
SARCLISA infuusiokonsentraatti, liuosta varten 20 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Vaikuttavat aineet ja niiden määrät
Yksi millilitra infuusiokonsentraattia liuosta varten sisältää 20 mg isatuksimabia.
Yksi injektiopullo sisältää 100 mg isatuksimabia 5 ml:ssa konsentraattia (100 mg/5 ml).
Yksi injektiopullo sisältää 500 mg isatuksimabia 25 ml:ssa konsentraattia (500 mg / 25 ml).
Isatuksimabi on nisäkässolulinjassa (kiinanhamsterin munasarjasoluissa, CHO) tuotettu immunoglobuliini G1 ‑luokan (IgG1) monoklonaalinen vasta-aine.
Apuaine, jonka vaikutus tunnetaan
Yksi injektiopullo, jossa on 5 ml isatuksimabi-infuusiokonsentraattia liuosta varten, sisältää 1 mg polysorbaatti 80:tä.
Yksi injektiopullo, jossa on 25 ml isatuksimabi-infuusiokonsentraattia liuosta varten, sisältää 5 mg polysorbaatti 80:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten
Kliiniset tiedot
Käyttöaiheet
Isatuksimabi on tarkoitettu
- yhdistelmänä pomalidomidin ja deksametasonin kanssa sellaisten relapsoitunutta ja refraktorista multippelia myeloomaa sairastavien aikuispotilaiden hoitoon, jotka ovat saaneet vähintään kahta aiempaa, mukaan lukien lenalidomidia ja proteasomin estäjää sisältänyttä hoitoa, ja joilla sairaus on edennyt viimeisimmän hoidon aikana
- yhdistelmänä karfiltsomibin ja deksametasonin kanssa sellaisten multippelia myeloomaa sairastavien aikuispotilaiden hoitoon, jotka ovat saaneet vähintään yhtä aiempaa hoitoa (ks. kohta Farmakodynamiikka).
- yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa sellaisten aikuispotilaiden hoitoon, joilla on äskettäin todettu multippeli myelooma ja joille autologinen kantasolusiirto ei sovellu.
- yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa sellaisten aikuispotilaiden induktiohoitoon, joilla on äskettäin todettu multippeli myelooma ja joille autologinen kantasolusiirto soveltuu.
Ehto
Valmistetta saavat määrätä ja antaa vain sellaiset lääkärit ja terveydenhuollon ammattilaiset, jotka ovat perehtyneet antineoplastisten valmisteiden käyttöön. Valmistetta saa antaa vain sairaalassa, jossa on asianmukaiset elvytyslaitteet.
Annostus ja antotapa
Isatuksimabihoidon saa määrätä vain syöpätautien hoitoon perehtynyt erikoislääkäri. Isatuksimabin antaa laskimoon terveydenhuollon ammattilainen hoitopaikassa, jossa on käytettävissä elvytysvälineistö.
Esilääkitys
Infuusioreaktion ehkäiseminen
Infuusioreaktioiden riskin ja vaikeusasteen pienentämiseksi ennen isatuksimabi-infuusiota on annettava esilääkityksenä seuraavia lääkevalmisteita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet):
-
deksametasoni 40 mg suun kautta tai laskimoon (tai 20 mg suun kautta tai laskimoon, jos potilas on ≥ 75-vuotias): kun annetaan yhdistelmänä isatuksimabin ja pomalidomidin kanssa;
deksametasoni 20 mg (laskimoon isatuksimabi- ja/tai karfiltsomibi-infuusioiden antopäivinä ja suun kautta muina päivinä): kun annetaan yhdistelmänä isatuksimabin ja karfiltsomibin kanssa
deksametasoni 20 mg (laskimoon isatuksimabi‑infuusion antopäivinä ja suun kautta muina päivinä): kun annetaan yhdistelmänä isatuksimabin, bortetsomibin ja lenalidomidin kanssa - montelukasti 10 mg suun kautta (tai vastaava lääkitys) ainakin hoitosyklissä 1
- parasetamoli 650 – 1 000 mg suun kautta (tai vastaava lääkitys)
- H2‑antagonistit (ranitidiini 50 mg laskimoon tai vastaava lääkitys [kuten simetidiini]) tai suun kautta annettavat protonipumpun estäjät (kuten omepratsoli tai esomepratsoli).
- difenhydramiini 25–50 mg laskimoon tai suun kautta (tai vastaava lääkitys [kuten setiritsiini, prometatsiini tai dekskloorifeniramiini]). Ainakin ensimmäiset neljä infuusiota on suositeltavaa antaa laskimoon.
Edellä mainittu suositeltu deksametasoniannos (suun kautta tai laskimoon) vastaa kokonaisannosta, joka annetaan vain kerran infuusiota edeltävänä esilääkityksen ja perushoidon osana ennen isatuksimabin ja pomalidomidin, ennen isatuksimabin ja karfiltsomibin ja ennen isatuksimabin, bortetsomibin ja lenalidomidin antamista.
Suositellut esilääkkeet on annettava 15–60 minuuttia ennen isatuksimabi-infuusion aloittamista. Jos potilaalla ei ilmene infuusioreaktiota isatuksimabin ensimmäisten neljän antokerran yhteydessä, esilääkityksen tarvetta jatkossa voidaan arvioida uudelleen.
Neutropenian hoito
Kantasoluryhmiä stimuloivien kasvutekijöiden (kuten G-CSF:n) käyttöä on harkittava neutropeniariskin pienentämiseksi. Jos potilaalla ilmenee vaikeusasteen 3 tai 4 neutropenia tai kuumeinen neutropenia ja/tai neutropeeninen infektio, isatuksimabin antamista on lykättävä tai hoito on lopetettava, kunnes potilas on toipunut (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Infektioiden ehkäiseminen
Hoidon aikana on harkittava profylaksiaa (kuten vyöruusun profylaksiaa) bakteeri- ja viruslääkkeillä hoitosuositusten mukaisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Suositeltu annos Sarclisa 20 mg/ml infuusiokonsentraatti, liuosta varten -valmistetta on 10 mg/kg, joka annetaan infuusiona laskimoon yhdistelmänä pomalidomidin ja deksametasonin kanssa (Isa Pd), yhdistelmänä karfiltsomibin ja deksametasonin kanssa (Isa Kd) tai yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa (Isa VRd).
Potilaat voivat aloittaa hoidon laskimoon tai ihon alle annettavalla isatuksimabilla. Laskimoon annettavaa isatuksimabia saavat potilaat voivat siirtyä käyttämään ihon alle annettavaa isatuksimabia milloin tahansa hoidon aikana hoitosyklin 1 päättymisen jälkeen.
Laskimoon annettavan isatuksimabin annostusohjelmat on esitetty taulukoissa 1, 2 ja 3:
Taulukko 1 – Annostusohjelma yhdistelmänä pomalidomidin ja deksametasonin kanssa tai yhdistelmänä karfiltsomibin ja deksametasonin kanssa
| Hoitosyklit | Annostusohjelma |
| Hoitosykli 1 (28 päivän hoitosykli) | 10 mg/kg laskimoon kerran viikossa; päivät 1, 8, 15 ja 22 |
| Hoitosykli 2 ja sen jälkeen (28 päivän hoitosyklit) | 10 mg/kg laskimoon kahden viikon välein; päivät 1 ja 15 |
Jokainen hoitosykli koostuu 28 päivän jaksosta. Hoito toistetaan, kunnes tauti etenee tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä.
Taulukko 2 – Annostusohjelma yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa potilaille, joilla on äskettäin todettu multippeli myelooma ja joille autologinen kantasolusiirto ei sovellu
| Hoitosyklit | Annostusohjelma |
| Hoitosykli 1 (42 päivän hoitosykli) | 10 mg/kg laskimoon; päivät 1, 8, 15, 22 ja 29 |
| Hoitosyklit 2–4 (42 päivän hoitosyklit) | 10 mg/kg laskimoon kahden viikon välein; päivät 1, 15 ja 29 |
| Hoitosyklit 5–17 (28 päivän hoitosyklit) | 10 mg/kg laskimoon kahden viikon välein; päivät 1 ja 15 |
| Hoitosykli 18 ja sen jälkeen (28 päivän hoitosyklit) | 10 mg/kg laskimoon neljän viikon välein; päivä 1 |
Hoitosykleissä 1–4 jokainen hoitosykli koostuu 42 päivän jaksosta ja hoitosyklistä 5 alkaen 28 päivän jaksosta. Hoito toistetaan, kunnes tauti etenee tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä.
Taulukko 3: Annostusohjelma yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa potilaille, joilla on äskettäin todettu multippeli myelooma ja joille autologinen kantasolusiirto soveltuu
| Hoitosyklit | Annostusohjelma |
| Induktiohoito | |
| Hoitosykli 1 (42 päivän hoitosykli) | 10 mg/kg laskimoon; päivät 1, 8, 15, 22 ja 29 |
| Hoitosyklit 2–3 (42 päivän hoitosyklit) | 10 mg/kg laskimoon kahden viikon välein); päivät 1, 15 ja 29 |
| Lopetetaan ennen intensiivihoitoa (suuriannoksinen solunsalpaajahoito ja autologinen kantasolusiirto), jonka jälkeen annetaan tavanomaista ylläpitohoitoa | |
Jokainen hoitosykli koostuu 42 päivän jaksosta.
Väliin jäänyt annos
Annostusohjelmaa täytyy noudattaa huolellisesti. Jos suunniteltu isatuksimabiannos jää antamatta, annos on annettava mahdollisimman pian ja hoito-ohjelmaa on säädettävä sen mukaisesti niin, että hoitojen väli säilyy ennallaan.
Annoksen muuttaminen
Isatuksimabiannoksen pienentämistä ei suositella.
Annostusta on muutettava, jos potilaalla ilmenee infuusioreaktioita (ks. "Antotapa" jäljempänä) tai vaikeusasteen 3 tai 4 neutropenia tai kuumeinen neutropenia ja/tai neutropeeninen infektio (ks. ”Neutropenian hoito” edellä).
Isatuksimabin kanssa annettavien muiden lääkevalmisteiden voimassa oleviin valmisteyhteenvetojen suositukset tulee huomioida.
Erityisryhmät
Iäkkäät potilaat
Annoksen muuttamista iäkkäillä potilailla ei suositella (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset ja Farmakokinetiikka).
Munuaisten vajaatoimintaa sairastavat potilaat
Annoksen muuttamista ei suositella lievää munuaisten vajaatoimintaa (GFR ≥ 60 – < 90 ml/min/1,73 m2), kohtalaista munuaisten vajaatoimintaa tai vaikeaa munuaisten vajaatoimintaa (GFR < 30 ml/min/1,73 m2), mukaan lukien loppuvaiheen munuaissairautta (GFR < 15 ml/min/1,73 m2), sairastavilla potilailla (ks. kohta Farmakokinetiikka).
Maksan vajaatoimintaa sairastavat potilaat
Annoksen muuttamista ei suositella lievää maksan vajaatoimintaa (kokonaisbilirubiiniarvo > 1‑kertainen – 1,5-kertainen viitealueen ylärajaan nähden tai aspartaattiaminotransferaasiarvo [ASAT] viitealueen ylärajaa suurempi) sairastavilla potilailla. Kohtalaista maksan vajaatoimintaa (kokonaisbilirubiiniarvo > 1,5-kertainen – 3-kertainen viitealueen ylärajaan nähden ja mikä tahansa ASAT-arvo) tai vaikeaa maksan vajaatoimintaa (kokonaisbilirubiiniarvo > 3-kertainen viitealueen ylärajaan nähden ja mikä tahansa ASAT-arvo) sairastavista potilaista on vain vähän tietoja (ks. kohta Farmakokinetiikka), mutta ei ole näyttöä, joka viittaisi siihen, että annoksen muuttaminen tällaisilla potilailla olisi tarpeen.
Pediatriset potilaat
Hyväksyttyjen käyttöaiheiden ulkopuolella laskimoon annettavaa isatuksimabia on tutkittu 28 päivän – alle 18 vuoden ikäisillä lapsilla, joilla on relapsoitunut tai refraktorinen akuutti lymfoblastileukemia tai akuutti myelooinen leukemia. Hoidon tehoa ei kuitenkaan ole varmistettu. Saatavissa oleva tieto on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka.
Antotapa
Sarclisa 20 mg/ml infuusiokonsentraatti, liuosta varten annetaan laskimoon. Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Infuusionopeudet
Laimentamisen jälkeen isatuksimabi-infuusio annetaan laskimoon infuusionopeudella, joka on esitetty seuraavassa taulukossa 4 (ks. kohta Farmakodynamiikka). Infuusionopeuden asteittaista suurentamista voidaan harkita ainoastaan siinä tapauksessa, että infuusioreaktioita ei ilmene (ks. kohta Haittavaikutukset).
Taulukko 4 – Isatuksimabin antamisen infuusionopeudet
| Laimennustilavuus | Aloitusnopeus | Ei infuusioreaktioita | Nopeuden suurentaminen | Enimmäisnopeus | |
| Ensimmäinen infuusio | 250 ml | 25 ml/tunti | 60 minuutin aikana | 25 ml/tunti 30 minuutin välein | 150 ml/tunti |
| Toinen infuusio | 250 ml | 50 ml/tunti | 30 minuutin aikana | 50 ml/tunti 30 minuutin ajan, sitten nopeutta suurennetaan 100 ml/tunti | 200 ml/tunti |
| Seuraavat infuusiot | 250 ml | 200 ml/tunti | 200 ml/tunti |
Annostelua on muutettava, jos potilaalla ilmenee infuusioreaktioita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Jos potilaan tila edellyttää hoitotoimenpiteitä (vaikeusasteen 2 eli kohtalainen infuusioreaktio), infuusion tilapäistä keskeyttämistä on harkittava ja potilaalle voidaan antaa lisäksi oireita lievittäviä lääkevalmisteita. Kun oireet ovat lievittyneet vaikeusasteeseen ≤ 1 (lievä), isatuksimabi-infuusiota voidaan jatkaa niin, että infuusionopeus on puolet aloitusnopeudesta ja potilasta tarkkaillaan huolellisesti ja hän saa tukihoitoa tarpeen mukaan. Jos oireet eivät ole uusiutuneet 30 minuutin kuluttua, infuusionopeus voidaan suurentaa takaisin aloitusnopeuteen ja sen jälkeen sitä voidaan asteittain suurentaa taulukossa 3 esitetyllä tavalla.
- Jos oireet eivät häviä nopeasti tai lievity vaikeusasteeseen ≤ 1 isatuksimabi-infuusion keskeyttämisen jälkeen tai jos ne pitkittyvät tai pahenevat, vaikka niitä on hoidettu asianmukaisilla lääkevalmisteilla, tai jos ne edellyttävät sairaalahoitoa tai ovat hengenvaarallisia, Sarclisa-hoito on lopetettava pysyvästi ja potilaalle on annettava muuta tukihoitoa tarpeen mukaan.
- Jos potilaalla ilmenee vaikeusasteen ≥ 3 yliherkkyysreaktio tai infuusioreaktio, isatuksimabihoito on lopetettava pysyvästi.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infuusioreaktiot
Infuusioreaktioita, jotka ovat olleet enimmäkseen lieviä tai kohtalaisia, on todettu 38,2 %:lla isatuksimabia saaneista potilaista ICARIA‑MM-tutkimuksessa, 45,8 %:lla Isa‑Kd-hoitoa saaneista potilaista IKEMA-tutkimuksessa, 24,0 %:lla Isa‑VRd-hoitoa saaneista potilaista IMROZ-tutkimuksessa ja 12,7 %:lla induktiojakson aikana Isa-VRd-hoitoa saaneista potilaista GMMG HD7-tutkimuksessa (ks. kohta Haittavaikutukset). ICARIA‑MM-tutkimuksessa kaikki infuusioreaktiot alkoivat ensimmäisen isatuksimabi-infuusion aikana ja 98 %:ssa infuusioista ne hävisivät samana päivänä. Yleisimpiä infuusioreaktion oireita olivat hengenahdistus, yskä, vilunväristykset ja pahoinvointi. Yleisimpiä vaikea-asteisia oireita olivat kohonnut verenpaine, hengenahdistus ja bronkospasmi. IKEMA-tutkimuksessa 99,2 % infuusioreaktioista ilmeni infuusiopäivänä. Niistä Isa‑Kd-hoitoa saaneista potilaista, joilla ilmeni infuusioreaktio, 94,4 %:lle se kehittyi ensimmäisen hoitosyklin aikana. Kaikki infuusioreaktiot hävisivät. Yleisimpiä infuusioreaktion oireita olivat yskä, hengenahdistus, nenän tukkoisuus, oksentelu ja pahoinvointi. Yleisimpiä vaikea-asteisia oireita olivat kohonnut verenpaine ja hengenahdistus. IMROZ‑tutkimuksessa infuusioreaktiot alkoivat kaikilla potilailla infuusiopäivänä, useimmiten ensimmäisen isatuksimabi‑infuusion aikana, ja 97,3 %:lla potilaista ne hävisivät samana päivänä. Kaikki infuusioreaktiot hävisivät. Yleisimpiä infuusioreaktion oireita olivat hengenahdistus ja vilunväristykset. Yleisin vaikea‑asteinen oire oli kohonnut verenpaine. GMMG-HD7-tutkimuksessa niistä induktiojakson aikana Isa VRd-hoitoa saaneista potilaista, joilla ilmeni infuusioreaktio, 88,1 %:lle reaktio kehittyi ensimmäisen infuusion yhteydessä ja 21,4 %:lle myöhempien infuusioiden yhteydessä. Kaikki infuusioreaktiot hävisivät. (Ks. kohta Haittavaikutukset.) Myös vakavia infuusioreaktioita, mukaan lukien vaikeat anafylaktiset reaktiot, on kuitenkin havaittu isatuksimabin antamisen jälkeen. Kuolemaan johtaneita anafylaktisia reaktioita on ilmoitettu (ks. kohta Haittavaikutukset).
Infuusioreaktioiden riskin ja vaikeusasteen pienentämiseksi potilaille tulee antaa ennen isatuksimabi-infuusiota esilääkityksenä montelukastia (ainakin hoitosyklissä 1), parasetamolia, difenhydramiinia tai vastaavaa lääkettä. Deksametasonia käytetään sekä esilääkityksenä että myeloomahoitona (ks. kohta Annostus ja antotapa). Vitaalitoiminnot on tarkistettava säännöllisesti koko isatuksimabi-infuusion ajan. Tarvittaessa isatuksimabi-infuusio on keskeytettävä ja on ryhdyttävä asianmukaisiin hoito- ja tukitoimiin (ks. kohta Annostus ja antotapa). Jos oireet eivät lievity vaikeusasteeseen ≤ 1 isatuksimabi-infuusion keskeyttämisen jälkeen, pitkittyvät tai pahenevat, vaikka niitä on hoidettu asianmukaisilla lääkevalmisteilla, edellyttävät sairaalahoitoa tai ovat hengenvaarallisia, isatuksimabihoito on lopetettava pysyvästi ja on aloitettava asianmukainen hoito. Jos potilaalla ilmenee anafylaktinen reaktio tai henkeä uhkaava (vaikeusasteen 4) infuusioreaktio, Sarclisa hoito on lopetettava pysyvästi ja on aloitettava asianmukainen hoito.
