
IMFINZI infuusiokonsentraatti, liuosta varten 50 mg/ml
Vaikuttavat aineet ja niiden määrät
Yksi millilitra infuusiokonsentraattia liuosta varten sisältää 50 mg durvalumabia.
Yksi injektiopullo, jossa on 2,4 ml konsentraattia, sisältää 120 mg durvalumabia.
Yksi injektiopullo, jossa on 10 ml konsentraattia, sisältää 500 mg durvalumabia.
Durvalumabia tuotetaan nisäkkään soluissa (kiinanhamsterin munasarjasoluissa) yhdistelmä-DNA-tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Ei-pienisoluinen keuhkosyöpä
IMFINZI on tarkoitettu leikkaukseen soveltuvan ei-pienisoluisen keuhkosyövän hoitoon aikuisille, joilla taudin uusiutumisen riski on suuri ja joilla ei ole EGFR-mutaatioita eikä ALK-uudelleenjärjestymiä. Tällöin annetaan ensin IMFINZI-valmistetta yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa esiliitännäishoitona ja myöhemmin IMFINZI-monoterapiaa liitännäishoitona. (Ks. valintakriteerit kohdasta Farmakodynamiikka).
IMFINZI monoterapiana on tarkoitettu paikallisesti edenneen, leikkaukseen soveltumattoman ei‑pienisoluisen keuhkosyövän hoitoon aikuisille, joilla kasvaimet ilmentävät PD‑L1-ligandia ≥ 1 %:ssa kasvainsoluista ja joiden sairaus ei ole edennyt platinapohjaisen kemosädehoidon jälkeen (ks. kohta Farmakodynamiikka).
IMFINZI on tarkoitettu yhdistelmänä tremelimumabin ja platinapohjaisen solunsalpaajahoidon kanssa ensilinjan hoitoon aikuisille, joilla on metastasoitunut ei-pienisoluinen keuhkosyöpä ilman herkistäviä epidermaalisen kasvutekijäreseptorin (EGFR) mutaatioita tai anaplastisen lymfoomakinaasin (ALK) mutaatioita.
Pienisoluinen keuhkosyöpä
IMFINZI monoterapiana on tarkoitettu rajoittuneen pienisoluisen keuhkosyövän hoitoon aikuisille, joiden sairaus ei ole edennyt platinapohjaisen kemosädehoidon jälkeen.
IMFINZI yhdistelmänä etoposidin ja joko karboplatiinin tai sisplatiinin kanssa on tarkoitettu ensilinjan hoitoon aikuisille, joilla on levinnyt pienisoluinen keuhkosyöpä.
Sappitiesyöpä
IMFINZI yhdistelmänä gemsitabiinin ja sisplatiinin kanssa on tarkoitettu ensilinjan hoitoon aikuisille, joilla on leikkaukseen soveltumaton tai etäpesäkkeinen sappitiesyöpä.
Maksasolusyöpä
IMFINZI monoterapiana on tarkoitettu ensilinjan hoitoon aikuisille, joilla on pitkälle edennyt tai leikkaushoitoon soveltumaton maksasolusyöpä.
IMFINZI on tarkoitettu yhdistelmänä tremelimumabin kanssa ensilinjan hoitoon aikuisille, joilla on pitkälle edennyt tai leikkaushoitoon soveltumaton maksasolusyöpä.
Kohdun limakalvon syöpä
IMFINZI on tarkoitettu yhdistelmänä karboplatiinin ja paklitakselin kanssa ensilinjan hoitoon aikuisille, joilla on primaari edennyt tai uusiutunut kohdun limakalvon syöpä ja joille systeeminen hoito soveltuu, minkä jälkeen annetaan ylläpitohoitona
- IMFINZI-valmistetta monoterapiana, jos kohdun limakalvon syöpään liittyy puutteellinen DNA:n kahdentumisvirheiden korjausmekanismi (dMMR; deficient mismatch repair)
- IMFINZI-valmistetta yhdistelmänä olaparibin kanssa, jos kohdun limakalvon syöpään liittyy toimiva DNA:n kahdentumisvirheiden korjausmekanismi (pMMR; proficient mismatch repair).
Lihasinvasiivinen rakkosyöpä
IMFINZI on tarkoitettu leikkaukseen soveltuvan lihasinvasiivisen rakkosyövän hoitoon aikuisille. Tällöin annetaan ensin esiliitännäishoitona IMFINZI-valmistetta yhdistelmänä gemsitabiinin ja sisplatiinin kanssa, ja radikaalin kystektomian jälkeen annetaan liitännäishoitona IMFINZI-monoterapiaa.
Mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooma
IMFINZI on tarkoitettu leikkaukseen soveltuvan mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooman hoitoon aikuisille. Tällöin IMFINZI-valmistetta annetaan ensin esiliitännäishoitona ja liitännäishoitona yhdistelmänä FLOT-solunsalpaajahoidon kanssa, minkä jälkeen annetaan liitännäishoitona IMFINZI-monoterapiaa.
Ehto
Valmistetta tulee käyttää vain syövän hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Syövän hoitoon perehtyneen lääkärin on aloitettava hoito ja valvottava sen toteuttamista.
PD-L1-määritys paikallisesti edennyttä ei‑pienisoluista keuhkosyöpää sairastavilla potilailla
Paikallisesti edennyttä ei pienisoluista keuhkosyöpää sairastavien potilaiden hoitoon soveltuvuuden arvioinnin on perustuttava PD‑L1-ligandin ilmentymiseen kasvaimissa, mikä varmistetaan validoidulla testillä (ks. kohta Farmakodynamiikka).
MMR-määritys potilailla, joilla on kohdun limakalvon syöpä
Kohdun limakalvon syöpää sairastavien potilaiden hoitoon soveltuvuuden arvioinnin on perustuttava MMR-mekanismin statukseen kasvaimessa (DNA:n kahdentumisvirheiden korjausmekanismin puutteellisuus tai toimivuus), mikä varmistetaan validoidulla testillä (ks. kohta Farmakodynamiikka).
Annostus
Taulukossa 1 esitetään IMFINZI-valmisteen suositeltu annos, kun valmistetta käytetään monoterapiana tai yhdistelmähoidossa. IMFINZI annetaan infuusiona laskimoon 1 tunnin aikana.
Kun IMFINZI annetaan yhdistelmänä muiden lääkeaineiden kanssa, katso lisätietoja näiden lääkeaineiden valmisteyhteenvedoista.
Taulukko 1. Suositeltu IMFINZI-annos monoterapiaa ja yhdistelmähoitoa varten
| Käyttöaihe | Suositeltu IMFINZI-annos | Hoidon kesto |
| Monoterapia | ||
| Paikallisesti edennyt ei-pienisoluinen keuhkosyöpä | 10 mg/kg 2 viikon välein tai 1 500 mg 4 viikon väleina | Kunnes tauti etenee tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 12 kuukauden ajanb |
| Rajoittunut pienisoluinen keuhkosyöpä | 1 500 mg 4 viikon väleina | Kunnes tauti etenee tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan |
| Maksasolusyöpä | 1 500 mg 4 viikon väleina | Kunnes tauti etenee tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä |
| Yhdistelmähoito | ||
| Leikkaukseen soveltuva ei-pienisoluinen keuhkosyöpä | 1 500 mgc yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa 3 viikon välein enintään 4 hoitosyklin ajan ennen leikkausta, minkä jälkeen 1 500 mg monoterapiana 4 viikon välein enintään 12 hoitosyklin ajan leikkauksen jälkeen. | Esiliitännäishoidon vaihe: kunnes taudin eteneminen sulkee pois definitiivisen leikkaushoidon tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä. Liitännäishoidon vaihe: kunnes tauti uusiutuu tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 12 hoitosyklin ajan leikkauksen jälkeen. |
| Metastasoitunut ei-pienisoluinen keuhkosyöpä | Platinapohjaisen solunsalpaajahoidon aikana: Platinapohjaisen solunsalpaajahoidon jälkeen: Tremelimumabia annetaan viides 75 mg:n annosf,g viikolla 16 IMFINZI-valmisteen kanssa | Kunnes tauti etenee tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä |
| Levinnyt pienisoluinen keuhkosyöpä | 1 500 mgh yhdistelmänä solunsalpaajahoidon kanssa 3 viikon (21 päivän) välein 4 hoitosyklin ajan, minkä jälkeen 1 500 mg 4 viikon välein monoterapiana | Kunnes tauti etenee tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä |
| Sappitiesyöpä | 1 500 mgi yhdistelmänä solunsalpaajahoidon kanssa 3 viikon (21 päivän) välein enintään 8 hoitosyklin ajan, minkä jälkeen 1 500 mg 4 viikon välein monoterapiana | Kunnes tauti etenee tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä |
| Maksasolusyöpä | IMFINZI 1 500 mgj yhdistelmänä tremelimumabin 300 mg:nj kerta-annoksen kanssa hoitosyklin 1 päivänä 1, minkä jälkeen annetaan IMFINZI-valmistetta monoterapiana 4 viikon välein | Kunnes tauti etenee tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä |
| Kohdun limakalvon syöpä | 1 120 mg yhdistelmänä karboplatiinin ja paklitakselin kanssa 3 viikon (21 päivän) välein vähintään 4 hoitosyklin ja enintään 6 hoitosyklin ajan, | Kunnes tauti etenee tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä |
| Lihasinvasiivinen rakkosyöpä | 1 500 mgl yhdistelmänä solunsalpaajahoidon kanssa 3 viikon välein 4 hoitosyklin ajan ennen leikkausta,
minkä jälkeen 1 500 mgl monoterapiana 4 viikon välein enintään 8 hoitosyklin ajan leikkauksen jälkeen.
| Esiliitännäishoidon vaihe: kunnes taudin eteneminen sulkee pois definitiivisen leikkaushoidon tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä.
Liitännäishoidon vaihe: kunnes tauti uusiutuu tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 8 hoitosyklin ajan leikkauksen jälkeen. |
| Mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooma | 1 500 mgm yhdistelmänä FLOT-solunsalpaajahoidon kanssa 4 viikon välein enintään 2 hoitosyklin ajan ennen leikkausta, minkä jälkeen 1 500 mgm yhdistelmänä FLOT-solunsalpaajahoidon kanssa 4 viikon välein enintään 2 hoitosyklin ajan ja tämän jälkeen 1 500 mg monoterapiana 4 viikon välein enintään 10 hoitosyklin ajan, yhteensä enintään 12 hoitosyklin ajan leikkauksen jälkeen. | Esiliitännäishoidon vaihe: kunnes taudin eteneminen sulkee pois definitiivisen leikkaushoidon tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä. Liitännäishoidon vaihe: kunnes tauti etenee tai uusiutuu tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 12 hoitosyklin ajan leikkauksen jälkeen. |
a Jos potilas painaa enintään 30 kg, on annostuksen perustuttava painoon. IMFINZI-hoitoa on annettava 10 mg/kg 2 viikon välein tai 20 mg/kg 4 viikon välein monoterapiana, kunnes paino suurenee yli 30 kg:n.
b Jos potilaan kliininen tila on vakaa, kun ensimmäisiä viitteitä taudin etenemisestä havaitaan, suositellaan hoidon jatkamista, kunnes taudin eteneminen on varmistunut.
c Jos leikkaukseen soveltuvaa ei-pienisoluista keuhkosyöpää sairastava potilas painaa enintään 30 kg, IMFINZI-annostuksen on perustuttava painoon (20 mg/kg). Hoitoa annetaan yhdistelmänä platinapohjaisen solunsalpaajan kanssa annoksella 20 mg/kg 3 viikon (21 päivän) välein ennen leikkausta, minkä jälkeen annetaan 20 mg/kg 4 viikon välein monoterapiana leikkauksen jälkeen, kunnes paino suurenee yli 30 kg:n.
d Jos metastasoitunutta ei-pienisoluista keuhkosyöpää sairastava potilas painaa enintään 30 kg, IMFINZI-annostuksen on perustuttava painoon. Tällöin annostus on 20 mg/kg IMFINZI-valmistetta, kunnes paino suurenee yli 30 kg:n. Jos potilas painaa enintään 34 kg, tremelimumabin annostuksen on perustuttava painoon. Tällöin annostus on 1 mg/kg tremelimumabia, kunnes paino suurenee yli 34 kg:n.
e Harkitse pemetreksediylläpitohoitoa potilaille, joiden kasvaimet eivät ole levyepiteeliperäisiä ja jotka ovat saaneet pemetreksedi- ja karboplatiini-/sisplatiinihoitoa platinapohjaisen solunsalpaajahoidon vaiheen aikana.
f Jos annoksen (annosten) antaminen viivästyy, tremelimumabin viides annos voidaan antaa viikon 16 jälkeen IMFINZI-valmisteen kanssa.
g Jos potilaat saavat platinapohjaista solunsalpaajahoitoa vähemmän kuin 4 hoitosykliä, jäljellä olevat hoitosyklit tremelimumabilla (yhteensä enintään 5 hoitosykliä) yhdessä IMFINZI-valmisteen kanssa annetaan platinapohjaisen solunsalpaajahoidon jälkeen.
h Jos levinnyttä pienisoluista keuhkosyöpää sairastava potilas painaa enintään 30 kg, IMFINZI-annostuksen on perustuttava painoon (20 mg/kg). Hoitoa annetaan yhdistelmänä solunsalpaaja-annoksen kanssa 3 viikon (21 päivän) välein, minkä jälkeen annetaan 20 mg/kg 4 viikon välein monoterapiana, kunnes paino suurenee yli 30 kg:n.
i Jos sappitiesyöpää sairastava potilas painaa enintään 36 kg, IMFINZI-annostuksen on perustuttava painoon (20 mg/kg). Hoitoa annetaan yhdistelmänä solunsalpaaja-annoksen kanssa 3 viikon (21 päivän) välein, minkä jälkeen annetaan 20 mg/kg 4 viikon välein monoterapiana, kunnes paino suurenee yli 36 kg:n.
j Jos maksasolusyöpää sairastava potilas painaa enintään 30 kg, IMFINZI-annostuksen on perustuttava painoon. Tällöin annostus on 20 mg/kg IMFINZI-valmistetta, kunnes paino suurenee yli 30 kg:n. Jos potilas painaa enintään 40 kg, tremelimumabin annostuksen on perustuttava painoon. Tällöin annostus on 4 mg/kg tremelimumabia, kunnes paino suurenee yli 40 kg:n.
kJos kohdun limakalvon syöpää sairastava potilas painaa ylläpitovaiheen aikana enintään 30 kg, IMFINZI-annostuksen on perustuttava painoon. Tällöin annostus on 20 mg/kg IMFINZI-valmistetta, kunnes paino suurenee yli 30 kg:n.
l Jos lihasinvasiivista rakkosyöpää sairastava potilas painaa enintään 30 kg, IMFINZI-annostuksen on perustuttava painoon (20 mg/kg).
m Jos mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinoomaa sairastava potilas painaa enintään 30 kg, IMFINZI-annostuksen on perustuttava painoon (20 mg/kg).
Annoksen suurentaminen tai pienentäminen ei ole suositeltavaa. Yksilöllinen turvallisuus ja siedettävyys saattaa edellyttää hoidosta pidättäytymistä tai hoidon lopettamista, ks. taulukko 2.
Immuunivälitteisten ja ei-immuunivälitteisten haittavaikutusten hoito-ohjeet on kuvattu taulukossa 2 (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet, jossa on lisätietoa hoitoa koskevista suosituksista, seurannasta ja haittavaikutusten arvioinnista).
Taulukko 2. Muutokset hoidossa IMFINZI-valmisteelle tai IMFINZI-valmisteen ja muiden lääkevalmisteiden yhdistelmälle
| Haittavaikutus | Vaikeusastea | Muutos hoidossa |
| Immuunivälitteiset haittavaikutukset | ||
| Immuunivälitteinen pneumoniitti / interstitiaalinen keuhkosairaus | Aste 2 | Hoidosta pidättäytyminen |
| Aste 3 tai 4 | Pysyvä lopettaminen | |
| Immuunivälitteinen maksatulehdus | ALAT tai ASAT > 3− ≤ 5 x ULN tai kokonaisbilirubiini > 1,5−≤ 3 x ULN | Hoidosta pidättäytyminen |
| ALAT tai ASAT > 5 − ≤ 10 x ULN | IMFINZI-valmisteen annosta pidättäytyminen ja tremelimumabihoidon pysyvä lopettaminen (tarvittaessa) | |
| Samanaikaisesti ALAT tai ASAT > 3 x ULN ja kokonaisbilirubiini > 2 x ULNb | Pysyvä lopettaminen | |
| ALAT tai ASAT > 10 x ULN tai kokonaisbilirubiini > 3 x ULN | ||
| Immuunivälitteinen maksatulehdus maksasolusyövän yhteydessä (tai maksaa sekundaarisesti affisioiva kasvain ja poikkeavat lähtöarvot)c | ALAT tai ASAT > 2,5 – ≤ 5 x lähtöarvo ja ≤ 20 x ULN | Hoidosta pidättäytyminen |
| ALAT tai ASAT > 5 – 7 x lähtöarvo ja ≤ 20 x ULN tai samanaikaisesti ALAT tai ASAT 2,5 – 5 x lähtöarvo ja ≤ 20 x ULN ja kokonaisbilirubiini > 1,5 – < 2 x ULNb | IMFINZI-valmisteen annosta pidättäytyminen ja tremelimumabihoidon pysyvä lopettaminen (tarvittaessa). | |
| ALAT tai ASAT > 7 x lähtöarvo tai > 20 x ULN sen mukaan, mikä tapahtuu ensin tai bilirubiini > 3 x ULN | Pysyvä lopettaminen | |
| Immuunivälitteinen paksusuolitulehdus tai ripuli | Aste 2 | Hoidosta pidättäytyminen |
| Aste 3, kun käytetään IMFINZI-valmistetta monoterapiana | Hoidosta pidättäytyminen | |
| Aste 3, kun käytetään IMFINZI-valmisteen ja tremelimumabin yhdistelmää | Tremelimumabihoidon pysyvä lopettaminend | |
| Aste 4 | Pysyvä lopettaminen | |
| Suolen puhkeamae | Mikä tahansa vaikeusaste | Pysyvä lopettaminen |
| Immuunivälitteinen hypertyreoosi, tyreoidiitti | Asteet 2–4 | Hoidosta pidättäytyminen, kunnes potilaan kliininen tila on vakaa |
| Immuunivälitteinen hypotyreoosi | Asteet 2–4 | Ei muutoksia |
| Immuunivälitteinen lisämunuaisten vajaatoiminta tai hypofysiitti/hypopituitarismi | Asteet 2–4 | Hoidosta pidättäytyminen, kunnes potilaan kliininen tila on vakaa |
| Immuunivälitteinen tyypin 1 diabetes | Asteet 2–4 | Ei muutoksia |
| Immuunivälitteinen munuaistulehdus | Aste 2, seerumin kreatiniini > 1,5−3 x (ULN tai lähtöarvo) | Hoidosta pidättäytyminen |
| Aste 3, seerumin kreatiniini > 3 x lähtöarvo tai > 3−6 x ULN; aste 4, seerumin kreatiniini > 6 x ULN | Pysyvä lopettaminen | |
| Immuunivälitteinen ihottuma tai dermatiitti (pemfigoidi mukaan lukien) | Aste 2 yli 1 viikon ajan | Hoidosta pidättäytyminen |
| Aste 3 | ||
| Aste 4 | Pysyvä lopettaminen | |
| Immuunivälitteinen sydänlihastulehdus | Aste 2–4 | Pysyvä lopettaminen |
| Immuunivälitteinen myosiitti tai polymyosiitti/rabdomyolyysi | Aste 2 tai 3 | Hoidosta pidättäytyminenf |
| Aste 4 | Pysyvä lopettaminen | |
| Infuusioon liittyvät reaktiot | Aste 1 tai 2 | Infuusio keskeytettävä tai infuusionopeutta pienennettävä |
| Aste 3 tai 4 | Pysyvä lopettaminen | |
| Infektio | Aste 3 tai 4 | Hoidosta pidättäytyminen, kunnes potilaan kliininen tila on vakaa |
| Immuunivälitteinen myasthenia gravis | Aste 2-4 | Pysyvä lopettaminen |
| Immuunivälitteinen transversaalimyeliitti | Mikä tahansa vaikeusaste | Pysyvä lopettaminen |
| Immuunivälitteinen meningiitti | Aste 2 | Hoidosta pidättäytyminen |
| Asteet 3 ja 4 | Pysyvä lopettaminen | |
| Immuunivälitteinen enkefaliitti | Aste 2–4 | Pysyvä lopettaminen |
| Immuunivälitteinen Guillain‑Barrén oireyhtymä | Aste 2–4 | Pysyvä lopettaminen |
| Muut immuunivälitteiset haittavaikutuksetg | Aste 2 tai 3 | Hoidosta pidättäytyminen |
| Aste 4 | Pysyvä lopettaminen | |
| Muut ei-immuunivälitteiset haittavaikutukset | ||
| Puhdas punasoluaplasia (PRCA)h | Mikä tahansa vaikeusaste | Pysyvä lopettaminen |
| Muut ei-immuunivälitteiset haittavaikutukset | Asteet 2 ja 3 | Hoidosta pidättäytyminen, kunnes aste on ≤ 1 tai palaa lähtötasolle |
| Aste 4 | Pysyvä lopettamineni | |
a CTCAE-luokitus (Common Terminology Criteria for Adverse Events), versio 4.03. ALAT: alaniiniaminotransferaasi; ASAT: aspartaattiaminotransferaasi; ULN: viitealueen yläraja.
b Potilailla, joilla on todettu jokin vaihtoehtoinen syy, noudatetaan samoja suosituksia kuin tilanteessa, jossa ASAT- tai ALAT-arvot ovat suurentuneet ilman samanaikaista bilirubiiniarvojen suurenemista.
c Jos ASAT- ja ALAT-arvot ovat lähtötilanteessa korkeintaan viitealueen ylärajalla ja potilaalla on maksa-affisiota, durvalumabin antamisesta on pidättäydyttävä tai se on lopetettava pysyvästi noudattaen samoja suosituksia kuin tilanteessa, jossa potilaalla on maksatulehdus mutta ei maksa-affisiota.
d Tremelimumabihoito on lopetettava pysyvästi asteen 3 haittavaikutuksen ilmetessä. Durvalumabihoitoa voidaan kuitenkin jatkaa, kun haittavaikutus on hävinnyt.
e Haittavaikutus liittyy vain IMFINZI-valmisteen käyttöön yhdistelmänä tremelimumabin kanssa.
f IMFINZI-hoito lopetetaan pysyvästi, jos haittavaikutus ei lievity 30 päivän kuluessa vaikeusasteeseen, joka on enintään 1, tai jos potilaalla ilmenee hengitysvajauksen merkkejä.
g Sisältää seuraavat: immuunitrombosytopenia, haimatulehdus, immuunivälitteinen niveltulehdus, uveiitti, ei-infektiivinen virtsarakkotulehdus ja polymyalgia rheumatica.
h Haittavaikutus liittyy vain IMFINZI-valmisteen käyttöön yhdistelmänä olaparibi-ylläpitohoidon kanssa, kun potilaalle on ensin annettu IMFINZI-valmistetta yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa.
i Lukuun ottamatta vaikeusastetta 4 olevia laboratorioarvojen poikkeavuuksia, joiden kohdalla päätös hoidon keskeyttämisestä perustuu muihin kliinisiin löydöksiin ja oireisiin sekä kliiniseen arvioon.
Haittavaikutuksen vaikeusasteen mukaisesti IMFINZI-hoidosta ja/tai tremelimumabihoidosta on pidättäydyttävä ja potilaalle on annettava kortikosteroideja (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Hoidosta pidättäytymisen jälkeen IMFINZI-hoitoa ja/tai tremelimumabihoitoa voidaan jatkaa 12 viikon kuluessa, jos haittavaikutukset ovat lievittyneet vaikeusasteen 1 tasolle tai alle ja kortikosteroidiannos on pienennetty korkeintaan 10 mg:aan prednisonia vuorokaudessa tai vastaavalle tasolle. IMFINZI-hoito ja tremelimumabihoito on lopetettava pysyvästi, jos todetaan uusiutuneita asteen 3 (vakavia) immuunivälitteisiä haittavaikutuksia tai mikä tahansa asteen 4 (henkeä uhkaava) immuunivälitteinen haittavaikutus, ellei kyseessä ole endokrinopatia, joka on saatu hyvään hoitotasapainoon hormonikorvaushoidolla.
Erityisryhmät
Iäkkäät
Annoksen muuttaminen ei ole tarpeen iäkkäillä (vähintään 65-vuotiailla) potilailla (ks. kohta Farmakodynamiikka).
Munuaisten vajaatoiminta
IMFINZI-annoksen muuttamista ei suositella potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta. Vaikeaa munuaisten vajaatoimintaa sairastavista potilaista on liian vähän tietoja, jotta tästä potilasryhmästä voitaisiin tehdä johtopäätöksiä (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
IMFINZI-annoksen muuttamista ei suositella potilailla, joilla on lievä tai keskivaikea maksan vajaatoiminta. Vaikeaa maksan vajaatoimintaa sairastavista potilaista on liian vähän tietoja, jotta tästä populaatiosta voitaisiin tehdä johtopäätöksiä (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
IMFINZI-valmisteen turvallisuutta ja tehoa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole varmistettu ei-pienisoluisen keuhkosyövän, pienisoluisen keuhkosyövän, sappitiesyövän, maksasolusyövän, kohdun limakalvon syövän eikä mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooman kohdalla. Tietoja ei ole saatavilla. Hyväksyttyjen käyttöaiheiden lisäksi IMFINZI-valmistetta yhdistelmänä tremelimumabin kanssa on tutkittu 1–17‑vuotiailla lapsilla, joilla on neuroblastooma, kiinteä kasvain tai sarkooma, mutta tutkimuksen tulosten perusteella ei voitu tehdä johtopäätöstä, että tällaisen käytön hyödyt olisivat suurempia kuin sen riskit. Saatavissa oleva tieto on kuvattu kohdissa Farmakodynamiikka ja Farmakokinetiikka.
Antotapa
IMFINZI annetaan laskimoon. IMFINZI annetaan laskimoinfuusioliuoksena 1 tunnin aikana (ks. kohta Käyttö- ja käsittelyohjeet).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
IMFINZI yhdistelmänä solunsalpaajahoidon kanssa
Kun IMFINZI-valmistetta annetaan yhdistelmänä solunsalpaajahoidon kanssa, IMFINZI annetaan ennen solunsalpaajahoitoa samana päivänä.
IMFINZI yhdistelmänä tremelimumabin ja platinapohjaisen solunsalpaajahoidon kanssa
Kun IMFINZI-valmistetta annetaan yhdistelmänä tremelimumabin ja platinapohjaisen solunsalpaajahoidon kanssa, tremelimumabi annetaan ensin, sen jälkeen IMFINZI ja sitten platinapohjainen solunsalpaajahoito, kaikki samana antopäivänä.
Kun IMFINZI-valmistetta annetaan yhdistelmänä tremelimumabin viidennen annoksen ja pemetreksediylläpitohoidon kanssa viikolla 16, tremelimumabi annetaan ensin, sen jälkeen IMFINZI ja sitten pemetreksediylläpitohoito, kaikki samana antopäivänä.
IMFINZI, tremelimumabi ja platinapohjainen solunsalpaajahoito annetaan erillisinä infuusioina laskimoon. Sekä IMFINZI että tremelimumabi annetaan 1 tunnin aikana. Katso valmisteyhteenvedosta platinapohjaisen solunsalpaajahoidon antamista koskevat tiedot. Katso valmisteyhteenvedosta pemetreksediylläpitohoidon antamista koskevat tiedot. Jokaiseen infuusioon on käytettävä eri infuusiopusseja ja suodattimia.
Hoitosyklin 1 aikana IMFINZI-valmisteen antaminen tremelimumabin jälkeen aloitetaan noin 1 tunnin (enintään 2 tunnin) kuluttua tremelimumabi-infuusion päättymisestä. Platinapohjaisen solunsalpaajahoidon infuusio aloitetaan noin 1 tunnin (enintään 2 tunnin) kuluttua IMFINZI-infuusion päättymisestä. Jos hoitosyklin 1 aikana ei ilmene kliinisesti merkittäviä huolenaiheita, seuraavien hoitosyklien yhteydessä IMFINZI voidaan lääkärin harkinnan mukaan antaa välittömästi tremelimumabin jälkeen ja IMFINZI-infuusion päättymisen ja solunsalpaajahoidon aloittamisen välinen aika voidaan lyhentää 30 minuuttiin.
IMFINZI yhdistelmänä tremelimumabin kanssa
Kun IMFINZI-valmistetta annetaan yhdistelmänä tremelimumabin kanssa, tremelimumabi annetaan ennen IMFINZI-valmistetta samana päivänä. IMFINZI ja tremelimumabi annetaan erillisinä infuusioina laskimoon. Katso valmisteyhteenvedosta tremelimumabin antamista koskevat tiedot.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Suositellut hoidon muutokset, ks. kohta Annostus ja antotapa, taulukko 2.
Epäillyt immuunivälitteiset haittavaikutukset on tutkittava asianmukaisesti niiden etiologian varmistamiseksi tai muiden syiden poissulkemiseksi. Haittavaikutuksen vaikeusasteen mukaan IMFINZI-valmisteen tai IMFINZI-valmisteen ja tremelimumabin yhdistelmän antamisesta on pidättäydyttävä tai hoito on lopetettava pysyvästi. Potilaalle on aloitettava kortikosteroidi- tai hormonihoito. Kun kortikosteroidihoitoa vaatineen tapahtuman oireet ovat lievittyneet niin, että niiden vaikeusaste on enintään 1, aloitetaan kortikosteroidiannoksen asteittainen pienentäminen, jota jatketaan vähintään 1 kuukauden ajan. Jos potilaan tila pahenee tai ei parane, on harkittava kortikosteroidiannoksen suurentamista ja/tai muiden systeemisten immuunisalpaajien lisäämistä hoitoon.
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Immuunivälitteinen pneumoniitti
IMFINZI-valmistetta, IMFINZI-valmisteen ja tremelimumabin yhdistelmää, IMFINZI-valmisteen ja platinapohjaisen solunsalpaajahoidon yhdistelmää ja tämän jälkeen IMFINZI-valmisteen ja olaparibin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla on ilmennyt immuunivälitteistä pneumoniittia ja interstitiaalista keuhkosairautta, jonka hoito edellyttää systeemisten kortikosteroidien käyttöä ja jolle ei ole muita selviä syitä (ks. kohta Haittavaikutukset). Vaikeusasteen 2 tapahtumien hoitoon aloitetaan prednisoni aloitusannoksella 1–2 mg/kg vuorokaudessa tai vastaava hoito. Annosta pienennetään asteittain. Vaikeusasteiden 3 ja 4 tapahtumien hoitoon aloitetaan metyyliprednisoloni aloitusannoksella 2–4 mg/kg vuorokaudessa tai vastaava hoito. Annosta pienennetään asteittain.
Pneumoniitti ja sädepneumoniitti
Keuhkoihin sädehoitoa saaneilla potilailla todetaan usein sädepneumoniittia. Pneumoniitin kliininen ilmenemismuoto on hyvin samankaltainen kuin sädepneumoniitilla. PACIFIC-tutkimuksessa pneumoniittia tai sädepneumoniittia ilmeni 161 potilaalla (33,9 %) IMFINZI-ryhmässä ja 58 potilaalla (24,8 %) lumeryhmässä niistä potilaista, jotka olivat jatkaneet hoidon loppuun ja saaneet ainakin kaksi hoitosykliä samanaikaista kemosädehoitoa 1–42 päivän sisällä ennen tutkimushoidon aloittamista. Vaikeusasteen 3 pneumoniittia tai sädepneumoniittia ilmeni 3,4 %:lla potilaista IMFINZI-ryhmässä ja 3,0 %:lla lumeryhmässä ja vaikeusasteen 5 haittaa 1,1 %:lla ja 1,7 %:lla, vastaavasti. AEGEAN-tutkimuksessa niistä potilaista, jotka olivat saaneet leikkauksenjälkeistä sädehoitoa, pneumoniittia tai sädepneumoniittia ilmeni 10 potilaalla (33,3 %) IMFINZI-ryhmässä ja 3 potilaalla (11,1 %) lumeryhmässä. IMFINZI-ryhmässä oli 2 potilasta (6,7 %), joilla pneumoniitin tai sädepneumoniitin vaikeusaste oli enintään 3.
ADRIATIC-tutkimuksessa pneumoniittia tai sädepneumoniittia ilmeni 100 potilaalla (38,2 %) IMFINZI-ryhmässä ja 80 potilaalla (30,2 %) lumeryhmässä niistä potilaista, jotka olivat jatkaneet kemosädehoidon loppuun ja joiden kemosädehoito oli päättynyt 1–42 päivän sisällä ennen tutkimushoidon aloittamista. Vaikeusasteen 3 pneumoniittia tai sädepneumoniittia ilmeni 3,1 %:lla potilaista IMFINZI-ryhmässä ja 2,3 %:lla lumeryhmässä ja vastaavasti vaikeusasteen 5 haittaa 0,4 %:lla ja 0,0 %:lla.
Potilaita pitää tarkkailla pneumoniittiin tai sädepneumoniittiin viittaavien oireiden varalta. Pneumoniittiepäily on varmistettava röntgenkuvauksella ja muut tulehdukselliset ja sairauteen liittyvät etiologiat on suljettava pois ja hoidettava kohdan Annostus ja antotapa suositusten mukaisesti.
Immuunivälitteinen maksatulehdus
Immuunivälitteistä maksatulehdusta, jonka hoito määritelmän mukaan edellyttää systeemisten kortikosteroidien käyttöä ja jolle ei ole muita selviä syitä, on esiintynyt IMFINZI-valmistetta, IMFINZI-valmisteen ja tremelimumabin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla (ks. kohta Haittavaikutukset). Alaniiniaminotransferaasi- ja aspartaattiaminotransferaasiarvo sekä kokonaisbilirubiini ja alkalisen fosfataasin pitoisuus on tutkittava ennen hoidon aloittamista ja hoidon aikana ennen jokaista infuusiota. Lisäseurantaa on harkittava kliinisen arvioinnin perusteella. Immuunivälitteinen maksatulehdus on hoidettava kohdan Annostus ja antotapa suositusten mukaisesti. Kaikkien vaikeusasteiden tapahtumien hoitoon annetaan kortikosteroideja (prednisoni aloitusannoksella 1–2 mg/kg vuorokaudessa tai vastaava hoito). Annosta pienennetään asteittain.
Immuunivälitteinen paksusuolitulehdus
Immuunivälitteistä paksusuolitulehdusta ja ripulia, joiden hoito määritelmän mukaan edellyttää systeemisten kortikosteroidien käyttöä ja joille ei ole muita selviä syitä, on esiintynyt IMFINZI-valmistetta, IMFINZI-valmisteen ja tremelimumabin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla (ks. kohta Haittavaikutukset). IMFINZI-valmistetta yhdistelmänä tremelimumabin kanssa saaneilla potilailla on ilmoitettu haittavaikutuksina suolen puhkeamia ja paksusuolen puhkeamia. Potilaita on tarkkailtava paksusuolitulehdukseen, ripuliin ja suolen puhkeamaan viittaavien oireiden varalta ja potilasta on hoidettava kohdan Annostus ja antotapa suositusten mukaisesti. Vaikeusasteiden 2–4 tapahtumien hoitoon annetaan kortikosteroideja (prednisoni aloitusannoksella 1–2 mg/kg vuorokaudessa tai vastaava hoito). Annosta pienennetään asteittain. Kirurgia on konsultoitava välittömästi, jos epäillään suolen puhkeamaa, olipa tilan vaikeusaste MIKÄ TAHANSA.
Immuunivälitteiset umpierityssairaudet
Immuunivälitteinen hypotyreoosi, hypertyreoosi ja tyreoidiitti
IMFINZI-valmistetta, IMFINZI-valmisteen ja tremelimumabin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla on ilmennyt immuunivälitteistä hypotyreoosia, hypertyreoosia ja tyreoidiittia, ja hypertyreoosin jälkeen saattaa ilmetä hypotyreoosia (ks. kohta Haittavaikutukset). Potilaiden kilpirauhasen toimintaa on seurattava muutosten varalta ennen hoitoa, säännöllisesti hoidon aikana ja kliiniseen arvioon perustuvan tarpeen mukaan. Immuunivälitteinen hypotyreoosi, hypertyreoosi ja tyreoidiitti on hoidettava kohdan Annostus ja antotapa suositusten mukaisesti. Vaikeusasteiden 2–4 immuunivälitteisen hypotyreoosin hoitoon aloitetaan kilpirauhashormonikorvaushoito kliinisen tarpeen mukaan. Vaikeusasteiden 2–4 immuunivälitteisen hypertyreoosin tai tyreoidiitin hoitoon voidaan antaa oireenmukaista hoitoa.
Immuunivälitteinen lisämunuaisten vajaatoiminta
IMFINZI-valmistetta, IMFINZI-valmisteen ja tremelimumabin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla on ilmennyt immuunivälitteistä lisämunuaisten vajaatoimintaa (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava lisämunuaisten vajaatoimintaan viittaavien kliinisten oireiden ja merkkien varalta. Oireinen lisämunuaisten vajaatoiminta on hoidettava kohdan Annostus ja antotapa suositusten mukaisesti. Vaikeusasteiden 2–4 tapahtumien hoitoon annetaan kortikosteroideja (prednisoni aloitusannoksella 1–2 mg/kg vuorokaudessa tai vastaava hoito). Annosta pienennetään asteittain. Lisäksi annetaan hormonikorvaushoitoa kliinisen tarpeen mukaan.
Immuunivälitteinen tyypin 1 diabetes
IMFINZI-valmistetta, IMFINZI-valmisteen ja tremelimumabin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla on ilmennyt immuunivälitteistä tyypin 1 diabetesta; se voi ensiksi ilmetä diabeettisena ketoasidoosina, joka voi johtaa kuolemaan, ellei sitä todeta varhain (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava tyypin 1 diabetekseen viittaavien kliinisten oireiden ja merkkien varalta. Oireinen tyypin 1 diabetes on hoidettava kohdan Annostus ja antotapa suositusten mukaisesti. Vaikeusasteiden 2–4 tapahtumien hoitoon voidaan aloittaa insuliinihoito kliinisen tarpeen mukaan.
