
DARZALEX injektioneste, liuos 1800 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Terveydenhuollon ammattilainen
Verikeskuksille: Tietoa daratumumabin vaikutuksista veren yhteensopivuustestaukseen ja toimenpiteet näiden vaikutusten vähentämiseen
Daratumumabin vaikutus veren yhteensopivuustestaukseen
Vaikuttavat aineet ja niiden määrät
Yksi 15 ml:n injektiopullo injektionestettä, liuosta, sisältää 1 800 mg daratumumabia (120 mg daratumumabia per ml).
Daratumumabi on ihmisen monoklonaalinen IgG1κ-vasta-aine CD38-antigeenia vastaan. Se tuotetaan rekombinantti-DNA-tekniikalla nisäkässolulinjassa (kiinanhamsterin munasarja).
Apuaineet, joiden vaikutus tunnetaan
Yksi 15 ml:n injektiopullo injektionestettä, liuosta, sisältää 735,1 mg sorbitolia (E420).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
Multippeli myelooma
Darzalex on tarkoitettu
- yhdistelmänä lenalidomidin ja deksametasonin tai bortetsomibin, melfalaanin ja prednisonin kanssa äskettäin diagnosoidun multippelin myelooman hoitoon aikuispotilaille, jotka eivät sovellu autologiseen kantasolusiirtoon
- yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa äskettäin diagnosoidun multippelin myelooman hoitoon aikuispotilaille
- yhdistelmänä bortetsomibin, talidomidin ja deksametasonin kanssa äskettäin diagnosoidun multippelin myelooman hoitoon aikuispotilaille, jotka soveltuvat autologiseen kantasolusiirtoon
- yhdistelmänä lenalidomidin ja deksametasonin tai bortetsomibin ja deksametasonin kanssa multippelin myelooman hoitoon aikuispotilaille, jotka ovat saaneet vähintään yhtä aiempaa hoitoa
- yhdistelmänä pomalidomidin ja deksametasonin kanssa multippelin myelooman hoitoon aikuispotilaille, jotka ovat saaneet yhtä aiempaa proteasomin estäjää ja lenalidomidia sisältävää hoitoa eikä tauti reagoinut lenalidomidiin, tai jotka ovat saaneet vähintään kahta aiempaa hoitoa, joihin kuului lenalidomidi ja jokin proteasomin estäjä, ja potilaan taudin on osoitettu edenneen viimeisimmän hoidon aikana tai sen jälkeen (ks. kohta Farmakodynamiikka)
- monoterapiana relapsoituneen ja hoitoon reagoimattoman multippelin myelooman hoitoon aikuispotilaille, joiden aiempi hoito on sisältänyt proteasomin estäjää ja immunomodulatiivista ainetta ja joiden taudin on osoitettu edenneen viimeisimmän hoidon aikana.
Indolentti multippeli myelooma
DARZALEX on tarkoitettu monoterapiana aikuispotilaille, joiden indolentin multippelin myelooman etenemisen riski multippeliksi myeloomaksi on suuri (ks. kohta Farmakodynamiikka).
Kevytketjuamyloidoosi (AL-amyloidoosi)
Darzalex on tarkoitettu yhdistelmänä syklofosfamidin, bortetsomibin ja deksametasonin kanssa äskettäin diagnosoidun systeemisen AL-amyloidoosin hoitoon aikuispotilaille.
Ehto
Valmisteen saa antaa kokenut terveydenhuollon ammattilainen ja ensimmäinen annos tulee antaa hoitopaikassa, jossa on elvytysvälineistö saatavissa.
Annostus ja antotapa
Darzalex-valmisteen ihon alle annettava koostumus ei ole tarkoitettu annettavaksi laskimoon, vaan sen saa antaa vain injektiona ihon alle mainittuina annoksina.
Ensimmäiset neljä DARZALEX-annosta antaa terveydenhuollon ammattilainen, ja ensimmäinen annos pitää antaa hoitopaikassa, jossa on elvytysvälineistö saatavissa.
DARZALEX on tarkoitettu annettavaksi terveydenhuollon ammattilaisen ohjauksessa. Ensimmäisten neljän annoksen jälkeen asianmukaisen opastuksen valmisteen valmisteluun ja ihon alle annettavan injektion pistostekniikkaan saatuaan potilas voi pistää seuraavat DARZALEX-annokset itse tai potilasta hoitava henkilö voi antaa pistokset potilaalle, jos terveydenhuollon ammattilainen katsoo sen asianmukaiseksi ja kun hoito tapahtuu tarvittaessa lääkärin seurannassa. Potilasta pitää ohjeistaa ottamaan yhteyttä terveydenhuollon ammattilaiseen, jos hänelle ilmaantuu ihon alle annettavasta injektiosta tai sairaudesta johtuvia oireita.
Injektiopullon etiketistä on tärkeää varmistaa, että potilaalle annetaan asianmukaista valmistetta (laskimoon tai ihon alle annettava valmiste) ja että potilaalle annetaan lääkemääräyksen mukainen annos.
Jos potilas saa parhaillaan hoitoa laskimoon annettavalla daratumumabivalmisteella, ihon alle annettavaa Darzalex-injektionestettä voidaan antaa seuraavasta hoitoaikataulun mukaisesta hoitokerrasta lähtien vaihtoehtona laskimoon annettavalle daratumumabivalmisteelle.
Potilaalle pitää antaa lääkehoitoa ennen injektiota ja sen jälkeen daratumumabista aiheutuvien infuusioon liittyvien reaktioiden riskin vähentämiseksi. Ks. jäljempänä Samanaikaisesti suositellut lääkkeet ja kohta Varoitukset ja käyttöön liittyvät varotoimet.
Annostus
Multippeli myelooma
Hoitoaikataulu yhdistelmähoidossa lenalidomidin ja deksametasonin tai pomalidomidin ja deksametasonin kanssa (4 viikon hoitosyklit) ja monoterapiassa
Suositeltu annos on 1 800 mg ihon alle annettavaa Darzalex-injektionestettä noin 3–5 minuutin kestoisena injektiona taulukossa 1 esitetyn hoitoaikataulun mukaisesti.
| Taulukko 1: Darzalex-hoitoaikataulu yhdistelmähoidossa lenalidomidin ja deksametasonin (Rd), pomalidomidin ja deksametasonin (Pd) kanssa (neljän viikon hoitosykli) ja monoterapiassa | |
| Viikot | Ajankohta |
| Viikot 1–8 | viikoittain (yhteensä 8 annosta) |
| Viikot 9–24a | joka toinen viikko (yhteensä 8 annosta) |
| Viikosta 25 alkaen, kunnes tauti eteneeb | joka neljäs viikko |
a Siirryttäessä joka toinen viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 9 b Siirryttäessä joka neljäs viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 25 | |
Deksametasonia annetaan 40 mg/viikko (tai > 75-vuotiaille potilaille pienennetty annos 20 mg/viikko).
Ihon alle annettavan Darzalex-injektionesteen kanssa annettavien lääkevalmisteiden annos ja hoitoaikataulu, ks. kohta Farmakodynamiikka ja kyseisen valmisteen valmisteyhteenveto.
Hoitoaikataulu yhdistelmähoidossa bortetsomibin, melfalaanin ja prednisonin kanssa (6 viikon hoitosyklit)
Suositeltu annos on 1800 mg ihon alle annettavaa Darzalex-injektionestettä noin 3–5 minuutin kestoisena injektiona taulukossa 2 esitetyn hoitoaikataulun mukaisesti.
| Taulukko 2: Darzalex-hoitoaikataulu yhdistelmähoidossa bortetsomibin, melfalaanin ja prednisonin kanssa ([VMP]; kuuden viikon hoitosykli) | |
| Viikot | Ajankohta |
| Viikot 1–6 | viikoittain (yhteensä 6 annosta) |
| Viikot 7–54a | joka kolmas viikko (yhteensä 16 annosta) |
| Viikosta 55 alkaen, kunnes tauti eteneeb | joka neljäs viikko |
a Siirryttäessä joka kolmas viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 7 b Siirryttäessä joka neljäs viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 55 | |
Bortetsomibia annetaan ensimmäisen 6 viikon hoitosyklin aikana kaksi kertaa viikossa viikoilla 1, 2, 4 ja 5, minkä jälkeen seuraavien kahdeksan 6 viikon hoitosyklin aikana bortetsomibia annetaan kerran viikossa viikoilla 1, 2, 4 ja 5. Lisätietoja ihon alle annettavan Darzalex-injektionesteen kanssa annettavan VMP-hoidon annoksesta ja hoitoaikataulusta, ks. kohta Farmakodynamiikka.
Hoitoaikataulu käytettäessä yhdistelmänä bortetsomibin, talidomidin ja deksametasonin kanssa (neljän viikon hoitosyklit) äskettäin diagnosoidun multippelin myelooman hoitoon potilaille, jotka soveltuvat autologiseen kantasolusiirtoon
Suositeltu annos on 1 800 mg ihon alle annettavaa Darzalex-injektionestettä noin 3–5 minuutin kestoisena injektiona taulukossa 3 esitetyn hoitoaikataulun mukaisesti.
| Taulukko 3: Darzalex-hoitoaikataulu käytettäessä yhdistelmänä bortetsomibin, talidomidin ja deksametasonin kanssa ([VTd]; neljän viikon hoitosykli) | ||
| Hoitovaihe | Viikot | Ajankohta |
| Induktio | Viikot 1–8 | viikoittain (yhteensä 8 annosta) |
| Viikot 9–16a | joka toinen viikko (yhteensä 4 annosta) | |
| Lopeta suuriannoksisen solunsalpaajahoidon ja autologisen kantasolusiirron ajaksi | ||
| Konsolidaatio | Viikot 1–8b | joka toinen viikko (yhteensä 4 annosta) |
a Siirryttäessä joka toinen viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 9 b Jatkettaessa hoitoa autologisen kantasolusiirron jälkeen joka toinen viikko tapahtuvan annostelun ensimmäinen annos annetaan viikolla 1 | ||
Deksametasonia annetaan 40 mg hoitosyklien 1 ja 2 päivinä 1, 2, 8, 9, 15, 16, 22 ja 23. Hoitosykleissä 3-4 annetaan 40 mg päivinä 1–2 ja 20 mg seuraavina antopäivinä (päivät 8, 9, 15, 16). Hoitosykleissä 5 ja 6 deksametasonia annetaan 20 mg päivinä 1, 2, 8, 9, 15 ja 16.
Ihon alle annettavan Darzalex-injektionesteen kanssa annettavien lääkevalmisteiden annokset ja hoitoaikataulut, ks. kohta Farmakodynamiikka ja kyseisten valmisteiden valmisteyhteenvedot.
Hoitoaikataulu käytettäessä yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa (neljän viikon hoitosyklit) äskettäin diagnosoidun multippelin myelooman hoitoon potilaille, jotka soveltuvat autologiseen kantasolusiirtoon
Suositeltu annos on 1 800 mg ihon alle annettavaa DARZALEX-injektionestettä noin 3–5 minuutin kestoisena injektiona taulukossa 4 esitetyn hoitoaikataulun mukaisesti.
| Taulukko 4: DARZALEX-hoitoaikataulu käytettäessä yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa ([VRd]; neljän viikon hoitosykli) | ||
| Hoitovaihe | Viikot | Ajankohta |
| Induktio | Viikot 1–8 | viikoittain (yhteensä 8 annosta) |
| Viikot 9–16a | joka toinen viikko (yhteensä 4 annosta) | |
| Lopeta suuriannoksisen solunsalpaajahoidon ja autologisen kantasolusiirron ajaksi | ||
| Konsolidaatio | Viikot 17–24b | joka toinen viikko (yhteensä 4 annosta) |
| Ylläpito | Viikosta 25 alkaen, kunnes tauti eteneec | joka neljäs viikko |
a Siirryttäessä joka toinen viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 9. b Viikko 17 vastaa hoidon jatkamista autologisesta kantasolusiirrosta toipumisen jälkeen. c DARZALEX-hoito voidaan lopettaa, jos potilaan todetaan olleen MRD-negatiivinen 12 kuukauden ajan ja jos potilas on saanut ylläpitohoitoa vähintään 24 kuukauden ajan. | ||
Induktio- ja konsolidaatiohoidon (hoitosyklit 1–6) aikana deksametasonia annetaan 40 mg kunkin 28 päivän pituisen hoitosyklin päivinä 1–4 ja päivinä 9–12.
Ihon alle annettavan DARZALEX-injektionesteen kanssa annettavien lääkevalmisteiden annokset ja hoitoaikataulut, ks. kohta Farmakodynamiikka ja kyseisten valmisteiden valmisteyhteenvedot.
Hoitoaikataulu käytettäessä yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa (3 viikon hoitosykli) äskettäin diagnoosin saaneille potilaille, jotka eivät sovellu autologiseen kantasolusiirtoon
Suositeltu annos on 1 800 mg ihon alle annettavaa DARZALEX-injektionestettä noin 3–5 minuutin kestoisena injektiona taulukossa 5 esitetyn hoitoaikataulun mukaisesti.
| Taulukko 5: DARZALEX-hoitoaikataulu käytettäessä yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa ([VRd]; 3 viikon hoitosykli) | |
| Viikot | Ajankohta |
| Viikot 1–6 | viikoittain (yhteensä 6 annosta) |
| Viikot 7–24a | joka kolmas viikko (yhteensä 6 annosta) |
| Viikosta 25 alkaen, kunnes tauti eteneeb | joka neljäs viikko |
a Siirryttäessä joka kolmas viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 7. b Siirryttäessä joka neljäs viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 25. | |
Deksametasonia annetaan 20 mg hoitosykleissä 1–8 kunkin 21 päivän pituisen hoitosyklin päivinä 1, 2, 4, 5, 8, 9, 11 ja 12. Potilaille, jotka ovat > 75-vuotiaita tai alipainoisia (painoindeksi [BMI] < 18,5), deksametasonia voidaan antaa 20 mg päivinä 1, 4, 8 ja 11.
Ihon alle annettavan DARZALEX-injektionesteen kanssa annettavien lääkevalmisteiden annokset ja hoitoaikataulut, ks. kohta Farmakodynamiikka ja kyseisten valmisteiden valmisteyhteenvedot.
Hoitoaikataulu käytettäessä yhdistelmänä bortetsomibin ja deksametasonin kanssa (kolmen viikon hoitosykli)
Suositeltu annos on 1 800 mg ihon alle annettavaa Darzalex-injektionestettä noin 3–5 minuutin kestoisena injektiona taulukossa 6 esitetyn hoitoaikataulun mukaisesti.
| Taulukko 6: Darzalex-hoitoaikataulu käytettäessä yhdistelmänä bortetsomibin ja deksametasonin (Vd) kanssa (kolmen viikon hoitosykli) | |
| Viikot | Ajankohta |
| Viikot 1–9 | viikoittain (yhteensä 9 annosta) |
| Viikot 10–24a | joka kolmas viikko (yhteensä 5 annosta) |
| Viikosta 25 alkaen, kunnes tauti eteneeb | joka neljäs viikko |
a Siirryttäessä joka kolmas viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 10 b Siirryttäessä joka neljäs viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 25 | |
Deksametasonia annetaan 20 mg bortetsomibihoidon ensimmäisten kahdeksan hoitosyklin päivinä 1, 2, 4, 5, 8, 9, 11 ja 12 tai pienennetty annos 20 mg/viikko, jos potilas on > 75-vuotias tai alipainoinen (painoindeksi < 18,5), potilaalla on huonossa hoitotasapainossa oleva diabetes mellitus tai potilas ei ole aiemmin sietänyt steroidihoitoa.
Ihon alle annettavan Darzalex-injektionesteen kanssa annettavien lääkevalmisteiden annos ja hoitoaikataulu, ks. kohta Farmakodynamiikka ja kyseisen valmisteen valmisteyhteenveto.
Indolentti multippeli myelooma
Hoitoaikataulu käytettäessä monoterapiana (4 viikon hoitosyklien hoito-ohjelma)
Suositeltu annos on 1 800 mg ihon alle annettavaa DARZALEX-injektionestettä noin 3–5 minuutin kestoisena injektiona taulukossa 7 esitetyn hoitoaikataulun mukaisesti.
| Taulukko 7: DARZALEX-hoitoaikataulu indolentin multippelin myelooman hoidossa käytettäessä monoterapiana (4 viikon hoitosyklien hoito-ohjelma)a | |
| Viikot | Ajankohta |
| Viikot 1–8 | viikoittain (yhteensä 8 annosta) |
| Viikot 9–24a | joka toinen viikko (yhteensä 8 annosta) |
| Viikosta 25 alkaen, kunnes tauti etenee, tai enintään 3 vuoden ajanb | joka neljäs viikko |
a Siirryttäessä joka toinen viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 9. b Siirryttäessä joka neljäs viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 25. | |
AL-amyloidoosi
Hoitoaikataulu käytettäessä yhdistelmänä bortetsomibin, syklofosfamidin ja deksametasonin kanssa (neljän viikon hoitosykli)
Suositeltu annos on 1 800 mg ihon alle annettavaa Darzalex-injektionestettä noin 3–5 minuutin kestoisena injektiona taulukossa 8 esitetyn hoitoaikataulun mukaisesti.
Taulukko 8: DARZALEX-hoitoaikataulu AL-amyloidoosin hoidossa käytettäessä yhdistelmänä bortetsomibin, syklofosfamidin ja deksametasonin (VCd) kanssa (neljän viikon hoitosykli)a | |
| Viikot | Ajankohta |
| Viikot 1–8 | viikoittain (yhteensä 8 annosta) |
| Viikot 9–24b | joka toinen viikko (yhteensä 8 annosta) |
| Viikosta 25 alkaen, kunnes tauti eteneec | joka neljäs viikko |
a Darzalex-valmistetta annettiin kliinisessä tutkimuksessa, kunnes tauti eteni tai hoitoa oli annettu enintään 24 hoitosykliä (~2 vuotta) ensimmäisestä tutkimushoitoannoksesta alkaen. b Siirryttäessä joka toinen viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 9. c Siirryttäessä joka neljäs viikko tapahtuvaan annosteluun ensimmäinen annos annetaan viikolla 25. | |
Ihon alle annettavan Darzalex-injektionesteen kanssa annettavien lääkevalmisteiden annos ja hoitoaikataulu, ks. kohta Farmakodynamiikka ja kyseisen valmisteen valmisteyhteenveto.
Annoksen/annosten antamatta jääminen
Jos suunniteltu Darzalex-annos jää antamatta, annos pitää antaa mahdollisimman pian ja hoitoaikataulua pitää muuttaa vastaavasti siten, että annosväli säilyy ennallaan.
Annosmuutokset
Darzalex-annoksen pienentämistä ei suositella. Jos potilaalla on hematologista toksisuutta, annoksen antamista saattaa olla tarpeen siirtää myöhempään ajankohtaan, jotta verisolujen määrät voivat korjautua (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Tiedot Darzalex-hoidon kanssa yhdistelmänä annettavista lääkevalmisteista, ks. kyseisen valmisteen valmisteyhteenveto.
Kliinisissä tutkimuksissa ihon alle annettavan Darzalex-injektionesteen antonopeutta tai annosta ei tarvinnut muuttaa infuusioon liittyvien reaktioiden hoitamiseksi.
Samanaikaisesti suositellut lääkkeet
Ennen injektiota annettava lääkitys
Kaikille potilaille pitää antaa lääkitys (suun kautta tai laskimoon) 1–3 tuntia ennen jokaista ihon alle annettavaa Darzalex-injektiota infuusioon liittyvien reaktioiden riskin vähentämiseksi seuraavasti:
-
kortikosteroidia (pitkä- tai keskipitkävaikutteista)
- Monoterapia:
100 mg metyyliprednisolonia tai vastaavaa. Kortikosteroidiannos voidaan pienentää 60 mg:aan metyyliprednisolonia toisen injektion jälkeen. - Yhdistelmähoito:
20 mg deksametasonia (tai vastaavaa) ennen jokaista ihon alle annettavaa Darzalex-injektiota. Kun deksametasoni on peruslääkityksenä käytettävä runkohoitospesifinen kortikosteroidi, deksametasonihoidon annos toimii esilääkityksenä Darzalex-injektion antopäivinä (ks. kohta Farmakodynamiikka).
Jos potilas on saanut deksametasonia (tai vastaavaa) esilääkityksenä, Darzalex-injektion antopäivinä ei pidä ottaa lisäksi peruslääkityksenä runkohoitospesifistä kortikosteroidia (esim. prednisonia).
- Monoterapia:
- antipyreettejä (650–1 000 mg parasetamolia)
- antihistamiinia (25–50 mg difenhydramiinia suun kautta tai laskimoon tai vastaavaa)
- leukotrieenireseptorin salpaajaa (10 mg montelukastia suun kautta tai vastaavaa) suositellaan indolenttia multippelia myeloomaa sairastaville potilaille 1. hoitosyklin 1. päivänä.
Injektion jälkeen annettava lääkitys
Injektion jälkeen on annettava lääkitystä viivästyneiden infuusioon liittyvien reaktioiden riskin vähentämiseksi seuraavasti:
-
Monoterapia:
Kortikosteroidia suun kautta (20 mg metyyliprednisolonia tai vastaava annos keskipitkä- tai pitkävaikutteista kortikosteroidia paikallisen hoitokäytännön mukaan) kahtena injektion jälkeisenä päivänä (injektion antoa seuraavasta päivästä alkaen). -
Yhdistelmähoito:
Harkitse pientä metyyliprednisoloniannosta suun kautta (≤ 20 mg) tai vastaavaa Darzalex-injektion jälkeisenä päivänä. Jos Darzalex-injektion jälkeisenä päivänä annetaan peruslääkityksenä runkohoitospesifistä kortikosteroidia (esim. deksametasonia, prednisonia), muuta injektion jälkeen annettavaa lääkitystä ei välttämättä tarvita (ks. kohta Farmakodynamiikka).
Jos potilaalla ei esiinny vakavia infuusioon liittyviä reaktioita kolmen ensimmäisen injektion jälkeen, kortikosteroidien antaminen injektion jälkeen (paitsi mahdollinen runkohoitona käytettävä kortikosteroidi) voidaan lopettaa.
Jos potilaalla on aiemmin ollut krooninen ahtauttava keuhkosairaus, lyhyt- ja pitkävaikutteista keuhkoputkia laajentavaa lääkitystä ja inhaloitavia kortikosteroideja pitää lisäksi harkita osaksi injektion jälkeistä lääkitystä. Jos potilaalle ei neljän ensimmäisen injektion jälkeen ilmaannu vakavia infuusioon liittyviä reaktioita, näiden injektioiden jälkeen käytettyjen inhaloitavien lääkkeiden käyttö voidaan lääkärin harkinnan mukaan lopettaa.
Herpes zoster ‑viruksen reaktivaation estohoito
Herpes zoster ‑viruksen reaktivaation estämiseksi pitää harkita estohoitoa viruslääkkeillä.
Erityiset potilasryhmät
Munuaisten vajaatoiminta
Daratumumabin käyttöä munuaisten vajaatoimintaa sairastaville potilaille ei ole tutkittu varsinaisissa tutkimuksissa. Populaatiofarmakokineettisten analyysien perusteella munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Daratumumabin käyttöä maksan vajaatoimintaa sairastaville potilaille ei ole tutkittu varsinaisissa tutkimuksissa.
Maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Iäkkäät
Annosmuutoksia ei katsota tarpeellisiksi (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Darzalex-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu.
Tietoja ei ole saatavilla.
Paino (> 120 kg)
Ihon alle annettavaa Darzalex-injektionestettä on tutkittu vakioannoksina (1 800 mg) pienellä joukolla > 120 kg:n painoisia potilaita, eikä tehoa ole näillä potilailla varmistettu. Painoon perustuvia annosmuutoksia ei tällä hetkellä voida suositella (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Antotapa
Ihon alle annettavaa Darzalex-valmistetta ei ole tarkoitettu annettavaksi laskimoon, joten sen saa antaa vain injektiona ihon alle mainittuina annoksina. Käytä DARZALEX-valmisteen injektiopullosta vetämiseen asianmukaista tekniikkaa. Vältä paksujen tai tylppäkärkisten siirtoneulojen käyttöä tai tulpan lävistämistä moneen kertaan vähentääksesi tulpan materiaalin irtoamisen todennäköisyyttä. Ks. kohdasta Käyttö- ja käsittelyohjeet erityiset varotoimet ennen lääkkeen antoa.
Kiinnitä hypoderminen injektioneula tai ihon alle antoon käytettävä infuusiolaite ruiskuun juuri ennen injektion antamista, jotta vältetään neulan tukkeutuminen.
Injisoi 15 ml ihon alle annettavaa Darzalex-injektionestettä vatsan ihonalaiskudokseen noin 7,5 cm navan oikealle tai vasemmalle puolelle noin 3–5 minuutin kestoisena injektiona. Älä injisoi ihon alle annettavaa Darzalex-injektionestettä muualle kehoon, sillä siitä ei ole tietoja saatavissa.
Peräkkäisissä injektioissa on käytettävä eri injektiokohtaa.
Ihon alle annettavaa Darzalex-injektionestettä ei pidä koskaan injisoida ihoalueille, joilla on punoitusta, mustelma, aristusta, kovettuma tai arpia.
Jos injektiosta aiheutuu potilaalle kipua, sen antoa voidaan tauottaa tai hidastaa. Jos kipu ei lievity injektion antoa hidastamalla, loput annoksesta voidaan antaa toiseen injektiokohtaan vatsan toiselle puolelle.
