
RINVOQ depottabletti 15 mg, 30 mg, 45 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Lääkäri
Terveydenhuollon ammattilaisten koulutusopas/Utbildningsguide för vårdpersonal
Potilas
Potilaskortti/Patientkort/Patient Card
Vaikuttavat aineet ja niiden määrät
RINVOQ 15 mg depottabletit
Yksi depottabletti sisältää upadasitinibihemihydraattia vastaten 15 mg upadasitinibia.
RINVOQ 30 mg depottabletit
Yksi depottabletti sisältää upadasitinibihemihydraattia vastaten 30 mg upadasitinibia.
RINVOQ 45 mg depottabletit
Yksi depottabletti sisältää upadasitinibihemihydraattia vastaten 45 mg upadasitinibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Depottabletti
Kliiniset tiedot
Käyttöaiheet
Nivelreuma
RINVOQ on tarkoitettu keskivaikean tai vaikean aktiivisen nivelreuman hoitoon aikuispotilaille, joilla vähintään yksi tautiprosessiin vaikuttava reumalääke on tuottanut riittämättömän vasteen tai ollut huonosti siedetty. RINVOQ-valmistetta voidaan käyttää ainoana lääkkeenä tai yhdessä metotreksaatin kanssa.
Nivelpsoriaasi
RINVOQ on tarkoitettu aktiivisen nivelpsoriaasin hoitoon aikuispotilaille, joilla vähintään yksi tautiprosessiin vaikuttava reumalääke on tuottanut riittämättömän vasteen tai ollut huonosti siedetty. RINVOQ-valmistetta voidaan käyttää ainoana lääkkeenä tai yhdessä metotreksaatin kanssa.
Aksiaalinen spondylartriitti
Röntgennegatiivinen aksiaalinen spondylartriitti (nr-axSpA)
RINVOQ on tarkoitettu aktiivisen röntgennegatiivisen aksiaalisen spondylartriitin hoitoon aikuispotilaille, joilla on objektiivisia inflammaation merkkejä todettavissa kohonneena C-reaktiivisen proteiinin arvona (CRP) ja/tai magneettikuvissa ja joilla tulehduskipulääkkeet (NSAID-lääkkeet) ovat tuottaneet riittämättömän vasteen.
Selkärankareuma (AS, röntgenpositiivinen aksiaalinen spondylartriitti)
RINVOQ on tarkoitettu aktiivisen selkärankareuman hoitoon aikuispotilaille, joilla tavanomainen hoito on tuottanut riittämättömän vasteen.
Jättisoluarteriitti
RINVOQ on tarkoitettu jättisoluarteriitin hoitoon aikuispotilaille.
Atooppinen ihottuma
RINVOQ on tarkoitettu keskivaikean tai vaikean atooppisen ihottuman hoitoon aikuisille ja vähintään 12-vuotiaille nuorille, joille harkitaan systeemistä hoitoa.
Haavainen paksusuolitulehdus
RINVOQ on tarkoitettu keskivaikean tai vaikean aktiivisen haavaisen paksusuolitulehduksen hoitoon aikuispotilaille, joilla tavanomainen hoito tai biologinen lääke on tuottanut riittämättömän vasteen, ollut huonosti siedetty tai vaste on menetetty.
Crohnin tauti
RINVOQ on tarkoitettu keskivaikean tai vaikean aktiivisen Crohnin taudin hoitoon aikuispotilaille, joilla tavanomainen hoito tai biologinen lääke on tuottanut riittämättömän vasteen, ollut huonosti siedetty tai vaste on menetetty.
Ehto
Valmisteen käyttöaiheissa mainittujen sairauksien hoitoon perehtyneen lääkärin on aloitettava hoito ja valvottava sitä.
Annostus ja antotapa
Upadasitinibihoidon aloittaa ja sitä valvoo upadasitinibin käyttöaiheiksi määriteltyjen sairauksien toteamiseen ja hoitoon perehtynyt lääkäri.
Annostus
Nivelreuma, nivelpsoriaasi ja aksiaalinen spondylartriitti
Suositeltu upadasitinibiannos on 15 mg kerran vuorokaudessa.
Hoidon lopettamista on harkittava aksiaalista spondylartriittia sairastavilla potilailla, jos 16 hoitoviikon jälkeen ei todeta kliinistä vastetta. Joillakin potilailla alkuun saavutettu osittainen vaste saattaa parantua, kun hoitoa jatketaan yli 16 viikon ajan.
Jättisoluarteriitti
Suositeltu upadasitinibiannos on 15 mg kerran vuorokaudessa yhdessä asteittain lopetettavan kortikosteroidihoidon kanssa. Upadasitinibia ei saa käyttää monoterapiana akuuttien pahenemisvaiheiden hoitoon (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jättisoluarteriitin kroonisen luonteen vuoksi upadasitinibin käyttöä voidaan jatkaa monoterapiana annoksella 15 mg kerran vuorokaudessa kortikosteroidihoidon päättymisen jälkeen.
Atooppinen ihottuma
Suositeltu upadasitinibiannos on 15 mg tai 30 mg kerran vuorokaudessa potilaan yksilöllisen tarpeen mukaan:
-
15 mg:n annosta suositellaan potilaille, joilla on suurentunut tromboembolisten laskimotapahtumien, merkittävien sydän- ja verisuonitapahtumien ja maligniteetin riski (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
-
Kerran vuorokaudessa annettava 30 mg:n annos saattaa sopia potilaille, joilla on suuri tautitaakka, mutta joilla tromboembolisten laskimotapahtumien, merkittävien sydän- ja verisuonitapahtumien ja maligniteetin riski ei ole suurentunut (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), tai potilaille, joille 15 mg kerran vuorokaudessa ei ole tuottanut riittävää vastetta.
-
Vähintään 30 kg painaville nuorille (12–17-vuotiaat) suositeltu annos on 15 mg. Jos 15 mg kerran vuorokaudessa ei tuota riittävää vastetta potilaalle, annos voidaan suurentaa 30 mg:aan kerran vuorokaudessa.
-
Vasteen ylläpitämiseen on käytettävä pienintä tehokasta annosta.
65 vuotta täyttäneille potilaille suositeltu annos on 15 mg kerran vuorokaudessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Samanaikaiset paikallishoidot
Upadasitinibia voidaan käyttää paikalliskortikosteroidien kanssa tai ilman niitä. Kalsineuriinin estäjiä voidaan käyttää paikallisesti herkillä alueilla, kuten kasvoilla, kaulalla sekä taive- ja genitaalialueilla.
Jos 12 hoitoviikon jälkeen ei todeta hoidon tuottamaa hyötyä, upadasitinibihoidon lopettamista on harkittava.
Haavainen paksusuolitulehdus
Induktiohoito
Induktiohoitoon suositeltu upadasitinibiannos on 45 mg kerran vuorokaudessa 8 viikon ajan. Jos potilas ei saa riittävää hyötyä hoidosta viikkoon 8 mennessä, upadasitinibihoitoa annoksella 45 mg kerran vuorokaudessa voidaan jatkaa vielä toisen 8 viikon jakson ajan (ks. kohta Farmakodynamiikka). Jos 16 viikon jälkeen ei todeta hoidon tuottamaa hyötyä, upadasitinibihoito on lopetettava.
Ylläpitohoito
Ylläpitohoitoon suositeltu upadasitinibiannos on 15 mg tai 30 mg kerran vuorokaudessa potilaan yksilöllisen tarpeen mukaan:
-
15 mg:n annosta suositellaan potilaille, joilla on suurentunut tromboembolisten laskimotapahtumien, merkittävien sydän- ja verisuonitapahtumien ja maligniteetin riski (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
-
Kerran vuorokaudessa annettava 30 mg:n annos saattaa sopia joillekin potilaille, esimerkiksi niille, joilla on suuri tautitaakka tai jotka tarvitsevat 16 viikkoa kestävän induktiohoidon, mutta joilla tromboembolisten laskimotapahtumien, merkittävien sydän- ja verisuonitapahtumien ja maligniteetin riski ei ole suurentunut (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), tai potilaille, joille 15 mg kerran vuorokaudessa ei ole tuottanut riittävää vastetta.
-
Vasteen ylläpitämiseen on käytettävä pienintä tehokasta annosta.
65 vuotta täyttäneille potilaille suositeltu annos on 15 mg kerran vuorokaudessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Upadasitinibihoitoon vastanneiden potilaiden kortikosteroidiannosta voidaan pienentää ja/tai kortikosteroidihoito voidaan lopettaa tavanomaisen hoitokäytännön mukaisesti.
Crohnin tauti
Induktiohoito
Induktiohoitoon suositeltu upadasitinibiannos on 45 mg kerran vuorokaudessa 12 viikon ajan. Jos potilas ei ole saanut riittävää hyötyä hoidosta ensimmäisen 12 viikon induktiohoidon jälkeen, induktiohoidon jatkamista toiset 12 viikkoa annoksella 30 mg kerran vuorokaudessa voidaan harkita. Upadasitinibi on lopetettava näillä potilailla, jos 24 hoitoviikon jälkeen ei todeta hoidon tuottamaa hyötyä.
Ylläpitohoito
Ylläpitohoitoon suositeltu upadasitinibiannos on 15 mg tai 30 mg kerran vuorokaudessa potilaan yksilöllisen tarpeen mukaan:
-
15 mg:n annosta suositellaan potilaille, joilla on suurentunut tromboembolisten laskimotapahtumien, merkittävien sydän- ja verisuonitapahtumien ja maligniteetin riski (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
-
Kerran vuorokaudessa annettava 30 mg:n annos saattaa sopia potilaille, joilla on suuri tautitaakka, mutta joilla tromboembolisten laskimotapahtumien, merkittävien sydän- ja verisuonitapahtumien ja maligniteetin riski ei ole suurentunut (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), tai potilaille, joille 15 mg kerran vuorokaudessa ei ole tuottanut riittävää vastetta.
-
Vasteen ylläpitämiseen on käytettävä pienintä tehokasta annosta.
65 vuotta täyttäneille potilaille suositeltu ylläpitoannos on 15 mg kerran vuorokaudessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Upadasitinibihoitoon vastanneiden potilaiden kortikosteroidiannosta voidaan pienentää ja/tai kortikosteroidihoito voidaan lopettaa tavanomaisen hoitokäytännön mukaisesti.
Yhteisvaikutukset
Haavaista paksusuolitulehdusta tai Crohnin tautia sairastavilla potilailla, jotka saavat voimakkaita sytokromi P450 (CYP) 3A4:n estäjiä (esim. ketokonatsolia, klaritromysiiniä), induktiohoitoon suositeltu annos on 30 mg kerran vuorokaudessa ja ylläpitohoitoon suositeltu annos on 15 mg kerran vuorokaudessa (ks. kohta Yhteisvaikutukset).
Hoidon aloittaminen
Hoitoa ei pidä aloittaa, jos potilaan absoluuttinen lymfosyyttiarvo on < 0,5 x 109 solua/l, absoluuttinen neutrofiiliarvo < 1 x 109 solua/l tai hemoglobiiniarvo < 8 g/dl (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Hoidon tauottaminen
Jos potilaalle kehittyy vakava infektio, hoito on tauotettava, kunnes infektio saadaan hallintaan.
Hoidon tauottaminen voi olla tarpeen, jotta taulukossa 1 kuvatut laboratorioarvojen poikkeavuudet saadaan hallintaan.
Taulukko 1 Laboratorioarvot ja seurantaohjeet
Laboratorioarvo | Toimintaohje | Seurantaohje |
Absoluuttinen neutrofiiliarvo | Hoito on tauotettava, jos absoluuttinen neutrofiiliarvo on < 1 x 109 solua/l. Hoito voidaan aloittaa uudelleen, kun absoluuttinen neutrofiiliarvo korjautuu tätä raja-arvoa suuremmaksi. | Arvioidaan lähtötilanteessa ja sitten viimeistään 12 viikon kuluttua hoidon aloittamisesta. Tämän jälkeen arvioidaan yksilöllisen seurannan mukaisesti. |
Absoluuttinen lymfosyyttiarvo | Hoito on tauotettava, jos absoluuttinen lymfosyyttiarvo on < 0,5 x 109 solua/l. Hoito voidaan aloittaa uudelleen, kun absoluuttinen lymfosyyttiarvo korjautuu tätä raja-arvoa suuremmaksi. | |
Hemoglobiini (Hb) | Hoito on tauotettava, jos hemoglobiinipitoisuus on < 8 g/dl. Hoito voidaan aloittaa uudelleen, kun hemoglobiinipitoisuus korjautuu tätä raja-arvoa suuremmaksi. | |
Maksan transaminaasiarvot | Hoito on tauotettava, jos epäillään lääkkeen aiheuttamaa maksavauriota. | Arvioidaan lähtötilanteessa ja tämän jälkeen rutiiniseurannan mukaisesti. |
Lipidit | Potilaita hoidetaan hyperlipidemian kansainvälisten kliinisten hoitosuositusten mukaisesti. | Arvioidaan 12 viikon kuluttua hoidon aloituksesta ja tämän jälkeen hyperlipidemian kansainvälisten kliinisten hoitosuositusten mukaisesti |
Erityisryhmät
Iäkkäät
Nivelreuma, nivelpsoriaasi ja aksiaalinen spondylartriitti
75 vuotta täyttäneiden potilaiden hoidosta on vain vähän tietoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Atooppinen ihottuma
Atooppista ihottumaa sairastaville 65 vuotta täyttäneille potilaille ei suositella suurempaa annosta kuin 15 mg kerran vuorokaudessa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Haavainen paksusuolitulehdus ja Crohnin tauti
Haavaista paksusuolitulehdusta tai Crohnin tautia sairastaville 65 vuotta täyttäneille potilaille ei suositella suurempaa ylläpitoannosta kuin 15 mg kerran vuorokaudessa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Upadasitinibin turvallisuutta ja tehoa 75 vuotta täyttäneiden potilaiden hoidossa ei ole vielä varmistettu.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa lievää eikä keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla. Upadasitinibin käytöstä vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla on vain vähän tietoa (ks. kohta Farmakokinetiikka). Upadasitinibin käytössä on noudatettava varovaisuutta, jos potilaalla on vaikea munuaisten vajaatoiminta, kuten taulukossa 2 on kuvattu. Upadasitinibia ei ole tutkittu henkilöillä, joilla on loppuvaiheen munuaisten vajaatoiminta, eikä sen käyttöä siksi suositella näille potilaille.
Taulukko 2 Suositeltu annos vaikeassa munuaisten vajaatoiminnassaa
Käyttöaihe | Suositeltu annos kerran vuorokaudessa |
Nivelreuma, nivelpsoriaasi, aksiaalinen spondylartriitti, jättisoluarteriitti, atooppinen ihottuma | 15 mg |
Haavainen paksusuolitulehdus, Crohnin tauti | Induktiohoito: 30 mg |
Ylläpitohoito: 15 mg | |
aglomerulusten laskennallinen suodatusnopeus (eGFR) 15 – < 30 ml/min/1,73 m2 | |
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen, jos potilaalla on lievä (Child–Pugh-luokka A) tai keskivaikea (Child–Pugh-luokka B) maksan vajaatoiminta (ks. kohta Farmakokinetiikka). Upadasitinibia ei pidä käyttää potilailla, joilla on vaikea (Child–Pugh-luokka C) maksan vajaatoiminta (ks. kohta Vasta-aiheet).
Pediatriset potilaat
RINVOQ-valmisteen turvallisuutta ja tehoa alle 12 vuoden ikäisten atooppista ihottumaa sairastavien lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
RINVOQ-valmisteen turvallisuutta ja tehoa 0 – alle 18 vuoden ikäisten nivelreumaa, nivelpsoriaasia, aksiaalista spondylartriittia, haavaista paksusuolitulehdusta tai Crohnin tautia sairastavien lasten ja nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Ei ole asianmukaista käyttää RINVOQ-valmistetta pediatrisille potilaille jättisoluarteriitin hoitoon.
Antotapa
RINVOQ otetaan suun kautta kerran vuorokaudessa ruoan kanssa tai ilman ruokaa. Se voidaan ottaa mihin kellonaikaan tahansa. Tabletit niellään kokonaisina, eikä niitä saa pilkkoa, murskata eikä pureskella, jotta potilas saa varmasti koko annoksen oikein.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Aktiivinen tuberkuloosi tai aktiivinen vakava infektio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Vaikea maksan vajaatoiminta (ks. kohta Annostus ja antotapa).
- Raskaus (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Upadasitinibia saa käyttää seuraaville potilaille vain, jos sopivaa hoitovaihtoehtoa ei ole:
|
Käyttö 65 vuotta täyttäneillä potilailla
Kun otetaan huomioon merkittävien sydän- ja verisuonitapahtumien, maligniteettien, vakavien infektioiden ja mistä tahansa syystä aiheutuvan kuolleisuuden suurentunut riski, joka havaittiin 65 vuotta täyttäneillä potilailla tofasitinibia (toinen Janus‑kinaasin [JAK] estäjä) koskeneessa laajassa satunnaistetussa tutkimuksessa, upadasitinibia saa käyttää näillä potilailla vain, jos sopivia hoitovaihtoehtoja ei ole.
Haittavaikutusten riski on suurentunut, jos 65 vuotta täyttäneiden potilaiden upadasitinibiannos on 30 mg kerran vuorokaudessa. Näin ollen tälle potilasryhmälle pitkäkestoiseen käyttöön suositeltu annos on 15 mg kerran vuorokaudessa (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Immunosuppressoivat lääkevalmisteet
Yhdistelmäkäyttöä muiden voimakkaiden immunosuppressanttien kuten atsatiopriinin, 6‑merkaptopuriinin, siklosporiinin, takrolimuusin ja tautiprosessiin vaikuttavien biologisten reumalääkkeiden kanssa tai muiden JAK:n estäjien kanssa ei ole arvioitu kliinisissä tutkimuksissa. Tällaista käyttöä ei suositella, sillä additiivisen immunosuppression riskiä ei voida sulkea pois.
Vakavat infektiot
Upadasitinibia saaneilla potilailla on ilmoitettu vakavia ja joskus kuolemaan johtaneita infektioita. Upadasitinibihoidon yhteydessä yleisimmin ilmoitettuja vakavia infektioita olivat keuhkokuume (ks. kohta Haittavaikutukset) ja ihonalaiskudoksen tulehdus. Upadasitinibia saaneilla potilailla on ilmoitettu bakteerimeningiitti- ja sepsistapauksia. Opportunisti-infektioista upadasitinibihoidon yhteydessä ilmoitettiin tuberkuloosia, useiden ihojaokkeiden vyöruusua, suun/ruokatorven kandidiaasia ja kryptokokkoosia.
Upadasitinibihoitoa ei pidä aloittaa potilaille, joilla on jokin aktiivinen, vakava infektio, mukaan lukien paikalliset infektiot (ks. kohta Vasta-aiheet).
Hoidon riskejä ja hyötyjä on punnittava ennen upadasitinibihoidon aloittamista seuraavissa tapauksissa:
- jos potilaalla on krooninen tai toistuva infektio
- jos potilas on altistunut tuberkuloosille
- jos potilaalla on anamneesissa vakava infektio tai opportunisti-infektio
- jos potilas on asunut tai matkustellut alueilla, joilla esiintyy endeemisesti tuberkuloosia tai sienitauteja; tai
- jos potilaan perussairaus voi altistaa tämän infektiolle.
Potilaita on seurattava tarkkaan infektion oireiden ja löydösten varalta upadasitinibihoidon aikana ja sen jälkeen. Jos potilaalle kehittyy vakava infektio tai opportunisti-infektio, upadasitinibihoito on tauotettava. Jos potilaalle kehittyy uusi infektio upadasitinibihoidon aikana, pikaiset ja täydelliset immuunipuutteisille potilaille sopivat diagnostiset tutkimukset ovat tarpeen; asianmukainen mikrobilääkehoito on aloitettava ja potilasta on seurattava tiiviisti. Jos mikrobilääkehoidolle ei saada vastetta, upadasitinibihoito on tauotettava. Kun infektio on hallinnassa, upadasitinibihoito voidaan aloittaa uudelleen.
30 mg:n upadasitinibiannokseen liittyi enemmän vakavia infektioita kuin 15 mg:n upadasitinibiannokseen.
Infektioiden ilmaantuvuus on iäkkäillä ja diabetespotilailla yleensä tavanomaista suurempi, joten iäkkäiden ja diabetespotilaiden hoidossa on noudatettava varovaisuutta. Upadasitinibia saa käyttää 65 vuotta täyttäneillä potilailla vain, jos sopivia hoitovaihtoehtoja ei ole käytettävissä (ks. kohta Annostus ja antotapa).
Tuberkuloosi
Potilaat on seulottava tuberkuloosin varalta ennen upadasitinibihoidon aloittamista. Upadasitinibia ei saa antaa potilaille, joilla on aktiivinen tuberkuloosi (ks. kohta Vasta-aiheet). Tuberkuloosilääkitystä on harkittava ennen upadasitinibihoidon aloittamista, jos potilaalla on aiemmin hoitamaton, latentti tuberkuloosi tai tuberkuloosi-infektion riskitekijöitä.
Päätettäessä tuberkuloosihoidon aloittamisesta yksittäiselle potilaalle on suositeltavaa konsultoida tuberkuloosin hoitoon perehtynyttä lääkäriä.
Potilaita on seurattava tuberkuloosin oireiden ja löydösten ilmaantumisen varalta. Tämä koskee myös potilaita, joilla latentin tuberkuloosin testitulos oli negatiivinen ennen hoidon aloittamista.
Virusten reaktivaatio
Kliinisissä tutkimuksissa ilmoitettiin virusten reaktivaatiota, mm. herpesvirusten reaktivaatiota (esim. vyöruusu) (ks. kohta Haittavaikutukset). Vyöruusun riski näyttää olevan suurempi japanilaisilla potilailla, joita hoidetaan upadasitinibilla. Jos potilaalle kehittyy vyöruusu, on harkittava upadasitinibihoidon tauottamista, kunnes vyöruusu paranee.
Virushepatiittia on seulottava ja reaktivaatiota on seurattava ennen upadasitinibihoidon aloittamista ja hoidon aikana. Potilaat, joilla hepatiitti C -viruksen vasta-ainetesti ja hepatiitti C -viruksen RNA-testi olivat positiiviset, suljettiin pois kliinisistä tutkimuksista. Potilaat, joilla hepatiitti B -viruksen pinta-antigeeni tai hepatiitti B -viruksen DNA-testi olivat positiiviset, suljettiin pois kliinisistä tutkimuksista. Jos hepatiitti B -viruksen DNA:ta havaitaan upadasitinibihoidon aikana, on konsultoitava maksatauteihin erikoistunutta lääkäriä.
Rokotukset
Upadasitinibihoitoa saavien potilaiden reagoinnista elävillä rokotteilla toteutettuihin rokotuksiin ei ole tietoa. Elävien, heikennettyjen rokotteiden anto upadasitinibihoidon aikana tai juuri ennen sitä ei ole suositeltavaa. On suositeltavaa varmistaa ennen upadasitinibihoidon aloittamista, että potilas on saanut kaikki ajankohtaisten rokotussuositusten mukaiset rokotukset, myös profylaktiset vyöruusurokotukset (ks. kohta Farmakodynamiikka).
Maligniteetit
JAK:n estäjiä, mukaan lukien upadasitinibia, saaneilla potilailla on ilmoitettu lymfoomaa ja muita maligniteetteja.
Laajassa, satunnaistetussa, aktiivikontrolloidussa tutkimuksessa arvioitiin tofasitinibia (toinen JAK:n estäjä) 50 vuotta täyttäneillä nivelreumapotilailla, joilla oli lisäksi vähintään yksi sydän- ja verisuonitautien riskitekijä. Tutkimuksessa havaittiin, että tofasitinibiin liittyi enemmän maligniteetteja, etenkin keuhkosyöpää, lymfoomaa ja ei-melanoottisia ihosyöpiä, kuin tuumorinekroositekijän (TNF) estäjiin.
30 mg:n upadasitinibiannokseen liittyi enemmän maligniteetteja kuin 15 mg:n upadasitinibiannokseen.
65 vuotta täyttäneillä potilailla, tupakoivilla tai aiemmin pitkään tupakoineilla sekä potilailla, joilla on muita maligniteettien riskitekijöitä (esim. nykyinen tai aiempi maligniteetti), upadasitinibia saa käyttää vain, jos sopivia hoitovaihtoehtoja ei ole.
Ei-melanoottinen ihosyöpä (NMSC)
Upadasitinibia saaneilla tutkittavilla on ilmoitettu ei-melanoottista ihosyöpää (ks. kohta Haittavaikutukset). 30 mg:n upadasitinibiannokseen liittyi enemmän ei‑melanoottista ihosyöpää kuin 15 mg:n upadasitinibiannokseen. Säännöllistä ihon tutkimista suositellaan kaikille potilaille ja erityisesti niille, joilla on ihosyövälle altistavia tekijöitä.
Veriarvojen poikkeavuudet
Kliinisissä tutkimuksissa ≤ 1 %:lla potilaista ilmoitettiin absoluuttisen neutrofiiliarvon pienenemistä tasolle < 1 x 109/l, absoluuttisen lymfosyyttiarvon pienenemistä tasolle < 0,5 x 109/l ja hemoglobiinipitoisuuden pienenemistä tasolle < 8 g/dl (ks. kohta Haittavaikutukset). Hoitoa ei saa aloittaa tai hoito on tauotettava, jos rutiiniseurannassa todetaan, että potilaan absoluuttinen neutrofiiliarvo on < 1 x 109/l, absoluuttinen lymfosyyttiarvo < 0,5 x 109/l tai hemoglobiiniarvo < 8 g/dl (ks. kohta Annostus ja antotapa).
Ruoansulatuskanavan perforaatiot
Divertikuliitti- ja ruoansulatuskanavan perforaatiotapahtumia on raportoitu kliinisissä tutkimuksissa ja markkinoille tulon jälkeen (ks. kohta Haittavaikutukset).
Upadasitinibia tulee käyttää varoen potilailla, joilla voi olla ruoansulatuskanavan perforaation riski (esim. potilaat, joilla on divertikuliitti, anamneesissa divertikuliitti tai jotka ottavat ei-steroidisia tulehduskipulääkkeitä (NSAID-lääkkeitä), kortikosteroideja tai opioideja). Aktiivista Crohnin tautia sairastavilla potilailla on suurentunut riski suoliston perforaation kehittymiselle. Potilaat, joille ilmaantuu uusia vatsaoireita, on tutkittava viipymättä divertikuliitin tai ruoansulatuskanavan perforaation tunnistamiseksi varhaisessa vaiheessa.
Merkittävät sydän- ja verisuonitapahtumat
Kliinisissä upadasitinibitutkimuksissa havaittiin merkittäviä sydän- ja verisuonitapahtumia.
Laajassa, satunnaistetussa, aktiivikontrolloidussa tutkimuksessa arvioitiin tofasitinibia (toinen JAK:n estäjä) 50 vuotta täyttäneillä nivelreumapotilailla, joilla oli lisäksi vähintään yksi sydän- ja verisuonitautien riskitekijä. Tutkimuksessa havaittiin, että tofasitinibiin liittyi enemmän merkittäviä sydän- ja verisuonitapahtumia, joiksi määriteltiin sydän- ja verisuoniperäinen kuolema, ei‑fataali sydäninfarkti ja ei‑fataali aivohalvaus, kuin TNF:n estäjiin.
Tämän vuoksi 65 vuotta täyttäneillä potilailla, tupakoivilla tai aiemmin pitkään tupakoineilla sekä potilailla, joilla on anamneesissa ateroskleroottinen sydän- ja verisuonitauti tai muita sydän- ja verisuonitautien riskitekijöitä, upadasitinibia saa käyttää vain, jos sopivia hoitovaihtoehtoja ei ole.
Lipidit
Upadasitinibihoitoon liittyi lipidiarvojen kuten kokonaiskolesterolin, LDL-kolesterolin ja HDL-kolesterolin annosriippuvaista suurenemista (ks. kohta Haittavaikutukset). Statiinihoito pienensi suurentuneet LDL-kolesteroliarvot hoitoa edeltävälle tasolle, vaikkakin tästä on rajallisesti näyttöä. Lipidiarvojen suurenemisen vaikutusta sydän- ja verisuonisairastavuuteen ja -kuolleisuuteen ei ole selvitetty (seurantaohjeet, ks. kohta Annostus ja antotapa).
Maksan transaminaasiarvojen suureneminen
Upadasitinibihoitoon liittyi maksaentsyymiarvojen suurenemista useammin kuin lumehoitoon (ks. kohta Haittavaikutukset).