Neutropenia
Isa‑Pd-hoitoa saaneista potilaista 96,1 %:lla ilmoitettiin neutropeniaa laboratorioarvojen poikkeavuutena ja 46,7 %:lla haittavaikutuksena(1), ja vaikeusasteen 3–4 neutropeniaa ilmoitettiin laboratorioarvojen poikkeavuutena 84,9 %:lla potilaista ja haittavaikutuksena 45,4 %:lla potilaista. Neutropeenisia komplikaatioita on todettu 30,3 %:lla potilaista; 11,8 %:lla todettiin kuumeinen neutropenia ja 25,0 %:lla neutropeenisia infektioita. Isa‑Kd-hoitoa saaneista potilaista 54,8 %:lla ilmoitettiin neutropeniaa laboratorioarvojen poikkeavuutena ja 4,5 %:lla haittavaikutuksena(1), ja vaikeusasteen 3–4 neutropeniaa ilmoitettiin laboratorioarvojen poikkeavuutena 19,2 %:lla potilaista (17,5 %:lla neutropenia oli vaikeusastetta 3 ja 1,7 %:lla vaikeusastetta 4) ja haittavaikutuksena 4,0 %:lla potilaista. Neutropeenisia komplikaatioita on todettu 2,8 %:lla potilaista; 1,1 %:lla todettiin kuumeinen neutropenia ja 1,7 %:lla neutropeenisia infektioita. IMROZ-tutkimuksessa Isa‑VRd‑hoitoa saaneista potilaista 87,5 %:lla ilmoitettiin neutropeniaa laboratorioarvojen poikkeavuutena ja 30 %:lla haittavaikutuksena, ja vaikeusasteen 3–4 neutropeniaa ilmoitettiin laboratorioarvojen poikkeavuutena 54,4 %:lla potilaista (35,7 %:lla neutropenia oli vaikeusastetta 3 ja 18,6 %:lla vaikeusastetta 4) ja haittavaikutuksena 30 %:lla potilaista. Neutropeenisia komplikaatioita on todettu 12,5 %:lla potilaista; 2,3 %:lla todettiin kuumeinen neutropenia ja 10,6 %:lla neutropeeninen infektio. GMMG-HD7-tutkimuksessa induktiojakson aikana Isa-VRd-hoitoa saaneista potilaista 30,9 %:lla ilmoitettiin neutropeniaa laboratorioarvojen poikkeavuutena ja 16,1 %:lla haittavaikutuksena ja vaikeusasteen 3–4 neutropeniaa ilmoitettiin laboratorioarvojen poikkeavuutena 5,9 %:lla potilaista (3,1 %:lla neutropenia oli vaikeusastetta 3 ja 2,8 %:lla vaikeusastetta 4) ja haittavaikutuksena 16,1 %:lla potilaista (ks. kohta Haittavaikutukset).
Täydellinen verenkuva on tutkittava säännöllisesti hoidon aikana. Neutropeniapotilaita on tarkkailtava infektio-oireiden varalta. Isatuksimabiannoksen pienentämistä ei suositella. Isatuksimabiannostelun lykkäämistä ja kantasoluryhmiä stimuloivien kasvutekijöiden (kuten G-CSF:n) käyttöä on harkittava neutropeniariskin pienentämiseksi (ks. kohta Annostus ja antotapa).
(1) Hematologiset laboratoriotulokset kirjattiin haittavaikutuksiksi vain, jos ne johtivat hoidon keskeytykseen ja/tai annosmuutokseen ja/tai täyttivät vakavan haitan kriteerit.
Infektiot
Isatuksimabia saaneilla potilailla infektioiden, mukaan lukien vähintään vaikeusasteen 3 infektiot, ilmaantuvuus oli suurentunut, ja suurin osa niistä oli keuhkokuumeita, ylähengitystieinfektioita ja keuhkoputkitulehduksia (ks. kohta Haittavaikutukset). Isatuksimabia saavia potilaita on tarkkailtava huolellisesti infektio-oireiden varalta ja tarvittaessa on aloitettava asianmukainen hoito.
Hoidon aikana on harkittava profylaksiaa (kuten vyöruusun profylaksiaa) bakteeri- ja viruslääkkeillä hoitosuositusten mukaisesti (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Sekundaarimaligniteetit
ICARIA-MM-tutkimuksessa ilmoitettiin sekundaarimaligniteetteja kymmenellä (6,6 %) Isa‑Pd-hoitoa saaneella potilaalla ja kolmella (2 %) Pd‑hoitoa saaneella potilaalla, kun seuranta-ajan mediaani oli 52,44 kuukautta. Sekundaarimaligniteettina oli ihosyöpä kuudella Isa‑Pd-hoitoa saaneella potilaalla ja kolmella Pd‑hoitoa saaneella potilaalla, muu kiinteä kasvain kuin ihosyöpä kolmella Isa‑Pd-hoitoa saaneella potilaalla (yhdellä näistä potilaista oli myös ihosyöpä) sekä hematologinen maligniteetti (myelodysplastinen oireyhtymä) yhdellä Isa‑Pd-hoitoa saaneella potilaalla (ks. kohta Haittavaikutukset). Potilaat jatkoivat hoitoa uuden maligniteetin poistoleikkauksen jälkeen lukuun ottamatta kahta potilasta, jotka saivat Isa‑Pd-hoitoa. Toiselle näistä potilaista kehittyi metastaattinen melanooma ja toiselle myelodysplastinen oireyhtymä. IKEMA-tutkimuksessa ilmoitettiin sekundaarimaligniteetteja 18:lla (10,2 %) Isa‑Kd-hoitoa saaneella potilaalla ja 10:llä (8,2 %) Kd‑hoitoa saaneella potilaalla, kun seuranta-ajan mediaani oli 56,61 kuukautta. Sekundaarimaligniteettina oli ihosyöpä 13:lla (7,3 %) Isa‑Kd-hoitoa saaneella potilaalla ja neljällä (3,3 %) Kd‑hoitoa saaneella potilaalla, muu kiinteä kasvain kuin ihosyöpä seitsemällä (4,0 %) Isa‑Kd-hoitoa saaneella potilaalla ja kuudella (4,9 %) Kd‑hoitoa saaneella potilaalla ja pahanlaatuinen veritauti (akuutti myelooinen leukemia) yhdellä (0,8 %) Kd‑hoitoa saaneella potilaalla. Yhdellä (0,6 %) Isa‑Kd-ryhmän potilaalla sekundaarimaligniteetin etiologia oli tuntematon. Kahdella (1,1 %) Isa-Kd-ryhmän potilaalla ja yhdellä (0,8 %) Kd‑ryhmän potilaalla oli sekä ihosyöpä että muu kiinteä kasvain kuin ihosyöpä (ks. kohta Haittavaikutukset). Ihosyöpäpotilaat jatkoivat hoitoa ihosyövän poistoleikkauksen jälkeen. Muita kiinteitä kasvaimia kuin ihosyöpää todettiin 3 kuukauden kuluessa hoidon aloittamisesta kolmella (1,7 %) Isa‑Kd-hoitoa saaneella potilaalla ja kahdella (1,6 %) Kd‑hoitoa saaneella potilaalla. IMROZ‑tutkimuksessa sekundaarimaligniteetteja ilmoitettiin 42:lla (16,0 %) Isa‑VRd‑hoitoa saaneella potilaalla (0,041 tapahtumaa potilasvuotta kohti) ja 16:lla (8,8 %) VRd‑hoitoa saaneella potilaalla (0,026 tapahtumaa potilasvuotta kohti), kun seuranta‑ajan mediaani oli 59,73 kuukautta. Sekundaarimaligniteettina oli ihosyöpä 22:lla (8,4 %) Isa‑VRd‑hoitoa saaneella potilaalla ja seitsemällä (3,9 %) VRd‑hoitoa saaneella potilaalla, muu kiinteä kasvain kuin ihosyöpä 17:llä (6,5 %) Isa‑VRd‑hoitoa saaneella potilaalla ja seitsemällä (3,9 %) VRd‑hoitoa saaneella potilaalla ja hematologinen maligniteetti kolmella (1,1 %) Isa‑VRd‑hoitoa saaneella potilaalla ja kahdella (1,1 %) VRd‑hoitoa saaneella potilaalla. Potilaat, joille kehittyi sekundaarimaligniteettina ihosyöpä, jatkoivat hoitoa ihosyövän poistoleikkauksen jälkeen lukuun ottamatta yhtä potilasta kussakin hoitoryhmässä. Kuolemaan johtaneita sekundaarimaligniteetteja ilmoitettiin kuudella (2,3 %) Isa‑VRd‑hoitoa saaneella potilaalla (ihon neuroendokriininen karsinooma, pahanlaatuinen melanooma, ihon okasolusyöpä, keuhkon levyepiteelikarsinooma, kolorektaalisyöpä ja peräsuolen adenokarsinooma) ja kahdella (1,1 %) VRd‑hoitoa saaneella potilaalla (metastaasit peritoneumissa ja paksusuolen adenokarsinooma). GMMG-HD7-tutkimuksessa induktio-, intensiivi- ja seurantajaksojen aikana potilailla, joita ei ollut satunnaistettu toiseen kertaan, sekundaarimaligniteetteja ilmoitettiin kahdella (0,6 %) Isa-VRd-hoitoa saaneella potilaalla ja neljällä (1,2 %) VRd-hoitoa saaneella potilaalla. Sekundaarimaligniteettina oli ihosyöpä yhdellä (0,3 %) VRd-hoitoa saaneella potilaalla, muu kiinteä kasvain kuin ihosyöpä yhdellä (0,3 %) Isa-VRd-hoitoa saaneella potilaalla ja kahdella (0,6 %) VRd-hoitoa saaneella potilaalla ja hematologinen maligniteetti yhdellä (0,3 %) Isa-VRd-hoitoa saaneella potilaalla ja yhdellä (0,3 %) VRd-hoitoa saaneella potilaalla. Sekundaarimaligniteettien kokonaisilmaantuvuus isatuksimabia saaneilla potilailla oli 6,1 %. Lääkäreiden on arvioitava potilaita sekundaarimaligniteettien kehittymisen varalta IMWG-työryhmän (International Myeloma Working Group) antamien suositusten mukaan ennen hoitoa ja hoidon aikana ja aloitettava tarpeenmukainen hoito.
Tuumorilyysioireyhtymä
Isatuksimabia saaneilla potilailla on ilmoitettu tuumorilyysioireyhtymää. Potilaita on seurattava tarkasti, ja asianmukaisiin varotoimiin on ryhdyttävä.
Häiriövaikutukset serologisissa tutkimuksissa (epäsuora antiglobuliinikoe)
Isatuksimabi sitoutuu punasolujen CD38:aan, mikä saattaa johtaa virheelliseen positiiviseen tulokseen epäsuorassa antiglobuliinikokeessa (epäsuorassa Coombsin kokeessa). Tämä epäsuoraan Coombsin kokeeseen kohdistuva häiriövaikutus voi kestää ainakin 6 kuukautta viimeisen isatuksimabi-infuusion jälkeen. Punasolujen siirtoon liittyvien mahdollisten ongelmien välttämiseksi isatuksimabihoitoa saavilta potilailta on määritettävä veriryhmä ja heille on tehtävä seulontatestit ennen ensimmäistä infuusiota. Ennen isatuksimabihoidon aloittamista voidaan harkita fenotyypitystä paikallisen käytännön mukaisesti. Jos isatuksimabihoito on jo aloitettu, tästä on ilmoitettava verikeskukselle. Potilaita on tarkkailtava hemolyysin teoreettisen riskin vuoksi. Jos kiireellinen verensiirto on tarpeen, voidaan antaa ABO ja RhD -ryhmien mukaisia punasoluja ilman sopivuustutkimusta paikallisen verikeskuksen käytännön mukaisesti (ks. kohta Yhteisvaikutukset).
Häiriövaikutukset täydellisen vasteen arvioinnissa
Isatuksimabi on monoklonaalinen IgG-kappa-vasta-aine, joka saatetaan todeta sekä seerumin proteiinielektroforeesimäärityksissä (SPE) että immunofiksaatiomäärityksissä (IFE), joita käytetään endogeenisen M-proteiinin kliiniseen seurantaan (ks. kohta Yhteisvaikutukset). Nämä häiriövaikutukset voivat muuttaa täydellisen vasteen arviointitarkkuutta joillakin potilailla, joilla on IgG-kappa-myeloomaproteiinia. Nämä häiriövaikutukset voivat jatkua ainakin 6 kuukautta isatuksimabin viimeisen antokerran jälkeen. Häiriövaikutukset tutkittiin Isa Pd-haaran 22 potilaalta, jotka täyttivät erittäin hyvän osittaisen vasteen (VGPR) kriteerit ja joilla todettiin vain residuaalisia positiivisia tuloksia immunofiksaatiomäärityksessä. Näiltä potilailta saadut plasmanäytteet tutkittiin massaspektrometrialla isatuksimabisignaalin erottamiseksi myelooma-M proteiinisignaalista. Isa Kd-haarassa 27 potilaalla todettiin mahdollinen häiriövaikutus ja heiltä saadut näytteet tutkittiin massaspektrometrialla käyttäen immunofiksaatiomäärityksen herkkyystasoa (25 mg/dl). Heistä 15 potilaalla, joilla riippumattoman vasteita arvioivan komitean (Independent Response Committee, IRC) mukaan ei ollut täydellistä vastetta, ei todettu havaittavaa residuaalista myelooma-M proteiinia. Näistä 15 potilaasta 11 potilaalla oli luuytimessä < 5 % plasmasoluja. Tämä viittaa siihen, että 179:stä Isa Kd-hoitoa saaneesta potilaasta vielä 11 potilaalla (6,1 %) voisi olla parhaana vasteena täydellinen vaste, jolloin täydellisten vasteiden mahdollinen osuus olisi 45,8 % (ks. kohta Yhteisvaikutukset).
Koulutusmateriaali
Kaikkien lääkärien, jotka aikovat määrätä isatuksimabia, sekä verikeskuksissa tai verensiirtokeskuksissa työskentelevien terveydenhuollon ammattilaisten on varmistettava, että he ovat saaneet serologisiin tutkimuksiin liittyvien häiriövaikutusten riskin hallintaa varten laaditun, terveydenhuollon ammattilaisille tarkoitetun koulutusmateriaalin ja perehtyneet siihen. Lääkärien on kerrottava potilaille, että isatuksimabi saattaa vaikuttaa serologisten tutkimusten tuloksiin ainakin 6 kuukauden ajan viimeisen isatuksimabiannoksen saamisen jälkeen. Tämä on myös kerrottu potilaskortissa, joka lääkärin on annettava potilaalle, kun potilas saa ensimmäisen annoksen isatuksimabia. Potilaita on kehotettava pitämään korttia mukanaan hoidon ajan ja ainakin 6 kuukautta hoidon päättymisen jälkeen ja näyttämään se heitä hoitaville terveydenhuollon ammattilaisille.
Iäkkäät potilaat
Iäkkäistä, ≥ 85-vuotiaista potilaista on vain vähän tietoa (ks. kohta Haittavaikutukset).
Apuaine, jonka vaikutus tunnetaan
Tämä lääkevalmiste sisältää 0,2 mg polysorbaatti 80:tä per millilitra isatuksimabi-infuusiokonsentraattia liuosta varten, mikä vastaa 0,1 mg:aa/kg.
Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Isatuksimabi ei vaikuta pomalidomidin, karfiltsomibin, bortetsomibin eikä lenalidomidin farmakokinetiikkaan eikä päinvastoin.
Häiriövaikutukset serologisissa tutkimuksissa
CD38-proteiinia ilmentyy punasolujen pinnalla, joten isatuksimabi voi CD38-vasta-aineena aiheuttaa häiriöitä verikeskuksen serologisissa tutkimuksissa tuottamalla mahdollisia virheellisiä positiivisia reaktioita epäsuorissa antiglobuliinikokeissa (epäsuorissa Coombsin kokeissa), (seulonta)kokeissa, joilla todetaan vasta-aineet, vasta-aineiden tunnistuspaneeleissa ja antihumaaniglobuliinin (AHG) sopivuuskokeissa isatuksimabia saaneilla potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Nämä häiriövaikutukset voivat jatkua ainakin 6 kuukautta isatuksimabin viimeisen antokerran jälkeen. Menetelmiä, joilla häiriövaikutuksia voidaan vähentää, ovat reagenssipunasolujen käsittely ditiotreitolilla (DTT) isatuksimabin sidosten katkaisemiseksi tai muut paikallisesti validoidut menetelmät. Kell-veriryhmäjärjestelmä on myös herkkä DTT-käsittelylle, joten kun allovasta-aineet on suljettu pois tai tunnistettu käyttämällä DTT-käsiteltyjä punasoluja, potilaalle on annettava K-negatiivisia yksiköitä.
Häiriövaikutukset seerumin proteiinielektroforeesi- ja immunofiksaatiomäärityksissä
Isatuksimabi voidaan todeta seerumin proteiinielektroforeesimäärityksissä (SPE) ja immunofiksaatiomäärityksissä (IFE), joita käytetään tautiin liittyvien monoklonaalisten immunoglobuliinien (M-proteiinin) seurantaan, ja se saattaa aiheuttaa häiriöitä International Myeloma Working Group (IMWG) kriteerien mukaisessa vasteen tarkassa luokittelussa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Nämä häiriövaikutukset voivat jatkua ainakin 6 kuukautta isatuksimabin viimeisen antokerran jälkeen. Potilailla, joilla todetaan jatkuvasti erittäin hyvä osittainen vaste ja epäillään isatuksimabin häiriövaikutusta, täydellisen vasteen arvioinnin helpottamiseksi kannattaa harkita validoidun isatuksimabi-spesifisen IFE-määrityksen (Sebia Hydrashift) käyttöä isatuksimabin erottamiseksi mahdollisesti jäljellä olevasta endogeenisesta M-proteiinista potilaan plasmassa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta raskauden ehkäisyä isatuksimabihoidon aikana ja 7 kuukauden ajan hoidon päättymisen jälkeen.
Raskaus
Ei ole olemassa tietoja isatuksimabin käytöstä raskaana oleville naisille. Isatuksimabilla ei ole tehty lisääntymistoksisuutta koskevia eläinkokeita. Immunoglobuliini G1 ‑luokan monoklonaalisten vasta-aineiden tiedetään läpäisevän istukan ensimmäisen raskauskolmanneksen jälkeen. Isatuksimabin käyttöä ei suositella raskaana oleville naisille.
Imetys
Ei tiedetä, erittyykö isatuksimabi ihmisen rintamaitoon. Ihmisen IgG-vasta-aineiden tiedetään erittyvän rintamaitoon muutamien vuorokausien ajan synnytyksen jälkeen, minkä jälkeen niiden erittynyt määrä pienenee pian pieniksi pitoisuuksiksi, mutta imetettävään lapseen tällä lyhyellä ajanjaksolla heti syntymän jälkeen kohdistuvia riskejä ei voida poissulkea. Tällä nimenomaisella ajanjaksolla on päätettävä, lopetetaanko rintaruokinta vai lopetetaanko isatuksimabihoito, ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille. Myöhemmin isatuksimabia voidaan käyttää rintaruokinnan aikana, jos se on kliinisesti tarpeen.
Hedelmällisyys
Ihmisistä tai eläimistä ei ole saatavilla tietoja isatuksimabin hedelmällisyyteen kohdistuvien mahdollisten vaikutusten määrittämiseksi miehillä ja naisilla (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Isatuksimabilla on vähäinen tai kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Isatuksimabia saaneilla potilailla on ilmoitettu uupumusta ja heitehuimausta, ja tämä on otettava huomioon ajettaessa tai koneita käytettäessä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
ICARIA‑MM-tutkimuksessa yleisimpiä haittavaikutuksia ovat neutropenia (46,7 %), infuusioreaktiot (38,2 %), keuhkokuume (30,9 %), ylähengitystieinfektio (28,3 %), ripuli (25,7 %) ja keuhkoputkentulehdus (23,7 %). Vakavia haittavaikutuksia ilmeni 61,8 %:lla Isa‑Pd-hoitoa saaneista potilaista. Yleisimpiä vakavia haittavaikutuksia ovat keuhkokuume (25,7 %) ja kuumeinen neutropenia (6,6 %). Hoidon lopettamisesta pysyvästi haittavaikutusten vuoksi ilmoitettiin 7,2 %:lla Isa‑Pd-hoitoa saaneista potilaista. Hoidon aikana kuolemaan johtaneita haittavaikutuksia ilmoitettiin 7,9 %:lla Isa‑Pd-hoitoa saaneista potilaista (yli 1 %:lla potilaista todettuja tällaisia haittavaikutuksia olivat keuhkokuume, jota ilmeni 1,3 %:lla potilaista, ja muut infektiot, joita ilmeni 2,0 %:lla potilaista).
