
JIVI injektiokuiva-aine ja liuotin, liuosta varten 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU
Vaikuttavat aineet ja niiden määrät
Jivi 250 IU injektiokuiva-aine ja liuotin, liuosta varten
Mukana toimitettuun liuottimeen liuottamisen jälkeen yksi ml Jivi 250 IU -valmistetta sisältää noin 100 IU (250 IU / 2,5 ml) ihmisen hyytymistekijä VIII:aa, damoktokogi alfa pegolia.
Jivi 500 IU injektiokuiva-aine ja liuotin, liuosta varten
Mukana toimitettuun liuottimeen liuottamisen jälkeen yksi ml Jivi 500 IU -valmistetta sisältää noin 200 IU (500 IU / 2,5 ml) ihmisen hyytymistekijä VIII:aa, damoktokogi alfa pegolia.
Jivi 1000 IU injektiokuiva-aine ja liuotin, liuosta varten
Mukana toimitettuun liuottimeen liuottamisen jälkeen yksi ml Jivi 1000 IU -valmistetta sisältää noin 400 IU (1 000 IU / 2,5 ml) ihmisen hyytymistekijä VIII:aa, damoktokogi alfa pegolia.
Jivi 2000 IU injektiokuiva-aine ja liuotin, liuosta varten
Mukana toimitettuun liuottimeen liuottamisen jälkeen yksi ml Jivi 2000 IU -valmistetta sisältää noin 800 IU (2 000 IU / 2,5 ml) ihmisen hyytymistekijä VIII:aa, damoktokogi alfa pegolia.
Jivi 3000 IU injektiokuiva-aine ja liuotin, liuosta varten
Mukana toimitettuun liuottimeen liuottamisen jälkeen yksi ml Jivi 3000 IU -valmistetta sisältää noin 1 200 IU (3 000 IU / 2,5 ml) ihmisen hyytymistekijä VIII:aa, damoktokogi alfa pegolia.
Jivi 4000 IU injektiokuiva-aine ja liuotin, liuosta varten
Mukana toimitettuun liuottimeen liuottamisen jälkeen yksi ml Jivi 4000 IU -valmistetta sisältää noin 800 IU (4 000 IU / 5 ml) ihmisen hyytymistekijä VIII:aa, damoktokogi alfa pegolia.
Vahvuuden kansainvälinen yksikkö (IU) määritetään käyttämällä Euroopan farmakopean kromogeenista määritystä.
Jivi-valmisteen spesifinen aktiivisuus on noin 10 000 IU/mg proteiinia.
Vaikuttava aine, damoktokogi alfa pegoli, on aluespesifisesti pegyloitu ihmisen rekombinantti hyytymistekijä VIII, jonka B-domeeni on poistettu ja joka on tuotettu hamsterin munuaissoluissa (BHK, baby hamster kidney -solut) käyttäen 60 kDa:n haaroittunutta polyeteeniglykolia (kaksi 30 kDa:n PEG-osaa). Proteiinin molekyylipaino on noin 234 kDa.
Jivi tuotetaan siten, että solunviljelyprosessissa, puhdistamisessa tai lopullisessa valmisteessa ei ole käytetty lainkaan ihmisestä tai eläimistä peräisin olevaa proteiinia.
Apuaine, jonka vaikutus tunnetaan
Yksi ml injektionestettä sisältää 0,08 mg polysorbaattia 80 (E 433).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Verenvuodon hoito ja ehkäisy hemofilia A:ta (synnynnäinen hyytymistekijä VIII:n puute) sairastavilla, aiemmin hoitoa saaneilla ≥ 7 vuoden ikäisillä potilailla.
Ehto
Hoito pitää toteuttaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito on suoritettava hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Hoidon seuranta
Hoidon aikana suositellaan hyytymistekijä VIII:n pitoisuuden asiamukaista määrittämistä, jotta varmistetaan riittävä hyytymistekijä VIII ‑pitoisuuksien saavuttaminen. Yksittäisten potilaiden vaste hyytymistekijä VIII:lle voi vaihdella johtuen erilaisista puoliintumisajoista ja palautumisesta. Ylipainoisten potilaiden kehonpainoon perustuvaa annosta voi olla tarpeen muuttaa. Erityisesti suurten kirurgisten toimenpiteiden yhteydessä korvaushoidon tarkka seuranta koagulaatioanalyysin (plasman hyytymistekijä VIII:n aktiivisuuden) avulla on välttämätöntä.
Kun hyytymistekijä VIII:n aktiivisuuden määrittämiseen potilaiden verinäytteistä käytetään in vitro aktivoituun partiaaliseen tromboplastiiniaikaan (aPTT) perustuvaa yksivaiheista hyytymismääritystä, sekä käytetty aPTT-reagenssi että määrityksessä käytetty viitestandardi voivat vaikuttaa merkittävästi plasman hyytymistekijä VIII:n aktiivisuuden tuloksiin, jolloin seurauksena voi olla hyytymistekijä VIII:n aktiivisuuden yli- tai aliarviointi. On huomattava, että aPTT-pohjaisessa yksivaiheisessa hyytymismäärityksessä ja kromogeenisessa määrityksessä käytetyillä spesifeillä reagensseilla saatujen määritystulosten välillä voi olla merkitseviä eroja. Tällä on merkitystä seurattaessa Jivi-valmisteen hyytymistekijä VIII -aktiivisuutta ja vaihdettaessa määrityksessä käytettyä laboratoriota ja/tai reagensseja. Tämä koskee myös muokattuja, pitkävaikutteisia hyytymistekijä VIII -valmisteita.
Laboratorioiden, jotka aikovat mitata Jivi-valmisteen aktiivisuutta, on tarkistettava menetelmiensä täsmällisyys. Kenttätutkimuksessa on osoitettu, että Jivi-valmisteen hyytymistekijä VIII:n aktiivisuus voidaan mitata täsmällisesti plasmasta käyttäen joko validoitua kromogeenista substraattimääritystä tai yksivaiheista hyytymismääritystä, jossa käytetään tiettyjä reagensseja. Jivi-valmisteen kohdalla joissakin piioksidipohjaisissa yksivaiheisissa määrityksissä (esim. APTTSP, STA‑PTT) hyytymistekijä VIII:n aktiivisuus plasmanäytteissä voi tulla aliarvioiduksi, kun taas joitakin reagensseja (esim. kaoliinipohjaisia aktivaattoreita sisältäviä) käytettäessä on olemassa yliarvioinnin mahdollisuus.
Hoidon vaikuttavuutta arvioitaessa tärkein elementti on hyytymistekijä VIII:n kliininen vaikutus. Tyydyttävien kliinisten tulosten saavuttamiseksi voi olla tarpeen säätää yksilöllistä annostusta potilastasolla. Jos laskennallisella annoksella ei saavuteta odotettuja hyytymistekijä VIII -pitoisuuksia, tai jos verenvuoto ei ole hallinnassa laskennallisen annoksen annon jälkeen, on syytä epäillä, että potilaan verenkierrossa on hyytymistekijä VIII:n inhibiittoreita tai PEG-vasta-aineita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Korvaushoidon annostus ja kesto riippuvat hyytymistekijä VIII:n puutteen vaikeusasteesta, verenvuodon määrästä ja vuotokohdasta sekä potilaan kliinisestä tilasta.