Maksan transaminaasiarvot on arvioitava lähtötilanteessa ja tämän jälkeen rutiiniseurannan mukaisesti. Maksaentsyymiarvojen suurenemisen syyn pikainen selvittäminen on suositeltavaa, jotta mahdolliset lääkkeestä johtuvat maksavauriotapaukset voidaan tunnistaa.
Jos rutiiniseurannassa todetaan ALAT- tai ASAT-arvojen suurenemista ja epäillään lääkkeen aiheuttamaa maksavauriota, upadasitinibihoito on tauotettava, kunnes tämä diagnoosi on suljettu pois.
Tromboemboliset laskimotapahtumat
Kliinisissä upadasitinibitutkimuksissa havaittiin syviä laskimotromboosi- ja keuhkoemboliatapahtumia.
Laajassa, satunnaistetussa, aktiivikontrolloidussa tutkimuksessa arvioitiin tofasitinibia (toinen JAK:n estäjä) 50 vuotta täyttäneillä nivelreumapotilailla, joilla oli lisäksi vähintään yksi sydän- ja verisuonitautien riskitekijä. Tutkimuksessa havaittiin, että tofasitinibiin liittyi annosriippuvaisesti enemmän tromboembolisia laskimotapahtumia, mukaan lukien syviä laskimotrombooseja ja keuhkoembolioita, kuin TNF:n estäjiin.
Jos potilaalla on sydän- ja verisuonitautien tai maligniteettien riskitekijöitä (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet ”Merkittävät sydän- ja verisuonitapahtumat” ja “Maligniteetit”), upadasitinibia saa käyttää vain, jos sopivia hoitovaihtoehtoja ei ole.
Upadasitinibia tulee käyttää varoen potilailla, joilla on jokin muu tunnettu tromboembolisten laskimotapahtumien riskitekijä kuin sydän- ja verisuonitautien tai maligniteettien riskitekijät. Muita tromboembolisten laskimotapahtumien riskitekijöitä kuin sydän- ja verisuonitautien tai maligniteettien riskitekijät ovat mm. aiempi laskimotromboembolia, potilaalle tehtävä suuri leikkaus, immobilisaatio, yhdistelmäehkäisyvalmisteen tai hormonikorvaushoidon käyttö ja perinnöllinen hyytymishäiriö. Potilaiden tila on arvioitava uudelleen säännöllisesti upadasitinibihoidon aikana tromboembolisten laskimotapahtumien riskin muutosten varalta. Tromboembolisten laskimotapahtumien oireet ja löydökset on arvioitava viipymättä ja hoito on lopetettava annoksesta riippumatta, jos potilaalla epäillään tromboembolista laskimotapahtumaa.
Verkkokalvon laskimotukos
Verkkokalvon laskimotukoksia on raportoitu potilailla, joita on hoidettu JAK:n estäjillä, mukaan lukien upadasitinibilla. Potilaita on neuvottava hakeutumaan viipymättä lääkärin hoitoon, jos heille ilmaantuu verkkokalvon laskimotukokseen viittaavia oireita.
Yliherkkyysreaktiot
Upadasitinibia saaneilla potilailla on ilmoitettu vakavia yliherkkyysreaktioita (kuten anafylaksia ja angioedeemaa). Jos potilaalle ilmaantuu kliinisesti merkittävä yliherkkyysreaktio, upadasitinibihoito on lopetettava ja asianmukainen hoito aloitettava (ks. kohdat Vasta-aiheet ja Haittavaikutukset).
Hypoglykemia diabetekseen hoitoa saavilla potilailla
Diabetekseen hoitoa saavilla potilailla on raportoitu hypoglykemiaa JAK:n estäjien, mukaan lukien upadasitinibin, käytön aloittamisen jälkeen. Diabeteslääkevalmisteen annoksen muuttaminen voi olla tarpeen, jos hypoglykemiaa ilmenee.
Lääkejäämät ulosteessa
Upadasitinibia käyttävillä potilailla on raportoitu lääkejäämiä ulosteessa tai avannepussin sisällössä. Useimmissa raporteissa kuvattiin anatomisia (esim. ileostomia, kolostomia, suolen resektio) tai toiminnallisia ruoansulatuskanavan tiloja, joissa ruoansulatuskanavan läpikulkuajat olivat lyhentyneet. Potilaita on neuvottava ottamaan yhteyttä terveydenhuollon ammattilaiseen, jos lääkejäämiä havaitaan toistuvasti. Potilaita on seurattava kliinisesti, ja vaihtoehtoista hoitoa on harkittava, jos hoitovaste on riittämätön.
Jättisoluarteriitti
Upadasitinibia ei saa käyttää monoterapiana akuuttien pahenemisvaiheiden hoitoon, koska sen tehoa niiden hoidossa ei ole varmistettu. Kortikosteroideja tulee antaa lääkärin harkinnan ja hoitokäytäntöjen mukaisesti.
Yhteisvaikutukset
Muiden lääkevalmisteiden mahdollinen vaikutus upadasitinibin farmakokinetiikkaan
Upadasitinibi metaboloituu lähinnä CYP3A4:n välityksellä. Tästä syystä CYP3A4-entsyymiä voimakkaasti estävät tai indusoivat lääkevalmisteet voivat vaikuttaa upadasitinibipitoisuuteen plasmassa.
Samanaikainen käyttö CYP3A4:n estäjien kanssa
Upadasitinibialtistus suurenee, kun valmistetta käytetään samanaikaisesti voimakkaiden CYP3A4:n estäjien (kuten ketokonatsoli, itrakonatsoli, posakonatsoli, vorikonatsoli, klaritromysiini ja greippi) kanssa. Kliinisessä tutkimuksessa upadasitinibin samanaikainen anto ketokonatsolin kanssa suurensi upadasitinibin Cmax-arvoa 70 % ja AUC-arvoa 75 %. Upadasitinibiannosta 15 mg kerran vuorokaudessa on käytettävä varoen potilailla, jotka saavat pitkäkestoista hoitoa voimakkaalla CYP3A4:n estäjällä. Upadasitinibiannosta 30 mg kerran vuorokaudessa ei suositella atooppista ihottumaa sairastaville potilaille, jotka saavat pitkäkestoista hoitoa voimakkaalla CYP3A4:n estäjällä. Haavaista paksusuolitulehdusta tai Crohnin tautia sairastavilla potilailla, jotka saavat voimakkaita CYP3A4:n estäjiä, induktiohoitoon suositeltu annos on 30 mg kerran vuorokaudessa ja ylläpitohoitoon suositeltu annos on 15 mg kerran vuorokaudessa (ks. kohta Annostus ja antotapa). Pitkäkestoisessa käytössä voimakkaille CYP3A4:n estäjille on harkittava vaihtoehtoja. Upadasitinibihoidon aikana on vältettävä greippiä sisältäviä ruokia ja juomia.
Samanaikainen käyttö CYP3A4:n indusorien kanssa
Upadasitinibialtistus pienenee, kun valmistetta käytetään samanaikaisesti voimakkaiden CYP3A4:n indusorien kanssa (kuten rifampisiini ja fenytoiini). Tämä voi heikentää upadasitinibin hoitovastetta. Kliinisessä tutkimuksessa upadasitinibin samanaikainen anto toistuvien rifampisiiniannosten (voimakas CYP3A:n indusori) kanssa pienensi upadasitinibin Cmax-arvoa noin 50 % ja AUC-arvoa noin 60 %. Tautiaktiivisuuden muutoksia on tarkkailtava, jos upadasitinibia käytetään samanaikaisesti voimakkaiden CYP3A4:n indusorien kanssa.
Metotreksaatti ja pH-arvoon vaikuttavat lääkevalmisteet (esim. antasidit tai protonipumpun estäjät) eivät vaikuta upadasitinibialtistukseen plasmassa.
Upadasitinibin mahdollinen vaikutus muiden lääkevalmisteiden farmakokinetiikkaan
Toistuvien upadasitinibiannosten (30 mg tai 45 mg kerran vuorokaudessa) anto terveille tutkimushenkilöille vaikutti rajallisessa määrin midatsolaamialtistukseen (herkkä CYP3A:n substraatti) plasmassa (midatsolaamin AUC ja Cmax pienenivät 24–26 %), minkä perusteella upadasitinibiannoksella 30 mg tai 45 mg kerran vuorokaudessa voi olla heikko induktiovaikutus CYP3A-entsyymiin. Kliinisessä tutkimuksessa, jossa toistuvia upadasitinibiannoksia (30 mg kerran vuorokaudessa) annettiin terveille tutkimushenkilöille, rosuvastatiinin AUC pieneni 33 %, atorvastatiinin AUC pieneni 23 % ja rosuvastatiinin Cmax pieneni 23 %. Upadasitinibi ei vaikuttanut merkittävästi atorvastatiinin Cmax-arvoon eikä ortohydroksiatorvastatiinialtistukseen (atorvastatiinin pääasiallinen aktiivinen metaboliitti) plasmassa. Toistuvien upadasitinibiannosten (45 mg kerran vuorokaudessa) anto terveille tutkimushenkilöille suurensi rajallisesti dekstrometorfaanin (herkkä CYP2D6:n substraatti) AUC-arvoa 30 % ja Cmax-arvoa 35 %, minkä perusteella upadasitinibiannoksella 45 mg kerran vuorokaudessa on heikko estovaikutus CYP2D6-entsyymiin. Annoksen muuttamista ei suositella käytettäessä CYP3A:n substraatteja, CYP2D6:n substraatteja, rosuvastatiinia tai atorvastatiinia samanaikaisesti upadasitinibin kanssa.
Upadasitinibi ei vaikuta merkittävästi etinyyliestradiolin, levonorgestreelin, metotreksaatin eikä sellaisten lääkevalmisteiden, jotka ovat CYP1A2-, CYP2B6-, CYP2C9- tai CYP2C19-välitteisen metabolian substraatteja, altistukseen plasmassa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 4 viikon ajan viimeisen upadasitinibiannoksen jälkeen. Tytöille ja/tai heidän vanhemmilleen/huoltajilleen on kerrottava, että heidän on otettava yhteys lääkäriin, kun tytön kuukautiset alkavat upadasitinibihoidon aikana.
Raskaus
Upadasitinibin käytöstä raskaana oleville naisille on olemassa vain vähän tai ei lainkaan tietoja. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Upadasitinibi oli teratogeeninen rotalla ja kanilla. In utero altistuksen jälkeen vaikutukset kohdistuivat rotan sikiöillä luustoon ja kanin sikiöillä sydämeen.
Upadasitinibi on vasta-aiheinen raskauden aikana (ks. kohta Vasta-aiheet).
Jos potilas tulee raskaaksi upadasitinibihoidon aikana, vanhemmille on kerrottava sikiöön mahdollisesti kohdistuvasta riskistä.
Imetys
Ei tiedetä, erittyvätkö upadasitinibi tai sen metaboliitit ihmisen rintamaitoon. Olemassa olevat farmakodynaamiset/toksikologiset tiedot koe-eläimistä ovat osoittaneet upadasitinibin erittyvän rintamaitoon (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea.
Upadasitinibia ei pidä käyttää rintaruokinnan aikana. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko upadasitinibihoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Upadasitinibin vaikutusta ihmisen hedelmällisyyteen ei ole arvioitu. Eläinkokeissa ei ole havaittu vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Upadasitinibilla voi olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn, koska heitehuimausta ja kiertohuimausta voi esiintyä RINVOQ-hoidon aikana (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Nivelreumaa, nivelpsoriaasia ja aksiaalista spondylartriittia koskevissa kliinisissä lumekontrolloiduissa tutkimuksissa yleisimmin ilmoitettuja haittavaikutuksia (≥ 2 %:lla potilaista vähintään yhdessä käyttöaiheista ja suurin esiintyvyys esitetyissä käyttöaiheissa) 15 mg:n upadasitinibiannoksella olivat ylähengitystieinfektiot (19,5 %), veren kreatiinikinaasipitoisuuden suureneminen (8,6 %), alaniinitransaminaasiarvojen suureneminen (4,3 %), bronkiitti (3,9 %), pahoinvointi (3,5 %), neutropenia (2,8 %), yskä (2,2 %), aspartaattitransaminaasiarvojen suureneminen (2,2 %) ja hyperkolesterolemia (2,2 %).
Atooppista ihottumaa koskevissa lumekontrolloiduissa kliinisissä tutkimuksissa yleisimmin ilmoitettuja haittavaikutuksia (≥ 2 %:lla potilaista) 15 mg:n tai 30 mg:n upadasitinibiannoksella olivat ylähengitystieinfektio (25,4 %), akne (15,1 %), herpes simplex (8,4 %), päänsärky (6,3 %), veren kreatiinikinaasipitoisuuden suureneminen (5,5 %), yskä (3,2 %), karvatuppitulehdus (3,2 %), vatsakipu (2,9 %), pahoinvointi (2,7 %), neutropenia (2,3 %), kuume (2,1 %) ja influenssa (2,1 %).
Haavaisen paksusuolitulehduksen ja Crohnin taudin induktio- ja ylläpitohoitoa koskevissa lumekontrolloiduissa kliinisissä tutkimuksissa yleisimmin ilmoitettuja haittavaikutuksia (≥ 3 %:lla potilaista) 45 mg:n, 30 mg:n tai 15 mg:n upadasitinibiannoksella olivat ylähengitystieinfektio (19,9 %), kuume (8,7 %), veren kreatiinikinaasipitoisuuden suureneminen (7,6 %), anemia (7,4 %), päänsärky (6,6 %), akne (6,3 %), vyöruusu (6,1 %), neutropenia (6,0 %), ihottuma (5,2 %), keuhkokuume (4,1 %), hyperkolesterolemia (4,0 %), bronkiitti (3,9 %), aspartaattitransaminaasiarvojen suureneminen (3,9 %), uupumus (3,9 %), karvatuppitulehdus (3,6 %), alaniinitransaminaasiarvojen suureneminen (3,5 %), herpes simplex (3,2 %) ja influenssa (3,2 %).
Yleisimpiä vakavia haittavaikutuksia olivat vakavat infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pitkäkestoisen upadasitinibihoidon turvallisuusprofiili vastasi yleisesti ottaen lumekontrolloidun jakson aikaista turvallisuusprofiilia kaikissa käyttöaiheissa.
Haittavaikutustaulukko
Seuraava haittavaikutusluettelo perustuu kliinisten tutkimusten ja kauppaantulon jälkeisiin kokemuksiin. Alla lueteltujen haittavaikutusten esiintymistiheys on määritelty seuraavan luokittelun mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000). Taulukon 3 haittavaikutusten yleisyydet perustuvat suurempiin raportoituihin esiintyvyyksiin RINVOQ-valmisteella tehdyissä reumatauteja koskevissa kliinisissä tutkimuksissa (15 mg), atooppista ihottumaa koskevissa kliinisissä tutkimuksissa (15 mg ja 30 mg), haavaista paksusuolitulehdusta koskevissa kliinisissä tutkimuksissa (15 mg, 30 mg ja 45 mg) tai Crohnin tautia koskevissa kliinisissä tutkimuksissa (15 mg, 30 mg ja 45 mg). Jos yleisyydessä havaittiin huomattavia eroja eri käyttöaiheiden välillä, nämä esitetään taulukon alla olevissa alaviitteissä.
Taulukko 3 Haittavaikutukset
Elinjärjestelmäluokka | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen |
Infektiot | Ylähengitystieinfektiota | Bronkiittia,b Vyöruusua Herpes simplexa Karvatuppitulehdus Influenssa Virtsatietulehdus Keuhkokuumea,h | Suun hiivatulehdus Divertikuliitti Sepsis |
|
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) |
| Ei‑melanoottinen ihosyöpäf |
|
|
Veri ja imukudos |
| Anemiaa Neutropeniaa Lymfopenia |
|
|
Immuunijärjestelmä |
| Nokkosihottumac,g | Vakavat yliherkkyysreaktiota,e |
|
Aineenvaihdunta ja ravitsemus |
| Hyperkolesterolemiaa,b Hyperlipidemiaa,b | Hypertriglyseridemia |
|
Hermosto |
| Päänsärkya,j Heitehuimaus |
|
|
Kuulo ja tasapainoelin |
| Kiertohuimausa |
|
|
Hengityselimet, rintakehä ja välikarsina |
| Yskä |
|
|
Ruoansulatuselimistö |
| Vatsakipua Pahoinvointi | Ruoansulatuskanavan perforaatioi |
|
Iho ja ihonalainen kudos | Aknea,c,d,g | Ihottumaa |
|
|
Sukupuolielimet ja rinnat |
|
|
| Siemennesteen värimuutosl |
Yleisoireet ja antopaikassa todettavat haitat |
| Uupumus Kuume Perifeerinen edeemaa,k |
|
|
Tutkimukset |
| Veren kreatiinikinaasiarvon suureneminen ALAT-arvojen suureneminenb ASAT-arvojen suureneminenb Painonnousug |
|
|
a Esitetty ryhmiteltynä terminä. b Atooppista ihottumaa koskevissa tutkimuksissa bronkiitin, hyperkolesterolemian, hyperlipidemian, ALAT-arvon suurenemisen ja ASAT-arvon suurenemisen yleisyys oli melko harvinainen. c Reumatauteja koskevissa tutkimuksissa aknen yleisyys oli yleinen ja nokkosihottuman melko harvinainen. d Haavaista paksusuolitulehdusta koskevissa tutkimuksissa aknen yleisyys oli yleinen. e Vakavat yliherkkyysreaktiot, mukaan lukien anafylaktinen reaktio ja angioedeema. f Valtaosa ilmoitetuista tapahtumista oli ihon tyvisolusyöpää ja okasolusyöpää. g Crohnin taudissa aknen yleisyys oli yleinen ja nokkosihottuman ja painonnousun yleisyys oli melko harvinainen. h Keuhkokuume oli yleinen Crohnin taudissa ja melko harvinainen muissa käyttöaiheissa. i Yleisyys perustuu Crohnin tautia koskeviin kliinisiin tutkimuksiin. jPäänsärky oli hyvin yleinen haittavaikutus jättisoluarteriittia koskevassa tutkimuksessa. kYleisyys perustuu jättisoluarteriittia koskevaan tutkimukseen. l Siemennesteen värimuutoksia, enimmäkseen siniseksi ja harvemmin vihreäksi, on raportoitu pääasiassa potilailla, jotka käyttävät upadasitinibia haavaiseen paksusuolitulehdukseen tai Crohnin tautiin. Siemennesteen värimuutoksen kliininen merkitys on tuntematon. | ||||
Valikoitujen haittavaikutusten kuvaus
Nivelreuma
Infektiot
Lumekontrolloiduissa kliinisissä tutkimuksissa, joissa taustahoitona käytettiin tautiprosessiin vaikuttavia reumalääkkeitä, infektioiden yleisyys 12/14 viikon aikana oli upadasitinibi 15 mg ryhmässä 27,4 % verrattuna lumeryhmän lukuun 20,9 %. Metotreksaattikontrolloiduissa tutkimuksissa infektioiden yleisyys 12/14 viikon aikana oli upadasitinibi 15 mg -monoterapiaryhmässä 19,5 % verrattuna metotreksaattiryhmän lukuun 24,0 %. Infektioiden pitkän aikavälin kokonaisesiintyvyys upadasitinibi 15 mg -ryhmässä oli kaikkien viiden vaiheen 3 kliinisen tutkimuksen (2 630 potilasta) osalta 93,7 tapahtumaa 100 potilasvuotta kohti.
Lumekontrolloiduissa kliinisissä tutkimuksissa, joissa taustahoitona käytettiin tautiprosessiin vaikuttavia reumalääkkeitä, vakavien infektioiden yleisyys 12/14 viikon aikana oli upadasitinibi 15 mg -ryhmässä 1,2 % verrattuna lumeryhmän lukuun 0,6 %. Metotreksaattikontrolloiduissa tutkimuksissa vakavien infektioiden yleisyys 12/14 viikon aikana oli upadasitinibi 15 mg -monoterapiaryhmässä 0,6 % verrattuna metotreksaattiryhmän lukuun 0,4 %. Vakavien infektioiden pitkän aikavälin kokonaisesiintyvyys upadasitinibi 15 mg -ryhmässä oli kaikkien viiden vaiheen 3 kliinisen tutkimuksen osalta 3,8 tapahtumaa 100 potilasvuotta kohti. Yleisin vakava infektio oli keuhkokuume. Vakavien infektioiden esiintymistiheys pysyi vakaana pitkäaikaisen altistuksen aikana.
Opportunisti-infektiot (paitsi tuberkuloosi)
Lumekontrolloiduissa kliinisissä tutkimuksissa, joissa taustahoitona käytettiin tautiprosessiin vaikuttavia reumalääkkeitä, opportunisti-infektioiden yleisyys 12/14 viikon aikana oli upadasitinibi 15 mg -ryhmässä 0,5 % verrattuna lumeryhmän lukuun 0,3 %. Metotreksaattikontrolloiduissa tutkimuksissa ei esiintynyt yhtään opportunisti-infektiota 12/14 viikon aikana upadasitinibi 15 mg monoterapiaryhmässä, kun taas metotreksaattiryhmän luku oli 0,2 %. Opportunisti-infektioiden pitkän aikavälin kokonaisesiintyvyys upadasitinibi 15 mg -ryhmässä oli kaikkien viiden vaiheen 3 kliinisen tutkimuksen osalta 0,6 tapahtumaa 100 potilasvuotta kohti.
Vyöruusun pitkän aikavälin esiintyvyys upadasitinibi 15 mg -ryhmässä oli kaikkien viiden vaiheen 3 kliinisen tutkimuksen osalta 3,7 tapahtumaa 100 potilasvuotta kohti. Useimmat vyöruusutapaukset affisioivat vain yhtä ihojaoketta ja olivat ei-vakavia.
Maksan transaminaasiarvojen suureneminen
Lumekontrolloiduissa tutkimuksissa, joissa taustahoitona käytettiin tautiprosessiin vaikuttavia reumalääkkeitä, todettiin 12/14 viikon aikana alaniinitransaminaasiarvojen (ALAT) ja aspartaattitransaminaasiarvojen (ASAT) suurenemista vähintään yhdellä mittauskerralla ≥ 3 x viitealueen ylärajan (ULN) suuruisiksi 2,1 %:lla ja 1,5 %:lla upadasitinibi 15 mg -hoitoa saaneista ja 1,5 %:lla ja 0,7 %:lla lumehoitoa saaneista. Todetuissa 22:ssa tapauksessa maksan transaminaasiarvojen suureneminen oli useimmiten oireetonta ja ohimenevää.
Metotreksaattikontrolloiduissa tutkimuksissa todettiin 12/14 viikon aikana ALAT- ja ASAT-arvojen suurenemista ≥ 3 x viitealueen ylärajan suuruisiksi 0,8 %:lla ja 0,4 %:lla upadasitinibi 15 mg -hoitoa saaneista ja 1,9 %:lla ja 0,9 %:lla metotreksaattia saaneista.
ALAT/ASAT-arvojen suurenemisen profiili ja ilmaantuvuus pysyivät vakaana ajan mittaan, myös pitkäkestoisissa jatkotutkimuksissa.
Lipidiarvojen suureneminen
Upadasitinibi 15 mg -hoidon yhteydessä esiintyi lipidiarvojen kuten kokonaiskolesteroli-, triglyseridi-, LDL-kolesteroli- ja HDL-kolesteroliarvojen suurenemista. LDL/HDL-suhde ei muuttunut. Arvojen suurenemista havaittiin 2–4 hoitoviikon kohdalla, ja pitkäaikaishoidon aikana ne pysyivät vakaina. Kontrolloitujen tutkimusten potilailla, joiden lähtöarvot alittivat määritellyt rajat, arvot suurenivat määriteltyjen rajojen yläpuolelle vähintään yhdellä mittauskerralla 12/14 viikon aikana seuraavilla yleisyyksillä (mukaan lukien potilaat, joilla havaittiin yksittäinen arvon suureneminen):
-
Kokonaiskolesteroli ≥ 5,17 mmol/l (200 mg/dl): upadasitinibi 15 mg -ryhmässä 62 % ja lumeryhmässä 31 %
-
LDL-kolesteroli ≥ 3,36 mmol/L (130 mg/dl): upadasitinibi 15 mg -ryhmässä 42 % ja lumeryhmässä 19 %
-
HDL-kolesteroli ≥ 1,03 mmol/l (40 mg/dl): upadasitinibi 15 mg -ryhmässä 89 % ja lumeryhmässä 61 %
-
Triglyseridit ≥ 2,26 mmol/l (200 mg/dl): upadasitinibi 15 mg -ryhmässä 25 % ja lumeryhmässä 15 %
Kreatiinifosfokinaasi
Lumekontrolloiduissa tutkimuksissa, joissa taustahoitona käytettiin tautiprosessiin vaikuttavia reumalääkkeitä, esiintyi kreatiinikinaasiarvojen suurenemista 12/14 viikon aikana. Kreatiinikinaasiarvojen suurenemista > 5 x viitealueen ylärajan suuruisiksi ilmoitettiin upadasitinibi 15 mg -ryhmässä 1,0 %:lla ja lumeryhmässä 0,3 %:lla 12/14 viikon aikana. Arvojen suureneminen > 5 x viitealueen ylärajan suuruiseksi oli useimmiten ohimenevää eikä edellyttänyt hoidon lopettamista. Kreatiinikinaasikeskiarvot suurenivat 4 viikkoon mennessä; 12 viikon kohdalla suurenemisen keskiarvo oli 60 U/l, ja arvot pysyivät sen jälkeen vakaina ja suurentuneina myös pitkäaikaishoidossa.
Neutropenia
Lumekontrolloiduissa tutkimuksissa, joissa taustahoitona käytettiin tautiprosessiin vaikuttavia reumalääkkeitä, esiintyi neutrofiiliarvojen pienenemistä alle tason 1 x 109 solua/l vähintään yhdellä mittauskerralla upadasitinibi 15 mg -ryhmässä 1,1 %:lla ja lumeryhmässä < 0,1 %:lla 12/14 viikon aikana. Kliinisissä tutkimuksissa hoito tauotettiin, jos absoluuttinen neutrofiiliarvo oli < 1 x 109 solua/l (ks. kohta Annostus ja antotapa). Neutrofiilimäärän keskiarvo pieneni 4–8 viikon ajan. Neutrofiiliarvojen pieneneminen pysyi stabiilina ja arvot lähtötasoa pienempinä ajan mittaan, myös pitkäaikaistutkimuksessa.
Nivelpsoriaasi
Turvallisuusprofiili oli upadasitinibi 15 mg -hoitoa saaneilla aktiivista nivelpsoriaasia sairastavilla potilailla yleisesti ottaen vastaavanlainen kuin nivelreumaa sairastavilla. Vakavia infektioita esiintyi enemmän (2,6 tapahtumaa 100 potilasvuotta kohden vs. 1,3 tapahtumaa 100 potilasvuotta kohden) ja maksan transaminaasiarvot kohosivat useammalla (asteen 3 tai suurempi ALAT:n nousu 1,4 %:lla vs. 0,4 %:lla), kun potilaat saivat upadasitinibia yhdessä metotreksaattihoidon kanssa verrattuna monoterapiaa saaneisiin potilaisiin.
Aksiaalinen spondylartriitti
Turvallisuusprofiili oli upadasitinibi 15 mg -hoitoa saavilla aktiivista aksiaalista spondylartriittia sairastavilla potilailla yleisesti ottaen vastaavanlainen kuin nivelreumaa sairastavilla. Uusia turvallisuuslöydöksiä ei havaittu.
Jättisoluarteriitti
Turvallisuusprofiili oli upadasitinibi 15 mg -hoitoa saavilla, jättisoluarteriittia sairastavilla potilailla yleisesti ottaen vastaavanlainen kuin upadasitinibin tunnettu turvallisuusprofiili.
Vakavat infektiot
Lumekontrolloidussa kliinisessä tutkimuksessa vakavien infektioiden yleisyys 52 viikon aikana oli upadasitinibi 15 mg -ryhmässä 5,7 % ja lumelääkeryhmässä 10,7 %. Vakavien infektioiden pitkän aikavälin esiintyvyys oli 5,9 tapahtumaa 100 potilasvuotta kohti upadasitinibi 15 mg -ryhmässä ja 10,5 tapahtumaa 100 potilasvuotta kohti lumelääkeryhmässä.