Immuunivälitteinen hypofysiitti/hypopituitarismi
IMFINZI-valmistetta, IMFINZI-valmisteen ja tremelimumabin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla on ilmennyt immuunivälitteistä hypofysiittia ja hypopituitarismia (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava hypofysiittiin tai hypopituitarismiin viittaavien kliinisten oireiden ja merkkien varalta. Oireinen hypofysiitti tai hypopituitarismi on hoidettava kohdan Annostus ja antotapa suositusten mukaisesti. Vaikeusasteiden 2–4 tapahtumien hoitoon annetaan kortikosteroideja (prednisoni aloitusannoksella 1–2 mg/kg vuorokaudessa tai vastaava hoito). Annosta pienennetään asteittain. Lisäksi annetaan hormonikorvaushoitoa kliinisen tarpeen mukaan.
Immuunivälitteinen munuaistulehdus
Immuunivälitteistä munuaistulehdusta, jonka hoito määritelmän mukaan edellyttää systeemisten kortikosteroidien käyttöä ja jolle ei ole muita selviä syitä, on esiintynyt IMFINZI-valmistetta, IMFINZI-valmisteen ja tremelimumabin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla (ks. kohta Haittavaikutukset). Potilaiden munuaisten toimintaa on seurattava muutosten varalta ennen hoitoa IMFINZI-valmisteella tai IMFINZI-valmisteen ja tremelimumabin yhdistelmällä ja säännöllisesti hoidon aikana ja potilasta on hoidettava kohdan Annostus ja antotapa suositusten mukaisesti. Vaikeusasteiden 2–4 tapahtumien hoitoon annetaan kortikosteroideja (prednisoni aloitusannoksella 1–2 mg/kg vuorokaudessa tai vastaava hoito). Annosta pienennetään asteittain.
Immuunivälitteinen ihottuma
Immuunivälitteistä ihottumaa ja dermatiittia (pemfigoidi mukaan lukien), joiden hoito määritelmän mukaan edellyttää systeemisten kortikosteroidien käyttöä ja joille ei ole muita selviä syitä, on esiintynyt IMFINZI-valmistetta, IMFINZI-valmisteen ja tremelimumabin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla. (ks. kohta Haittavaikutukset). PD‑1:n estäjiä saaneilla potilailla on ilmoitettu Stevens-Johnsonin oireyhtymän ja toksisen epidermaalisen nekrolyysin tapauksia. Potilaita on tarkkailtava ihottumaan tai dermatiittiin viittaavien oireiden varalta ja potilasta on hoidettava kohdan Annostus ja antotapa. suositusten mukaisesti. Yli viikon kestäneiden vaikeusasteen 2 tapahtumien sekä vaikeusasteiden 3 ja 4 tapahtumien hoitoon annetaan kortikosteroideja (prednisoni aloitusannoksella 1–2 mg/kg vuorokaudessa tai vastaava hoito). Annosta pienennetään asteittain.
Immuunivälitteinen sydänlihastulehdus
IMFINZI-valmistetta, IMFINZI-valmisteen ja tremelimumabin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla on ilmennyt immuunivälitteistä sydänlihastulehdusta, joka voi johtaa kuolemaan (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava immuunivälitteiseen sydänlihastulehdukseen viittaavien oireiden ja merkkien varalta ja potilasta on hoidettava kohdan Annostus ja antotapa suositusten mukaisesti. Vaikeusasteiden 2–4 tapahtumien hoitoon annetaan kortikosteroideja (prednisoni aloitusannoksella 2–4 mg/kg vuorokaudessa tai vastaava hoito). Annosta pienennetään asteittain. Jos potilaan tila ei parane 2–3 vuorokauden kuluessa kortikosteroidihoidosta huolimatta, on viipymättä aloitettava lisäksi toinen immunosuppressiivinen hoito. Kun haittavaikutus on hävinnyt (vaikeusaste 0), aloitetaan kortikosteroidiannoksen asteittainen pienentäminen, jota jatketaan vähintään 1 kuukauden ajan.
Immuunivälitteinen haimatulehdus
IMFINZI-valmisteen, tremelimumabin ja solunsalpaajahoidon yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla on ilmennyt immuunivälitteistä haimatulehdusta (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava immuunivälitteiseen haimatulehdukseen viittaavien oireiden ja löydösten varalta ja heitä on hoidettava kohdan Annostus ja antotapa suositusten mukaisesti.
Muut immuunivälitteiset haittavaikutukset
IMFINZI-valmisteen tai yhdistelmänä tremelimumabin kanssa käytetyn IMFINZI-valmisteen vaikutusmekanismin vuoksi muita mahdollisia immuunivälitteisiä haittavaikutuksia saattaa ilmetä. Seuraavia immuunivälitteisiä haittavaikutuksia on havaittu IMFINZI-monoterapiaa, IMFINZI-valmisteen ja tremelimumabin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla: myasthenia gravis, transversaalimyeliitti, myosiitti, polymyosiitti, rabdomyolyysi, meningiitti, enkefaliitti, Guillain‑Barrén oireyhtymä, immuunitrombosytopenia, immuunivälitteinen niveltulehdus, uveiitti, ei-infektiivinen virtsarakkotulehdus ja polymyalgia rheumatica (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava oireiden varalta ja potilasta on hoidettava kohdan Annostus ja antotapa suositusten mukaisesti. Vaikeusasteiden 2–4 tapahtumien hoitoon annetaan kortikosteroideja (prednisoni aloitusannoksella 1–2 mg/kg vuorokaudessa tai vastaava hoito). Annosta pienennetään asteittain.
Infuusioon liittyvät reaktiot
Potilaita on tarkkailtava infuusioon liittyvien reaktioiden varalta. IMFINZI-valmistetta, IMFINZI-valmisteen ja tremelimumabin yhdistelmää tai IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla potilailla on ilmoitettu vakavia infuusioon liittyviä reaktioita (ks. kohta Haittavaikutukset). Infuusioon liittyvät reaktiot on hoidettava kohdan Annostus ja antotapa suositusten mukaisesti. Vaikeusasteiden 1 ja 2 tapahtumien kohdalla voidaan harkita infuusioreaktioita ennaltaehkäisevää esilääkitystä. Vaikeusasteiden 3 ja 4 vaikeat infuusioon liittyvät reaktiot hoidetaan hoitoyksikön vakiokäytäntöjen, asianmukaisten kliinisten hoitosuositusten ja/tai lääketieteen alan seurojen hoitosuositusten mukaisesti.
Potilaat, joilla on entuudestaan autoimmuunisairaus
Tiedot havainnoivista tutkimuksista potilailla, joilla on entuudestaan autoimmuunisairaus, viittaavat immuunijärjestelmään liittyvien haittavaikutusten riskin suurenemiseen immuuniaktivaation vapauttajalla toteutetun hoidon jälkeen verrattuna potilaisiin, joilla ei ole entuudestaan autoimmuunisairautta. Myös taustalla olevan autoimmuunisairauden pahenemisjaksoja ilmeni usein, mutta suurin osa niistä oli lieviä ja hoidettavissa.
Tautikohtainen varotoimi (sappitiesyöpä)
Sappitietulehdus ja sappiteiden infektiot
Sappitietulehdus ja sappiteiden infektiot eivät ole harvinaisia pitkälle edennyttä sappitiesyöpää sairastavilla potilailla. TOPAZ-1-tutkimuksessa ilmoitettiin sappitietulehdustapahtumia kummassakin hoitoryhmässä (14,5 % [IMFINZI + solunsalpaajahoito] vs. 8,2 % [lumelääke + solunsalpaajahoito]); ne liittyivät enimmäkseen sappitiestentteihin eivätkä olleet etiologialtaan immuunivälitteisiä. Sappitiesyöpää sairastavia potilaita (etenkin potilaita, joilla on sappitiestentti) on seurattava tiiviisti sappitietulehduksen tai sappiteiden infektioiden kehittymisen varalta ennen hoidon aloittamista ja säännöllisesti sen jälkeen.
Hoitokohtainen varotoimenpide (IMFINZI yhdistelmänä olaparibin kanssa kohdun limakalvon syövässä)
Hematologinen toksisuus
Punasoluaplasiaa (ks. kohta Haittavaikutukset) on ilmoitettu, kun olaparibiylläpitohoitoa on käytetty yhdessä IMFINZI-valmisteen kanssa potilailla, joille oli ensin annettu IMFINZI-valmistetta yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa. Jos potilaalla vahvistetaan olevan punasoluaplasia, IMFINZI- ja olaparibihoito on lopetettava.
Autoimmuunihemolyyttistä anemiaa on ilmoitettu, kun olaparibiylläpitohoitoa on käytetty yhdessä IMFINZI-valmisteen kanssa potilailla, joille oli ensin annettu IMFINZI-valmistetta yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa. Jos potilaalla vahvistetaan olevan autoimmuunihemolyyttinen anemia, IMFINZI- ja olaparibihoito on lopetettava.
Metastasoitunut ei-pienisoluinen keuhkosyöpä
Iäkkäistä (vähintään 75-vuotiaista) potilaista, jotka saavat IMFINZI-hoitoa yhdistelmänä tremelimumabin ja platinapohjaisen solunsalpaajahoidon kanssa, on vain vähän tietoja (ks. kohta Haittavaikutukset ja Farmakodynamiikka). Tähän hoito-ohjelmaan liittyvien mahdollisten hyötyjen ja riskien huolellista potilaskohtaista arviointia suositellaan.
Kliinisistä tutkimuksista pois suljetut potilaat
Seuraavanlaiset potilaat suljettiin pois kliinisistä tutkimuksista: ECOG-suorituskykypistemäärä lähtötilanteessa ≥ 2; aktiivinen tai aiemmin dokumentoitu autoimmuunisairaus kahden vuoden sisällä tutkimuksen aloittamisesta; aiempi immuunipuutos; aiemmin vaikeita immuunivälitteisiä haittavaikutuksia; sairaudet, joiden hoito edellytti systeemistä immunosuppressiota, lukuun ottamatta systeemistä kortikosteroidihoitoa fysiologisella annoksella (prednisoni ≤ 10 mg vuorokaudessa tai vastaava hoito); kontrolloimattomat samanaikaiset sairaudet; aktiivinen tuberkuloosi tai hepatiitti B- tai hepatiitti C -infektio tai HIV-infektio ja potilaat, jotka olivat saaneet 30 päivän kuluessa ennen IMFINZI-hoidon aloittamista tai IMFINZI-hoidon aloittamisen jälkeen eläviä heikennettyjä taudinaiheuttajia sisältävän rokotteen. Koska durvalumabin käytöstä näille potilasryhmille ei ole tietoja, sen käytössä on noudatettava varovaisuutta, ja käytön on perustuttava mahdollisten hyötyjen ja riskien potilaskohtaiseen huolelliseen arviointiin.
Kallon ennaltaehkäisevän sädehoidon (PCI) turvallisuus samanaikaisesti IMFINZI-hoidon kanssa käytettynä levinnyttä pienisoluista keuhkosyöpää sairastavilla potilailla on tuntematon.
Lisätietoja kunkin tutkimuksen poissulkukriteereistä on kohdassa Farmakodynamiikka.
Yhteisvaikutukset
Systeemisten kortikosteroidien fysiologista annosta (prednisoni ≤ 10 mg vuorokaudessa tai vastaava hoito) lukuun ottamatta systeemisten kortikosteroidien tai immuunisalpaajien käyttöä ei suositella ennen durvalumabihoidon aloittamista, koska ne saattavat heikentää durvalumabin farmakodynaamista vaikutusta ja tehoa. Systeemisiä kortikosteroideja tai muita immuunisalpaajia voidaan kuitenkin käyttää durvalumabihoidon aloittamisen jälkeen immuunijärjestelmään liittyvien haittavaikutusten hoitoon (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Durvalumabilla ei ole tehty varsinaisia farmakokineettisiä yhteisvaikutustutkimuksia. Durvalumabin pääasialliset eliminaatioreitit ovat proteiinikatabolia retikuloendoteliaalijärjestelmän kautta ja kohdevälitteinen jakautuminen, joten metabolisia yhteisvaikutuksia ei ole odotettavissa. Durvalumabin farmakokineettisiä yhteisvaikutuksia yhdistelmänä muiden lääkkeiden kanssa verrattiin durvalumabiin CASPIAN‑, POSEIDON‑, HIMALAYA- ja DUO‑E-tutkimuksissa; kliinisesti merkittäviä farmakokineettisiä yhteisvaikutuksia ei todettu.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä durvalumabihoidon aikana ja vähintään 3 kuukauden ajan viimeisen durvalumabiannoksen saamisen jälkeen.
Raskaus
Durvalumabin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Vaikutusmekanisminsa perusteella durvalumabi saattaa vaikuttaa raskauden jatkumiseen. Tiineiden hiirten allogeenisessa mallissa PD-L1-signaalinvälityksen salpauksen on osoitettu lisäävän sikiönmenetyksiä. Durvalumabilla tehdyt eläinkokeet eivät viittaa lisääntymistoksisuuteen (ks. kohta Prekliiniset tiedot turvallisuudesta). Ihmisen IgG1:n tiedetään läpäisevän veri-istukkaesteen, ja eläinkokeissa on varmistettu, että durvalumabi läpäisee veri-istukkaesteen. Jos durvalumabia annetaan raskaana olevalle naiselle, se saattaa vahingoittaa sikiötä, eikä durvalumabin käyttöä suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä tehokasta ehkäisyä hoidon aikana ja vähintään 3 kuukauden ajan viimeisen annoksen saamisen jälkeen.
Imetys
Ei tiedetä, erittyykö durvalumabi ihmisillä äidinmaitoon. Saatavilla olevat toksikologiset tiedot cynomolgus-apinoista ovat osoittaneet, että emon maidossa on pieniä määriä durvalumabia 28. päivänä synnytyksen jälkeen (ks. kohta Prekliiniset tiedot turvallisuudesta). Ihmisillä vasta-aineet saattavat siirtyä rintamaitoon, mutta imeytymisen ja vastasyntyneelle aiheutuvan haitan todennäköisyyttä ei tiedetä. Imetettävään lapseen kohdistuvia mahdollisia riskejä ei voida kuitenkaan sulkea pois. On päätettävä, lopetetaanko imetys vai pidättäydytäänkö durvalumabihoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Ei ole olemassa tietoja durvalumabin mahdollisista vaikutuksista ihmisten tai eläinten hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
IMFINZI-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
IMFINZI monoterapiana
Tiedot IMFINZI-valmisteen turvallisuudesta monoterapiana perustuvat yhdistettyihin tietoihin 4 642 potilaasta, joilla oli erityyppisiä syöpiä. IMFINZI annettiin 10 mg/kg:n annoksella 2 viikon välein, 20 mg/kg:n annoksella 4 viikon välein tai 1 500 mg:n annoksella 4 viikon välein. Yleisimpiä (> 10 %) haittavaikutuksia olivat yskä/limaa tuottava yskä (18,1 %), ripuli (15,1 %), ihottuma (15,0 %), nivelkipu (12,4 %), kuume (12,5 %), vatsakipu (11,8 %), ylähengitystieinfektiot (11,8 %), kutina (11,1 %) ja hypotyreoosi (11,6 %). Yleisimpiä (> 2 %) NCI:n CTCAE-luokituksen mukaisen vaikeusasteen ≥ 3 haittavaikutuksia olivat keuhkokuume (3,4 %) ja kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (2,5 %).
IMFINZI-hoito lopetettiin haittavaikutusten vuoksi 3,9 %:lla potilaista. Yleisimpiä hoidon lopettamiseen johtaneita haittavaikutuksia olivat pneumoniitti (1,1 %) ja keuhkokuume (0,8 %).
IMFINZI-hoitoa lykättiin tai se keskeytettiin haittavaikutusten vuoksi 13,1 %:lla potilaista. Yleisimpiä annoksen antamisen lykkäämiseen tai hoidon keskeyttämiseen johtaneita haittavaikutuksia olivat keuhkokuume (2,3 %) ja kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (2,0 %).
Tiedot IMFINZI-valmisteen turvallisuudesta monoterapiana maksasolusyöpää sairastavien potilaiden hoidossa perustuvat tietoihin 492 potilaasta. Tiedot vastasivat IMFINZI-monoterapian yhdistetyssä tietoaineistossa (N = 4 642) todettua kokonaisturvallisuusprofiilia. Yleisimpiä (> 10 %) haittavaikutuksia olivat kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (20,3 %), vatsakipu (17,9 %), ripuli (15,9 %), kutina (15,4 %) ja ihottuma (15,2 %). Yleisimmät (> 2 %) asteen ≥ 3 haittavaikutukset olivat kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (8,1 %) ja vatsakipu (2,2 %).
IMFINZI-hoito lopetettiin haittavaikutusten vuoksi 3,7 %:lla potilaista. Yleisimpiä hoidon lopettamiseen johtaneita haittavaikutuksia olivat kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (0,8 %) ja maksatulehdus (0,6 %).
IMFINZI-hoitoa lykättiin tai se keskeytettiin haittavaikutusten vuoksi 11,6 %:lla potilaista. Yleisin annoksen antamisen lykkäämiseen tai hoidon keskeyttämiseen johtanut haittavaikutus oli kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (5,9 %).
IMFINZI yhdistelmänä solunsalpaajahoidon kanssa
Tiedot IMFINZI-valmisteen turvallisuudesta, kun sitä annetaan yhdistelmänä solunsalpaajahoidon kanssa, perustuvat 2 244 potilaan yhdistettyihin tietoihin kuudesta tutkimuksesta (TOPAZ-1, CASPIAN, DUO-E, AEGEAN, NIAGARA ja MATTERHORN). Yleisimmät (> 10 %) haittavaikutukset olivat neutropenia (44,6 %), pahoinvointi (42,4 %), väsymys (41,2 %), anemia (37,4 %), ripuli (27,5 %), ummetus (26,7 %), ruokahalun väheneminen (24,0 %), hiustenlähtö (22,0 %), perifeerinen neuropatia (21,6 %), trombosytopenia (19,7 %), ihottuma (19,7 %), oksentelu (18,8 %), vatsakipu (17,9 %), kuume (15,2 %), leukopenia (14,9 %), kutina (12,5 %), hypotyreoosi (11,1 %), nivelkipu (10,9 %), yskä / limaa tuottava yskä (10,8 %), ja kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (10,5 %). Yleisimmät (> 2 %) NCI:n CTCAE-luokituksen asteen ≥ 3 haittavaikutukset olivat neutropenia (28,5 %), anemia (11,9 %), trombosytopenia (5,6 %), leukopenia (4,9 %), väsymys (3,3 %), keuhkokuume (2,5 %), kuumeinen neutropenia (2,4 %) ja ripuli (2,3 %).
IMFINZI-hoito lopetettiin haittavaikutusten vuoksi 6,2 %:lla potilaista. Yleisimmät hoidon lopettamiseen johtaneet haittavaikutukset olivat pneumoniitti (0,8 %), ihottuma (0,6 %), maksatulehdus (0,5 %) ja väsymys (0,5 %).
IMFINZI-hoitoa lykättiin tai se keskeytettiin haittavaikutusten vuoksi 32,4 %:lla potilaista. Yleisimpiä annoksen antamisen lykkäämiseen tai hoidon keskeyttämiseen johtaneita haittavaikutuksia olivat neutropenia (15,5 %), trombosytopenia (4,1 %), anemia (3,3 %), leukopenia (2,0 %), väsymys (2,0 %) ja kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (2,0 %).
IMFINZI yhdistelmänä tremelimumabin (75 mg) ja platinapohjaisen solunsalpaajahoidon kanssa
Tiedot IMFINZI-valmisteen turvallisuudesta, kun sitä annetaan yhdistelmänä tremelimumabin (75 mg) ja solunsalpaajahoidon kanssa, perustuvat 330:n metastasoitunutta ei-pienisoluista keuhkosyöpää sairastaneen potilaan tietoihin. Yleisimpiä (> 20 %) haittavaikutuksia olivat anemia (49,7 %), pahoinvointi (41,5 %), neutropenia (41,2 %), väsymys (36,1 %), ihottuma (25,8 %), trombosytopenia (24,5 %) ja ripuli (21,5 %). Yleisimpiä (> 2 %) NCI:n CTCAE-luokituksen vaikeusasteen ≥ 3 haittavaikutuksia olivat neutropenia (23,9 %), anemia (20,6 %), keuhkokuume (9,4 %), trombosytopenia (8,2 %), leukopenia (5,5 %), väsymys (5,2 %), kohonnut lipaasipitoisuus (3,9 %), kohonnut amylaasipitoisuus (3,6 %), kuumeinen neutropenia (2,4 %), paksusuolitulehdus (2,1 %) ja kohonneet aspartaattiaminotransferaasiarvot tai kohonneet alaniiniaminotransferaasiarvot (2,1 %).
IMFINZI-hoito lopetettiin haittavaikutusten vuoksi 8,5 %:lla potilaista. Yleisimpiä hoidon lopettamiseen johtaneita haittavaikutuksia olivat keuhkokuume (2,1 %) ja paksusuolitulehdus (1,2 %).
IMFINZI-hoito keskeytettiin haittavaikutusten vuoksi 49,4 %:lla potilaista. Yleisimpiä hoidon keskeyttämiseen johtaneita haittavaikutuksia olivat neutropenia (16,1 %), anemia (10,3 %), trombosytopenia (7,3 %), leukopenia (5,8 %), keuhkokuume (5,2 %), kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (4,8 %), paksusuolitulehdus (3,3 %) ja pneumoniitti (3,3 %).
IMFINZI yhdistelmänä tremelimumabin (300 mg) kanssa
Tiedot IMFINZI-valmisteen turvallisuudesta, kun sitä annetaan yhdistelmänä 300 mg:n tremelimumabikerta-annoksen kanssa, perustuvat yhdistettyihin tietoihin 462 potilaasta, joilla oli maksasolusyöpä (maksasolusyöpää koskeva yhdistetty tietoaineisto) ja jotka osallistuivat HIMALAYA-tutkimukseen tai toiseen maksasolusyöpää sairastavilla potilailla tehtyyn tutkimukseen eli tutkimukseen 22. Yleisimpiä (> 10 %) haittavaikutuksia olivat ihottuma (32,5 %), kutina (25,5 %), ripuli (25,3 %), vatsakipu (19,7 %), kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (18,0 %), kuume (13,9 %), hypotyreoosi (13,0 %), yskä / limaa irrottava yskä (10,8 %), perifeerinen turvotus (10,4 %) ja suurentunut lipaasipitoisuus (10,0 %) (ks. taulukko 4). Yleisimpiä vaikeita (NCI:n CTCAE-luokituksen mukainen vaikeusaste ≥ 3) haittavaikutuksia olivat kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (8,9 %), suurentunut lipaasipitoisuus (7,1 %), suurentunut amylaasipitoisuus (4,3 %) ja ripuli (3,9 %).
Yleisimpiä vakavia haittavaikutuksia olivat paksusuolitulehdus (2,6 %), ripuli (2,4 %), keuhkokuume (2,2 %) ja maksatulehdus (1,7 %).
Haittavaikutuksista johtuneen tutkimushoidon lopettamisen yleisyys oli 6,5 %. Yleisimpiä hoidon lopettamiseen johtaneita haittavaikutuksia olivat maksatulehdus (1,5 %) ja kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (1,3 %).
Haittavaikutusten vaikeusasteet arvioitiin CTCAE-luokituksen (Common Terminology Criteria for Adverse Events) perusteella siten, että aste 1 = lievä, aste 2 = keskivaikea, aste 3 = vaikea, aste 4 = henkeä uhkaava ja aste 5 = kuolema.
IMFINZI yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa ja tämän jälkeen IMFINZI yhdistelmänä olaparibin (300 mg kahdesti vuorokaudessa) kanssa
Tiedot IMFINZI-valmisteen turvallisuudesta, kun sitä annetaan yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa ja tämän jälkeen annetaan IMFINZI-valmistetta yhdistelmänä olaparibin (300 mg kahdesti vuorokaudessa) kanssa, perustuvat tietoihin 238 potilaasta, joilla oli kohdun limakalvon syöpä. Yleisimmät (> 20 %) haittavaikutukset olivat anemia (61,8 %), pahoinvointi (54,6 %), väsymys (54,2 %), perifeerinen neuropatia (51,7 %), hiustenlähtö (50,8 %), neutropenia (39,5 %), ummetus (32,8 %), trombosytopenia (29,8 %), ripuli (28,2 %), oksentelu (25,6 %), nivelkipu (24,4 %), ihottuma (23,5 %), vatsakipu (23,5 %), ruokahalun väheneminen (23,1 %) ja leukopenia (20,2 %).
Yleisimpiä (> 2 %) NCI:n CTCAE-luokituksen vaikeusasteen ≥ 3 haittavaikutuksia olivat neutropenia (25,2 %), anemia (23,5 %), leukopenia (6,7 %), trombosytopenia (5,9 %), väsymys (5,5 %), kuumeinen neutropenia (3,4 %), pahoinvointi (2,9 %), kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo (2,9 %) ja perifeerinen neuropatia (2,5 %).
IMFINZI-hoito lopetettiin 4,6 %:lla potilaista. Yleisin hoidon lopettamiseen johtanut haittavaikutus oli pneumoniitti (1,7 %).
IMFINZI-hoito keskeytettiin 38,2 %:lla potilaista. Yleisimpiä hoidon keskeyttämiseen johtaneita haittavaikutuksia olivat anemia (13,4 %), trombosytopenia (11,8 %), neutropenia (10,1 %), leukopenia (2,9 %), hypotyreoosi (2,1 %) ja ylähengitystieinfektio (2,1 %).
Haittavaikutustaulukko
Taulukossa 3 esitetään haittavaikutusten ilmaantuvuus IMFINZI-monoterapian yhdistetyssä turvallisuustietoaineistossa (N = 4 642), potilailla, jotka saivat IMFINZI-hoitoa yhdistelmänä solunsalpaajahoidon kanssa (N = 2 244) ja potilailla, jotka saivat IMFINZI-hoitoa yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa ja tämän jälkeen IMFINZI-hoitoa yhdistelmänä olaparibin kanssa (platinapohjainen solunsalpaajahoito + IMFINZI + olaparibi) (N = 238). Ellei toisin mainita, taulukossa 4 luetellaan haittavaikutusten ilmaantuvuus potilailla, jotka saivat IMFINZI-valmistetta yhdistelmänä tremelimumabin (75 mg) ja platinapohjaisen solunsalpaajahoidon kanssa POSEIDON-tutkimuksessa (N = 330), ja potilailla, jotka saivat IMFINZI-valmistetta yhdistelmänä 300 mg:n tremelimumabikerta-annoksen kanssa maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa (N = 462). Haittavaikutukset on lueteltu MedDRA:n elinjärjestelmäluokituksen mukaan. Haittavaikutukset on esitetty kussakin elinjärjestelmäluokassa haittavaikutuksen esiintymistiheyden mukaan alenevassa järjestyksessä. Lääkkeen kaikkien haittavaikutusten vastaavat esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1 / 1 000, < 1/100), harvinainen (≥ 1 / 10 000, < 1 / 1000), hyvin harvinainen (< 1 / 10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Lääkkeen haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3. Haittavaikutukset potilailla, jotka saivat IMFINZI-hoitoa
| IMFINZI monoterapiana | IMFINZI yhdistelmänä solunsalpaajahoidon kanssa | Platinapohjainen solunsalpaajahoito + IMFINZI + olaparibi* | |
|---|---|---|---|
| Infektiot | |||
| Hyvin yleinen | Ylähengitystieinfektiota | Ylähengitystieinfektioa | |
| Yleinen | Keuhkokuumeb,c, influenssa, suun kandidiaasi, hampaisiin tai suun pehmytkudoksiin liittyvät infektiotd | Keuhkokuumeb,c, ylähengitystieinfektiota, hampaisiin tai suun pehmytkudoksiin liittyvät infektiotd | Keuhkokuume, suun kandidiaasi, hampaisiin tai suun pehmytkudoksiin liittyvät infektiotd |
| Melko harvinainen | Suun kandidiaasi, influenssa | Influenssa | |
| Veri ja imukudos | |||
| Hyvin yleinen | Anemia, leukopeniae, neutropeniaf, trombosytopeniag | Anemiah, leukopeniah, neutropeniah, trombosytopeniah | |
| Yleinen | Kuumeinen neutropenia | Punasoluaplasia, kuumeinen neutropeniah, lymfopeniai | |
| Melko harvinainen | Immuunitrombosytopeniac | Pansytopeniac | Pansytopeniah |
| Harvinainen | Immuunitrombosytopenia | ||
| Immuunijärjestelmä | |||
| Yleinen | Yliherkkyysi,j | ||
| Umpieritys | |||
| Hyvin yleinen | Hypotyreoosik | Hypotyreoosik | Hypotyreoosi |
| Yleinen | Hypertyreoosil | Hypertyreoosil | Hypertyreoosi, tyreoidiitti |
| Melko harvinainen | Tyreoidiittim, lisämunuaisten vajaatoiminta, hypofysiitti/hypopituitarismi, tyypin 1 diabetes | Lisämunuaisten vajaatoiminta, tyypin 1 diabetes, hypofysiitti/hypopituitarismi, tyreoidiittim | |
| Harvinainen | Diabetes insipidus | ||
| Silmät | |||
| Melko harvinainen | Uveiitti | ||
| Harvinainen | Uveiitti | Uveiitti | |
| Aineenvaihdunta ja ravitsemus | |||
| Hyvin yleinen | Ruokahalun väheneminen | Ruokahalun väheneminenh | |
| Hermosto | |||
| Hyvin yleinen | Perifeerinen neuropatian | Perifeerinen neuropatia, huimausi, päänsärkyi, makuhäiriöi,o | |
| Melko harvinainen | Myasthenia gravis, enkefaliittic,p | Myasthenia gravis | |
| Harvinainen | Meningiitti | Enkefaliittip, Guillain–Barrén oireyhtymä | |
| Tuntematon | Guillain–Barrén oireyhtymä, transversaalimyeliittiq | ||
| Verisuonisto | |||
| Yleinen | Tromboemboliset laskimotapahtumati,r | ||
| Harvinainen | Syvä laskimotromboosis | ||
| Sydän | |||
| Melko harvinainen | Sydänlihastulehdus | Akuutti sydäninfarktit,u, sydänlihastulehdusc | |
| Hengityselimet, rintakehä ja välikarsina | |||
| Hyvin yleinen | Yskä / limaa tuottava yskä | Yskä / limaa tuottava yskä | Yskä / limaa tuottava yskä, hengenahdistusi,v |
| Yleinen | Pneumoniittic,w, dysfonia | Pneumoniittic,w, dysfonia | Pneumoniitti, dysfonia |
| Melko harvinainen | Interstitiaalinen keuhkosairaus | Interstitiaalinen keuhkosairausc, keuhkoembolias | Interstitiaalinen keuhkosairaus |
| Ruoansulatuselimistö | |||
| Hyvin yleinen | Ripuli, vatsakipux | Ripuli, vatsakipux, ummetus, pahoinvointi, oksentelu | Ripuli, vatsakipux, ummetush, pahoinvointih, oksenteluh, stomatiittih |
| Yleinen | Stomatiittiy, paksusuolitulehdusz | Dyspepsiai, paksusuolitulehdusz | |
| Melko harvinainen | Paksusuolitulehdusc,z, haimatulehdusaa | Haimatulehdusaa, haiman eksokriininen vajaatoiminta | |
| Harvinainen | Keliakiaq, haiman eksokriininen vajaatoiminta | Keliakiaq | |
| Maksa ja sappi | |||
| Hyvin yleinen | Kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvobb | Kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvo | |
| Yleinen | Maksatulehdusc,cc, kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvoc,bb | Maksatulehdusc,cc | |
| Melko harvinainen | Maksatulehduscc | ||
| Iho ja ihonalainen kudos | |||
| Hyvin yleinen | Ihottumadd, kutina | Ihottumadd, hiustenlähtö, kutina | Ihottumadd, hiustenlähtöh, kutina |
| Yleinen | Yöhikoilu | Dermatiitti | Dermatiittiee |
| Melko harvinainen | Dermatiitti, psoriaasi, pemfigoidiff | Pemfigoidiff, yöhikoilu, psoriaasi | Yöhikoilu |
| Luusto, lihakset ja sidekudos | |||
| Hyvin yleinen | Nivelkipu | Nivelkipu | Nivelkipuh, lihassärky |
| Yleinen | Lihassärky | Lihassärky | |
| Melko harvinainen | Myosiittigg, immuunivälitteinen niveltulehdushh | Immuunivälitteinen niveltulehdushh, myosiittigg | Myosiitti |
| Harvinainen | Polymyosiittiii,, polymyalgia rheumatica | Polymyalgia rheumaticajj | Polymyalgia rheumaticajj |
| Munuaiset ja virtsatiet | |||
| Hyvin yleinen | Kohonnut veren kreatiniiniarvo | ||
| Yleinen | Kohonnut veren kreatiniiniarvo, dysuria | Kohonnut veren kreatiniiniarvo, dysuria | Dysuria |
| Melko harvinainen | Munuaistulehduskk, ei-infektiivinen virtsarakkotulehdus | Ei-infektiivinen virtsarakkotulehdus, munuaistulehduskk | Ei-infektiivinen virtsarakkotulehdush |
| Yleisoireet ja antopaikassa todettavat haitat | |||
| Hyvin yleinen | Kuume | Kuume, väsymysll | Kuume, väsymysh, perifeerinen turvotusmm |
| Yleinen | Perifeerinen turvotusmm | Perifeerinen turvotusmm | |
| Vammat, myrkytykset ja hoitokomplikaatiot | |||
| Yleinen | Infuusioon liittyvä reaktionn | Infuusioon liittyvä reaktionn | Infuusioon liittyvä reaktio |
Haittavaikutusten esiintymistiheyksien ei välttämättä voida täysin katsoa johtuvan pelkästään durvalumabista, vaan ne saattavat liittyä osaltaan perussairauteen tai muihin yhdistelmänä käytettyihin lääkevalmisteisiin.
* kokonaisarviointi hoidosta, johon kuuluu enintään kuusi 21 päivän hoitosykliä platinapohjaista solunsalpaajahoitoa yhdistelmänä IMFINZI-valmisteen kanssa ja sen jälkeen IMFINZI-valmistetta yhdistelmänä olaparibin kanssa.
a sisältää seuraavat: kurkunpäätulehdus, nasofaryngiitti, peritonsillaaripaise, nielutulehdus, riniitti, sinuiitti, tonsilliitti, trakeobronkiitti ja ylähengitystieinfektio
b sisältää seuraavat: Pneumocystis jiroveci -keuhkokuume, keuhkokuume, adenoviruksen aiheuttama keuhkokuume, bakteerikeuhkokuume, sytomegaloviruksen aiheuttama keuhkokuume, Haemophilus-keuhkokuume, pneumokokkikeuhkokuume, streptokokkikeuhkokuume, kandidiaasiin liittyvä keuhkokuume, Klebsiella-keuhkokuume ja Legionella-keuhkokuume.
c mukaan lukien kuolemaan johtaneet tapaukset.
d sisältää seuraavat: ientulehdus, suun infektio, parodontiitti, pulpiitti, hammasabsessi ja hampaan infektio.
e sisältää seuraavat: leukopenia ja valkosolujen niukkuus.
f sisältää seuraavat: neutropenia ja neutrofiilien niukkuus.
g sisältää seuraavat: trombosytopenia ja verihiutaleniukkuus.
h haittavaikutus koskee vain solunsalpaajahoidon haittavaikutuksia DUO-E-tutkimuksessa.
i haittavaikutus koskee vain olaparibin haittavaikutuksia DUO-E-tutkimuksessa.
j sisältää seuraavat: lääkeyliherkkyys ja yliherkkyys.
k sisältää seuraavat: autoimmuunihypotyreoosi, hypotyreoosi, immuunivälitteinen hypotyreoosi, veren suurentunut tyreotropiiniarvo (TSH-arvo).
l sisältää seuraavat: hypertyreoosi, Basedowin tauti, immuunivälitteinen hypertyreoosi ja veren pienentynyt tyreotropiiniarvo (TSH-arvo).
m sisältää seuraavat: autoimmuunityreoidiitti, immuunivälitteinen tyreoidiitti, tyreoidiitti ja subakuutti tyreoidiitti.
n sisältää seuraavat: perifeerinen neuropatia, parestesia ja perifeerinen sensorinen neuropatia.
o sisältää seuraavat: dysgeusia ja makuhäiriö.
p sisältää seuraavat: enkefaliitti, autoimmuunienkefaliitti, immuunivälitteinen enkefaliitti ja ei-infektiivinen enkefaliitti.
q tapaukset ilmoitettu markkinoille tulon jälkeen.
r sisältää seuraavat: syvä laskimotromboosi, embolia, laskimoembolia, lantion laskimotromboosi, pinnallinen laskimotromboosi ja tromboosi.
s haittavaikutus koskee vain oksaliplatiinin haittavaikutuksia MATTERHORN-tutkimuksessa.
t haittavaikutus koskee vain 5‑fluorourasiilin haittavaikutuksia MATTERHORN-tutkimuksessa.
u sisältää seuraavat: äkillinen sepelvaltimo-oireyhtymä, akuutti sydäninfarkti, sydäninfarkti, sydänlihasiskemia ja sepelvaltimotauti.
v sisältää seuraavat: hengenahdistus ja rasitushengenahdistus.
w sisältää seuraavat: pneumoniitti ja immuunivälitteinen keuhkosairaus.
x sisältää seuraavat: vatsakipu, alavatsakipu, ylävatsakipu ja kipu kyljessä.
y sisältää seuraavat: stomatiitti ja limakalvotulehdus.
z sisältää seuraavat: koliitti, enteriitti, enterokoliitti, immuunivälitteinen enterokoliitti ja proktiitti.
aa sisältää seuraavat: haimatulehdus, akuutti haimatulehdus, nekrotisoiva haimatulehdus ja immuunivälitteinen haimatulehdus.
bb sisältää seuraavat: kohonnut alaniiniaminotransferaasiarvo, kohonnut aspartaattiaminotransferaasiarvo, kohonneet maksaentsyymiarvot ja kohonneet transaminaasiarvot.
cc sisältää seuraavat: maksatulehdus, autoimmuunimaksatulehdus, toksinen maksatulehdus, äkillinen maksatulehdus, maksatoksisuus, immuunivälitteinen hepatiitti ja maksasolujen sytolyysi.
dd sisältää seuraavat: erytematoottinen ihottuma, täpläinen ihottuma, makulopapulaarinen ihottuma, näppyläinen ihottuma, kutiava ihottuma, märkärakkulaihottuma, punoitus, ekseema ja ihottuma.
ee sisältää seuraavat: dermatiitti ja immuunivälitteinen dermatiitti.
ff sisältää seuraavat: pemfigoidi, rakkulainen ihottuma ja pemfigus. Loppuun saatetuissa ja meneillään olevissa tutkimuksissa ilmoitettu esiintymistiheys on melko harvinainen.
gg sisältää seuraavat: myosiitti ja rabdomyolyysi.
hh sisältää seuraavat: autoimmuuniniveltulehdus, immuunivälitteinen niveltulehdus, moniniveltulehdus ja nivelreuma.
ii meneillään olevassa, sponsoroidussa kliinisessä tutkimuksessa yhdistetyn tietoaineiston ulkopuolella havaittiin polymyosiitti (kuolemaan johtanut) IMFINZI-hoitoa saaneella potilaalla.
jj ei havaittu IMFINZI-valmisteen ja solunsalpaajahoidon yhdistelmää koskevassa yhdistetyssä tietoaineistossa eikä platinapohjaisen solunsalpaajahoidon, IMFINZI-valmisteen ja olaparibin yhdistelmää koskevassa tietoaineistossa, mutta havaittiin muissa AstraZeneca-yhtiön toimeksiantamissa kliinisissä tutkimuksissa.
kk sisältää seuraavat: autoimmuuninefriitti, tubulointerstitiaalinen nefriitti, munuaistulehdus, munuaiskerästulehdus, membranoosi munuaiskerästulehdus ja immuunivälitteinen munuaistulehdus.
ll sisältää seuraavat: väsymys ja astenia.
mm sisältää seuraavat: perifeerinen edeema ja perifeerinen turvotus.
nn sisältää seuraavat: infuusioon liittyvä reaktio ja nokkosihottuma, joka alkaa annostelupäivänä tai annostelua seuraavana päivänä.
