
PREVYMIS tabletti, kalvopäällysteinen 240 mg, 480 mg
Vaikuttavat aineet ja niiden määrät
PREVYMIS 240 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 240 mg letermoviiria.
PREVYMIS 480 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 480 mg letermoviiria.
Apuaineet, joiden vaikutus tunnetaan
Yksi 240 mg:n kalvopäällysteinen tabletti sisältää 4 mg laktoosia (monohydraattina).
Yksi 480 mg:n kalvopäällysteinen tabletti sisältää 6,4 mg laktoosia (monohydraattina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti)
Kliiniset tiedot
Käyttöaiheet
PREVYMIS on tarkoitettu sytomegaloviruksen (CMV) uudelleenaktivoitumisen ja viruksen aiheuttaman taudin ehkäisyyn allogeenisen kantasolusiirron (HSCT) saaneille CMV-seropositiivisille aikuisille ja vähintään 15 kg painaville pediatrisille potilaille.
PREVYMIS on tarkoitettu sytomegaloviruksen aiheuttaman taudin ehkäisyyn CMV-seronegatiivisilla aikuisilla ja vähintään 40 kg painavilla pediatrisilla potilailla, jotka ovat saaneet munuaissiirteen CMV-seropositiiviselta luovuttajalta.
Viruslääkkeiden tarkoituksenmukaista käyttöä koskevat viralliset ohjeet on otettava huomioon.
Ehto
Hoidon saavat aloittaa valmisteyhteenvedossa mainittuun indikaatioon perehtyneet lääkärit.
Annostus ja antotapa
Letermoviirihoidon aloittavan lääkärin pitää olla perehtynyt allogeenisen kantasolusiirron tai munuaissiirteen saaneiden potilaiden hoitoon.
Annostus
Letermoviiria on saatavana myös rakeina annospussissa (20 mg ja 120 mg) ja infuusiokonsentraattina, liuosta varten (240 mg ja 480 mg).
Letermoviiritabletteja, letermoviirirakeita annospussissa ja letermoviiri‑infuusiokonsentraattia, liuosta varten, voidaan käyttää toistensa vaihtoehtoina lääkärin harkinnan mukaan. Annoksen muuttaminen saattaa olla tarpeen alle 30 kg painavilla pediatrisilla potilailla, kun vaihdetaan suun kautta annettavasta lääkemuodosta laskimoon annettavaan lääkemuotoon tai päinvastoin. Katso annostustiedot letermoviiri‑infuusiokonsentraatin, liuosta varten, valmistetiedoista.
Kantasolusiirto
Letermoviirihoito aloitetaan kantasolusiirron jälkeen. Letermoviirihoito voidaan aloittaa samana päivänä, kun kantasolusiirto tehdään, mutta kuitenkin viimeistään 28 vuorokauden kuluttua kantasolusiirrosta. Letermoviirihoito voidaan aloittaa ennen siirteen tarttumista tai sen jälkeen. Profylaktista letermoviirihoitoa jatketaan 100 vuorokauden ajan kantasolusiirron jälkeen.
Jotkut potilaat, joilla sytomegaloviruksen myöhäisen uudelleenaktivoitumisen riski on suuri, voivat hyötyä pitkäaikaisesta, yli 100 vuorokautta kantasolusiirron jälkeen kestävästä letermoviiriprofylaksista (ks. kohta Farmakodynamiikka). Letermoviirin yli 200 päivää jatkuvan käytön turvallisuutta ja tehoa ei ole tutkittu kliinisissä tutkimuksissa.
Aikuiset ja vähintään 30 kg painavat pediatriset potilaat, jotka ovat saaneet kantasolusiirron
Suositeltu letermoviiriannos on 480 mg kerran vuorokaudessa. Annos voidaan antaa joko yhtenä 480 mg:n tablettina tai kahtena 240 mg:n tablettina.
Jos potilas ei pysty nielemään tabletteja, katso annostustiedot letermoviirirakeiden, annospussi, valmistetiedoista.
Annoksen säätäminen aikuisilla ja vähintään 30 kg painavilla pediatrisilla potilailla, jotka ovat saaneet kantasolusiirron
Jos letermoviirihoitoa annetaan samanaikaisesti siklosporiinin kanssa, letermoviiriannos on laskettava tasolle 240 mg kerran vuorokaudessa (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
- Jos siklosporiini aloitetaan letermoviirihoidon aloittamisen jälkeen, letermoviiriannostusta on pienennettävä seuraavasta annoksesta lähtien tasolle 240 mg kerran vuorokaudessa.
- Jos siklosporiini lopetetaan letermoviirihoidon aloittamisen jälkeen, letermoviiriannostus on nostettava seuraavasta annoksesta lähtien tasolle 480 mg kerran vuorokaudessa.
- Jos siklosporiinihoito keskeytetään tilapäisesti suurten siklosporiinipitoisuuksien vuoksi, letermoviiriannosta ei tarvitse muuttaa.
Vähintään 15 kg – alle 30 kg painavat pediatriset potilaat, jotka ovat saaneet kantasolusiirron
Suositeltu letermoviiriannos on 240 mg kerran vuorokaudessa. Annos voidaan antaa yhtenä 240 mg:n tablettina (ks. myös kohta Farmakokinetiikka).
Jos pediatrinen potilas ei pysty nielemään tabletteja, katso annostustiedot letermoviirirakeiden, annospussi, valmistetiedoista.
Annoksen säätäminen vähintään 15 kg – alle 30 kg painavilla pediatrisilla potilailla, jotka ovat saaneet kantasolusiirron
Jos suun kautta annettavaa letermoviiria annetaan samanaikaisesti siklosporiinin kanssa, letermoviiriannos on laskettava tasolle 120 mg kerran vuorokaudessa (ks. myös kohdat Yhteisvaikutukset ja Farmakokinetiikka). Jos potilas tarvitsee 120 mg:n annoksen, katso annostustiedot letermoviirirakeiden, annospussi, valmistetiedoista.
-
Jos siklosporiinihoito aloitetaan letermoviirihoidon aloittamisen jälkeen, letermoviiriannostusta on pienennettävä seuraavasta annoksesta lähtien tasolle 120 mg kerran vuorokaudessa.
-
Jos siklosporiinihoito lopetetaan letermoviirihoidon aloittamisen jälkeen, letermoviiriannostus on nostettava seuraavasta annoksesta lähtien tasolle 240 mg kerran vuorokaudessa.
-
Jos siklosporiinihoito keskeytetään tilapäisesti suurten siklosporiinipitoisuuksien vuoksi, letermoviiriannosta ei tarvitse muuttaa.
Munuaisensiirto
Letermoviirihoito aloitetaan siirtopäivänä tai viimeistään 7 vuorokauden kuluttua munuaisensiirrosta, ja sitä jatketaan 200 vuorokauden ajan siirron jälkeen.
Aikuiset ja vähintään 40 kg painavat pediatriset potilaat, jotka ovat saaneet munuaissiirteen
Suositeltu letermoviiriannos on 480 mg kerran vuorokaudessa. Annos voidaan antaa joko yhtenä 480 mg:n tablettina tai kahtena 240 mg:n tablettina.
Jos potilas ei pysty nielemään tabletteja, katso annostustiedot letermoviirirakeiden, annospussi, valmistetiedoista.
Annoksen säätäminen aikuisilla ja vähintään 40 kg painavilla pediatrisilla potilailla, jotka ovat saaneet munuaissiirteen
Jos letermoviiria annetaan samanaikaisesti siklosporiinin kanssa, letermoviiriannos on laskettava tasolle 240 mg kerran vuorokaudessa (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
-
Jos siklosporiinihoito aloitetaan letermoviirihoidon aloittamisen jälkeen, letermoviiriannostusta on pienennettävä seuraavasta annoksesta lähtien tasolle 240 mg kerran vuorokaudessa.
-
Jos siklosporiinihoito lopetetaan letermoviirihoidon aloittamisen jälkeen, letermoviiriannostus on nostettava seuraavasta annoksesta lähtien tasolle 480 mg kerran vuorokaudessa.
-
Jos siklosporiinihoito keskeytetään tilapäisesti suurten siklosporiinipitoisuuksien vuoksi, letermoviiriannosta ei tarvitse muuttaa.
Jos annos jää väliin
Potilaille on kerrottava, että unohtunut letermoviiriannos on otettava heti, kun se muistetaan. Jos se muistetaan vasta, kun on jo seuraavan annoksen aika, on unohtunut annos jätettävä väliin ja hoitoa jatkettava normaalin aikataulun mukaan. Potilaat eivät saa ottaa seuraavaa annosta kaksinkertaisena eivätkä ottaa suurempaa annosta kuin heille on määrätty.
Erityisryhmät
Iäkkäät
Letermoviiriannosta ei tarvitse muuttaa iän perusteella (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Maksan vajaatoiminta
Lievä (Child-Pugh-luokka A) tai kohtalainen (Child-Pugh-luokka B) maksan vajaatoiminta ei vaadi letermoviiriannoksen muuttamista. Letermoviirihoitoa ei suositella potilaille, joilla on vaikea maksan vajaatoiminta (Child-Pugh-luokka C) (ks. kohta Farmakokinetiikka).
Sekä maksan että munuaisten vajaatoiminta
Letermoviirihoitoa ei suositella potilaille, joilla on sekä kohtalainenmaksan vajaatoiminta että kohtalainen tai vaikea munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Letermoviiriannoksenmuuttamista ei suositella lievässä, kohtalaisessa eikä vaikeassa munuaisten vajaatoiminnassa. Dialyysihoitoa tarvitseville ja muille loppuvaiheen munuaistautia sairastaville potilaille ei voida antaa annostussuosituksia. Tehoa ja turvallisuutta ei ole osoitettu loppuvaiheen munuaistautia sairastavien potilaiden hoidossa.
Pediatriset potilaat
Letermoviirin turvallisuutta ja tehoa kantasolusiirron saaneiden alle5 kg painavien potilaiden tai munuaissiirteen saaneiden alle 40 kg painavien potilaiden hoidossa ei ole varmistettu. Tietoja ei ole saatavilla. Farmakokineettisestä/farmakodynaamisesta ekstrapoloinnista ei saatu tietoja, jotka tukisivat annossuosituksia munuaissiirteen saaneille alle 40 kg painaville potilaille.
Antotapa
Suun kautta.
Tabletti niellään kokonaisena, ja se voidaan ottaa aterian yhteydessä tai tyhjään mahaan. Tablettia ei saa jakaa, murskata eikä pureskella, koska tällaisia toimintatapoja ei ole tutkittu.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Samanaikainen käyttö pimotsidin kanssa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Samanaikainen käyttö torajyväalkaloidien kanssa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Samanaikainen käyttö mäkikuisman (Hypericum perforatum) kanssa (ks. kohta Yhteisvaikutukset).
Kun letermoviiria käytetään yhdessä siklosporiinin kanssa:
- dabigatraania, atorvastatiinia, simvastatiinia, rosuvastatiinia tai pitavastatiinia ei saa käyttää samanaikaisesti (ks. kohta Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Sytomegaloviruksen DNA:n (CMV-DNA:n) seuranta kantasolusiirron saaneilla potilailla
Letermoviirin turvallisuus ja teho on osoitettu vaiheen 3 tutkimuksessa (P001) kantasolusiirron saaneilla potilailla, joilla CMV-DNA-määrityksen tulos oli negatiivinen ennen profylaktisen hoidon aloittamista. CMV-DNA-arvoa seurattiin viikoittain viikolle 14 kantasolusiirron jälkeen ja sen jälkeen kahden viikon välein viikolle 24 asti. Jos veren CMV-DNA-määrä oli kliinisesti merkittävä tai potilaalla oli viruksen aiheuttama tauti, letermoviiriprofylaksi lopetettiin ja aloitettiin tavanomainen ennakoiva hoito tai muu tavanomainen hoito. Jos potilaalle oli aloitettu letermoviiriprofylaksi ja myöhemmin todettiin, että lähtötilanteen CMV-DNA-arvo oli positiivinen, profylaktista hoitoa voitiin jatkaa, elleivät ennakoivan hoidon kriteerit täyttyneet (ks. kohta Farmakodynamiikka).
Lääkeaineiden yhteisvaikutuksiin liittyvä haittavaikutusten riski tai hoitotehon heikkeneminen
Letermoviirin yhteiskäyttö tiettyjen muiden lääkevalmisteiden kanssa voi johtaa tunnettuihin tai mahdollisesti merkittäviin yhteisvaikutuksiin, joista jotkin voivat aiheuttaa:
- mahdollisesti kliinisesti merkittäviä haittavaikutuksia, jotka johtuvat samanaikaisesti annetun toisen lääkevalmisteen tai letermoviirin pitoisuuden suurenemisesta
- samanaikaisesti annetun lääkevalmisteen pitoisuuden merkittävän pienenemisen plasmassa, mikä voi heikentää kyseisen samanaikaisesti annetun valmisteen hoitotehoa.
Taulukossa 1 on vaiheittaiset ohjeet näiden tunnettujen tai mahdollisesti merkittävien yhteisvaikutusten ehkäisystä tai hoidosta sekä annostussuositukset (ks. kohdat Vasta-aiheet ja Yhteisvaikutukset).
Yhteisvaikutukset muiden lääkkeiden kanssa
Varovaisuutta on noudatettava, jos letermoviiria käytetään yhdessä sellaisten CYP3A:n substraattien kanssa, joilla on kapea terapeuttinen pitoisuusalue (esim. alfentaniili, fentanyyli ja kinidiini), sillä yhteiskäyttö saattaa suurentaa CYP3A:n substraattien pitoisuutta plasmassa. On suositeltavaa seurata potilaiden tilaa tarkoin ja/tai muuttaa samanaikaisesti käytettävien CYP3A:n substraattien annosta (ks. kohta Yhteisvaikutukset).
Yleisesti suositellaan, että siklosporiinin, takrolimuusin ja sirolimuusin pitoisuuksia seurataan tavanomaista tiheämmin 2 ensimmäisen viikon ajan letermoviirihoidon aloittamisen ja lopettamisen jälkeen (ks. kohta Yhteisvaikutukset) ja sen jälkeen, kun letermoviirin antoreittiä on muutettu.
Letermoviiri on entsyymien ja kuljettajaproteiinien kohtalainen induktori. Induktio saattaa pienentää joidenkin sellaisten lääkeaineiden pitoisuuksia plasmassa, joiden metaboliaan ja kuljetukseen nämä proteiinit osallistuvat (ks. kohta Yhteisvaikutukset). Siksi suositellaan vorikonatsolin terapeuttisen tason seurantaa (Therapeutic Drug Monitoring (TDM)). Dabigatraanin samanaikaista käyttöä on vältettävä, koska se voi johtaa dabigatraanin tehon heikkenemiseen.