Ihon alle annettavan Darzalex-injektionesteen käytön aikana Darzalex-valmisteen antokohtiin ei saa antaa ihon alle muita lääkevalmisteita.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infuusioon liittyvät reaktiot
Ihon alle annettava DARZALEX-injektioneste voi aiheuttaa vaikea-asteisia ja/tai vakavia infuusioon liittyviä reaktioita, mukaan lukien anafylaktisia reaktioita. Kliinisissä tutkimuksissa noin 8,5 %:lle (134 / 1 573) potilaista ilmaantui infuusioon liittyvä reaktio. Suurin osa infuusioon liittyvistä reaktioista ilmeni ensimmäisen injektion jälkeen, ja niiden vaikeusaste oli gradus 1–2. Seuraavien injektioiden yhteydessä infuusioon liittyviä reaktioita esiintyi 1 %:lla potilaista (ks. kohta Haittavaikutukset).
Infuusioon liittyvien reaktioiden ilmaantumiseen DARZALEX-injektion jälkeen kuluneen ajan mediaani oli 3,3 tuntia (vaihteluväli 0,08–83 tuntia). Valtaosa infuusioon liittyneistä reaktioista ilmeni hoitopäivänä. Viivästyneitä infuusioon liittyneitä reaktioita ilmeni 1 %:lla potilaista.
Infuusioon liittyvien reaktioiden oireita ja löydöksiä voivat olla mm. hengityselinoireet, kuten nenän tukkoisuus, yskä, kurkun ärsytys, allerginen nuha, hengityksen vinkuminen, sekä kuume, rintakipu, kutina, vilunväristykset, oksentelu, pahoinvointi, hypotensio ja hämärtynyt näkö. Myös vaikea-asteisia reaktioita on esiintynyt, mm. bronkospasmeja, hypoksiaa, hengenahdistusta, hypertensiota, takykardiaa ja silmiin liittyviä haittavaikutuksia (mukaan lukien silmän suonikalvon effuusiota, akuuttia likitaittoisuutta ja akuuttia ahdaskulmaglaukoomaa) (ks. kohta Haittavaikutukset).
Potilaille pitää antaa esilääkityksenä antihistamiineja, antipyreettejä ja kortikosteroideja, ja potilas on pidettävä seurannassa. Potilaalle pitää kertoa infuusioon liittyvistä reaktioista etenkin ensimmäisen ja toisen injektion yhteydessä ja niiden jälkeen. Indolenttia multippelia myeloomaa sairastaville potilaille pitää harkita esilääkityksenä leukotrieenireseptorin salpaajaa 1. hoitosyklin 1. päivänä. Jos potilaalle ilmenee anafylaktinen reaktio tai hengenvaarallinen (gradus 4) reaktio, asianmukaiset kiireelliset elvytystoimenpiteet pitää käynnistää heti. DARZALEX-hoito pitää lopettaa heti ja pysyvästi (ks. kohdat Annostus ja antotapa ja Vasta-aiheet).
Kaikille potilaille pitää antaa Darzalex-injektion jälkeen kortikosteroideja suun kautta viivästyneiden infuusioon liittyvien reaktioiden riskin vähentämiseksi (ks. kohta Annostus ja antotapa). Jos potilaalla on aiemmin ollut krooninen ahtauttava keuhkosairaus, lisälääkitys hengityselinkomplikaatioiden hoitoon voi olla tarpeen injektion jälkeen. Injektion jälkeistä lääkitystä (esim. lyhyt- ja pitkävaikutteisia keuhkoputkia laajentavia lääkkeitä sekä inhaloitavia kortikosteroideja) pitää harkita, jos potilaalla on krooninen ahtauttava keuhkosairaus. Jos potilaalle ilmaantuu silmäoireita, keskeytä Darzalex-hoito ja pyydä heti silmälääkärin arvio ennen kuin Darzalex-hoitoa jatketaan (ks. kohta Annostus ja antotapa).
Neutropenia/trombosytopenia
Darzalex saattaa lisätä runkohoidosta aiheutuvaa neutropeniaa ja trombosytopeniaa (ks. kohta Haittavaikutukset).
Täydellistä verenkuvaa pitää seurata hoidon aikana säännöllisin väliajoin runkohoitoon kuuluvien valmisteiden valmistetietojen mukaisesti. Neutropeenisia potilaita pitää tarkkailla infektion oireiden havaitsemiseksi. Darzalex-hoidon siirtäminen myöhempään ajankohtaan saattaa olla tarpeen, jotta verisolujen määrät voivat korjautua. Neutropeniaa on havaittu yleisemmin potilaalla, joiden paino on alempi ja jotka ovat saaneet ihon alle annettavaa Darzalex-valmistetta, mutta tähän ei liittynyt vakavien infektioiden esiintyvyyden lisääntymistä. Darzalex-annoksen pienentämistä ei suositella. Harkitse verensiirtoja tai kasvutekijöitä tukihoitona.
Infektiot
DARZALEX-hoidosta voi aiheutua vakavia, henkeä uhkaavia tai kuolemaan johtavia infektioita (ks. kohta Haittavaikutukset).
Potilaita on ennen DARZALEX-hoitoa ja hoidon aikana seurattava tarkoin infektion oireiden ja löydösten varalta ja ne on hoidettava asianmukaisesti. Ennen hoitoa, hoidon aikana ja sen jälkeen pitää harkita estohoitoa mikrobilääkkeillä hoitosuositusten mukaisesti (ks. kohta Annostus ja antotapa).
Vaikutukset epäsuoraan antiglobuliinikokeeseen (epäsuoraan Coombsin kokeeseen)
Daratumumabi sitoutuu CD38-proteiiniin, jota on pieninä pitoisuuksina veren punasolujen pinnalla, ja se saattaa aiheuttaa epäsuoran Coombsin kokeen positiivisen testituloksen. Epäsuoran Coombsin kokeen tulokset voivat olla daratumumabin vaikutuksesta positiivisia enimmillään kuuden kuukauden ajan viimeisen daratumumabin antokerran jälkeen. On syytä huomioida, että veren punasoluihin sitoutunut daratumumabi saattaa häiritä minor-antigeeneihin kohdistuvien vasta-aineiden havaitsemista potilaan seerumista. Potilaan ABO- ja Rh-veriryhmien määritys ei häiriinny.
Potilaalle pitää tehdä tyypitys ja seulonta ennen daratumumabihoidon aloittamista. Ennen daratumumabihoidon aloittamista pitää harkita fenotyypitystä paikallisen käytännön mukaisesti. Daratumumabi ei vaikuta veren punasolujen genotyypitykseen, joten se voidaan tehdä milloin tahansa.
Jos potilaalle suunnitellaan verensiirtoa, veripalveluyksikölle pitää kertoa epäsuorien antiglobuliinitestien tällaisesta häiriintymisestä (ks. kohta Yhteisvaikutukset). Jos verensiirto on tarpeen hätätilanteessa, voidaan antaa ABO/RhD-yhteensopivia punasoluja ilman sopivuuskoetta paikallisen veripalveluyksikön käytännön mukaan.
Täydellisen vasteen määrittämisen häiriintyminen
Daratumumabi on ihmisen monoklonaalinen IgG-kappa-vasta-aine, joka voidaan havaita sekä seerumin proteiinielektroforeesi- (SPE) että immunofiksaatio- (IFE) määrityksellä, joita käytetään endogeenisen M-proteiinin kliiniseen seurantaan (ks. kohta Yhteisvaikutukset). Tämä voi häiritä täydellisen vasteen ja taudin etenemisen määrittämistä joillakin potilailla, joilla on IgG-kappamyeloomaproteiinia.
Hepatiitti B ‑viruksen (HBV) reaktivaatio
Darzalex-hoitoa saaneilla potilailla on raportoitu hepatiitti B ‑viruksen reaktivaatiota, joka on joissakin tapauksissa johtanut potilaan kuolemaan. Kaikille potilaille pitää tehdä hepatiitti B ‑viruksen seulonta ennen Darzalex-hoidon aloittamista.
Jos potilas todetaan serologisesti HBV-positiiviseksi, seuraa hepatiitti B ‑viruksen reaktivaatioon viittaavia kliinisiä oireita ja laboratoriokoetuloksia Darzalex-hoidon aikana ja vähintään kuuden kuukauden ajan hoidon päättymisen jälkeen. Hoida potilasta voimassa olevien kliinisten ohjeistojen mukaisesti. Harkitse hepatiittiin perehtyneen asiantuntijan konsultoimista kliinisen tarpeen mukaan.
Jos potilaalle kehittyy hepatiitti B ‑viruksen reaktivaatio Darzalex-hoidon aikana, keskeytä Darzalex-hoito, ja aloita tilanteen edellyttämä hoito. Darzalex-hoidon jatkamisesta potilailla, joilla hepatiitti B ‑viruksen reaktivaatio on riittävästi hallinnassa, pitää keskustella sellaisen lääkärin kanssa, jolla on asiantuntemusta hepatiitti B ‑virusinfektion hoitamisesta.
Paino (> 120 kg)
Potilailla, joiden paino on > 120 kg, ihon alle annettavan Darzalex-injektionesteen teho voi olla muita potilaita heikompi (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Apuaineet
Tämä lääkevalmiste sisältää sorbitolia (E420). Potilaille, joilla on perinnöllinen fruktoosi-intoleranssi (HFI), ei saa antaa tätä lääkevalmistetta.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Daratumumabi on monoklonaalinen IgG1қ-vasta-aine, joten erittyminen munuaisten kautta tai maksaentsyymivälitteinen metabolia eivät ole muuttumattoman daratumumabin todennäköisiä pääasiallisia eliminaatioreittejä. Lääkkeitä metaboloivissa entsyymeissä esiintyvän vaihtelun ei sinänsä oleteta vaikuttavan daratumumabin eliminaatioon. Daratumumabi sitoutuu suurella affiniteetilla CD38-proteiinin yksilölliseen epitooppiin, joten se ei oletettavasti muuta lääkkeitä metaboloivia entsyymejä.
Daratumumabin laskimoon tai ihon alle annettavien lääkemuotojen sekä lenalidomidin, pomalidomidin, talidomidin, bortetsomibin, melfalaanin, prednisonin, karfiltsomibin, syklofosfamidin ja deksametasonin yhdistelmän kliininen farmakokineettinen arviointi ei osoittanut kliinisesti oleellisia yhteisvaikutuksia daratumumabin ja näiden pienimolekyylisten lääkevalmisteiden välillä.
Vaikutukset epäsuoraan antiglobuliinikokeeseen (epäsuoraan Coombsin kokeeseen)
Daratumumabi sitoutuu veren punasolujen pinnalla CD38-proteiiniin ja häiritsee yhteensopivuustestausta, vasta-aineiden seulonta ja veriryhmien sopivuuskoe (ristikoe) mukaan lukien (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Daratumumabin testejä häiritsevää vaikutusta voidaan vähentää mm. käsittelemällä reagenssin punasolut ditiotreitolilla (DTT), jotta daratumumabin sitoutuminen häiriintyy, tai muilla paikallisesti validoiduilla menetelmillä. Koska Kell-veriryhmäjärjestelmä on myös herkkä DTT-käsittelylle, potilaalle pitää antaa Kell-negatiivisia yksikköjä sen jälkeen, kun allovasta-aineet on suljettu pois tai tunnistettu DTT-käsiteltyjen punasolujen avulla. Vaihtoehtoisesti voidaan harkita myös fenotyypitystä tai genotyypitystä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vaikutukset seerumin proteiinielektroforeesi- ja immunofiksaatiokokeisiin
Daratumumabi voidaan havaita sekä seerumin proteiinielektroforeesi- (SPE) että immunofiksaatio (IFE) ‑määrityksellä, joita käytetään sairauden monoklonaalisten immunoglobuliinien (M-proteiinin) seurantaan. Se voi aiheuttaa SPE- ja IFE-määritysten virheellisesti positiivisen tuloksen, jos potilaalla on IgG-kappamyeloomaproteiinia, mikä vaikuttaa International Myeloma Working Group (IMWG) ‑kriteerien mukaiseen täydellisen vasteen arviointiin. Jos potilaalla on pitkään erittäin hyvä osittainen vaste ja daratumumabin epäillään häiritsevän määritystä, on harkittava validoitua daratumumabispesifistä immunofiksaatiomääritystä, jotta daratumumabi ja potilaan seerumissa mahdollisesti jäljellä oleva endogeeninen M-proteiini voidaan erottaa toisistaan ja täydellinen vaste saadaan siten paremmin määritetyksi.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy
Naisten, jotka voivat tulla raskaaksi, pitää käyttää tehokasta ehkäisyä daratumumabihoidon aikana ja kolmen kuukauden ajan hoidon päättymisen jälkeen.
Raskaus
Daratumumabin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja. Eläimillä tehdyistä tutkimuksista saadut tiedot eivät riitä lisääntymistoksisuuden selvittämiseen (ks. kohta Prekliiniset tiedot turvallisuudesta). Darzalex-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyykö daratumumabi ihmisillä äidinmaitoon.
Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. On päätettävä, lopetetaanko imetys vai pidättäydytäänkö Darzalex-hoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Daratumumabista miehen tai naisen hedelmällisyyteen mahdollisesti aiheutuvien vaikutusten arvioimiseksi ei ole tietoja saatavissa (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Darzalex-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Daratumumabia käyttäneillä potilailla on kuitenkin raportoitu uupumusta, mikä pitää ottaa huomioon autoa ajettaessa ja koneita käytettäessä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Monoterapiana tai yhdistelmähoitona käytetyn daratumumabin (laskimoon tai ihon alle annettavan valmisteen) yleisimpiä minkä tahansa vaikeusasteen haittavaikutuksia (≥ 20 % potilaista) olivat infuusioreaktiot, uupumus, pahoinvointi, ripuli, ummetus, kuume, yskä, neutropenia, trombosytopenia, anemia, raajojen turvotus, perifeerinen neuropatia, ylähengitysteiden infektio, muskuloskeletaalinen kipu ja Covid-19-tauti. Vakavia haittavaikutuksia olivat keuhkokuume, keuhkoputkitulehdus, ylähengitysteiden infektio, sepsis, keuhkoedeema, influenssa, kuume, elimistön kuivuminen, ripuli, eteisvärinä ja pyörtyminen.
Ihon alle annettavan Darzalex-valmisteen turvallisuusprofiili oli samankaltainen kuin laskimoon annettavan lääkemuodon turvallisuusprofiili infuusioon liittyviä reaktioita lukuun ottamatta, joiden esiintyvyys oli pienempi. Neutropenia oli vaiheen III MMY3012-tutkimuksessa ainoa haittavaikutus, jota raportoitiin ihon alle annettavan Darzalex-valmisteen käytössä ≥ 5 % yleisemmin kuin laskimoon annettavan daratumumabin käytössä (Gradus 3/4: pistos ihon alle: 13 %; infuusio laskimoon: 8 %).
Haittavaikutustaulukko
Taulukossa 9 esitetään yhteenveto ihon alle annettavaa Darzalex-valmistetta tai laskimoon annettavaa daratumumabia saaneilla potilailla esiintyneistä haittavaikutuksista.
Tiedot kuvastavat altistusta ihon alle annettavalle Darzalex-valmisteelle (1 800 mg) 1 187:llä multippelia myeloomaa sairastavalla potilaalla. Tiedoissa on mukana 260 potilasta, jotka saivat vaikuttavalla aineella kontrolloidussa vaiheen III tutkimuksessa (MMY3012) ihon alle annettavaa Darzalex-injektionestettä monoterapiana, 149 potilasta, jotka saivat vaikuttavalla aineella kontrolloidussa vaiheen III tutkimuksessa (MMY3013) ihon alle annettavaa Darzalex-injektionestettä yhdistelmänä pomalidomidin ja deksametasonin kanssa (D-Pd), 351 potilasta, jotka saivat vaikuttavalla aineella kontrolloidussa vaiheen III tutkimuksessa (MMY3014) ihon alle annettavaa DARZALEX-injektionestettä yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin (D‑VRd) kanssa, ja 197 äskettäin diagnosoitua multippelia myeloomaa sairastavaa potilasta, joille ei suunniteltu siirtoa alkuvaiheen hoitona tai jotka eivät soveltuneet siirtoon ja jotka saivat vaikuttavalla aineella kontrolloidussa vaiheen III tutkimuksessa (MMY3019) ihon alle annettavaa DARZALEX-injektionestettä yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin (D-VRd) kanssa. Tiedot kuvastavat myös kolmea avointa kliinistä tutkimusta, joissa potilaat saivat ihon alle annettavaa Darzalex-injektionestettä monoterapiana (n = 31, MMY1004 ja MMY1008), ja tutkimuksessa MMY2040, jossa potilaat saivat ihon alle annettavaa Darzalex-injektionestettä yhdistelmänä joko bortetsomibin, melfalaanin ja prednisonin (D-VMP, n = 67), lenalidomidin ja deksametasonin (D-Rd, n = 65) tai bortetsomibin, lenalidomidin ja deksametasonin (D-VRd, n = 67) kanssa. Tiedot kuvastavat altistusta 193 potilaalla, joilla oli indolentti multippeli myelooma, jonka multippeliksi myeloomaksi etenemisen riski on suuri. Nämä potilaat olivat mukana vaiheen III satunnaistetussa tutkimuksessa (SMM3001), jossa potilaat saivat ihon alle annettavaa DARZALEX-injektionestettä monoterapiana. Tiedot kuvastavat lisäksi 193:n äskettäin diagnosoitua AL-amyloidoosia sairastavan potilaan altistusta. Nämä potilaat olivat mukana vaiheen III vaikuttavalla aineella kontrolloidussa tutkimuksessa (AMY3001), jossa potilaat saivat ihon alle annettavaa Darzalex-injektionestettä yhdistelmänä bortetsomibin, syklofosfamidin ja deksametasonin (D-VCd) kanssa.
Turvallisuutta koskevat tiedot kuvastavat myös altistusta laskimoon annettavalle daratumumabille (16 mg/kg) 2 324:lla multippelia myeloomaa sairastavalla potilaalla, joista 1 910 potilasta sai daratumumabia laskimoon yhdistelmänä runkohoidon kanssa ja 414 potilasta sai daratumumabia laskimoon monoterapiana. Myös valmisteen markkinoille tulon jälkeiset haittavaikutukset on otettu mukaan.
Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (> 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000).
| Taulukko 9: Haittavaikutukset laskimoon annettavaa daratumumabia tai ihon alle annettavaa daratumumabia saaneilla multippelia myeloomaa, mukaan lukien indolenttia multippelia myeloomaa, jonka multippeliksi myeloomaksi etenemisen riski on suuri, ja AL-amyloidoosia sairastavilla potilailla | ||||
| Elinjärjestelmä | Haittavaikutus | Esiintyvyys | Ilmaantuvuus (%) | |
| Kaikki gradukset | Gradus 3–4 | |||
| Infektiot | Ylähengitystieinfektioa | Hyvin yleinen | 46 | 3 |
| Covid-19-tautia, g | 23 | 6 | ||
| Keuhkokuumea | 19 | 11 | ||
| Keuhkoputkitulehdusa | 14 | 1 | ||
| Virtsatieinfektio | Yleinen | 7 | 1 | |
| Sepsisa | 4 | 4 | ||
| Sytomegalovirusinfektioa | Melko harvinainen | < 1 | < 1# | |
| Hepatiitti B ‑viruksen reaktivaatioa | < 1 | < 1 | ||
| Veri ja imukudos | Neutropeniaa | Hyvin yleinen | 42 | 36 |
| Trombosytopeniaa | 30 | 18 | ||
| Anemiaa | 26 | 11 | ||
| Lymfopeniaa | 12 | 10 | ||
| Leukopeniaa | 11 | 6 | ||
| Immuunijärjestelmä | Hypogammaglobulinemiaa | Yleinen | 3 | < 1# |
| Anafylaktinen reaktiob | Harvinainen | - | - | |
| Aineenvaihdunta ja ravitsemus | Hypokalemiaa | Hyvin yleinen | 10 | 3 |
| Heikentynyt ruokahalu | 10 | < 1 | ||
| Hyperglykemia | Yleinen | 6 | 3 | |
| Hypokalsemia | 6 | 1 | ||
| Elimistön kuivuminen | 2 | 1# | ||
| Psyykkiset häiriöt | Unettomuus | Hyvin yleinen | 17 | 1# |
| Hermosto | Perifeerinen neuropatia | Hyvin yleinen | 31 | 4 |
| Päänsärky | 11 | < 1# | ||
| Heitehuimaus | Yleinen | 9 | < 1# | |
| Parestesiat | 9 | < 1 | ||
| Pyörtyminen | 3 | 2# | ||
| Sydän | Eteisvärinä | Yleinen | 4 | 1 |
| Verisuonisto | Hypertensioa | Yleinen | 9 | 4 |
| Hengityselimet, rintakehä ja välikarsina | Yskäa | Hyvin yleinen | 22 | < 1# |
| Hengenahdistusa | 18 | 2 | ||
| Keuhkoedeemaa | Yleinen | 1 | < 1 | |
| Ruoansulatuselimistö | Ripuli | Hyvin yleinen | 33 | 5 |
| Ummetus | 28 | 1 | ||
| Pahoinvointi | 22 | 1# | ||
| Vatsakipua | 14 | 1 | ||
| Oksentelu | 13 | 1# | ||
| Haimatulehdusa | Yleinen | 1 | < 1 | |
| Iho ja ihonalainen kudos | Ihottuma | Hyvin yleinen | 12 | 1# |
| Kutina | Yleinen | 6 | < 1# | |
| Luusto, lihakset ja sidekudos | Muskuloskeletaalinen kipua,h | Hyvin yleinen | 35 | 3 |
| Nivelkipu | 14 | 1 | ||
| Lihasspasmit | 12 | < 1# | ||
| Yleisoireet ja antopaikassa todettavat haitat | Uupumus | Hyvin yleinen | 24 | 4 |
| Raajojen turvotusa | 24 | 1 | ||
| Kuume | 22 | 1 | ||
| Astenia | 19 | 2 | ||
| Injektiokohdan reaktiotd,e | 10 | 0 | ||
| Vilunväristykset | Yleinen | 8 | < 1# | |
| Vammat, myrkytykset ja hoitokomplikaatiot | Infuusioon liittyvät reaktiotc | |||
| Laskimoon annettava daratumumabif | Hyvin yleinen | 39 | 5 | |
| Ihon alle annettava daratumumabie | Yleinen | 9 | 1 | |
# Ei gradus 4 a Viittaa termien yhdistelmään. b Markkinoille tulon jälkeinen haittavaikutus. c Infuusioon liittyvät reaktiot sisältävät termit, jotka tutkijoiden määrityksen mukaan liittyvät daratumumabi-infuusioon/injektioon. d Injektiokohdan reaktiot sisältävät termit, jotka tutkijoiden määrityksen mukaan liittyvät daratumumabi-injektioon. e Esiintyvyys perustuu vain ihon alle annettavalla daratumumabilla tehtyihin tutkimuksiin (n = 1 573). f Esiintyvyys perustuu vain laskimoon annettavalla daratumumabilla tehtyihin tutkimuksiin (n = 2 324). g Ilmaantuvuus perustuu niiden potilaiden alaryhmään, jotka saivat tutkimuksissa MMY3003, MMY3006, MMY3008 ja MMY3013 vähintään yhden tutkimushoitoannoksen 1. helmikuuta 2020 tai sen jälkeen (covid-19-pandemian alku), sekä kaikkiin daratumumabihoitoa tutkimuksissa MMY3014, MMY3019 ja SMM3001 saaneisiin potilaisiin (N = 1 177). h Muskuloskeletaalinen kipu sisältää seuraavat: selkäkipu, kylkikipu, nivuskipu, muskuloskeletaalinen rintakipu, muskuloskeletaalinen kipu, muskuloskeletaalinen jäykkyys, lihaskipu, niskakipu, ei-sydänperäinen rintakipu ja raajakipu. Huom.: Perustuu 3 897:ään multippelia myeloomaa ja AL-amyloidoosia sairastavaan potilaaseen, jotka saivat laskimoon annettavaa daratumumabia tai ihon alle annettavaa daratumumabia. | ||||
Valikoitujen haittavaikutusten kuvaus
Infuusioon liittyvät reaktiot
Ihon alle annettavalla DARZALEX-valmisteella tehdyissä kliinisissä tutkimuksissa (monoterapia ja yhdistelmähoidot; n = 1 573) kaikkien vaikeusasteiden infuusioon liittyvien reaktioiden ilmaantuvuus oli ensimmäisen DARZALEX-injektion (1 800 mg, viikko 1) yhteydessä 7,5 %, viikon 2 injektion yhteydessä 0,5 % ja seuraavien injektioiden yhteydessä 1,3 %. Graduksen 3 infuusioon liittyviä reaktioita havaittiin 0,8 %:lla potilaista, ja graduksen 4 infuusioon liittyviä reaktioita havaittiin 0,1 %:lla potilaista.
Infuusioon liittyvien reaktioiden oireita ja löydöksiä voivat olla mm. hengityselinoireet, kuten nenän tukkoisuus, yskä, kurkun ärsytys, allerginen nuha, hengityksen vinkuminen, sekä kuume, rintakipu, kutina, vilunväristykset, oksentelu, pahoinvointi, hämärtynyt näkö ja hypotensio. Myös vaikea-asteisia reaktioita on esiintynyt, mm. bronkospasmeja, hypoksiaa, hengenahdistusta, hypertensiota, takykardiaa ja silmiin liittyviä haittavaikutuksia (mukaan lukien silmän suonikalvon effuusiota, akuuttia likitaittoisuutta ja akuuttia ahdaskulmaglaukoomaa) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Injektiokohdan reaktiot
Ihon alle annettavalla DARZALEX-valmisteella tehdyissä kliinisissä tutkimuksissa (n = 1 573) kaikkien vaikeusasteiden injektiokohdan reaktioiden ilmaantuvuus oli 10,2 %. Graduksen 3 tai 4 injektiokohdan reaktioita ei esiintynyt. Yleisimmät (> 1 %) injektiokohdan reaktiot injektiokohdassa olivat punoitus ja ihottuma.