Taulukko 4. Haittavaikutukset potilailla, jotka saivat IMFINZI-valmistetta yhdistelmänä tremelimumabin kanssa
| IMFINZI yhdistelmänä tremelimumabin (75 mg) ja platinapohjaisen solunsalpaajahoidon kanssa | IMFINZI yhdistelmänä tremelimumabin (300 mg) kanssa | |
|---|---|---|
| Infektiot | ||
| Hyvin yleinen | Ylähengitystieinfektiota, keuhkokuumeb | |
| Yleinen | Influenssa, suun kandidiaasi | Ylähengitystieinfektiota, keuhkokuumeb, influenssa, hampaisiin tai suun pehmytkudoksiin liittyvät infektiotc |
| Melko harvinainen | Hampaisiin tai suun pehmytkudoksiin liittyvät infektiotc | Suun kandidiaasi |
| Veri ja imukudos | ||
| Hyvin yleinen | Anemiad, neutropeniad,e, trombosytopeniad,f, leukopeniad,g | |
| Yleinen | Kuumeinen neutropeniad, pansytopeniad | |
| Melko harvinainen | Immuunitrombosytopenia | |
| Tuntematon | Immuunitrombosytopeniah | |
| Umpieritys | ||
| Hyvin yleinen | Hypotyreoosii | Hypotyreoosii |
| Yleinen | Hypertyreoosij, lisämunuaisten vajaatoiminta, hypopituitarismi/hypofysiitti, tyreoidiittik | Hypertyreoosij, tyreoidiittik, lisämunuaisten vajaatoiminta
|
| Melko harvinainen | Diabetes insipidus, tyypin 1 diabetes | Hypopituitarismi/hypofysiitti |
| Tuntematon | Diabetes insipidush, tyypin 1 diabetesh | |
| Silmät | ||
| Melko harvinainen | Uveiitti | |
| Harvinainen | Uveiittih | |
| Aineenvaihdunta ja ravitsemus | ||
| Hyvin yleinen | Vähentynyt ruokahalud | |
| Hermosto | ||
| Yleinen | Perifeerinen neuropatiad,l | |
| Melko harvinainen | Enkefaliittim, | Myasthenia gravis, meningiitti |
| Tuntematon | Myasthenia gravisn, Guillain–Barrén oireyhtymän, meningiittin, transversaalimyeliittio | Guillain–Barrén oireyhtymäh, enkefaliittih, transversaalimyeliittio
|
| Sydän | ||
| Melko harvinainen | Sydänlihastulehdusp | Sydänlihastulehdus |
| Hengityselimet, rintakehä ja välikarsina | ||
| Hyvin yleinen | Yskä / limaa irrottava yskä | Yskä / limaa irrottava yskä |
| Yleinen | Pneumoniittiq, dysfonia | Pneumoniittiq |
| Melko harvinainen | Interstitiaalinen keuhkosairaus | Dysfonia, interstitiaalinen keuhkosairaus |
| Ruoansulatuselimistö | ||
| Hyvin yleinen | Pahoinvointid, ripuli, ummetusd, oksentelud | Ripuli, vatsakipur |
| Yleinen | Stomatiittid,s, kohonnut amylaasipitoisuus, vatsakipur, kohonnut lipaasipitoisuus, paksusuolitulehdust, haimatulehdusu | Kohonnut lipaasipitoisuus, kohonnut amylaasipitoisuus, paksusuolitulehdust, haimatulehdusu
|
| Harvinainen | Keliakian | Keliakiah |
| Tuntematon | Suolen puhkeaman, paksusuolen puhkeaman | Suolen puhkeamah, paksusuolen puhkeamah |
| Maksa ja sappi | ||
| Hyvin yleinen | Kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvov | Kohonnut aspartaattiaminotransferaasiarvo tai kohonnut alaniiniaminotransferaasiarvov |
| Yleinen | Maksatulehdusw | Maksatulehdusw |
| Iho ja ihonalainen kudos | ||
| Hyvin yleinen | Hiustenlähtöd, ihottumax, kutina | Ihottumax, kutina |
| Yleinen | Dermatiittiy, yöhikoilu, | |
| Melko harvinainen | Dermatiitti, yöhikoilu, pemfigoidi | Pemfigoidi |
| Luusto, lihakset ja sidekudos | ||
| Hyvin yleinen | Nivelkipu | |
| Yleinen | Lihassärky | Lihassärky |
| Melko harvinainen | Myosiittiz, polymyosiittiz, immuunivälitteinen niveltulehdusn | Myosiittiz, polymyosiittiz, immuunivälitteinen niveltulehdus, polymyalgia rheumatica |
| Tuntematon | Polymyalgia rheumatican | |
| Munuaiset ja virtsatiet | ||
| Yleinen | Kohonnut veren kreatiniiniarvo, dysuria | Kohonnut veren kreatiniiniarvo, dysuria |
| Melko harvinainen | Munuaistulehdus, ei-infektiivinen virtsarakkotulehdus | Munuaistulehdusaa |
| Tuntematon | Ei-infektiivinen virtsarakkotulehdush | |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Hyvin yleinen | Väsymysd, kuume | Kuume, perifeerinen turvotusbb |
| Yleinen | Perifeerinen turvotusbb | |
| Vammat, myrkytykset ja hoitokomplikaatiot | ||
| Yleinen | Infuusioon liittyvä reaktiocc | Infuusioon liittyvä reaktiocc |
a Sisältää seuraavat: kurkunpäätulehdus, nasofaryngiitti, nielutulehdus, riniitti, sinuiitti, tonsilliitti, trakeobronkiitti ja ylähengitystieinfektio.
b Sisältää seuraavat: Pneumocystis jiroveci -keuhkokuume, keuhkokuume ja bakteerikeuhkokuume.
c Sisältää seuraavat: parodontiitti, pulpiitti, hammasabsessi ja hampaan infektio.
d Tällä haittavaikutuksella tarkoitetaan ainoastaan solunsalpaajahoidon haittavaikutuksia POSEIDON-tutkimuksessa.
e Sisältää seuraavat: neutropenia ja neutrofiilien niukkuus.
f Sisältää seuraavat: verihiutaleniukkuus ja trombosytopenia.
g Sisältää seuraavat: leukopenia ja valkosolujen niukkuus.
h Haittavaikutusta ei havaittu maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa, mutta sitä ilmoitettiin AstraZeneca-yhtiön toimeksiantamissa kliinisissä tutkimuksissa potilailla, jotka olivat saaneet IMFINZI-valmistetta tai IMFINZI-valmistetta ja tremelimumabia.
i Sisältää seuraavat: veren suurentunut tyreotropiiniarvo (TSH-arvo), hypotyreoosi ja immuunivälitteinen hypotyreoosi.
j Sisältää seuraavat: veren pienentynyt tyreotropiiniarvo (TSH-arvo) ja hypertyreoosi.
k Sisältää seuraavat: autoimmuunityreoidiitti, immuunivälitteinen tyreoidiitti, tyreoidiitti ja subakuutti tyreoidiitti.
l Sisältää seuraavat: perifeerinen neuropatia, parestesia ja perifeerinen sensorinen neuropatia.
m Sisältää seuraavat: enkefaliitti ja autoimmuunienkefaliitti.
n Haittavaikutusta ei havaittu POSEIDON-tutkimuksessa, mutta sitä ilmoitettiin kliinisissä tutkimuksissa POSEIDON-tietoaineiston ulkopuolella potilailla, jotka olivat saaneet IMFINZI-valmistetta tai IMFINZI-valmistetta ja tremelimumabia.
oIlmoitettiin tutkimuksissa POSEIDON-tutkimuksen ja maksasolusyöpää koskevan yhdistetyn tietoaineiston ulkopuolella.
p Sisältää autoimmuunimyokardiitin.
q Sisältää seuraavat: immuunivälitteinen pneumoniitti ja pneumoniitti.
r Sisältää seuraavat: vatsakipu, alavatsakipu, ylävatsakipu ja kipu kyljessä.
s Sisältää seuraavat: limakalvotulehdus ja stomatiitti.
t Sisältää seuraavat: koliitti, enteriitti ja enterokoliitti.
u Sisältää seuraavat: autoimmuunihaimatulehdus, haimatulehdus ja akuutti haimatulehdus.
v Sisältää seuraavat: kohonnut alaniiniaminotransferaasiarvo, kohonnut aspartaattiaminotransferaasiarvo, kohonneet maksaentsyymiarvot ja kohonneet transaminaasiarvot.
w Sisältää seuraavat: autoimmuunimaksatulehdus, maksatulehdus, maksasoluvaurio, maksatoksisuus, äkillinen maksatulehdus ja immuunivälitteinen hepatiitti.
x Sisältää seuraavat: ekseema, punoitus, ihottuma, makulaarinen ihottuma, makulopapulaarinen ihottuma, papulaarinen ihottuma, kutiava ihottuma ja märkärakkulainen ihottuma.
y Sisältää seuraavat: dermatiitti ja immuunivälitteinen dermatiitti.
z Sisältää seuraavat: rabdomyolyysi, myosiitti ja polymyosiitti.
aa Sisältää seuraavat: autoimmuunimunuaistulehdus ja immuunivälitteinen munuaistulehdus.
bb Sisältää seuraavat: perifeerinen edeema ja perifeerinen turvotus.
cc Sisältää seuraavat: infuusioon liittyvä reaktio ja nokkosihottuma.
Valikoitujen haittavaikutusten kuvaus
IMFINZI-hoitoon liittyy immuunivälitteisiä haittavaikutuksia. Useimmat niistä, vaikeat reaktiot mukaan lukien, häviävät asianmukaisen hoidon aloittamisen ja/tai IMFINZI-hoidon muuttamisen jälkeen. Seuraavia immuunivälitteisiä haittavaikutuksia koskevat tiedot ovat IMFINZI-monoterapiaa koskevasta yhdistetystä turvallisuustietokannasta, joka sisälsi tiedot PACIFIC‑, HIMALAYA- ja ADRIATIC-tutkimuksiin sekä lisätutkimuksiin osallistuneista 4 642 potilaasta. Lisätutkimusten potilailla oli erilaisia kiinteitä kasvaimia, ja niissä arvioitiin käyttöaiheita, joihin durvalumabia ei ole hyväksytty. IMFINZI-valmistetta annettiin kaikissa tutkimuksissa 10 mg/kg kahden viikon välein, 20 mg/kg 4 viikon välein tai 1 500 mg 3 tai 4 viikon välein. Solunsalpaajahoidon kanssa yhdistelmänä annetun IMFINZI-hoidon merkittävien haittavaikutusten tiedot esitetään tapauksissa, joissa havaittiin kliinisesti oleellisia eroja IMFINZI-monoterapiaan nähden.
Tiedot seuraavista immuunivälitteisistä haittavaikutuksista perustuvat myös tietoihin 2 280 potilaasta, jotka saivat joko IMFINZI-valmistetta (20 mg/kg) 4 viikon välein yhdistelmänä tremelimumabin (1 mg/kg) kanssa tai IMFINZI-valmistetta (1 500 mg) yhdistelmänä tremelimumabin (75 mg) kanssa 4 viikon välein. Tremelimumabin ja platinapohjaisen solunsalpaajahoidon kanssa yhdistelmänä annetun IMFINZI-hoidon merkittävien haittavaikutusten tiedot on esitetty tapauksissa, joissa havaittiin kliinisesti oleellisia eroja verrattuna IMFINZI-valmisteen käyttöön yhdistelmänä tremelimumabin kanssa.
Seuraavia immuunivälitteisiä haittavaikutuksia koskevat tiedot perustuvat myös tremelimumabin (300 mg) kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskevaan yhdistettyyn turvallisuustietokantaan, johon kuului 462 potilasta, joilla oli maksasolusyöpä (maksasolusyöpää koskeva yhdistetty tietoaineisto). Näissä kahdessa tutkimuksessa IMFINZI-valmistetta annettiin 1 500 mg:n annoksina yhdistelmänä tremelimumabin (300 mg) kanssa 4 viikon välein.
Näiden haittavaikutusten hoitosuositukset on kuvattu kohdissa Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.
Immuunivälitteinen pneumoniitti
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa (n = 4 642, useita kasvaintyyppejä) immuunivälitteistä pneumoniittia oli todettu 147 potilaalla (3,2 %). Näistä vaikeusasteen 3 tapahtumia oli 37 potilaalla (0,8 %), vaikeusasteen 4 tapahtumia 2 potilaalla (< 0,1 %) ja vaikeusasteen 5 tapahtumia 10 potilaalla (0,2 %). Mediaaniaika immuunivälitteisen pneumoniitin ilmaantumiseen oli 56 päivää (vaihteluväli: 1 – 1 308 päivää). 147 potilaasta 114 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa) ja 4 potilasta sai myös muita immuunisalpaajia, kuten infliksimabia tai siklosporiinia. IMFINZI-hoito lopetettiin 60 potilaalla. Pneumoniitti parani 85 potilaalla.
Immuunivälitteistä pneumoniittia ilmeni PACIFIC-tutkimuksessa enemmän potilailla, joilla samanaikaisen kemosädehoidon päättymisestä oli 1–42 päivää ennen tutkimushoidon aloittamista (10,7 %), kuin muilla yhdistetyn turvallisuustietokannan potilailla (1,0 %).
PACIFIC-tutkimuksessa (n = 475 IMFINZI-hoitohaarassa ja n = 234 lumehaarassa) ilmeni immuunivälitteistä pneumoniittia 47 potilaalla (9,9 %) IMFINZI-valmistetta saaneiden ryhmässä ja 14 potilaalla (6,0 %) lumeryhmässä. Näistä vaikeusasteen 3 tapahtumia oli 9 potilaalla (1,9 %) IMFINZI-ryhmässä ja 6 potilaalla (2,6 %) lumeryhmässä sekä vaikeusasteen 5 (kuolemaan johtaneita) tapahtumia 4 potilaalla (0,8 %) IMFINZI-ryhmässä ja 3 potilaalla (1,3 %) lumeryhmässä. Mediaaniaika pneumoniitin ilmaantumiseen oli IMFINZI-ryhmässä 46 päivää (vaihteluväli: 2–342 päivää) ja lumeryhmässä 57 päivää (vaihteluväli: 26–253 päivää). IMFINZI-valmistetta saaneiden ryhmässä kaikki potilaat saivat systeemistä kortikosteroidia, ja näistä 30 potilasta sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa) ja 2 potilasta sai myös infliksimabia. Lumeryhmässä kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja näistä 12 potilasta sai suuriannoksista kortikosteroidihoitoa ja 1 potilas sai myös syklofosfamidia ja takrolimuusia. Pneumoniitti parani 29 potilaalla IMFINZI-ryhmässä ja 6 potilaalla lumeryhmässä.
ADRIATIC-tutkimuksessa rajoittunutta pienisoluista keuhkosyöpää sairastavilla potilailla (n = 262 IMFINZI-hoitohaarassa ja n = 265 lumehaarassa) ilmeni immuunivälitteistä pneumoniittia 31 potilaalla (11,8 %) IMFINZI-valmistetta saaneiden ryhmässä ja 8 potilaalla (3,0 %) lumeryhmässä. Näistä vaikeusasteen 3 tapahtumia oli 5 potilaalla (1,9 %) IMFINZI-ryhmässä ja 1 potilaalla (0,4 %) lumeryhmässä. Yhdellä potilaalla (0,4 %) IMFINZI-ryhmässä ilmeni vaikeusasteen 5 (kuolemaan johtanut) tapahtuma. Mediaaniaika immuunivälitteisen pneumoniitin ilmaantumiseen oli IMFINZI-ryhmässä 55 päivää (vaihteluväli: 1–375 päivää) ja lumeryhmässä 65,5 päivää (vaihteluväli: 24–124 päivää). IMFINZI-valmistetta saaneiden ryhmässä kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja näistä 25 potilasta sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa) ja 1 potilas sai myös infliksimabia. Lumeryhmässä kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja näistä 7 potilasta sai suuriannoksista kortikosteroidihoitoa. Pneumoniitti parani 18 potilaalla IMFINZI-ryhmässä ja 3 potilaalla lumeryhmässä.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) immuunivälitteistä pneumoniittia oli todettu 86 potilaalla (3,8 %). Näistä vaikeusasteen 3 tapahtumia oli 30 potilaalla (1,3 %), vaikeusasteen 4 tapahtuma 1 potilaalla (< 0,1 %) ja vaikeusasteen 5 (eli kuolemaan johtaneita) tapahtumia 7 potilaalla (0,3 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 57 päivää (vaihteluväli: 8–912 päivää). Kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja 86 potilaasta 79 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Seitsemän potilasta sai myös muita immuunisalpaajia. Hoito lopetettiin 39 potilaalla. Immuunivälitteinen pneumoniitti parani 51 potilaalla.
Maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa (n = 462) immuunivälitteistä pneumoniittia oli todettu 6 potilaalla (1,3 %). Näistä vaikeusasteen 3 tapahtumia oli 1 potilaalla (0,2 %) ja vaikeusasteen 5 (eli kuolemaan johtaneita) tapahtumia 1 potilaalla (0,2 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 29 päivää (vaihteluväli: 5–774 päivää). Kuusi potilasta sai systeemistä kortikosteroidihoitoa, ja näistä kuudesta potilaasta viisi sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg vuorokaudessa tai vastaavaa hoitoa). Yksi potilas sai myös muita immuunisalpaajia. Hoito lopetettiin 2 potilaalla. Immuunivälitteinen pneumoniitti parani 3 potilaalla.
DUO-E-tutkimuksessa 238 potilasta sai platinapohjaista solunsalpaajahoitoa yhdistelmänä IMFINZI-valmisteen kanssa ja tämän jälkeen IMFINZI-valmistetta yhdistelmänä olaparibin kanssa (platinapohjainen solunsalpaajahoito + IMFINZI + olaparibi -hoitohaara), ja heistä 5:llä (2,1 %) ilmeni immuunivälitteistä pneumoniittia. Näistä vaikeusasteen 3 tapahtumia oli 3 potilaalla (1,3 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 85 päivää (vaihteluväli: 65–321 päivää). 5 potilasta sai systeemistä kortikosteroidihoitoa, ja heistä 4 potilasta sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg vuorokaudessa tai vastaavaa hoitoa). Immuunivälitteinen pneumoniitti parani kaikilla 5 potilaalla.
Immuunivälitteinen maksatulehdus
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa immuunivälitteistä maksatulehdusta oli todettu 120 potilaalla (2,6 %). Näistä vaikeusasteen 3 tapahtumia oli 70 potilaalla (1,5 %), vaikeusasteen 4 tapahtumia 9 potilaalla (0,2 %) ja vaikeusasteen 5 (kuolemaan johtaneita) tapahtumia 6 potilaalla (0,1 %). Mediaaniaika immuunivälitteisen maksatulehduksen ilmaantumiseen oli 36 päivää (vaihteluväli: 1–644 päivää). 120 potilaasta 94 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Yhdeksän potilasta sai myös muita immuunisalpaajia, kuten mykofenolaattia. IMFINZI-hoito lopetettiin 30 potilaalla. Immuunivälitteinen maksatulehdus parani 56 potilaalla.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) immuunivälitteistä maksatulehdusta oli todettu 80 potilaalla (3,5 %). Näistä vaikeusasteen 3 tapahtumia oli 48 potilaalla (2,1 %), vaikeusasteen 4 tapahtumia 8 potilaalla (0,4 %) ja vaikeusasteen 5 (eli kuolemaan johtaneita) tapahtumia 2 potilaalla (< 0,1 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 36 päivää (vaihteluväli: 1–533 päivää). Kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja 80 potilaasta 68 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Kahdeksan potilasta sai myös muita immuunisalpaajia. Hoito lopetettiin 27 potilaalla. Immuunivälitteinen maksatulehdus parani 47 potilaalla.
Maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa (n = 462) immuunivälitteistä maksatulehdusta oli todettu 34 potilaalla (7,4 %). Näistä vaikeusasteen 3 tapahtumia oli 20 potilaalla (4,3 %), vaikeusasteen 4 tapahtumia 1 potilaalla (0,2 %) ja vaikeusasteen 5 (eli kuolemaan johtaneita) tapahtumia 3 potilaalla (0,6 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 29 päivää (vaihteluväli: 13–313 päivää). Kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja 34 potilaasta 32 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg vuorokaudessa tai vastaavaa hoitoa). Yhdeksän potilasta sai myös muita immuunisalpaajia. Hoito lopetettiin 10 potilaalla. Immuunivälitteinen maksatulehdus parani 13 potilaalla.
Immuunivälitteinen paksusuolitulehdus
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa immuunivälitteistä paksusuolitulehdusta tai ripulia oli todettu 79 potilaalla (1,7 %). Näistä vaikeusasteen 3 tapahtumia oli 15 potilaalla (0,3 %) ja vaikeusasteen 4 tapahtumia 2 potilaalla (< 0,1 %). Mediaaniaika immuunivälitteisen paksusuolitulehduksen ilmaantumiseen oli 72 päivää (vaihteluväli: 1–920 päivää). 79 potilaasta 55 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Viisi potilasta sai myös muita immuunisalpaajia, kuten infliksimabia tai mykofenolaattia. IMFINZI-hoito lopetettiin 15 potilaalla. Immuunivälitteinen paksusuolitulehdus parani 54 potilaalla.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) immuunivälitteistä paksusuolitulehdusta tai ripulia oli todettu 167 potilaalla (7,3 %). Näistä vaikeusasteen 3 tapahtumia oli 76 potilaalla (3,3 %) ja vaikeusasteen 4 tapahtumia 3 potilaalla (0,1 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 57 päivää (vaihteluväli: 3–906 päivää). Kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja 167 potilaasta 151 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). 22 potilasta sai myös muita immuunisalpaajia. Hoito lopetettiin 54 potilaalla. Immuunivälitteinen paksusuolitulehdus tai ripuli parani 141 potilaalla.
IMFINZI-valmistetta yhdistelmänä tremelimumabin kanssa saaneilla potilailla on ilmoitettu melko harvinaisina haittavaikutuksina suolen puhkeamia ja paksusuolen puhkeamia.
Maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa (n = 462) immuunivälitteistä paksusuolitulehdusta tai ripulia oli todettu 31 potilaalla (6,7 %). Näistä vaikeusasteen 3 tapahtumia oli 17 potilaalla (3,7 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 23 päivää (vaihteluväli: 2–479 päivää). Kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja 31 potilaasta 28 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg vuorokaudessa tai vastaavaa hoitoa). Neljä potilasta sai myös muita immuunisalpaajia. Hoito lopetettiin 5 potilaalla. Immuunivälitteinen paksusuolitulehdus tai ripuli parani 29 potilaalla.
IMFINZI-valmistetta yhdistelmänä tremelimumabin kanssa saaneilla potilailla on havaittu (harvinaisena haittavaikutuksena) suolen puhkeamia tutkimuksissa, jotka eivät sisältyneet maksasolusyöpää koskevaan tietoaineistoon.
Immuunivälitteiset umpierityssairaudet
Immuunivälitteinen hypotyreoosi
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa immuunivälitteistä hypotyreoosia ilmeni 384 potilaalla (8,3 %), mukaan lukien asteen 3 haitat 7 potilaalla (0,2 %). Mediaaniaika haitan ilmaantumiseen oli 90,5 päivää (vaihteluväli: 1–951 päivää). 384 potilaasta 379 sai hormonikorvaushoitoa ja 7 potilasta sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa) immuunivälitteisen hypotyreoosin hoitoon. Yksi potilas lopetti IMFINZI-hoidon immuunivälitteisen hypotyreoosin vuoksi. Immuunivälitteinen hypotyreoosi parani 79 potilaalla.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) immuunivälitteistä hypotyreoosia oli todettu 209 potilaalla (9,2 %). Näistä vaikeusasteen 3 tapahtumia oli 6 potilaalla (0,3 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 85 päivää (vaihteluväli: 1–624 päivää). 13 potilasta sai systeemistä kortikosteroidihoitoa, ja näistä 13 potilaasta 8 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Hoito lopetettiin 3 potilaalla. Immuunivälitteinen hypotyreoosi parani 52 potilaalla. Immuunivälitteistä hypotyreoosia edelsi immuunivälitteinen hypertyreoosi 25 potilaalla ja immuunivälitteinen tyreoidiitti 2 potilaalla.
Maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa (n = 462) immuunivälitteistä hypotyreoosia oli todettu 46 (10,0 %) potilaalla. Mediaaniaika haittavaikutuksen ilmaantumiseen oli 85 päivää (vaihteluväli: 26–763 päivää). Yksi potilas sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg vuorokaudessa tai vastaavaa hoitoa). Kaikki potilaat tarvitsivat muuta hoitoa, kuten hormonikorvaushoitoa. Immuunivälitteinen hypotyreoosi parani 6 potilaalla. Immuunivälitteistä hypotyreoosia edelsi immuunivälitteinen hypertyreoosi 4 potilaalla.
Immuunivälitteinen hypertyreoosi
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa immuunivälitteistä hypertyreoosia ilmeni 76 potilaalla (1,6 %). Mediaaniaika haitan ilmaantumiseen oli 43 päivää (vaihteluväli: 1–253 päivää). 76 potilaasta 71 sai lääkehoitoa (tiamatsolia, karbimatsolia, propyylitiourasiilia, perkloraattia, kalsiuminestäjää tai beetasalpaajaa), 15 potilasta sai systeemistä kortikosteroidihoitoa, ja näistä 15 potilaasta 8 sai suuriannoksista systeemistä kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Yksi potilas lopetti IMFINZI-hoidon immuunivälitteisen hypertyreoosin vuoksi. Immuunivälitteinen hypertyreoosi parani 62 potilaalla. 31 potilaalla todettiin hypotyreoosi hypertyreoosin jälkeen.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) immuunivälitteistä hypertyreoosia oli todettu 62 potilaalla (2,7 %). Näistä vaikeusasteen 3 tapahtumia oli 5 potilaalla (0,2 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 33 päivää (vaihteluväli: 4–176 päivää). 18 potilasta sai systeemistä kortikosteroidihoitoa, ja näistä 18 potilaasta 11 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). 53 potilasta tarvitsi muuta hoitoa (tiamatsolia, karbimatsolia, propyylitiourasiilia, perkloraattia, kalsiuminestäjää tai beetasalpaajaa). Yksi potilas lopetti hoidon hypertyreoosin vuoksi. Immuunivälitteinen hypertyreoosi parani 47 potilaalla.
Maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa (n = 462) immuunivälitteistä hypertyreoosia oli todettu 21 potilaalla (4,5 %). Näistä 1 potilaalla (0,2 %) oli todettu vaikeusasteen 3 tapahtuma. Mediaaniaika haittavaikutuksen ilmaantumiseen oli 30 päivää (vaihteluväli: 13–60 päivää). 4 potilasta sai systeemistä kortikosteroidihoitoa, ja kaikki 4 potilasta saivat suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg vuorokaudessa tai vastaavaa hoitoa). 20 potilasta tarvitsi muuta hoitoa (tiamatsolia, karbimatsolia, propyylitiourasiilia, perkloraattia, kalsiuminestäjää tai beetasalpaajaa). Yksi potilas lopetti hoidon hypertyreoosin vuoksi. Immuunivälitteinen hypertyreoosi parani 17 potilaalla.
Immuunivälitteinen tyreoidiitti
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa immuunivälitteinen tyreoidiitti oli todettu 21 potilaalla (0,5 %). Näistä 2 potilaalla (< 0,1 %) oli todettu vaikeusasteen 3 tapahtuma. Mediaaniaika haitan ilmaantumiseen oli 57 päivää (vaihteluväli: 14–217 päivää). 21 potilaasta 18 sai hormonikorvaushoitoa ja 3 potilasta sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Yksi potilas lopetti IMFINZI-hoidon immuunivälitteisen tyreoidiitin vuoksi. Immuunivälitteinen tyreoidiitti parani 8 potilaalla. Viidellä potilaalla todettiin hypotyreoosi tyreoidiitin jälkeen.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) immuunivälitteistä tyreoidiittia oli todettu 15 potilaalla (0,7 %). Näistä 1 potilaalla (< 0,1 %) oli todettu vaikeusasteen 3 tapahtuma. Mediaaniaika haittavaikutuksen ilmaantumiseen oli 57 päivää (vaihteluväli: 22–141 päivää). 5 potilasta sai systeemistä kortikosteroidihoitoa, ja näistä 5 potilaasta 2 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). 13 potilasta tarvitsi muuta hoitoa, kuten hormonikorvaushoitoa, tiamatsolia, karbimatsolia, propyylitiourasiilia, perkloraattia, kalsiuminestäjää tai beetasalpaajaa. Yksikään potilas ei lopettanut hoitoa immuunivälitteisen tyreoidiitin vuoksi. Immuunivälitteinen tyreoidiitti parani 5 potilaalla.
Maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa (n = 462) immuunivälitteistä tyreoidiittia oli todettu 6 potilaalla (1,3 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 56 päivää (vaihteluväli: 7–84 päivää). 2 potilasta sai systeemistä kortikosteroidihoitoa, ja näistä kahdesta potilaasta toinen sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg vuorokaudessa tai vastaavaa hoitoa). Kaikki potilaat tarvitsivat muuta hoitoa, kuten hormonikorvaushoitoa. Immuunivälitteinen tyreoidiitti parani 2 potilaalla.
Immuunivälitteinen lisämunuaisten vajaatoiminta
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa immuunivälitteistä lisämunuaisten vajaatoimintaa ilmeni 24 potilaalla (0,5 %), mukaan lukien asteen 3 haitat 8 potilaalla (0,2 %). Mediaaniaika haitan ilmaantumiseen oli 157,5 päivää (vaihteluväli: 20–547 päivää). Kaikki 24 potilasta saivat systeemistä kortikosteroidihoitoa ja 24 potilaasta 8 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Yksi potilas lopetti IMFINZI-hoidon immuunivälitteisen lisämunuaisten vajaatoiminnan vuoksi. Immuunivälitteinen lisämunuaisten vajaatoiminta parani 6 potilaalla.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) immuunivälitteistä lisämunuaisten vajaatoimintaa oli todettu 33 potilaalla (1,4 %). Näistä vaikeusasteen 3 tapahtumia oli 16 potilaalla (0,7 %) ja vaikeusasteen 4 tapahtuma 1 potilaalla (< 0,1 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 105 päivää (vaihteluväli: 20–428 päivää). 32 potilasta sai systeemistä kortikosteroidihoitoa, ja näistä 32 potilaasta 10 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Hoito lopetettiin 1 potilaalla. Immuunivälitteinen lisämunuaisten vajaatoiminta parani 11 potilaalla.
Maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa (n = 462) immuunivälitteistä lisämunuaisten vajaatoimintaa oli todettu 6 potilaalla (1,3 %). Näistä 1 potilaalla (0,2 %) oli todettu vaikeusasteen 3 tapahtuma. Mediaaniaika haittavaikutuksen ilmaantumiseen oli 64 päivää (vaihteluväli: 43–504 päivää). Kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja näistä kuudesta potilaasta yksi sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg vuorokaudessa tai vastaavaa hoitoa). Immuunivälitteinen lisämunuaisten vajaatoiminta parani 2 potilaalla.
Immuunivälitteinen tyypin 1 diabetes
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa immuunivälitteinen tyypin 1 diabetes ilmeni 5 potilaalla (0,1 %). Näistä 3 potilaalla (0,1 %) todettiin vaikeusasteen 3 tapahtuma ja 1 potilaalla (< 0,1 %) vaikeusasteen 4 tapahtuma. Aika tyypin 1 diabeteksen ilmaantumiseen oli 43 päivää (vaihteluväli: 29–631 päivää). Kaikki viisi potilasta tarvitsivat insuliinihoitoa. IMFINZI-hoito lopetettiin pysyvästi yhdellä potilaalla. Yksi potilas toipui, ja yhden potilaan toipumiseen liittyi jälkiseurauksia.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) immuunivälitteinen tyypin 1 diabetes oli todettu 6 potilaalla (0,3 %). Näistä 1 potilaalla (< 0,1 %) oli todettu vaikeusasteen 3 tapahtuma ja 2 potilaalla (< 0,1 %) vaikeusasteen 4 tapahtuma. Mediaaniaika haittavaikutuksen ilmaantumiseen oli 58 päivää (vaihteluväli: 7–220 päivää). Kaikki potilaat tarvitsivat insuliinia. Hoito lopetettiin 1 potilaalla. Immuunivälitteinen tyypin 1 diabetes parani 1 potilaalla.
Immuunivälitteinen hypofysiitti tai hypopituitarismi
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa immuunivälitteinen hypofysiitti/hypopituitarismi ilmeni 6 potilaalla (0,1 %), mukaan lukien vaikeusasteen 3 tapahtuma 5 potilaalla (0,1 %). Aika haittatapahtumien ilmaantumiseen oli 85 päivää (vaihteluväli: 44–225 päivää). Kolme potilasta sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa), kolme potilasta lopetti IMFINZI-hoidon immuunivälitteisen hypofysiitin/hypopituitarismin vuoksi, ja immuunivälitteinen hypofysiitti/hypopituitarismi parani 1 potilaalla.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) immuunivälitteistä hypofysiittia tai hypopituitarismia oli todettu 16 potilaalla (0,7 %). Näistä vaikeusasteen 3 tapahtumia oli 8 potilaalla (0,4 %). Mediaaniaika tapahtuman ilmaantumiseen oli 123 päivää (vaihteluväli: 63–388 päivää). Kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja 16 potilaasta 8 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Neljä potilasta tarvitsi myös hormonihoitoa. Hoito lopetettiin 2 potilaalla. Immuunivälitteinen hypofysiitti tai hypopituitarismi parani 7 potilaalla.
Maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa (n = 462) immuunivälitteistä hypofysiittia tai hypopituitarismia oli todettu 5 potilaalla (1,1 %). Mediaaniaika tapahtuman ilmaantumiseen oli 149 päivää (vaihteluväli: 27–242 päivää). 4 potilasta sai systeemistä kortikosteroidihoitoa, ja näistä neljästä potilaasta yksi sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg vuorokaudessa tai vastaavaa hoitoa). Kolme potilasta tarvitsi myös hormonihoitoa. Immuunivälitteinen hypofysiitti tai hypopituitarismi parani 2 potilaalla.
Immuunivälitteinen munuaistulehdus
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa immuunivälitteistä munuaistulehdusta ilmeni 17 potilaalla (0,4 %), mukaan lukien asteen 3 haitat 4 potilaalla (0,1 %) ja asteen 4 haitta 1 potilaalla (< 0,1 %). Mediaaniaika haitan ilmaantumiseen oli 84 päivää (vaihteluväli: 4–393 päivää). Kaksitoista potilasta sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa) ja yksi potilas sai myös mykofenolaattia. IMFINZI-hoito lopetettiin 7 potilaalla. Immuunivälitteinen munuaistulehdus parani 8 potilaalla.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) immuunivälitteistä munuaistulehdusta oli todettu 9 potilaalla (0,4 %). Näistä 1 potilaalla (< 0,1 %) oli todettu vaikeusasteen 3 tapahtuma. Mediaaniaika haittavaikutuksen ilmaantumiseen oli 79 päivää (vaihteluväli: 39–183 päivää). Kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja 7 potilasta sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Hoito lopetettiin 3 potilaalla. Immuunivälitteinen munuaistulehdus parani 5 potilaalla.
Maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa (n = 462) immuunivälitteistä munuaistulehdusta oli todettu 4 potilaalla (0,9 %). Näistä vaikeusasteen 3 tapahtumia oli 2 potilaalla (0,4 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 53 päivää (vaihteluväli: 26–242 päivää). Kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja näistä 4 potilaasta 3 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Hoito lopetettiin 2 potilaalla. Immuunivälitteinen munuaistulehdus parani 3 potilaalla.
Immuunivälitteinen ihottuma
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa immuunivälitteistä ihottumaa tai dermatiittia (pemfigoidi mukaan lukien) ilmeni 74 potilaalla (1,6 %), mukaan lukien asteen 3 haitat 20 potilaalla (0,4 %). Mediaaniaika haitan ilmaantumiseen oli 56 päivää (vaihteluväli: 4–600 päivää). 74 potilaasta 37 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). IMFINZI-hoito lopetettiin 5 potilaalla. Immuunivälitteinen ihottuma tai dermatiitti parani 46 potilaalla.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) immuunivälitteistä ihottumaa tai dermatiittia (pemfigoidi mukaan lukien) oli todettu 112 potilaalla (4,9 %). Näistä vaikeusasteen 3 tapahtumia oli 17 potilaalla (0,7 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 35 päivää (vaihteluväli: 1–778 päivää). Kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja 112 potilaasta 57 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Hoito lopetettiin 10 potilaalla. Immuunivälitteinen ihottuma tai dermatiitti (pemfigoidi mukaan lukien) parani 65 potilaalla.