Opportunisti-infektiot (paitsi tuberkuloosi)
Lumekontrolloidussa kliinisessä tutkimuksessa opportunisti-infektioiden (paitsi tuberkuloosin ja vyöruusun) yleisyys 52 viikon aikana oli upadasitinibi 15 mg -ryhmässä 1,9 % ja lumelääkeryhmässä 0,9 %. Opportunisti-infektioiden (paitsi tuberkuloosin ja vyöruusun) pitkän aikavälin esiintyvyys oli 1,8 tapahtumaa 100 potilasvuotta kohti upadasitinibi 15 mg -ryhmässä ja 1,5 tapahtumaa 100 potilasvuotta kohti lumelääkeryhmässä.
Lumekontrolloidussa kliinisessä tutkimuksessa vyöruusun yleisyys 52 viikon aikana oli upadasitinibi 15 mg -ryhmässä 5,3 % ja lumelääkeryhmässä 2,7 %. Vyöruusun pitkän aikavälin esiintyvyys oli 5,9 tapahtumaa 100 potilasvuotta kohti upadasitinibi 15 mg -ryhmässä ja 3,0 tapahtumaa 100 potilasvuotta kohti lumelääkeryhmässä.
Atooppinen ihottuma
Infektiot
Kliinisten tutkimusten lumekontrolloidussa jaksossa infektioiden yleisyys 16 viikon aikana oli upadasitinibi 15 mg -ryhmässä 39 % ja upadasitinibi 30 mg ryhmässä 43 % verrattuna lumeryhmän lukuun 30 %. Infektioiden pitkän aikavälin esiintyvyys oli upadasitinibi 15 mg -ryhmässä 98,5 tapahtumaa ja upadasitinibi 30 mg ryhmässä 109,6 tapahtumaa 100 potilasvuotta kohti.
Lumekontrolloiduissa kliinisissä tutkimuksissa vakavien infektioiden yleisyys 16 viikon aikana oli upadasitinibi 15 mg -ryhmässä 0,8 % ja upadasitinibi 30 mg ryhmässä 0,4 % verrattuna lumelääkeryhmän lukuun 0,6 %. Vakavien infektioiden pitkän aikavälin esiintyvyys oli upadasitinibi 15 mg -ryhmässä 2,3 tapahtumaa ja upadasitinibi 30 mg ryhmässä 2,8 tapahtumaa 100 potilasvuotta kohti.
Opportunisti-infektiot (paitsi tuberkuloosi)
Kliinisten tutkimusten lumekontrolloidussa jaksossa kaikki raportoidut opportunisti-infektiot (paitsi tuberkuloosi ja vyöruusu) olivat herpeettisiä ekseemoja. Herpeettisen ekseeman yleisyys 16 viikon aikana oli upadasitinibi 15 mg -ryhmässä 0,7 % ja upadasitinibi 30 mg ryhmässä 0,8 % verrattuna lumelääkeryhmän lukuun 0,4 %. Herpeettisen ekseeman pitkän aikavälin esiintyvyys oli upadasitinibi 15 mg -ryhmässä 1,6 tapahtumaa ja upadasitinibi 30 mg ryhmässä 1,8 tapahtumaa 100 potilasvuotta kohti. Upadasitinibi 30 mg ryhmässä ilmoitettiin yksi ruokatorven kandidiaasitapaus.
Vyöruusun pitkän aikavälin esiintyvyys oli upadasitinibi 15 mg -ryhmässä 3,5 tapahtumaa ja upadasitinibi 30 mg ryhmässä 5,2 tapahtumaa 100 potilasvuotta kohti. Useimmat vyöruusutapaukset affisioivat vain yhtä ihojaoketta ja olivat ei-vakavia.
Poikkeavat laboratorioarvot
Upadasitinibihoitoon liittyvät annosriippuvaiset muutokset (ALAT- ja/tai ASAT-arvon suureneminen ≥ 3 x viitealueen ylärajan suuruisiksi, lipidiarvojen muutokset, kreatiinikinaasiarvojen suureneminen > 5 x viitealueen ylärajan suuruisiksi ja neutropenia eli absoluuttinen neutrofiiliarvo < 1 x 109 solua/l) vastasivat havaintoja reumatauteja koskevissa kliinisissä tutkimuksissa.
LDL-kolesteroliarvon vähäistä suurenemista havaittiin 16 viikon kuluttua atooppista ihottumaa koskevissa tutkimuksissa. Viikolla 52 LDL-kolesterolin keskimääräinen suureneminen lähtötilanteesta oli 0,41 mmol/l upadasitinibiannoksella 15 mg ja 0,56 mmol/l upadasitinibiannoksella 30 mg.
Haavainen paksusuolitulehdus
Haavaista paksusuolitulehdusta sairastavien potilaiden turvallisuusprofiili vastasi yleisesti ottaen nivelreumaa sairastavilla potilailla todettua turvallisuusprofiilia.
Vyöruusun esiintyvyys oli suurempi 16 viikon kuin 8 viikon induktiohoidon yhteydessä.
Infektiot
Induktiohoitoa koskeneissa lumekontrolloiduissa tutkimuksissa infektioiden yleisyys 8 viikon aikana oli upadasitinibi 45 mg -ryhmässä 20,7 % verrattuna lumeryhmän lukuun 17,5 %. Ylläpitohoitoa koskeneessa lumekontrolloidussa tutkimuksessa infektioiden yleisyys 52 viikon aikana oli upadasitinibi 15 mg -ryhmässä 40,4 % ja upadasitinibi 30 mg -ryhmässä 44,2 % verrattuna lumeryhmän lukuun 38,8 %. Infektioiden pitkän aikavälin esiintyvyys upadasitinibi 15 mg -ryhmässä oli 64,5 tapahtumaa 100 potilasvuotta kohti ja upadasitinibi 30 mg -ryhmässä 77,8 tapahtumaa 100 potilasvuotta kohti.
Induktiohoitoa koskeneissa lumekontrolloiduissa tutkimuksissa vakavien infektioiden yleisyys 8 viikon aikana oli 1,3 % sekä upadasitinibi 45 mg -ryhmässä että lumeryhmässä. Enempää vakavia infektioita ei todettu 8 viikkoa kestäneen jatkohoidon aikana upadasitinibiannoksella 45 mg. Ylläpitohoitoa koskeneessa lumekontrolloidussa tutkimuksessa vakavien infektioiden yleisyys 52 viikon aikana oli upadasitinibi 15 mg -ryhmässä 3,6 % ja upadasitinibi 30 mg -ryhmässä 3,2 % verrattuna lumeryhmän lukuun 3,3 %. Vakavien infektioiden pitkän aikavälin esiintyvyys upadasitinibi 15 mg -ryhmässä oli 3,0 tapahtumaa 100 potilasvuotta kohti ja upadasitinibi 30 mg ‑ryhmässä 4,6 tapahtumaa 100 potilasvuotta kohti. Yleisimmin ilmoitettu vakava infektio induktio- ja ylläpitovaiheissa oli COVID‑19-keuhkokuume.
Opportunisti-infektiot (paitsi tuberkuloosi)
Induktiohoitoa koskeneissa lumekontrolloiduissa tutkimuksissa opportunisti-infektioiden (paitsi tuberkuloosin ja vyöruusun) yleisyys 8 viikon aikana oli upadasitinibi 45 mg ‑ryhmässä 0,4 % verrattuna lumeryhmän lukuun 0,3 %. Enempää opportunisti-infektioita (paitsi tuberkuloosia ja vyöruusua) ei todettu 8 viikkoa kestäneen jatkohoidon aikana upadasitinibiannoksella 45 mg. Ylläpitohoitoa koskeneessa lumekontrolloidussa tutkimuksessa opportunisti-infektioiden (paitsi tuberkuloosin ja vyöruusun) yleisyys 52 viikon aikana oli upadasitinibi 15 mg -ryhmässä 0,8 % ja upadasitinibi 30 mg -ryhmässä 0,8 % verrattuna lumeryhmän lukuun 0,8 %. Opportunisti-infektioiden (paitsi tuberkuloosin ja vyöruusun) pitkän aikavälin esiintyvyys upadasitinibi 15 mg -ryhmässä oli 0,3 tapahtumaa 100 potilasvuotta kohti ja upadasitinibi 30 mg -ryhmässä 0,6 tapahtumaa 100 potilasvuotta kohti.
Induktiohoitoa koskeneissa lumekontrolloiduissa tutkimuksissa vyöruusun yleisyys 8 viikon aikana oli upadasitinibi 45 mg -ryhmässä 0,6 % verrattuna lumeryhmän lukuun 0 %. Vyöruusun yleisyys 16 viikkoa kestäneen upadasitinibi 45 mg -hoidon aikana oli 3,9 %. Ylläpitohoitoa koskeneessa lumekontrolloidussa tutkimuksessa vyöruusun yleisyys 52 viikon aikana oli upadasitinibi 15 mg ‑ryhmässä 4,8 % ja upadasitinibi 30 mg -ryhmässä 5,6 % verrattuna lumeryhmän lukuun 0 %. Vyöruusun pitkän aikavälin esiintyvyys upadasitinibi 15 mg -ryhmässä oli 4,5 tapahtumaa 100 potilasvuotta kohti ja upadasitinibi 30 mg -ryhmässä 7,2 tapahtumaa 100 potilasvuotta kohti.
Ruoansulatuskanavan perforaatiot
Lumelääkekontrolloidun ylläpitohoitojakson aikana ruoansulatuskanavan perforaatiota raportoitiin yhdellä lumelääkettä saaneella potilaalla (1,5 tapahtumaa 100 potilasvuotta kohti), mutta ei yhdelläkään upadasitinibia 15 mg tai 30 mg saaneella potilaalla. Pitkäkestoisessa jatkotutkimuksessa yhdellä 15 mg upadasitinibia saaneella potilaalla (0,1 tapahtumaa 100 potilasvuotta kohti), ja yhdellä 30 mg upadasitinibia saaneella potilaalla (< 0,1 tapahtumaa 100 potilasvuotta kohti) raportoitiin tapahtumia.
Poikkeavat laboratorioarvot
Induktiohoitoa ja ylläpitohoitoa koskeneissa kliinisissä tutkimuksissa todetut upadasitinibihoitoon liittyneet laboratorioarvojen poikkeavuudet, eli ALAT-arvon ja/tai ASAT-arvon suureneminen (≥ 3 x viitealueen yläraja), kreatiinikinaasiarvon suureneminen (> 5 x viitealueen yläraja) ja neutropenia (absoluuttinen neutrofiiliarvo < 1 x 109 solua/l), olivat yleisesti ottaen samankaltaisia kuin reumasairauksia ja atooppista ihottumaa koskeneissa kliinisissä tutkimuksissa. Näissä laboratorioarvoissa todettiin annoksesta riippuvia muutoksia, jotka liittyivät 15 mg:n ja 30 mg:n upadasitinibiannoksiin.
Induktiohoitoa koskeneissa, enintään 8 viikon pituisissa lumekontrolloiduissa tutkimuksissa lymfosyyttiarvon laskua alle 0,5 x 109 solua/l vähintään yhdellä mittauskerralla esiintyi 2,0 %:lla upadasitinibi 45 mg -ryhmän potilaista ja 0,8 %:lla lumeryhmän potilaista. Ylläpitohoitoa koskeneessa, enintään 52 viikon pituisessa lumekontrolloidussa tutkimuksessa lymfosyyttiarvon laskua alle 0,5 x 109 solua/l vähintään yhdellä mittauskerralla esiintyi 1,6 %:lla upadasitinibi 15 mg -ryhmän potilaista, 1,2 %:lla upadasitinibi 30 mg -ryhmän potilaista ja 0,8 %:lla lumeryhmän potilaista. Kliinisissä tutkimuksissa hoito tauotettiin, jos potilaan absoluuttinen lymfosyyttiarvo laski alle < 0,5 x 109 solua/l (ks. kohta Annostus ja antotapa). Keskimääräisissä lymfosyyttiarvoissa ei todettu upadasitinibihoidon aikana huomattavia muutoksia ajan mittaan.
Upadasitinibi 45 mg -hoidon yhteydessä todettiin lipidiarvojen nousua 8 hoitoviikon kohdalla, ja arvot pysyivät yleensä vakaina pidempikestoisessa upadasitinibihoidossa annoksilla 15 mg ja 30 mg. Induktiohoitoa koskeneiden lumekontrolloitujen tutkimusten potilailla, joiden lähtöarvot alittivat määritellyt rajat, arvot suurenivat määriteltyjen rajojen yläpuolelle vähintään yhdellä mittauskerralla 8 viikon aikana seuraavilla yleisyyksillä (mukaan lukien potilaat, joilla havaittiin yksittäinen arvon suureneminen):
-
Kokonaiskolesteroli ≥ 5,17 mmol/l (200 mg/dl): upadasitinibi 45 mg -ryhmässä 49 % ja lumeryhmässä 11 %
-
LDL-kolesteroli ≥ 3,36 mmol/l (130 mg/dl): upadasitinibi 45 mg -ryhmässä 27 % ja lumeryhmässä 9 %
-
HDL-kolesteroli ≥ 1,03 mmol/l (40 mg/dl): upadasitinibi 45 mg -ryhmässä 79 % ja lumeryhmässä 36 %
-
Triglyseridit ≥ 2,26 mmol/l (200 mg/dl): upadasitinibi 45 mg -ryhmässä 6 % ja lumeryhmässä 4 %
Crohnin tauti
Turvallisuusprofiili oli upadasitinibihoitoa saavilla Crohnin tautia sairastavilla potilailla yleisesti ottaen vastaavanlainen kuin upadasitinibin tunnettu turvallisuusprofiili.
Vakavat infektiot
Induktiohoitoa koskeneissa lumekontrolloiduissa tutkimuksissa vakavien infektioiden yleisyys 12 viikon aikana oli upadasitinibi 45 mg -ryhmässä 1,9 % ja lumeryhmässä 1,7 %. Ylläpitohoitoa koskeneessa lumekontrolloidussa tutkimuksessa vakavien infektioiden yleisyys 52 viikon aikana oli upadasitinibi 15 mg -ryhmässä 3,2 % ja upadasitinibi 30 mg -ryhmässä 5,7 % verrattuna lumeryhmän lukuun 4,5 %. Vakavien infektioiden pitkän aikavälin esiintyvyys upadasitinibi 15 mg -ryhmässä oli 5,1 tapahtumaa 100 potilasvuotta kohti ja upadasitinibi 30 mg ‑ryhmässä 7,3 tapahtumaa 100 potilasvuotta kohti potilailla, jotka saavuttivat hoitovasteen upadasitinibi 45 mg -induktiohoidolla. Yleisimmin ilmoitettu vakava infektio induktio- ja ylläpitovaiheissa oli maha-suolikanavan infektio.
Ruoansulatuskanavan perforaatiot
Induktiohoitoa koskeneissa lumekontrolloiduissa vaiheen kolme tutkimuksissa ruoansulatuskanavan perforaatiota raportoitiin yhdellä potilaalla (0,1 %), joka sai 45 mg upadasitinibia, eikä yhdelläkään lumelääkettä saaneella potilaalla 12 viikon aikana. Induktiohoitoa koskeneissa tutkimuksissa kaikista 45 mg upadasitinibia saaneista potilaista (n = 938) ruoansulatuskanavan perforaatioita raportoitiin neljällä potilaalla (0,4 %).
Pitkäkestoisen lumekontrolloidun jakson aikana ruoansulatuskanavan perforaatiota raportoitiin yhdellä lumelääkettä saaneella (0,7 sataa potilasvuotta kohti), yhdellä 15 mg upadasitinibia saaneella (0,4 sataa potilasvuotta kohti) ja yhdellä 30 mg upadasitinibia saaneella potilaalla (0,4 sataa potilasvuotta kohti). Kaikista pelastushoitona 30 mg upadasitinibia saaneista potilaista (n = 336) ruoansulatuskanavan perforaatiota raportoitiin kolmella potilaalla (0,8 sataa potilasvuotta kohti) pitkäkestoisen hoidon aikana.
Poikkeavat laboratorioarvot
Induktiohoitoa ja ylläpitohoitoa koskeneissa kliinisissä tutkimuksissa todetut upadasitinibihoitoon liittyneet laboratorioarvojen poikkeavuudet, eli ALAT-arvon ja/tai ASAT-arvon suureneminen (≥ 3 x viitealueen yläraja), kreatiinikinaasiarvon suureneminen (> 5 x viitealueen yläraja), neutropenia (absoluuttinen neutrofiiliarvo < 1 x 109 solua/l) ja lipidiarvot, olivat yleisesti ottaen samankaltaisia kuin reumasairauksia, atooppista ihottumaa ja haavaista paksusuolitulehdusta koskeneissa kliinisissä tutkimuksissa. Näissä laboratorioarvoissa todettiin annoksesta riippuvia muutoksia, jotka liittyivät 15 mg:n ja 30 mg:n upadasitinibiannoksiin.
Induktiohoitoa koskeneissa, enintään 12 viikon pituisissa lumekontrolloiduissa tutkimuksissa lymfosyyttiarvon laskua alle 0,5 x 109 solua/l vähintään yhdellä mittauskerralla esiintyi 2,2 %:lla upadasitinibi 45 mg -ryhmän potilaista ja 2,0 %:lla lumeryhmän potilaista. Ylläpitohoitoa koskeneessa, enintään 52 viikon pituisessa lumekontrolloidussa tutkimuksessa lymfosyyttiarvon laskua alle 0,5 x 109 solua/l vähintään yhdellä mittauskerralla esiintyi 4,6 %:lla upadasitinibi 15 mg ‑ryhmän potilaista, 5,2 %:lla upadasitinibi 30 mg -ryhmän potilaista ja 1,8 %:lla lumeryhmän potilaista. Kliinisissä tutkimuksissa hoito tauotettiin, jos potilaan absoluuttinen lymfosyyttiarvo laski alle 0,5 x 109 solua/l (ks. kohta Annostus ja antotapa). Keskimääräisissä lymfosyyttiarvoissa ei todettu upadasitinibihoidon aikana huomattavia muutoksia ajan mittaan.
Induktiohoitoa koskeneissa, enintään 12 viikon pituisissa lumekontrolloiduissa tutkimuksissa hemoglobiinipitoisuuden laskua alle 8 g/dl vähintään yhdellä mittauskerralla esiintyi 2,7 %:lla upadasitinibi 45 mg -ryhmän potilaista ja 1,4 %:lla lumeryhmän potilaista. Ylläpitohoitoa koskeneessa, enintään 52 viikon pituisessa lumekontrolloidussa tutkimuksessa hemoglobiinipitoisuuden laskua alle 8 g/dl vähintään yhdellä mittauskerralla esiintyi 1,4 %:lla upadasitinibi 15 mg -ryhmän potilaista, 4,4 %:lla upadasitinibi 30 mg -ryhmän potilaista ja 2,8 %:lla lumeryhmän potilaista. Kliinisissä tutkimuksissa hoito tauotettiin, jos potilaan hemoglobiiniarvo laski alle 8 g/dl (ks. kohta Annostus ja antotapa). Keskimääräisissä hemoglobiinipitoisuuksissa ei todettu upadasitinibihoidon aikana huomattavia muutoksia ajan mittaan.
Iäkkäät
Atooppista ihottumaa, haavaista paksusuolitulehdusta tai Crohnin tautia sairastavista, 65 vuotta täyttäneistä potilaista saatujen vähäisten tietojen perusteella haittavaikutusten kokonaisesiintyvyys oli suurempi 30 mg:n upadasitinibiannoksella kuin 15 mg:n annoksella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Yhteensä 541 atooppista ihottumaa sairastavaa 12–17-vuotiasta nuorta sai hoitoa maailmanlaajuisissa vaiheen 3 tutkimuksissa (n = 343) ja täydentävissä nuorilla tehdyissä alatutkimuksissa (n = 198), joissa 264 nuorta altistettiin 15 mg:n annokselle ja 265 nuorta altistettiin 30 mg:n annokselle. Upadasitinibin 15 mg:n ja 30 mg:n annosten turvallisuusprofiili nuorilla oli vastaavanlainen kuin aikuisilla. Pitkäaikaisen altistuksen yhteydessä ilmoitettiin haittavaikutuksena papilloomaviruksen aiheuttamia ihomuutoksia 3,4 %:lla 15 mg:n annosta saaneista ja 6,8 %:lla 30 mg:n annosta saaneista atooppista ihottumaa sairastavista nuorista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 Fimea
Yliannostus
Upadasitinibia annettiin kliinisissä tutkimuksissa enintään annoksina, jotka vastasivat päivittäisen AUC-arvon suhteen depotmuotoista kerran vuorokaudessa annettavaa 60 mg hoitoa. Haittavaikutukset vastasivat pienemmillä annoksilla todettuja haittatapahtumia, eikä spesifistä toksisuutta todettu. Noin 90 % systeemisen verenkierron upadasitinibista eliminoituu 24 tunnin kuluessa lääkkeenannosta (kliinisissä tutkimuksissa arvioitujen annosrajojen puitteissa). Yliannostustapauksessa on suositeltavaa seurata potilaan vointia haittavaikutusten oireiden ja löydösten varalta. Jos haittavaikutuksia kehittyy, potilasta hoidetaan asianmukaisesti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, januskinaasin (JAK) estäjät, ATC-koodi: L04AF03
Vaikutusmekanismi
Upadasitinibi on selektiivinen ja reversiibeli Janus-kinaasin (JAK) estäjä. JAK:t ovat solunsisäisiä entsyymejä, joiden välittämät sytokiini- tai kasvutekijäsignaalit ovat mukana monenlaisissa solun prosesseissa kuten tulehdusvasteissa, hematopoieesissa ja immuunivalvonnassa. JAK-entsyymiperheessä on neljä jäsentä, JAK1, JAK2, JAK3 ja TYK2, jotka toimivat pareittain ja fosforyloivat ja aktivoivat signaaleja välittäviä ja transkriptiota sääteleviä tekijöitä (STAT). Fosforylaatio puolestaan säätelee geenien ilmentymistä ja solun toimintoja. JAK1 on tärkeä inflammatorisessa sytokiinisignaloinnissa, kun taas JAK2 on tärkeä punasolujen kypsymiselle ja JAK3-signaaleilla on tehtäviä immuunivalvonnassa ja lymfosyyttien toiminnassa.
Ihmissoluilla tehdyissä määrityksissä upadasitinibi estää ensisijaisesti JAK1- tai JAK1/3-signalointia, ja se on funktionaalisesti selektiivinen näitä reseptoreita kohtaan JAK2-parien kautta signaloivien sytokiinireseptorien sijaan. Atooppisen ihottuman taustalla vaikuttavat proinflammatoriset sytokiinit (mukaan lukien IL-4, IL-13, IL-22, TSLP, IL-31 ja IFN-γ), jotka välittävät signaaleja JAK1-reitin kautta. JAK1‑reitin esto upadasitinibin avulla vähentää monien sellaisten välittäjien signalointia, jotka aiheuttavat atooppisen ihottuman oireita ja löydöksiä, kuten eksemaattisia ihomuutoksia ja kutinaa. Tulehduksellisen suolistosairauden patologiaan vaikuttavat proinflammatoriset sytokiinit (pääasiassa IL-6, IL-7, IL-15 ja IFN-γ), jotka välittävät signaaleja JAK1-reitin kautta. JAK1-reitin esto upadasitinibin avulla säätelee sellaisten JAK-riippuvaisten sytokiinien signaalinvälitystä, jotka aiheuttavat tulehdustaakkaa sekä tulehduksellisen suolistosairauden oireita ja löydöksiä.
Farmakodynaamiset vaikutukset
IL-6:n indusoiman STAT3:n fosforylaation ja IL-7:n indusoiman STAT5:n fosforylaation esto
Terveillä koehenkilöillä (välittömästi vapautuvan) upadasitinibin anto johti annos- ja pitoisuusriippuvaiseen IL-6:n (JAK1/JAK2) indusoiman STAT3:n fosforylaation estoon ja IL-7:n (JAK1/JAK3) indusoiman STAT5:n fosforylaation estoon kokoveressä. Maksimaalinen estovaikutus havaittiin 1 tunnin kuluttua annostelun jälkeen, ja annosteluvälin loppuun mennessä estovaikutus oli palautunut lähelle lähtötilannetta.
Lymfosyytit
Nivelreumaa sairastavilla upadasitinibihoitoon liittyi absoluuttisen lymfosyyttimäärän keskiarvon pieni, ohimenevä suureneminen lähtötilanteesta viikolle 36. Hoidon jatkuessa arvo palasi vähitellen lähtötasolle tai lähelle sitä.
Erittäin herkkä CRP (hs-CRP)
Nivelreumaa sairastavilla upadasitinibihoitoon liittyi hs-CRP-keskiarvon pienenemistä lähtötilanteesta jo viikolla 1, ja vaikutus säilyi hoidon jatkuessa.
Rokotetutkimukset
Upadasitinibin vaikutusta humoraaliseen vasteeseen adjuvantoidun rekombinantti glykoproteiini E vyöruusurokotteen annon jälkeen arvioitiin 93:lla nivelreumapotilaalla, jotka saivat vakaa-annoksista upadasitinibihoitoa 15 mg:n annoksella. Potilaista 98 % sai samanaikaisesti metotreksaattia. Potilaista 49 % sai kortikosteroideja suun kautta lähtötilanteessa. Ensisijainen päätetapahtuma oli niiden potilaiden osuus, joiden humoraalinen vaste oli tyydyttävä. Tyydyttävän vasteen määritelmä oli ≥ 4-kertainen suurenema rokotusta edeltäneessä pitoisuudessa anti-glykoproteiini E -titterissä viikolla 16 (4 viikkoa toisen rokoteannoksen jälkeen). Rokottamisella saavutettiin tyydyttävä humoraalinen vaste 79:llä 90:stä (88 % [95 %:n lv: 81,0; 94,5]) 15 mg:n upadasitinibiannoksella hoidetusta potilaasta viikolla 16.
Upadasitinibin vaikutusta humoraaliseen vasteeseen (13-valenttisen, adsorboidun) inaktivoidun pneumokokkipolysakkaridikonjugaattirokotteen annon jälkeen arvioitiin 111:llä nivelreumapotilaalla, jotka saivat vakaa-annoksista upadasitinibihoitoa 15 mg:n (n = 87) tai 30 mg:n (n = 24) annoksella. Potilaista 97 % (n = 108) sai samanaikaisesti metotreksaattihoitoa. Ensisijainen päätetapahtuma oli niiden potilaiden osuus, joiden humoraalinen vaste oli tyydyttävä. Tyydyttävän vasteen määritelmä oli ≥ 2-kertainen vasta-ainepitoisuuksien suurenema lähtötilanteesta viikolle 4 vähintään 6:ssa 12 pneumokokkiantigeenista (1, 3, 4, 5, 6B, 7F, 9V, 14, 18C, 19A, 19F ja 23F). Tulokset viikon 4 kohdalta osoittivat, että 15 mg:n upadasitinibiannoksella hoidetuista potilaista tyydyttävä humoraalinen vaste saavutettiin 67,5 %:lla (95 %:n lv: 57,4; 77,5) ja 30 mg:n annoksella hoidetuista potilaista 56,5 %:lla (95 %:n lv: 36,3; 76,8).
Kliininen teho ja turvallisuus
Nivelreuma
Kerran vuorokaudessa otettavan upadasitinibi 15 mg -hoidon tehoa ja turvallisuutta arvioitiin viidessä vaiheen 3 satunnaistetussa, kaksoissokkoutetussa monikeskustutkimuksessa, joiden potilailla oli vuoden 2010 ACR/EULAR-kriteerit täyttävä keskivaikea tai vaikea, aktiivinen nivelreuma (ks. taulukko 4). Tutkimukseen voitiin ottaa 18 vuotta täyttäneitä potilaita. Heillä tuli olla lähtötilanteessa vähintään 6 aristavaa ja 6 turvonnutta niveltä sekä näyttöä systeemisestä inflammaatiosta suurentuneen hs-CRP-arvon perusteella. Neljään tutkimukseen kuului enintään 5 vuoden pituinen pitkäkestoinen jatkovaihe ja yhteen tutkimukseen (SELECT‑COMPARE) enintään 10 vuoden pituinen pitkäkestoinen jatkovaihe.
Kunkin tutkimuksen ensisijaiseen analyysiin otettiin kaikki satunnaistetut tutkittavat, jotka saivat vähintään yhden upadasitinibi- tai lumelääkeannoksen. Kategoristen päätetapahtumien kohdalla käytettiin NRI-imputointia (non-responder imputation).
Vaiheen 3 tutkimuksissa upadasitinibi 15 mg x 1 -hoidon teho oli yleensä ottaen samanlainen kuin upadasitinibi 30 mg x 1 -hoidon teho.
