
ONDEXXYA infuusiokuiva-aine, liuosta varten 200 mg
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 200 mg andeksaneetti alfaa*.
Käyttökuntoon saattamisen jälkeen yksi ml liuosta sisältää 10 mg andeksaneetti alfaa.
* Andeksaneetti alfa tuotetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa (CHO-soluissa).
Apuaine, jonka vaikutus tunnetaan
Yksi Ondexxya‑injektiopullo sisältää 2 mg polysorbaatti 80:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokuiva-aine, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Tekijän Xa (FXa:n) suoraa estäjää (apiksabaania tai rivaroksabaania) saaville aikuisille potilaille, kun antikoagulaatio on kumottava hengenvaarallisen tai hallitsemattoman verenvuodon takia.
Ehto
Vain sairaalakäyttöön.
Annostus ja antotapa
Rajoitettu vain sairaalakäyttöön.
Annostus
Andeksaneetti alfa annetaan boluksena laskimoon tavoitenopeudella noin 30 mg/min 15 minuutin aikana (pieni annos) tai 30 minuutin aikana (suuri annos), minkä jälkeen antamista jatketaan jatkuvana infuusiona, jonka nopeus on 4 mg/min (pieni annos) tai 8 mg/min (suuri annos), 120 minuutin ajan (ks. taulukko 1). Andeksaneetti alfan annostus perustuu farmakokineettiseen/farmakodynaamiseen mallinnukseen ja simulaatioihin (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Taulukko 1: Annostusohjeet
| Aloitusbolus laskimoon | Jatkuva infuusio laskimoon | Tarvittavien 200 mg:n injektiopullojen kokonaismäärä | |
| Pieni annos | 400 mg tavoitenopeudella 30 mg/min | 4 mg/min 120 minuutin ajan (480 mg) | 5 |
| Suuri annos | 800 mg tavoitenopeudella 30 mg/min | 8 mg/min 120 minuutin ajan (960 mg) | 9 |
Apiksabaanin vaikutuksen kumoaminen
Ondexxya-valmisteen suositeltu annostus perustuu siihen apiksabaaniannokseen, jota potilas käyttää antikoagulaation kumoamisajankohtana, sekä aikaan, joka potilaan viimeisen apiksabaaniannoksen ottamisesta on kulunut (ks. taulukko 2). Tilanteisiin, joissa viimeisimmän antikoagulanttiannoksen vahvuus tai viimeisimmän lääkkeenannon ja verenvuototapahtuman välinen aika ei ole tiedossa, ei ole saatavilla annossuositusta. Lähtötilanteen anti-FXa-tason mittauksen tulee tukea kliinistä päätöstä aloittaa hoito (jos taso on saatavana hyväksyttävällä aikavälillä).
Taulukko 2: Yhteenveto apiksabaanin vaikutusten kumoamiseksi käytettävästä annostuksesta
| FXa:n estäjä | Viimeinen annos | Viimeisen annoksen ottamisesta kulunut aika ennen Ondexxya-hoidon aloittamista | |
| < 8 tuntia | ≥ 8 tuntia | ||
| Apiksabaani | ≤ 5 mg | Pieni annos | Pieni annos |
| > 5 mg | Suuri annos | ||
Rivaroksabaanin vaikutuksen kumoaminen
Ondexxya-valmisteen suositeltu annostus perustuu siihen rivaroksabaaniannokseen, jota potilas käyttää antikoagulaation kumoamisajankohtana, sekä aikaan, joka potilaan viimeisen rivaroksabaaniannoksen ottamisesta on kulunut (ks. taulukko 3). Tilanteisiin, joissa viimeisimmän antikoagulanttiannoksen vahvuus tai viimeisimmän lääkkeenannon ja verenvuototapahtuman välinen aika ei ole tiedossa, ei ole saatavilla annossuositusta. Lähtötilanteen anti-FXa-tason mittauksen tulee tukea kliinistä päätöstä aloittaa hoito (jos taso on saatavana hyväksyttävällä aikavälillä).
Taulukko 3: Yhteenveto rivaroksabaanin vaikutusten kumoamiseksi käytettävästä annostuksesta
| FXa:n estäjä | Viimeinen annos | Viimeisen annoksen ottamisesta kulunut aika ennen Ondexxya-hoidon aloittamista | |
| < 8 tuntia | ≥ 8 tuntia | ||
| Rivaroksabaani | ≤ 10 mg | Pieni annos | Pieni annos |
| > 10 mg | Suuri annos | ||
Antitromboottisen hoidon aloittaminen uudelleen
Sen jälkeen, kun Ondexxya-valmiste on annettu ja suuri verenvuoto tyrehtynyt, on harkittava antikoagulaatiohoidon aloittamista uudelleen potilaan perussairauteen liittyvien tromboottisten tapahtumien ehkäisemiseksi. Antitromboottinen hoito voidaan aloittaa uudelleen heti, kun se on hoidon jälkeen lääketieteellisesti aiheellista, jos potilaan tila on kliinisesti vakaa ja riittävä hemostaasi on saavutettu. Antitromboottisella hoidolla aikaansaatavan normaalin antikoagulaatioasteen saavuttamiseen kuluvaa aikaa ei ole vielä selvitetty. Lääkärin on hoitopäätöksessään otettava huomioon antikoagulaatiohoidon edut ja uuden verenvuodon riskit (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Erityisryhmät
Iäkkäät potilaat (65-vuotiaat ja sitä vanhemmat): Annoksen muuttaminen iäkkäillä potilailla ei ole tarpeen (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta: Munuaisten vajaatoiminnan vaikutusta altistuksiin andeksaneetti alfalle ei ole tutkittu. Nykyisten puhdistumatietojen perusteella annoksen muuttamista ei suositella.
Maksan vajaatoiminta: Andeksaneetti alfan nykyisten puhdistumatietojen perusteella annoksen muuttamista ei suositella. Lääkkeen tehoa ja turvallisuutta ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla (ks. kohta Farmakokinetiikka).
Pediatriset potilaat: Andeksaneetti alfan turvallisuutta ja tehoa lasten ja nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Anto laskimoon
Sen jälkeen kun sopiva määrä Ondexxya-injektiopulloja on saatettu käyttökuntoon, käyttökuntoon saatettu liuos (10 mg/ml) siirretään ilman jatkolaimennusta suuritilavuuksisiin steriileihin ruiskuihin, jos antamisessa käytetään ruiskupumppua, tai sopiviin, polyolefiinista (PO) tai polyvinyylikloridista (PVC) valmistettuihin tyhjiin infuusiopusseihin (ks. kohta Käyttö- ja käsittelyohjeet). Ennen valmisteen antoa infuusiona laskimoon on käytettävä 0,2 tai 0,22 mikronin polyeetterisulfonista (PES) tai vastaavasta valmistettua, heikosti proteiineja sitovaa kiinteää (in-line) suodatinta.
Ondexxya annetaan boluksena laskimoon tavoitenopeudella noin 30 mg/min 15 minuutin (pieni annos) tai 30 minuutin (suuri annos) aikana, minkä jälkeen antamista jatketaan jatkuvana infuusiona, jonka nopeus on 4 mg (pieni annos) tai 8 mg (suuri annos), minuutissa 120 minuutin ajan (ks. taulukko 1).
Ks. kohdasta Käyttö- ja käsittelyohjeet. ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aiemmin todettu allerginen reaktio hamsterin proteiineille.
Varoitukset ja käyttöön liittyvät varotoimet
Käytön rajoitukset
Tämän lääkkeen kliininen teho perustuu anti-FXa-aktiivisuuden kumoutumiseen terveillä vapaaehtoisilla ja hemostaattisen tehon saavuttamiseen verenvuotopotilailla, joille oli annettu apiksabaania tai rivaroksabaania. Kliinistä hyötyä pienentyneen sairastavuuden tai kuolleisuuden suhteen ei ole osoitettu (ks. kohta Farmakodynamiikka). Andeksaneetti alfa ei sovellu kiireellisten leikkausten esihoidoksi. Tietojen puuttumisen vuoksi käyttöä edoksabaanin tai enoksapariinin vaikutuksen kumoamiseen ei suositella. Andeksaneetti alfa ei kumoa muiden estäjien kuin FXa:n estäjien vaikutuksia (ks. kohta Farmakodynamiikka).