Letermoviiri saattaa suurentaa OATP1B1/3:n kuljettamien lääkeaineiden, kuten monien statiinien, pitoisuuksia plasmassa (ks. kohta Yhteisvaikutukset ja taulukko 1).
Apuaineet
PREVYMIS sisältää laktoosimonohydraattia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkevalmistetta.
Tämä lääkevalmiste sisältää alle 1 mmol (23 mg) natriumia per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Tietoa lääkeainealtistuksen eroista letermoviirin eri hoito-ohjelmien välillä
- Arvioitu plasman letermoviirialtistus vaihtelee käytetystä annostuksesta riippuen (ks. taulukko kohdassa Farmakokinetiikka). Siksi letermoviirin ja muiden lääkeaineiden yhteisvaikutusten kliiniset seurausvaikutukset riippuvat letermoviirin annostuksesta ja siitä, annetaanko letermoviiria yhdessä siklosporiinin kanssa.
- Siklosporiinin ja letermoviirin yhteiskäyttö saattaa voimistaa tai lisätä muihin samanaikaisesti käytettyihin lääkevalmisteisiin kohdistuvia vaikutuksia verrattuna yksinään annettuun letermoviiriin (ks. taulukko 1).
Muiden lääkevalmisteiden vaikutus letermoviiriin
Letermoviiri eliminoituu in vivo erittymällä sappeen ja glukuronidaation kautta. Näiden eliminoitumisteiden suhteellista merkitystä ei tunneta. Molempiin eliminoitumisteihin liittyy aktiivinen siirtyminen maksasoluun lääkeaineita maksaan kuljettavien OATP1B1/3-proteiinien välityksellä. Maksasoluihin siirtymisen jälkeen letermoviirin glukuronidaatio tapahtuu UGT1A1:n ja ‑3:n välityksellä. Lisäksi suolistossa ja maksassa näyttää tapahtuvan letermoviirin uloskuljetusta P-gp:n ja BCRP:n välittämänä (ks. kohta Farmakokinetiikka).
Lääkeaineita metaboloivien entsyymien tai kuljettajaproteiinien induktorit
Letermoviirin yhteiskäyttö (yhdessä siklosporiinin kanssa tai ilman sitä) voimakkaiden ja kohtalaisten kuljettajaproteiinien (kuten P-gp:n) ja/tai entsyymien (kuten UGT-entsyymien) induktoreiden kanssa ei ole suositeltavaa, koska se voi johtaa letermoviirialtistuksen pienenemiseen terapeuttisen tason alapuolelle (ks. taulukko 1).
- Voimakkaita induktoreita ovat esimerkiksi rifampisiini, fenytoiini, karbamatsepiini, rifabutiini ja fenobarbitaali.
- Kohtalaisia induktoreita ovat esimerkiksi tioridatsiini, modafiniili, ritonaviiri, lopinaviiri, efavirentsi ja etraviriini.
Rifampisiinin samanaikainen käyttö suurensi aluksi letermoviirin pitoisuuksia plasmassa (OATP1B1/3:n ja/tai P‑gp:n estymisen vuoksi), mikä ei ollut kliinisesti merkityksellistä, ja sen jälkeen letermoviirin pitoisuudet plasmassa pienenivät kliinisesti merkityksellisesti (P‑gp:n/UGT:n induktion vuoksi), kun rifampisiinin samanaikaista käyttöä jatkettiin (ks. taulukko 1).
Muiden lääkevalmisteiden muut vaikutukset letermoviiriin, kun letermoviiria annetaan yhdessä siklosporiinin kanssa
OATP1B1:n tai ‑3:n estäjät
Letermoviirin yhteiskäyttö sellaisten lääkevalmisteiden kanssa, jotka estävät OATP1B1/3-kuljettajien toimintaa, voi johtaa letermoviiripitoisuuden suurenemiseen plasmassa. Jos letermoviiria annetaan samanaikaisesti siklosporiinin (OATP1B1/3:n voimakkaan estäjän) kanssa, suositeltu letermoviiriannos on 240 mg kerran vuorokaudessa aikuisilla ja vähintään 30 kg painavilla pediatrisilla potilailla (ks. taulukko 1 ja kohdat Annostus ja antotapa ja Farmakokinetiikka). Jos suun kautta annettavaa letermoviiria annetaan samanaikaisesti siklosporiinin kanssa alle 30 kg painaville pediatrisille potilaille, annosta on pienennettävä (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka). Varovaisuutta on noudatettava, jos muita OATP1B1/3:n estäjiä annetaan samanaikaisesti letermoviirin ja siklosporiinin kanssa.
- OATP1B1:n estäjiä ovat esimerkiksi gemfibrotsiili, erytromysiini, klaritromysiini ja monet proteaasinestäjät (atatsanaviiri, simepreviiri).
P-gp:n/BCRP:n estäjät
In vitro ‑tulokset osoittavat, että letermoviiri on P-gp:n/BCRP:n substraatti. Itrakonatsolin aikaansaamasta P-gp:n/BCRP:n toiminnan estymisestä johtuvat plasman letermoviiripitoisuuden muutokset eivät olleet kliinisesti merkittäviä.
Letermoviirin vaikutus muihin lääkevalmisteisiin
Lääkevalmisteet, jotka eliminoituvat pääasiassa metaboloitumalla tai aktiivisen kuljetuksen välityksellä
Letermoviiri on entsyymien ja kuljettajaproteiinien yleinen induktori in vivo. Ellei tietyn entsyymin tai kuljettajan toiminta ole myös estynyt (ks. jäljempänä), induktiovaikutus on todennäköinen. Siksi letermoviiri saattaa pienentää samanaikaisesti käytettävien pääasiassa metaboloitumalla tai aktiivisen kuljetuksen välityksellä eliminoituvien lääkevalmisteiden pitoisuutta plasmassa ja mahdollisesti heikentää niiden tehoa.
Induktiovaikutuksen voimakkuus riippuu letermoviirin antoreitistä ja siitä, käytetäänkö samanaikaisesti siklosporiinia. Induktiovaikutuksen arvellaan olevan suurimmillaan, kun letermoviirihoito on kestänyt 10−14 vuorokautta. Kohteena olevan lääkkeen vakaan tilan saavuttamiseen kuluva aika vaikuttaa myös siihen, milloin plasman lääkeainepitoisuuteen kohdistuva vaikutus on suurimmillaan.
Letermoviiri on CYP3A:n, CYP2C8:n, CYP2B6:n, BCRP:n, UGT1A1:n, OATP2B1:n ja OAT3:n estäjä in vitro pitoisuuksina, joilla on merkitystä in vivo. Saatavilla on in vivo ‑tutkimuksia, joissa on tarkasteltu CYP3A4:ään, P-gp:hen, OATP1B1/3:een ja lisäksi CYP2C19:ään kohdistuvaa nettovaikutusta. Muihin lueteltuihin entsyymeihin ja kuljettajaproteiineihin kohdistuvaa nettovaikutusta in vivo ei tunneta. Yksityiskohtaiset tiedot on esitetty alla.
Ei tiedetä, vaikuttaako letermoviiri piperasilliini/tatsobaktaamin, amfoterisiini B:n ja mikafungiinin pitoisuuksiin. Letermoviirin ja näiden lääkeaineiden mahdollisia yhteisvaikutuksia ei ole tutkittu. Altistuksen pieneneminen induktion seurauksena on teoriassa mahdollista, mutta tämän vaikutuksen voimakkuutta ja siten sen kliinistä merkitystä ei toistaiseksi tiedetä.
CYP3A-entsyymin välityksellä metaboloituvat lääkevalmisteet
Letermoviiri on CYP3A:n kohtalainen estäjä in vivo. Letermoviirin ja suun kautta annettavan midatsolaamin (CYP3A:n substraatin) yhteiskäyttö johtaa plasman midatsolaamipitoisuuden suurenemiseen 2–3-kertaiseksi. Letermoviirin yhteiskäyttö saattaa suurentaa samanaikaisesti annettujen CYP3A:n substraattien pitoisuuksia plasmassa kliinisesti merkittävässä määrin (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
- Tällaisia lääkevalmisteita ovat esimerkiksi tietyt immunosuppressiiviset valmisteet (esim. siklosporiini, takrolimuusi, sirolimuusi), HMG-CoA-reduktaasin estäjät ja amiodaroni (ks. taulukko 1). Pimotsidia ja torajyväalkaloideja ei saa käyttää yhdessä PREVYMIS-valmisteen kanssa (ks. kohta Vasta-aiheet).
CYP3A:n toimintaa estävän vaikutuksen voimakkuus riippuu letermoviirin antoreitistä ja siitä, käytetäänkö samanaikaisesti siklosporiinia.
Ajasta riippuvan estovaikutuksen ja samanaikaisen induktiovaikutuksen vuoksi entsyymin toimintaa estävä nettovaikutus saavutetaan ehkä vasta 10–14 vuorokauden kuluttua. Kohteena olevan lääkkeen vakaan tilan saavuttamiseen kuluva aika vaikuttaa myös siihen, milloin plasman lääkeainepitoisuuteen kohdistuva vaikutus on suurimmillaan. Kun hoito lopetetaan, kestää 10–14 vuorokautta ennen kuin estovaikutus häviää. Jos pitoisuuksia seurataan, niiden seuraamista suositellaan 2 ensimmäisen viikon ajan letermoviirihoidon aloittamisen ja lopettamisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) ja sen jälkeen, kun letermoviirin antoreittiä on muutettu.
OATP1B1/3-proteiinien kuljettamat lääkevalmisteet
Letermoviiri on OATP1B1/3-kuljettajapolypeptidien estäjä. Letermoviiri saattaa suurentaa samanaikaisesti annettujen OATP1B1/3:n substraattien pitoisuuksia plasmassa kliinisesti merkittävässä määrin.
- Tällaisia lääkevalmisteita ovat esimerkiksi HMG-CoA-reduktaasin estäjät, feksofenadiini, repaglinidi ja glyburidi (ks. taulukko 1). Kun verrataan ilman siklosporiinia annettuja letermoviirin hoito-ohjelmia keskenään, vaikutus on voimakkaampi laskimoon annetun kuin suun kautta annetun letermoviirin jälkeen.
Muihin samanaikaisesti annettuihin lääkevalmisteisiin kohdistuva OATP1B1/3:n toimintaa estävä vaikutus on todennäköisesti voimakkaampi, kun letermoviiri annetaan yhdessä siklosporiinin (voimakkaan OATP1B1/3:n estäjän) kanssa. Tämä on otettava huomioon, kun letermoviirin hoito-ohjelmaa muutetaan OATP1B1/3:n substraatin käytön aikana.
CYP2C9- ja/tai CYP2C19-entsyymin välityksellä metaboloituvat lääkevalmisteet
Letermoviirin yhteiskäyttö vorikonatsolin (CYP2C19:n substraatin) kanssa pienentää merkittävästi plasman vorikonatsolipitoisuuksia, mikä viittaa siihen, että letermoviiri on CYP2C19:n induktori. Todennäköisesti myös CYP2C9 indusoituu. Letermoviiri saattaa pienentää CYP2C9:n ja/tai CYP2C19:n substraattien pitoisuuksia, ja nämä pitoisuudet voivat laskea terapeuttisen tason alapuolelle.
- Tällaisia lääkevalmisteita ovat esimerkiksi varfariini, vorikonatsoli, diatsepaami, lansopratsoli, omepratsoli, esomepratsoli, pantopratsoli, tilidiini, tolbutamidi (ks. taulukko 1).
Vaikutuksen arvellaan olevan vähäisempi, kun letermoviiri annetaan suun kautta ilman siklosporiinia, kuin jos letermoviiri annetaan laskimoon siklosporiinin kanssa tai ilman siklosporiinia tai jos se annetaan suun kautta siklosporiinin kanssa. Tämä on otettava huomioon, kun letermoviirin hoito-ohjelmaa muutetaan CYP2C9:n tai CYP2C19:n substraatin käytön aikana. Kehotetaan tutustumaan yllä oleviin induktiota koskeviin yleisiin tietoihin ja yhteisvaikutuksen ajankohtiin.
CYP2C8-entsyymin välityksellä metaboloituvat lääkevalmisteet
Letermoviiri estää CYP2C8-entsyymin toimintaa in vitro, mutta induktiopotentiaalinsa perusteella se voi myös indusoida CYP2C8-entsyymiä. Nettovaikutusta in vivo ei tunneta.
- Repaglinidi on esimerkki lääkevalmisteesta, joka metaboloituu pääasiassa CYP2C8-entsyymin välityksellä (ks. taulukko 1). Repaglinidin ja letermoviirin samanaikaista käyttöä ei suositella yhdessä siklosporiinin kanssa eikä ilman siklosporiinia.
Suoliston P-gp:n kuljettamat lääkevalmisteet
Letermoviiri on suoliston P-gp:n induktori. Letermoviiri saattaa pienentää samanaikaisesti annettujen suoliston P-gp:n merkittävässä määrin kuljettamien lääkevalmisteiden, kuten dabigatraanin ja sofosbuviirin, pitoisuuksia plasmassa kliinisesti merkittävästi.
CYP2B6:n tai UGT1A1:n välityksellä metaboloituvat tai BCRP:n tai OATP2B1:n kuljettamat lääkevalmisteet
Letermoviiri on yleinen induktori in vivo, mutta sen on myös havaittu estävän CYP2B6:n, UGT1A1:n, BCRP:n ja OATP2B1:n toimintaa in vitro. Nettovaikutusta in vivo ei tunneta. Siksi näiden entsyymien tai kuljettajien substraatteina toimivien lääkevalmisteiden pitoisuus plasmassa voi suurentua tai pienentyä, kun niitä annetaan yhdessä letermoviirin kanssa. Lisäseurantaa saatetaan suositella. Tämä on tarkistettava näiden lääkevalmisteiden valmistetiedoista.
- Esimerkki CYP2B6-entsyymin välityksellä metaboloituvista lääkevalmisteista on bupropioni.
- Esimerkkejä UGT1A1:n välityksellä metaboloituvista lääkevalmisteista ovat raltegraviiri ja dolutegraviiri.
- Esimerkkejä BCRP:n kuljettamista lääkevalmisteista ovat rosuvastatiini ja sulfasalatsiini.
- Esimerkki OATP2B1:n kuljettamista lääkevalmisteista on seliprololi.
Munuaisten orgaanisten anionien kuljettajaproteiini 3:n (OAT3:n) kuljettamat lääkevalmisteet
In vitro ‑tiedot osoittavat, että letermoviiri on OAT3:n estäjä, joten letermoviiri voi olla OAT3:n estäjä in vivo. OAT3:n kuljettamien lääkevalmisteiden pitoisuudet plasmassa saattavat suurentua.
