulehdus | Yleinen | 9 (1,7) | 1 (0,2) |
| Iho ja ihonalainen kudos | |||
| Kuiva ihok | Yleinen | 8 (1,5) | 0 (0,0) |
| Munuaiset ja virtsatiet | |||
| Virtsatieinfektiol | Hyvin yleinen | 63 (11,9) | 20 (3,8) |
| Akuutti munuaisvauriom | Yleinen | 48 (9,1) | 18 (3,4) |
| Yleisoireet ja antopaikassa todettavat haitat | |||
| Uupumusn | Hyvin yleinen | 254 (48,0) | 35 (6,6) |
| Ruokahalun heikkeneminen | Hyvin yleinen | 113 (21,4) | 10 (1,9) |
| Painon lasku | Hyvin yleinen | 58 (11,0) | 2 (0,4) |
| Perifeerinen ödeemao | Hyvin yleinen | 53 (10,0) | 2 (0,4) |
| Kuume | Yleinen | 37 (7,0) | 2 (0,4) |
Lyhenne: BSoC, paras standardihoito. a NCI CTCAE -kriteerit (National Cancer Institute Common Terminology Criteria for Adverse Events), versio 5.0. b Sisältää vain asteen 3–4 haittavaikutukset, poikkeuksena pansytopenia ja luuytimen vajaatoiminta. Asteen 5 (kuolemaan johtanutta) pansytopeniaa ilmoitettiin 2 potilaalla, jotka saivat Pluvicto-hoitoa yhdessä BSoC-hoidon kanssa. Asteen 5 (kuolemaan johtanutta) luuytimen vajaatoimintaa ilmoitettiin 1 potilaalla, joka sai Pluvicto-hoitoa yhdessä BSoC-hoidon kanssa. c Suun sieni-infektio sisältää suun kandidiaasin, candida-infektion, suun sieni-infektion, suunielun sienitulehduksen ja kielen sieni-infektion. d Leukopenia sisältää leukopenian ja neutropenian. e Pansytopenia sisältää pansytopenian ja bisytopenian. f Dysgeusia sisältää dysgeusian ja makuhäiriöt. g Suun kuivuus sisältää suun kuivuuden, huulten kuivumisen, syljen vajaaerityksen ja kurkun kuivuuden. h Oksentelu sisältää oksentelun ja yökkäilyn. j Vatsakipu sisältää vatsakivun, ylävatsakivun, epämiellyttävän tunteen vatsassa, alavatsakivun, vatsan arkuuden ja ruoansulatuskanavan kivun. j Ruokatorven häiriöihin kuuluvat gastroesofageaalinen refluksitauti, dysfagia ja ruokatorvitulehdus. k Kuiva iho sisältää kuivan ihon ja kseroderman. l Virtsatieinfektio sisältää virtsatieinfektion, kystiitin ja bakteerikystiitin. m Akuutti munuaisvaurio sisältää veren kreatiniinipitoisuuden suurenemisen, akuutin munuaisvaurion, munuaisten vajaatoiminnan ja veren ureapitoisuuden suurenemisen. n Väsymys sisältää väsymyksen ja voimattomuuden. o Perifeerinen ödeema sisältää perifeerisen ödeeman, nesteretention ja hypervolemian. | |||
Valikoitujen haittavaikutusten kuvaus
Myelosuppressio
VISION-tutkimuksessa myelosuppressiota esiintyi useammin Pluvicto- ja BSoC-hoitoa saaneilla kuin pelkkää BSoC-hoitoa saaneilla (kaikki asteet / aste ≥ 3): anemia (31,9 % / 12,9 %) vs (13,2 % / 4,9 %); trombosytopenia (17,2 % / 7,9 %) vs (4,4 % / 1,0 %); leukopenia (12,5 % / 2,5 %) vs (2,0 % / 0,5 %); lymfopenia (14,2 % / 7,8 %) vs (3,9 % / 0,5 %); neutropenia (8,5 % / 3,4 %) vs (1,5 % / 0,5 %); pansytopenia (1,5 % / 1,1 %) vs (0 % / 0 %), mukaan lukien kaksi kuolemaan johtanutta pansytopeniatapahtumaa Pluvicto-hoitoa yhdessä BSoC-hoidon kanssa saaneilla; bisytopenia (0,2 % / 0,2 %) vs (0 % / 0 %); ja luuytimen vajaatoiminta (0,2 % / 0,2 %) vs (0 % / 0 %), mukaan lukien yksi kuolemaan johtanut luuytimen vajaatoimintatapahtuma Pluvicto-hoitoa yhdessä BSoC-hoidon kanssa saaneella potilaalla.
Hoidon pysyvään lopettamiseen johtaneita myelosuppressiohaittoja, joita esiintyi ≥ 0,5 %:lla Pluvicto-hoitoa yhdessä BSoC-hoidon kanssa saaneista olivat anemia (2,8 %), trombosytopenia (2,8 %), leukopenia (1,3 %), neutropenia (0,8 %) ja pansytopenia (0,6 %). Hoidon tauottamiseen / annoksen pienentämiseen johtaneita myelosuppressiohaittoja, joita esiintyi ≥ 0,5 %:lla Pluvicto-hoitoa yhdessä BSoC-hoidon kanssa saaneista olivat anemia (5,1 % / 1,3 %), trombosytopenia (3,6 % / 1,9 %), leukopenia (1,5 % / 0,6 %) ja neutropenia (0,8 % / 0,6 %).
Munuaistoksisuus
VISION-tutkimuksessa munuaistoksisuutta esiintyi useammin Pluvicto- ja BSoC-hoitoa saaneilla kuin pelkkää BSoC-hoitoa saaneilla (kaikki asteet / asteet 3–4): veren kreatiniinipitoisuuden suureneminen (5,7 % / 0,2 %) vs (2,4 % / 0,5 %); akuutti munuaisvaurio (3,8 % / 3,2 %) vs (3,9 % / 2,4 %); munuaisten vajaatoiminta (0,2 % / 0 %) vs (0 % / 0 %); ja veren ureapitoisuuden suureneminen (0,2 % / 0 %) vs (0 % / 0 %).