Hoidon seurannan pitää perustua pääasiassa kliinisiin parametreihin, jotka kuvaavat asianmukaista vastetta (ts. hemostaasin saavuttaminen), tehon puutetta (ts. verenvuodon uusiutuminen) ja haittatapahtumia (ts. tromboemboliset tapahtumat). Andeksaneetti alfalla annetun hoidon seurannan ei tule perustua anti-FXa-aktiivisuuteen. Kaupalliset anti-FXa-aktiivisuuden määrittämiseen käytettävät testit eivät sovellu anti-FXa-aktiivisuuden mittaamiseen andeksaneetti alfan antamisen jälkeen, koska näillä testeillä tulokseksi saatavat anti-FXa-aktiivisuuden arvot ovat virheellisesti liian suuria, minkä vuoksi andeksaneetti alfan kumoava aktiivisuus arvioidaan merkittävästi liian pieneksi.
Annossuositus perustuu terveistä vapaaehtoisista saatujen tietojen mallintamiseen. Verenvuotopotilaita koskevia tietoja on niukasti, eikä validointi ole toistaiseksi onnistunut. Tiedot viittaavat suurempaan tromboosiriskiin potilailla, jotka saavat hoitoa suuremmalla andeksaneetti alfa -annoksella, sekä potilailla, jotka saavat rivaroksabaanihoitoa.
Kliinisiin tutkimuksiin osallistui potilaita, joilla oli kallonsisäinen verenvuoto (ICrH) (GCS > 7 ja hematooman tilavuus ≤ 60 ml). Andeksaneetti alfa ‑hoitoa ei ole tutkittu potilailla, joiden ICrH on tätä vaikeampi.
Tromboottiset tapahtumat
Vakavia tromboembolisia valtimo‑ ja laskimotapahtumia on raportoitu esiintyneen andeksaneetti alfa -hoidon jälkeen, ja tapahtumien on usein raportoitu ilmaantuneen varhain (72 tunnin kuluessa) antikoagulaation kumoamisen jälkeen. Tromboottisen tapahtumisen riski voi olla suurempi potilailla, joilla on anamneesissa aivohalvaus, sydäninfarkti tai sydämen vajaatoiminta (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). FXa:n estäjiä saavilla potilailla on perussairauksia ja -tiloja, jotka altistavat heidät tromboottisille tapahtumille. FXa:n estäjällä annetun hoidon vaikutuksen kumoaminen altistaa potilaat heidän perussairauteensa liittyvälle tromboosiriskille. Lisäksi andeksaneetti alfalla on osoitettu olevan riippumaton, kudostekijätien estäjän (TFPI) estoon perustuva prokoagulanttivaikutus, joka voi myös muodostaa tromboosiriskin. Tämän vaikutuksen kestoa verenvuotopotilailla ei tunneta. Laboratorioarvot, kuten anti-FXa-aktiivisuus, endogeeninen trombiini potentiaali (ETP) tai tromboosin markkerit, eivät välttämättä anna luotettavia tietoja. Tämän riskin vähentämiseksi on antikoagulaatiohoidon uudelleen aloittamista harkittava hoidon päätyttyä heti, kun se on lääketieteellisesti asianmukaista (ks. kohta Annostus ja antotapa).
Terveillä vapaaehtoisilla ei ilmoitettu tromboottisia tapahtumia mutta havaittiin koagulaation markkereiden (F1+2, TAT ja D-dimeeri) annosriippuvaista nousua ja TFPI:n annosriippuvaista laskua andeksaneetti alfan antamisen jälkeen. Näitä markkereita ei mitattu tutkimuksiin 14-505 ja 18‑513 osallistuneilla potilailla, mutta tromboembolisia tapahtumia on havaittu (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). Tämän vuoksi tromboosin merkkien ja oireiden tarkkailu on erittäin suositeltavaa, ja se on aloitettava varhain hoidon antamisen jälkeen.
Andeksaneetti alfan käyttö yhdessä muiden tukitoimenpiteiden kanssa
Andeksaneetti alfaa voidaan käyttää yhdessä tavanomaisten, lääketieteellisesti asianmukaisina pidettävien hemostaattisten tukitoimenpiteiden kanssa.
Andeksaneetti alfan turvallisuutta ei ole arvioitu potilailla, jotka saivat protrombiinikompleksikonsentraatteja, rekombinantti tekijä VIIa -valmistetta tai kokoverta verenvuototapahtumaa edeltävien seitsemän päivän aikana, sillä nämä potilaat suljettiin pois kliinisistä tutkimuksista. Prokoagulanttihoitoja (esim. kolmea tai neljää tekijää sisältävä protrombiinikompleksikonsentraatti [PCC] / aktivoitu PCC, rekombinantti tekijä VIIa, jääplasma) ja kokoverta on vältettävä, ellei niiden käyttö ole ehdottomasti tarpeen, koska andeksaneetti alfan käytöstä yhdessä näiden hoitojen kanssa ei ole tietoja.
Yhteisvaikutukset hepariinin kanssa
Andeksaneetti alfan käyttöä ennen heparinisaatiota esimerkiksi leikkausten tai toimenpiteiden aikana pitää välttää, sillä andeksaneetti alfa aiheuttaa reagoimattomuuden hepariinille. Andeksaneetti alfan käyttöä hepariinin tai pienimolekyylisen hepariinin vastalääkkeenä ei ole arvioitu eikä sitä suositella (ks. kohta Yhteisvaikutukset).
Infuusioon liittyvät reaktiot
Jos potilaalla esiintyy lieviä tai kohtalaisia infuusioon liittyviä reaktioita, tarkka seuranta voi riittää. Kohtalaisten oireiden kohdalla voidaan harkita infuusion lyhytkestoista keskeyttämistä tai hidastamista, kunnes oireet lievittyvät, minkä jälkeen hoitoa jatketaan. Difenhydramiinia voidaan antaa.
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Apuaine, jonka vaikutus tunnetaan
Tämä lääkevalmiste sisältää 2 mg polysorbaatti 80:tä per injektiopullo. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Andeksaneetti alfalla ei ole tehty yhteisvaikutustutkimuksia.
In vitro‑tiedot viittaavat andeksaneetti alfan yhteisvaikutukseen hepariini - antitrombiini III (ATIII) ‑kompleksin kanssa sekä hepariinin antikoagulanttivaikutuksen neutralisoitumiseen. Andeksaneetti alfan käyttöaiheista poikkeavaan (off-label) käyttöön ennen leikkausta, leikkauksen aikana tai toimenpiteiden aikana, kun heparinisaatio on tarpeen, on raporttien mukaan liittynyt reagoimattomuutta hepariinille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Andeksaneetti alfan käyttöä hepariinin tai pienimolekyylisen hepariinin vastalääkkeenä ei ole arvioitu eikä sitä suositella.
Raskaus ja imetys
Raskaus
Andeksaneetti alfan käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyistä tutkimuksista saadut tiedot eivät riitä lisääntymistoksisuuden selvittämiseen (ks. kohta Prekliiniset tiedot turvallisuudesta). Andeksaneetti alfan käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyykö andeksaneetti alfa ihmisillä äidinmaitoon. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. Imetys on lopetettava andeksaneetti alfa -hoidon ajaksi.
Hedelmällisyys
Ei ole olemassa tietoja andeksaneetti alfan vaikutuksista ihmisen hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Andeksaneetti alfalla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Terveillä vapaaehtoisilla useimmin havaittuja haittavaikutuksia olivat lievät tai kohtalaiset infuusioon liittyvät reaktiot kuten punoitus, kuumotus, yskä, dysgeusia ja dyspnea, ja niitä ilmaantui infuusion jälkeen aikavälillä, joka vaihteli muutamasta minuutista muutamaan tuntiin. Tutkituista terveistä tutkittavista naisilla ilmeni enemmän haittavaikutuksia (lähinnä infuusioon liittyviä reaktioita) kuin miehillä.
Kliinisissä tutkimuksissapotilailla, joilla oli akuutti suuri verenvuoto ja joita hoidettiin FXa:n estäjällä (apiksabaanilla tai rivaroksabaanilla), yleisimmin havaittuja haittavaikutuksia olivat kuume (8,8 %), iskeeminen aivohalvaus (6,7 %) ja sydäninfarkti (4,6 %).