Hyytymistekijä VIII:n yksikköinä on käytetty kansainvälistä yksikköä (IU), joka on suhteutettu voimassaolevaan hyytymistekijä VIII -valmisteita koskevaan WHO:n konsentraattistandardiin. Plasman hyytymistekijä VIII -aktiivisuus ilmaistaan joko prosentteina (suhteessa normaaliin ihmisen plasmaan) tai mieluummin IU-yksikköinä (suhteessa plasman hyytymistekijä VIII:n kansainväliseen standardiin).
Yksi IU hyytymistekijä VIII:n aktiivisuutta vastaa hyytymistekijä VIII:n määrää 1 ml:ssa normaalia ihmisen plasmaa.
Hoito tarvittaessa
Vaadittavan hyytymistekijä VIII -annoksen laskeminen perustuu kokemusperäiseen havaintoon siitä, että 1 kansainvälinen yksikkö (IU) hyytymistekijä VIII:aa painokiloa kohti nostaa plasman hyytymistekijä VIII -aktiivisuutta 1,5–2,5 % normaalista aktiivisuudesta.
Tarvittava Jivi-annos määritetään seuraavan kaavan avulla:
Tarvittavat yksiköt = paino (kg) x haluttu hyytymistekijä VIII -tason nousu (% tai IU/dl) x havaitun palautumisen käänteisluku (ts. 0,5, jos palautuminen on 2 %).
Annettavan määrän ja antotiheyden määrittämisessä on tavoitteena oltava aina kliinisen tehon saavuttaminen kussakin yksittäisessä tapauksessa.
Hyytymistekijä VIII:n aktiivisuus ei saisi laskea seuraavissa vuototyypeissä taulukossa esitetyn tason alapuolelle (% normaalista) vastaavan hoitojakson aikana. Seuraavasta taulukosta voidaan katsoa ohjeita annostukseen vuotoepisodien aikana ja leikkauksissa:
Taulukko 1: Ohjeita annostukseen vuotoepisodien aikana ja leikkauksissa
| Vuodon voimakkuus / kirurginen toimenpide | Tarvittava hyytymistekijä VIII:n pitoisuus (%) (IU/dl) | Antotiheys (tuntia) / Hoidon kesto (päivää) |
Verenvuoto Varhaisvaiheen nivelvuoto, lihaksen tai suuontelon vuoto | 20 ‑ 40 | Injektio toistetaan 24 ‑ 48 tunnin välein. Vähintään vuorokauden ajan, kunnes kipuna ilmenevä vuotoepisodi on mennyt ohi tai paraneminen tapahtunut |
| Suurehko nivelvuoto, lihaksen vuoto tai hematooma | 30 ‑ 60 | Injektio toistetaan 24 ‑ 48 tunnin välein 3 ‑ 4 vuorokauden ajan tai pitempään, kunnes kipu ja akuutit vaivat lakkaavat. |
Hengenvaaralliset Verenvuodot | 60 ‑ 100 | Injektio toistetaan 8 ‑ 24 tunnin välein, kunnes uhka poistuu. |
Leikkaus Pieni leikkaus mukaan lukien hampaanpoisto | 30 ‑ 60 | 24 tunnin välein vähintään vuorokauden ajan, kunnes paraneminen on tapahtunut. |
| Suuri leikkaus | 80 ‑ 100 (pre- ja postoperatiivisesti) | Injektio toistetaan 12 ‑ 24 tunnin välein, kunnes haava on parantunut riittävästi. Hoitoa jatketaan sen jälkeen vielä vähintään 7 vuorokauden ajan, jotta hyytymistekijä VIII:n aktiivisuus pysyy 30 ‑ 60 %:n (IU/dl) tasolla. |
Profylaksi
Kaikkien hoitopäätösten, joiden avulla pyritään löytämään sopivia profylaktinen annostelu, on perustuttava potilaan yksilöllisten ominaisuuksien ja hoitovasteen pohjalta tehtävään kliiniseen harkintaan. Annoksen ja antotiheyden muutoksia voidaan harkita saavutettujen hyytymistekijä VIII:n pitoisuuksien ja yksilöllisen vuotoalttiuden perusteella. Lisätietoja on kohdissa Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka.
Aikuiset ja vähintään 12-vuotiaat nuoret:
Annos on 45 ‑ 60 IU/kg 5 vuorokauden välein. Potilaan kliinisistä ominaisuuksista riippuen annos voi olla myös 60 IU/kg 7 vuorokauden välein tai 30 ‑ 40 IU/kg kaksi kertaa viikossa (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
7 – < 12-vuotiaat lapset:
Annos on 40 ‑ 60 IU/kg kahdesti viikossa (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Suositeltu aloitusannos on 60 IU/kg kahdesti viikossa.
Ylipainoisilla potilailla suurin sallittu annos/injektio estohoidossa saa olla enintään noin 6 000 IU.
Erityisryhmät
Pediatriset potilaat
Jivi-valmistetta ei ole tarkoitettu aiemmin hoitamattomille potilaille eikä alle 7-vuotiaille potilaille.
Nuoret
Tarvittaessa annettavan hoidon ja estohoidon annostus nuorille potilaille on sama kuin aikuisille potilaille.
Iäkkäät
Käytöstä ≥ 65-vuotiaille potilaille on vain vähän kokemusta.
Antotapa
Jivi on tarkoitettu annettavaksi laskimoon.
Jivi injisoidaan laskimoon 2 ‑ 5 minuutin aikana annettavasta kokonaismäärästä riippuen. Antonopeus on määritettävä potilaan tuntemuksien mukaan (suurin injektionopeus: 2,5 ml/min).
Ks. kohdasta Käyttö- ja käsittelyohjeet ja pakkausselosteesta ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Tunnettu allerginen reaktio hiiren tai hamsterin proteiineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yliherkkyys
Allergiset yliherkkyysreaktiot ovat mahdollisia Jivi-valmistetta käytettäessä. Tämä lääkevalmiste saattaa sisältää hiiri- ja hamsteriproteiinien jäämiä. Yliherkkyysreaktiot voivat liittyä myös PEG-vasta-aineisiin (ks. kappale ”Immuunivaste polyeteeniglykolille (PEG:ille)”). Jos yliherkkyysoireita ilmenee, potilasta on kehotettava keskeyttämään lääkevalmisteen käyttö välittömästi ja ottamaan yhteyttä lääkäriin. Potilaille on kerrottava yliherkkyysreaktioiden varhaisista merkeistä, kuten nokkosihottumasta, yleistyneestä nokkosihottumasta, puristavasta tunteesta rinnassa, vinkuvasta hengityksestä, alentuneesta verenpaineesta ja anafylaksiasta. Tarvittaessa on ryhdyttävä antamaan oireenmukaista hoitoa yliherkkyyteen. Mahdollisen anafylaksian tai sokin tapauksessa hoito on toteutettava sitä koskevien voimassa olevien hoito-ohjeiden mukaisesti.
Inhibiittorit
Hyytymistekijä VIII:aa neutraloivien vasta-aineiden (inhibiittorien) muodostuminen on tunnettu komplikaatio hemofilia A -potilaiden hoidossa. Nämä inhibiittorit ovat yleensä IgG-immunoglobuliineja, jotka estävät hyytymistekijä VIII:n hyytymistoiminnan aktivoitumisen ja joiden määrä ilmaistaan Bethesda-yksikköinä (BU) millilitrassa plasmaa käyttämällä muunneltua Bethesda-määritystä. Inhibiittoreiden muodostumisen riski riippuu taudin vaikeusasteesta ja altistumisesta tekijä VIII:lle. Riski on suurin 50 ensimmäisen altistuspäivän aikana, mutta jatkuu koko eliniän, vaikka riski onkin melko harvinainen. Joskus harvoin inhibiittoreita voi muodostua ensimmäisten 50 altistuspäivän jälkeen.