- Esimerkkejä OAT3:n kuljettamista lääkevalmisteista ovat siprofloksasiini, tenofoviiri, imipeneemi ja silastatiini.
Yleistä
Jos samanaikaisesti käytettävien muiden lääkkeiden annoksia muutetaan letermoviirihoidon vuoksi, annoksia on säädettävä uudelleen, kun letermoviirihoito päättyy. Annoksen säätö saattaa olla tarpeen myös, jos antoreittiä muutetaan tai immunosuppressiivista lääkettä vaihdetaan.
Taulukossa 1 luetellaan varmistetut tai mahdollisesti kliinisesti merkittävät lääkevalmisteiden yhteisvaikutukset. Kuvatut yhteisvaikutukset perustuvat aikuisilla tehtyihin letermoviiritutkimuksiin tai ne ovat ennakoituja yhteisvaikutuksia, joita saattaa esiintyä letermoviirihoidon aikana (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet, Farmakodynamiikka ja Farmakokinetiikka).
Taulukko 1:Yhteisvaikutukset muiden lääkevalmisteiden kanssa ja annossuositukset yhteiskäytössä. On huomioitava, että taulukko ei ole kattava vaan antaa esimerkkejä kliinisesti merkityksellisistä yhteisvaikutuksista. Tietoja lääkeaineiden yhteisvaikutuksista on myös yleisessä tekstiosuudessa edellä.
| Ellei toisin mainita, yhteisvaikutustutkimukset on tehty aikuisilla käyttäen suun kautta annettavaa letermoviiria ilman siklosporiinia. On otettava huomioon, että yhteisvaikutusten mahdollisuus ja kliiniset seurausvaikutukset saattavat olla erilaisia riippuen siitä, annetaanko letermoviiri suun kautta vai laskimoon ja käytetäänkö samanaikaisesti siklosporiinia. Jos antoreittiä muutetaan tai jos immunosuppressiivista lääkettä vaihdetaan, on perehdyttävä uudelleen yhteiskäyttöä koskeviin suosituksiin. |
| Samanaikaisesti annettu lääkevalmiste | Vaikutus pitoisuuteen† AUC- ja Cmax-keskiarvojen suhde (90 %:n luottamusväli) (todennäköinen vaikutusmekanismi) | Suositukset yhteiskäytöstä letermoviirin kanssa | |
|---|---|---|---|
| Antibiootit | |||
| nafsilliini | Yhteisvaikutuksia ei ole tutkittu. Odotetut: (P‑gp:n/UGT:n induktio) | Nafsilliini saattaa pienentää letermoviirin pitoisuuksia plasmassa. Letermoviirin ja nafsilliinin samanaikaista käyttöä ei suositella. | |
| Sienilääkkeet | |||
flukonatsoli (400 mg kerta-annoksena) / letermoviiri (480 mg kerta-annoksena) | ↔ flukonatsoli ↔ letermoviiri
Yhteisvaikutuksia vakaassa tilassa ei ole tutkittu. | Annosta ei tarvitse muuttaa.
| |
itrakonatsoli (200 mg kerran vuorokaudessa suun kautta) / letermoviiri (480 mg kerran vuorokaudessa suun kautta) | ↔ itrakonatsoli ↔ letermoviiri | Annosta ei tarvitse muuttaa. | |
posakonatsoli‡ (300 mg kerta-annoksena) / letermoviiri (480 mg/vrk) | ↔ posakonatsoli AUC 0,98 (0,82; 1,17) Cmax 1,11 (0,95; 1,29) | Annosta ei tarvitse muuttaa.
| |
vorikonatsoli‡ (200 mg kahdesti vuorokaudessa) / letermoviiri (480 mg/vrk) | ↓ vorikonatsoli (CYP2C9/19:n induktio)
| Jos yhteiskäyttö on välttämätöntä, suositellaan vorikonatsolin terapeuttisen tason seurantaa 2 ensimmäisen viikon ajan letermoviirihoidon aloittamisen tai lopettamisen jälkeen tai kun letermoviirin antoreittiä on muutettu tai immunosuppressiivinen lääke on vaihdettu. | |
| Mykobakteerilääkkeet | |||
| rifabutiini | Yhteisvaikutuksia ei ole tutkittu. (P-gp:n/UGT:n induktio) | Rifabutiini saattaa pienentää letermoviirin pitoisuuksia plasmassa. Letermoviirin ja rifabutiinin samanaikaista käyttöä ei suositella. | |
| rifampisiini | Rifampisiinin käyttö toistuvina annoksina pienentää letermoviirin pitoisuuksia plasmassa. Letermoviirin ja rifampisiinin samanaikaista käyttöä ei suositella. | ||
| (600 mg kerta-annoksena suun kautta) / letermoviiri (480 mg kerta-annoksena suun kautta) | ↔ letermoviiri (OATP1B1/3:n ja/tai P‑gp:n esto) | ||
| (600 mg kerta-annoksena laskimoon) / letermoviiri (480 mg kerta-annoksena suun kautta) | ↔ letermoviiri (OATP1B1/3:n ja/tai P‑gp:n esto) | ||
| (600 mg kerran vuorokaudessa suun kautta) / letermoviiri (480 mg kerran vuorokaudessa suun kautta) | ↓ letermoviiri (OATP1B1/3:n ja/tai P‑gp:n eston ja P‑gp:n/UGT:n induktion summa) | ||
| (600 mg kerran vuorokaudessa suun kautta (24 tuntia rifampisiinin jälkeen))§ / letermoviiri (480 mg kerran vuorokaudessa suun kautta) | ↓ letermoviiri (P‑gp:n/UGT:n induktio) | ||
| Psykoosilääkkeet | |||
| tioridatsiini | Yhteisvaikutuksia ei ole tutkittu. (P‑gp:n/UGT:n induktio) | Tioridatsiini saattaa pienentää letermoviirin pitoisuuksia plasmassa. Letermoviirin ja tioridatsiinin samanaikaista käyttöä ei suositella. | |
| Endoteliiniantagonistit | |||
| bosentaani | Yhteisvaikutuksia ei ole tutkittu. (P-gp:n/UGT:n induktio) | Bosentaani saattaa pienentää letermoviirin pitoisuuksia plasmassa. Letermoviirin ja bosentaanin samanaikaista käyttöä ei suositella. | |
| Viruslääkkeet | |||
asikloviiri‡ (400 mg kerta-annoksena) / letermoviiri (480 mg/vrk) | ↔ asikloviiri AUC 1,02 (0,87; 1,2) Cmax 0,82 (0,71; 0,93) | Annosta ei tarvitse muuttaa.
| |
valasikloviiri
| Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↔ valasikloviiri | Annosta ei tarvitse muuttaa. | |
| Rohdosvalmisteet | |||
| Mäkikuisma (Hypericum perforatum) | Yhteisvaikutuksia ei ole tutkittu. (P‑gp:n/UGT:n induktio) | Mäkikuisma saattaa pienentää letermoviirin pitoisuuksia plasmassa. Letermoviirin ja mäkikuisman samanaikainen käyttö on vasta-aiheista. | |
| HIV-lääkevalmisteet | |||
| efavirentsi | Yhteisvaikutuksia ei ole tutkittu. ↑ tai ↓ efavirentsi | Efavirentsi saattaa pienentää letermoviirin pitoisuuksia plasmassa. Letermoviirin ja efavirentsin samanaikaista käyttöä ei suositella. | |
| etraviriini, nevirapiini, ritonaviiri, lopinaviiri | Yhteisvaikutuksia ei ole tutkittu. (P‑gp:n/UGT:n induktio) | Nämä viruslääkkeet saattavat pienentää letermoviirin pitoisuuksia plasmassa. Letermoviirin samanaikaista käyttöä näiden viruslääkkeiden kanssa ei suositella. | |
| HMG‑CoA-reduktaasin estäjät | |||
atorvastatiini‡ (20 mg kerta-annoksena) / letermoviiri (480 mg/vrk) | ↑ atorvastatiini (CYP3A:n, OATP1B1/3:n esto) | Statiiniin liittyvien haittatapahtumien, kuten myopatian, mahdollista ilmaantumista on seurattava tarkoin. Atorvastatiinin annos ei saa olla yli 20 mg/vrk, kun sitä annetaan yhdessä letermoviirin kanssa#. Asiaa ei ole tutkittu, mutta plasman atorvastatiinipitoisuuden arvellaan suurenevan enemmän, jos letermoviiria annetaan yhdessä siklosporiinin kanssa kuin jos letermoviiria annetaan ilman siklosporiinia. Atorvastatiinia ei saa käyttää, jos letermoviiria annetaan yhdessä siklosporiinin kanssa. | |
| simvastatiini, pitavastatiini, rosuvastatiini | Yhteisvaikutuksia ei ole tutkittu. (CYP3A:n, OATP1B1/3:n esto) | Letermoviiri saattaa suurentaa olennaisesti näiden statiinien pitoisuuksia plasmassa. Yhteiskäyttöä pelkän letermoviirin kanssa ei suositella. Näitä statiineja ei saa käyttää, jos letermoviiria annetaan yhdessä siklosporiinin kanssa. | |
| fluvastatiini, pravastatiini | Yhteisvaikutuksia ei ole tutkittu. (OATP1B1/3:n ja/tai BCRP:n esto) | Letermoviiri saattaa suurentaa statiinin pitoisuutta plasmassa. Statiiniannoksen pienentäminen saattaa olla tarpeen, kun letermoviiria annetaan yhtaikaa näiden statiinien kanssa#. Statiiniin liittyvien haittatapahtumien, kuten myopatian, mahdollista ilmaantumista on seurattava tarkoin. Jos letermoviiria annetaan yhdessä siklosporiinin kanssa, pravastatiinin käyttöä ei suositella ja fluvastatiinin annosta voi olla tarpeen pienentää#. Statiiniin liittyvien haittatapahtumien, kuten myopatian, mahdollista ilmaantumista on seurattava tarkoin. | |
| Immunosuppressiiviset aineet | |||
siklosporiini (50 mg kerta-annoksena) / letermoviiri (240 mg/vrk) | ↑ siklosporiini AUC 1,66 (1,51; 1,82) Cmax 1,08 (0,97; 1,19) (CYP3A:n esto) | Jos letermoviirihoitoa annetaan samanaikaisesti siklosporiinin kanssa, letermoviiriannos on laskettava tasolle 240 mg kerran vuorokaudessa aikuisilla (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka) ja vähintään 30 kg painavilla pediatrisilla potilailla (ks. kohta Annostus ja antotapa). Jos suun kautta annettavaa letermoviiria annetaan samanaikaisesti siklosporiinin kanssa alle 30 kg painaville pediatrisille potilaille, annosta on pienennettävä (ks. kohta Annostus ja antotapa). Siklosporiinin pitoisuutta kokoveressä on seurattava tiheästi hoidon aikana, kun letermoviirin antoreittiä muutetaan ja letermoviirihoitoa lopetettaessa, ja siklosporiinin annosta on muutettava sen mukaisesti#. | |
siklosporiini (200 mg kerta-annoksena) / letermoviiri (240 mg/vrk) | ↑ letermoviiri (OATP1B1/3:n esto) | ||
mykofenolaattimofetiili (1 g kerta-annoksena) / letermoviiri (480 mg/vrk) | ↔mykofenolihappo AUC 1,08 (0,97; 1,20) Cmax 0,96 (0,82; 1,12) ↔ letermoviiri AUC 1,18 (1,04; 1,32) Cmax 1,11 (0,92; 1,34) | Annosta ei tarvitse muuttaa.
| |
sirolimuusi‡ (2 mg kerta-annoksena) / letermoviiri (480 mg/vrk) | ↑ sirolimuusi AUC 3,40 (3,01; 3,85) Cmax 2,76 (2,48; 3,06) (CYP3A:n esto) Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↔ letermoviiri | Sirolimuusin pitoisuutta kokoveressä on seurattava tiheästi hoidon aikana, kun letermoviirin antoreittiä muutetaan ja letermoviirihoitoa lopetettaessa, ja sirolimuusin annosta on muutettava sen mukaisesti#. Sirolimuusipitoisuuden tiheää seurantaa suositellaan, kun siklosporiini lisätään letermoviirihoitoon tai kun siklosporiinin käyttö lopetetaan. Kun letermoviiria annetaan yhdessä siklosporiinin kanssa, myös sirolimuusin valmistetiedoista on tarkistettava annostussuositukset, jotka koskevat sirolimuusin käyttöä yhdessä siklosporiinin kanssa. Sirolimuusin pitoisuus saattaa suurentua enemmän, jos letermoviiria annetaan yhdessä siklosporiinin kanssa, kuin jos letermoviiria annetaan ilman siklosporiinia. | |
takrolimuusi (5 mg kerta-annoksena) / letermoviiri (480 mg/vrk) | ↑ takrolimuusi
| Takrolimuusin pitoisuutta kokoveressä on seurattava tiheästi hoidon aikana, kun letermoviirin antoreittiä muutetaan ja letermoviirihoitoa lopetettaessa, ja takrolimuusin annosta on muutettava sen mukaisesti#. | |
takrolimuusi (5 mg kerta-annoksena) / letermoviiri (80 mg kahdesti vuorokaudessa) | ↔ letermoviiri AUC 1,02 (0,97; 1,07) Cmax 0,92 (0,84; 1,00) | ||
| Ehkäisytabletit | |||
etinyyliestradioli (EE) (0,03 mg) / levonorgestreeli (LNG)‡ (0,15 mg) kerta-annoksena / letermoviiri (480 mg/vrk) | ↔ EE AUC 1,42 (1,32; 1,52) Cmax 0,89 (0,83; 0,96) ↔ LNG AUC 1,36 (1,30; 1,43) Cmax 0,95 (0,86; 1,04) | Annosta ei tarvitse muuttaa.
| |
| Muut systeemisesti vaikuttavat ehkäisytableteissa käytettävät steroidit | Riskinä ↓ ehkäisytableteissa käytettävien steroidien kohdalla | Letermoviiri saattaa pienentää muiden ehkäisytableteissa käytettävien steroidien pitoisuuksia plasmassa, mikä vaikuttaa niiden tehoon. Suun kautta otettavia ehkäisyvalmisteita käytettäessä on valittava etinyyliestradiolia ja levonorgestreelia sisältäviä valmisteita riittävän ehkäisytehon varmistamiseksi. | |
| Diabeteslääkkeet | |||
| repaglinidi | Yhteisvaikutuksia ei ole tutkittu. (CYP2C8:n induktio, CYP2C8:n ja OATP1B:n esto) | Letermoviiri saattaa suurentaa tai pienentää repaglinidin pitoisuuksia plasmassa. (Nettovaikutusta ei tunneta.) Yhteiskäyttöä ei suositella. Plasman repaglinidipitoisuuden arvellaan suurenevan, jos letermoviiria annetaan yhdessä siklosporiinin kanssa, mikä johtuu siklosporiinin OATP1B:n estoa voimistavasta vaikutuksesta. Yhteiskäyttöä ei suositella#. | |
| glyburidi | Yhteisvaikutuksia ei ole tutkittu. (OATP1B1/3:n esto, CYP3A:n esto, CYP2C9:n induktio) | Letermoviiri saattaa suurentaa glyburidin pitoisuuksia plasmassa.
Glukoosipitoisuuksien tiheää seurantaa suositellaan 2 ensimmäisen viikon ajan letermoviirihoidon aloittamisen tai lopettamisen jälkeen ja kun letermoviirin antoreittiä on muutettu.
Jos letermoviiria annetaan yhdessä siklosporiinin kanssa, annostussuositukset on tarkistettava myös glyburidin valmistetiedoista. | |
| Epilepsialääkkeet (ks. myös teksti) | |||
| karbamatsepiini, fenobarbitaali | Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↓ letermoviiri (P‑gp:n/UGT:n induktio) | Karbamatsepiini ja fenobarbitaali saattavat pienentää letermoviirin pitoisuuksia plasmassa. Letermoviirin ja karbamatsepiinin tai fenobarbitaalin samanaikaista käyttöä ei suositella. | |
fenytoiini
| Yhteisvaikutuksia ei ole tutkittu. ↓ fenytoiini (CYP2C9/19:n induktio)
| Fenytoiini saattaa pienentää letermoviirin pitoisuuksia plasmassa.
Letermoviiri saattaa pienentää fenytoiinin pitoisuuksia plasmassa.
Letermoviirin ja fenytoiinin samanaikaista käyttöä ei suositella. | |
| Oraaliset antikoagulantit | |||
| varfariini | Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↓ varfariini (CYP2C9:n induktio) | Letermoviiri saattaa pienentää varfariinin pitoisuuksia plasmassa. INR-arvoja (International Normalised Ratio) on seurattava tiheästi, kun varfariinia annetaan samanaikaisesti letermoviirin kanssa#. Seurantaa suositellaan 2 ensimmäisen viikon ajan letermoviirihoidon aloittamisen tai lopettamisen jälkeen ja kun letermoviirin antoreittiä on muutettu tai immunosuppressiivinen lääke on vaihdettu. | |
| dabigatraani | Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↓ dabigatraani (suoliston P-gp:n induktio) | Letermoviiri saattaa pienentää dabigatraanin pitoisuuksia plasmassa ja heikentää dabigatraanin tehoa. Dabigatraanin samanaikaista käyttöä on vältettävä, koska se voi johtaa dabigatraanin tehon heikkenemiseen. Dabigatraania ei saa käyttää, jos letermoviiria annetaan yhdessä siklosporiinin kanssa. | |
| Rauhoittavat lääkkeet | |||
midatsolaami (1 mg kerta-annoksena laskimoon) / letermoviiri (240 mg kerran vuorokaudessa suun kautta)
midatsolaami (2 mg kerta-annoksena suun kautta) / letermoviiri (240 mg kerran vuorokaudessa suun kautta)
| ↑ midatsolaami
Suun kautta: | Potilaan kliinistä tilaa on seurattava tarkoin hengityslaman ja/tai pitkittyneen sedaation havaitsemiseksi, kun letermoviiria käytetään yhtaikaa midatsolaamin kanssa. Midatsolaamiannoksen säätämistä on harkittava #. Midatsolaamin pitoisuus plasmassa saattaa suurentua enemmän, kun midatsolaamia annetaan suun kautta yhdessä letermoviirin kanssa kliinisenä annoksena, kuin tutkittua annosta käytettäessä. | |
| Opioidiagonistit | |||
| Esimerkiksi: alfentaniili, fentanyyli | Yhteisvaikutuksia ei ole tutkittu. (CYP3A:n esto) | Näiden lääkevalmisteiden käyttöön liittyviä haittavaikutuksia on suositeltavaa tarkkailla tiheästi yhteiskäytön aikana. CYP3A:n metaboloimien opioidien annosta voi olla tarpeen muuttaa# (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Tarkkailua suositellaan myös, jos antoreittiä muutetaan. CYP3A:n metaboloimien opioidien pitoisuudet plasmassa saattavat suurentua enemmän, jos letermoviiria annetaan yhdessä siklosporiinin kanssa. Potilaan kliinistä tilaa on seurattava tarkoin hengityslaman ja/tai pitkittyneen sedaation havaitsemiseksi, kun letermoviiria käytetään yhtaikaa siklosporiinin ja alfentaniilin tai fentanyylin kanssa. Kehotetaan perehtymään asianomaisten valmisteiden valmistetietoihin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). | |
| Rytmihäiriölääkkeet | |||
| amiodaroni | Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↑ amiodaroni (ensisijaisesti CYP3A:n esto ja CYP2C8:n esto tai induktio) | Letermoviiri saattaa suurentaa amiodaronin pitoisuuksia plasmassa. Amiodaroniin liittyviä haittavaikutuksia on suositeltavaa tarkkailla tiheästi yhteiskäytön aikana. Kun amiodaronia annetaan yhdessä letermoviirin kanssa, amiodaronin pitoisuuksia on seurattava säännöllisesti#. | |
| kinidiini | Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↑ kinidiini | Letermoviiri saattaa suurentaa kinidiinin pitoisuuksia plasmassa. Potilaan kliinistä tilaa on seurattava tarkoin, kun letermoviiria annetaan yhtaikaa kinidiinin kanssa. Kehotetaan perehtymään asianomaisten valmisteiden valmistetietoihin#. | |
| Sydän- ja verisuonisairauksien lääkkeet | |||
digoksiini‡ (0,5 mg kerta-annoksena) / letermoviiri (240 mg kahdesti vuorokaudessa) | ↔ digoksiini AUC 0,88 (0,80; 0,96) Cmax 0,75 (0,63; 0,89) (P-gp:n induktio) | Annosta ei tarvitse muuttaa.
| |
| Protonipumpun estäjät | |||
omepratsoli
| Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↓ omepratsoli (CYP2C19:n induktio) Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↔ letermoviiri | Letermoviiri saattaa pienentää CYP2C19:n substraattien pitoisuuksia plasmassa. Kliinisen tilan seuranta ja annoksen säätäminen saattavat olla tarpeen. | |
| pantopratsoli | Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↓ pantopratsoli (todennäköinen syy CYP2C19:n induktio) Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↔ letermoviiri | Letermoviiri saattaa pienentää CYP2C19:n substraattien pitoisuuksia plasmassa. Kliinisen tilan seuranta ja annoksen säätäminen saattavat olla tarpeen. | |
| Valvetilaa ylläpitävät lääkkeet | |||
| modafiniili | Yhteisvaikutuksia ei ole tutkittu. Odotetut: ↓ letermoviiri (P‑gp:n/UGT:n induktio) | Modafiniili saattaa pienentää letermoviirin pitoisuuksia plasmassa. Letermoviirin ja modafiniilin samanaikaista käyttöä ei suositella. | |
| * Tämä taulukko ei kata kaikkia mahdollisia yhteisvaikutuksia. †↓ = pienenee, ↑ = suurenee ↔ = ei kliinisesti merkittävää muutosta ‡ Yksisuuntainen yhteisvaikutustutkimus, jossa arvioitiin letermoviirin vaikutusta samanaikaisesti annettuihin lääkevalmisteisiin. § Nämä tiedot koskevat rifampisiinin vaikutusta letermoviiriin 24 tunnin kuluttua viimeisen rifampisiiniannoksen ottamisesta. # Kehotetaan tutustumaan näiden valmisteiden valmistetietoihin. | |||
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisilla tehdyissä tutkimuksissa.
Raskaus ja imetys
Raskaus
Letermoviirin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Letermoviirin käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyykö letermoviiri ihmisillä äidinmaitoon. Olemassa olevat farmakodynaamiset/toksikologiset tiedot koe-eläimistä ovat osoittaneet letermoviirin erittyvän maitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida sulkea pois. On päätettävä, lopetetaanko imetys vai pidättäydytäänkö letermoviirihoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Naarasrottien hedelmällisyyteen kohdistuvia vaikutuksia ei havaittu. Urosrotilla havaittiin korjautumattomia kiveksiin kohdistuvia haittavaikutuksia ja hedelmällisyyden heikentymistä, mutta samaa vaikutusta ei havaittu uroshiirillä eikä urosapinoilla (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Letermoviirilla saattaa olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Joillakin potilailla on esiintynyt letermoviirihoidon aikana väsymystä ja huimausta, jotka voivat vaikuttaa potilaan ajokykyyn ja koneidenkäyttökykyyn (ks. kohta Haittavaikutukset).
Haittavaikutukset
Tiivistelmä turvallisuustiedoista
Letermoviirin turvallisuusarvio perustui kolmeen vaiheen 3 kliiniseen tutkimukseen.
Kantasolusiirto
P001-tutkimuksessa 565:lle kantasolusiirron saaneelle aikuiselle potilaalle annettiin joko letermoviiria tai lumevalmistetta viikolle 14 asti kantasolusiirron jälkeen ja hoidon turvallisuutta seurattiin viikolle 24 kantasolusiirron jälkeen (ks. kohta Farmakodynamiikka). Yleisimmin raportoidut haittavaikutukset, joita esiintyi letermoviiriryhmässä vähintään 1 prosentilla potilaista ja yleisemmin kuin lumeryhmässä, olivat: pahoinvointi (7,2 %), ripuli (2,4 %) ja oksentelu (1,9 %). Yleisimmin raportoidut letermoviirihoidon keskeyttämiseen johtaneet haittavaikutukset olivat: pahoinvointi (1,6 %), oksentelu (0,8 %) ja vatsakipu (0,5 %).
P040-tutkimuksessa 218:lle kantasolusiirron saaneelle aikuiselle potilaalle annettiin joko letermoviiria tai lumevalmistetta viikosta 14 (noin 100 vuorokauden kohdalta) viikolle 28 asti (noin 200 vuorokauteen asti) kantasolusiirron jälkeen ja hoidon turvallisuutta seurattiin viikolle 48 kantasolusiirron jälkeen (ks. kohta Farmakodynamiikka). Ilmoitetut haittavaikutukset vastasivat P001-tutkimuksessa kuvattua letermoviirin turvallisuusprofiilia.
Munuaisensiirto
P002-tutkimuksessa 292 aikuiselle munuaisensiirtopotilaalle annettiin letermoviirihoitoa viikolle 28 asti (noin 200 vuorokauteen asti) siirron jälkeen (ks. kohta Farmakodynamiikka).
Haittavaikutustaulukko
Seuraavia haittavaikutuksia havaittiin aikuisilla potilailla, jotka saivat letermoviirihoitoa kliinisissä tutkimuksissa. Haittavaikutukset on lueteltu alla elinjärjestelmäluokan ja yleisyyden mukaan. Yleisyysluokat ovat: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1 / 1 000, < 1/100), harvinainen (≥ 1 / 10 000, < 1 / 1 000) tai hyvin harvinainen (< 1 / 10 000).
Taulukko 2: Letermoviirihoidon yhteydessä todetut haittavaikutukset
| Yleisyys | Haittavaikutukset | |
| Immuunijärjestelmä | ||
| Melko harvinainen | yliherkkyys | |
| Aineenvaihdunta ja ravitsemus | ||
| Melko harvinainen | heikentynyt ruokahalu | |
| Hermosto | ||
| Melko harvinainen | makuhäiriö, päänsärky | |
| Kuulo ja tasapainoelin | ||
| Melko harvinainen | huimaus | |
| Ruoansulatuselimistö | ||
| Yleinen | pahoinvointi, ripuli, oksentelu | |
| Melko harvinainen | vatsakipu | |
| Maksa ja sappi | ||
| Melko harvinainen | kohonnut alaniiniaminotransferaasiarvo, kohonnut aspartaattiaminotransferaasiarvo | |
| Luusto, lihakset ja sidekudos | ||
| Melko harvinainen | lihaskouristukset | |
| Munuaiset ja virtsatiet | ||
| Melko harvinainen | kohonnut veren kreatiniiniarvo | |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Melko harvinainen | väsymys, perifeerinen edeema | |
Pediatriset potilaat
Letermoviirin turvallisuusarviointi pediatrisilla potilailla (vastasyntyneistä enintään 18-vuotiaisiin) perustui vaiheen 2b kliiniseen tutkimukseen (P030). P030-tutkimuksessa 63:lle kantasolusiirron saaneelle potilaalle annettiin letermoviiria viikkoon 14 asti kantasolusiirron jälkeen. Potilaiden iät jakautuivat niin, että mukana oli 28 nuorta, 14 iältään 7 – < 12-vuotiasta lasta, 13 iältään 2 – < 7-vuotiasta lasta ja 8 alle 2-vuotiasta lasta (jälkimmäisistä 5 oli alle 1-vuotiaita). Haittavaikutukset vastasivat aikuisilla tehdyissä kliinisissä letermoviiritutkimuksissa havaittuja haittavaikutuksia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Letermoviirin yliannostuksesta ei ole kokemusta ihmisillä. Vaiheen 1 kliinisissä tutkimuksissa 86 terveelle aikuiselle tutkittavalle annettiin letermoviiria 720 – 1 440 mg/vrk enintään 14 vuorokauden ajan. Haittavaikutusprofiili oli samanlainen kuin käytettäessä kliinistä annostusta 480 mg/vrk. Letermoviirin yliannostukseen ei ole spesifistä vastalääkettä. Yliannostustapauksissa on tarkkailtava potilaan tilaa haittavaikutusten havaitsemiseksi ja aloitettava sopiva oireenmukainen hoito.
Ei tiedetä, poistuuko letermoviiri merkittävässä määrin systeemisestä verenkierrosta dialyysissä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Systeemiset viruslääkkeet, virukseen vaikuttavat lääkeaineet, ATC-koodi: J05AX18
Vaikutusmekanismi
Letermoviiri estää sytomegaloviruksen DNA:n terminaasikompleksin toimintaa, jota tarvitaan muodostuvien uusien virusten DNA:n pilkkoutumisessa ja pakkautumisessa. Letermoviiri häiritsee oikeanpituisten genomien muodostumista ja virionien kypsymistä.
Antiviraalinen teho
Letermoviirin EC50-arvojen mediaani sytomegaloviruksen kliinisiä isolaatteja vastaan infektion soluviljelmämallissa oli 2,1 nM (vaihteluväli = 0,7–6,1 nM, n = 74).
Viruksen resistenssi
Soluviljelmässä
Sytomegaloviruksen geenit UL51, UL56 ja UL89 koodaavat sytomegaloviruksen DNA:n terminaasientsyymin alayksiköitä. CMV-mutantteja, joiden herkkyys letermoviirille on alentunut, on todettu soluviljelmässä. Rekombinanteilla CMV-mutanteilla, jotka ilmentävät substituutioita, jotka paikantuvat pUL51-geeniin (P91S), pUL56-geeniin (C25F, S229F, V231A, V231L, V236A, T244K, T244R, L254F, L257F, L257I, F261C, F261L, F261S, Y321C, L328V, M329T, A365S, N368D) ja pUL89-geeniin (N320H, D344E), EC50-arvot olivat 1,6 – < 10 kertaa suuremmat kuin vastaavat arvot villin tyypin vertailuvirusta vastaan. Näillä substituutioilla ei todennäköisesti ole kliinistä merkitystä. Rekombinanteilla CMV-mutanteilla, jotka ilmentävät pUL51-substituutiota A95V tai pUL56-substituutioita N232Y, V236L, V236M, E237D, E237G, L241P, K258E, C325F, C325R, C325W, C325Y, R369G, R369M, R369S ja R369T, EC50-arvot olivat 10 – 9 300 kertaa suuremmat kuin vastaavat arvot villin tyypin vertailuvirusta vastaan. Osa näistä substituutioista on havaittu potilailla, joilla ehkäisevä hoito kliinisissä tutkimuksissa oli epäonnistunut (ks. seuraava kohta).
Kliinisissä tutkimuksissa
Vaiheen 2b tutkimuksessa letermoviiria verrattiin lumevalmisteeseen annostuksina 60, 120 tai 240 mg/vrk enintään 84 vuorokauden ajan 131:llä kantasolusiirron saaneella aikuisella potilaalla. Tässä tutkimuksessa analysoitiin DNA-sekvenssi UL56-geenin tietyltä alueelta (aminohapot 231–369) näytteistä, jotka saatiin 12:lta letermoviirihoitoa saaneelta potilaalta, joilla profylaksi oli epäonnistunut ja joilta saatiin näytteet analyysiä varten. Yhdellä potilaalla (joka sai annostusta 60 mg/vrk) oli letermoviirille resistentti genotyyppivariantti (GV) (V236M).
Vaiheen 3 tutkimuksessa (P001) tehtiin UL56- ja UL89-geenien koko koodaavien alueiden DNA-sekvenssianalyysit näytteistä, jotka saatiin koko analyysijoukon 40:ltä letermoviirihoitoa saaneelta aikuiselta potilaalta, joilla ehkäisevä hoito epäonnistui ja joilta saatiin näytteet analyysiä varten. Kahdella potilaalla todettiin letermoviirille resistenttejä genotyyppivariantteja; molemmilla oli pUL56-geeniin paikantuvia substituutioita. Toisella potilaalla oli V236M-substituutio ja toisella E237G-substituutio. Lisäksi yhdellä potilaalla, jolla oli havaittavissa olevaa sytomegaloviruksen DNA:ta lähtötilanteessa (ja jonka tietoja ei sen vuoksi sisällytetty koko analyysijoukkoon), oli pUL56-geenin substituutioita (C325W ja R369T), jotka havaittiin letermoviirihoidon lopettamisen jälkeen.
Vaiheen 3 tutkimuksessa (P040) tehtiin UL51-, UL56- ja UL89-geenien koko koodaavien alueiden DNA-sekvenssianalyysit näytteistä, jotka saatiin 32 aikuiselta tutkittavalta (hoitoryhmästä riippumatta), joilla ehkäisevä hoito epäonnistui tai jotka keskeyttivät hoidon varhain ja joiden veressä oli hoidon keskeyttämisen yhteydessä sytomegalovirusta (CMV-viremia). Tutkimuksessa ei todettu letermoviiriresistenssiin liittyviä substituutioita, jotka olisivat ylittäneet validoidun 5 %:n testausrajan.
Vaiheen 3 tutkimuksessa (P002) tehtiin UL51-, UL56- ja UL89-geenien koko koodaavien alueiden DNA-sekvenssianalyysit näytteistä, jotka saatiin 52:lta letermoviirihoitoa saaneelta aikuiselta tutkittavalta, joilla oli todettu sytomegaloviruksen aiheuttama tauti tai jotka keskeyttivät hoidon varhain ja joilla oli hoidon keskeyttämisen yhteydessä CMV-viremia. Tutkimuksessa ei todettu letermoviiriresistenssiin liittyviä substituutioita, jotka olisivat ylittäneet validoidun 5 %:n testausrajan.
Vaiheen 2b tutkimuksessa (P030) tehtiin UL51-, UL56- ja UL89-geenien koko koodaavien alueiden DNA-sekvenssianalyysit näytteistä, jotka saatiin 10:ltä letermoviirihoitoa saaneelta pediatriselta potilaalta vastaanottokäynnillä, jolla sytomegalovirusinfektio arvioitiin. Kahdella tutkittavalla todettiin yhteensä kaksi letermoviiriresistenssiin liittyvää substituutiota, jotka molemmat paikantuivat pUL56-geeniin. Toisella tutkittavalla oli substituutio R369S ja toisella substituutio C325W.
Ristiresistenssi
Ristiresistenssi ei ole todennäköistä sellaisten lääkevalmisteiden kanssa, joilla on erilainen vaikutusmekanismi. Letermoviiri tehoaa hyvin viruspopulaatioihin, joissa esiintyy substituutioita, jotka aiheuttavat resistenssiä sytomegaloviruksen DNA-polymeraasin estäjiä (gansikloviiria, sidofoviiria ja foskarneettia) kohtaan. Joukko rekombinantteja sytomegaloviruskantoja, joilla oli letermoviiriresistenssiä aiheuttavia substituutioita, oli hyvin herkkä sidofoviirille, foskarneetille ja gansikloviirille, lukuun ottamatta rekombinanttia kantaa, jossa oli pUL56 E237G -substituutio, jonka vaikutuksesta gansikloviiriherkkyys pienenee 2,1-kertaisesti villiin tyyppiin verrattuna.
Sydämen sähköinen toiminta
Enintään 960 mg:n annoksina laskimoon annetun letermoviirin vaikutusta QTc-aikaan verrattiin lumevalmisteeseen ja vaikuttavaan vertailuaineeseen (moksifloksasiini 400 mg suun kautta) kerta-annoksilla tehdyssä satunnaistetussa 4-jaksoisessa vaihtovuoroisessa perusteellisessa QT-tutkimuksessa, jossa oli mukana 38 tervettä aikuista tutkittavaa. Letermoviiri ei pidennä QTc-aikaa kliinisesti merkittävässä määrin laskimoon annetun 960 mg:n annoksen jälkeen, kun pitoisuus plasmassa on noin 2 kertaa suurempi kuin laskimoon annetun 480 mg:n annoksen jälkeen.
Kliininen teho ja turvallisuus
CMV-seropositiiviset allogeenisen kantasolusiirteen saaneet aikuiset
P001: Profylaktinen hoito viikolle 14 asti (noin 100 vuorokauteen asti) kantasolusiirron jälkeen
Letermoviirin tehoa sytomegalovirusinfektion taisytomegaloviruksen aiheuttaman taudin ehkäisyssä arvioitiin vaiheen 3 kaksoissokkoutetussa monikeskus-lumevertailututkimuksessa (P001) aikuisilla allogeenisen kantasolusiirteen saaneilla CMV-seropositiivisilla potilailla. Potilaat saivat satunnaistetusti (2:1) joko letermoviiria 480 mg kerran vuorokaudessa (annos laskettiin 240 mg:aan, jos potilas sai samanaikaisesti siklosporiinia) tai lumevalmistetta. Satunnaistaminen ositettiin tutkimuspaikan ja tutkimukseen otettaessa arvioidun sytomegaloviruksen uudelleenaktivoitumisriskin (suuri tai pieni riski) mukaan. Letermoviirihoito aloitettiin kantasolusiirron jälkeen (0.–28. päivänä kantasolusiirron jälkeen) ja sitä jatkettiin viikolle 14 kantasolusiirron jälkeen. Letermoviiria annettiin joko suun kautta tai laskimoon, ja letermoviiriannos oli sama antoreitistä riippumatta. Potilaiden tilaa seurattiin ensisijaisen tehoa mittaavan päätetapahtuman osalta viikolle 24 kantasolusiirron jälkeen, ja seurantaa jatkettiin vielä viikolle 48 asti.
Sytomegaloviruksen DNA-määriä seurattiin viikoittain viikolle 14 kantasolusiirron jälkeen ja sen jälkeen kahden viikon välein viikolle 24 kantasolusiirron jälkeen, ja jos veren CMV-DNA-määrän katsottiin olevan kliinisesti merkittävä, aloitettiin tavanomainen CMV-infektion ennakoiva hoito. Potilaiden seurantaa jatkettiin viikolle 48 kantasolusiirron jälkeen.
Tutkimukseen osallistuneista 565 potilaasta 373 sai letermoviiria (mukaan lukien 99 potilasta, jotka saivat vähintään yhden annoksen laskimoon) ja 192 sai lumevalmistetta (mukaan lukien 48 potilasta, jotka saivat vähintään yhden annoksen laskimoon). Letermoviirihoito aloitettiin 9 vuorokauden (mediaani) kuluttua kantasolusiirrosta. Lähtötilanteessa siirre oli tarttunut 37 prosentilla potilaista. Potilaiden mediaani-ikä oli 54 vuotta (vaihteluväli: 18–78 vuotta), 65 vuotta täyttäneitä potilaita oli 56 (15,0 %); 58 % oli miehiä, 82 % valkoihoisia, 10 % aasialaisia, 2 % mustaihoisia tai afrikkalaisia ja 7 % latinalaisamerikkalaisia. Lähtötilanteessa 50 % potilaista sai myeloablatiivista hoitoa, 52 % sai siklosporiinia ja 42 % takrolimuusia. Kantasolusiirron yleisimmät ensisijaiset syyt olivat akuutti myelooinen leukemia (38 %), myelodysplastinen oireyhtymä (15 %) ja lymfooma (13 %). Potilaista 12 % oli CMV-DNA-positiivisia lähtötilanteessa.
Lähtötilanteessa 31 prosentilla potilaista uudelleenaktivoitumisen riski oli suuri, mikä perustui vähintään yhteen seuraavista kriteereistä: HLA (human leukocyte antigen) ‑sopiva (sisarus)luovuttaja, jolla on vähintään yksi epäsopivuus jossakin seuraavista kolmesta HLA-geenilokuksesta: HLA-A, -B tai -DR; haploidenttinen luovuttaja; vapaaehtoinen rekisteriluovuttaja, jolla on vähintään yksi epäsopivuus jossakin seuraavista neljästä HLA-geenilokuksesta: HLA-A, -B, -C ja -DRB1; napaveren kantasoluilla tehty siirto; siirre, josta T-solut on poistettu ex vivo; systeemistä kortikosteroidihoitoa vaativa 2. asteen tai vaikeampi käänteishyljintä (GVHD).
Ensisijainen tehoa mittaava päätetapahtuma
P001-tutkimuksessa ensisijaisen tehoa mittaavan päätetapahtuman eli kliinisesti merkittävän sytomegalovirusinfektion kriteerit olivat sytomegalovirusinfektion ennakoivan hoidon aloittamista vaativa CMV-DNA-määrä veressä tai sytomegaloviruksen kohde-elimen sairauden ilmaantuminen. Käytettiin menettelyä, jossa keskeyttäminen luokiteltiin hoitotehon puuttumiseksi (Non-Completer = Failure (NC=F)). Tämä tarkoittaa, että jos potilas keskeytti tutkimukseen osallistumisen ennen kuin kantasolusiirrosta oli kulunut 24 viikkoa tai potilaasta ei saatu tulosta viikolla 24 kantasolusiirron jälkeen, hoito katsottiin tehottomaksi.
Letermoviiri oli tehokkaampi kuin lume ensisijaisen päätetapahtuman analyysissä, kuten taulukko 3 osoittaa. Arvioitu hoitojen välinen ero (−23,5 %) oli tilastollisesti merkitsevä (yksisuuntainen p-arvo < 0,0001).
Taulukko 3:P001: Tehon osoittavat tulokset kantasolusiirron saaneilla potilailla (keskeyttäminen luokiteltiin hoitotehon puuttumiseksi, koko analyysijoukko)
| Letermoviiri (N = 325) n (%) | Lume (N = 170) n (%) | |
| Parametri | ||
| Ensisijainen tehoa mittaava päätetapahtuma (Niiden potilaiden osuus, joilla profylaksi epäonnistui viikkoon 24 mennessä) | 122 (37,5) | 103 (60,6) |
| Epäonnistumisen syyt† | ||
| Kliinisesti merkittävä CMV-infektio | 57 (17,5) | 71 (41,8) |
| 52 (16,0) | 68 (40,0) |
| 5 (1,5) | 3 (1,8) |
| Keskeyttänyt tutkimukseen osallistumisen | 56 (17,2) | 27 (15,9) |
| Tulokset puuttuvat | 9 (2,8) | 5 (2,9) |
| Ositteen mukaan korjattu hoitojen välinen ero (letermoviiri – lume)§ | ||
| Ero (95 %:n luottamusväli) | −23,5 (−32,5; −14,6) | |
| p-arvo | < 0,0001 | |
| † Luokat ovat toisensa poissulkevia ja perustuvat luokkien väliseen hierarkiaan mainitussa järjestyksessä. § Hoitojen välisen prosentuaalisen vasteen eron 95 %:n luottamusvälit ja p-arvo laskettiin käyttäen ositteen mukaan korjattua Mantel–Haenszelin menetelmää, jossa ero painotettiin kummankin hoitohaaran otoskoon harmonisen keskiarvon mukaan ositekohtaisesti (suuri tai pieni riski). Tilastollisen merkitsevyyden kriteerinä käytettiin yksisuuntaista p-arvoa ≤ 0,0249. Koko analyysijoukko (FAS). Koko analyysijoukkoon kuuluvat satunnaistetut potilaat, jotka saivat vähintään yhden annoksen tutkimusvalmistetta, ja siitä on suljettu pois potilaat, joilla oli havaittavia määriä CMV-DNA:ta lähtötilanteessa. Puuttuvien arvojen käsitteleminen: Keskeyttäminen luokiteltiin hoitotehon puuttumiseksi. Tämä menettely tarkoittaa sitä, että hoito katsottiin tehottomaksi kaikissa tapauksissa, joissa potilaalla oli kliinisesti merkittävä CMV-infektio tai potilas keskeytti osallistumisensa tutkimukseen ennenaikaisesti tai hänestä ei saatu tuloksia viikon 24 käyntiin mennessä kantasolusiirron jälkeen. N = potilaiden lukumäärä kummassakin hoitoryhmässä. n (%) = potilaiden lukumäärä (prosenttiosuus) kussakin alaryhmässä. Huom.: Niiden potilaiden osuus, joilla sytomegaloviruksen DNA oli määritysrajan yläpuolella 1. päivänä ja joille kehittyi kliinisesti merkittävä CMV-infektio, oli letermoviiriryhmässä 64,6 % (31/48) ja lumeryhmässä 90,9 % (20/22) viikkoon 24 mennessä kantasolusiirron jälkeen. Arvioitu ero (eron 95 %:n luottamusväli) oli −26,1 % (−45,9 %; −6,3 %), ja nimellinen yksisuuntainen p-arvo oli < 0,0048. | ||
Tekijöitä, jotka liittyivät CMV-DNA:n esiintymiseen letermoviiria saaneiden potilaiden veressä, kun kantasolusiirrosta oli kulunut yli 14 viikkoa, olivat korkea sytomegaloviruksen uudelleenaktivoitumisen riski lähtötilanteessa, käänteishyljintä (GVHD), kortikosteroidien käyttö ja CMV-seronegatiivinen luovuttaja.
Kuva 1:P001: Kaplan–Meier-kuvaaja CMV-infektion ennakoivan hoidon aloittamisajankohdasta tai CMV:n kohde-elimen sairauden ilmaantumisesta kantasolusiirron saaneilla potilailla 24 viikon aikana kantasolusiirron jälkeen (koko analyysijoukko)
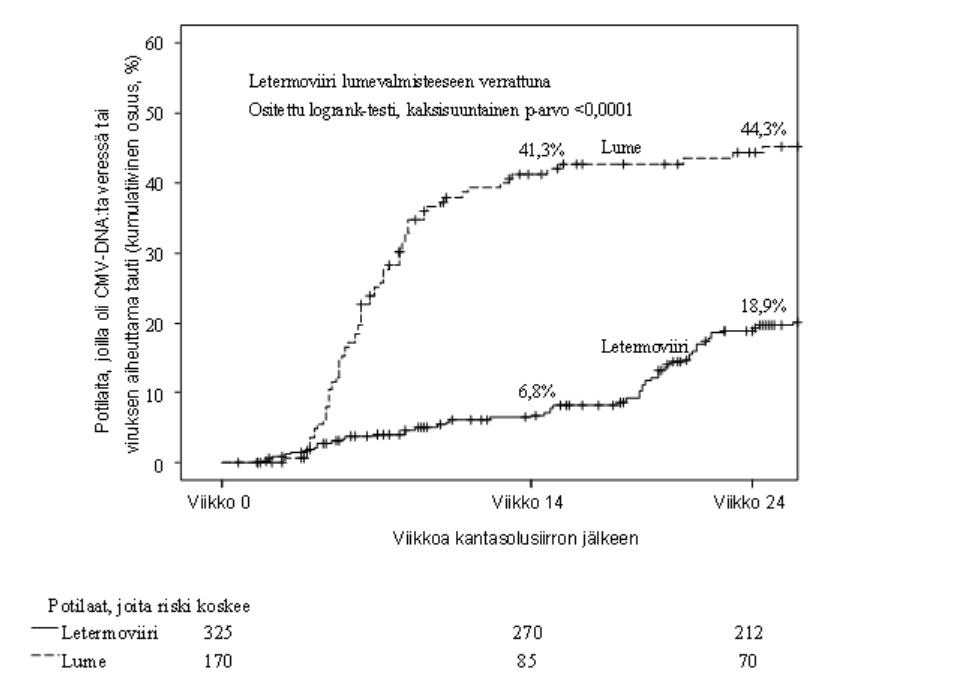
Siirteen tarttumisessa ja tarttumiseen kuluvassa ajassa ei ollut eroa letermoviiriryhmän ja lumeryhmän välillä.
Teho oli johdonmukaisesti parempi letermoviiria saaneilla potilailla kaikissa alaryhmissä, jotka perustuivat pieneen tai suureen sytomegaloviruksen uudelleenaktivoitumisen riskiin, esihoitoihin ja samanaikaisiin immunosuppressiivisiin hoitoihin (ks. kuva 2).
Kuva 2:P001: Forest plot ‑diagrammi niiden potilaiden osuudesta, joille aloitettiin CMV-infektion ennakoiva hoito tai joilla oli CMV:n kohde-elimen sairaus 24 viikon aikana kantasolusiirron jälkeen, valikoitujen alaryhmien mukaan (NC=F-menettely, koko analyysijoukko)
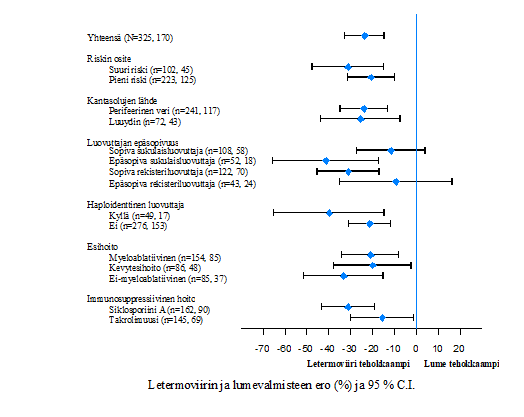
NC=F (Non-Completer=Failure). NC=F-menettely tarkoittaa, että jos potilas keskeytti tutkimukseen osallistumisen ennen kuin kantasolusiirrosta oli kulunut 24 viikkoa tai potilaasta ei saatu tulosta viikolla 24 kantasolusiirron jälkeen, hoito katsottiin tehottomaksi.
P040: Profylaktinen hoito viikosta 14 (noin 100 vuorokauden kohdalta) viikolle 28 asti (noin 200 vuorokauteen asti) kantasolusiirron jälkeen
Profylaktisen letermoviirihoidon pidentämisen (viikosta 14 [noin 100 vuorokaudesta] viikolle 28 asti [noin 200 vuorokauteen asti] kantasolusiirron jälkeen) tehoa potilailla, joilla oli myöhäisen sytomegalovirusinfektion ja viruksen aiheuttaman taudin riski, arvioitiin vaiheen 3 kaksoissokkoutetussa monikeskus-lumevertailututkimuksessa (P040) aikuisilla allogeenisen kantasolusiirteen saaneilla CMV-seropositiivisilla potilailla. Soveltuvat tutkittavat, jotka saivat profylaktisen letermoviirihoidon kokonaan eli noin 100 vuorokauteen asti kantasolusiirron jälkeen, satunnaistettiin (2:1) saamaan letermoviiria tai lumevalmistetta viikosta 14 viikolle 28 kantasolusiirron jälkeen. Tutkittavia seurattiin ensisijaisen tehoa mittaavan päätetapahtuman osalta viikolle 28 asti kantasolusiirron jälkeen, ja hoidon päättymisen jälkeen potilaiden seurantaa jatkettiin viikolle 48 kantasolusiirron jälkeen.
Hoidetuista 218 potilaasta 144 sai letermoviiria ja 74 sai lumevalmistetta. Tutkittavien mediaani-ikä oli 55 vuotta (vaihteluväli: 20–74 vuotta); 62 % oli miehiä, 79 % valkoihoisia, 11 % aasialaisia, 2 % mustaihoisia ja 10 % latinalaisamerikkalaisia. Siirron yleisimmät syyt olivat akuutti myelooinen leukemia (42 %), akuutti lymfaattinen leukemia (15 %) ja myelodysplastinen oireyhtymä (11 %).
Tutkimukseen otettaessa kaikilla tutkittavilla oli myöhäisen sytomegalovirusinfektion ja viruksen aiheuttaman taudin riskitekijöitä, ja 64 %:lla oli vähintään kaksi riskitekijää. Riskitekijöihin kuuluivat seuraavat: HLA-sopiva (sisarus)luovuttaja, jolla on vähintään yksi epäsopivuus jossakin seuraavista kolmesta HLA-geenilokuksesta: HLA-A, -B tai -DR; haploidenttinen luovuttaja; vapaaehtoinen rekisteriluovuttaja, jolla on vähintään yksi epäsopivuus jossakin seuraavista neljästä HLA-geenilokuksesta: HLA-A, -B, -C ja -DRB1; napaveren kantasoluilla tehty siirto; siirre, josta T-solut on poistettu ex vivo; potilaalle annettu antitymosyyttiglobuliini; potilaalle annettu alemtutsumabi; käytössä systeeminen prednisonihoito (tai vastaava), jonka annos oli ≥ 1 mg/painokilo vuorokaudessa.
Ensisijainen tehoa mittaava päätetapahtuma
Tutkimuksen P040 ensisijainen tehoa mittaava päätetapahtuma oli kliinisesti merkittävän sytomegalovirusinfektion ilmaantuvuus viikkoon 28 mennessä kantasolusiirron jälkeen. Kliinisesti merkittäväksi sytomegalovirusinfektioksi määriteltiin joko sytomegaloviruksen kohde-elimen sairauden ilmaantuminen tai sytomegalovirusinfektion ennakoivan hoidon aloittaminen veressä dokumentoidusti todetun sytomegaloviruksen sekä potilaan kliinisen tilan perusteella. Käytettiin hoitotehon havaitun puuttumisen (Observed Failure, OF) menettelyä, jossa hoito katsottiin tehottomaksi, jos potilaalle kehittyi kliinisesti merkittävä sytomegalovirusinfektio tai hän keskeytti osallistumisensa tutkimukseen suunniteltua aiemmin ja keskeyttämisen yhteydessä hänen veressään oli virusta.
Letermoviiri oli tehokkaampi kuin lume ensisijaisen päätetapahtuman analyysissä, kuten taulukko 4 osoittaa. Arvioitu hoitojen välinen ero (-16,1 %) oli tilastollisesti merkitsevä (yksisuuntainen p-arvo = 0,0005). Teho oli johdonmukaisesti parempi letermoviiria saaneilla potilailla eri alaryhmissä, jotka perustuivat potilaiden ominaisuuksiin (ikä, sukupuoli, etninen tausta) ja myöhäisen sytomegalovirusinfektion ja viruksen aiheuttaman taudin riskitekijöihin.
Taulukko 4: P040: Tehoon liittyvät tulokset kantasolusiirron saaneilla potilailla, joilla oli myöhäisen sytomegalovirusinfektion ja viruksen aiheuttaman taudin riski (hoitotehon havaitun puuttumisen menettely, koko analyysijoukko)
| Parametri | Letermoviiri (noin 200 vrk:n letermoviirihoito) (N = 144) n (%) | Lume (noin 100 vrk:n letermoviirihoito) (N = 74) n (%) |
| Hoitotehon havaittu puuttuminen* | 4 (2,8) | 14 (18,9) |
| Kliinisesti merkittävä CMV-infektio viikkoon 28 mennessä† | 2 (1,4) | 13 (17,6) |
| Ennakoivan hoidon aloittaminen veressä dokumentoidusti todetun CMV:n perusteella | 1 (0,7) | 11 (14,9) |
| CMV:n kohde-elimen sairaus | 1 (0,7) | 2 (2,7) |
| Keskeytti osallistumisensa tutkimukseen ennen viikkoa 28; keskeyttämisen yhteydessä veressä CMV:tä | 2 (1,4) | 1 (1,4) |
| Ositteen mukaan korjattu hoitojen välinen ero (letermoviiri [noin 200 vrk:n letermoviirihoito] - lume [noin 100 vrk:n letermoviirihoito])‡ | ||
| Ero (95 %:n luottamusväli) | -16,1 (-25,8, -6,5) 0,0005 | |
| p-arvo | ||
| * Luokat ovat toisensa poissulkevia ja perustuvat luokkien väliseen hierarkiaan mainitussa järjestyksessä. † Kliinisesti merkittäväksi sytomegalovirusinfektioksi määriteltiin joko sytomegaloviruksen kohde-elimen sairauden ilmaantuminen (dokumentoidusti tai todennäköisesti) tai sytomegalovirusinfektion ennakoivan hoidon aloittaminen veressä dokumentoidusti todetun sytomegaloviruksen sekä potilaan kliinisen tilan perusteella. ‡ Hoitojen välisen prosentuaalisen vasteen eron 95 %:n luottamusvälit ja p-arvo laskettiin käyttäen ositteen mukaan korjattua Mantel–Haenszelin menetelmää, jossa ero painotettiin kummankin hoitohaaran otoskoon harmonisen keskiarvon mukaan ositekohtaisesti (haploidenttinen tai ei-haploidenttinen luovuttaja). Tilastollisen merkitsevyyden kriteerinä käytettiin yksisuuntaista p-arvoa ≤ 0,0249. Puuttuvien arvojen käsitteleminen: Hoitotehon havaitun puuttumisen menettely. Tämä menettely tarkoittaa sitä, että hoito katsottiin tehottomaksi kaikissa tapauksissa, joissa potilaalla oli kliinisesti merkittävä CMV-infektio tai potilas keskeytti osallistumisensa tutkimukseen ennenaikaisesti ja hänen veressään oli keskeyttämisen yhteydessä CMV:tä viikon 14 jälkeen (noin 100 vuorokauden jälkeen) viikkoon 28 mennessä (noin 200 vuorokauteen mennessä) kantasolusiirron jälkeen. N = potilaiden lukumäärä kummassakin hoitoryhmässä. n (%) = potilaiden lukumäärä (prosenttiosuus) kussakin alaryhmässä. | ||
P002: CMV-seronegatiiviset aikuiset, jotka olivat saaneet munuaissiirteen CMV-seropositiiviselta luovuttajalta
Letermoviiriprofylaksin tehoa sytomegaloviruksen aiheuttaman taudin ehkäisyssä munuaissiirteen saaneilla potilailla arvioitiin vaiheen 3 kaksoissokkoutetussa, aktiivikontrolloidussa, vähintään samanveroisuutta arvioineessa monikeskustutkimuksessa (P002) aikuisilla munuaissiirteen saaneilla potilailla, jotka kuuluivat suuren riskin ryhmään (siirteen vastaanottaja oli CMV-seronegatiivinen ja luovuttaja CMV-seropositiivinen). Potilaat satunnaistettiin (1:1) saamaan joko letermoviiria tai valgansikloviiria. Letermoviiri annettiin samanaikaisesti asikloviirin kanssa. Valgansikloviiri annettiin samanaikaisesti asikloviiria muistuttavan lumelääkkeen kanssa. Satunnaistaminen ositettiin sen mukaan, oliko potilas saanut induktiohoidon aikana voimakkaasti sytolyyttistä, lymfosyyttejä tuhoavaa immunoterapiaa vai ei. Letermoviiri- tai valgansikloviirihoito aloitettiin munuaisensiirron jälkeen päivänä 0–7, ja sitä jatkettiin viikolle 28 asti (noin 200 vuorokauteen asti) siirron jälkeen. Potilaita seurattiin viikolle 52 siirron jälkeen.
Hoidetuista 589 potilaasta 292 sai letermoviiria ja 297 sai valgansikloviiria. Tutkittavien mediaani-ikä oli 51 vuotta (vaihteluväli: 18–82 vuotta); 72 % oli miehiä, 84 % valkoihoisia, 2 % aasialaisia, 9 % mustaihoisia ja 17 % latinalaisamerikkalaisia, ja 60 % oli saanut munuaissiirteen kuolleelta luovuttajalta. Siirron yleisimmät syyt olivat synnynnäinen kystinen munuaissairaus (17 %), hypertensio (16 %) ja diabetes / diabeettinen nefropatia (14 %).
Ensisijainen tehoa mittaava päätetapahtuma
P002-tutkimuksen ensisijainen tehoa mittaava päätetapahtuma oli sytomegaloviruksen aiheuttaman taudin (sytomegaloviruksen kohde-elimen sairaus tai sytomegalovirusoireyhtymä, jonka riippumaton arviointitoimikunta vahvisti) ilmaantuvuus viikkoon 52 mennessä siirron jälkeen. Tutkimuksessa käytettiin hoitotehon havaitun puuttumisen menettelyä eli hoitoa ei katsottu epäonnistuneeksi tilanteissa, joissa potilas keskeytti osallistumisensa tutkimukseen ennenaikaisesti mistä tahansa syystä tai potilasta koskevia tietoja puuttui joltakin ajankohdalta.
Letermoviiri oli vähintään samanveroinen kuin valgansikloviiri ensisijaisen päätetapahtuman analyysissä, kuten taulukko 5 osoittaa.
Taulukko 5: P002: Tehoon liittyvät tulokset munuaissiirteen saaneilla potilailla (hoitotehon havaitun puuttumisen menettely, koko analyysijoukko)
| Parametri | Letermoviiri (N = 289) n (%) | Valgansikloviiri (N = 297) n (%) |
| CMV:n aiheuttama tauti* viikkoon 52 mennessä | 30 (10,4) | 35 (11,8) |
| Ositteen mukaan korjattu hoitojen välinen ero (letermoviiri - valgansikloviiri)† Ero (95 %:n luottamusväli) | -1,4 (-6,5, 3,8)‡ | |
| * Riippumaton arviointitoimikunta vahvisti CMV:n aiheuttaman taudin tapaukset. † Hoitojen välisen prosentuaalisen vasteen eron 95 %:n luottamusvälit laskettiin käyttäen ositteen mukaan korjattua Mantel–Haenszelin menetelmää, jossa ero painotettiin kummankin hoitohaaran otoskoon harmonisen keskiarvon mukaan ositekohtaisesti (sen mukaan, oliko potilas saanut induktiohoidon aikana voimakkaasti sytolyyttistä, lymfosyyttejä tuhoavaa immunoterapiaa vai ei). ‡ Letermoviiri on vähintään samanveroinen kuin valgansikloviiri, kun vähintään samanveroisuuden marginaali on 10 %. Puuttuvien arvojen käsitteleminen: Hoitotehon havaitun puuttumisen menettely. Tämä menettely tarkoittaa sitä, että hoitoa ei katsota tehottomaksi, jos potilas keskeyttää osallistumisensa tutkimukseen ennenaikaisesti mistä tahansa syystä. Huom.: Letermoviiriryhmään satunnaistetuille potilaille annettiin asikloviiria herpes simplex ‑viruksen (HSV) ja varicella zoster ‑viruksen (VZV) aiheuttamien infektioiden ehkäisyyn. Valgansikloviiriryhmään satunnaistetut potilaat saivat asikloviiria muistuttavaa lumevalmistetta. N = potilaiden lukumäärä kummassakin hoitoryhmässä. n (%) = potilaiden lukumäärä (prosenttiosuus) kussakin alaryhmässä. | ||
Teho oli verrannollinen kaikissa alaryhmissä, jotka perustuivat sukupuoleen, ikään, etniseen taustaan, alueeseen ja siihen, oliko potilas saanut induktiohoidon aikana voimakkaasti sytolyyttistä, lymfosyyttejä tuhoavaa immunoterapiaa vai ei.
Pediatriset potilaat
P030: Allogeenisen kantasolusiirteen saaneet pediatriset potilaat
Letermoviirin tehoa sytomegalovirusinfektion tai sytomegaloviruksen aiheuttaman taudin ehkäisyssä siirteen saaneilla pediatrisilla potilailla arvioitiin vaiheen 2b avoimessa yksihaaraisessa monikeskustutkimuksessa (P030) allogeenisen kantasolusiirteen saaneilla pediatrisilla potilailla. Tutkimushoito aloitettiin kantasolusiirron jälkeen (0.–28. päivänä kantasolusiirron jälkeen), ja sitä jatkettiin viikolle 14 kantasolusiirron jälkeen. Tutkimuslääke annettiin joko suun kautta tai laskimoon, ja letermoviiriannos määräytyi iän, potilaan painon ja lääkemuodon mukaan.
Hoitoa saaneista 63 tutkittavasta 8 oli 0 – < 2‑vuotiaita, 27 oli 2 – < 12‑vuotiaita ja 28 oli 12 – < 18‑vuotiaita. Lähtötilanteessa 87 % tutkittavista oli saanut myeloablatiivisen esihoidon, 67 % sai siklosporiinia ja 27 % sai takrolimuusia. Kantasolusiirron yleisimmät ensisijaiset syyt olivat koko tutkimuspopulaatiossa akuutti myelooinen leukemia (18 %) ja aplastinen anemia (10 %) ja alle 2‑vuotiailla lapsilla sekamuotoinen immuunivajavuus (37,5 %) ja familiaalinen hemofagosyyttinen lymfohistiosytoosi (25,0 %).
Toissijainen tehoa mittaava päätetapahtuma
P030-tutkimuksen tehoa mittaavat päätetapahtumat olivat toissijaisia, ja niihin kuuluivat kliinisesti merkittävän sytomegalovirusinfektion ilmaantuvuudet viikkoon 14 mennessä kantasolusiirron jälkeen ja viikkoon 24 mennessä kantasolusiirron jälkeen. Kliinisesti merkittäväksi sytomegalovirusinfektioksi määriteltiin joko sytomegaloviruksen kohde-elimen sairauden ilmaantuminen tai sytomegalovirusinfektion ennakoivan hoidon aloittaminen veressä dokumentoidusti todetun sytomegaloviruksen sekä tutkittavan kliinisen tilan perusteella. Kliinisesti merkittävän sytomegalovirusinfektion ilmaantuvuus oli 7,1 % viikkoon 14 mennessä kantasolusiirron jälkeen ja 10,7 % viikkoon 24 mennessä kantasolusiirron jälkeen.
Farmakokinetiikka
Terveillä aikuisilla tutkittavilla letermoviirin farmakokinetiikkaa on kuvattu suun kautta ja laskimoon annettuna. Letermoviirialtistus suureni enemmän kuin suhteessa annokseen sekä suun kautta että laskimoon annettaessa. Todennäköinen mekanismi on OATP1B1/3:n kyllästyminen/autoinhibitio. Letermoviirin farmakokinetiikkaa on myös kuvattu suun kautta ja laskimoon annettuna aikuisilla kantasolusiirron saaneilla potilailla (ks. taulukko 6) ja pediatrisilla kantasolusiirron saaneilla potilailla (ks. taulukko 8 ja taulukko 9) sekä suun kautta annettuna aikuisilla munuaissiirteen saaneilla potilailla (ks. taulukko 7).
Terveet aikuiset tutkittavat
Vakaan tilan aikainen AUC-arvojen geometrinen keskiarvo oli 71 500 ng•h/ml ja Cmax-arvojen geometrinen keskiarvo oli 13 000 ng/ml, kun letermoviiria annettiin suun kautta 480 mg kerran vuorokaudessa.
Letermoviiri saavutti vakaan tilan 9–10 vuorokaudessa, ja kumuloitumissuhde oli AUC-arvojen osalta 1,2 ja Cmax-arvojen osalta 1.
Kantasolusiirron saaneet aikuiset potilaat
Letermoviirin AUC arvioitiin vaiheen 3 tutkimuksen (P001) tiedoista populaatiofarmakokineettisiä analyysejä käyttäen (ks. taulukko 6). Lääkeainealtistuksen erot hoito-ohjelmien välillä eivät ole kliinisesti merkittäviä. Teho oli yhdenmukainen kaikilla tutkimuksessa P001 todetuilla altistustasoilla.
Taulukko 6: Letermoviirin AUC-arvot (ng•h/ml) kantasolusiirron saaneilla aikuisilla potilailla
| Hoito-ohjelma | Mediaani (90 %:n ennusteväli)* |
| 480 mg suun kautta, ei siklosporiinia | 34 400 (16 900, 73 700) |
| 480 mg laskimoon, ei siklosporiinia | 100 000 (65 300, 148 000) |
| 240 mg suun kautta, siklosporiinin kanssa | 60 800 (28 700, 122 000) |
| 240 mg laskimoon, siklosporiinin kanssa | 70 300 (46 200, 106 000) |
| *Potilasjoukon post hoc ‑ennusteet populaatiofarmakokineettisen analyysin perusteella vaiheen 3 tutkimustietoja käyttäen | |
Munuaissiirteen saaneet aikuiset potilaat
Letermoviirin AUC arvioitiin vaiheen 3 tutkimuksen (P002) tiedoista populaatiofarmakokineettisiä analyysejä käyttäen (ks. taulukko 7). Teho oli yhdenmukainen kaikilla P002-tutkimuksessa todetuilla altistustasoilla.
Taulukko 7: Letermoviirin AUC-arvot (ng•h/ml) munuaissiirteen saaneilla aikuisilla potilailla
| Hoito-ohjelma | Mediaani (90 %:n ennusteväli)* |
| 480 mg suun kautta, ei siklosporiinia | 62 200 (28 900, 145 000) |
| 240 mg suun kautta, siklosporiinin kanssa | 57 700 (26 900, 135 000) |
| * Mediaanit ja 90 %:n ennustevälit perustuvat simulointeihin, joissa käytettiin vaiheen 3 tutkimustietoihin perustuvaa populaatiofarmakokineettistä mallia ja otettiin huomioon yksilöiden välinen vaihtelu. Huom.: Laskimoon annetun letermoviirin farmakokinetiikkaa ei tutkittu munuaissiirteen saaneilla potilailla. Laskimoon antamisen jälkeen AUC-arvon oletetaan kuitenkin olevan samanlainen kuin mallin avulla ennustettu AUC-arvo tilanteessa, jossa letermoviiria on annettu laskimoon kantasolusiirron saaneille potilaille (ks. taulukko 6). | |
Imeytyminen
Terveillä aikuisilla tutkittavilla letermoviiri imeytyi nopeasti ja saavutti huippupitoisuuden plasmassa 1,5–3,0 tunnin kuluttua (tmax) (mediaani), ja pitoisuus pieneni kaksivaiheisesti. Kantasolusiirron saaneilla aikuisilla potilailla letermoviirin hyötyosuuden arvioitiin olevan noin 35 %, kun letermoviiria annettiin 480 mg suun kautta kerran vuorokaudessa ilman siklosporiinia. Hyötyosuuden yksilöiden välisen vaihtelun arvioitiin olevan noin 37 %. Munuaissiirteen saaneilla aikuisilla potilailla letermoviirin hyötyosuuden arvioitiin olevan noin 60 %, kun letermoviiria annettiin 480 mg kerran vuorokaudessa suun kautta ilman siklosporiinia.
Siklosporiinin vaikutus
Kantasolusiirron saaneilla aikuisilla potilailla siklosporiinin samanaikainen käyttö suurensi letermoviirin pitoisuutta plasmassa, mikä johtui OATP1B:n toiminnan estymisestä. Letermoviirin hyötyosuuden arvioitiin olevan noin 85 %, kun potilaille annettiin letermoviiria 240 mg suun kautta kerran vuorokaudessa yhdessä siklosporiinin kanssa.
Jos letermoviiria annetaan siklosporiinin kanssa, suositeltu letermoviiriannos on 240 mg kerran vuorokaudessa aikuisilla ja vähintään 30 kg painavilla pediatrisilla potilailla (ks. kohta Annostus ja antotapa). Jos suun kautta annettavaa letermoviiria annetaan siklosporiinin kanssa alle 30 kg painaville pediatrisille potilaille, annosta on pienennettävä (ks. kohta Annostus ja antotapa).
Ruoan vaikutus
Kun terveille aikuisille tutkittaville annettiin suun kautta 480 mg:n letermoviiritabletti kerta-annoksena standardin runsasrasvaisen ja runsaskalorisen aterian yhteydessä, letermoviirin kokonaisaltistus (AUC) ei muuttunut ja huippupitoisuus (Cmax) suureni noin 30 %. Letermoviiritabletit voidaan antaa suun kautta joko aterian yhteydessä tai tyhjään mahaan, kuten kliinisissä tutkimuksissa on tehty (ks. kohta Annostus ja antotapa).
Kun terveille aikuisille tutkittaville annettiin suun kautta letermoviirirakeita 240 mg:n kerta-annoksena pehmeiden ruokien (vanukkaan tai omenasoseen) kanssa, letermoviirin kokonaisaltistus (AUC) suureni noin 13 % (vanukas) ja 20 % (omenasose) ja huippupitoisuudet (Cmax) suurenivat noin 25 % (vanukas) ja 33 % (omenasose). Letermoviirirakeet voidaan antaa pehmeiden ruokien kanssa, kuten pediatrisessa tutkimuksessa on tehty (ks. kohta Annostus ja antotapa).
Jakautuminen
Populaatiofarmakokineettisten analyysien perusteella laskimoon annetun letermoviirin vakaan tilan aikaisen jakautumistilavuuden keskiarvon arvioidaan olevan 45,5 litraa kantasolusiirron saaneilla aikuisilla potilailla.
Letermoviiri sitoutuu huomattavassa määrin (98,2 %) ihmisen plasman proteiineihin, arvioidusta pitoisuusalueesta (3–100 mg/l) riippumatta, in vitro. Pienemmillä pitoisuuksilla havaittiin kyllästymistä. Vereen ja plasmaan jakautuneen letermoviirin suhde on 0,56 ja riippumaton arvioidusta pitoisuusalueesta (0,1–10 mg/l) in vitro.
Prekliinisissä jakautumistutkimuksissa letermoviiri jakautuu eri elimiin ja kudoksiin siten, että suurimmat pitoisuudet todetaan ruoansulatuskanavassa, sapenjohtimessa ja maksassa ja pienet pitoisuudet aivoissa.
Biotransformaatio
Letermoviirista peräisin olevat aineosat plasmassa ovat suurimmaksi osaksi muuttumatonta lähtöainetta (96,6 %). Plasmassa ei havaita merkittäviä metaboliitteja. Letermoviiri eliminoituu osittain UGT1A1/1A3:n välityksellä glukuronidaatioreitin kautta.
Eliminaatio
Letermoviirin laskennallisen terminaalisen puoliintumisajan keskiarvo on noin 12 tuntia, kun letermoviiria annetaan terveille aikuisille tutkittaville 480 mg laskimoon. Letermoviiri eliminoituu pääasiassa erittymällä sappeen sekä suoran glukuronidaation kautta. Tähän prosessiin osallistuvat lääkeaineita maksasoluihin kuljettavat proteiinit OATP1B1 ja ‑3, joiden toiminnan jälkeen tapahtuu UGT1A1/3:n katalysoima glukuronidaatio.
Populaatiofarmakokineettisten analyysien perusteella letermoviirin vakaan tilan aikaisen näennäisen puhdistuman arvioidaan olevan 4,84 l/h laskimoon annetun 480 mg:n annoksen jälkeen kantasolusiirron saaneilla aikuisilla potilailla. Puhdistuman yksilöiden välisen vaihtelun arvioidaan olevan 24,6 %.
Erittyminen
Kun radioaktiivisesti merkittyä letermoviiria annettiin suun kautta, 93,3 % radioaktiivisesta annoksesta erittyi ulosteeseen. Suurin osa letermoviirista erittyi sappeen muuttumattomana lähtöaineena ja vain pieni osa (6 % annoksesta) asyyliglukuronidimetaboliittina ulosteeseen. Asyyliglukuronidi on epästabiili ulosteessa. Letermoviirin erittyminen virtsaan oli hyvin vähäistä (< 2 % annoksesta).
Farmakokinetiikka erityisryhmissä
Maksan vajaatoiminta
Sitoutumattoman letermoviirin AUC oli kohtalaista maksan vajaatoimintaa (Child–Pugh-luokka B [CP-B], 7−9 pistettä) sairastavilla aikuisilla tutkittavilla noin 81 % suurempi ja vaikeaa maksan vajaatoimintaa (Child–Pugh-luokka C [CP-C], 10–15 pistettä) sairastavilla aikuisilla tutkittavilla noin 4 kertaa suurempi kuin terveillä aikuisilla tutkittavilla. Letermoviirialtistuksen muutokset eivät ole kliinisesti merkittäviä kohtalaista maksan vajaatoimintaa sairastavilla aikuisilla potilailla.
Sitoutumattoman letermoviirin pitoisuus saattaa suurentua huomattavasti, jos potilaalla on kohtalainen maksan vajaatoiminta ja samanaikaisesti kohtalainen tai vaikea munuaisten vajaatoiminta (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Kliininen tutkimus potilailla, joilla oli munuaisten vajaatoiminta
Sitoutumattoman letermoviirin AUC oli kohtalaista munuaisten vajaatoimintaa (eGFR 31,0–56,8 ml/min/1,73 m2) sairastavilla aikuisilla tutkittavilla noin 115 % suurempi ja vaikeaa munuaisten vajaatoimintaa (eGFR 11,9–28,1 ml/min/1,73 m2) sairastavilla aikuisilla tutkittavilla noin 81 % suurempi kuin terveillä aikuisilla tutkittavilla. Kohtalaisesta tai vaikeasta munuaisten vajaatoiminnasta johtuvien letermoviirialtistuksen muutosten ei katsota olevan kliinisesti merkittäviä. Loppuvaiheen munuaistautia sairastavia potilaita ei ole tutkittu.
Munuaisensiirron jälkeen (P002-tutkimus)
Populaatiofarmakokineettisen analyysin perusteella letermoviirin AUC oli noin 12 % suurempi lievää munuaisten vajaatoimintaa (kreatiniinipuhdistuma vähintään 60 ml/min mutta alle 90 ml/min) sairastavilla aikuisilla tutkittavilla, 27 % suurempi kohtalaista munuaisten vajaatoimintaa (kreatiniinipuhdistuma vähintään 30 ml/min mutta alle 60 ml/min) sairastavilla aikuisilla tutkittavilla ja 35 % suurempi vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma vähintään 15 ml/min mutta alle 30 ml/min) sairastavilla aikuisilla tutkittavilla kuin aikuisilla henkilöillä, joilla kreatiniinipuhdistuma oli vähintään 90 ml/min. Näiden muutosten ei katsota olevan kliinisesti merkittäviä.
Paino
Terveisiin aikuisiin tutkittaviin perustuvien populaatiofarmakokineettisten analyysien perusteella letermoviirin AUC:n arvioidaan olevan 18,7 % pienempi 80–100 kg painavilla kuin 67 kg painavilla henkilöillä. Munuaissiirteen saaneisiin aikuisiin potilaisiin perustuvan populaatiofarmakokineettisen analyysin perusteella (P002-tutkimus) letermoviirin AUC:n arvioidaan olevan 26 % pienempi yli 80 kg painavilla kuin enintään 80 kg painavilla henkilöillä. Nämä erot eivät ole kliinisesti merkittäviä.
Etninen tausta
Terveisiin aikuisiin tutkittaviin perustuvien populaatiofarmakokineettisten analyysien perusteella letermoviirin AUC:n arvioidaan olevan 33,2 % suurempi aasialaisilla kuin valkoihoisilla. Tämä muutos ei ole kliinisesti merkittävä.
Sukupuoli
Populaatiofarmakokineettisten analyysien perusteella letermoviirin farmakokinetiikassa ei ole eroa aikuisten naisten ja miesten välillä.
Iäkkäät
Populaatiofarmakokineettisten analyysien perusteella ikä ei vaikuta letermoviirin farmakokinetiikkaan. Annoksen muuttaminen ei ole tarpeen iän perusteella.
Pediatriset potilaat
Letermoviirin AUC kantasolusiirron saaneilla pediatrisilla potilailla arvioitiin populaatiofarmakokineettisellä analyysillä tutkimuksessa 030 havaituista farmakokinetiikkaa koskevista tiedoista (ks. taulukko 8 ja taulukko 9). Kantasolusiirron saaneiden pediatristen potilaiden altistukset eri painoluokissa olivat kantasolusiirron saaneilla aikuisilla potilailla saavutettujen vertailualtistusten alueella (ks. taulukko 6).
Taulukko 8: Suun kautta annetun letermoviirin AUC-arvot (ng•h/ml) kantasolusiirron saaneilla pediatrisilla potilailla
Paino | Suun kautta annettu annos, ei siklosporiinia | Mediaani (90 %:n ennusteväli)* | Suun kautta annettu annos, siklosporiinin kanssa | Mediaani (90 %:n ennusteväli)* |
Vähintään 30 kg | 480 mg | 39 100 | 240 mg | 49 100 |
15 kg – alle 30 kg | 240 mg | 38 900 | 120 mg | 51 000 |
7,5 kg – alle 15 kg | 120 mg | 32 000 | 60 mg | 41 600 |
5 kg – alle 7,5 kg | 80 mg | 30 600 | 40 mg | 39 000 |
* Mediaanit ja 90 %:n ennustevälit perustuvat simulointeihin, joissa käytettiin kantasolusiirron saaneita pediatrisia potilaita koskevaa populaatiofarmakokineettistä mallia ja otettiin huomioon yksilöiden välinen vaihtelu. | ||||
Taulukko 9: Laskimoon annetun letermoviirin AUC-arvot (ng•h/ml) kantasolusiirron saaneilla pediatrisilla potilailla
Paino | Laskimoon annettu annos, ei siklosporiinia | Mediaani (90 %:n ennusteväli)* | Laskimoon annettu annos, siklosporiinin kanssa | Mediaani (90 %:n ennusteväli)* |
Vähintään 30 kg | 480 mg | 111 000 | 240 mg | 59 800 |
15 kg – alle 30 kg | 120 mg | 57 200 | 120 mg | 61 100 |
7,5 kg – alle 15 kg | 60 mg | 46 000 | 60 mg | 49 200 |
5 kg – alle 7,5 kg | 40 mg | 43 400 | 40 mg | 45 900 |
* Mediaanit ja 90 %:n ennustevälit perustuvat simulointeihin, joissa käytettiin kantasolusiirron saaneita pediatrisia potilaita koskevaa populaatiofarmakokineettistä mallia ja otettiin huomioon yksilöiden välinen vaihtelu. | ||||
Prekliiniset tiedot turvallisuudesta
Yleinen toksisuus
Korjautumattomia kiveksiin kohdistuvia haittavaikutuksia havaittiin vain rotilla, kun systeeminen altistus (AUC) oli ≥ 3‑kertainen verrattuna ihmisen altistukseen suositeltuja annoksia käytettäessä. Näitä haittavaikutuksia olivat siementiehyiden degeneraatio sekä oligotsoospermia ja soludebris lisäkiveksissä ja kivesten ja lisäkivesten painon lasku. Rotilla ei esiintynyt kiveksiin kohdistuvia haittavaikutuksia altistustasolla, joka vastasi ihmisen altistusta (AUC) suositeltua annostusta käytettäessä. Kiveksiin kohdistuvia haittavaikutuksia ei havaittu hiirillä eikä apinoilla suurimmilla testatuilla annostasoilla, joiden aikaansaama altistus oli hiirillä enintään 4-kertainen ja apinoilla enintään 2-kertainen verrattuna ihmisen altistukseen suositeltua annosta käytettäessä. Tämän havainnon merkitystä ihmisille ei tunneta.
Karsinogeneesi
6 kuukauden pituisessa karsinogeenisuustutkimuksessa, jossa valmistetta annettiin suun kautta RasH2-siirtogeenisille (Tg.RasH2) hiirille, ei todettu näyttöä ihmisen kannalta merkityksellisestä kasvainten muodostumisesta suurimmillakaan tutkituilla annoksilla (150 mg/kg/vrk uroksilla ja 300 mg/kg/vrk naarailla).
Mutageneesi
Letermoviiri ei ollut genotoksinen in vitro- eikä in vivo ‑tutkimuksissa, joita olivat mikrobeilla tehdyt mutageenisuustutkimukset, kiinanhamsterin munasarjasoluissa tehty kromosomipoikkeavuustutkimus ja hiiren mikrotumatutkimus in vivo.
Lisääntyminen
Hedelmällisyys
Rotilla tehdyissä hedelmällisyystutkimuksissa ja varhaisen alkionkehityksen tutkimuksissa letermoviiri ei vaikuttanut naaraiden hedelmällisyyteen. Urosrotilla havaittiin siittiöiden vähenemistä, siittiöiden liikkuvuuden heikkenemistä ja hedelmällisyyden laskua, kun systeeminen altistus oli ≥ 3‑kertainen verrattuna ihmisen AUC-arvoon suositeltua annosta käytettäessä (ks. Yleinen toksisuus).
Letermoviiria saaneilla apinoilla ei havaittu näyttöä kiveksiin kohdistuvista haittavaikutuksista histopatologisen arvioinnin, kivesten koon mittauksen, veren hormonimääritysten (follikkelia stimuloiva hormoni, inhibiini B ja testosteroni) ja siemennestetutkimuksen (siittiöiden määrä, liikkuvuus ja morfologia) perusteella, kun systeeminen altistus oli noin 2-kertainen verrattuna ihmisen AUC-arvoon suositeltua annosta käytettäessä.
Yksilönkehitys
Rotilla emoon kohdistuvia haittavaikutuksia (myös painonnousun hidastumista) havaittiin annostasolla 250 mg/kg/vrk (noin 11-kertainen altistus verrattuna ihmisen AUC-arvoon suositeltua annosta käytettäessä). Jälkeläisillä havaittuja vaikutuksia olivat sikiöiden painon lasku ja hidastunut luutuminen, vähäinen sikiöiden turvotus, lyhyen napanuoran yleisempi esiintyminen ja nikamien, kylkiluiden ja lantion muutosten ja epämuodostumien lisääntyminen. Emoon tai yksilönkehitykseen kohdistuneita vaikutuksia ei havaittu annostasolla 50 mg/kg/vrk (noin 2,5-kertainen altistus verrattuna ihmisen AUC-arvoon suositeltua annosta käytettäessä).
Kaniineilla emoon kohdistuvia haittavaikutuksia (myös kuolleisuutta ja keskenmenoja) havaittiin annostasolla 225 mg/kg/vrk (noin 2-kertainen altistus verrattuna ihmisen AUC-arvoon suositeltua annosta käytettäessä). Jälkeläisillä havaittiin nikamien ja kylkiluiden epämuodostumien ja muutosten lisääntymistä.
Pre- ja postnataalista kehitystä koskevassa tutkimuksessa letermoviiria annettiin suun kautta tiineille rotille. Yksilönkehitykseen kohdistuneita haittavaikutuksia ei havaittu suurimmillakaan testatuilla altistustasoilla (2-kertainen altistus verrattuna ihmisen AUC-arvoon suositeltua annosta käytettäessä).
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa (E460)
Kroskarmelloosinatrium (E468)
Povidoni (E1201)
Vedetön kolloidinen piidioksidi (E551)
Magnesiumstearaatti (E470b)
Kalvopäällyste
Laktoosimonohydraatti
Hypromelloosi (E464)
Titaanidioksidi (E171)
Triasetiini
Keltainen rautaoksidi (E172)
Punainen rautaoksidi (vain 480 mg:n tabletit) (E172)
Karnaubavaha (E903)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
PREVYMIS tabletti, kalvopäällysteinen
240 mg (L:kyllä) 28 fol (4652,22 €)
480 mg (L:kyllä) 28 fol (9123,85 €)
PF-selosteen tieto
Pakkauksissa on 28x1 tablettia yksittäispakatuissa polyamidi/alumiini/PVC – alumiini-läpipainopakkauksissa.
Valmisteen kuvaus:
PREVYMIS 240 mg kalvopäällysteiset tabletit
Keltainen, soikea 16,5 mm x 8,5 mm tabletti, jossa on toisella puolella merkintä "591" ja toisella puolella yhtiön logo.
PREVYMIS 480 mg kalvopäällysteiset tabletit
Vaaleanpunainen, kaksoiskupera 21,2 mm x 10,3 mm tabletti, jossa on toisella puolella merkintä "595" ja toisella puolella yhtiön logo.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
PREVYMIS tabletti, kalvopäällysteinen
240 mg 28 fol
480 mg 28 fol
- Rajoitettu peruskorvaus reseptimerkinnällä (40 %). Merkintä lääkemääräyksen 'Perustelut'-kohdassa: Allogeeninen kantasolusiirto, lääkehoidon aloitus (E022).
ATC-koodi
J05AX18
Valmisteyhteenvedon muuttamispäivämäärä
25.04.2025
Yhteystiedot
 MSD FINLAND OY
MSD FINLAND OY Keilaniementie 1, PL 46
02151 Espoo
09 804 650
www.msd.fi
info@msd.fi