IKEMA-tutkimuksessa yleisimpiä haittavaikutuksia ovat infuusioreaktiot (45,8 %), kohonnut verenpaine (36,7 %), ripuli (36,2 %), ylähengitystieinfektio (36,2 %), keuhkokuume (28,8 %), uupumus (28,2 %), hengenahdistus (27,7 %), unettomuus (23,7 %), keuhkoputkitulehdus (22,6 %) ja selkäkipu (22,0 %). Vakavia haittavaikutuksia ilmeni 59,3 %:lla Isa‑Kd-hoitoa saaneista potilaista. Yleisin vakava haittavaikutus on keuhkokuume (21,5 %). Hoidon lopettamisesta pysyvästi haittavaikutusten vuoksi ilmoitettiin 8,5 %:lla Isa‑Kd-hoitoa saaneista potilaista. Hoidon aikana kuolemaan johtaneita haittavaikutuksia ilmoitettiin 3,4 %:lla Isa‑Kd-hoitoa saaneista potilaista (yli 1 %:lla potilaista todettuja tällaisia haittavaikutuksia olivat keuhkokuume ja sydämen vajaatoiminta, joita molempia ilmeni 1,1 %:lla potilaista).
IMROZ‑tutkimuksessa yleisimpiä haittavaikutuksia ovat ripuli (54,8 %), perifeerinen sensorinen neuropatia (54,4 %), keuhkokuume (39,9 %), kaihi (38,0 %), ummetus (35,7 %), uupumus (34,6 %), ylähengitystieinfektiot (34,2 %), perifeerinen turvotus (32,7 %), neutropenia (30,0 %:lla haittavaikutuksena), infuusioreaktio (23,6 %), unettomuus (22,4 %), COVID‑19 (22,4 %), selkäkipu (22,1 %), keuhkoputkitulehdus (22,1 %) ja astenia (21,7 %). Vakavia haittavaikutuksia ilmeni 70,7 %:lla Isa‑VRd‑hoitoa saaneista potilaista. Yleisin vakava haittavaikutus on keuhkokuume (29,7 %, mukaan lukien COVID‑19‑keuhkokuume). Hoidon aikaisia kuolemaan johtaneita haittatapahtumia (vaikeusaste 5) ilmoitettiin 11 %:lla Isa‑VRd‑hoitoa saaneista potilaista. Näihin sisältyivät myös hoidon aikaiset vaikeusasteen 5 infektiohaittatapahtumat, joita ilmeni 6,5 %:lla potilaista. Hoidon lopettamisesta pysyvästi haittavaikutusten vuoksi ilmoitettiin 22,8 %:lla Isa‑VRd‑hoitoa saaneista potilaista.
GMMG-HD7-tutkimuksessa induktiojakson aikana yleisimpiä haittavaikutuksia ovat polyneuropatia (18,8 %), neutropenia (16,1 %) ja infuusioon liittyvät reaktiot (12,4 %). Vakavia haittavaikutuksia ilmeni 35,2 %:lla Isa-VRd-hoitoa saaneista potilaista. Yleisimpiä vakavia haittavaikutuksia ovat keuhkokuume (3,6 %), kuume (3,3 %) ja ripuli (2,1 %). Hoidon aikaisia kuolemaan johtaneita haittavaikutuksia (vaikeusaste 5) ilmoitettiin 1,2 %:lla Isa-VRd-hoitoa saaneista potilaista (syinä COVID-19, influenssaan liittyvä keuhkokuume, septinen sokki ja kallonsisäinen verenvuoto, joista kutakin ilmoitettiin 0,3 %:lla potilaista). Hoidon lopettamisesta pysyvästi haittavaikutusten vuoksi ilmoitettiin 3 %:lla Isa-VRd-hoitoa saaneista potilaista.
Haittavaikutustaulukko
Haittavaikutukset on kuvattu käyttämällä NCI:n (National Cancer Institute) yleisiä toksisuuskriteerejä ja COSTART- ja MedDRA-termejä. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1 / 1 000, < 1/100), harvinainen (≥ 1 / 10 000, < 1 / 1 000), hyvin harvinainen (< 1 / 10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Haittavaikutuksia ilmoitettiin kliinisissä tutkimuksissa (ks. kohta Farmakodynamiikka) ja lääkkeen markkinoilletulon jälkeen.
Taulukko 5 – Isatuksimabia yhdistelmänä pomalidomidin ja deksametasonin kanssa saaneilla, multippelia myeloomaa sairastavilla potilailla ilmoitetut haittavaikutukset
Elinjärjestelmä Suositeltu termi | Haittavaikutus | Esiintymistiheys | Ilmaantuvuus (N = 244) | |
Mikä tahansa vaikeusaste (%) | Vaikeusaste ≥ 3 | |||
Infektiot | Keuhkokuumea b | Hyvin yleinen | 34,8 % | 27,9 % |
Ylähengitystieinfektio | Hyvin yleinen | 40,2 % | 3,3 % | |
Keuhkoputkentulehdus | Hyvin yleinen | 20,9 % | 3,7 % | |
Vyöruusu | Yleinen | 2,5 % | 0,4 % | |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit)c | Ihosyöpä | Yleinen | 4,9 % | 1,6 % |
Kiinteä kasvain (muu kuin ihosyöpä) | Yleinen | 2,9 % | 1,6 % | |
Hematologinen maligniteetti | Melko harvinainen | 0,4 % | 0,4 % | |
Veri ja imukudos | Neutropenia | Hyvin yleinen | 52,5 % | 51,6 % |
Trombosytopenia | Hyvin yleinen | 12,7 % | 11,9 % | |
Kuumeinen neutropenia | Yleinen | 7,4 % | 7,4 % | |
Anemia | Yleinen | 6,1 % | 4,5 % | |
Lymfopenia | Tuntematon |
|
| |
Immuunijärjestelmä | Anafylaktinen reaktiod | Melko harvinainen | 0,3 % | 0,3 % |
Aineenvaihdunta ja ravitsemus | Heikentynyt ruokahalu | Hyvin yleinen | 11,5 % | 1,2 % |
Sydän | Eteisvärinä | Yleinen | 5,7 % | 2,5 % |
Hengityselimet, rintakehä ja välikarsina | Hengenahdistus | Hyvin yleinen | 25,8 % | 5,7 % |
Ruoansulatuselimistö | Ripuli | Hyvin yleinen | 34,0 % | 2,5 % |
Pahoinvointi | Hyvin yleinen | 22,1 % | 0 % | |
Oksentelu | Hyvin yleinen | 14,8 % | 0,8 % | |
Tutkimukset | Painon lasku | Yleinen | 4,9 % | 0 % |
Vammat, myrkytykset ja hoitokomplikaatiot | Infuusioreaktiob | Hyvin yleinen | 39,3 % | 2,0 % |
a Termi "keuhkokuume" käsittää seuraavat termit: atyyppinen keuhkokuume, bronkopulmonaalinen aspergilloosi, keuhkokuume, Haemophilus-keuhkokuume, influenssaan liittyvä keuhkokuume, pneumokokin aiheuttama keuhkokuume, streptokokin aiheuttama keuhkokuume, viruskeuhkokuume, bakteerikeuhkokuume, hemofilusinfektio, keuhkoinfektio, sienen aiheuttama keuhkokuume ja Pneumocystis jirovecin aiheuttama keuhkokuume.
b Ks. kohta ”Valikoitujen haittavaikutusten kuvaus”.
c Perustuu sekundaarimaligniteetteihin, jotka ilmoitettiin tutkimushoitojakson aikana ja hoidon jälkeisenä ajanjaksona.
d Markkinoilletulon jälkeen on ilmoitettu anafylaktisia reaktioita, myös kuolemaan johtaneita tapauksia.
Taulukko 6a: Isatuksimabia yhdistelmänä karfiltsomibin ja deksametasonin kanssa saaneilla, multippelia myeloomaa sairastavilla potilailla ilmoitetut haittavaikutukset
Elinjärjestelmä Suositeltu termi | Haittavaikutus | Esiintymistiheys | Ilmaantuvuus (N = 177) | |
Mikä tahansa vaikeusaste | Vaikeusaste ≥ 3 | |||
Infektiot | Keuhkokuumeb c | Hyvin yleinen | 28,8 % | 20,9 % |
Ylähengitystieinfektio | Hyvin yleinen | 36,2 % | 3,4 % | |
Keuhkoputkentulehdus | Hyvin yleinen | 22,6 % | 2,3 % | |
Vyöruusu | Yleinen | 2,3 % | 0,6 % | |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit)d | Ihosyövät | Yleinen | 7,3 % | 1,7 % |
Kiinteät kasvaimet (muut kuin ihosyövät) | Yleinen | 4,0 % | 3,4 % | |
Veri ja imukudos | Anemia | Yleinen | 5,1 % | 4,5 % |
Neutropenia | Yleinen | 4,5 % | 4,0 % | |
Trombosytopenia | Yleinen | 2,8 % | 2,3 % | |
Lymfopenia | Tuntematon |
|
| |
Immuunijärjestelmä | Anafylaktinen reaktioe | Melko harvinainen | 0,3 % | 0,3 % |
Verisuonisto | Kohonnut verenpaine | Hyvin yleinen | 36,7 % | 20,3 % |
Hengityselimet, rintakehä ja välikarsina | Hengenahdistus | Hyvin yleinen | 27,7 % | 5,1 % |
Yskä | Hyvin yleinen | 19,8 % | 0 % | |
Ruoansulatuselimistö | Ripuli | Hyvin yleinen | 36,2 % | 2,8 % |
Oksentelu | Hyvin yleinen | 15,3 % | 1,1 % | |
Yleisoireet ja antopaikassa todettavat haitat | Uupumus | Hyvin yleinen | 28,2 % | 3,4 % |
Vammat, myrkytykset ja hoitokomplikaatiot | Infuusioreaktioc | Hyvin yleinen | 45,8 % | 0,6 % |
a Tiedonkeruun päättymispäivä 7.2.2020. Seuranta-ajan mediaani = 20,73 kuukautta.
b Termi ”keuhkokuume” käsittää seuraavat termit: atyyppinen keuhkokuume, Pneumocystis jirovecin aiheuttama keuhkokuume, keuhkokuume, influenssaan liittyvä keuhkokuume, legionellan aiheuttama keuhkokuume, streptokokin aiheuttama keuhkokuume, viruskeuhkokuume ja keuhkojen sepsis.
c Ks. kohta ”Valikoitujen haittavaikutusten kuvaus”.
d Tiedonkeruun päättymispäivä 7.2.2023. Seuranta-ajan mediaani = 56,61 kuukautta. Perustuu sekundaarimaligniteetteihin, joita ilmoitettiin tutkimushoitojakson aikana ja hoidon jälkeisenä ajanjaksona.
e Markkinoilletulon jälkeen on ilmoitettu anafylaktisia reaktioita, myös kuolemaan johtaneita tapauksia.
Taulukko 7: Haittavaikutukset, joita ilmoitettiin isatuksimabia yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa saaneilla potilailla, joilla oli äskettäin todettu multippeli myelooma ja jotka eivät soveltuneet siirteen saamiseen
Elinjärjestelmä Suositeltu termi | Haittavaikutus | Esiintymistiheys | Ilmaantuvuus (N = 336) | |
Mikä tahansa vaikeusaste (%) | Vaikeusaste ≥ 3 | |||
Infektiot | Keuhkokuumea | Hyvin yleinen | 34,2 % | 24,1 % |
Keuhkoputkitulehdus | Hyvin yleinen | 22,6 % | 3,0 % | |
COVID‑19 | Hyvin yleinen | 19,9 % | 1,2 % | |
Hyvän‑ ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Ihosyöpä | Yleinen | 8,0 % | 2,7 % |
Kiinteä kasvain (muu kuin ihosyöpä) | Yleinen | 5,7 % | 3,6 % | |
Hematologinen maligniteetti | Melko harvinainen | 0,9 % | 0,3 % | |
Veri ja imukudos | Neutropenia | Hyvin yleinen | 28,0 % | 27,1 % |
Trombosytopenia | Hyvin yleinen | 13,4 % | 10,7 % | |
Anemia | Yleinen | 6,3 % | 2,7 % | |
Lymfopenia | Tuntematon | _____ | _____ | |
Immuunijärjestelmä | Anafylaktinen reaktiob | Melko harvinainen | 0,3 % | 0,3 % |
Silmät | Kaihi | Hyvin yleinen | 36,0 % | 13,1 % |
Ruoansulatuselimistö | Ripuli | Hyvin yleinen | 56,8 % | 8,3 % |
Oksentelu | Yleinen | 9,5 % | 0,3 % | |
Yleisoireet ja antopaikassa todettavat haitat | Uupumus | Hyvin yleinen | 32,7 % | 6,5 % |
Vammat, myrkytykset ja hoitokomplikaatiot | Infuusioreaktio | Hyvin yleinen | 27,4 % | 0,6 % |
a Termi ”keuhkokuume” käsittää seuraavat termit: atyyppinen keuhkokuume, bronkopulmonaalinen aspergilloosi, COVID‑19‑keuhkokuume, Pneumocystis jirovecin aiheuttama keuhkokuume, keuhkokuume, bakteerikeuhkokuume, Haemophilus‑keuhkokuume, influenssaan liittyvä keuhkokuume, Klebsiella‑keuhkokuume, legionellan aiheuttama keuhkokuume, pneumokokin aiheuttama keuhkokuume, Pseudomonas‑keuhkokuume, RS‑viruksen aiheuttama keuhkokuume, viruskeuhkokuume, keuhkojen sepsis ja tuberkuloosi.
b Markkinoilletulon jälkeen on ilmoitettu anafylaktisia reaktioita, myös kuolemaan johtaneita tapauksia.
MedDRA 26.0
Taulukko 8: Haittavaikutukset, joita ilmoitettiin induktiojakson aikana isatuksimabia yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa saaneilla potilailla, joilla oli äskettäin todettu multippeli myelooma ja jotka soveltuivat siirteen saamiseen
Elinjärjestelmä Suositeltu termi | Haittavaikutus | Esiintymistiheys | Ilmaantuvuus | |
Mikä tahansa vaikeusaste | Vaikeusaste ≥ 3 | |||
Infektiot | Keuhkokuumea b | Yleinen | 5,5 % | 4,8 % |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Kiinteät kasvaimet (muut kuin ihosyövät) | Melko harvinainen | 0,3 % | 0,3 % |
Veri ja imukudos | Neutropenia | Hyvin yleinen | 16,1 % | 16,1 % |
Anemia | Yleinen | 3,6 % | 3,6 % | |
Trombosytopenia | Yleinen | 4,5 % | 4,5 % | |
Lymfopenia | Yleinen | 3,3 % | 3,3 % | |
Immuunijärjestelmä | Anafylaktinen reaktioc | Melko harvinainen | 0,3 % | 0,3 % |
Tutkimukset | Neutrofiilien määrän pieneneminen | Yleinen | 7,3 % | 7,3 % |
Vammat, myrkytykset ja hoitokomplikaatiot | Infuusioreaktiob | Hyvin yleinen | 12,7 % | 0,9 % |
a Termi ”keuhkokuume” käsittää seuraavat termit: keuhkokuume, influenssaan liittyvä keuhkokuume, atyyppinen keuhkokuume ja sienen aiheuttama keuhkokuume.
b Ks. kohta ”Valikoitujen haittavaikutusten kuvaus”.
c Markkinoilletulon jälkeen on ilmoitettu anafylaktisia reaktioita, myös kuolemaan johtaneita tapauksia.
Valikoitujen haittavaikutusten kuvaus
Infuusioreaktiot
ICARIA-MM-tutkimuksessa ilmoitettiin infuusioreaktioita 58 potilaalla (38,2 %), jotka olivat saaneet isatuksimabia. Kaikilla potilailla, joilla infuusioreaktioita ilmeni, ne ilmenivät ensimmäisen isatuksimabi-infuusion aikana, ja kolmella potilaalla (2,0 %) ilmeni infuusioreaktioita myös toisen infuusion aikana ja kahdella potilaalla (1,3 %) neljännen infuusion yhteydessä. Vaikeusasteen 1 infuusioreaktioita ilmoitettiin 3,9 %:lla, vaikeusasteen 2 reaktioita 31,6 %:lla, vaikeusasteen 3 reaktioita 1,3 %:lla ja vaikeusasteen 4 reaktioita 1,3 %:lla potilaista. Kaikki infuusioreaktiot olivat ohimeneviä, ja infuusioreaktiot hävisivät samana päivänä 98 %:ssa infuusioista. Vaikeusastetta 3 tai 4 olevien infuusioreaktioiden oireita olivat hengenahdistus, kohonnut verenpaine ja bronkospasmi.
Infuusioreaktioista johtuvan infuusion keskeyttämisen ilmaantuvuus oli 28,9 %. Mediaaniaika infuusion keskeyttämiseen oli 55 minuuttia.
Hoidon lopettamisesta infuusioreaktion vuoksi ilmoitettiin 2,6 %:lla Isa Pd-ryhmän potilaista.
IKEMA-tutkimuksessa ilmoitettiin infuusioreaktioita 81 potilaalla (45,8 %), jotka olivat saaneet Isa Kd-hoitoa. Vaikeusasteen 1 infuusioreaktioita ilmoitettiin 13,6 %:lla, vaikeusasteen 2 reaktioita 31,6 %:lla ja vaikeusasteen 3 reaktioita 0,6 %:lla Isa Kd-hoitoa saaneista potilaista. Kaikki infuusioreaktiot olivat ohimeneviä, ja infuusioreaktiot hävisivät samana päivänä 73,8 %:ssa tapauksista Isa Kd-hoitoa saaneilla potilailla ja yli 2 päivän kuluessa 2,5 %:ssa tapauksista Isa Kd-hoitoa saaneilla potilailla. Vaikeusastetta 3 olevien infuusioreaktioiden oireita olivat hengenahdistus ja kohonnut verenpaine. Infuusioreaktioiden vuoksi isatuksimabi-infuusion keskeyttäneiden potilaiden ilmaantuvuus oli 29,9 %. Mediaaniaika isatuksimabi-infuusion keskeyttämiseen oli 63 minuuttia. Isatuksimabihoito lopetettiin infuusioreaktion vuoksi 0,6 %:lla potilaista.
IMROZ tutkimuksessa ilmoitettiin infuusioreaktioita 63 potilaalla (24,0 %), jotka olivat saaneet Isa VRd hoitoa. Vaikeusasteen 1 infuusioreaktioita ilmoitettiin 1,9 %:lla, vaikeusasteen 2 reaktioita 21,3 %:lla, vaikeusasteen 3 reaktioita 0,4 %:lla ja vaikeusasteen 4 reaktioita 0,4 %:lla potilaista, jotka olivat saaneet Isa VRd hoitoa. Infuusioreaktiot alkoivat kaikilla potilailla infuusiopäivänä, useimmiten ensimmäisen isatuksimabi-infuusion aikana, ja 97,3 %:lla potilaista ne hävisivät samana päivänä. Kaikki infuusioreaktiot hävisivät. Vaikeusastetta 3 tai 4 olevien infuusioreaktioiden oireita olivat kohonnut verenpaine, bronkospasmi ja hypoksia. Infuusioreaktioiden vuoksi isatuksimabi infuusion keskeyttäneiden potilaiden ilmaantuvuus oli 20,9 %. Mediaaniaika isatuksimabi infuusion keskeyttämiseen oli 66,0 minuuttia. Isatuksimabihoito lopetettiin infuusioreaktion vuoksi 0,8 %:lla potilaista.
