
BRAFTOVI kapseli, kova 50 mg, 75 mg
Vaikuttavat aineet ja niiden määrät
Braftovi 50 mg kapseli, kova
Yksi kova kapseli sisältää 50 mg enkorafenibia.
Braftovi 75 mg kapseli, kova
Yksi kova kapseli sisältää 75 mg enkorafenibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, kova (kapseli).
Kliiniset tiedot
Käyttöaiheet
Melanooma
Enkorafenibi on tarkoitettu käytettäväksi yhdistelmähoitona binimetinibin kanssa leikkaukseen soveltumattoman tai etäpesäkkeisen melanooman hoitoon aikuisille potilaille, joiden kasvaimessa on BRAF V600 -mutaatio.
Kolorektaalisyöpä
-
Enkorafenibi on tarkoitettu käytettäväksi yhdistelmähoitona setuksimabin kanssa etäpesäkkeisen kolorektaalisyövän hoitoon aikuisille potilaille, joiden kasvaimessa on BRAF V600E ‑mutaatio ja jotka ovat aikaisemmin saaneet systeemistä hoitoa.
-
Enkorafenibi on tarkoitettu käytettäväksi yhdessä setuksimabin ja FOLFOXin kanssa etäpesäkkeisen kolorektaalisyövän ensilinjan hoitona aikuisille potilaille, joiden kasvaimessa on BRAF V600E ‑mutaatio.
Ei-pienisoluinen keuhkosyöpä
Enkorafenibi on tarkoitettu käytettäväksi yhdistelmähoitona binimetinibin kanssa pitkälle edenneen ei-pienisoluisen keuhkosyövän hoitoon aikuisille potilaille, joiden kasvaimessa on BRAF V600E -mutaatio.
Potilaiden valinta biomarkkeriperusteella, ks. kohta Annostus ja antotapa.
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Enkorafenibihoidon aloittaa ja sitä valvoo syövän lääkehoitoon perehtynyt lääkäri.
Potilaiden valinta
Ennen enkorafenibin käytön aloittamista potilaan BRAF V600E -mutaatio on vahvistettava tähän käyttötarkoitukseen tarkoitetulla CE-merkityllä in vitro -diagnostiikkaan (IVD) tarkoitetulla lääkinnällisellä laitteella. Jos CE-merkittyä in vitro -diagnostiikkaan tarkoitettua lääkinnällistä laitetta ei ole saatavilla, on käytettävä muuta validoitua testiä.
Enkorafenibin teho ja turvallisuus on varmistettu vain BRAF V600E- ja BRAF V600K -mutaatiota ilmentävien melanoomakasvainten, BRAF V600E -mutaatiota ilmentävien kolorektaalisyöpäkasvainten ja BRAF V600E -mutaatiota ilmentävien ei-pienisoluisten keuhkosyöpäkasvainten hoidossa. Enkorafenibia ei saa käyttää maligniin melanoomaan, kolorektaalisyöpään eikä ei-pienisoluiseen keuhkosyöpään, jos BRAF-geeni on villiä tyyppiä.
Annostus
Melanooma ja ei-pienisoluinen keuhkosyöpä
Enkorafenibin suositusannos on 450 mg (kuusi 75 mg kapselia) kerran vuorokaudessa käytettäessä yhdessä binimetinibin kanssa.
Kolorektaalisyöpä
Enkorafenibin suositusannos on 300 mg (neljä 75 mg kapselia) kerran vuorokaudessa käytettäessä yhdessä setuksimabin kanssa (anto kerran viikossa) tai yhdessä setuksimabin (anto 2 viikon välein) ja FOLFOXin kanssa. Samanaikaisesti käytettävien solunsalpaajien ja setuksimabin annostus ja suositellut annosmuutokset, ks. näiden lääkevalmisteiden valmistetiedot.
Annosmuutokset
Melanooma ja ei-pienisoluinen keuhkosyöpä
Haittavaikutusten hoito saattaa vaatia annoksen pienentämistä tai enkorafenibihoidon tilapäistä keskeyttämistä tai lopettamista (ks. taulukot 1, 3 ja 4).
Tiedot binimetinibin annostuksesta ja annosmuutossuosituksista, ks. binimetinibin valmisteyhteenvedon kohta Annostus ja antotapa.
Enkorafenibin annosmuutossuositukset esitetään taulukossa 1.
Taulukko 1: Enkorafenibin annosmuutossuositukset yhdistelmähoidossa binimetinibin kanssa, kun käyttöaiheena on melanooma tai ei-pienisoluinen keuhkosyöpä
Annostaso | Enkorafenibiannos enkorafenibin ja binimetinibin yhdistelmän käytön aikana |
Aloitusannos | Kuusi 75 mg:n (450 mg) kapselia kerran vuorokaudessa |
1. kertaa pienennetty annos | Neljä 75 mg:n (300 mg) kapselia kerran vuorokaudessa |
2. kertaa pienennetty annos | Kolme 75 mg:n (225 mg) kapselia kerran vuorokaudessa |
Seuraavat muutokset | Käyttöaiheena melanooma: Annoksen pienentämisestä tasolle 100 mg kerran vuorokaudessa on vain vähän tietoa. Enkorafenibihoito on lopetettava pysyvästi, jos potilas ei siedä 100 mg:n Käyttöaiheena ei-pienisoluinen keuhkosyöpä: Enkorafenibihoito on lopetettava pysyvästi, jos potilas ei siedä 225 mg:n |
Enkorafenibin käyttöä annoksella 450 mg kerran vuorokaudessa ei suositella monoterapiana.
Jos binimetinibihoito keskeytetään tilapäisesti, enkorafenibiannos on pienennettävä tasolle 300 mgkerran vuorokaudessa binimetinibihoidon keskeyttämisen ajaksi (ks. binimetinibin valmisteyhteenvedon kohta Annostus ja antotapa), sillä 450 mg enkorafenibiannos ei ole hyvin siedetty ainoana lääkkeenä. Jos binimetinibihoito lopetetaan pysyvästi, enkorafenibihoito on lopetettava.
Jos enkorafenibihoito keskeytetään väliaikaisesti (ks. taulukot 3 ja 4), binimetinibihoito on keskeytettävä. Jos enkorafenibihoito lopetetaan pysyvästi, binimetinibihoitokin on lopetettava.
Jos hoitoon liittyviä haittoja esiintyy, enkorafenibin ja binimetinibin annosta on pienennettävä tai hoidot on keskeytettävä tai lopetettava. Seuraavissa tapauksissa vain binimetinibiannoksen pienentäminen on välttämätöntä (haittavaikutukset liittyvät ensisijaisesti binimetinibiin): verkkokalvon pigmenttiepiteelin irtauma, verkkokalvon laskimotukos, interstitiaalinen keuhkosairaus /pneumoniitti, sydäntoiminnan häiriö, kreatiinikinaasipitoisuuden suureneminen ja rabdomyolyysi, sekä tromboemboliset laskimotapahtumat.
Jos jokin näistä haitoista ilmenee, binimetinibiannoksen muuttamisohjeet on tarkistettava binimetinibin valmisteyhteenvedon kohdasta Annostus ja antotapa.
Kolorektaalisyöpä
Haittavaikutusten hoito saattaa vaatia annoksen pienentämistä tai enkorafenibihoidon tilapäistä keskeyttämistä tai lopettamista (ks. taulukot 2, 3 ja 4).
Tiedot setuksimabin ja FOLFOXin (5‑fluorourasiili/foliinihappo/oksaliplatiini) annostuksesta ja annosmuutossuosituksista, ks. näiden lääkevalmisteiden valmisteyhteenvedot.
Enkorafenibin annosmuutossuositukset esitetään taulukossa 2.
Taulukko 2: Enkorafenibin annosmuutossuositukset yhdistelmähoidossa setuksimabin tai setuksimabin ja FOLFOXin kanssa, kun käyttöaiheena on kolorektaalisyöpä
Annostaso | Enkorafenibiannos enkorafenibin ja setuksimabin yhdistelmän käytön aikana tai enkorafenibin, setuksimabin ja FOLFOXin yhdistelmän käytön aikana |
Aloitusannos | Neljä 75 mg:n (300 mg) kapselia kerran vuorokaudessa |
1. kertaa pienennetty annos | Kolme 75 mg:n (225 mg) kapselia kerran vuorokaudessa |
2. kertaa pienennetty annos | Kaksi 75 mg:n (150 mg) kapselia kerran vuorokaudessa Enkorafenibi on lopetettava pysyvästi, jos potilas ei siedä annosta 150 mg (kaksi 75 mg:n kapselia) kerran vuorokaudessa. |
Jos enkorafenibihoito lopetetaan pysyvästi, setuksimabihoito on lopetettava. Pelkkää FOLFOX-hoitoa voidaan jatkaa tavanomaisen hoitokäytännön mukaisesti.
Jos setuksimabihoito lopetetaan pysyvästi, enkorafenibihoito on lopetettava. Pelkkää FOLFOX-hoitoa voidaan jatkaa tavanomaisen hoitokäytännön mukaisesti.
Jos FOLFOX-hoito lopetetaan pysyvästi, enkorafenibin ja setuksimabin yhdistelmähoitoa voidaan jatkaa.
Melanooma, kolorektaalisyöpä ja ei-pienisoluinen keuhkosyöpä
Annosmuutokset haittavaikutusten varalle esitetään jäljempänä sekä taulukoissa 3 ja 4.
Ihon uudet primaarimaligniteetit: Enkorafenibiannosta ei tarvitse muuttaa.
Uudet, muut kuin ihon RAS-mutaatiopositiiviset primaarimaligniteetit: Enkorafenibihoidon lopettamista pysyvästi on harkittava.
Taulukko 3: Enkorafenibin annosmuutossuositukset tiettyjen haittavaikutusten yhteydessä yhdistelmähoidossa binimetinibin tai setuksimabin tai setuksimabin ja FOLFOXin kanssa
Haittavaikutuksen vaikeusastea | Enkorafenibi |
Ihoreaktiot |
|
| Enkorafenibihoitoa ei muuteta. Jos ihottuma pahenee tai ei lievity 2 hoitoviikon kuluessa, enkorafenibihoito on keskeytettävä, kunnes haitta on lievittynyt asteeseen 0 tai 1. Tämän jälkeen hoito aloitetaan uudelleen samalla annoksella. |
| Enkorafenibihoito on keskeytettävä, kunnes haitta on lievittynyt asteeseen 0 tai 1. Tämän jälkeen hoito aloitetaan uudelleen samalla annoksella, jos kyseessä on haitan ensimmäinen esiintymiskerta, tai pienennetyllä annoksella, jos kyseessä on uusiutunut asteen 3 haitta. |
| Enkorafenibihoito on lopetettava pysyvästi. |
Käsi-jalkaoireyhtymä | |
| Enkorafenibihoitoa ei muuteta. Tukitoimet, kuten paikallishoito, on aloitettava. Jos haitta ei lievity tukihoidosta huolimatta 2 viikon kuluessa, enkorafenibihoito on keskeytettävä, kunnes haitta on lievittynyt asteeseen 0 tai 1. Tämän jälkeen hoito aloitetaan uudelleen samalla annoksella tai pienennetyllä annoksella. |
| Enkorafenibihoito on keskeytettävä. Tukitoimet, kuten paikallishoito, on aloitettava, ja potilaan tila on arvioitava uudelleen viikoittain. Kun haitta on lievittynyt asteeseen 0 tai 1, enkorafenibihoito aloitetaan uudelleen samalla annoksella tai pienennetyllä annoksella. |
Uveiitti, mukaan lukien iriitti jairidosykliitti | |
| Jos asteen 1 tai 2 uveiittiin ei saada vastetta spesifisellä silmähoidolla (esim. paikallishoidolla) tai jos uveiitti on astetta 3, enkorafenibihoito on keskeytettävä ja silmien tila on arvioitava uudelleen 2 viikon kuluessa.
|
| Enkorafenibihoito on lopetettava pysyvästi. Silmälääkärin seuranta on tarpeen. |
QTc-ajan piteneminen |
|
| Enkorafenibihoito on keskeytettävä (seuranta, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kun QTcF on ≤ 500 ms, enkorafenibihoito aloitetaan uudelleen pienennetyllä annoksella. Enkorafenibihoito on lopetettava, jos haitta uusiutuu useammin kuin kerran. |
| Enkorafenibihoito on lopetettava pysyvästi (seuranta, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). |
Maksa-arvojen poikkeavuudet | |
| Enkorafenibihoitoa ei muuteta. Jos haitta ei lievity 4 viikon kuluessa, enkorafenibihoito on keskeytettävä, kunnes haitta on lievittynyt asteeseen 0 tai 1 tai hoitoa edeltävälle tasolle/lähtötasolle. Tämän jälkeen hoito aloitetaan uudelleen samalla annoksella. |
| Enkorafenibihoito on keskeytettävä enintään 4 viikoksi.
|
| Enkorafenibihoito on keskeytettävä enintään 4 viikoksi.
Tai enkorafenibihoito lopetetaan pysyvästi. |
| Enkorafenibihoidon lopettamista pysyvästi on harkittava. |
| Enkorafenibihoito on lopetettava pysyvästi. |
a NCI CTCAE -kriteerit (National Cancer Institute Common Terminology Criteria for Adverse Events), versio 4.03
Taulukko 4: Enkorafenibin annosmuutossuositukset muiden haittavaikutusten yhteydessä yhdistelmähoidossa binimetinibin tai setuksimabin tai setuksimabin ja FOLFOXin kanssa
Haittavaikutuksen vaikeusaste | Enkorafenibi |
| Enkorafenibihoito on keskeytettävä enintään 4 viikoksi.
|
| Enkorafenibihoito on keskeytettävä enintään 4 viikoksi.
Tai enkorafenibihoito lopetetaan pysyvästi. |
| Enkorafenibihoidon lopettamista pysyvästi on harkittava. |
| Enkorafenibihoito on lopetettava pysyvästi. |
Hoidon kesto
Hoitoa on jatkettava, kunnes potilas ei enää hyödy siitä tai ilmaantuu haittavaikutuksia, jotka eivät ole hyväksyttävissä.
Annoksen unohtuminen
Jos enkorafenibiannos unohtuu, unohtunut annos voidaan ottaa vain, jos seuraavaan suunniteltuun annokseen on yli 12 tuntia.
Oksentelu
Jos potilas oksentaa enkorafenibin annon jälkeen, uutta annosta ei saa ottaa, vaan seuraava annos otetaan suunniteltuun aikaan.
Erityisryhmät
Iäkkäät potilaat
65‑vuotiaiden ja sitä iäkkäämpien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Enkorafenibialtistus saattaa olla suurentunut, jos potilaalla on lievä, keskivaikea tai vaikea maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Jos potilaalla on lievä maksan vajaatoiminta (Child–Pugh-luokka A), enkorafenibia on annettava varoen annoksena 300 mg kerran vuorokaudessa.
Potilaille, joilla on keskivaikea maksan vajaatoiminta (Child–Pugh-luokka B) tai vaikea maksan vajaatoiminta (Child–Pugh-luokka C), ei voida antaa annossuosituksia.
Munuaisten vajaatoiminta
Populaatiofarmakokineettisen analyysin perusteella annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta. Enkorafenibin käytöstä vaikeaa munuaisten vajaatoimintaa sairastavilla ei ole kliinisiä tietoja. Tästä syystä mahdollista annosmuutosten tarvetta ei voida määrittää. Enkorafenibin käytössä on noudatettava varovaisuutta, jos potilaalla on vaikea munuaisten vajaatoiminta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Enkorafenibin turvallisuutta ja tehoa lasten ja nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Braftovi otetaan suun kautta. Kapselit niellään kokonaisina veden kera. Ne voidaan ottaa ruoan kanssa tai ilman. Enkorafenibin ja greippimehun samanaikaista käyttöä on vältettävä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Potilaille, jotka eivät pysty nielemään, kapselit voidaan avata ja niiden sisältö liuottaa pieneen määrään (noin 20 ml) omenasosetta ja ottaa välittömästi.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Enkorafenibi on tarkoitettu annettavaksi yhdessä binimetinibin kanssa (potilaille, joilla on BRAF V600 -mutaatiopositiivinen leikkaukseen soveltumaton tai etäpesäkkeinen melanooma ja potilaille, joilla on BRAF V600E -mutaatiopositiivinen pitkälle edennyt ei-pienisoluinen keuhkosyöpä) tai yhdessä setuksimabin kanssa (potilaille, joilla on etäpesäkkeinen BRAF V600E ‑mutaatiopositiivinen kolorektaalisyöpä ja jotka ovat aiemmin saaneet systeemistä hoitoa) tai yhdessä setuksimabin ja FOLFOXin kanssa (ensilinjan hoitona potilaille, joilla on etäpesäkkeinen BRAF V600E -mutaatiopositiivinen kolorektaalisyöpä). Lisätiedot binimetinibi-, setuksimabi- ja 5‑fluorourasiili/foliinihappo/oksaliplatiinihoitoon liittyvistä varoituksista ja varotoimista, ks. näiden lääkevalmisteiden valmisteyhteenvedot, kohta Varoitukset ja käyttöön liittyvät varotoimet.
Enkorafenibi yhdessä binimetinibin kanssa potilailla, joiden tauti on edennyt BRAF:n estäjähoidon aikana
Enkorafenibin ja binimetinibin yhdistelmän käyttöä koskevia tietoja on rajallisesti potilaista, joiden tauti on edennyt aiemman BRAF:n estäjähoidon aikana, kun on hoidettu leikkaukseen soveltumatonta tai etäpesäkkeistä melanoomaa ja kasvaimessa on ollut BRAF V600 ‑mutaatio. Näiden tietojen perusteella yhdistelmän teho on heikompi kyseisillä potilailla.
Enkorafenibi yhdessä binimetinibin kanssa potilailla, joilla on etäpesäkkeitä aivoissa
Enkorafenibin ja binimetinibin yhdistelmän tehoa koskevaa tietoa on rajallisesti potilaista, joilla on BRAF V600 -mutaatiopositiivinen melanooma tai BRAF V600E -mutaatiopositiivinen ei-pienisoluinen keuhkosyöpä ja sen etäpesäkkeitä aivoissa (ks. kohta Farmakodynamiikka).
Vasemman kammion toimintahäiriö
Vasemman kammion toimintahäiriötä eli oireista tai oireetonta ejektiofraktion pienenemistä on ilmoitettu enkorafenibin ja binimetinibin yhdistelmän käytön aikana.
Vasemman kammion ejektiofraktion arviointi kaikukardiografialla tai MUGA-tutkimuksella on suositeltavaa ennen enkorafenibi- ja binimetinibihoidon aloittamista, kuukauden kuluttua hoidon aloittamisesta ja tämän jälkeen noin 3 kuukauden välein tai tiheämmin kliinisen tarpeen mukaan, kunnes hoito lopetetaan. Jos vasemman kammion toimintahäiriötä esiintyy hoidon aikana, ks. binimetinibin valmisteyhteenvedon kohta Annostus ja antotapa.