Hoidon pysyvään lopettamiseen johtaneita munuaishaittoja, joita esiintyi ≥ 0,2 %:lla Pluvicto-hoitoa yhdessä BSoC-hoidon kanssa saaneista olivat: veren kreatiniinipitoisuuden suureneminen (0,2 %). Hoidon tauottamiseen / annoksen pienentämiseen johtaneita munuaishaittoja, joita esiintyi ≥ 0,2 %:lla Pluvicto-hoitoa yhdessä BSoC-hoidon kanssa saaneista olivat veren kreatiniinipitoisuuden suureneminen (0,2 % / 0,4 %) ja akuutti munuaisvaurio (0,2 % / 0 %).
Sekundaarisyöpä
Ionisoivalle säteilylle altistuminen on yhteydessä syövän kehittymiseen ja voi aiheuttaa perinnöllisiä poikkeavuuksia. Terapeuttisen altistuksen aiheuttama säteilyannos saattaa suurentaa syöpien ja mutaatioiden ilmaantuvuutta. Kaikissa tapauksissa on varmistettava, että säteilyn aiheuttamat riskit ovat pienempiä kuin itse taudin aiheuttama riski. Pluvicto lisää potilaan pitkäaikaista kokonaissäteilyaltistusta, jolla on yhteys syöpäriskin suurenemiseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Siksi radiofarmaseuttisten lääkevalmisteiden, kuten Pluvicton, kohdalla ei voida sulkea pois sekundaarisyöpäriskiä. VISION-tutkimuksen primaarianalyysin tietojenkeräyksen päättymispäivään 27.01.2021 mennessä oli Pluvicto-hoitoa yhdessä BSoC-hoidon kanssa saaneilla potilailla ilmoitettu levyepiteelisyöpätapauksia (4 potilaalla; 0,8 %) ja yksi tapaus kutakin (1 potilas; 0,2 %) seuraavaa syöpätyyppiä: tyvisolusyöpä, pahanlaatuinen melanooma, ihon levyepiteelisyöpä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Jos potilas saa Pluvicto-valmisteesta yliannostuksen säteilyä, elimistöön absorboituvaa annosta on pyrittävä pienentämään mahdollisuuksien mukaan tehostamalla radionuklidin eliminaatiota elimistöstä tiheän virtsaamisen tai tehostetun diureesin ja tiheästi toistuvan virtsarakon tyhjentämisen avulla. Annetun efektiivisen annoksen arvioinnista voi olla hyötyä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Terapeuttiset radioaktiiviset lääkevalmisteet, Muut terapeuttiset radioaktiiviset lääkevalmisteet, ATC-koodi: V10XX05
Vaikutusmekanismi
Pluvicto-valmisteen aktiivinen osa on radionuklidi lutetium‑177, joka on yhdistetty pienimolekyyliseen ligandiin. Se kohdistuu ja sitoutuu suurella affiniteetilla PSMA:han, joka on eturauhassyövässä (myös mCRPC-syövässä) yli-ilmentynyt kalvoproteiini. Pluvicto-valmisteen sitoutuessa PSMA:ta ilmentäviin syöpäsoluihin lutetium-177 säteilee terapeuttista β–-säteilyä kohdesoluihin ja ympäröiviin soluihin ja indusoi DNA-vaurioita, jotka voivat johtaa solukuolemaan.
Farmakodynaamiset vaikutukset
Vipivotiditetraksetaanilla ei ole farmakodynaamista aktiivisuutta.
Kliininen teho ja turvallisuus
VISION
Pluvicto-valmisteen tehoa arvioitiin edennyttä, PSMA-positiivista mCRPC-syöpää sairastavilla potilailla VISION-tutkimuksessa, joka oli satunnaistettu, avoin, vaiheen III monikeskustutkimus. Tutkimuksessa 831 aikuispotilasta (N = 831) satunnaistettiin suhteessa 2:1 saamaan joko Pluvicto-hoitoa 7 400 MBq 6 viikon välein (yhteensä enintään 6 annosta) yhdistettynä BSoC-hoitoon (N = 551) tai pelkkää BSoC-hoitoa (N = 280). Neljä Pluvicto-annosta saaneilla potilailla arvioitiin uudelleen vastetta koskevaa näyttöä, jäännöstaudin merkkejä ja valmisteen siedettävyyttä, minkä jälkeen heille voitiin antaa vielä kaksi lisäannosta lääkärin harkinnan mukaan.
Kastraatiostatuksen ylläpitämiseksi kaikilla potilailla jatkettiin gonadotropiinia vapauttavan hormonin analogihoitoa tai heille tehtiin bilateraalinen orkiektomia. Tutkimukseen soveltuvien potilaiden oli täytettävä seuraavat kriteerit: edennyt, PSMA-positiivinen mCRPC, ECOG-toimintakykyluokka 0–2, vähintään yksi etäpesäkemuutos tietokonetomografiassa, magneettikuvauksessa tai luustokartoituksessa ja riittävä munuaisten ja maksan toiminta sekä hematologinen toiminta.
Tutkimukseen soveltuvien potilaiden oli täytettävä myös seuraavat kriteerit: anamneesissa vähintään yksi androgeenireseptorireitin estäjähoito, kuten abirateroniasetaatti tai entsalutamidi ja 1–2 taksaanipohjaista solunsalpaajahoito-ohjelmaa (yhden hoito-ohjelman oli sisällettävä vähintään 2 taksaanihoitojaksoa). Jos anamneesissa oli vain yksi aikaisempi taksaanipohjainen solunsalpaajahoito-ohjelma ja potilas oli kieltäytynyt toisesta hoito-ohjelmasta tai se ei lääkärin mukaan soveltunut potilaalle, potilas katsottiin soveltuvaksi tutkimukseen. Tutkimukseen eivät soveltuneet potilaat, joilla oli epästabiileja oireisia keskushermostometastaaseja tai oireinen tai kliinisesti/radiologisesti todennettu lähestyvä selkäydinkompressio. Potilaille tehtiin gallium(68Ga)gotsetotidi-PET-kuvaus, jolla arvioitiin PSMA:n ilmentymistä syöpämuutoksissa keskitetysti tulkituin kriteerein. Soveltuvilla potilailla oli oltava PSMA-positiivinen mCRPC, eli vähintään yksi kasvainmuutos, johon kertyi gallium(68Ga)gotsetotidia enemmän kuin normaaliin maksaan. Potilas suljettiin pois tutkimuksesta, jos jossain lyhyellä akselilla kokokriteerit ylittäneessä muutoksessa (elimet ≥ 1 cm, imusolmukkeet ≥ 2,5 cm, luusto [pehmytkudoskomponentti] ≥ 1 cm) kertymä oli vähäisempi tai samaa luokkaa kuin normaalissa maksassa.