Taulukoitu luettelo haittavaikutuksista
Taulukossa 4 esitetään luettelo haittavaikutuksista, joita on todettu kliinisissä tutkimuksissa andeksaneetti alfalla hoidetuilla verenvuotopotilailla. Haittavaikutukset on luokiteltu elinjärjestelmäluokkien (SOC) ja esiintymistiheyden perusteella seuraavan käytännön mukaisesti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tai tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 4: Luettelo kliinisissä tutkimuksissa verenvuotopotilailla esiintyneistä haittavaikutuksista
| Elinjärjestelmäluokka | Yleinen ≥ 1/100, < 1/10 | Melko harvinainen ≥ 1 / 1 000, < 1/100 |
| Hermosto | Iskeeminen aivohalvausb | Ohimenevä aivoverenkiertohäiriö (TIA) |
| Sydän | Sydäninfarktic | Sydänpysähdys |
| Verisuonisto | Syvä laskimotukos | Valtimoemboliad |
| Hengityselimet, rintakehä ja välikarsina | Keuhkoembolia
| |
| Yleisoireet ja antopaikassa todettavat haitat | Kuume | |
| Vammat, myrkytykset ja hoitokomplikaatiot | Infuusioon liittyvä reaktioa |
a Ilmoitetut merkit ja oireet (vilunpuistatukset, vilunväristykset, kohonnut verenpaine, happidesaturaatio, kiihtyneisyys ja sekavuus) olivat ohimeneviä ja vaikeusasteeltaan lieviä tai kohtalaisia.
b Iskeeminen aivohalvaus sisältää esimerkiksi seuraavat suositellut termit: aivoverisuonitapahtuma, pikkuaivoinfarkti ja aivoinfarkti.
c Sydäninfarkti sisältää esimerkiksi seuraavan suositellun termin: akuutti sydäninfarkti.
d Valtimoembolia sisältää esimerkiksi seuraavat suositellut termit: lonkkavaltimon tukos, munuaisinfarkti ja reisivaltimoembolia.
Valikoitujen haittavaikutusten kuvaus
Tromboottiset tapahtumat
Kliinisissä tutkimuksissa on havaittu tromboottisia valtimo‑ ja laskimotapahtumia, mukaan lukien iskeeminen aivohalvaus, sydäninfarkti, keuhkoembolia, syvä laskimotukos, systeeminen valtimoembolia ja ohimenevä aivoverenkiertohäiriö, ja tapahtumien on usein ilmoitettu ilmaantuneen varhain (72 tunnin kuluessa) andeksaneetti alfa ‑hoidon jälkeen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
FXa:n estäjähoidon vaikutuksen kumoaminen altistaa potilaat heidän perussairauteensa liittyvälle tromboosiriskille. Lisäksi andeksaneetti alfan anti‑FXa‑vaikutuksesta riippumaton prokoagulanttivaikutus saattaa myös muodostaa hoidon jälkeisen tromboosin kehittymisen riskin.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Andeksaneetti alfan yliannostuksesta ei ole kliinistä kokemusta. Kliinisten tutkimusten aikana ei ole havaittu annosta rajoittavia toksisia vaikutuksia.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut lääkevalmisteet, myrkytysten hoitoon käytettävät lääkeaineet, ATC‑koodi: V03AB38
Vaikutusmekanismi
Andeksaneetti alfa on ihmisen FXa-proteiinin rekombinantti muoto, jota on muokattu siten, että siitä puuttuu FXa:n entsyymiaktiivisuus. Aktiivisen kohdan seriini on korvattu alaniinilla, joten molekyyli ei pysty pilkkomaan ja aktivoimaan protrombiinia, ja gamma-karboksiglutamiinihapon (Gla) domeeni on poistettu, jotta proteiini ei pysty kasautumaan protrombinaasikompleksiksi, eli kaikki antikoaguloivat vaikutukset on poistettu.
Andeksaneetti alfa on spesifinen aine FXa:n estäjien vaikutuksen kumoamiseen. Sen vaikutusmekanismiin sisältyy FXa:n estäjän sitominen ja sekvestraatio. Lisäksi andeksaneetti alfan on havaittu sitoutuvan kudostekijätien estäjään (TFPI) ja estävän sitä. TFPI-aktiivisuuden esto voi lisätä kudostekijän käynnistämää trombiinimuodostusta, millä on veren hyytymistä edistävä vaikutus.
Farmakodynaamiset vaikutukset
Andeksaneetti alfan vaikutusten mittareina voidaan käyttää farmakodynaamisia markkereita, muun muassa käytettävissä olevan FXa:n estäjän vapaata fraktiota, samoin kuin trombiinimuodostuksen palautumista. Lisäksi andeksaneetti alfan on osoitettu estävän TFPI-aktiivisuutta.
Kaupalliset anti-FXa-aktiivisuuden määrittämiseen käytettävät testit eivät sovellu anti-FXa-aktiivisuuden mittaamiseen andeksaneetti alfan antamisen jälkeen. Koska andeksaneetti alfan sitoutuminen FXa:n estäjään on palautuvaa, näissä testeissä nykyään käytetty näytteen suuri laimennus johtaa siihen, että estäjä dissosioituu andeksaneetti alfasta. Tämä puolestaan johtaa siihen, että anti-FXa-aktiivisuuden arvojen havaitaan olevan virheellisesti liian suuria. Tämän vuoksi andeksaneetti alfan kumoava aktiivisuus arvioidaan merkittävästi liian pieneksi.
Terveillä tutkimushenkilöillä tehdyissä prospektiivisissa, satunnaistetuissa, lumekontrolloiduissa annosmääritystutkimuksissa määritettiin FXa:n estäjien (apiksabaani tai rivaroksabaani) anti-FXa-aktiivisuuden kumoamiseen ja trombiinimuodostuksen palauttamiseen tarvittava andeksaneetti alfan annos ja annostusohjelma muunnelluilla testeillä, jotka eivät ole kaupallisesti saatavilla.
Anti-FXa-aktiivisuuden maksimaalinen kumoutuminen saavutettiin kahden minuutin kuluessa boluksen annon lopettamisesta. Kun andeksaneetti alfaa annettiin boluksena ja sen jälkeen jatkuvana infuusiona, tuloksena oli, että anti-FXa-aktiivisuus pysyi vähentyneenä. Anti-FXa-aktiivisuus palautui lumeryhmän tuloksia vastaaville tasoille ja niiden yläpuolelle noin kahden tunnin kuluttua boluksen annon tai infuusion lopettamisen jälkeen annostuksesta riippuen.
Kun andeksaneetti alfaa annettiin boluksena, minkä jälkeen antoa jatkettiin jatkuvana infuusiona, sitoutumattomien FXa:n estäjien määrä pieneni minimiin nopeasti (kahden minuutin kuluessa boluksen annon lopettamisesta), pysyi pienentyneenä infuusion aikana ja nousi sitten vähittäin ajan myötä, saavuttaen maksimimäärän noin kaksi tuntia infuusion päättymisen jälkeen.
Trombiinimuodostuksen palautuminen annon jälkeen riippui annoksesta ja annostusohjelmasta eikä korreloinut anti-FXa-aktiivisuuden kanssa enää noin neljän tunnin jälkeen (ks. jäljempänä kohta ”Trombiinimuodostuksen palautuminen”).
Plasman TFPI-aktiivisuuden on osoitettu estyvän terveillä koehenkilöillä täysin ajanjaksolla 2 minuuttia – 14,5 tuntia andeksaneetti alfan boluksena annon jälkeen ja palautuvan lähtötilanteen tasolle 3 vuorokauden kuluessa. Kudostekijän käynnistämä trombiinimuodostus lisääntyi välittömästi lähtötilanteen tasoa (ennen antikoagulaatiota) korkeammaksi ja pysyi kohonneena > 20 tunnin ajan, toisin kuin lumelääkettä annettaessa. D-dimeeri-, TAT- ja F1+2-pitoisuuksien peräkkäinen suureneminen ja pysyminen koholla tukee TFPI:n eston prokoagulanttivaikutuksen todennäköisyyttä.
Immunogeenisuus
Lääkevasta‑aineita havaittiin harvoin. Lääkevasta‑aineiden vaikutuksista farmakokinetiikkaan, tehoon tai turvallisuuteen ei havaittu näyttöä. Tietoja on kuitenkin vasta vähän.