Infektiot
Daratumumabia monoterapiana saaneilla multippelia myeloomaa sairastavilla potilailla infektioiden kokonaisilmaantuvuus oli samankaltainen ihon alle annettavaa Darzalex-valmistetta saaneiden (52,9 %) ja laskimoon annettavaa daratumumabia saaneiden ryhmien (50,0 %) välillä. Myös graduksen 3 tai 4 infektioita esiintyi samankaltaisina esiintyvyyksinä ihon alle annettavaa Darzalex-valmistetta saaneilla (11,7 %) ja laskimoon annettavaa daratumumabia saaneilla (14,3 %). Useimmat infektiot olivat hoidettavissa ja johtivat harvoin hoidon keskeyttämiseen. Tutkimuksissa yleisimmin raportoitu graduksen 3 tai 4 infektio oli keuhkokuume. Aktiivisella aineella kontrolloiduissa tutkimuksissa 1–4 % potilaista keskeytti hoidon infektioiden vuoksi. Kuolemaan johtaneita infektioita olivat pääasiassa keuhkokuume ja sepsis.
Laskimoon annettavaa daratumumabia yhdistelmähoitona saaneilla multippelia myeloomaa sairastavilla potilailla raportoitiin seuraavia:
graduksen 3 tai 4 infektiot:
Tutkimukset potilailla, joilla oli relapsoitunut tai hoitoon reagoimaton tauti: DVd: 21 %, Vd: 19 %; DRd: 28 %, Rd: 23 %; DPd: 28 %.
Tutkimukset potilailla, joilla oli äskettäin diagnosoitu tauti: D‑VMP: 23 %, VMP: 15 %; DRd: 32 %, Rd: 23 %; D-VTd: 22 %, VTd: 20 %.
Kuolemaan johtaneet (gradus 5) infektiot:
Tutkimukset potilailla, joilla oli relapsoitunut tai hoitoon reagoimaton tauti: DVd: 1 %, Vd: 2 %; DRd: 2 %, Rd: 1 %; DPd: 2 %.
Tutkimukset potilailla, joilla oli äskettäin diagnosoitu tauti: D-VMP: 1 %, VMP: 1 %; DRd: 2 %, Rd: 2 %; DVTd: 0 %, VTd: 0 %.
Ihon alle annettavaa Darzalex-injektionestettä yhdistelmähoitona saaneilla multippelia myeloomaa sairastavilla potilailla raportoitiin seuraavia:
graduksen 3 tai 4 infektiot: DPd: 28 %, Pd: 23 %; D‑VRd (siirtoon soveltuneet): 35 %, VRd (siirtoon soveltuneet): 27 %; D-VRd (siirtoon soveltumattomat): 40 %; VRd (siirtoon soveltumattomat): 32 %
graduksen 5 (kuolemaan johtaneet) infektiot: DPd: 5 %, Pd: 3 %; D‑VRd (siirtoon soveltuneet): 2 %; VRd (siirtoon soveltuneet): 3 %; D-VRd (siirtoon soveltumattomat): 8 %; VRd (siirtoon soveltumattomat): 6 %.
Lyhenteet: D = daratumumabi; Vd = bortetsomibi-deksametasoni; Rd = lenalidomidi-deksametasoni; Pd = pomalidomidi-deksametasoni; VMP = bortetsomibi-melfalaani-prednisoni; VTd = bortetsomibi-talidomidi-deksametasoni; VRd = bortetsomibi-lenalidomidi-deksametasoni.
Ihon alle annettavaa DARZALEX-injektionestettä monoterapiana saaneilla indolenttia multippelia myeloomaa, jonka multippeliksi myeloomaksi etenemisen riski on suuri, sairastavilla potilailla raportoitiin seuraavia:
graduksen 3 tai 4 infektiot: ihon alle annettava DARZALEX-injektioneste: 16 %
graduksen 5 infektiot: ihon alle annettava DARZALEX-injektioneste: 1 %.
Ihon alle annettavaa Darzalex-injektionestettä yhdistelmähoitoa saaneilla AL-amyloidoosia sairastavilla potilailla raportoitiin seuraavia:
graduksen 3 tai 4 infektiot: D-VCd: 17 %, VCd: 10 %
graduksen 5 infektiot: D-VCd: 1 %, VCd: 1 %
Lyhenteet: D = daratumumabi; VCd = bortetsomibi-syklofosfamidi-deksametasoni
Hemolyysi
Hemolyysin riski on teoriassa olemassa. Tätä turvallisuutta koskevaa signaalia seurataan jatkuvasti kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeisistä turvallisuutta koskevista tiedoista.
Sydänsairaudet ja AL-amyloidoosiin liittyvä sydänlihassairaus
Valtaosalla AMY3001-tutkimuksen potilaista oli lähtötilanteessa AL-amyloidoosiin liittyvä sydänlihassairaus (D-VCd 72 % vs. VCd 71 %). Graduksen 3 tai 4 sydänsairauksia oli 11 %:lla D‑VCd-hoitoa saaneista potilaista verrattuna 10 %:iin VCd-hoitoa saaneista potilaista, kun taas vakavia sydänsairauksia oli 16 %:lla D-VCd-hoitoa saaneista potilaista vs. 13 %:lla VCd-hoitoa saaneista potilaista. ≥ 2 %:lla potilaista esiintyneitä vakavia sydänsairauksia olivat sydämen vajaatoiminta (D-VCd 6,2 % vs. VCd 4,3 %), sydämenpysähdys (D-VCd 3,6 % vs. VCd 1,6 %) ja eteisvärinä (D-VCd 2,1 % vs. VCd 1,1 %). Kaikilla D-VCd-hoitoa saaneilla potilailla, jolla oli vakava tai kuolemaan johtanut sydänsairaus, oli lähtötilanteessa AL-amyloidoosiin liittynyt sydänlihassairaus. Sydänsairauksien esiintyvyyden vertailussa näiden kahden hoitoryhmän välillä pitää ottaa huomioon hoidon pidempi kesto (mediaani) D-VCd-ryhmässä kuin VCd-ryhmässä (D-VCd-ryhmä 9,6 kuukautta vs. VCd-ryhmä 5,3 kuukautta). Altistuksen suhteen korjatut ilmaantuvuudet (niiden potilaiden lukumäärä, joilla tapahtuma ilmeni, riskiryhmän 100 potilaskuukautta kohden) olivat kaikkiaan verrannolliset D-VCd-ryhmän ja VCd-ryhmän välillä: graduksen 3 tai 4 sydänsairaudet (1,2 vs. 2,3), sydämen vajaatoiminta (0,5 vs. 0,6), sydämenpysähdys (0,1 vs. 0,0) ja eteisvärinä (0,2 vs. 0,1).
Tutkimuksen AMY3001 seuranta-ajan mediaani oli 11,4 kuukautta. Tutkimuksessa todetut kuolemat (D-VCd 14 % vs. VCd 15 %) johtuivat kummassakin hoitoryhmässä pääasiassa AL-amyloidoosiin liittyvästä sydänlihassairaudesta.
Muut erityispotilasjoukot
Vaiheen III tutkimuksessa MMY3007 verrattiin D-VMP-hoitoa VMP-hoitoon potilailla, joilla oli äskettäin diagnosoitu multippeli myelooma ja jotka eivät soveltuneet autologiseen kantasolusiirtoon. Hoidon turvallisuutta koskeva analyysi niiden potilaiden alaryhmästä, joiden ECOG-toimintakykyluokka oli 2 (D-VMP: n = 89, VMP: n = 84), oli yhdenmukainen koko potilasjoukon kanssa (ks. kohta Farmakodynamiikka).
Iäkkäät potilaat
Suositeltuja daratumumabiannoksia saaneista 4 553 potilaasta (ihon alle n = 1 615; laskimoon n = 2 938) 38 % oli 65- – alle 75-vuotiaita ja 15 % oli 75‑vuotiaita tai vanhempia. Tehossa ei yleisesti havaittu ikään liittyviä eroja. Vakavien haittavaikutusten ilmaantuvuus oli iäkkäillä potilailla suurempi kuin nuoremmilla potilailla. Relapsoitunutta ja hoitoon reagoimatonta multippelia myeloomaa sairastavilla potilailla (n = 1 976) yleisimmät vakavat haittavaikutukset, jotka olivat yleisempiä iäkkäillä (≥ 65-vuotiailla), olivat keuhkokuume ja sepsis. Äskettäin diagnosoitua multippelia myeloomaa sairastavilla potilailla, jotka eivät soveltuneet autologiseen kantasolusiirtoon (n = 777), yleisin vakava haittavaikutus, joka oli yleisempi iäkkäillä (≥ 75-vuotiailla), oli keuhkokuume. Äskettäin diagnosoitua multippelia myeloomaa sairastavilla potilailla, jotka soveltuivat autologiseen kantasolusiirtoon (n = 351), yleisin vakava haittavaikutus, joka oli yleisempi iäkkäillä (≥ 65-vuotiailla), oli keuhkokuume. Äskettäin diagnosoitua multippelia myeloomaa sairastavilla potilailla, joille ei suunniteltu siirtoa alkuvaiheen hoitona tai jotka eivät soveltuneet autologiseen kantasolusiirtoon (n = 197), yleisin iäkkäillä (≥ 65-vuotiailla) ilmennyt vakava haittavaikutus oli keuhkokuume. Indolenttia multippelia myeloomaa, jonka multippeliksi myeloomaksi etenemisen riski on suuri, sairastavilla potilailla (n = 193) yleisin vakava haittavaikutus, joka oli iäkkäillä (≥ 65-vuotiailla) yleisempi, oli keuhkokuume. Äskettäin diagnosoitua AL-amyloidoosia sairastavilla potilailla (n = 193) yleisin vakava haittavaikutus, joka oli iäkkäillä (≥ 65-vuotiailla) yleisempi, oli keuhkokuume.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet ja löydökset
Yliannostuksesta ei ole kliinisiin tutkimuksiin perustuvaa kokemusta.
Hoito
Daratumumabiyliannoksen hoitoon ei tunneta spesifistä vastalääkettä. Yliannoksen yhteydessä potilasta on seurattava haittavaikutusten oireiden ja löydösten havaitsemiseksi ja oireiden tarkoituksenmukainen hoito on aloitettava heti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, monoklonaaliset vasta-aineet ja vasta‑ainekonjugoidut lääkkeet, ATC-koodi: L01FC01.
Ihon alle annettava Darzalex-injektioneste sisältää rekombinanttia ihmisen hyaluronidaasia (rHuPH20). rHuPH20 vaikuttaa paikallisesti ja ohimenevästi hajottamalla hyaluronaania ([HA], joka on kaikkialla elimistössä luontaisesti esiintyvä glykoaminoglykaani) ihonalaistilan solunulkoisessa matriksissa siten, että se katkaisee sidoksen hyaluronaasin muodostavan kahden sokerin (N‑asetyyliglykosamiinin ja glukuronihapon) välillä. rHuPH20:n puoliintumisaika ihossa on alle 30 minuuttia. Hyaluronaanin pitoisuus ihonalaisessa kudoksessa palautuu normaaliksi 24–48 tunnin kuluessa, sillä hyaluronaanin biosynteesi on nopea.
Vaikutusmekanismi
Daratumumabi on ihmisen monoklonaalinen IgG1κ-vasta-aine, joka sitoutuu CD38-proteiiniin. CD38-proteiini ilmentyy monenlaisissa hematologisissa syövissä solujen pinnalla, mukaan lukien multippelissa myeloomassa ja AL-amyloidoosissa klonaalisten plasmasolujen pinnalla, sekä muissa solutyypeissä ja kudoksissa. CD38-proteiinilla on useita tehtäviä, kuten reseptorivälitteinen adheesio, signaalinvälitys ja entsymaattinen aktiivisuus.
Daratumumabin on osoitettu estävän in vivo tehokkaasti CD38-proteiinia ilmentävien kasvainsolujen kasvua. Daratumumabi saattaa in vitro ‑tutkimusten perusteella hyödyntää useita efektoritoimintoja, jotka aiheuttavat immuunivälitteisen kasvainsolukuoleman. Nämä tutkimukset viittaavat siihen, että daratumumabi voi komplementtiriippuvaisen sytotoksisuuden, vasta-aineriippuvaisen soluvälitteisen sytotoksisuuden ja vasta-aineriippuvaisen solujen fagosytoosin kautta indusoida kasvainsolulyysin CD38-proteiinia ilmentävissä syöpäkasvaimissa. Daratumumabivälitteinen solulyysi vähentää joidenkin myelooisten suppressorisolujen (CD38+MDSCs), säätelijä-T-solujen (CD38+Tregs) ja ‑B-solujen (CD38+Bregs) määrää. Myös T-solujen (CD3+, CD4+ ja CD8+) tiedetään kehitysvaiheesta ja aktivaatiotasosta riippuen ilmentävän CD38-proteiinia. Daratumumabihoidon havaittiin lisäävän huomattavasti ääreisosien kokoveressä ja luuytimessä olevaa T-solujen (CD4+ ja CD8+) absoluuttista määrää ja lymfosyyttien prosenttiosuutta. T-solureseptorien DNA-sekvensointi varmisti lisäksi, että daratumumabihoito lisäsi T-solujen klonaalisuutta. Tämä osoittaa, että daratumumabivalmisteella on immuniteettia muuntavia vaikutuksia, jotka saattavat edistää kliinisen vasteen saamista.
Daratumumabi indusoi in vitro apoptoosia Fc‑välitteisen silloittumisen (cross-linking) jälkeen. Daratumumabi muunsi lisäksi CD38-proteiinin entsymaattista aktiivisuutta. Se esti siten syklaasientsyymin aktiivisuutta ja stimuloi hydrolaasin aktiivisuutta. Näiden in vitro ‑vaikutusten kliininen merkitys ja merkitys kasvaimen kasvun kannalta tunnetaan huonosti.
Farmakodynaamiset vaikutukset
Luonnolliset tappajasolut (NK-solut) ja T-solumäärä
NK-solujen tiedetään ilmentävän suuria CD38-proteiinipitoisuuksia ja olevan herkkiä daratumumabivälitteiselle solulyysille. Daratumumabihoidon havaittiin vähentävän kaikkien NK-solujen (CD16+CD56+) sekä aktivoitujen NK-solujen (CD16+CD56dim) absoluuttista määrää ja prosenttiosuutta sekä ääreisosien kokoveressä ja luuytimessä. Lähtötilanteen NK-solupitoisuudella ei kuitenkaan osoitettu olevan yhteyttä kliiniseen vasteeseen.
Immunogeenisuus
Ihon alle annettavaa daratumumabia monoterapiana ja yhdistelmähoitona kliinisissä tutkimuksissa saaneista multippelia myeloomaa, mukaan lukien indolenttia multippelia myeloomaa, jonka multippeliksi myeloomaksi etenemisen riski on suuri, ja AL-amyloidoosia sairastavista potilaista alle 1 %:lle kehittyi hoidon aikana daratumumabin vasta-aineita ja 8 potilaalla todettiin testissä neutraloivia vasta-aineita.
Multippelia myeloomaa, mukaan lukien indolenttia multippelia myeloomaa, jonka multippeliksi myeloomaksi etenemisen riski on suuri, ja AL-amyloidoosia sairastavilla potilailla, jotka saivat joko ihon alle annettavaa DARZALEX-valmistetta monoterapiana tai ihon alle annettavaa DARZALEX-valmistetta yhdistelmähoitona, hoidonaikaisten rHuPH20:n vasta-aineiden ilmaantuvuus oli 8,9 % (133 / 1 491), ja yhdellä potilaalla todettiin testissä neutraloivia vasta-aineita. rHuPH20:n vasta-aineet eivät näyttäneet vaikuttavan daratumumabialtistuksiin. Ihon alle annettavan DARZALEX-hoidon jälkeisen daratumumabin tai rHuPH20:n vasta-aineiden kehittymisen kliinistä merkitystä ei tiedetä.
Kliininen kokemus Darzalex-injektionesteen antamisesta injektioina ihon alle (ihon alle annettava valmiste)
Monoterapia – relapsoitunut/hoitoon reagoimaton multippeli myelooma
MMY3012 oli avoin, satunnaistettu, vaiheen III non‑inferioriteettitutkimus, jossa ihon alle annettavan Darzalex-injektionesteen (1 800 mg) tehoa ja turvallisuutta verrattiin laskimoon annettavaan daratumumabiin (16 mg/kg) relapsoitunutta tai hoitoon reagoimatonta multippelia myeloomaa sairastavilla potilailla, jotka olivat saaneet aiemmin vähintään kolme hoitolinjaa, mukaan lukien proteasomin estäjää ja immuniteettia muuntavaa lääkeainetta, tai jotka eivät reagoineet proteasomin estäjällä eivätkä immuniteettia muuntavalla aineella annettuun hoitoon. Hoitoa jatkettiin, kunnes ilmaantui toksisuutta, joka ei ollut hyväksyttävissä, tai tauti eteni.
Yhteensä 522 potilasta satunnaistettiin: 263 satunnaistettiin ihon alle annettavaa Darzalex-valmistetta ryhmään ja 259 satunnaistettiin laskimoon annettavaa daratumumabia saavaan ryhmään. Demografiset ja taudin ominaisuudet olivat lähtötilanteessa samankaltaiset näiden kahden hoitoryhmän välillä. Potilaiden iän mediaani oli 67 vuotta (vaihteluväli: 33–92 vuotta), 55 % oli miehiä, ja 78 % oli valkoihoisia. Potilaiden painon mediaani oli 73 kg (vaihteluväli: 29–138 kg). Potilaat olivat aiemmin saaneet neljä hoitolinjaa (mediaani). Yhteensä 51 % potilaista oli saanut aiemmin autologisen kantasolusiirron, 100 % potilaista oli aiemmin saanut hoitoa sekä proteasomin estäjällä/estäjillä että immuniteettia muuntavalla aineella / muuntavilla aineilla, ja valtaosa potilaista ei reagoinut aiempaan systeemiseen hoitoon, mukaan lukien sekä proteasomin estäjään ja immuniteettia muuntavaan aineeseen (49 %).
Tutkimuksessa saavutettiin ensisijaiset yhdistelmäpäätetapahtumat, jotka olivat kokonaisvasteluku (ORR) IMWG-vastekriteerien perusteella (taulukko 10) ja hoitosyklin 3 päivän 1 annosta edeltävä suurin Ctrough-arvo (ks. kohta Farmakokinetiikka).
| Taulukko 10: Tutkimuksen MMY3012 keskeiset tulokset | ||
Ihon alle annettava daratumumabi (n = 263) | Laskimoon annettava daratumumabi (n = 259) | |
| Ensisijainen päätetapahtuma | ||
| Kokonaisvasteluku (sCR+CR+VGPR+PR), n (%)a | 108 (41,1 %) | 96 (37,1 %) |
| 95 %:n luottamusväli (%) | (35,1 %; 47,3 %) | (31,2 %; 43,3 %) |
| Vastelukujen suhde (95 %:n luottamusväli)b | 1,11 (0,89; 1,37) | |
| Täydellinen vaste (CR) tai parempi, n (%) | 5 (1,9 %) | 7 (2,7 %) |
| Erittäin hyvä osittainen vaste (VGPR) | 45 (17,1 %) | 37 (14,3 %) |
| Osittainen vaste (PR) | 58 (22,1 %) | 52 (20,1 %) |
| Toissijainen päätetapahtuma | ||
| Infuusioon liittyvien reaktioiden määrä, n (%)c | 33 (12,7 %) | 89 (34,5 %) |
| Etenemättömyysaika, kuukautta | ||
| Mediaani (95 %:n luottamusväli) | 5,59 (4,67; 7,56) | 6,08 (4,67; 8,31) |
| Riskisuhde (95 %:n luottamusväli) | 0,99 (0,78; 1,26) | |
a Perustuu hoitoaikeen mukaiseen potilasjoukkoon (intent-to-treat population). b Vertailukelpoisuutta (non-inferiority) koskevan hypoteesin Farrington-Manningin testillä saatu p-arvo < 0,0001. c Perustuu turvallisuuspopulaatioon. Cochran-Mantel-Haenszelin khiin neliötestiin perustuva p-arvo < 0,0001. | ||
Kokonaiselossaolon mediaani oli 29,3 kuukauden (mediaani) seurannan jälkeen ihon alle annettavaa Darzalex-valmistetta saaneessa ryhmässä 28,2 kuukautta (95 %:n luottamusväli: 22,8; ei arvioitavissa) ja laskimoon annettavaa daratumumabia saaneessa ryhmässä 25,6 kuukautta (95 %:n luottamusväli: 22,1; ei arvioitavissa).
Turvallisuutta ja siedettävyyttä koskevat tulokset olivat myös pienipainoisilla potilailla yhdenmukaiset ihon alle annettavan Darzalex-valmisteen ja laskimoon annettavan daratumumabin tunnetun turvallisuusprofiilin kanssa.
Potilaiden tyytyväisyyttä hoitoonsa selvittävän muokatun kyselyn (modified‑CTSQ) tulokset osoittivat, että ihon alle annettavaa Darzalex-valmistetta saaneet potilaat olivat hoitoonsa tyytyväisempiä kuin laskimoon annettavaa daratumumabia saaneet potilaat. Avoimiin tutkimuksiin voi kuitenkin liittyä harhaa.
Multippelin myelooman yhdistelmähoidot
Yhdistelmähoito bortetsomibin, lenalidomidin ja deksametasonin kanssa (VRd) äskettäin diagnosoitua multippelia myeloomaa sairastavilla potilailla, jotka soveltuivat autologiseen kantasolusiirtoon
Tutkimus MMY3014 oli avoin, satunnaistettu, vaikuttavalla aineella kontrolloitu vaiheen III tutkimus, jossa induktio- ja konsolidaatiohoitoa ihon alle annettavalla DARZALEX-injektionesteellä (1800 mg) yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin (D‑VRd) kanssa ja sen jälkeen annettua ylläpitohoitoa DARZALEX-valmisteella yhdistelmänä lenalidomidin kanssa verrattiin hoitoon bortetsomibin, lenalidomidin ja deksametasonin yhdistelmällä (VRd), jonka jälkeen annettiin ylläpitohoitoa lenalidomilla. Vertailu tehtiin 70-vuotiailla ja sitä nuoremmilla äskettäin diagnosoitua multippelia myeloomaa sairastavilla potilailla, jotka soveltuivat autologiseen kantasolusiirtoon, kunnes todettiin taudin dokumentoitu eteneminen tai toksisuutta, joka ei ollut hyväksyttävissä. Ennen hoitoa oli sallittua antaa hätätilanteessa lyhyt kortikosteroidikuuri (joka vastasi deksametasoniannosta 40 mg/vrk enintään 4 päivän ajan). Potilaat saivat ihon alle annettavaa DARZALEX-valmistetta (1 800 mg) hoitosykleissä 1–2 kerran viikossa (päivinä 1, 8, 15 ja 22) ja sen jälkeen hoitosykleissä 3–6 kerran joka toinen viikko (päivinä 1 ja 15). Ylläpitohoidossa (hoitosyklistä 7 alkaen) potilaat saivat ihon alle annettavaa DARZALEX-valmistetta (1 800 mg) kerran joka neljäs viikko. Potilailla, joiden todettiin olleen MRD-negatiivisia 12 kuukauden ajan ja jotka olivat saaneet ylläpitohoitoa vähintään 24 kuukauden ajan, hoito ihon alle annettavalla DARZALEX-valmisteella (1 800 mg) lopetettiin. Bortetsomibi annettiin injektioina ihon alle (s.c.) kehon pinta-alan mukaisena annoksena 1,3 mg/m2 toistuvien 28 päivän (4 viikon) pituisten hoitosyklien 1–6 aikana kaksi kertaa viikossa kahden viikon ajan (päivinä 1, 4, 8 ja 11). Lenalidomidi annettiin suun kautta annoksena 25 mg päivittäin hoitosyklien 1–6 päivinä 1–21. Ylläpitohoidossa (hoitosyklistä 7 alkaen) potilaat saivat 10 mg lenalidomidia päivittäin kunkin hoitosyklin päivinä 1–28 (jatkuvasti), kunnes todettiin taudin dokumentoitu eteneminen tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Deksametasoni (suun kautta tai laskimoon) annettiin annoksena 40 mg hoitosyklien 1–6 päivinä 1–4 ja päivinä 9–12. Ihon alle annettavalla DARZALEX-valmisteella toteutettujen injektioiden (1 800 mg) antopäivinä deksametasoniannos annettiin suun kautta tai laskimoon injektion esilääkityksenä. Bortetsomibin, lenalidomidin ja deksametasonin annosmuutokset tehtiin valmistajan valmistetietojen mukaisesti.
Yhteensä 709 potilasta satunnaistettiin: 355 potilasta D‑VRd-ryhmään ja 354 potilasta VRd-ryhmään. Lähtötilanteen demografiset ja sairauden ominaisuudet olivat näiden kahden hoitoryhmän välillä samankaltaiset. Iän mediaani oli 60 vuotta (vaihteluväli: 31–70 vuotta). Valtaosa potilaista oli miehiä (59 %); ECOG-toimintakykyluokka oli 64 %:lla potilaista 0, ECOG-toimintakykyluokka oli 31 %:lla potilaista 1 ja ECOG-toimintakykyluokka oli 5 %:lla potilaista 2. Lisäksi ISS-levinneisyysluokkaan I kuului 51 %, ISS-levinneisyysluokkaan II 34 %, ISS-levinneisyysluokkaan III 15 % potilaista, 75 %:lla oli tavanomainen sytogeneettinen riski, 22 %:lla oli suuri sytogeneettinen riski (del17p, t[4;14], t[14;16]) ja 3 %:lla sytogeneettinen riski oli määrittelemätön.
Kun seuranta-ajan mediaani oli 47,5 kuukautta, tutkimuksen MMY3014 etenemättömyysajan primaarianalyysi osoitti, että etenemättömyysaika oli pidentynyt D- VRd-ryhmässä VRd-ryhmään verrattuna (riskisuhde = 0,42; 95 %:n luottamusväli: 0,30; 0,59; p < 0,0001). Kummassakaan ryhmässä ei saavutettu etenemättömyysajan mediaania.