Lääkärin harkinnan mukaan annettuja BSoC-hoitoja olivat seuraavat: tukihoito (kuten kivun hoito, nesteytys, verensiirto), ketokonatsoli, paikallisiin eturauhassyöpämuutoksiin kohdennettu sädehoito (mukaan lukien brakyterapia tai mikä tahansa ulkoinen sädehoito [mukaan lukien stereotaktinen kehon sädehoito ja palliatiivinen ulkoinen sädehoito]), luustoon kohdistettu lääkehoito (kuten tsoledronihappo, denosumabi ja mikä tahansa bisfosfonaatti), androgeenia vähentävät aineet (kuten gonadotropiinia vapauttavan hormonin analogit, mitkä tahansa kortikosteroidit ja 5-alfareduktaasit) ja androgeenireseptorireitin estäjät. BSoC-hoidoksi ei käynyt mikään seuraavista: tutkimusvaiheessa olevat lääkeaineet, sytotoksiset solunsalpaahoidot, immunoterapiat, muut systeemiset radioisotoopit ja puolikehon sädetys.
Potilaat jatkoivat satunnaistettua hoitoa, kunnes ilmeni näyttöä syövän etenemisestä (perusteena Prostate Cancer Working Group 3 [PCWG3] -työryhmän kriteereihin perustuva tutkijan arvio), sietämätön toksisuus, kielletyn hoidon käyttö, huono hoitomyöntyvyys, hoidon lopetus tai kliinisen hyödyn puute.
Ensisijaiset tehon päätetapahtumat olivat kokonaiselossaolo (OS) ja radiologinen etenemättömyys (rPFS), joita arvioitiin sokkoutetusti, riippumattomasti ja keskitetysti (BICR-arviointi) PCWG3-kriteerein. Toissijaisia tehon päätetapahtumia olivat kokonaisvasteprosentti (ORR) (BICR-arviointi RECIST [Response Evaluation Criteria in Solid Tumors] v1.1-kriteerein) ja aika ensimmäiseen oireiseen luustotapahtumaan (SSE), jonka määritelmänä oli ensimmäinen uusi oireinen patologinen luunmurtuma, selkäydinkompressio, kasvaimeen liittyvä ortopedinen kirurgia, luustokivun lievittämiseksi välttämätön sädehoito tai kuolema mistä tahansa syystä (ensin tapahtunut valittiin). Kasvainta arvioitiin radiologisesti (tehosteaineella toteutettu TT-kuvaus / magneettikuvaus ja luustokartoitus) 8 viikon välein (± 4 vrk) ensimmäisen annoksen jälkeen ensimmäisten 24 viikon ajan (riippumatta annosviivästyksistä) ja tämän jälkeen 12 viikon välein (± 4 vrk).
Hoitoryhmien väliset demografiset tiedot ja taudin lähtötilannetiedot olivat tasapainossa. Mediaani-ikä oli 71 vuotta (vaihteluväli 40–94 vuotta); 86,8 % oli valkoihoisia, 6,6 % mustia/afroamerikkalaisia ja 2,4 % aasialaisia; 92,4 %:lla oli ECOG-toimintakykyluokka 0–1; 7,6 %:lla oli ECOG-toimintakykyluokka 2. Satunnaistaminen stratifioitiin seuraavien tekijöiden perusteella: lähtötilanteen laktaattidehydrogenaasipitoisuus (LDH ≤ 260 IU/l vs > 260 IU/l), maksametastaasien esiintyminen (kyllä vs ei), ECOG-toimintakykyluokka (0 tai 1 vs 2), ja androgeenireseptorireitin estäjän käyttö BSoC-hoidon osana satunnaistamishetkellä (kyllä vs ei). Satunnaistamishetkellä kaikilla potilailla (100,0 %) oli anamneesissa vähintään yksi taksaanipohjainen solunsalpaajahoito-ohjelma ja 41,2 %:lla kaksi; 97,1 % potilaista oli saanut dosetakselia ja 38,0 % kabatsitakselia. Satunnaistamishetkellä 51,3 % potilaista oli saanut aiemmin yhtä androgeenireseptorireitin estäjää, 41,0 % potilaista kahta androgeenireseptorireitin estäjää ja 7,7 % kolmea tai useampaa androgeenireseptorireitin estäjää. Satunnaistetun hoitojakson aikana 52,6 % Pluvicto- ja BSoC-hoitoa saaneista potilaista ja 67,8 % pelkkää BSoC-hoitoa saaneista potilaista sai vähintään yhtä androgeenireseptorireitin estäjää.
VISION-tutkimuksen tehotulokset esitetään taulukossa 3 ja kuvissa 1 ja 2. OS- ja rPFS-tulosten lopulliset analyysit olivat tapahtumalähtöisiä. OS-analyysi tehtiin 530 kuoleman ja rPFS-analyysi 347 tapahtuman esiinnyttyä.