Maksasolusyöpää koskevassa yhdistetyssä tietoaineistossa (n = 462) immuunivälitteistä ihottumaa tai dermatiittia (pemfigoidi mukaan lukien) oli todettu 26 potilaalla (5,6 %). Näistä vaikeusasteen 3 tapahtumia oli 9 potilaalla (1,9 %) ja vaikeusasteen 4 tapahtumia 1 potilaalla (0,2 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 25 päivää (vaihteluväli: 2–933 päivää). Kaikki potilaat saivat systeemistä kortikosteroidihoitoa, ja näistä 26 potilaasta 14 sai suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg:n vuorokausiannoksella tai vastaavaa hoitoa). Yksi potilas sai muita immuunisalpaajia. Hoito lopetettiin 3 potilaalla. Immuunivälitteinen ihottuma tai dermatiitti (pemfigoidi mukaan lukien) parani 19 potilaalla.
DUO-E-tutkimuksessa 238 potilasta sai platinapohjaista solunsalpaajahoitoa yhdistelmänä IMFINZI-valmisteen kanssa ja tämän jälkeen IMFINZI-valmistetta yhdistelmänä olaparibin kanssa (platinapohjainen solunsalpaajahoito + IMFINZI + olaparibi ‑hoitohaara), ja heistä 8:lla (3,4 %) ilmeni immuunivälitteistä ihottumaa. Näistä vaikeusasteen 3 tapahtumia oli 2 potilaalla (0,8 %). Mediaaniaika haittavaikutuksen ilmaantumiseen oli 155 päivää (vaihteluväli: 2–308 päivää). Kaikki potilaat saivat suuriannoksista kortikosteroidihoitoa (prednisonia vähintään 40 mg vuorokaudessa tai vastaavaa hoitoa). Immuunivälitteinen ihottuma parani kaikilla 8 potilaalla.
Infuusioon liittyvät reaktiot
IMFINZI-monoterapiaa koskeneessa yhdistetyssä turvallisuustietokannassa infuusioon liittyviä reaktioita ilmeni 70 potilaalla (1,5 %), mukaan lukien asteen 3 haitat 6 potilaalla (0,1 %). Vaikeusasteen 4 tai 5 tapahtumia ei ilmennyt.
Tremelimumabin kanssa yhdistelmänä käytettyä IMFINZI-valmistetta koskeneessa yhdistetyssä turvallisuustietokannassa (n = 2 280) infuusioon liittyviä reaktioita oli todettu 45 potilaalla (2,0 %). Näistä vaikeusasteen 3 tapahtumia oli 2 potilaalla (< 0,1 %). Vaikeusasteen 4 tai 5 tapahtumia ei ilmennyt.
DUO-E-tutkimuksessa 238 potilasta sai platinapohjaista solunsalpaajahoitoa yhdistelmänä IMFINZI-valmisteen kanssa ja tämän jälkeen IMFINZI-valmistetta yhdistelmänä olaparibin kanssa (platinapohjainen solunsalpaajahoito + IMFINZI + olaparibi ‑hoitohaara), ja heistä 13:lla (5,5 %) ilmeni infuusioon liittyviä reaktioita. Näistä vaikeusasteen 3 tapahtumia oli 1 potilaalla (0,4 %). Vaikeusasteen 4 tai 5 tapahtumia ei ilmennyt.
Punasoluaplasia
Punasoluaplasiaa on ilmoitettu, kun IMFINZI-valmistetta on käytetty yhdessä olaparibin kanssa. Kliinisessä tutkimuksessa, jossa kohdun limakalvon syöpää sairastaville potilaille annettiin IMFINZI-valmistetta yhdessä olaparibin kanssa, punasoluaplasian ilmaantuvuus oli 1,6 %. Kaikki tapahtumat olivat CTCAE-astetta 3 tai 4. Tapahtumat olivat hoidettavissa, kun sekä IMFINZI-hoito että olaparibihoito lopetettiin. Suurin osa tapahtumista hoidettiin verensiirrolla ja immunosuppressiolla, ja nämä potilaat toipuivat. Yksikään tapahtumista ei johtanut kuolemaan. Hoito, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Laboratorioarvojen poikkeavuudet
IMFINZI-monoterapiaa saaneista potilaista niiden potilaiden osuudet, joiden laboratorioarvot muuttuivat lähtötilanteesta vaikeusasteen 3 tai 4 poikkeavuuksiksi, olivat seuraavat: kohonneet alaniiniaminotransferaasiarvot 3,7 %, kohonneet aspartaattiaminotransferaasiarvot 5,7 %, kohonnut veren kreatiniiniarvo 0,9 %, kohonnut amylaasipitoisuus 4,8 % ja kohonnut lipaasipitoisuus 8,2 %. Niiden potilaiden osuus, joiden TSH-arvo oli lähtötilanteessa korkeintaan viitealueen ylärajalla (≤ ULN) ja kohosi ylärajan yläpuolelle (kaikki vaikeusasteet), oli 20 %. Niiden potilaiden osuus, joiden TSH-arvo oli lähtötilanteessa vähintään viitealueen alarajalla (≥ LLN) ja laski alarajan alapuolelle (kaikki vaikeusasteet), oli 18,2 %.
IMFINZI-valmistetta yhdistelmänä solunsalpaajahoidon kanssa saaneilla potilailla niiden potilaiden osuudet, joiden laboratorioarvot muuttuivat lähtötilanteesta vaikeusasteen 3 tai 4 poikkeavuuksiksi, olivat seuraavat: kohonneet alaniiniaminotransferaasiarvot 4,2 %, kohonneet aspartaattiaminotransferaasiarvot 3,4 %, kohonnut veren kreatiniiniarvo 4,0 %, suurentunut amylaasipitoisuus 6,0 %, suurentunut lipaasipitoisuus 12,1 % ja suurentunut bilirubiinipitoisuus 2,3 %. Niiden potilaiden osuus, joiden TSH-arvo oli lähtötilanteessa korkeintaan viitealueen ylärajalla (≤ ULN) ja kohosi ylärajan yläpuolelle (kaikki vaikeusasteet), oli 22,8 %. Niiden potilaiden osuus, joiden TSH-arvo oli lähtötilanteessa vähintään viitealueen alarajalla (≥ LLN) ja laski alarajan alapuolelle (kaikki vaikeusasteet), oli 22,6 %.
IMFINZI-valmistetta yhdistelmänä tremelimumabin ja platinapohjaisen solunsalpaajahoidon kanssa saaneilla potilailla niiden potilaiden osuudet, joiden laboratorioarvot muuttuivat lähtötilanteesta vaikeusasteen 3 tai 4 poikkeavuuksiksi, olivat seuraavat: kohonneet alaniiniaminotransferaasiarvot, 6,2 %; kohonneet aspartaattiaminotransferaasiarvot, 5,2 %; kohonnut veren kreatiniiniarvo, 4,0 %; kohonnut amylaasipitoisuus, 9,4 %; ja kohonnut lipaasipitoisuus, 13,6 %. Niiden potilaiden osuus, joiden TSH-arvo oli lähtötilanteessa korkeintaan viitealueen ylärajalla (≤ ULN) ja kohosi ylärajan yläpuolelle, oli 24,8 %. Niiden potilaiden osuus, joiden TSH-arvo oli lähtötilanteessa vähintään viitealueen alarajalla (≥ LLN) ja laski alarajan alapuolelle, oli 32,9 %.
IMFINZI-valmistetta yhdistelmänä tremelimumabin kanssa saaneilla potilailla niiden potilaiden osuudet, joiden laboratorioarvot muuttuivat lähtötilanteesta vaikeusasteen 3 tai 4 poikkeavuuksiksi, olivat seuraavat: kohonneet alaniiniaminotransferaasiarvot, 5,1 %; kohonneet aspartaattiaminotransferaasiarvot, 5,8 %; kohonnut veren kreatiniiniarvo, 1,0 %; kohonnut amylaasipitoisuus, 5,9 %; ja kohonnut lipaasipitoisuus, 11,3 %. Niiden potilaiden osuus, joiden TSH-arvo oli lähtötilanteessa korkeintaan viitealueen ylärajalla (≤ ULN) ja kohosi ylärajan yläpuolelle, oli 4,2 %. Niiden potilaiden osuus, joiden TSH-arvo oli lähtötilanteessa vähintään viitealueen alarajalla (≥ LLN) ja laski alarajan alapuolelle, oli 17,2 %.
Potilailla, jotka saivat platinapohjaista solunsalpaajahoitoa yhdistelmänä IMFINZI-valmisteen kanssa ja tämän jälkeen IMFINZI-valmistetta yhdistelmänä olaparibin kanssa (platinapohjainen solunsalpaajahoito + IMFINZI + olaparibi ‑hoitohaara), niiden potilaiden osuudet, joiden laboratorioarvot muuttuivat lähtötilanteesta vaikeusasteen 3 tai 4 poikkeavuuksiksi, olivat seuraavat: kohonneet alaniiniaminotransferaasiarvot 3,8 %, kohonneet aspartaattiaminotransferaasiarvot 3,4 % ja kohonnut veren kreatiniiniarvo 1,7 %. Niiden potilaiden osuus, joiden TSH-arvo oli lähtötilanteessa korkeintaan viitealueen ylärajalla (≤ ULN) ja kohosi ylärajan yläpuolelle, oli 28,6 %. Niiden potilaiden osuus, joiden TSH-arvo oli lähtötilanteessa vähintään viitealueen alarajalla ja laski alarajan alapuolelle, oli 20,1 %.
Immunogeenisuus
Tiedot monoterapiana annetun IMFINZI-hoidon immunogeenisuudesta perustuvat yhdistettyihin tietoihin 3 069 potilaasta, jotka saivat IMFINZI-hoitoa 10 mg/kg 2 viikon välein tai 20 mg/kg 4 viikon välein ainoana lääkkeenä ja joilta voitiin määrittää lääkevasta-aineet (ADA). Hoidon aikana ilmeneviä lääkevasta-aineita todettiin testeissä 84 potilaalla (2,7 %). Durvalumabia neutraloivia vasta-aineita (nAb) todettiin 0,5 %:lla (16/3 069) potilaista. Lääkevasta-aineiden läsnäolo ei vaikuttanut kliinisesti merkittävästi farmakokinetiikkaan tai turvallisuuteen. Potilaiden määrä ei ole riittävä, jotta voitaisiin määrittää lääkevasta-aineiden vaikutukset tehoon.
Useissa vaiheen III tutkimuksissa 0–10,1 %:lle potilaista, jotka saivat IMFINZI-valmistetta yhdistelmänä muiden lääkeaineiden kanssa, kehittyi hoidon aikana lääkevasta-aineita. Durvalumabia neutraloivia vasta-aineita todettiin 0–1,7%:lla potilaista, jotka saivat IMFINZI-valmistetta yhdistelmänä muiden lääkeaineiden kanssa. Lääkevasta-aineiden kehittymisellä ei ollut havaittavia vaikutuksia farmakokinetiikkaan tai turvallisuuteen.
Iäkkäät
Iäkkäiden (≥ 65‑vuotiaiden) ja nuorempien potilaiden välillä ei yleisesti ilmoitettu olevan turvallisuuteen liittyviä eroja.
PACIFIC-, ADRIATIC‑, CASPIAN-, TOPAZ-1-, HIMALAYA-, NIAGARA- ja MATTERHORN-tutkimuksissa vähintään 75-vuotiaiden potilaiden turvallisuudesta on liian vähän tietoa, jotta tästä populaatiosta voitaisiin tehdä johtopäätöksiä.
Metastasoitunutta ei-pienisoluista keuhkosyöpää sairastavien potilaiden ensilinjan hoidossa POSEIDON-tutkimuksessa iäkkäiden (≥ 65-vuotiaiden) ja nuorempien potilaiden välillä ilmoitettiin olevan joitakin turvallisuuteen liittyviä eroja. Turvallisuustiedot vähintään 75‑vuotiaista potilaista rajoittuvat yhteensä 74 potilaaseen. Niillä 35 potilaalla, jotka olivat vähintään 75‑vuotiaita ja jotka olivat saaneet IMFINZI-valmistetta yhdistelmänä tremelimumabin ja platinapohjaisen solunsalpaajahoidon kanssa, ilmeni enemmän vakavia haittavaikutuksia (45,7 %) ja jonkin tutkimushoidon keskeyttämistä haittavaikutusten vuoksi (28,6 %) kuin niillä 39 potilaalla, jotka olivat vähintään 75‑vuotiaita ja jotka olivat saaneet vain platinapohjaista solunsalpaajahoitoa (näistä potilaista 35,9 %:lla ilmeni vakavia haittavaikutuksia ja 20,5 %:lla tutkimushoito keskeytettiin haittavaikutusten vuoksi).
Leikkaukseen soveltuvaa ei-pienisoluista keuhkosyöpää sairastavilla potilailla tehdyssä AEGEAN-tutkimuksessa iäkkäiden (≥ 65‑vuotiaiden) ja nuorempien potilaiden välillä ilmoitettiin olevan joitakin turvallisuuteen liittyviä eroja. Turvallisuustiedot vähintään 75‑vuotiaista potilaista rajoittuvat 86 potilaaseen molemmissa hoitohaaroissa. Potilailla, jotka olivat vähintään 75‑vuotiaita ja jotka olivat saaneet IMFINZI-valmistetta yhdistelmänä solunsalpaajahoidon kanssa, ilmeni enemmän vakavia haittavaikutuksia (26,5 %) kuin potilailla, jotka olivat saaneet vain solunsalpaajahoitoa (10,8 %). Potilailla, jotka olivat vähintään 75‑vuotiaita ja jotka olivat saaneet IMFINZI-valmistetta yhdistelmänä solunsalpaajahoidon kanssa, ilmeni enemmän jonkin tutkimushoidon keskeyttämistä haittavaikutusten vuoksi (16,3 %) kuin potilailla, jotka olivat saaneet vain solunsalpaajahoitoa (8,1 %).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Durvalumabin yliannostuksesta ei ole tietoja. Yliannostustapauksissa potilaan tilaa on seurattava tarkoin haittavaikutuksiin viittaavien oireiden tai löydösten havaitsemiseksi, ja oireenmukainen hoito on aloitettava välittömästi.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, PD-1/PDL-1 (ohjelmoituneen solukuoleman proteiinin 1 / ligandin 1) estäjät. ATC-koodi: L01FF03.
Vaikutusmekanismi
PD‑L1 (programmed cell death ligand-1, ohjelmoituneen solukuoleman ligandi-1) -proteiinin ilmentyminen on adaptiivinen immuunivaste, jonka seurauksena immuunijärjestelmä ei pysty havaitsemaan ja eliminoimaan kasvaimia. Tulehdussignaalit (kuten IFN‑gamma) voivat indusoida PD‑L1:tä ja sitä voi ilmentyä sekä kasvainsoluissa että kasvaimeen liittyvissä immuunisoluissa kasvaimen mikroympäristössä. PD‑L1 salpaa T‑solujen toimintaa ja aktivaatiota vuorovaikutuksella PD‑1:n ja CD80:n (B7.1) kanssa. PD‑L1 sitoutuu reseptoreihinsa ja vähentää siten sytotoksista T‑soluaktiivisuutta, proliferaatiota ja sytokiinituotantoa.
Durvalumabi on täysin humanisoitu monoklonaalinen immunoglobuliinin G1 kappa (IgG1κ) vasta-aine, joka salpaa selektiivisesti PD‑L1:n vuorovaikutusta PD‑1:n ja CD80:n (B7.1) kanssa. Durvalumabi ei indusoi vasta-aineesta riippuvaista soluvälitteistä sytotoksisuutta (antibody dependent cell-mediated cytotoxicity, ADCC). Selektiivinen PD‑L1/PD‑1- ja PD‑L1/CD80-vuorovaikutusten salpaaminen tehostaa kasvaimen kasvua ehkäiseviä immuunivasteita ja lisää T‑soluaktivaatiota.
CTLA-4:n estäjä tremelimumabin ja PD-L1:n estäjä durvalumabin yhdistelmä tehostaa kasvaimen kasvua estävää T-solujen aktivaatiota ja toimintaa immuunivasteen eri vaiheissa, minkä ansiosta aikaansaadaan parempi vaste kasvainta vastaan. Hiiren syngeenisissä kasvainmalleissa PD-L1:n ja CTLA-4:n kaksoissalpaus tehosti vaikutusta kasvaimia vastaan.
Kliininen teho ja turvallisuus
Durvalumabin annoksia 10 mg/kg 2 viikon välein, 1 120 mg 3 viikon välein tai 1 500 mg 4 viikon välein arvioitiin ei‑pienisoluista keuhkosyöpää, levinnyttä pienisoluista keuhkosyöpää ja kohdun limakalvon syöpää koskevissa kliinisissä tutkimuksissa. Farmakokineettisen mallinnuksen ja simulaation sekä altistus-teho- ja altistus-turvallisuusanalyysien (altistus-vastesuhteet) perusteella durvalumabin tehossa ja turvallisuudessa ei ole odotettavissa kliinisesti merkittäviä eroja annoksilla 10 mg/kg 2 viikon välein, 1 120 mg 3 viikon välein ja 1 500 mg 4 viikon välein.
Leikkaukseen soveltuva ei-pienisoluinen keuhkosyöpä – AEGEAN-tutkimus
AEGEAN oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen III monikeskustutkimus, joka oli suunniteltu arvioimaan IMFINZI-valmisteen tehoa leikkaukseen soveltuvaa ei-pienisoluista keuhkosyöpää sairastavilla potilailla, kun sitä annettiin ensin yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa esiliitännäishoitona ja sitten jatkettiin IMFINZI-monoterapiana leikkauksen jälkeen.
Potilaat, joilla on käyttöaiheen mukainen tauti ja suuri taudin uusiutumisen riski, määritellään seuraavien kriteerien perusteella; kriteerit kuvastavat AJCC/UICC-luokituskäsikirjan 8. painoksen mukaan määriteltyä potilaspopulaatiota, jonka potilailla on levinneisyysasteen IIA – IIIB (IIIB vain valikoiduissa tapauksissa) tauti:
- kuka tahansa potilas, jolla kasvaimen koko on ≥ 4 cm
- kuka tahansa potilas, jolla taudin status on N1 tai N2 (primaarikasvaimen koosta riippumatta), mukaan lukien potilaat, joilla on useita N2‑luokan mukaisia imusolmukkeita
- potilaat, joilla on useita kasvainnoduluksia samassa lohkossa tai pääkeuhkoputkea affisioivia kasvaimia tai kasvaimia, jotka ovat tunkeutuneet viskeraaliseen pleuraan, rintakehän seinämään (mukaan lukien parietaalisen pleuran ja superiorisen sulcusalueen kasvaimet), palleahermoon tai parietaaliseen perikardiumiin; tai kasvaimia, joihin liittyy atelektaasi tai obstruktiivinen pneumoniitti, joka ulottuu keuhkoportin seutuun tai affisioi osaa keuhkosta tai koko keuhkoa.
Tutkimukseen otettiin mukaan potilaita, jotka eivät olleet aiemmin saaneet hoitoa; joilla oli dokumentoitu levyepiteeliperäinen tai ei-levyepiteeliperäinen ei-pienisoluinen keuhkosyöpä; jotka eivät olleet aiemmin saaneet immuunivälitteistä hoitoa; joiden WHO/ECOG-suorituskykyluokka oli 0 tai 1; ja joilla oli ainakin yksi RECIST 1.1 ‑kriteerien mukainen kohdeleesio. Ennen satunnaistamista kasvainten PD‑L1:n ilmentymisstatus varmistettiin VENTANA PD‑L1 (SP263) ‑analyysillä.
Tutkimuksesta suljettiin pois potilaat, joilla oli aktiivinen tai aiemmin dokumentoitu autoimmuunisairaus tai jotka olivat saaneet immunosuppressiivista lääkitystä 14 päivän sisällä ennen ensimmäisen durvalumabiannoksen saamista. Tehoa koskevan analyysin tutkimuspopulaatiosta (muokattu hoitoaiepopulaatio [mITT]) suljettiin pois potilaat, joilla tiedettiin olevan EGFR-mutaatioita tai ALK-uudelleenjärjestymiä. Tutkimussuunnitelman muutoksen jälkeen vaadittiin paikalliset ALK-määritykset (ellei kyseessä ollut histologialtaan levyepiteeliperäinen kasvain) ja keskitetyt EGFR-määritykset. Tutkimuksessa satunnaistetuista ja tutkimuksessa hoitoa saaneista potilaista 51:llä oli EGFR-mutaatioita ja 11:llä ALK-uudelleenjärjestymiä. Nämä potilaat eivät kuitenkaan olleet mukana tehoa koskevan analyysin mITT-populaatiossa, eikä ole mahdollista tehdä luotettavia johtopäätöksiä potilaista, joilla on EGFR-mutaatioita tai ALK-uudelleenjärjestymiä.
Satunnaistamisessa ositustekijöinä olivat taudin levinneisyysaste (levinneisyysaste II vs. levinneisyysaste III) ja PD‑L1:n ilmentymisstatus (osuus kasvainsoluista [TC] < 1 % vs. ≥ 1 %).
Leikkauksenjälkeinen sädehoito sallittiin potilaille, joille se oli paikallisten ohjeiden mukaan aiheellista. Leikkauksenjälkeinen sädehoito oli aloitettava 8 viikon kuluessa leikkauksesta, ja liitännäishoito durvalumabilla/lumelääkkeellä oli sitten aloitettava 3 viikon kuluessa leikkauksenjälkeisen sädehoidon päättymisestä.
AEGEAN-tutkimuksessa satunnaistettiin 802 potilasta suhteessa 1:1 saamaan perioperatiivisesti IMFINZI-valmistetta (hoitohaara 1) tai lumelääkettä (hoitohaara 2) yhdistelmänä esiliitännäishoitona annetun solunsalpaajahoidon kanssa. Siirtyminen hoitohaarasta toiseen ei ollut sallittua.
- Hoitohaara 1: IMFINZI 1 500 mg + solunsalpaajahoito 3 viikon välein enintään 4 hoitosyklin ajan ennen leikkausta ja sitten IMFINZI 1 500 mg 4 viikon välein enintään 12 hoitosyklin ajan leikkauksen jälkeen.
- Hoitohaara 2: Lumelääke + solunsalpaajahoito 3 viikon välein enintään 4 hoitosyklin ajan ennen leikkausta ja sitten lumelääke 4 viikon välein enintään 12 hoitosyklin ajan leikkauksen jälkeen.
Näissä kahdessa hoitohaarassa potilaat saivat histologisten tietojen perusteella jotakin seuraavista solunsalpaajahoidoista:
-
Levyepiteeliperäinen ei-pienisoluinen keuhkosyöpä
- Karboplatiini + paklitakseli: karboplatiini AUC 6 ja paklitakseli 200 mg/m2 laskimoinfuusiona kunkin 3 viikon pituisen hoitosyklin päivänä 1, 4 hoitosyklin ajan.
-
Levyepiteeliperäinen ei-pienisoluinen keuhkosyöpä
- Sisplatiini + gemsitabiini: sisplatiini 75 mg/m2 laskimoinfuusiona kunkin 3 viikon pituisen hoitosyklin päivänä 1, 4 hoitosyklin ajan, ja gemsitabiini 1 250 mg/m2 laskimoinfuusiona kunkin 3 viikon pituisen hoitosyklin päivänä 1 ja päivänä 8, 4 hoitosyklin ajan.
-
Ei-levyepiteeliperäinen ei-pienisoluinen keuhkosyöpä
- Pemetreksedi + sisplatiini: pemetreksedi 500 mg/m2 ja sisplatiini 75 mg/m2 laskimoinfuusiona kunkin 3 viikon pituisen hoitosyklin päivänä 1, 4 hoitosyklin ajan.
-
Ei-levyepiteeliperäinen ei-pienisoluinen keuhkosyöpä
- Pemetreksedi + karboplatiini: pemetreksedi 500 mg/m2 ja karboplatiini AUC 5 laskimoinfuusiona kunkin 3 viikon pituisen hoitosyklin päivänä 1, 4 hoitosyklin ajan.
Siedettävyysongelmien ilmetessä potilaan sisplatiinihoito voitiin vaihtaa karboplatiinihoidoksi milloin tahansa, ja jos potilaalla oli samanaikaisia sairauksia tai hän ei tutkijalääkärin arvion mukaan olisi sietänyt sisplatiinia, hänelle voitiin antaa karboplatiinia (AUC 5) hoitosyklistä 1 lähtien.
Kasvaimet arvioitiin RECIST 1.1 ‑kriteerien mukaan lähtötilanteessa ja esiliitännäishoitojakson päätyttyä (ennen leikkausta). Ensimmäinen leikkauksenjälkeinen rintakehän ja vatsan TT- tai magneettikuvaus (mukaan lukien koko maksa ja molemmat lisämunuaiset) tehtiin 5 viikkoa ± 2 viikkoa leikkauksen jälkeen ja ennen liitännäishoidon aloittamista, kuitenkin mahdollisimman lähellä liitännäishoidon aloittamisajankohtaa. Sen jälkeen kasvaimet arvioitiin 12 viikon välein (leikkauspäivästä laskien) viikkoon 48 asti, 24 viikon välein (leikkauspäivästä laskien) viikkoon 192 asti (noin 4 vuoteen asti) ja sitten 48 viikon välein (leikkauspäivästä laskien), kunnes todettiin radiologisesti RECIST 1.1 ‑kriteerien mukaisesti taudin edenneen tai potilas peruutti suostumuksensa osallistua tutkimukseen tai kuoli. Elossaoloarvioinnit tehtiin kuukausina 2, 3 ja 4 hoidon lopettamisen jälkeen, sitten 2 kuukauden välein kuukauteen 12 asti ja sen jälkeen 3 kuukauden välein.
Tutkimuksen ensisijaiset päätetapahtumat olivat patologisesti todettu täydellinen vaste (pathological complete response, pCR) sokkoutetun keskitetyn patologisen arvion mukaan ja tapahtumavapaa elossaolo (event-free survival, EFS) sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) tekemän arvion mukaan. Kokonaiselossaolo oli keskeinen toissijainen päätetapahtuma.
Tehoa koskevassa analyysissä oli mukana mITT-populaation 740 potilasta: 366 potilasta hoitohaarassa 1 ja 374 potilasta hoitohaarassa 2. Populaation demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat: miehiä (71,6 %), naisia (28,4 %), ikä vähintään 65 vuotta (51,6 %), iän mediaani 65 vuotta (vaihteluväli: 30–88), WHO/ECOG-suorituskykypistemäärä 0 (68,4 %), WHO/ECOG-suorituskykypistemäärä 1 (31,6 %), valkoihoisia (53,6 %), aasialaisia (41,5 %), mustaihoisia tai afrikkalaisamerikkalaisia (0,9 %), Amerikan tai Alaskan alkuperäisväestöön kuuluvia (1,4 %), muun etnisen taustan omaavia (2,6 %), taustaltaan espanjankielisiä tai latinalaisamerikkalaisia (16,1 %), muita kuin taustaltaan espanjankielisiä tai latinalaisamerikkalaisia (83,9 %), edelleen tupakoivia tai aiemmin tupakoineita (85,5 %), ei milloinkaan tupakoineita (14,5 %), histologialtaan levyepiteeliperäinen kasvain (48,6 %) ja histologialtaan ei-levyepiteeliperäinen kasvain (50,7 %), levinneisyysaste II (28,4 %), levinneisyysaste III (71,6 %), PD‑L1:n ilmentymisstatus TC ≥ 1 % (66,6 %), PD‑L1:n ilmentymisstatus TC < 1 % (33,4 %).
Leikkaus, jossa pyrittiin kuratiiviseen tulokseen, tehtiin mITT-populaation hoitohaaran 1 potilaista 295 potilaalle (80,6 %) ja hoitohaaran 2 potilaista 302 potilaalle (80,7 %). Leikkauksenjälkeistä sädehoitoa saaneita potilaita oli 26 (7,1 %) hoitohaarassa 1 ja 24 (6,4 %) hoitohaarassa 2.
Tapahtumavapaata elossaoloa koskevassa (ennalta määritellyssä) primaarianalyysissä (viimeinen tiedonkeruupäivä 10.11.2022), kun maturiteetti oli 31,9 % ja tapahtumavapaan elossaolon seuranta-ajan mediaani sensuroiduilla potilailla 11,7 kuukautta, todettiin tilastollisesti merkitsevästi parempi tulos IMFINZI-hoitohaarassa verrattuna lumehaaraan [HR = 0,68 (95 %:n luottamusväli: 0,53, 0,88), p = 0,003902].
Päivitetyssä (ennalta määritellyssä) tapahtumavapaata elossaoloa koskevassa analyysissä (viimeinen tiedonkeruupäivä 10.5.2024) tapahtumavapaan elossaolon seuranta-ajan mediaani oli sensuroiduilla potilailla 25,9 kuukautta. Tässä analyysissä kokonaiselossaolon tietoja ei testattu muodollisesti tilastollisen merkitsevyyden suhteen; kokonaiselossaolon HR oli 0,89 (95 %:n luottamusväli: 0,70, 1,14) IMFINZI-hoitohaarassa verrattuna lumehaaraan.
Taulukko 5. AEGEAN-tutkimuksen tehoa koskevat tulokset (mITT)
| IMFINZI + solunsalpaajahoito (N = 366) | Lumelääke + solunsalpaajahoito (N = 374) | |
|---|---|---|
| Tapahtumavapaa elossaoloa,c | ||
| Tapahtumien määrä, n (%) | 124 (33,9) | 165 (44,1) |
| Tapahtumavapaan elossaoloajan mediaani (95 %:n luottamusväli) (kuukausina) | Ei saavutettu (42,3, ei saavutettu) | 30 (20,6, ei saavutettu) |
| Riskisuhde (95 %:n luottamusväli) | 0,69 (0,55, 0,88) | |
| Patologisesti todettu täydellinen vastea,b,c | ||
| Vasteen saavuttaneiden potilaiden määrä | 63 | 16 |
| Vasteprosentti, % (95 %:n luottamusväli) | 17,21 (13,49, 21,48) | 4,28 (2,46, 6,85) |
| Osuuksien ero, % (95 %:n luottamusväli) | 12,96 (8,67, 17,57) | |
a Tulokset perustuvat päivitettyyn (ennalta määriteltyyn) tapahtumavapaata elossaoloa koskevaan analyysiin (viimeinen tiedonkeruupäivä 10.5.2024) ja patologisesti todettua täydellistä vastetta koskevaan lopulliseen analyysiin (viimeinen tiedonkeruupäivä 10.11.2022).
b Ennalta määritellyn, patologisesti todettua täydellistä vastetta koskevan välianalyysin perusteella (viimeinen tiedonkeruupäivä 14.1.2022, n = 402), patologisesti todettujen täydellisten vasteiden osuus oli tilastollisesti merkitsevä (p = 0,000036) verrattuna merkitsevyystasoon 0,0082 %.
c Kaksitahoinen p‑arvo patologisesti todetulle täydelliselle vasteelle laskettiin stratifioidun Cochran–Mantel–Haenszelin (CMH‑) testin perusteella. Kaksitahoinen p‑arvo tapahtumavapaalle elossaololle laskettiin stratifioidun log-rank-testin perusteella. Ositustekijöinä olivat lähtötilanteen PD‑L1 ja taudin levinneisyysaste. Tilastollisen merkitsevyyden toteamiseen vaadittavat raja-arvot kullekin tehoa koskevalle päätetapahtumalle määritettiin Lan–DeMetsin alfavirheen korjausfunktiolla, joka approksimoi O’Brien–Flemingin lähestymistapaa (tapahtumavapaa elossaolo = 0,9899 %, patologisesti todettu täydellinen vaste = 0,0082 %, kaksitahoinen).
Kuva 1. Päivitetyn tapahtumavapaata elossaoloa koskevan analyysin Kaplan–Meier-käyrä (viimeinen tiedonkeruupäivä 10.5.2024)
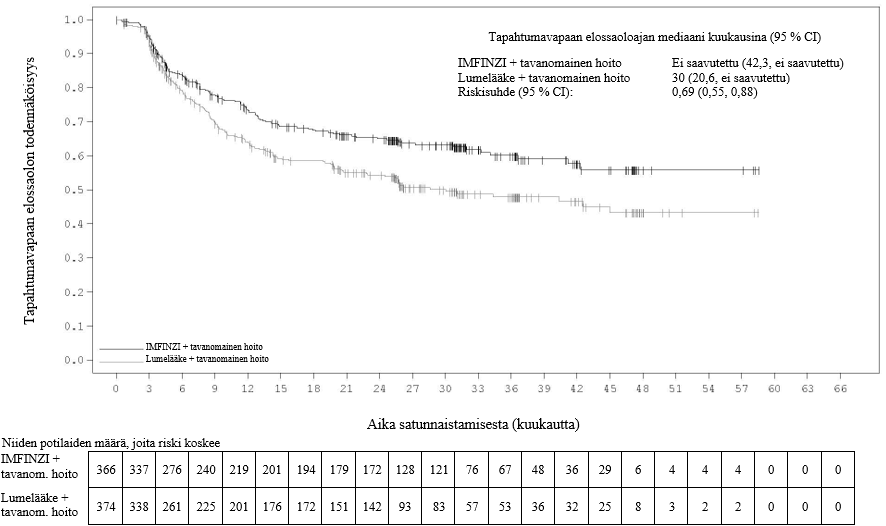
Ei-pienisoluinen keuhkosyöpä – PACIFIC-tutkimus
IMFINZI-valmisteen tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa PACIFIC-monikeskustutkimuksessa 713 potilaalla, joilla oli paikallisesti edennyt ja leikkaukseen soveltumaton ei-pienisoluinen keuhkosyöpä. Potilaat olivat jatkaneet loppuun asti ainakin kaksi definitiivisen platinapohjaisen solunsalpaajahoidon ja samanaikaisen sädehoidon hoitosykliä 1−42 päivän sisällä ennen tutkimuksessa aloittamista, ja heidän ECOG-suorituskykypistemääränsä oli 0 tai 1. 92 %:lla potilaista kokonaissäteilyannos oli 54–66 Gy. Tutkimuksesta suljettiin pois potilaat, joiden tauti oli edennyt kemosädehoidon jälkeen; potilaat, jotka olivat aiemmin altistuneet jollekin PD-1- tai PD-L1-vasta-aineelle; potilaat, joilla oli aktiivinen tai aiemmin dokumentoitu autoimmuunisairaus kahden vuoden sisällä tutkimuksen aloittamisesta; potilaat, joilla oli aiemmin ollut immuunipuutos; potilaat, joilla oli aiemmin ollut vaikeita immuunivälitteisiä haittavaikutuksia; potilaat, joilla oli sairaus, jonka hoito oli edellyttänyt systeemistä immunosuppressiota, lukuun ottamatta systeemistä kortikosteroidihoitoa fysiologisella annoksella; potilaat, joilla oli aktiivinen tuberkuloosi tai hepatiitti B- tai hepatiitti C -infektio tai HIV-infektio; ja potilaat, jotka olivat saaneet eläviä heikennettyjä taudinaiheuttajia sisältävän rokotteen 30 päivän kuluessa ennen IMFINZI-hoidon aloittamista tai IMFINZI-hoidon aloittamisen jälkeen. Potilaat satunnaistettiin suhteessa 2:1 saamaan IMFINZI-valmistetta 10 mg/kg (n = 476) tai lumelääkettä 10 mg/kg (n = 237) infuusiona laskimoon kahden viikon välein enintään 12 kuukauden ajan tai kunnes ilmaantuu toksisia vaikutuksista, joita ei voida hyväksyä, tai taudin eteneminen vahvistetaan. Satunnaistaminen stratifioitiin sukupuolen, iän (< 65 vuotta tai ≥ 65 vuotta) ja tupakoinnin (tupakoijat ja tupakoimattomat) mukaan. Potilaille, joilla tauti oli hoitotasapainossa 12 kuukauden kohdalla, annettiin mahdollisuus hoidon uudelleen aloittamiseen taudin edetessä. Kasvaimet arvioitiin ensimmäisten 12 kuukauden aikana 8 viikon välein ja sen jälkeen 12 viikon välein.
Potilaita otettiin tutkimukseen heidän kasvaimensa PD‑L1-ligandin ilmentymisen tasosta riippumatta. Jos kasvainkudoksesta oli saatavilla arkistonäyte, joka oli otettu ennen kemosädehoitoa, siitä tutkittiin retrospektiivisesti PD‑L1:n ilmentyminen kasvainsoluissa (TC) käyttämällä VENTANA PD‑L1 (SP263) IHC ‑analyysiä. 713 satunnaistetusta potilaasta 63 %:lta potilaista saatiin kudosnäyte, jonka laatu ja määrä riitti PD‑L1:n ilmentymisen määrittämiseen, ja 37 %:lla potilaista PD‑L1-status oli tuntematon.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat hyvin samankaltaiset eri tutkimushaaroissa. Koko tutkimuspopulaation demografiset tiedot lähtötilanteessa olivat: miehiä (70 %), ikä ≥ 65 vuotta (45 %), ikä ≥ 75 vuotta (8 %), valkoihoisia (69 %), aasialaisia (27 %), muita (4 %), tupakoijia (16 %), aiemmin tupakoineita (75 %), ei milloinkaan tupakoineita (9 %), ECOG-suorituskykypistemäärä 0 (49 %), ECOG-suorituskykypistemäärä 1 (51 %). Sairauden ominaispiirteet olivat: aste IIIA (53 %), aste IIIB (45 %), histologinen alatyyppi levyepiteelisyöpä (46 %), muu kuin levyepiteelisyöpä (54 %). 451 potilaasta, joista oli saatavilla PD‑L1:n ilmentymistä koskevat tiedot, 67 %:lla TC oli ≥ 1 % [PD-L1 TC 1–24 % (32 %), PD‑L1 TC ≥ 25 % (35 %)] ja 33 %:lla TC oli < 1 %.