Kuvio 1: Tutkimuksen MMY3014 etenemättömyysajan (PFS) Kaplan–Meierin käyrä
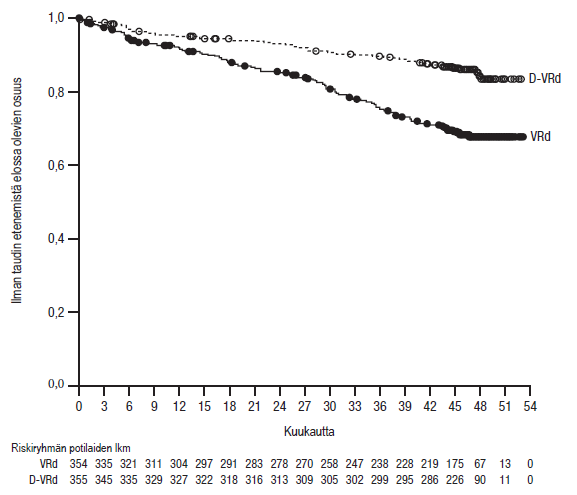
Tutkimuksen MMY3014 muut tehoa koskevat tulokset esitetään alla olevassa taulukossa 11.
| Taulukko 11: Tutkimuksen MMY3014 tehoa koskevat tulokseta | |||
| D-VRd (n = 355) | VRd (n = 354) | Kerroinsuhde (95 %:n luottamusväli)d | |
| Kokonaisvaste (sCR+CR+VGPR+PR) n (%)a | 343 (96,6 %) | 332 (93,8 %) | |
| Täydellinen vaste lisäehdoin (stringent complete response, sCR) | 246 (69,3 %) | 158 (44,6 %) | |
| Täydellinen vaste (complete response, CR) | 66 (18,6 %) | 90 (25,4 %) | |
| Erittäin hyvä osittainen vaste (very good partial response, VGPR) | 26 (7,3 %) | 68 (19,2 %) | |
| Osittainen vaste (partial response, PR) | 5 (1,4 %) | 16 (4,5 %) | |
| Täydellinen vaste tai parempi (sCR+CR) | 312 (87,9 %) | 248 (70,1 %) | 3,13 (2,11; 4,65) |
| 95 %:n luottamusväli (%) | (84,0 %; 91,1 %) | (65,0 %; 74,8 %) | |
| p-arvob | < 0,0001 | ||
| MRD-negatiivisten kokonaislukumääräa,c | 267 (75,2 %) | 168 (47,5 %) | 3,40 (2,47; 4,69) |
| 95 %:n luottamusväli (%) | (70,4 %; 79,6 %) | (42,2 %; 52,8 %) | |
| p-arvob | < 0,0001 | ||
D-VRd = daratumumabi-bortetsomibi-lenalidomidi-deksametasoni; VRd = bortetsomibi-lenalidomidi-deksametasoni; MRD = minimaalinen jäännöstauti a Perustuu hoitoaikeen mukaiseen potilasjoukkoon b p-arvo saatu Cochran Mantel-Haenszelin khi-neliötestillä c Potilailla todettiin sekä MRD-negatiivisuus (kynnysarvo 10-5) ja täydellinen vaste tai parempi d Ositetuissa taulukoissa käytetään yleisen kerroinsuhteen Mantel-Haenszelin estimaattia | |||
Yhdistelmähoito bortetsomibin, lenalidomidin ja deksametasonin kanssa (VRd) äskettäin diagnosoitua multippelia myeloomaa sairastavilla potilailla, joille ei suunnitella autologista kantasolusiirtoa alkuvaiheen hoitona tai jotka eivät sovellu autologiseen kantasolusiirtoon
Tutkimus MMY3019 oli avoin, satunnaistettu, vaikuttavalla aineella kontrolloitu vaiheen III tutkimus, jossa ihon alle annettavan DARZALEX-injektionesteen (1 800 mg) ja bortetsomibin, lenalidomidin ja deksametasonin yhdistelmää (D‑VRd) verrattiin hoitoon bortetsomibin, lenalidomidin ja deksametasonin yhdistelmällä (VRd) potilailla, joilla oli äskettäin diagnosoitu multippeli myelooma ja joille ei suunniteltu autologista kantasolusiirtoa alkuvaiheen hoitona tai jotka eivät soveltuneet autologiseen kantasolusiirtoon. Ennen hoitoa oli sallittua antaa hätätilanteessa lyhyt kortikosteroidikuuri (joka vastasi deksametasoniannosta 40 mg/vrk enintään 4 päivän ajan). Potilaat saivat ihon alle annettavaa DARZALEX-valmistetta (1 800 mg) hoitosykleissä 1–2 ihon alle kerran viikossa (päivinä 1, 8 ja 15) ja sen jälkeen hoitosykleissä 3–8 kerran joka kolmas viikko sekä hoitosyklistä 9 alkaen kerran joka neljäs viikko, kunnes sairaus dokumentoidusti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Bortetsomibi annettiin injektiona ihon alle kehon pinta-alan mukaisena annoksena 1,3 mg/m2 toistuvien 21 päivän (3 viikon) pituisten hoitosyklien 1–8 aikana kaksi kertaa viikossa (päivinä 1, 4, 8 ja 11). Lenalidomidi annettiin suun kautta annoksena 25 mg päivittäin hoitosyklien 1–8 päivinä 1–14 ja hoitosyklistä 9 alkaen päivinä 1–21. Deksametasoni annettiin suun kautta annoksena 20 mg 21 päivän (3 viikon) pituisten hoitosyklien 1–8 päivinä 1, 2, 4, 5, 8, 9, 11 ja 12 ja hoitosyklistä 9 alkaen 28 päivän (4 viikon) pituisten hoitosyklien päivinä 1, 8, 15 ja 22. Ihon alle annettavalla DARZALEX-valmisteella toteutettujen injektioiden (1 800 mg) antopäivinä deksametasoniannos annettiin suun kautta tai laskimoon injektion esilääkityksenä. Bortetsomibin, lenalidomidin ja deksametasonin annosmuutokset tehtiin valmistajan valmistetietojen mukaisesti.
Yhteensä 395 potilasta satunnaistettiin: 197 potilasta D‑VRd-ryhmään ja 198 potilasta VRd-ryhmään. Lähtötilanteen demografiset ja sairauden ominaisuudet olivat näiden kahden hoitoryhmän välillä samankaltaiset. Iän mediaani oli 70 vuotta (vaihteluväli: 31–80 vuotta). Viisikymmentä prosenttia oli miehiä, ECOG-toimintakykyluokka oli 39 %:lla potilaista 0, ECOG-toimintakykyluokka oli 51 %:lla potilaista 1 ja ECOG-toimintakykyluokka oli 9 %:lla potilaista 2. Kahdeksantoista prosenttia oli alle 70-vuotiaita , jotka eivät soveltuneet siirtoon, ja 27 % oli alle 70-vuotiaita, joilla siirto oli siirretty myöhempään ajankohtaan. Lisäksi ISS-levinneisyysluokkaan I kuului 34 %, ISS-levinneisyysluokkaan II 38 %, ISS-levinneisyysluokkaan III 28 %, 75 %:lla oli tavanomainen sytogeneettinen riski, 13 %:lla oli suuri sytogeneettinen riski (del17p, t[4;14], t[14;16]) ja 11 %:lla sytogeneettinen riski oli määrittelemätön.
Kun seuranta-ajan mediaani oli 22,3 kuukautta, ensisijainen MRD-analyysi tutkimuksessa MMY3019 osoitti D-VRd-ryhmän vertailussa VRd-ryhmään paranemisen niiden MRD-negatiivisten (rinnakkaissekvensoinnissa arvo enintään 10-5) potilaiden kokonaislukumäärässä, joilla todettiin täydellinen vaste tai parempi. MRD-negatiivisten kokonaisosuus oli D-VRd-ryhmässä 53,3 % (95 %:n luottamusväli: 46,1; 60,4) ja VRd-ryhmässä 35,4 % (95 %:n luottamusväli: 28,7; 42,4) (kerroinsuhde [D‑VRd vs. VRd] 2,07 ja 95 %:n luottamusväli: 1,38; 3,10; p = 0,0004).
Ensisijaisen MRD-analyysin ajankohtana todettiin D-VRd-ryhmän vertailussa VRd-ryhmään paraneminen täydellisen vasteen tai paremman saaneiden kokonaisosuudessa. Täydellisen vasteen tai paremman saaneiden kokonaisosuus oli D-VRd-ryhmässä 76,6 % (95 %:n luottamusväli: 70,1; 82,4) ja VRd-ryhmässä 59,1 % (95 %:n luottamusväli: 51,9; 66,0) (kerroinsuhde [D‑VRd vs. VRd] 2,31; 95 %:n luottamusväli: 1,48; 3,60; p = 0,0002).
Kun seuranta-ajan mediaani oli 39 kuukautta, tutkimuksen MMY3019 etenemättömyysajan (PFS) välianalyysissa osoitettiin etenemättömyysajan pidentyneen D-VRd-ryhmässä VRd-ryhmään verrattuna (riskisuhde = 0,61; 95 %:n luottamusväli: 0,42; 0,90; p = 0,0104). Kummassakaan ryhmässä ei ollut saavutettu etenemättömyysajan mediaania. Kun etenemättömyysaikaa koskevat tiedot olivat etenemättömyysajan loppuanalyysissa valmiimmat, etenemättömyysaikaa koskeva hoidon teho oli parempi ja riskisuhde oli 0,57 (95 %:n luottamusväli: 0,41; 0,79). D-VRd-ryhmässä ei ollut saavutettu etenemättömyysajan mediaania, ja VRd-ryhmässä se oli 52,6 kuukautta.
Kuvio 2: Tutkimuksen MMY3019 loppuanalyysiin perustuvan etenemättömyysajan (PFS) Kaplan–Meierin käyrä
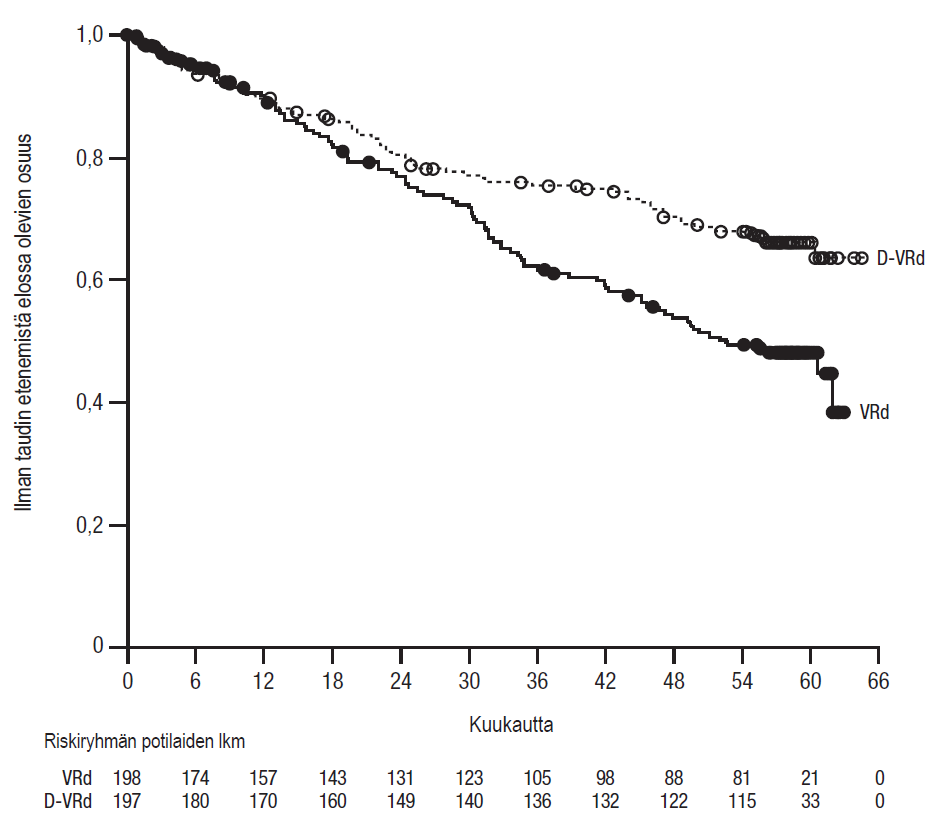
Etenemättömyysajan välianalyysin ajankohtana 1 vuoden ajan MRD-negatiivisina (rinnakkaissekvensoinnissa arvo enintään 10-5) säilyneiden lukumäärässä havaittiin paraneminen potilailla, joilla todettiin täydellinen vaste tai parempi, vertailtaessa D-VRd-ryhmää VRd-ryhmään. MRD-negatiivisina säilyneiden osuus oli D-VRd-ryhmässä 42,6 % (95 %:n luottamusväli: 35,6; 49,9) ja VRd-ryhmässä 25,3 % (95 %:n luottamusväli: 19,4; 31,9) (kerroinsuhde [D‑VRd vs. VRd] 2,18 ja 95 %:n luottamusväli: 1,42; 3,34; p = 0,0003).
Tutkimuksen MMY3019 muut tehoa koskevat tulokset esitetään jäljempänä taulukossa 12.
| Taulukko 12: Tutkimuksen MMY3019 etenemättömyysajan loppuanalyysin tehoa koskevat tulokseta | ||
| D-VRd (n = 197) | VRd (n = 198) | |
| MRD-negatiivisten kokonaislukumääräb | 120 (60,9 %) | 78 (39,4 %) |
| Kerroinsuhde (95 %:n luottamusväli)c | 2,37 (1,58; 3,55) | |
| MRD-negatiivisina säilyneiden lukumääräd | 96 (48,7 %) | 52 (26,3 %) |
| Kerroinsuhde (95 %:n luottamusväli)c | 2,63 (1,73; 4,00) | |
| Täydellinen vaste tai parempi (sCR+CR) | 160 (81,2 %) | 122 (61,6 %) |
| Kerroinsuhde (95 %:n luottamusväli)c | 2,73 (1,71; 4,34) | |
| Kokonaisvaste (sCR+CR+VGPR+PR) n (%)a | 191 (97,0 %) | 184 (92,9 %) |
| Täydellinen vaste lisäehdoin (sCR) | 128 (65,0 %) | 88 (44,4 %) |
| Täydellinen vaste (CR) | 32 (16,2 %) | 34 (17,2 %) |
| Erittäin hyvä osittainen vaste (VGPR) | 23 (11,7 %) | 50 (25,3 %) |
| Osittainen vaste (PR) | 8 (4,1 %) | 12 (6,1 %) |
D‑VRd = daratumumabi‑bortetsomibi‑lenalidomidi‑deksametasoni; VRd = bortetsomibi‑lenalidomidi‑deksametasoni; MRD = minimaalinen jäännöstauti a Perustuu hoitoaikeen mukaiseen potilasjoukkoon, seurannan mediaani 59 kuukautta b Potilailla todettiin sekä MRD-negatiivisuus (kynnysarvo enintään 10-5) että täydellinen vaste tai parempi c Ositetuissa taulukoissa käytetään suhdeluvun Mantel–Haenszelin estimaattia. Ositustekijät ovat: ISS-levinneisyysluokka (I, II, III), ikä/soveltuvuus siirtoon (< 70 vuotta soveltumaton tai ikä < 70 vuotta ja kieltäytyy siirrosta tai ikä ≥ 70 vuotta) satunnaistettuna. Kerroinsuhde > 1 osoittaa edun D‑VRd-hoidon suhteen. d MRD-negatiivisena säilymiseksi on määritelty MRD-negatiivinen ja MRD-negatiivisuus varmistettu vähintään 1 vuoden välein eikä varmistusten välillä MRD-positiivisutta. | ||
Multippelin myelooman yhdistelmähoidot
MMY2040 oli avoin tutkimus, jossa arvioitiin ihon alle annettavan Darzalex-valmisteen 1 800 mg:n annosten tehoa ja turvallisuutta
- yhdistelmänä bortetsomibin, melfalaanin ja prednisonin kanssa (D‑VMP) multippelin myelooman diagnoosin äskettäin saaneilla potilailla, jotka eivät soveltuneet siirtoon. Bortetsomibia annettiin injektioina ihon alle kehon pinta-alaan suhteutettuina annoksina 1,3 mg/m2 kaksi kertaa viikossa ensimmäisen 6 viikon pituisen hoitosyklin viikoilla 1, 2, 4 ja 5 (sykli 1; 8 annosta), jonka jälkeen annoksia annettiin kerran viikossa vielä kahdeksan 6 viikon pituisen hoitosyklin ajan viikoilla 1, 2, 4 ja 5 (syklit 2–9; 4 annosta per sykli). Melfalaania annettiin annoksina 9 mg/m2 ja prednisonia annettiin annoksina 60 mg/m2 suun kautta yhdeksän 6 viikon pituisen hoitosyklin ajan päivinä 1–4 (syklit 1–9). Ihon alle annettavan Darzalex-valmisteen käyttöä jatkettiin, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä.
- yhdistelmänä lenalidomidin ja deksametasonin kanssa (D‑Rd) potilailla, joilla oli relapsoitunut tai hoitoon reagoimaton multippeli myelooma. Lenalidomidia (25 mg suun kautta kerran päivässä 28 päivän [4 viikon] pituisten toistettujen hoitosyklien päivinä 1–21) annettiin pienten deksametasoniannosten 40 mg/viikko kanssa (tai pienennetty annos 20 mg/viikko > 75-vuotiaille potilaille tai potilaille, joiden painoindeksi [BMI] oli < 18,5). Ihon alle annettavaa Darzalex-hoitoa jatkettiin, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä.
- yhdistelmänä bortetsomibin, lenalidomidin ja deksametasonin kanssa (D‑VRd) multippelin myelooman diagnoosin äskettäin saaneilla potilailla, jotka soveltuivat siirtoon. Bortetsomibia annettiin injektiona ihon alle kehon pinta-alaan suhteutettuina annoksina 1,3 mg/m2 kaksi kertaa viikossa viikkoina 1 ja 2. Lenalidomidia annettiin suun kautta annoksina 25 mg kerran päivässä päivinä 1–14; pieniä deksametasoniannoksia 40 mg/viikko annettiin 3 viikon hoitosykleissä. Hoidon koko kesto oli 4 hoitosykliä.
Mukaan otettiin yhteensä 199 potilasta (D‑VMP: 67; D‑Rd: 65; D‑VRd: 67). Tehoa koskevat tulokset saatiin tietokonealgoritmilla IMWG-kriteerien mukaisesti. Tutkimuksessa saavutettiin sen ensisijainen päätetapahtuma, joka D‑VMP- ja D‑Rd-hoidoilla oli kokonaisvasteluku, sekä ensisijainen päätetapahtuma, joka oli erittäin hyvä osittainen vaste (VGPR) tai parempi vaste D‑VRd-hoitoon (ks. taulukko 13).
| Taulukko 13: Tutkimuksen MMY2040 tehoa koskevat tulokset | |||
| D-VMP (n = 67) | D-Rd (n = 65) | D-VRd (n = 67) | |
| Kokonaisvaste (sCR+CR+VGPR+PR), n (%)a | 60 (89,6 %) | 61 (93,8 %) | 65 (97,0 %) |
| 90 %:n luottamusväli (%) | (81,3 %; 95,0 %) | (86,5 %, 97,9 %) | (90,9 %; 99,5 %) |
| Täydellinen vaste lisäehdoin (sCR) | 13 (19,4 %) | 12 (18,5 %) | 6 (9,0 %) |
| Täydellinen vaste (CR) | 19 (28,4 %) | 13 (20,0 %) | 5 (7,5 %) |
| Erittäin hyvä osittainen vaste (VGPR) | 20 (29,9 %) | 26 (40,0 %) | 37 (55,2 %) |
| Osittainen vaste (PR) | 8 (11,9 %) | 10 (15,4 %) | 17 (25,4 %) |
| Erittäin hyvä osittainen vaste tai parempi (sCR + CR + VGPR) | 52 (77,6 %) | 51 (78,5 %) | 48 (71,6 %) |
| 90 %:n luottamusväli (%) | (67,6 %; 85,7 %) | (68,4 %, 86,5 %) | (61,2 %; 80,6 %) |
D-VMP = daratumumabi-bortetsomibi-melfalaani-prednisoni; D-Rd = daratumumabi-lenalidomidi-deksametasoni; D‑VRd = daratumumabi-bortetsomibi-lenalidomidi-deksametasoni; daratumumabi = ihon alle annettava Darzalex-valmiste. a Perustuu hoidettuihin tutkittaviin | |||
Yhdistelmähoito pomalidomidin ja deksametasonin (Pd) kanssa:
Tutkimus MMY3013 oli avoin, satunnaistettu, aktiivisella aineella kontrolloitu vaiheen III tutkimus, jossa ihon alle annettavaa Darzalex-valmistetta (1 800 mg) yhdistelmänä pomalidomidin ja pienten deksametasoniannosten (D-Pd) kanssa verrattiin pomalidomidin ja pienten deksametasoniannosten (Pd) yhdistelmään multippelia myeloomaa sairastavilla potilailla, jotka olivat saaneet vähintään yhtä aiemman linjan hoitoa lenalidomidilla ja jollakin proteasomin estäjällä. Pomalidomidi (4 mg kerran päivässä suun kautta toistuvien 28 päivän [4 viikon] pituisten hoitosyklien päivinä 1–21) annettiin suun kautta otettavan tai laskimoon annettavan pienen deksametasoniannoksen 40 mg/viikko (tai > 75-vuotiaille potilaille pienennetyn annoksen 20 mg/viikko) kanssa. Ihon alle annettavan Darzalex-valmisteen antopäivinä 20 mg deksametasoniannoksesta annettiin esilääkityksenä ja loput antoa seuraavana päivänä. Pienennettyjä deksametasoniannoksia saaville potilaille annettiin koko 20 mg:n annos ihon alle annettavaa Darzalex-valmistetta esilääkityksenä. Pomalidomidi- ja deksametasoniannoksia annettiin valmistajien laatimien valmistetietojen mukaisesti. Hoitoa jatkettiin kummassakin hoitoryhmässä, kunnes sairaus eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä.
Yhteensä 304 potilasta satunnaistettiin; 151 satunnaistettiin D-Pd-ryhmään, ja 153 satunnaistettiin Pd-ryhmään. Tutkimukseen otettiin mukaan potilaita, joiden sairaus oli dokumentoidusti edennyt viimeisimmän hoidon aikana tai sen jälkeen. Niitä potilaita, joilla oli edellisen hoidon aikana ollut graduksen ≥ 3 ihottumaa, ei pomalidomidin valmisteyhteenvedon perusteella otettu tutkimukseen mukaan. Lähtötilanteen demografiset ja taudin ominaisuuksia koskevat tiedot olivat näiden kahden hoitoryhmän välillä samankaltaiset. Potilaiden iän mediaani oli 67 vuotta (vaihteluväli 35–90 vuotta), 18 % oli ≥ 75-vuotiaita, 53 % oli miehiä ja 89 % oli valkoihoisia. Potilaat olivat saaneet kahden (mediaani) aiemman hoitolinjan hoitoa. Kaikki potilaat olivat aiemmin saaneet jotakin proteasomin estäjää ja lenalidomidia, ja 56 % potilaista oli saanut kantasolusiirron. Yhdeksänkymmentäkuusi prosenttia (96 %) potilaista oli saanut aiempana hoitona bortetsomibia. Valtaosalla potilaista tauti ei ollut reagoinut lenalidomidihoitoon (80 %), proteasomin estäjään (48 %) tai sekä immuniteettia muuntavaan valmisteeseen ja proteasomin estäjään (42 %). Yksitoista prosenttia potilaista oli saanut yhden aiemman linjan hoitoa; yhdelläkään potilaalla tauti ei ollut reagoinut lenalidomidihoitoon eikä 32,4 %:lla tauti ollut reagoinut sekä lenalidomidiin että proteaasin estäjään. Hoidon tehoa arvioitiin IMWG (International Myeloma Working Group) ‑kriteerien mukaisella etenemättömyysajalla.
Tutkimuksen MMY3013 etenemättömyysajan ensisijainen analyysi osoitti 16,9 kuukauden (mediaani) seurannan jälkeen etenemättömyysajan pidentyneen D-Pd-ryhmässä tilastollisesti merkitsevästi Pd-ryhmään verrattuna; etenemättömyysajan mediaani oli D-Pd-ryhmässä 12,4 kuukautta ja Pd-ryhmässä 6,9 kuukautta (riskisuhde [95 %:n luottamusväli]: 0,63 [0,47, 0,85]; p-arvo = 0,0018), mikä tarkoittaa, että D-Pd-hoitoa saaneilla potilailla sairauden etenemisen tai kuoleman riski väheni 37 % Pd-hoitoa saaneisiin potilaisiin verrattuna.
Kuvio 3: Tutkimuksen MMY3013 etenemättömyysajan (PFS) Kaplan–Meierin käyrä
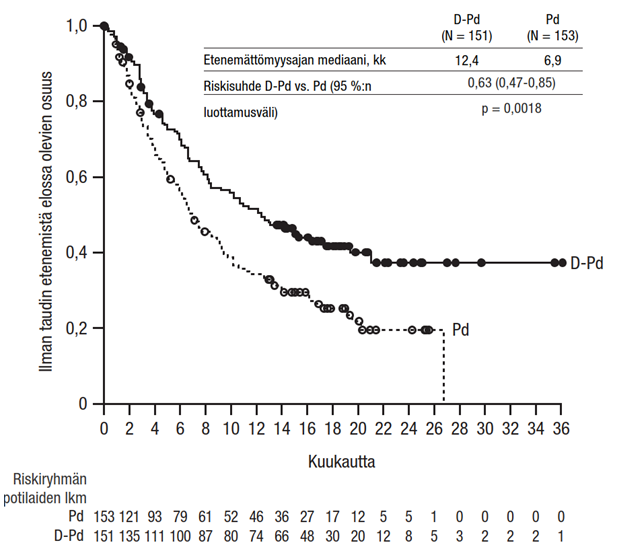
Lisäksi suunniteltu kokonaiselinajan seuranta-analyysi tehtiin, kun seuranta-ajan mediaani oli 39,6 kuukautta. Kun kokonaiselinajan maturiteetti oli 57 %, kokonaiselinajan mediaani oli D‑Pd-ryhmässä 34,4 kuukautta ja Pd-ryhmässä 23,7 kuukautta (riskisuhde [95 %:n luottamusväli]: 0,82 [0,61; 1,11]).