Taulukko 4 Kliinisten tutkimusten yhteenveto
Tutkimuksen nimi | Tutkimuspopulaatio (n) | Hoitoryhmät | Keskeiset tulosmuuttujat |
SELECT-EARLY | Ei aiempaa MTX-hoitoaa (947) |
Monoterapia |
|
SELECT-MONOTHERAPY | MTX-IRb (648) |
Monoterapia |
|
SELECT-NEXT | csDMARD-IRc (661) |
Taustahoitona csDMARD |
|
SELECT-COMPARE | MTX-IRd (1 629) |
Taustahoitona MTX |
|
SELECT-BEYOND | bDMARD-IRe (499) |
Taustahoitona csDMARD |
|
Lyhenteet: ACR20 (tai 50) = ≥ 20 % (tai ≥ 50 %) koheneminen American College of Rheumatology kriteereillä, bDMARD = tautiprosessiin vaikuttava biologinen reumalääke, CRP = C-reaktiivinen proteiini, DAS28 = Disease Activity Score pisteet (28 niveltä), mTSS = muokatut Sharpin kokonaispisteet, csDMARD = tavanomainen synteettinen tautiprosessiin vaikuttava reumalääke, HAQ-DI = Health Assessment Questionnaire Disability Index mittari, SF-36 PCS = Short Form (36) Health Survey (SF-36), fyysisen osion yhteispisteet, CDAI = Clinical Disease Activity Index, FACIT-F = Functional Assessment of Chronic Illness Therapy-Fatigue -pisteet, IR = riittämättömän vasteen saaneet, MTX = metotreksaatti, n = satunnaistettujen potilaiden määrä a. Ei aiempaa metotreksaattihoitoa tai korkeintaan 3 viikoittaista metotreksaattiannosta b Riittämätön vaste metotreksaattihoidolle c Riittämätön vaste csDMARD-hoidolle; potilaat, jotka olivat aiemmin altistuneet enintään yhdelle bDMARD-hoidolle, soveltuivat tutkimukseen (enintään 20 % potilaiden kokonaismäärästä) jos altistus oli rajallinen (< 3 kk) tai bDMARD-hoito oli jouduttu lopettamaan huonon siedettävyyden vuoksi d Riittämätön vaste metotreksaatille; potilaat, jotka olivat aiemmin altistuneet enintään yhdelle bDMARD-hoidolle (lukuun ottamatta adalimumabia) soveltuivat tutkimukseen (enintään 20 % tutkimuspotilaiden kokonaismäärästä) jos altistus oli rajallinen (< 3 kk) tai bDMARD-hoito oli jouduttu lopettamaan huonon siedettävyyden vuoksi e Riittämätön vaste vähintään yhdelle bDMARD-valmisteelle tai vähintään yhden bDMARD-valmisteen puutteellinen siedettävyys | |||
Kliininen vaste
Remissio ja vähäinen tautiaktiivisuus
Tutkimuksissa vähäisen tautiaktiivisuuden (DAS28-CRP ≤ 3,2) ja kliinisen remission (DAS28-CRP < 2,6) saavutti merkitsevästi suurempi osa upadasitinibi 15 mg -hoitoa saaneista potilaista kuin lumetta, metotreksaattia tai adalimumabia saaneista (taulukko 5). SELECT-COMPARE-tutkimuksessa viikon 12 kohdalla saavutettiin merkitsevästi suuremmat vähäisen tautiaktiivisuuden osuudet adalimumabiin verrattuna. Kaikkiaan sekä vähäisen tautiaktiivisuuden että kliinisen remission osuudet olivat johdonmukaiset eri potilaspopulaatioissa riippumatta metotreksaattihoidon käytöstä. Kolmen vuoden kohdalla SELECT‑COMPARE‑tutkimuksessa 297/651 potilasta (45,6 %) jatkoi heille alun perin satunnaistettua upadasitinibi 15 mg -hoitoa ja 111/327 potilasta (33,9 %) jatkoi heille alun perin satunnaistettua adalimumabihoitoa ja SELECT-EARLY‑tutkimuksessa 216/317 potilasta (68,1 %) jatkoi heille alun perin satunnaistettua upadasitinibi 15 mg -hoitoa ja 149/315 potilasta (47,3 %) jatkoi heille alun perin satunnaistettua metotreksaattimonoterapiaa. Vähäinen tautiaktiivisuus ja kliininen remissio säilyivät 3 vuoteen asti potilailla, jotka jatkoivat heille alun perin satunnaistettua hoitoa.
ACR-vaste
Kaikissa tutkimuksissa ACR20-, ACR50- tai ACR70-vasteen 12 viikon kohdalla saavuttaneiden osuus oli upadasitinibi 15 mg -ryhmässä suurempi kuin lumelääkettä, metotreksaattia tai adalimumabia saaneilla (taulukko 5). Tehon alkamiseen kulunut aika oli lyhyt kaikilla mittareilla mitattuna, ja jo viikolla 1 saavutettiin paremmat ACR20-vasteet. Alkuperäistä satunnaistettua hoitoaan jatkaneilla potilailla todettiin pitkäkestoiset vasteprosentit (riippumatta metotreksaatin käytöstä) ja ACR20/50/70-vasteet säilyivät 3 vuoteen asti.
Upadasitinibi 15 mg -hoito kohensi ACR-vasteen yksittäisten osa-alueiden tuloksia (mm. aristavien ja turvonneiden nivelten määrät, potilaan ja lääkärin yleisarviot, HAQ-DI, kivun arviointi ja hs-CRP) sekä ainoana lääkkeenä että yhdessä tavanomaisten synteettisten reumalääkkeiden kanssa käytettynä.
Taulukko 5 Vaste ja remissio
Tutkimus | SELECT- EARLY Ei aiempaa MTX-hoitoa | SELECT- MONO MTX-IR | SELECT- NEXT csDMARD-IR | SELECT- COMPARE MTX-IR | SELECT- BEYOND bDMARD-IR | ||||||||
| MTX | UPA 15 mg | MTX | UPA 15 mg | Lume | UPA 15 mg | Lume | UPA 15 mg | ADA 40mg | Lume | UPA 15 mg | ||
N | 314 | 317 | 216 | 217 | 221 | 221 | 651 | 651 | 327 | 169 | 164 | ||
Viikko |
| ||||||||||||
Vähäinen tautiaktiivisuus, DAS28-CRP ≤ 3,2 (% potilaista) | |||||||||||||
12a/14b | 28 | 53g | 19 | 45e | 17 | 48e | 14 | 45e,h | 29 | 14 | 43e | ||
24c/26d | 32 | 60f |
|
|
|
| 18 | 55g,h | 39 |
|
| ||
48 | 39 | 59g |
|
|
|
|
| 50h | 35 |
|
| ||
CR DAS28-CRP < 2,6 (% potilaista) | |||||||||||||
12a/14b | 14 | 36g | 8 | 28e | 10 | 31e | 6 | 29e,h | 18 | 9 | 29g | ||
24c/26d | 18 | 48e |
|
|
|
| 9 | 41g,h | 27 |
|
| ||
48 | 29 | 49g |
|
|
|
|
| 38i | 28 |
|
| ||
ACR20 (% potilaista) | |||||||||||||
12a/14b | 54 | 76g | 41 | 68e | 36 | 64e | 36 | 71e,j | 63 | 28 | 65e | ||
24c/26d | 59 | 79g |
|
|
|
| 36 | 67g,i | 57 |
|
| ||
48 | 57 | 74g |
|
|
|
|
| 65i | 54 |
|
| ||
ACR50 (% potilaista) | |||||||||||||
12a/14b | 28 | 52g | 15 | 42g | 15 | 38g | 15 | 45g,h | 29 | 12 | 34g | ||
24c/26d | 33 | 60e |
|
|
|
| 21 | 54g,h | 42 |
|
| ||
48 | 43 | 63g |
|
|
|
|
| 49i | 40 |
|
| ||
ACR70 (% potilaista) | |||||||||||||
12a/14b | 14 | 32g | 3 | 23g | 6 | 21g | 5 | 25g,h | 13 | 7 | 12 | ||
24c/26d | 18 | 44g |
|
|
|
| 10 | 35g,h | 23 |
|
| ||
48 | 29 | 51g |
|
|
|
|
| 36h | 23 |
|
| ||
CDAI ≤ 10 (% potilaista) | |||||||||||||
12a/14b | 30 | 46g | 25 | 35l | 19 | 40e | 16 | 40e,h | 30 | 14 | 32g | ||
24c/26d | 38 | 56g |
|
|
|
| 22 | 53g,h | 38 |
|
| ||
48 | 43 | 60g |
|
|
|
|
| 47h | 34 |
|
| ||
Lyhenteet: ACR20 (tai 50 tai 70) = ≥ 20 % (tai ≥ 50 % tai ≥ 70 %) koheneminen American College of Rheumatology -kriteereillä; ADA = adalimumabi; CDAI = Clinical Disease Activity Index; CR = kliininen remissio; CRP = C-reaktiivinen proteiini, DAS28 = Disease Activity Score tautiaktiivisuuspisteet 28 nivelen perusteella arvioituna; IR = riittämättömän vasteen saaneet; MTX = metotreksaatti; UPA= upadasitinibi a SELECT-NEXT, SELECT-EARLY, SELECT-COMPARE, SELECT-BEYOND b SELECT-MONOTHERAPY c SELECT-EARLY d SELECT-COMPARE e upadasitinibin multiplisiteetin suhteen kontrolloitu p-arvo ≤ 0,001 vs. lume tai metotreksaatti f upadasitinibin multiplisiteetin suhteen kontrolloitu p-arvo ≤ 0,01 vs. lume tai metotreksaatti g upadasitinibin nimellinen p-arvo ≤ 0,001 vs. lume tai metotreksaatti h upadasitinibin nimellinen p-arvo ≤ 0,001 vs. adalimumabi i upadasitinibin nimellinen p-arvo ≤ 0,01 vs. adalimumabi j upadasitinibin nimellinen p-arvo < 0,05 vs. adalimumabi k upadasitinibin nimellinen p-arvo ≤ 0,01 vs. lume tai metotreksaatti l upadasitinibin nimellinen p-arvo < 0,05 vs. metotreksaatti Huom. Viikon 48 tiedot ovat peräisin koko analyysipopulaation (FAS) analyysista satunnaistettujen ryhmien mukaisesti, käyttäen vasteettomuusimputointia | |||||||||||||
Radiografinen vaste
Rakenteellisten nivelvaurioiden etenemisen estymistä arvioitiin muokatuilla Sharpin kokonaispisteillä (mTSS) ja mTSS-pisteiden osatekijöillä eli eroosiopisteiden ja nivelraon kaventumispisteiden avulla viikkojen 24/26 kohdalla ja viikon 48 kohdalla SELECT-EARLY- ja SELECT-COMPARE-tutkimuksissa.
Upadasitinibi 15 mg -hoito esti rakenteellisten nivelvaurioiden etenemistä merkitsevästi enemmän kuin lumelääke yhdistelmänä metotreksaatin kanssa SELECT-COMPARE-tutkimuksessa ja ainoana lääkkeenä metotreksaattiin verrattuna SELECT-EARLY-tutkimuksessa (taulukko 6). Eroosiopisteiden ja nivelraon kaventumispisteiden analyysitulokset vastasivat kokonaispistemäärien tuloksia. Niiden potilaiden osuus, joilla ei todettu lainkaan radiografista taudin etenemistä (mTSS-pisteiden muutos ≤ 0), oli kummassakin tutkimuksessa merkitsevästi suurempi upadasitinibi 15 mg -ryhmässä. Rakenteellisten nivelvaurioiden etenemisen estyminen säilyi molemmissa tutkimuksissa viikolle 96 asti potilailla, jotka jatkoivat heille alun perin satunnaistettua upadasitinibi 15 mg -hoitoa (perustuu saatavilla oleviin tuloksiin 327 potilaalta SELECT-COMPARE‑tutkimuksesta ja 238 potilaalta SELECT-EARLY‑tutkimuksesta).
Taulukko 6 Radiografiset muutokset
Tutkimus | SELECT- EARLY Ei aiempaa MTX-hoitoa | SELECT- COMPARE MTX-IR | ||||
Hoitoryhmä | MTX | UPA 15 mg | Lumea | UPA 15 mg | ADA 40 mg | |
mTSS-pisteet, keskimuutos lähtötilanteesta | ||||||
Viikko 24b/26c | 0,7 | 0,1f | 0,9 | 0,2g | 0,1 | |
Viikko 48 | 1,0 | 0,03e | 1,7 | 0,3e | 0,4 | |
Potilaat, joilla ei radiografista taudin etenemistäd | ||||||
Viikko 24b/26c | 77,7 | 87,5f | 76,0 | 83,5f | 86,8 | |
Viikko 48 | 74,3 | 89,9e | 74,1 | 86,4e | 87,9 | |
Lyhenteet: ADA = adalimumabi; IR = riittämätön vaste; MTX = metotreksaatti; UPA= upadasitinibi a Kaikki lumehoitojen tiedot viikolta 48 perustuvat lineaariseen ekstrapolointiin b SELECT-EARLY c SELECT-COMPARE d ”Ei taudin etenemistä” määriteltiin tilanteeksi, jossa mTSS-pisteiden muutos oli ≤ 0 e upadasitinibin nimellinen p-arvo ≤ 0,001 vs. lume tai metotreksaatti f upadasitinibin multiplisiteetin suhteen kontrolloitu p-arvo ≤ 0,01 vs. lume tai metotreksaatti g upadasitinibin multiplisiteetin suhteen kontrolloitu p-arvo ≤ 0,001 vs. lume tai metotreksaatti | ||||||
Fyysisen toimintakyvyn vaste ja terveyteen liittyvät hoitotulokset
Upadasitinibi 15 mg -hoidon käyttö joko ainoana hoitona tai yhdessä tavanomaisten synteettisten tautiprosessiin vaikuttavien reumalääkkeiden kanssa johti fyysisen toimintakyvyn merkitsevästi suurempaan kohenemiseen verrattuna kaikkiin vertailuhoitoihin, kun mittausperusteena käytettiin HAQ-DI-arvoja (ks. taulukko 7). SELECT‑COMPARE- ja SELECT‑EARLY-tutkimusten saatavilla olevien tulosten perusteella HAQ‑DI-arvon paranema säilyi 3 vuoteen asti potilailla, jotka jatkoivat heille alun perin satunnaistettua upadasitinibi 15 mg ‑hoitoa.
Taulukko 7 HAQ-DI-pisteiden keskimuutos lähtötilanteestaa,b
Tutkimus | SELECT- EARLY Ei aiempaa MTX-hoitoa | SELECT- MONO MTX-IR | SELECT- NEXT csDMARD-IR | SELECT- COMPARE MTX-IR | SELECT- BEYOND BIO-IR | ||||||
Hoitoryhmä | MTX | UPA 15 mg | MTX | UPA 15 mg | Lume | UPA 15 mg | Lume | UPA 15 mg | ADA 40 mg | Lume | UPA 15 mg |
N | 313 | 317 | 216 | 216 | 220 | 216 | 648 | 644 | 324 | 165 | 163 |
Lähtötasopisteet, keskiarvo | 1,6 | 1,6 | 1,5 | 1,5 | 1,4 | 1,5 | 1,6 | 1,6 | 1,6 | 1,6 | 1,7 |
Viikko 12c/14d | -0,5 | -0,8h | -0,3 | -0,7g | -0,3 | -0,6g | -0,3 | -0,6g,h | -0,5 | -0,2 | -0,4g |
Viikko 24e /26f | -0,6 | -0,9g |
|
|
|
| -0,3 | -0,7h,i | -0,6 |
|
|
Lyhenteet: ADA = adalimumabi; HAQ-DI = Health Assessment Questionnaire-Disability Index -mittari; IR = riittämätön vaste; MTX = metotreksaatti; UPA = upadasitinibi a Esitetyt tiedot ovat keskiarvoja b Health Assessment Questionnaire-Disability Index -mittari: 0 = paras, 3 = huonoin; 20 kysymystä; 8 luokkaa: pukeutuminen ja siistiytyminen, ylös nouseminen, syöminen, käveleminen, hygienia, kurottuminen, tarttuminen ja päivittäistoimet. c SELECT-EARLY, SELECT-NEXT, SELECT-COMPARE, SELECT-BEYOND d SELECT-MONOTHERAPY e SELECT-EARLY f SELECT-COMPARE g multiplisiteetin suhteen kontrolloitu upadasitinibin p-arvo ≤ 0,001 vs. lume tai metotreksaatti h upadasitinibin nimellinen p-arvo ≤ 0,001 vs. lume tai metotreksaatti i upadasitinibin nimellinen p-arvo ≤ 0,01 vs. adalimumabi | |||||||||||
SELECT-MONOTHERAPY-, SELECT-NEXT- ja SELECT-COMPARE-tutkimuksissa upadasitinibi 15 mg -hoito johti merkitsevästi parempaan aamuisen niveljäykkyyden keston keskiarvojen kohenemiseen verrattuna lume- tai metotreksaattihoitoon.
Upadasitinibihoitoa saaneet potilaat ilmoittivat kliinisissä tutkimuksissa potilaan raportoiman elämänlaadun merkitsevää paranemista, jonka mittarina käytettiin SF36-mittarin (Short Form (36) Health Survey (SF-36)) fyysisen osion yhteispisteitä verrattuna lumelääkkeeseen ja metotreksaattiin. Lisäksi upadasitinibihoitoa saaneet potilaat ilmoittivat merkitsevää uupumuksen lievittymistä, jonka mittarina käytettiin FACIT-F-pisteitä (Functional Assessment of Chronic Illness Therapy-Fatigue), verrattuna lumelääkkeeseen.
Nivelpsoriaasi
Kerran vuorokaudessa otettavan upadasitinibi 15 mg -hoidon tehoa ja turvallisuutta arvioitiin kahdessa vaiheen 3 satunnaistetussa, kaksoissokkoutetussa ja lumekontrolloidussa monikeskustutkimuksessa, joissa tutkittavat olivat iältään vähintään 18-vuotiaita ja sairastivat keskivaikeaa tai vaikeaa aktiivista nivelpsoriaasia. Kaikilla potilailla oli ollut aktiivinen nivelpsoriaasi vähintään 6 kuukauden ajan CASPAR-kriteerien (Classification Criteria for Psoriatic Arthritis) mukaan, vähintään kolme aristavaa niveltä ja vähintään kolme turvonnutta niveltä sekä aktiivinen läiskäpsoriaasi tai anamneesissa läiskäpsoriaasi. Molemmissa tutkimuksissa ensisijainen päätetapahtuma oli ACR20-vasteen saavuttaneiden potilaiden osuus viikolla 12.
SELECT-PsA 1 oli 24 viikkoa kestänyt tutkimus, johon osallistui 1 705 potilasta, jotka olivat saaneet riittämättömän vasteen vähintään yhdelle ei-biologiselle tautiprosessiin vaikuttavalle reumalääkkeelle tai eivät sietäneet sitä. Lähtötilanteessa 1 393 (82 %) potilaista sai samanaikaisesti vähintään yhtä ei‑biologista tautiprosessiin vaikuttavaa reumalääkettä; 1 084 (64 %) potilaista sai samanaikaisesti ainoastaan metotreksaattia; ja 311 (18 %) potilaista sai vain yhtä lääkettä. Potilaat saivat upadasitinibia 15 mg tai 30 mg kerran vuorokaudessa, adalimumabia tai lumelääkettä. Viikolla 24 kaikki lumelääkeryhmään satunnaistetut potilaat vaihdettiin ryhmään, jossa he saivat sokkoutetusti upadasitinibia joko 15 mg tai 30 mg kerran vuorokaudessa. SELECT-PsA 1 tutkimukseen kuului pitkäaikainen jatkovaihe, enintään 5 vuotta.
SELECT-PsA 2 oli 24 viikkoa kestänyt tutkimus, johon osallistui 642 potilasta, jotka olivat saaneet riittämättömän vasteen vähintään yhdelle biologiselle tautiprosessiin vaikuttavalle reumalääkkeelle tai eivät sietäneet sitä. Lähtötilanteessa 296 (46 %) potilaista sai samanaikaisesti vähintään yhtä ei-biologista tautiprosessiin vaikuttavaa reumalääkettä; 222 (35 %) potilaista sai samanaikaisesti ainoastaan metotreksaattia; ja 345 (54 %) potilaista sai vain yhtä lääkettä. Potilaat saivat upadasitinibia 15 mg tai 30 mg kerran vuorokaudessa tai lumelääkettä. Viikolla 24 kaikki lumelääkeryhmään satunnaistetut potilaat vaihdettiin ryhmään, jossa he saivat sokkoutetusti upadasitinibia joko 15 mg tai 30 mg kerran vuorokaudessa. SELECT-PsA 2 -tutkimukseen kuului pitkäaikainen jatkovaihe, enintään 3 vuotta.
Kliininen vaste
Molemmissa tutkimuksissa upadasitinibi 15 mg -hoitoa saaneista potilaista tilastollisesti merkitsevästi suurempi osuus saavutti ACR20-vasteen verrattuna lumelääkettä saaneisiin viikolla 12 (taulukko 8). Aika tehon alkamiseen oli lyhyt kaikilla mittareilla mitattuna, ja jo viikolla 2 saavutettiin paremmat ACR20-vasteet.
Upadasitinibi 15 mg -hoito kohensi ACR-vasteen yksittäisten ACR-osa-alueiden tuloksia (mm. aristavien/kipeiden ja turvonneiden nivelten määrät, potilaan ja lääkärin yleisarviot, HAQ-DI, kivun arviointi ja hs-CRP) lumelääkkeeseen verrattuna.
SELECT-PsA 1 -tutkimuksessa upadasitinibi 15 mg -hoito oli vähintään yhdenveroinen adalimumabiin verrattuna, kun arvioitiin ACR20-vasteen saaneiden potilaiden osuutta viikolla 12; ylivertaisuutta adalimumabiin nähden ei kuitenkaan voitu osoittaa.
Molemmissa tutkimuksissa havaittiin johdonmukaiset vasteet ensisijaisen päätetapahtuman ja keskeisten toissijaisten päätetapahtumien suhteen yksinään tai yhdessä metotreksaatin kanssa.
Upadasitinibi 15 mg -hoidon teho pystyttiin osoittamaan riippumatta arvioiduista alaryhmistä ml. lähtötilanteen painoindeksi, lähtötilanteen hs-CRP ja aiempien ei-biologisten tautiprosessiin vaikuttavien reumalääkkeiden lukumäärä (≤ 1 tai > 1).
Taulukko 8 Kliininen vaste SELECT-PsA 1- ja SELECT-PsA 2 -tutkimuksissa
Tutkimus | SELECT-PsA 1 ei-biologinen DMARD-IR | SELECT-PsA 2 bDMARD-IR | |||
Hoitoryhmä | Lume | UPA 15 mg | ADA 40 mg | Lume | UPA 15 mg |
N | 423 | 429 | 429 | 212 | 211 |
ACR20, potilaiden prosenttiosuus (95 %:n lv) | |||||
Viikko 12 | 36 (32; 41) | 71 (66; 75)f | 65 (61; 70) | 24 (18; 30) | 57 (50; 64) |
Ero lumelääkkeeseen (95 %:n lv) | 35 (28; 41)d,e | - | 33 (24; 42)d,e | ||
Viikko 24 | 45 (40; 50) | 73 (69; 78) | 67 (63; 72) | 20 (15; 26) | 59 (53; 66) |
Viikko 56 |
| 74 (70; 79) | 69 (64; 73) |
| 60 (53; 66) |
ACR50, potilaiden prosenttiosuus (95 %:n lv) | |||||
Viikko 12 | 13 (10; 17) | 38 (33; 42) | 38 (33; 42) | 5 (2; 8) | 32 (26; 38) |
Viikko 24 | 19 (15; 23) | 52 (48; 57) | 44 (40; 49) | 9 (6; 13) | 38 (32; 45) |
Viikko 56 |
| 60 (55; 64) | 51 (47; 56) |
| 41 (34; 47) |
ACR70, potilaiden prosenttiosuus (95 %:n lv) | |||||
Viikko 12 | 2 (1; 4) | 16 (12; 19) | 14 (11; 17) | 1 (0; 1) | 9 (5; 12) |
Viikko 24 | 5 (3; 7) | 29 (24; 33) | 23 (19; 27) | 1 (0; 2) | 19 (14; 25) |
Viikko 56 |
| 41 (36; 45) | 31 (27; 36) |
| 24 (18; 30) |
MDA, potilaiden prosenttiosuus (95 %:n lv) | |||||
Viikko 12 | 6 (4; 9) | 25 (21; 29) | 25 (21; 29) | 4 (2; 7) | 17 (12; 22) |
Viikko 24 | 12 (9; 15) | 37 (32; 41)e | 33 (29; 38) | 3 (1; 5) | 25 (19; 31)e |
Viikko 56 |
| 45 (40; 50) | 40 (35; 44) |
| 29 (23; 36) |
Entesiittien häviäminen (LEI=0), potilaiden prosenttiosuus (95 %:n lv)a | |||||
Viikko 12 | 33 (27; 39) | 47 (42; 53) | 47 (41; 53) | 20 (14; 27) | 39 (31; 47) |
Viikko 24 | 32 (27; 39) | 54 (48; 60)e | 47 (42; 53) | 15 (9; 21) | 43 (34; 51) |
Viikko 56 |
| 59 (53; 65) | 54 (48; 60) |
| 43 (34; 51) |
Daktyliittien häviäminen (LDI=0), potilaiden prosenttiosuus (95 %:n lv)b | |||||
Viikko 12 | 42 (33; 51) | 74 (66; 81) | 72 (64; 80) | 36 (24; 48) | 64 (51; 76) |
Viikko 24 | 40 (31; 48) | 77 (69; 84) | 74 (66; 82) | 28 (17; 39) | 58 (45; 71) |
Viikko 56 |
| 75 (68; 82) | 74 (66; 82) |
| 51 (38; 64) |
PASI75, potilaiden prosenttiosuus (95 %:n lv)c | |||||
Viikko 16 | 21 (16; 27) | 63 (56; 69)e | 53 (46; 60) | 16 (10; 22) | 52 (44; 61)e |
Viikko 24 | 27 (21; 33) | 64 (58; 70) | 59 (52; 65) | 19 (12; 26) | 54 (45; 62) |
Viikko 56 |
| 65 (59; 72) | 61 (55; 68) |
| 52 (44; 61) |
PASI90, potilaiden prosenttiosuus (95 %:n lv)c | |||||
Viikko 16 | 12 (8; 17) | 38 (32; 45) | 39 (32; 45) | 8 (4; 13) | 35 (26; 43) |
Viikko 24 | 17 (12; 22) | 42 (35; 48) | 45 (38; 52) | 7 (3; 11) | 36 (28; 44) |
Viikko 56 |
| 49 (42; 56) | 47 (40; 54) |
| 41 (32; 49) |
Lyhenteet: ACR20 (tai 50 tai 70) = ≥ 20 %:n (tai ≥ 50 %:n tai ≥ 70 %:n) koheneminen American College of Rheumatology -kriteereillä; ADA = adalimumabi; bDMARD = biologinen tautiprosessiin vaikuttava reumalääke; IR = riittämättömän vasteen saaneet; MDA = minimal disease activity, vähäinen tautiaktiivisuus; PASI75 (tai 90) = ≥ 75 %:n (tai ≥ 90 %:n) koheneminen Psoriasis Area and Severity Index -kriteereillä; Lume = lumelääke; UPA= upadasitinibi Potilaat, jotka keskeyttivät satunnaistetun hoidon tai joilta puuttui tietoja arviointiviikolla, imputoitiin analyyseissä potilaiksi, jotka eivät saavuttaneet vastetta. Viikolla 16 rescue-hoitoa saaneet tutkittavat imputoitiin analyyseissä potilaiksi, jotka eivät saavuttaneet vastetta MDA:ssa, entesiittien häviämisessä eikä daktyliittien häviämisessä viikoilla 24/56. a Potilaat, joilla oli entesiittejä lähtötilanteessa (n = 241, 270 ja 265 SELECT-PsA 1:ssä ja n = 144 ja 133 SELECT-PsA 2:ssa) b Potilaat, joilla oli daktyliittejä lähtötilanteessa (n = 126, 136 ja 127 SELECT-PsA 1:ssä ja n = 64 ja 55 SELECT-PsA 2:ssa) c Potilaat, joilla oli psoriaasia ≥ 3 % kehon pinta-alasta (BSA) lähtötilanteessa (n = 211, 214 ja 211 SELECT-PsA 1:ssä ja n = 131 ja 130 SELECT-PsA 2:ssa) d ensisijainen päätetapahtuma e multiplisiteetin suhteen kontrolloitu p-arvo ≤ 0,001 vs. lume f upadasitinibin multiplisiteetin suhteen kontrolloitu p-arvo ≤ 0,001 vs. adalimumabi (noninferioriteettitesti) | |||||
Radiografinen vaste
SELECT-PsA 1 -tutkimuksessa rakenteellisten vaurioiden etenemisen estäminen arvioitiin radiografisesti, ja se ilmaistiin mTSS-pisteiden (modified Total Sharp Score) ja mTSS:n osa-alueiden (eroosiopisteet ja nivelrakojen kaventumispisteet) muutoksena lähtötilanteesta viikolla 24.