Tutkimuksen kaksi ensisijaista päätemuuttujaa olivat etenemättömyysaika (Progression Free Survival, PFS) ja kokonaiselossaoloaika (Overall Survival, OS) IMFINZI-valmistetta saaneilla verrattuna lumelääkettä saaneisiin. Toissijaiset tehoa mittaavat päätemuuttujat olivat etenemättömyysaika 12 kuukauden kohdalla (PFS 12) ja 18 kuukauden kohdalla (PFS 18) satunnaistamisesta sekä aika satunnaistamisesta taudin toiseen etenemisjaksoon (PFS2). Sokkoutettu riippumaton keskitetty arvioijataho (Blinded Independent Central Review, BICR) arvioi etenemättömyysajan RECIST v1.1 -kriteerien mukaisesti.
Tutkimuksessa osoitettiin etenemättömyysajan tilastollisesti merkitsevä piteneminen IMFINZI-ryhmässä verrattuna lumeryhmään [riskisuhde (HR) = 0,52 (95 %:n luottamusväli: 0,42, 0,65); p < 0,0001]. Tutkimuksessa osoitettiin, että kokonaiselossaoloaika piteni tilastollisesti merkitsevästi IMFINZI-ryhmässä verrattuna lumeryhmään [riskisuhde = 0,68 (95 %:n luottamusväli: 0,53, 0,87), p = 0,00251].
Viiden vuoden seuranta-analyysissä, jossa seuranta-ajan mediaani oli 34,2 kuukautta, IMFINZI-valmistetta saaneiden potilaiden kokonaiselossaoloaika ja etenemättömyysaika olivat edelleen paremmat kuin lumelääkettä saaneilla potilailla. Primaarianalyysin ja seuranta-analyysin kokonaiselossaoloaikaa ja etenemisvapaata elinaikaa koskevista tuloksista on esitetty yhteenveto taulukossa 6.
Taulukko 6. PACIFIC-tutkimuksen tehoa koskevat tulokset
| Primaarianalyysia | 5 vuoden seuranta‑analyysib | |||
IMFINZI (n = 476) | Lumelääke (n = 237) | IMFINZI (n = 476) | Lumelääke (n = 237) | |
| Kokonaiselossaoloaika | ||||
| Kuolemantapausten määrä (%) | 183 (38,4 %) | 116 (48,9 %) | 264 (55,5 %) | 155 (65,4 %) |
| Mediaani (kuukausina) (95 %:n luottamusväli) | Ei saavutettu (34,7, ei saavutettu) | 28,7 (22,9, ei saavutettu) | 47,5 (38,1, 52,9) | 29,1 (22,1, 35,1) |
| Riskisuhde (95 %:n luottamusväli) | 0,68 (0,53, 0,87) | 0,72 (0,59, 0,89) | ||
| Kaksitahoinen p‑arvo | 0,00251 | |||
| Kokonaiselossaoloaika 24 kuukauden kohdalla (%) (95 %:n luottamusväli) | 66,3 % (61,7 %, 70,4 %) | 55,6 % (48,9 %, 61,3 %) | 66,3 % (61,8 %, 70,4 %) | 55,3 % (48,6 %, 61,4 %) |
| p-arvo | 0,005 | |||
| Kokonaiselossaoloaika 48 kuukauden kohdalla (%) (95 %:n luottamusväli) | 49,7 % (45,0 %, 54,2 %) | 36,3 % (30,1 %, 42,6 %) | ||
| Kokonaiselossaoloaika 60 kuukauden kohdalla (%) (95 %:n luottamusväli) | 42,9 % (38,2 %, 47,4 %) | 33,4 % (27,3 %, 39,6 %) | ||
| Etenemättömyysaika | ||||
| Tapahtuminen määrä (%) | 214 (45,0 %) | 157 (66,2 %) | 268 (56,3 %) | 175 (73,8 %) |
| Etenemättömyysajan mediaani (kuukausina) (95 %:n luottamusväli) | 16,8 (13,0, 18,1) | 5,6 (4,6, 7,8) | 16,9 (13,0, 23,9) | 5,6 (4,8, 7,7) |
| Riskisuhde (95 %:n luottamusväli) | 0,52 (0,42, 0,65) | 0,55 (0,45, 0,68) | ||
| p‑arvo | p < 0,0001 | |||
| Etenemättömyysaika 12 kuukauden kohdalla (%) (95 %:n luottamusväli) | 55,9 % (51,0 %, 60,4 %) | 35,3 % (29,0 %, 41,7 %) | 55,7 % (51,0 %, 60,2 %) | 34,5 % (28,3 %, 40,8 %) |
| Etenemättömyysaika 18 kuukauden kohdalla (%) (95 %:n luottamusväli) | 44,2 % (37,7 %, 50,5 %) | 27,0 % 19,9 %, 34,5 %) | 49,1 % (44,2 %, 53,8 %) | 27,5 % (21,6 %, 33,6 %) |
| Etenemättömyysaika 48 kuukauden kohdalla (%) (95 %:n luottamusväli) | 35,0 % (29,9 %, 40,1 %) | 19,9 % (14,4 %, 26,1 %) | ||
| Etenemättömyysaika 60 kuukauden kohdalla (%) (95 %:n luottamusväli) | 33,1 % (28,0 %, 38,2 %) | 19,0 % (13,6 %, 25,2 %) | ||
| Aika satunnaistamisesta taudin toiseen etenemisjaksoonc | ||||
| Aika satunnaistamisesta taudin toiseen etenemisjaksoon, mediaani (kuukausina) (95 %:n luottamusväli) | 28,3 (25,1, 34,7) | 17,1 (14,5, 20,7) | ||
| Riskisuhde (95 %:n luottamusväli) | 0,58 (0,46, 0,73) | |||
| p-arvo | p < 0,0001 | |||
a Etenemättömyysajan primaarianalyysi viimeisen tiedonkeruupäivän (13.2.2017) kohdalla. Kokonaiselinajan ja PFS2-ajan (aika satunnaistamisesta taudin toiseen etenemisjaksoon) primaarianalyysi viimeisen tiedonkeruupäivän (22.3.2018) kohdalla.
b Kokonaiselossaoloajan ja etenemättömyysajan seuranta-analyysi viimeisen tiedonkeruupäivän (11.1.2021) kohdalla.
c Aika satunnaistamisesta taudin toiseen etenemisjaksoon on määritelty ajaksi satunnaistamispäivästä päivään, jolloin taudin toinen etenemisjakso ilmenee (määritellään paikallisen tavanomaisen kliinisen käytännön mukaisesti), tai kuolemaan.
Kokonaiselossaoloajan ja etenemättömyysajan Kaplan–Meier‑käyrät viiden vuoden seuranta-analyysistä on esitetty kuvissa 2 ja 3.
Kuva 2. Kokonaiselossaoloajan Kaplan-Meier-käyrä
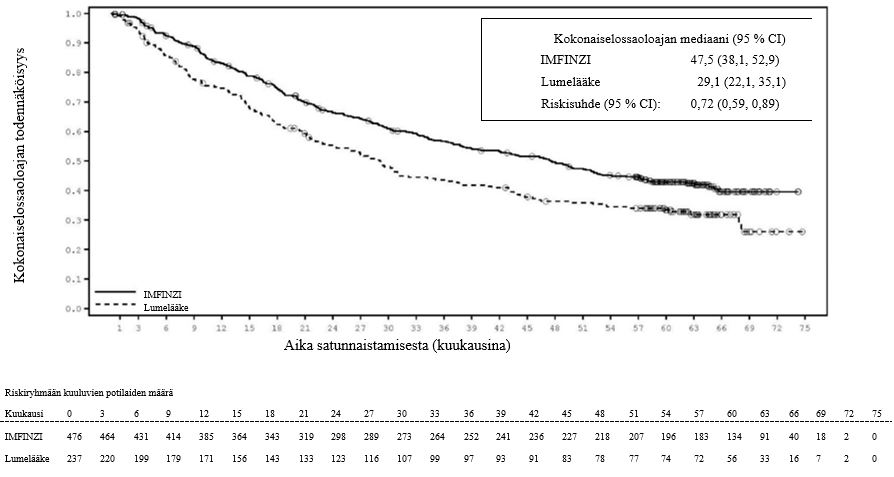
Kuva 3. Etenemättömyysajan Kaplan‑Meier-käyrä
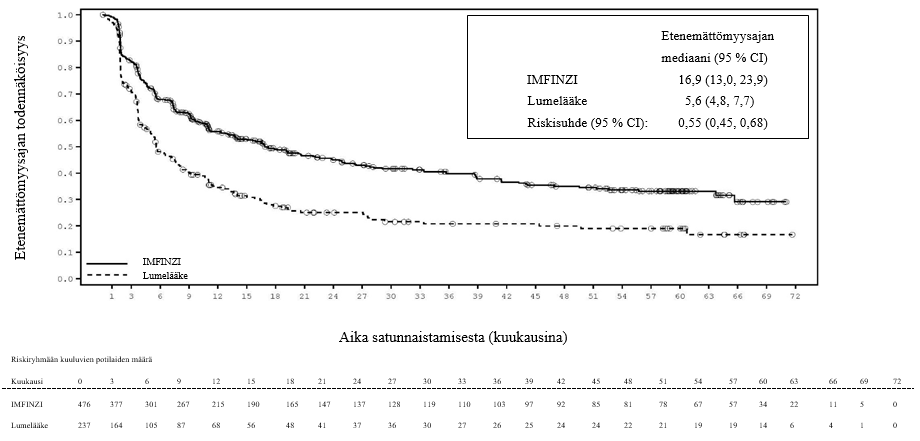
Etenemättömyysajan ja kokonaiselossaoloajan piteneminen, joka suosi IMFINZI-valmistetta saaneita potilaita verrattuna lumelääkettä saaneisiin, havaittiin johdonmukaisesti kaikissa analysoiduissa alaryhmissä, jotka oli ennalta määritelty etnisen taustan, iän, sukupuolen, tupakointihistorian, EGFR-mutaatiostatuksen ja histologian mukaan.
Post hoc ‑alaryhmäanalyysi PD‑L1:n ilmentymisen mukaan
Muita alaryhmäanalyysejä tehtiin, jotta voitiin arvioida tehoa kasvaimen PD‑L1-ligandin ilmentymisen (≥ 25 %, 1–24 %, ≥ 1 % tai < 1 %) mukaan ja potilailla, joiden PD‑L1-statusta ei ollut varmistettu (PD‑L1 tuntematon). Viiden vuoden seuranta-analyysin etenemisvapaata elinaikaa ja kokonaiselossaoloaikaa koskevista tuloksista on esitetty yhteenvedot kuvissa 4, 5, 6 ja 7.
Kuva 4. Kokonaiselossaoloajan Kaplan-Meier-käyrä potilailla, joiden PD‑L1 TC oli ≥ 1 %
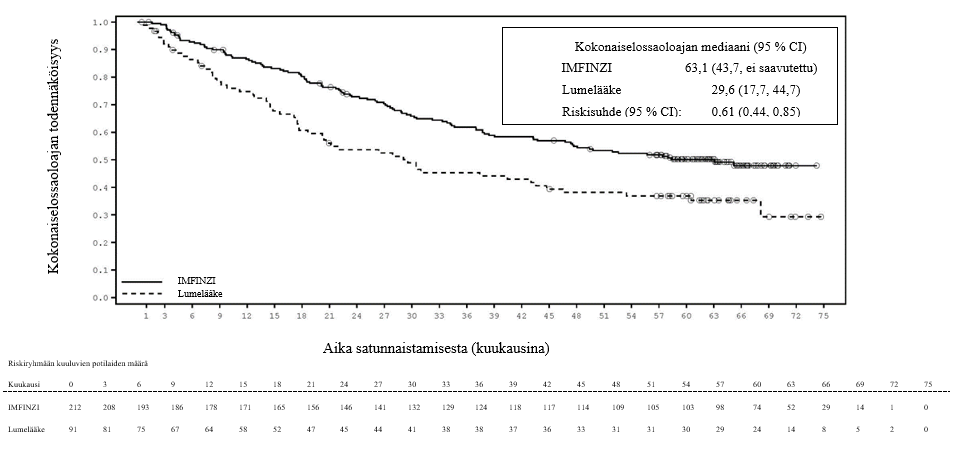
Kuva 5. Etenemättömyysajan Kaplan‑Meier-käyrä potilailla, joiden PD-L1 TC oli ≥1 %
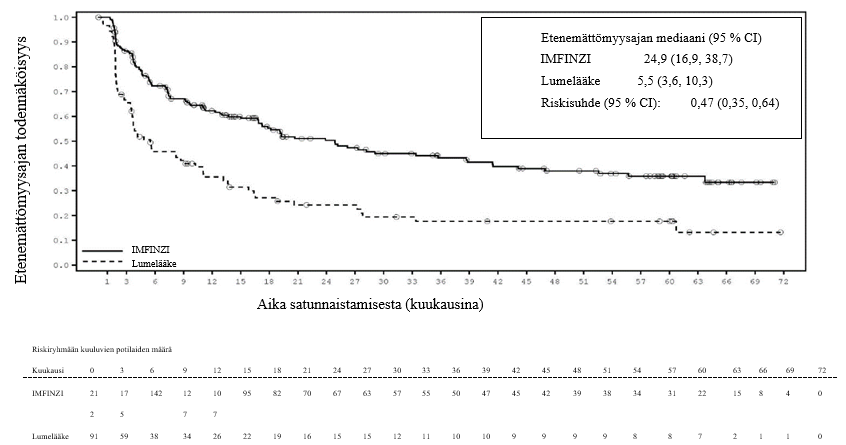
Kuva 6. Kokonaiselossaoloajan Forest plot-kuvaaja PD‑L1:n ilmentymisen mukaan
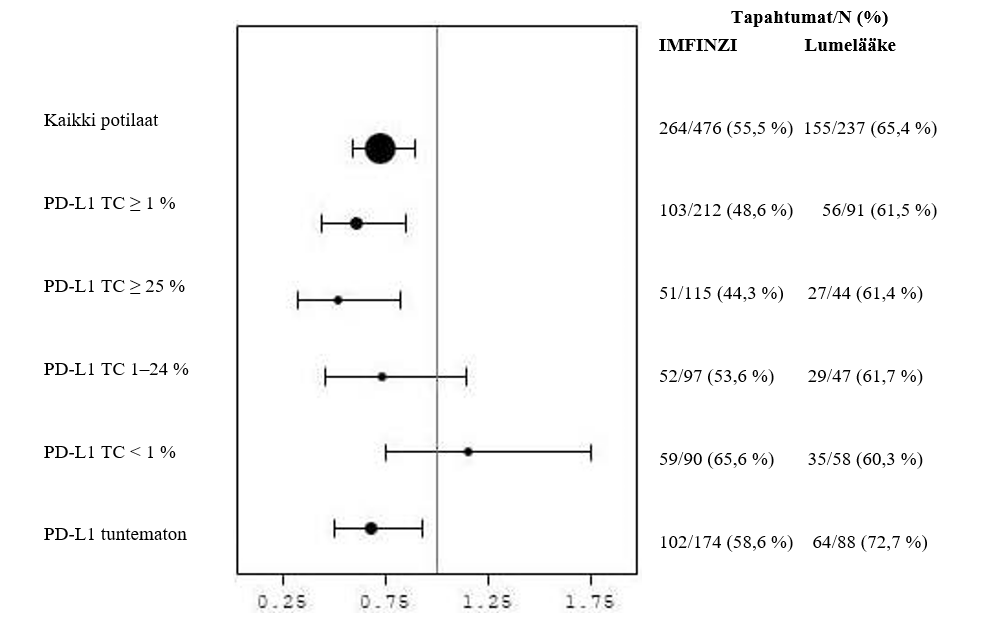
Kuva 7. Etenemättömyysajan Forest plot-kuvaaja PD‑L1:n ilmentymisen mukaan
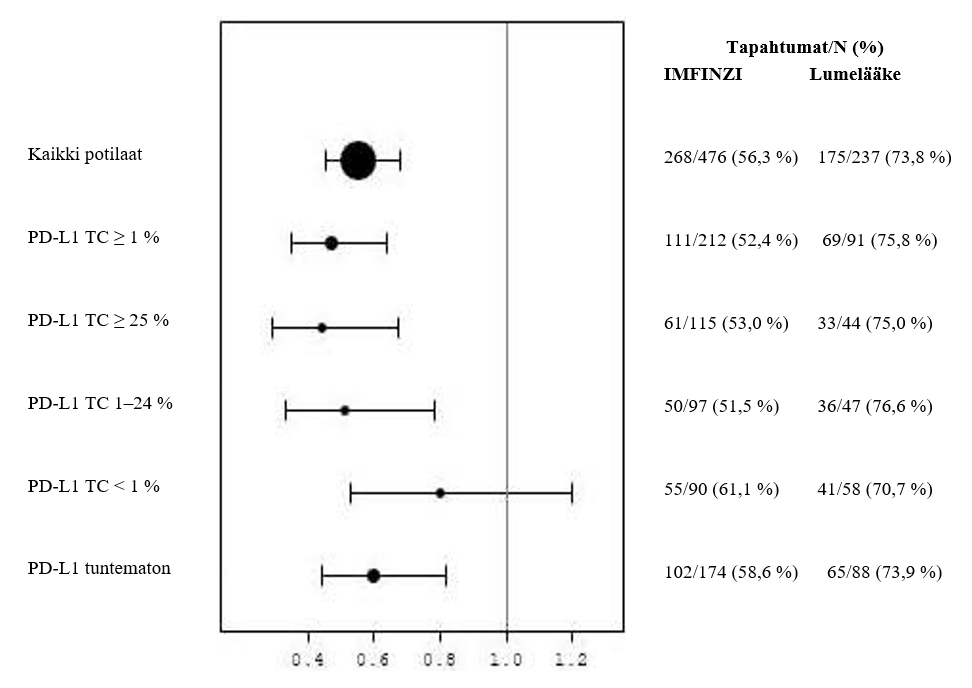
Alaryhmässä, jossa PD‑L1 TC oli ≥ 1 %, durvalumabin turvallisuusprofiili oli kaiken kaikkiaan vastaavanlainen kuin hoitoaiepopulaatiossa samoin kuin alaryhmässä, jossa PD‑L1 TC oli < 1 %.
Potilaiden ilmoittamat hoitotulokset (Patient-reported outcomes, PRO)
Tiedot potilaiden ilmoittamista oireista, toimintakyvystä ja terveyteen liittyvästä elämänlaadusta kerättiin EORTC QLQ‑C30 -kyselyllä ja sen keuhkosyöpämoduulilla (EORTC QLQ‑LC13). LC13- ja C30-kyselyt tehtiin lähtötilanteessa, 4 viikon välein ensimmäisten 8 viikon ajan ja sen jälkeen 8 viikon välein joko hoitojakson päättymiseen, toksisista vaikutuksista johtuvan IMFINZI-hoidon lopettamiseen tai taudin etenemiseen saakka. Kyselyihin vastanneiden tutkittavien määrät olivat ryhmissä samankaltaiset (IMFINZI-ryhmässä kaiken kaikkiaan 83 % ja lumeryhmässä 85,1 % sellaisista täytetyistä lomakkeista, jotka voitiin arvioida).
Lähtötilanteessa IMFINZI- ja lumeryhmien välillä ei havaittu eroja potilaiden ilmoittamista oireissa, toimintakyvyssä tai terveyteen liittyvässä elämänlaadussa. Tutkimuksen keston aikana viikkoon 48 mennessä IMFINZI- ja lumeryhmien välillä ei todettu kliinisesti merkityksellisiä (vähintään 10 pisteen) eroja oireissa, toimintakyvyssä tai terveyteen liittyvässä elämänlaadussa.
Ei-pienisoluinen keuhkosyöpä – POSEIDON-tutkimus
POSEIDON-tutkimuksen tarkoituksena oli arvioida IMFINZI-valmisteen tehoa, kun IMFINZI-valmistetta annettiin tremelimumabin kanssa tai ilman sitä, yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa. POSEIDON oli satunnaistettu, avoin monikeskustutkimus, johon osallistuneet 1 013 potilasta sairastivat metastasoitunutta ei-pienisoluista keuhkosyöpää eikä heillä ollut herkistävää epidermaalisen kasvutekijän reseptorin (EGFR) mutaatiota eikä anaplastisen lymfoomakinaasin (ALK) poikkeavuuksia kasvaimen genomissa. Tutkimukseen otettiin potilaita, joilla oli histologisesti tai sytologisesti dokumentoitu metastasoitunut ei-pienisoluinen keuhkosyöpä. Potilaat eivät olleet aiemmin saaneet solunsalpaajahoitoa tai muuta systeemistä hoitoa metastasoituneeseen ei-pienisoluiseen keuhkosyöpään. Ennen satunnaistamista kasvainten PD‑L1-status varmistettiin Ventana PD‑L1 (SP263) ‑analyysillä. Potilaiden WHO/ECOG (World Health Organization / Eastern Cooperative Oncology Group) ‑suorituskykypistemäärä oli 0 tai 1 heidän aloittaessaan tutkimuksessa.
Tutkimuksesta suljettiin pois potilaat, joilla oli aktiivinen tai aiemmin dokumentoitu autoimmuunisairaus; aktiivisia ja/tai hoitamattomia aivometastaaseja; anamneesissa immuunipuutos; systeemisten immuunisalpaajien käyttöä 14 päivän kuluessa ennen IMFINZI- tai tremelimumabihoidon aloittamista, lukuun ottamatta systeemistä kortikosteroidihoitoa fysiologisella annoksella; aktiivinen tuberkuloosi tai hepatiitti B- tai hepatiitti C ‑infektio tai HIV-infektio, sekä potilaat, jotka saivat eläviä heikennettyjä taudinaiheuttajia sisältävän rokotteen 30 päivän kuluessa ennen IMFINZI- ja/tai tremelimumabihoidon aloittamista tai sen aloittamisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Satunnaistamisessa ositustekijöinä olivat kasvainsolujen PD‑L1:n ilmentyminen (≥ 50 % kasvainsoluista vs. < 50 % kasvainsoluista), taudin levinneisyysaste (levinneisyysaste IVA vs. levinneisyysaste IVB American Joint Committee on Cancer ‑organisaation julkaiseman luokituskäsikirjan 8. painoksen mukaan) ja histologia (ei-levyepiteeliperäinen vs. levyepiteeliperäinen).
Potilaat satunnaistettiin suhteessa 1:1:1 seuraavasti:
- Hoitohaara 1: IMFINZI 1 500 mg sekä tremelimumabi 75 mg ja platinapohjainen solunsalpaajahoito 3 viikon välein 4 hoitosyklin ajan ja sen jälkeen IMFINZI 1 500 mg 4 viikon välein monoterapiana. Tremelimumabia annettiin viides 75 mg:n annos viikolla 16 IMFINZI-annoksen 6 kanssa.
- Hoitohaara 2: IMFINZI 1 500 mg ja platinapohjainen solunsalpaajahoito 3 viikon välein 4 hoitosyklin ajan ja sen jälkeen IMFINZI 1 500 mg 4 viikon välein monoterapiana.
- Hoitohaara 3: Platinapohjainen solunsalpaajahoito 3 viikon välein 4 hoitosyklin ajan. Tutkijalääkärin harkinnan ja kliinisen tarpeen mukaan hoitoon saatettiin lisätä vielä kaksi hoitosykliä (yhteensä 6 hoitosykliä satunnaistamisen jälkeen).
Näissä kolmessa hoitohaarassa potilaat saivat histologisten tietojen perusteella jotakin seuraavista solunsalpaajahoidoista:
-
Ei-levyepiteeliperäinen ei-pienisoluinen keuhkosyöpä
- Pemetreksedi 500 mg/m2 sekä karboplatiini AUC 5–6 tai sisplatiini 75 mg/m2 kolmen viikon välein. Pemetreksediylläpitohoitoa voitiin antaa, ellei tutkijalääkäri katsonut sen olevan vasta-aiheista.
-
Levyepiteeliperäinen ei-pienisoluinen keuhkosyöpä
- Gemsitabiini 1 000 tai 1 250 mg/m2 päivinä 1 ja 8 sekä sisplatiini 75 mg/m2 tai karboplatiini AUC 5–6 päivänä 1 kolmen viikon välein.
-
Ei-levyepiteeliperäinen tai levyepiteeliperäinen ei-pienisoluinen keuhkosyöpä
- Nab-paklitakseli 100 mg/m2 päivinä 1, 8 ja 15 sekä karboplatiini AUC 5–6 päivänä 1 kolmen viikon välein.
Jos tauti ei edennyt eikä ilmennyt toksisia vaikutuksia, joita ei voitu hyväksyä, tremelimumabia voitiin antaa enintään 5 annosta. IMFINZI-hoitoa ja histologisiin tietoihin perustuvaa pemetreksediylläpitohoitoa (jos sen käyttö oli soveltuvaa) jatkettiin, kunnes tauti eteni tai ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä.
Kasvaimet arvioitiin viikon 6 ja viikon 12 kohdalla satunnaistamispäivästä lukien ja sen jälkeen 8 viikon välein, kunnes taudin vahvistettiin objektiivisesti edenneen. Elossaoloarvioinnit tehtiin 2 kuukauden välein hoidon lopettamisen jälkeen.
Tutkimuksen kaksi ensisijaista päätetapahtumaa olivat etenemättömyysaika ja kokonaiselossaoloaika, kun verrattiin IMFINZI-valmisteen ja platinapohjaisen solunsalpaajahoidon yhdistelmää pelkkään platinapohjaiseen solunsalpaajahoitoon. Tutkimuksen keskeiset toissijaiset päätetapahtumat olivat etenemättömyysaika ja kokonaiselossaoloaika IMFINZI-valmisteen, tremelimumabin ja platinapohjaisen solunsalpaajahoidon yhdistelmällä sekä pelkällä platinapohjaisella solunsalpaajahoidolla. Toissijaisia päätetapahtumia olivat objektiivinen vasteprosentti (ORR) ja vasteen kesto. Sokkoutettu riippumaton keskitetty arvioijataho arvioi etenemättömyysajan, objektiivisen vasteprosentin ja vasteen keston RECIST v1.1 ‑kriteerien mukaisesti.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat hyvin samankaltaiset eri tutkimushaaroissa. Koko tutkimuspopulaation demografiset tiedot lähtötilanteessa olivat: miehiä (76,0 %), ikä vähintään 65 vuotta (47,1 %), ikä vähintään 75 vuotta (11,3 %), iän mediaani 64 vuotta (vaihteluväli: 27–87 vuotta), valkoihoisia (55,9 %), aasialaisia (34,6 %), mustaihoisia tai afrikkalaisamerikkalaisia (2,0 %), muita (7,6 %), muita kuin taustaltaan espanjankielisiä tai latinalaisamerikkalaisia (84,2 %), edelleen tupakoivia tai aiemmin tupakoineita (78,0 %), WHO/ECOG-suorituskykypistemäärä 0 (33,4 %), WHO/ECOG-suorituskykypistemäärä 1 (66,5 %). Sairauden ominaispiirteet olivat: levinneisyysaste IVA (50,0 %), levinneisyysaste IVB (49,6 %), histologinen alatyyppi levyepiteeliperäinen (36,9 %), ei-levyepiteeliperäinen (62,9 %), aivometastaaseja (10,5 %), PD‑L1:n ilmentyminen kasvainsoluissa ≥ 50 % (28,8 %), PD‑L1:n ilmentyminen kasvainsoluissa < 50 % (71,1 %).
Tutkimuksessa osoitettiin kokonaiselossaoloajan olevan tilastollisesti merkitsevästi pidempi IMFINZI-valmisteen, tremelimumabin ja platinapohjaisen solunsalpaajahoidon yhdistelmää saaneilla verrattuna platinapohjaista solunsalpaajahoitoa saaneisiin. IMFINZI-valmisteen, tremelimumabin ja platinapohjaisen solunsalpaajahoidon yhdistelmää saaneilla potilailla havaittiin etenemättömyysajan olevan tilastollisesti merkitsevästi pidempi verrattuna pelkkää platinapohjaista solunsalpaajahoitoa saaneisiin. Tulokset on esitetty yhteenvetona alla.
Taulukko 7. POSEIDON-tutkimuksen tehoa koskevat tulokset
| Hoitohaara 1: IMFINZI + tremelimumabi + platinapohjainen solunsalpaajahoito (n = 338) | Hoitohaara 3: Platinapohjainen solunsalpaajahoito (n = 337) | |
| Kokonaiselossaoloaikaa | ||
| Kuolemantapausten määrä (%) | 251 (74,3) | 285 (84,6) |
| Kokonaiselossaoloajan mediaani (kuukausina) (95 %:n luottamusväli) | 14,0 (11,7; 16,1) | 11,7 (10,5; 13,1) |
| Riskisuhde (95 %:n luottamusväli)b | 0,77 (0,650; 0,916) | |
| p‑arvoc | 0,00304 | |
| Etenemättömyysaikaa | ||
| Tapahtumien lukumäärä (%) | 238 (70,4) | 258 (76,6) |
| Etenemättömyysajan mediaani (kuukausina) (95 %:n luottamusväli) | 6,2 (5,0; 6,5) | 4,8 (4,6; 5,8) |
| Riskisuhde (95 %:n luottamusväli)b | 0,72 (0,600; 0,860) | |
| p‑arvoc | 0,00031 | |
| Objektiiviset vasteet, n (%)d,e | 130 (38,8) | 81 (24,4) |
| Täydellinen vaste, n (%) | 2 (0,6) | 0 |
| Osittainen vaste, n (%) | 128 (38,2) | 81 (24,4) |
| Vasteen keston mediaani (kuukausina) (95 %:n luottamusväli)d,e | 9,5 (7,2; ei saavutettu) | 5,1 (4,4; 6,0) |
a Etenemättömyyden analyysi tiedonkeruun katkaisupisteen 24.7.2019 kohdalla (seuranta-ajan mediaani 10,15 kuukautta). Kokonaiselossaolon analyysi tiedonkeruun katkaisupisteen 12.3.2021 kohdalla (seuranta-ajan mediaani 34,86 kuukautta). Tehon toteamiseen vaadittavat raja-arvot (hoitohaara 1 verrattuna hoitohaaraan 3: etenemättömyysaika 0,00735, kokonaiselossaoloaika 0,00797; kaksitahoinen) määritettiin Lan–DeMetsin alfavirheen korjausfunktiolla, joka approksimoi O’Brien–Flemingin lähestymistapaa. Sokkoutettu riippumaton keskitetty arvioijataho arvioi etenemättömyysajan RECIST v1.1 ‑kriteerien mukaisesti.
b Riskitiheyssuhteet saadaan käyttämällä PD‑L1-statuksen, histologian ja taudin levinneisyysasteen mukaan stratifioitua Coxin pH‑mallia.
c Kaksitahoinen p-arvo, joka perustuu PD‑L1-statuksen, histologian ja taudin levinneisyysasteen mukaan stratifioituun log rank ‑testiin.
d Vahvistettu objektiivinen vaste.
e Post hoc ‑analyysi.
Kuva 8. Kokonaiselossaoloajan Kaplan–Meier-käyrä
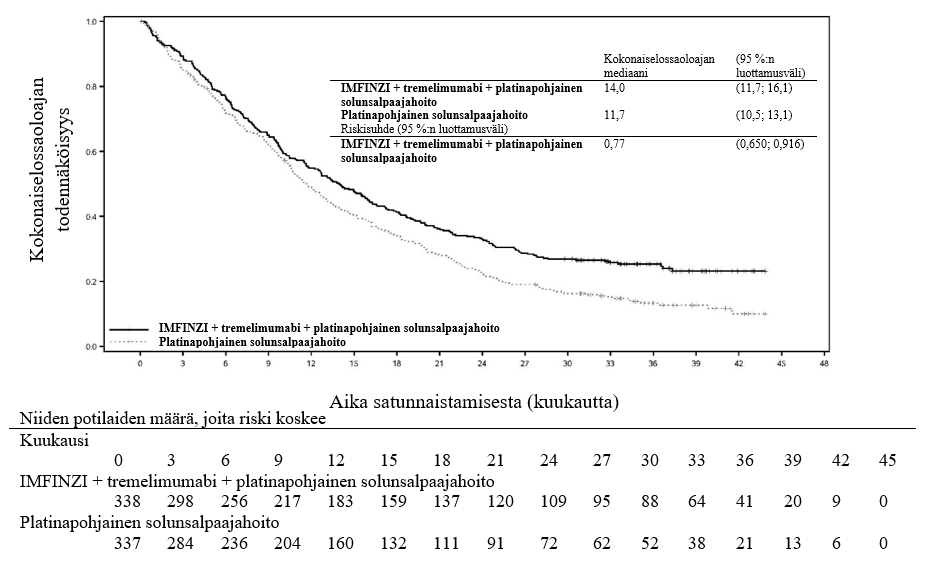
Kuva 9. Etenemättömyysajan Kaplan–Meier-käyrä
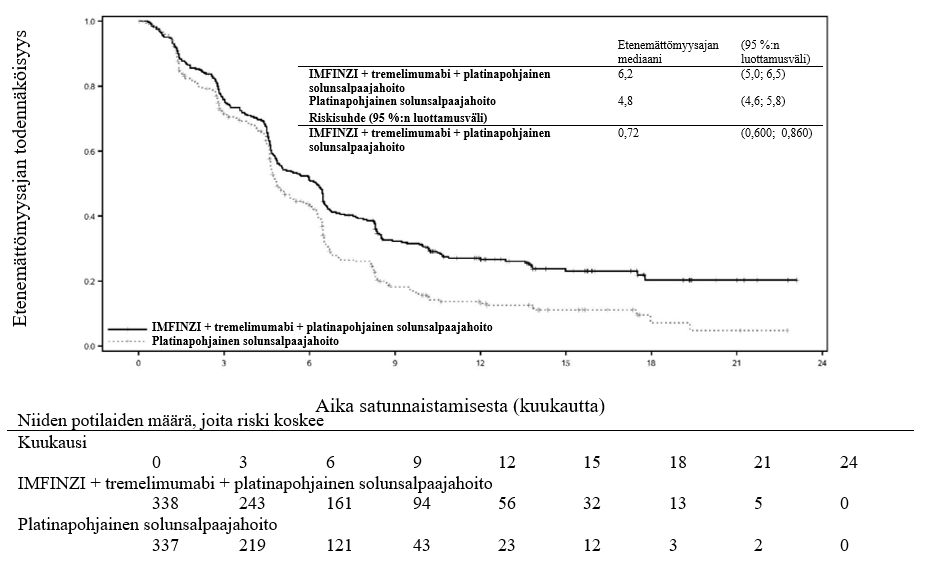
Kuvassa 10 on esitetty yhteenvedot tuloksista, jotka koskevat tehoa kokonaiselossaoloajan suhteen kasvaimen PD‑L1:n ilmentymisen mukaan ennalta määritellyissä alaryhmäanalyyseissä.
Kuva 10. Kokonaiselossaoloajan Forest plot ‑kuvaaja PD‑L1:n ilmentymisen mukaan IMFINZI-valmisteen, tremelimumabin ja platinapohjaisen solunsalpaajahoidon yhdistelmää saaneilla verrattuna platinapohjaista solunsalpaajahoitoa saaneisiin
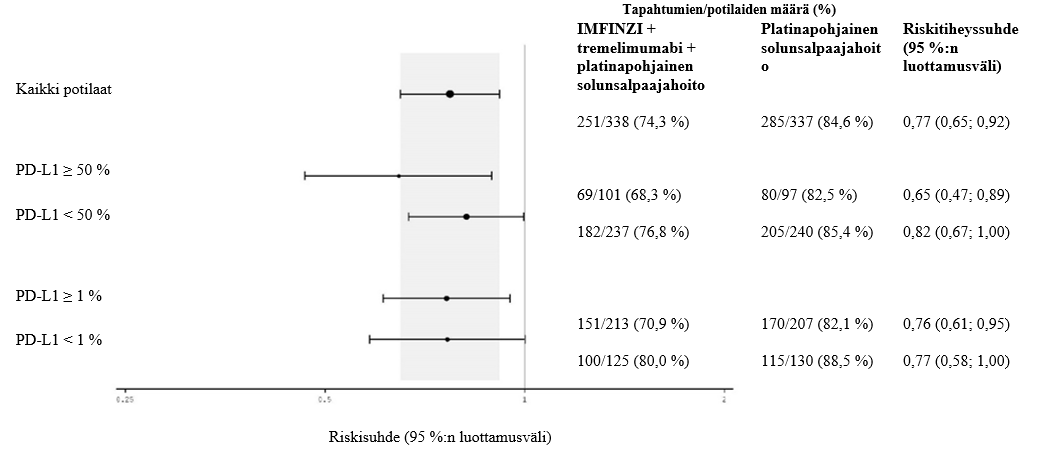
Iäkkäät henkilöt
Yhteensä 75 potilasta, jotka olivat vähintään 75‑vuotiaita, otettiin POSEIDON-tutkimuksen hoitohaaroihin, joissa tutkittavat saivat IMFINZI-valmistetta yhdistelmänä tremelimumabin ja solunsalpaajahoidon kanssa (n = 35) tai pelkkää platinapohjaista solunsalpaajahoitoa (n = 40). Kokonaiselossaoloajalle havaittu eksploratiivinen riskisuhde oli 1,05 (95 %:n luottamusväli: 0,64, 1,71) IMFINZI-valmistetta yhdistelmänä tremelimumabin ja platinapohjaisen solunsalpaajahoidon kanssa saaneilla verrattuna platinapohjaiseen solunsalpaajahoitoon tässä tutkimuksen alaryhmässä. Koska tämä alaryhmäanalyysi oli luonteeltaan eksploratiivinen, lopullisia johtopäätöksiä ei voida tehdä. Varovaisuutta kuitenkin suositellaan noudatettavan, kun tätä hoito-ohjelmaa harkitaan iäkkäille potilaille.
Pienisoluinen keuhkosyöpä –ADRIATIC-tutkimus
ADRIATIC-tutkimuksen tarkoituksena oli arvioida IMFINZI-valmisteen tehoa, kun IMFINZI-valmistetta annettiin joko tremelimumabin kanssa tai ilman sitä. ADRIATIC oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu monikeskustutkimus, johon osallistuneilla 730 potilaalla oli histologisesti tai sytologisesti varmistettu rajoittunut pienisoluinen keuhkosyöpä (levinneisyysaste I–III AJCC-luokituskäsikirjan 8. painoksen mukaan), eikä heidän sairautensa ollut edennyt samanaikaisen kemosädehoidon jälkeen. Levinneisyysasteiden I ja II kohdalla edellytettiin, että tutkijalääkäri oli arvioinut syövän olevan lääketieteellisistä syistä soveltumaton leikkaushoitoon. Potilaat olivat jatkaneet loppuun asti neljä definitiivisen platinapohjaisen kemosädehoidon hoitosykliä, 60–66 Gy kerran vuorokaudessa 6 viikon ajan tai 45 Gy kahdesti vuorokaudessa 3 viikon ajan, siten, että viimeinen hoitokerta oli ollut 1–42 päivän sisällä ennen tutkimusvalmisteen ensimmäisen annoksen saamista. Kallon ennaltaehkäisevää sädehoitoa (PCI) voitiin antaa tutkijalääkärin harkinnan mukaan kemosädehoidon jälkeen ja 1–42 päivän sisällä ennen tutkimusvalmisteen ensimmäisen annoksen saamista. Potilaiden ECOG-suorituskykypistemäärä oli 0 tai 1 heidän aloittaessaan tutkimuksessa.