Enkorafenibin ja binimetinibin yhdistelmän turvallisuutta ei ole varmistettu potilailla, joilla vasemman kammion ejektiofraktio on lähtötilanteessa alle 50 % tai alle laitoksen viitealueen alarajan. Tällaisissa tilanteissa binimetinibiä on siis käytettävä varoen, ja binimetinibi- ja enkorafenibihoito on lopetettava, jos potilaalla ilmenee oireinen vasemman kammion toimintahäiriö, asteen 3–4 vasemman kammion ejektiofraktion pienenemä tai vasemman kammion ejektiofraktion ≥ 10 % absoluuttinen pienenemä lähtötilanteesta, ja vasemman kammion ejektiofraktio on arvioitava 2 viikon välein, kunnes haitta on korjautunut.
Verenvuoto
Verenvuotoa, mukaan lukien suuria verenvuototapahtumia, voi esiintyä enkorafenibihoidon aikana (ks. kohta Haittavaikutukset). Samanaikainen antikoagulaatio- tai antitromboottinen hoito saattaa suurentaa verenvuotoriskiä. Verenvuototapahtumat, joiden aste on ≥ 3, on hoidettava keskeyttämällä tai lopettamalla hoito (ks. kohta Annostus ja antotapa, taulukko 4) ja kliinisen tarpeen mukaan.
Silmähaitat
Silmähaittoja, mukaan lukien uveiitti, iriitti ja iridosykliitti, voi esiintyä enkorafenibin annon aikana. Myös verkkokalvon pigmenttiepiteelin irtaumaa on ilmoitettu enkorafenibin ja binimetinibin yhdistelmää käyttäneillä potilailla (ks. kohta Haittavaikutukset).
Potilaat on arvioitava jokaisella käynnillä uusien tai pahenevien näköhäiriöiden oireiden varalta. Silmälääkärin on suositeltavaa tehdä silmätutkimus viipymättä, jos uusien tai pahenevien näköhäiriöiden oireita havaitaan, mukaan lukien keskeisen näön heikentyminen, näön hämärtyminen tai näön menetys.
Jos hoidon aikana esiintyy uveiittia, mukaan lukien iridosykliittiä ja iriittiä, ks. kohta Annostus ja antotapa.
Jos potilaalle kehittyy hoidon aikana verkkokalvon pigmenttiepiteelin irtauma tai verkkokalvon laskimotukos, ks. binimetinibin valmisteyhteenveto, kohta Annostus ja antotapa.
QT‑ajan piteneminen
BRAF:n estäjähoitoa saaneilla potilailla on havaittu QT‑ajan pitenemistä. Perusteellista QT‑tutkimusta, jossa arvioitaisiin enkorafenibin vaikutusta QT‑ajan pitenemiseen, ei ole toteutettu.
Tulokset viittaavat kokonaisuudessaan siihen, että enkorafenibimonoterapia saattaa aiheuttaa lievää syketiheyden kohoamista. Enkorafenibin ja binimetinibin suositusannosten käyttöä koskevien yhdistettyjen yhdistelmätutkimusten ja enkorafenibimonoterapiaa koskevan tutkimuksen tulokset viittaavat siihen, että enkorafenibi saattaa hieman pidentää QTc‑aikaa (ks. kohta Farmakodynamiikka).
Tietoja ei ole riittävästi, jotta kliinisesti merkittävä altistuksesta riippuvainen QT-ajan pidentyminen voitaisiin sulkea pois.
Mahdollisen QT‑ajan pitenemisriskin vuoksi on suositeltavaa korjata seerumin elektrolyyttihäiriöt, mukaan lukien magnesium- ja kaliumhäiriöt, sekä kontrolloida QT‑ajan pitenemisen riskitekijät (esim. kongestiivinen sydämen vajaatoiminta, bradyarytmiat) ennen hoidon aloittamista ja hoidon aikana.
EKG-tutkimuksen tekeminen on suositeltavaa ennen enkorafenibihoidon aloittamista, kuukauden kuluttua hoidon aloittamisesta ja tämän jälkeen noin 3 kuukauden välein tai tiheämmin kliinisen tarpeen mukaan, kunnes hoito lopetetaan. QTc‑ajan pitenemistä voidaan hoitaa pienentämällä annosta tai keskeyttämällä tai lopettamalla hoito sekä korjaamalla elektrolyyttihäiriöt ja kontrolloimalla riskitekijöitä (ks. kohta Annostus ja antotapa).
Uudet primaarimaligniteetit
Uusia ihon ja muita kuin ihon primaarimaligniteetteja on havaittu BRAF:n estäjillä hoidetuilla potilailla, ja niitä voi esiintyä enkorafenibin annon yhteydessä (ks. kohta Haittavaikutukset).
Ihon maligniteetit
Ihon maligniteetteja, kuten ihon okasolusyöpää (keratoakantooma mukaan lukien), on havaittu potilailla, joita on hoidettu BRAF:n estäjillä, mukaan lukien enkorafenibilla.
Uusia primaarimelanoomia on todettu potilailla, joita on hoidettu BRAF:n estäjillä, mukaan lukien enkorafenibilla (ks. kohta Haittavaikutukset).
Dermatologiset arvioinnit on tehtävä ennen enkorafenibin käytön aloittamista, 2 kuukauden välein hoidon aikana ja 6 kuukauden ajan enkorafenibihoidon päättymisen jälkeen. Epäilyttävät ihomuutokset on poistettava kirurgisesti ja arvioitava dermatopatologisesti. Potilasta kehotetaan ilmoittamaan lääkärille heti, jos uusia ihomuutoksia kehittyy. Enkorafenibin käyttöä jatketaan muuttamatta annosta.
Muut kuin ihon maligniteetit
Vaikutusmekanisminsa perusteella enkorafenibi voi edistää maligniteettien kehittymistä. Ilmiö liittyy RAS:n aktivaatioon mutaation tai muiden mekanismien välityksellä. Enkorafenibia käyttäville potilaille on tehtävä pään ja kaulan alueen tutkimus, rintakehän / vatsan alueen TT-kuvaus, anaalinen tutkimus ja gynekologinen sisätutkimus (naiset), ja heiltä on tutkittava täydellinen verenkuva ennen hoidon aloittamista, hoidon aikana ja hoidon lopussa kliinisen tarpeen mukaan. Enkorafenibihoidon lopettamista pysyvästi on harkittava, jos potilaalle kehittyy RAS-mutaatiopositiivinen maligniteetti muualle kuin ihoon. Hyödyt ja riskit on punnittava tarkoin ennen enkorafenibin antoa potilaalle, jolla on tai on ollut RAS-mutaatioon liittyvä syöpä.
Tuumorilyysioireyhtymä
Enkorafenibin käyttöön yhdessä binimetinibin kanssa on liittynyt tuumorilyysioireyhtymää, joka voi johtaa kuolemaan (ks. kohta Haittavaikutukset). Tuumorilyysioireyhtymän riskitekijöitä ovat mm. suuri kasvainkuorma, krooninen munuaisten vajaatoiminta, oliguria, nestehukka, hypotensio ja virtsan happamuus. Näitä potilaita on seurattava tarkoin, ja kliinisen tarpeen mukaista hoitoa on annettava viipymättä. Lisäksi on harkittava profylaktista nesteytystä.
Maksa-arvojen poikkeavuudet
Maksan toimintaa mittaavien laboratoriokoearvojen poikkeavuuksia (mukaan lukien ASAT- ja ALAT-arvon kohoaminen) on havaittu enkorafenibihoidon yhteydessä (ks. kohta Haittavaikutukset). Maksa-arvoja on seurattava ennen enkorafenibin käytön aloittamista ja vähintään kuukausittain ensimmäisten 6 hoitokuukauden aikana ja sen jälkeen kliinisen tarpeen mukaan. Maksa-arvojen poikkeavuudet on hoidettava keskeyttämällä hoito, pienentämällä annosta tai lopettamalla hoito (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Enkorafenibi metaboloituu ja eliminoituu pääasiassa maksan kautta. Näin ollen lievässä, keskivaikeassa ja vaikeassa maksan vajaatoiminnassa enkorafenibialtistus voi suurentua yksilöllistä vaihtelualuetta suuremmaksi (ks. kohta Farmakokinetiikka).
Kliinisten tietojen puuttuessa enkorafenibin käyttö ei ole suositeltavaa, jos potilaalla on keskivaikea tai vaikea maksan vajaatoiminta.
Enkorafenibia on annosteltava annoksella 300 mg kerran vuorokaudessa varovaisuutta noudattaen, jos potilaalla on lievä maksan vajaatoiminta (ks. kohta Annostus ja antotapa).
Lievää maksan vajaatoimintaa sairastavia potilaita on suositeltavaa seurata tavanomaista tarkemmin enkorafenibiin liittyvien haittojen varalta mm. kliinisten tutkimustoimenpiteiden avulla ja tutkimalla maksatoiminta, ja EKG on tutkittava kliinisen tarpeen mukaan hoidon aikana.
Munuaisten vajaatoiminta
Valmisteen käytöstä vaikeaa munuaisten vajaatoimintaa sairastavien hoidossa ei ole tietoja (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Enkorafenibin käytössä on noudatettava varovaisuutta, jos potilaalla on vaikea munuaisten vajaatoiminta. Kreatiniinipitoisuuden suurenemista on ilmoitettu yleisesti, kun enkorafenibia on annettu monoterapiana tai yhdessä binimetinibin tai setuksimabin kanssa. Todetut munuaisten vajaatoimintatapaukset (mukaan lukien akuutti munuaisvaurio ja munuaisten vajaatoiminta) liittyivät yleensä oksenteluun ja nestehukkaan. Muita vaikuttavia tekijöitä olivat esimerkiksi diabetes ja hypertensio. Veren kreatiniinipitoisuutta on seurattava kliinisen tarpeen mukaan ja suurentunutta kreatiniinipitoisuutta hoidettava muuttamalla annosta tai lopettamalla hoito (ks. kohta Annostus ja antotapa, taulukko 4). Potilaiden on varmistettava hoidon aikana riittävä nesteensaanti.
Muiden lääkevalmisteiden vaikutukset enkorafenibiin
Vahvojen CYP3A:n estäjien käyttöä enkorafenibihoidon aikana on vältettävä. Jos vahvan CYP3A:n estäjän samanaikainen käyttö on välttämätöntä, potilaita on seurattava tarkoin turvallisuussyistä (ks. kohta Yhteisvaikutukset).
Varovaisuutta on noudatettava, jos enkorafenibin kanssa annetaan samanaikaisesti keskivahvaa CYP3A:n estäjää.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset enkorafenibiin
Enkorafenibi metaboloituu pääosin CYP3A4‑välitteisesti.
CYP3A4:n estäjät
Keskivahvojen CYP3A4:n estäjien (diltiatseemi) ja vahvojen CYP3A4:n estäjien (posakonatsoli) samanaikainen käyttö enkorafenibin kerta-annosten kanssa terveillä tutkittavilla johti pitoisuus-aikakäyrän alle jäävän pinta-alan (AUC) suurenemiseen 2-kertaiseksi keskivahvojen CYP3A4:n estäjien käytössä ja 3-kertaiseksi vahvojen estäjien käytössä sekä enkorafenibin maksimipitoisuuden (Cmax) suurenemiseen keskivahvojen CYP3A4:n estäjien käytössä 44,6 % ja vahvojen estäjien käytössä 68,3 %.
Malleihin perustuvat ennustukset viittaavat siihen, että posakonatsolin vaikutus enkorafenibin AUC-arvoon voi olla toistuvan annon jälkeen samaa luokkaa (3-kertainen suurenema) ja vaikutus enkorafenibin Cmax‑arvoon hieman suurempi (2,7‑kertainen suurenema). Malleihin perustuvat ennustukset ketokonatsolin vaikutuksesta viittaavat noin viisinkertaiseen enkorafenibin AUC‑suurenemaan, kun enkorafenibiä annosteltiin annoksella 450 mg kerran vuorokaudessa, ja 3‑4‑kertaiseen enkorafenibin Cmax‑suurenemaan, kun enkorafenibiä annosteltiin annoksella 300 mg kerran vuorokaudessa.
Tästä syystä enkorafenibin ja vahvojen CYP3A4:n estäjien samanaikaista käyttöä on vältettävä (suurentuneen enkorafenibialtistuksen ja mahdollisen haittojen voimistumisen vuoksi, ks. kohta Farmakokinetiikka). Vahvoja CYP3A4:n estäjiä ovat mm. ritonaviiri, itrakonatsoli, klaritromysiini, telitromysiini, posakonatsoli ja greippimehu. Jos vahvan CYP3A:n estäjän samanaikainen käyttö on välttämätöntä, potilaita on seurattava tarkoin turvallisuussyistä.
Keskivahvojen CYP3A4:n estäjien samanaikaisessa käytössä on noudatettava varovaisuutta. Keskivahvoja CYP3A4:n estäjiä ovat mm. amiodaroni, erytromysiini, flukonatsoli, diltiatseemi, amprenaviiri ja imatinibi. Kun enkorafenibia annetaan samanaikaisesti keskivahvan CYP3A:n estäjän kanssa, potilaita on seurattava tarkoin turvallisuussyistä.
CYP3A4:n induktorit
Enkorafenibin ja vahvojen CYP3A4:n induktorien samanaikaista käyttöä ei ole arvioitu kliinisessä tutkimuksessa, mutta enkorafenibialtistuksen pieneneminen on todennäköistä ja se voi johtaa enkofarenibin tehon heikkenemiseen. Vahvoja CYP3A4:n induktoreita ovat mm. karbamatsepiini, rifampisiini, fenytoiini ja mäkikuisma. On harkittava muita lääkevaihtoehtoja, jotka eivät aiheuta lainkaan CYP3A:n induktiota tai aiheuttavat sitä korkeintaan kohtalaisissa määrin.
Enkorafenibin vaikutukset muihin lääkevalmisteisiin
CYP:n substraatit
Enkorafenibi on vahva CYP3A4:n induktori. Samanaikainen CYP3A4:n substraattien (esim. hormonaalisten ehkäisyvalmisteiden) käyttö voi johtaa näiden lääkeaineiden tehon heikkenemiseen. Jos enkorafenibin kanssa on välttämätöntä antaa samanaikaisesti CYP3A4:n substraatteja, joiden terapeuttinen leveys on pieni, näiden substraattien annosta on muutettava niiden hyväksytyn valmisteyhteenvedon mukaisesti.
Enkorafenibi on UGT1A1:n estäjä. Samanaikaisesti käytettävien UGT1A1:n substraattien (esim. raltegraviiri, atorvastatiini, dolutegraviiri) altistus saattaa suurentua, ja siksi niiden käytössä on noudatettava varovaisuutta.
Enkorafenibin vaikutus binimetinibiin
Enkorafenibi on suhteellisen vahva reversiibeli UGT1A1:n estäjä, mutta binimetinibialtistuksessa ei ole kliinisesti havaittu eroja, kun binimetinibiä on annettu yhdessä enkorafenibin kanssa.
Kuljettajaproteiinien substraatit
Enkorafenibi on OATP1B1:n, OATP1B3:n ja/tai BCRP:n estäjä in vivo. Enkorafenibin ja OATP1B1:n, OATP1B3:n ja BCRP:n substraattien (kuten rosuvastatiini, atorvastatiini ja metotreksaatti) samanaikainen käyttö saattaa suurentaa näiden substraattien pitoisuuksia (ks. kohta Farmakokinetiikka).
Enkorafenibi saattaa estää lukuisien muiden kuljettajaproteiinien toimintaa in vitro. Munuaisten OAT1-, OAT3- ja OCT2-kuljettajaproteiinien substraattien (kuten furosemidi ja penisilliini), maksan OCT1-kuljettajaproteiinien substraattien (kuten bosentaani) tai P-gp:n substraattien (esim. posakonatsoli) altistus saattaa myös suurentua.
Siksi näiden kuljettajaproteiinien substraattien samanaikaisessa käytössä on noudatettava varovaisuutta.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi/raskauden ehkäisy naisilla
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä enkorafenibihoidon aikana ja vähintään 1 kuukauden ajan viimeisen annoksen jälkeen. Enkorafenibi saattaa heikentää hormonaalisten ehkäisyvalmisteiden tehoa (ks. kohta Yhteisvaikutukset). Siksi hormonaalista ehkäisyä käyttävien naispotilaiden on suositeltavaa käyttää ylimääräistä tai muunlaista ehkäisymenetelmää kuten estemenetelmää (esim. kondomia) enkorafenibihoidon aikana ja vähintään 1 kuukauden ajan viimeisen annoksen jälkeen.
Raskaus
Enkorafenibin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Enkorafenibin käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä ehkäisyä. Jos enkorafenibia käytetään raskauden aikana tai raskaus alkaa enkorafenibihoidon aikana, potilaalle on kerrottava sikiöön mahdollisesti kohdistuvista riskeistä.
Imetys
Ei tiedetä, erittyvätkö enkorafenibi tai sen metaboliitit ihmisillä äidinmaitoon. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. On päätettävä, lopetetaanko imetys vai pidättäydytäänkö
enkorafenibihoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Enkorafenibin vaikutuksista ihmisen hedelmällisyyteen ei ole tietoa. Eläinkoelöydösten perusteella enkorafenibin käyttö voi heikentää lisääntymiskykyisten miesten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta). Tämän kliinistä merkitystä ei tunneta, joten miespotilaille on kerrottava mahdollisesta spermatogeneesin heikentymisriskistä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Enkorafenibilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Näköhäiriöitä on ilmoitettu joillakin enkorafenibihoitoa saaneilla potilailla kliinisissä tutkimuksissa. Potilaita on kehotettava olemaan ajamatta tai käyttämättä koneita, jos heillä on näköhäiriöitä tai muita ajokykyyn ja koneidenkäyttökykyyn vaikuttavia haittavaikutuksia (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Enkorafenibin (450 mg suun kautta kerran vuorokaudessa) ja binimetinibin (45 mg suun kautta kahdesti vuorokaudessa) yhdistelmän turvallisuutta arvioitiin yhdistetyssä turvallisuuspopulaatiossa (Combo 450 ISP), johon kuului 372 potilasta, joilla oli BRAF V600 -mutaatiopositiivinen leikkaukseen soveltumaton tai etäpesäkkeinen melanooma tai BRAF V600E -mutaatiopositiivinen pitkälle edennyt ei-pienisoluinen keuhkosyöpä. Combo 450 ISP:n potilaista 274 potilasta sai yhdistelmähoitoa BRAF V600 -mutaatiopositiiviseen, leikkaukseen soveltumattomaan tai etäpesäkkeiseen melanoomaan (kahdessa vaiheen II tutkimuksessa [CMEK162X2110 ja CLGX818X2109] ja yhdessä vaiheen III tutkimuksessa [CMEK162B2301, osa 1]) ja 98 potilasta sai yhdistelmähoitoa BRAF V600E -mutaatiopositiiviseen pitkälle edenneeseen ei-pienisoluiseen keuhkosyöpään (yhdessä vaiheen II tutkimuksessa [ARRAY-818-202]) (ks. kohta Farmakodynamiikka).