Taulukko 3 VISION-tutkimuksen tehotulokset
| Tehoparametrit | Pluvicto + BSoC | BSoC |
| Ensisijaiset vaihtoehtoiset tehon päätetapahtumat | ||
| Kokonaiselossaolo (OS)a | N = 551 | N = 280 |
| Kuolemat, n (%) | 343 (62,3 %) | 187 (66,8 %) |
| Mediaani, kk (95 % lv)b | 15,3 (14,2–16,9) | 11,3 (9,8–13,5) |
| Riskitiheyssuhde (95 % lv)c | 0,62 (0,52–0,74) | |
| P‑arvod | < 0,001 | |
| Radiologinen etenemättömyys (rPFS)e,f | N = 385 | N = 196 |
| Tapahtumia (taudin eteneminen tai kuolema), n (%) | 254 (66,0 %) | 93 (47,4 %) |
| Radiologinen eteneminen, n (%) | 171 (44,4 %) | 59 (30,1 %) |
| Kuolemat, n (%) | 83 (21,6 %) | 34 (17,3 %) |
| Mediaani, kk (99,2 % lv)b | 8,7 (7,9–10,8) | 3,4 (2,4–4,0) |
| Riskitiheyssuhde (99,2 % lv)c | 0,40 (0,29–0,57) | |
| P‑arvod | < 0,001 | |
| Toissijaiset tehon päätetapahtumat | ||
| Aika ensimmäiseen oireiseen luustotapahtumaan (SSE)f | N = 385 | N = 196 |
| Tapahtumia (SSE tai kuolema), n (%) | 256 (66,5 %) | 137 (69,9 %) |
| SSE, n (%) | 60 (15,6 %) | 34 (17,3 %) |
| Kuolemat, n (%) | 196 (50,9 %) | 103 (52,6 %) |
| Mediaani, kk (95 % lv)b | 11,5 (10,3–13,2) | 6,8 (5,2–8,5) |
| Riskitiheyssuhde (95 % lv)c | 0,50 (0,40–0,62) | |
| P‑arvog | < 0,001 | |
| Paras kokonaisvaste (BOR) | ||
| Potilaat, joilla lähtötilanteessa arvioitavissa oleva tauti | N = 319 | N = 120 |
| Täydellinen vaste (CR), n (%) | 18 (5,6 %) | 0 (0 %) |
| Osittainen vaste (PR), n (%) | 77 (24,1 %) | 2 (1,7 %) |
| Kokonaisvasteprosentti (ORR)h,i | 95 (29,8 %) | 2 (1,7 %) |
| P‑arvoj | < 0,001 | |
| Vasteen kesto (DOR)h | ||
| Mediaani, kk (95 % lv)b | 9,8 (9,1–11,7) | 10,6 (NE; NE)k |
BSoC: paras standardihoito; Lv: luottamusväli; NE: ei arvioitavissa; BICR: sokkoutettu, riippumaton keskitetty arviointi; PCWG3: Prostate Cancer Working Group 3 -työryhmä; RECIST: Response Evaluation Criteria in Solid Tumors -kriteerit. a Analysoitu lähtöryhmien mukaisten ryhmien perusteella (intent‑to‑treat, ITT) kaikilla satunnaistetuilla potilailla. b Perustuu Kaplan–Meier-estimaattiin. c Riskitiheyssuhde perustuu stratifioituun Coxin suhteellisten riskitiheyksien malliin. Riskitiheyssuhde < 1 suosii Pluvicto- ja BSoC-hoitoa. d Stratifioidun log-rank-testin yksitahoinen p-arvo. e BICR-arviointi PCWG3-kriteerein. Ensisijaisesta rPFS-analyysistä sensuroitiin potilaat, joilla oli jäänyt väliin vähintään 2 peräkkäistä kasvainarviointia juuri ennen taudin etenemistä tai kuolemaa. rPFS-tulokset olivat sensuroinnista riippumatta johdonmukaiset. f Analysoitu lähtöryhmien mukaisten ryhmien (intent‑to‑treat, ITT) perusteella kaikkien 5.3.2019 tai sen jälkeen satunnaistettujen potilaiden osalta, sillä tällöin BSoC-ryhmässä toteutettiin toimia varhaisen keskeyttämisen vähentämiseksi. g Stratifioidun log-rank-testin kaksitahoinen p-arvo. h BICR-arviointi RECIST v1.1 -kriteerein. i ORR: CR + PR. Vahvistetut CR- ja PR-vasteet. j Stratifioidun Waldin Khii-neliö-testin kaksitahoinen p-arvo. k DOR-mediaani pelkkää BSoC-hoitoa saaneessa ryhmässä ei ollut luotettava, sillä vain yksi potilas kahdesta hoitoon vastanneesta koki RECIST v1.1 -kriteerien mukaisen radiologisen etenemisen tai kuoleman. | ||
Kuva 1 Kaplan–Meier-kuvaaja kokonaiselossaolosta (OS) VISION-tutkimuksessa

Stratifioitu log-rank-testi ja stratifioitu Coxin malli, jossa käytettiin ositteita IRT-teknologian (Interactive Response Technology) mukaisesti; määritelmänä LDH-arvo, maksametastaasien esiintyminen, ECOG-toimintakykyluokka ja androgeenireseptorireitin estäjän käyttö BSoC-hoitona satunnaistamishetkellä.
n/N: Tapahtumien/potilaiden määrä hoitoryhmässä.
Kuva 2 Kaplan–Meier-kuvaaja BICR-arvioidusta radiologisesta etenemättömyydestä (rPFS) VISION-tutkimuksessa
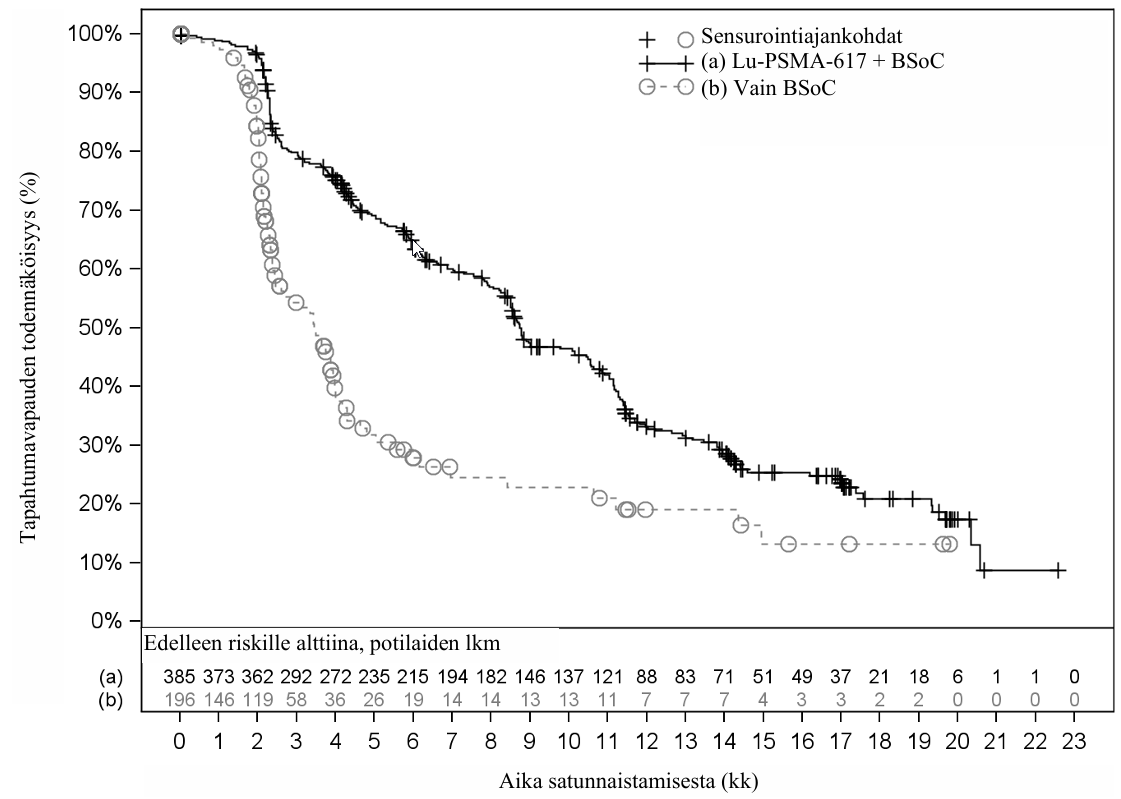
Stratifioitu log-rank-testi ja stratifioitu Coxin malli, joissa käytettiin ositteita IRT-teknologian mukaisesti; määritelmänä LDH-arvo, maksametastaasien esiintyminen, ECOG-toimintakykyluokka ja androgeenireseptorireitin estäjän käyttö BSoC-hoitona satunnaistamishetkellä.