Tutkimuksesta suljettiin pois potilaat, joilla oli aktiivinen autoimmuunisairaus tai joilla oli ollut dokumentoitu autoimmuunisairaus viiden vuoden sisällä ennen tutkimuksen aloittamisesta; potilaat, joilla oli tai oli aiemmin ollut aktiivinen, primaarinen immuunipuutos; potilaat, joilla oli tai oli aiemmin ollut vaikeusasteen ≥ 2 pneumoniitti tai aktiivinen tuberkuloosi tai hepatiitti B- tai hepatiitti C ‑infektio tai HIV-infektio, sekä potilaat, joilla oli aktiivinen interstitiaalinen keuhkosairaus. Tutkimuksesta suljettiin pois myös potilaat, joilla keuhkosyöpä oli histologialtaan sekamuotoinen (pienisoluinen/ei-pienisoluinen).
Satunnaistamisessa ositustekijöinä olivat levinneisyysaste (I/II vs. III) ja kallon ennaltaehkäisevän sädehoidon saaminen (kyllä vs. ei). Potilaat satunnaistettiin suhteessa 1:1:1 seuraavasti:
- Hoitohaara 1: IMFINZI 1 500 mg + lumelääke 4 viikon välein 4 hoitosyklin ajan ja sen jälkeen IMFINZI 1 500 mg 4 viikon välein.
- Hoitohaara 2: Lumelääke + toinen lumelääke 4 viikon välein 4 hoitosyklin ajan ja sen jälkeen yksi lumelääke 4 viikon välein.
- Hoitohaara 3: IMFINZI 1 500 mg + tremelimumabi 75 mg 4 viikon välein 4 hoitosyklin ajan ja sen jälkeen IMFINZI 1 500 mg 4 viikon välein.
Kun kaikkiin kolmeen hoitohaaraan oli satunnaistettu yhteensä 600 potilasta, satunnaistaminen hoitohaaraan 3 päättyi. Seuraavat 130 potilasta satunnaistettiin suhteessa 1:1 joko hoitohaaraan 1 tai 2, ja he saivat joko IMFINZI-valmistetta 1 500 mg 4 viikon välein tai lumelääkettä 4 viikon välein.
Hoitoa jatkettiin, kunnes tauti eteni tai ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä, tai enintään 24 kuukauden ajan. Kasvaimet arvioitiin ensimmäisten 72 viikon aikana 8 viikon välein, sitten enintään 96 viikon aikana 12 viikon välein ja sen jälkeen 24 viikon välein.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat hyvin samankaltaiset eri tutkimushaaroissa. IMFINZI- ja lumehaarojen demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat: miehiä (69,1 %), ikä vähintään 65 vuotta (39,2 %), valkoihoisia (50,4 %), mustaihoisia tai afrikkalaisamerikkalaisia (0,8 %), aasialaisia (47,5 %), muita (1,3 %), taustaltaan espanjankielisiä tai latinalaisamerikkalaisia (4,2 %), edelleen tupakoivia (22,3 %), aiemmin tupakoineita (68,5 %), ei milloinkaan tupakoineita (9,2 %), WHO/ECOG-suorituskykypistemäärä 0 (48,7 %), WHO/ECOG-suorituskykypistemäärä 1 (51,3 %), levinneisyysaste I (3,6 %), levinneisyysaste II (9,1 %) ja levinneisyysaste III (87,4 %).
Ennen satunnaistamista kaikki potilaat saivat platinapohjaista solunsalpaajahoitoa (66,2 % sai sisplatiinia ja etoposidia, 33,8 % karboplatiinia ja etoposidia); 72,1 % potilaista sai sädehoitoa kerran vuorokaudessa (näistä 92,4 %:lla annos oli ≥ 60 – ≤ 66 Gy kerran vuorokaudessa); 27,9 % sai sädehoitoa kaksi kertaa vuorokaudessa (näistä 96,6 %:lla annos oli 45 Gy kahdesti vuorokaudessa) ja 53,8 % potilaista sai kallon ennaltaehkäisevää sädehoitoa. Vasteet kemosädehoitoon olivat seuraavanlaiset: täydellinen vaste (12,3 %), osittainen vaste (73,8 %), vakaa tauti (14,0 %).
Tutkimuksen kaksi ensisijaista päätetapahtumaa olivat kokonaiselossaoloaika ja etenemättömyysaika IMFINZI-valmistetta saaneilla verrattuna lumelääkettä saaneisiin. Toissijaisiin tehoa koskeviin päätetapahtumiin sisältyi muun muassa objektiivinen vasteprosentti IMFINZI-valmistetta saaneilla verrattuna lumelääkettä saaneisiin. Sokkoutettu riippumaton keskitetty arvioijataho arvioi etenemättömyysajan ja objektiivisen vasteprosentin RECIST v1.1 ‑kriteerien mukaisesti.
Suunnitellussa välianalyysissä tutkimus osoitti, että IMFINZI-valmistetta saaneilla kokonaiselossaoloaika ja etenemättömyysaika pitenivät tilastollisesti merkitsevästi verrattuna lumelääkettä saaneisiin. Ks. taulukko 8 ja kuvat 11 ja 12.
Taulukko 8. ADRIATIC-tutkimuksen tehoa koskevat tulokset
| Hoitohaara 1: IMFINZI (n = 264) | Hoitohaara 2: Lumelääke (n = 266) | |
|---|---|---|
Kokonaiselossaoloaikaa | ||
| Kuolemantapausten määrä (%) | 115 (43,6) | 146 (54,9) |
| Kokonaiselossaoloajan mediaani (kuukausina) (95 %:n luottamusväli)b | 55,9 (37,3; ei saavutettu) | 33,4 (25,5; 39,9) |
| Riskisuhde (95 %:n luottamusväli)c | 0,73 (0,569; 0,928) | |
| p‑arvod | 0,01042 | |
| Etenemättömyysaikae | ||
| Tapahtumien määrä (%) | 139 (52,7) | 169 (63,5) |
| Etenemättömyysajan mediaani (kuukausina) (95 %:n luottamusväli)b | 16,6 (10,2; 28,2) | 9,2 (7,4; 12,9) |
| Riskisuhde (95 %:n luottamusväli)f | 0,76 (0,606; 0,950) | |
| p‑arvod | 0,01608 | |
a Kokonaiselossaoloajan seurannan keston mediaani sensuroiduilla potilailla oli IMFINZI-valmisteen hoitohaarassa 37,19 kuukautta ja lumehaarassa 37,24 kuukautta.
b Laskettu Kaplan–Meier-tekniikalla. Mediaanin luottamusväli laskettiin Brookmeyer–Crowley-menetelmällä.
c Riskisuhdeanalyysi tehtiin käyttäen ositettua Coxin suhteellisten riskitiheyksien mallia, ja kaksitahoinen p-arvo perustuu stratifioituun log-rank-testiin; molemmat on korjattu kallon ennaltaehkäisevän sädehoidon saamisen suhteen.
d p‑arvo perustui ennalta suunnitellusta välianalyysistä saatuihin tuloksiin. Tilastollisen merkitsevyyden raja oli kokonaiselossaoloajalle 0,01679, kun kokonaisalfavirhe oli 4,5 %, ja etenemättömyysajalle 0,02805, kun kokonaisalfavirhe oli 5 %, perustuen Lan–DeMetsin alfavirheen korjausfunktioon, O'Brien–Flemingin tyyppiseen rajaan ja havaittujen tapahtumien todelliseen lukumäärään (Lan ja DeMets 1983).
e Sokkoutettu riippumaton keskitetty arvioijataho arvioi RECIST v1.1 ‑kriteerien mukaisesti.
f Riskisuhdeanalyysi tehtiin käyttäen ositettua Coxin suhteellisten riskitiheyksien mallia, ja kaksitahoinen p-arvo perustuu stratifioituun log-rank-testiin; molemmat on korjattu TNM-luokituksen mukaisen levinneisyysasteen ja kallon ennaltaehkäisevän sädehoidon saamisen suhteen.
Kuva 11: Kokonaiselossaoloajan Kaplan–Meier -käyrä
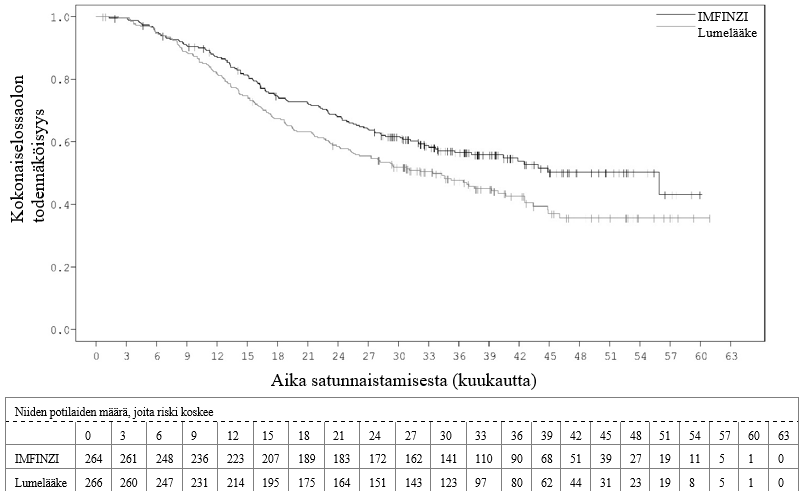
Kuva 12: Etenemättömyysajan Kaplan–Meier-käyrä
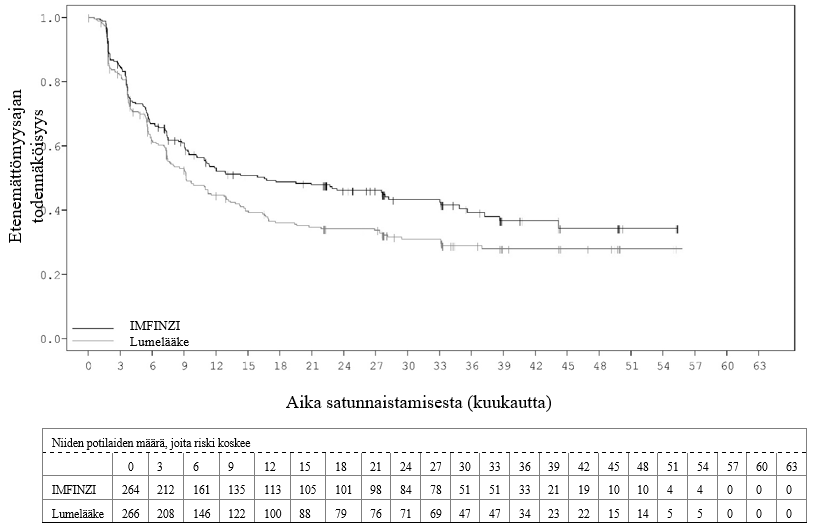
Pienisoluinen keuhkosyöpä – CASPIAN-tutkimus
CASPIAN-tutkimus oli suunniteltu arvioimaan tremelimumabin kanssa tai ilman sitä annetun IMFINZI-valmisteen tehoa, kun sitä annettiin yhdistelmänä etoposidin ja joko karboplatiinin tai sisplatiinin kanssa. CASPIAN oli satunnaistettu, avoin monikeskustutkimus, johon osallistui 805 aiemmin hoitamatonta levinnyttä pienisoluista keuhkosyöpää sairastavaa potilasta. Potilaiden WHO/ECOG-suorituskykyluokka oli 0 tai 1 ja paino > 30 kg, he soveltuivat saamaan platinapohjaista solunsalpaajahoitoa pienisoluisen keuhkosyövän ensilinjan hoitona, heidän elinajanodotteensa oli ≥ 12 viikkoa, ja heillä oli vähintään yksi RECIST 1.1. -kriteerien mukainen kohdeleesio ja riittävä sisäelinten ja luuytimen toimintakyky. Potilaat, joilla oli oireettomia tai hoidettuja aivometastaaseja, soveltuivat tutkimukseen. Tutkimuksesta suljettiin pois potilaat, joilla oli anamneesissa rintakehän sädehoitoa; anamneesissa aktiivinen, primaarinen immuunipuutos; autoimmuunisairaus mukaan lukien paraneoplastinen oireyhtymä; aktiivinen tai aiempi dokumentoitu autoimmuuni- tai tulehdussairaus; systeemisten immunosuppressanttien käyttöä 14 päivän sisällä ennen ensimmäistä hoitoannosta, lukuun ottamatta fysiologisina annoksina käytettyjä systeemisiä kortikosteroideja; aktiivinen tuberkuloosi tai hepatiitti B tai C tai HIV-infektio; tai jotka olivat saaneet eläviä, heikennettyjä rokotteita 30 päivän kuluessa ennen IMFINZI-hoidon aloitusta tai sen jälkeen.
Satunnaistaminen stratifioitiin suunnitellun platinapohjaisen hoidon (karboplatiini- tai sisplatiinihoidon) mukaan hoitosyklissä 1.
Potilaat satunnaistettiin suhteessa 1:1:1 saamaan:
- Hoitohaara 1: IMFINZI 1 500 mg + tremelimumabi 75 mg + etoposidi ja joko karboplatiini tai sisplatiini.
- Hoitohaara 2: IMFINZI 1 500 mg + etoposidi ja joko karboplatiini tai sisplatiini.
- Hoitohaara 3: Joko karboplatiini (AUC 5 tai 6 mg/ml/min) tai sisplatiini (75–80 mg/m2) päivänä 1 ja etoposidi (80–100 mg/m2) laskimoon kunkin 21 päivän pituisen hoitosyklin päivinä 1, 2 ja 3 neljän–kuuden hoitosyklin ajan.
Hoitohaaroihin 1 ja 2 satunnaistettujen potilaiden osalta etoposidi- ja karboplatiini- tai sisplatiinihoidon kesto rajoitettiin 4 hoitosykliin 3 viikon välein satunnaistamisen jälkeen. IMFINZI-monoterapiaa jatkettiin 4 viikon välein, kunnes tauti eteni tai ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä. IMFINZI-monoterapiaa voitiin jatkaa taudin etenemisenkin jälkeen, jos potilaan kliininen tila oli vakaa ja tutkijalääkäri katsoi, että hoidosta oli kliinistä hyötyä.
Hoitohaaraan 3 satunnaistetut potilaat saattoivat saada tarvittaessa yhteensä enintään 6 hoitosyklin verran etoposidia ja joko karboplatiinia tai sisplatiinia. Etoposidi- ja platinahoidon loppuun suorittamisen jälkeen ennaltaehkäisevä kallon sädehoito PCI oli sallittua vain hoitohaarassa 3 tutkijalääkärin harkinnan mukaan.
Kasvaimet arvioitiin viikon 6 ja viikon 12 kohdalla satunnaistamispäivästä lukien ja sen jälkeen 8 viikon välein, kunnes taudin vahvistettiin objektiivisesti edenneen. Elossaoloarvioinnit tehtiin 2 kuukauden välein hoidon lopettamisen jälkeen.
Tutkimuksen ensisijaiset päätetapahtumat olivat kokonaiselossaoloaika yhdistelmällä IMFINZI + etoposidi + platina (hoitohaara 2) vs. yhdistelmällä pelkkä etoposidi + platina (hoitohaara 3) sekä kokonaiselossaoloaika yhdistelmällä IMFINZI + tremelimumabi + etoposidi + platina (hoitohaara 1) vs. yhdistelmällä pelkkä etoposidi + platina (hoitohaara 3). Keskeinen toissijainen päätetapahtuma oli etenemättömyysaika. Muut toissijaiset päätetapahtumat olivat objektiivinen vasteprosentti, kokonaiselossaoloajan ja etenemättömyysajan merkkipaalut ja potilaan ilmoittamat vaikutukset. Etenemisvapaata elinaikaa ja objektiivista vasteprosenttia arvioitiin tutkijan RECIST v1.1 -kriteerien mukaisten arviointien avulla.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat hyvin samankaltaiset kahdessa hoitohaarassa (hoitohaarassa 2 oli 268 potilasta ja hoitohaarassa 3 oli 269 potilasta). Koko tutkimuspopulaation demografiset tiedot lähtötilanteessa olivat: miehiä (69,6 %), ikä ≥ 65 vuotta (39,6 %), iän mediaani 63 vuotta (vaihteluväli: 28–82 vuotta), valkoihoisia (83,8 %), aasialaisia (14,5 %), tummaihoisia tai afroamerikkalaisia (0,9 %), muita (0,6 %), muita kuin taustaltaan espanjankielisiä tai latinalaisamerikkalaisia (96,1 %), tupakoitsijoita tai aiemmin tupakoineita (93,1 %), tupakoimattomia (6,9 %), WHO/ECOG-toimintakykyluokka 0 (35,2 %), WHO/ECOG-toimintakykyluokka 1 (64,8 %), levinneisyysaste IV 90,3 %, 24,6 % potilaista sai sisplatiinihoitoa ja 74,1 % karboplatiinihoitoa. Hoitohaarassa 3 potilaista 56,8 % sai 6 hoitosykliä etoposidin ja platinahoidon yhdistelmää ja 7,8 % potilaista sai ennaltaehkäisevää kallon sädehoitoa.
Suunnitellussa (ensisijaisessa) välianalyysissä tutkimus osoitti, että IMFINZI-valmisteen, etoposidin ja platinahoidon yhdistelmää saaneessa hoitohaarassa (hoitohaara 2) kokonaiselossaoloaika koheni tilastollisesti merkitsevästi verrattuna pelkkää etoposidia ja platinahoitoa saaneeseen hoitohaaraan (hoitohaara 3) [HR = 0,73 (95 %:n luottamusväli: 0,591, 0,909); p = 0,0047]. Vaikkakaan merkitsevyyttä ei testattu muodollisesti, IMFINZI-valmisteen, etoposidin ja platinahoidon yhdistelmää saaneilla potilailla havaittiin etenemättömyysajan pitenemistä verrattuna pelkkää etoposidia ja platinahoitoa saaneisiin potilaisiin [HR = 0,78 (95 %:n luottamusväli: 0,645, 0,936)].
Suunnitellun lopullisen analyysin (viimeinen tiedonkeruupäivä 27.1.2020) etenemisvapaata elinaikaa, objektiivista vasteprosenttia ja vasteen kestoa koskevat tulokset esitetään yhteenvetona taulukossa 9. Etenemättömyysajan Kaplan–Meier-käyrä esitetään kuvassa 14.
Kokonaiselossaoloajan suunnitellun pitkän aikavälin seuranta-analyysin (viimeinen tiedonkeruupäivä 22.3.2021) (seuranta-ajan mediaani: 39,3 kuukautta) kokonaiselossaoloaikaa koskevat tulokset esitetään taulukossa 9. IMFINZI-valmisteen, etoposidin ja platinahoidon yhdistelmää saaneiden potilaiden (hoitohaara 2) kokonaiselossaoloaika oli edelleen pitkäkestoisesti parempi kuin pelkkää etoposidia ja platinahoitoa saaneiden potilaiden (hoitohaara 3). Kokonaiselossaoloajan Kaplan–Meier-käyrä esitetään kuvassa 13.
Taulukko 9. CASPIAN-tutkimuksen tehoa koskevat tulokset
| Lopullinen analyysia | Pitkän aikavälin seuranta-analyysib | |||
| Hoitohaara 2: IMFINZI + etoposidi ja joko karboplatiini tai sisplatiini (n = 268) | Hoitohaara 3: etoposidi ja joko karboplatiini tai sisplatiini (n = 269) | Hoitohaara 2: IMFINZI + etoposidi ja joko karboplatiini tai sisplatiini (n = 268) | Hoitohaara 3: etoposidi ja joko karboplatiini tai sisplatiini (n = 269) | |
| Kokonaiselossaoloaika | ||||
| Kuolemantapausten määrä (%) | 210 (78,4) | 231 (85,9) | 221 (82,5) | 248 (92,2) |
| Kokonaiselossaoloajan mediaani (kuukausina) (95 %:n CI) | 12,9 (11,3, 14,7) | 10,5 (9,3, 11,2) | 12,9 (11,3, 14,7) | 10,5 (9,3, 11,2) |
| Riskisuhde (95 %:n luottamusväli)c | 0,75 (0,625, 0,910) | 0,71 (0,595, 0,858) | ||
| p-arvod | 0,0032 | 0,0003 | ||
| Kokonaiselossaoloaika 18 kuukauden kohdalla (%) (95 %:n CI) | 32,0 (26,5, 37,7) | 24,8 (19,7, 30,1) | 32,0 (26,5, 37,7) | 24,8 (19,7, 30,1) |
| Kokonaiselossaoloaika 36 kuukauden kohdalla (%) (95 %:n CI) | 17,6 (13,3, 22,4) | 5,8 (3,4, 9,1) | ||
| Etenemättömyysaika | ||||
| Tapahtumien lukumäärä (%) | 234 (87,3) | 236 (87,7) | ||
| Etenemättömyysajan mediaani (kuukausina) (95 %:n CI) | 5,1 (4,7, 6,2) | 5,4 (4,8, 6,2) | ||
| Riskisuhde (95 %:n luottamusväli)c | 0,80 (0,665, 0,959) | |||
| Etenemättömyysaika 6 kuukauden kohdalla (%) (95 %:n CI) | 45,4 (39,3, 51,3) | 45,8 (39,5, 51,9) | ||
| Etenemättömyysaika 12 kuukauden kohdalla (%) (95 %:n CI) | 17,9 (13,5, 22,8) | 5,3 (2,9, 8,8) | ||
| Objektiivinen vasteprosentti n (%) (95 %:n CI)e | 182 (67,9) (62,0, 73,5) | 156 (58,0) (51,8, 64,0) | ||
| Täydellinen vaste, n (%) | 7 (2,6) | 2 (0,7) | ||
| Osittainen vaste, n (%) | 175 (65,3) | 154 (57,2) | ||
| Vasteen keston mediaani (kuukausina) (95 %:n CI)e,f | 5,1 (4,9, 5,3) | 5,1 (4,8, 5,3) | ||
a Etenemättömyysajan, objektiivisen vasteprosentin ja vasteen keston lopullinen analyysi viimeisen tiedonkeruupäivän (27.1.2020) kohdalla.
b Kokonaiselossaoloajan pitkän aikavälin seuranta-analyysi viimeisen tiedonkeruupäivän (22.3.2021) kohdalla.
c Analyysissa käytettiin stratifioitua log rank -testiä, jonka tulokset korjattiin hoitosyklin 1 suunnitellun platinahoidon (karboplatiini tai sisplatiini) mukaan käyttäen sijalukujen riippuvuustestejä (rank tests of association).
d Välianalyysissa (viimeinen tiedonkeruupäivä 11.3.2019) kokonaiselossaoloajan p-arvo oli 0,0047. Se alitti tilastollisen merkitsevyyden rajan, joka on 0,0178, kun kaksipuolinen kokonaisalfa on 4 %. Tämä perustuu Lan–DeMetsin alfakäytön funktioon ja käyttäen O’Brien–Flemingin tyyppistä rajaa ja havaittujen tapahtumien todellista lukumäärää.
e Vahvistettu objektiivinen vaste.
f Jälkianalyysi (post-hoc).
Kuva 13. Kokonaiselossaoloajan Kaplan–Meier-käyrä
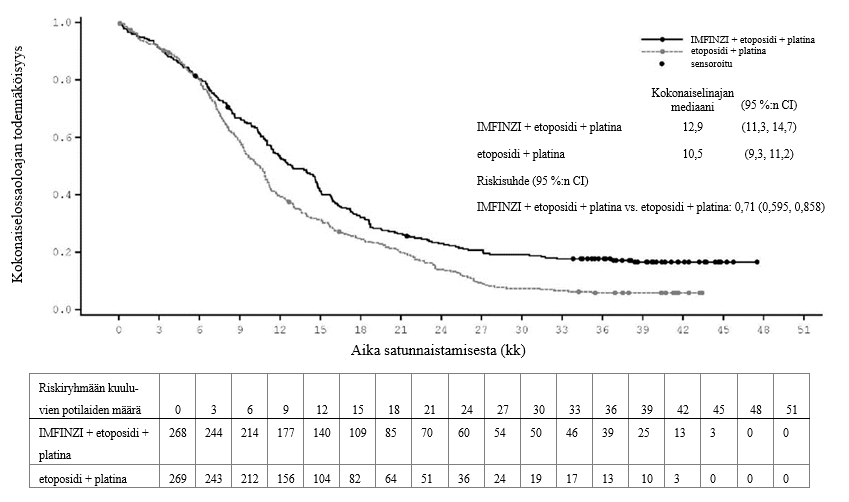
Kuva 14. Etenemättömyysajan Kaplan–Meier-käyrä
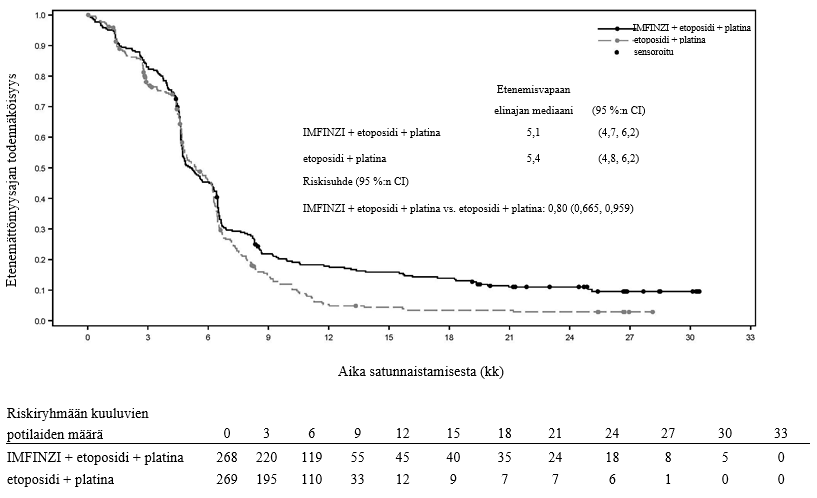
Alaryhmäanalyysi
Kokonaiselossaoloajan pitenemistä IMFINZI-valmisteen, etoposidin ja platinahoidon yhdistelmää saaneilla potilailla verrattuna pelkkää etoposidia ja platinahoitoa saaneisiin havaittiin johdonmukaisesti kaikissa demografisten tietojen, maantieteellisen alueen, karboplatiinin tai sisplatiinin käytön ja sairauden ominaispiirteiden mukaan ennalta määritellyissä alaryhmissä.
Sappitiesyöpä – TOPAZ-1-tutkimus
TOPAZ-1-tutkimus oli suunniteltu arvioimaan IMFINZI-valmisteen tehoa, kun sitä annettiin yhdistelmänä gemsitabiinin ja sisplatiinin kanssa. TOPAZ-1 oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu monikeskustutkimus, johon osallistui 685 potilasta, joilla oli leikkaukseen soveltumaton tai etäpesäkkeinen sappitiesyöpä (mukaan lukien intrahepaattinen ja ekstrahepaattinen kolangiokarsinooma ja sappirakon karsinooma) ja joiden ECOG-suorituskykyluokka oli 0 tai 1. Potilaat eivät olleet saaneet aiempaa hoitoa syövän ollessa pitkälle edennyt tai leikkaukseen soveltumaton. Mukaan otettiin potilaita, joiden tauti uusiutui yli 6 kuukauden kuluttua leikkauksesta ja/tai liitännäishoidon päättymisestä. Potilailla täytyi olla riittävä sisäelinten ja luuytimen toimintakyky ja hyväksyttävä seerumin bilirubiinipitoisuus (enintään 2,0 kertaa viitealueen yläraja [ULN]), ja jos heillä oli kliinisesti merkittävä sappitiehyiden ahtauma, sen oli korjaannuttava ennen satunnaistamista.
Tutkimuksesta suljettiin pois potilaat, joilla oli ampullaarinen karsinooma, aivometastaaseja, aktiivinen tai aiemmin dokumentoitu autoimmuunisairaus tai tulehdussairaus, HIV-infektio tai aktiivinen infektio, mukaan lukien tuberkuloosi tai C-hepatiitti, ja potilaat, jotka saivat immunosuppressiivista lääkitystä tai olivat saaneet sitä 14 päivän sisällä ennen ensimmäisen IMFINZI-annoksen saamista. Potilaat, joilla oli aktiivinen HBV, saivat osallistua tutkimukseen, jos he saivat viruslääkitystä.
Satunnaistaminen ositettiin tautitilanteen (alkuvaiheessa leikkaukseen soveltumaton vs. uusiutunut) ja primaarikasvaimen sijainnin (intrahepaattinen kolangiokarsinooma vs. ekstrahepaattinen kolangiokarsinooma vs. sappirakon karsinooma) perusteella.
Potilaat satunnaistettiin suhteessa 1:1 saamaan:
- Hoitohaara 1: IMFINZI 1 500 mg päivänä 1 + gemsitabiini 1 000 mg/m2 ja sisplatiini 25 mg/m2 (kaikki päivinä 1 ja 8) 3 viikon (21 päivän) välein enintään 8 hoitosyklin ajan, ja sen jälkeen IMFINZI 1 500 mg 4 viikon välein, kunnes tauti eteni tai ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä, tai
- Hoitohaara 2: Lumelääke päivänä 1 + gemsitabiini 1 000 mg/m2 ja sisplatiini 25 mg/m2 (kaikki päivinä 1 ja 8) 3 viikon (21 päivän) välein enintään 8 hoitosyklin ajan, ja sen jälkeen lumelääke 4 viikon välein, kunnes tauti eteni tai ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä.
Kasvaimet arvioitiin 6 viikon välein ensimmäisten 24 viikon ajan satunnaistamispäivästä lukien ja sen jälkeen 8 viikon välein, kunnes taudin vahvistettiin objektiivisesti edenneen.
Tutkimuksen ensisijainen päätemuuttuja oli kokonaiselossaoloaika, ja keskeinen toissijainen päätemuuttuja oli etenemättömyysaika. Muita toissijaisia päätemuuttujia olivat objektiivinen vasteprosentti, vasteen kesto ja potilaan ilmoittamat vaikutukset. Tutkija arvioi etenemättömyysaika, objektiivista vasteprosenttia ja vasteen kestoa RECIST v1.1 -kriteerien perusteella.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat hyvin samankaltaiset molemmissa hoitohaaroissa (hoitohaarassa 1 oli 341 potilasta ja hoitohaarassa 2 oli 344 potilasta). Koko tutkimuspopulaation demografiset tiedot lähtötilanteessa olivat: miehiä (50,4 %), ikä < 65 vuotta (53,3 %), valkoihoisia (37,2 %), aasialaisia (56,4 %), mustaihoisia tai afrikkalaisamerikkalaisia (2,0 %), muita (4,2 %), muita kuin taustaltaan espanjankielisiä tai latinalaisamerikkalaisia (93,1 %), ECOG-toimintakykyluokka 0 (49,1 %) vs. luokka 1 (50,9 %), primaarikasvaimen sijainti (intrahepaattinen sappitiehyt 55,9 %, ekstrahepaattinen sappitiehyt 19,1 % ja sappirakko 25,0 %), tautitilanne [uusiutunut (19,1 %) vs. leikkaukseen soveltumaton (80,7 %), etäpesäkkeinen (86,0 %) vs. paikallisesti edennyt (13,9 %)]. PD-L1:n ilmentyminen kasvain- ja immuunisoluissa tutkittiin käyttämällä Ventana PD-L1 (SP263) -analyysiä ja TAP (tumour area positivity) -algoritmia; 58,7 %:lla potilaista TAP oli ≥ 1 % ja 30,1 %:lla < 1 %.
Kokonaiselossaoloaika ja etenemättömyysaika testattiin muodollisesti ennalta suunnitellussa välianalyysissa (viimeinen tiedonkeruupäivä 11.8.2021) 9,8 kuukauden seuranta-ajan (mediaani) jälkeen. Tehoa koskevat tulokset on esitetty taulukossa 10 ja kuvassa 16. Kokonaiselossaoloajan maturiteetti oli 62 % ja etenemättömyysajan maturiteetti 84 %. IMFINZI + solunsalpaajahoito (hoitohaara 1) paransi kokonaiselossaoloaikaa ja etenemättömyysajan tilastollisesti merkitsevästi lumelääkkeeseen ja solunsalpaajahoitoon (hoitohaara 2) verrattuna.
Taulukko 10. TOPAZ-1-tutkimuksen tehoa koskevat tulokseta
| IMFINZI + gemsitabiini ja sisplatiini (n = 341) | Lumelääke + gemsitabiini ja sisplatiini (n = 344) | |
| Kokonaiselossaoloaika | ||
| Kuolemantapausten määrä (%) | 198 (58,1) | 226 (65,7) |
| Kokonaiselossaoloajan mediaani (kuukausina) (95 %:n CI)b | 12,8 (11,1, 14,0) | 11,5 (10,1, 12,5) |
| Riskisuhde (95 %:n luottamusväli)c | 0,80 (0,66, 0,97) | |
| p-arvoc,d | 0,021 | |
| Kaikkien potilaiden seuranta-ajan mediaani (kuukausina) | 10,2 | 9,5 |
| Etenemättömyysaika | ||
| Tapahtumien lukumäärä (%) | 276 (80,9) | 297 (86,3) |
| Etenemättömyysajan mediaani (kuukausina) (95 %:n CI)b | 7,2 (6,7, 7,4) | 5,7 (5,6, 6,7) |
| Riskisuhde (95 %:n luottamusväli)c | 0,75 (0,63, 0,89) | |
| p-arvoc,e | 0,001 | |
| Kaikkien potilaiden seuranta-ajan mediaani (kuukausina) | 7,2 | 5,6 |
| Objektiivinen vasteprosenttif | 91 (26,7) | 64 (18,7) |
| Täydellinen vaste, n (%) | 7 (2,1) | 2 (0,6) |
| Osittainen vaste, n (%) | 84 (24,6) | 62 (18,1) |
| Vasteen kesto | ||
| Vasteen keston mediaani (kuukausina) (95 %:n CI)b | 6,4 (5,9, 8,1) | 6,2 (4,4, 7,3) |
a Analyysi viimeisen tiedonkeruupäivän (11.8.2021) kohdalla.
b Laskettu Kaplan–Meier-tekniikalla. Mediaanin luottamusväli laskettiin Brookmeyer–Crowley-menetelmällä.
c Riskisuhdeanalyysi tehtiin käyttäen ositettua Coxin suhteellisten riskitiheyksien mallia, ja kaksitahoinen p-arvo perustuu stratifioituun log-rank-testiin; molemmat on korjattu tautitilanteen ja primaarikasvaimen sijainnin suhteen.
d Välianalyysissa (viimeinen tiedonkeruupäivä 11.8.2021) kokonaiselinajan p-arvo oli 0,021. Se alitti tilastollisen merkitsevyyden rajan, joka on 0,03, kun kaksipuolinen kokonaisalfa on 4,9 %. Tämä perustuu Lan–DeMetsin alfavirheen korjausfunktioon, O’Brien–Flemingin tyyppiseen rajaan ja havaittujen tapahtumien todelliseen lukumäärään.
e Välianalyysissa (viimeinen tiedonkeruupäivä 11.8.2021) etenemisvapaan elinajan p-arvo oli 0,001. Se alitti tilastollisen merkitsevyyden rajan, joka on 0,0481, kun kaksipuolinen kokonaisalfa on 4,9 %. Tämä perustuu Lan–DeMetsin alfavirheen korjausfunktioon, Pocockin tyyppiseen rajaan ja havaittujen tapahtumien todelliseen lukumäärään.
f Vahvistettu objektiivinen vaste
6,5 kuukautta välianalyysin jälkeen, kun kokonaiselossaoloajan maturiteetti oli 77 %, tehtiin suunniteltu kokonaiselossaoloajan seurannan lisäanalyysi (viimeinen tiedonkeruupäivä 25.2.2022). IMFINZI + solunsalpaajahoito paransi edelleen kokonaiselossaoloaikaa pelkkään solunsalpaajahoitoon verrattuna [riskisuhde = 0,76, (95 %:n luottamusväli: 0,64, 0,91)], ja seuranta-ajan mediaani piteni 12 kuukauteen.
Kuva 15: Kokonaiselossaoloajan Kaplan–Meier-käyrä, Kokonaiselossaoloajan seurannan analyysi viimeisen tiedonkeruupäivän kohdalla 25.2.2022
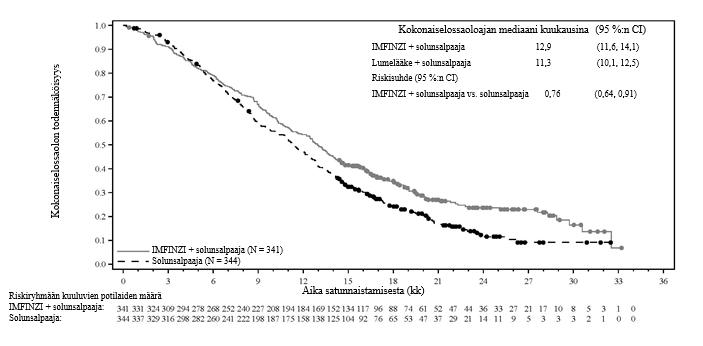
Kuva 16: Etenemättömyysajan Kaplan–Meier-käyrä, inferentiaalinen (primaari)analyysi viimeisen tiedonkeruupäivän kohdalla 11.8.2021
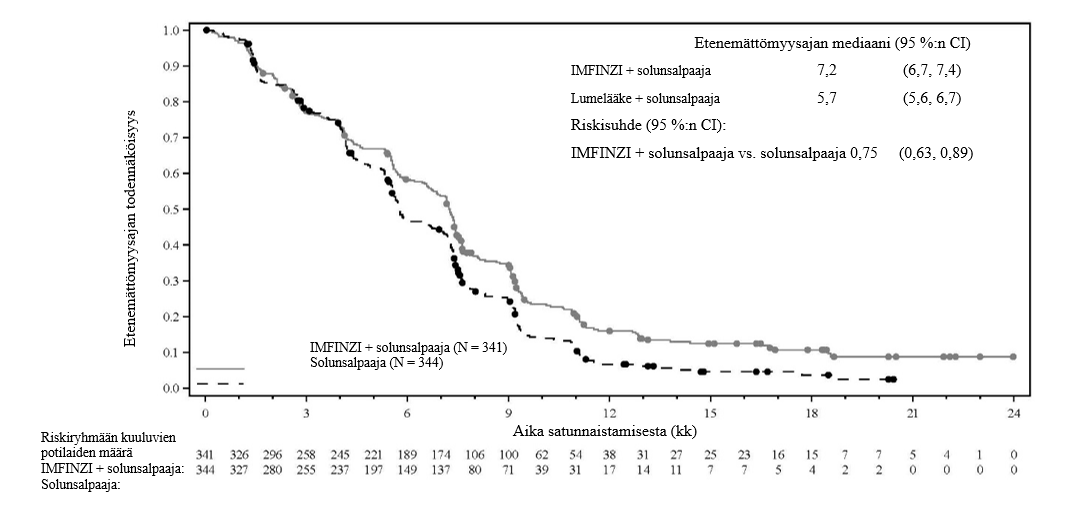
Maksasolusyöpä – HIMALAYA-tutkimus
IMFINZI-valmisteen tehoa monoterapiana ja silloin, kun sitä annettiin yhdistelmänä 300 mg:n tremelimumabikerta-annoksen kanssa, arvioitiin HIMALAYA-tutkimuksessa, joka oli satunnaistettu, avoin monikeskustutkimus potilailla, joilla oli vahvistetusti leikkaushoitoon soveltumaton maksasolusyöpä ja joiden maksasolusyöpää ei ollut aiemmin hoidettu systeemisesti. Tutkimukseen osallistui potilaita, joilla taudin BCLC (Barcelona Clinic Liver Cancer) -levinneisyysaste oli C tai B (lokoregionaalinen hoito ei soveltunut potilaalle) ja Child–Pugh-luokka A.
Tutkimuksesta suljettiin pois potilaat, joilla oli aivometastaaseja sillä hetkellä tai anamneesissa; samanaikaisesti sekä hepatiitti B- että hepatiitti C -virusinfektio; aktiivinen tai aiemmin dokumentoitu maha-suolikanavan verenvuoto 12 kuukauden sisällä; ei-lääkkeellistä hoitointerventiota edellyttänyt askites 6 kuukauden sisällä; maksaenkefalopatia 12 kuukauden sisällä ennen hoidon aloittamista; tai aktiivinen tai aiemmin dokumentoitu autoimmuuni- tai tulehdussairaus.
Potilaat, joilla oli ruokatorven suonikohjuja, otettiin mukaan tutkimukseen, jos heillä ei ollut aktiivista tai edeltävien 12 kuukauden sisällä dokumentoitua aiempaa maha-suolikanavan verenvuotoa ennen tutkimuksessa aloittamista.
Satunnaistamisessa ositustekijöinä olivat makrovaskulaarinen invaasio (kyllä vs. ei), maksasairauden etiologia (vahvistettu hepatiitti B -virusinfektio vs. vahvistettu hepatiitti C -virusinfektio vs. muut) ja ECOG-suorituskykypistemäärä (0 vs. 1). HIMALAYA-tutkimuksessa satunnaistettiin 1 171 potilasta suhteessa 1:1:1 saamaan
- IMFINZI-valmistetta: 1 500 mg durvalumabia 4 viikon välein.
- 300 mg tremelimumabia kerta-annoksena + 1 500 mg IMFINZI-valmistetta; minkä jälkeen 1 500 mg IMFINZI-valmistetta 4 viikon välein.
- 400 mg sorafenibia kahdesti päivässä.
Kasvaimet arvioitiin ensimmäisten 12 kuukauden aikana 8 viikon välein ja sen jälkeen 12 viikon välein. Elossaoloarvioinnit tehtiin kuukauden välein ensimmäisten 3 kuukauden aikana hoidon lopettamisen jälkeen ja sen jälkeen 2 kuukauden välein.
Ensisijainen päätetapahtuma oli kokonaiselossaoloajan paremmuus IMFINZI-valmistetta yhdistelmänä tremelimumabikerta-annoksen kanssa saaneilla verrattuna sorafenibia saaneisiin. Keskeiset toissijaiset tavoitteet olivat kokonaisselossaoloajan vähintään samanveroisuus ja tämän jälkeen paremmuus IMFINZI-valmistetta saaneilla verrattuna sorafenibia saaneisiin. Muita toissijaisia päätetapahtumia olivat etenemättömyysaika, tutkijan arvioima objektiivinen vasteprosentti ja vasteen kesto RECIST v1.1 -kriteerien mukaisesti.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat hyvin samankaltaiset eri tutkimushaaroissa. Koko tutkimuspopulaation demografiset tiedot lähtötilanteessa olivat: miehiä (83,7 %), ikä alle 65 vuotta (50,4 %), valkoihoisia (44,6 %), aasialaisia (50,7 %), mustaihoisia tai afrikkalaisamerikkalaisia (1,7 %), muun etnisen taustan omaavia (2,3 %), ECOG-suorituskykypistemäärä 0 (62,6 %); Child–Pugh-luokka A (99,5 %), makrovaskulaarinen invaasio (25,2 %), leviäminen maksan ulkopuolelle (53,4 %), lähtötilanteen AFP-arvo (alfafetoproteiinipitoisuus) < 400 ng/ml (63,7 %), lähtötilanteen AFP-arvo ≥ 400 ng/ml (34,5 %), virusetiologia: hepatiitti B (30,6 %), hepatiitti C (27,2 %), ei infektiota (42,2 %), arvioitavissa olevia PD-L1-tietoja (86,3 %), PD-L1 TAP (Tumor Area Positivity eli PD-L1-positiivisten solujen osuus kasvaimen pinta-alasta) ≥ 1 % (38,9 %), PD-L1 TAP < 1 % (48,3 %) [Ventana PD-L1 (SP263) -analyysi].
Tulokset on esitetty taulukossa 11, kuvassa 17 ja kuvassa 18.
Taulukko 11. HIMALAYA-tutkimuksen tehoa koskevat tulokset IMFINZI-hoidolle, jota annettiin yhdistelmänä 300 mg:n tremelimumabikerta-annoksen kanssa, ja monoterapiana annetulle IMFINZI-hoidolle verrattuna sorafenibiin
| IMFINZI + tremelimumabi 300 mg(n = 393) | Sorafenibi (n = 389) | IMFINZI (n = 389) | |
|---|---|---|---|
| Seurannan kesto | |||
| Seurannan keston mediaani (kuukausina)a | 33,2 | 32,2 | 32,6 |
| Kokonaiselossaoloaika | |||
| Kuolemantapausten määrä (%) | 262 (66,7) | 293 (75,3) | 280 (72,0) |
| Kokonaiselossaoloajan mediaani (kuukausina) (95 %:n luottamusväli) | 16,4(14,2, 19,6) | 13,8 (12,3, 16,1) | 16,6 (14,1, 19,1) |
| Riskisuhde (95 %:n luottamusväli)b,c | 0,78 (0,66, 0,92) | - | |
| p-arvod | 0,0035 | - | |
| Riskisuhde (95 %:n luottamusväli)b,c,e | - | 0,86 (0,73, 1,03) | |
| Etenemättömyysaika | |||
| Tapahtumien määrä (%) | 335 (85,2) | 327 (84,1) | 345 (88,7) |
| Etenemättömyysajan mediaani (kuukausina) (95 %:n luottamusväli) | 3,78 (3,68–5,32) | 4,07 (3,75–5,49) | 3,65 (3,19–3,75) |
| Riskisuhde (95 %:n luottamusväli) | 0,90 (0,77, 1,05) | - | |
| Riskisuhde (95 %:n luottamusväli) | - | 1,02 (0,88, 1,19) | |
| Objektiiviset vasteet | |||
| Objektiiviset vasteet, n (%)f | 79 (20,1) | 20 (5,1) | 66 (17,0) |
| Täydellinen vaste, n (%) | 12 (3,1) | 0 | 6 (1,5) |
| Osittainen vaste, n (%) | 67 (17,0) | 20 (5,1) | 60 (15,4) |
| Vasteen kesto | |||
| Vasteen keston mediaani (kuukausina) | 22,3 | 18,4 | 16,8 |
a Laskettu käänteisellä Kaplan–Meier-tekniikalla (käyttäen käänteistä sensurointiosoitinta).
b Perustuu ositettuun Coxin malliin, jonka tulokset korjattiin hoidon, maksasairauden etiologian (HBV vs. HCV vs. muut) ja ECOG-suorituskykypistemäärän (0 vs. 1) mukaan.
c Analyysissa käytettiin stratifioitua log rank ‑testiä, jonka tulokset korjattiin maksasairauden etiologian (HBV vs. HCV vs. muut), ECOG-suorituskykypistemäärän (0 vs. 1) ja makrovaskulaarisen invaasion (kyllä vs. ei) mukaan.
d Tilastollisen merkitsevyyden raja IMFINZI + tremelimumabi 300 mg ‑hoidolle verrattuna sorafenibiin oli 0,0398, kun käytössä oli Lan–DeMetsin alfavirheen korjausfunktio, O’Brien–Flemingin tyyppinen raja ja havaittujen tapahtumien todellinen lukumäärä (Lan ja DeMets 1983).
e Riskisuhteen (IMFINZI vs. sorafenibi) vähintään samanveroisuuden marginaali oli 1,08, kun käytössä oli 95,67 %:n luottamusväli, Lan–DeMetsin alfavirheen korjausfunktio, O’Brien–Flemingin tyyppinen raja ja havaittujen tapahtumien todellinen lukumäärä (Lan ja DeMets 1983). Kun testattiin IMFINZI-valmisteen paremmuutta verrattuna sorafenibiin, p-arvo oli 0,0674 eikä tilastollista merkitsevyyttä saavutettu.
f Vahvistettu objektiivinen vaste.
Kuva 17. Kokonaiselossaoloajan Kaplan–Meier-käyrä, kun IMFINZI-valmistetta annettiin yhdistelmänä 300 mg:n tremelimumabikerta-annoksen kanssa
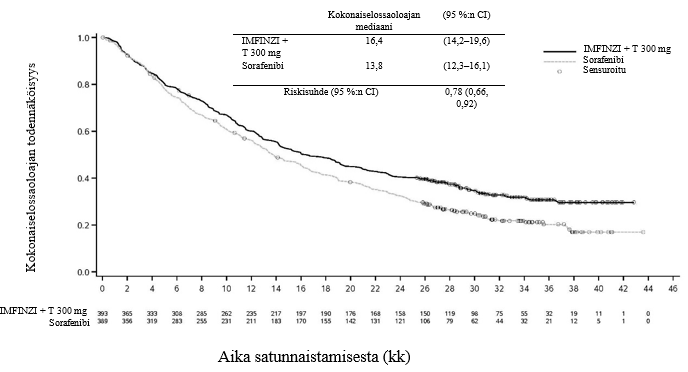
Kuva 18. Kokonaiselossaoloajan Kaplan–Meier-käyrä, kun IMFINZI-valmistetta annettiin monoterapiana
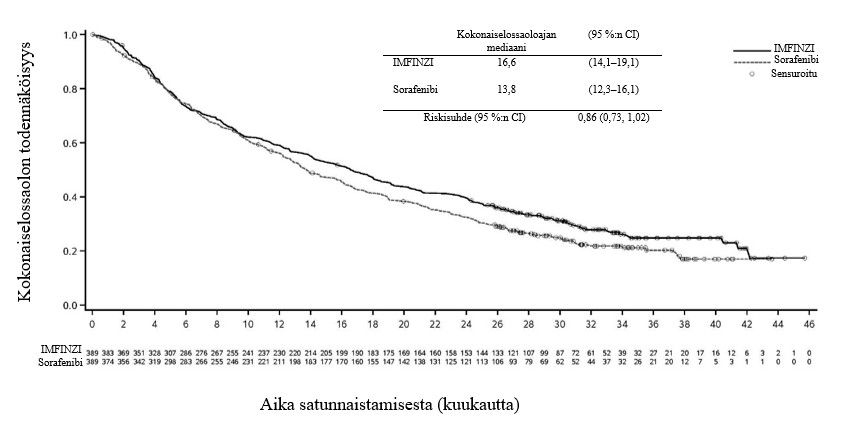
Kohdun limakalvon syöpä – DUO-E-tutkimus
DUO-E oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen III monikeskustutkimus, jossa pitkälle edennyttä tai uusiutunutta kohdun limakalvon syöpää sairastaville potilaille annettiin ensilinjan hoitona platinapohjaista solunsalpaajahoitoa yhdistelmänä IMFINZI-valmisteen kanssa ja tämän jälkeen IMFINZI-valmistetta joko yhdistelmänä olaparibin kanssa tai ilman olaparibia. Potilailla oli oltava kohdun limakalvon syöpä, joka vastasi jotakin seuraavista: äskettäin todettu levinneisyysasteen III tauti (RECIST v1.1 ‑kriteerien mukaisesti mitattavissa oleva tauti leikkaushoidon tai diagnostisen biopsian jälkeen), äskettäin todettu levinneisyysasteen IV tauti (riippumatta siitä, oliko tautia jäljellä leikkauksen tai diagnostisen biopsian jälkeen) tai taudin uusiutuminen (RECIST v1.1 ‑kriteerien mukaisesti joko mitattavissa tai ei-mitattavissa oleva tauti), jossa mahdollisuudet saavuttaa kuratiivinen tulos pelkällä leikkaushoidolla tai yhdistelmähoidolla olivat huonot. Jos potilaalla oli uusiutunut tauti, aiempi solunsalpaajahoito katsottiin hyväksyttäväksi vain, jos se oli annettu liitännäishoitona ja jos viimeisen solunsalpaaja-annoksen antopäivän ja taudin myöhemmän uusiutumisen välillä oli kulunut vähintään 12 kuukautta. Tutkimukseen otetuilla potilailla oli kohdun limakalvon epiteliaalisia karsinoomia; kaikki histologiset muodot sallittiin, mukaan lukien karsinosarkoomat. Potilaat, joilla oli kohdun limakalvon sarkooma, suljettiin pois.
Satunnaistaminen stratifioitiin kasvainkudoksen DNA:n kahdentumisvirheiden korjausmekanismin (MMR-mekanismin) statuksen (toimiva vs. puutteellinen), tautitilanteen (uusiutunut vs. äskettäin todettu) ja maantieteellisen alueen (Aasia vs. muu maailma) mukaan. Potilaat satunnaistettiin suhteessa 1:1:1 johonkin seuraavista hoitohaaroista:
- Hoitohaara 1 (platinapohjainen solunsalpaajahoito): Platinapohjaista solunsalpaajahoitoa (paklitakselia ja karboplatiinia) 3 viikon välein enintään 6 hoitosyklin ajan ja durvalumabia muistuttavaa lumelääkettä 3 viikon välein. Solunsalpaajahoidon päätyttyä potilaat, joiden taudin ei todettu objektiivisesti edenneen, saivat durvalumabia muistuttavaa lumelääkettä 4 viikon välein ja olaparibia muistuttavia lumelääketabletteja kahdesti vuorokaudessa ylläpitohoitona taudin etenemiseen saakka.
- Hoitohaara 2 (platinapohjainen solunsalpaajahoito + IMFINZI): Platinapohjaista solunsalpaajahoitoa (paklitakselia ja karboplatiinia) 3 viikon välein enintään 6 hoitosyklin ajan ja 1 120 mg durvalumabia 3 viikon välein. Solunsalpaajahoidon päätyttyä potilaat, joiden taudin ei todettu objektiivisesti edenneen, saivat 1 500 mg durvalumabia 4 viikon välein ja olaparibia muistuttavia lumelääketabletteja kahdesti vuorokaudessa ylläpitohoitona taudin etenemiseen saakka.
- Hoitohaara 3 (platinapohjainen solunsalpaajahoito + IMFINZI + olaparibi): Platinapohjaista solunsalpaajahoitoa (paklitakselia ja karboplatiinia) 3 viikon välein enintään 6 hoitosyklin ajan ja 1 120 mg durvalumabia 3 viikon välein. Solunsalpaajahoidon päätyttyä potilaat, joiden taudin ei todettu objektiivisesti edenneen, saivat 1 500 mg durvalumabia 4 viikon välein ja 300 mg:n olaparibitabletteja kahdesti vuorokaudessa ylläpitohoitona taudin etenemiseen saakka.
Potilaat, jotka lopettivat jommankumman valmisteen (IMFINZI/lumelääke tai olaparibi/lumelääke) käytön muista syistä kuin taudin etenemisen vuoksi, saattoivat jatkaa toisen valmisteen käyttöä, jos tämä oli asianmukaista toksisuusnäkökohtien ja tutkijan harkinnan mukaan.
Hoitoa jatkettiin, kunnes tauti eteni RECIST v1.1 ‑kriteerien mukaisesti tai ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä. Kasvaimen status arvioitiin ensimmäisten 18 viikon aikana satunnaistamisen jälkeen 9 viikon välein ja sen jälkeen 12 viikon välein.
Ensisijainen päätetapahtuma oli etenemättömyysaika, jonka tutkijalääkäri arvioi RECIST v1.1 ‑kriteerien mukaisesti. Toissijaisia tehoa koskevia päätetapahtumia olivat kokonaiselossaoloaika, objektiivinen vasteprosentti ja vasteen kesto.
Tutkimuksessa osoitettiin etenemättömyysajan tilastollisesti merkitsevä piteneminen lähtöryhmien mukaisessa populaatiossa (ITT-populaatiossa) potilailla, joiden hoitona oli platinapohjainen solunsalpaajahoito + IMFINZI + olaparibi, verrattuna platinapohjaista solunsalpaajahoitoa saaneisiin [HR = 0,55 (95 %:n luottamusväli: 0,43, 0,69), p =< 0,0001], ja potilailla, joiden hoitona oli platinapohjainen solunsalpaajahoito + IMFINZI, verrattuna platinapohjaista solunsalpaajahoitoa saaneisiin [HR = 0,71 (95 %:n luottamusväli: 0,57, 0,89), p = 0,003]. Etenemättömyysajan analyysin ajankohtana kokonaiselossaoloaikaa koskevien alustavien tietojen maturiteetti oli 28 % ja tapahtumia oli todettu 199:llä 718 potilaasta.
DNA:n kahdentumisvirheiden korjausmekanismin (mismatch repair; MMR-mekanismin) status määritettiin keskitetysti käyttäen immunohistokemiallista MMR-testipaneelia. Tutkimukseen satunnaistettiin yhteensä 718 potilasta, joista 575:llä (80 %) kasvaimeen liittyi toimiva MMR-mekanismi (pMMR) ja 143:lla (20 %) kasvaimeen liittyi puutteellinen MMR-mekanismi (dMMR).
Potilaat, joiden kohdun limakalvon syöpään liittyi puutteellinen MMR-mekanismi (dMMR)
Potilailla, joiden MMR-mekanismi oli puutteellinen (kasvaimen status oli dMMR), demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat yleisesti hyvin samankaltaiset eri hoitohaaroissa. Kaikkien kolmen hoitohaaran demografiset tiedot lähtötilanteessa olivat: iän mediaani 62 vuotta (vaihteluväli: 34–85), 41 %:lla ikä vähintään 65 vuotta, 1,5 %:lla ikä vähintään 75 vuotta, valkoihoisia 62 %, aasialaisia 29 % ja tummaihoisia tai afroamerikkalaisia 2 %. Sairauden ominaispiirteet olivat: ECOG-toimintakykyluokka 0 (58 %) tai 1 (42 %), 46 % tapauksista äskettäin todettuja ja 54 % tapauksista uusiutuneita tauteja. Histologiset alatyypit olivat endometrioidi (83 %), sekamuotoinen epiteliaalinen (5 %), seroosi (3 %), karsinosarkooma (3 %), erilaistumaton (2 %) ja muu (3 %).
Tulokset potilailla, joiden kasvaimen status on dMMR, esitetään yhteenvetona taulukossa 12 ja kuvassa 19. Etenemättömyysajan seuranta-ajan mediaani sensuroiduilla potilailla, joiden kasvaimen status oli dMMR, oli platinapohjaisen solunsalpaajahoidon ja IMFINZI-valmisteen hoitohaarassa 15,5 kuukautta ja platinapohjainen solunsalpaajahoidon hoitohaarassa 10,2 kuukautta. Etenemättömyysajan analyysin ajankohtana kokonaiselossaoloaikaa koskevien alustavien tietojen maturiteetti oli 26 % ja tapahtumia oli todettu 25:llä 95 potilaasta, joiden hoitona oli ollut platinapohjainen solunsalpaajahoito + IMFINZI tai platinapohjainen solunsalpaajahoito.
Taulukko 12. DUO-E-tutkimuksen tehoa koskevat tulokset (potilaat, joiden kasvaimen status oli dMMR)
| Platinapohjainen solunsalpaajahoito + IMFINZI N = 46 | Platinapohjainen solunsalpaajahoito N = 49 | |
| Etenemättömyysaikaa,b | ||
| Tapahtumien määrä (%) | 15 (32,6) | 25 (51,0) |
| Etenemättömyysajan mediaani (kuukausina) (95 %:n luottamusväli)c | Ei saavutettu (ei saavutettu, ei saavutettu) | 7,0 (6,7, 14,8) |
| HR (95 %:n luottamusväli) | 0,42 (0,22, 0,80) | - |
| Kokonaiselossaoloaikab | ||
| Tapahtumien määrä (%) | 7 (15,2) | 18 (36,7) |
| Kokonaiselossaoloajan mediaani (kuukausina) (95 %:n luottamusväli)c | Ei saavutettu (ei saavutettu, ei saavutettu) | 23,7 (16,9, ei saavutettu) |
| HR (95 %:n luottamusväli) | 0,34 (0,13, 0,79) | - |
| Objektiiviset vasteetb | ||
| Objektiiviset vasteetd, n (%) | 30 (71,4) | 17 (40,5) |
| Vasteen kestob | ||
| Vasteen keston mediaani (kuukausina) (95 %:n luottamusväli)c | Ei saavutettu (ei saavutettu, ei saavutettu) | 10,5 (4,3, ei saavutettu) |
a Tutkijalääkärin arvioima.
b Tulokset perustuvat ensimmäiseen välianalyysiin (viimeinen tiedonkeruupäivä 12.4.2023).
c Laskettu Kaplan–Meier-tekniikalla.
d Vaste: Paras objektiivinen vaste eli vahvistettu täydellinen vaste tai osittainen vaste. Perustuu kussakin hoitohaarassa niiden potilaiden määrään, joiden tauti oli lähtötilanteessa mitattavissa (N = 42 platinapohjaisen solunsalpaajahoidon ja IMFINZI-valmisteen hoitohaarassa, N = 42 platinapohjaisen solunsalpaajahoidon hoitohaarassa).
HR = riskisuhde.
Kuva 19. Etenemättömyysajan Kaplan–Meier-käyrä DUO-E-tutkimuksessa (potilaat, joiden kasvaimen status oli dMMR)
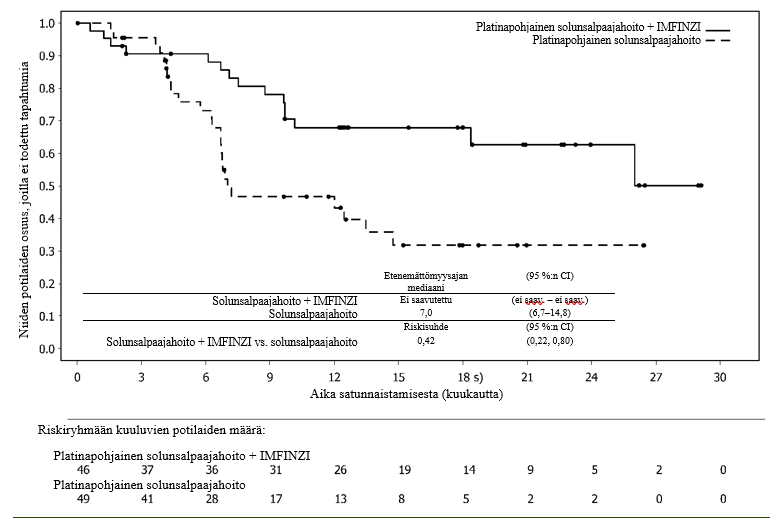
Potilaat, joiden kohdun limakalvon syöpään liittyi toimiva MMR-mekanismi (pMMR)
Potilailla, joiden MMR-mekanismi oli toimiva (kasvaimen status oli pMMR), demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat yleisesti hyvin samankaltaiset eri hoitohaaroissa. Kaikkien kolmen hoitohaaran demografiset tiedot lähtötilanteessa olivat: iän mediaani 64 vuotta (vaihteluväli: 22–86), 48 %:lla ikä vähintään 65 vuotta, 8,1 %:lla ikä vähintään 75 vuotta, valkoihoisia 56 %, aasialaisia 30 % ja tummaihoisia tai afroamerikkalaisia 6 %. Sairauden ominaispiirteet olivat: ECOG-toimintakykyluokka 0 (69 %) tai 1 (31 %), 47 % tapauksista äskettäin todettuja ja 53 % tapauksista uusiutuneita tauteja. Histologiset alatyypit olivat endometrioidi (54 %), seroosi (26 %), karsinosarkooma (8 %), sekamuotoinen epiteliaalinen (4 %), kirkassoluinen (3 %), erilaistumaton (2 %), musinoosi (< 1 %) ja muu (3 %).
Tulokset potilailla, joiden kasvaimen status oli pMMR, esitetään yhteenvetona taulukossa 13 ja kuvassa 20. Seuranta-ajan mediaani sensuroiduilla potilailla, joiden kasvaimen status oli pMMR, oli platinapohjaisen solunsalpaajahoidon, IMFINZI-valmisteen ja olaparibin hoitohaarassa 15,2 kuukautta ja platinapohjainen solunsalpaajahoidon hoitohaarassa 12,8 kuukautta.
Etenemättömyysajan analyysin ajankohtana kokonaiselossaoloaikaa koskevien alustavien tietojen maturiteetti oli 29 % ja tapahtumia oli todettu 110:llä 383 potilaasta, joiden hoitona oli ollut platinapohjainen solunsalpaajahoito + IMFINZI + olaparibi tai platinapohjainen solunsalpaajahoito.
Taulukko 13. Tehoa koskevat tulokset DUO-E-tutkimuksessa (potilaat, joiden kasvaimen status oli pMMR)
| Platinapohjainen solunsalpaajahoito + IMFINZI + olaparibi N = 191 | Platinapohjainen solunsalpaajahoito N = 192 | |
| Etenemättömyysaikaa,b | ||
| Tapahtumien määrä (%) | 108 (56,5) | 148 (77,1) |
| Etenemättömyysajan mediaani (kuukausina) (95 %:n luottamusväli)c | 15,0 (12,4, 18,0) | 9,7 (9,2, 10,1) |
| HR (95 %:n luottamusväli) | 0,57 (0,44, 0,73) | - |
| Kokonaiselossaoloaikab | ||
| Tapahtumien määrä (%) | 46 (24,1) | 64 (33,3) |
| Kokonaiselossaoloajan mediaani (kuukausina) (95 %:n luottamusväli)c | Ei saavutettu (ei saavutettu, ei saavutettu) | 25,9 (25,1, ei saavutettu) |
| HR (95 %:n luottamusväli) | 0,69 (0,47, 1,00) | - |
| Objektiiviset vasteetb | ||
| Objektiiviset vasteetd, n (%) | 90 (61,2) | 92 (59,0) |
| Vasteen kestob | ||
| Vasteen keston mediaani (kuukausina) (95 %:n luottamusväli)c | 18,7 (10,5, ei saavutettu) | 7,6 (7,1, 10,2) |
a Tutkijalääkärin arvioima.
b Tulokset perustuvat ensimmäiseen välianalyysiin (viimeinen tiedonkeruupäivä 12.4.2023).
c Laskettu Kaplan–Meier-tekniikalla.
d Vaste: Paras objektiivinen vaste eli vahvistettu täydellinen vaste tai osittainen vaste. Perustuu kussakin hoitohaarassa niiden potilaiden määrään, joiden tauti oli lähtötilanteessa mitattavissa (N = 147 platinapohjaisen solunsalpaajahoidon, IMFINZI-valmisteen ja olaparibin hoitohaarassa, N = 156 platinapohjaisen solunsalpaajahoidon hoitohaarassa).
HR = riskisuhde
Kuva 20. Etenemättömyysajan Kaplan–Meier-käyrä DUO-E-tutkimuksessa (potilaat, joiden kasvaimen status oli pMMR)
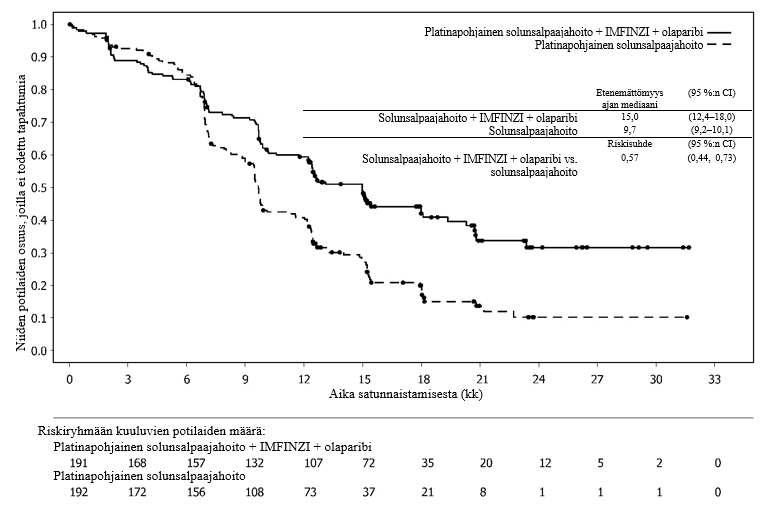
Potilailla, joiden kasvaimen status oli pMMR, etenemättömyysajan riskisuhteet (HR) olivat 0,44 (95 %:n luottamusväli: 0,31, 0,61) potilailla, jotka olivat positiivisia PD-L1:n ilmentymisen suhteen (236/383; 62 %), ja 0,87 (95 %:n luottamusväli: 0,59, 1,28) potilailla, jotka olivat negatiivisia PD-L1:n ilmentymisen suhteen (140/383; 37 %), platinapohjainen solunsalpaajahoito + IMFINZI+ olaparibi ‑hoitohaarassa verrattuna platinapohjaisen solunsalpaajahoidon hoitohaaraan. Positiivisuus PD-L1:n ilmentymisen suhteen määriteltiin tilanteeksi, jossa PD-L1-positiivisten solujen osuus kasvaimen pinta-alasta (tumour area positive, TAP) oli ≥ 1 %.
Lihasinvasiivinen rakkosyöpä – NIAGARA-tutkimus
NIAGARA oli satunnaistettu, avoin vaiheen III monikeskustutkimus, joka oli suunniteltu arvioimaan IMFINZI-valmisteen tehoa lihasinvasiivista rakkosyöpää sairastavilla potilailla, joille annettiin ensin IMFINZI-valmistetta yhdistelmänä gemsitabiinin ja sisplatiinin kanssa esiliitännäishoitona ja sen jälkeen IMFINZI-monoterapiaa liitännäishoitona. Tutkimuksessa satunnaistettiin 1 063 potilasta, joille radikaali kystektomia soveltui ja jotka eivät olleet saaneet aiemmin systeemistä solunsalpaajahoitoa eivätkä immuunivälitteistä hoitoa kliinisen levinneisyysasteen T2–T4aN0/1M0 lihasinvasiiviseen rakkosyöpään. Tutkimuksesta suljettiin pois potilaat, joiden syöpä oli histologialtaan puhtaasti ei-uroteliaalinen, joiden syövän histologiassa oli mikä tahansa pienisoluisen syövän histologia tai joiden uroteelisyöpä oli primaaristi lähtöisin muualta kuin virtsarakosta (eli virtsanjohtimesta, virtsaputkesta tai munuaisaltaasta), joilla oli aktiivinen tai aiemmin dokumentoitu autoimmuunisairaus, joilla oli aktiivinen tuberkuloosi tai hepatiitti B- tai hepatiitti C ‑infektio tai HIV-infektio tai jotka olivat saaneet immunosuppressiivista lääkitystä 14 päivän sisällä ennen ensimmäisen durvalumabiannoksen saamista, lukuun ottamatta fysiologisilla annoksilla tai esilääkityksenä käytettyä systeemistä kortikosteroidihoitoa.
Satunnaistamisessa ositustekijöinä olivat kasvaimen kliininen levinneisyysaste (T2N0 vs. > T2N0 [johon sisältyivät T2N1, T3 ja T4a]), munuaisten toiminta (riittävän hyvä munuaisten toiminta [kreatiniinipuhdistuma ≥ 60 ml/min] vs. riittävyyden rajoilla oleva [borderline] munuaisten toiminta [kreatiniinipuhdistuma ≥ 40 ml/min – < 60 ml/min]) ja PD‑L1:n ilmentymisstatus (runsas vs. niukka tai olematon). Potilaat satunnaistettiin suhteessa 1:1 saamaan perioperatiivisesti joko IMFINZI-valmistetta yhdistelmänä esiliitännäishoitona annetun solunsalpaajahoidon kanssa (hoitohaara 1) tai pelkkää esiliitännäishoitona annettua solunsalpaajahoitoa (hoitohaara 2):
- Hoitohaara 1 (IMFINZI + solunsalpaajahoito): IMFINZI 1 500 mg + gemsitabiini 1 000 mg/m2 ja sisplatiini 70 mg/m2 3 viikon välein 4 hoitosyklin ajan ennen leikkausta ja sitten IMFINZI 1 500 mg 4 viikon välein enintään 8 hoitosyklin ajan leikkauksen jälkeen.
- Hoitohaara 2 (solunsalpaajahoito): gemsitabiini 1 000 mg/m2 ja sisplatiini 70 mg/m2 3 viikon välein 4 hoitosyklin ajan ennen leikkausta; ei hoitoa leikkauksen jälkeen.
Potilailla, joiden munuaisten toiminta oli riittävyyden rajoilla, sisplatiiniannos jaettiin kahteen osaan niin, että he saivat sisplatiinia 35 mg/m2 kunkin hoitosyklin päivinä 1 ja 8.
Kasvaimet arvioitiin RECIST 1.1 ‑kriteerien mukaan lähtötilanteessa ja esiliitännäishoidon päättyessä (ennen leikkausta). Leikkauksen jälkeen kasvaimia arvioitiin RECIST 1.1 ‑kriteerien mukaan 12 viikon välein ensimmäisten 24 kuukauden ajan, sen jälkeen 24 viikon välein 36 kuukauden ajan ja sen jälkeen 52 viikon välein taudin etenemiseen, tutkimuksen päättymiseen tai potilaan kuolemaan asti.
Ensisijaiset päätetapahtumat olivat patologisesti todettu täydellinen vaste (pathological complete response, pCR) sokkoutetun keskitetyn patologisen arvion mukaan sekä tapahtumavapaa elossaoloaika (event-free survival, EFS), joka perustui muun muassa sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) tekemään arvioon. Kokonaiselossaoloaika oli keskeinen toissijainen päätetapahtuma.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat yleisesti ottaen hyvin samankaltaiset molemmissa hoitohaaroissa. Hoitohaarassa 1 oli 533 potilasta ja hoitohaarassa 2 oli 530 potilasta. Demografiset tiedot lähtötilanteessa olivat: miehiä (81,8 %), ikä < 65 vuotta (46,9 %), valkoihoisia (67 %), aasialaisia (27,9 %), mustaihoisia tai afrikkalaisamerikkalaisia (0,9 %), muita (0,8 %), taustaltaan espanjankielisiä tai latinalaisamerikkalaisia (8,0 %) ja ECOG-suorituskykypistemäärä 0 (78 %) vs. ECOG-suorituskykypistemäärä 1 (22 %). Sairauden ominaispiirteet olivat: kasvaimen levinneisyysaste T2N0 (40,3 %) ja > T2N0a (59,7 %), alueelliset imusolmukkeet N0 (94,5 %) ja N1 (5,5 %), riittävän hyvä munuaisten toiminta (81,1 %) ja riittävyyden rajoilla oleva munuaisten toiminta (18,9 %) sekä PD‑L1:n ilmentymisstatus runsas (73,1 %) vs. niukka tai olematon (26,9 %). Histologiset alatyypit olivat uroteelikarsinooma (84,5 %), uroteelikarsinooma, johon liittyi erilaistuneita levyepiteelirakenteita (8,2 %), johonkin histologiseen varianttiin kuuluva uroteelikarsinooma (5,0 %) ja uroteelikarsinooma, johon liittyi erilaistuneita rauhasrakenteita (2,4 %).
Koko tutkimuspopulaatiossa 469:lle (88,0 %) potilaalle hoitohaarassa 1 ja 441:lle (83,2 %) potilaalle hoitohaarassa 2 tehtiin radikaali kystektomia.
Tulokset on esitetty taulukossa 14, kuvassa 21 ja kuvassa 22.