GMMG HD7-tutkimuksessa induktiojakson aikana ilmoitettiin infuusioreaktioita 42 potilaalla (12,7 %:lla), jotka olivat saaneet Isa VRd-hoitoa. Tutkimuksessa ei kerätty tietoa vaikeusasteen 1 infuusioreaktioista. Vaikeusasteen 2 infuusioreaktioita ilmoitettiin 11,8 %:lla, vaikeusasteen 3 reaktioita 0,6 %:lla ja vaikeusasteen 4 reaktioita 0,3 %:lla potilaista, jotka olivat saaneet Isa VRd-hoitoa. Kaikki infuusioreaktiot hävisivät. Infuusioreaktioiden vuoksi isatuksimabi-infuusion keskeyttäneiden potilaiden ilmaantuvuus oli 7,6 %. Isatuksimabihoito lopetettiin infuusioreaktion vuoksi 0,3 %:lla potilaista (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Infektiot
ICARIA-MM-tutkimuksessa vähintään vaikeusasteen 3 infektioiden ilmaantuvuus oli 42,8 %. Keuhkokuume oli yleisimmin ilmoitettu vaikea infektio, jonka ilmoitettiin olevan vaikeusastetta 3 kaikkiaan 21,7 %:lla Isa‑Pd-ryhmän potilaista ja 16,1 %:lla Pd‑ryhmän potilaista ja vaikeusastetta 4 kaikkiaan 3,3 %:lla Isa‑Pd-ryhmän potilaista ja 2,7 %:lla Pd‑ryhmän potilaista. Hoidon lopettamisesta infektion vuoksi ilmoitettiin 2,6 %:lla Isa‑Pd-ryhmän potilaista ja 5,4 %:lla Pd‑ryhmän potilaista. Kuolemaan johtaneita infektioita ilmoitettiin 3,3 %:lla Isa‑Pd-ryhmän potilaista ja 4,0 %:lla Pd‑ryhmän potilaista. IKEMA-tutkimuksessa vähintään vaikeusasteen 3 infektioiden ilmaantuvuus oli 38,4 %. Keuhkokuume oli yleisimmin ilmoitettu vaikea infektio, jonka ilmoitettiin olevan vaikeusastetta 3 kaikkiaan 15,8 %:lla Isa‑Kd-ryhmän potilaista ja 10,7 %:lla Kd‑ryhmän potilaista ja vaikeusastetta 4 kaikkiaan 3,4 %:lla Isa‑Kd-ryhmän potilaista ja 2,5 %:lla Kd‑ryhmän potilaista. Hoito lopetettiin infektion vuoksi 2,8 %:lla Isa‑Kd-ryhmän potilaista ja 4,9 %:lla Kd‑ryhmän potilaista. Kuolemaan johtaneita infektioita ilmoitettiin 2,3 %:lla Isa‑Kd-ryhmän potilaista ja 0,8 %:lla Kd‑ryhmän potilaista. IMROZ‑tutkimuksessa vähintään vaikeusasteen 3 infektioiden ilmaantuvuus oli 44,9 % Isa‑VRd‑ryhmässä ja 38,1 % VRd‑ryhmässä. Keuhkokuume oli yleisimmin ilmoitettu vaikea infektio, jonka ilmoitettiin olevan vaikeusastetta 3 kaikkiaan 25,1 %:lla Isa‑VRd‑ryhmän potilaista ja 15,5 %:lla VRd‑ryhmän potilaista ja vaikeusastetta 4 kaikkiaan 2,3 %:lla Isa‑VRd‑ryhmän potilaista ja 3,9 %:lla VRd‑ryhmän potilaista. Vaikeusasteen 5 keuhkokuumetta (suositeltuun termiin perustuva) ilmeni 1,5 %:lla Isa‑VRd‑ryhmän potilaista ja 1,1 %:lla VRd‑ryhmän potilaista. Hoidon lopettamisesta infektion vuoksi ilmoitettiin 8,4 %:lla Isa‑VRd‑ryhmän potilaista ja 9,4 %:lla VRd‑ryhmän potilaista. Kuolemaan johtaneita infektioita ilmoitettiin 6,5 %:lla Isa‑VRd‑ryhmän potilaista ja 4,4 %:lla VRd‑ryhmän potilaista. GMMG-HD7-tutkimuksessa induktiojakson aikana vähintään vaikeusasteen 3 infektioiden ilmaantuvuus oli 13 % Isa-VRd-ryhmässä ja 10,1 % VRd-ryhmässä. Keuhkokuume oli yleisimmin ilmoitettu vaikea infektio, jonka ilmoitettiin olevan vaikeusastetta ≥ 3 kaikkiaan 3,9 %:lla Isa-VRd-ryhmän potilaista ja 2,1 %:lla VRd-ryhmän potilaista. Hoidon lopettamisesta infektion vuoksi ilmoitettiin 0,3 %:lla Isa-VRd-ryhmän potilaista ja 0,9 %:lla VRd-ryhmän potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Relapsoitunutta ja refraktorista multippelia myeloomaa koskeneissa kliinisissä tutkimuksissa ilmoitettiin vyöruusua 2,0 %:lla potilaista. ICARIA‑MM-tutkimuksessa vyöruusun ilmaantuvuus oli 4,6 % Isa‑Pd-ryhmässä ja 0,7 % Pd‑ryhmässä, ja IKEMA-tutkimuksessa ilmaantuvuus oli 2,3 % Isa‑Kd-ryhmässä ja 1,6 % Kd‑ryhmässä. Äskettäin todettua multippelia myeloomaa koskeneissa kliinisissä tutkimuksissa ilmoitettiin vyöruusua 3,3 %:lla potilaista. IMROZ‑tutkimuksessa vyöruusun ilmaantuvuus oli 5,7 % Isa‑VRd‑ryhmässä ja 5,5 % VRd‑ryhmässä. GMMG-HD7-tutkimuksessa induktiojakson aikana vyöruusun ilmaantuvuus oli 0,9 % Isa-VRd-ryhmässä ja 0,3 % VRd-ryhmässä.
Sydämen vajaatoiminta
IKEMA-tutkimuksessa ilmoitettiin sydämen vajaatoimintaa (mukaan lukien sydämen vajaatoiminta, kongestiivinen sydämen vajaatoiminta, akuutti sydämen vajaatoiminta, krooninen sydämen vajaatoiminta, sydämen vasemman kammion vajaatoiminta ja keuhkopöhö) 7,3 %:lla Isa‑Kd-ryhmän potilaista (4,0 % oli vaikeusastetta ≥ 3) ja 6,6 %:lla Kd‑ryhmän potilaista (4,1 % oli vaikeusastetta ≥ 3). Vakavaa sydämen vajaatoimintaa ilmoitettiin 4,0 %:lla Isa‑Kd-ryhmän potilaista ja 3,3 %:lla Kd‑ryhmän potilaista. Hoidon aikana kuolemaan johtanutta sydämen vajaatoimintaa ilmoitettiin 1,1 %:lla Isa‑Kd-ryhmän potilaista, Kd‑ryhmässä sitä ei ilmoitettu lainkaan (ks. voimassa oleva karfiltsomibin valmisteyhteenveto).
Hematologiset laboratorioarvot
Taulukko 9 – Hematologisten laboratoriotulosten poikkeavuudet isatuksimabia yhdistelmänä pomalidomidin ja deksametasonin kanssa saaneilla potilailla verrattuna pomalidomidia ja deksametasonia saaneisiin potilaisiin (ICARIA-MM)
Laboratorioparametri | Isatuksimabi + pomalidomidi + deksametasoni | Pomalidomidi + deksametasoni (N = 147) | ||||
Kaikki vaikeusasteet | Vaikeusaste 3 | Vaikeusaste 4 | Kaikki vaikeusasteet | Vaikeusaste 3 | Vaikeusaste 4 | |
Anemia | 99,3 % | 31,6 % | 0 % | 98,6 % | 27,9 % | 0 % |
Neutropenia | 96,1 % | 24,3 % | 60,5 % | 93,2 % | 38,8 % | 31,3 % |
Lymfosytopenia | 92,1 % | 42,1 % | 12,5 % | 93,2 % | 35,4 % | 8,2 % |
Trombosytopenia | 83,6 % | 14,5 % | 16,4 % | 80,3 % | 9,5 % | 15,0 % |
Prosentuaalisten osuuksien laskennassa käytetty nimittäjä oli sellaisten potilaiden määrä, joilta oli arvioitu ainakin yksi laboratoriokoetulos tarkastelujaksolla.
Taulukko 10 – Hematologisten laboratoriotulosten poikkeavuudet isatuksimabia yhdistelmänä karfiltsomibin ja deksametasonin kanssa saaneilla potilailla verrattuna karfiltsomibia ja deksametasonia saaneisiin potilaisiin (IKEMA)
Laboratorioparametri | Isatuksimabi + karfiltsomibi + deksametasoni (N = 177) | Karfiltsomibi + deksametasoni (N = 122) | ||||
| Kaikki vaikeusasteet | Vaikeusaste 3 | Vaikeusaste 4 | Kaikki vaikeusasteet | Vaikeusaste 3 | Vaikeusaste 4 |
Anemia | 99,4 % | 22,0 % | 0 % | 99,2 % | 19,7 % | 0 % |
Neutropenia | 54,8 % | 17,5 % | 1,7 % | 43,4 % | 6,6 % | 0,8 % |
Lymfosytopenia | 94,4 % | 52,0 % | 16,9 % | 95,1 % | 43,4 % | 13,9 % |
Trombosytopenia | 94,4 % | 18,6 % | 11,3 % | 87,7 % | 15,6 % | 8,2 % |
Prosentuaalisten osuuksien laskennassa käytetty nimittäjä oli sellaisten potilaiden määrä, joilta oli arvioitu ainakin yksi laboratoriokoetulos tarkastelujaksolla.
Taulukko 11: Hematologisten laboratoriotulosten poikkeavuudet isatuksimabia yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa saaneilla potilailla verrattuna bortetsomibia, lenalidomidia ja deksametasonia saaneisiin potilaisiin (IMROZ ja TCD13983)
Laboratorioparametri | Isatuksimabi + bortetsomibi+ lenalidomidi + deksametasoni (N = 336) | Bortetsomibi+ lenalidomidi + deksametasoni (N = 181) | ||||
Kaikki vaikeusasteet | Vaikeusaste 3 | Vaikeusaste 4 | Kaikki vaikeusasteet | Vaikeusaste 3 | Vaikeusaste 4 | |
Anemia | 99,1 % | 15,8 % | 0 % | 97,8 % | 16,0 % | 0 % |
Lymfosytopenia | 96,1 % | 45,5 % | 18,5 % | 92,3 % | 37,6 % | 15,5 % |
Trombosytopenia | 94,6 % | 16,7 % | 14,6 % | 84,5 % | 19,3 % | 8,3 % |
Neutropenia | 86,9 % | 35,4 % | 17,3 % | 80,1 % | 28,2 % | 8,8 % |
Prosentuaalisten osuuksien laskennassa käytetty nimittäjä oli sellaisten potilaiden määrä, joilta oli arvioitu ainakin yksi laboratoriokoetulos tarkastelujaksolla.
CTCAE‑luokituksen versio: 4.03.
Taulukko 12: Hematologisten laboratoriotulosten poikkeavuudet induktiojakson aikana isatuksimabia yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa saaneilla potilailla verrattuna bortetsomibia, lenalidomidia ja deksametasonia saaneisiin potilaisiin (GMMG-HD7)
Laboratorioparametri | Isatuksimabi + bortetsomibi+ lenalidomidi + deksametasoni (N = 330) | Bortetsomibi+ lenalidomidi + deksametasoni (N = 328) | ||||
Kaikki vaikeusasteet | Vaikeusaste 3 | Vaikeusaste 4 | Kaikki vaikeusasteet | Vaikeusaste 3 | Vaikeusaste 4 | |
Anemia | 80,0 % | 1,6 % | 0 % | 79,7 % | 0,9 % | 0 % |
Neutropenia | 30,9 % | 3,1 % | 2,8 % | 22,9 % | 2,5 % | 1,9 % |
Lymfopenia | 54,4 % | 15,6 % | 2,2 % | 67,4 % | 18,7 % | 5,1 % |
Trombosytopenia | 21,6 % | 0,6 % | 0,9 % | 20,3 % | 0,3 % | 0 % |
Prosentuaalisten osuuksien laskennassa käytetty nimittäjä oli sellaisten potilaiden määrä, joilta oli arvioitu ainakin yksi laboratoriokoetulos tarkastelujaksolla.
Iäkkäät potilaat
Isatuksimabia koskeneiden kliinisten tutkimusten kaikista potilaista 42,7 % (763 potilasta) oli alle 65‑vuotiaita, 43,2 % (772 potilasta) 65–74‑vuotiaita ja 14,1 % (252 potilasta) vähintään 75‑vuotiaita. Turvallisuudessa havaittiin eroja iäkkäämpien ja nuorempien ikäryhmien välillä. Hoidon aikaisia haittatapahtumia, joiden vaikeusaste oli > 3, ilmoitettiin 64,7 %:lla alle 65‑vuotiaista potilaista, 79,7 %:lla 65–74‑vuotiaista potilaista ja 76,6 %:lla vähintään 75‑vuotiaista potilaista. Hoidon aikaisia haittatapahtumia, joiden vaikeusaste oli 5, ilmoitettiin 5,6 %:lla alle 65‑vuotiaista potilaista, 7,5 %:lla 65–74‑vuotiaista potilaista ja 12,3 %:lla vähintään 75‑vuotiaista potilaista. Vakavia hoidon aikaisia haittatapahtumia ilmoitettiin 46,7 %:lla alle 65‑vuotiaista potilaista, 59,3 %:lla 65–74‑vuotiaista potilaista ja 61,1 %:lla vähintään 75‑vuotiaista potilaista. Hoidon lopulliseen lopettamiseen johtaneita hoidon aikaisia haittatapahtumia ilmoitettiin 6,4 %:lla alle 65‑vuotiaista potilaista, 14,4 %:lla 65–74‑vuotiaista potilaista ja 15,9 %:lla vähintään 75‑vuotiaista potilaista.
IMROZ‑tutkimuksessa ei ilmoitettu hoidon aikaisia vaikeusasteen 5 haittatapahtumia alle 65‑vuotiailla potilailla, mutta niitä ilmoitettiin 10,7 %:lla 65–74‑vuotiaista potilaista ja 13,2 %:lla vähintään 75‑vuotiaista potilaista. GMMG-HD7-tutkimukseen otettiin vain enintään 70-vuotiaita potilaita. Hoidon aikaisia haittatapahtumia, joiden vaikeusaste oli ≥ 3, ilmoitettiin induktiojakson aikana 59,7 %:lla alle 65-vuotiaista potilaista ja 77,8 %:lla vähintään 65-vuotiaista potilaista. Hoidon lopulliseen lopettamiseen johtaneita hoidon aikaisia haittatapahtumia ilmoitettiin 1,6 %:lla alle 65-vuotiaista potilaista ja 8,3 %:lla vähintään 65-vuotiaista potilaista. Hoidon aikaisia vaikeusasteen 5 haittatapahtumia ilmoitettiin 0,8 %:lla alle 65-vuotiaista potilaista ja 2,8 %:lla vähintään 65-vuotiaista potilaista.
Immunogeenisuus
Kaikissa yhdeksässä relapsoitunutta tai refraktorista multippelia myeloomaa koskeneessa kliinisessä tutkimuksessa (N = 1 023), joissa käytettiin isatuksimabia ainoana lääkkeenä ja yhdistelmähoitona, ICARIA-MM ja IKEMA mukaan lukien, hoidon aiheuttamien lääkevasta-aineiden ilmaantuvuus oli < 2 %. Lääkevasta-aineilla ei havaittu olevan vaikutuksia isatuksimabin farmakokinetiikkaan, turvallisuuteen tai tehoon. Kaikissa kolmessa äskettäin todettua multippelia myeloomaa koskeneessa kliinisessä tutkimuksessa (N = 383), joissa käytettiin isatuksimabia yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa, IMROZ ja GMMG-HD7 mukaan lukien, lääkevasta aineiden ilmaantuvuuden vaihteluväli oli 9,1–21,6 %. IMROZ tutkimuksessa 60 % (15/25) ja GMMG-HD7-tutkimuksessa 50 % (4/8) hoidon indusoimista lääkevasta-aineista oli neutraloivia. Äskettäin todettua multippelia myeloomaa sairastavilla lääkevasta ainepositiivisilla potilailla havaittiin trendinomaisesti pienempiä altistuksia. Lääkevasta aineilla ei havaittu olevan vaikutuksia isatuksimabin tehoon. Turvallisuudesta ei voida tehdä johtopäätöksiä lääkevasta ainepositiivisten potilaiden pienen alaryhmän vuoksi.
Pediatriset potilaat
Yksihaaraiseen vaiheen 2 tutkimukseen osallistui 67 pediatrista potilasta, joilla oli relapsoitunut tai refraktorinen akuutti lymfoblastileukemia tai akuutti myelooinen leukemia. Kaikki potilaat voitiin arvioida turvallisuuden suhteen, ja hoidon aikaisia haittatapahtumia, joiden vaikeusaste oli ≥ 3, ilmoitettiin 79,1 %:lla potilaista. Yleisimmät hoidon aikaiset haittatapahtumat, joiden vaikeusaste oli ≥ 3 ja joita ilmeni yli 10 %:lla potilaista, olivat kuumeinen neutropenia (41,8 %), septinen sokki (11,9 %) ja stomatiitti (10,4 %). Isatuksimabin lisääminen tavanomaisiin solunsalpaajahoitoihin ei muuttanut näiden solunsalpaajahoitojen odotettavissa olevaa turvallisuusprofiilia tässä pediatrisessa potilasjoukossa. Turvallisuusprofiili myös vastasi ICARIA- ja IKEMA-tutkimuksissa multippelia myeloomaa sairastavilla aikuisilla todettua isatuksimabin turvallisuusprofiilia (ks. kohta Annostus ja antotapa).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet ja merkit
Kliinisissä tutkimuksissa ei ole ilmennyt isatuksimabin yliannostustapauksia. Kliinisissä tutkimuksissa laskimoon annetut isatuksimabiannokset ovat olleet enintään 20 mg/kg.
Hoito
Isatuksimabin yliannostukseen ei ole tiedossa spesifistä vastalääkettä. Yliannostustapauksessa potilasta on tarkkailtava haittavaikutusten oireiden tai merkkien varalta, ja kaikkiin asianmukaisiin toimenpiteisiin on ryhdyttävä välittömästi.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, ATC-koodi: L01FC02.
Vaikutusmekanismi
Isatuksimabi on IgG1-peräinen monoklonaalinen vasta-aine, joka sitoutuu CD38-reseptorin spesifiseen ekstrasellulaariseen epitooppiin. CD38 on transmembraaniglykoproteiini, jota ilmentyy runsaasti multippelin myelooman soluissa.
Isatuksimabin vaikutus in vitro perustuu IgG:n Fc-osasta riippuvaisiin mekanismeihin, joita ovat: vasta-aineesta riippuvainen soluvälitteinen sytotoksisuus (ADCC), vasta-aineesta riippuvainen sellulaarinen fagosytoosi (ADCP) ja komplementista riippuvainen sytotoksisuus (CDC). Lisäksi isatuksimabi voi myös laukaista kasvainsolun kuoleman indusoimalla apoptoosia Fc-osasta riippumattomalla mekanismilla.