n/N: Tapahtumia/potilaita hoitoryhmässä.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Pluvicto-valmisteen käytöstä PSMA:ta ilmentävän eturauhassyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Lutetium(177Lu)vipivotiditetraksetaanin farmakokinetiikkaa karakterisoitiin 30 potilaalla vaiheen III VISION-osatutkimuksessa.
Imeytyminen
Pluvicto annetaan laskimoon, ja sen biologinen hyötyosuus on välitön ja täydellinen.
Lutetium(177Lu)vipivotiditetraksetaanialtistuksen (pitoisuus-aikakäyrän alle jäävä pinta-ala [AUCinf]) geometrinen keskiarvo veressä on suositellulla annoksella 52,3 ng.h/ml (geometrinen variaatiokerroin, keskiarvo [CV] 31,4 %). Lutetium(177Lu)vipivotiditetraksetaanin enimmäispitoisuuden (Cmax) geometrinen keskiarvo veressä on 6,58 ng/ml (CV 43,5 %) kun käytetään suositeltua annosta.
Jakautuminen
Lutetium(177Lu)vipivotiditetraksetaanin jakautumistilavuuden (Vz) geometrinen keskiarvo on 123 l (CV 78,1 %).
Vipivotiditetraksetaani ja ei-radioaktiivinen lutetium(175Lu)vipivotiditetraksetaani sitoutuvat ihmisellä plasman proteiineihin 60–70-prosenttisesti.
Kertyminen elimiin
Lutetium(177Lu)vipivotiditetraksetaanin biojakautuvuuden perusteella suurimmat kertymät esiintyvät kyynelrauhasissa, sylkirauhasissa, munuaisissa, virtsarakon seinämässä, maksassa, ohutsuolessa ja paksusuolessa (vasemman- ja oikeanpuoleinen koolon).
Eliminaatio
Lutetium(177Lu)vipivotiditetraksetaanin puhdistuman (CL) geometrinen keskiarvo on 2,04 l/h (CV 31,5 %).
Lutetium(177Lu)vipivotiditetraksetaani erittyy pääasiassa munuaisteitse.
Puoliintumisaika
Pluvicto eliminoituu bieksponentiaalisesti, ja terminaalisen eliminaation puoliintumisajan (t½) geometrinen keskiarvo on 41,6 tuntia (CV 68,8 %).
Biotransformaatio
Lutetium(177Lu)vipivotiditetraksetaani ei metaboloidu maksassa tai munuaisissa.
Lääkeyhteisvaikutusten mahdollisuuden arviointi in vitro
CYP450-entsyymit
Vipivotiditetraksetaani ei ole CYP450-entsyymien substraatti. Se ei indusoi CYP450-entsyymejä 1A2, 2B6 tai 3A4, eikä estä CYP450-entsyymejä 1A2, 2B6, 2C8, 2C9, 2C19, 2D6 tai 3A4/5 in vitro.
Kuljettajaproteiinit
Vipivotiditetraksetaani ei ole BCRP:n, P‑gp:n, MATE1:n, MATE2‑K:n, OAT1:n, OAT3:n tai OCT2:n substraatti, eikä BCRP:n, P‑gp:n, BSEP:n, MATE1:n, MATE2‑K:n, OAT1:n, OAT3:n, OATP1B1:n, OATP1B3:n, OCT1:n tai OCT2:n estäjä in vitro.
Erityisryhmät
Iän ja painon vaikutukset
Kliinisesti merkitseviä vaikutuksia lutetium(177Lu)vipivotiditetraksetaanin farmakokineettisiin parametreihin ei havaittu seuraavien kovariaattien osalta, joita arvioitiin 30 potilaalla vaiheen III VISION-osatutkimuksessa: ikä (mediaani: 67 vuotta; vaihteluväli 52–80 vuotta) ja kehon paino (mediaani: 88,8 kg; vaihteluväli 63,8–143,0 kg).
Munuaisten vajaatoiminta
Lievää munuaisten vajaatoimintaa sairastavilla potilailla lutetium(177Lu)vipivotiditetraksetaanialtistus (AUC) oli 20 % suurempi verrattuna normaaliin munuaistoimintaan. Munuaisdosimetrian puoliintumisaika oli lievää munuaisten vajaatoimintaa sairastavilla potilailla pidempi (51 tuntia) verrattuna normaaliin munuaistoimintaan (37 tuntia). Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla voi olla suurempi riksi haittavaikutuksille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Farmakokineettisiä tietoja ei ole saatavilla potilaille, joilla on keskivaikea tai vaikea munuaisten vajaatoiminta (lähtötilanteen kreatiniinipuhdistuma < 50 ml/min) tai loppuvaiheen munuaissairaus.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta ja kerta-altistuksen aiheuttamaa toksisuutta koskeneissa tutkimuksissa ei havaittu toksikologisia vaikutuksia rotilla ja minisioilla annettaessa niille ei-radioaktiivista formulaatiota, joka sisälsi vipivotiditetraksetaania ja lutetium(175Lu)vipivotiditetraksetaania, eikä toistuvan altistuksen aiheuttamaa toksisuutta koskeneissa tutkimuksissa rotilla annettaessa niille vipivotiditetraksetaania.
Karsinogeenisuus ja mutageenisuus
Lutetium(177Lu)vipivotiditetraksetaania koskevia mutageenisuustutkimuksia ja pitkän aikavälin karsinogeenisuustutkimuksia ei ole tehty; säteily kuitenkin on karsinogeenista ja mutageenista.