Inhibiittoreiden muodostumisen kliininen merkitys riippuu inhibiittorin pitoisuudesta. Riittämättömän kliinisen vasteen riski on pienempi, jos inhibiittoreiden pitoisuus on matala.
Hyytymistekijä VIII -valmisteilla hoidettavien potilaiden inhibiittorien esiintyvyyttä on seurattava tarkkaan asianmukaisin kliinisin havainnoin ja laboratoriokokein.
Jos odotettuja hyytymistekijä VIII:n aktiivisuuden plasmapitoisuuksia ei saavuteta, tai jos verenvuotoa ei saada hallintaan asianmukaisella annoksella, on potilaalta testattava hyytymistekijä VIII:n inhibiittorin esiintyminen. Jos potilaalla on korkea inhibiittoripitoisuus, hyytymistekijä VIII -hoito ei ehkä ole tehokasta ja on harkittava muita terapeuttisia vaihtoehtoja. Näiden potilaiden hoidon on tapahduttava sellaisten lääkärien valvonnassa, joilla on kokemusta hemofiliasta ja hyytymistekijä VIII:n inhibiittoreista.
Immuunivaste polyeteeniglykolille (PEG:ille)
PEG-vasta-aineisiin liittyvää kliinistä immuunivastetta, joka ilmenee akuutteina yliherkkyysoireina ja/tai lääkkeen vaikutuksen häviämisenä, on havaittu ensisijaisesti ensimmäisten neljän altistuspäivän aikana. Injektion jälkeiset hyytymistekijä VIII:n matalat pitoisuudet havaittavissa olevien hyytymistekijä VIII:n inhibiittorien puuttuessa osoittavat, että lääkkeen vaikutuksen häviäminen johtuu todennäköisesti PEG-vasta-aineista; sellaisissa tapauksissa Jivi-valmisteen käyttö on lopetettava ja potilaiden on palattava käyttämään aiemmin tehonnutta hyytymistekijä VIII -valmistetta.
Jos terapeuttisen tehon häviämistä epäillään, on potilaalta suositeltavaa testata hyytymistekijä VIII:n inhibiittorit ja hyytymistekijä VIII:n saanto.
PEG:in immuunivasteen riskin havaittiin laskevan huomattavasti iän myötä. Tämä ilmiö voi liittyä immuniteetin kehityksen myötä tapahtuvaan muutokseen, ja vaikka selkeää ikärajapyykkiä riskin muuttumiselle on vaikea määrittää, ilmiötä esiintyy pääasiassa hemofiliaa sairastavilla pienillä lapsilla.
Mahdollisia riskejä potilaille, joilla ilmenee yliherkkyysreaktio pegyloiduille proteiineille, ei tiedetä. Reaktion saaneilla potilailla PEG IgM ‑vasta‑aineiden pitoisuus laski Jivi-valmisteen annon lopettamisen jälkeen ja pitoisuudet laskivat ajan myötä alle määritysrajan. PEG IgM -vasta-aineiden ja muiden muuntelematonta hyytymistekijä VIII:aa sisältävien valmisteiden välillä ei havaittu ristireaktiivisuutta. Kaikkia potilaita pystyttiin hoitamaan onnistuneesti niillä hyytymistekijä VIII ‑valmisteilla, joita he olivat käyttäneet ennen tätä valmistetta.
Koska tämä immuunivaste on ohimenevä ja PEG IgM -vasta-aineet häviävät 4‑6 viikon kuluessa, Jivi-hoidon uudelleenaloitusta voidaan harkita, jos saanto normalisoituu (ks. kohta Annostus ja antotapa). Saantoa on seurattava hoidon uudelleenaloituksen yhteydessä.
Vähentynyt hyytymistekijä VIII:n inkrementaalinen saanto
Hoidon aloittamisen jälkeen on havaittu saannon vähäistä alenemaa (saanto noin 1 IU/dl per IU/kg), mikä saattaa johtua ohimenevistä matalan tiitterin PEG IgM -vasta-aineista pääasiassa lapsilla. Alhainen inkrementaalinen saanto voi mahdollisesti liittyä tehon heikkenemiseen tänä aikana. Pediatristen potilaiden seurantaa, annoksen jälkeisen hyytymistekijä VIII:n aktiivisuuden seuranta mukaan lukien, suositellaan. Jos verenvuotoa ei saada hallintaan suositellulla annoksella ja/tai odotettua hyytymistekijä VIII:n aktiivisuustasoa ei saavuteta ilman FVIII-inhibiittoreita, harkitse annoksen tai antotiheyden muuttamista tai valmisteen käytön lopettamista.
Kardiovaskulaariset tapahtumat
Jos potilaalla on sydämeen ja verisuoniin liittyviä riskitekijöitä, korvaushoito hyytymistekijä VIII:lla voi lisätä sydän- ja verisuonitapahtumien riskiä.
Katetreihin liittyvät komplikaatiot
Jos toimenpide edellyttää keskuslaskimokatetria, on huomioitava keskuslaskimokatetriin liittyvät komplikaatiot, mukaan lukien paikalliset infektiot, bakteremia ja katetrointikohdan tromboosi.
Pediatriset potilaat
Luetellut varoitukset ja varotoimet koskevat aikuisia, nuoria ja 7 – < 12-vuotiaita lapsia.
Jivi-valmistetta ei ole tarkoitettu < 7 vuoden ikäisille eikä aiemmin hoitamattomille potilaille.
Tietoa apuaineista
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Polysorbaatti 80 (E 433)
Tämä lääkevalmiste sisältää 0,2 mg polysorbaattia 80 per 250/500/1 000/2 000/3 000 IU:n injektiopullo ja 0,4 mg polysorbaattia 80 per 4 000 IU:n injektiopullo, joka vastaa 0,08 mg/ml injektioneste liuosta. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Ihmisen hyytymistekijä VIII (rDNA) -valmisteilla ei ole raportoitu olevan yhteisvaikutuksia muiden lääkevalmisteiden kanssa.
Raskaus ja imetys
Raskaus ja imetys
Hyytymistekijä VIII:lla ei ole tehty lisääntymistä koskevia tutkimuksia eläimillä. Koska hemofilia A:n esiintyminen naisilla on harvinaista, ei kokemusta hyytymistekijä VIII:n käytöstä raskauden ja imetyksen aikana ole. Siksi hyytymistekijä VIII:aa tulisi käyttää raskauden ja imetyksen aikana vain silloin, kun se on ehdottoman välttämätöntä.
Hedelmällisyys
Kaneilla ja rotilla tehdyissä Jivi-valmisteen toistuvan annoksen systeemisen toksisuuden tutkimuksissa ei havaittu hoitoon liittyviä vaikutuksia urosten sukuelimissä (ks. kohta Prekliiniset tiedot turvallisuudesta). Vaikutusta ihmisten hedelmällisyyteen ei tiedetä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Jivi-valmisteella ei ole vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Yliherkkyys- ja allergisia reaktioita (esim. angioedeema, pistoskohdan polte ja kirvely, vilunväristykset, punoitus, yleistynyt nokkosihottuma, päänsärky, nokkosihottuma, hypotensio, letargia, pahoinvointi, levottomuus, takykardia, puristava tunne rintakehässä, kihelmöinti, oksentelu, vinkuva hengitys) on havaittu, ja ne voivat joissain tapauksissa johtaa vaikeaan anafylaksiaan (mukaan lukien sokki).