Upadasitinibi 15 mg -hoito sai aikaan tilastollisesti merkitsevästi suuremman nivelvaurioiden rakenteellista etenemistä estävän vaikutuksen verrattuna lumelääkkeeseen viikolla 24 (taulukko 9). Eroosio- ja nivelraon kaventumispisteet olivat johdonmukaiset kokonaispistemäärien tulosten kanssa. Niiden potilaiden osuus, joilla ei todettu lainkaan radiografista taudin etenemistä (mTSS‑pisteiden muutos ≤ 0,5), oli suurempi upadasitinibi 15 mg ‑ryhmässä kuin lumelääkeryhmässä viikolla 24.
Taulukko 9 Radiografiset muutokset SELECT-PsA 1 -tutkimuksessa
Hoitoryhmä | Lume | UPA 15 mg | ADA 40 mg |
mTSS-pisteet, keskimääräinen muutos lähtötilanteesta (95 %:n lv) | |||
Viikko 24 | 0,25 (0,13; 0,36) | -0,04 (-0,16; 0,07)c | 0,01 (-0,11; 0,13) |
Viikko 56a | 0,44 (0,29; 0,59) | -0,05 (-0,20; 0,09) | -0,06 (-0,20; 0,09) |
Potilaat, joilla ei radiografista taudin etenemistäb, % (95 %:n lv) | |||
Viikko 24 | 92 (89; 95) | 96 (94; 98) | 95 (93; 97) |
Viikko 56a | 89 (86; 92) | 97 (96; 99) | 94 (92; 97) |
Lyhenteet: ADA = adalimumabi; Lume = lumelääke; UPA= upadasitinibi a Kaikki lumehoitojen tiedot viikolta 56 perustuvat lineaariseen ekstrapolointiin b ”Ei taudin etenemistä” määriteltiin tilanteeksi, jossa mTSS-pisteiden muutos oli ≤ 0,5 c Multiplisiteetin suhteen kontrolloitu p-arvo ≤ 0,001 vs. Lume | |||
Fyysisen toimintakyvyn vaste ja terveyteen liittyvät hoitotulokset
SELECT-PsA 1 -tutkimuksessa upadasitinibi 15 mg -hoitoa saaneilla potilailla todettiin tilastollisesti merkitsevästi parantunut fyysinen toimintakyky lähtötilanteeseen verrattuna HAQ‑DI-mittarilla arvioituna viikolla 12 (-0,42 [95 %:n lv: -0,47; -0,37]) lumelääkkeeseen verrattuna (-0,14 [95 %:n lv: -0,18; -0,09]); adalimumabihoitoa saaneilla potilailla paranema oli -0,34 (95 %:n lv: -0,38; -0,29). SELECT‑PsA 2 -tutkimuksessa upadasitinibi 15 mg -hoitoa saaneilla potilailla todettiin tilastollisesti merkitsevä paranema lähtötilanteeseen verrattuna HAQ‑DI-mittarilla viikolla 12 (-0,30 [95 %:n lv: -0,37; -0,24]) lumelääkkeeseen verrattuna (-0,10 [95 %:n lv: -0,16; -0,03]). Paranema fyysisessä toimintakyvyssä säilyi viikkoon 56 asti molemmissa tutkimuksissa.
Terveyteen liittyvä elämänlaatu arvioitiin SF-36v2-mittarilla. Molemmissa tutkimuksissa upadasitinibi 15 mg -hoitoa saavilla potilailla todettiin tilastollisesti merkitsevästi suurempi paranema lähtötilanteesta fyysisen osion yhteispisteissä lumelääkkeeseen verrattuna viikolla 12. Paranemat lähtötilanteeseen nähden säilyivät viikkoon 56 asti molemmissa tutkimuksissa.
Molemmissa tutkimuksissa upadasitinibi 15 mg -hoitoa saavien potilaiden väsymyksessä havaittiin tilastollisesti merkitsevä paranema lähtötilanteeseen verrattuna FACIT-F-pisteytyksellä lumelääkkeeseen verrattuna viikolla 12. Paranemat lähtötilanteeseen nähden säilyivät viikkoon 56 asti molemmissa tutkimuksissa.
Psoriaasispondyliittiä raportoitiin lähtötilanteessa 31 %:lla (SELECT‑PsA 1) ja 34 %:lla potilaista (SELECT‑PsA 2). Upadasitinibi 15 mg -hoitoa saaneilla psoriaasispondyliittipotilailla todettiin BASDAI-pisteissä (Bath Ankylosing Spondylitis Disease Activity Index) paranema lähtötilanteesta lumelääkkeeseen verrattuna viikolla 24. Paranemat lähtötilanteeseen nähden säilyivät viikkoon 56 asti molemmissa tutkimuksissa.
Aksiaalinen spondylartriitti
Röntgennegatiivinen aksiaalinen spondylartriitti
Kerran vuorokaudessa otettavan upadasitinibi 15 mg ‑hoidon tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa ja lumekontrolloidussa monikeskustutkimuksessa, jossa tutkittavat olivat iältään vähintään 18-vuotiaita ja sairastivat aktiivista röntgennegatiivista aksiaalista spondylartriittia. SELECT-AXIS 2 (nr-axSpA) oli 52 viikkoa kestänyt lumekontrolloitu tutkimus, johon osallistui 314 potilasta, joilla oli aktiivinen röntgennegatiivinen aksiaalinen spondylartriitti ja jotka olivat joko saaneet riittämättömän vasteen vähintään kahdelle tulehduskipulääkkeelle tai eivät sietäneet tulehduskipulääkkeitä tai joille ne olivat vasta-aiheisia. Potilailla tuli olla objektiivisia inflammaation merkkejä todettavissa kohonneena C‑reaktiivisen proteiinin arvona (CRP) (määritelty viitealueen ylärajan ylittämiseksi), ja/tai sakroiliittinä magneettikuvissa kuitenkin ilman selkeää radiografista näyttöä risti-suoliluunivelten rakenteellisista vaurioista. Potilailla oli aktiivinen tauti, eli määritelmän mukaisesti BASDAI-pisteinä (Bath Ankylosing Spondylitis Disease Activity Index) ≥ 4 sekä potilaan arvioiman selkäkivun pisteinä ≥ 4 numeerisella arviointiasteikolla (Numerical Rating Scale, NRS) 0–10 seulonta- ja lähtötasokäynneillä tarkasteltuna. Lähtötilanteessa potilailla oli ollut röntgennegatiivisen aksiaalisen spondylartriitin oireita keskimäärin 9,1 vuoden ajan, ja 29,1 % potilaista sai samanaikaisesti tavanomaista synteettistä tautiprosessiin vaikuttavaa reumalääkettä. Potilaista 32,9 %:lla oli riittämätön vaste hoitoon tautiprosessiin vaikuttavalla biologisella reumalääkkeellä tai he eivät sietäneet sitä. Potilaat saivat kerran vuorokaudessa joko 15 mg upadasitinibia tai lumelääkettä. Viikolla 52 kaikki lumelääkeryhmään satunnaistetut potilaat siirtyivät saamaan upadasitinibia 15 mg kerran vuorokaudessa. Ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saavuttivat ASAS40-vasteen (Assessment of SpondyloArthritis international Society 40) viikolla 14. Tutkimukseen kuului pitkäaikainen jatkovaihe, enintään 2 vuotta. Tutkimuksen alussa upadasitinibiryhmään satunnaistetuista potilaista 75 % (117/156) SELECT-AXIS 2 (nr-axSpA) -tutkimuksessa jatkoi hoidolla 2 vuoden ajan.
Kliininen vaste
SELECT-AXIS 2 (nr-axSpA) ‑tutkimuksessa upadasitinibi 15 mg ‑hoitoa saaneista potilaista tilastollisesti merkitsevästi suurempi osuus saavutti ASAS40-vasteen lumelääkettä saaneisiin verrattuna viikolla 14 (taulukko 10). Hoitoryhmien välillä havaittiin numeerinen ero jokaisella tarkasteluhetkellä viikosta 2 viikkoon 14.
Upadasitinibi 15 mg ‑hoito sai aikaan paraneman yksittäisissä ASAS-vasteen osa-alueissa (potilaan yleisarvio taudin aktiivisuudesta, selkäkivun kokonaisarvio, inflammaatio ja toimintakyky) sekä muissa sairauden aktiivisuutta kuvaavissa mittareissa (mm. hs-CRP) lumelääkkeeseen verrattuna viikolla 14.
Upadasitinibi 15 mg -hoidon teho pystyttiin osoittamaan kaikissa alaryhmissä, joita olivat sukupuoli, lähtötilanteen painoindeksi, röntgennegatiivisen aksiaalisen spondylartriitin oireiden kesto, lähtötilanteen hs-CRP, sakroiliitti magneettikuvissa sekä aiempi tautiprosessiin vaikuttavien biologisten reumalääkkeiden käyttö.
Taulukko 10. Kliininen vaste SELECT-AXIS 2 (nr-axSpA) ‑tutkimuksessa
Hoitoryhmä | Lume | UPA 15 mg |
N | 157 | 156 |
ASAS40, potilaiden prosenttiosuus (95 %:n lv) a | ||
Viikko 14 | 22,5 (16,0; 29,1) | 44,9 (37,1; 52,7) |
Ero lumelääkkeeseen (95 %:n lv) | 22,2 (12,1; 32,3)b | |
Viikko 52 | 42,7 (34,9; 50,4) | 62,8 (55,2; 70,4)d |
ASAS20, potilaiden prosenttiosuus (95 %:n lv) a | ||
Viikko 14 | 43,8 (36,0; 51,5) | 66,7 (59,3; 74,1)b |
ASAS osittainen remissio, potilaiden prosenttiosuus (95 %:n lv) | ||
Viikko 14 | 7,6 (3,5; 11,8) | 18,6 (12,5; 24,7)c |
BASDAI 50, potilaiden prosenttiosuus (95 %:n lv) | ||
Viikko 14 | 22,1 (15,5; 28,6) | 42,3 (34,6; 50,1)b |
Muutos lähtötilanteesta ASDAS-CRP:ssä (95 %:n lv) | ||
Viikko 14 | -0,71 (-0,85; -0,56) | -1,36 (-1,50; -1,21)b |
ASDAS inaktiivinen tauti, potilaiden prosenttiosuus (95 %:n lv) | ||
Viikko 14 | 5,2 (1,7; 8,7) | 14,1 (8,6; 19,6)c |
ASDAS matala tautiaktiivisuus, potilaiden prosenttiosuus (95 %:n lv) | ||
Viikko 14 | 18,3 (12,2; 24,4) | 42,3 (34,6; 50,1)b |
Lyhenteet: ASAS20 (tai ASAS40) = Assessment of SpondyloArthritis international Society ≥ 20 %:n (tai ≥ 40 %:n) paranema; ASDAS-CRP = Ankylosing Spondylitis Disease Activity Score C-Reactive Protein; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; UPA = upadasitinibi a ASAS20 (tai ASAS40) ‑vaste on määritelty ≥ 20 %:n (≥ 40 %:n) paranemana sekä ≥ 1 (≥ 2) yksikön absoluuttisena paranemana lähtötilanteesta (vaihteluväli 0–10) ≥ 3 osa alueessa 4:stä (potilaan yleisarvio, selkäkivun kokonaisarvio, toimintakyky ja inflammaatio) ilman pahenemista neljännessä osa-alueessa (paheneminen määriteltynä ≥ 20 %:n ja ≥ 1 yksikön pahenemisena ASAS20:ssä tai > 0 yksikön pahenemisena ASAS40:ssä). b multiplisiteetin suhteen kontrolloitu p-arvo ≤ 0,001 upadasitinibi vs. lume c multiplisiteetin suhteen kontrolloitu p-arvo ≤ 0,01 upadasitinibi vs. lume d nimellinen p-arvo ≤ 0,001 upadasitinibi vs lume, ennalta määritellyn multiplisiteetin suhteen kontrolloidun koesekvenssin mukaisesti Binääristen päätetapahtumien osalta tulokset perustuvat NRI-imputointiin (non-responder imputation) yhdessä moni-imputoinnin kanssa. Jatkuvien päätetapahtumien osalta tulokset perustuvat pienimmän neliösumman keskimääräiseen muutokseen lähtötilanteesta, ja menetelmässä käytetään sekamallia toistuvien mittausten analyysissä. | ||
Teho säilyi 2 vuoden ajan arvioituna taulukossa 10 esitettyjen päätetapahtumien perusteella.
Fyysisen toimintakyvyn vaste ja terveyteen liittyvät hoitotulokset
Upadasitinibi 15 mg ‑hoitoa saaneilla potilailla todettiin merkitsevä fyysisen toimintakyvyn paranema lähtötilanteesta lumelääkkeeseen verrattuna BASFI-pisteiden perusteella viikolla 14.
Upadasitinibi 15 mg ‑hoitoa saaneilla potilailla todettiin merkitsevä paraneminen sekä kokonaisselkäkivussa että yöllisessä selkäkivussa lumelääkkeeseen verrattuna viikolla 14.
Upadasitinibi 15 mg ‑hoitoa saaneilla potilailla todettiin merkitsevä paraneminen terveyteen liittyvässä elämänlaadussa, jota mitattiin ASQoL-terveysindeksillä, sekä kokonaisterveydessä, jota mitattiin ASAS-terveysindeksillä, lumelääkkeeseen verrattuna viikolla 14.
Paranemat BASFI-pisteissä, kokonaisselkäkivussa ja yöllisessä selkäkivussa sekä ASQoL- ja ASAS-terveysindekseissä säilyivät 2 vuoden ajan.
Objektiivinen inflammaation arviointi
Inflammaation merkkejä arvioitiin magneettikuvauksella ja ilmaistiin muutoksena lähtötilanteesta risti-suoliluunivelten SPARCC-pisteissä (Spondyloarthritis Research Consortium of Canada). Viikolla 14 havaittiin merkitsevä paranema risti-suoliluunivelten inflammaation merkeissä upadasitinibi 15 mg ‑hoitoa saaneilla potilailla lumelääkkeeseen verrattuna. Magneettikuvauksella todettu inflammaation paranema säilyi 2 vuoden ajan.
Selkärankareuma (AS, röntgenpositiivinen aksiaalinen spondylartriitti)
Kerran vuorokaudessa otettavan upadasitinibi 15 mg -hoidon tehoa ja turvallisuutta arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa, jossa tutkittavat olivat iältään vähintään 18‑vuotiaita ja sairastivat aktiivista selkärankareumaa. Aktiivinen sairaus määriteltiin BASDAI-pisteinä ≥ 4 ja potilaan arvioiman selkäkivun pisteinä ≥ 4. Kumpaankin tutkimukseen kuului pitkäaikainen jatkovaihe, enintään 2 vuotta.
SELECT-AXIS 1 oli 14 viikkoa kestänyt lumekontrolloitu tutkimus, jonka 187 selkärankareumapotilasta olivat saaneet riittämättömän vasteen vähintään kahdelle tulehduskipulääkkeelle tai eivät sietäneet niitä tai joilla oli vasta-aihe niille, ja jotka eivät olleet aiemmin saaneet biologisia tautiprosessiin vaikuttavia reumalääkkeitä. Lähtötilanteessa potilailla oli ollut selkärankareuman oireita keskimäärin 14,4 vuoden ajan, ja noin 16 % potilaista sai samanaikaisesti tavanomaista synteettistä tautiprosessiin vaikuttavaa reumalääkettä. Potilaat saivat upadasitinibi 15 mg -hoitoa kerran vuorokaudessa tai lumelääkettä. Viikolla 14 kaikki lumelääkeryhmään satunnaistetut potilaat vaihdettiin ryhmään, jossa he saivat upadasitinibia 15 mg kerran vuorokaudessa. Ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saavuttivat ASAS40-vasteen (Assessment of SpondyloArthritis international Society) viikolla 14.
SELECT-AXIS 2 (AS) oli 14 viikon lumekontrolloitu tutkimus 420 selkärankareumapotilaalla, jotka olivat aiemmin saaneet biologisia tautiprosessiin vaikuttavia reumalääkkeitä (77,4 %:lla TNF:n estäjä tai interleukiini-17:n estäjä (IL-17i) ei ollut tehonnut, 30,2 % potilaista ei ollut sietänyt niitä ja 12,9 %:lla oli aiempaa kokemusta kahdesta biologisesta tautiprosessiin vaikuttavasta reumalääkkeestä ilman tehon puutetta). Lähtötilanteessa potilailla oli ollut selkärankareuman oireita keskimäärin 12,8 vuoden ajan, ja noin 31 % potilaista sai samanaikaisesti myös tavanomaista synteettistä tautiprosessiin vaikuttavaa reumalääkettä. Potilaat saivat kerran vuorokaudessa joko 15 mg upadasitinibia tai lumelääkettä. Viikolla 14 kaikki lumelääkeryhmään satunnaistetut potilaat siirtyivät saamaan upadasitinibia 15 mg kerran vuorokaudessa. Ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saavuttivat ASAS40-vasteen (Assessment of SpondyloArthritis international Society 40) viikolla 14.
Tutkimuksen alussa upadasitinibiryhmään satunnaistetuista potilaista 72 % (67/93) SELECT-AXIS 1 ja 77 % (163/211) SELECT-AXIS 2 (AS) -tutkimuksissa jatkoi hoidolla 2 vuoden ajan.
Kliininen vaste
Kummassakin tutkimuksessa upadasitinibi 15 mg -hoitoa saaneista potilaista tilastollisesti merkitsevästi suurempi osuus saavutti ASAS40-vasteen lumelääkettä saaneisiin verrattuna viikolla 14 (taulukko 11). Hoitoryhmien välillä havaittiin SELECT-AXIS 1 ‑tutkimuksessa viikosta 2 alkaen ja SELECT-AXIS 2 (AS) ‑tutkimuksessa viikosta 4 alkaen numeerinen ero ASAS40-vasteessa.
Upadasitinibi 15 mg -hoito sai aikaan paraneman yksittäisissä ASAS-vasteen osa-alueissa (potilaan yleinen sairauden aktiivisuuden arvio, selkäkivun kokonaisarvio, inflammaatio ja toimintakyky) sekä muissa sairauden aktiivisuutta kuvaavissa mittareissa (mm. hs-CRP) lumelääkkeeseen verrattuna viikolla 14.
Upadasitinibi 15 mg -hoidon teho pystyttiin osoittamaan riippumatta arvioiduista alaryhmistä ml. sukupuoli, lähtötilanteen painoindeksi, selkärankareuman oireiden kesto, lähtötilanteen hs-CRP ja aiempi biologisten tautiprosessiin vaikuttavien reumalääkkeiden käyttö.
Taulukko 11 Kliininen vaste
Tutkimus | SELECT-AXIS 1 bDMARD-naiivi | SELECT-AXIS 2 (AS) bDMARD-IR | ||
Hoitoryhmä | Lume | UPA 15 mg | Lume | UPA 15 mg |
N | 94 | 93 | 209 | 211 |
ASAS40, potilaiden prosenttiosuus (95 %:n lv)a, b | ||||
Viikko 14 | 25,5 (16,7; 34,3) | 51,6 (41,5; 61,8) | 18,2 (13,0; 23,4) | 44,5 (37,8; 51,3) |
Ero lumelääkkeeseen (95 %:n lv) | 26,1 (12,6; 39,5)c | 26,4 (17,9; 34,9)c | ||
ASAS20, potilaiden prosenttiosuus (95 %:n lv)a | ||||
Viikko 14 | 40,4 (30,5; 50,3) | 64,5 (54,8; 74,2)e | 38,3 (31,7; 44,9) | 65,4 (59,0; 71,8)c |
ASAS osittainen remissio, potilaiden prosenttiosuus (95 %:n lv) | ||||
Viikko 14 | 1,1 (0,0; 3,1) | 19,4 (11,3; 27,4)c | 4,3 (1,6; 7,1) | 17,5 (12,4; 22,7)c |
BASDAI 50, potilaiden prosenttiosuus (95 %:n lv) | ||||
Viikko 14 | 23,4 (14,8; 32,0) | 45,2 (35,0; 55,3)d | 16,7 (11,7; 21,8) | 43,1 (36,4; 49,8)c |
Muutos lähtötilanteesta ASDAS-CRP:ssä (95 %:n lv) | ||||
Viikko 14 | -0,54 (-0,71; -0,37) | -1,45 (-1,62; -1,28)c | -0,49 (-0,62; -0,37) | -1,52 (-1,64; -1,39)c |
ASDAS inaktiivinen tauti, potilaiden prosenttiosuus (95 %:n lv) | ||||
Viikko 14 | 0 | 16,1 (8,7; 23,6)e | 1,9 (0,1; 3,8) | 12,8 (8,3; 17,3)c |
ASDAS matala tautiaktiivisuus, potilaiden prosenttiosuus (95 %:n lv) | ||||
Viikko 14 | 10,6 (4,4; 16,9) | 49,5 (39,3; 59,6)f | 10,1 (6,0; 14,2) | 44,1 (37,4; 50,8)c |
ASDAS merkittävä paranema, potilaiden prosenttiosuus (95 %:n lv) | ||||
Viikko 14 | 5,3 (0,8; 9,9) | 32,3 (22,8; 41,8)e | 4,8 (1,9; 7,7) | 30,3 (24,1; 36,5)e |
a ASAS20 (tai ASAS40) -vaste on määritelty ≥ 20 %:n (≥ 40 %:n) paranemana sekä ≥ 1 (≥ 2) yksikön absoluuttisena paranemana lähtötilanteesta (vaihteluväli 0–10) ≥ 3 osa‑alueessa 4:stä (potilaan yleinen arvio, selkäkivun kokonaisarvio, toimintakyky ja inflammaatio) ilman pahenemista neljännessä osa-alueessa (määriteltynä ≥ 20 %:n ja ≥ 1 yksikön pahenemisena ASAS20:ssä tai > 0 yksikön pahenemisena ASAS40:ssä). b ensisijainen päätetapahtuma c upadasitinibin multiplisiteetin suhteen kontrolloitu p-arvo ≤ 0,001 vs. lume d upadasitinibin multiplisiteetin suhteen kontrolloitu p-arvo ≤ 0,01 vs. lume e vertailu ei multiplisiteetin suhteen kontrolloitu f SELECT-AXIS 1 ‑tutkimuksen post-hoc-analyysi, ei multiplisiteetin suhteen kontrolloitu Binääristen päätetapahtumien osalta viikon 14 tulokset perustuvat SELECT-AXIS 1 ‑tutkimuksessa NRI-imputaatioon (non-responder imputation) ja SELECT-AXIS 2 (AS) ‑tutkimuksessa NRI-imputaatioon yhdessä moni-imputoinnin kanssa. Jatkuvien päätetapahtumien osalta viikon 14 tulokset perustuvat pienimmän neliösumman keskimääräiseen muutokseen lähtötilanteesta käyttäen sekamallia toistuvien mittausten analyysissä. | ||||
Molemmissa tutkimuksissa teho säilyi 2 vuoden ajan arvioituna taulukossa 11 esitettyjen päätetapahtumien perusteella.
Fyysisen toimintakyvyn vaste ja terveyteen liittyvät hoitotulokset
Kummassakin tutkimuksessa upadasitinibi 15 mg -hoitoa saaneilla potilailla todettiin merkitsevä fyysisen toimintakyvyn paranema lumelääkkeeseen verrattuna arvioituna BASFI-pisteiden (Bath Ankylosing Spondylitis Functional Index) muutoksena lähtötilanteesta viikolla 14. Paranema BASFI-pisteissä säilyi 2 vuoden ajan.
SELECT-AXIS 2 (AS) ‑tutkimuksessa upadasitinibi 15 mg ‑hoitoa saaneilla potilailla todettiin merkitsevä paraneminen sekä kokonaisselkäkivussa että yöllisessä selkäkivussa lumelääkkeeseen verrattuna viikolla 14. Paranemat kokonaisselkäkivussa ja yöllisessä selkäkivussa säilyivät 2 vuoden ajan.
SELECT-AXIS 2 (AS) ‑tutkimuksessa upadasitinibi 15 mg ‑hoitoa saaneilla potilailla todettiin merkitsevä paraneminen terveyteen liittyvässä elämänlaadussa, jota mitattiin ASQoL-terveysindeksillä, sekä kokonaisterveydessä, jota mitattiin ASAS-terveysindeksillä, lumelääkkeeseen verrattuna viikolla 14. ASQoL- ja ASAS-terveysindekseillä mitatut paranemat säilyivät 2 vuoden ajan.
Entesiitit
SELECT-AXIS 2 (AS) ‑tutkimuksessa upadasitinibi 15 mg ‑hoitoa saaneilla potilailla, joilla oli ennestään entesiittejä (n=310), todettiin lumelääkkeeseen verrattuna viikolla 14 entesiittien merkitsevä paraneminen, jota mitattiin muutoksena lähtötason MASES-arvoon (Maastricht Ankylosing Spondylitis Enthesitis Score). Paranema entesiiteissä säilyi 2 vuoden ajan.
Selkärangan liikkuvuus
SELECT-AXIS 2 (AS) ‑tutkimuksessa upadasitinibi 15 mg ‑hoitoa saaneilla potilailla todettiin lumelääkkeeseen verrattuna viikolla 14 selkärangan liikkuvuudessa merkitsevä paraneminen, jota mitattiin muutoksena lähtötason BASMI-pisteisiin (Bath Ankylosing Spondylitis Metrology Index). Paranema BASMI-pisteissä säilyi 2 vuoden ajan.
Objektiivinen inflammaation arviointi
Inflammaation merkkejä arvioitiin magneettikuvauksella ja ilmaistiin muutoksena lähtötilanteesta selkärangan SPARCC-pisteissä. Kummassakin tutkimuksessa viikolla 14 havaittiin merkitsevä paranema selkärangan inflammaation merkeissä upadasitinibi 15 mg -hoitoa saaneilla potilailla lumelääkkeeseen verrattuna. Magneettikuvista todettu paranema inflammaatiossa säilyi 2 vuoden ajan.
Jättisoluarteriitti
Kerran vuorokaudessa otettavan upadasitinibi 15 mg -hoidon tehoa ja turvallisuutta arvioitiin vaiheen 3 satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa SELECT-GCA-monikeskustutkimuksessa, jossa tutkittavat olivat iältään vähintään 50-vuotiaita ja sairastivat uutena ilmaantunutta tai uusiutunutta jättisoluarteriittia. SELECT-GCA (tutkimusjakso 1) oli 52 viikkoa kestänyt tutkimus, johon osallistuneet 428 potilasta satunnaistettiin suhteessa 2:1:1 saamaan kerran vuorokaudessa 15 mg upadasitinibia, 7,5 mg upadasitinibia tai lumelääkettä. Kaikki potilaat saivat perushoitona kortikosteroideja (prednisoni tai prednisoloni). Upadasitiniryhmissä noudatettiin ennalta määritettyä hoito-ohjelmaa kortikosteroidien asteittaisesta lopettamisesta, jolla pyrittiin saavuttamaan taso 0 mg viikkoon 26 mennessä; lumeryhmässä noudatettiin ennalta määritettyä hoito-ohjelmaa kortikosteroidien asteittaisesta lopettamisesta, jolla pyrittiin saavuttamaan taso 0 mg viikkoon 52 mennessä. Ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saavuttivat pysyvän remission viikolla 52 eli joilla ei ollut jättisoluarteriitin oireita ja löydöksiä viikon 12 ja viikon 52 välisenä aikana ja jotka noudattivat tutkimussuunnitelman mukaista hoito-ohjelmaa kortikosteroidien asteittaisesta lopettamisesta. Potilaat, jotka keskeyttivät tutkimushoidon (upadasitinibi tai lumelääke) ennenaikaisesti tai joista ei ollut arviota, luokiteltiin potilaiksi, jotka eivät saaneet vastetta. Tutkimukseen sisältyi 52 viikon pituinen sokkoutettu jatkovaihe (tutkimusjakso 2) eli tutkimuksen kokonaiskesto oli enintään 2 vuotta.