Farmaseuttiset tiedot
Apuaineet
Etikkahappo
Natriumasetaatti
Gentisiinihappo
Natriumaskorbaatti
Dietyleenitriamiinipentaetikkahappo
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdissa Annostus ja antotapa ja 12.
Kestoaika
120 tuntia (5 vrk) kalibrointiajankohdasta.
Säilytys
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa ionisoivalta säteilyltä suojaamiseksi (lyijysuoja).
Radiofarmaseuttiset valmisteet on säilytettävä radioaktiivisia aineita koskevien kansallisten määräysten mukaisesti.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
PLUVICTO injektio-/infuusioneste, liuos
1000 MBq/ml (L:ei) 1 kpl (24015,60 €)
PF-selosteen tieto
Kirkas, väritön, tyypin I lasista valmistettu injektiopullo, joka on suljettu bromobutyylikumitulpalla ja alumiinisinetillä.
Yksi injektiopullo sisältää liuosta 7,5–12,5 ml:n tilavuuden, mikä vastaa radioaktiivisuutta 7 400 MBq ± 10 % valmisteen antoajankohtana.
Injektiopullo on suljettu suojaavaan lyijysäiliöön.
Valmisteen kuvaus:
Kirkas, väritön tai hiukan kellertävä liuos, pH: 4,5–7,0.
Käyttö- ja käsittelyohjeet
Yleinen varoitus
Vain valtuutetut henkilöt saavat vastaanottaa, käyttää ja antaa radiofarmaseuttisia valmisteita, ja käsittelyn on tapahduttava tähän tarkoitukseen varatuissa kliinisissä tiloissa. Valmisteiden vastaanotossa, säilytyksessä, käytössä, kuljetuksessa ja hävittämisessä on noudatettava toimivaltaisen viranomaisorganisaation antamia määräyksiä ja/tai asianmukaisia lupia.
Radiofarmaseuttisten valmisteiden valmistuksessa on huomioitava sekä säteilyturvallisuus että farmaseuttiset laatuvaatimukset. Asianmukaisia aseptisia varotoimia on noudatettava.
Ks. kohdasta 12 ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Jos lyijysuojan tai injektiopullon eheys vaarantuu missä tahansa vaiheessa lääkevalmisteen valmistelua, valmistetta ei saa käyttää.
Valmistetta annettaessa on minimoitava lääkevalmisteen kontaminaatioriski ja antajaan kohdistuva säteilyriski. Riittävän säteilysuojauksen käyttö on pakollista.
Radiofarmaseuttisten valmisteiden anto voi aiheuttaa ulkopuolisille henkilöille kontaminaatioriskin ulkoisen säteilyn tai eriteroiskeiden (esim. virtsa, oksennus) vuoksi. Tämän vuoksi on noudatettava kansallisten määräysten mukaisia säteilysuojaustoimenpiteitä.
Tämä lääkevalmiste todennäköisesti aiheuttaa suurimmalle osalle potilaista suhteellisen suuren säteilyannoksen. Pluvicto-valmisteen anto voi aiheuttaa merkittävän ympäristöriskin. Riski voi koskea hoitoa saavan potilaan lähipiiriä tai väestöä yleisesti riippuen potilaalle annetusta aktiivisuudesta. Potilaasta eliminoituvaa aktiivisuutta vastaan on suojauduttava kansallisen määräyksen mukaisin varotoimin kontaminaation välttämiseksi.
Lutetium-177 Pluvicto-valmistetta varten voidaan valmistaa käyttämällä kahta eri vakaan nuklidin lähdettä (lutetium-176 tai ytterbium-176). Jos Lutetium-177 Pluvicto-valmistetta varten valmistetaan käyttämällä vakaata lutetium-176-isotooppia (”kantoaine lisätty”), on valmisteen hävittämiseen kiinnitettävä erityistä huomiota sen sisältämän pitkäikäisen (puoliintumisaika 160,4 vrk) metastabiilin lutetium-177 (177mLu) epäpuhtauden vuoksi. Lutetium-177 Pluvicto-valmistetta varten valmistetaan käyttämällä ytterbium-176:ta (”ei kantoainetta”), ellei erävapautustodistuksessa (batch release certificate) toisin mainita. Asianmukaisen jätteenkäsittelyn varmistamiseksi käyttäjän on ennen Pluvicto-valmisteen käyttöä tutustuttava mukana toimitettavaan erävapautustodistukseen.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
PLUVICTO injektio-/infuusioneste, liuos
1000 MBq/ml 1 kpl
- Ei korvausta.
ATC-koodi
V10XX05
Valmisteyhteenvedon muuttamispäivämäärä
17.02.2026
Dosimetria
Tautiprosessin aikaansaamat patofysiologiset muutokset voivat merkittävästi vaikuttaa eri elinten säteilyannoksiin (mukaan lukien elimet, joihin hoito ei kohdistu). Tämä on otettava huomioon hyödynnettäessä oheisia tietoja.
Lutetium(177Lu)vipivotiditetraksetaania koskevaa dosimetriatietoa kerättiin 29 potilaalta vaiheen III VISION-osatutkimuksessa, jotta voitaisiin laskea eri elimissä ja koko elimistössä esiintyvän säteilyn dosimetriaa. Eri elimiin absorboituvan arvioidun annoksen keskiarvo ja keskihajonta Pluvicto-hoitoa saavilla aikuispotilailla esitetään taulukossa 4. Suurimmat elimiin absorboituvat annokset esiintyvät kyynelrauhasissa ja sylkirauhasissa.
Lutetium-177:n enimmäispenetraatio kudokseen on noin 2 mm ja keskipenetraatio 0,67 mm.