Yleisimmät haittavaikutukset (≥ 25 %) enkorafenibin ja binimetinibin yhdistelmää saaneilla potilailla olivat väsymys, pahoinvointi, ripuli, oksentelu, vatsakipu, myopatia/lihashäiriöt ja nivelkipu.
Enkorafenibin (300 mg suun kautta kerran vuorokaudessa) turvallisuutta käytettynä yhdessä binimetinibin kanssa (45 mg suun kautta kahdesti vuorokaudessa) arvioitiin 257 potilaalla, joilla oli BRAF V600 -mutaatiopositiivinen leikkaukseen soveltumaton tai etäpesäkkeinen melanooma (Combo 300 -populaatio). Hoitoa arvioitiin vaiheen III tutkimuksessa (CMEK162B2301, osa 2). Yleisimpiä (≥ 25 %) haittavaikutuksia potilailla, jotka saivat enkorafenibia 300 mg annettuna binimetinibin kanssa, olivat väsymys, pahoinvointi ja ripuli.
Enkorafenibimonoterapian (300 mg suun kautta kerran vuorokaudessa) turvallisuusprofiili perustuu 217 potilaaseen, joilla oli leikkaukseen soveltumaton tai etäpesäkkeinen BRAF V600 ‑mutaatiopositiivinen melanooma (yhdistetty enkorafenibi 300 ‑populaatio). Yleisimmät enkorafenibi 300 -hoidon yhteydessä ilmoitetut haittavaikutukset (≥ 25 %) olivat hyperkeratoosi, hiustenlähtö, käsi-jalkaoireyhtymä, väsymys, ihottuma, nivelkipu, ihon kuivuus, pahoinvointi, lihaskipu, päänsärky, oksentelu ja kutina.
Enkorafenibin (300 mg suun kautta kerran vuorokaudessa) ja setuksimabin (annosteltu sen valmisteyhteenvedon mukaan) yhdistelmän turvallisuutta arvioitiin 216 potilaalla, joilla oli BRAF V600E ‑mutaatiopositiivinen etäpesäkkeinen kolorektaalisyöpä. Hoitoa arvioitiin vaiheen III tutkimuksessa (ARRAY-818-302). Yleisimmät haittavaikutukset (> 25 %), joita ilmoitettiin tällä populaatiolla, olivat: väsymys, pahoinvointi, ripuli, aknetyyppinen dermatiitti, vatsakipu, nivelkipu/tuki- ja liikuntaelinkipu, ruokahalun heikkeneminen, ihottuma ja oksentelu.
Haittavaikutuksista johtunut tutkimuslääkkeen käytön keskeyttämisprosentti oli 1,9 % potilailla, jotka saivat enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa.
Enkorafenibin (300 mg suun kautta kerran vuorokaudessa), setuksimabin ja FOLFOXin yhdistelmän turvallisuutta arvioitiin 259 potilaalla, joilla oli BRAF V600E ‑mutaatiopositiivinen etäpesäkkeinen kolorektaalisyöpä. Hoitoa arvioitiin tutkimuksessa C4221015 – BREAKWATER (jäljempänä ”yhdistetty ES + FOLFOX ‑populaatio”, joka sisältää kaikki potilaat, jotka saivat enkorafenibiä ja setuksimabia (ES) sekä FOLFOXia tutkimuksen eri osissa). Yleisimmät haittavaikutukset (≥ 25 %), joita ilmoitettiin tällä populaatiolla, olivat: perifeerinen neuropatia, neutropenia, pahoinvointi, väsymys, anemia, ripuli, oksentelu, ruokahalun heikkeneminen, ihottuma, trombosytopenia, vatsakipu, verenvuoto, nivelkipu/tuki- ja liikuntaelinkipu, kuume, ummetus, limakalvotulehdus ja infektio. Haittavaikutuksista johtunut tutkimuslääkkeen käytön keskeyttämisprosentti oli 3,5 % potilailla, jotka saivat enkorafenibin (300 mg), setuksimabin ja FOLFOXin yhdistelmähoitoa.
Haittavaikutustaulukko
Haittavaikutukset luetellaan alla MedDRA-elinjärjestelmäluokituksen ja yleisyyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 5: Haittavaikutukset
Yleisyys | Enkorafenibimonoterapia 300 mg (n = 217) | Enkorafenibi 450 mg yhdessä binimetinibin kanssa (n = 372) | Enkorafenibi 300 mg yhdessä setuksimabin kanssa (n = 216) | Enkorafenibi 300 mg yhdessä setuksimabin ja FOLFOXin kanssa (n = 259) |
Hyvän- ja pahanlaatuiset kasvaimet | ||||
Hyvin yleinen | Ihon papillooma* Melanosyyttiluomet |
| Melanosyyttiluomet |
|
Yleinen | Ihon okasolusyöpäa Uusi primaarimelanooma* | Ihon okasolusyöpäa Ihon papillooma* | Ihon okasolusyöpäa Ihon papillooma* Uusi primaarimelanooma* | Melanosyyttiluomet Ihon papillooma* Tyvisolusyöpä Ihon okasolusyöpäa |
Melko harvinainen | Tyvisolusyöpä | Tyvisolusyöpä* | Tyvisolusyöpä* | Uusi primaarimelanooma* |
Veri ja imukudos | ||||
Hyvin yleinen |
| Anemia |
| Neutropenia* Anemia* Trombosytopenia* Leukopenia* |
Immuunijärjestelmä | ||||
Hyvin yleinen |
|
|
| Yliherkkyysb |
Yleinen | Yliherkkyysb | Yliherkkyysb | Yliherkkyysb |
|
Aineenvaihdunta ja ravitsemus | ||||
Hyvin yleinen | Ruokahalun heikkeneminen |
| Ruokahalun heikkeneminen | Ruokahalun heikkeneminen Hypokalemia Hypomagnesemia Hypoalbuminemia* |
Tuntematon |
| Tuumorilyysioireyhtymä |
|
|
Psyykkiset häiriöt | ||||
Hyvin yleinen | Unettomuus |
| Unettomuus | Unettomuus* |
Hermosto | ||||
Hyvin yleinen | Päänsärky* Perifeerinen neuropatia* Makuaistin häiriöt* | Perifeerinen neuropatia* Huimaus* Päänsärky* | Perifeerinen neuropatia* Päänsärky* | Perifeerinen neuropatian Makuaistin häiriöt* Päänsärky* |
Yleinen | Kasvopareesic | Makuaistin häiriöt* | Huimaus* Makuaistin häiriöt | Huimaus* |
Melko harvinainen |
| Kasvopareesic |
|
|
Silmät | ||||
Hyvin yleinen |
| Näköhäiriöt* Verkkokalvon pigmenttiepiteelin irtauma* |
|
|
Yleinen |
| Uveiitti* |
|
|
Melko harvinainen | Uveiitti* |
|
|
|
Sydän | ||||
Yleinen | Supraventrikulaarinen takykardiad | Vasemman kammion toimintahäiriöh | Supraventrikulaarinen takykardiad | Supraventrikulaarinen takykardiad |
Verisuonisto | ||||
Hyvin yleinen |
| Verenvuotoi Hypertensio* | Verenvuotoi | Verenvuotoi |
Yleinen |
| Tromboembolisetlaskimotapahtumatj |
|
|
Ruoansulatuselimistö | ||||
Hyvin yleinen | Pahoinvointi Oksentelu* Ummetus | Pahoinvointi Oksentelu* Ummetus Vatsakipu* Ripuli* | Pahoinvointi Oksentelu* Ummetus Vatsakipu* Ripuli* | Pahoinvointi Ripuli* Oksentelu* Vatsakipu* Ummetus Limakalvotulehdus* |
Yleinen |
| Koliittik |
|
|
Melko harvinainen | Haimatulehdus* | Haimatulehdus* | Haimatulehdus* | Haimatulehdus* |
Iho ja ihonalainen kudos | ||||
Hyvin yleinen | Käsi-jalkaoireyhtymä Hyperkeratoosi* Ihottuma* Ihon kuivuus* Kutina* Hiustenlähtö* Punoituse Ihon hyperpigmentaatio* | Hyperkeratoosi* Ihottuma* Ihon kuivuus* Kutina* Hiustenlähtö* | Aknetyyppinen dermatiitti* Ihottuma* Ihon kuivuus* Kutina* | Ihottuma* Ihon hyperpigmentaatio* Aknetyyppinen dermatiitti* Ihon kuivuus* Hiustenlähtö Käsi-jalkaoireyhtymä Kutina |
Yleinen | Aknetyyppinen dermatiitti* Ihon kesiminenf Valoherkkyys* | Aknetyyppinen dermatiitti* Käsi-jalkaoireyhtymä Punoitus* Pannikuliitti* Valoherkkyys* | Ihon hyperpigmentaatio Käsi-jalkaoireyhtymä Hyperkeratoosi* Hiustenlähtö* Punoituse | Punoitus Hyperkeratoosi* Ihon kesiminen |
Melko harvinainen |
|
| Ihon kesiminenf |
|
Luusto, lihakset ja sidekudos | ||||
Hyvin yleinen | Nivelkipu* Lihaskipug Ääreisosien kipu Selkäkipu | Nivelkipu* Myopatia/lihashäiriötl Ääreisosien kipu Selkäkipu* | Nivelkipu/tuki- ja liikuntaelinkipu* Myopatia/lihashäiriöt* Ääreisosien kipu Selkäkipu | Nivelkipu/tuki- ja liikuntaelinkipu* Myopatia/lihashäiriötl Selkäkipu* |
Yleinen | Niveltulehdus* |
|
| Ääreisosien kipu* |
Melko harvinainen |
| Rabdomyolyysi |
|
|
Munuaiset ja virtsatiet | ||||
Yleinen | Munuaisten vajaatoiminta* | Munuaisten vajaatoiminta* | Munuaisten vajaatoiminta* | Munuaisten vajaatoiminta* |
Yleisoireet ja antopaikassa todettavat haitat | ||||
Hyvin yleinen | Väsymys* Kuume* | Väsymys* Kuume* Ääreisosien turvotusm | Väsymys* Kuume* | Väsymys* Kuume* |
Infektiot | ||||
Hyvin yleinen |
|
|
| Infektioto |
Yleinen |
|
|
| Ylähengitystieinfektio* Sepsis* |
Tutkimukset | ||||
Hyvin yleinen | Gammaglutamyylitransferaasiarvon suureneminen* | Veren kreatiinikinaasiarvon suureneminen Gammaglutamyylitransferaasiarvon suureneminen* Transaminaasiarvojen suureneminen* |
| Lipaasiarvon suureneminen* Painonlasku Transaminaasiarvojen suureneminen* |
Yleinen | Transaminaasiarvojen suureneminen* Veren kreatiniiniarvon suureneminen* Lipaasiarvon suureneminen | AFOS-arvon suureneminen Veren kreatiniiniarvon suureneminen* Amylaasiarvon suureneminen Lipaasiarvon suureneminen | Veren kreatiniiniarvon suureneminen* Transaminaasiarvojen suureneminen* | Veren kreatiniiniarvon suureneminen* Amylaasiarvon suureneminen* |
Melko harvinainen | Amylaasiarvon suureneminen |
| Amylaasiarvon suureneminen Lipaasiarvon suureneminen |
|
* yhdistelmätermi, joka sisältää useamman kuin yhden termin
a sisältää muun muassa keratoakantooman, levyepiteelikarsinooman ja ihon okasolusyövän
b sisältää muun muassa angioedeeman, anafylaktisen reaktion, lääkeyliherkkyyden, yliherkkyyden, yliherkkyysverisuonitulehduksen ja nokkosihottuman
c sisältää kasvohermohäiriön, kasvohalvauksen, kasvopareesin ja Bellin pareesin
d sisältää muun muassa lisälyönnit, supraventrikulaarisen takykardian, eteistakykardian ja sinustakykardian
e sisältää punoituksen, yleistyneen punoituksen ja plantaarisen punoituksen
f sisältää eksfoliatiivisen dermatiitin, ihon kesimisen ja eksfoliatiivisen ihottuman
g sisältää lihaskivun, lihasten väsymisen, lihasvaurion, lihasspasmit ja lihasheikkouden
h sisältää vasemman kammion toimintahäiriön, ejektiofraktion pienenemisen, sydämen vajaatoiminnan ja ejektiofraktion poikkeavuuden
i sisältää verenvuodon eri alueilla, muun muassa aivoverenvuodon, kallonsisäisen verenvuodon, emätinverenvuodon, runsaan kuukautisvuodon, kuukautisten välisen vuodon, veriulosteen, nenäverenvuodon, veriyskökset, veririnnan, ruoansulatuskanavan verenvuodon ja hematurian
j sisältää muun muassa keuhkoembolian, syvän laskimotukoksen, embolian, tromboflebiitin, pinnallisen tromboflebiitin, tromboosin, flebiitin, yläonttolaskimo-oireyhtymän, suolilievelaskimon tromboosin ja onttolaskimon tromboosin
k sisältää koliitin, haavaisen koliitin, enterokoliitin ja proktiitin
l sisältää muun muassa lihaskivun, lihasheikkouden, lihasspasmit, lihasvaurion, myopatian ja myosiitin
m sisältää muun muassa nesteretention, ääreisosien turvotuksen, paikallisen turvotuksen ja yleistyneen turvotuksen
n sisältää muun muassa kylmään liittyvän dysestesian, dysestesian, hyperestesian, hypestesian, neuralgian, perifeerisen neuropatian, neurotoksisuuden, parestesian, perifeerisen sensorisen neuropatian, polyneuropatian
o sisältää muun muassa virtsatieinfektion, gastroenteriitin, peritoniitin, selluliitin, vatsaontelon infektion, ruoansulatuskanavan infektion, infektion, alempien hengitysteiden infektion, virus- ja bakteeriperäisen keuhkokuumeen, virusinfektion
Kun enkorafenibia käytettiin annoksella 300 mg kerran vuorokaudessa yhdistelmänä binimetinibin kanssa (45 mg kahdesti vuorokaudessa) (Combo 300) tutkimuksen CMEK162B2301 osassa 2, seuraavien haittavaikutusten yleisyysluokka oli pienempi kuin yhdistetyssä Combo 450 -populaatiossa: anemia, perifeerinen neuropatia, verenvuoto, hypertensio, kutina (yleinen); sekä koliitti, amylaasiarvon suureneminen ja lipaasiarvon suureneminen (melko harvinainen).
Valikoitujen haittavaikutusten kuvaus
Ihon maligniteetit
Ihon okasolusyöpä
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa ihon okasolusyöpää, mukaan lukien keratoakantoomaa, todettiin 3,0 %:lla (11/372) potilaista. Ensimmäisen ihon okasolusyöpätapahtuman (kaikki asteet) alkamiseen kuluneen ajan mediaani oli 6,5 kk (1,0–22,8 kk).
Yhdistetyssä enkorafenibi 300 -populaatiossa ihon okasolusyöpää ilmoitettiin 7,4 %:lla (16/217) potilaista. Vaiheen III tutkimuksen (CMEK162B2301) potilailla, joilla esiintyi ihon okasolusyöpää, ensimmäisen okasolusyöpätapahtuman (kaikki asteet) alkamiseen kuluneen ajan mediaani oli 2,3 kk (0,3–12,0 kk).
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneilla potilailla ihon okasolusyöpää, mukaan lukien keratoakantoomaa, todettiin 1,4 %:lla (3/216) potilaista. Ensimmäisen ihon okasolusyöpätapahtuman (kaikki asteet) alkamiseen kulunut aika oli 0,5, 0,6 ja 3,6 kuukautta näillä kolmella potilaalla.
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa ihon okasolusyöpää, mukaan lukien keratoakantoomaa, todettiin 1,2 %:lla (3/259) potilaista. Ensimmäisen ihon okasolusyöpätapahtuman (kaikki asteet) alkamiseen kulunut aika oli 1,9, 2,9 ja 6,1 kuukautta näillä kolmella potilaalla. Hoidon lopettamisia ei ilmoitettu.
Uusi primaarimelanooma
Melanooma
Yhdistetyssä enkorafenibi 300 -populaatiossa uusia primaarimelanoomatapahtumia esiintyi 4,1 %:lla (9/217) potilaista: asteen 1 tapahtumia ilmoitettiin 1,4 %:lla (3/217) potilaista, asteen 2 tapahtumia 2,1 %:lla (4/217) potilaista, asteen 3 tapahtumia 0,5 %:lla (1/217) potilaista ja asteen 4 tapahtumia 0,5 %:lla (1/217) potilaista.
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneilla potilailla uusia primaarimelanoomatapahtumia esiintyi 1,9 %:lla (4/216) potilaista: asteen 2 tapahtumia ilmoitettiin 0,9 %:lla (2/216) potilaista ja asteen 3 tapahtumia 0,9 %:lla (2/216) potilaista.
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa uusia primaarimelanoomatapahtumia esiintyi 0,8 %:lla (2/259) potilaista ja kaikki tapaukset olivat asteen 3 tapahtumia. Hoidon keskeyttäminen tai annoksen pienentäminen oli tarpeen 0,4 %:lla (1/259) potilaista. Hoidon lopettamisia ei ilmoitettu.
Silmätapahtumat
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa uveiittia ilmoitettiin 3,5 %:lla (13/372) potilaista: asteen 1 uveiittia 0,5 %:lla (2/372), asteen 2 uveiittia 2,7 %:lla (10/372) ja asteen 3 uveiittia 0,3 %:lla (1/372). Näköhäiriöitä, näön hämärtyminen ja näöntarkkuuden heikentyminen mukaan lukien, esiintyi 23,1 %:lla (86/372) potilaista. Uveiitti ja näköhäiriöt olivat yleensä korjautuvia.
Verkkokalvon pigmenttiepiteelin irtaumaa esiintyi 22,3 %:lla (83/372) potilaista, joista useimmilla oli asteen 1–2 tapahtuma, ja 1,6 %:lla (6/372) oli asteen 3 tapahtuma.
Tutkimuksen CMEK162B2301 osan 2 Combo 300 -ryhmässä verkkokalvon pigmenttiepiteelin irtauma havaittiin 12,5 %:lla (32/257) potilaista, ja 0,4 %:lla (1/257) oli asteen 4 tapahtuma.
Vasemman kammion toimintahäiriö
Vasemman kammion toimintahäiriötä on ilmoitettu enkorafenibin ja binimetinibin samanaikaisen käytön yhteydessä melanoomaa tai ei-pienisoluista keuhkosyöpää sairastavilla potilailla (ks. binimetinibin valmisteyhteenvedon kohta Haittavaikutukset).
Verenvuoto
Melanooma ja ei-pienisoluinen keuhkosyöpä
Verenvuototapahtumia havaittiin 16,7 %:lla (62/372) Combo 450 ISP -populaation potilaista. Useimmat tapahtumat olivat astetta 1 tai 2 (13,2 %; 49/372), ja 3,5 % (13/372) oli astetta ≥ 3. Hoidon keskeyttäminen tai annoksen pienentäminen oli tarpeen vain harvoilla potilailla (2,4 % eli 9/372). Verenvuototapahtumat johtivat hoidon lopettamiseen 0,8 %:lla (3/372) potilaista. Yleisimmät verenvuototapahtumat olivat hematuria 2,7 %:lla (10/372) potilaista, veriuloste 2,7 %:lla (10/372) ja peräsuolen verenvuoto 2,2 %:lla (8/372) potilaista. Yhdellä potilaalla esiintyi kuolemaan johtanut mahahaavan verenvuoto ja rinnakkaisena kuolinsyynä monielinvaurio.
Aivoverenvuoto / kallonsisäinen verenvuoto ilmoitettiin 1,6 %:lla (6/372) potilaista, ja se johti kuolemaan 4 potilaalla.
Tutkimuksen CMEK162B2301 osan 2 Combo 300 -ryhmässä verenvuototapahtumia havaittiin 6,6 %:lla (17/257) potilaista, ja asteen 3–4 tapahtumia esiintyi 1,6 %:lla (4/257) potilaista.
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Verenvuototapahtumia havaittiin 21,3 %:lla (46/216) potilaista, jotka saivat enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa: asteen 3 tapahtumia havaittiin 1,4 %:lla (3/216) potilaista ja yhden tapahtuman ilmoitettiin johtaneen kuolemaan. Hoidon keskeyttäminen tai annoksen pienentäminen oli tarpeen 1,9 %:lla (4/216) potilaista. Verenvuototapahtumat johtivat hoidon lopettamiseen yhdellä potilaalla (0,5 %).
Yleisimmät verenvuototapahtumat olivat nenäverenvuoto 6,9 %:lla (15/216) potilaista, veriuloste 2,8 %:lla (6/216), peräsuolen verenvuoto 2,8 %:lla (6/216) potilaista ja hematuria 2,8 %:lla (6/216) potilaista.
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa verenvuototapahtumia havaittiin 32,8 %:lla (85/259) potilaista; asteen 3 tapahtuma havaittiin 1,5 %:lla (4/259) potilaista ja asteen 4 tapahtuma 0,8 %:lla (2/259) potilaista. Hoidon keskeyttäminen tai annoksen pienentäminen oli tarpeen 3,9 %:lla (10/259) potilaista. Hoidon lopettamisia ei ilmoitettu.
Yleisimmät verenvuototapahtumat olivat nenäverenvuoto 18,5 %:lla (48/259) potilaista, hematuria 4,2 %:lla (11/259), ienverenvuoto 3,9 %:lla (10/259), peräsuolen verenvuoto 3,5 %:lla (9/259) ja peräpukamien verenvuoto 3,5 %:lla (9/259) potilaista.
Hypertensio
Hypertensiota on ilmoitettu enkorafenibin ja binimetinibin samanaikaisen käytön yhteydessä melanoomaa tai ei-pienisoluista keuhkosyöpää sairastavilla potilailla (ks. binimetinibin valmisteyhteenvedon kohta Haittavaikutukset).
Tromboemboliset laskimotapahtumat
Tromboembolisia laskimotapahtumia on ilmoitettu enkorafenibin ja binimetinibin samanaikaisen käytön yhteydessä melanoomaa tai ei-pienisoluista keuhkosyöpää sairastavilla potilailla (ks. binimetinibin valmisteyhteenvedon kohta Haittavaikutukset).
Haimatulehdus
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa ilmoitettiin haimaentsyymiarvojen suurenemista, joka oli pääasiassa oireetonta. Amylaasiarvojen suurenemista ilmoitettiin 4,0 %:lla (15/372) potilaista ja lipaasiarvojen suurenemista 7,8 %:lla (29/372) potilaista. Haimatulehdusta ilmoitettiin 0,8 %:lla (3/372) potilaista. Kaikilla näillä potilailla kyseessä oli asteen 3 tapahtuma. Haimatulehdus johti hoidon keskeyttämiseen 0,3 %:lla (1/372) potilaista.
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneessa populaatiossa asteen 3 haimatulehdusta, johon liittyi lipaasi- ja amylaasiarvojen suurenemista ja joka johti hoidon keskeyttämiseen, ilmoitettiin 1 potilaalla (0,5 %).
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa lipaasiarvon suurenemista (pääasiassa oireetonta) ilmoitettiin 23,6 %:lla potilaista. Asteen 3 haimatulehdus ilmoitettiin 2 potilaalla (0,8 %), joista toisella siihen liittyi lipaasiarvon suurenemista. Hoidon keskeyttäminen ilmoitettiin molempien potilaiden kohdalla, ja hoito lopetettiin toisen potilaan kohdalla.
Ihoreaktiot
Ihottuma
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa ihottumaa esiintyi 20,4 %:lla (76/372) potilaista. Useimmat tapahtumat olivat lieviä; asteen 3 tai 4 tapahtumia ilmoitettiin 1,1 %:lla (4/372) potilaista. Ihottuma johti hoidon
lopettamiseen 0,8 %:lla (3/372) potilaista ja hoidon keskeyttämiseen tai annoksen muuttamiseen 2,4 %:lla (9/372) potilaista.
Yhdistetyssä enkorafenibi 300 -populaatiossa ihottumaa ilmoitettiin 43,3 %:lla (94/217) potilaista. Useimmat tapahtumat olivat lieviä; asteen 3 ja 4 tapahtumia ilmoitettiin 4,6 %:lla (10/217) potilaista. Ihottuma johti hoidon lopettamiseen 0,5 %:lla (1/217) potilaista ja hoidon keskeyttämiseen tai annoksen muuttamiseen 7,4 %:lla (16/217) potilaista.
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneilla potilailla ihottumaa esiintyi 30,6 %:lla (66/216) potilaista. Useimmat tapahtumat olivat lieviä, ja asteen 3 tapahtuma ilmoitettiin 0,5 %:lla (1/216) potilaista. Ihottuma johti hoidon keskeyttämiseen 0,5 %:lla (1/216) potilaista.
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa ihottumaa esiintyi 35,9 %:lla (93/259) potilaista. Useimmat tapahtumat olivat lieviä, ja asteen 3 tapahtuma ilmoitettiin 1,2 %:lla (3/259) potilaista. Ihottuma johti hoidon lopettamiseen 0,8 %:lla (2/259) potilaista ja hoidon keskeyttämiseen tai annoksen pienentämiseen 4,2 %:lla (11/259) potilaista.
Käsi-jalkaoireyhtymä
Melanooma ja ei-pienisoluinen keuhkosyöpä
Käsi-jalkaoireyhtymää ilmoitettiin 5,1 %:lla (19/372) Combo 450 ISP -populaation potilaista. Kaikki käsi-jalkaoireyhtymähaittavaikutukset olivat joko astetta 1 (2,7 %) tai astetta 2 (2,4 %). Hoito keskeytettiin tai annosta muutettiin 1,1 %:lla (4/372) potilaista.
Avaintutkimuksen osan 2 Combo 300 -ryhmässä käsi-jalkaoireyhtymää havaittiin 3,9 %:lla (10/257) potilaista, asteen 3 haittana 0,4 %:lla (1/257) potilaista.
Yhdistetyssä enkorafenibi 300 -populaatiossa käsi-jalkaoireyhtymää ilmoitettiin 51,6 %:lla (112/217) potilaista. Useimmat tapahtumat olivat lieviä tai keskivaikeita: asteen 1 tapahtumia oli 12,4 %:lla (27/217) potilaista, asteen 2 tapahtumia 26,7 %:lla (58/217) potilaista ja asteen 3 tapahtumia 12,4 %:lla (27/217) potilaista. Käsi-jalkaoireyhtymä johti hoidon lopettamiseen 4,1 %:lla (9/217) potilaista ja hoidon keskeyttämiseen tai annoksen muuttamiseen 23,0 %:lla (50/217) potilaista.
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneessa populaatiossa käsi-jalkaoireyhtymää ilmoitettiin 5,1 %:lla (11/216) potilaista. Suurin osa käsi-jalkaoireyhtymähaittavaikutuksista oli astetta 1 (3,7 %:lla (8/216) potilaista). Asteen 2 tapahtumia ilmoitettiin 0,9 %:lla (2/216) potilaista ja asteen 3 tapahtumia 0,5 %:lla (1/216) potilaista. Hoidon keskeyttäminen, annoksen muuttaminen tai hoidon lopettaminen eivät olleet tarpeen.
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa käsi- ja jalkaoireyhtymää ilmoitettiin 15,8 %:lla (41/259) potilaista. Suurin osa tapahtumista oli astetta 1 tai 2, ja 2,7 % (7/259) oli astetta 3. Käsi- ja jalkaoireyhtymä johti hoidon lopettamiseen 0,4 %:lla (1/259) potilaista ja hoidon keskeyttämiseen tai annoksen pienentämiseen 3,5 %:lla (9/259) potilaista.
Aknetyyppinen dermatiitti
Melanooma ja ei-pienisoluinen keuhkosyöpä
Aknetyyppistä dermatiittia on ilmoitettu enkorafenibin ja binimetinibin samanaikaisen käytön yhteydessä (ks. binimetinibin valmisteyhteenvedon kohta Haittavaikutukset).
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneilla potilailla aknetyyppistä dermatiittia esiintyi 33,3 %:lla (72/216) potilaista, ja se oli pääasiassa astetta 1 (25,5 %:lla (55/216) potilaista) tai astetta 2 (6,9 %:lla (15/216) potilaista). Annoksen pienentämistä tai hoidon keskeyttämistä ilmoitettiin 2,3 %:lla (5/216) potilaista. Hoidon lopettamisesta ei raportoitu. Aknetyyppinen dermatiitti oli yleisesti ottaen korjautuvaa.
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa aknetyyppistä dermatiittia esiintyi 22,4 %:lla (58/259) potilaista. Suurin osa tapahtumista oli astetta 1 tai 2, ja 0,8 % (2/259) tapahtumista oli astetta 3. Annoksen pienentämistä tai hoidon keskeyttämistä ilmoitettiin 1,9 %:lla (5/259) potilaista. Hoidon lopettamisia ei raportoitu.
Valoherkkyys
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa valoherkkyyttä havaittiin 4,3 %:lla (16/372) potilaista. Useimmat tapahtumat olivat astetta 1–2, ja astetta 3 ilmoitettiin 0,3 %:lla (1/372) potilaista. Yksikään tapahtumista ei johtanut hoidon lopettamiseen. Hoidon keskeyttäminen tai annosmuutos ilmoitettiin 0,3 %:lla (1/372) potilaista.
Yhdistetyssä enkorafenibi 300 ‑populaatiossa valoherkkyyttä havaittiin 4,1 %:lla (9/217) potilaista. Kaikki tapahtumat olivat astetta 1–2. Yksikään tapahtuma ei vaatinut hoidon lopettamista, annoksen muuttamista eikä hoidon keskeyttämistä.
Kasvopareesi
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa kasvopareesia esiintyi 0,8 %:lla (3/372) potilaista, 0,3 %:lla (1/372) haitta oli astetta 3. Tapahtumat olivat korjautuvia, eikä yksikään tapahtumista johtanut hoidon lopettamiseen. Hoidon keskeyttäminen tai annosmuutos ilmoitettiin 0,3 %:lla (1/372) potilaista.
Yhdistetyssä enkorafenibi 300 ‑populaatiossa kasvopareesia havaittiin 7,4 %:lla (16/217) potilaista. Useimmat tapahtumat olivat lieviä tai keskivaikeita: 2,3 %:lla (5/217) potilaista astetta 1, 3,7 %:lla (8/217) astetta 2 ja 1,4 %:lla (3/217) astetta 3. Ensimmäisen kasvopareesitapahtumaan kuluneen ajan mediaani oli 0,3 kk (vaihteluväli 0,1–12,1 kk). Kasvopareesi oli yleensä korjautuva ja johti hoidon lopettamiseen 0,9 %:lla (2/217). Hoidon keskeyttäminen tai annosmuutos ilmoitettiin 3,7 %:lla (8/217) oireenmukaisen hoidon (mukaan lukien kortikosteroidien) anto 5,1 %:lla (11/217) potilaista.
CK-pitoisuuden suureneminen ja rabdomyolyysi
CK-pitoisuuden suurenemista ja rabdomyolyysia on esiintynyt enkorafenibin ja binimetinibin samanaikaisen käytön yhteydessä melanoomaa tai ei-pienisoluista keuhkosyöpää sairastavilla potilailla (ks. binimetinibin valmisteyhteenvedon kohta Haittavaikutukset).
Munuaisten vajaatoiminta
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa lievää, yleensä asteen 1 oireetonta veren kreatiniiniarvon suurenemista havaittiin 9,4 %:lla (35/372) Combo 450 -hoitoa saaneista potilaista. Asteen 3 tai 4 tapahtumien ilmaantuvuus oli 0,8 % (3/372). Munuaisten vajaatoimintatapahtumia (mukaan lukien akuutti munuaisvaurio, munuaisten vajaatoiminta ja munuaistoiminnan heikentyminen) ilmoitettiin 3,5 %:lla (13/372) enkorafenibin ja binimetinibin yhdistelmää saaneista potilaista. Asteen 3 tai 4 tapahtumia oli 1,9 %:lla (7/372) potilaista. Munuaisten vajaatoiminta yleensä korjautui, kun hoito keskeytettiin ja potilaalle annettiin nesteytystä ja tehtiin muita yleisiä tukitoimenpiteitä.
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Veren kreatiniiniarvon suurenemista raportoitiin 2,8 %:lla (6/216) potilaista, jotka saivat enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa. Kaikki tapahtumat yhtä asteen 4 tapahtumaa lukuun ottamatta olivat lieviä. Munuaisten vajaatoimintatapahtumat olivat astetta 3 tai 4, ja ne raportoitiin akuuttina munuaisvauriona 1,9 %:lla (4/216) potilaista ja munuaisten vajaatoimintana 0,5 %:lla (1/216) potilaista.
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa veren kreatiniiniarvon suurenemista raportoitiin 3,9 %:lla (10/259) potilaista. Kaikki tapahtumat olivat joko asteen 1 (2,7 %:lla [7/259] potilaista) tai asteen 2 (1,2 %:lla [3/259] potilaista) tapahtumia.
Munuaisten vajaatoimintaa, mukaan lukien akuutti munuaisvaurio ja munuaisten toiminnan heikentyminen, raportoitiin 3,5 %:lla (9/259) potilaista (asteen 3 tapahtumia 1,2 %:lla potilaista ja asteen 4 tapahtumia 0,4 %:lla potilaista). Munuaisten vajaatoimintatapahtumat johtivat hoidon keskeyttämiseen tai annoksen pienentämiseen 2,7 %:lla potilaista, ja tila korjautui 90 %:ssa tapauksista. Hoidon lopettamisia ei ilmoitettu.
Maksa-arvojen poikkeavuudet
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa ilmoitettujen maksa-arvojen poikkeavuuksien ilmaantuvuudet olivat:
-
Transaminaasiarvojen suureneminen: 16,4 % (61/372) kokonaisuudessaan; aste 3–4: 6,5 %
(24/372) -
GGT-arvon suureneminen: 11,3 % (42/372) kokonaisuudessaan; aste 3–4: 6,7 % (25/372)
Tutkimuksen CMEK162B230 osan 2 Combo 300 -ryhmässä maksa-arvojen poikkeavuuksien ilmaantuvuudet olivat:
-
Transaminaasiarvojen suureneminen: 13,2 % (34/257) kokonaisuudessaan; aste 3–4: 5,4 %
(14/257) -
GGT-arvon suureneminen: 14,0 % (36/257) kokonaisuudessaan; aste 3–4: 4,7 % (12/257)
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Transaminaasiarvojen suurenemisen ilmaantuvuus potilailla, jotka saivat enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa, oli 8,8 % (19/216), asteen 3 tapahtumien ilmaantuvuuden ollessa 1,4 % (3/216).
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa transaminaasiarvojen suurenemisen ilmaantuvuus potilailla oli 17,0 % (44/259) ja asteen 3 tapahtuma ilmaantui 1,5 %:lle (4/259) potilaista.
Ruoansulatuselimistö
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa ripulia havaittiin 41,7 %:lla (155/372) potilaista ja asteen 3–4 ripulia 3,8 %:lla (14/372) potilaista. Ripuli johti hoidon lopettamiseen 0,8 %:lla potilaista ja hoidon keskeyttämiseen tai annoksen muuttamiseen 8,1 %:lla potilaista.
Ummetusta esiintyi 24,7 %:lla (92/372) potilaista, ja se oli astetta 1 tai 2. Vatsakipua ilmoitettiin 28,5 %:lla (106/372) potilaista ja asteen 3 vatsakipua 2,2 %:lla (8/372) potilaista. Pahoinvointia esiintyi 46,0 %:lla (171/372) potilaista ja asteen 3 pahoinvointia 3,0 %:lla (11/372) potilaista.
Oksentelua esiintyi 31,2 %:lla (116/372) potilaista, ja asteen 3 oksentelua ilmoitettiin 1,9 %:lla (7/372) potilaista.
Tutkimuksen CMEK162B2301 osan 2 Combo 300 -ryhmässä pahoinvointia havaittiin 27,2 %:lla (70/257) potilaista, asteen 3 haittana 1,6 %:lla (4/257) potilaista. Oksentelua esiintyi 15,2 %:lla (39/257) potilaista, asteen 3 haittana 0,4 %:lla (1/257) potilaista. Ripulia esiintyi 28,4 %:lla (73/257) potilaista, asteen 3 haittana 1,6 %:lla (4/257) potilaista.
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneilla potilailla ripulia havaittiin 38,4 %:lla (83/216) potilaista ja asteen 3 ripulia 2,8 %:lla (6/216) potilaista. Ripuli johti hoidon lopettamiseen 0,5 %:lla (1/216) potilaista ja hoidon keskeyttämiseen tai annoksen muuttamiseen 3,7 %:lla (8/216) potilaista.
Vatsakipua ilmoitettiin 36,6 %:lla (79/216) potilaista ja asteen 3 vatsakipua 5,1 %:lla (11/216) potilaista. Pahoinvointia esiintyi 38,0 %:lla (82/216) potilaista ja asteen 3 pahoinvointia 0,5 %:lla (1/216) potilaista. Oksentelua esiintyi 27,3 %:lla (59/216) potilaista, ja asteen 3 oksentelua ilmoitettiin 1,4 %:lla (3/216) potilaista. Ummetusta esiintyi 18,1 %:lla (39/216) potilaista, ja se oli astetta 1 tai 2.
