
BAVENCIO infuusiokonsentraatti, liuosta varten 20 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Potilaan turvallisuuskortti Bavencio 20 mg/ml infuusiokonsentraatti.
Vaikuttavat aineet ja niiden määrät
Yksi ml konsentraattia sisältää 20 mg avelumabia.
Yksi 10 ml:n injektiopullo sisältää 200 mg avelumabia.
Avelumabi on ihmisen monoklonaalinen IgG1-vasta-aine, joka kohdistuu solun pinnalla olevaan immunomodulatoriseen PD-L1-ligandiproteiiniin ja joka on tuotettu kiinanhamsterin munasarjasoluissa yhdistelmä-DNA-tekniikalla.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää 5 mg polysorbaattia 20 per injektiopullo.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Bavencio on tarkoitettu käytettäväksi monoterapiana metastasoituneen merkelinsolukarsinooman (MCC) hoitoon aikuispotilailla.
Bavencio on tarkoitettu käytettäväksi monoterapiana ensilinjan ylläpitohoitoon aikuispotilailla, joilla on paikallisesti edennyt tai metastasoitunut uroteelikarsinooma (UC) ja joilla tauti ei ole edennyt platinapohjaisen kemoterapian jälkeen.
Bavencio on tarkoitettu käytettäväksi yhdessä aksitinibin kanssa ensilinjan hoitona edenneen munuaissolukarsinooman (RCC) hoitoon aikuispotilailla (ks. kohta Farmakodynamiikka).
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Hoidon aloittavan ja hoitoa valvovan lääkärin on oltava perehtynyt syövän hoitoon.
Annostus
Suositeltu Bavencio-annos monoterapiana on 800 mg annosteltuna laskimoon 60 minuutissa kahden viikon välein.
Bavencio-valmisteen antoa jatketaan suositeltua annostusta noudattaen, kunnes tauti etenee tai potilaalla ilmenee haittavaikutuksia, joita ei voida hyväksyä
Suositeltu Bavencio-annos annettuna yhdessä aksitinibin kanssa on 800 mg annosteltuna laskimoon 60 minuutissa 2 viikon välein ja aksitinibiannos 5 mg suun kautta otettuna kahdesti vuorokaudessa (12 tunnin välein) aterian yhteydessä tai tyhjään mahaan, kunnes tauti etenee tai potilaalla ilmenee haittavaikutuksia, joita ei voida hyväksyä.
Katso aksitinibin annostusta koskevia tietoja aksitinibin valmisteyhteenvedosta.
Esilääkitys
Ennen neljää ensimmäistä Bavencio-infuusiota potilaille on annettava esilääkityksenä antihistamiinia ja parasetamolia. Jos neljännen infuusion jälkeen ei ilmene infuusioon liittyviä reaktiota, lääkäri päättää seuraavien annosten esilääkityksestäharkintansa mukaan.
Hoidon muutokset
Annoksen suurentaminen tai pienentäminen ei ole suositeltavaa. Annoksen siirtäminen tai hoidon keskeytys voi olla tarpeen yksilöllisen turvallisuuden ja siedettävyyden perusteella, ks. taulukko 1.
Yksityiskohtaiset ohjeet immuunivälitteisten haittavaikutuksien hallintaan on annettu kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Taulukko 1: Suositukset Bavencio-hoidon annostuksen siirtämiseen tai lopettamiseen
Hoitoon liittyvä haittavaikutus | Vaikeusaste* | Hoidon muutos |
|---|---|---|
Infuusioon liittyvät reaktiot | Infuusioon liittyvä asteen 1 reaktio | Hidasta infuusionopeutta 50 % |
Infuusioon liittyvä asteen 2 reaktio | Keskeytä, kunnes haittavaikutukset lieventyvät tasolle 0–1; aloita infuusio uudelleen 50 % hitaammalla nopeudella | |
Infuusioon liittyvä asteen 3 tai 4 reaktio | Lopeta pysyvästi | |
Pneumoniitti | Asteen 2 pneumoniitti | Siirrä antamista, kunnes haittavaikutukset lievittyvät asteen 0‑1 tasolle |
Asteen 3 tai 4 pneumoniitti tai uusiutuva asteen 2 pneumoniitti | Lopeta pysyvästi | |
Hepatiitti
Bavencio annettuna yhdessä aksitinibin kanssa, ks. jäljempänä | Aspartaattiaminotransferaasi (ASAT) tai alaniiniaminotransferaasi (ALAT) 3–5 kertaa normaaliarvon yläraja (ULN) tai kokonaisbilirubiini 1,5–3 kertaa ULN | Siirrä antamista, kunnes haittavaikutukset lievittyvät asteen 0‑1 tasolle |
ASAT tai ALAT yli 5 kertaa ULN tai kokonaisbilirubiini yli 3 kertaa ULN | Lopeta pysyvästi | |
Koliitti | Asteen 2 tai 3 koliitti tai ripuli | Siirrä antamista, kunnes haittavaikutukset lievittyvät asteen 0‑1 tasolle |
Asteen 4 koliitti tai ripuli tai uusiutuva asteen 3 koliitti | Lopeta pysyvästi | |
Pankreatiitti | Epäilty pankreatiitti | Siirrä antamista |
Varmistettu pankreatiitti | Lopeta pysyvästi | |
Myokardiitti | Epäilty myokardiitti | Siirrä antamista |
Varmistettu myokardiitti | ||
Umpierityssairaudet (hypotyreoosi, hypertyreoosi, lisämunuaisen vajaatoiminta, hyperglykemia) | Asteen 3 tai 4 umpierityssairaus | Siirrä antamista, kunnes haittavaikutukset lievittyvät asteen 0‑1 tasolle |
Munuaistulehdus ja munuaisten vajaatoiminta | Seerumin kreatiniini 1,5–6 kertaa ULN | Siirrä antamista, kunnes haittavaikutukset lievittyvät asteen 0‑1 tasolle |
Seerumin kreatiniini yli 6 kertaa ULN | Lopeta pysyvästi | |
Ihoreaktiot | Asteen 3 ihottuma | Siirrä antamista, kunnes haittavaikutukset lievittyvät asteen 0‑1 tasolle |
| Asteen 4 ihottuma tai uusiutuva asteen 3 ihottuma tai varmistettu Stevens-Johnsonin oireyhtymä tai toksinen epidermaalinen nekrolyysi | Lopeta pysyvästi | |
| Muut immuunivälitteiset haittavaikutukset (mukaan lukien muut kliinisesti merkittävät immuunivälitteiset haittavaikutukset, jotka luetellaan kohdassa Muut immuunivälitteiset haittavaikutukset, ks. kohta 4. | Seuraavissa tapauksissa:
| Siirrä antamista, kunnes haittavaikutukset lievittyvät asteen 0‑1 tasolle |
Seuraavissa tapauksissa:
| Lopeta pysyvästi | |
| Myokardiitti-myosiitti-myasthenia gravis -limittymisoireyhtymäa | Asteen 2 myosiitti, asteen 2 myasthenia gravis, epäilty myokardiitti | Siirrä antamista |
| Asteen 3 myosiitti, asteen 3 myasthenia gravis, vahvistettu myokardiitti | Lopeta pysyvästi |
* Toksisuuden luokittelu perustuu Yhdysvaltain syöpäinstituutin (National Cancer Institute) Common Terminology Criteria for Adverse Events -luokituksen versioon 4.0 (NCI‑CTCAE v4.03)
a Esiintyy joko kahden tai kaikkien kolmen sairauden limittymisenä. Vakavin CTCAE-aste yksittäisistä tapahtumista on otettava huomioon, kun suositeltua avelumabin hoidon muutosta arvioidaan.
Hoidon muutokset kun Bavencio-valmistetta käytetään yhdessä aksitinibin kanssa
Jos ALAT tai ASAT ≥ 3 kertaa ULN, mutta < 5 kertaa ULN tai kokonaisbilirubiini ≥ 1,5 kertaa ULN mutta < 3 kertaa ULN, sekä Bavencio- että aksitinibihoito tulisi toistaiseksi keskeyttää, kunnes nämä haittavaikutukset lievittyvät asteen 0‑1 tasolle. Jos ne pitkittyvät (yli 5 vuorokautta), on harkittava kortikosteroidihoitoa prednisonilla tai vastaavalla ja sen jälkeen kortikosteroidiannoksen purkamista asteittain. Haittavaikutusten palautumisen jälkeen on harkittava Bavencio- tai aksitinibihoidon jatkamista tai sekventiaalista hoitoa, jossa käytetään sekä Bavencio-valmistetta että aksitinibia. Mikäli aksitinibihoitoa jatketaan, on annoksen pienentämistä harkittava aksitinibin valmisteyhteenvedon mukaisesti.
Jos ALAT tai ASAT ≥ 5 kertaa ULN tai > 3 kertaa ULN kun samanaikaisesti kokonaisbilirubiini ≥ 2 kertaa ULN tai kokonaisbilirubiini ≥ 3 kertaa ULN, sekä Bavencio- että aksitinibihoito on lopetettava pysyvästi ja harkittava kortikosteroidihoitoa.
Ohjeet koskien aksitinibin annosmuutoksia, kun sitä käytetään yhdessä Bavencio-valmisteen kanssa
Kun Bavencio-valmistetta annetaan yhdessä aksitinibin kanssa, katso aksitinibin annosmuutoksia koskevat suositukset aksitinibin valmisteyhteenvedosta.
Erityisryhmät
Iäkkäät potilaat
Annosta ei tarvitse muuttaa hoidettaessa iäkkäitä potilaita (≥ 65-vuotiaat) (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Pediatriset potilaat
Bavencio-valmisteen turvallisuutta ja tehoa alle 18-vuotiaiden lasten ja nuorten hoidossa ei ole varmistettu. Bavencio-valmistetta koskevan saatavissa olevan tiedon perusteella, joka on kuvattu kohdassa Farmakodynamiikka, ei voida antaa suosituksia annostuksesta.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa hoidettaessa potilaita, joilla on lievä tai kohtalainen munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka). Vaikeaa munuaisten vajaatoimintaa sairastavista potilaista ei ole riittävästi tietoja, jotta annossuosituksia voitaisiin antaa.
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa hoidettaessa potilaita, joilla on lievä maksan vajaatoiminta (ks. kohta Farmakokinetiikka). Kohtalaista tai vaikeaa maksan vajaatoimintaa sairastavista potilaista ei ole riittävästi tietoja, jotta annossuosituksia voitaisiin antaa.
Antotapa
Bavencio on tarkoitettu annettavaksi vain laskimonsisäisenä infuusiona. Sitä ei saa antaa laskimoon nopeana injektiona tai boluksena.
Bavencio on laimennettava joko natriumkloridi 9 mg/ml (0,9 %)- tai natriumkloridi 4,5 mg/ml (0,45 %) liuoksella. Bavencio annetaan infuusiona laskimoon 60 minuutin aikana käyttäen steriiliä, pyrogeenitonta, heikosti proteiinia sitovaa 0,2 mikrometrin kiinteää (in‑line) tai irrallista (add‑on) suodatinta.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen valmistamisesta ja antamisesta.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infuusioon liittyvät reaktiot
Avelumabia saavilla potilailla on raportoitu infuusioon liittyviä reaktioita, jotka voivat olla vakavia (ks. kohta Haittavaikutukset).
Potilailla tulee tarkkailla infuusioreaktioon viittaavia oireita tai löydöksiä, kuumetta, vilunväristyksiä, punoitusta, alhaista verenpainetta, hengenahdistusta, hengityksen vinkumista, selkäkipua, vatsakipua ja nokkosihottumaa.
Jos potilaalla ilmenee infuusioon liittyvä asteen 3 tai 4 reaktio, infuusio on keskeytettävä ja avelumabihoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Jos potilaalla ilmenee infuusioon liittyvä asteen 1 reaktio, kesken olevan infuusion nopeutta on hidastettava 50 %. Jos potilaalla on infuusioon liittyvä asteen 2 reaktio, infuusio tulisi keskeyttää toistaiseksi, kunnes reaktio lievittyy asteen 1 tasolle tai väistyy, jolloin infuusion antamista voidaan jatkaa 50 % pienemmällä nopeudella (ks. kohta Annostus ja antotapa).
Jos asteen 1 tai 2 infuusioon liittyvä reaktio uusiutuu, avelumabin antamista potilaalle voidaan jatkaa, mikäli potilaan tilaa seurataan tarkasti, infuusionopeutta muokataan tilanteen mukaan ja potilas saa esilääkitystä parasetamolilla ja antihistamiinilla (ks. kohta Annostus ja antotapa).
Kliinisissä tutkimuksissa 98,6 prosentilla (433/439) potilaista, joilla ilmeni infuusioon liittyviä reaktioita, ensimmäinen infuusioon liittyvä reaktio esiintyi ensimmäisten 4 infuusion aikana. Reaktioista 2,7 % (12/439) oli vaikeusasteeltaan ≥ 3. Lopuilla 1,4 prosentilla (6/439) potilaista infuusioon liittyvät reaktiot ilmenivät ensimmäisten 4 infuusion jälkeen ja olivat kaikki asteen 1 tai 2 reaktioita.
Immuunivälitteiset haittavaikutukset
Immuunivälitteisiä haittavaikutuksia, jotka saattavat olla vakavia tai kuolemaan johtavia, voi esiintyä yhdessä tai useassa elinjärjestelmässä tai kudoksessa samanaikaisesti avelumabihoitoa saavilla potilailla. Aloita lääketieteellinen hallinta viipymättä, mukaan lukien erikoisalakonsultaatio tarpeen mukaan (ks. kohta Annostus ja antotapa) tilaan liittyvän morbiditeetin ja kuolemanriskin hoitamiseksi.
Vaikka immuunivälitteiset haittavaikutukset ilmenevät yleensä hoidon aikana, oireet voivat ilmaantua myös hoidon lopettamisen jälkeen.
Immuunivälitteisten haittavaikutusten varhainen tunnistaminen ja hoitaminen ovat erittäin tärkeitä avelumabin turvallisen käytön varmistamiseksi. Tarkkaile potilaita immuunivälitteisten haittavaikutusten oireiden ja löydösten varalta.
Avelumabin aiheuttamat immuunivälitteiset haittavaikutukset ovat olleet tavallisesti palautuvia ja niitä hoidettiin keskeyttämällä avelumabin anto toistaiseksi tai pysyvästi ja käyttämällä kortikosteroideja ja/tai muita tukihoitoja.
Epäillyt immuunivälitteiset haittavaikutukset tulee tutkia asianmukaisesti etiologian varmistamiseksi ja muiden syiden poissulkemiseksi. Haittavaikutuksen vaikeusasteen perusteella tulisi avelumabi-hoito toistaiseksi keskeyttää ja potilaalle tulisi antaa kortikosteroideja. Jos haittavaikutuksen hoitoon käytetään kortikosteroideja, kortikosteroidien asteittainen vähentäminen aloitetaan haitan lievittymisen jälkeen ja sitä jatketaan vähintään yhden kuukauden ajan.
Potilailla, joiden immuunivälitteisiä haittavaikutuksia ei saada hallintaan käyttämällä kortikosteroideja, voidaan harkita muiden systeemisten immunosuppressiivisten lääkeaineiden antamista.
Havainnointitutkimuksista saadut tiedot viittaavat siihen, että potilailla, joilla on ennestään jokin autoimmuunisairaus (AID), immuunivälitteisten haittavaikutusten riski saattaa olla suurentunut immuunivasteen tarkistuspisteen estäjähoidon jälkeen verrattuna potilaisiin, joilla ei ole ennestään mitään autoimmuunisairautta. Lisäksi taustalla olevan autoimmuunisairauden pahenemisvaiheet olivat yleisiä, mutta suurin osa niistä oli lieviä ja hoidettavissa.
Immuunivälitteinen pneumoniitti
Avelumabihoitoa saavilla potilailla on raportoitu immuunivälitteistä pneumoniittia. Yksi kuolemaan johtanut tapaus on raportoitu avelumabia saaneilla potilailla (ks. kohta Haittavaikutukset).
Potilaita on tarkkailtava immuunivälitteiseen pneumoniittiin viittaavien oireiden ja löydösten varalta ja muut syyt pneumoniitille on suljettava pois. Pneumoniittiepäily tulee varmistaa kuvantamistutkimuksella.
Kortikosteroidihoitoa tulee antaa, jos potilaalla todetaan vähintään vaikeusasteen 2 pneumoniitti (prednisonia tai vastaavaa 1‑2 mg/kg/vrk aloitusannoksena, ja annoksen purku tapahtuu asteittain).
Avelumabihoito tulisi keskeyttää toistaiseksi, jos immuunivälitteisen pneumoniitin vaikeusaste on 2, ja avelumabihoito tulisi lopettaa pysyvästi, jos immuunivälitteisen pneumoniitin vaikeusaste on 3 tai 4 tai jos vaikeusaste 2 uusiutuu (ks. kohta Annostus ja antotapa).
Immuunivälitteinen hepatiitti
Avelumabihoitoa saavilla potilailla on raportoitu immuunivälitteistä hepatiittia. Kaksi kuolemaan johtanutta tapausta on raportoitu avelumabia saaneilla potilailla (ks. kohta Haittavaikutukset).
Potilaita tulee tarkkailla maksassa tapahtuvien muutosten ja immuunivälitteiseen hepatiittiin viittaavien oireiden ja löydösten varalta ja muut syyt hepatiitille on suljettava pois.
Kortikosteroidihoitoa tulee antaa, jos potilaalla todetaan vähintään vaikeusasteen 2 hepatiitti (prednisonia tai vastaavaa 1‑2 mg/kg/vrk aloitusannoksena, ja annoksen purku tapahtuu asteittain).
Avelumabihoito tulisi keskeyttää toistaiseksi, jos immuunivälitteisen hepatiitin vaikeusaste on 2, ja avelumabihoito tulisi lopettaa pysyvästi, jos immuunivälitteisen hepatiitin vaikeusaste on 3 tai 4 (ks. kohta Annostus ja antotapa).
Immuunivälitteinen koliitti
Avelumabihoitoa saavilla potilailla on raportoitu immuunivälitteistä koliittia (ks. kohta Haittavaikutukset).
Potilaita tulee tarkkailla immuunivälitteiseen koliittiin viittaavien oireiden ja löydösten varalta ja muut syyt on suljettava pois. Kortikosteroidihoitoa tulee antaa, jos potilaalla todetaan vähintään vaikeusasteen 2 koliitti (prednisonia tai vastaavaa 1‑2 mg/kg/vrk aloitusannoksena, ja annoksen purku tapahtuu asteittain).
Avelumabihoito tulisi keskeyttää toistaiseksi, jos immuunivälitteisen koliitin vaikeusaste on 2 tai 3, ja avelumabihoito tulisi lopettaaa pysyvästi, jos immuunivälitteisen koliitin vaikeusaste on 4 tai jos vaikeusaste 3 uusiutuu (ks. kohta Annostus ja antotapa).
Immuunivälitteinen pankreatiitti
Avelumabihoitoa saavilla potilailla on raportoitu immuunivälitteistä pankreatiittia. Kaksi kuolemaan johtanutta tapausta on raportoitu avelumabia yhdessä aksitinibin kanssa saaneilla potilailla (ks. kohta Haittavaikutukset).
Potilaita tulee tarkkailla immuunivälitteisen pankreatiitin oireiden ja löydösten varalta. Jotta asianmukaiset toimenpiteet voidaan aloittaa varhaisessa vaiheessa, pyydä oireileville potilaille gastroenterologinen konsultaatio ja laboratoriokokeet (mukaan lukien kuvantaminen). Kortikosteroidihoitoa tulee antaa, jos potilaalla todetaan immuunivälitteinen pankreatiitti (prednisonia tai vastaavaa 1‑2 mg/kg/vrk aloitusannoksena, ja annoksen purku tapahtuu asteittain).
Jos epäillään immuunivälitteistä pankreatiittia, avelumabihoito on toistaiseksi keskeytettävä. Jos varmistuu, että potilaalla on immuunivälitteinen pankreatiitti, avelumabihoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Immuunivälitteinen myokardiitti
Avelumabihoitoa saavilla potilailla on raportoitu immuunivälitteistä myokardiittia. Kaksi kuolemaan johtanutta tapausta on raportoitu avelumabia yhdessä aksitinibin kanssa saaneilla potilailla (ks. kohta Haittavaikutukset).
Potilaita tulee tarkkailla immuunivälitteisen myokardiitin oireiden ja löydösten varalta. Jotta asianmukaiset toimenpiteet voidaan aloittaa varhaisessa vaiheessa, pyydä oireileville potilaille kardiologinen konsultaatio ja laboratoriokokeet. Kortikosteroidihoitoa tulee antaa, jos potilaalla todetaan immuunivälitteinen myokardiitti (prednisonia tai vastaavaa 1–2 mg/kg/vrk aloitusannoksena, ja annoksen purku tapahtuu asteittain). Jos kortikosteroidihoitoa saavan potilaan tila ei kohennu 24 tunnin kuluessa, on harkittava lisäksi immunosuppressiivista valmistetta (esim. mykofenolaatti, infliksimabi, antitymosyyttiglobuliini).
Jos epäillään immuunivälitteistä myokardiittia, avelumabihoito on toistaiseksi keskeytettävä. Jos varmistuu, että potilaalla on immuunivälitteinen myokardiitti, avelumabihoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Immuunivälitteiset umpierityssairaudet
Avelumabihoitoa saavilla potilailla on raportoitu immuunivälitteisiä kilpirauhasen toimintahäiriöitä, immuunivälitteistä lisämunuaisen vajaatoimintaa ja tyypin 1 diabetes (ks. kohta Haittavaikutukset). Potilaita tulee tarkkailla umpierityssairauksiin viittaavien oireiden ja löydösten varalta. Avelumabihoito tulisi keskeyttää toistaiseksi, jos umpierityssairauksien vaikeusaste on 3 tai 4 (ks. kohta Annostus ja antotapa).
Kilpirauhasen toimintahäiriöt (hypotyreoosi/hypertyreoosi)
Kilpirauhasen toimintahäiriöitä voi esiintyä hoidon missä tahansa vaiheessa (ks. kohta Haittavaikutukset).
Potilaita tulee tarkkailla kilpirauhasen toiminnan muutosten (hoidon alkaessa, säännöllisesti hoitojakson aikana ja lisäksi tarvittaessa kliinisen tarpeen mukaan) ja kilpirauhasen toimintahäiriöiden kliinisten oireiden ja löydösten varalta. Hypotyreoosiin tulee antaa hormonikorvaushoitoa ja hypertyreoosiin tarvittaessa tyroksiinin tuotantoa jarruttavaa lääkettä.
Avelumabihoito tulisi keskeyttää toistaiseksi, jos kilpirauhasen toimintahäiriöiden vaikeusaste on 3 tai 4 (ks. kohta Annostus ja antotapa).
Lisämunuaisen vajaatoiminta
Potilaita tulee tarkkailla lisämunuaisen vajaatoimintaan viittaavien oireiden ja löydösten varalta hoitojakson aikana sekä sen jälkeen. Lisämunuaisen vajaatoiminnan vaikeusasteen ollessa ≥ 3 potilaalle tulee antaa kortikosteroideja (1–2 mg/kg/vrk prednisonia suonensisäisesti tai vastaava määrä suun kautta) ja annosta on pienennettävä asteittain annokseen ≤ 10 mg/vrk saakka.
Avelumabihoito tulisi keskeyttää toistaiseksi, jos sekundaarisen lisämunuaisten vajaatoiminnan vaikeusaste on 3 tai 4 (ks. kohta Annostus ja antotapa).
Tyypin 1 diabetes
Avelumabi voi aiheuttaa tyypin 1 diabetesta ja myös diabeettisen ketoasidoosin (ks. kohta Haittavaikutukset).
Potilaita tulee tarkkailla hyperglykemian ja muiden diabetekseen viittaavien oireiden ja löydösten varalta. Tyypin 1 diabetekseen aloitetaan insuliinihoito. Avelumabihoito tulisi keskeyttää toistaiseksi ja potilaille tulisi antaa verensokeria alentavaa lääkettä, jos hyperglykemian vaikeusaste on ≥ 3. Avelumabihoitoa jatketaan, kun aineenvaihdunta on saatu tasapainoon insuliinihoidolla.
Immuunivälitteinen munuaistulehdus ja munuaisten vajaatoiminta
Avelumabi voi aiheuttaa immuunivälitteistä munuaistulehdusta (ks. kohta Haittavaikutukset).
Potilasta tulee tarkkailla seerumin kreatiniiniarvon kohoamisen varalta ennen hoidon aloittamista ja säännöllisesti hoitojakson aikana. Jos munuaistulehduksen vaikeusaste on ≥ 2, potilaalle annetaan kortikosteroideja (prednisonia tai vastaavaa 1–2 mg/kg/vrk aloitusannoksena, ja annoksen purku tapahtuu asteittain). Jos munuaistulehduksen vaikeusaste on 2 tai 3, avelumabihoito tulisi keskeyttää, kunnes munuaishaitta on lievittynyt vaikeusasteeseen ≤ 1. Jos munuaistulehduksen vaikeusaste on 4, avelumabihoito tulisi lopettaa pysyvästi.
Muut immuunivälitteiset haittavaikutukset
Kliinisissä tutkimuksissa tai avelumabin markkinoille saattamisen jälkeisessä käytössä on raportoitu muita kliinisesti merkittäviä immuunivälitteisiä haittavaikutuksia kuten myosiitti, aivolisäkkeen etulohkon vajaatoiminta, uveiitti, myasthenia gravis, myasteeninen oireyhtymä, myokardiitti-myosiitti-myasthenia gravis -limittymisoireyhtymä, ei infektiivinen virtsarakkotulehdus, sarkoidoosi,, Guillain-Barrén oireyhtymä, sklerosoiva kolangiitti, niveltulehdus, polymyalgia rheumatica, Sjögrenin oireyhtymä ja mahatulehdus (ks. kohta Haittavaikutukset).
Epäillyt immuunivälitteiset haittavaikutukset tulee tutkia asianmukaisesti etiologian varmistamiseksi tai muiden syiden poissulkemiseksi. Haittavaikutuksen vaikeusasteesta riippuen avelumabihoito tulisi toistaiseksi keskeyttää ja potilaalle tulee antaaa kortikosteroideja. Avelumabihoitoa voidaan jatkaa, kun immuunivälitteiset haittavaikutukset ovat lieventyneet vaikeusasteeseen ≤ 1 ja kortikosteroidiannos on asteittain purettu. Avelumabihoito tulisi lopettaa pysyvästi, jos vaikeusasteen 3 immuunivälitteinen haittavaikutus uusiutuu tai jos potilaalla ilmenee vaikeusasteen 4 immuunivälitteinen haittavaikutus (ks. kohta Annostus ja antotapa).
Avelumabihoidon yhteydessä on raportoitu myokardiitti-myosiitti-myasthenia gravis -limittymisoireyhtymätapauksia (esiintyy joko kahden tai kaikkien kolmen sairauden limittymisenä), joista osa on johtanut kuolemaan. Varhainen tunnistaminen ja aggressiivinen hallinta ovat erittäin tärkeitä tilaan liittyvän morbiditeetin ja kuolemanriskin hoidossa (ks. kohta Annostus ja antotapa).
Maksatoksisuus (yhdessä aksitinibin kanssa)
Maksatoksisuutta esiintyi potilailla, jotka saivat avelumabia yhdessä aksitinibin kanssa, odotettua enemmän vaikeusasteen 3 tai 4 ALAT- ja ASAT-arvojen nousun kanssa verrattuna avelumabia monoterapiana saaneisiin potilaisiin (ks. kohta Haittavaikutukset).
Potilaiden maksan toiminnan muutoksia ja oireita on tarkkailtava useammin kuin avelumabimonoterapiaa käytettäessä.
Avelumabihoito tulisi keskeyttää toistaiseksi, jos maksatoksisuuden vaikeusaste on 2, toksisuuden häviämiseen saakka ja avelumabihoito tulisi lopettaa pysyvästi, jos maksatoksisuuden vaikeusaste on 3 tai 4. Kortikosteroidihoitoa tulee harkita, jos potilaalla todetaan vähintään vaikeusasteen 2 tapahtumia (ks. kohta Annostus ja antotapa).
Kliinisistä tutkimuksista pois suljetut potilaat
Kliinisistä tutkimuksista suljettiin pois potilaat, joilla oli seuraavia sairauksia: aktiivisia etäpesäkkeitä keskushermostossa, aktiivinen tai aiemmin sairastettu autoimmuunisairaus, syöpä viimeisten viiden vuoden aikana, elinsiirto, immunosuppressiivista hoitoa vaativa sairaus tai aktiivinen HIV-, hepatiitti B- tai hepatiitti C -infektio.
Avelumabia on käytettävä näille potilasryhmille varoen ja vasta yksilöllisten hyötyjen/riskien huolellisen arvioinnin jälkeen.
Natriumpitoisuus
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”. Bavencio on laimennettava joko natriumkloridi 9 mg/ml (0,9 %)- tai natriumkloridi 4,5 mg/ml (0,45 %) -liuoksella. Tämä on huomioitava hoidettaessa potilaita, joilla on ruokavalion natriumrajoitus (ks. kohta Käyttö- ja käsittelyohjeet).
Polysorbaattipitoisuus
Tämä lääkevalmiste sisältää 5 mg polysorbaattia 20 per injektiopullo. Polysorbaatit saattavat aiheuttaa allergisia reaktioita. Tämä on huomioitava hoidettaessa potilaita Bavencio-valmisteella.
Potilaskortti
Lääkkeen määrääjän on keskusteltava Bavencio-hoidon riskeistä potilaan kanssa. Potilaalle annetaan potilaskortti, jossa kehotetaan pitämään kortti aina mukana.
Yhteisvaikutukset
Avelumabilla ei ole tehty yhteisvaikutustutkimuksia.
Koska avelumabi metaboloituu pääasiallisesti katabolian kautta, farmakokineettisiä yhteisvaikutuksia muiden lääkeaineiden kanssa ei ole odotettavissa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi/Ehkäisy
Naisia, jotka voivat tulla raskaaksi, on neuvottava välttämään raskaaksi tulemista avelumabihoidon aikana käyttämällä tehokasta ehkäisyä hoidon aikana sekä vähintään yhden kuukauden ajan viimeisen avelumabiannoksen jälkeen.
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja avelumabin käytöstä raskaana olevilla naisille.
Avelumabilla ei ole tehty lisääntymistutkimuksia eläimillä. Tiineiden hiirten eläinmalleissa PD-L1:n signaalinvälityksen salpauksen on kuitenkin todettu häiritsevän äidin immunologista toleranssia sikiötä kohtaan ja lisäävän keskenmenoja (ks. kohta Prekliiniset tiedot turvallisuudesta). Nämä tulokset viittaavat siihen, että avelumabin vaikutusmekanismin vuoksi valmisteen käyttö raskauden aikana saattaa olla haitallista sikiölle, ja se voi lisätä keskenmenon tai sikiökuoleman riskiä.
Ihmisen IgG1-immunoglobuliinin tiedetään läpäisevän istukan. Näin ollen avelumabi saattaa kulkeutua äidistä kehittyvään sikiöön. Ei ole suositeltavaa käyttää avelumabia raskauden aikana, ellei raskaana olevan potilaan kliininen tila edellytä hoitoa avelumabilla.
Imetys
Ei tiedetä, erittyykö avelumabi ihmisen rintamaitoon. Vasta-aineiden tiedetään voivan erittyä ihmisen rintamaitoon, joten vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea.
Imeväiseen kohdistuvien mahdollisesti vakavien haittavaikutusten riskin vuoksi imettäviä naisia on kehotettava lopettamaan rintaruokinta avelumabihoidon ajaksi ja vähintään yhden kuukauden ajaksi viimeisen annoksen jälkeen.
Hedelmällisyys
Avelumabin vaikutusta miesten ja naisten hedelmällisyyteen ei tunneta.
Vaikka tutkimuksia avelumabin vaikutuksesta hedelmällisyyteen ei ole tehty, 1 kuukauden ja 3 kuukauden toistuvilla annoksilla tehdyissä toksisuustutkimuksissa ei havaittu merkittäviä naarasapinoiden lisääntymiselimiin kohdistuvia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Avelumabi ei vaikuta ajokykyyn ja koneidenkäyttökykyyn. Väsymystä on raportoitu avelumabin antamisen jälkeen (ks. kohta Haittavaikutukset). Potilaita on neuvottava varovaisuuteen ajaessaan tai käyttäessään koneita, kunnes he ovat varmoja, että avelumabilla ei ole heihin haitallista vaikutusta.
Haittavaikutukset
Tiivistelmä turvallisuustiedoista
Avelumabihoidon yhteydessä ilmenevät haittavaikutukset liittyvät immuunijärjestelmään. Useimmat näistä, mukaan lukien vakavat haittavaikutukset, korjaantuivat asianmukaisen hoidon aloittamisen tai avelumabihoidon lopettamisen jälkeen (ks. ”Valikoitujen haittavaikutusten kuvaus” jäljempänä).
Avelumabin yleisimmät haittavaikutukset olivat väsymys (30,0 %), pahoinvointi (23,6 %), ripuli (18,5 %), ummetus (18,1 %), heikentynyt ruokahalu (17,6 %) infuusioon liittyvät reaktiot (15,9 %), oksentelu (15,6 %) ja painon lasku (14,5 %).
Yleisimpiä vaikeusasteen ≥ 3 haittavaikutuksia olivat anemia (5,6 %), hypertensio (3,9 %), hyponatremia (3,6 %), hengenahdistus (3,5 %) ja vatsakipu (2,6 %). Vakavia haittavaikutuksia olivat immuunivälitteiset haittavaikutukset ja infuusioon liittyvät reaktiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Monoterapiana annetun avelumabin turvallisuutta on arvioitu 2 082 potilaalla, joilla oli kiinteitä kasvaimia, mukaan lukien metastasoitunut MCC tai paikallisesti edennyt tai metastasoitunut UC, ja jotka saivat kliinisissä tutkimuksissa avelumabia 10 mg/kg kahden viikon välein, ja avelumabin markkinoille saattamisen jälkeisestä käytöstä saatujen raporttien perusteella (ks. taulukko 2).
Haittavaikutukset on esitetty elinjärjestelmän ja yleisyyden mukaan. Yleisyysluokat on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Kunkin yleisyysluokan haittavaikutukset on esitetty alenevassa järjestyksessä vakavimmasta alkaen.
Taulukko 2: Avelumabia monoterapiana saaneilla potilailla todetut haittavaikutukset
Esiintyvyys | Haittavaikutukset |
|---|---|
Veri ja imukudos | |
Hyvin yleinen | Anemia |
Yleinen | Lymfopenia, trombosytopenia |
Melko harvinainen | Eosinofilia§ |
Immuunijärjestelmä | |
| Hyvin yleinen | Infuusioon liittyvä reaktio# |
| Melko harvinainen | Yliherkkyys, lääkeyliherkkyys |
| Harvinainen | Anafylaktinen reaktio, tyypin I yliherkkyys, sarkoidoosi* |
Umpieritys | |
Yleinen | Hypotyreoosi*, hypertyreoosi* |
Melko harvinainen | Lisämunuaisten vajaatoiminta*, autoimmuunityreoidiitti*, tyreoidiitti*, autoimmuunihypotyreoosi* |
Harvinainen | Äkillinen lisämunuaiskuoren vajaatoiminta*, aivolisäkkeen etulohkon vajaatoiminta* |
Aineenvaihdunta ja ravitsemus | |
Hyvin yleinen | Heikentynyt ruokahalu |
Yleinen | Hyponatremia |
Melko harvinainen | Hyperglykemia* |
Harvinainen | Diabetes*, tyypin 1 diabetes* |
Hermosto | |
| Yleinen | Päänsärky, huimaus, perifeerinen neuropatia |
| Melko harvinainen | Myasthenia gravis*,†,ϕ, myasteeninen oireyhtymä*,†, ϕ |
| Harvinainen | Guillain-Barrén oireyhtymä*, Miller-Fisherin oireyhtymä* |
| Silmät | |
Harvinainen | Uveiitti* |
Sydän | |
Harvinainen | Sydänlihastulehdus*,ϕ |
Verisuonisto | |
Yleinen | Korkea verenpaine |
Melko harvinainen | Matala verenpaine, punastelu |
Hengityselimet, rintakehä ja välikarsina | |
Hyvin yleinen | Yskä, hengenahdistus |
Yleinen | Pneumoniitti* |
Harvinainen | Interstitiaalinen keuhkosairaus* |
Ruoansulatuselimistö | |
Hyvin yleinen | Pahoinvointi, ripuli, ummetus, oksentelu, vatsakipu |
Yleinen | Suun kuivuminen |
Melko harvinainen | Suolitukos, koliitti* |
| Harvinainen | Pankreatiitti*, autoimmuunikoliitti*, enterokoliitti*, autoimmuunipankreatiitti*, enteriitti*, proktiitti* |
Tuntematon | Mahatulehdus* |
Maksa ja sappi | |
Melko harvinainen | Autoimmuunihepatiitti* |
Harvinainen | Maksan äkillinen vajaatoiminta*, maksan vajaatoiminta*, hepatiitti*, maksatoksisuus* |
| Tuntematon | Sklerosoiva kolangiitti* |
Iho ja ihonalainen kudos | |
Yleinen | Kutina*, ihottuma*, ihon kuivuminen, makulopapulaarinen ihottuma* |
| Melko harvinainen | Ekseema, dermatiitti, kutiava ihottuma*, psoriaasi*, punoitus*, punoittava ihottuma*, yleistynyt ihottuma*, makulaarinen ihottuma*, näppyläinen ihottuma* |
| Harvinainen | Monimuotoinen punavihoittuma*, purppura*, vitiligo*, yleistynyt kutina*, hilseilevä ihottuma*, pemfigoidi*, psoriaasin kaltainen ihottuma*, lääkeihottuma*, lichen planus* |
Luusto, lihakset ja sidekudos | |
Hyvin yleinen | Selkäkipu, nivelsärky |
Yleinen | Lihaskipu |
Melko harvinainen | Myosiitti*, ϕ, nivelreuma* |
Harvinainen | Niveltulehdus*, moniniveltulehdus*, oligoartriitti*, Sjögrenin oireyhtymä* |
| Tuntematon | Polymyalgia rheumatica |
Munuaiset ja virtsatiet | |
| Melko harvinainen | Munuaisten vajaatoiminta*, munuaistulehdus* |
| Harvinainen | Tubulointerstitiaalinen munuaistulehdus*, ei-infektiivinen virtsarakkotulehdus* |
Yleisoireet ja antopaikassa todettavat haitat | |
Hyvin yleinen | Uupumus, kuume, perifeerinen turvotus |
| Yleinen | Astenia, vilunväristykset, flunssantapaiset oireet |
| Harvinainen | Systeeminen tulehdusreaktio* |
Tutkimukset | |
Hyvin yleinen | Painon lasku |
Yleinen | Kohonnut veren kreatiniiniarvo, kohonnut veren alkalinen fosfataasiarvo, kohonnut lipaasiarvo, kohonnut gammaglutamyylitransferaasiarvo, kohonnut amylaasiarvo |
Melko harvinainen | Kohonnut alaniiniaminotransferaasiarvo (ALAT)*, kohonnut aspartaattiaminotransferaasiarvo (ASAT)*, kohonnut veren kreatiniinifosfokinaasiarvo* |
Harvinainen | Kohonneet transaminaasiarvot*, alentunutvapaan tyroksiinin pitoisuus*, kohonnut veren tyreotropiiniarvo* |
* Lääketieteelliseen tutkimukseen perustuvat immuunivälitteiset haittavaikutukset
§ Reaktiota todettiin vain EMR100070-003 -tutkimuksessa (osa B) poolatun analyysin tiedonkeruun päättymiseen mennessä, joten sen yleisyydestä on annettu arvio
# Mukaan lukien sytokiinioireyhtymä, jonka esiintyvyys on melko harvinainen
† Haittavaikutuksia esiintyi avelumabimonoterapialle altistetuilla arviolta 4 000 potilaalla poolatun analyysin ulkopuolella
Munuaissolukarsinooma
Tiivistelmä turvallisuustiedoista
Avelumabin turvallisuutta yhdessä aksitinibin kanssa annettaessa on arvioitu kahdessa kliinisessä tutkimuksessa 489 potilaalla, joilla oli edennyt RCC ja jotka saivat avelumabia 10 mg/kg kahden viikon välein ja aksitinibia 5 mg suun kautta kahdesti vuorokaudessa.
Tässä potilasjoukossa yleisimmät haittavaikutukset olivat ripuli (62,8 %), korkea verenpaine (49,3 %), uupumus (42,9 %), pahoinvointi (33,5 %), dysfonia (32,7 %), heikentynyt ruokahalu (26,0 %), hypotyreoosi (25,2 %), yskä (23,7 %), päänsärky (21,3 %), hengenahdistus (20,9 %) ja nivelsärky (20,9 %).
Haittavaikutustaulukko
Taulukossa 3 esitetään haittavaikutukset, jotka on raportoitu 489 potilaalla, joilla oli edennyt RCC ja jotka saivat kahdessa kliinisessä tutkimuksessa avelumabia yhdessä aksitinibin kanssa, tai avelumabin markkinoille saattamisen jälkeisestä käytöstä saatujen raporttien perusteella.
Haittavaikutukset on esitetty elinjärjestelmän ja yleisyyden mukaan. Yleisyysluokat on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Kunkin yleisyysluokan haittavaikutukset on esitetty alenevassa järjestyksessä vakavimmasta alkaen.
ϕ Voivat esiintyä samanaikaisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Taulukko 3: Avelumabia yhdessä aksitinibin kanssa saaneilla potilailla todetut haittavaikutukset
| Esiintyvyys | Haittavaikutukset |
|---|---|
| Infektiot | |
| Melko harvinainen | Pustulaarinen ihottuma |
| Veri ja imukudos | |
| Yleinen | Anemia, trombosytopenia |
| Melko harvinainen | Lymfopenia, eosinofilia |
| Immuunijärjestelmä | |
| Hyvin yleinen | Infuusioon likittyvä reaktio# |
| Yleinen | Yliherkkyys |
| Tuntematon | Sarkoidoosi* |
| Umpieritys | |
| Hyvin yleinen | Hypotyreoosi |
| Yleinen | Hypertyreoosi, lisämunuaisen vajaatoiminta, tyreoidiitti |
| Melko harvinainen | Autoimmuunityreoidiitti, hypofysiitti |
| Aineenvaihdunta ja ravitsemus | |
| Hyvin yleinen | Heikentynyt ruokahalu |
| Yleinen | Hyperglykemia |
| Melko harvinainen | Diabetes, tyypin 1 diabetes |
| Hermosto | |
| Hyvin yleinen | Päänsärky, heitehuimaus |
| Yleinen | Perifeerinen neuropatia |
| Melko harvinainen | Myasthenia gravis, myasteeninen oireyhtymä |
| Sydän | |
| Melko harvinainen | Sydänlihastulehdus |
| Verisuonisto | |
| Hyvin yleinen | Korkea verenpaine |
| Yleinen | Matala verenpaine, punastelu |
| Hengityselimet, rintakehä ja välikarsina | |
| Hyvin yleinen | Dysfonia, yskä, hengenahdistus |
| Yleinen | Pneumoniitti |
| Ruoansulatuselimistö | |
| Hyvin yleinen | Ripuli, pahoinvointi, ummetus, oksentelu, vatsakipu |
| Yleinen | Suun kuivuminen, koliitti |
| Melko harvinainen | Autoimmuunikoliitti, autoimmuunipankreatiitti, enterokoliitti, suolitukos, nekrotisoiva pankreatiitti |
| Tuntematon | Mahatulehdus* |
| Maksa ja sappi | |
| Yleinen | Maksan poikkeava toiminta |
| Melko harvinainen | Hepatiitti, maksatoksisuus, immuunivälitteinen hepatiitti, maksasairaus |
| Tuntematon | Sklerosoiva kolangiitti* |
| Iho ja ihonalainen kudos | |
| Hyvin yleinen | Ihottuma, kutina |
| Yleinen | Kutiava ihottuma, makulopapulaarinen ihottuma, yleistynyt kutina, aknea muistuttava dermatiitti, punoitus, makulaarinen ihottuma, papulaarinen ihottuma, punoittava ihottuma, dermatiitti, ekseema, yleistynyt ihottuma |
| Melko harvinainen | Lääkeaineihottuma, monimuotoinen punavihoittuma, psoriaasi |
| Luusto, lihakset ja sidekudos | |
| Hyvin yleinen | Nivelsärky, selkäkipu, lihaskipu |
| Melko harvinainen | Niveltulehdus* |
| Tuntematon | Polymyalgia rheumatica* ja Sjögrenin oireyhtymä* |
| Munuaiset ja virtsatiet | |
| Yleinen | Äkillinen munuaisvaurio |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Hyvin yleinen | Uupumus, vilunväristykset, astenia, kuume |
| Yleinen | Perifeerinen turvotus, flunssantapaiset oireet |
| Tutkimukset | |
| Hyvin yleinen | Painon lasku, kohonnut alaniiniaminotransferaasiarvo (ALAT), kohonnut aspartaattiaminotransferaasiarvo (ASAT) |
| Yleinen | Kohonnut veren kreatiniiniarvo, kohonnut amylaasiarvo, kohonnut lipaasiarvo, kohonnut gamma‑glutamyylitransferaasiarvo, kohonnut veren alkalinen fosfataasi, kohonnut veren kreatiniinifosfokinaasi, alentunut veren tyreotropiiniarvo, kohonneet transaminaasiarvot |
| Melko harvinainen | Kohonneet maksan toimintakoearvot |
* Lääketieteelliseen tutkimukseen perustuvat immuunivälitteiset haittavaikutukset
# Mukaan lukien sytokiinioireyhtymä, jonka esiintyvyys on melko harvinainen
Valikoitujen haittavaikutusten kuvaus
Tiedot immuunivälitteisistä haittavaikutuksista perustuvat monoterapiana annetun avelumabin osalta tietoihin 2 082 potilaasta, mukaan lukien vaiheen I EMR100070‑001-tutkimuksen 1 650 potilasta, joilla oli kiinteä kasvain, EMR100070‑003-tutkimuksen 88 potilasta, joilla oli MCC, sekä B9991001-tutkimuksen 344 potilasta, joilla oli UC, ja yhdessä aksitinibin kanssa annetun avelumabin osalta tietoihin B9991002- ja B9991003-tutkimusten 489 RCC-potilaasta (ks. kohta Farmakodynamiikka).
Näiden haittavaikutusten hoito-ohjeet on annettu kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Immuunivälitteinen pneumoniitti
Avelumabia monoterapiana saaneista potilaista 1,3 %:lle (28/2 082) kehittyi immuunivälitteinen pneumoniitti. Näistä 1 potilaalla (alle 0,1 %) immuunivälitteinen pneumoniitti johti kuolemaan, 1 potilaalla (alle 0,1 %) immuunivälitteisen pneumoniitin vaikeusaste oli 4 ja 6 potilaalla (0,3 %) vaikeusaste oli 3.
Mediaaniaika immuunivälitteisen pneumoniitin ilmaantumiseen oli 2,5 kuukautta (vaihteluväli: 3 vuorokautta– 13,8 kuukautta). Keston mediaani oli 8,1 viikkoa (vaihteluväli: 4 vuorokautta– yli 4,9 kuukautta).
Immuunivälitteinen pneumoniitti johti avelumabihoidon lopettamiseen 0,4 %:lla (9/2 082) potilaista. Kaikki 28 potilasta, jotka sairastivat immuunivälitteistä pneumoniittia, saivat kortikosteroideja ja 21/28 potilasta (75 %) sai korkea-annoksista kortikosteroidia hoitojakson mediaanin ollessa 9 vuorokautta (vaihteluväli: 1 vuorokausi – 2,3 kuukautta). Immuunivälitteinen pneumoniitti parani 18/28 potilaalla (64,3 %) tiedonkeruun päättymiseen mennessä.
Avelumabia yhdessä aksitinibin kanssa saaneista potilaista 0,6 %:lle (3/489) kehittyi immuunivälitteinen pneumoniitti. Näistä potilaista yhdelläkään ei ollut vaikeusasteen ≥ 3 immuunivälitteistä pneumoniittia.
Mediaaniaika immuunivälitteisen pneumoniitin ilmaantumiseen oli 3,7 kuukautta (vaihteluväli: 2,7‑8,6 kuukautta). Keston mediaani oli 2,6 kuukautta (vaihteluväli: 3,3 viikkoa – yli 7,9 kuukautta).
Immuunivälitteinen pneumoniitti ei johtanut avelumabihoidon lopettamiseen yhdelläkään potilaalla. Kaikki kolme immuunivälitteistä pneumoniittia sairastanutta potilasta sai suuriannoksista kortikosteroidia hoitojakson mediaanin ollessa 3,3 kuukautta (vaihteluväli: 3 viikkoa – 22,3 kuukautta). Immuunivälitteinen pneumoniitti parani 2/3 potilaalla (66,7 %) tiedonkeruun päättymiseen mennessä.
Immuunivälitteinen hepatiitti
Avelumabia monoterapiana saaneista potilaista 1,0 %:lle (21/2 082) kehittyi immuunivälitteinen hepatiitti. Näistä potilaista 2 potilaalla (0,1 %) immuunivälitteinen hepatiitti johti kuolemaan ja 16 potilaalla (0,8 %) immuunivälitteisen hepatiitin vaikeusaste oli 3.
Mediaaniaika immuunivälitteisen hepatiitin ilmaantumiseen oli 3,3 kuukautta (vaihteluväli: 9 vuorokautta – 14,8 kuukautta). Keston mediaani oli 2,5 kuukautta (vaihteluväli: 1 vuorokausi – yli 7,4 kuukautta).
Immuunivälitteinen hepatiitti johti avelumabihoidon lopettamiseen 0,6 %:lla (13/2 082) potilaista. Kaikki 21 potilasta, jotka sairastivat immuunivälitteistä hepatiittia, saivat kortikosteroideja ja 20/21 potilasta (95,2 %) sai korkea-annoksista kortikosteroidia hoitojakson mediaanin ollessa 17 vuorokautta (vaihteluväli: 1 vuorokausi – 4,1 kuukautta). Immuunivälitteinen hepatiitti parani 12/21 potilaalla (57,1 %) tiedonkeruun päättymiseen mennessä.
Avelumabia yhdessä aksitinibin kanssa saaneista potilaista 6,3 %:lle (31/489) kehittyi immuunivälitteinen hepatiitti. Näistä potilaista 18 potilaalla (3,7 %) immuunivälitteisen hepatiitin vaikeusaste oli 3 ja 3 potilaalla (0,6 %) immuunivälitteisen hepatiitin vaikeusaste oli 4.
Immuunivälitteinen hepatiitti johti avelumabihoidon lopettamiseen 4,7 %:lla (23/489) potilaista. Kaikki 31 potilasta, joilla oli immuunivälitteinen hepatiitti, saivat hoitoa hepatiittiin. Näistä 30 (96,8 %) potilasta sai kortikosteroidihoitoa ja yhtä potilasta hoidettiin ei-steroidaalisella immuunisalpaajalla. Kaksikymmentäkahdeksan potilasta (90,3 %) 31:stä sai suuriannoksisia kortikosteroideja hoitojakson mediaanin ollessa 2,4 viikkoa (vaihteluväli: 1 vuorokausi ‑ 10,2 kuukautta). Immuunivälitteinen hepatiitti parani 27/31 potilaalla (87,1 %) tiedonkeruun päättymiseen mennessä.
Immuunivälitteinen koliitti
Avelumabia monoterapiana saaneista potilaista 1,5 %:lle (31/2 082) kehittyi immuunivälitteinen koliitti. Näistä 10 potilaalla (0,5 %) immuunivälitteisen koliitin vaikeusaste oli 3.
Mediaaniaika immuunivälitteisen koliitin ilmaantumiseen oli 2,0 kuukautta (vaihteluväli: 2 vuorokautta – 11,5 kuukautta). Keston mediaani oli 5,9 viikkoa (vaihteluväli: 1 vuorokausi – yli 14 kuukautta).
Immuunivälitteinen koliitti johti avelumabihoidon lopettamiseen 0,5 %:lla (11/2 082) potilaista. Kaikki 31 potilasta, jotka sairastivat immuunivälitteistä koliittia, saivat kortikosteroideja ja 19/31 potilasta (61,3 %) sai korkea-annoksista kortikosteroidia hoitojakson mediaanin ollessa 19 vuorokautta (vaihteluväli: 1 vuorokausi – 2,3 kuukautta). Immuunivälitteinen koliitti parani 22/31 potilaalla (71 %) tiedonkeruun päättymiseen mennessä.
Avelumabia yhdessä aksitinibin kanssa saaneista potilaista 2,7 %:lle (13/489) kehittyi immuunivälitteinen koliitti. Näistä 9 potilaalla (1,8 %) immuunivälitteisen koliitin vaikeusaste oli 3.
Mediaaniaika immuunivälitteisen koliitin ilmaantumiseen oli 5,1 kuukautta (vaihteluväli: 2,3 viikkoa - 14 kuukautta). Keston mediaani oli 1,6 viikkoa (vaihteluväli: 1 vuorokausi ‑ yli 9 kuukautta).
Immuunivälitteinen koliitti johti avelumabihoidon lopettamiseen 0,4 %:lla (2/489) potilaista. Kaikki 13 potilasta, jotka sairastivat immuunivälitteistä koliittia, saivat kortikosteroideja ja 12/13 potilasta (92,3 %) sai suuriannoksista kortikosteroidia hoitojakson mediaanin ollessa 2,3 viikkoa (vaihteluväli: 5 vuorokautta - 4,6 kuukautta). Immuunivälitteinen koliitti parani 10/13 potilaalla (76,9 %) tiedonkeruun päättymiseen mennessä.
Immuunivälitteinen pankreatiitti
Kliinisissä, useita tuumorityyppejä käsittäneissä tutkimuksissa avelumabia monoterapiana saaneista potilaista immuunivälitteistä pankreatiittia esiintyi alle 1 %:lla (1/4 000) potilaista, avelumabia yhdessä aksitinibin kanssa saaneilla potilailla sitä esiintyi 0,6 %:lla (3/489) potilaista, mukaan lukien 2 (0,4 %) potilasta, joilla sairaus johti kuolemaan.
Immuunivälitteinen myokardiitti
Kliinisissä, useita tuumorityyppejä käsittäneissä tutkimuksissa avelumabia monoterapiana saaneista potilaista, immuunivälitteistä myokardiittia esiintyi alle 1 %:lla (5/4 000) potilaista, ja avelumabia yhdessä aksitinibin kanssa saaneista potilaista sitä esiintyi 0,6 %:lla (3/489) potilaista, mukaan lukien 2 (0,4 %) potilasta, joilla sairaus johti kuolemaan.
Immuunivälitteiset umpierityssairaudet
Kilpirauhasen toimintahäiriöt
Avelumabia monoterapiana saaneista potilaista 6,7 %:lle (140/2 082) kehittyi kilpirauhasen immuunivälitteinen toimintahäiriö. Näistä 127 potilaalla (6,1 %) kyseessä oli hypotyreoosi, 23 potilaalla (1,1 %) kyseessä oli hypertyreoosi ja 7 potilaalla (0,3 %) tyreoidiitti. Näistä 4 potilaalla (0,2 %) kilpirauhasen immuunivälitteisen toimintahäiriön vaikeusaste oli 3.
Mediaaniaika kilpirauhasen immuunivälitteisen toimintahäiriön ilmaantumiseen oli 2,8 kuukautta (vaihteluväli: 2 viikkoa – 12,8 kuukautta). Keston mediaani ei ollut arvioitavissa (vaihteluväli: 3 vuorokautta – yli 27,6 kuukautta).
Kilpirauhasen immuunivälitteinen toimintahäiriö johti avelumabihoidon lopettamiseen 0,2 %:lla (4/2 082) potilaista. Kilpirauhasen immuunivälitteinen toimintahäiriö parani 14/140 potilaalla (10 %) tiedonkeruun päättymiseen mennessä.
Avelumabia yhdessä aksitinibin kanssa saaneista potilaista 24,7 %:lle (121/489) kehittyi kilpirauhasen immuunivälitteinen toimintahäiriö. Näistä 111 potilaalla (22,7 %) kyseessä oli hypotyreoosi, 17 potilaalla (3,5 %) kyseessä oli hypertyreoosi ja 7 potilaalla (1,4 %) tyreoidiitti. Näistä 2 potilaalla (0,4 %) kilpirauhasen immuunivälitteisen toimintahäiriön vaikeusaste oli 3.
Mediaaniaika kilpirauhasen immuunivälitteisen toimintahäiriön ilmaantumiseen oli 2,8 kuukautta (vaihteluväli: 3,6 viikkoa ‑ 19,3 kuukautta). Mediaanikesto ei ollut arvioitavissa (vaihteluväli: 8 vuorokautta ‑ yli 23,9 kuukautta).
Kilpirauhasen immuunivälitteinen toimintahäiriö johti avelumabihoidon lopettamiseen 0,2 %:lla (1/489) potilaista. Kilpirauhasen immuunivälitteinen toimintahäiriö parani 15/121 potilaalla (12,4 %) tiedonkeruun päättymiseen mennessä.
Lisämunuaisen vajaatoiminta
Avelumabia monoterapiana saaneista potilaista 0,5 %:lle (11/2 082) potilaista kehittyi lisämunuaisen immuunivälitteinen vajaatoiminta. Näistä 1 potilaalla (alle 0,1 %) lisämunuaisen immuunivälitteisen vajaatoiminnan vaikeusaste oli 3.
Mediaaniaika lisämunuaisen immuunivälitteisen vajaatoiminnan ilmaantumiseen oli 3,3 kuukautta (vaihteluväli: 1 vuorokausi – 7,6 kuukautta). Keston mediaani ei ollut arvioitavissa (vaihteluväli: 2 vuorokautta – yli 10,4 kuukautta).
Lisämunuaisten immuunivälitteinen vajaatoiminta johti avelumabihoidon lopettamiseen 0,1 %:lla potilaista (2/2 082). Kaikki 11 potilasta, jotka sairastivat lisämunuaisten immuunivälitteistä vajaatoimintaa, saivat kortikosteroideja ja 5/11 potilasta (45,5 %) sai korkea-annoksista systeemistä kortikosteroidia (≥ 40 mg prednisonia tai vastaavaa) hoitojakson mediaanin ollessa 2 vuorokautta (vaihteluväli: 1 vuorokausi – 24 vuorokautta). Lisämunuaisten immuunivälitteinen vajaatoiminta parani 3 (27,3 %) potilaalla tiedonkeruun päättymiseen mennessä.
Avelumabia yhdessä aksitinibin kanssa saaneista potilaista 1,8 %:lle (9/489) kehittyi immuunivälitteinen lisämunuaisen vajaatoiminta. Näistä 2 potilaalla (0,4 %) lisämunuaisen immuunivälitteisen vajaatoiminnan vaikeusaste oli 3.
Mediaaniaika lisämunuaisen immuunivälitteisen vajaatoiminnan ilmaantumiseen oli 5,5 kuukautta (vaihteluväli: 3,6 viikkoa ‑ 8,7 kuukautta). Mediaanikesto oli 2,8 kuukautta (vaihteluväli: 3 vuorokautta ‑ yli 15,5 kuukautta).
Lisämunuaisen immuunivälitteinen vajaatoiminta ei johtanut avelumabihoidon lopettamiseen yhdelläkään potilaalla. Kahdeksan (88,9 %) potilasta, jotka sairastivat lisämunuaisten immuunivälitteistä vajaatoimintaa, saivat kortikosteroideja ja 2/8 potilasta (25 %) sai suuriannoksista systeemistä kortikosteroidia (≥ 40 mg prednisonia tai vastaavaa) hoitojakson mediaanin ollessa 8 vuorokautta (vaihteluväli: 5 vuorokautta ‑ 11 vuorokautta). Lisämunuaisen vajaatoiminta parani 4/9 potilaalla (44,4 %) tiedonkeruun päättymiseen mennessä.
Tyypin 1 diabetes
Avelumabia monoterapiana saaneista potilaista tyypin I diabetes ilman vaihtoehtoista etiologiaa esiintyi 0,2 %:lla potilaista (5/2 082). Kaikilla 5 potilaalla tyypin I diabeteksen vaikeusaste oli 3.
Mediaaniaika tyypin I diabeteksen ilmaantumiseen oli 3,3 kuukautta (vaihteluväli: 1 vuorokausi – 18,7 kuukautta). Keston mediaani ei ollut arvioitavissa (vaihteluväli: 14 vuorokautta – yli 4,8 kuukautta).
Tyypin I diabetes johti avelumabihoidon lopettamiseen 0,1 %:lla (2/2 082) potilaista. Tyypin I diabetes parani 2 potilaalla (40 %) tiedonkeruun päättymiseen mennessä.
Avelumabia yhdessä aksitinibin kanssa saaneista potilaista 1,0 %:lla (5/489) ilmeni tyypin 1 diabetes mellitus ilman vaihtoehtoista etiologiaa. Näistä 1 potilaalla (0,2 %) tyypin I diabetes mellituksen vaikeusaste oli 3.
Mediaaniaika tyypin I diabetes mellituksen ilmaantumiseen oli 1,9 kuukautta (vaihteluväli: 1,1‑7,3 kuukautta).
Tyypin I diabetes johti avelumabihoidon lopettamiseen 0,2 %:lla (1/489) potilaista. Kaikkia 5 potilasta, joilla oli tyypin 1 diabetes mellitus, hoidettiin insuliinilla. Tyypin 1 diabetes mellitus ei parantunut yhdelläkään potilaalla tiedonkeruun päättymiseen mennessä.
Immuunivälitteinen munuaistulehdus ja munuaisten vajaatoiminta
Immuunivälitteistä munuaistulehdusta esiintyi avelumabia monoterapiana saaneista potilaista 0,3 %:lla (7/2 082). Näistä 1 potilaalla (alle 0,1 %) immuunivälitteisen munuaistulehduksen vaikeusaste oli 3.
Mediaaniaika immuunivälitteisen munuaistulehduksen ilmaantumiseen oli 2,4 kuukautta (vaihteluväli: 7,1 viikkoa – 21,9 kuukautta). Keston mediaani oli 6,1 kuukautta (vaihteluväli: 9 vuorokautta – 6,1 kuukautta).
Immuunivälitteinen munuaistulehdus johti avelumabihoidon lopettamiseen 0,2 %:lla (4/2 082) potilaista. Kaikki 7 immuunivälitteistä munuaistulehdusta sairastanutta potilasta saivat kortikosteroidihoitoa. Näistä 7:stä immuunivälitteistä munuaistulehdusta sairastaneesta potilaasta 6 (85,7 %) sai suuriannoksista kortikosteroidia hoitojakson mediaanin ollessa 2,5 viikkoa (vaihteluväli: 6 vuorokautta – 2,8 kuukautta). Immuunivälitteinen munuaistulehdus parani 4 potilaalla (57,1 %) tiedonkeruun päättymiseen mennessä.
Avelumabia yhdessä aksitinibin kanssa saaneista potilaista 0,4 %:lla (2/489) ilmeni immuunivälitteinen munuaistulehdus. Näistä 2 potilaalla (0,4 %) immuunivälitteisen munuaistulehduksen vaikeusaste oli 3.
Mediaaniaika immuunivälitteisen munuaistulehduksen ilmaantumiseen oli 1,2 kuukautta (vaihteluväli: 2,9 viikkoa ‑ 1,8 kuukautta). Keston mediaani oli 1,3 viikkoa (vaihteluväli: yli 4 vuorokautta - 1,3 viikkoa).
Immuunivälitteinen munuaistulehdus ei johtanut avelumabihoidon lopettamiseen yhdelläkään potilaalla. Kaikki kaksi immuunivälitteistä munuaistulehdusta sairastanutta potilasta sai suuriannoksista kortikosteroidia hoitojakson mediaanin ollessa 1,1 viikkoa (vaihteluväli: 3 vuorokautta - 1,9 viikkoa). Immuunivälitteinen munuaistulehdus parani 1/2 potilaalla (50 %) tiedonkeruun päättymiseen mennessä.
Maksatoksisuus (yhdessä aksitinibin kanssa)
Avelumabia yhdessä aksitinibin kanssa saaneista potilailla on raportoitu vaikeusasteen 3 ja vaikeusasteen 4 kohonneita ALAT-arvoja 9 %:lla potilaista ja kohonneita ASAT-arvoja 7 %:lla potilaista.
Potilailla, joilla ALAT ≥ 3 kertaa ULN (vaikeusasteet 2–4, n = 82), ALAT korjaantui 92 %:lla potilaista vaikeusasteille 0–1.
Niistä 73 potilaasta, joilla uusittiin joko avelumabimonoterapia (59 %) tai aksitinibimonoterapia (85 %) tai molemmat (55 %), 66 %:lla ALAT ei kohonnut uudestaan tasolle ≥ 3 kertaa ULN.
Immuunijärjestelmän tarkistuspisteen estäjien luokkavaikutukset
Muilla immuunijärjestelmän tarkistuspisteen estäjillä annetun hoidon aikana on havaittu seuraavia haittavaikutuksia, joita voi ilmaantua myös avelumabihoidon aikana: haiman eksokriininen vajaatoiminta, keliakia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista liitteessä V luetellun kansallisen
Yliannostus
Kolmella potilaalla raportoitiin yliannostus, joka oli 5–10 % suurempi kuin suositeltu avelumabiannos. Potilailla ei ilmennyt oireita, yliannostus ei vaatinut hoitoa ja potilaat jatkoivat avelumabihoitoa.
Yliannostustapauksissa potilaan tilaa on seurattava tarkoin haittavaikutuksiin viittaavien oireiden ja löydösten havaitsemiseksi, ja oireenmukainen hoito on aloitettava tarvittaessa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut solunsalpaajatAntineoplastiset lääkeaineet, monoklonaaliset vasta-aineet, PD 1/PDL 1 (ohjelmoitu solukuolemaproteiini 1/kuolemaligandi 1) estäjät, ATC-koodi: L01FF04.
Vaikutusmekanismi
Avelumabi on humaani monoklonaalinen G1-immunoglobuliini (IgG1) -vasta-aine, jonka vaikutus kohdistuu PD-L1 (programmed death ligand 1) -reseptoriin. Avelumabi sitoutuu PD-L1-reseptoriin ja estää sen vuorovaikutuksen PD-1- ja B7-1-reseptorien kanssa. Tämä poistaa PD-L1:n sytotoksisiin CD8+-T-soluihin kohdistuvat estävät vaikutukset, mikä johtaa T-solujen antituumorivasteen palautumiseen. Avelumabin on osoitettu myös indusoivan luonnollisten tappajasolujen soluvälitteistä suoraa tuumorisolujen solulyysiä vasta-aineesta riippuvaisen soluvälitteisen sytotoksisuuden (ADCC, Antibody-dependent cellular cytotoxicity) kautta.
Kliininen teho ja turvallisuus
Merkelinsolukarsinooma (tutkimus EMR100070‑003)
Avelumabin tehoa ja turvallisuutta tutkittiin yksihaaraisessa EMR100070‑003-monikeskustutkimuksessa kahdessa osassa. Osassa A tutkittiin potilaita, joilla oli histologisesti varmistettu metastasoitunut MCC ja joiden tauti oli edennyt metastasoituneen taudin hoitoon annetun kemoterapian aikana tai sen jälkeen elinajanodotteen ollessa yli 3 kuukautta. Osassa B tutkittiin potilaita, joilla oli histologisesti varmistettu metastasoitunut MCC, ja jotka eivät olleet saaneet systeemistä hoitoa metastaattisessa vaiheessa.
Tutkimuksesta suljettiin pois potilaat, joilla oli tai oli ollut aktiivisia etäpesäkkeitä keskushermostossa, aktiivinen tai aiemmin sairastettu autoimmuunisairaus, syöpä viimeisten viiden vuoden aikana, elinsiirto, immunosuppressiivista hoitoa vaativa sairaus tai aktiivinen HIV-, hepatiitti B- tai hepatiitti C -infektio.
Potilaat saivat avelumabia 10 mg/kg kahden viikon välein, kunnes tauti eteni tai potilaalla ilmeni sellaisia haittavaikutuksia, joita ei voitu hyväksyä. Jos potilaalla havaittiin taudin radiologista etenemistä, johon ei kuitenkaan liittynyt merkittävää kliinisen tilan huonontumista eli ei todettu uusia tai pahenevia oireita, ei yli kaksi viikkoa kestäviä toimintakyvyn muutoksia eikä tarvetta salvage‑hoitoon, hoitoa voitiin jatkaa.
Riippumaton päätetapahtumien arviointilautakunta arvioi kasvaimen vasteen 6 viikon välein käyttäen RECIST 1.1 -kriteeristöä (Response Evaluation Criteria in Solid Tumours).
Tutkimus 003 Osa A – aiemmin hoitoa saaneet potilaat
Ensisijainen tehoa mittaava päätetapahtuma oli vahvistettu paras kokonaisvaste (BOR, best overall response); toissijaiset tehoa mittaavat päätetapahtumat olivat vasteen kesto (DOR, duration of response), elinaika ilman taudin etenemistä (PFS, progression free survival) ja kokonaiselinaika (OS, overall survival). Tehon analyysi suoritettiin kaikilla 88 potilaalla vähintään 36 kuukauden mittaisen seurantajakson päätteeksi. Potilaat saivat keskimäärin 7 annosta avelumabia (vaihteluväli: 1–95 annosta) ja hoidon mediaanikesto oli 17 viikkoa (vaihteluväli: 2–208 viikkoa).
Tutkimuksen 88 potilaasta 65 (74 %) oli miehiä, mediaani-ikä oli 73 vuotta (ikäjakauma 33–88 vuotta), 81 potilasta (92 %) oli valkoihoisia, 49 potilaan (56 %) ECOG (Eastern Cooperative Oncology Group) -toimintakykyluokitus oli 0 ja 39 potilaan (44 %) EOCG-toimintakykyluokitus oli 1.
Tutkimuksen mukaan 52 potilasta (59 %) oli saanut yhtä aikaisempaa syöpähoitoa MCC:hen, 26 potilasta (30 %) oli saanut kahta aikaisempaa hoitoa, 10 potilasta (11 %) oli saanut vähintään kolmea aikaisempaa hoitoa. Yhteensä 47 potilaalla (53 %) oli viskeraalisia etäpesäkkeitä.
Taulukko 4 sisältää yhteenvedon EMR100070-003-tutkimuksen osan A tehoa mittaavista päätetapahtumista potilailla, jotka saivat suositellun annoksen avelumabia. Taulukossa on vähintään 36 kuukauden mittaisesta seurantajaksosta saadut tiedot. Kokonaiselinaika arvioitiin analyysissa, jonka tiedot saatiin vähintään 44 kuukauden mittaisesta seurantajaksosta. OS mediaani oli 12,6 kuukautta (95 % CI: 7,5; 17,1).
Taulukko 4: Metastasoitunutta MCC:tä sairastavien potilaiden hoitovaste EMR100070-003-tutkimuksen osassa A, kun avelumabia annettiin 10 mg/kg kahden viikon välein*
Tehon päätetapahtumat (osa A) (RECIST v1.1, IERC) | Tulokset (N=88) |
|---|---|
Objektiivinen vasteosuus (ORR) Vasteosuus, CR+PR** n (%) (95 % CI) |
29 (33,0 %) (23,3; 43,8) |
Vahvistettu paras kokonaisvaste (BOR) Täydellinen vaste (CR)** n (%) Osittainen vaste (PR)** n (%) |
10 (11,4 %) 19 (21,6 %) |
Vasteen kesto (DOR)a Mediaani, kuukautta (95 % CI) Minimi, maksimi (kuukautta) ≥ 6 kuukautta, KM, (95 % CI) ≥ 12 kuukautta, KM, (95 % CI) ≥ 24 kuukautta, KM, (95 % CI) ≥ 36 kuukautta, KM, (95 % CI) |
40,5 (18, ei arvioitavissa) 2,8; 41,5+ 93 % (75; 98) 71 % (51; 85) 67 % (47; 82) 52 % (26; 73) |
Elinaika ilman taudin etenemistä (PFS) Mediaani PFS, kuukautta (95 % CI) 6 kuukauden PFS-osuus, K-M, (95 % CI) 12 kuukauden PFS-osuus, K-M, (95 % CI) 24 kuukauden PFS-osuus, K-M, (95 % CI) 36 kuukauden PFS-osuus, K-M, (95 % CI) |
2,7 (1,4; 6,9) 40 % (29; 50) 29 % (19; 39) 21 % (12; 32) |
CI: luottamusväli; RECIST: Hoitovaste kasvaimeen perustuen vastearvioon kiinteissä kasvaimissa (Response Evaluation Criteria in Solid Tumours); IERC: riippumaton päätetapahtumien arviointikomitea (Independent Endpoint Review Committee); KM: Kaplan-Meier; + ilmaisee sensuroidun arvon
* Tehotiedot sisältävät vähintään 36 kuukauden mittaisesta seurantajaksosta saaduilla tiedoilla (tiedonkeruun katkaisupäivä 14.9.2018)
** CR tai PR vahvistettiin seuraavassa kasvaimen määrityksessä
a Perustuu niiden potilaiden lukumäärään, joiden hoitovaste on vahvistettu (CR tai PR)
Mediaani aika vasteen saavuttamiseen oli 6 viikkoa (vaihteluväli: 6–36 viikkoa) ensimmäisestä avelumabiannoksesta laskettuna. Vasteen saavuttaneista 29 potilaasta 22 potilasta (76 %) saavutti vasteen 7 viikon kuluessa ensimmäisen avelumabiannoksen saamisesta.
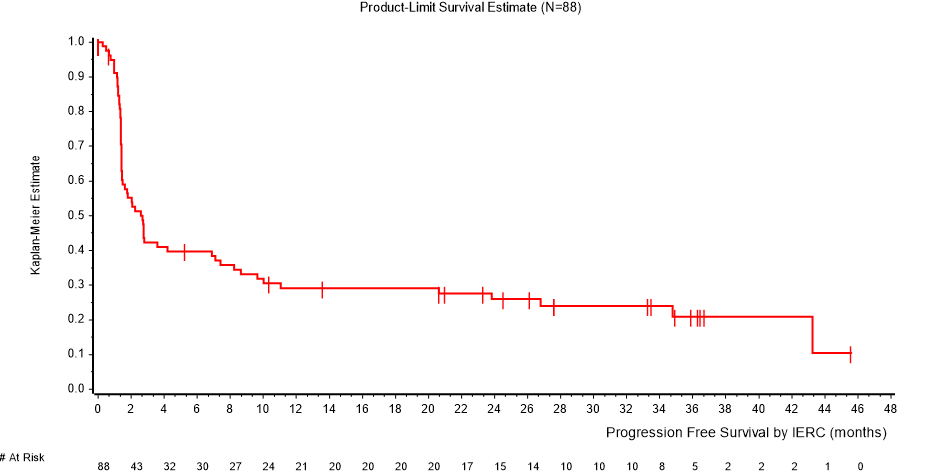
Kaplan-Meierin estimaatit elinajalle ilman taudin etenemistä niillä 88 potilaalla (osa A), joilla on metastasoitunut MCC, on esitetty kuvassa 1.
Kuva 1: Kaplan-Meierin estimaatit elinajalle ilman taudin etenemistä RECIST 1.1 -kriteeristöä käyttäen, IERC (osa A, vähintään 36 kuukauden mittainen seurantajakso)
Kasvainnäytteistä arvioitiin PD-L1:n ilmentyminen ja merkelinsolupolyoomavirus (MCV) käyttäen immunohistokemiallista tutkimuksellista menetelmää. Taulukossa 5 esitetään yhteenveto objektiivisista vasteosuuksista PD-L1:n ilmentymisen ja MCV-statuksen mukaan EMR100070‑003-tutkimuksen osassa A, jossa potilailla oli metastasoitunut MCC.
Taulukko 5: Objektiiviset vasteosuudet kasvaimen PD-L1:n ilmentymisen ja MCV-statuksen mukaan EMR100070-003-tutkimuksen osassa A, jossa potilailla oli metastasoitunut MCC
| Avelumabin ORR (95 % CI)* |
PD-L1:n ilmentyminen ≥ 1 %:n katkaisukohdassa | N=74a |
Positiivinen (n=58) | 36,2 % (24,0; 49,9) |
Negatiivinen (n=16) | 18,8 % (4,0; 45,6) |
(Kasvaimen) IHC-MCV-status | N=77b |
Positiivinen (n=46) | 28,3 % (16,0; 43,5) |
Negatiivinen (n=31) | 35,5 % (19,2; 54,6) |
IHC: immunohistokemia; MCV: merkelinsolupolyoomavirus; ORR: objektiivinen vasteosuus
* ORR (tiedonkeruun katkaisupäivä 14.9.2018)
a Perustuu tietoihin potilaista, joiden PD-L1 oli arvioitavissa
b Perustuu tietoihin potilaista, joiden MCV-status oli arvioitavissa immunohistokemiallisesti
Tutkimus 003 Osa B – potilaat, jotka eivät olleet saaneet systeemistä hoitoa metastaattiseen tautiin
Ensisijainen tehoa mittaava päätetapahtuma osassa B oli kestävä vaste, joka määritettiin vähintään 6 kuukauden mittaiseksi objektiiviseksi vasteeksi (joko täydellinen (CR) tai osittainen vaste (PR)); toissijaiset tehoa mittaavat päätetapahtumat olivat BOR, DOR, PFS ja OS.
Osassa B suoritettuun ensisijaiseen arviointiin osallistui 116 potilasta, jotka saivat vähintään yhden annoksen avelumabia ja joiden seurantajakso oli jatkunut vähintään 15 kuukauden ajan tiedonkeruun katkaisupäivämääränä (katkaisupäivä 2.5.2019).
116 potilaasta 81 (70 %) oli miehiä, mediaani-ikä oli 74 vuotta (ikäjakauma: 41–93 vuotta), 75 (65 %) oli valkoihoisia, 72 (62 %) potilaan ECOG (Eastern Cooperative Oncology Group) -toimintakykyluokitus oli 0 ja 44 (38 %) potilaan ECOG‑toimintakykyluokitus oli 1.
Taulukossa 6 esitetään yhteenveto tehoa mittaavien päätetapahtumien ensisijaisesta arvioinnista EMR100070‑003-tutkimuksen osassa B, kun avelumabia annettiin suositusannostuksen mukaan. Väliarviointiin sisältyvät DOR- ja PFS-estimaatit 24 kuukauden kohdalla Kaplan-Meierin perusteella.
Taulukko 6: Metastasoitunutta MCC:tä sairastavien potilaiden hoitovaste EMR100070-003-tutkimuksen osan B ensisijaisessa arvioinnissa, kun avelumabia annettiin 10 mg/kg kahden viikon välein*
Tehon päätetapahtuma (Osa B) (RECIST v1.1, IERC) | Tulokset (N=116) |
|---|---|
Kestävä vaste ≥ 6 kuukautta (95 % CI) |
30,2 % (22,0; 39,4) |
Objektiivinen vasteosuus (ORR) Vasteosuus, CR+PR** n (%) (95 % CI) |
46 (39,7 %) (30,7; 49,2) |
Vahvistettu paras kokonaisvaste (BOR) Täydellinen vaste (CR)** n (%) Osittainen vaste (PR)** n (%) |
19 (16,4 %) 27 (23,3 %) |
Vasteen kesto (DOR)a Mediaani, kuukautta (95 % CI) Minimi, maksimi (kuukautta) ≥ 3 kuukautta, K‑M, (95 % CI) ≥ 6 kuukautta, K‑M, (95 % CI) ≥ 12 kuukautta, K‑M, (95 % CI) ≥ 18 kuukautta, K-M, (95 % CI) ≥ 24 kuukautta, K-M, (95 % CI) |
18,2 (10,3; ei arvioitavissa) 1,2; 28,3 89 % (75; 95) 78 % (63; 87) 66 % (50; 78) 52 % (34; 67) 45 % (25; 63) |
Elinaika ilman taudin etenemistä (PFS) Mediaani PFS, kuukautta (95 % CI) 3 kuukauden PFS-osuus, K‑M, (95 % CI) 6 kuukauden PFS-osuus, K‑M, (95 % CI) 12 kuukauden PFS-osuus, K‑M, (95 % CI) 24 kuukauden PFS-osuus, K-M, (95 % CI) |
4,1 (1,4; 6,1) 51 % (42; 60) 41 % (32; 50) 31 % (23; 40) 20 % (12; 30) |
CI: luottamusväli; RECIST: hoitovaste kasvaimeen perustuen vastearvioon kiinteissä kasvaimissa (Response Evaluation Criteria in Solid Tumours); IERC: riippumaton päätetapahtumien arviointikomitea (Independent Endpoint Review Committee); KM: Kaplan-Meier
* Tehotiedot vähintään 15 kuukauden mittaisen seurantajakson ajalta (katkaisupäivämäärä 2.5.2019)
** CR tai PR vahvistettiin seuraavassa kasvaimen määrityksessä
a Perustuu niiden potilaiden lukumäärään, joiden hoitovaste on vahvistettu (CR tai PR)
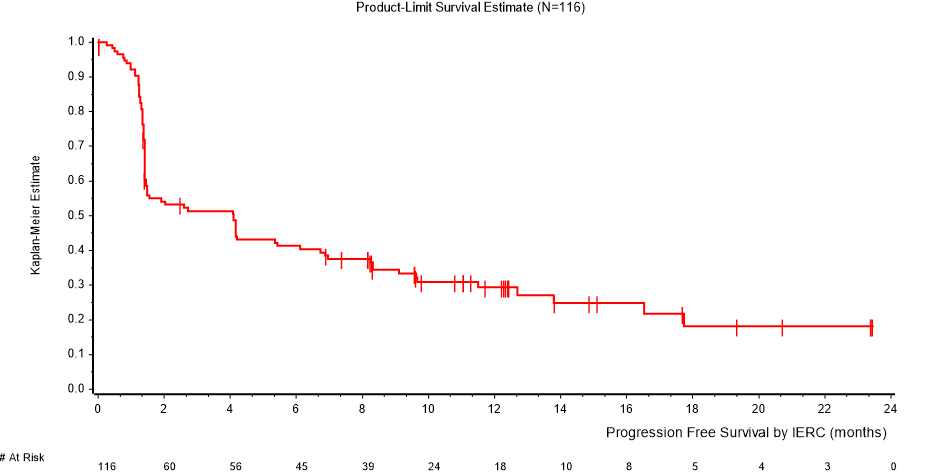
Kuvassa 2 esitetään Kaplan-Meierin estimaatit elinajalle ilman taudin etenemistä niillä 116 potilaalla, jotka osallistuivat osan B ensisijaiseen arviointiin ja joiden seurantajakso oli jatkunut vähintään 15 kuukauden ajan.
Kuva 2: Kaplan-Meierin estimaatit elinajalle ilman taudin etenemistä RECIST 1.1 -kriteeristöä käyttäen, IERC (osa B, N=116)
Kasvainnäytteistä arvioitiin PD-L1:n ilmentyminen ja MCV käyttäen immunohistokemiallista tutkimuksellista menetelmää. Taulukossa 7 esitetään yhteenveto objektiivisista vasteosuuksista PD‑L1:n ilmentymisen ja MCV-statuksen mukaan EMR100070-003-tutkimuksen osassa B, jossa potilailla oli metastasoitunut MCC.
Taulukko 7: Objektiiviset vasteosuudet kasvaimen PD-L1:n ilmentymisen ja MCV-statuksen mukaan EMR100070-003-tutkimuksen osassa B, jossa potilailla oli metastasoitunut MCC
Avelumabi ORR (95 % CI)* | |
|---|---|
| PD-L1:n ilmentyminen ³ 1 %:n katkaisukohdassa | N=108a |
| Positiivinen (n=21) | 61,9 % (38,4; 81,9) |
| Negatiivinen (n=87) | 33,3 % (23,6, 44,3) |
| (Kasvaimen) IHC-MCV-status | N=107b |
| Positiivinen (n=70) | 34,3 % (23,3; 46,6) |
| Negatiivinen (n=37) | 48,6 % (31,9; 65,6) |
IHC: immunohistokemia; MCV: merkelinsolupolyoomavirus; ORR: objektiivinen vasteosuus
* ORR (tiedonkeruun katkaisupäivä 2.5.2019)
a Perustuu tietoihin potilaista, joiden PD-L1 oli arvioitavissa
b Perustuu tietoihin potilaista, joiden MCV-status oli arvioitavissa immunohistokemiallisesti
Paikallisesti edennyt tai metastasoitunut uroteelikarsinooma (tutkimus B9991001)
Avelumabin teho ja turvallisuus osoitettiin satunnaistetussa avoimessa monikeskustutkimuksessa B9991001, jossa 700 potilaalla oli leikkaushoitoon soveltumaton, paikallisesti edennyt tai metastasoitunut uroteelikarsinooma, joka ei ollut edennyt platinapohjaisen ensilinjan induktiokemoterapian 4‑6 syklin aikana. Tutkimukseen ei otettu potilaita, joilla oli jokin autoimmuunisairaus tai immunosuppressiivista hoitoa vaativa lääketieteellinen tila.
Satunnaistaminen stratifioitiin kemoterapialla saavutetun parhaan vasteen (CR/PR vs. vakaa tauti [SD]) perusteella ja sen perusteella, missä etäpesäkkeet sijaitsivat (viskeraalinen vs. ei-viskeraalinen) ensilinjan induktiokemoterapian aloitusvaiheessa. Potilaat satunnaistettiin (1:1) saamaan joko avelumabia 10 mg/kg infuusiona laskimoon 2 viikon välein ja parasta tukihoitoa (BSC) tai pelkkää parasta tukihoitoa.
Avelumabin anto oli sallittua myös sen jälkeen, kun potilaalla havaittiin RECIST-kriteerien (Response Evaluation Criteria in Solid Tumours, versio 1.1) mukainen sairauden eteneminen BICR-arvioinnin (Blinded Independent Central Review) perusteella, mikäli potilaan tila oli vakaa ja hoidosta oli tutkijan arvion mukaan hänelle kliinistä hyötyä. Kasvainstatus arvioitiin lähtötilanteessa, satunnaistamisen jälkeen viikolla 8, ja sitten 8 viikon välein, kunnes satunnaistamisen ajankohdasta oli kulunut 12 kuukautta, ja tämän jälkeen 12 viikon välein, kunnes RECIST v1.1 -kriteerien mukainen sairauden eteneminen oli todistetusti varmistettu BICR-arvioinnilla.
Avelumabia ja parasta tukihoitoa saaneen hoitohaaran ja pelkkää parasta tukihoitoa saaneen hoitohaaran potilaiden demografiset ja lähtötilanteen piirteet olivat yleisesti ottaen hyvin tasapainossa. Lähtötilanteen piirteitä olivat: mediaani-ikä 69 vuotta (vaihteluväli: 32–90), 66 % potilaista oli 65‑vuotiaita tai vanhempia, 77 % oli miespuolisia, 67 % oli valkoihoisia ja ECOG-toimintakykyluokka oli molemmissa hoitohaaroissa 0 (61 %) tai 1 (39 %).
Ensilinjan induktiokemoterapiassa 56 % potilaista sai sisplatiinia ja gemsitabiinia, 38 % sai karboplatiinia ja gemsitabiinia ja 6 % sai sisplatiinia ja gemsitabiinia sekä karboplatiinia ja gemsitabiinia (nämä potilaat saivat siis vähintään yhden syklin kumpaakin yhdistelmää). Paras vaste ensilinjan induktiokemoterapialle oli CR tai PR (72 %) tai SD (28 %). Ennen kemoterapiaa potilaiden etäpesäkkeet olivat sijainniltaan viskeraalisia (55 %) tai ei-viskeraalisia (45 %). Potilaista 51 %:lla oli PD-L1-positiivisia kasvaimia. Avelumabia ja parasta tukihoitoa saaneen hoitohaaran potilaista 6 % ja pelkkää tukihoitoa saaneen hoitohaaran potilaista 44 % sai PD-1/PD-L1-tarkistuspisteen estäjää hoidon lopettamisen jälkeen.
Ensisijainen tehon päätetapahtuma oli kokonaiselinaika (OS, overall survival) kaikilla satunnaistetuilla potilailla ja potilailla, joilla on PD‑L1-positiivisia kasvaimia. RECIST v.1.1 ‑kriteeristön mukaan tehtyyn BICR-arviointiin perustuva elinaika ilman taudin etenemistä (PFS, progression-free survival) oli toinen tehon päätetapahtuma. Tehotulokset mitattiin satunnaistamishetkestä alkaen, eli sen jälkeen kun platinapohjaista induktiokemoterapiaa oli annettu 4–6 sykliä.
Kasvaimen PD-L1-status arvioitiin Ventana PD-L1 (SP263) -määrityksellä. PD-L1-positiivisuus määritettiin seuraavasti: ≥ 25 % kasvainsoluista PD-L1-positiivisia värjäyksessä; tai ≥ 25 % immuunisoluista PD‑L1-positiivisia värjäyksessä, jos > 1 % kasvainalueesta sisälsi immuunisoluja; tai 100 % immuunisoluista PD‑L1-positiivisia värjäyksessä, jos = 1 % kasvainalueesta sisälsi immuunisoluja.
Ennalta määritetyn väliarvioinnin (tiedonkeruun katkaisupäivä 21. lokakuuta 2019) kohdalla tutkimuksen B9991001 ensisijainen päätetapahtuma kokonaiselinajan (OS) osalta saavutettiin molemmissa ensisijaisissa populaatioissa: kaikilla satunnaistetuilla potilailla avelumabia ja parasta tukihoitoa saaneiden haarassa mediaani kokonaiselinaika (OS) oli 21,4 kuukautta (95 % CI: 18,9; 26,1; riskisuhde 0,69; 95 % CI: 0,556; 0,863) ja pelkkää tukihoitoa saaneiden haarassa mediaani kokonaiselinaika (OS) oli 14,3 kuukautta (95 % CI: 12,9; 17,8). Potilailla, joilla oli PD‑L1‑positiivisia kasvaimia, mediaania elossaoloaikaa (OS) ei saavutettu avelumabia ja parasta tukihoitoa saaneiden haarassa (95 % CI: 20,3; ei saavutettu; riskisuhde 0,56; 95 %, CI: 0,404; 0,787), ja mediaani kokonaiselinaika (OS) pelkkää tukihoitoa saaneiden haarassa oli 17,1 kuukautta (95 % CI: 13,5; 23,7). Päivitetyt kokonaiselinaikaa (OS) koskevat tulokset, joiden tiedonkeruun katkaisupäivä oli 19 tammikuuta 2020, sekä tiedot koskien elinaikaa ilman taudin etenemistä (PFS), joiden tiedonkeruun katkaisupäivä oli 21 lokakuuta 2019, esitetään jäljempänä taulukossa 8 sekä kuvassa 3 ja kuvassa 4.
Taulukko 8: Tehotulokset PD-L1-ilmentymisen mukaan tutkimuksessa B9991001
| Tehon päätetapahtumat | Avelumabi + paras tukihoito | Paras tukihoito | Avelumabi + paras tukihoito | Paras tukihoito | Avelumabi + paras tukihoito | Paras tukihoito |
|---|---|---|---|---|---|---|
| (N = 350) | (N = 350) | (N = 189) | (N = 169) | (N = 139) | (N = 131) | |
| Kaikki satunnaistetut potilaat | PD-L1‑positiiviset kasvaimet | PD-L1‑negatiiviset kasvaimetc | ||||
| Kokonaiselinaika (OS)a | ||||||
| Tapahtumat (%) | 156 (44,6) | 190 (54,3) | 68 (36,0) | 85 (50,3) | 80 (57,6) | 80 (61,1) |
| Mediaani kuukausina | 22,1 | 14,6 | NE | 17,5 | 18,9 | 13,4 |
| (95 % CI) | (19,0; 26,1) | (12,8; 17,8) | (20,6; NE) | (13,5; 31,6) | (13,3; 22,1) | (10,4; 17,3) |
| Riskisuhde | 0,70 | 0,60 | 0,83 | |||
| (95 % CI) | (0,564; 0,862) | (0,439; 0,833) | (0,603; 1,131) | |||
| 2‑puolinen p‑arvod | 0,0008 | 0,0019 | - | |||
| Elinaika ilman taudin etenemistä (PFS)b, e, f | ||||||
| Tapahtumat (%) | 225 (64,3) | 260 (74,3) | 109 (57,7) | 130 (76,9) | 103 (74,1) | 99 (75,6) |
| Mediaani kuukausina | 3,7 | 2,0 | 5,7 | 2,1 | 3,0 | 1,9 |
| (95 % CI) | (3,5; 5,5) | (1,9; 2,7) | (3,7; 7,4) | (1,9; 3,5) | (2,0; 3,7) | (1,9; 2,1) |
| Riskisuhde | 0,62 | 0,56 | 0,63 | |||
| (9 5% CI) | (0,519; 0,751) | (0,431; 0,728) | (0,474; 0,847) | |||
| 2‑puolinen p‑arvod | < 0,0001 | < 0,0001 | - | |||
CI: luottamusväli (confidence interval); K‑M: Kaplan‑Meier, NE: ei arvioitavissa (not estimable)
Huom: 72 potilaalla (22 potilaalla avelumabia ja parasta tukihoitoa saaneessa hoitohaarassa ja 50 potilaalla pelkkää parasta tukihoitoa saaneessa hoitohaarassa) oli kasvain, jonka PD‑L1-status ei ollut tiedossa
a OS-tietojen keruu päättyi 19. tammikuuta 2020
b PFS-tietojen keruu päättyi 21. lokakuuta 2019
c PD-L1-negatiivisten potilaiden analyysit olivat eksploratiivisia, eikä muodollista testiä tehty
d p‑arvo perustuu stratifioituun log-rank-testiin
e Perustuu RECIST v1.1 -kriteerien mukaiseen BICR-arviointiin
fPFS-tietojen sensuroinnin syyt hierarkian mukaisessa peräkkäisessä järjestyksessä: ei kelvollista lähtötilanteen määritystä, uuden syöpähoidon aloittaminen, tapahtuma kahden tai useamman puuttuvan määrityksen jälkeen, suostumuksen peruuttaminen, katoaminen seurannasta, ei kelvollista kasvainarviointia lähtötilanteen jälkeen, jatkuu ilman tapahtumia
Kuva 3: Kaplan‑Meierin estimaatit kokonaiselinajalle (OS) PD-L1-ilmentymisen mukaan (tiedonkeruun katkaisupäivä 19. tammikuuta 2020) – koko analysoitava populaatio
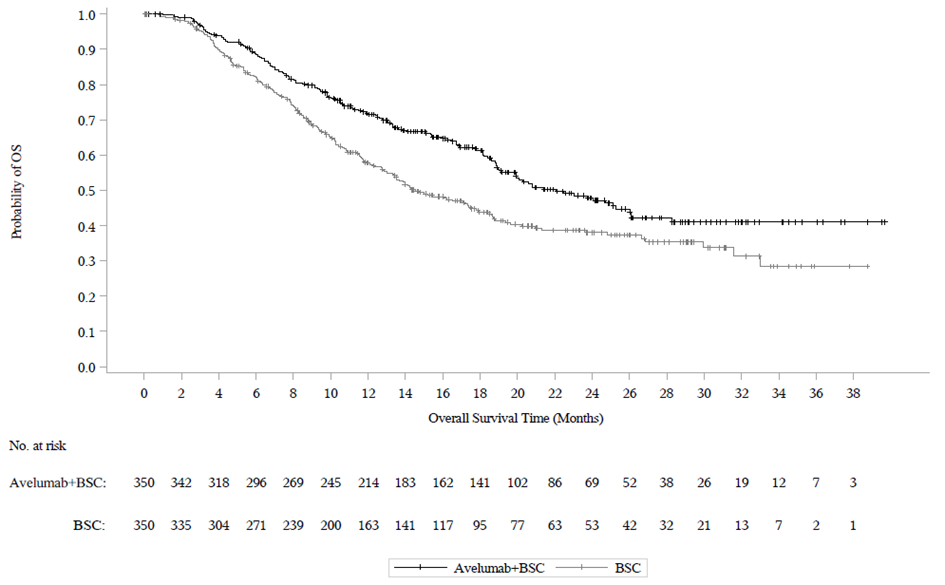
(A): Kaikki satunnaistetut potilaat
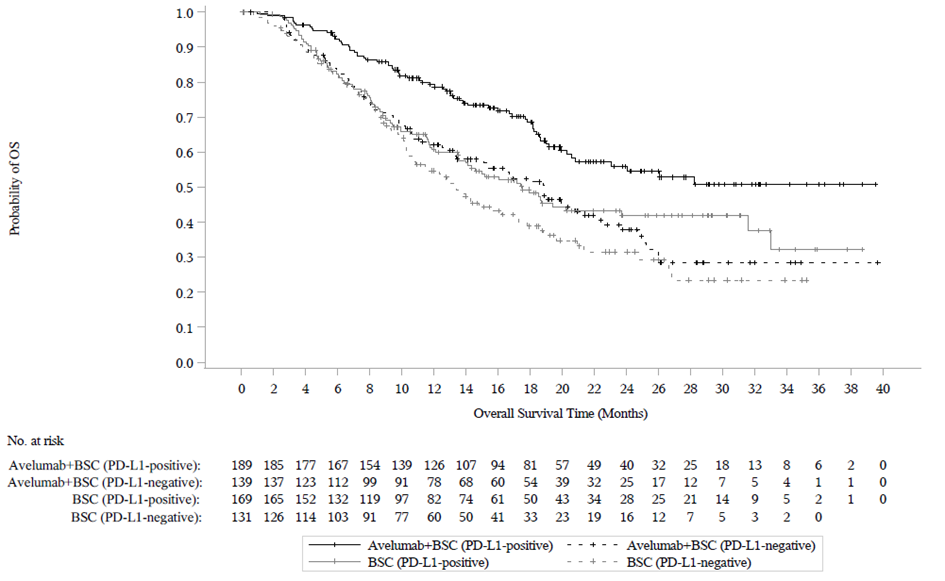
(B): Potilaat, ryhmiteltynä PD-L1-ilmentymisen mukaan
Kuva 4: Kaplan‑Meierin estimaatit elinajalle ilman taudin etenemistä (PFS) PD-L1-ilmentymisen mukaan BICR-arvioinnin (RECIST v1.1) perusteella (tiedonkeruun katkaisupäivä 21. lokakuuta 2019) – koko analysoitava populaatio
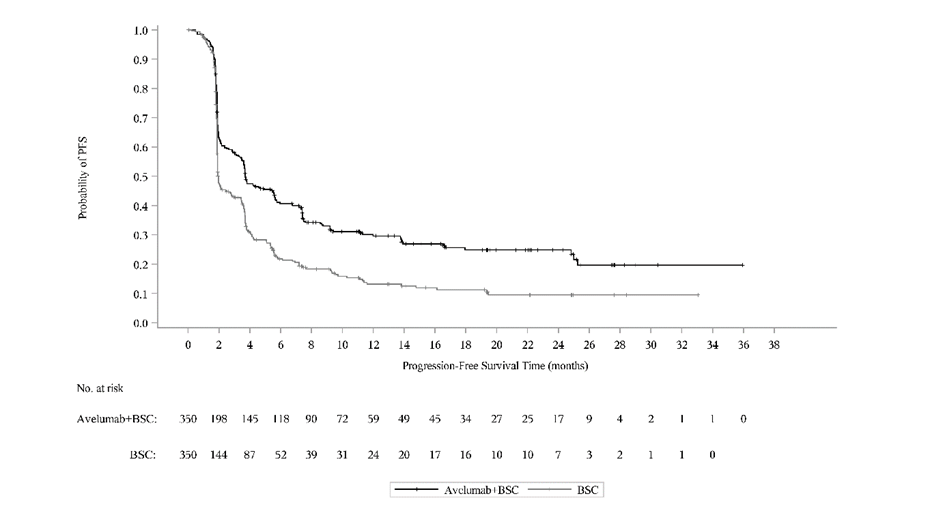
(A): Kaikki satunnaistetut potilaat
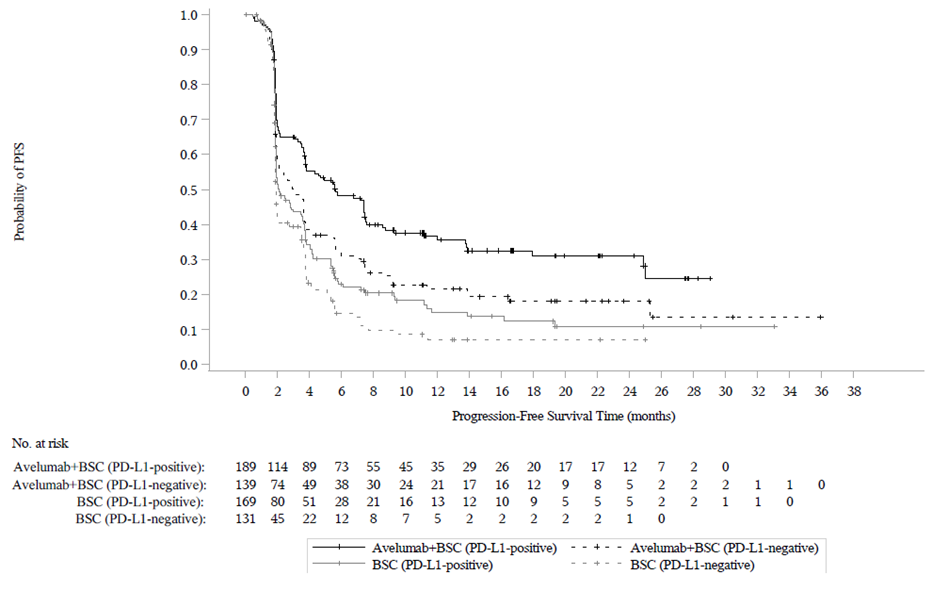
(B): Potilaat, ryhmiteltynä PD-L1-ilmentymisen mukaan
Munuaissolukarsinooma (tutkimus B9991003)
Avelumabin teho ja turvallisuus yhdessä aksitinibin kanssa osoitettiin satunnaistetussa avoimessa monikeskustutkimuksessa B9991003, jossa 886 potilaalla oli aiemmin hoitamaton edennyt tai metastaattinen kirkassoluinen RCC.
Potilaat otettiin tutkimukseen riippumatta prognostisesta riskiryhmästä tai kasvaimen PD‑L1:n ilmentymisestä, ja heillä oli oltava vähintään yksi mitattavissa oleva, RECIST-kriteerien (Response Evaluation Criteria in Solid Tumours, versio 1.1) mukainen leesio, jota ei ollut aikaisemmin sädetetty. Tutkimukseen ei otettu potilaita, jotka olivat aiemmin saaneet systeemistä hoitoa edenneeseen tai metastaattiseen RCC:hen; jotka olivat aiemmin saaneet systeemistä immunoterapiaa, joissa oli käytetty IL‑2-, IFN-α-, anti-PD‑1-, anti-PD‑L1- tai anti-CTLA‑4-vasta-aineita; joilla oli aktiivinen aivometastaasi; joilla oli aktiivinen autoimmuunisairaus, jota immuunijärjestelmää stimuloivat aineet saattaisivat pahentaa; joilla oli ollut muita syöpäsairauksia viimeisten 5 vuoden aikana ja joille oli tehty elinsiirto.
Satunnaistaminen stratifioitiin ECOG (Eastern Cooperative Oncology Group) -toimintakykyluokituksen (PS, performance status) (0 vs. 1) ja alueen (Yhdysvallat vs. Kanada/Länsi-Eurooppa vs. muu maailma) mukaisesti. Potilaat satunnaistettiin (1:1) yhteen seuraavista hoitohaaroista:
- Avelumabi 10 mg/kg infuusiona laskimoon kahden viikon välein ja samanaikaisesti aksitinibi 5 mg kahdesti päivässä suun kautta (N = 442). Potilailla, jotka sietivät aksitinibia 5 mg kahdesti päivässä ilman vaikeusasteen 2 tai vaikeampia aksitinibiin liittyviä haittavaikutuksia 2 peräkkäisen viikon ajan, annosta voitiin nostaa kahdesti päivässä annettavaan 7 mg:aan ja sen jälkeen 10 mg:aan. Toksisuuden hallitsemiseksi aksitinibihoito voitiin keskeyttää tai annos vähentää 3 mg:aan kahdesti päivässä ja sen jälkeen vielä 2 mg:aan kahdesti päivässä.
- Sunitinibi 50 mg kerran päivässä suun kautta 4 viikon ajan, minkä jälkeen pidettiin 2 viikon tauko (N = 444), kunnes taudin eteneminen havaittiin kuvantamalla tai kliinisesti tai kunnes potilaalla ilmeni sellaisia haittavaikutuksia, joita ei voitu hyväksyä.
Avelumabi- ja aksitinibihoito jatkui siihen asti, kunnes havaittiin RECIST v1.1-kriteeristön mukainen sairauden eteneminen BICR-arvioinnin (Blinded Independent Central Review) avulla tai kunnes toksisuus oli liiallista. Avelumabin ja aksitinibin anto oli sallittua RECIST-kriteeristön mukaisen sairauden etenemisen jälkeenkin perustuen tutkijan arvioon siitä, mikä oli potilaan hyöty/riski-suhde ja kliininen tila, mukaan lukien toimintakykyluokka, kliiniset oireet, haittatapahtumat ja laboratoriokokeiden tulokset. Suurimmalla osalla (n = 160, 71,4 %) potilaista, joilla oli etenevä sairaus, hoitoa jatkettiin etenemisen jälkeen käyttäen molempia lääkevalmisteita. Kasvainstatus arvioitiin lähtötilanteessa, satunnaistamisen jälkeen viikolla 6, ja sitten 6 viikon välein, kunnes satunnaistamisen ajankohdasta oli kulunut 18 kuukautta, ja tämän jälkeen 12 viikon välein, kunnes sairauden eteneminen oli todistetusti varmistettu BICR-arvioinnilla.
Tehon ensisijaiset päätetapahtumat olivat elinaika ilman taudin etenemistä (PFS, progression-free survival) arvioituna sokkoutetun riippumattoman keskitetyn arvioinnin (BICR) avulla käyttäen RECIST v1.1 -kriteeristöä ja kokonaiselinaika (OS, overall survival) ensilinjan hoidossa edennyttä RCC:tä sairastavilla potilailla, joilla on PD‑L1-positiivisia kasvaimia (PD‑L1:n ilmentymisaste ≥ 1 %). Tärkeimmät toissijaiset päätetapahtumat olivat RESIST v.1.1 -kriteeristön mukaan tehtyyn BICR:in arviointiin perustuva PFS sekä OS riippumatta PD‑L1:n ilmenemisestä. PD‑L1-status määritettiin immunohistokemiallisesti. Muita toissijaisia päätetapahtumia olivat objektiivinen vaste (OR, objective response), aika vasteeseen (TTR, time to response) ja vasteen kesto (DOR, duration of response).
Tutkimuspotilasjoukon piirteet: mediaani-ikä 61 vuotta (vaihteluväli: 27,0 - 88,0), 38 % potilaista oli 65‑vuotiaita tai vanhempia, 75 % oli miespuolisia, 75 % oli valkoihoisia ja ECOG-toimintakykyluokka oli 0 (63 %) tai 1 (37 %).
Potilaat jakautuivat IMDC (International Metastatic Renal Cell Carcinoma Consortium Database) ‑kriteerien perusteella seuraavasti: 21 % hyvän, 62 % kohtalaisen ja 16 % huonon ennusteen ryhmiin. Potilaat jakautuivat MSKCC (Memorial Sloan–Kettering Cancer Center) -kriteerien mukaisesti riskiryhmiin seuraavasti: 22 % hyvän, 65 % kohtalaisen ja 11 % huonon ennusteen ryhmiin.
Tehoa koskevat tulokset esitetään taulukossa 9 ja kuvassa 5 perustuen tiedonkeruun katkaisukohdan, 28.1.2019, tietoihin. Kokonaiselinajan (OS) seurannan mediaani oli 19 kuukautta ja sen OS-tiedot olivat epäkypsiä sisältäen 27 % kuolemia. Havaittu kokonaiselinajan riskisuhde (HR) oli 0,80 (95 % CI: 0,616 1,027) yhdessä aksitinibin kanssa annetun avelumabin osalta verrattuina sunitinibiin.
Taulukko 9: Tehoa koskevat tulokset tutkimuksesta B9991003 riippumatta PD-L1:n ilmenemisestä potilailla
Tehon päätetapahtumat (BICR-arvioinnin perusteella) | Avelumabi + aksitinibi (N=442) | Sunitinibi (N=444) |
|---|---|---|
| Elinaika ilman taudin etenemistä (PFS) | ||
| Tapahtumat (%) | 229 (52) | 258 (58) |
| Mediaani kuukausina (95 % CI) | 13,3 (11,1; 15,3) | 8,0 (6,7; 9,8) |
| Riskisuhde (95 % CI) | 0,69 (0,574; 0,825) | |
| p-arvo* | < 0,0001 | |
| PFS 12 kk:n kohdalla, K‑M, (95 % CI)** | 52,4 % (47,4; 57,2) | 39,2 % (34.1; 44,2) |
| PFS 18 kk:n kohdalla, K‑M, (95 % CI)** | 43,9 % (38,8; 49,0) | 29,3 % (24,2; 34,6) |
| Vahvistettu objektiivinen vasteosuus (ORR) | ||
| Objektiivinen vasteosuus (ORR) n (%) | 232 (52,5) | 121 (27,3) |
| (95 % CI) | 47,7; 57,2 | 23,2; 31,6 |
| Täydellinen vaste (CR) n (%) | 17 (3,8) | 9 (2,0) |
| Osittainen vaste (PR) n (%) | 215 (48,6) | 112 (25,2) |
| Aika vasteeseen (TTR) | ||
| Mediaani kuukausina (vaihteluväli) | 2,7 (1,2; 20,7) | 4,0 (1,2; 18,0) |
| Vasteen kesto (DOR) | ||
| Mediaani kuukausina (95 % CI) | 18,5 (17,8; NE) | NE (16,4; NE) |
BICR: sokkoutettu riippumaton keskitetty arviointi (Blinded Independent Central Review); CI: luottamusväli (confidence interval); K-M: Kaplan-Meier; NE: ei arvioitavissa (not estimable).
* 1-puolinen p-arvo perustuen stratifioituun log-rank-testiin.
** Luottamusvälit (CI) johdetaan käyttäen log-log-muunnosta sekä takaisinmuunnosta muuntamattomaan asteikkoon.
Kuva 5: Kaplan-Meierin estimaatit elinajalle ilman taudin etenemistä perustuen BICR-arviointiin riippumatta PD-L1:n ilmenemisestä potilailla
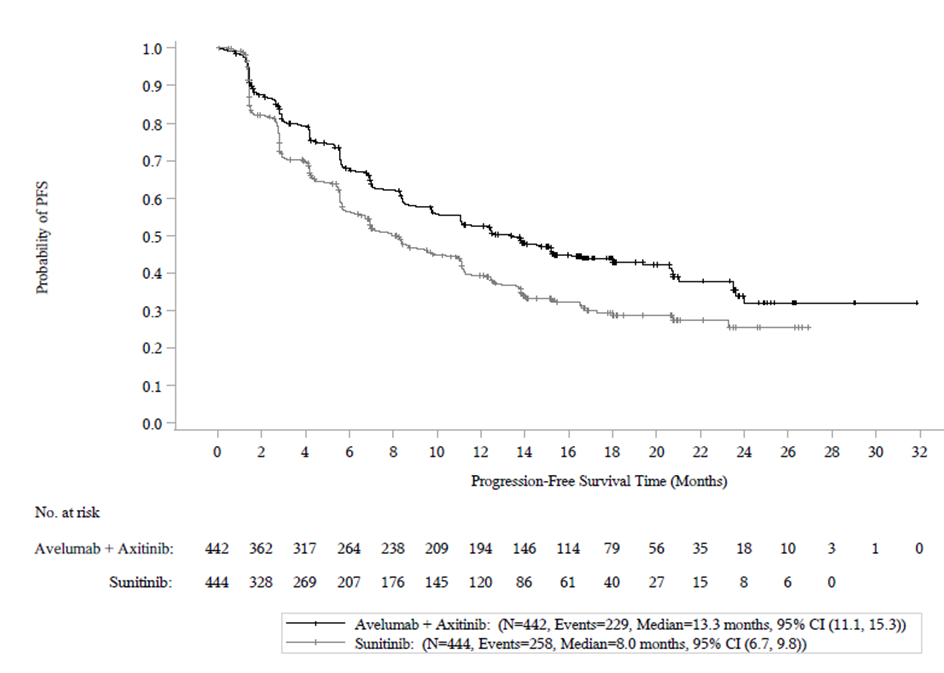
PFS hyöty havaittiin kaikissa ennalta määrätyissä alaryhmissä.
Kuva 6: Elinaikaa ilman taudin etenemistä kuvaava forest plot -diagrammi, joka perustuu potilailla tehtyyn BICR-arviointiin riippumatta PD‑L1:n ilmenemisestä
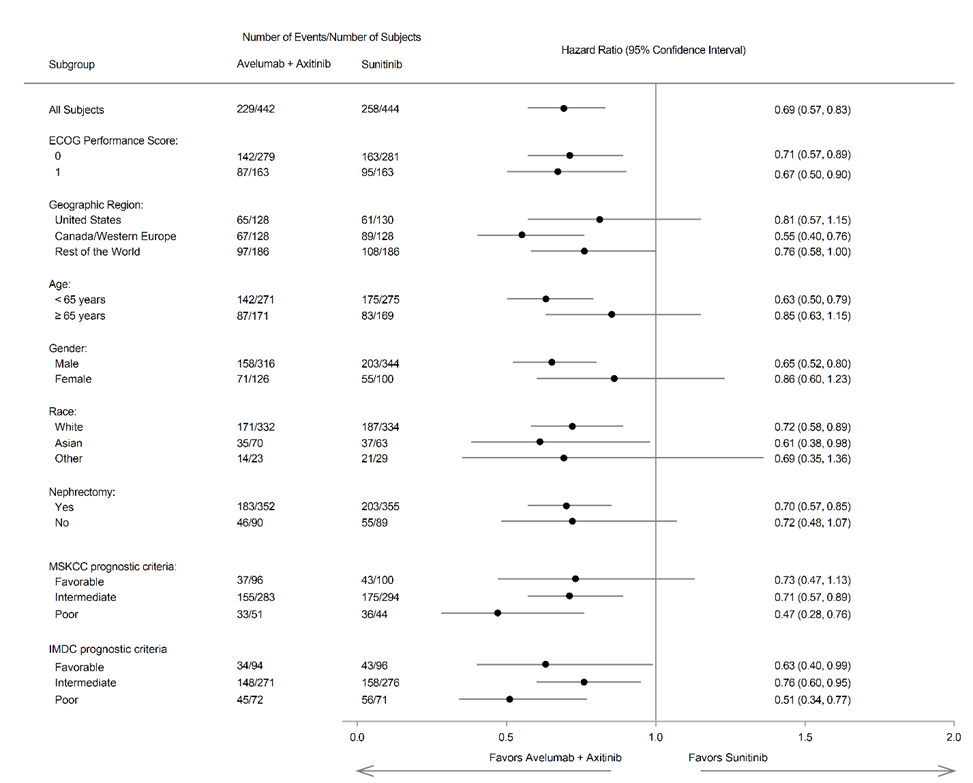
Immunogeenisuus
Hoidon aikana ilmaantuneita lääkevasta-aineita (ADA, anti drug antibodies) havaittiin 8,5 %:lla MCC-potilaista (EMR107000 003 tutkimus, 8,9 % osassa A ja 8,2 % osassa B), 19 %:lla UC-potilaista (B9991001 tutkimus) ja 16 %:lla RCC-potilaista (B9991003 tutkimus). Suurin osa ADA-aineista oli luonteeltaan neutraloivia. ADA-aineiden tai neutraloivien vasta aineiden (nAb) vaikutuksesta farmakokinetiikkaan, tehoon tai turvallisuuteen ei havaittu viitteitä.
Pediatriset potilaat
Tutkimus MS100070-0306 oli avoin vaiheen I/II monikeskustutkimus, jossa avelumabin annosta, turvallisuutta ja siedettävyyttä, kasvaimia torjuvaa vaikutusta, farmakokinetiikkaa ja farmakodynamiikkaa arvioitiin pediatrisilla potilailla syntymästä alle 18 vuoden ikään. Potilailla oli hoitoon huonosti reagoivia tai uusiutuneita kiinteitä kasvaimia, mukaan lukien keskushermoston kasvaimia ja lymfoomia, joihin ei joko ollut mitään standardihoitoa tai joihin saatavilla olevaa hoitoa potilas ei voinut saada.
Tutkimukseen otettiin 21 pediatrista potilasta, joiden ikä oli 3–17 vuotta (11 potilasta oli ≤ 12‑vuotiaita ja 10 potilasta oli > 12‑vuotiaita) ja jotka saivat avelumabia joko 10 mg/kg (N = 6) tai 20 mg/kg (N = 15) laskimoon 2 viikon välein vahvistettuun taudin etenemiseen, kuolemaan tai sietämättömään toksisuuteen asti.
Primaariset kasvainluokat olivat pehmytkudos-/luusarkooma (N = 12), keskushermoston pahanlaatuiset kasvaimet (N = 8) ja ruoansulatuskanavan karsinooma (N = 1).
Tässä tutkimuksessa ei todettu täydellisiä vasteita (CR) eikä osittaisia vasteita (PR) RECIST 1.1 -kriteerien mukaan arvioituna.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Bavencio-valmisteen käytöstä merkelinsolukarsinooman, uroteelikarsinooman ja munuaissolukarsinooman hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Monoterapiana annetun avelumabin ja yhdessä aksitinibin kanssa annetun avelumabin populaatiofarmakokineettisen analyysin perusteella avelumabin altistuksessa ei odoteta olevan kliinisesti merkittäviä eroja potilasryhmien välillä annettuna kahden viikon välein annoksella 800 mg tai 10 mg/kg.
Jakautuminen
Avelumabin oletetaan jakautuvan systeemiseen verenkiertoon sekä vähemmässä määrin solunulkoiseen tilaan. Jakautumistilavuus vakaan tilan aikana oli 4,72 l.
Koska avelumabin ekstravaskulaarinen jakautuminen on vähäistä, sen jakautumistilavuus vakaan tilan aikana on pieni. Koska avelumabi on vasta-aine, se ei sitoudu spesifisesti plasman proteiineihin.
Eliminaatio
Avelumabin systeeminen kokonaispuhdistuma (CL) on 0,59 l/vrk perustuen 1 629 potilaan populaatiofarmakokineettiseen analyysiin. Täydentävässä analyysissa avelumabin puhdistuma laski ajan myötä: suurin keskimääräinen maksimaalinen lasku (variaatiokerroin% [CV%]) lähtötilanteeseen verrattuna eri kasvaintyypeillä oli noin 32,1 % (CV 36,2 %).
Avelumabin vakaa pitoisuus saavutettiin noin 4–6 viikon (2–3 syklin) kuluttua, kun toistuva annos oli 10 mg/kg kahden viikon välein, ja systeeminen kumuloituminen oli noin 1,25-kertainen.
Suositeltua annosta käytettäessä eliminaation puoliintumisaika (t½) on 6,1 vuorokautta populaatiofarmakokineettisen analyysin perusteella.
Lineaarisuus/ei-lineaarisuus
Avelumabialtistus suureni suhteessa annokseen annosalueella 10–20 mg/kg kahden viikon välein.
Kun avelumabia annettiin 10 mg/kg yhdessä 5 mg:n aksitinibiannoksen kanssa, avelumabin ja aksitinibin altistukset säilyivät kumpikin muuttumattomina verrattuna näihin lääkeaineisiin yksin annettuina. Tutkimuksissa ei ilmennyt näyttöä, joka viittaisi avelumabin kliinisesti merkittävään puhdistuman muutokseen edennyttä RCC:tä sairastavilla potilailla.
Erityisryhmät
Populaatiofarmakokineettisen analyysin perusteella ikä, sukupuoli, rotu, PDL1-status, kasvaintaakka, munuaisten vajaatoiminta ja lievä tai kohtalainen maksan vajaatoiminta eivät vaikuttaneet avelumabin systeemiseen kokonaispuhdistumaan.
Systeeminen kokonaispuhdistuma suurenee potilaan painon lisääntyessä. Painon mukaan normalisoitu annostus sai aikaan suunnilleen samansuuruisen vakaan tilan altistuksen hyvin vaihtelevanpainoisilla (30–204 kg) ihmisillä.
Munuaisten vajaatoiminta
Avelumabin puhdistumassa ei ilmennyt kliinisesti merkittäviä eroja niiden potilaiden välillä, joilla oli lievä munuaisten vajaatoiminta (glomerulusten laskennallinen suodatusnopeus (GFR) 60–89 ml/min, Cockcroft-Gaultin menetelmällä mitattu kreatiniinipuhdistuma (CrCL); n=623), kohtalainen munuaisten vajaatoiminta (GFR 30–59 ml/min, n=320) ja normaali munuaisten toiminta (GFR ≥ 90 ml/min, n=671).
Avelumabia ei ole tutkittu potilailla, joilla on vaikea munuaisten vajaatoiminta (GFR 15–29 ml/min).
Maksan vajaatoiminta
Populaatiofarmakokineettisessä analyysissa avelumabin puhdistumassa ei ilmennyt kliinisesti merkittäviä eroja niiden potilaiden välillä, joilla oli lievä maksan vajaatoiminta (bilirubiini ≤ ULN ja ASAT > ULN tai bilirubiini 1–1,5 kertaa ULN, n=217) ja normaali maksan toiminta (bilirubiini ja ASAT ≤ ULN, n=1 388). Maksan vajaatoiminta määritettiin Yhdysvaltain syöpäinstituutin (NCI, National Cancer Institute) maksan toimintahäiriön kriteerien perusteella.
Avelumabia ei ole tutkittu potilailla, joilla on kohtalainen maksan vajaatoiminta (bilirubiini 1,5–3 kertaa ULN) tai vaikea maksan vajaatoiminta (bilirubiini > 3 kertaa ULN).
Pediatriset potilaat
Avelumabin farmakokinetiikkaa arvioitiin tutkimuksessa MS100070-0306 yhteensä 21 lapsella ja nuorella, joiden ikä oli 3–17 vuotta ja jotka saivat avelumabia joko 10 mg/kg (N = 6) tai 20 mg/kg (N = 15) laskimoon 2 viikon välein vahvistettuun taudin etenemiseen, kuolemaan tai sietämättömään toksisuuteen asti.
Pediatriset farmakokineettiset parametrit ja vastaavat farmakokineettiset profiilit arvioitiin kaikkien potilaiden osalta annoksen mukaan ja stratifioitiin painon perusteella.
Avelumabia 20 mg/kg saaneiden pediatristen potilaiden altistus oli samaa luokkaa tai suurempi kuin avelumabia 10 mg/kg tai 800 mg saaneilla aikuisilla. Avelumabia 10 mg/kg saaneiden pediatristen potilaiden altistus oli pienempi kuin aikuisilla.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Toistuvan altistuksen aiheuttamaa toksisuutta selvitettiin jaavanmakakeilla, joille annettiin suonensisäisesti 20, 60 tai 140 mg/kg viikon välein 1 kuukauden tai 3 kuukauden ajan; 3 kuukauden antojaksoa seurasi 2 kuukauden palautumisjakso. Perivaskulaarista mononukleaarista solujen paksuuntumista havaittiin niiden apinoiden aivoissa ja selkäytimessä, jotka saivat avelumabia ≥ 20 mg/kg 3 kuukauden ajan. Vaikka annoksen ja vasteen välillä ei ollut selkeää yhteyttä, löydöksen yhteyttä avelumabihoitoon ei voida kuitenkaan sulkea pois.
Avelumabilla ei ole tehty lisääntymistutkimuksia eläimillä. Anti-PD-1/PD-L1-reitin ajatellaan olevan osallisena toleranssin ylläpitämisessä sikiötä kohtaan koko raskauden ajan. Tiineiden hiirten eläinmalleissa PD-L1-signaalinvälityksen eston on osoitettu heikentävän toleranssia sikiötä kohtaan ja lisäävän keskenmenoja. Nämä tulokset viittaavat siihen, että avelumabin käyttö raskauden aikana saattaa olla haitallista sikiölle, ja se voi lisätä keskenmenon tai sikiökuoleman riskiä.
Tutkimuksia ei ole tehty avelumabin karsinogeenisuuden tai genotoksisuuden selvittämiseksi.
Avelumabilla ei ole tehty hedelmällisyystutkimuksia. Apinoilla tehdyissä yhden ja kolmen kuukauden toistuvien annosten toksisuustutkimuksissa ei ilmennyt merkittäviä naaraiden lisääntymiselimiin kohdistuvia vaikutuksia. Monet näissä tutkimuksissa käytetyistä urosapinoista olivat seksuaalisesti epäkypsiä ja näin ollen ei voida tehdä selviä päätelmiä valmisteen urosten lisääntymiselimiin kohdistuvista vaikutuksista.
Farmaseuttiset tiedot
Apuaineet
Mannitoli (E421)
Väkevä etikkahappo
Polysorbaatti 20 (E432)
Natriumhydroksidi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta
Avaamisen jälkeen
Mikrobiologiselta kannalta lääkevalmiste tulisi laimentaa ja infusoida heti avaamisen jälkeen.
Infuusion valmistamisen jälkeen
Laimennetun liuoksen kemiallisen ja fysikaalisen käytönaikaisen säilyvyyden on osoitettu olevan seuraava:
| Infuusion valmistelu | Säilytys 2 ºC – 8 ºC:ssa valolta suojattuna | Säilytys 20 °C – 25 °C:ssa ja huonevalaistuksessa |
| Natriumkloridi 9 mg/ml (0,9 %) | 96 tuntia | 72 tuntia |
| Natriumkloridi 4,5 mg/ml (0,45 %) | 24 tuntia | 24 tuntia |
Mikrobiologiselta kannalta laimennettu liuos tulisi infusoida välittömästi, ellei laimentamista suoriteta tavalla, jossa mikrobisaastuminen ei ole mahdollista. Jos liuosta ei käytetä välittömästi, käytönaikaiset säilytysajat ja -olosuhteet ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2°C ‑ 8°C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BAVENCIO infuusiokonsentraatti, liuosta varten
20 mg/ml (L:ei) 10 ml (1220,43 €)
PF-selosteen tieto
10 ml konsentraattia injektiopullossa (tyypin I lasia), jossa on halobutyylitulppa ja irrotettavalla muovikorkilla varustettu alumiinisuljin.
Pakkauksessa on 1 injektiopullo.
Valmisteen kuvaus:
Kirkas, väritön tai kellertävä liuos. Liuoksen pH on 5,0–5,6 ja sen osmolaliteetti on 285‑350 mOsm/kg.
Käyttö- ja käsittelyohjeet
Bavencio-valmisteen kanssa voidaan käyttää polyetyleenistä, polypropyleenistä ja etyleenivinyyliasetaatista valmistettuja infuusiopusseja, lasipulloja, polyvinyylikloridista valmistettuja infuusiosarjoja sekä polyeteerisulfonista valmistettuja in-line-suodattimia, joiden huokoskoko on 0,2 mikrometriä.
Käsittelyohjeet
Käytä infuusioliuoksen valmistamisessa aseptista tekniikkaa.
- Tarkasta injektiopullo silmämääräisesti mahdollisten hiukkasten ja värimuutosten havaitsemiseksi. Bavencio on kirkas, väritön tai kellertävä liuos. Jos liuos on sameaa tai siinä on värimuutoksia tai hiukkasia, hävitä injektiopullo.
- Käytä sopivankokoista (mieluiten 250 ml) infuusiopussia, joka sisältää joko natriumkloridi 9 mg/ml (0,9 %)- tai natriumkloridi 4,5 mg/ml (0,45 %) liuosta. Vedä injektiopullo(i)sta ruiskuun tarvittava määrä Bavencio-valmistetta ja ruiskuta se infuusiopussiin. Hävitä osittain käytetyt ja tyhjät injektiopullot.
- Sekoita laimennettua liuosta kääntelemällä pussia varovasti vaahtoamisen ja liiallisten sekoitusvoimien estämiseksi.
- Tarkasta liuos varmistaaksesi, että se on kirkasta ja väritöntä ja että siinä ei ole näkyviä hiukkasia. Käytä laimennettu liuos heti valmistamisen jälkeen.
- Älä anna muita lääkevalmisteita saman infuusioletkun kautta. Anna infuusioliuos, käyttäen steriiliä, pyrogeenitonta, heikosti proteiinia sitovaa 0,2 mikrometrin kiinteää (in‑line) tai irrallista (add‑on) suodatinta kohdassa Annostus ja antotapa annetun kuvauksen mukaisesti.
Huuhtele letku Bavencio-valmisteen antamisen jälkeen joko natriumkloridi 9 mg/ml (0,9 %)- tai natriumkloridi 4,5 mg/ml (0,45 %) liuoksella.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BAVENCIO infuusiokonsentraatti, liuosta varten
20 mg/ml 10 ml
- Ei korvausta.
ATC-koodi
L01FF04
Valmisteyhteenvedon muuttamispäivämäärä
25.05.2026
Yhteystiedot
Keilaranta 6
02150 Espoo
09 867 8700
www.merck.fi