Taulukko 4 Arvioidut absorboituvat Pluvicto-annokseta VISION-osatutkimuksessa
| Absorboituva annos aktiivisuusyksikköä kohti (mGy/MBq) (N = 29) | Laskennallinen absorboituva annos annettaessa 7 400 MBq (Gy) | Laskennallinen absorboituva annos annettaessa 6 x 7 400 MBq (kumulatiivinen aktiivisuus 44 400 MBq) (Gy) | ||||
| Elin | Keskiarvo | Keskihajonta | Keskiarvo | Keskihajonta | Keskiarvo | Keskihajonta |
| Lisämunuaiset | 0,033 | 0,026 | 0,25 | 0,19 | 1,5 | 1,1 |
| Aivot | 0,0069 | 0,0051 | 0,051 | 0,038 | 0,30 | 0,23 |
| Silmät | 0,022 | 0,025 | 0,16 | 0,18 | 0,98 | 1,1 |
| Sappirakon seinämä | 0,028 | 0,026 | 0,21 | 0,19 | 1,3 | 1,2 |
| Sydämen seinämä | 0,17 | 0,12 | 1,3 | 0,88 | 7,8 | 5,3 |
| Munuaiset | 0,42 | 0,19 | 3,1 | 1,4 | 19 | 8,4 |
| Kyynelrauhaset | 2,1 | 0,48 | 15 | 3,6 | 92 | 21 |
| Vasemmanpuoleinen koolon | 0,58 | 0,14 | 4,3 | 1,0 | 26 | 6,1 |
| Maksa | 0,093 | 0,050 | 0,69 | 0,37 | 4,1 | 2,2 |
| Keuhkot | 0,11 | 0,11 | 0,78 | 0,83 | 4,7 | 5,0 |
| Ruokatorvi | 0,025 | 0,026 | 0,18 | 0,19 | 1,1 | 1,2 |
| Osteogeeniset solut | 0,036 | 0,029 | 0,26 | 0,21 | 1,6 | 1,3 |
| Haima | 0,027 | 0,026 | 0,20 | 0,19 | 1,2 | 1,2 |
| Eturauhanen | 0,027 | 0,026 | 0,20 | 0,19 | 1,2 | 1,2 |
| Peräsuoli | 0,56 | 0,14 | 4,1 | 1,1 | 25 | 6,3 |
| Punainen luuydin | 0,035 | 0,020 | 0,26 | 0,15 | 1,5 | 0,91 |
| Oikeanpuoleinen koolon | 0,32 | 0,079 | 2,4 | 0,58 | 14 | 3,5 |
| Sylkirauhaset | 0,63 | 0,37 | 4,6 | 2,7 | 28 | 16 |
| Ohutsuoli | 0,071 | 0,031 | 0,52 | 0,23 | 3,1 | 1,4 |
| Perna | 0,061 | 0,028 | 0,45 | 0,21 | 2,7 | 1,2 |
| Mahalaukun seinämä | 0,025 | 0,026 | 0,19 | 0,19 | 1,1 | 1,2 |
| Kivekset | 0,023 | 0,025 | 0,17 | 0,19 | 1,0 | 1,1 |
| Kateenkorva | 0,024 | 0,026 | 0,18 | 0,19 | 1,1 | 1,2 |
| Kilpirauhanen | 0,26 | 0,37 | 1,9 | 2,8 | 11 | 17 |
| Koko elimistö | 0,037 | 0,027 | 0,28 | 0,20 | 1,7 | 1,2 |
| Virtsarakon seinämä | 0,32 | 0,025 | 2,4 | 0,19 | 14 | 1,1 |
| Efektiivinen annosb | 0,12 mSv/MBq | 0,043 mSv/MBq | 0,89 Sv | 0,315 Sv | 5,3 Sv | 1,9 Sv |
a Arviot absorboituvista annoksista johdettiin OLINDA v2.4 -ohjelmistoa käyttäen. Arvot on laskettu täydellä tarkkuudella dosimetria-arvioiden perusteella ja pyöristetty asianmukaisesti. b Johdettu ICRP Publication 103 -julkaisun mukaisesti. | ||||||
Radiofarmaseuttisten valmisteiden valmistusohjeet
Asianmukaisen jätteenkäsittelyn varmistamiseksi käyttäjän on ennen Pluvicto-valmisteen käyttöä tutustuttava mukana toimitettavaan erävapautustodistukseen (ks. kohta Käyttö- ja käsittelyohjeet).
Valmiste vedetään injektiopullosta ruiskuun aseptisesti. Injektiopullon saa avata vasta kun tulppa on desinfioitu. Liuos vedetään injektiopullosta tulpan läpi kerta-annosruiskulla, johon on liitetty sopiva suojus ja kertakäyttöinen, steriili neula. Myös hyväksyttyä automaattista antolaitetta voidaan käyttää.
Valmisteluohjeet
- Käytä aseptista tekniikkaa ja säteilysuojausta Pluvicto-valmisteen käsittelyssä ja annossa. Tarvittaessa voidaan käyttää pihtejä säteilyaltistuksen minimoimiseksi.
- Tarkasta valmiste silmämääräisesti suojaavan lasin takaa hiukkasten ja värimuutosten varalta ennen antoa. Hävitä injektiopullo jos hiukkasia ja/tai värimuutoksia esiintyy.
- Älä injisoi Pluvicto-liuosta suoraan toiseen laskimonsisäisesti annettavaan liuokseen.
- Vahvista potilaalle annettava radioaktiivisuusmäärä asianmukaisesti kalibroidulla annoskalibraattorilla ennen Pluvicto-valmisteen jokaista antoa ja jokaisen annon jälkeen.
Antotavat laskimoon
Ohjeet antoon ruiskumenetelmällä
- Vedä tarvittavan radioaktiivisuuden mukainen tilavuus Pluvicto-liuosta kertakäyttöruiskuun, jossa on suojus ja johon on liitetty 9 cm pitkä, 18 G steriili kertakäyttöneula (pitkä neula). Liuoksen ruiskuun vetämisen helpottamiseksi voidaan käyttää 2,5 cm 20 G suodatinneulaa (lyhyt ilmausneula) paineistetun injektiopullon vastuksen vähentämiseksi. Varmista, että lyhyt neula ei kosketa injektiopullossa olevaa Pluvicto-liuosta.
- Jos käytät ruiskupumppua, aseta ruisku suojattuun pumppuun ja aseta kolmitiehana ruiskun ja Pluvicton antoon käytettävän steriilillä 9 mg/ml (0,9 %) NaCl-injektionesteellä esitäytetyn laskimokanyylin väliin.
- Anna Pluvicto potilaalle hitaana injektiona laskimoon noin 1–10 minuutin kuluessa (joko ruiskupumpulla tai manuaalisesti ilman ruiskupumppua) laskimokanyylin kautta, joka on esitäytetty steriilillä 9 mg/ml (0,9 %) NaCl-injektionesteellä ja jota käytetään ainoastaan Pluvicto-valmisteen antoon potilaalle.