Ruoansulatuskanavan häiriöitä hoidettiin tyypillisesti tavanomaisella hoidolla.
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa ripulia havaittiin 41,3 %:lla (107/259) potilaista ja asteen 3 ripulia 1,9 %:lla (5/259) potilaista. Ripuli johti hoidon lopettamiseen 1,2 %:lla (3/259) potilaista ja hoidon keskeyttämiseen tai annoksen muuttamiseen 8,9 %:lla (23/259) potilaista.
Vatsakipua ilmoitettiin 32,8 %:lla (85/259) potilaista ja asteen 3 vatsakipua 5,4 %:lla (14/259) potilaista. Pahoinvointia esiintyi 56,0 %:lla (145/259) potilaista ja asteen 3 pahoinvointia 2,7 %:lla (7/259) potilaista. Oksentelua esiintyi 36,7 %:lla (95/259) potilaista ja asteen 3 oksentelua 3,9 %:lla (10/259) potilaista. Ummetusta esiintyi 27,0 %:lla (70/259) potilaista ja asteen 3 ummetusta 0,4 %:lla (1/259) potilaista.
Ruoansulatuskanavan häiriöitä hoidettiin tyypillisesti tavanomaisella hoidolla.
Anemia
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa anemiaa ilmoitettiin 23,1 %:lla (86/372) potilaista; 7,0 %:lla (26/372) potilaista haitta oli astetta 3 tai 4. Kukaan potilaista ei lopettanut hoitoa anemian takia; 3,2 %:lla (12/372) hoito oli keskeytettävä tai annosta muutettava.
Tutkimuksen CMEK162B2301 osan 2 Combo 300 -ryhmässä anemiaa havaittiin 9,7 %:lla (25/257) potilaista, asteen 3–4 haittana 2,7 %:lla (7/257) potilaista.
Kolorektaalisyöpä
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa anemiaa ilmoitettiin 43,6 %:lla (113/259) potilaista; 14,3 %:lla (37/259) potilaista haitta oli astetta 3. Anemia johti hoidon lopettamiseen 2,3 %:lla (6/259) potilaista ja hoidon keskeyttämiseen tai annoksen muuttamiseen 13,9 %:lla (36/259) potilaista.
Päänsärky
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa päänsärkyä esiintyi 18,8 %:lla (70/372) potilaista, asteen 3 haittana 1,1 %:lla (4/372) potilaista.
Tutkimuksen CMEK162B2301 osan 2 Combo 300 -ryhmässä päänsärkyä ilmoitettiin 12,1 %:lla (31/257) potilaista, asteen 3 haittana 0,4 %:lla (1/257) potilaista.
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneilla potilailla päänsärkyä esiintyi 20,4 %:lla (44/216) potilaista, ja se oli astetta 1 tai 2.
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa päänsärkyä esiintyi 15,1 %:lla (39/259) potilaista ja asteen 3 päänsärkyä 0,4 %:lla (1/259) potilaista. Hoidon keskeyttäminen tai annoksen muuttaminen oli tarpeen 0,4 %:lla (1/259) potilaista. Hoidon lopettamisia ei raportoitu.
Väsymys
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa väsymystä esiintyi 48,1 %:lla (179/372) potilaista, asteen 3 tai 4 haittana 4,3 %:lla (16/372) potilaista.
Tutkimuksen CMEK162B2301 osan 2 Combo 300 -ryhmässä väsymystä havaittiin 33,5 %:lla (86/257) potilaista, asteen 3–4 haittana 1,6 %:lla (4/257) potilaista.
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneilla potilailla väsymystä ilmoitettiin 56,9 %:lla (123/216) potilaista mukaan lukien asteen 3 väsymys 7,9 %:lla (17/216) potilaista.
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa väsymystä ilmoitettiin 54,1 %:lla (140/259) potilaista mukaan lukien asteen 3 väsymys 6,6 %:lla (17/259) potilaista. Väsymys johti hoidon lopettamiseen 3,5 %:lla (9/259) potilaista ja hoidon keskeyttämiseen tai annoksen muuttamiseen 16,6 %:lla (43/259) potilaista.
Erityisryhmät
Iäkkäät
Melanooma ja ei-pienisoluinen keuhkosyöpä
Combo 450 ISP -populaatiossa (n = 372) 230 potilasta (61,8 %) oli < 65-vuotiaita, 107 potilasta (28,8 %) oli 65–74-vuotiaita ja 35 potilasta (9,4 %) oli > 75-vuotiaita. Turvallisuus ja teho eivät kokonaisuutena ajatellen olleet erilaisia iäkkäiden (≥ 65 v) ja nuorempien potilaiden välillä, lukuun ottamatta sitä, että ripulia ja kutinaa ilmoitettiin enemmän vanhemmilla potilailla.
≥ 75-vuotiaiden alaryhmässä ilmoitettiin < 75-vuotiaisiin verrattuna useammin asteen ≥ 3 haittavaikutuksia (62,9 % vs. 47,5 %), minkä tahansa tutkimuslääkkeen (kaikki tasot) annoksen muuttamista vaatineita haittavaikutuksia (60,0 % vs. 48,1 %) tai hoidon lopettamiseen johtaneita haittavaikutuksia (25,7 % vs. 7,4 %) ilmoitettiin useammin kuin < 75-vuotiailla potilailla. Yleisimmät ilmoitetut haittavaikutukset, joiden ilmaantuvuus oli suurempi ≥ 75-vuotiailla < 75-vuotiaisiin verrattuna, olivat väsymys, pahoinvointi, ripuli, oksentelu ja anemia.
Kolorektaalisyöpä
Enkorafenibin ja setuksimabin yhdistelmä
Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneista potilaista (n = 216) 134 potilasta (62 %) oli < 65-vuotiaita, 62 potilasta (28,7 %) oli 65–74-vuotiaita ja 20 potilasta (9,3 %) oli ≥ 75‑vuotiaita. Yleisimmät haittavaikutukset, joiden ilmoitettu ilmaantuvuus oli suurempi ≥ 65-vuotiailla kuin < 65‑vuotiailla, olivat väsymys, ruokahalun heikkeneminen ja verenvuoto.
Enkorafenibin, setuksimabin ja FOLFOXin yhdistelmä
Yhdistetyssä ES + FOLFOX ‑populaatiossa (n = 259) 166 potilasta (64,1 %) oli < 65‑vuotiaita ja 93 potilasta (35,9 %) oli ≥ 65‑vuotiaita. Yleisimmät ilmoitetut haittavaikutukset, joiden ilmaantuvuus oli suurempi ≥ 65‑vuotiailla kuin < 65‑vuotiailla, olivat neutropenia, anemia, infektio, painonlasku ja hypomagnesemia.
Kolorektaalisyöpää sairastavassa populaatiossa eroavaisuuksia haittavaikutusten ilmaantuvuudessa ≥ 75-vuotiailla potilailla verrattuna < 75-vuotiaisiin potilaisiin ei voitu arvioida, koska ikäryhmään ≥ 75 vuotta kuuluvien hoitoa saavien potilaiden lukumäärä oli hyvin pieni.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet
Enkorafenibiannoksilla 600–800 mg kerran vuorokaudessa munuaisten vajaatoimintaa (asteen 3 hyperkreatinemia) todettiin 3 potilaalla 14:stä. Suurin enkorafenibiannos johtui annosteluvirheestä: yksi potilas otti enkorafenibia 600 mg kahdesti vuorokaudessa yhden vuorokauden ajan (kokonaisannos 1 200 mg). Potilaan ilmoittamat haittavaikutukset olivat asteen 1 tapahtumia: pahoinvointi, oksentelu ja näön hämärtyminen. Kaikki tapahtumat korjautuivat myöhemmin.
Hoito
Yliannostukseen ei ole spesifistä hoitoa.
Enkorafenibi sitoutuu kohtalaisesti plasman proteiineihin, joten hemodialyysistä ei todennäköisesti ole hyötyä enkorafenibiyliannostuksen hoidossa. Enkorafenibille ei tunneta vastalääkettä. Yliannostustapauksissa enkorafenibihoito on keskeytettävä ja munuaistoimintaa ja haittavaikutuksia on seurattava. Oireenmukaista hoitoa ja elintoimintoja tukevaa hoitoa on annettava tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: muut syöpälääkkeet, proteiinikinaasin estäjät, ATC-koodi: L01EC03
Vaikutusmekanismi
Enkorafenibi on vahva, erittäin selektiivinen, ATP:n kanssa kilpaileva pienimolekyylinen RAF-kinaasin estäjä. Enkorafenibin puolet maksimiestotehosta tuottava pitoisuus (IC50) BRAF V600E ‑entsyymien suhteen oli 0,35 nM, BRAF-entsyymien suhteen 0,47 nM ja CRAF-entsyymien suhteen 0,30 nM. Enkorafenibin dissosiaation puoliintumisaika oli > 30 tuntia, mikä pidensi pERK-eston kestoa. Enkorafenibi lamaa RAF/MEK/ERK-reittiä kasvainsoluissa, jotka ilmentävät useita BRAF-kinaasin mutaatiomuotoja (V600E, V600D ja V600K). Enkorafenibi estää spesifisesti BRAF V600E/V600D/V600K -mutaatiopositiivisten melanoomasolujen ja BRAF V600E ‑mutaatiopositiivisten kolorektaalisyöpäsolujen kasvua in vitro ja in vivo. Enkorafenibi ei estä RAF/MEK/ERK-signalointia villin tyypin BRAF-geeniä ilmentävissä soluissa.
Käyttö yhdessä binimetinibin kanssa
Sekä enkorafenibi että binimetinibi (MEK-estäjä, ks. binimetinibin valmisteyhteenvedon kohta Farmakodynamiikka) estävät MAPK-reittiä, mikä tehostaa kasvaimeen kohdistuvaa vaikutusta verrattuna kumpaankaan lääkkeeseen yksinään.
Käyttö yhdessä setuksimabin kanssa yhdistettynä FOLFOXiin tai ilman sitä
EGFR:n uudelleenaktivaatio ja BRAF-reitin kautta tapahtuvan signaloinnin ohittaminen on tunnistettu yhdeksi BRAF-mutaatiopositiivisen kolorektaalisyövän pääasialliseksi resistenssimekanismiksi RAF-estäjiä vastaan. BRAF-estäjän, esim. enkorafenibin, ja epidermaalisen kasvutekijän reseptoriin (EGFR) kohdistuvien lääkeaineiden, esim. setuksimabin, yhdistelmän on osoitettu parantavan antitumorista tehokkuutta non-kliinisissä malleissa. Sytostaattisen aineen, kuten 5‑fluorourasiilin, ja sytotoksisen aineen, kuten oksaliplatiinin, lisäämisen on osoitettu parantavan antituumoritehoa kliinisissä tutkimuksissa.
Kliininen teho ja turvallisuus
BRAF V600 -mutaatiopositiivinen leikkaukseen soveltumaton tai etäpesäkkeinen melanooma
Enkorafenibin ja binimetinibin yhdistelmän tehoa ja turvallisuutta arvioitiin kaksiosaisessa vaiheen III satunnaistetussa (1:1:1), aktiivikontrolloidussa, avoimessa monikeskustutkimuksessa (tutkimus CMEK162B2301) potilailla, joilla oli leikkaukseen soveltumaton tai etäpesäkkeinen BRAF V600E- tai BRAF V600K -mutaatiopositiivinen melanooma (tutkittu BRAF-määrityksellä). Potilailla oli histologisesti vahvistettu ihon tai tuntemattoman alueen primaarimelanooma, mutta tutkimuksesta suljettiin pois potilaat, joilla oli uvean tai limakalvon melanooma. Potilaille oli sallittu aiempi liitännäishoito ja yksi aiempi immunoterapialinja leikkaukseen soveltumattomaan paikallisesti levinneeseen tai etäpesäkkeiseen tautiin. Aiempi BRAF:n / MEK:n estäjähoito ei ollut sallittu.
Tutkimus CMEK162B2301, osa 1
Osassa 1 tutkimukseen osallistuvat potilaat satunnaistettiin saamaan 450 mg enkorafenibia vuorokaudessa suun kautta sekä 45 mg binimetinibiä kahdesti vuorokaudessa suun kautta (Combo 450, n = 192), 300 mg enkorafenibia vuorokaudessa suun kautta (Enco 300, n = 194) tai 960 mg vemurafenibia kahdesti vuorokaudessa suun kautta (Vem, n = 191). Hoitoa jatkettiin, kunnes tauti eteni tai ilmeni sietämätöntä toksisuutta. Satunnaistaminen stratifioitiin AJCC‑luokan (American Joint Committee on Cancer; IIIB, IIIC, IVM1a tai IVM1b vs. IVM1c) ja ECOG-toimintakykyluokan (Eastern Cooperative Oncology Group; 0 vs. 1) mukaan sekä leikkaukseen soveltumattoman tai etäpesäkkeisen taudin aiemman immunoterapian (kyllä vs. ei) mukaan.
Ensisijainen tehon tulosmuuttuja oli Combo 450 -ryhmän etenemisvapaa elinaika (PFS) vemurafenibiin verrattuna. Tämän arvioi sokkoutetusti riippumaton arviointitoimikunta (blinded independent review committee, BIRC). Tutkijoiden arvioima etenemisvapaa elinaika oli supportiivinen analyysi. Ylimääräinen toissijainen päätetapahtuma oli Combo 450 -ryhmän etenemisvapaa elinaika Enco 300 -ryhmään verrattuna. Muita Combo 450 -hoidon ja joko vemurafenibin tai Enco 300 -hoidon toissijaisia tehon vertailumuuttujia olivat kokonaiselinaika (OS), objektiivinen kokonaisvaste (ORR), vasteen kesto (DoR) ja taudin hallintaprosentti (DCR) riippumattoman arviointitoimikunnan (BIRC) ja tutkijan arvioimina.
Potilaiden mediaani-ikä oli 56 vuotta (vaihteluväli 20–89), 58 % oli miehiä, 90 % oli valkoihoisia ja 72 %:lla potilaista ECOG-toimintakykyluokka oli lähtötilanteessa 0. Valtaosalla oli etäpesäkkeinen tauti (95 %) ja tautiluokka oli IVM1c (64 %); 27 %:lla potilaista oli lähtötilanteessa kohonnut seerumin laktaattidehydrogenaasipitoisuus (LDH) ja 45 %:lla potilaista oli lähtötilanteessa kasvaimia vähintään 3 elimessä ja 3,5 %:lla etäpesäkkeitä aivoissa. 27 potilasta (5 %) oli saanut aiemmin immuunivasteen säätelijämolekyylien estäjää eli checkpoint-estäjää (anti-PD1/PDL1 tai ipilimumabi) (8 potilasta Combo 450 -ryhmässä [4 %]; 7 potilasta vemurafenibiryhmässä [4 %]; 12 potilasta Enco 300 -ryhmässä [6 %]), mukaan lukien 22 potilasta etäpesäkkeisen taudin hoitoon (6 potilasta Combo 450 ‑ryhmässä; 5 potilasta vemurafenibiryhmässä; 11 potilasta Enco 300 ‑ryhmässä), ja 5 potilasta liitännäishoitona (2 potilasta Combo 450 ‑ryhmässä; 2 potilasta vemurafenibiryhmässä; 1 potilas Enco 300 ‑ryhmässä).
Altistuksen keston mediaani oli 11,7 kk Combo 450 -hoitoa saaneilla potilailla, 7,1 kk Enco 300 ‑hoitoa saaneilla ja 6,2 kk vemurafenibia saaneilla. Suhteellisen annosintensiteetin mediaani oli Combo 450 -ryhmässä 100 % enkorafenibille ja 99,6 % binimetinibille. Enco 300 -hoidon suhteellisen annosintensiteetin mediaani oli 86,2 % ja vemurafenibin 94,5 %.
CMEK162B2301-tutkimuksen osassa 1 etenemisvapaa elinaika (PFS) parani tilastollisesti merkitsevästi Combo 450 -hoitoa saaneilla potilailla vemurafenibia saaneisiin verrattuna. Taulukossa 6 on yhteenveto etenemisvapaasta elinajasta ja muista tehotuloksista. Tulokset perustuvat sokkoutetun riippumattoman radiologitoimikunnan tekemään tietojen keskitettyyn arviointiin.
Tutkijan arvioon perustuvat tehotulokset vastasivat riippumattoman, keskitetyn arvioinnin tuloksia. Stratifioimattomien alaryhmäanalyysien piste-estimaatit, mukaan lukien lähtötilanteen LDH-arvo, ECOG-toimintakykyluokka ja AJCC-luokka, suosivat Combo 450 -hoitoa.
Taulukko 6: Tutkimus CMEK162B2301, osa 1: Etenemisvapaa elinaika ja vahvistetut kokonaisvastetulokset (riippumaton keskitetty arviointi)
| Enkorafenibi + binimetinibi N = 192 (Combo 450) | Enkorafenibi N = 194 (Enco 300) | Vemurafenibi N = 191 (Vem) |
Tiedonkeruun katkaisupäivä: 19.5.2016 | |||
Etenemisvapaa elinaika (PFS) (ensisijainen analyysi) | |||
Tapahtumia (%) | 98 (51,0) | 96 (49,5) | 106 (55,5) |
Mediaani, kk (95 % lv) | 14,9 (11,0; 18,5) | 9,6 (7,5; 14,8) | 7,3 (5,6; 8,2) |
HRa (95 % lv) (vs. Vem) p-arvo (stratifioitu log-rank-testi)b | 0,54 (0,41; 0,71) < 0,0001 |
|
|
HRa (95 % lv) (vs. Vem) Nimellinen p-arvo |
| 0,68 (0,52, 0,90) 0,007 |
|
HRa (95 % lv) (vs. Enco 300) p-arvo (stratifioitu log-rank-testi)b | 0,75 (0,56; 1,00) 0,051 |
|
|
Vahvistetut kokonaisvasteet | |||
Kokonaisvaste, n (%) | 121 (63,0) | 98 (50,5) | 77 (40,3) |
(95 % lv) | (55,8; 69,9) | (43,3; 57,8) | (33,3; 47,6) |
CR, n (%) | 15 (7,8) | 10 (5,2) | 11 (5,8) |
PR, n (%) | 106 (55,2) | 88 (45,4) | 66 (34,6) |
SD, n (%) | 46 (24,0) | 53 (27,3) | 73 (38,2) |
DCR, n (%) | 177 (92,2) | 163 (84,0) | 156 (81,7) |
(95 % lv) | (87,4; 95,6) | (78,1; 88,9) | (75,4; 86,9) |
Vasteen kesto | |||
Mediaani, kk (95 % lv) | 16,6 (12,2; 20,4) | 14,9 (11,1; Ei arv.) | 12,3 (6,9; 16,9) |
lv = luottamusväli; CR = täydellinen vaste; DCR = taudin hallintaprosentti (CR + PR + SD + Non-CR/Non-PD; Non-CR/Non-PD koskee vain potilaita, joilla ei ollut kohdemuutosta ja joilla ei saavutettu täydellistä vastetta tai joilla ei ollut etenevää tautia); HR = riskisuhde; Ei arv. = ei arvioitavissa; PFS = etenemisvapaa elinaika; PR = osittainen vaste; SD = stabiili tauti; Vem = vemurafenibi.
a Riskisuhde perustuu stratifioituun Coxin suhteellisen riskin malliin.
b Log-rank-testin p-arvo (2‑tahoinen)
Elämänlaatu (QoL) (tiedonkeruun katkaisupäivä: 19.5.2016)
Terveyteen liittyvää elämänlaatua, toimintakykyä, melanoomaoireita ja hoitoon liittyviä haittavaikutuksia koskevia potilaiden raportoimia tulosmuuttujia arvioitiin FACT‑M-kyselylomakkeella (Functional Assessment of Cancer Therapy-Melanoma), EORTC QLQ‑C30 ‑kyselylomakkeella (European Organization for Research and Treatment of Cancer’s core quality of life questionnaire) ja EQ‑5D‑5L-kyselylomakkeella (EuroQoL‑5 Dimension‑5 Level). FACT‑M- ja EORTC QLQ‑C30-lomakkeilla arvioitu selvä 10 %:n heikentyminen viivästyi merkitsevästi Combo 450 -hoitoa saaneilla potilailla suhteessa muihin hoitoihin. FACT‑M-pisteiden selvään 10 %:n heikentymiseen kuluvan ajan mediaania ei saavutettu Combo 450 -ryhmässä, ja vemurafenibiryhmässä mediaani oli 22,1 kk (95 % lv: 15,2, ei arvioitavissa). Eron riskisuhde (HR) oli 0,46 (95 % lv: 0,29; 0,72). EORTC QLQ‑C30 -pisteiden selvään 10 %:n heikentymiseen kuluvan ajan analyysistä saadut tulokset olivat samaa luokkaa.
Combo 450 -hoitoa saaneiden potilaiden ilmoitusten perusteella EQ‑5D‑5L-indeksiarvon keskimuutos ei muuttunut tai kohentui hieman lähtötilanteesta kaikilla käynneillä, kun sen sijaan vemurafenibia tai enkorafenibia saaneet potilaat puolestaan ilmoittivat vähentymisestä kaikilla käynneillä (tilastollisesti merkitsevät erot). Ajan myötä tapahtuvan pistemäärän muutoksen arviointi tuotti saman trendin EORTC QLQ‑C30 -lomakkeella arvioituna ja kaikilla käynneillä FACT‑M-lomakkeella arvioituna.
Tutkimus CMEK162B2301, osa 2
Tutkimuksen CMEK162B2301 osassa 2 oli tarkoitus arvioida binimetinibin merkitystä enkorafenibin ja binimetinibin yhdistelmän käytössä.
Etenemisvapaata elinaikaa (PFS) enkorafenibin (300 mg/vrk suun kautta) ja binimetinibin (45 mg kahdesti vuorokaudessa suun kautta) yhdistelmää käyttävillä (Combo 300, n = 258) verrattiin Enco 300 -ryhmän PFS-tuloksiin (n = 280, mukaan lukien 194 potilasta osasta 1 ja 86 potilasta osasta 2). Tutkittavien otto osaan 2 alkoi, kun kaikki osan 1 potilaat oli satunnaistettu.
Tutkimuksen CMEK162B2301 osien 1 ja 2 lopullinen tehoanalyysi (tiedonkeruun katkaisupäivä: 31.3.2023)
Lopullinen tehoanalyysi oli yhtäpitävä välianalyysin tulosten kanssa, ja siinä osoitettiin hyöty kokonaiselinajassa Combo 450 ‑hoitoa saaneilla verrattuna vemurafenibihoitoa saaneisiin (HR 0,67 [95 % lv: 0,53, 0,84], kokonaiselossaolon mediaani 33,6 kuukautta vs. 16,9 kuukautta). Myös etenemisvapaan elinajan ja sokkoutetun riippumattoman arviointitoimikunnan vahvistaman objektiivisen kokonaisvasteen tulokset vahvistivat Combo 450 ‑hoidon numeerisen paremmuuden: 7,6 kuukautta pitempi etenemisvapaan elinajan mediaani Combo 450 ‑ryhmässä vemurafenibiryhmään verrattuna. Lopullisen tehoanalyysin yksityiskohtaisemmat tiedot esitetään alla taulukossa 7 sekä kuvissa 1 ja 2.
Lisäksi osan 2 lopullinen analyysi osoitti numeerisen eron kokonaiselinajassa Combo 300 ‑hoidolla (osa 2) verrattuna Enco 300 ‑monoterapiaan (osat 1+2) (HR 0,89 [95 % lv: 0,72, 1,09], kokonaiselinajan mediaani 27,1 kuukautta [95 % lv: 21,6, 33,3] vs. 22,7 kuukautta [95 % lv: 19,3, 29,3]). Etenemisvapaan elinajan mediaani oli pitempi Combo 300 ‑ryhmässä (osa 2) verrattuna Enco 300 ‑ryhmään (osat 1+2); PFS-mediaaniestimaatit 12,9 kuukautta (95 % lv: 10,9, 14,9) vs. 9,2 kuukautta (95 % lv: 7,4, 11,1). Sokkoutetun riippumattoman arviointitoimikunnan vahvistama objektiivinen kokonaisvaste oli Combo 300 ‑ryhmässä (osa 2) 67,8 % (95 % lv: 61,8, 73,5) ja Enco 300 -ryhmässä (osat 1+2) 51,4 % (95 % lv: 45,4, 57,4). Tutkijan arvioinnissa tulokset olivat samankaltaisia.
Taulukko 7: Tutkimus CMEK162B2301: Etenemisvapaan elinajan (PFS), kokonaiselinajan (OS) ja vahvistetun objektiivisen kokonaisvasteen (ORR) lopulliset tulokset (tiedonkeruun katkaisupäivä: 31.3.2023)
| Enkorafenibi + binimetinibi N = 192 (Combo 450) | Enkorafenibi N = 194 (Enco 300) | Vemurafenibi N = 191 (Vem) |
|---|---|---|---|
Lopullinen analyysi, tiedonkeruun katkaisupäivä: 31.3.2023 | |||
Etenemisvapaa elinaika (PFS) (sokkoutetun riippumattoman arviointitoimikunnan vahvistama) | |||
Tapahtumia (%) | 123 (64,1) | 119 (61,3) | 121 (63,4) |
Mediaania, kk (95% lv) | 14,9 (11,0, 20,2) | 9,6 (7,4, 14,8) | 7,3 (5,6, 7,9) |
HR (95 % lv) (vs. Vem) p-arvo (log-rank-testi) (1-tahoinen)* | 0,51 (0,39, 0,66) < 0,0001 | 0,68 (0,53 – 0,88) 0,0017 |
|
HR(95 % lv) (vs. Enco 300) p-arvo (log-rank-testi) (1-tahoinen)* | 0,77 (0,60 – 0,99) 0,0214 |
|
|
Kokonaiselinaika (OS) | |||
Tapahtumia (%) | 139 (72,4) | 125 (64,4) | 147 (77,0) |
Mediaania, kk (95% lv) | 33,6 (24,4, 39,2) | 23,5 (19,6, 33,6) | 16,9 (14,0, 24,5) |
Eloonjäämisen todennäköisyysb |
|
|
|
1 vuoden kohdalla % (95 % lv) | 75,5 (68,8, 81,0) | 74,6 (67,6, 80,3) | 63,1 (55,7, 69,7) |
2 vuoden kohdalla % (95 % lv) | 57,7 (50,3, 64,3) | 49,1 (41,5, 56,2) | 43,2 (35,9, 50,2) |
3 vuoden kohdalla % (95 % lv) | 46,5 (39,3, 53,4) | 40,9 (33,6, 48,1) | 31,4 (24,8, 38,2) |
5 vuoden kohdalla % (95 % lv) | 34,7 (28,0, 41,5) | 34,9 (27,9, 42,0) | 21,4 (15,7, 27,8) |
9 vuoden kohdalla % (95 % lv) | 26,0 (19,8, 32,5) | 27,8 (21,1, 34,8) | 18,2 (12,8, 24,3) |
HRc (95 % lv) (vs. Vem) p-arvo (log-rank-testi) (1-tahoinen)* | 0,67 (0,53, 0,84) 0,0003 | 0,74 (0,58; 0,94) 0,0063 |
|
HRc (95 % lv) (vs. Enco 300) p-arvo (log-rank-testi) (1-tahoinen)* | 0,93 (0,73, 1,19) 0,2821 |
|
|
Vahvistettu paras kokonaisvaste (sokkoutetun riippumattoman arviointitoimikunnan vahvistama) | |||
Vahvistettu ORRd, n (%) (95 % lv) | 123 (64,1) (56,8, 70,8) | 100 (51,5) (44,3, 58,8) | 78 (40,8) (33,8, 48,2) |
CR, n (%) | 29 (15,1) | 17 (8,8) | 16 (8,4) |
PR, n (%) | 94 (49,0) | 83 (42,8) | 62 (32,5) |
SD, n (%) | 44 (22,9) | 52 (26,8) | 71 (37,2) |
DCRd, n (%) (95 % lv) | 177 (92,2) (87,4, 95,6) | 163 (84,0) (78,1, 88,9) | 155 (81,2) (74,8, 86,4) |
Vasteen kesto (sokkoutetun riippumattoman arviointitoimikunnan vahvistama) | |||
Mediaani, kk (95 % lv) | 18,6 (12,7, 27,6) | 15,5 (11,1, 29,5) | 12.3 (6,9, 14,5) |
lv = luottamusväli; CR = täydellinen vaste; PR = osittainen vaste; SD = stabiili tauti; DCR = taudin hallintaprosentti (CR+PR+SD+Non-CR/Non-PD); HR = riskisuhde; ORR = objektiivinen kokonaisvaste (CR+PR); PR ja CR on vahvistettu toistetuilla arvioinneilla, jotka on suoritettu aikaisintaan 4 viikon kuluttua siitä, kun vasteen kriteerit ovat täyttyneet ensimmäisen kerran. a Mediaani (aika tapahtumaan) ja sen 95 % luottamusvälit on laskettu käyttämällä Brookmeyer & Crowley -menetelmän mukaista KM-estimointia. b Eloonjäämisen todennäköisyys (saatu KM-elossaoloestimaateista, luottamusväleihin käytetty Greenwoodin kaavaa) c Sekä Log-rank-testi että Coxin PH-malli on stratifioitu IVRS AJCC stage- ja ECOG Performance status -menetelmillä. d estimoidut 95 % lv:t on saatu käyttämällä Clopper-Pearsonin eksaktia menetelmää. * Nimellinen p-arvo | |||
Kuva 1: Tutkimus CMEK162B2301: Kaplan–Meier-kaavio etenemisvapaasta elinajasta; sokkoutettu riippumaton arviointi (tiedonkeruun katkaisupäivä: 31.3.2023)
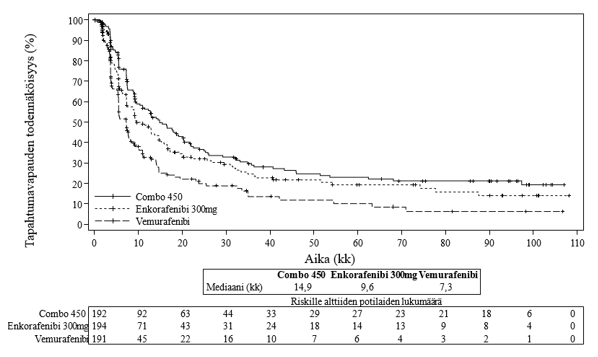
Kuva 2:Tutkimus CMEK162B2301: Kaplan-Meier-kaavio kokonaiselinajasta (tiedonkeruun katkaisupäivä: 31.3.2023)
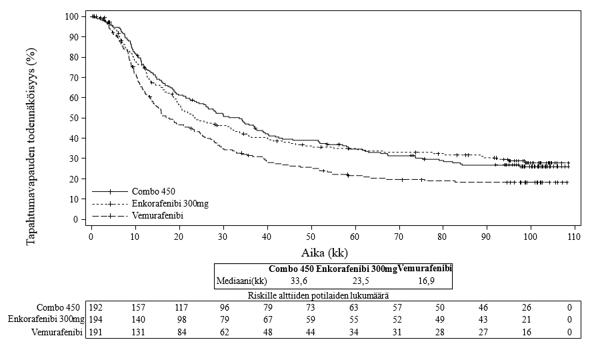
BRAF V600E ‑mutaatiopositiivinen etäpesäkkeinen kolorektaalisyöpä – enkorafenibi yhdistettynä setuksimabiin – Tutkimus ARRAY-818-302
Enkorafenibin ja setuksimabin yhdistelmää arvioitiin satunnaistetussa, aktiivikontrolloidussa, avoimessa monikeskustutkimuksessa (ARRAY 818-302 BEACON CRC). Tutkimukseen soveltuvilla potilailla oli oltava BRAF V600E ‑mutaatiopositiivinen etäpesäkkeinen kolorektaalisyöpä, joka oli edennyt aikaisemmasta yhdestä tai kahdesta hoitokerrasta huolimatta. Mukaan otetut potilaat soveltuivat kasvaimensa RAS-tilanteen perusteella saamaan setuksimabia paikallisen valmisteyhteenvedon mukaisesti. Aiempi RAF-, MEK- tai EGFR-estäjien käyttö ei ollut sallittu. Satunnaistaminen stratifioitiin ECOG-toimintakykyluokan (Eastern Cooperative Oncology Group) mukaan sekä aiemman irinotekaanin ja setuksimabin käytön mukaan.
665 potilasta satunnaistettiin (1:1:1) saamaan joko 300 mg enkorafenibia vuorokaudessa suun kautta yhdessä setuksimabin kanssa, jota annosteltiin sen hyväksytyn valmisteyhteenvedon mukaan (n = 200) tai 300 mg enkorafenibia vuorokaudessa suun kautta ja 45 mg binimetinibiä kahdesti vuorokaudessa suun kautta sekä setuksimabia, jota annosteltiin sen hyväksytyn valmisteyhteenvedon mukaan (n = 224) tai kontrollia (irinotekaania ja setuksimabia tai irinotekaania, 5-fluorourasiilia ja foliinihappoa sisältävää valmistetta (FOLFIRI) ja setuksimabia, n = 221). Hoitoa jatkettiin, kunnes tauti eteni tai ilmeni sietämätöntä toksisuutta.
Tehon tulosmuuttujia olivat kokonaiselinaika (OS) ja kokonaisvaste (ORR) sokkoutetusti riippumattoman arviointitoimikunnan (blinded independent review committee, BIRC) tekemän arvioinnin mukaan, jossa vertailtiin enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa ja kontrollia toisiinsa. Taulukossa 8 on yhteenveto muista tehomuuttujista.
Potilaiden mediaani-ikä oli 61 vuotta (vaihteluväli 26–91), 47 % oli miehiä ja 83 % oli valkoihoisia. 51 %:lla potilaista ECOG-toimintakykyluokka oli lähtötilanteessa 0, ja 51 % oli saanut aiemmin irinotekaania ja 46,8 %:lla potilaista oli lähtötilanteessa kasvaimia vähintään 3 elimessä.
Altistuksen keston mediaani oli 3,2 kk enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneilla potilailla ja 1,4 kk joko irinotrekaanin ja setuksimabin yhdistelmää tai FOLFIRI-valmisteen ja setuksimabin yhdistelmää saaneilla potilailla (kontrolliryhmä). Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoitoa saaneilla potilailla suhteellisen annosintensiteetin mediaani oli 98 % enkorafenibille ja 93,5 % setuksimabille. Kontrolliryhmässä suhteellisen annosintensiteetin mediaani oli 85,4 % setuksimabille ja 75,7 % irinotekaanille sekä foliinihappoa ja 5-fluorourasiilia saaneessa potilasryhmässä suhteellisen annosintensiteetin mediaani oli 75,2 % foliinihapolle ja 75 % 5‑fluorourasiilille.
Enkorafenibin (300 mg) ja setuksimabin yhdistelmähoito paransi tilastollisesti merkitsevästi kokonaiselinaikaa (OS), kokonaisvastetta (ORR) ja etenemisvapaata elinaikaa (PFS) kontrolliin verrattuna. Taulukossa 8 sekä kuvassa 3 on yhteenveto tehotuloksista.
Tutkijan arvioon perustuvat tehotulokset vastasivat riippumattoman, keskitetyn arvioinnin tuloksia.