Neutraloivia vasta-aineita (inhibiittoreita) voi kehittyä hemofilia A ‑potilaille, jotka saavat hyytymistekijä VIII ‑hoitoa, kuten Jivi-valmistetta (ks. kohta Farmakodynamiikka). Mikäli tällaisia inhibiittoreita esiintyy, se näkyy riittämättömänä kliinisenä vasteena hoidolle. Tällaisissa tapauksissa on suositeltavaa ottaa yhteyttä erikoistuneeseen hemofiliakeskukseen.
Kliinisissä tutkimuksissa yleisimmin raportoituja haittavaikutuksia aiemmin hoidetuilla potilailla (PTP) olivat päänsärky, yskä ja kuume.
Haittavaikutustaulukko
Turvallisuuspopulaatio koostui kaikkiaan 256 potilaasta, jotka osallistuivat neljään keskeiseen faasin I ja III tutkimukseen (yksi faasin I tutkimus ja kolme faasin III tutkimusta): 148 nuorta/aikuista ja 108 pediatrista potilasta < 12-vuotiaita.
Jivin altistuspäivien lukumäärän mediaani potilasta kohden oli 195 (min-max: 1-698) kaikilla potilailla kliinisissä tutkimuksissa.
Kokonaisuudessaan kaikissa tutkimuksissa 75 potilaan (joista 39 oli alle 12-vuotiaita) hoitoa seurattiin yli 5 vuoden ajan. Lisätietoja kliinisistä tutkimuksista on kohdassa Farmakodynamiikka.
Alla oleva taulukko on MedDRA-elinluokituksen mukainen (elinjärjestelmä ja suositeltava termi). Esiintyvyys on arvioitu seuraavan esitystavan mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000).
Haittavaikutukset esitetään kussakin esiintyvyysluokassa haittavaikutusten vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2: Haittavaikutusten esiintyvyys kliinisissä tutkimuksissa
| Elinjärjestelmä (MedDRA) | Haittavaikutukset | Esiintyvyys |
| Veri ja imukudos | Hyytymistekijä VIII:n inhibitio | Melko harvinainen (PTP)a |
| Immuunijärjestelmä | Yliherkkyys | yleinen |
| Psyykkiset häiriöt | Unettomuus | yleinen |
| Hermosto | Päänsärky | hyvin yleinen |
| Huimaus | yleinen | |
| Dysgeusia | melko harvinainen | |
| Verisuonisto | Punoitus | melko harvinainen |
| Hengityselimet, rintakehä ja välikarsina | Yskä | yleinen |
| Ruoansulatuselimistö | Vatsakipu, pahoinvointi, oksentelu | yleinen |
| Iho ja ihonalainen kudos | Eryteemac, ihottumad | yleinen |
| Kutina | melko harvinainen | |
| Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan reaktiot b, kuume | yleinen |
PTP = aiemmin hoidetut potilaat
aEsiintyvyys perustuu kaikilla hyytymistekijä VIII ‑valmisteilla tehtyihin tutkimuksiin, joihin osallistui vaikeaa hemofilia A:ta sairastavia potilaita.
bSisältää seuraavat: injektiokohdan kutina, injektiokohdan ihottuma, infuusiokohdan kipu ja suonipunktiokohdan kutina
cSisältää seuraavat: eryteema ja erythema multiforme
dSisältää seuraavat: ihottuma ja papulaarinen ihottuma
Turvallisuusprofiilissa ei tapahtunut muutoksia PROTECT VIII- ja PROTECT Kids ‑jatkotutkimusten aikana.
Valikoitujen haittavaikutusten kuvaus
Immunogeenisuus
Immunogeenisuutta arvioitiin Jivi-valmisteella tehtyjen kliinisten tutkimusten aikana 159 aiemmin hoidetulla (mukaan lukien leikkauspotilaat) nuorella (≥ 12 – < 18-vuotiaat, n = 14) ja aikuisella (n = 145), joilla oli diagnosoitu vaikea hemofilia A (FVIII:C < 1 %) ja jotka olivat aiemmin altistuneet hoidolle ≥ 150 päivän ajan, sekä 108 pediatrisella < 12-vuotiaalla PTP-potilaalla (n = 60 iältään 7 – < 12‑vuotiaita), jotka olivat aiemmin altistuneet hoidolle ≥ 50 päivän ajan.
Hyytymistekijä VIII:n inhibiittorit
Tutkimuksissa ei havaittu de novo tai vahvistettuja inhibiittoritapauksia hyytymistekijä VIII:aa vastaan. Yksi vahvistamaton positiivinen tulos, jossa havaittiin matala tiitteri hyytymistekijä VIII ‑inhibiittoria (1,7 BU/ml), raportoitiin eräällä aikuispotilaalla leikkauksen jälkeen.
PEG-vasta-aineet
Kahdella potilaalla havaittiin PEG:iä vastaan kohdistuvaa immunogeenisuutta, johon sisältyi spesifisten IgM PEG -vasta-aineiden kehittymistä. Yhdellä > 12-vuotiaalla potilaalla immuunivasteeseen liittyi kliininen yliherkkyysreaktio neljän Jivi-injektion jälkeen. PEG-vasta-aineet hävisivät, kun Jivi-hoito lopetettiin.
Yhdelle > 7-vuotiaalle lapselle kehittyi neljän ensimmäisen altistuspäivän aikana korkean tiitterin neutraloivia PEG IgM -vasta-aineita, joihin liittyi lääkkeen tehon häviäminen. Vasta-aineet hävisivät hoidon lopettamisen jälkeen, ja Jivi-hoito aloitettiin turvallisesti uudelleen 2 kuukautta myöhemmin.
Joillakin potilailla havaittiin neljän ensimmäisen altistuspäivän aikana ohimeneviä matalan tiitterin IgM-isotyypin PEG-vasta-aineita, joihin liittyi saannon lievää alenemaa.
Lääkkeen tehon heikkenemiseen tai yliherkkyyteen johtavaa kliinistä immuunivastetta PEG:lle ei havaittu 5. altistuspäivästä jatkotutkimuksien loppuun asti.
Pediatriset potilaat PROTECT Kids -tutkimuksessa
Päätökseen saadussa kliinisessä tutkimuksessa, johon osallistui 73 pediatrista, aiemmin hoitoa saanutta (PTP) < 12-vuotiasta (44 < 6‑vuotiasta PTP-potilasta, 29 6–< 12-vuotiasta PTP-potilasta), alle 6-vuotiailla lapsilla havaittiin haittavaikutuksia, jotka johtuivat PEG:in aikaansaamasta immuunivasteesta. Tästä ikäryhmästä eli alle 6-vuotiaiden potilaiden ikäryhmästä 10 potilaalla 44:stä (23 %:lla) havaittiin neljän ensimmäisen altistuspäivän aikana PEG-vasta-aineiden neutraloivasta vaikutuksesta johtuva lääkkeen vaikutuksen häviäminen. Kolmella 44:stä potilaasta (7 %:lla) lääkkeen vaikutuksen häviämiseen liittyi yliherkkyysreaktioita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Mitään PEG:in aiheuttaman immuunivasteen laukaisevia tekijöitä tai ennusmerkkejä ei pystytty tunnistamaan.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa oli yksi yliannostustapaus. Haittavaikutuksia ei raportoitu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: veren hyytymistekijät: veren hyytymistekijä VIII, ATC-koodi: B02BD02.