Taulukko 14: NIAGARA-tutkimuksen tehoa koskevat tulokset
| IMFINZI + solunsalpaajahoito (N = 533) | Solunsalpaajahoito (N = 530) | ||
|---|---|---|---|
| Tapahtumavapaa elossaoloaikaa | |||
| Tapahtumien määrä (%) | 187 (35,1) | 246 (46,4) | |
| Tapahtumavapaan elossaoloajan mediaani (kuukausina) (95 %:n luottamusväli)b | Ei saavutettu (ei saavutettu, ei saavutettu) | 46,1 (32,2, ei saavutettu) | |
| Riskisuhde (95 %:n luottamusväli)c | 0,68 (0,56, 0,82) | ||
| Kaksitahoinen p-arvod,e | < 0,0001 | ||
| Patologisesti todettu täydellinen vastef | |||
| Vasteen saavuttaneiden potilaiden määrä | 180 | 137 | |
| Vasteprosentti, % (95 %:n luottamusväli)g | 33,8 (29,8, 38,0) | 25,8 (22,2, 29,8) | |
| Kerroinsuhde (95 %:n luottamusväli)h | 1,49 (1,14, 1,96) | ||
| Kaksitahoinen p-arvoh | 0,0038 | ||
| Kokonaiselossaoloaikaa | |||
| Tapahtumien määrä (%) | 136 (25,5) | 169 (31,9) | |
| Kokonaiselossaoloajan mediaani (kuukausina) (95 %:n luottamusväli)b | Ei saavutettu (ei saavutettu, ei saavutettu) | Ei saavutettu (ei saavutettu, ei saavutettu) | |
| Riskisuhde (95 %:n luottamusväli)c | 0,75 (0,59, 0,93) | ||
| Kaksitahoinen p-arvod,e | 0,0106 | ||
a Tulokset perustuvat ennalta suunniteltuun välianalyysiin (viimeinen tiedonkeruupäivä 29.4.2024), joka tehtiin 68 kuukauden kuluttua tutkimuksen aloittamisesta.
b Laskettu Kaplan–Meier-tekniikalla.
c Perustuu ositettuun Coxin suhteellisten riskitiheyksien malliin, jossa ositustekijöinä olivat kasvaimen levinneisyysaste [T2N0 vs. > T2N0], munuaisten toiminta [riittävän hyvä vs. riittävyyden rajoilla] ja PD‑L1:n ilmentymisstatus [runsas vs. niukka tai olematon].
d Perustuu stratifioituun log-rank-testiin, jossa ositustekijöinä olivat kasvaimen levinneisyysaste [T2N0 vs. > T2N0], munuaisten toiminta [riittävän hyvä vs. riittävyyden rajoilla] ja PD‑L1:n ilmentymisstatus [runsas vs. niukka tai olematon].
e Tehoa koskevien ensisijaisten päätetapahtumien (patologisesti todettujen täydellisten vasteiden osuus ja tapahtumavapaa elossaolo) ja keskeisen toissijaisen päätetapahtuman (kokonaiselossaolo) tilastollisen merkitsevyyden rajat määriteltiin monivertailutestausmenettelyllä, jossa käytettiin aiemmat alfat huomioivaa uudelleenkäyttöstrategiaa (alpha-exhaustive recycling strategy). Tapahtumavapaalle elossaololle ja kokonaiselossaololle valittu alfa-arvo perustui Lan–DeMetsin alfavirheen korjausfunktioon, jossa käytettiin O’Brien–Flemingin lähestymistapaa (patologisesti todettu täydellinen vaste = 0,001, tapahtumavapaa elossaolo = 0,0412, kokonaiselossaolo = 0,0154, kaksitahoinen).
f Perustuu patologisesti todettujen täydellisten vasteiden lopulliseen analyysiin (viimeinen tiedonkeruupäivä 14.1.2022).
g Luottamusväli laskettiin Clopper–Pearsonin menetelmällä.
h Laskettu logistisella regressiolla, jota korjattiin ositustekijöiden (munuaisten toiminta [riittävän hyvä vs. riittävyyden rajoilla], kasvaimen levinneisyysaste [T2N0 vs. > T2N0] ja IVRS-järjestelmään kirjattu PD‑L1:n ilmentymisstatus [runsas vs. niukka tai olematon]) suhteen.
Kuva 21. Tapahtumavapaan elossaoloajan (EFS) Kaplan–Meier-käyrä
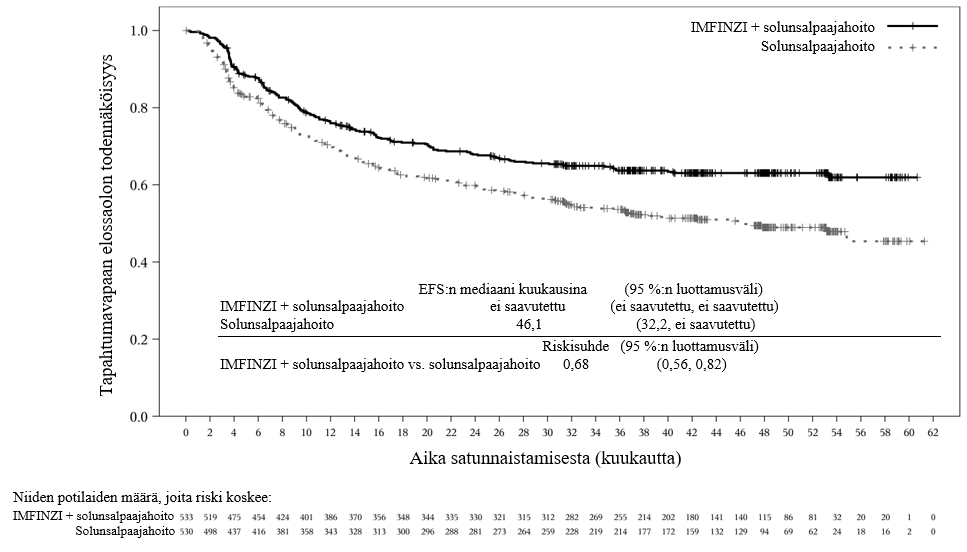
Kuva 22. Kokonaiselossaoloajan (OS) Kaplan–Meier-käyrä
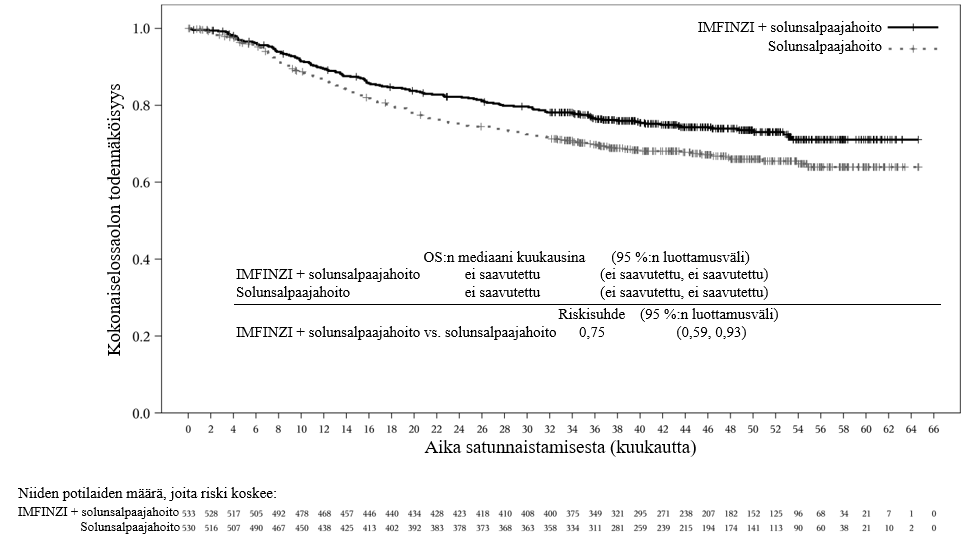
Alaryhmäanalyysi
Kasvaimen levinneisyysasteen mukaan tehdyssä eksploratiivisessa analyysissä tapahtumavapaan elossaoloajan riskisuhde (HR) oli 0,61 (95 %:n luottamusväli: 0,48, 0,78) alaryhmässä, jossa kasvainten kliininen levinneisyysaste oli > T2N0 (N = 635), ja 0,81 (95 %:n luottamusväli: 0,60, 1,10) alaryhmässä, jossa kasvainten kliininen levinneisyysaste oli T2N0 (N = 428). Kokonaiselossaolon riskisuhde oli 0,67 (95 %:n luottamusväli: 0,50, 0,89) alaryhmässä, jossa potilaiden kasvainten kliininen levinneisyysaste oli > T2N0, ja 0,89 (95 %:n luottamusväli: 0,62, 1,29) alaryhmässä, jossa potilaiden kasvainten kliininen levinneisyysaste oli T2N0.
Leikkaukseen soveltuva mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooma – MATTERHORN-tutkimus
MATTERHORN oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen III monikeskustutkimus, jossa arvioitiin IMFINZI-valmisteen tehoa yhdistelmänä FLOT-solunsalpaajahoidon (5‑fluorourasiili, leukovoriini, oksaliplatiini ja dosetakseli) kanssa leikkaukseen soveltuvaa mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinoomaa (levinneisyysaste IIA–IVA [AJCC-luokituskäsikirjan 8. painoksen mukaan]) sairastavilla potilailla, joille annettiin ensin IMFINZI-valmistetta yhdistelmänä FLOT-solunsalpaajahoidon kanssa esiliitännäishoitona ja liitännäishoitona ja sen jälkeen IMFINZI-monoterapiaa liitännäishoitona. Tutkimukseen otettiin potilaita, jotka eivät olleet aiemmin saaneet hoitoa; joilla oli dokumentoitu mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooma; jotka eivät olleet aiemmin saaneet immuunivälitteistä hoitoa; ja joiden WHO/ECOG-suorituskykyluokka oli 0 tai 1. Potilaita otettiin tutkimukseen PD‑L1:n ilmentymisen tasosta riippumatta. Ennen satunnaistamista kasvainten PD‑L1:n ilmentymisen taso varmistettiin TAP-pisteytyksellä Ventana PD‑L1 (SP263) ‑testillä. TAP-pisteytys määritellään kokonaisprosenttiosuudeksi kasvaimen pinta-alasta (kasvaimen ja mahdollisen desmoplastisen strooman pinta-alasta), jota peittävät kasvainsolut, joiden solukalvossa todetaan silmämääräisesti arvioituna minkä tahansa tasoista PD‑L1-värjäytymistä, ja kasvaimeen liittyvät immuunisolut, joissa todetaan silmämääräisesti arvioituna minkä tahansa tasoista PD‑L1-värjäytymistä. Tutkimuksesta suljettiin pois potilaat, joilla oli aktiivinen tai aiemmin dokumentoitu autoimmuuni- tai tulehdussairaus tai jotka olivat saaneet immunosuppressiivista lääkitystä 14 päivän sisällä ennen ensimmäisen durvalumabiannoksen saamista.
Satunnaistaminen stratifioitiin maantieteellisen alueen (Aasia vs. muu kuin Aasia), kliinisen imusolmukestatuksen (positiivinen vs. negatiivinen) ja PD‑L1:n ilmentymisen tason (TAP < 1 % vs. TAP ≥ 1 %) mukaan.
Potilaat satunnaistettiin suhteessa 1:1 jompaankumpaan seuraavista hoitohaaroista. Siirtyminen hoitohaarasta toiseen ei ollut sallittua.
- Hoitohaara 1: IMFINZI 1 500 mg päivänä 1 + FLOT-solunsalpaajahoito (5‑fluorourasiili, 2 600 mg/m2; leukovoriini, 200 mg/m2; oksaliplatiini, 85 mg/m2; dosetakseli, 50 mg/m2) päivinä 1 ja 15 aina 4 viikon välein 4 hoitosyklin ajan (1 annos IMFINZI-valmistetta ja 2 annosta FLOT-solunsalpaajahoitoa kunkin hoitosyklin aikana; 2 hoitosykliä esiliitännäishoidon vaiheessa + 2 hoitosykliä liitännäishoidon vaiheessa) ja sitten IMFINZI 1 500 mg päivänä 1 aina 4 viikon välein vielä enintään 10 hoitosyklin ajan leikkauksen jälkeen niin, että hoitosyklejä oli yhteensä 12 (1 annos kunkin hoitosyklin aikana).
- Hoitohaara 2: Lumelääke päivänä 1 + FLOT-solunsalpaajahoito (5‑fluorourasiili, 2 600 mg/m2; leukovoriini, 200 mg/m2; oksaliplatiini, 85 mg/m2; dosetakseli, 50 mg/m2) päivinä 1 ja 15 aina 4 viikon välein 4 hoitosyklin ajan (1 annos lumelääkettä ja 2 annosta FLOT-solunsalpaajahoitoa kunkin hoitosyklin aikana; 2 hoitosykliä esiliitännäishoidon vaiheessa + 2 hoitosykliä liitännäishoidon vaiheessa) ja sitten lumelääkettä päivänä 1 aina 4 viikon välein vielä enintään 10 hoitosyklin ajan leikkauksen jälkeen niin, että hoitosyklejä oli yhteensä 12 (1 annos kunkin hoitosyklin aikana).
Potilaat, jotka joko esiliitännäishoidon tai liitännäishoidon vaiheessa lopettivat FLOT-solunsalpaajahoidon muusta syystä kuin taudin etenemisen tai uusiutumisen vuoksi, saivat tutkijan harkinnan mukaan jatkaa IMFINZI-monoterapiaa edellä kuvattuun tapaan.
Kasvaimet arvioitiin RECIST 1.1 ‑kriteerien mukaan lähtötilanteessa ennen esiliitännäishoidon aloittamista, ja seurantaa varten tehtiin uusi kuvantamistutkimus ennen leikkausta 4 viikon kuluessa viimeisen solunsalpaaja-annoksen saamisesta. Potilaille tehtiin liitännäishoidon lähtötilanteen kuvantamistutkimus aikaisintaan 4 viikon kuluttua leikkauksen jälkeen ja ennen liitännäishoidon aloittamista. Kasvaimia arvioitiin 12 viikon välein (liitännäishoidon lähtötilanteen kuvantamistutkimuksesta laskettuna) 2 vuoden ajan ja sitten 24 viikon välein, kunnes todettiin radiologisesti RECIST 1.1 ‑kriteerien mukaisesti taudin edenneen tai potilas peruutti suostumuksensa osallistua tutkimukseen tai kuoli.
Tutkimuksen ensisijainen päätetapahtuma oli tapahtumavapaa elossaoloaika (EFS), jonka arvioi sokkoutettu riippumaton keskitetty arvioijataho (BICR) ja joka määriteltiin ajaksi satunnaistamisesta päivään, jolloin todettiin jokin seuraavista tapahtumista (sen mukaan, mikä tapahtui ensin): tauti eteni RECIST-kriteerien version 1.1 mukaisesti sokkoutetun riippumattoman keskitetyn arvioijatahon arvion mukaan niin, että leikkaushoito oli poissuljettu tai tutkimussuunnitelmasta poikkeava hoito oli tarpeen esiliitännäishoitovaiheen aikana; tauti eteni tai uusiutui RECIST-kriteerien version 1.1 mukaisesti liitännäishoitovaiheen aikana; tauti eteni muutoin kuin RECIST-kriteerien mukaisesti (tutkijan arvion mukaan tai biopsialla vahvistetusti) niin, että leikkaushoito oli poissuljettu tai tutkimussuunnitelmasta poikkeava hoito oli tarpeen esiliitännäishoitovaiheen aikana tai tällainen taudin eteneminen todettiin leikkauksen aikana; tauti eteni tai uusiutui, mikä vahvistettiin biopsialla leikkauksen jälkeen; tai potilas kuoli mistä tahansa syystä. Keskeiset toissijaiset päätetapahtumat olivat kokonaiselossaoloaika (OS) ja patologisesti todettujen täydellisten vasteiden (pCR) osuus sokkoutetun keskitetyn patologisen arvion mukaan.
Demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat yleisesti hyvin samankaltaiset molemmissa hoitohaaroissa (hoitohaarassa 1 oli 474 potilasta ja hoitohaarassa 2 oli 474 potilasta). Populaation demografiset tiedot lähtötilanteessa olivat: miehiä (71,9 %), ikä vähintään 65 vuotta (41,4 %), iän mediaani 62 vuotta (vaihteluväli: 26–84), valkoihoisia (67,8 %), aasialaisia (20,4 %), mustaihoisia tai afrikkalaisamerikkalaisia (1,1 %), Amerikan tai Alaskan alkuperäisväestöön kuuluvia (4,0 %), muun etnisen taustan omaavia (1,7 %), muita kuin taustaltaan espanjankielisiä tai latinalaisamerikkalaisia (80,0 %), WHO/ECOG-suorituskykypistemäärä 0 (74,2 %), WHO/ECOG-suorituskykypistemäärä 1 (25,8 %). Sairauden ominaispiirteet olivat: levinneisyysaste II (29,4 %), levinneisyysaste III (61,7 %), levinneisyysaste IVA (8,8 %), sijainti mahalaukussa (67,5 %), sijainti ruokatorvi-mahalaukkurajassa (32,5 %), Siewertin tyyppi 1 (10,4 %), Siewertin tyyppi 2 (14,8 %), Siewertin tyyppi 3 (7,3 %), intestinaalinen tyyppi (50,9 %), diffuusi tyyppi (26,3 %), määrittelemätön tyyppi (22,8 %), kliininen imusolmukestatus positiivinen (70,4 %), kliininen imusolmukestatus negatiivinen (29,2 %), PD‑L1:n ilmentymisstatus TAP ≥ 1 % (90,0 %), PD‑L1:n ilmentymisstatus TAP < 1 % (10,0 %).
Leikkausta, jossa pyrittiin kuratiiviseen tulokseen, yritettiin hoitohaaran 1 potilaista 431 potilaalla (90,9 %) ja hoitohaaran 2 potilaista 428 potilaalla (90,3 %). Leikkaus, jossa pyrittiin kuratiiviseen tulokseen, toteutettiin hoitohaaran 1 potilaista 412 potilaalle (86,9 %) ja hoitohaaran 2 potilaista 400 potilaalle (84,4 %).
Taulukko 15: MATTERHORN-tutkimuksen tehoa koskevat tulokset
| IMFINZI + FLOT-solunsalpaajahoito (N = 474) | Lumelääke + FLOT-solunsalpaajahoito (N = 474) | |
| Tapahtumavapaa elossaoloa | ||
| Tapahtumien määrä, n (%) | 167 (35,2 %) | 218 (46 %) |
| Tapahtumavapaan elossaoloajan mediaani (kuukausina) (95 %:n luottamusväli)b | Ei saavutettu (40,7, ei saavutettu) | 32,8 (27,9, ei saavutettu) |
| Riskitiheyssuhde (95 %:n luottamusväli)c | 0,71 (0,58, 0,86) | |
| Kaksitahoinen p-arvod,e | < 0,001 | |
| Tapahtumavapaa elossaolo 24 kuukauden kohdalla, % (95 %:n luottamusväli)b | 67,4 (62,9, 71,6) | 58,5 (53,8, 63,0) |
| Kokonaiselossaolof | ||
| Kuolemantapausten määrä (%) | 160 (33,8 %) | 192 (40,5 %) |
| Kokonaiselossaoloajan mediaani (kuukausina) (95 %:n luottamusväli)b | Ei saavutettu (ei saavutettu, ei saavutettu) | Ei saavutettu (ei saavutettu, ei saavutettu) |
| Riskitiheyssuhde (95 %:n luottamusväli)c | 0,78 (0,63, 0,96) | |
| Kaksitahoinen p-arvod,g | 0,021 | |
| Kokonaiselossaolo 24 kuukauden kohdalla, % (95 %:n luottamusväli)b | 75,5 (71,4, 79,1) | 70,4 (66,0, 74,3) |
| Kokonaiselossaolo 36 kuukauden kohdalla, % (95 %:n luottamusväli)b | 68,6 (64,2, 72,6) | 61,9 (57,3, 66,2) |
a Tulokset perustuvat ennalta määriteltyyn tapahtumavapaata elossaoloa koskevaan välianalyysiin (viimeinen tiedonkeruupäivä 20.12.2024).
b Laskettu Kaplan–Meier-tekniikalla.
c Perustuu ositettuun Coxin suhteellisten riskitiheyksien malliin, jossa ositustekijöinä olivat maantieteellinen alue, kliininen imusolmukestatus ja PD‑L1:n ilmentymisstatus satunnaistamishetkellä. Riskitiheyssuhde < 1 suosii IMFINZI-valmistetta. Luottamusväli laskettu profiiliuskottavuusmenetelmällä.
d Perustuu stratifioituun log rank ‑testiin, jonka tulokset korjattiin maantieteellisen alueen, kliinisen imusolmukestatuksen ja satunnaistamishetkellä todetun PD‑L1:n ilmentymisstatuksen suhteen.
e Kun perustana oli Lan–DeMetsin alfavirheen korjausfunktio, O’Brien–Flemingin tyyppinen raja ja havaittujen tapahtumien todellinen lukumäärä viimeisenä tiedonkeruupäivänä, tapahtumavapaata elossaoloa koskeva tilastollisen merkitsevyyden raja oli 0,0239, kun kokonaisalfavirhe oli 5 % (kaksitahoinen).
f Tulokset perustuvat ennalta määriteltyyn kokonaiselossaoloa koskevaan lopulliseen analyysiin (viimeinen tiedonkeruupäivä 1.9.2025).
g Kokonaiselossaoloa koskevalle lopulliselle analyysille valittiin alfa-arvo 4,99 % (kaksitahoinen), joka vastaa tilastollisen merkitsevyyden raja-arvoa 0,0499.
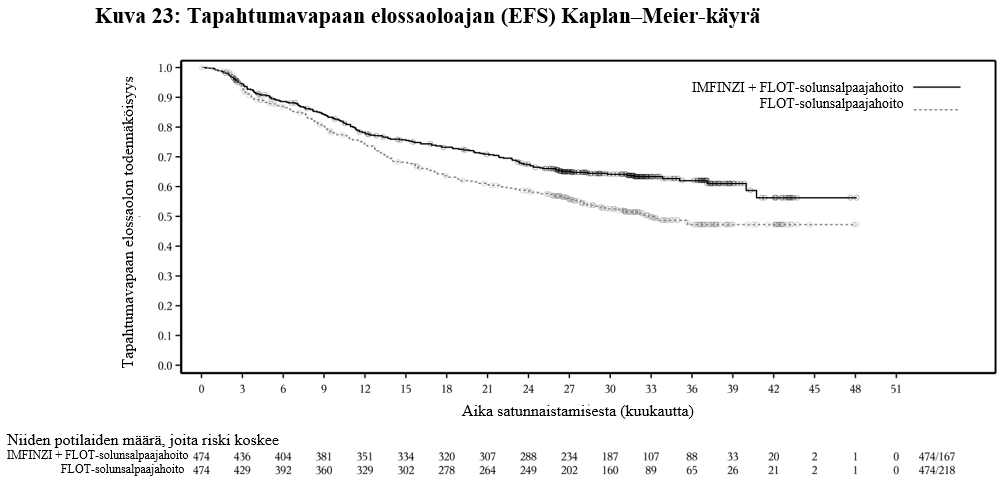
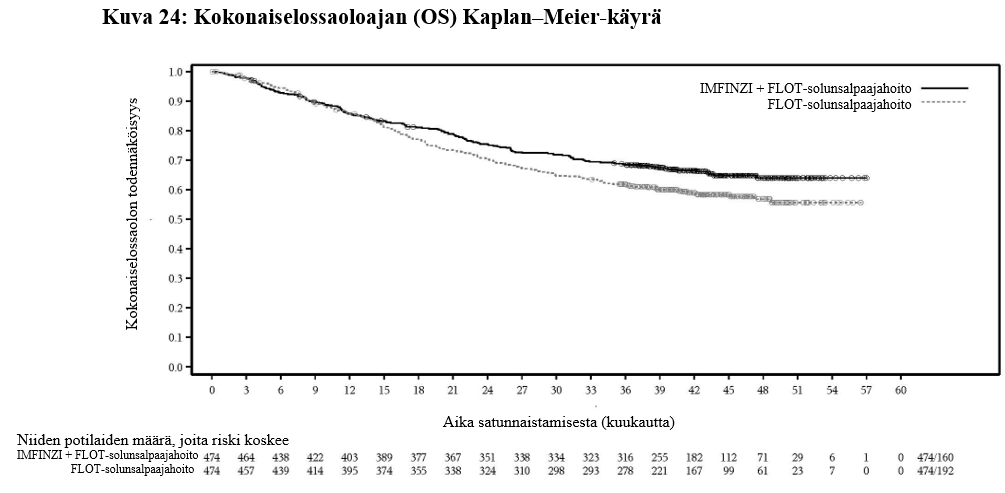
Alaryhmäanalyysi
Kliinisen imusolmukestatuksen mukaan tehdyssä ennalta määritellyssä eksploratiivisessa analyysissä tapahtumavapaan elossaoloajan riskitiheyssuhde (HR) oli 0,67 (95 %:n luottamusväli: 0,53, 0,84) alaryhmässä, jossa potilaiden kliininen imusolmukestatus oli positiivinen (N = 667), ja 0,85 (95 %:n luottamusväli: 0,57, 1,27) alaryhmässä, jossa potilaiden kliininen imusolmukestatus oli negatiivinen (N = 277). Kokonaiselinajan riskitiheyssuhde oli 0,73 (95 %:n luottamusväli: 0,57, 0,94) alaryhmässä, jossa potilaiden kliininen imusolmukestatus oli positiivinen, ja 1,01 (95 %:n luottamusväli: 0,67, 1,51) alaryhmässä, jossa potilaiden kliininen imusolmukestatus oli negatiivinen.
Pediatriset potilaat
IMFINZI-valmisteen turvallisuutta ja tehoa yhdistelmänä tremelimumabin kanssa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole varmistettu. D419EC00001-tutkimus oli avoin monikeskustutkimus, johon sisältyi annosmääritysvaihe ja tutkimuksen laajentamisvaihe. Tutkimuksessa arvioitiin IMFINZI-valmisteen turvallisuutta, alustavaa tehoa ja farmakokinetiikkaa, kun valmistetta annettiin ensin yhdistelmänä tremelimumabin kanssa ja sen jälkeen monoterapiana pediatrisille potilaille. Potilailla oli pitkälle edenneitä pahanlaatuisia kiinteitä kasvaimia (ei kuitenkaan primaarisia keskushermostokasvaimia), heidän tautinsa oli edennyt, eikä heille ollut olemassa tavanomaista hoitoa. Tutkimukseen otettiin mukaan 50 pediatrista potilasta, jotka olivat iältään 1–17-vuotiaita ja joiden primaarikasvain oli jokin seuraavista: neuroblastooma, kiinteä kasvain tai sarkooma. Potilaat saivat joko IMFINZI-valmistetta annoksella 20 mg/kg yhdistelmänä tremelimumabin (1 mg/kg) kanssa tai IMFINZI-valmistetta 30 mg/kg yhdistelmänä tremelimumabin (1 mg/kg) kanssa laskimoon 4 viikon välein 4 hoitosyklin ajan ja sen jälkeen IMFINZI-valmistetta monoterapiana 4 viikon välein. Annosmääritysvaiheessa annettiin yksi hoitosykli IMFINZI-monoterapiaa ennen IMFINZI-valmisteella ja tremelimumabilla toteutettua yhdistelmähoitoa; tämän vaiheen aikana 8 potilasta kuitenkin lopetti hoidon ennen kuin he saivat tremelimumabia. Näin ollen tutkimukseen otetuista 50 potilaasta 42 sai IMFINZI-hoitoa yhdistelmänä tremelimumabin kanssa ja 8 sai pelkkää IMFINZI-hoitoa. Vasteen suhteen arvioitavissa olevassa analyysipopulaatiossa tutkimuksen laajentamisvaiheen aikana ilmoitettu objektiivinen vasteprosentti oli 5,0 % (1 potilas 20:stä). Uusia turvallisuussignaaleja verrattuna IMFINZI-valmisteen ja tremelimumabin tunnettuihin turvallisuusprofiileihin aikuisilla ei havaittu. Ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Farmakokinetiikka
Durvalumabin farmakokinetiikkaa arvioitiin käytettäessä IMFINZI-valmistetta ainoana lääkkeenä, yhdistelmänä solunsalpaajahoidon kanssa, yhdistelmänä tremelimumabin ja platinapohjaisen solunsalpaajahoidon kanssa, yhdistelmänä tremelimumabin kanssa ja yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa niin, että tämän jälkeen annettiin IMFINZI-valmistetta yhdistelmänä olaparibin kanssa.
Durvalumabin farmakokinetiikkaa tutkittiin 2 903 potilaalla, joilla oli kiinteitä kasvaimia. Käytetyt annokset olivat 0,1–20 mg/kg ja ne annettiin laskimoon kahden, kolmen tai neljän viikon välein monoterapiana. Farmakokineettinen altistus suureni enemmän kuin suhteessa annokseen (ei‑lineaarinen farmakokinetiikka) < 3 mg/kg:n annoksilla ja suhteessa annokseen (lineaarinen farmakokinetiikka) ≥ 3 mg/kg:n annoksilla. Vakaa tila saavutettiin suunnilleen 16 viikon kohdalla. Populaatiofarmakokineettisessä analyysissä oli mukana 1 878 potilasta, jotka saivat durvalumabia monoterapiana ja joilla annostus oli ≥ 10 mg/kg 2 viikon välein, ja analyysin perusteella geometrinen keskiarvo vakaan tilan jakautumistilavuudelle (Vss) oli 5,64 l. Durvalumabin puhdistuma (CL) pieneni ajan myötä niin, että päivänä 365 geometrinen keskiarvo vakaan tilan puhdistumalle (CLss) oli 8,16 ml/h. CLss:n pienenemisen ei katsottu olevan kliinisesti merkityksellinen. Terminaalinen puoliintumisaika (t1/2) lähtötilanteen CL-arvosta laskettuna oli noin 18 päivää. Ainoana lääkkeenä käytetyn, yhdistelmänä solunsalpaajahoidon kanssa käytetyn, yhdistelmänä tremelimumabin ja platinapohjaisen solunsalpaajahoidon kanssa käytetyn, yhdistelmänä tremelimumabin kanssa käytetyn ja yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa (minkä jälkeen annettiin IMFINZI-valmistetta yhdistelmänä olaparibin kanssa) käytetyn durvalumabin farmakokinetiikassa ei ollut kliinisesti merkittävää eroa. Durvalumabin pääasialliset eliminaatioreitit ovat proteiinikatabolia retikuloendoteliaalijärjestelmän kautta ja kohdevälitteinen jakautuminen.
Erityisryhmät
Ikä (19–96 vuotta), kehon paino (31–149 kg), sukupuoli, positiivinen lääkevasta-ainestatus (ADA-status), albumiiniarvot, LDH-arvot, kreatiniiniarvot, liukoinen PD‑L1, kasvaintyyppi, rotu tai ECOG-pistemäärä eivät vaikuttaneet kliinisesti merkittävästi durvalumabin farmakokinetiikkaan.
Munuaisten vajaatoiminta
Lievällä munuaisten vajaatoiminnalla (kreatiniinipuhdistuma 60–89 ml/min) ja keskivaikealla munuaisten vajaatoiminnalla (kreatiniinipuhdistuma 30–59 ml/min) ei ollut kliinisesti merkittävää vaikutusta durvalumabin farmakokinetiikkaan. Vaikean munuaisten vajaatoiminnan (kreatiniinipuhdistuma 15–29 ml/min) vaikutusta durvalumabin farmakokinetiikkaan ei tiedetä. Monoklonaaliset IgG-vasta-aineet eivät kuitenkaan poistu ensisijaisesti munuaisten kautta, joten munuaisten toiminnan muutosten ei odoteta vaikuttavan durvalumabialtistukseen.
Maksan vajaatoiminta
Lievällä maksan vajaatoiminnalla (bilirubiini enintään viitealueen ylärajalla ja ASAT viitealueen ylärajan yläpuolella tai bilirubiini > 1,0 – 1,5 kertaa viitealueen yläraja ja mikä tahansa ASAT-arvo) tai keskivaikealla maksan vajaatoiminnalla (bilirubiini > 1,5 – 3 kertaa viitealueen yläraja ja ASAT mikä tahansa) ei ollut kliinisesti merkittävää vaikutusta durvalumabin farmakokinetiikkaan. Vaikean maksan vajaatoiminnan (bilirubiini > 3,0 kertaa viitealueen yläraja ja mikä tahansa ASAT-arvo) vaikutusta durvalumabin farmakokinetiikkaan ei tiedetä. Monoklonaaliset IgG-vasta-aineet eivät kuitenkaan poistu ensisijaisesti maksan kautta, joten maksan toiminnan muutosten ei odoteta vaikuttavan durvalumabialtistukseen.
Pediatriset potilaat
Durvalumabin farmakokinetiikkaa yhdistelmänä tremelimumabin kanssa arvioitiin D419EC00001-tutkimuksessa 50 pediatrisella potilaalla, jotka olivat 1–17-vuotiaita. Potilaat saivat joko durvalumabia annoksella 20 mg/kg yhdistelmänä tremelimumabin (1 mg/kg) kanssa tai durvalumabia 30 mg/kg yhdistelmänä tremelimumabin (1 mg/kg) kanssa laskimoon 4 viikon välein 4 hoitosyklin ajan ja sen jälkeen durvalumabia monoterapiana 4 viikon välein. Populaatiofarmakokineettisen analyysin perusteella systeeminen altistus durvalumabille ≥ 35 kg painavilla pediatrisilla potilailla, jotka saivat durvalumabia 20 mg/kg 4 viikon välein, oli samanlainen kuin altistus aikuisilla, jotka saivat durvalumabia 20 mg/kg 4 viikon välein, kun taas pediatrisilla potilailla (≥ 35 kg), jotka saivat durvalumabia 30 mg/kg 4 viikon välein, altistus oli noin 1,5 kertaa suurempi kuin aikuisilla, jotka saivat durvalumabia 20 mg/kg 4 viikon välein. Alle 35 kg painavilla pediatrisilla potilailla, jotka saivat durvalumabia 30 mg/kg 4 viikon välein, systeeminen altistus vastasi altistusta aikuisilla, jotka saivat durvalumabia 20 mg/kg 4 viikon välein.
Prekliiniset tiedot turvallisuudesta
Karsinogeenisuus ja mutageenisuus
Durvalumabin karsinogeenisuutta ja genotoksisuutta ei ole arvioitu.
Lisääntymistoksisuus
Kuten kirjallisuudessa on raportoitu, PD‑1/PD‑L1-reitillä on keskeinen merkitys raskauden jatkumisessa, koska se ylläpitää äidin immunologista toleranssia sikiötä kohtaan, ja tiineiden hiirten allogeenisissa malleissa PD‑L1-signaalinvälityksen salpauksen on osoitettu lisäävän sikiönmenetyksiä. Eläimillä tehdyissä lisääntymistutkimuksissa durvalumabia annettiin tiineille cynomolgus-apinoille tiineyden varmistamisesta synnytykseen asti altistuksilla, jotka olivat noin 18 kertaa suurempia kuin durvalumabin kliinisellä annoksella 10 mg/kg on havaittu (AUC-arvoista laskettuna), ja durvalumabin todettiin läpäisevän istukan, mutta ei havaittu maternaalista toksisuutta tai vaikutuksia alkion tai sikiön kehitykseen, tiineyden tulokseen tai postnataaliseen kehitykseen. Durvalumabia todettiin merkityksettöminä määrinä cynomolgus-apinoiden maidossa 28. päivänä synnytyksen jälkeen.
Farmaseuttiset tiedot
Apuaineet
Histidiini
Histidiinihydrokloridimonohydraatti
Trehaloosidihydraatti
Polysorbaatti 80 (E 433)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamaton injektiopullo
3 vuotta
Laimennettu liuos
Kemiallisen ja fysikaalisen käytönaikaisen säilyvyyden on osoitettu olevan enintään 30 päivää 2–8 °C:ssa ja enintään 24 tuntia huoneenlämpötilassa (enintään 25 °C:ssa) liuoksen valmistuksesta.
Mikrobiologiselta kannalta valmis infuusioliuos pitäisi käyttää välittömästi. Jos sitä ei käytetä välittömästi, käytönaikainen säilytysaika ja -olosuhteet ennen käyttöä ovat käyttäjän vastuulla eivätkä ne yleensä saa olla kuin enintään 24 tuntia 2–8 °C:ssa tai 12 tuntia huoneenlämpötilassa (enintään 25 °C:ssa), ellei laimennusta ole tehty valvotuissa, validoiduissa, aseptisissa olosuhteissa.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
IMFINZI infuusiokonsentraatti, liuosta varten
50 mg/ml (L:ei) 2,4 ml (725,15 €), 10 ml (2806,92 €)
PF-selosteen tieto
IMFINZI-valmisteesta on saatavilla kaksi pakkauskokoa:
2,4 ml (120 mg durvalumabia) konsentraattia tyypin I lasista valmistetussa injektiopullossa, jossa on elastomeeritulppa ja harmaa alumiininen repäisykorkki. Pakkaus sisältää 1 injektiopullon.
10 ml (500 mg durvalumabia) konsentraattia tyypin I lasista valmistetussa injektiopullossa, jossa on elastomeeritulppa ja valkoinen alumiininen repäisykorkki. Pakkaus sisältää 1 injektiopullon.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai opaalinhohtoinen, väritön tai kellertävä liuos, jossa ei ole näkyviä hiukkasia. Liuoksen pH on noin 6,0 ja osmolaliteetti noin 400 mOsm/kg.
Käyttö- ja käsittelyohjeet
Liuoksen valmistus
IMFINZI toimitetaan kerta-annosinjektiopullossa, joka ei sisällä säilöntäaineita. Aseptista tekniikkaa on noudatettava.
-
Tarkista lääkevalmiste silmämääräisesti, ettei siinä ole havaittavissa hiukkasia eikä värimuutoksia. IMFINZI on kirkas tai opaalinhohtoinen, väritön tai kellertävä liuos. Hävitä injektiopullo, jos liuos on sameaa tai siinä on havaittavissa värimuutoksia tai hiukkasia. Älä ravista injektiopulloa.
-
Vedä IMFINZI-injektiopullo(i)sta tarvittava määrä liuosta ruiskuun ja siirrä se laskimoon antamista varten tarkoitettuun infuusiopussiin, jossa on natriumkloridi-injektioliuosta (9 mg/ml, 0,9 %) tai glukoosi-injektioliuosta (50 mg/ml, 5 %). Sekoita laimennettu liuos kääntelemällä varovasti. Laimennetun liuoksen lopullinen pitoisuus on 1–15 mg/ml. Älä anna liuoksen jäätyä äläkä ravista liuosta.
- Hävitä injektiopulloon jäänyt käyttämätön lääke.
Antaminen
-
Anna infuusioliuos laskimoon 1 tunnin aikana laskimolinjalla, joka sisältää steriilin, niukasti proteiineja sitovan 0,2 tai 0,22 mikronin kiinteän (in-line) suodattimen.
- Älä anna muita lääkevalmisteita saman infuusioletkun kautta.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
IMFINZI infuusiokonsentraatti, liuosta varten
50 mg/ml 2,4 ml, 10 ml
- Ei korvausta.
ATC-koodi
L01FF03
Valmisteyhteenvedon muuttamispäivämäärä
12.03.2026
Yhteystiedot
Keilaranta 18
02150 Espoo
010 23 010
www.astrazeneca.fi