Kliininen vaste
Upadasitinibi 15 mg -hoidolla ja 26 viikkoa kestäneellä asteittain lopetettavalla kortikosteroidihoidolla saavutettiin superioriteetti viikolla 52 pysyvässä remissiossa ilman kortikosteroidihoitoa verrattuna lumelääkkeeseen ja 52 viikkoa kestäneeseen asteittain lopetettavaan kortikosteroidihoitoon (taulukko 12). Tulokset pysyvän remission ja pysyvän täydellisen remission kaikista osa-alueista viikolla 52 vastasivat yhdistetyistä päätetapahtumista saatuja tuloksia. Vastaavat prosenttiosuudet potilaista kummassakin hoitoryhmässä luokiteltiin potilaiksi, jotka eivät saavuttaneet pysyvää remissiota viikolla 52 (ensisijainen päätetapahtuma) joko ennenaikaisen tutkimushoidon keskeyttämisen vuoksi (lumelääke: 19,6 %; upadasitinibi 15 mg: 20,1 %) tai puuttuvan arvion vuoksi (lumelääke: 0,9 %; upadasitinibi 15 mg: 0,5%).
Hoitovaikutukset alaryhmissä (sukupuoli, ikä, etninen tausta, aiempi interleukiini 6:n estäjän käyttö, uutena ilmaantunut tai uusiutunut jättisoluarteriitti, kortikosteroidiannos lähtötilanteessa sekä jättisoluarteriitin esiintyminen polymyalgia rheumatican kanssa tai ilman sitä) vastasivat koko tutkimuspopulaation tuloksia.
Merkitsevästi pienemmällä osalla upadasitinibi 15 mg -hoitoa ja asteittain 26 viikon aikana lopetettavaa kortikosteroidihoitoa saaneista potilaista esiintyi vähintään yksi jättisoluarteriitin pahenemisvaihe viikkoon 52 mennessä verrattuna lumelääkettä saaneisiin potilaisiin, joiden kortikosteroidihoito lopetettiin asteittain 52 viikon aikana. Lisäksi pahenemisvaiheiden riski oli upadasitinibiryhmässä merkitsevästi pienempi kuin lumeryhmässä, kun mittarina oli ensimmäiseen pahenemisvaiheeseen kulunut aika viikolle 52 asti (taulukko 12).
Taulukko 12 Kliininen vaste SELECT-GCA-tutkimuksessa (tutkimusjakso 1)
Hoitoryhmä | Lume + 52 viikon aikana asteittain lopetettava kortikosteroidihoito N = 112 | UPA 15 mg + 26 viikon aikana asteittain lopetettava kortikosteroidihoito N = 209 | Hoitoero (95 %:n lv) |
Pysyvä remissio viikolla 52a | 29,0 % | 46,4 % | 17,1 %e (6,3; 27,8) |
Pysyvä täydellinen remissio viikolla 52b | 16,1 % | 37,1 % | 20,7 %f (11,3; 30,2) |
Täydellinen remissio viikolla 52c | 19,6 % | 50,2 % | 30,3 %f (20,4; 40,2) |
Täydellinen remissio viikolla 24c | 36,1 % | 57,2 % | 20,8 %f (9,7; 31,9) |
Aika ensimmäiseen GCA:n pahenemisvaiheeseen viikolle 52 astid |
|
| 0,57e,g (0,399; 0,826) |
Potilaat, joilla esiintyi vähintään yksi GCA:n pahenemisvaihe viikolle 52 astid | 55,6 % | 34,3 % | 0,47e,h (0,29; 0,74) |
Lyhenteet: ESR = lasko; GCA = jättisoluarteriitti; hsCRP = herkkä C-reaktiivinen proteiini; lume = lumelääke; UPA = upadasitinibi a Pysyvä remissio määritellään tilanteeksi, jossa potilaalla ei ole GCA:n merkkejä tai oireita viikon 12 ja viikon 52 välisenä aikana ja hän on noudattanut tutkimussuunnitelmassa määriteltyä hoito-ohjelmaa kortikosteroidien asteittaisesta lopettamisesta b Pysyvä täydellinen remissio määritellään tilanteeksi, jossa potilaalla ei ole GCA:n merkkejä tai oireita viikon 12 ja viikon 52 välisenä aikana, hänen ESR-arvonsa on normalisoitunut (tasolle ≤ 30 mm/h; kriteerin voidaan katsoa täyttyvän myös, jos ESR on > 30 mm/h, mutta nousu ei johdu GCA:sta) viikon 12 ja viikon 52 välisenä aikana ja hänen hsCRP-arvonsa on normalisoitunut tasolle < 1 mg/dl eikä nouse tasolle ≥ 1 mg/dl (kahdella peräkkäisellä käynnillä) viikon 12 ja viikon 52 välisenä aikana, ja hän on noudattanut tutkimussuunnitelmassa määriteltyä hoito-ohjelmaa kortikosteroidien asteittaisesta lopettamisesta c Täydellinen remissio määritellään tilanteeksi, jossa potilaalla ei ole GCA:n merkkejä tai oireita, hänen ESR-arvonsa on normalisoitunut (tasolle ≤ 30 mm/h; kriteerin voidaan katsoa täyttyvän myös, jos ESR on > 30 mm/h, mutta nousu ei johdu GCA:sta), hänen hsCRP-arvonsa on normalisoitunut tasolle < 1 mg/dl, ja hän on noudattanut tutkimussuunnitelmassa määriteltyä hoito-ohjelmaa kortikosteroidien asteittaisesta lopettamisesta d GCA:n pahenemisvaihe määritellään tapahtumaksi, joka merkitsee GCA:n merkkien tai oireiden uusiutumista, tai ESR-mittaustulokseksi > 30 mm/h (GCA:sta johtuva), joka vaatii kortikosteroidiannoksen suurentamista. Pahenemisvaiheen mahdollisuutta harkitaan vain, jos potilas on täyttänyt seuraavat 3 kriteeriä: GCA:n merkit ja oireet eivät ole uusiutuneet, ESR-arvo on normalisoitunut, eikä kortikosteroidiannosta ole suurennettu. Jos potilaasta ei ole arviota, jossa kaikki kolme kriteeriä täyttyvät, hänellä katsotaan olevan GCA:n pahenemisvaihe lähtötilanteessa. Aika ensimmäiseen GCA:n pahenemisvaiheeseen lasketaan ajankohdasta, jolloin potilas täyttää kaikki edellä mainitut kolme kriteeriä. Potilaat, jotka täyttävät kaikki edellä mainitut kolme kriteeriä, mutta joilla ei missään vaiheessa esiinny GCA:n pahenemisvaihetta, sensuroidaan viimeisen arvioinnin ajankohtana e p ≤ 0,01 f p ≤ 0,001 g Riskisuhde (Hazard ratio) h Kerroinsuhde (Odds ratio) | |||
Kumulatiivinen kortikosteroidiannos
Potilailla, jotka pysyivät 52 viikkoa seurannassa, kumulatiivinen kortikosteroidialtistus viikolla 52 oli merkitsevästi pienempi upadasitinibi 15 mg -hoitoa saaneilla potilailla, joiden kortikosteroidihoito lopetettiin asteittain 26 viikon aikana, kuin lumehoitoa saaneilla potilailla, joiden kortikosteroidihoito lopetettiin asteittain 52 viikon aikana (mediaanit 1 615 mg vs. 2 882 mg). Kumulatiivisen kortikosteroidiannoksen vertailuun upadasitinibiryhmän ja lumeryhmän välillä vaikuttaa erilaiset ennalta määritetyt hoito-ohjelmat kortikosteroidien asteittaisesta lopettamisesta upadasitinibi- ja lumeryhmässä.
Terveyteen liittyvät hoitotulokset
Uupumusta arvioitiin FACIT-Fatigue-pistearvon perusteella. Upadasitinibi 15 mg -hoitoa ja asteittain 26 viikon aikana lopetettavaa kortikosteroidihoitoa saaneilla potilailla FACIT-Fatigue-pistearvo oli parantunut merkitsevästi enemmän lähtötilanteesta viikolla 52 verrattuna lumehoitoa saaneisiin potilaisiin, joiden kortikosteroidihoito lopetettiin asteittain 52 viikon aikana (4,0; 95 %:n lv: 1,33; 6,76).
Terveyteen liittyvää elämänlaatua arvioitiin SF-36-asteikolla. Upadasitinibi 15 mg -hoitoa ja asteittain 26 viikon aikana lopetettavaa kortikosteroidihoitoa saaneilla potilailla SF-36-mittarin fyysisen osion yhteenvetopisteet olivat parantuneet merkitsevästi enemmän lähtötilanteesta viikolla 52 verrattuna lumehoitoa saaneisiin potilaisiin, joiden kortikosteroidihoito lopetettiin asteittain 52 viikon aikana (3,75; 95 %:n lv: 1,39; 6,11).
Vasteen säilyminen
Yhteensä 223 potilasta oli mukana SELECT-GCA-tutkimuksen sokkoutetussa jatkovaiheessa (tutkimusjakso 2). Potilaat, jotka oli satunnaistettu lähtötilanteessa saamaan 15 mg upadasitinibia tutkimusjaksossa 1, ja jotka olivat saavuttaneet pysyvän remission (ts. potilailla ei ollut jättisoluarteriitin oireita ja löydöksiä eivätkä he käyttäneet kortikosteroideja) vähintään 24 viikkoa ennen viikkoa 52, satunnaistettiin uudelleen joko jatkamaan upadasitinibi 15 mg -hoitoa (N = 68) tai saamaan lumelääkettä (N = 35).
Potilaista, jotka saavuttivat pysyvän remission viikolla 52, ja jotka satunnaistettiin uudelleen jatkamaan upadasitinibi 15 mg -hoitoa, 71,7 % säilytti kortikosteroidivapaan remission viikolle 104 verrattuna 31,4 %:iin potilaista, jotka satunnaistettiin uudelleen lumelääkeryhmään.
Lumelääkeryhmään uudelleen satunnaistetuista potilaista 59,5 %:lla esiintyi jättisoluarteriitin pahenemisvaihe verrattuna 7,4 %:iin potilaista, jotka satunnaistettiin uudelleen jatkamaan upadasitinibi 15 mg -hoitoa. Mediaaniaika pahenemisvaiheeseen oli 113 päivää potilailla, jotka satunnaistettiin uudelleen lumelääkeryhmään, ja se oli pidempi kuin 365 päivää (mediaania ei saavutettu) potilailla, jotka satunnaistettiin uudelleen saamaan 15 mg upadasitinibia.
Atooppinen ihottuma
Kerran vuorokaudessa otettavien upadasitinibi 15 mg ja 30 mg -annosten tehoa ja turvallisuutta arvioitiin kolmessa vaiheen 3 satunnaistetussa, kaksoissokkoutetussa monikeskustutkimuksessa (MEASURE UP 1, MEASURE UP 2 ja AD UP) yhteensä 2 782 potilaalla (vähintään 12-vuotiaita). Upadasitinibia arvioitiin 542 (344 ensisijaisessa analyysissä) nuorella ja 2 240 aikuisella keskivaikeaa tai vaikeaa atooppista ihottumaa sairastavalla potilaalla, joiden vaste paikallislääkitykseen oli riittämätön. Potilaiden oli täytettävä lähtötilanteessa kaikki seuraavista kriteereistä: tutkijan yleisarvion pistemäärä (vIGA‑AD) ≥ 3 atooppisen ihottuman yleisarvioinnissa (punoitus, kovettumat/näppylät ja vetistys/karstaisuus) vaikeusasteikolla 0–4 (pistemäärä suurenee vaikeusasteen myötä), ihottuman pinta-alaa ja vaikeusastetta kuvaavan EASI-mittarin (Eczema Area and Severity Index) pistemäärä ≥ 16 (yhteispistemäärä, jolla arvioidaan punoituksen, turvotuksen/näppylöiden, rikkoutumien ja jäkälöitymisen laajuutta ja vaikeusastetta 4:ssä eri kohdassa kehoa), ihottuma-alueen minimiosuus kehon pinta-alasta ≥ 10 % ja pahinta kutinaa kuvaavan numeerisen arviointiasteikon (Numerical Rating Scale, NRS) viikoittainen keskiarvo ≥ 4.
Kaikissa kolmessa tutkimuksessa potilaat saivat 15 mg tai 30 mg upadasitinibia tai vastaavaa lumelääkettä kerran vuorokaudessa 16 viikon ajan. AD UP -tutkimuksessa potilaat saivat myös samanaikaisia paikalliskortikosteroideja. Kaksoissokkoutetun jakson päättymisen jälkeen potilaat, jotka oli alun perin satunnaistettu saamaan upadasitinibia, saivat edelleen samaa annosta viikolle 260 asti. Lumeryhmän potilaat satunnaistettiin uudelleen suhteessa 1:1 saamaan 15 mg tai 30 mg upadasitinibia viikolle 260 asti.
Lähtötilanteen ominaisuudet
Monoterapiatutkimuksissa (MEASURE UP 1 ja 2) 50,0 %:lla potilaista lähtötilanteen vIGA-AD-pistemäärä oli 3 (keskivaikea) ja 50,0 %:lla potilaista 4 (vaikea). Lähtötilanteen keskimääräinen EASI-pistemäärä oli 29,3 ja lähtötilanteen keskimääräinen viikoittainen keskiarvo pahinta kutinaa kuvaavalla NRS-asteikolla oli 7,3. Tutkimuksessa, jossa potilaat käyttivät samanaikaisia paikalliskortikosteroideja (AD UP), 47,1 %:lla potilaista lähtötilanteen vIGA-AD-pistemäärä oli 3 (keskivaikea) ja 52,9 %:lla potilaista 4 (vaikea). Lähtötilanteen keskimääräinen EASI-pistemäärä oli 29,7 ja lähtötilanteen keskimääräinen viikoittainen keskiarvo pahinta kutinaa kuvaavalla NRS-asteikolla oli 7,2.
Kliininen vaste
Monoterapiatutkimukset (MEASURE UP 1 JA MEASURE UP 2) ja samanaikaisia paikalliskortikosteroideja sisältävä tutkimus (AD UP)
Merkittävästi suurempi osuus upadasitinibia 15 mg tai 30 mg saaneista potilaista saavutti vIGA-AD-pistemäärän 0 tai 1, EASI-75-vasteen tai ≥ 4 pisteen paraneman pahinta kutinaa kuvaavalla NRS-asteikolla lumelääkkeeseen verrattuna viikolla 16. Myös iho‑oireiden ja kutinan nopea lievittyminen saavutettiin (ks. taulukko 13).
Kuvassa 1 esitetään EASI-75-vasteen saavuttaneiden potilaiden osuus ja keskimääräinen prosenttimuutos lähtötilanteesta pahinta kutinaa kuvaavalla NRS-asteikolla viikkoon 16 mennessä MEASURE UP 1- ja MEASURE UP 2 -tutkimuksissa.
Taulukko 13 Upadasitinibin tehoa koskevat tulokset
Tutkimus | MEASURE UP 1 | MEASURE UP 2 | AD UP | ||||||
Hoitoryhmä | Lume | UPA 15 mg | UPA 30 mg | Lume | UPA 15 mg | UPA 30 mg | Lume + TCS | UPA 15 mg + TCS | UPA 30 mg + TCS |
Satunnaistettujen tutkittavien määrä | 281 | 281 | 285 | 278 | 276 | 282 | 304 | 300 | 297 |
Viikon 16 päätetapahtumat, vasteen saaneiden prosenttiosuus (95 %:n lv) | |||||||||
vIGA-AD 0/1a,b (rinnakkainen ensisijainen) | 8 (5,12) | 48d (42,54) | 62d (56,68) | 5 (2,7) | 39d (33,45) | 52d (46,58) | 11 (7,14) | 40d (34,45) | 59d (53,64) |
EASI-75a (rinnakkainen ensisijainen) | 16 (12,21) | 70d (64,75) | 80d (75,84) | 13 (9,17) | 60d (54,66) | 73d (68,78) | 26 (21,31) | 65d (59,70) | 77d (72,82) |
EASI-90a | 8 (5,11) | 53d (47,59) | 66d (60,71) | 5 (3,8) | 42d (37,48) | 58d (53,64) | 13 (9,17) | 43d (37,48) | 63d (58,69) |
EASI-100a | 2 (0,3) | 17d (12,21) | 27d (22,32) | 1 (0,2) | 14d (10,18) | 19d (14,23) | 1 (0,3) | 12e (8,16) | 23d (18,27) |
Pahinta kutinaa kuvaava NRS-asteikkoc (≥ 4 pisteen paranema) | 12 (8,16) | 52d (46,58) | 60d (54,66) | 9 (6,13) | 42d (36,48) | 60d (54,65) | 15 (11,19) | 52d (46,58) | 64d (58,69) |
Alkuvaiheen päätetapahtumat, vasteen saaneiden prosenttiosuus (95 %:n lv) | |||||||||
EASI-75a (Viikko 2) | 4 (1,6) | 38d (32,44) | 47d (42,53) | 4 (1,6) | 33d (27,39) | 44d (38,50) | 7 (4,10) | 31d (26,36) | 44d (38,50) |
Pahinta kutinaa kuvaava NRS-asteikko (≥ 4 pisteen paranema viikolla 1)c,f | 0 (0,1) | 15d (11,19) | 20d (15,24) | 1 (0,2) | 7d (4,11) | 16d (11,20) | 3 (1,5) | 12d (8,16) | 19d (15,24) |
Lyhenteet: UPA= upadasitinibi (RINVOQ); TCS = paikalliskortikosteroidi Potilaat, jotka saivat rescue-lääkettä tai joilta puuttui tietoja, laskettiin potilaiksi, jotka eivät saavuttaneet vastetta. Niiden tutkittavien määrä ja prosenttiosuus, jotka rescue-lääkityksen vuoksi imputoitiin viikolla 16 ei-vasteen saaneiksi EASI-75:n ja vIGA-AD 0/1:n osalta lumelääke-, upadasitinibi 15 mg – ja upadasitinibi 30 mg -ryhmässä, oli 132 (47,0 %), 31 (11,0 %), 16 (5.6 %) MEASURE UP 1 -tutkimuksessa, 119 (42,8 %), 24 (8,7 %), 16 (5,7 %) MEASURE UP 2 -tutkimuksessa ja 78 (25,7 %), 15 (5,0 %), 14 (4,7 %) AD UP -tutkimuksessa. a Perustuu satunnaistettujen tutkittavien määrään b Vasteen saamisen määritelmänä oli vIGA-AD-pistemäärä 0 tai 1 (”oireeton” tai ”lähes oireeton”) ja ≥ 2 pisteen vähenemä järjestysasteikolla 0–4 c Tulokset arviointikelpoisten potilaiden osajoukossa (potilaat, joiden pahinta kutinaa kuvaava NRS-pistemäärä lähtötilanteessa ≥ 4) d Tilastollisesti merkitsevä lumelääkkeeseen verrattuna ja p < 0,001 e p < 0,001 lumelääkkeeseen verrattuna, ilman multiplisiteettikontrollia f MEASURE UP 1- ja MEASURE UP 2 -tutkimuksissa havaittiin tilastollisesti merkitsevä paranema lumelääkkeeseen verrattuna jopa jo 1 päivän kuluttua upadasitinibi 30 mg -hoidon aloittamisesta ja 2 päivän kuluttua upadasitinibi 15 mg -hoidon aloittamisesta | |||||||||
Kuva 1 EASI-75-vasteen saavuttaneiden potilaiden osuus ja pahinta kutinaa kuvaavan NRS‑pistemäärän keskimääräinen prosenttimuutos lähtötilanteesta MEASURE UP 1- ja MEASURE UP 2 -tutkimuksissa
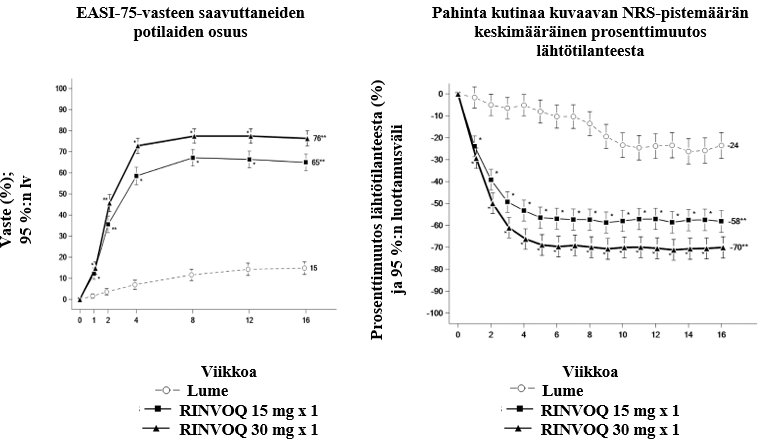
*: p < 0,001 lumelääkkeeseen verrattuna, ilman multiplisiteettikontrollia
**: Tilastollisesti merkitsevä lumelääkkeeseen verrattuna ja p < 0,001
Hoitovaikutukset alaryhmissä (paino, ikä, sukupuoli, etninen tausta ja aiempi systeeminen immunosuppressanttihoito) vastasivat koko tutkimuspopulaation tuloksia.
Viikon 16 tulokset säilyivät viikolle 52 asti upadasitinibia 15 mg tai 30 mg saaneilla potilailla.
Elämänlaatu / potilaiden ilmoittamat hoitotulokset
Taulukko 14 Upadasitinibiin liittyvät potilaiden ilmoittamat hoitotulokset viikolla 16
Tutkimus | MEASURE UP 1 | MEASURE UP 2 | ||||
Hoitoryhmä | Lume | UPA 15 mg | UPA 30 mg | Lume | UPA 15 mg | UPA 30 mg |
Satunnaistettujen tutkittavien määrä | 281 | 281 | 285 | 278 | 276 | 282 |
Vasteen saaneiden prosenttiosuus (95 %:n lv) | ||||||
ADerm-SS – ihon kipu (≥ 4 pisteen paranema)a | 15 (10,20) | 54e (47,60) | 63e (57,69) | 13 (9,18) | 49e (43,56) | 65e (59,71) |
ADerm-IS – nukkuminen (≥ 12 pisteen paranema)a,b | 13 (9,18) | 55e (48,62) | 66e (60,72) | 12 (8,17) | 50e (44,57) | 62e (56,69) |
DLQI 0/1c | 4 (2,7) | 30e (25,36) | 41e (35,47) | 5 (2,7) | 24e (19,29) | 38e (32,44) |
Ahdistuneisuus HADS-asteikolla < 8 ja masennus HADS-asteikolla < 8d | 14 (8,20) | 46e (37,54) | 49e (41,57) | 11 (6,17) | 46e (38,54) | 56e (48,64) |
Lyhenteet: UPA = upadasitinibi (RINVOQ); DLQI = terveyteen liittyvä elämänlaatu (Dermatology Life Quality Index); HADS = ahdistuneisuutta ja masennusta kuvaava asteikko (Hospital Anxiety and Depression Scale) Potilaat, jotka saivat rescue-lääkettä tai joilta puuttui tietoja, laskettiin potilaiksi, jotka eivät saavuttaneet vastetta Määritetyt kynnysarvot vastaavat pienintä kliinisesti merkittävää eroa (MCID), jota käytettiin vasteen määrittämiseen. a Tulokset arviointikelpoisten potilaiden osajoukossa (potilaat, joiden arvioinnin pistemäärä lähtötilanteessa > MCID). b ADerm-IS – nukkuminen -mittarilla arvioidaan atooppisesta ihottumasta johtuvia nukahtamisvaikeuksia, univaikutuksia ja yöheräämisiä. c Tulokset arviointikelpoisten potilaiden osajoukossa (potilaat, joiden DLQI-pistemäärä lähtötilanteessa > 1). d Tulokset arviointikelpoisten potilaiden osajoukossa (potilaat, joiden ahdistuneisuus HADS-asteikolla ≥ 8 ja masennus HADS-asteikolla ≥ 8 lähtötilanteessa) e Tilastollisesti merkitsevä lumelääkkeeseen verrattuna ja p < 0,001 | ||||||
Haavainen paksusuolitulehdus
Upadasitinibin tehoa ja turvallisuutta arvioitiin kolmessa vaiheen 3 kaksoissokkoutetussa, lumekontrolloidussa kliinisessä monikeskustutkimuksessa: kahdessa induktiohoitoa koskeneessa identtisessä tutkimuksessa, UC‑1 (U‑ACHIEVE Induction) ja UC‑2 (U‑ACCOMPLISH), sekä ylläpitohoitoa koskeneessa tutkimuksessa UC‑3 (U‑ACHIEVE Maintenance). Lisäksi upadasitinibin turvallisuutta ja tehoa arvioitiin pitkäkestoisessa UC-4 (U-ACTIVATE) -jatkotutkimuksessa.
Taudin aktiivisuus perustui mukautettuun Mayo-pisteytykseen (aMS, Mayo-pisteytys ilman lääkärin yleisarviota) asteikolla 0–9. Asteikkoon sisältyy kolme alaosiota, jotka kaikki pisteytettiin välillä 0 (normaali) ja 3 (vaikea-asteisin): ulostamistiheysosion pisteet (SFS), peräsuoliverenvuoto-osion pisteet (RBS) ja keskitetysti arvioidun endoskopiaosion pisteet (ES).
Induktiohoitoa koskevat tutkimukset (UC‑1 ja UC‑2)
Tutkimuksissa UC-1 ja UC-2 yhteensä 988 potilasta (473 ja 515 potilasta) satunnaistettiin saamaan upadasitinibia 45 mg kerran vuorokaudessa tai lumelääkettä 8 viikon ajan suhteessa 2:1. Nämä potilaat otettiin mukaan tehoanalyysiin. Kaikilla tutkimuksiin otetuilla potilailla oli keskivaikea tai vaikea aktiivinen haavainen paksusuolitulehdus, joka määriteltiin aMS-pistemääräksi 5–9, jossa ES-pistemäärä oli 2–3. Potilaiden aiempi tavanomainen hoito ja/tai aiempi hoito biologisella lääkkeellä oli epäonnistunut vasteen riittämättömyyden, vasteen menetyksen tai hoidon huonon siedettävyyden takia. Vähintään yhden aiemman biologisen hoidon epäonnistuminen (aiempi biologinen hoito epäonnistunut) todettiin 52 %:lla (246/473) ja vastaavasti 51 %:lla (262/515) potilaista. Aiemman tavanomaisen hoidon epäonnistuminen (ilman aiempaa biologisen hoidon epäonnistumista) todettiin 48 %:lla (227/473) ja vastaavasti 49 %:lla (253/515) potilaista.
Lähtötilanteessa 39 % tutkimuksen UC-1 potilaista ja 37 % tutkimuksen UC-2 potilaista sai kortikosteroideja: tutkimuksessa UC-1 metotreksaattia sai 1,1 % potilaista ja aminosalisylaatteja 68 % potilaista, ja tutkimuksessa UC-2 metotreksaattia sai 0,6 % potilaista ja aminosalisylaatteja 69 % potilaista. Tutkimuksissa ei sallittu tiopuriinien samanaikaista käyttöä. Taudin aktiivisuus oli keskivaikea (aMS ≥ 5, ≤ 7) 61 %:lla ja 60 %:lla potilaista ja vaikea (aMS > 7) 39 %:lla ja 40 %:lla potilaista.
Ensisijainen päätetapahtuma oli kliininen remissio aMS-pistemäärän perusteella viikolla 8. Taulukossa 15 esitetään ensisijaiset ja tärkeimmät toissijaiset päätetapahtumat, mukaan lukien kliininen vaste, limakalvon paraneminen, histologis-endoskooppinen limakalvon paraneminen sekä limakalvon syvä paraneminen.