Vaikutusmekanismi
Hyytymistekijä VIII / von Willebrandin tekijä -kompleksi käsittää kaksi molekyyliä (hyytymistekijä VIII ja von Willebrandin tekijä), joiden fysiologinen tehtävä on erilainen. Hemofiliapotilaalle annettaessa hyytymistekijä VIII sitoutuu potilaan von Willebrandin tekijään. Aktivoitunut hyytymistekijä VIII toimii aktivoituneen hyytymistekijä IX:n kofaktorina kiihdyttäen hyytymistekijä X:n muuttumista aktivoituneeksi hyytymistekijä X:ksi. Aktivoitunut hyytymistekijä X muuttaa protrombiinin trombiiniksi. Trombiini muuttaa puolestaan fibrinogeenin fibriiniksi ja hyytyminen voi tapahtua. Hemofilia A on sukupuoleen sidottu perinnöllinen veren hyytymishäiriö, joka johtuu hyytymistekijä VIII:C:n määrän vähenemisestä tai puutoksesta ja aiheuttaa verenvuotoa niveliin, lihaksiin ja sisäelimiin, joko spontaanisti tai vamman tai leikkauksen seurauksena. Korvaushoidon ansiosta hyytymistekijä VIII:n tasot kohoavat, jolloin hyytymistekijän puutos ja vuototaipumus tilapäisesti korjaantuvat.
Damoktokogi alfa pegoli on rFVIII:n pegyloitu muoto. Spesifinen pegylointi pienentää hyytymistekijä VIII:n puhdistumaa, mikä johtaa pidentyneeseen puoliintumisaikaan. Samalla rFVIII-molekyylin, jonka B-domeeni on poistettu, normaalit toiminnot säilyvät (ks. kohta Farmakokinetiikka).
Damoktokogi alfa pegoli ei sisällä von Willebrandin tekijää.
Kliininen teho ja turvallisuus
Kliiniset tutkimukset
Kaikkiaan 267 aiemmin hoitoa saanutta potilasta, joilla on vaikea hemofilia A, altistui hoidolle kliinisessä tutkimusohjelmassa, johon kuului yksi faasin I tutkimus ja kolme faasin II/III tutkimusta. Tutkimushenkilöistä 219 oli ≥ 7-vuotiaita.
Faasi II/III
PROTECT VIII: Jivi-valmisteen farmakokinetiikkaa, turvallisuutta ja tehoa arvioitiin tarvittaessa käytettävässä hoidossa, estohoidossa kolmella eri annostelulla (30–40 IU/kg kaksi kertaa viikossa, 45–60 IU/kg 5 vuorokauden välein ja 60 IU/kg 7 vuorokauden välein), sekä hemostaasissa suurten leikkausten aikana monikansallisessa, avoimessa, kontrolloimattomassa, osittain satunnaistetussa tutkimuksessa, joka suoritettiin sovitun lastenlääkkeitä koskevan tutkimussuunnitelman (Paediatric Investigation Plan) mukaisesti. Jatkotutkimukseen otettiin mukaan potilaita, jotka olivat olleet mukana päätutkimuksen loppuun asti. Ensisijainen tehoa mittaava muuttuja oli vuosittainen vuotomäärä (ABR, annualized bleed rate).
134 aiemmin hoitoa saanutta miespotilasta (joista 13 oli iältään 12–17-vuotiaita) sai vähintään yhden Jivi-injektion estohoitoon (n=114) tai tarvittaessa annettavaan hoitoon (n=20) 36 viikon ajan. Tutkimuksen jatko-osan aikana hoitoa sai kaikkiaan 121 potilasta, 107 potilasta saivat estohoitoa ja 14 potilasta tarvittaessa annettavaa hoitoa. 36 potilasta saivat estohoitoa > 5 vuotta 7,0 vuoteen asti. Hoidon mediaanikesto oli 3,9 vuotta (0,8–7,0 vuotta) kaikilla 121 potilaalla. Hemostaasi arvioitiin leikkausosassa 20 suuren leikkauksen aikana 17 potilaalla.
Faasi III
PROTECT Kids: Jivi-valmisteen farmakokinetiikkaa, turvallisuutta ja tehoa kolmella eri annostelulla (kaksi kertaa viikossa, 5 vuorokauden välein ja 7 vuorokauden välein) sekä vuotojen hoitoa arvioitiin monikansallisessa, kontrolloimattomassa, avoimessa tutkimuksessa 73 pediatrisella potilaalla (< 12-vuotiaat) 50 altistuspäivän ja vähintään 6 kuukauden ajan. Tämä tutkimus on tehty hyväksytyn lastenlääkkeitä koskevan tutkimussuunnitelman (Paediatric Investigation Plan) mukaisesti. 61 potilasta (83,6 %) oli mukana päätutkimuksen loppuun asti ja 59 potilasta osallistui myös valinnaiseen jatkotutkimukseen. Tutkimuksen kokonaismediaaniaika oli 5,8 vuotta (vaihteluväli 1,0-6,6 vuotta).
Alfa PROTECT: Yhdellä hoitoryhmällä toteutetussa avoimessa tutkimuksessa Jivi-infuusioiden turvallisuutta arvioitiin estohoidossa ja verenvuotojen hoidossa vaikeaa hemofilia A:ta sairastavilla, aiemmin hoitoa saaneilla 7 – < 12‑vuotiailla lapsilla. Tutkimuksessa selvitettiin polyetyleeniglykoliin (PEG) liittyvästä immuunireaktiosta johtuvan mahdollisen yliherkkyyden ja lääkkeen tehon häviämisen riskiä neljän ensimmäisen Jivi-altistuksen yhteydessä. Potilaat saivat estohoitoa 6 kuukauden ajan, ja heillä oli tilaisuus jatkaa hoitoa 18 kuukauden pituisessa jatkotutkimuksessa.
Estohoito ≥ 12-vuotiailla potilailla
Päätutkimusjakson aikana potilaille annettiin Jivi-estohoitoa 2 kertaa viikossa (n=24) tai heidät satunnaistettiin saamaan Jivi-hoitoa 5 vuorokauden välein (n=43) tai 7 vuorokauden välein (n=43) tai he saivat tarvittaessa annettavaa Jivi-hoitoa (n=20). 99 potilasta 110:sta (90 %) pysyi suunnitellussa annostelussa. 11 potilasta 7 vuorokauden välein annettavan hoidon tutkimushaarassa siirtyi tiheämmin annettavaan hoitoon. Mediaaniannos kaikissa estohoito-ohjelmissa oli 46,9 IU/kg/injektio. Mediaani (Q1; Q3) ABR estohoidon aikana oli 2,09 (0,0; 6,1) kaikkien verenvuotojen osalta ja 0,0 (0,0; 4,2) spontaanien verenvuotojen osalta verrattuna tarvittaessa annetun hoidon ryhmässä havaittuun kokonaisvuotomäärään 23,4 (18; 37). Estohoitoryhmissä 42:lla 110:stä (38,2 %) ei ollut mitään verenvuototapahtumaa.
Jatkotutkimuksen aikana (mediaanikesto 3,2 vuotta, välillä 0,1–6,3 vuotta), 23 potilasta sai hoitoa 2 kertaa viikossa, 33 potilasta 5 vuorokauden välein, 23 potilasta 7 vuorokauden välein koko jatkotutkimuksen ajan ja 28 potilasta vaihtoi annostelua. Estohoidon mediaaniannos oli 47,8 IU/kg. Vuotojen (ABR) kokonaismäärän mediaani (Q1; Q3) oli 1,49 (0,4; 4,8), spontaanien verenvuotojen määrä oli 0,75 (0,0; 2,9) estohoitoryhmissä yhteensä ja ABR-kokonaismäärä oli 34,1 tarvittaessa hoitoa saaneiden ryhmässä.
On huomioitava, että ABR-luku ei ole vertailukelpoinen eri hyytymistekijöiden konsentraattien ja eri kliinisten tutkimusten välillä.
Verenvuodon hoito
Päätutkimuksen aikana Jivi-valmisteella hoidetuista 702 verenvuototapahtumasta 636 (90,6 %) hoidettiin 1 tai 2 injektiolla, joista 81,1 % yhdellä injektiolla. Mediaaniannos/injektio oli 31,7 (vaihteluväli 14‑62) IU/kg. Jatkotutkimuksen aikana Jivi-valmisteella hoidettiin 1 902 verenvuotoa ja 94,0 % saatiin hallintaan 1 tai 2 injektiolla, joista 84,9 % yhdellä injektiolla. Mediaaniannos oli 37,9 IU/kg/injektio (vaihteluväli 15‑64 IU/kg/injektio).
Perioperatiivinen hallinta
Kaikkiaan 20 suurta kirurgista toimenpidettä suoritettiin ja arvioitiin 17 potilaalla. Suurten leikkausten mediaani kokonaisannos oli 219 IU/kg (vaihteluväli: 50–1 500 IU/kg, mukaan lukien enintään 3 viikon pituinen leikkauksen jälkeinen jakso). Perioperatiivinen hemostaattinen teho arvioitiin hyväksi tai erinomaiseksi kaikkien suurten leikkausten aikana.
Lisäksi suoritettiin 34 pientä leikkausta 19 potilaalle. Hemostaasi arvioitiin hyväksi tai erinomaiseksi kaikissa tapauksissa, jotka olivat käytettävissä tässä arvioinnissa.
< 12-vuotiaat pediatriset potilaat
Jivi-valmistetta ei ole tarkoitettu käytettäväksi alle 7-vuotiaille lapsille (ks. kohdasta Annostus ja antotapa tietoa käytöstä pediatrisille potilaille).
PROTECT Kids: Faasin III tutkimuksessa kaikkiaan 73 pediatrista, aiemmin hoitoa saanutta potilasta (44 < 6-vuotiasta ja 29 6– < 12‑vuotiasta potilasta) sai estohoitoa joko kaksi kertaa viikossa, 5 vuorokauden välein tai 7 vuorokauden välein. Niillä 53 potilaalla, jotka olivat mukana päätutkimuksen loppuun asti, vuosittaisten verenvuotojen mediaani (Q1, Q3) oli 2,87 (1,1; 6,1) ja spontaani ABR 0,0 (0,0; 2,6). Verenvuotojen hoidossa vuodoista 84,4 % saatiin hoidettua 1 injektiolla ja 91,9 % vuodoista saatiin hoidettua 1 tai 2 injektiolla.
11 potilasta < 6-vuotiaiden ikäryhmästä jätettiin kesken tutkimuksen pois, koska heille ilmaantui PEG-immuunivaste, joka liittyi tehon häviämiseen ja/tai yliherkkyysreaktion ilmenemiseen ensimmäisten neljän altistuspäivän aikana.
Jatkotutkimukseen jatkaneilla 59 potilaalla ABR-kokonaismediaani (Q1;Q3) jatkotutkimuksen aikana oli 1,64 (0,5;3,1). 30 ≥ 12-vuotilla potilailla ABR-mediaani (Q1;Q3) oli 1,76 (0,5;3,3) jatkotutkimuksen lopussa.
Alfa PROTECT: Kaikkiaan 35 aiemmin hoitoa saanutta potilasta (7 – < 12‑vuotiaita) sai estohoitoa kaksi kertaa viikossa (40 60 IU/kg); mediaaniannos oli 55 IU/kg. Vuosittaisen vuotomäärän mediaani (Q1, Q3) tehopopulaatiossa (32 potilasta) oli 0,0 (0,0; 1,9). Verenvuodoista 95,2 % saatiin hoidettua 1 tai 2 injektiolla. Yhdellä potilaalla ilmeni PEG-immuunivaste, johon liittyi lääkkeen tehon häviäminen, neljän ensimmäisen altistuspäivän aikana. Potilas keskeytti hoidon 2 kuukaudeksi, vasta-aineet hävisivät, ja hoito voitiin aloittaa uudelleen.
Farmakokinetiikka
Jivi-valmisteen farmakokinetiikkaa verrattiin hyytymistekijä VIII:n farmakokinetiikkaan vaihtovuoroisessa faasin I tutkimuksessa. Faasin II/III tutkimuksessa farmakokinetiikkaa arvioitiin myös 12 potilaalla, joiden ikä oli 7 – < 12 vuotta [PROTECT Kids] ja 22 potilaalla, joiden ikä oli ≥ 12 vuotta [PROTECT VIII]; näistä potilaista 16:lla arviointi tehtiin 6 kuukauden estohoidon jälkeen.
Farmakokineettisistä tiedoista (kromogeenisen määrityksen perusteella) ilmeni, että Jivi-valmisteella oli pienempi puhdistuma (CL), minkä seurauksena sen terminaalinen puoliintumisaika on 1,4-kertaa pidempi ja annosvakioitu AUC on 1,4-kertaisesti suurempi vertailussa käytettyyn hyytymistekijä VIII -valmisteeseen verrattuna. Annosriippuvaista kasvua havaittiin 25 ja 60 IU/kg:n annosten välillä, mikä osoittaa annoksen lineaarisuutta 25 IU/kg:n ja 60 IU/kg:n annosten välillä.
Taulukossa 3 esitetään yhteenveto farmakokineettisistä muuttujista, jotka todettiin 60 IU/kg suuruisen kerta-annoksen jälkeen 12 potilaalla, joiden ikä oli 7 – < 12 vuotta, ja 22 potilaalla, joiden ikä oli ≥ 12 vuotta. Toistetuissa farmakokineettisissä mittauksissa potilailla, joiden ikä oli ≥ 12 vuotta, ei ilmennyt merkityksellisiä farmakokineettisiä muutoksia pitkäkestoisen hoidon jälkeen.
Taulukko 3: Jivi-valmisteen farmakokineettiset parametrit (geometrinen keskiarvo (% CV) ja aritmeettinen keskiarvo (±SD)) 60 IU/kg:n kerta-annoksen antamisen jälkeen tehdyn kromogeenisen määrityksen perusteella
| Parametrit (yksiköt) | Jivi ≥ 12-vuotiaat potilaat N=22 PROTECT VIII | Jivi 7 – < 12-vuotiaat potilaat N = 12 PROTECT Kids |
| AUC (IU*h/dl) | 3 710 (33,8) 3 900 ± 1280 | 2 842 (20,3) 2 892,8 ± 546,83 |
| AUC, norm (h*kg/dl) | 62,5 (33,7) 65.7 ± 21.4 | 47,5 (19,6) 48,3 ± 8,9 |
| Cmax (IU/dl) | 163 (14,7) 164 ± 23.8 | 128 (19,9) 130 ± 25,0 |
| t½ (h) | 17,1 (27,1) 17.6 ± 4.26 | 15,6 (23,5) 16,0 ± 3,6 |
| MRTIV (h) | 24,4 (27,5) 25.2 ± 6.19 | 23,5 (24,4) 24,2 ± 5,7 |
| Vss (dl/kg) | 0,391 (16,3) 0,396 ± 0,0631 | 0,496 (20,2) 0,505 ± 0,099 |
| CL (dl/h/kg) | 0,0160 (33,7) 0,0168 ± 0,00553 | 0,0211 (19,6) 0,0214 ± 0,00433 |
AUC: käyrän alle jäävä pinta-ala; AUC, norm: annosvakioitu AUC; Cmax: lääkkeen maksimipitoisuus; t½: terminaalinen puoliintumisaika; MRT IV: keskimääräinen viipymäaika laskimoon annon jälkeen; VSS: näennäinen jakautumistilavuus vakaassa tilassa; CL: puhdistuma
Inkrementaalinen saanto määritettiin 131 potilaalta, joiden ikä oli ≥ 12 vuotta, useina ajankohtina. Mediaani (Q1; Q3) saanto oli 2,6 (2,3; 3,0) mitattuna kromogeenisella määrityksellä. Inkrementaalisen saannon mediaani (vaihteluväli) 57 potilaalla, joiden ikä oli 7 – < 12 vuotta, oli 1,9 (1,1‑3,8) mitattuna kromogeenisella määrityksellä.