Tutkimuksen MMY3013 muut tehoa koskevat tulokset esitetään jäljempänä taulukossa 14.
| Taulukko 14: Tutkimuksen MMY3013 tehoa koskevat tulokseta | ||
| D-Pd (n = 151) | Pd (n = 153) | |
| Kokonaisvasteluku (sCR+CR+VGPR+PR) n(%)a | 104 (68,9 %) | 71 (46,4 %) |
| p-arvob | < 0,0001 | |
| Täydellinen vaste lisäehdoin (stringent complete response, sCR) | 14 (9,3 %) | 2 (1,3 %) |
| Täydellinen vaste (complete response, CR) | 23 (15,2 %) | 4 (2,6 %) |
| Erittäin hyvä osittainen vaste (very good partial response, VGPR) | 40 (26,5 %) | 24 (15,7 %) |
| Osittainen vaste (partial response, PR) | 27 (17,9 %) | 41 (26,8 %) |
| MRD-negatiivisten lukumääräc n(%) | 13 (8,7 %) | 3 (2,0 %) |
| 95 %:n luottamusväli (%) | (4,7 %; 14,3 %) | (0,4 %; 5,6 %) |
| p-arvod | 0,0102 | |
D-Pd = daratumumabi-pomalidomidi-deksametasoni; Pd = pomalidomidi-deksametasoni; MRD = minimaalinen jäännöstauti (minimal residual disease) a Perustuu hoitoaikeen mukaiseen potilasjoukkoon (intent-to-treat population) b Cochran-Mantel-Haenszelin khiin neliötestillä saatu osituskertoimien suhteen korjattu p-arvo c Hoitoaikeen mukaiseen potilasjoukkoon ja raja-arvoon 10-5 perustuva MRD-negatiivisten lukumäärä d Fisherin eksaktiin testiin perustuva p-arvo. | ||
Vasteen saaneilla tutkittavilla ajan mediaani vasteeseen oli D-Pd-ryhmässä 1 kuukausi (vaihteluväli: 0,9–9,1 kuukautta) ja Pd-ryhmässä 1,9 kuukautta (vaihteluväli: 0,9–17,3 kuukautta). Vasteen keston mediaania ei ollut saavutettu D-Pd-ryhmässä (vaihteluväli: 1–34,9+ kuukautta) ja Pd-ryhmässä se oli 15,9 kuukautta (vaihteluväli: 1+ – 24,8 kuukautta).
Monoterapia – indolentti multippeli myelooma, jonka multippeliksi myeloomaksi etenemisen riski on suuri
Tutkimus SMM3001 oli avoin, satunnaistettu vaiheen III tutkimus, jossa ihon alle annettavalla DARZALEX-valmisteella (1 800 mg) annetun hoidon tehoa ja turvallisuutta verrattiin aktiiviseen seurantaan potilailla, joilla oli indolentti multippeli myelooma, jonka multippeliksi myeloomaksi etenemisen riski on suuri. Hoitoryhmään satunnaistetuille potilaille annettiin ihon alle annettavaa DARZALEX-valmistetta (1 800 mg) ihon alle hoitosykleissä 1–2 kerran viikossa (päivinä 1, 8, 15 ja 22), sen jälkeen hoitosykleissä 3–6 joka toinen viikko (päivinä 1 ja 15) ja sen jälkeen neljän viikon välein hoitosykliin 39 saakka tai enintään 36 kuukauden ajan tai kunnes sairauden eteneminen varmistui.
Yhteensä 390 potilasta satunnaistettiin: 194 satunnaistettiin ihon alle annettavaa DARZALEX-valmistetta saavaan ryhmään ja 196 satunnaistettiin aktiivisen seurannan ryhmään. Lähtötilanteen demografiset ja taudin ominaisuuksia koskevat tiedot olivat näiden kahden tutkimusryhmän välillä samankaltaiset. Potilaiden iän mediaani oli 64 vuotta (vaihteluväli: 31–86 vuotta); 12 % oli ≥ 75‑vuotiaita, 48 % oli miehiä, 83 % oli valkoihoisia, 8 % aasialaisia ja 3 % afroamerikkalaisia. ECOG-toimintakykypisteet olivat 83 %:lla tutkittavista 0 ja 17 %:lla tutkittavista 1. Plasmasolujen prosenttiosuus (mediaani) luuytimessä oli 20 %, ja indolentin multippelin myelooman ensimmäisestä diagnosointipäivämäärästä satunnaistamiseen kuluneen ajan mediaani oli 0,7 vuotta. Kahdeksallakymmenellä prosentilla potilaista oli alle kolme multippeliksi myeloomaksi etenemiseen liittyvää riskitekijää. Riskitekijöitä olivat seerumin M-proteiinipitoisuus ≥ 30 g/l; IgA SMM; immunopareesi, johon liittyi kahden normaalin immunoglobuliinin isotyypin vähenemä; seerumin normaalien ja monoklonaalisten vapaiden kevytketjujen suhde ≥ 8 ja < 100, luuytimessä klonaalisia plasmasoluja > 50 – < 60 % ja mitattavissa oleva tauti. Tutkimukseen SMM3001 mukaan otettaviksi soveltuneilla potilailla edellytettiin olevan vähintään yksi näistä riskitekijöistä ja luuytimen plasmasoluja ≥ 10 %. Yhdeksällätoista prosentilla potilaista seerumin M‑proteiinipitoisuus oli ≥ 30 g/l, 25 %:lla oli IgA SMM, 60 %:lla oli immunopareesi, johon liittyi kahden normaalin immunoglobuliinin isotyypin vähenemä, 72 %:lla oli seerumin normaalien ja monoklonaalisten vapaiden kevytketjujen suhde ≥ 8 ja < 100 ja 3 %:lla oli luuytimessä klonaalisia plasmasoluja > 50 – < 60 % ja mitattavissa oleva tauti.
Tutkimuksen ensisijainen päätetapahtuma oli riippumattoman arviointikomitean (Independent Review Committee, IRC) arvioima etenemättömyysaika. Etenemättömyysajan Kaplan–Meier-käyrä esitetään kuviossa 4, ja tutkimuksen SMM3001 tehoa koskevat tulokset esitetään jäljempänä taulukossa 15.
Kuvio 4: Tutkimuksen SMM3001 etenemättömyysajan Kaplan–Meier-käyrä
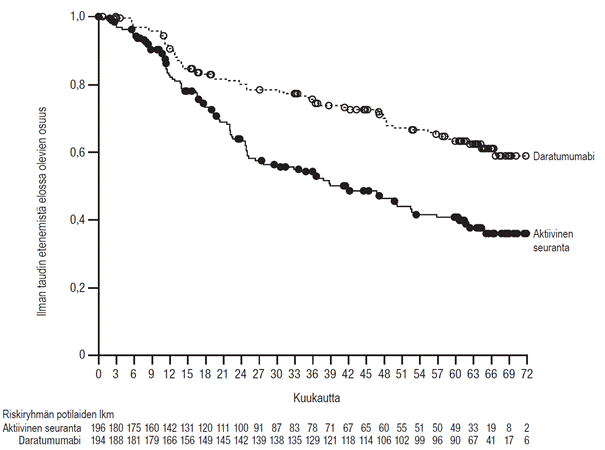
| Taulukko 15: Tutkimuksen SMM3001 tehoa koskevat tulokseta | |||
| Ihon alle annettava DARZALEX-valmiste (n = 194) | Aktiivinen seuranta (n = 196) | Kerroinsuhde (95 %:n luottamusväli)b |
Etenemättömyysaika, kuukauttac |
|
|
|
Mediaani (95 %:n luottamusväli) | NE (66,7; NE) | 41,5 (26,4; 53,3) |
|
Riskisuhde (95 %:n luottamusväli) | 0,49 (0,36; 0,67) |
| |
p-arvod | < 0,0001 |
| |
Kokonaisvasteluku (sCR+CR+VGPR+PR), n (%)a | 123 (63,4 %) | 4 (2,0 %) | 83,80 (29,69; 236,54), p < 0,0001 |
Täydellinen vaste lisäehdoin (stringent complete response, sCR) | 5 (2,6 %) | 0 |
|
Täydellinen vaste (complete response, CR) | 12 (6,2 %) | 0 |
|
Erittäin hyvä osittainen vaste (very good partial response, VGPR) | 41 (21,1 %) | 2 (1,0 %) |
|
Osittainen vaste (partial response, PR) | 65 (33,5 %) | 2 (1,0 %) |
|
NE = ei arvioitavissa (not evaluable). a Perustuu hoitoaikeen mukaiseen potilasjoukkoon (intent-to-treat population). b Ositetut taulukot yleisen kerroinsuhteen Mantel–Haenszelin estimaatin mukaan. c Seurannan keston mediaani oli 65,2 kuukautta. d p-arvo perustuu ositustekijällä ositettuun log-rank-testiin. | |||
AL-amyloidoosia sairastavien potilaiden yhdistelmähoito bortetsomibin, syklofosfamidin ja deksametasonin kanssa
Tutkimus AMY3001 oli avoin, satunnaistettu, aktiivisella aineella kontrolloitu vaiheen III tutkimus, jossa ihon alle annettavaa Darzalex-valmistetta (1 800 mg) yhdistelmänä bortetsomibin, syklofosfamidin ja deksametasonin (D-VCd) kanssa verrattiin pelkästään bortetsomibiin, syklofosfamidiin ja deksametasoniin (VCd) äskettäin diagnosoitua systeemistä AL-amyloidoosia sairastavilla potilailla. Satunnaistaminen ositettiin AL-amyloidoosia koskevan CSS-luokituksen (Cardiac Staging System) mukaan niissä maissa, joissa AL-amyloidoosia sairastaville potilaille tyypillisesti tarjotaan kantasolusiirtoa, sekä munuaisten toiminnan mukaan.
Kaikilla tutkimukseen AMY3001 mukaan otetuilla potilailla oli äskettäin diagnosoitu AL-amyloidoosi ja vähintään yksi affisioitunut elin, mitattavissa oleva hematologinen sairaus, sydäntä koskeva luokitus Cardiac Stage I-IIIA (European Modification of Mayo 2004 Cardiac Stage ‑luokituksen mukaan) ja NYHA-luokka I-IIIA. Potilaita, joiden NYHA-luokka oli IIIB tai IV, ei otettu tutkimukseen mukaan.
Bortetsomibia (s.c.; 1,3 mg/m2 kehon pinta-alan perusteella), syklofosfamidia (suun kautta tai i.v.; 300 mg/m2 kehon pinta-alan perusteella; maksimiannos 500 mg) ja deksametasonia (suun kautta tai i.v.; 40 mg tai pienennetty 20 mg:n annos, jos potilas oli > 70-vuotias tai potilaan painoindeksi [BMI] oli < 18,5 tai potilaalla oli hypervolemia, huonossa hoitotasapainossa oleva diabetes mellitus tai potilas ei ollut aiemmin sietänyt steroidihoitoa) annettiin toistetuissa 28 päivän (4 viikon) pituisissa hoitosykleissä viikoittain päivinä 1, 8, 15 ja 22. Darzalex-valmisteen antopäivinä injisoitiin deksametasoniannoksesta 20 mg esilääkityksenä ja loput annoksesta annettiin Darzalex-hoidon jälkeisenä päivänä. Bortetsomibia, syklofosfamidia ja deksametasonia annettiin kummassakin hoitoryhmässä kuuden 28 päivän [4 viikon] pituisen hoitosyklin ajan, kun taas Darzalex-hoitoa jatkettiin taudin etenemiseen tai seuraavan hoidon alkuun saakka tai kunnes tutkimushoidon ensimmäisen annoksen jälkeen oli annettu enintään 24 hoitosykliä (~2 vuotta). Bortetsomibin, syklofosfamidin ja deksametasonin annoksia muutettiin valmistajien laatimien valmistetietojen mukaisesti.
Yhteensä 388 potilasta satunnaistettiin; 195 satunnaistettiin D-VCd-ryhmään, ja 193 satunnaistettiin VCd-ryhmään. Lähtötilanteen demografiset ja taudin ominaisuuksia koskevat tiedot olivat näiden kahden ryhmän välillä samankaltaiset. Valtaosalla (79 %) potilaista kevytketjutautiin ei liittynyt lambdaketjuja. Potilaiden iän mediaani oli 64 vuotta (vaihteluväli: 34–87 vuotta); 47 % oli ≥ 65‑vuotiaita, 58 % oli miehiä, 76 % oli valkoihoisia, 17 % aasialaisia ja 3 % afroamerikkalaisia, 23 %:lla oli AL-amyloidoosin CCS (Clinical Cardiac Stage) ‑luokka I, 40 %:lla oli luokka II, 35 %:lla oli luokka IIIA ja 2 %:lla oli luokka IIIB. Kaikilla potilailla oli yksi tai useampi elinaffisio ja affisioituneiden elinten lukumäärän mediaani oli 2 (vaihteluväli: 1–6), ja 66 %:lla potilaista affisioituneita elimiä oli kaksi tai useampia. Affisioituneita elintärkeitä elimiä olivat 71 %:lla sydän, 59 %:lla munuaiset ja 8 %:lla maksa. Tutkimuksen ei otettu mukaan potilaita, joilla oli graduksen 2 sensorista tai graduksen 1 kivuliasta perifeeristä neuropatiaa. Ensisijainen tehon päätetapahtuma oli riippumattoman arviointikomitean (Independent Review Committee) kansainvälisten konsensuskriteerien (International Concensus Criteria) perusteella määrittelemä hematologinen kokonaisvasteluku (HemCR). Tutkimuksessa AMY3001 osoitettiin, että hematologinen vasteluku parani D-VCd-ryhmässä VCd-ryhmään verrattuna. Yhteenveto tehoa koskevista tuloksista on taulukossa 16.
| Taulukko 16: Tutkimuksen AMY3001 tehoa koskevat tulokseta | |||
D-VCd (n = 195) | VCd (n = 193) | p-arvo | |
| Täydellinen hematologinen vaste (hematologic complete response, HemCR), n (%) | 104 (53,3 %) | 35 (18,1 %) | < 0,0001b |
| Erittäin hyvä osittainen vaste (very good partial response, VGPR), n (%) | 49 (25,1 %) | 60 (31,1 %) | |
| Osittainen vaste (partial response, PR), n (%) | 26 (13,3 %) | 53 (27,5 %) | |
| Erittäin hyvä osittainen tai sitä parempi hematologinen vaste (HemCR + VGPR), n (%) | 153 (78,5 %) | 95 (49,2 %) | < 0,0001b |
| Tärkeitä elimiä koskeva etenemättömyysaika (major organ deterioration progression-free survival, MOD-PFS), riskisuhde ja 95 %:n luottamusvälic | 0,58 (0,36; 0,93) | 0,0211d
| |
D-VCd = daratumumabi-bortetsomibi-syklofosfamidi-deksametasoni; VCd = bortetsomibi-syklofosfamidi-deksametasoni a Kaikki suunnitellusta analyysista 11,4 kuukauden (mediaani) seurannan jälkeen saadut tulokset perustuvat hoitoaikeen mukaiseen potilasjoukkoon (intent-to-treat population) b Cochran-Mantel-Haenszelin khiin neliötestiin perustuva p-arvo. c Tärkeitä elimiä koskevaksi etenemättömyysajaksi määritelty hematologinen eteneminen, tärkeän elimen (sydämen tai munuaisten) heikkeneminen tai kuolema d Käänteisellä todennäköisyyden sensuroinnilla painotettuun log-rank-testiin perustuva nimellinen p-arvo | |||
11,4 kuukauden (mediaani) seurannan aikapisteessä vasteen saaneilla tutkittavilla ajan mediaani täydelliseen hematologiseen vasteeseen oli D-VCd-ryhmässä 60 päivää (vaihteluväli: 8–299 päivää) ja VCd-ryhmässä 85 päivää (vaihteluväli: 14–340 päivää). Ajan mediaani erittäin hyvään osittaiseen vasteeseen tai parempaan vasteeseen oli D‑VCd-ryhmässä 17 päivää (vaihteluväli: 5–336 päivää) ja VCd-ryhmässä 25 päivää (vaihteluväli: 8–171 päivää). Täydellisen hematologisen vasteen keston mediaania ei ollut saavutettu kummassakaan ryhmässä.
Seurannan kestettyä 61,4 kuukautta (mediaani) täydellisen hematologisen vasteen saaneiden kokonaisosuus oli D-VCd-ryhmässä 59,5 % (95 %:n luottamusväli: 52,2; 66,4) ja VCd-ryhmässä 19,2 % (95 %:n luottamusväli: 13,9; 25,4) (kerroinsuhde [D-VCd versus VCd] 6,03 ja 95 %:n luottamusväli: 3,80; 9,58).
Tärkeitä elimiä koskevan etenemättömyysajan (MOD-PFS) analyysin tulokset 61,4 kuukauden (mediaani) seurannan jälkeen osoittivat tärkeitä elimiä koskevan etenemättömyysajan pidentyneen D‑VCd-ryhmässä VCd-ryhmään verrattuna. Tärkeitä elimiä koskevan etenemättömyysajan riskisuhde (HR) oli 0,44 (95 %:n luottamusväli: 0,31; 0,63) ja p-arvo oli < 0,0001. Tärkeitä elimiä koskevan etenemättömyysajan mediaania ei saavutettu D-VCd-ryhmässä ja VCd-ryhmässä se oli 30,2 kuukautta. Kaplan–Meierin estimaatti 60 kuukauden pituisesta tärkeitä elimiä koskevasta etenemättömyysajasta oli D-VCd-ryhmässä 60 % (95 %:n luottamusväli: 52; 67) ja VCd-ryhmässä 33 % (95 %:n luottamusväli: 23; 44).
Kuvio 5: Tutkimuksen AMY3001 tärkeitä elimiä koskevan etenemättömyysajan Kaplan–Meierin käyrä
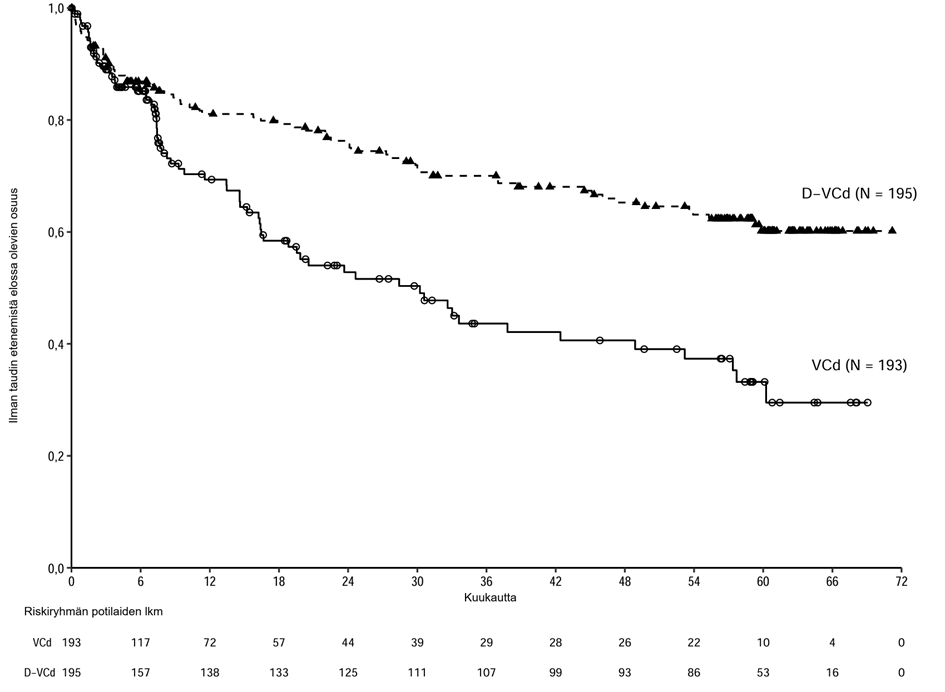
61,4 kuukauden (mediaani) seurannan jälkeen oli havaittu yhteensä 112 kuolemaa (D-VCd-ryhmän n = 46 [23,6 %] vs. VCd-ryhmän n = 66 [34,2 %]). Kummassakaan ryhmässä ei ollut saavutettu kokonaiselossaolon mediaania, mutta kokonaiselossaolon riskisuhde (HR) oli 0,62 (95 %:n luottamusväli: 0,42; 0,90) ja p-arvo oli 0,0121. 60 kuukauden kokonaiselossaololuku oli D-VCd-ryhmässä 76 % (95 %:n luottamusväli: 69; 82) ja VCd-ryhmässä 65 % (95 %:n luottamusväli: 57; 71).
Kuvio 6: Tutkimuksen AMY3001 kokonaiselossaolon Kaplan–Meierin käyrä
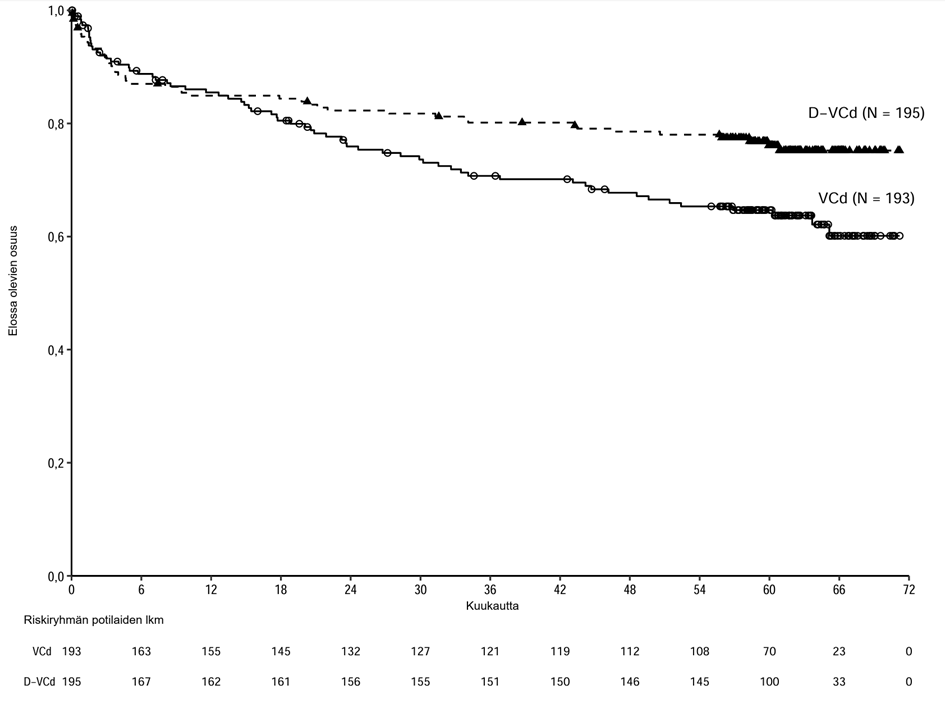
Kliininen kokemus daratumumabi-infuusiokonsentraatista, liuosta varten (laskimoon annettava valmiste)
Äskettäin diagnosoitu multippeli myelooma
Lenalidomidista ja deksametasonista koostuva yhdistelmähoito potilaille, jotka eivät sovellu autologiseen kantasolusiirtoon
Tutkimus MMY3008 oli avoin, satunnaistettu, aktiivisella aineella kontrolloitu vaiheen III tutkimus, jossa laskimoon annettavaa daratumumabia annoksina 16 mg/kg yhdistelmänä lenalidomidin ja pienen deksametasoniannoksen kanssa (DRd) verrattiin hoitoon lenalidomidin ja pienen deksametasoniannoksen yhdistelmällä (Rd) äskettäin diagnosoitua multippelia myeloomaa sairastavilla potilailla. Lenalidomidi (25 mg kerran päivässä suun kautta toistettujen 28 päivän [4 viikon] hoitosyklien päivinä 1–21) annettiin yhdessä suun kautta tai laskimoon annetun pienen deksametasoniannoksen 40 mg/viikko kanssa (tai pienennetyn annoksen 20 mg/viikko kanssa, jos potilas oli > 75-vuotias tai jos potilaan painoindeksi [body mass index, BMI] oli < 18,5). Laskimoon annettavan daratumumabin infuusiopäivinä deksametasoniannos annettiin infuusion esilääkityksenä. Lenalidomidin ja deksametasonin annosta muutettiin valmistajan valmistetietojen mukaisesti. Hoitoa jatkettiin kummassakin hoitoryhmässä, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä.
Yhteensä 737 potilasta satunnaistettiin: 368 potilasta satunnaistettiin DRd-ryhmään, ja 369 potilasta satunnaistettiin Rd-ryhmään. Lähtötilanteen demografiset ja taudin ominaisuudet olivat näissä kahdessa hoitoryhmässä samankaltaiset. Potilaiden iän mediaani oli 73 vuotta (vaihteluväli: 45–90), ja 44 % potilaista oli ≥ 75-vuotiaita. Suurin osa potilaista oli valkoihoisia (92 %) ja miehiä (52 %). ECOG-toimintakykyluokka (Eastern Cooperative Oncology Group) oli 34 %:lla potilaista 0, 49,5 %:lla potilaista 1 ja 17 %:lla potilaista ≥ 2. International Staging System (ISS) ‑levinneisyysluokkaan I kuului 27 %, ISS-levinneisyysluokkaan II 43 % ja ISS-levinneisyysluokkaan III 29 %. Hoidon tehoa arvioitiin IMWG-kriteerien (International Myeloma Working Group) mukaisella etenemättömyysajalla (Progression Free Survival, PFS) ja kokonaiselossaololla (overall survival, OS).
Tutkimuksessa MMY3008 etenemättömyysajan ensisijainen analyysi osoitti 28 kuukauden (mediaani) seurannan jälkeen etenemättömyysajan pidentyneen DRd-ryhmässä Rd-ryhmään verrattuna; etenemättömyysajan mediaania ei ollut saavutettu DRd-ryhmässä ja Rd-ryhmässä se oli 31,9 kuukautta (HR = 0,56; 95 %:n luottamusväli: 0,43; 0,73; p < 0,0001), mikä tarkoittaa, että DRd-hoitoa saaneiden potilaiden taudin etenemisen tai kuoleman riski väheni 44 %. Tulokset 64 kuukauden (mediaani) seuranta-ajan jälkeen päivitetystä etenemättömyysajan analyysista osoittivat edelleen, että etenemättömyysaika oli pidentynyt DRd-ryhmän potilailla verrattuna Rd‑ryhmään. Etenemättömyysajan mediaani oli DRd-ryhmässä 61,9 kuukautta ja Rd-ryhmässä 34,4 kuukautta (riskisuhde = 0,55; 95 %:n luottamusväli: 0,45; 0,67).
Kuvio 7: Tutkimuksen MMY3008 etenemättömyysajan (PFS) Kaplan–Meier-käyrä
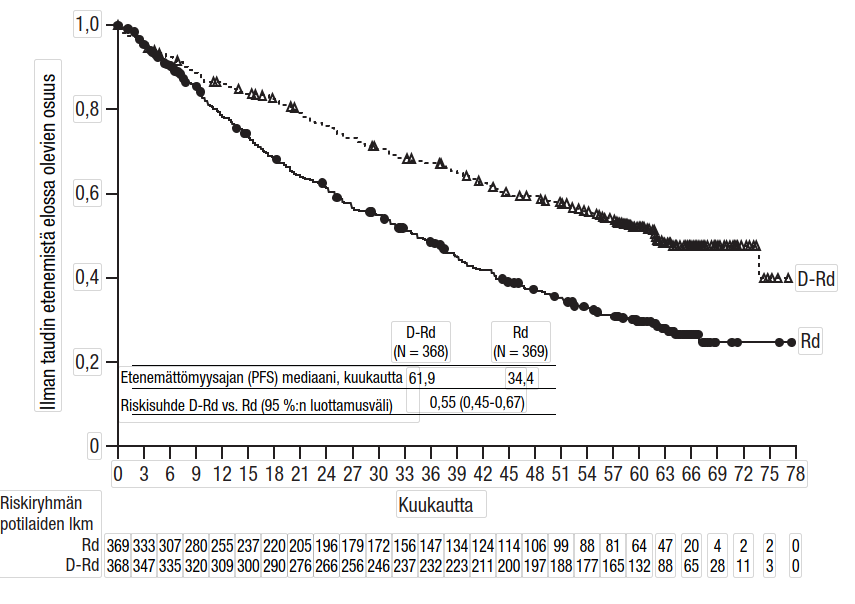
56 kuukauden (mediaani) seurannan jälkeen DRd-ryhmässä todettiin pidempi kokonaiselossaolo kuin Rd-ryhmässä (riskisuhde = 0,68; 95 %:n luottamusväli: 0,53; 0,86; p = 0,0013). Tulokset 89 kuukauden (mediaani) jälkeen päivitetystä kokonaiselossaolon analyysista osoittivat edelleen, että kokonaiselossaolo oli DRd-ryhmän potilailla pidentynyt verrattuna Rd-ryhmään. Kokonaiselossaolon mediaani oli DRd-ryhmässä 90,3 kuukautta, ja Rd-ryhmässä se oli 64,1 kuukautta (riskisuhde = 0,67; 95 %:n luottamusväli: 0,55, 0,82).
Kuvio 8: Tutkimuksen MMY3008 kokonaiselossaolon (OS) Kaplan–Meier-käyrä
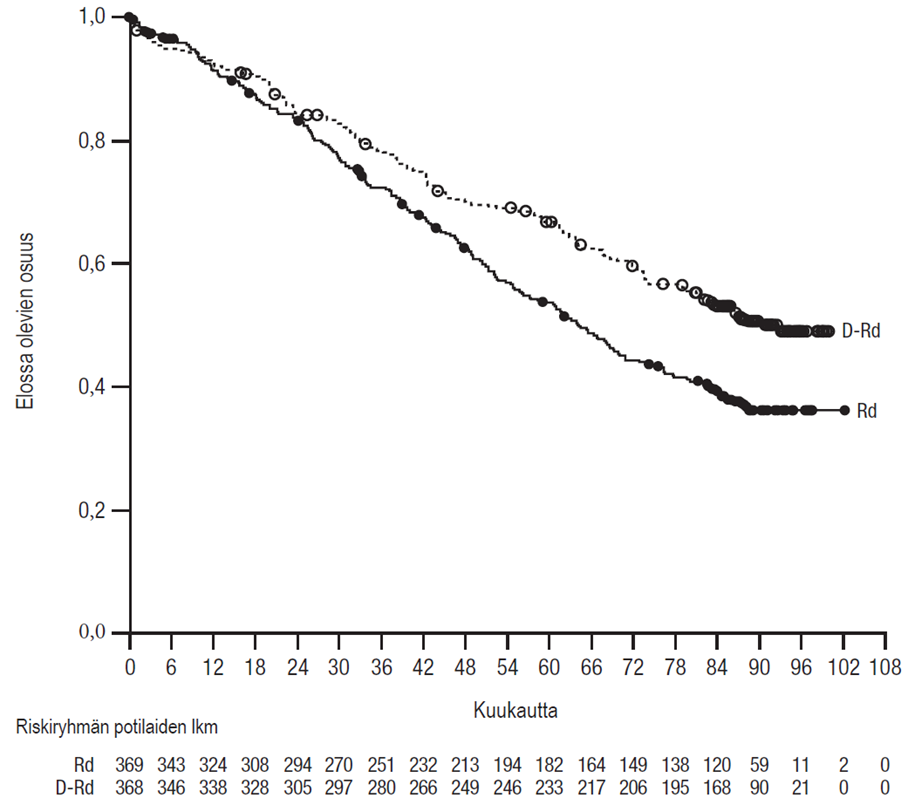
Tutkimuksen MMY3008 muut tehoa koskevat tulokset esitetään alla olevassa taulukossa 17.
| Taulukko 17: Tutkimuksen MMY3008 muut tehoa koskevat tulokseta | ||
| DRd (n = 368) | Rd (n = 369) | |
| Kokonaisvasteluku (sCR+CR+VGPR+PR) n (%)a | 342 (92,9 %) | 300 (81,3 %) |
| p-arvob | < 0,0001 | |
| Täydellinen vaste lisäehdoin (stringent complete response, sCR) | 112 (30,4 %) | 46 (12,5 %) |
| Täydellinen vaste (complete response, CR) | 63 (17,1 %) | 46 (12,5 %) |
| Erittäin hyvä osittainen vaste (very good partial response, VGPR) | 117 (31,8 %) | 104 (28,2 %) |
| Osittainen vaste (partial response, PR) | 50 (13,6 %) | 104 (28,2 %) |
| Täydellinen vaste tai parempi (sCR + CR) | 175 (47,6 %) | 92 (24,9 %) |
| p-arvob | < 0,0001 | |
| Erittäin hyvä osittainen vaste tai parempi (sCR + CR + VGPR) | 292 (79,3 %) | 196 (53,1 %) |
| p-arvob | < 0,0001 | |
| MRD-negatiivisten lukumääräa,c n (%) | 89 (24,2 %) | 27 (7,3 %) |
| 95 %:n luottamusväli (%) | (19,9 %; 28,9 %) | (4,9 %; 10,5 %) |
| Kerroinsuhde, 95 %:n luottamusvälid | 4,04 (2,55; 6,39) | |
| p-arvoe | < 0,0001 | |
DRd = daratumumabi-lenalidomidi-deksametasoni; Rd = lenalidomidi-deksametasoni; MRD = minimaalinen jäännöstauti (minimal residual disease) a Perustuu hoitoaikeen mukaiseen potilasjoukkoon (intent-to-treat population). b Cochran-Mantel-Haenszelin khiin neliötestiin perustuva p-arvo. c Perustuu raja-arvoon 10-5. d Osittamattomissa taulukoissa käytetään kerroinsuhteen Mantel-Haenszelin estimaattia. Kerroinsuhde > 1 osoittaa etua DRd-hoidon suhteen. e Fisherin eksaktiin testiin perustuva p-arvo. | ||
Vasteen saaneilla tutkittavilla ajan mediaani vasteeseen oli DRd-ryhmässä 1,05 kuukautta (vaihteluväli: 0,2–12,1 kuukautta) ja Rd-ryhmässä 1,05 kuukautta (vaihteluväli: 0,3–15,3 kuukautta). Vasteen keston mediaania ei ollut saavutettu DRd-ryhmässä ja Rd-ryhmässä se oli 34,7 kuukautta (95 %:n luottamusväli: 30,8; ei arvioitavissa).
Bortetsomibista, melfalaanista ja prednisonista (VMP) koostuva yhdistelmähoito potilaille, jotka eivät sovellu autologiseen kantasolusiirtoon
Tutkimus MMY3007 oli avoin, satunnaistettu, aktiivisella aineella kontrolloitu vaiheen III tutkimus, jossa laskimoon annettavaa daratumumabia annoksina 16 mg/kg yhdistelmänä bortetsomibin, melfalaanin ja prednisonin kanssa (D-VMP) verrattiin VMP-hoitoon äskettäin diagnosoitua multippelia myeloomaa sairastavilla potilailla. Bortetsomibi annettiin ensimmäisen 6 viikon hoitosyklin aikana (sykli 1; 8 annosta) injektioina ihon alle (s.c.) kehon pinta-alaan suhteutettuina annoksina 1,3 mg/m2 kaksi kertaa viikossa viikoilla 1, 2, 4 ja 5, minkä jälkeen annoksia annettiin kerran viikossa viikoilla 1, 2, 4 ja 5 vielä kahdeksan 6 viikon hoitosykliä (syklit 2–9; 4 annosta per sykli). Melfalaaniannoksia 9 mg/m2 ja prednisoniannoksia 60 mg/m2 annettiin suun kautta yhdeksän 6 viikon hoitosyklin (syklit 1–9) päivinä 1–4. Laskimoon annettavaa daratumumabihoitoa jatkettiin, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä.
Yhteensä 706 potilasta satunnaistettiin: 350 potilasta satunnaistettiin D-VMP-ryhmään, ja 356 potilasta satunnaistettiin VMP-ryhmään. Lähtötilanteen demografiset ja taudin ominaisuuksia koskevat parametrit olivat näissä kahdessa hoitoryhmässä samankaltaiset. Potilaiden iän mediaani oli 71 vuotta (vaihteluväli: 40–93 vuotta), ja 30 % potilaista oli ≥ 75-vuotiaita. Suurin osa potilaista oli valkoihoisia (85 %) ja naisia (54 %). ECOG-toimintakykyluokka oli 25 %:lla potilaista 0, 50 %:lla potilaista 1 ja 25 %:lla potilaista 2. Potilailla oli eri myeloomatyyppejä seuraavasti: IgG 64 %, IgA 22 % ja kevytketjumyelooma 10 %. International Staging System (ISS) ‑levinneisyysluokkaan I kuului 19 %, ISS-levinneisyysluokkaan II 42 % ja ISS-levinneisyysluokkaan III 38 % potilaista. 84 % kuului tavanomaisen riskin sytogeneettiseen kategoriaan. Hoidon tehoa arvioitiin IMWG-kriteerien mukaisella etenemättömyysajalla ja kokonaiselossaololla.
Tutkimuksessa MMY3007 etenemättömyysajan ensisijainen analyysi osoitti 16,5 kuukauden (mediaani) seurannan jälkeen etenemättömyysajan pidentyneen D‑VMP-ryhmässä verrattuna VMP-ryhmään; etenemättömyysajan mediaania ei ollut saavutettu D‑VMP-ryhmässä ja VMP-ryhmässä se oli 18,1 kuukautta (riskisuhde = 0,5; 95 %:n luottamusväli: 0,38; 0,65; p < 0,0001). Tulokset 40 kuukauden (mediaani) seuranta-ajan jälkeen päivitetystä etenemättömyysajan analyysista osoittivat edelleen, että etenemättömyysaika oli pidentynyt D-VMP-ryhmässä verrattuna VMP-ryhmään. Etenemättömyysajan mediaani oli D-VMP-ryhmässä 36,4 kuukautta ja VMP-ryhmässä 19,3 kuukautta (riskisuhde = 0,42; 95 %:n luottamusväli: 0,34; 0,51; p < 0,0001), mikä tarkoittaa, että D-VMP-hoitoa saaneiden potilaiden taudin etenemisen tai kuoleman riski oli vähentynyt 58 %.
Kuvio 9: Tutkimuksen MMY3007 etenemättömyysajan (PFS) Kaplan–Meier-käyrä
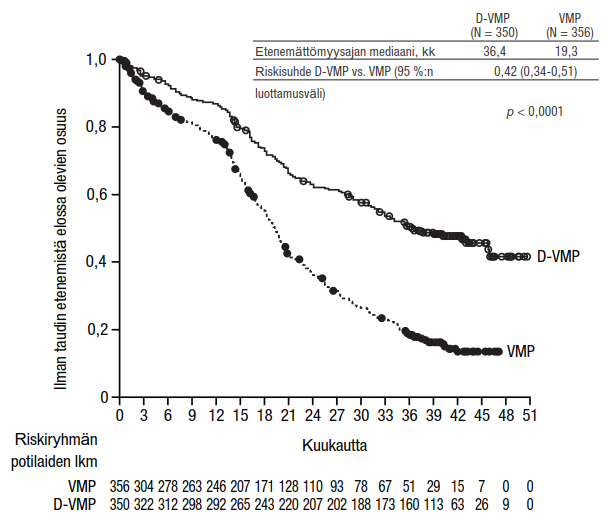
Kokonaiselossaolon todettiin olleen 40 kuukauden (mediaani) seurannan jälkeen D‑VMP-ryhmässä VMP-ryhmää parempi (riskisuhde = 0,60; 95 %:n luottamusväli: 0,46; 0,80; p = 0,0003), mikä tarkoittaa, että D-VMP-hoitoa saaneen ryhmän potilaiden kuoleman riski väheni 40 %. Kokonaiselossaolon mediaani oli 87 kuukauden (mediaani) seurannan jälkeen D‑VMP‑ryhmässä 83 kuukautta (95 %:n luottamusväli: 72,5; ei arvioitavissa) ja VMP‑ryhmässä 53,6 kuukautta (95 %:n luottamusväli: 46,3; 60,9).
Kuvio 10: Tutkimuksen MMY3007 kokonaiselossaolon (OS) Kaplan–Meier-käyrä
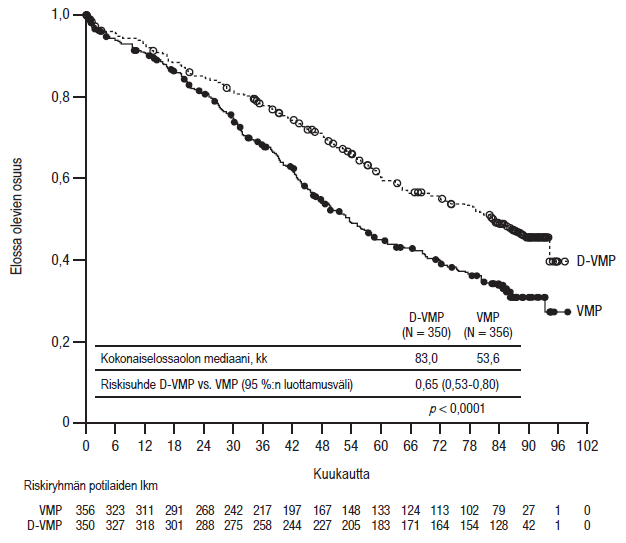
Tutkimuksen MMY3007 muut tehoa koskevat tulokset esitetään alla olevassa taulukossa 18.
| Taulukko 18: Tutkimuksen MMY3007 muut tehoa koskevat tulokseta | ||
| D-VMP (n = 350) | VMP (n = 356) | |
| Kokonaisvasteluku (sCR+CR+VGPR+PR) [n (%)] | 318 (90,9) | 263 (73,9) |
| p-arvob | < 0,0001 | |
| Täydellinen vaste lisäehdoin (stringent complete response, sCR) [n (%)] | 63 (18,0) | 25 (7,0) |
| Täydellinen vaste (complete response, CR) [n (%)] | 86 (24,6) | 62 (17,4) |
| Erittäin hyvä osittainen vaste (very good partial response, VGPR) [n (%)] | 100 (28,6) | 90 (25,3) |
| Osittainen vaste (partial response, PR) [n (%)] | 69 (19,7) | 86 (24,2) |
| MRD-negatiivisten lukumäärä (95 %:n luottamusväli) c (%) | 22,3 (18,0; 27,0) | 6,2 (3,9; 9,2) |
| Kerroinsuhde, 95 %:n luottamusvälid | 4,36 (2,64; 7,21) | |
| p-arvoe | < 0,0001 | |
D-VMP = daratumumabi-bortetsomibi-melfalaani-prednisoni; VMP = bortetsomibi-melfalaani-prednisoni; MRD = minimaalinen jäännöstauti (minimal residual disease) a Perustuu hoitoaikeen mukaiseen potilasjoukkoon (intent-to-treat population). b Cochran-Mantel-Haenszelin khiin neliötestiin perustuva p-arvo. c Perustuu raja-arvoon 10-5. d Ositetuissa taulukoissa käytetään yleisten kerroinsuhteiden Mantel-Haenszelin estimaattia. Kerroinsuhde > 1 osoittaa etua D-VMP-hoidon suhteen. e Fisherin eksaktiin testiin perustuva p-arvo. | ||
Vasteen saaneilla tutkittavilla ajan mediaani vasteeseen oli D-VMP-ryhmässä 0,79 kuukautta (vaihteluväli: 0,4–15,5 kuukautta) ja VMP-ryhmässä 0,82 kuukautta (vaihteluväli: 0,7–12,6 kuukautta). Vasteen keston mediaania ei ollut saavutettu D-VMP-ryhmässä ja VMP-ryhmässä se oli 21,3 kuukautta (vaihteluväli: 18,4 - ei arvioitavissa).
Alaryhmäanalyysi tehtiin seuraavista potilasryhmistä: vähintään 70-vuotiaat; 65–69-vuotiaat, joiden ECOG-toimintakykyluokka oli 2; alle 65-vuotiaat, joilla on samanaikaisesti muita merkittäviä sairauksia tai ECOG-toimintakykyluokka 2 (D-VMP: n = 273, VMP: n = 270). Hoidon tehoa koskevat tulokset olivat tutkittavien tässä alaryhmässä yhdenmukaiset koko potilasjoukon kanssa. Tutkittavien tässä osajoukossa etenemättömyysajan mediaania ei saavutettu D-VMP-ryhmässä ja VMP-ryhmässä se oli 17,9 kuukautta (riskisuhde = 0,56; 95 %:n luottamusväli: 0,42; 0,75; p < 0,0001). Kokonaisvasteluku oli D-VMP-ryhmässä 90 % ja VMP-ryhmässä 74 % (erittäin hyvän osittaisen vasteen (VGPR) osuus: D-VMP-ryhmässä 29 % ja VMP-ryhmässä 26 %; täydellisen vasteen (CR) osuus: D-VMP-ryhmässä 22 % ja VMP-ryhmässä 18 %; täydellisen vasteen lisäehdoin (sCR) osuus: D-VMP-ryhmässä 20 % ja VMP-ryhmässä 7 %). Myös hoidon turvallisuutta koskevat tulokset olivat tässä tutkittavien osajoukossa yhdenmukaiset koko potilasjoukon kanssa. Lisäksi hoidon turvallisuutta koskeva analyysi potilaiden osajoukosta, jonka ECOG-toimintakykyluokka oli 2 (D-VMP: n = 89, VMP: n = 84), oli myös yhdenmukainen koko potilasjoukon kanssa.
Bortetsomibista, talidomidista ja deksametasonista (VTd) koostuva yhdistelmähoito potilaille, jotka soveltuvat autologiseen kantasolusiirtoon
Tutkimus MMY3006 on kaksiosainen, avoin, satunnaistettu, aktiivisella aineella kontrolloitu vaiheen III tutkimus. Ensimmäisessä osassa laskimoon annettavaa daratumumabi-induktio- ja ‑konsolidaatiohoitoa annoksina 16 mg/kg yhdistelmänä bortetsomibin, talidomidin ja deksametasonin kanssa (D-VTd) verrattiin hoitoon bortetsomibin, talidomidin ja deksametasonin yhdistelmällä (VTd) äskettäin diagnosoitua multippelia myeloomaa sairastavilla potilailla, jotka soveltuivat autologiseen kantasolusiirtoon. Hoidon konsolidaatiovaihe alkoi aikaisintaan 30 päivän kuluttua autologisesta kantasolusiirrosta, kun potilaat olivat toipuneet riittävästi ja siirre oli täysin kiinnittynyt. Toisessa osassa tutkittavat, jotka olivat saaneet vähintään osittaisen vasteen (PR) kantasolusiirron jälkeen päivään 100 mennessä, satunnaistettiin uudelleen suhteessa 1:1 daratumumabiylläpitohoitoon tai pelkkään tarkkailuun. Tästä eteenpäin kuvataan vain ensimmäisen osan tuloksia.
Bortetsomibia annettiin injektioina ihon alle (s.c.) tai injektioina laskimoon (i.v.) kehon pinta-alaan suhteutettuina annoksina 1,3 mg/m2 kaksi kertaa viikossa kahden viikon ajan (päivinä 1, 4, 8 ja 11) 28 päivän (4 viikon) pituisina toistettuina induktiohoitosykleinä (syklit 1–4) sekä syklin 4 jälkeen tehdyn autologisen kantasolusiirron jälkeen kahtena konsolidaatiosyklinä (syklit 5 ja 6). Talidomidia annettiin kuuden bortetsomibisyklin aikana suun kautta 100 mg:n vuorokausiannoksina. Deksametasonia (suun kautta tai laskimoon) annettiin 40 mg:n annoksina syklien 1 ja 2 päivinä 1, 2, 8, 9, 15, 16, 22 ja 23 sekä sykleissä 3–4 annoksina 40 mg päivinä 1–2 ja annoksina 20 mg seuraavina hoitopäivinä (päivät 8, 9, 15, 16). Deksametasonia annettiin 20 mg:n annoksina syklien 5 ja 6 päivinä 1, 2, 8, 9, 15, 16. Laskimoon annettavan daratumumabi-infuusion antopäivinä deksametasoniannos annettiin laskimoon infuusion esilääkityksenä. Bortetsomibin, talidomidin ja deksametasonin annosmuutokset tehtiin valmistajan valmistetietojen mukaisesti.
Yhteensä 1 085 potilasta satunnaistettiin: 543 potilasta satunnaistettiin D-VTd-ryhmään, ja 542 potilasta satunnaistettiin VTd-ryhmään. Lähtötilanteen demografiset ja taudin ominaisuudet olivat näissä kahdessa hoitoryhmässä samankaltaiset. Potilaiden iän mediaani oli 58 vuotta (vaihteluväli: 22–65 vuotta). Kaikki potilaat olivat ≤ 65-vuotiaita: 43 % kuului ikäryhmään ≥ 60–65 vuotta, 41 % kuului ikäryhmään ≥ 50–60 vuotta ja 16 % oli alle 50-vuotiaita. Suurin osa potilaista oli miehiä (59 %). ECOG-toimintakykyluokka oli 48 %:lla potilaista 0, 42 %:lla potilaista 1 ja 10 %:lla potilaista 2. International Staging System (ISS) ‑levinneisyysluokkaan I kuului 40 %, ISS-levinneisyysluokkaan II 45 % ja ISS-levinneisyysluokkaan III 15 % potilaista.
Tehon arviointi perustui täydellisen vasteen lisäehdoin (sCR) saaneiden potilaiden lukumäärään 100 päivää kantasolusiirron jälkeen sekä etenemättömyysaikaan.
| Taulukko 19: Tutkimuksen MMY3006 tehoa koskevat tulokseta | |||
| D-VTd (n = 543) | VTd (n = 542) | p-arvob | |
| Vasteen arviointi 100 päivää kantasolusiirron jälkeen | |||
| Täydellinen vaste lisäehdoin (sCR) | 157 (28,9 %) | 110 (20,3 %) | 0,0010 |
| Täydellinen vaste tai parempi (sCR+CR) | 211 (38,9 %) | 141 (26,0 %) | < 0,0001 |
| Erittäin hyvä osittainen vaste tai parempi (sCR+CR+VGPR) | 453 (83,4 %) | 423 (78,0 %) | |
| MRD-negatiivisuusc, d, n (%) | 346 (63,7 %) | 236 (43,5 %) | < 0,0001 |
| 95 %:n luottamusväli (%) | (59,5 %; 67,8 %) | (39,3 %; 47,8 %) | |
| Kerroinsuhde, 95 %:n luottamusvälie | 2,27 (1,78; 2,90) | ||
| MRD-negatiivisuus sekä täydellinen vaste tai parempic, n (%) | 183 (33,7 %) | 108 (19,9 %) | < 0,0001 |
| 95 %:n luottamusväli (%) | (29,7 %; 37,9 %) | (16,6 %; 23,5 %) | |
| Kerroinsuhde, 95 %:n luottamusvälie | 2,06 (1,56; 2,72) | ||
D-VTd = daratumumabi-bortetsomibi-talidomidi-deksametasoni; VTd = bortetsomibi-talidomidi-deksametasoni; MRD = minimaalinen jäännöstauti (minimal residual disease) a Perustuu hoitoaikeen mukaiseen potilasjoukkoon (intent-to-treat population). b Cochran-Mantel-Haenszelin khiin neliötestiin perustuva p-arvo. c Perustuu raja-arvoon 10-5. d IMWG-kriteerien mukaisesta vasteesta riippumatta. e Ositetuissa taulukoissa käytetään yleisten kerroinsuhteiden Mantel-Haenszelin estimaattia. | |||
Etenemättömyysajan ensisijainen analyysi tehtiin 18,8 kuukauden (mediaani) seurannan jälkeen sensuroimalla potilaat, jotka satunnaistettiin toisessa satunnaistamisessa daratumumabiylläpitohoitoon; tulokset toisen satunnaistamisen päivämääränä olivat: riskisuhde = 0,50; 95 %:n luottamusväli: 0,34; 0,75; p = 0,0005. Etenemättömyysajan päivitetty analyysi tehtiin 44,5 kuukauden (mediaani) seurannan jälkeen ja siinä sensuroitiin potilaat, jotka satunnaistettiin toisessa satunnaistamisessa daratumumabiylläpitohoitoon. Päivitetyn analyysin tulokset olivat: riskisuhde = 0,43; 95 %:n luottamusväli: 0,33; 0,55; p < 0,0001. Etenemättömyysajan mediaania ei saavutettu D-VTd-ryhmässä ja VTd-ryhmässä se oli 37,8 kuukautta.
Kuvio 11: Tutkimuksen MMY3006 etenemättömyysajan (PFS) Kaplan–Meier-käyrä
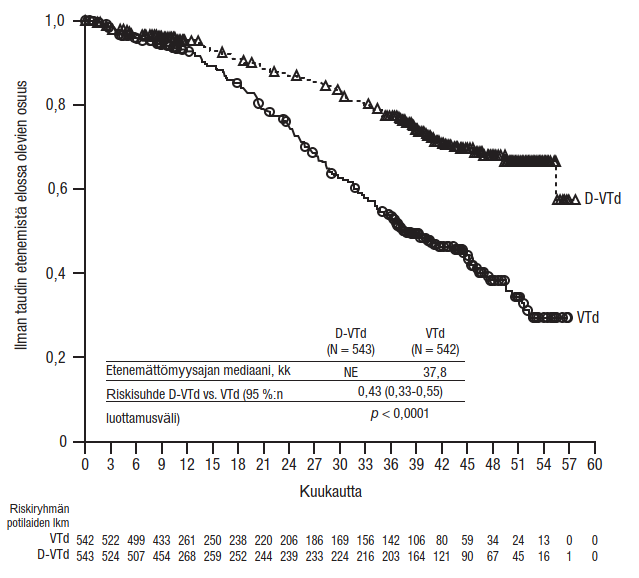
Relapsoitunut tai hoitoon reagoimaton multippeli myelooma
Monoterapia:
Laskimoon annettavan daratumumabimonoterapian kliininen teho ja turvallisuus relapsoitunutta ja hoitoon reagoimatonta multippelia myeloomaa sairastaville aikuispotilaille, joiden aiempaan hoitoon kuului jokin proteasomin estäjä ja jokin immunomodulatiivinen aine ja joiden taudin osoitettiin edenneen viimeisimmän hoidon aikana, osoitettiin kahdessa avoimessa tutkimuksessa.
Tutkimuksessa MMY2002 oli mukana 106 relapsoitunutta ja hoitoon reagoimatonta multippelia myeloomaa sairastavaa potilasta, ja he saivat laskimoon annettavaa daratumumabia annoksina 16 mg/kg, kunnes tauti eteni. Potilaiden iän mediaani oli 63,5 vuotta (vaihteluväli 31–84 vuotta), potilaista 11 % oli ≥ 75‑vuotiaita, miehiä oli 49 % potilaista, ja valkoihoisia 79 %. Potilaat olivat saaneet viisi (mediaani) aiempaa hoitolinjaa. Kahdeksankymmentä prosenttia potilaista oli saanut aiemmin autologisen kantasolusiirron. Potilaat olivat saaneet aiempina hoitoina bortetsomibia (99 %), lenalidomidia (99 %), pomalidomidia (63 %) ja karfiltsomibia (50 %). Lähtötilanteessa 97 %:lla potilaista tauti ei reagoinut viimeisimpään hoitoon, 95 %:lla tauti ei reagoinut proteasomin estäjähoitoon eikä immunomodulatiiviseen lääkehoitoon, 77 %:lla tauti ei reagoinut alkyloiviin aineisiin, 63 %:lla tauti ei reagoinut pomalidomidiin ja 48 %:lla tauti ei reagoinut karfiltsomibiin.
Riippumattoman arviointikomitean (Independent Review Committee, IRC) arvioon perustuvan etukäteen suunnitellun välianalyysin tehoa koskevat tulokset esitetään jäljempänä taulukossa 20.
| Taulukko 20: Riippumattoman arviointikomitean arvio tutkimuksen MMY2002 tehoa koskevista tuloksista | |
| Tehon päätetapahtuma | Laskimoon annettava daratumumabi 16 mg/kg n = 106 |
| Kokonaisvasteluku1 (Overall response rate, ORR: sCR+CR+VGPR+PR) [n (%)] | 31 (29,2) |
| 95 %:n luottamusväli (%) | (20,8; 38,9) |
| Täydellinen vaste lisäehdoin (stringent complete response, sCR) [n (%)] | 3 (2,8) |
| Täydellinen vaste (complete response, CR) [n] | 0 |
| Erittäin hyvä osittainen vaste (very good partial response, VGPR) [n (%)] | 10 (9,4) |
| Osittainen vaste (partial response, PR) [n (%)] | 18 (17,0) |
| Kliinistä hyötyä osoittava luku (ORR+MR) [n (%)] | 36 (34,0) |
| Vasteen keston mediaani [kuukautta (95 %:n luottamusväli)] | 7,4 (5,5; NE) |
| Ajan mediaani vasteeseen [kuukautta (vaihteluväli)] | 1 (0,9; 5,6) |
1 Ensisijainen tehon päätetapahtuma (International Myeloma Working Groupin kriteerit) NE = ei arvioitavissa (not estimable); MR = vähäinen vaste (minimal response) | |
Tutkimuksen MMY2002 kokonaisvasteluku (ORR) oli samankaltainen myelooman aiemmasta hoidosta riippumatta.
14,7 kuukauden (mediaani) seurannan jälkeen tehdyssä elossaolon päivityksessä kokonaiselossaolon mediaani oli 17,5 kuukautta (95 %:n luottamusväli; 13,7; ei arvioitavissa).
Tutkimuksessa GEN501 oli mukana 42 relapsoitunutta ja hoitoon reagoimatonta multippelia myeloomaa sairastavaa potilasta, jotka saivat laskimoon annettavaa daratumumabia annoksina 16 mg/kg, kunnes tauti eteni. Potilaiden iän mediaani oli 64 vuotta (vaihteluväli 44–76 vuotta), miehiä oli 64 % potilaista, ja valkoihoisia 76 %. Tutkimuksen potilaat olivat saaneet neljä (mediaani) aiempaa hoitolinjaa. Seitsemänkymmentäneljä prosenttia potilaista oli saanut aiemmin autologisen kantasolusiirron. Potilaat olivat saaneet aiempina hoitoina bortetsomibia (100 %), lenalidomidia (95 %), pomalidomidia (36 %) ja karfiltsomibia (19 %). Lähtötilanteessa 76 %:lla potilaista tauti ei reagoinut viimeisimpään hoitoon, 64 %:lla tauti ei reagoinut proteasomin estäjähoitoon eikä immunomodulatiiviseen lääkehoitoon, 60 %:lla tauti ei reagoinut alkyloiviin aineisiin, 36 %:lla tauti ei reagoinut pomalidomidiin ja 17 %:lla tauti ei reagoinut karfiltsomibiin.
Ennalta suunniteltu välianalyysi osoitti, että annoksina 16 mg/kg annetun daratumumabihoidon kokonaisvasteluku oli 36 % ja että täydellisen vasteen sai 5 % ja erittäin hyvän osittaisen vasteen sai 5 % potilaista. Ajan mediaani vasteen saamiseen oli 1 kuukausi (vaihteluväli: 0,5–3,2). Vasteen kestoajan mediaania ei saavutettu (95 %:n luottamusväli: 5,6 kuukautta; ei arvioitavissa).
15,2 kuukauden (mediaani) seurannan jälkeen tehdyssä elossaolon päivityksessä kokonaiselossaolon mediaania ei saavutettu (95 %:n luottamusväli: 19,9 kuukautta; ei arvioitavissa), ja 74 % tutkittavista oli edelleen elossa.
Yhdistelmähoito lenalidomidin kanssa
Tutkimus MMY3003 oli avoin, satunnaistettu, aktiivisella aineella kontrolloitu vaiheen III tutkimus, jossa laskimoon annettavaa daratumumabia annoksina 16 mg/kg yhdistelmänä lenalidomidin ja matala-annoksisen deksametasonin kanssa (DRd) verrattiin hoitoon lenalidomidilla ja matala-annoksisella deksametasonilla (Rd) relapsoitunutta tai hoitoon reagoimatonta multippelia myeloomaa sairastavilla potilailla, jotka olivat saaneet vähintään yhtä aiempaa hoitoa. Lenalidomidi (25 mg kerran päivässä suun kautta 28 päivän [4 viikon] pituisten toistuvien syklien päivinä 1–21) annettiin yhdessä matala-annoksisen deksametasonin 40 mg/viikko (tai pienennetty annos 20 mg/viikko, jos potilas oli > 75-vuotias tai potilaan painoindeksi [BMI] oli < 18,5) kanssa. Laskimoon annettavan daratumumabi-infuusion antopäivinä annettiin ennen infuusiota 20 mg:n deksametasoniannos ja loput annettiin infuusion jälkeisenä päivänä. Hoitoa jatkettiin kummassakin hoitoryhmässä, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä.
Yhteensä 569 potilasta satunnaistettiin; 286 satunnaistettiin DRd-ryhmään ja 283 satunnaistettiin Rd-ryhmään. Lähtötilanteen demografiset ja taudin ominaisuuksia koskevat tiedot olivat laskimoon annettavaa daratumumabia saaneessa ryhmässä ja vertailuryhmässä samankaltaiset. Potilaiden iän mediaani oli 65 vuotta (vaihteluväli 34−89 vuotta), ja 11 % potilaista oli ≥ 75-vuotiaita. Suurin osa potilaista (86 %) oli saanut aiemmin jotakin proteasomin estäjää, 55 % potilaista oli saanut aiemmin jotakin immunomodulatiivista lääkehoitoa, ja näistä potilaista 18 % oli saanut aiemmin lenalidomidia, ja 44 % oli saanut aiemmin sekä jotakin proteasomin estäjää että immunomodulatiivista lääkehoitoa. Lähtötilanteessa 27 % potilaista ei ollut reagoinut viimeiseen hoitolinjaan. Kahdeksantoista prosenttia (18 %) potilaista ei ollut reagoinut pelkästään johonkin proteasomin estäjään, ja 21 % potilaista ei ollut reagoinut bortetsomibiin. Lenalidomidihoitoon reagoimattomia potilaita ei otettu tutkimukseen mukaan.
Tutkimuksessa MMY3003 etenemättömyysajan ensisijainen analyysi osoitti 13,5 kuukauden (mediaani) seurannan jälkeen etenemättömyysajan pidentyneen DRd-ryhmässä Rd-ryhmään verrattuna; etenemättömyysajan mediaania ei saavutettu DRd-ryhmässä, ja se oli Rd-ryhmässä 18,4 kuukautta (riskisuhde = 0,37; 95 %:n luottamusväli: 0,27; 0,52; p < 0,0001). Etenemättömyysajan päivitetyn analyysin tulokset osoittivat 55 kuukauden (mediaani) seurannan jälkeen edelleen, että etenemättömyysaika oli pidentynyt DRd-ryhmässä verrattuna Rd-ryhmään. Etenemättömyysajan mediaani oli DRd-ryhmässä 45,0 kuukautta ja Rd-ryhmässä 17,5 kuukautta (riskisuhde = 0,44; 95 %:n luottamusväli: 0,35; 0,54; p < 0,0001), mikä tarkoittaa, että DRd-hoitoa saaneiden potilaiden taudin etenemisen tai kuoleman riski väheni 56 % (ks. kuvio 12).
Kuvio 12: Tutkimuksen MMY3003 etenemättömyysajan (PFS) Kaplan–Meier-käyrä
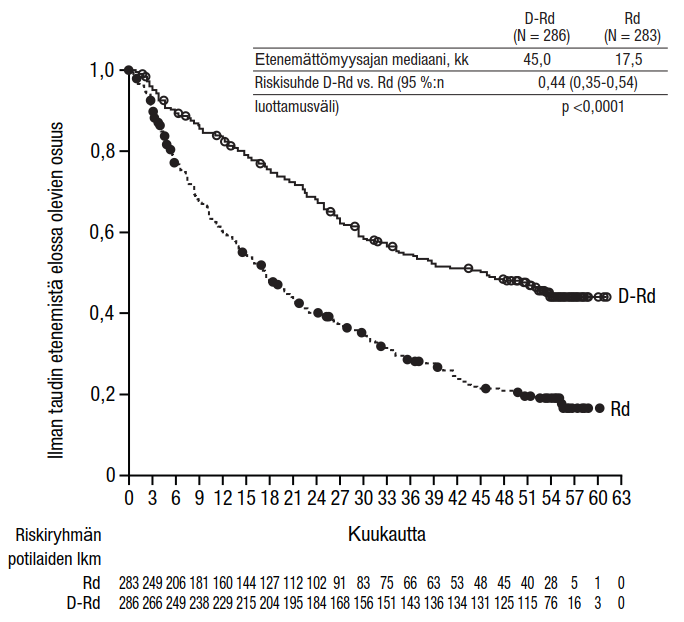
80 kuukauden (mediaani) seurannan jälkeen DRd-ryhmässä todettiin pidempi kokonaiselossaolo kuin Rd-ryhmässä (riskisuhde = 0,73; 95 %:n luottamusväli: 0,58; 0,91; p = 0,0044). Kokonaiselossaolon mediaani oli DRd-ryhmässä 67,6 kuukautta ja Rd-ryhmässä 51,8 kuukautta.
Kuvio 13: Tutkimuksen MMY3003 kokonaiselossaolon Kaplan–Meier-käyrä
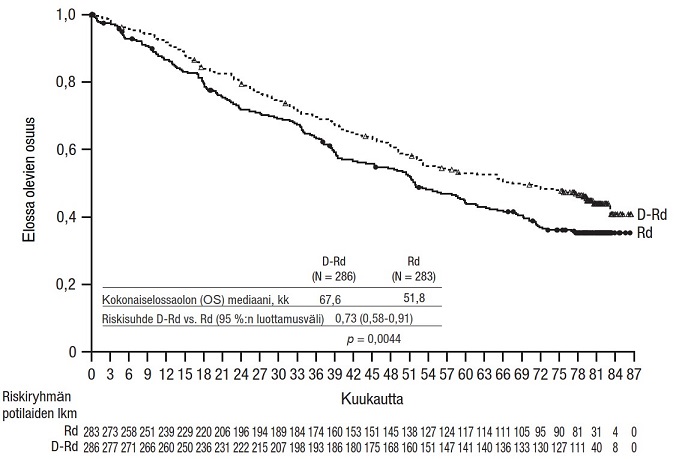
Tutkimuksen MMY3003 muut tehoa koskevat tulokset esitetään jäljempänä taulukossa 21.
| Taulukko 21: Tutkimuksen MMY3003 muut tehoa koskevat tulokset | ||
| Arvioitavissa olleiden potilaiden lukumäärä | DRd (n = 281) | Rd (n = 276) |
| Kokonaisvaste (sCR+CR+VGPR+PR) n(%) | 261 (92,9) | 211 (76,4) |
| p-arvoa | < 0,0001 | |
| Täydellinen vaste lisäehdoin (sCR) | 51 (18,1) | 20 (7,2) |
| Täydellinen vaste (CR) | 70 (24,9) | 33 (12,0) |
| Erittäin hyvä osittainen vaste (VGPR) | 92 (32,7) | 69 (25,0) |
| Osittainen vaste (PR) | 48 (17,1) | 89 (32,2) |
| Ajan mediaani vasteeseen [kuukautta (95 %:n luottamusväli)] | 1,0 (1,0; 1,1) | 1,3 (1,1; 1,9) |
| Vasteen keston mediaani [kuukautta (95 %:n luottamusväli))] | NE (NE; NE) | 17,4 (17,4; NE) |
| MRD-negatiivisten lukumäärä (95 %:n luottamusväli) b (%) | 21,0 (16,4; 26,2) | 2,8 (1,2; 5,5) |
| Kerroinsuhde, 95 %:n luottamusvälic | 9,31 (4,31; 20,09) | |
| P-arvod | < 0,0001 | |
DRd = daratumumabi-lenalidomidi-deksametasoni; Rd = lenalidomidi-deksametasoni; MRD = minimaalinen jäännöstauti (minimal residual disease); NE = ei arvioitavissa (not estimable). a Cochran Mantel-Haenszelin khiin neliötestin p-arvo. b Perustuu hoitoaikeen mukaiseen potilasjoukkoon (intent-to-treat population) ja raja-arvoon 10-5. c Yleisen kerroinsuhteen Mantel-Haenszelin estimaatin mukaan. Kerroinsuhde > 1 viittaa etuun DRd-hoidon suhteen. d p-arvo perustuu Fisherin eksaktiin testiin. | ||
Yhdistelmähoito bortetsomibin kanssa
Tutkimus MMY3004 oli avoin, satunnaistettu, aktiivisella aineella kontrolloitu vaiheen III tutkimus, jossa laskimoon annettavaa daratumumabia annoksina 16 mg/kg yhdistelmänä bortetsomibin ja deksametasonin kanssa (DVd) verrattiin bortetsomibin ja deksametasonin yhdistelmään (Vd) relapsoitunutta tai hoitoon reagoimatonta multippelia myeloomaa sairastavilla potilailla, jotka olivat saaneet vähintään yhtä aiempaa hoitoa. Bortetsomibi annettiin injektiona ihon alle tai injektiona laskimoon kehon pinta-alaan perustuvina annoksina 1,3 mg/m2 kaksi kertaa viikossa kahden viikon ajan 21 päivän (kolmen viikon) pituisten toistuvien hoitosyklien (päivinä 1, 4, 8 ja 11) ajan yhteensä 8 hoitosykliä. Deksametasoni annettiin suun kautta annoksina 20 mg kunkin kahdeksan bortetsomibihoitosyklin (80 mg/viikko kahdessa kolmesta bortetsomibihoitosyklistä) päivinä 1, 2, 4, 5, 8, 9, 11 ja 12 tai pienennettyinä annoksina 20 mg/viikko, jos potilas oli > 75-vuotias tai potilaan painoindeksi (BMI) oli < 18,5, potilaalla oli huonossa hoitotasapainossa oleva diabetes mellitus tai potilas ei ollut aiemmin sietänyt steroidihoitoa. Laskimoon annettavan daratumumabi-infuusion antopäivinä annettiin ennen infuusiota 20 mg:n annos deksametasonia. Laskimoon annettavaa daratumumabihoitoa jatkettiin, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä.
Yhteensä 498 potilasta satunnaistettiin; 251 satunnaistettiin DVd-ryhmään, ja 247 satunnaistettiin Vd-ryhmään. Lähtötilanteen demografiset ja taudin ominaisuuksia koskevat tiedot olivat laskimoon annettavaa daratumumabia saaneessa ryhmässä ja vertailuryhmässä samankaltaiset. Potilaiden iän mediaani oli 64 vuotta (vaihteluväli 30−88 vuotta), ja 12 % oli ≥ 75-vuotiaita. Kuusikymmentäyhdeksän prosenttia (69 %) potilaista oli aiemmin saanut jotakin proteasomin estäjää (66 % oli saanut bortetsomibia) ja 76 % potilaista oli saanut jotakin immunomodulatiivista lääkehoitoa (42 % oli saanut lenalidomidia). Lähtötilanteessa 32 % potilaista ei ollut reagoinut viimeiseen hoitolinjaan. Kolmekymmentäkolme prosenttia (33 %) potilaista ei ollut reagoinut immunomodulatiiviseen lääkehoitoon, ja 28 % ei ollut reagoinut lenalidomidiin. Bortetsomibihoitoon reagoimattomia potilaita ei otettu tutkimukseen mukaan.
Tutkimuksessa MMY3004 etenemättömyysajan ensisijainen analyysi osoitti 7,4 kuukauden (mediaani) seurannan jälkeen etenemättömyysajan pidentyneen DVd-ryhmässä Vd-ryhmään verrattuna; etenemättömyysajan mediaania ei saavutettu DVd-ryhmässä, ja se oli Vd-ryhmässä 7,2 kuukautta (riskisuhde [95 %:n luottamusväli]: 0,39 [0,28; 0,53]; p-arvo < 0,0001). Etenemättömyysajan päivitetyn analyysin tulokset osoittivat 50 kuukauden (mediaani) seurannan jälkeen edelleen, että etenemättömyysaika oli pidentynyt DVd-ryhmässä verrattuna Vd-ryhmään. Etenemättömyysajan mediaani oli DVd-ryhmässä 16,7 kuukautta ja Vd-ryhmässä 7,1 kuukautta (riskisuhde [95 %:n luottamusväli]: 0,31 [0,24; 0,39]; p-arvo < 0,0001), mikä tarkoittaa, että DVd-hoitoa saaneiden potilaiden taudin etenemisen tai kuoleman riski väheni 69 % Vd-hoitoon verrattuna (ks. kuvio 14).
Kuvio 14: Tutkimuksen MMY3004 etenemättömyysajan (PFS) Kaplan–Meier-käyrä
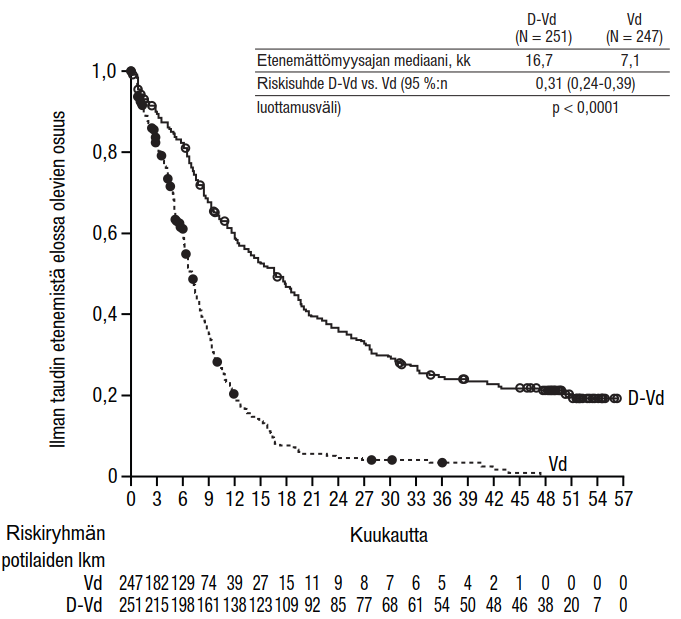
73 kuukauden (mediaani) seurannan jälkeen DVd-ryhmässä todettiin pidempi kokonaiselossaolo kuin Vd-ryhmässä (riskisuhde = 0,74; 95 %:n luottamusväli: 0,59; 0,92; p = 0,0075). Kokonaiselossaolon mediaani oli DVd-ryhmässä 49,6 kuukautta ja Vd-ryhmässä 38,5 kuukautta.
Kuvio 15: Tutkimuksen MMY3004 kokonaiselossaolon Kaplan–Meier-käyrä
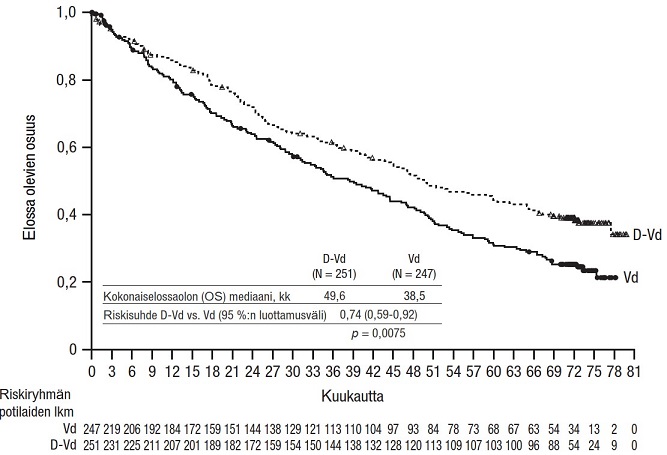
Tutkimuksen MMY3004 muut tehoa koskevat tulokset esitetään jäljempänä taulukossa 22.
| Taulukko 22: Tutkimuksen MMY3004 muut tehoa koskevat tulokset | ||
| Arvioitavissa olleiden potilaiden lukumäärä | DVd (n = 240) | Vd (n = 234) |
| Kokonaisvaste (sCR+CR+VGPR+PR) n(%) | 199 (82,9) | 148 (63,2) |
| P-arvoa | < 0,0001 | |
| Täydellinen vaste lisäehdoin (sCR) | 11 (4,6) | 5 (2,1) |
| Täydellinen vaste (CR) | 35 (14,6) | 16 (6,8) |
| Erittäin hyvä osittainen vaste (VGPR) | 96 (40,0) | 47 (20,1) |
| Osittainen vaste (PR) | 57 (23,8) | 80 (34,2) |
| Ajan mediaani vasteeseen [kuukautta (vaihteluväli)] | 0,9 (0,8; 1,4) | 1,6 (1,5; 2,1) |
| Vasteen keston mediaani [kuukautta (95 %:n luottamusväli)] | NE (11,5; NE) | 7,9 (6,7; 11,3) |
| MRD-negatiivisten lukumäärä (95 %:n luottamusväli)b | 8,8 % (5,6 %; 13,0 %) | 1,2 % (0,3 %; 3,5 %) |
| Kerroinsuhde, 95 %:n luottamusvälic | 9,04 (2,53; 32,21) | |
| P-arvod | 0,0001 | |
DVd = daratumumabi-bortetsomibi-deksametasoni; Vd = bortetsomibi-deksametasoni; MRD = minimaalinen jäännöstauti (minimal residual disease); NE = ei arvioitavissa (not estimable). a Cochran Mantel-Haenszelin khiin neliötestin p-arvo. b Perustuu hoitoaikeen mukaiseen potilasjoukkoon (intent-to-treat population) ja raja-arvoon 10-5. c Yleisen kerroinsuhteen Mantel–Haenszelin estimaatin mukaan. Kerroinsuhde > 1 viittaa etuun DVd-hoidon suhteen. d p-arvo perustuu Fisherin eksaktiin testiin. | ||
Sydämen elektrofysiologia
Daratumumabi on suurikokoinen proteiini, joten yhteisvaikutukset suoran ionikanavan kanssa ovat epätodennäköisiä. Avoimessa tutkimuksessa (GEN501), jossa oli mukana 83 relapsoitunutta ja hoitoon reagoimatonta multippelia myeloomaa sairastavaa potilasta, tutkittiin daratumumabin vaikutusta QTc-aikaan daratumumabi-infuusioiden (4–24 mg/kg) jälkeen. Lineaariset farmakokineettiset ja farmakodynaamiset seka-analyysit eivät osoittaneet daratumumabin huippupitoisuuksilla (Cmax) keskimääräisen QTcF-ajan merkittävää pidentymistä (eli yli 20 ms).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Darzalex-valmisteen käytöstä multippelin myelooman hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Monoterapiatutkimuksessa multippelia myeloomaa sairastaville potilaille ihon alle annetun Darzalex-valmisteen suositusannoksen 1800 mg jälkeen (viikoittain 8 viikon ajan, joka toinen viikko 16 viikon ajan, sen jälkeen kuukausittain) ja samana annostusohjelmana laskimoon annetun daratumumabiannoksen 16 mg/kg jälkeen daratumumabialtistus oli vertailukelpoinen (non‑inferior) toisen ensisijaisen päätetapahtuman osalta, joka oli suurin Ctrough-arvo (sykli 3 ennen päivän 1 annosta), ja keskiarvo ± keskihajonta oli 593 ± 306 mikrog/ml verrattuna laskimoon annetun daratumumabin altistukseen 522 ± 226 mikrog/ml, jolloin suhteen geometrinen keskiarvo oli 107,93 % (90 %:n luottamusväli: 95,74–121,67).
Yhdistelmätutkimuksessa AMY3001 AL-amyloidoosia sairastavien potilaiden Ctrough-maksimiarvo (ennen hoitosyklin 3 päivän 1 annosta) oli samankaltainen kuin multippelia myeloomaa sairastavilla potilailla: ihon alle annettavan Darzalex-valmisteen suositellun 1 800 mg:n annoksen (8 viikon ajan viikoittain, 16 viikon ajan kerran kahdessa viikossa ja sen jälkeen kuukausittain) jälkeen keskiarvo ± keskihajonta oli 597 ± 232 mikrog/ml.
Ihon alle annettavan Darzalex-liuoksen suositusannoksen 1 800 mg jälkeen huippupitoisuus (Cmax) suureni 4,8‑kertaiseksi ja kokonaisaltistus (AUC0‑7days) suureni 5,4‑kertaiseksi ensimmäisestä annoksesta viimeiseen viikoittaiseen annokseen (8. annos). Ihon alle annettavan Darzalex-valmisteen suurimmat jäännöspitoisuudet havaitaan sekä monoterapiassa että yhdistelmähoidossa tyypillisesti viikoittaisen anto-ohjelman lopussa.
Ihon alle annettavan Darzalex-valmisteen kuuden viikoittaisen 1 800 mg:n annoksen jälkeen multippelia myeloomaa sairastavilla potilaillasimuloidut jäännöspitoisuudet olivat yhdistelmähoidossa samankaltaisia kuin ihon alle annettavan Darzalex-valmisteen 1 800 mg:n annosten jälkeen monoterapiassa.
Äskettäin diagnosoitua multippelia myeloomaa sairastavilla potilailla, jotka soveltuivat autologiseen kantasolusiirtoon, daratumumabialtistus oli yhdistelmätutkimuksessa bortetsomibin, lenalidomidin ja deksametasonin kanssa yhdistelmänä annettuna (MMY3014) samankaltainen kuin monoterapiassa: ihon alle annettavan DARZALEX-valmisteen suositellun 1 800 mg:n annoksen (8 viikon ajan viikoittain, 16 viikon ajan kerran kahdessa viikossa ja sen jälkeen kuukausittain) jälkeen Ctrough-maksimiarvon (ennen hoitosyklin 3 päivän 1 annosta) keskiarvo ± keskihajonta oli 526 ± 209 mikrog/ml.
Äskettäin diagnosoitua multippelia myeloomaa sairastavilla potilailla, joille ei suunniteltu autologista kantasolusiirtoa alkuvaiheen hoitoon tai jotka eivät soveltuneet autologiseen kantasolusiirtoon, daratumumabialtistus oli yhdistelmätutkimuksessa bortetsomibin, lenalidomidin ja deksametasonin kanssa yhdistelmänä annettuna (MMY3019) samankaltainen kuin monoterapiassa ja muissa yhdistelmähoidoissa, joissa hoito-ohjelma oli samankaltainen: ihon alle annettavan DARZALEX-valmisteen suositellun 1 800 mg:n annoksen (6 viikon ajan viikoittain, 18 viikon ajan kolmen viikon välein ja sen jälkeen kuukausittain) Ctrough-maksimiarvon (ennen hoitosyklin 3 päivän 1 annosta) keskiarvo ± keskihajonta oli 407 ± 183 mikrog/ml.
Multippelia myeloomaa sairastavilla potilailla daratumumabialtistus oli yhdistelmätutkimuksessa pomalidomidin ja deksametasonin kanssa yhdistelmänä annettuna (tutkimus MMY3013) samankaltainen kuin monoterapiassa: ihon alle annettavan Darzalex-valmisteen suositellun 1 800 mg:n annoksen (8 viikon ajan viikoittain, 16 viikon ajan kerran kahdessa viikossa ja sen jälkeen kuukausittain) jälkeen Ctrough-maksimiarvon (ennen hoitosyklin 3 päivän 1 annosta) keskiarvo ± keskihajonta oli 537 ± 277 mikrog/ml).
Indolenttia multippelia myeloomaa, jonka multippeliksi myeloomaksi etenemisen riski on suuri, sairastavilla potilailla daratumumabialtistus oli monoterapiatutkimuksessa (SMM3001) samankaltainen kuin multippeliin myeloomaan annetussa monoterapiassa: ihon alle annettavan DARZALEX-valmisteen suositellun 1 800 mg:n annoksen (8 viikon ajan viikoittain, 16 viikon ajan kerran kahdessa viikossa ja sen jälkeen kuukausittain) jälkeen Ctrough-maksimiarvon (ennen hoitosyklin 3 päivän 1 annosta) keskiarvo ± keskihajonta oli 654 ± 243 mikrog/ml.
Imeytyminen ja jakautuminen
Ihon alle injektioina multippelia myeloomaa sairastaville potilaille annettavan Darzalex-valmisteen suositusannosten 1800 mg käytössä absoluuttinen biologinen hyötyosuus on 69 %, imeytymisnopeus on 0,012 tunnissa-1, ja huippupitoisuus todetaan 70–72 tunnissa (Tmax). AL-amyloidoosia sairastaville potilaille annetun suositellun annoksen 1800 mg absoluuttista biologista hyötyosuutta ei arvioitu, mutta imeytymisnopeuden vakio oli 0,77 vrk-1 (variaatiokerroin 8,31 %) ja huippupitoisuus todettiin 3 vuorokauden aikapisteessä.
Multippelia myeloomaa sairastavilla potilailla malli ennusti daratumumabimonoterapiassa jakautumistilavuuden keskiarvon estimaatiksi keskustilassa 5,25 l (variaatiokerroin 36,9 %) ja ääreistilassa (V2) 3,78 l ja annettaessa daratumumabi yhdistelmänä pomalidomidin ja deksametasonin kanssa jakautumistilavuuden mallinnetun estimaatin keskiarvo oli keskustilassa (V1) 4,36 l (variaatiokerrroin 28,0 %) ja ääreistilassa (V2) 2,80 l. AL-amyloidoosia sairastavilla potilailla mallin estimaatti laskennalliseksi jakautumistilavuudeksi ihon alle tapahtuneen annon jälkeen on 10,8 l (variaatiokerroin 3,1 %). Nämä tulokset viittavat siihen, että daratumumabia on pääasiassa verisuonistossa ja jakautuminen verisuonten ulkopuoliseen kudokseen on vähäistä.
Metabolia ja eliminaatio
Daratumumabilla on sekä pitoisuus- että aikariippuvainen farmakokinetiikka, jossa eliminaatio on rinnakkain lineaarista ja epälineaarista (saturoituvaa), mikä on tyypillistä kohdevälitteiselle puhdistumalle. Multippelia myeloomaa sairastavilla potilailla populaatiofarmakokineettinen malli arvioi daratumumabin puhdistuman keskiarvon estimaatiksi daratumumabimonoterapiassa 4,96 ml/h (variaatiokerroin 58,7 %) ja annettaessa daratumumabi yhdistelmänä pomalidomidin ja deksametasonin kanssa 4,32 ml/h (variaatiokerroin 43,5 %). AL-amyloidoosipotilailla laskennallinen puhdistuma ihon alle tapahtuneen annon jälkeen on 210 ml/vrk (variaatiokerroin 4,1 %). Lineaariseen eliminaatioon liittyvän puoliintumisajan malliin perustuva geometrinen keskiarvo on daratumumabimonoterapiassa 20,4 vuorokautta (variaatiokerroin 22,4 %) ja annettaessa daratumumabi yhdistelmänä pomalidomidin ja deksametasonin kanssa se on multippelia myeloomaa sairastavilla potilailla 19,7 vuorokautta (variaatiokerroin 15,3 %) ja AL-amyloidoosia sairastavilla potilailla 27,5 vuorokautta (variaatiokerroin 74,0 %). Monoterapiassa ja yhdistelmähoidossa vakaa tila saavutetaan noin 5 kuukauden hoidon jälkeen, kun suositeltua annosta ja hoito-ohjelmaa annetaan 4 viikon välein (1800 mg kerran viikossa 8 viikon ajan, joka toinen viikko 16 viikon ajan ja sen jälkeen 4 viikon välein).
Ihon alle injektioina multippelia myeloomaa, mukaan lukien indolenttia myeloomaa, sairastaville potilaille annettavasta DARZALEX-monoterapiasta ja ‑yhdistelmähoidosta saaduista tiedoista tehtiin populaatiofarmakokineettiset analyysit, ja yhteenveto ennustetuista farmakokineettisistä altistuksista esitetään taulukossa 23. Potilailla, jotka saivat ihon alle annettavaa DARZALEX-injektionestettä monoterapiana ja yhdistelmähoitona, daratumumabialtistukset olivat samankaltaiset.
| Taulukko 23: Daratumumabialtistus multippelia myeloomaa, mukaan lukien indolenttia multippelia myeloomaa, sairastavilla potilailla ihon alle annettavalla DARZALEX-valmisteella (1 800 mg) tai laskimoon annettavalla daratumumabilla (16 mg/kg) toteutetun monoterapian jälkeen | ||||
| Farmakokineettiset parametrit | Syklit | multippeli myelooma: ihon alle annettava daratumumabi mediaani (5.; 95. persentiili) | indolentti multippeli myelooma: ihon alle annettava daratumumabi mediaani (5.; 95. persentiili) | laskimoon annettava daratumumabi mediaani (5.; 95. persentiili) |
| Ctrough (mikrog/ml) | Sykli 1; 1. viikoittainen annos | 123 (36; 220) | 155 (104; 235) | 112 (43; 168) |
| Sykli 2; viimeinen viikoittainen annos (Sykli 3 Päivä 1 Ctrough) | 563 (177; 1 063) | 690 (269; 1 034) | 472 (144; 809) | |
| Cmax (mikrog/ml) | Sykli 1; 1. viikoittainen annos | 132 (54; 228) | 158 (106; 241) | 256 (173; 327) |
| Sykli 2; viimeinen viikoittainen annos | 592 (234; 1 114) | 780 (340; 1 152) | 688 (369; 1 061) | |
| AUC0‑7 days (mikrog/ml•vrk) | Sykli 1; 1. viikoittainen annos | 720 (293; 1 274) | 861 (529; 1 325) | 1 187 (773; 1 619) |
| Sykli 2; viimeinen viikoittainen annos | 4 017 (1 515; 7 564) | 5 043 (2 242; 7 426) | 4 019 (1 740; 6 370) | |
Yhteenveto ennustetuista farmakokineettisistä altistuksista 526 potilaalla, joilla oli siirtoon soveltuva multippeli myelooma ja jotka saivat ihon alle annettavaa DARZALEX-injektionestettä yhdistelmänä VRd-hoidon kanssa, on taulukossa 24.
| Taulukko 24: Daratumumabialtistus siirtoon soveltuvaa multippelia myeloomaa sairastavilla potilailla ihon alle annettavan DARZALEX-valmisteen (1 800 mg) ja VRd-hoidon yhdistelmän annon jälkeen | ||
| Farmakokineettiset parametrit | Syklit | ihon alle annettava daratumumabi mediaani (5.; 95. persentiili) |
| Ctrough (mikrog/ml) | Sykli 1; 1. viikoittainen annos | 113 (66; 171) |
| Sykli 2; viimeinen viikoittainen annos (sykli 3 päivä 1 Ctrough) | 651 (413; 915) | |
| Cmax (mikrog/ml) | Sykli 1; 1. viikoittainen annos | 117 (67; 179) |
| Sykli 2; viimeinen viikoittainen annos | 678 (431; 958) | |
| AUC0-7 days (mikrog/ml•vrk) | Sykli 1; 1. viikoittainen annos | 643 (322; 1 027) |
| Sykli 2; viimeinen viikoittainen annos | 4 637 (2 941; 6 522) | |
Ihon alle injektioina AL-amyloidoosia sairastaville potilaille annettavasta yhdistelmähoidosta saaduista tiedoista tehtiin populaatiofarmakokineettinen analyysi, johon otettiin mukaan 211 potilaan tiedot. Ennustetut daratumumabipitoisuudet olivat suositellulla annoksella 1 800 mg hieman suuremmat, mutta yleisesti samaa suuruusluokkaa kuin multippelia myeloomaa sairastavilla potilailla.
Taulukko 25: Daratumumabialtistus AL-amyloidoosia sairastavilla potilailla ihon alle annettavan Darzalex-valmisteen (1 800 mg) annon jälkeen | ||
Farmakokineettiset parametrit | Syklit | ihon alle annettava daratumumabi mediaani (5.; 95. persentiili) |
| Ctrough (mikrog/ml) | Sykli 1, 1. viikoittainen annos | 138 (86; 195) |
Sykli 2, viimeinen viikoittainen annos (sykli 3 Päivä 1 Ctrough) | 662 (315; 1 037) | |
| Cmax (mikrog/ml) | Sykli 1, 1. viikoittainen annos | 151 (88; 226) |
Sykli 2, viimeinen viikoittainen annos | 729 (390; 1 105) | |
| AUC0-7 days (mikrog/ml•vrk) | Sykli 1, 1. viikoittainen annos | 908 (482; 1 365) |
Sykli 2, viimeinen viikoittainen annos | 4 855 (2 562; 7 522) | |
Erityiset potilasryhmät
Ikä ja sukupuoli
Iällä (vaihteluväli: 33–92 vuotta) ei monoterapiaa tai erilaisia yhdistelmähoitoja saaneiden potilaiden populaatiofarmakokineettisen analyysin perusteella ollut tilastollisesti merkitsevää vaikutusta daratumumabin farmakokinetiikkaan. Hoitoa ei tarvitse säätää yksilöllisesti iän perusteella.
Sukupuolella oli tilastollisesti merkitsevä vaikutus farmakokineettisiin parametreihin multippelia myeloomaa sairastavilla potilailla, mutta ei AL-amyloidoosia sairastavilla potilailla. Naisilla havaittiin hieman suurempi altistus kuin miehillä, mutta eroa altistuksessa ei katsottu kliinisesti merkittäväksi. Hoitoa ei tarvitse säätää yksilöllisesti sukupuolen perusteella.
Munuaisten vajaatoiminta
Ihon alle annettavan Darzalex-valmisteen käytöstä munuaisten vajaatoimintaa sairastavilla potilailla ei ole tehty formaalisia tutkimuksia. Populaatiofarmakokineettiset analyysit tehtiin perustuen olemassa oleviin tietoihin munuaisten toiminnasta multippelia myeloomaa sairastavilla potilailla, jotka saivat ihon alle annettavaa Darzalex-valmistetta monoterapiana, tai multippelia myeloomaa tai AL-amyloidoosia sairastavilla potilailla, jotka saivat erilaisia yhdistelmähoitoja. Daratumumabialtistuksessa ei havaittu kliinisesti merkittäviä eroja sen mukaan, sairastiko potilas munuaisten vajaatoimintaa vai oliko potilaan munuaisten toiminta normaali.
Maksan vajaatoiminta
Ihon alle annettavan Darzalex-valmisteen käytöstä maksan vajaatoimintaa sairastavilla potilailla ei ole tehty formaalisia tutkimuksia.
Populaatiofarmakokineettiset analyysit tehtiin perustuen multippelia myeloomaa sairastaviin potilaisiin, jotka saivat ihon alle annettavaa Darzalex-valmistetta monoterapiana, tai multippelia myeloomaa tai AL-amyloidoosia sairastaviin potilaiisiin, jotka saivat erilaisia yhdistelmähoitoja. Daratumumabialtistuksessa ei havaittu kliinisesti merkittäviä eroja niiden potilaiden välillä, joiden maksan toiminta oli normaali tai joilla oli lievää maksan vajaatoimintaa. Keskivaikeaa ja vaikeaa maksan vajaatoimintaa sairastavia potilaita oli hyvin vähän, joten näistä potilasryhmistä ei voitu tehdä merkittäviä päätelmiä.
Rotu
Ihon alle annettavaa Darzalex-valmistetta monoterapiana tai erilaisina yhdistelmähoitoina saaneista potilaista tehtyjen populaatiofarmakokineettisten analyysien perusteella daratumumabialtistus oli kaikilla roduilla samankaltainen.
Paino
Monoterapiana ihon alle annettavan Darzalex-valmisteen 1 800 mg:n vakioannoksista saavutettiin riittävä altistus kaikissa painon mukaisissa alaryhmissä. Hoitosyklin 3 päivänä 1 Ctrough-arvon keskiarvo oli multippelia myeloomaa sairastavilla potilailla pienemmän painon alaryhmässä (≤ 65 kg) 60 % suurempi ja suuremman painon alaryhmässä (> 85 kg) 12 % pienempi kuin daratumumabia laskimoon saaneessa alaryhmässä. Joillakin > 120 kg:n painoisilla potilailla havaittiin pienempi altistus, joten teho voi olla heikentynyt. Tämä havainto perustuu kuitenkin pieneen potilasmäärään.
AL-amyloidoosia sairastavilla potilailla ei painosta riippumatta havaittu Ctrough-arvossa merkittäviä eroja.
Prekliiniset tiedot turvallisuudesta
Toksikologiset tiedot on saatu simpansseilla tehdyistä daratumumabitutkimuksista sekä cynomolgus-apinoiden korvaavasta anti-CD38-vasta-aineesta. Kroonista toksisuutta ei ole tutkittu.
Daratumumabin mahdollista karsinogeenisuutta ei ole selvitetty eläinkokeissa.
Daratumumabin mahdollisia vaikutuksia lisääntymiseen ja kehitykseen ei ole tutkittu eläinkokeissa, eikä mahdollisia vaikutuksia urosten tai naaraiden hedelmällisyyteen ole selvitetty.
Rekombinantilla ihmisen hyaluronidaasilla ei tehty karsinogeenisuus-, genotoksisuus- eikä hedelmällisyystutkimuksia. Apinoilla, jotka saivat 22 000 U/kg/viikossa ihon alle (12 kertaa suurempi ihmisen annokseen verrattuna) 39 viikon ajan, ei ollut vaikutuksia lisääntymiskudoksiin tai lisääntymistoimintoihin eikä hyaluronidaasin systeemiseen altistukseen. Hyaluronidaasi on ihmisen endogeenisen hyaluronidaasin rekombinantti muoto, joten karsinogeenisuutta, mutageenisuutta tai vaikutuksia hedelmällisyyteen ei oletettavasti esiinny.
Farmaseuttiset tiedot
Apuaineet
Rekombinantti ihmisen hyaluronidaasi (rHuPH20)
L‑histidiini
L‑histidiinihydrokloridimonohydraatti
L‑metioniini
Polysorbaatti 20 (E432)
Sorbitoli (E420)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa käyttää muiden materiaalien kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta.
Avaamattomia injektiopulloja voidaan säilyttää kestoajan puitteissa vallitsevassa lämpötilassa (≤ 30 °C) yhden enintään 24 tunnin pituisen jakson ajan. Kun valmiste on otettu jääkaapista, sitä ei saa enää laittaa jääkaappiin takaisin (ks. kohta Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet).
Käyttöön valmisteltu ruisku
Kemialliseksi ja fysikaaliseksi käytönaikaiseksi säilyvyydeksi ruiskussa on osoitettu 24 tuntia jääkaapissa (2 °C – 8 °C) ja sen jälkeen enintään 12 tuntia 15 °C – 25 °C:ssa ja vallitsevassa valossa. Ellei pakkauksen avaamistapa sulje pois mikrobikontaminaation riskiä, valmiste pitää mikrobiologiselta kannalta käyttää heti. Jos sitä ei käytetä heti, käytönaikaiset säilytysajat ja ‑olosuhteet ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Avatun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
DARZALEX injektioneste, liuos
1800 mg (L:ei) 15 ml (120 mg/ml) (5205,83 €)
PF-selosteen tieto
15 ml liuosta tyypin I lasisessa injektiopullossa, jossa on elastomeerisuljin ja alumiinisinetti sekä irti napsautettava korkki ja joka sisältää 1 800 mg daratumumabia. Pakkauskoko 1 injektiopullo.
Valmisteen kuvaus:
Liuos on kirkas tai opalisoiva, väritön tai keltainen, sen pH on 5,6 ja osmolaalisuus on 343–395 mOsm/kg.
Käyttö- ja käsittelyohjeet
Ihon alle annettava Darzalex-injektioneste on käyttövalmis ja tarkoitettu yhteen käyttökertaan.
Ihon alle annettavan Darzalex-injektionesteen pitää olla kirkas tai opalisoiva ja väritön tai keltainen liuos. Jos liuoksessa on läpinäkymättömiä hiukkasia, värimuutoksia tai muita vieraspartikkeleita, älä käytä sitä.
Ihon alle annettava Darzalex-injektioneste on yhteensopiva polypropeenista tai polyeteenista valmistettujen ruiskujen kanssa, polypropeenista, polyeteenista tai polyvinyylikloridista (PVC) valmistettujen ihon alle antoon tarkoitettujen infuusiovälineiden kanssa sekä ruostumattomasta teräksestä valmistettujen siirto- ja injektioneulojen kanssa.
Avaamaton injektiopullo
Ota ihon alle annettavan Darzalex-injektionesteen sisältävä injektiopullo jääkaappisäilytyksestä (2 °C – 8 °C), ja anna sen lämmetä vallitsevaan lämpötilaan (≤ 30 °C). Avaamatonta injektiopulloa voidaan säilyttää vallitsevassa lämpötilassa ja vallitsevassa valossa enintään 24 tunnin ajan alkuperäispakkauksessa. Herkkä valolle. Pidä poissa suorasta auringonvalosta. Ei saa ravistaa.
Valmisteltu ruisku
Valmistele annosteluruisku kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa. Vedä injektiopullosta 15 ml ruiskuun käyttämällä 18–22G:n siirtoneulaa, jossa on tavanomainen viistokärki, jotta tulpan materiaalin irtoamisen riski minimoituu. Työnnä neula injektiopulloon kohtisuoraan (90°:n kulmassa) tulpan renkaaseen nähden ja minimoi lävistyskerrat tulpan rikkoutumisen välttämiseksi. Tarkista ruiskun sisältö varmistaaksesi, ettei siinä ole hiukkasia, värimuutoksia eikä muita vierashiukkasia.
Kun ihon alle annettava Darzalex-injektioneste on siirretty injektiopullosta ruiskuun, säilytä sitä enintään 24 tuntia jääkaapissa ja sen jälkeen enintään 12 tuntia 15 °C – 25 °C:ssaja vallitsevassa valossa (ks. kohta Kestoaika). Anna jääkaapissa säilytetyn liuoksen lämmetä vallitsevaan lämpötilaan ennen antoa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
DARZALEX injektioneste, liuos
1800 mg 15 ml
- Ylempi erityiskorvaus (100 %). Daratumumabi: Multippelin myelooman ja AL-amyloidoosin hoito erityisin edellytyksin (1534).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Daratumumabi: Multippelin myelooman hoito erityisin edellytyksin (3060).
ATC-koodi
L01FC01
Valmisteyhteenvedon muuttamispäivämäärä
26.03.2026
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com