Populaatiofarmakokineettinen/farmakodynaaminen mallinnus ja simulaatio
Farmakokineettinen/farmakodynaaminen mallinnus ja simulaatiot perustuvat andeksaneetti alfan ja FXa:n estäjän farmakokinetiikan vuorovaikutuksiin sekä biomarkkerien, eli anti-FXa-aktiivisuuden, TFPI-aktiivisuuden ja endogeenisen trombiinin potentiaalin, välisiin suhteisiin. Eri antikoagulanttien apiksabaanin ja rivaroksabaanin vaikutuksesta TFPI-aktiivisuuden estoon perustuvan kumoamisvaikutuksen kestosta ja jatkuvan infuusion tarpeellisuudesta on vielä epävarmuutta. Yksilöiden välisen suuren vaihtelun vuoksi simulaatioiden tarkkuus on verenvuotopotilailla heikompi kuin terveillä vapaaehtoisilla.
Kliininen teho ja turvallisuus
Andeksaneetti alfan kliinistä tehoa ja turvallisuutta on arvioitu seuraavissa tutkimuksissa: 1) satunnaistetut, lumekontrolloidut faasin II annosmääritystutkimukset, joissa terveille vapaaehtoisille annettiin FXa:n estäjiä vaikutuksen kumoamiseen tarvittavien annosten selvittämiseksi; 2) kaksi faasin III tutkimusta, joista toisessa käytettiin apiksabaania ja toisessa rivaroksabaania, tavoitteena vahvistaa suuren ja pienen annoksen annostusohjelmien teho; 3) maailmanlaajuinen, prospektiivinen, avoin faasin IIIb/IV monikeskustutkimus (ANNEXA‑4) potilailla, joilla oli akuutti suuri verenvuotokohtaus, joka edellytti FXa-antikoagulaation kiireellistä kumoamista; ja 4) satunnaistettu, avoin faasin IV tutkimus (ANNEXA‑I) potilailla, joilla oli akuutti kallonsisäinen verenvuoto (ICrH).
Antikoagulaation kumoaminen 50-75-vuotiailla terveillä tutkittavilla (tutkimukset 14‑503 ja 14-504)
Prospektiivisessa, satunnaistetussa, lumekontrolloidussa tutkimuksessa terveille tutkittaville, joiden mediaani-ikä oli 56,5 vuotta ja jotka saivat apiksabaania 5 mg kahdesti vuorokaudessa, annettiin andeksaneetti alfaa (n = 24) 400 mg:n boluksena laskimoon ja välittömästi sen jälkeen 4 mg minuutissa 120 minuutin ajan (480 mg) infuusiona laskimoon tai lumelääkettä (n = 8).
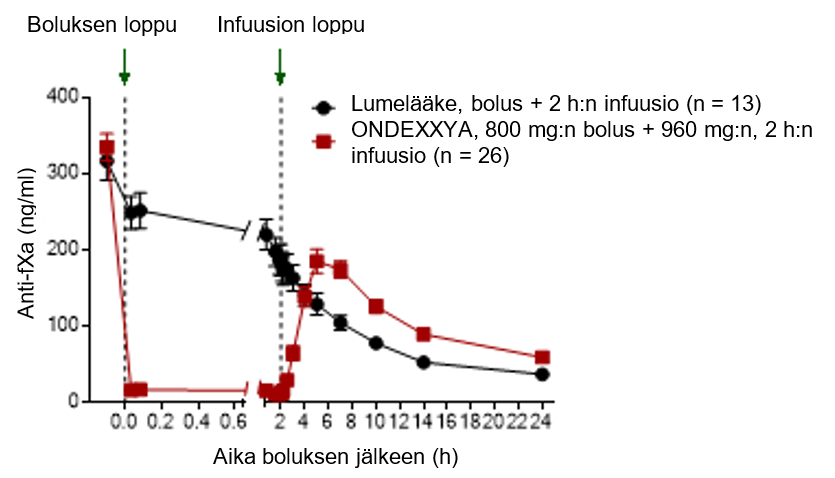
Samanlaisessa tutkimuksessa tutkittaville, joiden mediaani-ikä oli 57 vuotta ja jotka saivat rivaroksabaania 20 mg kerran vuorokaudessa, annettiin andeksaneetti alfaa (n = 26) 800 mg:n boluksena laskimoon ja välittömästi sen jälkeen 8 mg minuutissa 120 minuutin ajan (960 mg) infuusiona laskimoon tai lumelääkettä (n = 13).
Anti-FXa-aktiivisuuden väheneminen
Sekä tutkimuksen 14‑503 (apiksabaani) että tutkimuksen 14‑504 (rivaroksabaani) ensisijainen päätetapahtuma oli anti-FXa-aktiivisuuden prosentuaalinen muutos lähtötilanteesta infuusion jälkeiseen nadiiriin.
Potilailla, joita oli tutkimuksessa 14‑503 hoidettu apiksabaanilla, anti-FXa-aktiivisuuden prosentuaalinen muutos (± keskihajonta) oli ‑92,34 % (± 2,809 %) andeksaneetti alfaa saaneiden ryhmässä ja ‑32,70 % (± 5,578 %) lumelääkeryhmässä (p < 0,0001), jälkimmäisen kuvastaessa antikoagulantin luontaista puhdistumaa.
Potilailla, joita oli tutkimuksessa 14‑504 hoidettu rivaroksabaanilla, anti-FXa-aktiivisuuden prosentuaalinen muutos (± keskihajonta) oli ‑96,72 % (± 1,838 %) andeksaneetti alfaa saaneiden ryhmässä ja ‑44,75 % (± 11,749 %) lumelääkeryhmässä (p < 0,0001), jälkimmäisen kuvastaessa antikoagulantin luontaista puhdistumaa.
Anti-FXa-aktiivisuus eri ajankohtina ennen andeksaneetti alfan antoa ja sen jälkeen esitetään kuvassa 1. Anti-FXa-aktiivisuuden väheneminen korreloi trombiinimuodostuksen palautumisen kanssa. Anti-FXa-aktiivisuuden kynnysarvon trombiinimuodostuksen normalisoitumisen suhteen (keskimääräisen ETP:n ja keskihajontojen avulla määriteltynä) arvioitiin olevan 44,2 ng/ml (yhden keskihajonnan sisällä normaalista ETP:stä) tutkimuksista 14‑503 ja 14‑504 saatujen yhdistettyjen tietojen perusteella.
Kuva 1: Anti-FXa-aktiivisuuden (ng/ml) muutos terveillä tutkittavilla, jotka oli antikoaguloitu apiksabaanilla (A) tai rivaroksabaanilla (B)
(A)
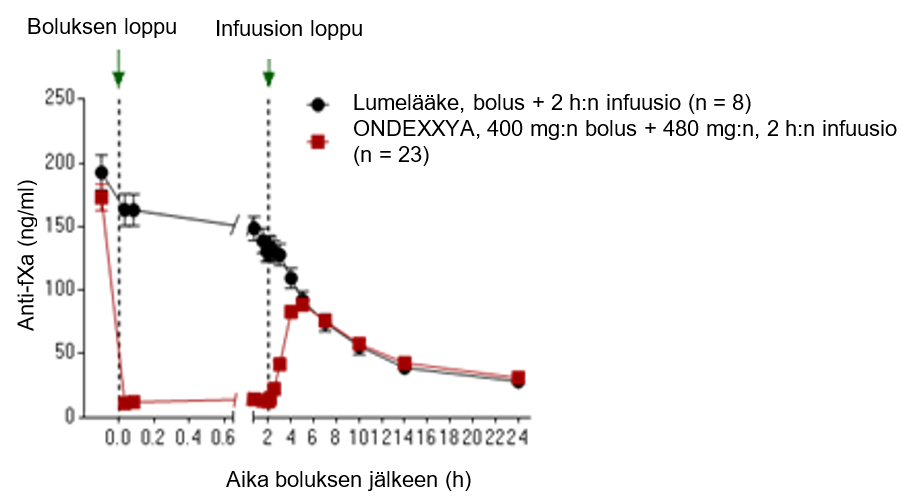
(B)
Trombiinimuodostuksen palautuminen
Sekä tutkimuksessa 14‑503 että tutkimuksessa 14‑504 andeksaneetti alfa -hoito johti myös tilastollisesti merkitsevään trombiinimuodostuksen nousuun terveillä, apiksabaanilla tai rivaroksabaanilla antikoaguloiduilla tutkittavilla lumeeseen verrattuna (p < 0,0001). Trombiinimuodostuksen palautuminen normaaliarvojen alueelle (määritelmän mukaan yksi keskihajonta lähtötilanteen pitoisuuksista) kahden minuutin kuluessa ja sen säilyminen 20 tunnin ajan saavutettiin pelkällä boluksella sekä boluksen ja infuusion yhdistelmällä pieniannoksisella andeksaneetti alfalla potilailla, jotka saivat apiksabaanihoitoa. Rivaroksabaanihoitoa saaneilla potilailla suuriannoksinen andeksaneetti alfa (bolus + infuusio) sai trombiinimuodostuksen, suurenemaan yli kahden keskihajonnan tasolle. Apiksabaanilla hoidetuille tutkittaville, jotka saivat suuriannoksista andeksaneetti alfaa, ei näissä tutkimuksissa tehty kliinistä arviointia. Rivaroksabaanilla hoidetuille tutkittaville, jotka saivat pieniannoksista andeksaneetti alfaa, ei näissä tutkimuksissa tehty arviointia.
Vapaan FXa:n estäjän pitoisuuden muutos lähtötilanteesta nadiirissa havaittuun pitoisuuteen
Apiksabaanin keskimääräinen sitoutumaton pitoisuus oli < 3,5 ng/ml ja rivaroksabaanin vastaavasti 4 ng/ml sen jälkeen, kun andeksaneetti alfa oli annettu boluksena, ja nämä pitoisuudet säilyivät koko jatkuvan infuusion ajan.
FXa:n estäjällä saavutetun antikoagulaation kumoaminen potilailla, joilla on akuutti suuri verenvuoto (tutkimus 14-505)
Faasin IIIb/IV monikansallisessa, prospektiivisessa, yksihaaraisessa avoimessa tutkimuksessa 14‑505 (ANNEXA‑4) Andeksaneetti alfaa annettiin 477:lle FXa:n estäjähoitoa saavalle potilaalle, joista 419 sai apiksabaania ja rivaroksabaania ja joilla oli akuutti suuri verenvuoto. Kaksi rinnakkaista ensisijaista päätetapahtumaa olivat seuraavat: a) anti-FXa-aktiivisuuden prosentuaalinen muutos lähtötilanteesta nadiiriin aikavälillä, joka alkoi viisi minuuttia boluksen annon päättymisen jälkeen ja loppui infuusion lopussa; ja b) ”hyvän” tai ”erinomaisen” (verrattuna ”heikkoon” tai "olemattomaan") hemostaattisen tehon osuus 12 tunnin sisällä infuusion jälkeen päätetapahtumien riippumattoman arviointitoimikunnan arvioimana.
Noin puolet potilaista oli miespuolisia ja keski-ikä oli 77,9 vuotta. Useimmat potilaat olivat aiemmin saaneet joko apiksabaania (245/477; 51,4 %) tai rivaroksabaania (174/477; 36,5 %) tai edoksabaania (36/477; 7,5 %) tai enoksapariinia (22/477; 4,6 %), ja heillä ilmeni joko kallonsisäinen verenvuoto (ICrH) (329/477; 69 %) tai maha-suolikanavan (GI) verenvuoto (109/477; 22,9 %). 381/477 (79,9 %) sai pieniä andeksaneetti alfa -annoksia ja 96/477 potilasta (20,1 %) sai suuria annoksia kohdan Annostus ja antotapa mukaisesti.
Anti‑FXa‑aktiivisuuden muutos lähtötilanteesta nadiiriin
Tutkimukseen otetuista 477 potilaasta 347 (73 %) oli arviointikelpoisia tehon suhteen, koska he saivat andeksaneetti alfaa vahvistettuun suureen verenvuotoon ja heidän lähtötilanteen anti-FXa-aktiivisuus oli yli 75 ng/ml. Tässä joukossa lähtötilanteen anti-FXa-aktiivisuus (mediaani) oli apiksabaania saaneilla potilailla 147 ng/ml ja rivaroksabaania saaneilla potilailla 214 ng/ml. Anti-FXa-aktiivisuuden osalta mediaani (95 %:n luottamusväli) lasku lähtötilanteesta nadiiriin oli apiksabaanilla ‑93,3 % (‑94,2 %; ‑92,5 %) ja rivaroksabaanilla ‑94,1 % (‑95,1 %; ‑93,0 %).
Hemostaattinen teho
Hemostaattinen teho luokiteltiin hyväksi tai erinomaiseksi 79 %:lla 169 potilaasta, jotka saivat apiksabaania, ja 80 %:lla 127 potilaasta, jotka saivat rivaroksabaania.
Tutkimuksen 14‑505 analyysi osoitti, että anti‑FXa‑aktiivisuuden muutos (korvikemuuttuja) ei ennustanut hemostaattisen tehon saavuttamista.
Anti-TFPI-vaikutus
Välitön ja pysyvä (noin 3 päivää infuusion jälkeen) TFPI:n eston prokoagulanttivaikutus vahvistettiin potilailla, joilla oli suuri verenvuoto. Tämä oli yhdenmukainen terveille vapaaehtoisille tehdyistä tutkimuksista saatujen vastaavien tulosten kanssa (14-503, 14-504, 16-508, 19-514).
Kuolemat
Turvallisuuspopulaatioon kuuluvista (N = 419) potilaista 75 potilasta (18 %) kuoli. Kuolleisuusluvut olivat 19,0 % (55/289) potilailla, joilla ilmeni kallonsisäinen verenvuoto (ICrH); 14,7 % (14/95) potilailla, joilla ilmeni maha-suolikanavan vuoto; ja 17,1 % (6/35) potilailla, joilla oli muuntyyppistä verenvuotoa. Kuoleman kardiovaskulaarisia syitä (n = 36) olivat mm. seuraavat: verenvuodosta johtuva aivohalvaus (n = 6), iskeeminen aivohalvaus (n = 10), äkillinen sydänkuolema (myös todistamaton) (n = 6), sydämen mekaaninen vajaatoiminta/pumppausvajaus (n = 4), sydäninfarkti (n = 2), muu verenvuoto kuin aivohalvaukseen liittyvä (n = 2) ja muut kardiovaskulaariset syyt (n = 6). Muita kuin kardiovaskulaarisia syitä (n = 39) olivat mm. seuraavat: infektio/sepsis (n = 11), hengitysvajaus (n = 6), onnettomuus/trauma (n = 2), syöpä (n = 2) ja jokin muu syy / muu kuin vaskulaarinen syy (n = 18). Keskimääräinen aika kuolemaan oli 15 päivää hoidon jälkeen. Kaikki kuolemat tapahtuivat ennen päivää 44.
Tromboemboliset tapahtumat
14-505-tutkimuksessa 45/419 potilaalla (11 %) esiintyi yksi tai useampi seuraavista tromboembolisista tapahtumista: aivoverisuonitapahtuma (CVA) (19/45; 42 %), syvä laskimotukos (11/45; 24 %), sydäninfarkti, mukaan lukien äkillinen sydäninfarkti ja sydänlihasiskemia (9/45; 20 %), keuhkoembolia (5/45; 11 %) ja ohimenevä aivoverenkiertohäiriö (TIA) (1/45; 2 %). Mediaaniaika ensimmäiseen tromboemboliseen tapahtumaan oli kymmenen vuorokautta. Kaikista potilaista, joilla esiintyi tromboembolisia tapahtumia, 38 %:lla (17/45) tromboembolinen tapahtuma ilmeni ensimmäisten kolmen päivän aikana. Andeksaneetti alfaa saaneesta 419 tutkittavasta 266 sai vähintään yhden antikoagulaatioannoksen 30 päivän kuluessa hoidon jälkeen ennaltaehkäisevänä toimenpiteenä kliinisen arvion perusteella.