- Kun tarvittava Pluvicto‑radioaktiivisuusannos on annettu, pysäytä ruiskupumppu ja muuta sitten kolmitiehanan asentoa ja huuhtele ruisku 25 ml:lla steriiliä 9 mg/ml (0,9 %) NaCl-injektionestettä. Käynnistä ruiskupumppu uudelleen.
- Kun ruiskun huuhtelu on päättynyt, huuhtele laskimokanyyli antamalla sen kautta potilaalle vähintään 10 ml steriiliä 9 mg/ml (0,9 %) NaCl-injektionestettä.
Ohjeet antoon painovoimamenetelmällä
- Työnnä Pluvicto-injektiopulloon 2,5 cm, 20 G neula (lyhyt neula) ja yhdistä injektiopullo kanyylin kautta steriiliin 500 ml:n 9 mg/ml (0,9 %) NaCl-injektionesteeseen (käytetään Pluvicto-liuoksen kuljettamiseen infuusion aikana). Varmista, ettei lyhyt neula kosketa injektiopullossa olevaa Pluvicto-liuosta. Älä liitä lyhyttä neulaa suoraan potilaaseen. Älä anna steriilin 9 mg/ml (0,9 %) NaCl-injektionesteen virrata Pluvicto-injektiopulloon ennen Pluvicto-infuusion aloitusta. Älä myöskään injisoi Pluvicto-liuosta suoraan steriiliin 9 mg/ml (0,9 %) NaCl-injektionesteeseen.
- Työnnä Pluvicto-injektiopulloon toinen, 9 cm, 18 G neula (pitkä neula). Varmista, että pitkä neula on tiiviisti kiinni Pluvicto-injektiopullon pohjassa koko infuusion ajan. Yhdistä pitkä neula potilaan laskimokanyyliin, joka on esitäytetty steriilillä 9 mg/ml (0,9 %) NaCl-injektionesteellä ja jota käytetään ainoastaan Pluvicto-infuusion antoon potilaalle.
- Käytä letkunpuristinta tai infuusiopumppua säätelemään steriilin 9 mg/ml (0,9 %) NaCl-injektionesteen virtausta lyhyen neulan kautta Pluvicto-injektiopulloon (steriili 9 mg/ml [0,9 %] NaCl-injektioneste virtaa injektiopulloon lyhyen neulan kautta ja kuljettaa injektiopullossa olevan Pluvicto-liuoksen potilaalle laskimokanyyliin yhdistetyn pitkän neulan kautta noin 30 minuutissa).
- Varmista, että Pluvicto-injektiopullossa olevan liuoksen määrä pysyy samana koko infuusion ajan.
- Kun radioaktiivisuuden taso on pysynyt vakaana vähintään viiden minuutin ajan, irrota pitkään neulaan yhdistetty letku injektiopullosta ja sulje steriilin 9 mg/ml (0,9 %) NaCl-injektionesteen letku puristimella.
- Huuhtele laskimokanyyli infuusion jälkeen antamalla sen kautta potilaalle vähintään 10 ml steriiliä 9 mg/ml (0,9 %) NaCl-injektionestettä.
Ohjeet antoon peristalttisen pumpun menetelmällä
- Työnnä Pluvicto-injektiopulloon 2,5 cm, 20 G suodatinneula (lyhyt ilmausneula). Varmista, ettei lyhyt neula kosketa injektiopullossa olevaa Pluvicto-liuosta. Älä liitä lyhyttä neulaa suoraan potilaaseen äläkä peristalttiseen pumppuun.
- Työnnä Pluvicto-injektiopulloon toinen, 9 cm, 18 G neula (pitkä neula). Varmista, että pitkä neula on tiiviisti kiinni Pluvicto-injektiopullon pohjassa koko infuusion ajan. Yhdistä pitkä neula ja steriili 9 mg/ml (0,9 %) NaCl-injektioneste asianmukaisilla letkuilla kolmitiehanaan.
- Yhdistä kolmitiehanan porttiin peristalttisen pumpun sisäänmenopuoleen liitetty letku pumpun valmistajan ohjeiden mukaisesti.
- Esitäytä linja avaamalla kolmitiehana ja pumppaamalla Pluvicto-liuosta letkuun, kunnes se saavuttaa kolmitiehanan ulosmenoaukon.
- Esitäytä potilaaseen yhdistettävä laskimokanyylijärjestelmä avaamalla kolmitiehana steriilin 9 mg/ml (0,9 %) NaCl-injektionesteen suuntaan ja pumppaamalla järjestelmään steriiliä 9 mg/ml (0,9 %) NaCl-injektionestettä, kunnes se poistuu järjestelmän ulosmenoaukosta.
- Yhdistä esitäytetty laskimokanyylijärjestelmä potilaaseen ja käännä kolmitiehana auki peristalttisen pumpun Pluvicto-liuoksen suuntaan.
- Infusoi haluttuun radioaktiivisuuteen tarvittava tilavuus Pluvicto-liuosta suunnilleen nopeudella 25 ml/h.
- Kun haluttu radioaktiivisuusannos on annettu, pysäytä peristalttinen pumppu ja käännä kolmitiehanaa niin, että se on auki peristalttisen pumpun steriilin 9 mg/ml (0,9 %) NaCl-injektionesteen suuntaan. Käynnistä peristalttinen pumppu uudelleen ja huuhtele laskimokanyyli antamalla sen kautta potilaalle vähintään 10 ml steriiliä 9 mg/ml (0,9 %) NaCl-injektionestettä.
Laadunvalvonta
Liuos on tarkastettava silmämääräisesti ennen käyttöä vahingoittumisen ja kontaminaation varalta. Liuoksen saa käyttää vain, jos se on kirkas eikä siinä ole näkyviä hiukkasia. Liuoksen silmämääräisen tarkastuksen on tapahduttava säteilyltä suojaavan lasin takana. Injektiopulloa ei saa avata.
Jos lyijysuojan tai injektiopullon eheys vaarantuu missä tahansa vaiheessa lääkevalmisteen valmistelua, valmistetta ei saa käyttää.
Injektiopullon radioaktiivisuusmäärä on mitattava asianmukaisella radioaktiivisuuden kalibraatiojärjestelmällä ennen valmisteen antoa, jotta varmistetaan, että potilaalle annettava radioaktiivisuuden todellinen määrä antohetkellä vastaa suunniteltua määrää.
Lisätietoa tästä lääkevalmisteesta on Euroopan lääkeviraston verkkosivulla https://www.ema.europa.eu.
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com