Isatuksimabi estää CD38-glykoproteiinin entsymaattista aktiivisuutta in vitro. CD38 katalysoi kalsiumia mobilisoivan syklisen ADP-riboosin (cADPR) synteesiä ja hydrolyysiä. Isatuksimabi estää cADPR:n muodostusta ekstrasellulaarisesta nikotiiniamidiadeniinidinukleotidistä (NAD) multippelin myelooman soluissa.
Isatuksimabi pystyy aktivoimaan NK-soluja (luonnollisia tappajasoluja) in vitro, kun läsnä ei ole CD38-positiivisia kohdekasvainsoluja.
Isatuksimabimonoterapiaa saaneilla potilailla havaittiin perifeerisessä veressä pienentyneitä CD16+- ja CD56+-NK-solujen, CD19+-B-solujen, CD4+-T-solujen ja säätelijä-T-solujen (TREG-solujen) (CD3+, CD4+, CD25+ ja CD127‑) absoluuttisia kokonaismääriä in vivo.
Multippelia myeloomaa sairastavilla potilailla isatuksimabimonoterapia indusoi lukuisten T-solureseptorien klonaalista ekspansiota, mikä viittaa adaptiiviseen immuunivasteeseen.
Pelkkään isatuksimabiin verrattuna isatuksimabin ja pomalidomidin yhdistelmä tehostaa in vitro CD38:aa ilmentävien multippelin myelooman solujen lyysiä efektorisolujen avulla (ADCC) ja suoraan tappamalla kasvainsoluja. In vivo ‑eläinkokeissa, joissa käytettiin ihmisen multippelin myelooman ksenograftimallia hiirillä, osoitettiin, että isatuksimabin ja pomalidomidin yhdistelmä tehostaa kasvaimen kasvua ehkäisevää vaikutusta verrattuna pelkän isatuksimabin tai pomalidomidin vaikutukseen.
Kliininen teho ja turvallisuus
Relapsoitunut ja/tai refraktorinen multippeli myelooma
ICARIA‑MM (EFC14335)
Pomalidomidin ja deksametasonin kanssa yhdistelmänä käytettävän isatuksimabin tehoa ja turvallisuutta arvioitiin relapsoitunutta ja/tai refraktorista multippelia myeloomaa sairastavilla potilailla kansainvälisessä, satunnaistetussa, avoimessa, kaksihaaraisessa vaiheen III monikeskustutkimuksessa ICARIA-MM (EFC14335). Potilaat olivat saaneet vähintään kahta aiempaa hoitoa, jotka sisälsivät lenalidomidia ja proteasomin estäjää, ja tauti oli edennyt aiemman hoidon aikana tai 60 päivän sisällä sen päättymisestä. Potilaat, joilla oli primaarinen refraktorinen tauti, suljettiin pois.
Yhteensä 307 potilasta satunnaistettiin suhteessa 1:1 saamaan joko isatuksimabia yhdistelmänä pomalidomidin ja deksametasonin kanssa (Isa‑Pd-hoito, 154 potilasta) tai pomalidomidia ja deksametasonia (Pd‑hoito, 153 potilasta). Hoitoa annettiin molemmissa ryhmissä 28 päivän hoitosykleinä, kunnes tauti eteni tai ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä. Isatuksimabi 10 mg/kg annettiin infuusiona laskimoon kerran viikossa ensimmäisen hoitosyklin aikana ja sen jälkeen kahden viikon välein. Pomalidomidi 4 mg otettiin suun kautta kerran vuorokaudessa kunkin 28 päivän pituisen hoitosyklin päivinä 1–21. Deksametasoni (suun kautta / laskimoon) 40 mg:n annoksella (20 mg ≥ 75-vuotiaille potilaille) annettiin kunkin 28 päivän pituisen hoitosyklin päivinä 1, 8, 15 ja 22.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat näiden kahden hoitoryhmän välillä kaiken kaikkiaan samankaltaiset, vaikka joitakin pieniä eroja oli. Potilaiden mediaani-ikä oli 67 vuotta (vaihteluväli 36–86), ja 19,9 % potilaista oli ≥ 75-vuotiaita. ECOG-suorituskykypistemäärä (Eastern Cooperative Oncology Group performance status) oli 0 isatuksimabihaarassa 35,7 %:lla ja vertailuhaarassa 45,1 %:lla potilaista, se oli 1 isatuksimabihaarassa 53,9 %:lla ja vertailuhaarassa 44,4 %:lla potilaista ja 2 isatuksimabihaarassa 10,4 %:lla ja vertailuhaarassa 10,5 %:lla potilaista. 10,4 %:lla tutkimukseen osallistuneista isatuksimabihaaran potilaista ja 10,5 %:lla vertailuhaaran potilaista oli aiemmin todettu keuhkoahtaumatauti tai astma, ja 38,6 %:lla isatuksimabihaaran potilaista ja 33,3 %:lla vertailuhaaran potilaista oli munuaisten vajaatoiminta (kreatiniinipuhdistuma < 60 ml/min/1,73 m²). Potilaiden aloittaessa tutkimuksessa heistä 37,5 %:lla (41,6 %:lla isatuksimabihaarassa ja 33,3 %:lla vertailuhaarassa) oli luokan I tauti, 35,5 %:lla (34,4 %:lla isatuksimabihaarassa ja 36,6 %:lla vertailuhaarassa) luokan II tauti ja 25,1 %:lla (22,1 %:lla isatuksimabihaarassa ja 28,1 %:lla vertailuhaarassa) luokan III tauti kansainvälisen luokitusjärjestelmän (International Staging System, ISS) mukaan. Yhteensä 19,5 %:lla potilaista (15,6 %:lla isatuksimabihaarassa ja 23,5 %:lla vertailuhaarassa) oli tutkimuksessa aloittaessaan kromosomipoikkeavuus, johon liittyi suuri riski: 12,1 %:lla potilaista (9,1 %:lla isatuksimabihaarassa ja 15,0 %:lla vertailuhaarassa) oli del(17p)-mutaatio, 8,5 %:lla potilaista (7,8 %:lla isatuksimabihaarassa ja 9,2 %:lla vertailuhaarassa) t(4;14)-mutaatio ja 1,6 %:lla (0,6 %:lla isatuksimabihaarassa ja 2,6 %:lla vertailuhaarassa) t(14;16)-mutaatio.
Aiempien linjojen hoitojen määrän mediaani oli 3 (vaihteluväli 2–11). Kaikki potilaat olivat saaneet aiemmin proteasomin estäjää, kaikki potilaat olivat saaneet aiemmin lenalidomidia ja 56,4 % oli saanut aiemmin kantasolusiirron. Suurin osa (92,5 %) potilaista oli refraktorisia lenalidomidille, 75,9 % proteasomin estäjälle ja 72,6 % sekä immuunivasteen muuntajalle että proteasomin estäjälle, ja 59 % potilaista oli ollut refraktorisia viimeisimmän linjan hoitona käytetylle lenalidomidille.
Hoidon keston mediaani oli Isa‑Pd-ryhmässä 41,0 viikkoa ja Pd‑ryhmässä 24,0 viikkoa.
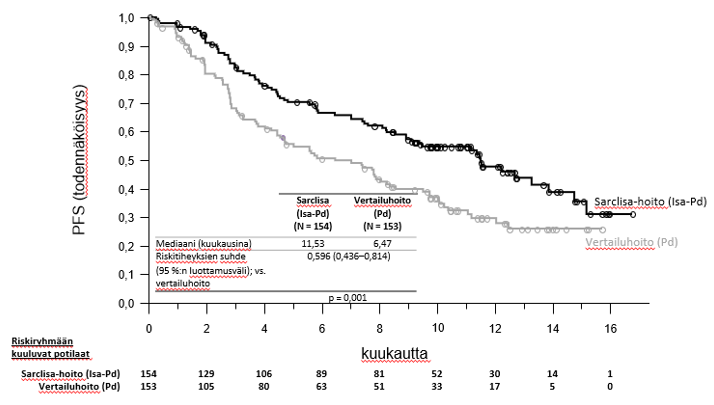
ICARIA-MM-tutkimuksen ensisijainen tehoa koskeva päätetapahtuma oli etenemättömyysaika (progression free survival, PFS). Isa‑Pd-hoitoa saaneilla potilailla etenemättömyysajan piteneminen merkitsi 40,4 %:n pienenemistä sairauden etenemisen tai kuoleman riskin suhteen.
Tehoa koskevat tulokset on esitetty taulukossa 13, ja etenemättömyysajan ja kokonaiselinajan (overall survival, OS) Kaplan-Meier-käyrät on esitetty kuvissa 1 ja 2:
Taulukko 13: Isatuksimabin teho yhdistelmänä pomalidomidin ja deksametasonin kanssa verrattuna pomalidomidiin ja deksametasoniin multippelin myelooman hoidossa (lähtöryhmien mukainen analyysi [intent-to-treat analysis, ITT analysis])
| Päätetapahtuma | Isatuksimabi + pomalidomidi + deksametasoni N = 154 | Pomalidomidi + deksametasoni N = 153 |
| Etenemättömyysaikaa b | ||
Mediaani (kuukausia) [95 %:n luottamusväli] | 11,53 [8,936–13,897] | 6,47 [4,468–8,279] |
| Riskitiheyksien suhde (HR)c [95 %:n luottamusväli] | 0,596 [0,436–0,814] | |
| p-arvo (ositettu log rank ‑testi)c | 0,0010 | |
Kokonaisvasteosuusd Vasteen saaneet (täydellinen vaste lisäehdoin [sCR] + täydellinen vaste [CR] + VGPR + osittainen vaste [PR]), n (%) [95 %:n luottamusväli]e | 93 (60,4) [0,5220–0,6817] | 54 (35,3) [0,2775–0,4342] |
| Kerroinsuhde vs. vertailu [95 %:n tarkka luottamusväli] | 2,795 [1,715–4,562] | |
| p-arvo (ositettu Cochran-Mantel-Haenszel)c | < 0,0001 | |
| sCR + CR, n (%) | 7 (4,5) | 3 (2,0) |
| VGPR, n (%) | 42 (27,3) | 10 (6,5) |
| PR, n (%) | 44 (28,6) | 41 (26,8) |
Vähintään VGPR, n (%) [95 %:n luottamusväli]e | 49 (31,8) [0,2455–0,3980] | 13 (8,5) [0,0460–0,1409] |
| Kerroinsuhde vs. vertailu [95 %:n tarkka luottamusväli] | 5,026 [2,514–10,586] | |
| p-arvo (ositettu Cochran-Mantel-Haenszel)c | < 0,0001 | |
Vasteen kestof* Mediaani kuukausina [95 %:n luottamusväli]g | 13,27 [10,612 – ei saavutettu] | 11,07 [8,542 – ei saavutettu] |
a Riippumaton vasteita arvioiva komitea (Independent Response Committee, IRC) arvioi etenemättömyysaikoja koskevat tulokset keskuslaboratoriosta saatujen M-proteiinia koskevien tietojen ja keskitetysti tehtyjen kuvantamistutkimusten tarkastelun perusteella käyttämällä International Myeloma Working Group (IMWG) ‑kriteerejä.
b Potilaat, joilla ei ollut etenevää tautia tai jotka eivät olleet kuolleet ennen analyysin tiedonkeruun päättymistä tai toisen myeloomahoidon aloituspäivää, sensuroitiin siitä päivästä, jolloin tehtiin viimeinen vaatimukset täyttävä taudin arviointi, jossa ei todettu taudin etenemistä ja joka tehtiin ennen mahdollisen toisen myeloomahoidon aloittamista tai analyysin tiedonkeruun päättymispäivää sen mukaan, mikä näistä toteutui ensin.
c Ositettu iän (< 75 vuotta vs. > 75 vuotta) ja aiempien hoitolinjojen määrän (2 tai 3 vs. > 3) mukaan interaktiivisen teknologian avulla saatujen vastausten (IRT) perusteella.
d IRC arvioi sCR:n, CR:n, VGPR:n ja PR:n käyttämällä IMWG-vastekriteerejä.
e Arvioitu Clopper-Pearsonin menetelmällä.
f Vasteen kesto määritettiin potilaille, jotka saavuttivat vähintään PR:n (93 potilasta isatuksimabihaarassa ja 54 potilasta vertailuhaarassa). Vasteen keston Kaplan-Meier-estimaatit.
g Kaplan-Meier-estimaattien luottamusväli lasketaan elossaolofunktion log-log-muunnoksella ja Brookmeyerin ja Crowleyn menetelmillä.
*Tiedonkeruun päättymispäivä 11.10.2018. Seuranta-ajan mediaani = 11,60 kuukautta. HR < 1 suosii Isa‑Pd-haaraa.
Potilailla, joilla oli suuri sytogeneettinen riski (keskuslaboratoriossa arvioitu), etenemättömyysajan mediaani oli 7,49 (95 %:n luottamusväli: 2,628 – ei laskettavissa) Isa‑Pd-ryhmässä ja 3,745 (95 %:n luottamusväli: 2,793–7,885) Pd‑ryhmässä (HR = 0,655; 95 %:n luottamusväli: 0,334–1,283). Etenemättömyysajan todettiin pidentyneen myös Isa‑Pd-ryhmän potilailla, jotka olivat > 75-vuotiaita (HR = 0,479; 95 %:n luottamusväli: 0,242–0,946), potilailla, joilla ISS-luokka oli III heidän aloittaessaan tutkimuksessa (HR = 0,635; 95 %:n luottamusväli: 0,363–1,110), potilailla, joilla lähtötilanteen kreatiniinipuhdistuma oli < 60 ml/min/1,73 m² (HR = 0,502; 95 %:n luottamusväli: 0,297–0,847), potilailla, joilla aiempien linjojen hoitojen määrä oli > 3 (HR = 0,590; 95 %:n luottamusväli: 0,356–0,977), potilailla, jotka olivat olleet refraktorisia aiemmalle lenalidomidihoidolle (HR = 0,593; 95 %:n luottamusväli: 0,431–0,816) tai hoidolle proteasomin estäjällä (HR = 0,578; 95 %:n luottamusväli: 0,405–0,824) sekä potilailla, jotka olivat olleet refraktorisia viimeisimmän linjan hoitona käytetylle lenalidomidille ennen aloittamistaan tutkimuksessa (HR = 0,601; 95 %:n luottamusväli: 0,436–0,828).
Ei ole saatavilla riittävästi tietoja, jotta voitaisiin tehdä johtopäätöksiä Isa‑Pd-hoidon tehosta potilailla, jotka ovat aiemmin saaneet daratumumabia (yksi potilas isatuksimabihaarassa eikä yhtään potilasta vertailuhaarassa).
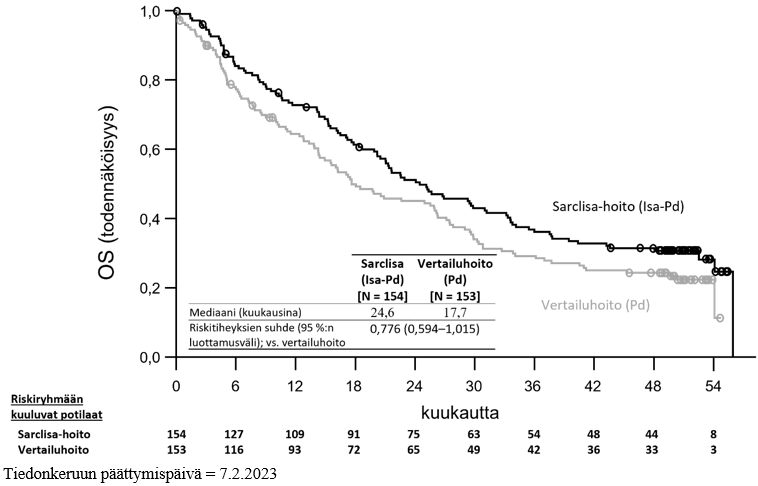
Mediaaniaika ensimmäiseen vasteeseen oli vasteen saaneilla 35 päivää Isa‑Pd-ryhmässä ja 58 päivää Pd‑ryhmässä. Kun seuranta-ajan mediaani oli 52,44 kuukautta, lopullinen kokonaiselinajan mediaani oli 24,57 kuukautta Isa‑Pd-ryhmässä ja 17,71 kuukautta Pd‑ryhmässä (HR = 0,776; 95 %:n luottamusväli: 0,594–1,015).
Kuva 1: Etenemättömyysajan Kaplan-Meier-käyrät – ITT-populaatio – ICARIA-MM (IRC arvioi)
Kuva 2 – Kokonaiselinajan Kaplan–Meier-käyrät – ITT-populaatio – ICARIA-MM
Tutkimuksessa ICARIA-MM (EFC14335) isatuksimabi-infuusiossa käytettiin painon mukaan määritettyä tilavuutta. Tutkimuksen TCD14079 osassa B arvioitiin vakioituun tilavuuteen perustuvaa infuusiomenetelmää, joka on kuvattu kohdassa Annostus ja antotapa, ja farmakokineettiset simulaatiot vahvistivat, että kun infuusiot annettiin potilaan painon mukaan määritettynä tilavuutena tai 250 ml:n vakioituna tilavuutena, erot farmakokinetiikassa olivat erittäin pienet (ks. kohta Farmakokinetiikka). Tutkimuksen TCD14079 osassa B ei havaittu uusia turvallisuussignaaleja eikä tehoon eikä turvallisuuteen liittyviä eroja ICARIA-MM-tutkimukseen verrattuna.
IKEMA (EFC15246)
Karfiltsomibin ja deksametasonin kanssa yhdistelmänä käytettävän isatuksimabin tehoa ja turvallisuutta arvioitiin relapsoitunutta ja/tai refraktorista multippelia myeloomaa sairastavilla potilailla kansainvälisessä, satunnaistetussa, avoimessa, kaksihaaraisessa vaiheen III monikeskustutkimuksessa IKEMA (EFC15246). Potilaat olivat saaneet yhdestä kolmeen aiempaa hoitoa. Potilaat, joilla oli primaarinen refraktorinen tauti tai jotka olivat aiemmin saaneet karfiltsomibia tai jotka olivat refraktorisia aiemmalle hoidolle monoklonaalisella CD38-vasta-aineella, suljettiin pois.
Yhteensä 302 potilasta satunnaistettiin suhteessa 3:2 saamaan joko isatuksimabia yhdistelmänä karfiltsomibin ja deksametasonin kanssa (Isa‑Kd-hoito, 179 potilasta) tai karfiltsomibia ja deksametasonia (Kd‑hoito, 123 potilasta). Hoitoa annettiin molemmissa ryhmissä 28 päivän hoitosykleinä, kunnes tauti eteni tai ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä. Isatuksimabi 10 mg/kg annettiin infuusiona laskimoon kerran viikossa ensimmäisen hoitosyklin aikana ja sen jälkeen kahden viikon välein. Karfiltsomibi annettiin infuusiona laskimoon annoksella 20 mg/m² päivinä 1 ja 2; annoksella 56 mg/m² hoitosyklin 1 päivinä 8, 9, 15 ja 16; ja annoksella 56 mg/m² kunkin myöhemmän 28 päivän pituisen hoitosyklin päivinä 1, 2, 8, 9, 15 ja 16. Deksametasoni (laskimoon isatuksimabi- ja/tai karfiltsomibi-infuusioiden antopäivinä, suun kautta muina päivinä) annettiin 20 mg:n annoksella kunkin 28 päivän pituisen hoitosyklin päivinä 1, 2, 8, 9, 15, 16, 22 ja 23.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat näiden kahden hoitoryhmän välillä kaiken kaikkiaan samankaltaiset. Potilaiden mediaani-ikä oli 64 vuotta (vaihteluväli 33–90), ja 8,9 % potilaista oli ≥ 75‑vuotiaita. ECOG-suorituskykypistemäärä oli 0 Isa‑Kd-ryhmässä 53,1 %:lla ja Kd‑ryhmässä 59,3 %:lla potilaista, se oli 1 Isa‑Kd-ryhmässä 40,8 %:lla ja Kd‑ryhmässä 36,6 %:lla potilaista, 2 Isa‑Kd-ryhmässä 5,6 %:lla ja Kd‑ryhmässä 4,1 %:lla potilaista ja 3 Isa‑Kd-ryhmässä 0,6 %:lla ja Kd‑ryhmässä 0 %:lla potilaista. Munuaisten vajaatoimintaa (eGFR < 60 ml/min/1,73 m2) sairastavien potilaiden osuus oli 24,0 % Isa‑Kd-ryhmässä ja 14,6 % Kd‑ryhmässä. Potilaiden aloittaessa tutkimuksessa heistä 53,0 %:lla oli luokan I tauti, 31,1 %:lla luokan II tauti ja 15,2 %:lla luokan III tauti kansainvälisen luokitusjärjestelmän (ISS) mukaan. Revised-ISS-luokitusjärjestelmän (R-ISS) mukaan 25,8 %:lla potilaista oli luokan I tauti, 59,6 %:lla luokan II tauti ja 7,9 %:lla luokan III tauti heidän aloittaessaan tutkimuksessa. Yhteensä 24,2 %:lla potilaista oli tutkimuksessa aloittaessaan kromosomipoikkeavuus, johon liittyi suuri riski: 11,3 %:lla potilaista oli del(17p)-mutaatio, 13,9 %:lla potilaista t(4;14)-mutaatio ja 2,0 %:lla t(14;16)-mutaatio. Lisäksi 42,1 %:lla potilaista oli gain(1q21)-poikkeavuus.
Aiempien linjojen hoitojen määrän mediaani oli 2 (vaihteluväli 1–4), ja 44,4 % potilaista oli saanut yhtä aiemman linjan hoitoa. Yhteensä 89,7 % potilaista oli saanut aiemmin proteasomin estäjää, 78,1 % oli saanut aiemmin immuunivasteen muuntajaa (mukaan lukien ne 43,4 % potilaista, jotka olivat saaneet aiemmin lenalidomidia) ja 61,3 % oli saanut aiemmin kantasolusiirron. Yhteensä 33,1 % potilaista oli refraktorisia aiemmin annetulle proteasomin estäjälle, 45,0 % oli refraktorisia aiemmin annetulle immuunivasteen muuntajalle (mukaan lukien ne 32,8 % potilaista, jotka olivat refraktorisia lenalidomidille) ja 20,5 % oli refraktorisia sekä proteasomin estäjälle että immuunivasteen muuntajalle.
Hoidon keston mediaani oli Isa‑Kd-ryhmässä 80,0 viikkoa ja Kd‑ryhmässä 61,4 viikkoa.
IKEMA-tutkimuksen ensisijainen tehoa koskeva päätetapahtuma oli etenemättömyysaika (PFS). Kun seuranta-ajan mediaani oli 20,73 kuukautta, etenemättömyysajan primaarianalyysissa Isa‑Kd-ryhmän potilailla todettiin tilastollisesti merkitsevä etenemättömyysajan piteneminen, mikä merkitsi sairauden etenemisen tai kuoleman riskin pienenemistä 46,9 %:lla verrattuna Kd‑ryhmän potilaisiin.
Tehoa koskevat tulokset on esitetty taulukossa 14, ja etenemättömyysajan Kaplan–Meier-käyrät on esitetty kuvassa 3 ja kokonaiselinajan Kaplan–Meier-käyrät kuvassa 4.
Taulukko 14: Isatuksimabin teho yhdistelmänä karfiltsomibin ja deksametasonin kanssa verrattuna karfiltsomibiin ja deksametasoniin multippelin myelooman hoidossa (lähtöryhmien mukainen analyysi)
| Päätetapahtuma | Isatuksimabi + karfiltsomibi + deksametasoni N = 179 | Karfiltsomibi + deksametasoni N = 123 |
Etenemättömyysaikaa Mediaani (kuukausia) [95 %:n luottamusväli] Riskitiheyksien suhde (HR)b [99 %:n luottamusväli] | Ei saavutettu [ei saavutettu – ei saavutettu] | 19,15 [15,77 – ei saavutettu] |
| 0,531 [0,318–0,889] | ||
| p‑arvo (ositettu log rank ‑testi)b | 0,0013 | |
Kokonaisvasteosuusc Vasteen saaneet (sCR + CR + VGPR + PR) [95 %:n luottamusväli]d | 86,6 % [0,8071–0,9122] | 82,9 % [0,7509–0,8911] |
| p‑arvo (ositettu Cochran–Mantel–Haenszel)b | 0,3859 | |
| CR | 39,7 % | 27,6 % |
| VGPR | 33,0 % | 28,5 % |
| PR | 14,0 % | 26,8 % |
Vähintään VGPR (sCR + CR +VGPR) [95 %:n luottamusväli]d | 72,6 % [0,6547–0,7901] | 56,1 % [0,4687–0,6503] |
| p‑arvo (ositettu Cochran–Mantel–Haenszel)b e | 0,0021 | |
CRf [95 %:n luottamusväli]d | 39,7 % [0,3244–0,4723] | 27,6 % [0,1996–0,3643] |
Minimaalisen jäännöstaudin suhteen negatiivisten potilaiden osuusg [95 %:n luottamusväli]d | 29,6 % [0,2303–0,3688] | 13,0 % [0,0762–0,2026] |
| p‑arvo (ositettu Cochran–Mantel–Haenszel)b e | 0,0008 | |
Vasteen kestoh* (vähintään PR) Mediaani kuukausina (95 %:n luottamusväli)i | Ei saavutettu [ei saavutettu – ei saavutettu] | Ei saavutettu [14,752 – ei saavutettu] |
| Riskitiheyksien suhde (HR)b [95 %:n luottamusväli] | 0,425 [0,269–0,672] | |
a Riippumaton vasteita arvioiva komitea arvioi etenemättömyysaikoja koskevat tulokset keskuslaboratoriosta saatujen M‑proteiinia koskevien tietojen ja keskitetysti tehdyn kuvantamistutkimusten tarkastelun perusteella käyttämällä IMWG‑kriteerejä.
b Ositettu aiempien hoitolinjojen määrän (1 vs. > 1) ja R‑ISS-luokan (I tai II vs. III vs. ei luokiteltu) mukaan interaktiivisen teknologian avulla saatujen vastausten perusteella.
c IRC arvioi sCR:n, CR:n, VGPR:n ja PR:n käyttämällä IMWG-vastekriteerejä.
d Arvioitu Clopper–Pearsonin menetelmällä.
e Nimellinen p‑arvo.
f CR tutkitaan lopullisen analyysin yhteydessä.
g Perustuu herkkyystasoon 10-5 NGS‑menetelmän mukaan ITT‑populaatiossa.
h Perustuu vasteen saaneisiin ITT‑populaatiossa. Vasteen keston Kaplan–Meier-estimaatit.
i Kaplan–Meier-estimaattien luottamusväli lasketaan elossaolofunktion log-log-muunnoksella ja Brookmeyerin ja Crowleyn menetelmillä.
* Tiedonkeruun päättymispäivä 7.2.2020. Seuranta-ajan mediaani = 20,73 kuukautta. HR < 1 suosii Isa‑Kd-haaraa.
Etenemättömyysajan todettiin parantuneen Isa‑Kd-ryhmässä potilailla, joilla oli suuri sytogeneettinen riski (keskuslaboratoriossa arvioitu; HR = 0,724; 95 %:n luottamusväli: 0,361–1,451), potilailla, joilla oli gain(1q21)-kromosomipoikkeavuus (HR = 0,569; 95 %:n luottamusväli: 0,330–0,981), ≥ 65‑vuotiailla potilailla (HR = 0,429; 95 %:n luottamusväli: 0,248–0,742), potilailla, joilla lähtötilanteen eGFR‑arvo (MDRD-kaavalla laskettuna) oli < 60 ml/min/1,73 m² (HR = 0,273; 95 %:n luottamusväli: 0,113–0,660), potilailla, joilla aiempien linjojen hoitojen määrä oli > 1 (HR = 0,479; 95 %:n luottamusväli: 0,294–0,778), potilailla, joilla ISS‑luokka oli III heidän aloittaessaan tutkimuksessa (HR = 0,650; 95 %:n luottamusväli: 0,295–1,434), sekä potilailla, jotka olivat olleet refraktorisia aiemmalle lenalidomidihoidolle (HR = 0,598; 95 %:n luottamusväli: 0,339–1,055).
Herkkyysanalyysissä, jossa tietoja ei sensuroitu toisen myeloomahoidon perusteella, etenemättömyysajan mediaania ei saavutettu Isa‑Kd-ryhmässä ja se oli 19,0 kuukautta (95 %:n luottamusväli: 15,38 – ei saavutettu) Kd‑ryhmässä (HR = 0,572; 99 %:n luottamusväli: 0,354–0,925; p = 0,0025).
Ei ole saatavilla riittävästi tietoja, jotta voitaisiin tehdä johtopäätöksiä Isa‑Kd-hoidon tehosta potilailla, jotka ovat aiemmin saaneet daratumumabia (yksi potilas isatuksimabihaarassa eikä yhtään potilasta vertailuhaarassa).
Mediaaniaika ensimmäiseen vasteeseen oli 1,08 kuukautta Isa‑Kd-ryhmässä ja 1,12 kuukautta Kd‑ryhmässä. Mediaaniaika seuraavaan myeloomahoitoon oli 43,99 kuukautta Isa‑Kd-ryhmässä ja 25,00 kuukautta Kd‑ryhmässä (HR = 0,583; 95 %:n luottamusväli: 0,429–0,792).
Kuva 3 –Etenemättömyysajan Kaplan–Meier-käyrät – ITT-populaatio – IKEMA (IRC arvioi)
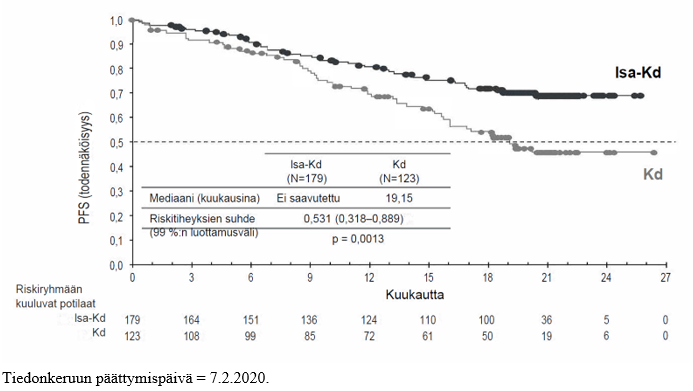
Kuva 4: Kokonaiselinajan Kaplan–Meier-käyrät – ITT-populaatio – IKEMA
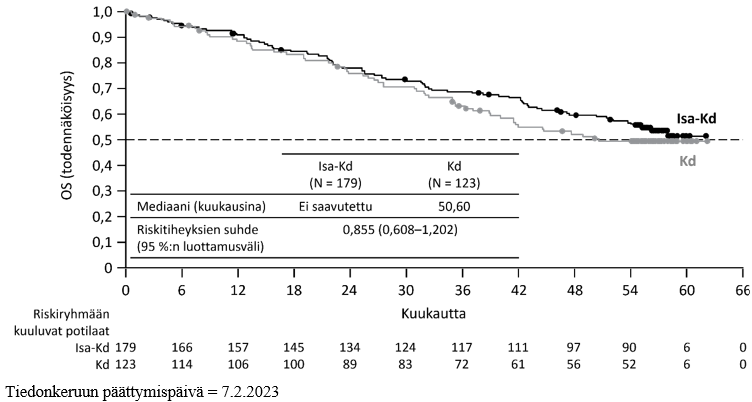
Potilaista, joilla eGFR (MDRD-kaavalla laskettuna) oli lähtötilanteessa < 50 ml/min/1,73 m2, todettiin täydellinen munuaisia koskeva vaste (≥ 60 ml/min/1,73 m2 ainakin yhdessä arvioinnissa lähtötilanteen jälkeen) 52,0 %:lla (13/25) Isa‑Kd-ryhmän potilaista ja 30,8 %:lla (4/13) Kd‑ryhmän potilaista. Pitkäkestoinen (≥ 60 päivää) täydellinen munuaisia koskeva vaste todettiin 32,0 %:lla (8/25) Isa‑Kd-ryhmän potilaista ja 7,7 %:lla (1/13) Kd‑ryhmän potilaista. Niistä neljästä Isa‑Kd-ryhmän potilaasta ja kolmesta Kd‑ryhmän potilaasta, joilla oli lähtötilanteessa vaikea munuaisten vajaatoiminta (MDRD-kaavalla laskettu eGFR > 15 – < 30 ml/min/1,73 m2), minimaalinen munuaisia koskeva vaste (≥ 30 – < 60 ml/min/1,73 m2 ainakin yhdessä arvioinnissa lähtötilanteen jälkeen) todettiin 100 %:lla potilaista Isa‑Kd-ryhmässä ja 33,3 %:lla potilaista Kd‑ryhmässä.
Seurannan keston mediaani oli 43,96 kuukautta etenemättömyysajan lopullisessa analyysissa. Tällöin etenemättömyysajan mediaani oli Isa-Kd-ryhmässä 35,65 kuukautta ja vastaavasti Kd-ryhmässä 19,15 kuukautta; riskitiheyksien suhde 0,576 (95,4 %:n luottamusväli: 0,418–0,792). Validoidulla isatuksimabi-spesifisellä IFE-määrityksellä (Sebia Hydrashift) lopullisessa analyysissa arvioitu täydellinen vaste (ks. kohta Yhteisvaikutukset) oli 44,1 % Isa-Kd-ryhmässä ja vastaavasti 28,5 % Kd-ryhmässä; kerroinsuhde 2,094 (95 %:n luottamusväli: 1,259–3,482, deskriptiivinen p = 0,0021). Isa-Kd-ryhmän potilaista 26,3 % saavutti sekä negatiivisen tuloksen minimaalisen jäännöstaudin suhteen että täydellisen vasteen. Vastaava osuus Kd-ryhmässä oli 12,2 %; kerroinsuhde 2,571 (95 %:n luottamusväli: 1,354–4,882, deskriptiivinen p = 0,0015).
Kun seurannan keston mediaani oli 56,61 kuukautta, kokonaiselinajan mediaania ei ollut saavutettu Isa-Kd-ryhmässä (95 %:n luottamusväli: 52,172 – ei saavutettu), ja Kd-ryhmässä se oli 50,60 kuukautta (95 %:n luottamusväli: 38,932 – ei saavutettu) (HR = 0,855; 95 %:n luottamusväli: 0,608–1,202).
Äskettäin todettu multippeli myelooma
IMROZ (EFC12522)
Bortetsomibin, lenalidomidin ja deksametasonin kanssa yhdistelmänä käytettävän isatuksimabin tehoa ja turvallisuutta arvioitiin kansainvälisessä, satunnaistetussa, avoimessa, kaksihaaraisessa vaiheen III monikeskustutkimuksessa IMROZ (EFC12522) potilailla, joilla oli äskettäin todettu multippeli myelooma ja joille kantasolusiirto ei soveltunut. Tutkimuksesta suljettiin pois potilaat, joilla oli äskettäin todettu multippeli myelooma ja jotka olivat yli 80‑vuotiaita tai joilla oli samanaikaisia sairauksia (esim. keuhko‑ tai sepelvaltimotauti), joiden vuoksi ei tutkijalääkärin lääketieteellisen arvioinnin perusteella voitu tehdä elinsiirtotoimenpiteitä.
Yhteensä 446 potilasta satunnaistettiin suhteessa 3:2 saamaan joko isatuksimabia yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa (Isa‑VRd‑hoito, 265 potilasta) tai bortetsomibia, lenalidomidia ja deksametasonia (VRd‑hoito, 181 potilasta). Hoitoa annettiin molemmissa ryhmissä neljän 42 päivän hoitosyklin aikana induktiojakson ajan. Kun hoitosykli 4 oli päättynyt, potilaat aloittivat jatkuvan hoitojakson hoitosyklistä 5 alkaen. 28 päivän hoitosyklejä jatkettiin, kunnes tauti eteni tai ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä. Jatkuvan hoitojakson aikana Isa‑VRd‑ryhmän potilaat saivat isatuksimabia yhdistelmänä lenalidomidin ja deksametasonin kanssa (Isa‑Rd‑hoito) ja VRd‑ryhmän potilaat saivat lenalidomidia ja deksametasonia (Rd‑hoito).
Induktiojakson aikana (hoitosyklit 1–4, 42 päivän pituiset hoitosyklit) isatuksimabi 10 mg/kg annettiin infuusiona laskimoon päivinä 1, 8, 15, 22 ja 29 ensimmäisen hoitosyklin ajan ja päivinä 1, 15 ja 29 hoitosyklien 2–4 ajan. Bortetsomibi annettiin ihon alle annoksella 1,3 mg/m2 kunkin hoitosyklin päivinä 1, 4, 8, 11, 22, 25, 29 ja 32. Lenalidomidi annettiin suun kautta annoksella 25 mg/vrk kunkin hoitosyklin päivinä 1–14 ja 22–35. Deksametasoni 20 mg/vrk (laskimoon isatuksimabi‑infuusioiden antopäivinä ja suun kautta muina päivinä) annettiin kunkin hoitosyklin päivinä 1, 2, 4, 5, 8, 9, 11, 12, 15, 22, 23, 25, 26, 29, 30, 32 ja 33 ja vähintään 75‑vuotiaille potilaille kunkin hoitosyklin päivinä 1, 4, 8, 11, 15, 22, 25, 29 ja 32.
Jatkuvan hoitojakson aikana (hoitosyklistä 5 alkaen, 28 päivän pituiset hoitosyklit) isatuksimabi 10 mg/kg annettiin infuusiona laskimoon hoitosyklien 5–17 päivinä 1 ja 15 ja päivänä 1 alkaen hoitosyklistä 18. Lenalidomidi annettiin suun kautta annoksella 25 mg/vrk kunkin hoitosyklin päivinä 1–21. Deksametasoni 20 mg/vrk (laskimoon isatuksimabi‑infuusioiden antopäivinä ja suun kautta muina päivinä) annettiin kunkin hoitosyklin päivinä 1, 8, 15 ja 22.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat näiden kahden hoitoryhmän välillä kaiken kaikkiaan samankaltaiset. Potilaiden mediaani‑ikä oli 72 vuotta (vaihteluväli 60–80), ja 26 % potilaista oli ≥ 75‑vuotiaita. ECOG‑suorituskykypistemäärä oli 0 Isa‑VRd‑ryhmässä 46,4 %:lla ja VRd‑ryhmässä 43,6 %:lla potilaista, 1 Isa‑VRd‑ryhmässä 42,3 %:lla ja VRd‑ryhmässä 45,9 %:lla potilaista, 2 Isa‑VRd‑ryhmässä 10,9 %:lla ja VRd‑ryhmässä 10,5 %:lla potilaista ja 3 Isa‑VRd‑ryhmässä 0,4 %:lla ja VRd‑ryhmässä 0 %:lla potilaista. Munuaisten vajaatoimintaa (eGFR < 60 ml/min/1,73 m2) sairastavien potilaiden osuus oli 24,9 % Isa‑VRd‑ryhmässä ja 34,3 % VRd‑ryhmässä. Potilaiden aloittaessa tutkimuksessa heistä 24,9 %:lla oli luokan I tauti, 61,5 %:lla luokan II tauti ja 10,2 %:lla luokan III tauti R‑ISS‑luokitusjärjestelmän mukaan. Yhteensä 15,1 %:lla potilaista oli tutkimuksessa aloittaessaan kromosomipoikkeavuus, johon liittyi suuri riski: 5,7 %:lla potilaista oli del(17p)‑mutaatio, 7,9 %:lla potilaista t(4;14)‑mutaatio ja 1,9 %:lla t(14;16)‑mutaatio. Lisäksi 35,8 %:lla potilaista oli 1q21+‑poikkeavuus.
Hoidon keston mediaani oli Isa‑VRd‑ryhmässä 53,2 kuukautta ja VRd‑ryhmässä 31,3 kuukautta.
IMROZ‑tutkimuksen ensisijainen tehoa koskeva päätetapahtuma oli etenemättömyysaika (PFS). Kun seuranta‑ajan mediaani oli 59,73 kuukautta, etenemättömyysajan etukäteen suunnitellussa toisessa välianalyysissä Isa‑VRd‑ryhmän potilailla todettiin tilastollisesti merkitsevä etenemättömyysajan piteneminen, mikä merkitsi sairauden etenemisen tai kuoleman riskin pienenemistä 40,4 %:lla verrattuna VRd‑ryhmän potilaisiin.
Tehoa koskevat tulokset on esitetty taulukossa 15, ja etenemättömyysajan Kaplan–Meier‑käyrät on esitetty kuvassa 5:
Taulukko 15*: Isatuksimabin teho yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa verrattuna bortetsomibiin, lenalidomidiin ja deksametasoniin äskettäin todetun multippelin myelooman hoidossa potilailla, jotka eivät soveltuneet siirteen saamiseen (lähtöryhmien mukainen analyysi)
| Päätetapahtuma | Isatuksimabi + bortetsomibi + lenalidomidi + deksametasoni N = 265 | Bortetsomibi + lenalidomidi + deksametasoni N = 181 |
Etenemättömyysaikaa Mediaani (kuukausia) [95 %:n luottamusväli] Riskitiheyksien suhde (HR)b [98,5 %:n luottamusväli] | Ei saavutettu [ei saavutettu – ei saavutettu] | 54,34 [45,21 – ei saavutettu] |
| 0,596 [0,406–0,876] | ||
| p‑arvo (ositettu log rank ‑testi)b | 0,0009 | |
Vähintään CR (sCR ja CR) [95 %:n luottamusväli]c | 74,7 % [0,6904–0,7984] | 64,1 % [0,5664–0,7107] |
| p‑arvo (ositettu log rank ‑testi)b | 0,0160 | |
Negatiivisuus minimaalisen jäännöstaudin suhteend ja CR [95 %:n luottamusväli]c | 55,5 % [0,4927–0,6155] | 40,9 % [0,3365–0,4842] |
| p‑arvo (ositettu Cochran–Mantel–Haenszel)b | 0,0026 | |
Kokonaisvasteosuuse Vasteen saaneet (sCR + CR + VGPR + PR) [95 %:n luottamusväli]c | 91,3 % [0,8726–0,9442] | 92,3 % [0,8736–0,9571] |
Täydellinen vaste lisäehdoin (Stringent Complete Response, sCR) Täydellinen vaste (Complete Response, CR) Erittäin hyvä ositteinen vaste (Very Good Partial Response, VGPR) Osittainen vaste (Partial Response, PR) | 10,9 % 63,8 % 14,3 % 2,3 % | 5,5 % 58,6 % 18,8 % 9,4 % |
a Riippumaton vasteita arvioiva komitea (Independent Response Committee, IRC) arvioi etenemättömyysaikoja koskevat tulokset keskuslaboratoriosta saatujen M‑proteiinia koskevien tietojen ja keskitetysti tehdyn kuvantamistutkimusten tarkastelun perusteella käyttämällä IMWG‑kriteerejä.
b Ositettu iän (< 70 vuotta vs. ≥ 70 vuotta) ja R‑ISS‑luokan (I tai II vs. III tai ei luokiteltu) mukaan interaktiivisen teknologian avulla saatujen vastausten perusteella.
c Arvioitu Clopper–Pearsonin menetelmällä.
d Perustuu herkkyystasoon 10‑5 NGS‑menetelmän mukaan ITT‑populaatiossa.
e IRC arvioi sCR:n, CR:n, VGPR:n ja PR:n käyttämällä IMWG‑vastekriteerejä. Tulokset on tulkittava deskriptiivisesti.
* Tiedonkeruun päättymispäivä 26.9.2023. Seuranta‑ajan mediaani = 59,73 kuukautta.
Etenemättömyysajan piteneminen vahvistettiin herkkyysanalyysien avulla. Etenemättömyysajan todettiin pidentyneen useimmissa alaryhmissä, myös potilailla, joilla oli 1q21+‑kromosomipoikkeavuus (HR = 0,407; 95 %:n luottamusväli: 0,253–0,653), ≥ 70‑vuotiailla potilailla (HR = 0,671; 95 %:n luottamusväli: 0,463–0,972), potilailla, joilla lähtötilanteen eGFR‑arvo (MDRD‑kaavalla laskettuna) oli < 60 ml/min/1,73 m2 (HR = 0,63; 95 %:n luottamusväli: 0,371–1,068), sekä potilailla, joilla ECOG‑suorituskykypistemäärä oli > 1 (HR = 0,606; 95 %:n luottamusväli: 0,246–1,493).
NGS‑menetelmällä määritetty negatiivisuus minimaalisen jäännöstaudin suhteen (herkkyyskynnys 10‑5) saavutettiin 58,1 %:lla Isa‑VRd‑ryhmän potilaista, ja mediaaniaika ensimmäisen tällaisen NGS‑menetelmällä määritetyn negatiivisuuden saavuttamiseen oli 196,5 päivää (vaihteluväli: 87 – 1 834). VRd‑ryhmässä NGS‑menetelmällä määritetty negatiivisuus minimaalisen jäännöstaudin suhteen (herkkyyskynnys 10‑5) saavutettiin 43,6 %:lla potilaista, joilla mediaaniaika ensimmäisen tällaisen NGS‑menetelmällä määritetyn negatiivisuuden saavuttamiseen oli 197,0 päivää (vaihteluväli: 107 – 1 512).
Pitkäkestoinen, vähintään 12 kuukautta jatkunut NGS‑menetelmällä määritetty negatiivisuus minimaalisen jäännöstaudin suhteen havaittiin 46,8 %:lla Isa‑VRd‑ryhmän potilaista ja 24,3 %:lla VRd‑ryhmän potilaista.
Etenemiseen kuluneen ajan mediaania ei saavutettu Isa‑VRd‑ryhmässä, ja VRd‑ryhmässä se oli 59,70 kuukautta (95 %:n luottamusväli: 48,164 – ei saavutettu) (HR = 0,414; 95 %:n luottamusväli: 0,286–0,598). Vasteen keston mediaania ei saavutettu Isa‑VRd‑ryhmässä, ja VRd‑ryhmässä se oli 58,25 kuukautta (95 %:n luottamusväli: 44,583 – ei saavutettu). Mediaaniaika ensimmäiseen vasteeseen oli 1,51 kuukautta Isa‑VRd‑ryhmässä ja 1,48 kuukautta VRd‑ryhmässä. Isa‑VRd‑ryhmässä tutkimushoidon lopetti 52,1 % potilaista; 14,3 % lopetti hoidon taudin etenemisen vuoksi. VRd‑ryhmässä tutkimushoidon lopetti 75,7 % potilaista; 37 % lopetti hoidon taudin etenemisen vuoksi. Seuraavaan myeloomahoitoon kuluneen ajan mediaania ei saavutettu Isa‑VRd‑ryhmässä, ja VRd‑ryhmässä se oli 63,57 kuukautta (HR = 0,376; 95 %:n luottamusväli: 0,265–0,534). Kokonaiselinajan mediaania ei saavutettu kummassakaan hoitoryhmässä. Kokonaiselinaikatietoja koskevan deskriptiivisen analyysin perusteella 26 % Isa‑VRd‑ryhmän potilaista ja 32,6 % VRd‑ryhmän potilaista oli kuollut (HR = 0,776; 99,97 %:n luottamusväli: 0,407–1,48).
Kuva 5 – Etenemättömyysajan Kaplan–Meier‑käyrät – ITT‑populaatio – IMROZ (IRC arvioi)
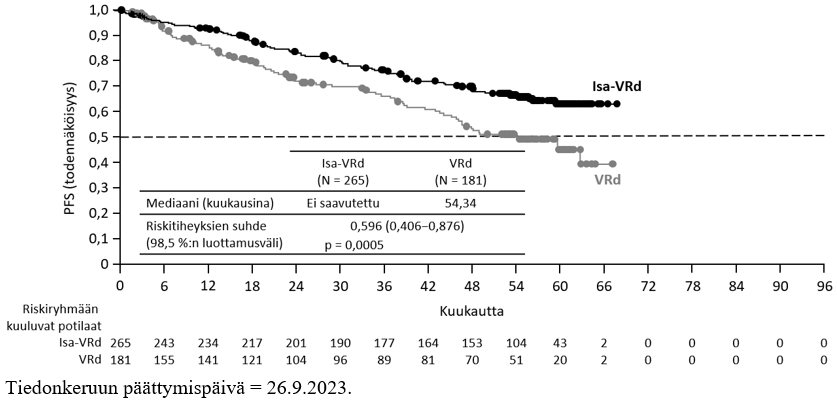
GMMG HD7 (IIT 15403)
Bortetsomibin, lenalidomidin ja deksametasonin kanssa yhdistelmänä käytettävän isatuksimabin tehoa ja turvallisuutta arvioitiin prospektiivisessa, satunnaistetussa, avoimessa, kaksihaaraisessa vaiheen III tutkimuksessa GMMG-HD7, jonka toteutti German-Speaking Myeloma Multicenter Group (GMMG) potilailla, joilla oli äskettäin todettu multippeli myelooma ja joille kantasolusiirto soveltui. Arvio kantasolusiirron soveltumisesta potilaalle perustui pääasiassa potilaan ikään (≤ 70 vuotta) tai samanaikaisiin sairauksiin, jotka olisivat voineet estää potilasta saamasta suuriannoksisia hoitoja yhdessä autologisen siirron kanssa. Tutkimus toteutettiin kahdessa osassa: osa 1 käsittää induktio- ja intensiivihoidot, ja osa 2 on ylläpitohoito (meneillään oleva vaihe). Osan 1 tulokset on kuvattu.
Yhteensä 662 potilasta satunnaistettiin suhteessa 1:1 saamaan joko isatuksimabia yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa (Isa-VRd-hoito, 331 potilasta) tai bortetsomibia, lenalidomidia ja deksametasonia (VRd-hoito, 331 potilasta). Hoitoa annettiin molemmissa ryhmissä kolmen 42 päivän hoitosyklin (hoitosyklit 1–3) aikana induktiohoidon ajan. Hoitosyklin 3 jälkeen potilaille annettiin tavanomaista intensiivihoitoa, autologinen kantasolusiirto mukaan lukien. Autologinen kantasolusiirto toistettiin 2–3 kuukauden kuluttua ensimmäisestä autologisesta kantasolusiirrosta, jos potilaalla ei ollut saavutettu täydellistä vastetta tai potilas kuului suuren riskin ryhmään. Osassa 2, ennen ylläpitohoidon aloittamista, potilaat satunnaistettiin uudelleen suhteessa 1:1 saamaan joko isatuksimabia yhdistelmänä lenalidomidin kanssa tai pelkkää lenalidomidia. Ylläpitohoitoa annetaan 28 päivän hoitosykleissä enintään 3 vuoden ajan, kunnes tauti etenee tai ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, sen mukaan, kumpi näistä toteutuu ensin. Potilaat saivat molemmissa hoitohaaroissa deksametasonia vain ensimmäisen ylläpitohoitosyklin ajan.
Induktiojakson aikana isatuksimabi 10 mg/kg annettiin infuusiona laskimoon hoitosyklin 1 päivinä 1, 8, 15, 22 ja 29 ja hoitosyklien 2–3 päivinä 1, 15 ja 29. Bortetsomibi annettiin ihon alle annoksella 1,3 mg/m² kunkin hoitosyklin päivinä 1, 4, 8, 11, 22, 25, 29 ja 32. Lenalidomidi annettiin suun kautta annoksella 25 mg/vrk kunkin hoitosyklin päivinä 1–14 ja 22–35. Deksametasoni 20 mg/vrk (laskimoon isatuksimabi-infuusioiden antopäivinä ja suun kautta muina päivinä) annettiin kunkin hoitosyklin päivinä 1, 2, 4, 5, 8, 9, 11, 12, 15, 22, 23, 25, 26, 29, 30, 32 ja 33.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat näiden kahden hoitoryhmän (Isa-VRd ja VRd) välillä kaiken kaikkiaan samankaltaiset. Potilaiden mediaani-ikä oli 59,5 vuotta (vaihteluväli 26–70), ja 23,9 % potilaista oli ≥ 65 vuotiaita. Munuaisten vajaatoimintaa (eGFR < 60 ml/min/1,73 m2) sairastavien potilaiden osuus oli 19,7 % Isa-VRd-ryhmässä ja 17,7 % VRd-ryhmässä. Potilaiden aloittaessa tutkimuksessa heistä 41,4 %:lla oli luokan I tauti, 36,9 %:lla luokan II tauti ja 21,8 %:lla luokan III tauti kansainvälisen luokitusjärjestelmän (ISS) mukaan. Revised-ISS-luokitusjärjestelmän (R ISS) mukaan 26,6 %:lla oli luokan I tauti, 61 %:lla luokan II tauti ja 8 %:lla luokan III tauti heidän aloittaessaan tutkimuksessa. Yhteensä 18,7 %:lla potilaista oli tutkimuksessa aloittaessaan kromosomipoikkeavuus, johon liittyi suuri riski: 8,9 %:lla potilaista oli del(17p)-mutaatio, 10,1 %:lla t(4;14)-mutaatio ja 2,6 %:lla t(14;16)-mutaatio. Lisäksi 32,9 %:lla potilaista oli 1q21+-poikkeavuus.
Induktiohoidon keston mediaani oli Isa-VRd- ja VRd-ryhmissä 18 viikkoa. Osan 1 (induktio- ja intensiivihoidot) keston mediaani oli Isa-VRd-ryhmässä 45,4 viikkoa ja VRd-ryhmässä 45,1 viikkoa.
Minimaalisen jäännöstaudin suhteen negatiivisten potilaiden osuus induktiohoidon jälkeen, GMMG-HD7-tutkimuksen ensisijainen tehoa koskeva päätetapahtuma, oli 50,5 % (95 %:n luottamusväli: 44,9–56 %) Isa-VRd-haarassa ja 35,6 % (95 %:n luottamusväli: 30,5–41,4 %) VRd-haarassa (kerroinsuhde 1,838, 95 %:n luottamusväli: 1,346–2,511, p-arvo 0,0001). Tulokset (NGF-menetelmä, herkkyystaso 10-5) osoittivat minimaalisen jäännöstaudin suhteen negatiivisten potilaiden osuuden suurentuneen; induktiohoidon jälkeen tämä osuus oli Isa-VRd-haarassa 14,8 % suurempi kuin VRd-haarassa.
Kun seuranta-ajan mediaani oli 49,31 kuukautta, etenemättömyysajan analyysissä ensimmäisestä satunnaistamisesta lähtien todettiin tilastollisesti merkitsevä paranema; Isa-VRd-hoitoa saaneilla potilailla sairauden etenemisen tai kuoleman riski pieneni 35,7 % verrattuna VRd-hoitoa saaneisiin potilaisiin induktiohoidon aikana, kun sen jälkeen tehtiin autologinen kantasolusiirto ja annettiin lenalidomidiylläpitohoitoa (HR = 0,643; 95 %:n luottamusväli: 0,456–0,907).
Tehoa koskevat tulokset on esitetty taulukossa 16, ja etenemättömyysajan Kaplan–Meier-käyrät on esitetty kuvassa 6.
Taulukko 16: Isatuksimabin teho yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa verrattuna bortetsomibiin, lenalidomidiin ja deksametasoniin äskettäin todetun multippelin myelooman hoidossa potilailla, jotka soveltuivat siirteen saamiseen (lähtöryhmien mukainen analyysi)
| Päätetapahtuma | Isatuksimabi + bortetsomibi + lenalidomidi + deksametasoni (N = 331) | Bortetsomibi + lenalidomidi + deksametasoni (N = 331) |
|---|---|---|
Etenemättömyysaika 1. satunnaistamisesta lähtien, satunnaistamisen jälkeen lenalidomidiylläpitohoito Mediaani (kuukausia) |
Ei saavutettu |
Ei saavutettu
|
| [95 %:n luottamusväli] | [Ei saavutettu – ei saavutettu] | [51,351 – ei saavutettu] |
| Riskitiheyksien suhde [95 %:n luottamusväli] | 0,643 [0,456–0,907] | |
| p‑arvoa (ositettu log rank ‑testi)b | 0,0111 | |
a Kaksitahoinen merkitsevyystaso on 0,028.
b Ositettu R-ISS-luokan (I tai II vs. III vs. ei luokiteltu) mukaan interaktiivisen teknologian avulla saatujen vastausten perusteella.
Induktiojakson päättyessä täydellisten vasteiden osuus oli Isa-VRd-ryhmässä 24,8 % (95 %:n luottamusväli: 20,2–29,8 %) ja VRd-ryhmässä 22,1 % (95 %:n luottamusväli: 17,7–26,9 %). Intensiivijakson päättyessä täydellisten vasteiden osuus oli Isa-VRd-ryhmässä 46,5 % (95 %:n luottamusväli: 41,1–52,1 %) ja VRd-ryhmässä 37,2 % (95 %:n luottamusväli: 31,9–42,6 %).
Isa-VRd-ryhmän potilaista 18,7 %:lla oli saavutettu sekä NGF-menetelmällä määritetty negatiivinen tulos minimaalisen jäännöstaudin suhteen (herkkyyskynnys 10 5) että täydellinen vaste induktiojakson päättyessä. VRd-ryhmässä vastaava osuus oli 14,5 %. Isa-VRd-ryhmän potilaista 40,5 %:lla oli saavutettu sekä NGF-menetelmällä määritetty negatiivinen tulos minimaalisen jäännöstaudin suhteen (herkkyyskynnys 10-5) että täydellinen vaste intensiivijakson päättyessä. VRd-ryhmässä vastaava osuus oli 26,6 %.
Induktiojakson aikana 5,4 % Isa-VRd-ryhmän potilaista ja 10,6 % VRd-ryhmän potilaista lopetti tutkimushoidon.
14,8 % Isa-VRd-haaran potilaista ja 12,7 % VRd-haaran potilaista oli kuollut, kun seuranta-ajan mediaani oli 49,31 kuukautta.
Kuva 6 – Etenemättömyysajan Kaplan–Meier-käyrät – ITT-populaatio – GMMG HD7
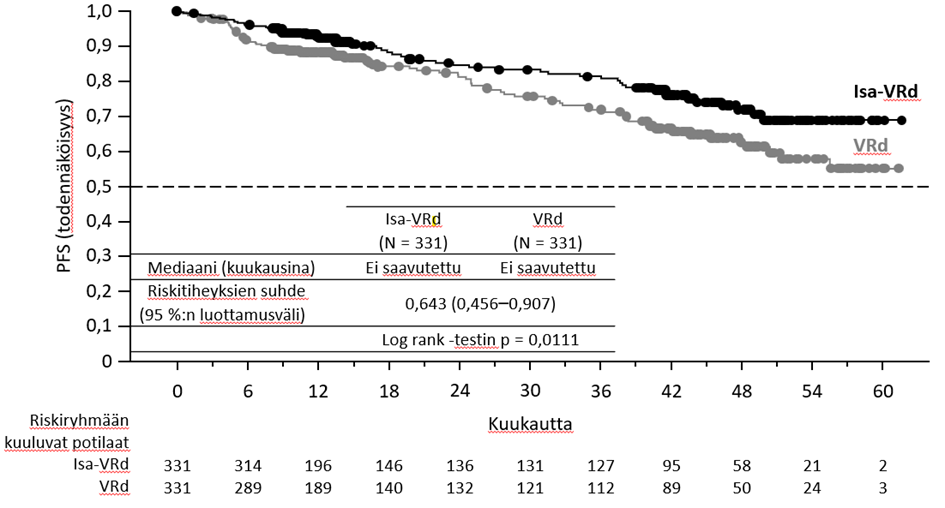
Tiedonkeruun päättymispäivä = 31.1.2024
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset laskimoon annettavan isatuksimabin käytöstä hematopoieettisen kudoksen ja imukudoksen pahanlaatuisten kasvainten hoidossa yhdessä tai useammissa pediatrisissa potilasryhmissä. Ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
67 pediatrista potilasta osallistui yksihaaraiseen vaiheen 2 tutkimukseen, joka toteutettiin kolmessa eri tautikohortissa. Tehon suhteen voitiin arvioida 59 potilasta. Näillä potilailla oli relapsoitunut tai refraktorinen T-solujen akuutti lymfoblastileukemia (T-ALL, 11 potilasta), B-solujen akuutti lymfoblastileukemia (B-ALL, 25 potilasta) tai akuutti myelooinen leukemia (AML, 23 potilasta). Potilailla, joilla oli T- tai B-solujen akuutti lymfoblastileukemia, hoito koostui yhdestä induktiohoitojaksosta ja yhdestä konsolidaatiohoitojaksosta. Akuuttia myelooista leukemiaa sairastavilla potilailla hoito koostui enintään kahdesta induktiohoitojaksosta. Potilaiden mediaani-ikä oli 8 vuotta (vaihteluväli 17 kuukautta – 17 vuotta). Potilaat saivat isatuksimabia yhdistelmänä tavanomaisten solunsalpaajahoitojen (esim. antimetaboliittien, antrasykliinien ja alkyloivien lääkeaineiden) kanssa. Välianalyysin kohdalla täydellisten vasteiden osuus (ensisijainen tehoa koskeva päätetapahtuma; määritelmänä täydellinen vaste [CR] tai täydellinen vaste ja epätäydellinen perifeerinen toipuminen [CRi]) ei yltänyt ennalta määriteltyyn tilastolliseen kynnysarvoon kolmessa kohortissa; 52,0 % B-ALL-potilaista, 45,5 % T-ALL-potilaista ja 60,9 % AML-potilaista saavutti täydellisen vasteen (CR + CRi). Tutkimus keskeytettiin ennalta määritellyn välianalyysin jälkeen.
Farmakokinetiikka
Isatuksimabin farmakokinetiikkaa arvioitiin 476:lla multippelia myeloomaa sairastavalla potilaalla, jotka saivat isatuksimabia infuusiona laskimoon monoterapiana tai yhdistelmänä pomalidomidin ja deksametasonin kanssa annoksella 1–20 mg/kg, joka annettiin joko kerran viikossa, kahden viikon välein, kahden viikon välein kahdeksan viikon ajan ja sen jälkeen neljän viikon välein tai kerran viikossa neljän viikon ajan ja sen jälkeen kahden viikon välein.
Isatuksimabin farmakokinetiikka on ei-lineaarinen ja sen dispositio on kohdevälitteinen, koska se sitoutuu CD38-reseptoriin.
Isatuksimabialtistus (pitoisuutta plasmassa ajan funktiona kuvaavan käyrän alla oleva pinta-ala annosvälillä, AUC) suurenee enemmän kuin suhteessa annokseen, kun annos on 1–20 mg/kg kahden viikon välein, kun taas suhteessa annokseen ei havaita poikkeamaa, kun annos on 5–20 mg/kg kerran viikossa neljän viikon ajan ja sen jälkeen kahden viikon välein. Tämä johtuu ei-lineaarisen kohdevälitteisen puhdistuman suuresta vaikutuksesta kokonaispuhdistumaan alle 5 mg/kg:n annoksilla, kun taas suurilla annoksilla tämä vaikutus muuttuu merkityksettömäksi. Kun isatuksimabia annettiin annoksena 10 mg/kg kerran viikossa neljän viikon ajan ja sen jälkeen kahden viikon välein, mediaaniaika vakaan tilan saavuttamiseen oli 18 viikkoa ja kumulaatio oli 3,1-kertainen.
Kliinisessä ICARIA‑MM-tutkimuksessa, jossa relapsoitunutta ja/tai refraktorista multippelia myeloomaa sairastavat potilaat saivat isatuksimabia yhdistelmänä pomalidomidin ja deksametasonin kanssa, vakaan tilan keskimääräinen (variaatiokerroin prosentteina) ennustettu huippupitoisuus plasmassa (Cmax) oli 351 µg/ml (36,0 %) ja AUC 72 600 µg·h/ml (51,7 %). Vaikka isatuksimabi-infuusion antamistapa vaihdettiin painon mukaan määritetyn tilavuuden antamiseen perustuvasta menetelmästä vakioituun tilavuuteen perustuvaan infuusiomenetelmään johtaen muutoksiin tmax-arvoissa, muutoksen vaikutus farmakokineettiseen altistukseen oli vähäinen: todettiin samansuuruiset simuloidut Cmax-arvot vakaassa tilassa (283 µg/ml vs. 284 µg/ml) ja Ctrough-arvot 4 viikon kohdalla (119 µg/ml vs. 119 µg/ml) keskipainoisilla (76 kg) potilailla. Cmax- ja Ctrough-arvot olivat vertailukelpoiset myös muissa painoryhmissä.
Kliinisessä IKEMA-tutkimuksessa, jossa relapsoitunutta ja/tai refraktorista multippelia myeloomaa sairastavat potilaat saivat isatuksimabia yhdistelmänä karfiltsomibin ja deksametasonin kanssa, vakaan tilan keskimääräinen (variaatiokerroin prosentteina) ennustettu huippupitoisuus plasmassa (Cmax) oli 637 µg/ml (30,9 %) ja AUC 152 000 µg·h/ml (37,8 %).
Kun isatuksimabia annettiin yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa suositellulla annoksella ja ohjelmalla, isatuksimabin vakaan tilan keskimääräinen (variaatiokerroin prosentteina) ennustettu Cmax oli 604 µg/ml (21,9 %) ja AUC 155 000 µg·h/ml (23,9 %). Äskettäin todettua multippelia myeloomaa koskeneissa kliinisissä tutkimuksissa lääkevasta ainepositiivisilla potilailla havaittiin trendinomaisesti pienempiä altistuksia; kumulatiiviset AUC arvot pienenivät ensimmäisten 4 tai 6 hoitoviikon aikana (hoitosykli 1) lääkevasta-ainepositiivilla potilailla 23–29 % verrattuna lääkevasta-ainenegatiivisiin potilaisiin. Lääkevasta ainevaste oli tilapäinen ja sen ilmaantumisajankohta oli ensisijaisesti isatuksimabihoidon alussa, joten vakaan tilan altistus pysyi kuitenkin samankaltaisena lääkevasta-ainepositiivisilla ja lääkevasta-ainenegatiivisilla potilailla.
Yhteisvaikutukset samanaikaisesti käytettävien lääkkeiden kanssa
Isatuksimabin ja pomalidomidin tai isatuksimabin ja karfiltsomibin tai isatuksimabin, bortetsomibin ja lenalidomidin samanaikainen käyttö ei vaikuttanut niiden farmakokinetiikkaan.
Jakautuminen
Isatuksimabin arvioitu kokonaisjakautumistilavuus on 8,75 l.
Metabolia
Isatuksimabi on suuri proteiini, joten sen odotetaan metaboloituvan saturoitumattomilla proteolyyttisillä katabolisilla prosesseilla.
Eliminaatio
Isatuksimabilla on kaksi rinnakkaista eliminaatioreittiä: ei-lineaarinen kohdevälitteinen reitti, joka on vallitsevana pienillä pitoisuuksilla, ja ei-spesifinen lineaarinen reitti, joka on vallitsevana suurilla pitoisuuksilla. Kun pitoisuus plasmassa on terapeuttisella alueella, lineaarinen reitti on vallitsevana, ja eliminaationopeus pienenee ajan myötä 50 % vakaan tilan arvoon 9,55 ml/h (0,229 l/vrk). Tähän liittyvä terminaalinen puoliintumisaika on 28 vuorokautta.
Terminaalinen puoliintumisaika vakaassa tilassa on 40 vuorokautta, kun se arvioidaan populaatiofarmakineettisellä analyysillä suuremmalla potilasjoukolla (N = 769), jossa potilaat saavat isatuksimabia joko laskimoon tai ihon alle yhdistelmänä Pd-, Kd- tai VRd-hoidon kanssa.
Erityisryhmät
Ikä
Populaatiofarmakokineettiset analyysit 476 potilaalla, jotka olivat 36–85-vuotiaita, osoittivat, että isatuksimabialtistus < 75-vuotiailla potilailla (n = 406) vastaa altistusta ≥ 75-vuotiailla (n = 70).
Sukupuoli
Populaatiofarmakokineettisessä analyysissä 207 naispotilaalla (43,5 %) ja 269 miespotilaalla (56,5 %) ei osoitettu, että sukupuolella olisi kliinisesti merkittäviä vaikutuksia isatuksimabin farmakokinetiikkaan.
Etninen tausta
Populaatiofarmakokineettisessä analyysissä 377 valkoihoisella (79 %), 25 aasialaisella (5 %), 18 mustaihoisella (4 %) ja 33:lla muihin etnisiin ryhmiin kuuluvalla potilaalla (7 %) ei osoitettu, että rodulla olisi kliinisesti merkittäviä vaikutuksia isatuksimabin farmakokinetiikkaan.
Paino
476 potilaalla tehdyssä populaatiofarmakokineettisessä analyysissä isatuksimabin puhdistuma suureni painon suurentuessa, mikä tukee valmisteen annostusta painon perusteella.
Maksan vajaatoiminta
Isatuksimabia koskevia virallisia tutkimuksia ei ole tehty potilailla, joilla on maksan vajaatoiminta. Populaatiofarmakokineettisten analyysien 476 potilaasta 65 potilaalla oli lievä maksan vajaatoiminta [kokonaisbilirubiiniarvo > 1‑kertainen – 1,5-kertainen viitealueen ylärajaan nähden tai aspartaattiaminotransferaasiarvo (ASAT) viitealueen ylärajaa suurempi] ja yhdellä potilaalla oli kohtalainen maksan vajaatoiminta (kokonaisbilirubiiniarvo > 1,5-kertainen – 3-kertainen viitealueen ylärajaan nähden ja mikä tahansa ASAT-arvo). Lievällä maksan vajaatoiminnalla ei ollut kliinisesti merkittäviä vaikutuksia isatuksimabin farmakokinetiikkaan. Kohtalaisen maksan vajaatoiminnan (kokonaisbilirubiiniarvo > 1,5-kertainen – 3-kertainen viitealueen ylärajaan nähden ja mikä tahansa ASAT-arvo) ja vaikean maksan vajaatoiminnan (kokonaisbilirubiiniarvo > 3-kertainen viitealueen ylärajaan nähden ja mikä tahansa ASAT-arvo) vaikutuksia isatuksimabin farmakokinetiikkaan ei tunneta. Koska isatuksimabi on monoklonaalinen vasta-aine, ei kuitenkaan ole odotettavissa, että se poistuisi elimistöstä metaboloitumalla maksaentsyymien välityksellä, ja siksi maksan toiminnan muutosten ei oleteta vaikuttavan isatuksimabin eliminaatioon (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Isatuksimabia koskevia virallisia tutkimuksia ei ole tehty potilailla, joilla on munuaisten vajaatoiminta. 476 potilaalla tehdyissä populaatiofarmakokineettisissä analyyseissä oli mukana 192 potilasta, joilla oli lievä munuaisten vajaatoiminta (glomerulusten laskennallinen suodatusnopeus (eGFR) ≥ 60 ml/min/1,73 m2 ja < 90 ml/min/1,73 m2), 163 potilasta, joilla oli kohtalainen munuaisten vajaatoiminta (eGFR ≥ 30 ml/min/1,73 m2 ja < 60 ml/min/1,73 m2), ja 12 potilasta, joilla oli vaikea munuaisten vajaatoiminta (eGFR < 30 ml/min/1,73 m2). Analyysit viittasivat siihen, että lievällä, kohtalaisella tai vaikealla munuaisten vajaatoiminnalla ei olisi kliinisesti merkittäviä vaikutuksia isatuksimabin farmakokinetiikkaan normaaliin munuaisten toimintaan verrattuna.
Farmakokineettinen analyysi 22:sta loppuvaiheen munuaissairautta sairastavasta potilaasta sisältäen dialyysihoidossa olevia potilaita (eGRF < 15 ml/min/1,73 m2) ei osoittanut loppuvaiheen munuaissairaudella olevan merkitsevää vaikutusta isatuksimabin farmakokinetiikkaan verrattuna potilaisiin, joiden munuaisten toiminta oli normaali, lievä tai kohtalainen.
Pediatriset potilaat
Pediatrisilla potilailla (17 kuukauden – 17 vuoden ikäisillä) kolmessa tutkitussa kohortissa Cmax-keskiarvo oli isatuksimabin ensimmäisen annon jälkeen 322–433 µg/ml ja AUC1week-keskiarvo 28 592 – 31 703 µg.h/ml. Kun isatuksimabia annettiin toistuvasti 3 viikon aikana, kumulatiivinen AUC-keskiarvo oli 130 862 – 148 397 µg.h/ml. Akuuttia myelooista leukemiaa ja akuuttia lymfoblastileukemiaa sairastavilla pediatrisilla potilailla ilmoitetut farmakokinetiikkaa koskevat tiedot vastasivat samaa isatuksimabiannosta saaneiden, akuuttia lymfaattista leukemiaa ja multippelia myeloomaa sairastavien aikuisten potilaiden tietoja.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille, joskaan valittu laji ei ole farmakologisesti herkkä, ja siksi tulosten merkitystä ihmisille ei tunneta. Genotoksisuutta, karsinogeenisuutta tai lisääntymis- ja kehitystoksisuutta koskevia tutkimuksia ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Sakkaroosi
Histidiinihydrokloridimonohydraatti
Histidiini
Polysorbaatti 80 (E433)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta
Laimentamisen jälkeen
Isatuksimabi-infuusioliuoksen kemiallisen ja fysikaalisen käytönaikaisen säilyvyyden on osoitettu olevan 48 tuntia 2–8 °C:ssa ja sen jälkeen 8 tuntia (infuusioaika mukaan lukien) huoneenlämmössä (15–25 °C:ssa).
Mikrobiologisesta näkökulmasta valmiste on käytettävä välittömästi. Jos valmistetta ei käytetä välittömästi, käytönaikaiset säilytysajat ja -olosuhteet ovat käyttäjän vastuulla, eivätkä ne yleensä saisi ylittää 24 tuntia 2–8 °C:n lämpötilassa, ellei laimennusta ole tehty kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Infuusiopussissa oleva valmiste ei ole herkkä valolle.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SARCLISA infuusiokonsentraatti, liuosta varten
20 mg/ml (L:ei) 5 ml (728,44 €), 25 ml (3347,46 €)
PF-selosteen tieto
5 ml konsentraattia, joka sisältää 100 mg isatuksimabia 6 ml:n injektiopullossa, joka on tyypin I väritöntä kirkasta lasia ja suljettu ETFE-kopolymeerillä (eteenin ja tetrafluorieteenin kopolymeerillä) pinnoitetulla bromibutyylitulpalla. Injektiopulloissa on alumiininen suljin, jossa on harmaa repäisykorkki. Täyttötilavuus (5,4 ml) on määritetty niin, että injektiopullosta pystyy ottamaan 5 ml. Pakkaukset sisältävät yhden kerta-antoon tarkoitetun injektiopullon tai kolme injektiopulloa.
25 ml konsentraattia, joka sisältää 500 mg isatuksimabia 30 ml:n injektiopullossa, joka on tyypin I väritöntä kirkasta lasia ja suljettu ETFE-kopolymeerillä (eteenin ja tetrafluorieteenin kopolymeerillä) pinnoitetulla bromibutyylitulpalla. Injektiopulloissa on alumiininen suljin, jossa on sininen repäisykorkki. Täyttötilavuus (26 ml) on määritetty niin, että injektiopullosta pystyy ottamaan 25 ml. Pakkaus sisältää yhden injektiopullon.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Väritön tai kellertävä liuos, jossa ei käytännössä ole lainkaan näkyviä hiukkasia (pH 6,0, osmolaalisuus 350–400 mOsm/kg).
Käyttö- ja käsittelyohjeet
Valmistelut ennen laskimoon antoa
Infuusioliuos täytyy valmistaa aseptisissa olosuhteissa.
- Sarclisa-konsentraattiannos (mg) lasketaan potilaan painon mukaan (potilas punnitaan aina ennen hoitosykliä ja annettava annos säädetään painon mukaan, ks. kohta Annostus ja antotapa). Potilaan tarvitseman annoksen aikaansaamiseksi saatetaan tarvita useampia kuin yksi injektiopullo.
- Sarclisa-konsentraattia sisältävistä injektiopulloista on ennen laimentamista tarkistettava silmämääräisesti, ettei niissä ole hiukkasia eikä värimuutoksia.
- Älä ravista injektiopulloja.
- 250 ml:n pussista, joka sisältää laimenninta, natriumkloridi-injektioliuosta 9 mg/ml (0,9 %) tai glukoosiliuosta (5 %), on poistettava tarvittavaa Sarclisa-konsentraattimäärää vastaava määrä laimenninta.
- Sarclisa-injektiopullosta otetaan oikea määrä Sarclisa-konsentraattia, joka laimennetaan 250 ml:n infuusiopussissa natriumkloridi-injektioliuoksella 9 mg/ml (0,9 %) tai glukoosiliuoksella (5 %).
- Infuusiopussin täytyy olla valmistettu polyolefiineista (PO), polyeteenistä (PE), polypropeenista (PP), polyvinyylikloridista (PVC), johon on lisätty di-(2-etyyliheksyyli)ftalaattia (DEHP), tai etyylivinyyliasetaatista (EVA).
- Pussia käännellään varovasti, jotta saadaan tasalaatuinen laimennettu liuos. Älä ravista.
Anto
- Infuusioliuos täytyy antaa infuusiona laskimoon käyttämällä laskimoinfuusioletkuja (PE, PVC DEHP:n kanssa tai ilman sitä, polybutadieeni (PBD) tai polyuretaani (PU)), joissa on 0,22 mikronin in-line-suodatin (polyeetterisulfoni (PES), polysulfoni tai nailon).
- Infuusioliuoksen antoaika riippuu infuusionopeudesta (ks. kohta Annostus ja antotapa).
- Käyttökuntoon saatettua infuusiopussia ei tarvitse suojata valolta tavanomaisessa keinovaloympäristössä.
- Älä anna Isatuksimabiliuosta infuusiona samanaikaisesti muiden lääkeaineiden kanssa käyttämällä samaa laskimoyhteyttä.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SARCLISA infuusiokonsentraatti, liuosta varten
20 mg/ml 5 ml, 25 ml
- Ei korvausta.
ATC-koodi
L01FC02
Valmisteyhteenvedon muuttamispäivämäärä
04.06.2026
Yhteystiedot
 SANOFI OY
SANOFI OY Revontulenkuja 1
02100 Espoo
0201 200 300
www.sanofi.fi