Hemostaattinen teho ja FXa‑aktiivisuuden kumoaminen potilailla, joilla oli kallonsisäinen verenvuoto (ICrH) (tutkimus 18‑513)
Tutkimus 18‑513 (ANNEXA‑I) oli satunnaistettu, avoin faasin IV tutkimus, jossa ensisijaisten tehoa ja turvallisuutta koskevien päätetapahtumien arviointi oli sokkoutettua. Tutkimuksessa arvioitiin andeksaneetti alfan tehoa ja turvallisuutta tavanomaiseen hoitoon verrattuna potilailla, joilla oli akuutti kallonsisäinen verenvuoto (ICrH) ja hematooman tilavuus ≥ 0,5 – ≤ 60 ml, kun oireiden alkamisen ja lähtötilanteen kuvantamistutkimuksen välillä oli kulunut enintään 6 tuntia ja FXa:n estäjän ottamisesta suun kautta oli kulunut enintään 15 tuntia.
Ensisijaisena päätetapahtumana oli arvioida andeksaneetti alfan vaikutusta tehokkaan hemostaasin saavuttaneiden osuuteen tavanomaiseen hoitoon verrattuna. Hemostaasi arvioitiin 12 tunnin kuluttua satunnaistamisesta, ja se määriteltiin tehokkaaksi, jos hematooman tilavuus suureni lähtötilanteeseen verrattuna ≤ 35 % JA lähtötilanteen NIHSS‑pistemäärässä todettiin alle 7 pisteen muutos EIKÄ potilas ollut saanut varahoitoja 3–12 tunnin kuluessa satunnaistamisesta (yhdistetty päätetapahtuma). Toissijainen päätetapahtuma oli anti‑FXa‑aktiivisuuden prosentuaalinen muutos lähtötilanteesta nadiiriin ensimmäisten 2 tunnin aikana satunnaistamisen jälkeen.
ANNEXA‑I‑tutkimuksessa soveltuvat potilaat satunnaistettiin suhteessa 1:1 saamaan andeksaneetti alfaa tai tavanomaista hoitoa. Tutkimukseen otettiin yhteensä 530 potilasta, joista 320 (60,4 %) oli saanut apiksabaania ja 154 (29,1 %) oli saanut rivaroksabaania. Näistä muodostui laajennettu populaatio, jota käytettiin turvallisuus‑ ja herkkyysanalyyseihin. Tehoa arvioitiin välianalyysissä 452 potilaalla (ensisijainen tehon arvioinnin populaatio), joista 275 (60,8 %) oli saanut apiksabaania ja 129 (28,5 %) oli saanut rivaroksabaania. Laajennetussa populaatiossa mediaani‑ikä oli 80 vuotta, 52,3 % oli miehiä ja 93,3 % oli valkoihoisia. FXa:n estäjien yleisin käyttöaihe oli eteisvärinä (84,0 %).
Kaiken kaikkiaan 76,8 % andeksaneetti alfaa saaneiden ryhmän potilaista sai pieniannoksista hoitoa ja 21,2 % suuriannoksista hoitoa. Tavanomaista hoitoa saaneiden ryhmässä 87,6 % potilaista sai PCC‑hoitoa, 10,3 % potilaista ei saanut hemostaattista hoitoa (verihiutaleet tai punasolutiiviste olivat sallittuja) ja 0,9 % potilaista sai muuta hoitoa.
Yleisin verenvuodon sijainti oli aivoverenvuoto (91,7 %), useimmat verenvuodot olivat spontaaneja (86,9 %) ja hematooman tilavuuden mediaani (kvartiiliväli) lähtötilanteessa oli 9,9 (3,6, 24,5) ml. Mediaaniaika oireiden alkamisesta hoitoon oli 4,1 tuntia.
Hemostaattinen teho
Ensisijaisessa tehon arvioinnin populaatiossa andeksaneetti alfa oli tavanomaista hoitoa tilastollisesti parempi tehokkaan hemostaasin saavuttamisessa 12 tunnin kohdalla potilailla, joilla oli akuutti kallonsisäinen verenvuoto (ICrH) ja jotka saivat FXa:n suoraa estäjää suun kautta (67,0 % vs. 53,1 %, ero 13,4 % [95 %:n luottamusväli 4,6 %, 22,2 %], p = 0,0032).
Anti‑FXa‑aktiivisuuden muutoslähtötilanteesta nadiiriin
Ensisijaisessa tehon arvioinnin populaatiossa andeksaneetti alfa oli tavanomaista hoitoa tilastollisesti parempi anti‑FXa‑aktiivisuuden vähentämisessä lähtötilanteesta nadiiriin ensimmäisten 2 tunnin aikana satunnaistamisen jälkeen potilailla, joilla oli akuutti kallonsisäinen verenvuoto (ICrH) ja jotka saivat FXa:n suoraa estäjää suun kautta (vähenemisen mediaani −94,4 % vs. −27,5 %, p < 0,0001). Anti‑FXa‑aktiivisuuden hoidonaikaisen nadiirin mediaani oli andeksaneetti alfaa saaneiden ryhmässä 5,1 ng/ml ja tavanomaista hoitoa saaneiden ryhmässä 80,9 ng/ml. Mediaani (95 %:n luottamusväli) anti‑FXa‑aktiivisuuden laskulle lähtötilanteesta nadiiriin oli −94,1 % (−95,1 %, −93,3 %) andeksaneetti alfaa saaneiden ryhmässä vs. −20,8 % (−28,4 %, −13,9 %) tavanomaista hoitoa saaneiden ryhmässä potilailla, jotka olivat aiemmin ottaneet apiksabaania, ja −96,4 % (−97,3 %, −94,9 %) andeksaneetti alfaa saaneiden ryhmässä vs. −46,8 % (−60,6 %, −35,5 %) tavanomaista hoitoa saaneiden ryhmässä potilailla, jotka olivat aiemmin ottaneet rivaroksabaania.
Tromboottiset tapahtumat
ANNEXA‑I‑tutkimuksessa vahvistettuja tromboottisia tapahtumia ilmoitettiin 30 päivän kuluessa satunnaistamisesta 26 potilaalla (10,9 %:lla) andeksaneetti alfaa saaneiden ryhmässä ja 13 potilaalla (5,6 %:lla) tavanomaisesta hoitoa saaneiden ryhmässä.
Kun otettiin huomioon perussairauksia koskeva anamneesi, todettiin, että andeksaneetti alfaa saaneilla potilailla, joilla oli anamneesissa aivohalvaus tai sydäninfarkti tai joilla oli aiemmin todettu sydämen vajaatoiminta, tromboottisten tapahtumien määrä oli numeerisesti suurempi kuin potilailla, joilla ei ollut anamneesissa näitä perussairauksia. Niistä 73 potilaasta, joilla oli anamneesissa aivohalvaus tai sydäninfarkti, 10 potilaalle (13,7 %:lle) ilmaantui tromboottinen tapahtuma, kun taas 166 potilaasta, joilla ei ollut näitä anamneesissa, tromboottinen tapahtuma ilmaantui 16 potilaalle (9,6 %:lle). Niistä 40 potilaasta, joilla oli aiemmin todettu sydämen vajaatoiminta, 8 potilaalle (20,0 %:lle) ilmaantui tromboottinen tapahtuma, kun taas 199 potilaasta, joilla ei ollut tällaista anamneesissa, tromboottinen tapahtuma ilmaantui 18 potilaalle (9,0 %:lle) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Tällaista numeerista suurenemista ei havaittu tavanomaista hoitoa saaneiden ryhmän vastaavissa alaryhmissä.
Andeksaneetti alfaa saaneiden ryhmän ja tavanomaista hoitoa saaneiden ryhmän potilaille ilmaantui yksi tai useampi seuraavista vahvistetuista tromboottisista tapahtumista: iskeeminen aivohalvaus (6,7 %:lle andeksaneetti alfaa saaneista potilaista vs. 1,3 %:lle tavanomaista hoitoa saaneista potilaista), sydäninfarkti (4,6 %lle vs. 1,3 %:lle), keuhkoembolia (0,4 %:lle vs. 2,6 %:lle), systeeminen valtimoembolia (1,3 %:lle vs. 0,4 %:lle) ja syvä laskimotukos (0,4 %:lle vs. 0,9 %:lle). Tromboottisen tapahtuman ilmaantumiseen kuluneen ajan mediaani oli andeksaneetti alfaa saaneiden ryhmässä 3 päivää ja tavanomaista hoitoa saaneiden ryhmässä 14 päivää. Andeksaneetti alfaa saaneiden ryhmässä tromboottisia tapahtumia ilmaantui 14 potilaalle ensimmäisten 3 päivän aikana, kun taas tavanomaista hoitoa saaneiden ryhmässä tromboottinen tapahtuma ilmaantui yhdelle potilaalle. Kaikki tromboottiset tapahtumat, jotka ilmaantuivat ensimmäisten 5 päivän aikana hoidon jälkeen, olivat valtimotapahtumia. Yksikään potilaista, joilla ilmeni tapahtuma, ei ollut saanut minkäänlaista antikoagulanttiannosta ennen tromboottista tapahtumaa. Kuolemaan johtaneita vahvistettuja tromboottisia tapahtumia ilmoitettiin 6 potilaalla (2,5 %:lla) andeksaneetti alfaa saaneiden ryhmässä ja 2 potilaalla (0,9 %:lla) tavanomaisesta hoitoa saaneiden ryhmässä.