Taulukko 15 Ensisijaiset ja tärkeimmät toissijaiset tehon päätetapahtumat täyttäneiden potilaiden osuudet viikolla 8 induktiohoitoa koskeneissa tutkimuksissa UC-1 ja UC-2
| UC-1 (U-ACHIEVE) | UC-2 (U-ACCOMPLISH) | ||||
Päätetapahtuma | Lume N = 154 | UPA 45 mg N = 319 | Hoitoero (95 %:n lv) | Lume N = 174 | UPA 45 mg N = 341 | Hoitoero (95 %:n lv) |
Kliininen remissioa | 4,8 % | 26,1 % | 21,6 %* (15,8; 27,4) | 4,1 % | 33,5 % | 29,0 %* (23,2; 34,7) |
Aiempi biologinen hoito epäonnistunut+ | 0,4 % | 17,9 % | 17,5 % | 2,4 % | 29,6 % | 27,1 % |
Ei aiempaa biologisen hoidon epäonnistumista+ | 9,2 % | 35,2 % | 26,0 % | 5,9 % | 37,5 % | 31,6 % |
Kliininen vasteb | 27,3 % | 72,6 % | 46,3 %* (38,4; 54,2) | 25,4 % | 74,5 % | 49,4 %* (41,7; 57,1) |
Aiempi biologinen hoito epäonnistunut+ | 12,8 % | 64,4 % | 51,6 % | 19,3 % | 69,4 % | 50,1 % |
Ei aiempaa biologisen hoidon epäonnistumista+ | 42,1 % | 81,8 % | 39,7 % | 31,8 % | 79,8 % | 48,0 % |
Limakalvon paraneminenc | 7,4 % | 36,3 % | 29,3 %* (22,6; 35,9) | 8,3 % | 44,0 % | 35,1 %* (28,6; 41,6) |
Aiempi biologinen hoito epäonnistunut+ | 1,7 % | 27,0 % | 25,3 % | 4,8 % | 37,1 % | 32,3 % |
Ei aiempaa biologisen hoidon epäonnistumista+ | 13,2 % | 46,8 % | 33,6 % | 12,0 % | 51,2 % | 39,2 % |
Histologis-endoskooppinen limakalvon paraneminend | 6,6 % | 30,1 % | 23,7 %* (17,5; 30,0) | 5,9 % | 36,7 % | 30,1 %* (24,1; 36,2) |
Aiempi biologinen hoito epäonnistunut+ | 1,4 % | 22,7 % | 21,3 % | 4,6 % | 30,7 % | 26,1 % |
Ei aiempaa biologisen hoidon epäonnistumista+ | 11,8 % | 38,2 % | 26,4 % | 7,2 % | 42,9 % | 35,7 % |
Limakalvon syvä paraneminene | 1,3 % | 10,7 % | 9,7 %* (5,7; 13,7) | 1,7 % | 13,5 % | 11,3 %* (7,2; 15,3) |
Aiempi biologinen hoito epäonnistunut+ | 0 | 6,5 % | 6,5 % | 1,1 % | 9,2 % | 8,1 % |
Ei aiempaa biologisen hoidon epäonnistumista+ | 2,6 % | 15,4 % | 12,8 % | 2,4 % | 17,9 % | 15,5 % |
Lyhenteet: Lume = lumelääke; UPA = upadasitinibi; aMS = mukautettu Mayo-pisteytys, perustuu Mayo-pisteytykseen (ilman lääkärin yleisarviota) asteikolla 0–9. Asteikkoon sisältyy kolme alaosiota, jotka kaikki pisteytettiin välillä 0 (normaali) ja 3 (vaikea-asteisin): ulostamistiheysosion pisteet (SFS), peräsuoliverenvuoto-osion pisteet (RBS) ja keskitetysti arvioidun endoskopiaosion pisteet (ES). +Potilaita, joilla aiempi biologinen hoito oli epäonnistunut, oli tutkimusten UC-1 ja UC-2 lumeryhmissä 78 ja 89 ja upadasitinibi 45 mg -ryhmissä 168 ja 173; potilaita, joilla ei ollut aiempaa biologisen hoidon epäonnistumista, oli tutkimusten UC-1 ja UC-2 lumeryhmissä 76 ja 85 ja upadasitinibi 45 mg -ryhmissä 151 ja 168. *p < 0,001; mukautettu hoitoero (95 %:n lv) a aMS-pisteytyksen mukaan: SFS ≤ 1 eikä lähtöarvoa suurempi, RBS = 0, ES ≤ 1 ilman kosketusverenvuotoa baMS-pisteytyksen mukaan: ≥ 2 pisteen vähenemä ja ≥ 30 % lähtöarvosta sekä RBS-pistemäärän vähenemä ≥ 1 pisteellä lähtöarvosta tai absoluuttinen RBS-pistemäärä ≤ 1. c ES ≤ 1 ilman kosketusverenvuotoa d ES ≤ 1 ilman kosketusverenvuotoa ja Geboesin pisteet ≤ 3,1 (neutrofiilien infiltraatio < 5 %:iin kryptista, ei kryptien tuhoutumista, eroosiota, haavaumia tai granulaatiokudosta) e ES = 0, Geboesin pisteet < 2 (ei neutrofiileja kryptissa tai lamina propriassa, ei eosinofiilimäärän nousua, ei kryptien tuhoutumista, eroosiota, haavaumia tai granulaatiokudosta) | ||||||
Taudin aktiivisuus ja oireet
Osittainen mukautettu Mayo-pisteytys (paMS) koostuu SFS- ja RBS-pisteistä. paMS-kriteeristöllä oireenmukaisen vasteen määritelmä on ≥ 1 pisteen vähenemä ja ≥ 30 % lähtöarvosta sekä RBS-pistemäärän vähenemä ≥ 1 pisteellä tai absoluuttinen RBS-pistemäärä ≤ 1. paMS-kriteerien mukaista tilastollisesti merkitsevää paranemista lumelääkkeeseen verrattuna oli nähtävissä jo viikolla 2 (UC-1: 60,1 % vs. 27,3 % ja UC-2: 63,3 % vs. 25,9 %).
Pidennetty induktiohoito
Tutkimuksissa UC-1 ja UC-2 yhteensä 125 potilasta, jotka eivät olleet saavuttaneet kliinistä vastetta saatuaan 8 viikon ajan upadasitinibia 45 mg kerran vuorokaudessa, siirtyivät 8 viikkoa kestäneeseen avoimeen induktiohoidon pidennysvaiheeseen. Kun kerran vuorokaudessa annettavaa upadasitinibi 45 mg -hoitoa oli jatkettu vielä toiset 8 viikkoa (yhteensä 16 viikkoa), 48,3 % potilaista oli saavuttanut aMS-kriteerien mukaisen kliinisen vasteen. Niistä potilaista, jotka saavuttivat hoitovasteen saatuaan upadasitinibia 45 mg kerran vuorokaudessa 16 viikon ajan, aMS-kriteerien mukainen vaste säilyi 35,7 %:lla upadasitinibia 15 mg kerran vuorokaudessa ylläpitohoitona saaneista potilaista ja 19,0 % heistä saavutti aMS-kriteerien mukaisen kliinisen remission viikolla 52, kun taas aMS-kriteerien mukainen vaste säilyi 66,7 %:lla upadasitinibia 30 mg kerran vuorokaudessa ylläpitohoitona saaneista potilaista ja 33,3 % heistä saavutti aMS-kriteerien mukaisen kliinisen remission viikolla 52.
Ylläpitohoitoa koskeva tutkimus (UC‑3)
Tutkimuksessa UC-3 tehoanalyysi tehtiin 451 potilaalla, jotka saavuttivat aMS-kriteerien mukaisen kliinisen vasteen saatuaan induktiohoitona upadasitinibia 45 mg kerran vuorokaudessa 8 viikon ajan. Potilaat satunnaistettiin saamaan 15 mg tai 30 mg upadasitinibia tai lumelääkettä kerran vuorokaudessa enintään 52 viikon ajan.
Ensisijainen päätetapahtuma oli aMS-kriteerien mukainen kliininen remissio viikolla 52. Taulukossa 16 esitetään tärkeimmät toissijaiset päätetapahtumat, mukaan lukien kliinisen remission säilyminen, kliininen remissio ilman kortikosteroidihoitoa, limakalvon paraneminen, histologis-endoskooppinen limakalvon paraneminen sekä limakalvon syvä paraneminen.
Taulukko 16 Ensisijaiset ja tärkeimmät toissijaiset tehon päätetapahtumat saavuttaneiden potilaiden osuudet viikolla 52 ylläpitohoitoa koskeneessa tutkimuksessa UC-3
| Lume N = 149 | UPA 15 mg N = 148 | UPA 30 mg N = 154 | Hoitoero 15 mg vs. lume (95 %:n lv) | Hoitoero 30 mg vs. lume (95 %:n lv) |
Kliininen remissioa | 12,1 % | 42,3 % | 51,7 % | 30,7 %* (21,7; 39,8) | 39,0 %* (29,7; 48,2) |
Aiempi biologinen hoito epäonnistunut+ | 7,5 % | 40,5 % | 49,1 % | 33,0 % | 41,6 % |
Ei aiempaa biologisen hoidon epäonnistumista+ | 17,6 % | 43,9 % | 54,0 % | 26,3 % | 36,3 % |
Kliinisen remission säilyminenb | N = 54 22,2 % | N = 47 59,2 % | N = 58 69,7 % | 37,4 %* (20,3; 54,6) | 47,0 %* (30,7; 63,3) |
Aiempi biologinen hoito epäonnistunut | N = 22 13,6 % | N = 17 76,5 % | N = 20 73,0 % | 62,8 % | 59,4 % |
Ei aiempaa biologisen hoidon epäonnistumista | N = 32 28,1 % | N = 30 49,4 % | N = 38 68,0 % | 21,3 % | 39,9 % |
Kliininen remissio ilman kortikosteroidihoitoac | N = 54 22,2 % | N = 47 57,1 % | N = 58 68,0 % | 35,4 %* (18,2; 52,7) | 45,1 %* (28,7; 61,6) |
Aiempi biologinen hoito epäonnistunut | N = 22 13,6 % | N = 17 70,6 % | N = 20 73,0 % | 57,0 % | 59,4 % |
Ei aiempaa biologisen hoidon epäonnistumista | N = 32 28,1 % | N = 30 49,4 % | N = 38 65,4 % | 21,3 % | 37,2 % |
Limakalvon paraneminend | 14,5 % | 48,7 % | 61,6 % | 34,4 %* (25,1; 43,7) | 46,3 %* (36,7; 55,8) |
Aiempi biologinen hoito epäonnistunut+ | 7,8 % | 43,3 % | 56,1 % | 35,5 % | 48,3 % |
Ei aiempaa biologisen hoidon epäonnistumista+ | 22,5 % | 53,6 % | 66,6 % | 31,1 % | 44,1 % |
Histologis-endoskooppinen limakalvon paraneminene | 11,9 % | 35,0 % | 49,8 % | 23,8 %* (14,8; 32,8) | 37,3 %* (27,8; 46,8) |
Aiempi biologinen hoito epäonnistunut+ | 5,2 % | 32,9 % | 47,6 % | 27,7 % | 42,4 % |
Ei aiempaa biologisen hoidon epäonnistumista+ | 20,0 % | 36,9 % | 51,8 % | 16,9 % | 31,8 % |
Limakalvon syvä paraneminenf | 4,7 % | 17,6 % | 19,0 % | 13,0 %* (6,0; 20,0) | 13,6 %* (6,6; 20,6) |
Aiempi biologinen hoito epäonnistunut+ | 2,5 % | 17,2 % | 16,1 % | 14,7 % | 13,6 % |
Ei aiempaa biologisen hoidon epäonnistumista+ | 7,5 % | 18,0 % | 21,6 % | 10,6 % | 14,2 % |
Lyhenteet: Lume = lumelääke; UPA = upadasitinibi; aMS = mukautettu Mayo-pisteytys, perustuu Mayo-pisteytykseen (ilman lääkärin yleisarviota) asteikolla 0–9. Asteikkoon sisältyy kolme alaosiota, jotka kaikki pisteytettiin välillä 0 (normaali) ja 3 (vaikea-asteisin): ulostamistiheysosion pisteet (SFS), peräsuoliverenvuoto-osion pisteet (RBS) ja keskitetysti arvioidun endoskopiaosion pisteet (ES) +Potilaita, joiden aiempi biologinen hoito oli epäonnistunut, oli lumeryhmässä 81, upadasitinibia 15 mg saaneessa ryhmässä 71 ja upadasitinibia 30 mg saaneessa ryhmässä 73. Potilaita, joilla ei ollut aiempaa biologisen hoidon epäonnistumista, oli lumeryhmässä 68, upadasitinibia 15 mg saaneessa ryhmässä 77 ja upadasitinibia 30 mg saaneessa ryhmässä 81. * p < 0,001; mukautettu hoitoero (95 %:n lv) a aMS-pisteytyksen mukaan: SFS ≤ 1 eikä lähtöarvoa suurempi, RBS = 0, ES ≤ 1 ilman kosketusverenvuotoa b aMS-kriteerien mukainen kliininen remissio viikolla 52 niillä potilailla, jotka olivat saavuttaneet kliinisen remission induktiohoidon päättyessä. c aMS-kriteerien mukainen kliininen remissio viikolla 52 ja ei kortikosteroidihoitoa viikkoa 52 välittömästi edeltävien ≥ 90 vuorokauden aikana niillä potilailla, jotka olivat saavuttaneet kliinisen remission induktiohoidon päättyessä. d ES ≤ 1 ilman kosketusverenvuotoa e ES ≤ 1 ilman kosketusverenvuotoa ja Geboesin pisteet ≤ 3,1 (neutrofiilien infiltraatio < 5 %:iin kryptista, ei kryptien tuhoutumista, eroosiota, haavaumia tai granulaatiokudosta). f ES = 0, Geboesin pisteet < 2 (ei neutrofiileja kryptissa tai lamina propriassa, ei eosinofiilimäärän nousua, ei kryptien tuhoutumista, eroosiota, haavaumia tai granulaatiokudosta). | |||||
Taudin oireet
paMS-kriteereillä oireenmukaisen remission, joka määriteltiin SFS-pistemääräksi ≤ 1 ja RBS-pistemääräksi 0, saavutti ajan mittaan viikkoon 52 mennessä suurempi osa upadasitinibia 15 mg tai 30 mg kerran vuorokaudessa saaneista potilaista kuin lumelääkettä saaneista potilaista (kuva 2).
Kuva 2 Osittaisella mukautetulla Mayo-pisteytyksellä oireenmukaisen remission ajan mittaan saavuttaneiden potilaiden osuus ylläpitohoitoa koskevassa tutkimuksessa UC-3
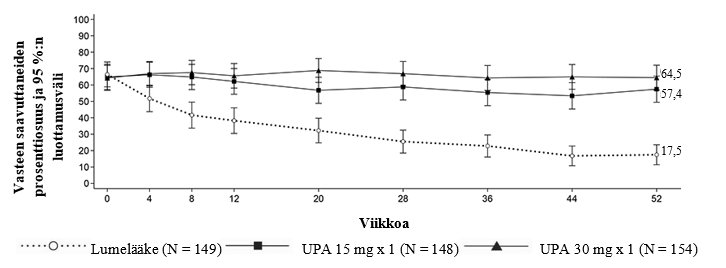
Endoskooppinen arviointi
Endoskooppinen remissio (limakalvon ulkonäön normalisoituminen endoskopiassa) määriteltiin ES-pistemääräksi 0. Viikolla 8 merkitsevästi suurempi osa upadasitinibia 45 mg kerran vuorokaudessa saaneista potilaista kuin lumelääkettä saaneista potilaista oli saavuttanut endoskooppisen remission (UC-1: 13,7 % ja 1,3 %, UC‑2: 18,2 % ja 1,7 %). Tutkimuksessa UC-3 merkitsevästi suurempi osuus upadasitinibia 15 mg tai 30 mg kerran vuorokaudessa saaneista potilaista saavutti endoskooppisen remission viikolla 52 lumelääkettä saaneisiin potilaisiin verrattuna (24,2 % ja 25,9 % vs. 5,6 %). Potilailla, joiden limakalvo oli parantunut induktiohoidon päättyessä, limakalvon paraneminen säilyi viikolla 52 (ES ≤ 1 ilman kosketusverenvuotoa) merkitsevästi suuremmalla osalla upadasitinibia 15 mg tai 30 mg kerran vuorokaudessa saaneista potilaista kuin lumelääkettä saaneista potilaista (61,6 % ja 69,5 % vs. 19,2 %).
Elämänlaatu
Lumelääkettä saaneisiin potilaisiin verrattuna upadasitinibia saaneilla potilailla todettiin merkitsevästi suurempaa ja kliinisesti merkittävää paranemista terveyteen liittyvässä elämänlaadussa, jota mitattiin tulehduksellisia suolistosairauksia koskevan kyselyn (IBDQ) kokonaispisteinä. Paranemista todettiin kaikkien neljän alaosion pisteinä, joita olivat: systeemiset oireet (mukaan lukien väsymys), sosiaalinen toimintakyky, tunne-elämän toimintakyky ja suolisto-oireet (mukaan lukien vatsakipu ja ulostamispakko). Viikolla 8 IBDQ-kokonaispistemäärän muutokset lähtöarvosta olivat upadasitinibia 45 mg kerran vuorokaudessa saaneilla 55,3 ja lumelääkettä saaneilla potilailla 21,7 tutkimuksessa UC‑1 sekä 52,2 ja 21,1 tutkimuksessa UC‑2. Viikolla 52 IBDQ-kokonaispistemäärän muutokset lähtöarvosta olivat 49,2 upadasitinibia 15 mg kerran vuorokaudessa saaneilla potilailla, 58,9 upadasitinibia 30 mg kerran vuorokaudessa saaneilla potilailla ja 17,9 lumelääkettä saaneilla potilailla.
Pitkäkestoinen jatkotutkimus (UC-4)
Potilaat, jotka saavuttivat UC-3 -tutkimuksessa aMS-kriteerien mukaisen kliinisen remission 1 vuoden kohdalla, saivat jatkaa samalla annoksella jatkotutkimuksessa (UC-4). UC-4 -tutkimuksen alkaessa 96 potilasta upadasitinibi 15 mg -ryhmässä ja 146 potilasta upadasitinibi 30 mg -ryhmässä oli kliinisessä remissiossa sekä vastaavasti 49 potilasta ja 82 potilasta endoskooppisessa remissiossa. Tämä populaatio on osittain, mutta ei täysin, päällekkäinen ylläolevassa taulukossa esitetyn populaation kanssa. Taulukko kuvaa niiden potilaiden osuutta, jotka saavuttivat päätetapahtumat viikolla 52 ylläpitohoitoa koskevassa UC-3 -tutkimuksessa. Potilaista, jotka saavuttivat remission UC-3 -tutkimuksessa aMS-kriteereiden mukaan 1 vuoden kohdalla ja joista oli saatavilla tiedot 96 viikon ajalta, 55/70 (78,6 %) upadasitinibi 15 mg -ryhmässä ja 75/89 (84,3 %) upadasitinibi 30 mg -ryhmässä säilytti kliinisen remission ja vastaavasti 22/34 (64,7 %) ja 40/54 (74,1 %) säilytti endoskooppisen remission 96 viikon jatkohoidon jälkeen.
Potilailla, jotka osallistuivat jatkotutkimukseen oltuaan mukana UC-3 -tutkimuksen loppuun (1 vuosi) ja joista oli saatavilla tiedot 96 viikon ajalta, paranemat IBDQ-kokonaispisteissä ja IBDQ-alaosioiden pisteissä säilyivät viikolle 96 asti UC-4 -tutkimuksessa.
Upadasitinibin turvallisuusprofiili pitkäaikaishoidossa oli yhdenmukainen lumekontrolloidun jakson turvallisuusprofiilin kanssa.
Crohnin tauti
Upadasitinibin tehoa ja turvallisuutta arvioitiin kolmessa vaiheen 3 kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa: kahdessa induktiohoitoa koskeneessa tutkimuksessa, CD-1 (U-EXCEED) ja CD-2 (U-EXCEL), ja niiden jälkeen 52 viikon ylläpitohoitoa koskeneessa pitkäkestoisessa jatkotutkimuksessa CD-3 (U-ENDURE). Rinnakkaiset ensisijaiset päätetapahtumat olivat kliininen remissio ja endoskooppinen vaste CD-1- ja CD-2-tutkimuksissa viikolla 12 ja CD-3-tutkimuksessa viikolla 52.
Tutkimuksiin otetut potilaat olivat 18–75-vuotiaita, ja heillä oli keskivaikea tai vaikea aktiivinen Crohnin tauti (CD), jonka määritelmä oli keskimääräinen päivittäinen erittäin pehmeiden tai nestemäisten ulosteiden ulostamistiheys (SF) ≥ 4 ja/tai keskimääräiset päivittäiset vatsakipupisteet (APS) ≥ 2 ja Crohnin taudin keskitetysti tarkistetut yksinkertaiset endoskooppiset pisteet (SES-CD) ≥ 6 tai ≥ 4, mikäli kyseessä oli ainoastaan ileumin alueella esiintyvä tauti ilman ahtaumia. Potilaat, joilla oli oireellisia suolen ahtaumia, suljettiin pois Crohnin taudin tutkimuksista.
Induktiohoitoa koskevat tutkimukset (CD‑1 ja CD‑2)
Tutkimuksissa CD-1 ja CD-2 yhteensä 1 021 potilasta (495 ja 526 potilasta) satunnaistettiin saamaan upadasitinibia 45 mg kerran vuorokaudessa tai lumelääkettä 12 viikon ajan suhteessa 2:1.
Tutkimuksessa CD-1 kaikki potilaat olivat saaneet riittämättömän vasteen vähintään yhdelle biologiselle hoidolle tai eivät sietäneet sitä (aiempi biologinen hoito epäonnistunut). Näistä potilaista 61 % (301/495) oli saanut riittämättömän vasteen vähintään kahdelle biologiselle hoidolle tai eivät sietäneet niitä.
Tutkimuksessa CD-2 45 % (239/526) potilaista oli saanut riittämättömän vasteen vähintään yhdelle biologiselle hoidolle tai eivät sietäneet sitä (aiempi biologinen hoito epäonnistunut) ja 55 % (287/526) potilaista oli saanut riittämättömän vasteen tavanomaisille hoidoille tai ei sietänyt niitä, mutta näin ei ollut tapahtunut biologiselle hoidolle (ei aiempaa biologisen hoidon epäonnistumista).
Lähtötilanteessa tutkimuksessa CD-1 34 % ja tutkimuksessa CD-2 36 % potilaista sai kortikosteroideja, tutkimuksessa CD-1 7 % ja tutkimuksessa CD-2 3 % potilaista sai immunomodulaattoreita ja tutkimuksessa CD-1 15 % ja tutkimuksessa CD-2 25 % potilaista sai aminosalisylaatteja.
Kummassakin tutkimuksessa kortikosteroideja lähtötilanteessa saaneilla potilailla aloitettiin kortikosteroidien purkamisohjelma viikolla 4.
Kumpikin tutkimus sisälsi 12 viikon jatkohoitojakson upadasitinibilla 30 mg kerran vuorokaudessa potilaille, jotka saivat upadasitinibia 45 mg kerran vuorokaudessa ja jotka eivät saavuttaneet SF-/APS-kriteerien mukaista kliinistä vastetta (keskimääräinen päivittäinen erittäin pehmeiden tai nestemäisten ulosteiden ulostamistiheys (SF) vähentynyt ≥ 30 % ja/tai keskimääräiset päivittäiset vatsakipupisteet (APS) vähentyneet ≥ 30 % eikä kumpikaan lähtöarvoa suurempi) viikolla 12.
Taudin kliininen aktiivisuus ja oireet
Tutkimuksissa CD-1 ja CD-2 upadasitinibi 45 mg ‑hoitoa saaneista potilaista merkitsevästi suurempi osuus saavutti rinnakkaisen ensisijaisen päätetapahtuman eli kliinisen remission lumelääkettä saaneisiin verrattuna viikolla 12 (taulukko 17). Hoito tehosi nopeasti, ja teho saavutettiin jo viikolla 2 (taulukko 17).
Kummassakin tutkimuksessa upadasitinibi 45 mg ‑hoitoa saaneet potilaat kokivat uupumuksen lievittyneen merkitsevästi enemmän lähtötilanteesta lumelääkettä saaneisiin verrattuna viikolla 12 FACIT-F-pisteillä mitattuna.
Endoskooppinen arviointi
Tutkimuksissa CD-1 ja CD-2 upadasitinibi 45 mg ‑hoitoa saaneista potilaista merkitsevästi suurempi osuus saavutti rinnakkaisen ensisijaisen päätetapahtuman eli endoskooppisen vasteen lumelääkettä saaneisiin verrattuna viikolla 12 (taulukko 17). Tutkimuksissa CD-1 ja CD-2 suurempi osuus upadasitinibi 45 mg ‑hoitoa saaneista potilaista (14 % ja 19 % vastaavasti) saavutti SES-CD-pisteet 0–2 lumelääkettä saaneisiin verrattuna (0 % ja 5 % vastaavasti).
Taulukko 17 Ensisijaiset ja muut tehon päätetapahtumat saavuttaneiden potilaiden osuudet induktiohoitoa koskeneissa tutkimuksissa CD-1 ja CD-2
Tutkimus | CD-1(U-EXCEED) | CD-2 (U-EXCEL) | ||||
Hoitoryhmä | LumeN = 171 | UPA45 mgN = 324 | Hoitoero(95 %:n lv) | LumeN = 176 | UPA45 mgN = 350 | Hoitoero(95 %:n lv) |
Rinnakkaiset ensisijaiset päätetapahtumat viikolla 12 | ||||||
Kliininen remissioa | 14 % | 40 % | 26 % (19, 33)* | 22 % | 51 % | 29 % (21, 36)* |
Aiempi biologinen hoito epäonnistunut |
|
|
| N = 78 14 % | N = 161 47 % | 33 % (22, 44) |
Ei aiempaa biologisen hoidon epäonnistumista |
|
|
| N = 98 29 % | N = 189 54 % | 26 % (14, 37) |
Endoskooppinen vasteb | 4 % | 35 % | 31 % (25, 37)* | 13 % | 46 % | 33 % (26, 40)* |
Aiempi biologinen hoito epäonnistunut |
|
|
| N = 78 9 % | N = 161 38 % | 29 % (19, 39) |
Ei aiempaa biologisen hoidon epäonnistumista |
|
|
| N = 98 16 % | N = 189 52 % | 36 % (25, 46) |
Muut päätetapahtumat viikolla 12 | ||||||
CDAI:n mukainen kliininen remissioc | 21 % | 39 % | 18 % (10, 26)* | 29 % | 49 % | 21 % (13, 29)* |
Kliininen vaste (CR-100)d | 27 % | 51 % | 23 % (14, 31)* | 37 % | 57 % | 20 % (11, 28)* |
Kliininen remissio ilman kortikosteroidi-hoitoaa,e | N = 60 7 % | N = 108 37 % | 30 % (19, 41)* | N = 64 13 % | N = 126 44 % | 33 % (22, 44)* |
Endoskooppinen remissiof | 2 % | 19 % | 17 % (12, 22)* | 7 % | 29 % | 22 % (16, 28)* |
Limakalvon paranemineng | N = 171 0 % | N = 322 17 % | 17 % (13, 21)*** | N = 174 5 % | N = 349 25 % | 20 % (14, 25)*** |
Alkuvaiheen päätetapahtumat | ||||||
Kliininen remissio viikolla 4a | 9 % | 32 % | 23 % (17, 30)* | 15 % | 36 % | 21 % (14, 28)* |
CR-100 viikolla 2d | 12 % | 33 % | 21 % (14, 28)* | 20 % | 32 % | 12 % (4, 19)** |
Lyhenteet: Lume = lumelääke, UPA = upadasitinibi * p < 0,001, mukautettu hoitoero (95 %:n lv) ** p < 0,01, mukautettu hoitoero (95 %:n lv) *** nimellinen p-arvo < 0,001 UPA vs. lume, mukautettu hoitoero (95 %:n lv) a Keskimääräinen päivittäinen SF-luku ≤ 2,8 ja keskimääräiset päivittäiset APS-pisteet ≤ 1,0 eikä kumpikaan lähtöarvoa suurempi b SES-CD:n vähenemä > 50 % induktiohoitoa koskevan tutkimuksen lähtöarvosta (tai mikäli potilaan SES-CD:n lähtöarvo oli 4 induktiohoitoa koskevassa tutkimuksessa, vähintään 2 pisteen vähenemä induktiohoitoa koskevan tutkimuksen lähtöarvosta) c CDAI < 150 d CDAI-pisteiden väheneminen vähintään 100 pistettä lähtötilanteesta e Steroidia lähtötilanteessa käyttävillä potilailla steroidin lopettaminen ja kliinisen remission saavuttaminen f SES-CD ≤ 4 ja vähintään 2 pisteen vähenemä lähtötilanteesta, eivätkä missään alaosiossa pisteet olleet > 1 yksittäisen muuttujan suhteen g SES-CD:n haavaista limakalvon pintaa kuvaavan alaosion pisteet 0 potilailla, joiden SES-CD:n haavaista limakalvon pintaa kuvaavan alaosion pisteiden lähtöarvo oli ≥ 1 | ||||||
Ylläpitohoitoa koskeva tutkimus (CD-3)
Tutkimuksessa CD-3 tehoanalyysi tehtiin 502 potilaalla, jotka saavuttivat SF-/APS-kriteerien mukaisen kliinisen vasteen saatuaan induktiohoitona upadasitinibia 45 mg kerran vuorokaudessa 12 viikon ajan. Potilaat satunnaistettiin uudelleen saamaan ylläpitohoitona 15 mg tai 30 mg upadasitinibia tai lumelääkettä kerran vuorokaudessa 52 viikon ajan.