Käytettävissä olleiden hyytymistekijä VIII:n mittausten (tiheästä farmakokineettisestä näytteenotosta ja kaikista saantonäytteistä) perusteella kehitettiin kaikkien 3 kliinisen tutkimuksen aikana populaatiofarmakokineettinen malli, jolloin voitiin laskea farmakokineettiset parametrit eri tutkimuksissa mukana olleille potilaille. Seuraavassa taulukossa 4 esitetään farmakokineettiset parametrit populaatiofarmakokineettiseen malliin perustuen.
Taulukko 4: Populaatiofarmakokineettiseen malliin perustuvat farmakokineettiset parametrit (geometrinen keskiarvo [% CV]), jotka on saatu kromogeenisen määrityksen avulla
| Farmakokineettinen parametri (yksikkö) | 7 – < 12-vuotiaat N = 25 | 12–< 18-vuotiaat N=12 | ≥ 18-vuotiaat N=133 | Yhteensä (≥ 12-vuotiaat) N=145 |
| AUC (IU*h/dl) | 2 694 (23) | 3 341 (34,2) | 4 052 (31,1) | 3 997 (31,6) |
| AUC, norm (h*kg/dl) | 44,9 (23) | 57,4 (32,6) | 67,5 (30,6) | 66,6 (31,0) |
| t1/2 (h) | 15,0 (19,3) | 16,8 (25,2) | 17,4 (28,8) | 17,4 (28,4) |
| Vss (dl/kg) | 0,481 (15,3) | 0,423 (15,5) | 0,373 (15,6) | 0,376 (15,9) |
| CL (dl/h/kg) | 0,0223 (22,9) | 0,0174 (34,2) | 0,0148 (31,1) | 0,0150 (31,6) |
*AUC laskettu annokselle 60 IU/kg
Prekliiniset tiedot turvallisuudesta
Jivi-valmistetta arvioitiin farmakologisissa, kerta- ja toistuvan annoksen tutkimuksissa sekä nuorille yksilöille tehdyissä toksisuustutkimuksissa rotilla ja kaneilla. Pitkäkestoisessa 6 kuukauden kroonista toksisuutta koskevassa tutkimuksessa ei havaittu viitteitä PEG:n kertymiseen tai muita Jivi-valmisteen antamiseen liittyviä vaikutuksia. Lisäksi kahdella lajilla tehtiin 4 viikon toksisuustutkimuksia, joissa käytettiin Jivi-valmisteen PEG-osaa. PEG-linkkeriosaa testattiin myös useissa vakiomallisissa in vivo ja in vitro ‑genotoksisuustutkimuksissa, joissa ei havaittu genotoksisuuspotentiaalia. Näissä tutkimuksissa ei ilmennyt mitään turvallisuusriskejä ihmisille.
Kerta-annostutkimuksissa rotilla, joissa käytettiin radioaktiivisesti merkittyä PEG-osaa, ei havaittu merkkejä radioaktiivisuuden jäämisestä tai irreversiibelistä sitoutumisesta eläimen ruumiiseen. Varsinkaan aivoissa ei havaittu radioaktiivisuuden jäämiä, mikä osoittaa, että radioaktiivisesti merkitty yhdiste ei ole läpäissyt veri‑aivoestettä. Rotilla tehdyissä jakautumis- ja erittymistutkimuksissa Jivi-valmisteen 60 kDa:n PEG-osan osoitettiin olevan laajasti jakautunut elimiin ja kudoksiin ja eliminoituvan niistä. Sen havaittiin erittyvän virtsaan (68,4 % 231 päivän kuluttua) ja ulosteeseen (13.8 % 168 päivän kuluttua).
Pitkäaikaisia eläinkokeita Jivi-valmisteen karsinogeenisuuden arvioimiseksi ei ole tehty. Myöskään tutkimuksia Jivi-valmisteen vaikutuksista lisääntymiseen ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Kuiva‑aine
Sakkaroosi
Histidiini
Glysiini (E 640)
Natriumkloridi
Kalsiumklorididihydraatti (E 509)
Polysorbaatti 80 (E 433)
Etikkahappo, väkevä (pH:n säätöön) (E 260)
Liuotin
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Vain pakkauksessa mukana olevia tarvikkeita saa käyttää käyttövalmiiksi saattamisessa ja injektiossa, koska hyytymistekijä VIII:n adsoptio joidenkin injektiolaitteiden sisäpintoihin voi aiheuttaa hoidon epäonnistumisen.
Kestoaika
Avaamaton injektiopullo
2 vuotta.
Käyttövalmiiksi saatettu liuos
Käyttövalmiiksi saattamisen jälkeen valmisteen kemialliseksi ja fysikaaliseksi säilyvyydeksi on osoitettu 3 tuntia huoneenlämmössä. Älä säilytä kylmässä käyttövalmiiksi saattamisen jälkeen.
Mikrobiologiselta kannalta valmiste on käytettävä heti sen käyttövalmiiksi saattamisen jälkeen. Ellei sitä käytetä välittömästi, käytönaikaiset säilytysajat ja olosuhteet ennen käyttöä ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2 °C ‑ 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ja esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
2 vuoden kestoajan voimassaoloaikana valmistetta (säilytettynä sen ulkopakkauksessa) voidaan säilyttää korkeintaan 25 °C:n lämpötilassa 6 kuukauden ajan. Tämän 6 kuukauden säilytysjakson päättymispäivä on merkittävä valmisteen ulkopakkaukseen. Tämä päivämäärä ei saa milloinkaan olla myöhäisempi kuin alun perin ulkopakkauksessa mainittu viimeinen käyttöpäivämäärä. Tämän ajanjakson lopussa valmistetta ei pidä laittaa takaisin jääkaappiin, vaan se on käytettävä tai hävitettävä.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
JIVI injektiokuiva-aine ja liuotin, liuosta varten
500 IU (L:ei) 1 pakkaus (200 IU/ml) (383,65 €)
1000 IU (L:ei) 1 pakkaus (400 IU/ml) (747,11 €)
2000 IU (L:ei) 1 pakkaus (800 IU/ml) (1449,83 €)
3000 IU (L:ei) 1 pakkaus (1200 IU/ml) (2132,59 €)
PF-selosteen tieto
Yksitääisen Jivi-pakkauksen sisältö:
- yksi injektiopullo, jossa on kuiva-ainetta (10 ml:n kirkas injektiopullo tyypin 1 lasia, harmaa bromobutyylikumitulppa sekä alumiinisinetti)
- yksi esitäytetty ruisku, jossa on 2,5 ml (250 IU, 500 IU, 1 000 IU, 2 000 IU ja 3 000 IU) tai 5 ml (4 000 IU) liuotinta (kirkas ruisku tyypin 1 lasia, harmaa bromobutyylikumitulppa)
- yksi ruiskun mäntä
- yksi injektiopullon liitinosa (jossa on integroitu suodatin)
- yksi injektiovälineistö.
Pakkauskoot
- 1 yksittäispakkaus
-
1 monipakkaus, joka sisältää 30 yksittäispakkausta.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Injektiokuiva-aine: kiinteä, valkoinen tai hieman kellertävä.
Liuotin: kirkas liuos.
Käyttö- ja käsittelyohjeet
Jivi-injektiokuiva-aine liuotetaan vain pakkauksen esitäytetyssä ruiskussa olevaan liuottimeen (2,5 ml tai 5 ml injektionesteisiin käytettävää vettä) ja käyttäen injektiopullon liitinosaa. Lääkevalmiste on valmisteltava injektiota varten vain aseptisissa olosuhteissa. Jos jokin pakkauksen välineistä on avattu tai vaurioitunut, kyseistä välinettä ei saa käyttää.
Käyttövalmiiksi sekoittamisen jälkeen liuos on kirkas ja väritön ja vedetään sitten takaisin ruiskuun. Parenteraaliset lääkevalmisteet on tarkastettava silmämääräisesti ennen antoa mahdollisten hiukkasten ja värimuutosten varalta.
Käyttövalmiiksi saatettu valmiste on suodatettava ennen antoa mahdollisten hiukkasten poistamiseksi liuoksesta. Valmiste suodatetaan injektiopullon liitinosaa käyttämällä.
Tarkat ohjeet Jivi-valmisteen käyttövalmiiksi saattamiseen ja antamiseen
Tarvitset alkoholipuhdistuslappuja, harsotaitoksia, laastareita ja kiristyssiteen. Nämä tarvikkeet eivät sisälly Jivi-pakkaukseen.
1. Pese kädet huolellisesti saippualla ja lämpimällä vedellä.
| |
2. Lämmitä avaamaton injektiopullo ja ruisku käsissäsi miellyttävän lämpöisiksi (ei yli 37 °C).
| |
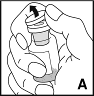
3. Poista suojakorkki injektiopullosta (A). Pyyhi injektiopullon kumitulppa alkoholiin kostutetulla lapulla ja anna tulpan kuivua ennen käyttöä.
|  |
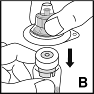
4. Aseta kuiva-ainetta sisältävä injektiopullo tukevalle, liukumattomalle alustalle. Poista suojapaperi injektiopullon liitinosan muovisen kotelon päältä. Älä poista liitinosaa muovisesta kotelostaan. Pidä kiinni liitinosan kotelosta, aseta se kuiva-ainetta sisältävän injektiopullon päälle ja paina voimakkaasti alaspäin (B).Liitinosa napsahtaa injektiopullon korkin päälle. Älä poista liitinosan koteloa tässä vaiheessa.
|  |
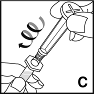
5. Pidä liuotinta sisältävää esitäytettyä ruiskua pystyasennossa. Ota kiinni ruiskun männästä kuvan osoittamalla tavalla ja kiinnitä mäntä kiertämällä sitä tiukasti myötäpäivään, kiinni kierteelliseen tulppaan (C).
|  |
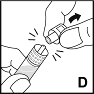
6. Pidä ruiskua sylinteristä ja irrota ruiskun korkki ruiskun kärjestä (D). Älä kosketa ruiskun kärkeä kädelläsi äläkä anna sen koskettaa mitään pintaa. Aseta ruisku syrjään myöhempää käyttöä varten.
|  |
7. Poista nyt liitinosan kotelo ja hävitä se (E).
| 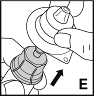 |
8. Kiinnitä esitäytetty ruisku injektiopullon kierteelliseen liitinosaan kiertämällä myötäpäivään (F).
| 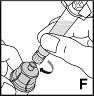 |
| 9. Injisoi liuotin painamalla mäntää hitaasti alas (G). | 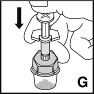 |
10. Pyörittele injektiopulloa varovasti kunnes kaikki jauhe on liuennut (H). Älä ravista injektiopulloa. Varmista, että kuiva-aine liukenee kokonaan. Tarkista ennen liuoksen antoa, ettei liuoksessa näy hiukkasia tai värimuutoksia. Älä käytä liuoksia, joissa on näkyviä hiukkasia tai jotka ovat sameita.
| 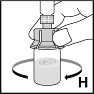 |
11. Pidä injektiopulloa ylösalaisin injektiopullon liitinosan ja ruiskun yläpuolella (I). Täytä ruisku vetämällä mäntä ulos hitaasti ja tasaisesti. Varmista, että injektiopullon koko sisältö on vedetty ruiskuun. Pidä ruiskua pystysuorassa ja paina mäntää, kunnes ruiskussa ei ole jäljellä enää yhtään ilmaa.
| 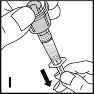 |
12. Kiinnitä kiristysside käsivarteesi.
| |
13. Valitse injektiokohta ja puhdista iho.
| |
14. Pistä laskimoon ja kiinnitä injektiokanyyli laastarilla.
| |
| 15. Pidä injektiopullon liitinosaa paikallaan ja poista ruisku injektiopullon liitinosasta (liitinosan pitäisi pysyä kiinni injektiopullossa). Liitä ruisku injektiokanyyliin (J). Varmista, ettei ruiskuun pääse verta. | 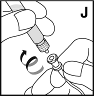 |
16. Poista kiristysside.
| |
17. Injisoi liuos laskimoon 2–5 minuutin aikana tarkkaillen samalla neulan asentoa. Injektionopeuden on oltava sellainen, että tunnet olosi mukavaksi, mutta anto ei saa olla nopeampaa kuin 2,5 ml minuutissa.
| |
18. Jos tarvitaan lisäannos, käytä uutta ruiskua, joka on täytetty yllä olevien valmistusohjeiden mukaisesti käyttökuntoon saatetulla kuiva-aineella.
| |
19. Jos lisäannosta ei tarvita, poista injektiovälineistö ja ruisku. Paina harsotaitosta injektiokohtaa vasten käsivarsi ojennettuna noin 2 minuutin ajan. Aseta lopulta pieni paineside injektiokohtaan ja tarvittaessa laastari.
| |
20. On suositeltavaa, että joka kerta kun käytät Jivi-valmistetta, kirjaat valmisteen nimen ja eränumeron ylös.
| |
| 21. Lääkkeitä ei pidä heittää viemäriin eikä hävittää talousjätteiden mukana. Kysy käyttämättömien lääkkeiden hävittämisestä apteekista tai lääkäriltä. Näin menetellen suojelet luontoa. | |
Jivi on tarkoitettu vain kertakäyttöön.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
JIVI injektiokuiva-aine ja liuotin, liuosta varten
250 IU 1 pakkaus
500 IU 1 pakkaus
1000 IU 1 pakkaus
2000 IU 1 pakkaus
3000 IU 1 pakkaus
- Ylempi erityiskorvaus (100 %). Krooniset hyytymishäiriöt (126).
- Peruskorvaus (40 %).
ATC-koodi
B02BD02
Valmisteyhteenvedon muuttamispäivämäärä
02.07.2025
Yhteystiedot
 BAYER OY
BAYER OY Tuulikuja 2, PL 73
02151 Espoo
020 785 21
www.bayer.fi