Kaiken kaikkiaan 182 potilasta (76,2 %) andeksaneetti alfaa saaneiden ryhmässä ja 168 potilasta (72,4 %) tavanomaista hoitoa saaneiden ryhmässä aloitti uudelleen jonkin antikoagulantin käytön 30 päivän kuluessa satunnaistamisesta kliinisen arvion perusteella.
Kuolleisuus
Yhteensä 67 potilasta (28,0 %) andeksaneetti alfaa saaneiden ryhmästä ja 61 potilasta (26,3 %) tavanomaista hoitoa saaneiden ryhmästä kuoli ennen kuin satunnaistamisesta oli kulunut 30 päivää. Kaiken kaikkiaan 54 potilasta (22,6 %) andeksaneetti alfaa saaneiden ryhmästä ja 51 potilasta (22,0 %) tavanomaista hoitoa saaneiden ryhmästä kuoli sairaalahoidon aikana. 72 tunnin kuluessa satunnaistamisesta tapahtuneita verenvuotoon liittyneitä kuolemantapauksia ilmoitettiin 12 potilaalla (5,0 %:lla) andeksaneetti alfaa saaneiden ryhmässä ja 16 potilaalla (6,9 %:lla) tavanomaista hoitoa saaneiden ryhmässä.
Toimintakykyyn liittyvät tulokset
Kun tarkasteltiin NIHSS‑pistemäärän muutosta lähtötilanteesta siihen asti, että satunnaistamisesta oli kulunut 72 tuntia, muutos oli andeksaneetti alfaa saaneiden ryhmässä numeerisesti parempi kuin tavanomaista hoitoa saaneiden ryhmässä; keskiarvojen ero 72 tunnin ajalta oli −1,2 (95 %:n luottamusväli −2,3 %, −0,2 %). Vaikutukset neurologiseen heikkenemiseen (NIHSS‑pistemäärän suureneminen ≥ 4 pisteellä tai GCS‑pistemäärän pieneneminen ≥ 2 pisteellä 24 tunnin kuluttua satunnaistamisesta), mRS‑pistemäärään ja GCS‑pistemääriin olivat samaa luokkaa molemmissa hoitoryhmissä. Kun andeksaneetti alfaa verrattiin tavanomaiseen hoitoon, itsenäisen toimintakyvyn vetosuhde (odds ratio) (mRS‑pistemäärä 0–3) 30 päivän kohdalla oli 1,23 (95 %:n luottamusväli 0,78, 1,92) ja GCS‑pisteiden keskiarvojen ero 72 tunnin ajalta oli 0,1 (95 %:n luottamusväli −0,4, 0,6).
Protromboottiset laboratoriomarkkerit
Andeksaneetti alfan annon jälkeen havaittiin koagulaatiomarkkereiden (F1+2, TAT ja D-dimeerit) pitoisuuksien annosriippuvaista suurenemista 223 terveellä vapaaehtoisella, jotka saivat FXa:n estäjiä ja joita hoidettiin andeksaneetti alfalla, mutta näillä terveillä vapaaehtoisilla ei ilmennyt tromboembolisia tapahtumia. Näitä markkereita (F1+2, TAT ja D-dimeerit) ei mitattu 14-505- ja 18‑513‑tutkimuksiin otetuilla potilailla, eikä niiden merkitystä verenvuotopotilailla tunneta.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset andeksaneetti alfan käytöstä FXa:n -estäjään liittyvien verenvuotojen hoidossa ja ehkäisyssä yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Ehdollinen myyntilupa
Tämä lääkevalmiste on saanut ns. ehdollisen myyntiluvan. Se tarkoittaa, että lääkevalmisteesta odotetaan uutta tietoa. Euroopan lääkevirasto arvioi vähintään kerran vuodessa tätä lääkevalmistetta koskevat uudet tiedot, ja tarvittaessa tämä valmisteyhteenveto päivitetään.
Farmakokinetiikka
Andeksaneetti alfan tutkimukset terveillä tutkittavilla FXa:n suorien estäjien käytön yhteydessä osoittivat farmakokinetiikan olevan suhteessa annokseen aiotuilla terapeuttisilla annoksilla, kun arvioitiin sekä Cmax että käyrän alle jäävä pinta-ala (AUC). Toteutettavuuteen liittyvistä syistä andeksaneetti alfan farmakokinetiikkaa ei ole tutkittu verenvuotopotilailla.
Taulukko 5: Farmakokineettiset parametrit 400 mg:n ja 800 mg:n andeksaneetti alfa -bolusinjektioiden jälkeen
| Farmakokineettinen parametri | 400 mg:n bolus | 800 mg:n bolus |
| AUC0-∞ (h*mikrog/ml) | 61,3 [43,8; 94,9] | 127 [57,5; 209] |
| Cmax (mikrog/ml) | 61,0 [40,3; 98,5] | 118 [50,2; 191] |
| Puhdistuma (l/h) | 6,52 [4,21; 9,13] | 6,29 [3,83; 13,9] |
| t1/2 (h) | 3,78 [2,59; 6,39] | 4,24 [2,47; 6,52] |
| Vss (vakaan tilan jakautumistilavuus) (l) | 9,47 [6,08; 15,3] | 8,94 [5,36; 23,1] |
Lähde: tutkimus 19-514
Esitetyt luvut ovat geometrisia keskiarvoja [pienin, suurin].
Farmakokinetiikka erityisryhmillä
Iäkkäät
Tutkimuksessa, jossa verrattiin andeksaneetti alfan farmakokinetiikkaa apiksabaania saaneilla iäkkäillä (65‑69-vuotiailla) ja nuoremmilla (26‑42-vuotiailla) terveillä tutkittavilla, andeksaneetti alfan farmakokinetiikassa ei ollut tilastollisia eroja iäkkäiden ja nuorempien tutkittavien välillä.
Munuaisten vajaatoiminta
Tutkimuksia andeksaneetti alfan farmakokinetiikasta munuaisten vajaatoimintaa sairastavilla potilailla ei ole tehty. Käytettävissä olevien farmakokineettisten tietojen perusteella andeksaneetti alfan munuaispuhdistuma on pieni tai olematon, joten annosta ei tarvitse muuttaa munuaisten vajaatoimintaa sairastavilla potilailla.
Maksan vajaatoiminta
Tutkimuksia andeksaneetti alfan farmakokinetiikasta maksan vajaatoimintaa sairastavilla potilailla ei ole tehty. Eliminaatio sapen ja/tai ulosteen mukana ei ole tunnettu proteiinilääkevalmisteiden eliminaatioreitti. Sen vuoksi annoksen muuttamista maksan vajaatoimintaa sairastaville potilaille ei pidetä tarpeellisena.
Sukupuoli
Populaatiofarmakokineettisen analyysin perusteella sukupuolella ei ole kliinisesti merkittävää vaikutusta andeksaneetti alfan farmakokinetiikkaan.
Pediatriset potilaat
Andeksaneetti alfan farmakokinetiikkaa ei ole tutkittu pediatrisilla potilailla.
Prekliiniset tiedot turvallisuudesta
Rotilla ja apinoilla tehtyjen, enintään kaksi viikkoa kestäneiden farmakologista turvallisuutta ja toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Andeksaneetti alfan mahdollista mutageenisuutta tai karsinogeenisuutta selvittäviä tutkimuksia ei ole tehty. Sen vaikutusmekanismin ja proteiineille tyypillisten ominaisuuksien perusteella karsinogeenisia tai genotoksisia vaikutuksia ei ole odotettavissa.
Andeksaneetti alfalla ei ole tehty lisääntymis- tai kehitystoksisuuteen liittyviä eläintutkimuksia.
Farmaseuttiset tiedot
Apuaineet
Tris-emäs
Trishydrokloridi
L-arginiinihydrokloridi
Sakkaroosi
Mannitoli
Polysorbaatti 80
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Injektiopullo (avaamaton)
5 vuotta varastoituna 2‑8 °C:ssa.
Käyttökuntoon saatettu lääkevalmiste
Valmisteen on osoitettu säilyvän kemiallisesti ja fysikaalisesti stabiilina käytön aikana 16 tunnin ajan 2-8°C:n lämpötilassa alkuperäisessä injektiopullopakkauksessa. Käyttökuntoon saatettua liuosta, joka on siirretty infuusiopussiin, voidaan tarvittaessa säilyttää vielä 8 tunnin ajan huoneenlämmössä. Mikrobiologiselta kannalta valmiste on käyttökuntoon saattamisen jälkeen käytettävä välittömästi. Jos valmistetta ei käytetä välittömästi, käytönaikaiset säilytysajat ja käyttöä edeltävät olosuhteet ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ONDEXXYA infuusiokuiva-aine, liuosta varten
200 mg (L:ei) 5 x 1 kpl (18340,60 €)
PF-selosteen tieto
Kuiva-aine 20 ml:n injektiopullossa (tyypin I lasia), joka on varustettu tulpalla (butyylikumia).
Pakkauskoko: neljä tai viisi injektiopulloa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen kylmäkuivattu jauhe.
Käyttö- ja käsittelyohjeet
Käyttökuntoon saattaminen
Välineet, jotka on otettava esiin ennen käyttökuntoon saattamisen aloittamista:
- laskettu määrä injektiopulloja (ks. kohta Annostus ja antotapa)
- sama määrä 20 ml:n (tai suurempia) liuotinruiskuja, jotka on varustettu 20 G:n (tai läpimitaltaan pienemmällä, esim. 21 G:n) neulalla
- alkoholilla kostutettuja puhdistuslappuja
- iso (50 ml:n tai suurempi) steriili ruisku. Jos antamisessa käytetään ruiskupumppua, käyttökuntoon saatetun valmisteen lopullinen tilavuus on jaettava useampaan kuin yhteen ruiskuun.
- polyolefiinista (PO) tai polyvinyylikloridista (PVC) valmistettuja infuusiopusseja (150 ml tai suurempia), joihin käyttökuntoon saatetun valmisteen lopullinen tilavuus mahtuu (jos annossa käytetään infuusiopussia)
- injektionesteisiin käytettävää vettä
- 0,2 tai 0,22 mikronin polyeetterisulfonista (PES) tai vastaavasta valmistettu, heikosti proteiineja sitova kiinteä (in-line) suodatin.
Andeksaneetti alfan ei tarvitse lämmetä huoneenlämpöiseksi ennen käyttökuntoon saattamista tai potilaalle antamista. Käyttökuntoon saattamisen aikana on noudatettava aseptisia tekniikoita.
Kukin injektiopullo saatetaan käyttökuntoon seuraavia ohjeita noudattaen:
- Poista repäisykansi jokaisesta injektiopullosta.
- Pyyhi jokaisen injektiopullon kumitulppa alkoholilla kostutetulla puhdistuslapulla.
- Käyttäen 20 ml:n (tai suurempaa) ruiskua ja 20 G:n (tai läpimitaltaan pienemmällä, esim. 21 G:n) neulaa vedä ruiskuun 20 ml injektionesteisiin käytettävää vettä.
- Työnnä ruiskun neula kumitulpan läpi tulpan keskeltä.
- Paina mäntä alas ja ruiskuta injektiopulloon hitaasti 20 ml injektionesteisiin käytettävää vettä siten, että suuntaat virtauksen injektiopullon sisäseinämää kohti vaahtoamisen välttämiseksi.
- Pyörittele jokaista injektiopulloa varovasti, kunnes kuiva-aine on täysin liuennut. Injektiopulloja EI SAA RAVISTAA, koska se voi aiheuttaa vaahtoamista. Liukenemisaika kunkin injektiopullon kohdalla on noin kolmesta viiteen minuuttia.
- Käyttökuntoon saatettu liuos on ennen antoa tarkistettava hiukkasmuodostuksen ja/tai värimuutosten varalta. Ei saa käyttää, jos liuoksessa havaitaan läpikuultamattomia hiukkasia tai värimuutoksia.
- Jotta tarvittavan annoksen käyttökuntoon saattaminen olisi mahdollisimman sujuvaa ja virheiltä vältyttäisiin, ruiskuta jokaiseen tarvittavaan injektiopulloon 20 ml injektionesteisiin käytettävää vettä ennen seuraavaan vaiheeseen siirtymistä.
- Käytä kahdeksan tunnin kuluessa käyttökuntoon saattamisesta, jos säilytys tapahtuu huoneenlämmössä.
Anto ruiskupumppua käyttäen
- Kun kaikki tarvittavat injektiopullot on saatettu käyttökuntoon, käyttökuntoon saatettu liuos vedetään jokaisesta injektiopullosta käyttäen suuritilavuuksista (50 ml:n tai suurempaa) ruiskua, joka on varustettu 20 G:n (tai läpimitaltaan pienemmällä, esim. 21 G:n) neulalla.
- Bolus ja infuusio valmistetaan erillisissä suuritilavuuksisissa ruiskuissa.
- Suuriannoksinen bolus ja infuusio on niiden suuremmasta tilavuudesta johtuen jaettava vielä ylimääräisiin ruiskuihin (sekä bolusta että infuusiota varten tarvitaan kaksi ruiskua).
- Jotta ilmaa ei tahattomasti pääsisi ruiskuihin, toimi huolellisesti pitäen ruiskun neulaa ylhäällä. Ruiskua ei saa laskea alas injektiopulloista vedettävien määrien ottamisen välillä.
- Kiinnitä lisävarusteet (esim. jatkoletku, 0,2 tai 0,22 mikronin polyeetterisulfonista (PES) tai vastaavasta valmistettu, heikosti proteiineja sitova kiinteä (in-line) suodatin, ruiskupumppu) ennen antamisen aloittamista.
- Anna käyttökuntoon saatettu liuos asianmukaisella nopeudella.
- Hävitä kaikki käytetyt ruiskut, neulat ja injektiopullot, myös mahdollinen käyttökuntoon saatetun liuoksen käyttämättä jäänyt osuus.
Anto infuusiopusseja käyttäen
- Kun kaikki tarvittavat injektiopullot on saatettu käyttökuntoon, vedä käyttökuntoon saatettu liuos jokaisesta injektiopullosta käyttäen suuritilavuuksista (50 ml:n tai suurempaa) ruiskua, joka on varustettu 20 G:n (tai läpimitaltaan pienemmällä, esim. 21 G:n) neulalla.
- Siirrä käyttökuntoon saatettu liuos ruiskusta asianmukaiseen infuusiopussiin.
- Toista tarvittaessa vaiheet 1 ja 2, jotta saat siirrettyä koko bolus- ja infuusiotilavuuden PO- tai PVC-infuusiopusseihin.
- On suositeltavaa jakaa bolus ja infuusio kahteen eri pussiin oikean antonopeuden takaamiseksi. Bolukseen ja infuusioon on sallittua käyttää myös yhtä PO- tai PVC-infuusiopussia, mutta siirryttäessä boluksesta infuusioon on varmistettava oikea infuusionopeus.
- Kiinnitä lisävarusteet (esim. jatkoletku, 0,2 tai 0,22 mikronin polyeetterisulfonista (PES) tai vastaavasta valmistettu, heikosti proteiineja sitova kiinteä (in-line) suodatin, infuusiopumppu) ennen antamisen aloittamista.
- Anna käyttökuntoon saatettu liuos asianmukaisella nopeudella.
Hävittäminen
Kaikki käytetyt ruiskut, neulat ja injektiopullot, mukaan lukien mahdollinen käyttökuntoon saatetun liuoksen käyttämättä jäänyt osuus, on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ONDEXXYA infuusiokuiva-aine, liuosta varten
200 mg 5 x 1 kpl
- Ei korvausta.
ATC-koodi
V03AB38
Valmisteyhteenvedon muuttamispäivämäärä
14.11.2025
Yhteystiedot
Keilaranta 18
02150 Espoo
010 23 010
www.astrazeneca.fi