Taulukko 8: Tutkimus ARRAY-818-302: Tehotulokset
| Enkorafenibi + setuksimabi | Irinotekaani + setuksimabi tai FOLFIRI + setuksimabi (kontrolli) |
Tiedonkeruun katkaisupäivä: 11.2.2019 (ensisijainen analyysi) | ||
Kokonaiselinaika (OS) | ||
Potilaitaa | 220 | 221 |
Tapahtumia (%) | 93 (42,3) | 114 (51,6) |
Mediaani, kk (95 % lv) | 8,4 (7,5-11,0) | 5,4 (4,8; 6,6) |
HR (95 % lv)b,c (vs. kontrolli) p-arvob,c | 0,60 (0,41-0,88) 0,0002 |
|
Seurannan keston mediaani, kk (95 % lv) | 7,6 (6,4; 9,20) | 7,2 (6,1; 8,1) |
Kokonaisvaste (ORR) (BIRC:n arvioimana) | ||
Potilaitae | 113 | 107 |
ORR, n (%) (95 % lv)f | 23 (20,4) (13,4; 29,0) | 2 (1,9) (0,2; 6,6) |
p-arvob,d,g | < 0,0001 |
|
Etenemisvapaa elinaika (PFS) (BIRC:n arvioimana) | ||
Potilaitaa | 220 | 221 |
Tapahtumia (%) | 133 (60,5) | 128 (57,9) |
PFS:n mediaani, kk (95 % lv) | 4,2 (3,7; 5,4) | 1,5 (1,5; 1,7) |
HR (95 % lv)b,c p-arvob,d | 0,40 (0,30; 0,55) < 0,0001 |
|
Päivitetty analyysi, tiedonkeruun katkaisupäivä: 15.8.2019 | ||
Kokonaiselinaika (OS) | ||
Potilaitaa | 220 | 221 |
Tapahtumia (%) | 128 (58,2) | 157 (71,0) |
Mediaani, kk (95 % lv) | 9,3 (8,0; 11,3) | 5,9 (5,1; 7,1) |
HR (95 % lv)b (vs. kontrolli) p-arvob,d,h | 0,61 (0,48; 0,77) < 0,0001 |
|
Jatkoseurannan keston mediaani, kk (95 % lv) | 12,3 (11,1; 14,1) | 12,9 (10,9; 14,6) |
Etenemisvapaa elinaika (PFS) (BIRC:n arvioimana) | ||
Potilaitaa | 220 | 221 |
Tapahtumia (%) | 167 (75,9) | 147 (66,5) |
PFS:n mediaani, kk (95 % lv) | 4,3 (4,1; 5,5) | 1,5 (1,5; 1,9) |
HR (95 % lv)b p-arvob,d,h | 0,44 (0,35; 0,55) < 0,0001 |
|
lv = luottamusväli; HR = riskisuhde; ORR = kokonaisvaste, OS = kokonaiselinaika
a Satunnaistettu faasin 3 tutkimus, täydellinen analyysisarja
b Satunnaistamisessa stratifioitu ECOG-toimintakykyluokan, setuksimabin lähteen ja aikaisemman irinotekaanin käytön mukaan
c Luottamusväli laskettiin toistuvasti käyttäen Lan–DeMets–O’Brien–Fleming‑raja-arvoja ja välianalyysin ajankohtana havaittua informaatiofraktiota
d 1-tahoinen
e Ensimmäisten 331 satunnaistetun potilaan joukossa
f Clopper-Pearsonin menetelmä
g Cochran Mantel-Haenszelin testi
h Nimellinen p-arvo
Kuva 3: TutkimusARRAY-818-302: Kaplan–Meier-kaavio kokonaiselinajan analyysistä (tiedonkeruun katkaisupäivä: 11.2.2019)
BRAF V600E ‑mutaatiopositiivinen etäpesäkkeinen kolorektaalisyöpä – enkorafenibi yhdistettynä setuksimabiin ja FOLFOXiin – Tutkimus C4221015
Enkorafenibin, setuksimabin ja mFOLFOX6:n yhdistelmää ensilinjan hoitona arvioitiin avoimessa ja satunnaistetussa faasin 3 monikeskustutkimuksessa (tutkimus C4221015, BREAKWATER). Tutkimukseen soveltuvilla potilailla oli oltava MSS/pMMR-muotoinen (lukuun ottamatta potilaita, joilla on varmistettu dMMR- tai MSI‑H-status, mutta joille ei voitu antaa immuuniaktivaation vapauttajaa), RAS-villityypin BRAF V600E -mutaatiopositiivinen etäpesäkkeinen kolorektaalisyöpä, jota ei ollut aiemmin hoidettu. Aiempi BRAF-estäjien tai EGFR-estäjien käyttö ei ollut sallittu. Satunnaistaminen stratifioitiin ECOG-toimintakykyluokan (Eastern Cooperative Oncology Group) ja maantieteellisen alueen mukaan.
Yhteensä 637 potilasta satunnaistettiin (1:1:1) saamaan joko 300 mg enkorafenibia vuorokaudessa suun kautta yhdessä 2 viikon välein annettavan setuksimabin kanssa, jota annosteltiin sen hyväksytyn valmisteyhteenvedon mukaan (n = 158; uusien tutkittavien rekrytointi keskeytettiin tutkimuksen aikana), tai 300 mg enkorafenibia vuorokaudessa suun kautta yhdessä setuksimabin ja mFOLFOX6:n (5‑fluorourasiili/foliinihappo/oksaliplatiini) kanssa, joita annosteltiin kunkin valmisteen hyväksytyn valmisteyhteenvedon mukaan (n = 236), tai kontrollia (mFOLFOX6, FOLFOXIRI tai CAPOX, joko bevasitsumabin kanssa tai ilman sitä, n = 243). Hoitoa jatkettiin, kunnes tauti eteni tai ilmeni sietämätöntä toksisuutta.
Ensisijaisia tehon tulosmuuttujia olivat etenemisvapaa elinaika (PFS) ja objektiivinen kokonaisvaste (ORR) sokkoutetusti riippumattoman arviointitoimikunnan (blinded independent review committee, BIRC) tekemän arvioinnin mukaan, jossa vertailtiin enkorafenibin (300 mg), setuksimabin ja mFOLFOX6:n yhdistelmähoitoa ja kontrollia toisiinsa. Keskeinen toissijainen päätetapahtuma oli kokonaiselinaika (OS). Taulukossa 9 on yhteenveto tehotuloksista.
Enkorafenibin (300 mg), setuksimabin ja mFOLFOX6:n yhdistelmähoitoa saaneiden potilaiden mediaani-ikä oli 61 vuotta (vaihteluväli 24–84); 50 % oli miehiä ja 59 % oli valkoihoisia. Lähtötilanteessa 54 %:lla potilaista ECOG-toimintakykyluokka oli 0 ja 48 %:lla potilaista kasvain oli levinnyt vähintään kolmeen elimeen.
Altistuksen keston mediaani oli 11,5 kk enkorafenibin (300 mg), setuksimabin ja mFOLFOX6:n yhdistelmähoitoa saaneilla potilailla ja 6,0 kk potilailla, jotka saivat mFOLFOX6-, FOLFOXIRI- tai CAPOX-hoitoa joko bevasitsumabin kanssa tai ilman sitä (kontrolliryhmä). Enkorafenibin (300 mg), setuksimabin ja mFOLFOX6:n yhdistelmähoitoa saaneilla potilailla suhteellisen annosintensiteetin mediaani oli 90 % enkorafenibille, 93 % setuksimabille, 80 % fluorourasiilille, 93 % foliinihapolle ja 83 % oksaliplatiinille.
Enkorafenibin (300 mg), setuksimabin ja mFOLFOX6:n yhdistelmähoito paransi tilastollisesti merkitsevästi etenemisvapaata elinaikaa (PFS), kokonaisvastetta (ORR) ja kokonaiselinaikaa (OS) kontrolliin verrattuna. Taulukossa 9 ja kuvassa 4 on yhteenveto tehotuloksista.
Taulukko 9:Tutkimus C4221015: Tehotulokset
| Enkorafenibi + setuksimabi ja mFOLFOX6 | mFOLFOX6, FOLFOXIRI tai CAPOX; bevasitsumabin kanssa tai ilman sitä (kontrolli) |
Tiedonkeruun katkaisupäivä: 22.12.2023 (kokonaisvasteen [ORR] ensisijainen analyysi) | ||
Kokonaisvaste (ORR) (BIRC:n arvioimana) | ||
Potilaitaa | 110 | 110 |
ORR, n (%) (95 % lv) | 67 (60,9) (51,6; 69,5) | 44 (40,0) (31,3; 49,3) |
p-arvob,c,d | 0,0008 |
|
Tiedonkeruun katkaisupäivä: 6.1.2025 (PFS:n ensisijainen analyysi) | ||
Etenemisvapaa elinaika (PFS) (BIRC:n arvioimana) | ||
Potilaitae | 236 | 243 |
Tapahtumia (%) | 122 (51,7) | 132 (54,3) |
PFS:n mediaani, kk (95 % lv) | 12,8 (11,2; 15,9) | 7,1 (6,8; 8,5) |
HR (95 %:n lv)c,f p-arvod,g | 0,53 (0,41; 0,68) < 0,0001 |
|
Kokonaiselinaika (OS) | ||
Potilaitae | 236 | 243 |
Tapahtumia (%) | 94 (39,8) | 148 (60,9) |
OS:n mediaani, kk (95 % lv) | 30,3 (21,7; Ei arv.) | 15,1 (13,7; 17,7) |
HR (95 % lv)c,f p-arvod,g | 0,49 (0,38; 0,63) < 0,0001 |
|
Jatkoseurannan keston mediaani, kk (95 % lv) | 21,8 (20,4; 23,4) | 22,2 (18,9; 23,5) |
a Kokonaisvasteen (ORR) alajoukko, koko analyysijoukko (FAS)
b p-arvo perustuu Cochran–Mantel–Haenszelin testiin
c Stratifioitu satunnaistamisessa ECOG-toimintakykyluokan ja maantieteellisen alueen mukaan
d 1-tahoinen
e Satunnaistettu faasin 3 tutkimus, koko analyysijoukko (FAS)
f Riskitiheyssuhde (HR) perustuu Coxin suhteellisten riskitiheyksien malliin. Suhteellisten riskitiheyksien mallissa HR < 1 viittaa pienempään riskitiheyteen ES + mFOLFOX6 ‑ryhmässä verrattuna kontrolliryhmään
g p-arvo perustuu log-rank-testiin
BIRC = sokkoutettu, riippumaton arviointikomitea (blinded independent central review committee); lv = luottamusväli; Ei arv. = Ei arvioitavissa; HR = riskisuhde; ORR = kokonaisvaste; OS = kokonaiselinaika; PFS = etenemisvapaa elinaika
Kuva 4: Tutkimus C4221015: Kaplan–Meier-kaavio kokonaiselinajan analyysistä (tiedonkeruun katkaisupäivä: 6.1.2025)
BRAF V600E -mutaatiopositiivinen pitkälle edennyt ei-pienisoluinen keuhkosyöpä – tutkimus
ARRAY-818-202
Enkorafenibin ja binimetinibin yhdistelmän turvallisuutta ja tehoa tutkittiin vaiheen II avoimessa, ei-vertailevassa monikeskustutkimuksessa (ARRAY-818-202, PHAROS). Potilailla oli oltava histologisesti varmistettu etäpesäkkeinen, BRAF V600E -mutaatiopositiivinen ei-pienisoluinen keuhkosyöpä, ECOG-toimintakykyluokka 0 tai 1 sekä mitattavissa oleva tauti. Potilaat eivät olleet saaneet lainkaan aikaisempaa systeemistä hoitoa tai olivat saaneet enintään yhden aikaisemman systeemisen hoidon etäpesäkkeisessä vaiheessa. Aiempi BRAF:n estäjien tai MEK:n estäjien käyttö oli kielletty.
Potilaat otettiin tutkimukseen paikallisen laboratorion toteuttaman kasvainkudoksen tai veren BRAF V600E -mutaatiomäärityksen perusteella (esim. ctDNA-geenitestaus). BRAF V600E -mutaatiostatus (mikä tahansa lyhyt variantti, jossa oleva mutaatio saa proteiinissa aikaan muutoksen V600E) vahvistettiin keskitetysti FoundationOne CDx – F1CDx -(kudos)määrityksellä joko arkistoidusta tai uudesta kasvainkudosnäytteestä, joka oli otettu tutkimukseenottovaiheessa.
F1CDx-määritysten herkkyys arvioitiin määrittämällä toteamisraja (Limit of Detection, LoD) löytyvyysarvomenetelmällä (alin taso, jolla havaitaan ≥ 95 %) arvioimalla lyhyiden varianttien alleelifrekvenssi (VAF). F1CDx-määrityksen LoD-arvon mediaaniksi substituutioille todettiin 3,2 %:n VAF.
Tutkimukseen otettiin yhteensä 98 potilasta, jotka saivat enkorafenibia 450 mg kerran vuorokaudessa suun kautta ja binimetinibia 45 mg kahdesti vuorokaudessa suun kautta. Hoitoa jatkettiin, kunnes tauti eteni tai kunnes ilmeni liiallista toksisuutta.
Tehon ensisijainen päätetapahtuma oli kokonaisvaste (ORR) RECIST v1.1:n mukaisesti riippumattomalla radiologisella arvioinnilla (IRR) arvioituna. Toissijaisia päätetapahtumia olivat vasteen kesto (DoR), taudin hallintaprosentti (DCR), etenemisvapaa elinaika ja kokonaiselinaika. Jäljempänä esitetään primaarianalyysin tulokset 18,2 kk:n kohdalta potilailta, jotka eivät olleet saaneet aiempaa hoitoa, ja 12,8 kk:n kohdalta aiemmin hoitoa saaneilta.
Tutkimukseen otetusta 98 potilaasta 59 (60,2 %) ei ollut saanut aiempaa hoitoa. Potilaiden iän mediaani oli 70 vuotta (vaihteluväli 47–86 vuotta), 53 % oli naisia, 88 % oli valkoihoisia ja 30 % ei ollut koskaan tupakoinut. 74 %:lla lähtötilanteen ECOG-toimintakykyluokka oli 1 (lähtötilanteessa luokka oli 1 67,8 %:lla osallistujista, jotka eivät olleet saaneet aiempaa hoitoa, ja 82,1 %:lla aiemmin hoitoa saaneista). Kaikilla potilailla oli etäpesäkkeinen tauti; lähtötilanteessa 8 %:lla oli aivometastaaseja ja 97 %:lla adenokarsinooma.
Primaarianalyysin ajankohtana altistumisen keston mediaani oli 15,1 kuukautta niillä potilailla, jotka eivät olleet saaneet aiempaa hoitoa, ja aiempaa hoitoa saaneilla 5,4 kuukautta. Koko populaatiossa suhteellisen annosintensiteetin mediaani oli enkorafenibilla 99,2 % ja binimetinibilla 95,4 %.
Primaarianalyysin ajankohtana ensisijainen päätetapahtuma, IRR-arvioitu kokonaisvaste, oli 74,6 % (95 %:n lv 61,6–85,0) niillä potilailla, jotka eivät olleet saaneet aiempaa hoitoa; heistä 9:llä (15,3 %) saavutettiin täydellinen vaste ja 35:llä (59,3 %) osittainen vaste.
Aiemmin hoitoa saaneiden IRR-arvioitu kokonaisvaste oli 46,2 % (95 %:n lv 30,1–62,8); heistä 4:llä (10,3 %) saavutettiin täydellinen vaste ja 14:llä (35,9 %) osittainen vaste.
Taulukossa 10 esitetään päivitetyt tulokset, joissa on huomioitu 10 kuukautta kestänyt lisäseuranta (altistuksen keston mediaani 16,3 kuukautta potilailla, jotka eivät olleet saaneet aiempaa hoitoa, ja 5,5 kuukautta aiemmin hoitoa saaneilla).
Taulukko 10: Tutkimus ARRAY-818-202: tehotulokset
| Enkorafenibi + binimetinibi | |
|---|---|---|
| Ei aiempaa hoitoa (N = 59) | Aiempi hoito (N = 39) |
IRR-arvioitu ORR |
|
|
ORR, % (95 % lv) | 75 % (62; 85) | 46 % (30; 63) |
CR, % | 15 % | 10 % |
PR, % | 59 % | 36 % |
IRR-arvioitu DoR | N = 44 | N = 18 |
DoR:n mediaani, kk (95 % lv) | 40,0 (23,1; NE)* | 16,7 (7,4; NE)* |
DoR ≥ 12 kk, % | 64 % | 44 % |
* Herkkyysanalyysissä, jossa taudin etenemisen ja kuoleman lisäksi käsitettiin tapahtumaksi uusi syöpähoito, tulokset olivat 23,1 kuukautta potilailla, jotka eivät olleet saaneet aiempaa hoitoa (14,8; NE), ja 12,0 kuukautta (6,3; NE) aiemmin hoitoa saaneilla potilailla.
CR = täydellinen vaste; DoR = vasteen kesto; IRR = riippumaton radiologinen arviointi; lv = luottamusväli; N = potilaiden lukumäärä; NE = ei arvioitavissa; ORR = kokonaisvaste; PR = osittainen vaste.
Sydämen elektrofysiologia
Yhdistettyjen tutkimusten turvallisuusanalyysin perusteella ensimmäistä kertaa ilmaantuneen QTcF-ajan pitenemisen (> 500 ms) ilmaantuvuus oli Combo 450 ISP -populaatiossa (n = 372) 1,1 % (4/363) ja enkorafenibia ainoana lääkkeenä käyttäneessä melanoomapotilaiden ryhmässä 2,5 % (5/203). QTcF-ajan pitenemistä > 60 ms:lla verrattuna hoitoa edeltäviin arvoihin havaittiin 6,0 %:lla (22/364) potilaista Combo 450 ISP -populaatiossa ja 3,4 %:lla (7/204) pelkkää enkorafenibia käyttäneessä ryhmässä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Kolorektaalisyövän indikaatiossa aiemmin systeemistä hoitoa saaneille potilaille toteutetun faasin 3 (ARRAY-818-302) turvallisuustutkimusten turvallisuusanalyysin perusteella ensimmäistä kertaa ilmaantuneen QTcF‑ajan pitenemisen (> 500 ms) ilmaantuvuus oli 2,8 % (6/216) enkorafenibi + setuksimabi -ryhmässä. Lisäksi QTcF‑ajan pitenemistä > 60 ms:lla verrattuna hoitoa edeltäviin arvoihin havaittiin 8,8 %:lla (19/216) potilaista enkorafenibi + setuksimabi -ryhmässä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Yhdistetyn ES + FOLFOX ‑populaation (tutkimus C4221015, BREAKWATER) kolorektaalisyövän ensilinjan hoidon indikaatiossa toteutettujen turvallisuustutkimusten turvallisuusanalyysin perusteella ensimmäistä kertaa ilmaantuneen QTcF‑ajan pitenemisen (> 500 ms) ilmaantuvuus oli 4 % (10/253). Lisäksi QTcF‑ajan pitenemistä > 60 ms:lla verrattuna hoitoa edeltäviin arvoihin havaittiin 11 %:lla (29/253) potilaista (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset enkorafenibin käytöstä kolorektaalisyövän ja keuhkosyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Enkorafenibin farmakokinetiikkaa tutkittiin terveillä tutkittavilla ja potilailla, joilla oli kiinteä kasvain. Enkorafenibin farmakokinetiikan on osoitettu olevan suunnilleen lineaarisesti riippuvainen annoksesta kerta-annoksen ja toistuvien annosten jälkeen. Toistuvan kerran vuorokaudessa tapahtuneen annon jälkeen vakaa tila saavutettiin 15 vuorokauden kuluessa. Kumulaatiokerroin oli noin 0,5, mikä todennäköisesti johtuu CYP3A4:n autoinduktiosta. AUC-vaihtelu (CV%) tutkittavien välillä oli 12,3–68,9 %.
Imeytyminen
Suun kautta otettu enkorafenibi imeytyy nopeasti, ja sen Tmax-ajan mediaani on 1,5–2 tuntia. Suun kautta annetun 100 mg [14C]-enkorafenibikerta-annoksen jälkeen vähintään 86 % enkorafenibiannoksesta imeytyi terveillä tutkittavilla. Kun 100 mg kerta-annos enkorafenibia annettiin yhdessä runsasrasvaisen, runsaskalorisen aterian kanssa, Cmax-arvo pieneni 36 %, mutta AUC-arvo pysyi muuttumattomana. Terveillä tutkittavilla toteutetussa yhteisvaikutustutkimuksessa todettiin, että mahan pH:ta muuttava aine (rabepratsoli) ei muuta altistusta enkorafenibille.
Jakautuminen
Enkorafenibi sitoutuu kohtalaisesti (86,1‑prosenttisesti) ihmisen plasman proteiineihin in vitro. Suun kautta annetun 100 mg [14C]-enkorafenibikerta-annoksen jälkeen veren ja plasman lääkepitoisuuksien suhteen keskiarvo (SD) on 0,58 (0,02) ja näennäisen jakautumistilavuuden (Vz/F) keskiarvo (CV %) 226 l (32,7 %) terveillä tutkittavilla.
Biotransformaatio
Suun kautta annetun 100 mg [14C]-enkorafenibikerta-annoksen jälkeen metabolian todettiin olevan enkorafenibin tärkein eliminaatioreitti (noin 88 % erittyneestä radioaktiivisesta annoksesta) terveillä tutkittavilla. Enkorafenibin pääasiallinen biotransformaatioreaktio oli N‑dealkylaatio. Muita merkittäviä metaboliareittejä olivat hydroksylaatio, karbamaattihydrolyysi, epäsuora glukuronidaatio ja glukoosikonjugaattimuodostus.
Eliminaatio
Suun kautta annetun 100 mg [14C]-enkorafenibikerta-annoksen jälkeen terveillä tutkittavilla radioaktiivisuutta poistui yhtä lailla sekä ulosteeseen että virtsaan (keskiarvo 47,2 %). Virtsaan radioaktiivisuudesta erittyi enkorafenibina 1,8 %. Enkorafenibin näennäisen puhdistuman (CL/F) keskiarvo (CV %) oli 27,9 l/h (9,15 %). Enkorafenibin terminaalisen puoliintumisajan (T1/2) mediaani (vaihteluväli) oli 6,32 h (3,74–8,09 h).
Lääkevalmisteiden yhteisvaikutukset
Enkorafenibin ja setuksimabin tai enkorafenibin ja FOLFOXin välillä ei ole todettu olevan lääkkeiden yhteisvaikutusta.
CYP-entsyymien vaikutus enkorafenibiin
Enkorafenibi metaboloituu CYP3A4-, CYP2C19- ja CYP2D6-välitteisesti. CYP3A4:n ennustettiin olevan pääasiallinen entsyymi, joka vaikuttaa enkorafenibin oksidatiiviseen kokonaispuhdistumaan ihmisen maksan mikrosomeissa in vitro (~83,3 %), ja seuraavaksi merkittävimmät ovat CYP2C19 (~16,0 %) ja CYP2D6 (0,71 %).
Vahvan CYP3A4:n induktorin samanaikaisen annon vaikutusta enkorafenibialtistukseen ei ole selvitetty spesifisissä tutkimuksissa. Kun melanoomapotilaille annettiin toistuvaisannoksina enkorafenibia (450 mg kerran vuorokaudessa) ja binimetinibiä (45 mg kahdesti vuorokaudessa) sekä modafiniilia, joka on keskivahva CYP3A4:n induktori, enkorafenibin vakaan tilan AUC-arvo pieneni 24 % ja Cmax-arvo 20 % verrattuna pelkän enkorafenibin antoon.
Enkorafenibin vaikutus CYP:n substraatteihin
In vitro -kokeet viittaavat siihen, että enkorafenibi on suhteellisen vahva reversiibeli UGT1A1:n, CYP2B6:n, CYP2C9:n ja CYP3A4/5:n estäjä sekä aikariippuvainen CYP3A4:n estäjä. Enkorafenibi indusoi CYP1A2-, CYP2B6-, CYP2C9- ja CYP3A4-toimintaa ihmisen primaarimaksasoluissa.
Kun melanoomapotilaille annettiin toistuvaisannoksina enkorafenibia (450 mg kerran vuorokaudessa) ja binimetinibiä (45 mg kahdesti vuorokaudessa) sekä kerta-annoksena useita CYP-selektiivisiä
koetinsubstraatteja sisältävä seos, midatsolaamin (2 mg; CYP3A4:n substraatti) AUC-arvo pieneni 82 % ja Cmax-arvo 74 %. Omepratsolin (20 mg; CYP2C19:n substraatti) AUC-arvo pieneni 17 % ja Cmax-arvo säilyi samana. Kofeiinin (50 mg; CYP1A2:n substraatti) AUC-arvo suureni 27 % ja Cmax-arvo 13 %. Losartaanin metaboliitin (E3174:n) pitoisuuden suhde losartaanipitoisuuteen virtsassa pieneni 28 % (losartaani on CYP2C9:n substraatti); dekstrometorfaanin metaboliitin (dekstrorfaanin) pitoisuuden suhde dekstrometorfaanipitoisuuteen virtsassa ei muuttunut (dekstrometorfaani on CYP2D6:n substraatti). Nämä tulokset viittavat voimakkaaseen CYP3A4:n induktioon ja lievään CYP1A2:n estoon sekä siihen, ettei enkorafenibilla ole vaikutusta CYP2C19:n substraattien farmakokinetiikkaan. Virtsan pitoisuuksia koskevien tietojen pohjalta ei voida tehdä lopullisia johtopäätöksiä CYP2C9:n ja CYP2D6:n eston voimakkuudesta. Hitaita CYP2D6-metaboloijia koskevia tietoja ei ole saatavilla.
Kerta-annos enkorafenibia (450 mg) ja binimetinibiä (45 mg) pienensi bupropionin (75 mg; CYP2B6:n substraatti) AUC-arvoa ja Cmax-arvoa ≤ 25 %. Kun enkorafenibia (450 mg kerran vuorokaudessa) ja binimetinibiä (45 mg kahdesti vuorokaudessa) annettiin toistuvaisannoksina, bupropionin AUC-arvo ja Cmax-arvo pienenivät ≤ 26 % ja sen aktiivisen metaboliitin hydroksibupropionin AUC-arvo suureni 49 %, mikä viittaa lievään induktioon.
Kun enkorafenibin kanssa annetaan suolessa metaboloituvia UGT1A1:n substraatteja, odotettavissa on lieviä tai kohtalaisia yhteisvaikutuksia. Binimetinibi on UGT1A1:n substraatti, mutta se ei metaboloidu suolessa, joten samanaikaisessa käytössä enkorafenibin kanssa ei ole odotettavissa yhteisvaikutuksia. Binimetinibin ja enkorafenibin yhdistelmän käytön yhteydessä ei ole myöskään kliinisesti havaittu altistuseroja.
Kuljettajaproteiinien vaikutus enkorafenibiin
Enkorafenibin on todettu olevan P-glykoproteiini (P-gp) -kuljettajaproteiinien substraatti. On epätodennäköistä, että P-gp:n esto johtaisi enkorafenibipitoisuuksien kliinisesti merkittävään suurenemiseen, sillä enkorafenibin läpäisevyys on luontaisesti suuri. Useiden soluunottokuljettajaproteiiniryhmien (OCT1, OATP1B1, OATP1B3 ja OATPB1) osallisuutta on tutkittu in vitro käyttämällä asiaankuuluvia kuljettajaproteiinien estäjiä. Tiedot viittaavat siihen, että maksan soluunottokuljettajaproteiinit eivät liity enkorafenibin jakautumiseen ihmisen primaarimaksasoluihin.
Enkorafenibin vaikutus kuljettajaproteiineihin
Enkorafenibin (450 mg kerran vuorokaudessa) ja binimetinibin (45 mg kahdesti vuorokaudessa) toistuva anto yhdessä rosuvastatiinin (OATP1B1:n, OATP1B3:n ja BCRP:n substraatti) kerta-annoksen kanssa suurensi rosuvastatiinin Cmax-arvoa 2,7-kertaisesti ja AUC-arvoa 1,6-kertaisesti, mikä on osoitus OATP1B1-, OATP1B3- ja/tai BCRP-kuljettajaproteiinien vähäisestä estovaikutuksesta. Enkorafenibi estää maksan kuljettajaproteiini OCT1:n toimintaa in vitro, mutta on epätodennäköistä, että se olisi kliinisesti tehokas estäjä. In vitro -tutkimusten perusteella enkorafenibi saattaa estää munuaisten kuljettajaproteiinien OCT2, OAT1 ja OAT3 toimintaa kliinisinä pitoisuuksina. Enkorafenibi saattaa myös estää P-gp:tä suolistossa odotettavissa olevilla kliinisillä pitoisuuksilla.
Erityisryhmät
Ikä
Populaatiofarmakokineettisen analyysin perusteella iän todettiin olevan merkitsevä kovariaatti enkorafenibin jakautumistilavuuden suhteen, mutta vaihtelu oli suurta. Kun otetaan huomioon, että muutokset olivat pieniä ja vaihtelu suurta, muutokset eivät todennäköisesti ole kliinisesti merkittäviä. Täten iäkkäiden potilaiden annosta ei tarvitse muuttaa.
Kliinistä tietoa ei ole saatavilla pediatrisista potilaista. Turvallisuusprofiili pediatrisilla potilailla on tuntematon. BRAF V600 ‑mutatoitunutta metastaattista melanoomaa sairastavilla 12 – < 18-vuotiailla nuorilla populaatiofarmakokineettiset simulaatiot osoittavat, että seuraavat annokset ja annoksen pienennykset haittavaikutusten hallintaan johtavat samanlaiseen plasman altistukseen kuin aikuisilla:
-
Vähintään 40 kg painavat: aikuisen annos 450 mg kerran vuorokaudessa samoilla annoksen pienennyksillä: 300 ja 225 mg kerran vuorokaudessa.
-
30 – < 40 kg:n painoiset: 300 mg kerran vuorokaudessa, annos pienennetään 225 mg:aan kerran vuorokaudessa ja sitten 150 mg:aan kerran vuorokaudessa.
-
Alle 30 kg:n painoiset: 225 mg kerran vuorokaudessa, annos pienennetään 150 mg:aan kerran vuorokaudessa.
Alle 12-vuotiaita lapsia ei tule hoitaa enkorafenibilla.
Sukupuoli
Populaatiofarmakokineettisen analyysin perusteella sukupuolen ei todettu olevan merkitsevä mallikovariaatti puhdistuman eikä jakautumistilavuuden suhteen. Täten suuria sukupuoleen liittyviä enkorafenibialtistuksen muutoksia ei ole odotettavissa.
Paino
Populaatiofarmakokineettisen analyysin perusteella painon todettiin olevan merkittävä mallikovariaatti puhdistuman ja jakautumistilavuuden suhteen. Mallissa puhdistumamuutokset olivat kuitenkin vähäisiä ja ennakoitavissa olevan jakautumistilavuuden vaihtelu suurta, joten painolla ei todennäköisesti ole aikuisilla kliinisesti merkittävää vaikutusta enkorafenibialtistukseen.
Etninen tausta
Enkorafenibin farmakokinetiikassa ei ole kliinisesti merkittäviä eroja aasialaisten ja ei-aasialaisten välillä. Tietoja ei ole riittävästi, jotta mahdollisia enkorafenibialtistuksen eroja muissa etnisissä väestöryhmissä voitaisiin arvioida.
Maksan vajaatoiminta
Spesifisen kliinisen tutkimuksen tulokset viittaavat siihen, että enkorafenibin kokonaisaltistus on 25 % suurempi lievässä maksan vajaatoiminnassa (Child‑Pugh‑luokka A) kuin normaalissa maksatoiminnassa. Tämä tarkoittaa 55 % suurempaa altistusta sitoutumattomalle enkorafenibille.
Enkorafenibin farmakokinetiikkaa ei ole arvioitu kliinisesti potilailla, joilla on keskivaikea (Child‑Pugh‑luokka B) tai vaikea (Child‑Pugh‑luokka C) maksan vajaatoiminta. Enkorafenibi metaboloituu ja eliminoituu ensisijaisesti maksassa, ja fysiologiaan perustuvan farmakokineettisen mallinnuksen perusteella altistus saattaa olla suurempi potilailla, joilla on keskivaikea tai vaikea maksan vajaatoiminta, kuin potilailla, joilla on lievä maksan vajaatoiminta. Potilaille, joilla on keskivaikea tai vaikea maksan vajaatoiminta, ei voida antaa annostussuosituksia (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Munuaisten vajaatoiminta
Enkorafenibi eliminoituu munuaisten kautta erittäin vähäisessä määrin. Munuaisten vajaatoiminnan vaikutusta enkorafenibin farmakokinetiikkaan ei ole arvioitu tätä tarkoitusta varten tehdyssä kliinisessä tutkimuksessa.
Populaatiofarmakokineettisessä analyysissa ei havaittu selkeää trendiä enkorafenibin näennäisessä puhdistumassa (CL/F) potilailla, joilla oli lievä (eGFR 60–90 ml/min/1,73 m2) tai keskivaikea (eGFR 30–59 ml/min/1,73 m2) munuaisten vajaatoiminta, verrattuna tutkittaviin, joiden munuaistoiminta oli normaali (eGFR ≥ 90 ml/min/1,73 m2). Potilailla, joilla oli lievä tai keskivaikea munuaisten vajaatoiminta, ennustettiin pientä CL/F‑arvon pienenemistä (≤ 5 %), joka ei todennäköisesti ole kliinisesti merkittävä. Enkorafenibin farmakokinetiikkaa ei ole tutkittu vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla.
Prekliiniset tiedot turvallisuudesta
Rotilla toteutetuissa 4 viikon ja 13 viikon pituisissa toksisuustutkimuksissa havaittiin kliinisiä löydöksiä, kuten ruumiin, lisäkivesten ja eturauhasen painon pienenemistä, sekä mikroskooppisia löydöksiä kiveksissä, lisäkiveksissä, mahassa ja ihossa. Löydösten havaittiin osin korjautuvan 4 viikon toipumisvaiheen jälkeen. NOAEL-arvoa (annostaso, jolla ei havaittu haittavaikutuksia) ei voitu määrittää 4 viikon tutkimuksessa. 13 viikon tutkimuksessa määritelty NOAEL-arvo oli yli 10-kertainen ihmisen terapeuttiseen altistukseen verrattuna.
Apinoilla toteutetuissa 4 viikon ja 13 viikon pituisissa toksisuustutkimuksissa havaittiin yksittäisiä/satunnaisia oksentelu- ja ripulitapauksia sekä silmämuutoksia terapeuttisten altistusten ollessa hieman ihmiselle tarkoitettuja suurempia. Silmämuutokset olivat osittain korjautuvia. Näitä olivat ulomman sauvasolu- ja tappisolukerroksen erkaneminen tai irtauma verkkokalvon pigmenttiepiteelistä makulan keskellä olevalla fovea-alueella. Tämä havainto vastaa ihmisillä kuvattua sentraalista seroosimaista korioretinopatiaa tai sentraalista seroosia retinopatiaa.
Enkorafenibi ei osoittautunut genotoksiseksi.
Enkorafenibin vaikutusta hedelmällisyyteen ei ole tutkittu. Kun enkorafenibia annettiin rotille 6 mg/kg/vrk (annostus yli 5‑kertainen verrattuna ihmisen altistukseen terapeuttisella annoksella) 13 viikon pituisessa toksikologiatutkimuksessa, todettiin kivesten ja lisäkivesten painon pienenemistä sekä siementiehyiden degeneraatiota ja oligospermiaa. 13 viikon pituisessa tutkimuksessa todettiin osittaista korjautuvuutta suurimmalla annostuksella (60 mg/kg/vrk).
Rotilla tehty alkion- ja sikiönkehitystutkimus viittasi siihen, että enkorafenibi aiheuttaa sikiötoksisuutta, kuten sikiöiden painon pienenemistä ja luuston kehityksen viivästymistä.
Kaniineilla tehty alkion- ja sikiönkehitystutkimus viittasi siihen, että enkorafenibi aiheuttaa sikiötoksisuutta, kuten sikiöiden painon pienenemistä ja luuston kehityksen ohimeneviä muutoksia. Joillain sikiöillä todettiin aortankaaren dilataatiota.
Enkorafenibi oli valotoksinen NRU-testissä (3T3 Neutral Red Uptake Test) in vitro. Enkorafenibi ei aiheuttanut herkistymistä herkistystestissä hiirillä in vivo. Nämä tiedot yhdessä viittaavat siihen, että enkorafenibin terapeuttisiin annoksiin liittyy potilailla valotoksisuuden riski ja erittäin pieni herkistymisriski.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Kopovidoni (E1208)
Poloksameeri 188
Mikrokiteinen selluloosa (E460i)
Meripihkahappo (E363)
Krospovidoni (E1202)
Vedetön kolloidinen piidioksidi (E551)
Magnesiumstearaatti (E470b)
Kapselin kuori
Liivate (E441)
Titaanidioksidi (E171)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Painomuste
Shellakka (E904)
Musta rautaoksidi (E172)
Propyleeniglykoli (E1520)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Säilytä alle 30 °C.
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BRAFTOVI kapseli, kova
50 mg (L:ei) 28 fol (794,26 €)
75 mg (L:ei) 42 fol (1479,10 €)
PF-selosteen tieto
Braftovi 50 mg kapseli, kova
Yksi pakkaus sisältää joko 28 x 1 tai 112 x 1 kovaa kapselia yksittäispakatussa polyamidi/alumiini/PVC/alumiini/PET/paperi-läpipainopakkauksessa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Braftovi 75 mg kapseli, kova
Yksi pakkaus sisältää joko 42 x 1 tai 168 x 1 kovaa kapselia yksittäispakatussa polyamidi/alumiini/PVC/alumiini/PET/paperi-läpipainopakkauksessa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Braftovi 50 mg kapseli, kova
Oranssi, läpinäkymätön yläosa ja ihonvärinen, läpinäkymätön alaosa. Yläosaan on painettu tyylitelty ”A” ja alaosaan ”LGX 50mg”. Kapselin pituus on noin 22 mm.
Braftovi 75 mg kapseli, kova
Ihonvärinen, läpinäkymätön yläosa ja valkoinen, läpinäkymätön alaosa. Yläosaan on painettu tyylitelty ”A” ja alaosaan ”LGX 75mg”. Kapselin pituus on noin 23 mm.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BRAFTOVI kapseli, kova
75 mg 42 fol
- Ylempi erityiskorvaus (100 %). Binimetinibi ja enkorafenibi (melanooma): Melanooman hoito erityisin edellytyksin (1513), Enkorafenibi (kolorektaalisyöpä): Aikuisten etäpesäkkeisen kolorektaalisyövän yhdistelmähoito setuksimabin kanssa erityisin edellytyksin (1526).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Binimetinibi ja enkorafenibi (melanooma): Melanooman hoito erityisin edellytyksin (3016), Enkorafenibi (kolorektaalisyöpä): Aikuisten etäpesäkkeisen kolorektaalisyövän yhdistelmähoito setuksimabin kanssa erityisin edellytyksin (3049).
BRAFTOVI kapseli, kova
50 mg 28 fol
- Ylempi erityiskorvaus (100 %). Binimetinibi ja enkorafenibi (melanooma): Melanooman hoito erityisin edellytyksin (1513).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Binimetinibi ja enkorafenibi (melanooma): Melanooman hoito erityisin edellytyksin (3016).
ATC-koodi
L01EC03
Valmisteyhteenvedon muuttamispäivämäärä
19.06.2026
Yhteystiedot
c/o Pierre Fabre Dermo-Cosmétique Nordic A/S Metsäneidonkuja 4
02130 Espoo
www.pierre-fabre.com