Taudin kliininen aktiivisuus ja oireet
Upadasitinibi 15 mg- tai 30 mg ‑hoitoa saaneista potilaista merkitsevästi suurempi osuus saavutti rinnakkaisen ensisijaisen päätetapahtuman eli kliinisen remission lumelääkettä saaneisiin verrattuna viikolla 52 (kuva 3, taulukko 18).
Kuva 3 Kliinisen remission saavuttaneiden potilaiden osuus ylläpitohoitoa koskevassa tutkimuksessa CD-3
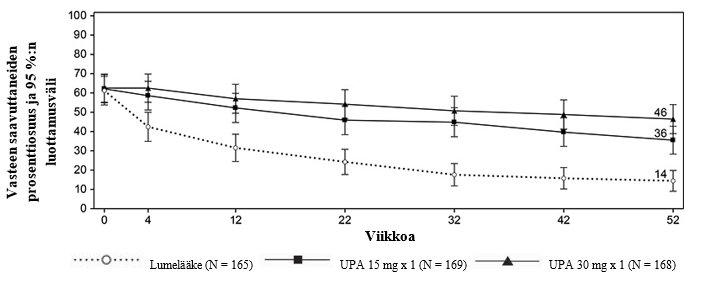
Potilaat, jotka saivat 30 mg upadasitinibia, kokivat uupumuksen lievittyneen merkitsevästi enemmän lähtötilanteesta FACIT-F-pisteillä mitattuna viikolla 52 verrattuna lumelääkettä saaneisiin.
Taulukko18 Ensisijaiset ja muut tehon päätetapahtumat saavuttaneiden potilaiden osuudet ylläpitohoitoa koskevassa tutkimuksessa CD-3
Hoitoryhmä | Lume+ N=165 | UPA 15 mg N = 169 | UPA 30 mg N = 168 | Hoitoero 15 mg vs. lume (95 %:n lv) | Hoitoero 30 mg vs. lume (95 %:n lv) |
Rinnakkaiset ensisijaiset päätetapahtumat | |||||
Kliininen remissioa | 14 % | 36 % | 46 % | 22 % (14, 30)* | 32 % (23, 40)* |
Aiempi biologinen hoito epäonnistunut | N = 126 9 % | N = 124 32 % | N = 127 43 % | 24 % (14, 33) | 34 % (24, 44) |
Ei aiempaa biologisen hoidon epäonnistumista | N = 39 33 % | N = 45 44 % | N = 41 59 % | 12 % (−9, 33) | 26 % (5, 47) |
Endoskooppinen vasteb | 7 % | 28 % | 40 % | 21 % (14, 28)* | 34 % (26, 41)* |
Aiempi biologinen hoito epäonnistunut | N = 126 4 % | N = 124 23 % | N = 127 39 % | 19 % (11, 27) | 35 % (26, 44) |
Ei aiempaa biologisen hoidon epäonnistumista | N = 39 18 % | N = 45 40 % | N = 41 44 % | 22 % (3, 41) | 26 % (7, 45) |
Muut päätetapahtumat | |||||
CDAI:n mukainen kliininen remissioc | 15 % | 37 % | 48 % | 24 % (15, 32)* | 33 % (24, 42)* |
Kliininen vaste (CR-100)d | 15 % | 41 % | 51 % | 27 % (18, 36)* | 36 % (28, 45)* |
Kliininen remissio ilman kortikosteroidihoitoaa,e | 14 % | 35 % | 45 % | 21 % (13, 30)* | 30 % (21, 39)* |
Kliinisen remission säilyminena,f | N = 101 20 % | N = 105 50 % | N = 105 60 % | 32 % (20, 44)* | 40 % (28, 52)* |
Endoskooppinen remissiog | 5 % | 19 % | 29 % | 14 % (8, 21)* | 24 % (16, 31)* |
Limakalvon paraneminenh | N = 164 4 % | N = 167 13 % | N = 168 24 % | 10 % (4, 16)*** | 21 % (14, 27)*** |
Syvä remissioa,i | 4 % | 14 % | 23 % | 10 % (4, 16)** | 18 % (11, 25)* |
Lyhenteet: Lume = lumelääke, UPA = upadasitinibi + Lumelääkeryhmä koostui potilaista, jotka saavuttivat SF-/APS-kriteerien mukaisen kliinisen vasteen upadasitinibi 45 mg -hoidolla induktiohoitoa koskevan tutkimuksen lopussa ja jotka satunnaistettiin saamaan lumelääkettä ylläpitohoidon alussa * p < 0,001, mukautettu hoitoero (95 %:n lv) ** p < 0,01, mukautettu hoitoero (95 %:n lv) *** nimellinen p-arvo < 0,001 UPA vs. lume, mukautettu hoitoero (95 %:n lv) a Keskimääräinen päivittäinen SF-luku ≤ 2,8 ja keskimääräiset päivittäiset APS-pisteet ≤ 1,0 eikä kumpikaan lähtöarvoa suurempi b SES-CD:n vähenemä > 50 % induktiohoitoa koskevan tutkimuksen lähtöarvosta (tai mikäli potilaan SES-CD:n lähtöarvo oli 4 induktiohoitoa koskevassa tutkimuksessa, vähintään 2 pisteen vähenemä induktiohoitoa koskevan tutkimuksen lähtöarvosta) c CDAI < 150 d CDAI-pisteiden väheneminen vähintään 100 pistettä lähtötilanteesta e Ilman kortikosteroidihoitoa 90 päivän ajan ennen viikkoa 52 ja kliinisen remission saavuttaminen. Induktiohoidon lähtötilanteessa kortikosteroidihoitoa saaneiden potilaiden osajoukossa 38 % (N = 63) upadasitinibi 15 mg -ryhmässä, 38 % (N = 63) upadasitinibi 30 mg -ryhmässä ja 5 % (N = 61) lumelääkeryhmässä oli ilman kortikosteroidihoitoa 90 päivän ajan ennen viikkoa 52 ja saavutti kliinisen remission f Määritellään kliinisen remission saavuttamiseksi viikolla 52 potilailla, jotka olivat saavuttaneet kliinisen remission ylläpitohoitoa koskevaan tutkimukseen siirtyessään g SES-CD ≤ 4 ja vähintään 2 pisteen vähenemä lähtötilanteesta, alaosioiden pisteet eivät olleet > 1 minkään yksittäisen muuttujan suhteen h SES-CD:n haavaista limakalvon pintaa kuvaavan alaosion pisteet 0 potilailla, joiden SES-CD:n haavaista limakalvon pintaa kuvaavan alaosion pisteiden lähtöarvo oli ≥ 1 i Kliininen remissio ja endoskooppinen remissio | |||||
Potilaat, jotka eivät saavuttaneet SF-/APS-kriteerien mukaista kliinistä vastetta upadasitinibin induktiohoidolla viikolla 12 CD-1- ja CD-2-tutkimuksissa (122 potilasta), saivat upadasitinibia 30 mg kerran vuorokaudessa toiset 12 viikkoa. Näistä potilaista 53 % saavutti kliinisen vasteen viikolla 24. Jatkohoitoon vastanneista ja upadasitinibi 30 mg -ylläpitohoitoa saaneista potilaista 25 % saavutti kliinisen remission ja 22 % saavutti endoskooppisen vasteen viikolla 52.
Endoskooppinen arviointi
Tutkimuksessa CD-3 merkitsevästi suurempi osuus upadasitinibi 15 mg- tai 30 mg -hoitoa saaneista potilaista saavutti rinnakkaisen ensisijaisen päätetapahtuman eli endoskooppisen vasteen viikolla 52 lumelääkettä saaneisiin potilaisiin verrattuna (taulukko 18). Taulukossa 18 kuvattujen endoskooppisten päätetapahtumien lisäksi suurempi osuus upadasitinibi 15 mg- tai 30 mg -hoitoa saaneista potilaista (11 % ja 21 % vastaavasti) saavutti SES-CD-pisteet 0–2 viikolla 52 lumelääkettä saaneisiin potilaisiin (3 %) verrattuna. Suurempi osuus upadasitinibi 15 mg- tai 30 mg -hoitoa saaneista potilaista (17 % ja 25 % vastaavasti), jotka käyttivät steroidia lähtötilanteessa, saavutti endoskooppisen remission ilman kortikosteroidihoitoa viikolla 52 lumelääkettä saaneisiin potilaisiin (3 %) verrattuna.
Suoliston ulkopuolisten oireiden häviäminen
Suoliston ulkopuolisten oireiden häviäminen havaittiin suuremmalla osuudella upadasitinibi 15 mg ‑hoitoa saaneista potilaista (25 %) ja merkitsevästi suuremmalla osuudella upadasitinibi 30 mg ‑hoitoa saaneista potilaista (36 %) viikolla 52 lumelääkettä saaneisiin potilaisiin (15 %) verrattuna.
Pelastushoito (Rescue treatment)
Tutkimuksessa CD-3 potilaat, joilla ylläpitohoito ei ollut tuottanut riittävää vastetta tai joilla vaste oli menetetty, soveltuivat saamaan upadasitinibi 30 mg -pelastushoitoa. Upadasitinibi 15 mg -ryhmään satunnaistetuista ja upadasitinibi 30 mg -pelastushoitoa vähintään 12 viikon ajan saaneista potilaista 84 % (76/90) saavutti SF-/APS-kriteerien mukaisen kliinisen vasteen ja 48 % (43/90) saavutti kliinisen remission 12 viikon kuluttua pelastushoidon aloittamisesta.
Terveyteen liittyvää elämänlaatua koskevat hoitotulokset
Lumelääkettä saaneisiin potilaisiin verrattuna upadasitinibia saaneet potilaat saavuttivat suuremman paranemisen terveyteen liittyvässä elämänlaadussa, jota mitatiin Inflammatory Bowel Disease Questionnaire (IBDQ) -kyselyn kokonaispisteinä. Paranemista todettiin kaikkien neljän alaosion pisteissä, joita olivat: systeemiset oireet (mukaan lukien väsymys), suolisto-oireet (mukaan lukien vatsakipu ja ulostamispakko), sosiaalinen toimintakyky ja tunne-elämän toimintakyky. Viikolla 12 IBDQ-kokonaispistemäärän muutokset lähtöarvosta olivat upadasitinibia 45 mg kerran vuorokaudessa saaneilla 46,0 ja lumelääkettä saaneilla potilailla 21,6 tutkimuksessa CD‑1 sekä 46,3 ja 24,4 tutkimuksessa CD‑2. Viikolla 52 IBDQ-kokonaispistemäärän muutokset lähtöarvosta olivat 59,3 upadasitinibia 15 mg kerran vuorokaudessa saaneilla potilailla, 64,5 upadasitinibia 30 mg kerran vuorokaudessa saaneilla potilailla ja 46,4 lumelääkettä saaneilla potilailla.
Pediatriset potilaat
Kolmeen maailmanlaajuiseen vaiheen 3 tutkimukseen satunnaistettiin yhteensä 542 iältään 12–17-vuotiasta, keskivaikeaa tai vaikeaa atooppista ihottumaa sairastavaa nuorta, joista 344 arvioitiin ensisijaista analyysiä varten. Ensisijaisessa analyysissä nuoret satunnaistettiin saamaan joko 15 mg (N = 114) tai 30 mg (N = 114) upadasitinibia tai vastaavaa lumelääkettä (N = 116) monoterapiana tai yhdessä paikalliskortikosteroidien kanssa. Teho oli yhdenmukainen nuorilla ja aikuisilla. Nuorten turvallisuusprofiili oli yleisesti ottaen vastaavanlainen kuin aikuisten, ja joidenkin haittatapahtumien, mukaan lukien neutropenian ja vyöruusun, esiintyvyyden lisääntyminen oli annosriippuvaista. Neutropenian esiintyvyys oli kummallakin annoksella hieman suurempaa nuorilla kuin aikuisilla. Vyöruusun esiintyvyys kummallakin annoksella oli aikuisilla suurempi kuin esiintyvyys nuorilla.
Taulukko 19 Upadasitinibin tehoa koskevat tulokset nuorilla viikolla 16
Tutkimus | MEASURE UP 1 | MEASURE UP 2 | AD UP | ||||||
Hoitoryhmä | Lume | UPA 15 mg | UPA 30 mg | Lume | UPA 15 mg | UPA 30 mg | Lume + TCS | UPA 15 mg + TCS | UPA 30 mg + TCS |
Satunnaistettujen nuorten tutkittavien määrä | 40 | 42 | 42 | 36 | 33 | 35 | 40 | 39 | 37 |
Vasteen saaneiden prosenttiosuus (95 %:n lv) | |||||||||
vIGA-AD 0/1a,b | 8 (0,16) | 38 (23,53) | 69 (55,83) | 3 (0,8) | 42 (26,59) | 62 (46,79) | 8 (0,16) | 31 (16,45) | 65 (50,80) |
EASI-75a | 8 (0,17) | 71 (58,85) | 83 (72,95) | 14 (3,25) | 67 (51,83) | 74 (59,90) | 30 (16,44) | 56 (41,72) | 76 (62,90) |
Pahinta kutinaa kuvaava NRS-asteikkoc (≥ 4 pisteen paranema) | 15 (4,27) | 45 (30,60) | 55 (40,70) | 3 (0,8) | 33 (16,50) | 50 (33,67) | 13 (2,24) | 42 (26,58) | 55 (38,72) |
Lyhenteet: UPA= upadasitinibi (RINVOQ); TCS = paikalliskortikosteroidi Potilaat, jotka saivat rescue-lääkettä tai joilta puuttui tietoja, laskettiin potilaiksi, jotka eivät saavuttaneet vastetta. a Perustuu satunnaistettujen tutkittavien määrään b Vasteen saamisen määritelmänä vIGA-AD-pistemäärä 0 tai 1 (”oireeton” tai ”lähes oireeton”) ja ≥ 2 pisteen vähenemä järjestysasteikolla 0–4. c Tulokset arviointikelpoisten potilaiden osajoukossa (potilaat, joiden pahinta kutinaa kuvaava NRS-pistemäärä lähtötilanteessa ≥ 4). | |||||||||
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset RINVOQ-valmisteen käytöstä kroonisen idiopaattisen artriitin (mukaan lukien nivelreuma, nivelpsoriaasi, spondylartriitti ja lastenreuma), atooppisen ihottuman, haavaisen paksusuolitulehduksen ja Crohnin taudin hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Upadasitinibipitoisuus plasmassa on terapeuttisella annosalueella suhteessa annokseen. Vakaan tilan pitoisuudet plasmassa saavutetaan 4 vrk kuluessa, ja kumulaatio on minimaalista toistuvien kerran vuorokaudessa annettujen annosten jälkeen.
Imeytyminen
Suun kautta otettu depotmuotoinen upadasitinibi imeytyy siten, että Tmax-ajan mediaani on 2–4 tuntia. Upadasitinibin anto samanaikaisesti runsasrasvaisen aterian kanssa ei vaikuttanut kliinisesti oleellisesti upadasitinibialtistukseen (AUC suureni 29 % ja Cmax 39–60 %). Kliinisissä tutkimuksissa upadasitinibia annettiin aterioista riippumatta (ks. kohta Annostus ja antotapa). In vitro upadasitinibi on P-gp- ja BCRP-effluksikuljettajien substraatti.
Jakautuminen
Upadasitinibi sitoutuu plasman proteiineihin 52-prosenttisesti. Upadasitinibi jakautuu yhtä lailla plasmaan ja verisolukomponentteihin, kuten veri–plasmasuhde 1,0 ilmaisee.
Metabolia
Upadasitinibin metabolia välittyy CYP3A4:n kautta, ja CYP2D6 saattaa osallistua metaboliaan vähäisessä määrin. Upadasitinibikanta-aineen katsotaan vastaavan farmakologisesta aktiivisuudesta. Ihmisillä toteutetussa tutkimuksessa, jossa käytettiin radioaktiivisesti merkittyä upadasitinibia, muuttumaton upadasitinibi vastasi 79 %:sta plasman kokonaisradioaktiivisuudesta, kun taas päämetaboliitti (mono-oksidaation ja sen jälkeisen glukuronidaation tulos) vastasi 13 %:sta plasman kokonaisradioaktiivisuudesta. Upadasitinibilla ei ole tunnistettu aktiivisia metaboliitteja.
Eliminaatio
Kun [14C]-merkittyä välittömästi vapautuvaa upadasitinibiliuosta annettiin kerta-annoksena, upadasitinibi erittyi lähinnä muuttumattomana kanta-aineena virtsaan (24 %) ja ulosteeseen (38 %). Noin 34 % upadasitinibiannoksesta erittyi metaboliitteina. Upadasitinibin terminaalisen eliminaation puoliintumisajan keskiarvo vaihteli 9–14 tunnin välillä.
Erityisryhmät
Munuaisten vajaatoiminta
Upadasitinibin AUC suureni lievää munuaisten vajaatoimintaa (glomerulusten laskennallinen suodatusnopeus 60–89 ml/min/1,73 m2) sairastavilla 18 %, keskivaikeaa vajaatoimintaa (glomerulusten laskennallinen suodatusnopeus 30–59 ml/min/1,73 m2) sairastavilla 33 % ja vaikeaa vajaatoimintaa (glomerulusten laskennallinen suodatusnopeus 15–29 ml/min/1,73 m2) sairastavilla 44 % verrattuna tutkittaviin, joiden munuaistoiminta oli normaali. Upadasitinibin Cmax oli normaalin munuaistoiminnan ja munuaisten vajaatoiminnan yhteydessä samaa luokkaa. Lievä tai keskivaikea munuaisten vajaatoiminta ei vaikuta kliinisesti oleellisesti upadasitinibialtistukseen (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Lievällä (Child–Pugh-luokka A) ja keskivaikealla (Child–Pugh-luokka B) maksan vajaatoiminnalla ei ole kliinisesti oleellista vaikutusta upadasitinibialtistukseen. Upadasitinibin AUC-arvo oli lievää maksan vajaatoimintaa sairastavilla 28 % suurempi ja keskivaikeaa maksan vajaatoimintaa sairastavilla 24 % suurempi kuin normaalissa maksatoiminnassa. Upadasitinibin Cmax-arvo pysyi muuttumattomana lievässä maksan vajaatoiminnassa, ja keskivaikeassa maksan vajaatoiminnassa se suureni 43 % verrattuna normaaliin maksatoimintaan. Upadasitinibia ei ole tutkittu vaikeaa (Child–Pugh-luokka C) maksan vajaatoimintaa sairastavilla potilailla.
Pediatriset potilaat
Upadasitinibin farmakokinetiikkaa ei ole vielä arvioitu nivelreumaa, nivelpsoriaasia, aksiaalista spondylartriittia, haavaista paksusuolitulehdusta ja Crohnin tautia sairastavilla pediatrisilla potilailla (ks. kohta Annostus ja antotapa).
Upadasitinibin farmakokinetiikka ja vakaan tilan pitoisuudet ovat samaa luokkaa atooppista ihottumaa sairastavilla aikuisilla ja 12–17-vuotiailla nuorilla. 30 kg – < 40 kg painavien nuorten annostus määritettiin populaatiofarmakokineettisen mallinnuksen ja simulaation avulla. Altistusta koskevia kliinisiä tietoja ei ole saatavilla alle 40 kg painaville nuorille.
Upadasitinibin farmakokinetiikkaa atooppista ihottumaa sairastavien pediatristen potilaiden (< 12‑vuotiaiden) hoidossa ei ole osoitettu.
Potilaiden ominaisuudet
Iällä, sukupuolella, painolla, rodulla ja etnisellä taustalla ei ollut kliinisesti merkittävää vaikutusta upadasitinibialtistukseen. Upadasitinibin farmakokinetiikka oli yhdenmukainen nivelreumaa, nivelpsoriaasia, aksiaalista spondylartriittia, jättisoluarteriittia, atooppista ihottumaa, haavaista paksusuolitulehdusta ja Crohnin tautia sairastavilla potilailla.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Upadasitinibi ei ollut karsinogeeninen Sprague-Dawley-rotilla toteutetussa 2 vuoden karsinogeenisuustutkimuksessa, jossa Sprague-Dawley-urosrotille koitui noin 4-kertainen ja Sprague-Dawley-naarasrotille noin 10-kertainen altistus (AUC-arvon perusteella) kliiniseen 15 mg annokseen nähden ja Sprague-Dawley-urosrotille noin 2-kertainen ja naarasrotille noin 5-kertainen altistus kliiniseen 30 mg annokseen nähden sekä Sprague-Dawley-urosrotille noin 1,7-kertainen ja naarasrotille noin 4-kertainen altistus kliiniseen 45 mg annokseen nähden. Upadasitinibi ei ollut karsinogeeninen 26 viikon karsinogeenisuustutkimuksessa CByB6F1-Tg(HRAS)2Jic -transgeenisilla hiirillä.
Upadasitinibi ei ollut mutageeninen eikä genotoksinen perustuen in vitro- ja in vivo -tutkimusten tuloksiin, joissa tarkkailtiin geenimutaatioita ja kromosomipoikkeavuuksia.
Upadasitinibi ei vaikuttanut uros- eikä naarasrottien hedelmällisyyteen altistuksilla, jotka olivat enintään 17-kertaisia urosrotilla ja 34-kertaisia naarasrotilla ihmiselle suositeltuun enimmäisannokseen (maximum recommended human dose, MRHD) 45 mg nähden AUC-arvojen perusteella hedelmällisyyttä ja alkioiden varhaiskehitystä koskeneessa tutkimuksessa. Tässä hedelmällisyystutkimuksessa esiintyneen sikiöiden resorptioiden annosriippuvaisen lisääntymisen ja implantaation jälkeisten alkiokuolemien katsottiin rotalla liittyvän upadasitinibin kehitykseen kohdistuviin/teratogeenisiin vaikutuksiin. Haittavaikutuksia ei havaittu kliinisen altistuksen alittavilla altistuksilla (AUC-arvojen perusteella). Implantaation jälkeisiä alkiokuolemia havaittiin 9-kertaisilla altistuksilla ihmisille suositeltuun enimmäisannokseen 45 mg nähden (AUC-arvojen perusteella).
Eläimillä tehdyissä alkion ja sikiön kehitystä koskevissa tutkimuksissa upadasitinibi oli teratogeeninen sekä rotalla että kanilla. Upadasitinibi lisäsi rotilla luuston epämuodostumia altistuksilla, jotka olivat 1,6-kertaisia, 0,8-kertaisia ja 0,6-kertaisia suhteessa (AUC-pohjaiseen) kliiniseen altistukseen 15 mg, 30 mg ja 45 mg (MRHD) annoksilla. Kaneilla kardiovaskulaaristen epämuodostumien ilmaantuvuuden lisääntymistä havaittiin 15-, 7,6- ja 6-kertaisilla altistuksilla suhteessa kliiniseen altistukseen 15 mg, 30 mg ja 45 mg (MRHD) annoksilla (AUC-arvojen perusteella).
Kun upadasitinibia annettiin imettäville rotille, upadasitinibin pitoisuudet maidossa ajan myötä seurasivat yleensä ottaen plasman pitoisuuksia, ja altistus maidossa oli noin 30 kertaa suurempi verrattuna emon plasmaan. Noin 97 % maidon upadasitinibiperäisestä materiaalista oli kantamolekyyliä upadasitinibia.
Farmaseuttiset tiedot
Apuaineet
Tabletin sisältö
Mikrokiteinen selluloosa
Hypromelloosi
Mannitoli
Viinihappo
Piidioksidi, kolloidinen, vedetön
Magnesiumstearaatti
Kalvopäällyste
Poly(vinyylialkoholi)
Makrogoli
Talkki
Titaanidioksidi (E171)
Musta rautaoksidi (E172) (vain 15 mg vahvuus)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172) (vain 45 mg vahvuus)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
RINVOQ 15 mg depottabletit
Depottabletit läpipainopakkauksessa: 2 vuotta
Depottabletit purkissa: 3 vuotta
RINVOQ 30 mg depottabletit
Depottabletit läpipainopakkauksessa: 2 vuotta
Depottabletit purkissa: 3 vuotta
RINVOQ 45 mg depottabletit
Depottabletit läpipainopakkauksessa: 2 vuotta
Depottabletit purkissa: 3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Säilytä alkuperäisessä läpipainopakkauksessa tai tablettipurkissa. Herkkä kosteudelle. Pidä purkki tiiviisti suljettuna.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
RINVOQ depottabletti
15 mg (L:ei) 28 fol (868,21 €)
30 mg (L:ei) 28 fol (1086,61 €)
45 mg (L:ei) 28 fol (1377,83 €)
PF-selosteen tieto
RINVOQ 15 mg depottabletit
Polyvinyylikloridi/polyeteeni/polyklooritrifluorieteeni/alumiini-kalenteriläpipainopakkaukset, joissa on 28 tai 98 depottablettia tai monipakkaukset, joissa on 84 depottablettia (3 kpl 28 depottabletin pakkauksia).
HDPE-purkit, joissa on kuivausainetta ja polypropeenikorkki, koteloissa, joissa on 30 depottablettia. Pakkauskoko: 1 tablettipurkki (30 depottablettia) tai 3 tablettipurkkia (90 depottablettia).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
RINVOQ 30 mg depottabletit
Polyvinyylikloridi/polyeteeni/polyklooritrifluorieteeni/alumiini-kalenteriläpipainopakkaukset, joissa on 28 tai 98 depottablettia.
HDPE-purkit, joissa on kuivausainetta ja polypropeenikorkki, koteloissa, joissa on 30 depottablettia.
Pakkauskoko: 1 tablettipurkki (30 depottablettia) tai 3 tablettipurkkia (90 depottablettia).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
RINVOQ 45 mg depottabletit
Polyvinyylikloridi/polyeteeni/polyklooritrifluorieteeni/alumiini-kalenteriläpipainopakkaukset, joissa on 28 depottablettia.
HDPE-purkit, joissa on kuivausainetta ja polypropeenikorkki, koteloissa, joissa on 28 depottablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
RINVOQ 15 mg depottabletit
Violetit, pitkänomaiset, kaksoiskuperat depottabletit, koko 14 x 8 mm, toisella puolella painatus ”a15”.
RINVOQ 30 mg depottabletit
Punaiset, pitkänomaiset, kaksoiskuperat depottabletit, koko 14 x 8 mm, toisella puolella painatus ”a30”.
RINVOQ 45 mg depottabletit
Keltaiset tai kirjavan keltaiset, pitkänomaiset, kaksoiskuperat depottabletit, koko 14 x 8 mm, toisella puolella painatus ”a45”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
RINVOQ depottabletti
15 mg 28 fol
30 mg 28 fol
- Ylempi erityiskorvaus (100 %). Abrositinibi ja upadasitinibi (vaikea atooppinen ihottuma): Vaikeaan atooppiseen ihottumaan liittyvän yleisen erytrodermian hoito erityisin edellytyksin (1543).
- Alempi erityiskorvaus (65 %). Upadasitinibi (tulehdukselliset reuma- ja suolistosairaudet): Aikuisten nivelreuman, nivelpsoriaasin, selkärankareuman, aksiaalisen spondylartriitin ja keskivaikean tai vaikean aktiivisen haavaisen paksusuolitulehduksen sekä Crohnin taudin hoito erityisin edellytyksin (298).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Upadasitinibi: Aikuisten nivelreuman, nivelpsoriaasin, selkärankareuman ja keskivaikean tai vaikean aktiivisen haavaisen paksusuolitulehduksen ja jättisoluarteriitin hoito sekä vaikean atooppisen ihottuman hoito erityisin edellytyksin (3029).
RINVOQ depottabletti
45 mg 28 fol
- Alempi erityiskorvaus (65 %). Upadasitinibi (tulehdukselliset reuma- ja suolistosairaudet): Aikuisten nivelreuman, nivelpsoriaasin, selkärankareuman, aksiaalisen spondylartriitin ja keskivaikean tai vaikean aktiivisen haavaisen paksusuolitulehduksen sekä Crohnin taudin hoito erityisin edellytyksin (298).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Upadasitinibi: Aikuisten nivelreuman, nivelpsoriaasin, selkärankareuman ja keskivaikean tai vaikean aktiivisen haavaisen paksusuolitulehduksen ja jättisoluarteriitin hoito sekä vaikean atooppisen ihottuman hoito erityisin edellytyksin (3029).
ATC-koodi
L04AF03
Valmisteyhteenvedon muuttamispäivämäärä
11.06.2026
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi