
EVRYSDI jauhe oraaliliuosta varten 0,75 mg/ml
Vaikuttavat aineet ja niiden määrät
Yksi pullo sisältää 60 mg risdiplaamia 2 g:ssa jauhetta oraaliliuosta varten.
Yksi ml käyttökuntoon saatettua liuosta sisältää 0,75 mg risdiplaamia.
Apuaineet, joiden vaikutus tunnetaan
Yksi ml sisältää 0,38 mg natriumbentsoaattia (E211) ja 2,97 mg isomaltia (E953).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Jauhe oraaliliuosta varten.
Kliiniset tiedot
Käyttöaiheet
Evrysdi on tarkoitettu kromosomiin 5q liittyvän spinaalisen lihasatrofian (SMA, spinal muscular atrophy) hoitoon potilaille, joilla on tyypin 1, tyypin 2 tai tyypin 3 spinaalisen lihasatrofian kliininen diagnoosi tai yhdestä neljään SMN2-kopiota.
Ehto
Hoidon aloittavan lääkärin tulee olla perehtynyt kyseessä olevan sairauden hoitoon.
Annostus ja antotapa
Risdiplaamihoito tulisi aloittaa sellaisen lääkärin toimesta, joka on perehtynyt spinaalisen lihasatrofian hoitoon.
Annostus
Suositeltu risdiplaamiannos otetaan kerran päivässä ja määräytyy iän ja painon mukaan (ks. taulukko 1).
Taulukko 1. Evrysdi- jauheen oraaliliuosta varten iän ja painon mukainen annostus
| Ikä* ja paino | Suositeltu vuorokausiannos |
| < 2 kuukautta | 0,15 mg/kg |
| 2 kuukautta – < 2 vuotta | 0,20 mg/kg |
| ≥ 2 vuotta (< 20 kg) | 0,25 mg/kg |
| ≥ 2 vuotta (≥ 20 kg) | 5 mg |
| * keskosvauvoilla perustuu korjattuun ikään | |
Potilaille, joiden ikä on ≥ 2 vuotta ja paino ≥ 20 kg, on saatavana vaihtoehtoinen valmistemuoto kalvopäällysteiset tabletit. Ks. Evrysdi- kalvopäällysteisten tablettien valmisteyhteenveto.
Lääkärin pitää määrätä sopiva lääkemuoto tarvittavan annoksen ja potilaan tarpeen mukaan huomioiden myös potilaan nielemiskyky. Jos potilaalla on vaikeuksia niellä kokonainen tabletti tai jos valmiste on annettava nenä-mahaletkun tai gastrostomialetkun kautta, kalvopäällysteinen tabletti voidaan dispergoida veteen tai potilaalle voidaan määrätä jauhetta oraaliliuosta varten.
Hoitoa yli 5 mg:n vuorokausiannoksilla ei ole tutkittu.
Annosten myöhästyminen tai unohtuminen
Jos suunniteltu annos unohtuu ja hoitoaikataulun mukaisesta ajankohdasta on kulunut alle 6 tuntia, annos pitää antaa mahdollisimman pian. Muussa tapauksessa unohtunut annos pitää jättää antamatta ja antaa seuraava annos seuraavana päivänä tavanomaisena hoitoaikataulun mukaisena ajankohtana.
Jos potilas ei niele risdiplaamiannosta kokonaan tai oksentaa risdiplaami-annoksen ottamisen jälkeen, uutta annosta ei pidä antaa vajaan annoksen korvaamiseksi. Seuraava annos pitää antaa tavanomaisena hoitoaikataulun mukaisena ajankohtana.
Iäkkäät
65-vuotiaista ja sitä vanhemmista tutkittavista saatujen suppeiden tietojen perusteella iäkkäiden potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Risdiplaamia ei ole tutkittu tässä potilasryhmässä. Munuaisten vajaatoimintaa sairastavien potilaiden annosta ei oletettavasti tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Lievää tai keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa. Vaikeaa maksan vajaatoimintaa sairastavia potilaita ei ole tutkittu, ja heillä altistus risdiplaamille voi olla suurentunut (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Pediatriset potilaat
Tietoja ei ole saatavilla risdiplaamin farmakokinetiikasta alle 16 vuorokauden ikäisillä potilailla.
Antotapa
Suun kautta.
Terveydenhuollon ammattilaisen (esim. apteekkihenkilökunnan) on saatettava Evrysdi- jauhe oraaliliuosta varten käyttökuntoon ennen sen toimittamista potilaalle.
Terveydenhuollon ammattilaisen on suositeltavaa kertoa potilaalle tai potilaan huoltajalle ennen ensimmäisen annoksen antamista, miten määrätty päivittäinen annos valmistellaan.
Evrysdi-valmistetta otetaan suun kautta kerran päivässä ruoan kanssa tai tyhjään mahaan joka päivä suunnilleen samaan aikaan päivästä käyttämällä pakkauksen sisältämää useaan käyttökertaan tarkoitettua mittaruiskua. Evrysdi-valmistetta ei pidä sekoittaa maitoon eikä äidinmaidonkorvikkeeseen.
Evrysdi pitää ottaa heti sen jälkeen, kun se on vedetty mittaruiskuun. Jos sitä ei oteta 5 minuutin kuluessa, mittaruiskun sisältö pitää hävittää ja valmistella uusi annos. Jos Evrysdi-valmistetta roiskuu tai joutuu iholle, alue pitää pestä vedellä ja saippualla.
Potilaan pitää juoda vettä Evrysdin ottamisen jälkeen sen varmistamiseksi, että koko lääkevalmisteannos on nielty. Jos potilas ei kykene nielemään tai jos hänellä on nenä-mahaletku tai gastrostomialetku kiinnitettynä, Evrysdi- jauhe oraaliliuosta varten voidaan antaa letkun kautta. Letku pitää huuhdella vedellä Evrysdin antamisen jälkeen.
Mittaruiskun valitseminen määrätyn vuorokausiannoksen mukaan:
| Ruiskun koko | Annostilavuus | Ruiskun merkinnät |
| 1 ml | 0,3 ml – 1 ml | 0,01 ml |
| 6 ml | 1 ml – 6 ml | 0,1 ml |
| 12 ml | 6,2 ml – 6,6 ml | 0,2 ml |
Annoksen tilavuuden laskemisessa pitää huomioida ruiskun merkinnät. Annoksen tilavuus pitää pyöristää valitun mittaruiskun lähimpään annosasteikkomerkintään.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Mahdollinen alkio- ja sikiötoksisuus
Eläinkokeissa on havaittu alkio- ja sikiötoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Lisääntymiskykyisille potilaille pitää kertoa riskeistä, ja heidän on käytettävä erittäin tehokasta ehkäisyä hoidon aikana sekä naispotilaiden vähintään 1 kuukauden ajan viimeisen annoksen jälkeen ja miespotilaiden 4 kuukauden ajan viimeisen annoksen jälkeen. Naispotilaista, jotka voivat tulla raskaaksi, pitää varmistaa ennen risdiplaamihoidon aloittamista, ovatko he raskaana (ks. kohta Raskaus ja imetys).
Mahdolliset vaikutukset miesten hedelmällisyyteen
Eläinkokeiden havaintojen vuoksi miespotilaiden ei pidä luovuttaa siemennestettä hoidon aikana eikä 4 kuukauteen viimeisen risdiplaamiannoksen jälkeen. Lisääntymiskykyisten miespotilaiden pitää keskustella hedelmällisyyden säilyttämismahdollisuuksista ennen hoidon aloittamista (ks. kohdat Raskaus ja imetys ja Prekliiniset tiedot turvallisuudesta). Risdiplaamin vaikutuksia miesten hedelmällisyyteen ei ole tutkittu ihmisillä.
Apuaineet
Isomalti
Evrysdi sisältää isomaltia (2,97 mg per ml). Potilaiden, joilla on harvinainen perinnöllinen fruktoosi-intoleranssi, ei pidä käyttää tätä lääkettä.
Natrium
Evrysdi sisältää 0,375 mg natriumbentsoaattia per ml. Natriumbentsoaatti voi lisätä vastasyntyneen (enintään 4 viikon ikäisen) ikterusta (ihon ja silmien keltaisuutta).
Evrysdi sisältää alle 1 mmol natriumia (23 mg) per 5 mg:n annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset risdiplaamiin
Risdiplaamin farmakokineettisissä parametreissa ei todettu kliinisesti oleellisia vaikutuksia, kun suun kautta otettavan 6 mg:n risdiplaamikerta-annoksen kanssa samaan aikaan otettiin 200 mg:n itrakonatsoliannos (voimakas CYP3A:n estäjä) kaksi kertaa päivässä (AUC-arvo suureni 11 %, Cmax-arvo pieneni 9 %). Annosta ei tarvitse muuttaa, kun risdiplaamia käytetään samaan aikaan jonkin CYP3A:n estäjän kanssa.
Yhteisvaikutuksia FMO1- ja FMO3-reittien välityksellä ei oletettavasti esiinny.
Risdiplaamin vaikutukset muihin lääkevalmisteisiin
Risdiplaami on CYP3A:n heikko estäjä. Suun kautta kerran päivässä 2 viikon ajan terveille aikuisille tutkittaville annettu risdiplaami lisäsi hieman altistusta (AUC-arvo suureni 11 %; Cmax-arvo suureni 16 %) midatsolaamille, joka on herkkä CYP3A:n substraatti. Yhteisvaikutuksen laajuutta ei katsota kliinisesti oleelliseksi, joten CYP3A:n substraattien annosta ei tarvitse muuttaa.
In vitro -tutkimukset ovat osoittaneet, että risdiplaami ja sen pääasiallinen metaboliitti ihmisellä M1 eivät ole ihmisen MDR1:n, orgaanisten anionin kuljettajapolypeptidien (OATP)1B1, OATP1B3, orgaanisten anionin kuljettajaproteiinien 1 ja 3 (OAT 1 ja 3) merkittäviä estäjiä. Risdiplaami ja sen metaboliitti ovat in vitro ihmisen orgaanisen kationin kuljettajaproteiinin 2 (OCT2) ja monilääke- ja toksiiniekstruusioproteiinien (MATE)1 ja MATE2-K kuljettajia. Terapeuttisilla lääkeainepitoisuuksilla ei oletettavasti esiinny yhteisvaikutuksia OCT2:n substraattien kanssa. Risdiplaamin samanaikaisen käytön vaikutusta MATE1:n ja MATE2-K:n substraattien farmakokinetiikkaan ihmisellä ei tunneta. In vitro -tietojen perusteella risdiplaami saattaa suurentaa MATE1:n tai MATE2-K:n välityksellä eliminoituvien lääkevalmisteiden, kuten metformiinin, pitoisuutta plasmassa. Jos samanaikaista käyttöä ei voida välttää, lääkevalmisteeseen liittyvää toksisuutta pitää seurata ja samanaikaisesti käytettävän lääkkeen annoksen pienentämistä pitää tarvittaessa harkita.
Risdiplaamin ja nusinerseenin samanaikaisen käytön tueksi ei ole tehoa tai turvallisuutta koskevia tietoja.
Raskaus ja imetys
Lisääntymiskykyiset potilaat
Ehkäisy mies- ja naispotilaille
Lisääntymiskykyisten mies- ja naispotilaiden pitää noudattaa seuraavia ehkäisyä koskevia vaatimuksia:
- Naispotilaiden, jotka voivat tulla raskaaksi, pitää käyttää erittäin tehokasta ehkäisyä hoidon aikana ja vähintään 1 kuukauden ajan viimeisen annoksen jälkeen.
- Miespotilaiden ja heidän naiskumppaniensa, jotka voivat tulla raskaaksi, pitää varmistaa erittäin tehokas ehkäisy hoidon aikana ja vähintään 4 kuukauden ajan viimeisen annoksen jälkeen.
Raskaustestit
Naispotilaista, jotka voivat tulla raskaaksi, pitää ennen risdiplaamihoidon aloittamista varmistaa, ovatko he raskaana. Raskaana oleville naisille pitää kertoa selkeästi sikiölle mahdollisesti aiheutuvasta riskistä.
Raskaus
Risdiplaamin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Risdiplaamin käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä ehkäisyä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Imetys
Ei tiedetä, erittyykö risdiplaami ihmisillä äidinmaitoon. Rotilla tehdyt tutkimukset osoittavat, että risdiplaami erittyy maitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Imetettävälle lapselle mahdollisesti aiheutuvaa haittaa ei tunneta, joten on suositeltavaa olla imettämättä hoidon aikana.
Hedelmällisyys
Miespotilaat
Prekliinisten havaintojen perusteella miesten hedelmällisyys saattaa heikentyä hoidon aikana. Rottien ja apinoiden lisääntymiselimissä havaittiin siemennesteen laadun heikkenemistä ja siittiöiden määrän vähenemistä (ks. kohta Prekliiniset tiedot turvallisuudesta). Eläinkokeiden havaintojen perusteella vaikutusten siittiösoluihin odotetaan kumoutuvan risdiplaamin käytön lopettamisen jälkeen.
Miespotilaat voivat harkita siemennesteen talteenottoa ennen hoidon aloittamista tai vähintään 4 kuukauden hoidottoman jakson jälkeen. Jos miespotilas haluaa siittää lapsen, hoito on lopetettava vähintään 4 kuukaudeksi. Hoitoa voidaan jatkaa hedelmöittymisen jälkeen.
Naispotilaat
Risdiplaami ei prekliinisten tietojen (ks. kohta Prekliiniset tiedot turvallisuudesta) perusteella oletettavasti vaikuta naisten hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Risdiplaamilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Imeväisiässä alkavaa spinaalista lihasatrofiaa sairastavilla potilailla risdiplaamin kliinisissä tutkimuksissa yleisimmin havaitut haittavaikutukset olivat kuume (54,8 %), ihottuma (29,0 %) ja ripuli (19,4 %).
Myöhemmin alkavaa spinaalista lihasatrofiaa sairastavilla potilailla risdiplaamin kliinisissä tutkimuksissa yleisimmin havaitut haittavaikutukset olivat kuume (21,7 %), päänsärky (20,0 %), ripuli (16,7 %) ja ihottuma (16,7 %).
Edellä mainituilla haittavaikutuksilla ei ollut tunnistettavissa olevaa kliinistä tai ajallista ilmenemistapaa, ja ne yleensä hävisivät, vaikka imeväisiässä alkavaa ja myöhemmin alkavaa spinaalista lihasatrofiaa sairastavien potilaiden hoito jatkui.
Haittavaikutustaulukko
Kunkin haittavaikutuksen esiintyvyysluokka perustuu seuraavaan esitystapaan: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Kliinisissä tutkimuksissa havaitut haittavaikutukset (taulukko 2) luetellaan MedDRA-elinjärjestelmäluokituksen mukaisesti.
Taulukko 2. Imeväisiässä alkavaa ja myöhemmin alkavaa spinaalista lihasatrofiaa sairastavilla potilailla risdiplaamin kliinisten tutkimusten perusteella esiintyneet haittavaikutukset ja markkinoille tulon jälkeiset kokemukset
| Elinjärjestelmäluokka | Imeväisiässä alkava spinaalinen lihasatrofia (tyyppi 1) | Myöhemmin alkava spinaalinen lihasatrofia (tyyppi 2 ja 3) |
| Infektiot | ||
| Virtsatieinfektio (mukaan lukien kystiitti) | Yleinen | Yleinen |
| Hermosto | ||
| Päänsärky | Ei sovellettavissa | Hyvin yleinen |
| Ruoansulatuselimistö | ||
| Ripuli | Hyvin yleinen | Hyvin yleinen |
| Pahoinvointi | Ei sovellettavissa | Yleinen |
| Suun haavaumat ja aftahaavat | Yleinen | Yleinen |
| Iho ja ihonalainen kudos | ||
| Ihottuma* | Hyvin yleinen | Hyvin yleinen |
| Kutaaninen vaskuliitti** | Tuntematon | |
| Luusto, lihakset ja sidekudos | ||
| Nivelsärky | Ei sovellettavissa | Yleinen |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Kuume (mukaan lukien hyvin korkea kuume) | Hyvin yleinen | Hyvin yleinen |
*Sisältää: dermatiitti, aknetyyppinen dermatiitti, allerginen dermatiitti, eryteema, follikuliitti, ihottuma, erytematoottinen ihottuma, makulopapulaarinen ihottuma, papulaarinen ihottuma
** Kutaanista vaskuliittia on raportoitu markkinoille tulon jälkeen. Oireet menivät ohi, kun risdiplaamihoito lopetettiin pysyvästi. Esiintyvyyttä ei voida arvioida saatavilla olevan tiedon perusteella.
Turvallisuusprofiili vielä oireettomilla potilailla
Evrysdi-valmisteen turvallisuusprofiili vielä oireettomilla potilailla on RAINBOWFISH-tutkimuksen ensisijaisen analyysin perusteella yhdenmukainen oireista imeväisiässä alkavaa ja myöhemmin alkavaa spinaalista lihasatrofiaa sairastavien potilaiden turvallisuusprofiilin kanssa. RAINBOWFISH-tutkimukseen otettiin mukaan 26 vielä oireetonta spinaalista lihasatrofiaa sairastavaa potilasta, joiden ikä ensimmäisen annoksen ajankohtana oli 16–41 vuorokautta (paino 3,1–5,7 kg). Altistuksen keston mediaani oli 20,4 kuukautta (vaihteluväli 10,6–41,9 kuukautta). Myyntiluvan myöntämisen jälkeisiä tietoja alle 20 vuorokauden ikäisistä vastasyntyneistä on rajallisesti saatavilla.
Turvallisuusprofiili potilailla, jotka ovat aiemmin saaneet muita spinaalisen lihasatrofian hoitoon käytettäviä lääkkeitä
Risdiplaamin turvallisuusprofiili spinaalisen lihasatrofian hoidossa potilailla, jotka olivat saaneet aiemmin hoitoa (mukaan lukien nusinerseeni- tai onasemnogeeniabeparvoveekkihoitoa aiemmin saaneet potilaat), on yhdenmukainen turvallisuusprofiilin kanssa potilailla, jotka eivät ole saaneet aiemmin hoitoa spinaaliseen lihasatrofiaan ja saivat risdiplaamihoitoa kliinisissä FIREFISH-, SUNFISH- ja RAINBOWFISH-tutkimuksissa (ks. kohta Farmakodynamiikka).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Risdiplaamin yliannostukseen ei tunneta vastalääkettä. Yliannoksen yhteydessä potilasta pitää seurata tarkoin, ja potilaalle pitää aloittaa tukihoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut tuki- ja liikuntaelinten sairauksien lääkkeet
ATC-koodi: M09AX10
Vaikutusmekanismi
Risdiplaami on survival motor neuron 2 -geenin (SMN2-geenin) lähetti-RNA:n (mRNA) esiasteen silmukoinnin muuntaja, joka on kehitetty 5q-kromosomin SMN-1-geenin mutaatioista johtuvan SMN-proteiinin puutoksen aiheuttaman spinaalisen lihasatrofian hoitoon. Toimivan SMN-proteiinin puutos on suoraan yhteydessä spinaalisen lihasatrofian patofysiologiaan, johon kuuluu mm. etenevä liikehermosolukato ja lihasheikkous. Risdiplaami korjaa SMN2-geenin silmukoinnin siten, että lähetti-RNA:n transkriptissa tasapaino siirtyy eksoni 7:n ekskluusiosta eksoni 7:n inkluusioon, jolloin toimivan ja stabiilin SMN-proteiinin tuotanto lisääntyy. Risdiplaami hoitaa siten spinaalista lihasatrofiaa suurentamalla ja ylläpitämällä toiminnallisen SMN-proteiinin pitoisuutta.
Farmakodynaamiset vaikutukset
Risdiplaami lisäsi imeväisiässä alkavaa spinaalista lihasatrofiaa ja myöhemmin alkavaa spinaalista lihasatrofiaa koskeneissa FIREFISH- (tutkimukseen tullessaan potilaat olivat 2–7 kuukauden ikäisiä), SUNFISH- (tutkimukseen tullessaan potilaat olivat 2–25 vuoden ikäisiä) ja JEWELFISH-tutkimuksissa (tutkimukseen tullessaan potilaat olivat 1–60 vuoden ikäisiä) SMN-proteiinin määrää veressä siten, että 4 viikon kuluessa hoidon aloittamisesta spinaalisen lihasatrofian kaikissa tutkituissa tyypeissä tapahtunut muutos lähtötilanteesta oli yli kaksinkertainen (mediaani). Lisäys säilyi koko hoitojakson ajan (vähintään 24 kuukautta).
Sydämen elektrofysiologia
Risdiplaamin vaikutusta QTc-aikaan arvioitiin tutkimuksessa, johon osallistui 47 tervettä aikuista. Terapeuttisella altistuksella risdiplaami ei pidentänyt QTc-aikaa.
Kliininen teho ja turvallisuus
Risdiplaamin tehoa imeväisiässä alkavaa spinaalista lihasatrofiaa (spinaalinen lihasatrofia tyyppi 1) ja myöhemmin alkavaa spinaalista lihasatrofiaa (spinaalinen lihasatrofia tyypit 2 ja 3) sairastavilla potilailla selvitettiin kahdessa kliinisessä pivotaalitutkimuksessa (FIREFISH ja SUNFISH). Risdiplaamin tehoa koskevat tiedot vielä oireettomien spinaalista lihasatrofiaa sairastavien potilaiden hoidossa arvioitiin kliinisessä RAINBOWFISH-tutkimuksessa. Tyypin 4 spinaalista lihasatrofiaa sairastavia potilaita ei ole tutkittu kliinisissä tutkimuksissa.
Imeväisiässä alkava spinaalinen lihasatrofia
BP39056-tutkimus (FIREFISH) on avoin, kaksiosainen tutkimus, jossa selvitetään risdiplaamin tehoa, turvallisuutta, farmakokinetiikkaa ja farmakodynamiikkaa oireista tyypin 1 spinaalista lihasatrofiaa sairastavilla potilailla (kaikilla potilailla oli geneettisesti varmistettu sairaus, johon liittyi kaksi SMN2-geenin kopiota). FIREFISH-tutkimuksen osa 1 oli suunniteltu tutkimuksen annoshakuosaksi. FIREFISH-tutkimuksen varmistavassa osassa 2 arvioitiin risdiplaamin tehoa. Osan 1 potilaat eivät osallistuneet osaan 2.
Keskeinen tehon päätetapahtuma oli kyky istua tuetta vähintään 5 sekuntia, joka mitattiin 12 kuukauden hoidon jälkeen BSID-III-asteikon (Bayley Scales of Infant and Toddler Development – Third Edition) karkeamotoriikkaa arvioivalla osiolla, kohta 22 (Item 22).
FIREFISH-tutkimuksen osa 2
FIREFISH-tutkimuksen osaan 2 otettiin mukaan 41 tyypin 1 spinaalista lihasatrofiaa sairastavaa potilasta. Tyypin 1 spinaalisen lihasatrofian kliinisten oireiden ja löydösten ilmaantuessa iän mediaani oli 1,5 kuukautta (vaihteluväli: 1,0–3,0 kuukautta); 54 % oli tyttöjä, 54 % oli valkoihoisia ja 34 % oli aasialaisia. Iän mediaani tutkimukseen tullessa oli 5,3 kuukautta (vaihteluväli: 2,2–6,9 kuukautta), ja ajan mediaani oireiden alkamisesta ensimmäiseen annokseen oli 3,4 kuukautta (vaihteluväli: 1,0–6,0 kuukautta). Lähtötilanteen CHOP-INTEND (Children's Hospital of Philadelphia Infant Test for Neuromuscular Disease) -pisteiden mediaani oli 22,0 pistettä (vaihteluväli: 8,0–37,0), ja HINE-2 (Hammersmith Infant Neurological Examination Module 2) -pisteiden mediaani oli 1,0 (vaihteluväli: 0,0–5,0).
Ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka kykenivät 12 kuukauden hoidon jälkeen istumaan tuetta vähintään 5 sekuntia (BSID-III-asteikon karkeamotoriikkaa arvioiva kohta 22). Risdiplaamihoitoa saaneiden potilaiden keskeiset tehon päätetapahtumat esitetään taulukossa 3.
Taulukko 3. Yhteenveto keskeisistä tehon tuloksista 12 kuukauden ja 24 kuukauden kohdalla (FIREFISH osa 2)
| Tehon päätetapahtumat | Potilaiden osuus N = 41 (90 %:n luottamusväli) | |
| Kuukausi 12 | Kuukausi 24 | |
| Motoriset toiminnot ja motorinen kehitystaso | ||
| BSID-III: istuu tuetta vähintään 5 sekuntia | 29,3 % (17,8 %; 43,1 %) p < 0,0001a | 61,0 % (46,9 %; 73,8 %) |
| CHOP-INTEND: pisteet 40 tai enemmän | 56,1 % (42,1 %; 69,4 %) | 75,6 % (62,2 %; 86,1 %) |
| CHOP-INTEND: ≥ 4 pisteen lisäys lähtötilanteesta | 90,2 % (79,1 %; 96,6 %) | 90,2 % (79,1 %; 96,6 %) |
| HINE-2: motorisen kehitystason suhteen vasteen saaneetb | 78,0 % (64,8 %; 88,0 %) | 85,4 % (73,2 %; 93,4 %) |
| HINE-2: istuu tuettac | 24,4 % (13,9 %; 37,9 %) | 53,7 % (39,8 %; 67,1 %) |
| Elossaolo ja elossaoloaika ilman tapahtumia | ||
| Elossaoloaika ilman tapahtumiad | 85,4 % (73,4 %; 92,2 %) | 82,9 % (70,5 %; 90,4 %) |
| Elossa | 92,7 % (82,2 %; 97,1 %) | 92,7 % (82,2 %; 97,1 %) |
| Nieleminen ja syöminen | ||
| Kykenee syömääne | 82,9 % (70,3 %; 91,7 %) | 85,4 % (73,2 %; 93,4 %) |
Lyhenteet: CHOP-INTEND = Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; HINE-2 = Module 2 of the Hammersmith Infant Neurological Examination.
a p-arvo perustuu yksitahoiseen eksaktiin binomitestiin. Tuloksia verrataan 5 %:n kynnysarvoon.
b HINE-2:n mukaan: tässä analyysissä vasteen saamiseksi määritellään ≥ 2 pisteen lisäys (tai maksimipisteet) kyvyssä potkia TAI ≥ 1 pisteen lisäys motorisessa kehitystasossa (pään hallinta, kääntyminen, istuminen, ryömiminen, seisominen tai käveleminen) JA useamman motorisen kehitystasoluokan paraneminen kuin huononeminen.
c Istuu tuetta, mukaan lukien potilaat, jotka kuukauden 24 aikapisteen HINE2-pisteiden perusteella ”istuvat tukevasti” (24 %, 10/41) ja ”kääntyvät istuessaan (pyörähtävät)” (29 %, 12/41).
d Tapahtuma vastaa pysyvän ventilaation päätetapahtumaa, joksi määriteltiin trakeostomia tai ≥ 16 tuntia ei-invasiivista ventilaatiota päivässä tai intubaatio > 21 peräkkäisen päivän ajan ilman akuuttia korjautuvaa tapahtumaa tai tällaisen tapahtuman häviämisen jälkeen. Kolme potilasta kuoli kolmen ensimmäisen kuukauden aikana tutkimukseen osallistumisesta ja neljällä potilaalla todettiin pysyvän ventilaation päätetapahtuma ennen kuukautta 24. Kaikkien näiden neljän potilaan CHOP-INTEND-pisteet suurenivat vähintään 4 pistettä lähtötilanteesta.
e Sisältää potilaat, jotka saivat ravintoa vain suun kautta (kaikkiaan 29 potilasta), ja potilaat, jotka saivat ravintoa suun ja ruokintaletkun kautta (kaikkiaan 6 potilasta), kuukautena 24.
Kuukauden 24 aikapisteessä 44 % potilaista istui tuetta 30 sekuntia (BSID-III-asteikon kohta 26). Potilaat jatkoivat HINE2-pisteillä mitattujen uusien motoristen kehitystasojen saavuttamista; 80,5 % osasi kääntyä ja 27 % potilaista saavutti seisomista osoittavan mittaustuloksen (12 % kannatteli painoa ja 15 % seisoi tuettuna).
Imeväisiässä alkavaa spinaalista lihasatrofiaa sairastavat hoitamattomat potilaat eivät koskaan kykenisi istumaan tuetta ja vain 25 % olisi oletettavasti elossa ilman jatkuvaa ventilaatiohoitoa 14 kuukauden ikää pidempään.
Kuva 1. Kaplan–Meierin kuvaaja elossaoloajasta ilman tapahtumia (FIREFISH-tutkimuksen osa 1 ja osa 2).
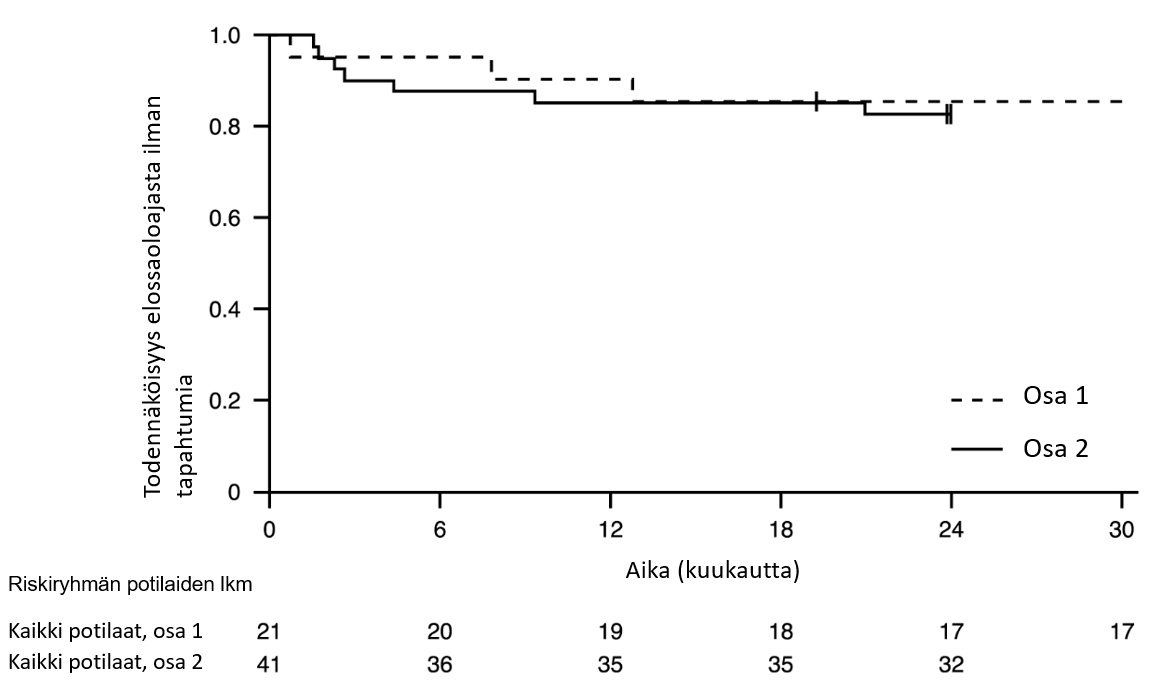
+ Sensuroitu: kaksi osan 2 potilasta sensuroitiin, sillä he olivat käyneet kuukauden 24 käynnillä liian aikaisin; yksi osan 1 potilas sensuroitiin hoidon lopettamisen jälkeen ja hän kuoli 3,5 kuukautta myöhemmin
Kuva 2. Keskimääräinen muutos lähtötilanteen CHOP-INTEND-kokonaispisteistä (FIREFISH-tutkimuksen osa 2)
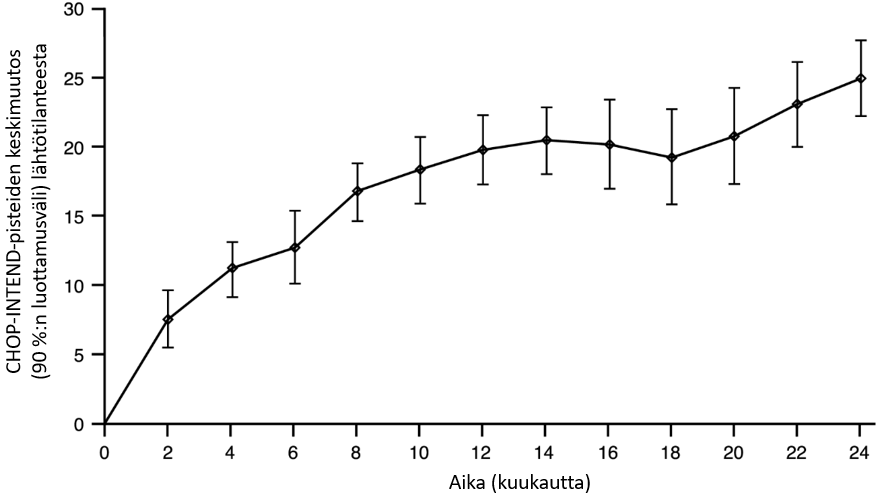
FIREFISH-tutkimuksen osa 1
FIREFISH-tutkimuksen osan 1 tulokset myös tukivat risdiplaamin tehoa tyypin 1 spinaalista lihasatrofiaa sairastavien potilaiden hoidossa. Osassa 1 mukana olleiden 21 potilaan lähtötilanteen ominaisuudet olivat yhdenmukaiset tyypin 1 spinaalista lihasatrofiaa sairastavien oireisten potilaiden kanssa. Iän mediaani tutkimukseen mukaan tullessa oli 6,7 kuukautta (vaihteluväli: 3,3–6,9 kuukautta), ja ajan mediaani oireiden alkamisen ja ensimmäisen annoksen välillä oli 4,0 kuukautta (vaihteluväli: 2,0–5,8 kuukautta).
Risdiplaamihoitoannoksen (osaan 2 valittu annos) sai yhteensä 17 potilasta. 12 hoitokuukauden jälkeen 41 % (7/17) näistä potilaista kykeni istumaan itsenäisesti vähintään 5 sekunnin ajan (BSID-III, osio 22). 24 hoitokuukauden jälkeen vielä 3 muuta hoitoannoksen saanutta potilasta kykeni istumaan itsenäisesti vähintään 5 sekuntia eli tämän motorisen kehitystason saavutti yhteensä 10 potilasta (59 %).
12 hoitokuukauden jälkeen 90 % (19/21) potilaista oli elossa eikä heillä ollut tapahtumia (ei jatkuvaa ventilaatiohoitoa) ja oli saavuttanut vähintään 15 kuukauden iän. Vähintään 33 hoitokuukauden jälkeen 81 % (17/21) potilaista oli elossa eikä heillä ollut tapahtumia ja oli saavuttanut vähintään 37 kuukauden iän (mediaani 41 kuukautta, vaihteluväli 37–53 kuukautta), ks. kuva 1. Kolme potilasta kuoli hoidon aikana, ja yksi potilas kuoli 3,5 kuukautta hoidon lopettamisen jälkeen.
Myöhemmin alkava spinaalinen lihasatrofia
BP39055-tutkimus (SUNFISH) on kaksiosainen monikeskustutkimus, jossa selvitetään risdiplaamin tehoa, turvallisuutta, farmakokinetiikkaa ja farmakodynamiikkaa tyypin 2 tai tyypin 3 spinaalista lihasatrofiaa sairastavilla 2–25-vuotiailla potilailla. Osa 1 oli eksploratiivinen annoshakuosio ja osa 2 oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu varmistava osio. Osan 1 potilaat eivät osallistuneet osaan 2.
Ensisijainen päätetapahtuma oli lähtötilanteen pisteiden muutos 12 kuukauden kohdalla MFM32 (Motor Function Measure-32) -mittarilla arvioituna. MFM32-mittarilla voidaan arvioida monia motorisia toimintoja monentyyppisillä spinaalista lihasatrofiaa sairastavilla potilailla. MFM32-kokonaispisteet ilmoitetaan suurimman mahdollisen pistemäärän prosenttiosuutena (vaihteluväli: 0–100), ja suuremmat pisteet osoittavat parempaa motorista toimintaa.
SUNFISH-tutkimuksen osa 2
SUNFISH-tutkimuksen osa 2 on SUNFISH-tutkimuksen satunnaistettu, kaksoissokkoutettu, lumekontrolloitu osio, jossa on mukana 180 tyypin 2 (71 %) tai tyypin 3 (29 %) spinaalista lihasatrofiaa sairastavaa kävelykyvytöntä potilasta. Potilaat satunnaistettiin suhteessa 2:1 joko risdiplaamihoitoon hoitoannoksilla (ks. kohta Annostus ja antotapa) tai lumehoitoon. Satunnaistaminen ositettiin ikäryhmittäin (2–5-vuotiaat, 6–11-vuotiaat, 12–17-vuotiaat, 18–25-vuotiaat).
Iän mediaani hoidon alussa oli 9,0 vuotta (vaihteluväli: 2–25 vuotta), ja ajan mediaani spinaalisen lihasatrofian ensimmäisten oireiden alkamisen ja ensimmäisen hoidon välillä oli 102,6 kuukautta (vaihteluväli: 1–275 kuukautta). Tutkimukseen mukaan tullessa 30 % oli 2–5-vuotiaita, 32 % oli 6–11-vuotiaita, 26 % oli 12–17-vuotiaita ja 12 % oli 18–25-vuotiaita. Tutkimuksessa mukana olleista 180 potilaasta 51 % oli tyttöjä/naisia, 67 % oli valkoihoisia ja 19 % oli aasialaisia. Lähtötilanteessa 67 %:lla potilaista oli skolioosi (32 %:lla potilaista oli vaikea-asteinen skolioosi). Potilaiden keskimääräiset lähtötilanteen MFM32-pisteet olivat 46,1 ja RULM (Revised Upper Limb Module) ‑pisteet olivat 20,1. Lähtötilanteen demografiset ominaisuudet olivat tasapainossa risdiplaami- ja lumehaarojen välillä, lukuun ottamatta potilaita, joilla oli skolioosi (risdiplaamihaarassa 63 %:lla potilaista ja lumehoitoa saaneessa verrokkihaarassa 73 %:lla potilaista).
MFM32-kokonaispisteiden muutoksessa lähtötilanteesta kuukauteen 12 todettiin SUNFISH-tutkimuksen osan 2 primaarianalyysissä kliinisesti merkittävä ja tilastollisesti merkitsevä ero risdiplaamihoitoa ja lumehoitoa saaneiden potilaiden välillä. Primaarianalyysin ja keskeisten toissijaisten päätetapahtumien tulokset esitetään taulukossa 4, kuvassa 3 ja kuvassa 4.
Taulukko 4. Yhteenveto 12 kuukauden hoidon tehosta myöhemmin alkavaa spinaalista lihasatrofiaa sairastavilla potilailla (SUNFISH-tutkimuksen osa 2)
| Päätetapahtuma | Risdiplaami (N = 120) | Lumehoito (N = 60) |
| Ensisijainen päätetapahtuma: | ||
MFM32-kokonaispisteiden muutos lähtötilanteesta1 12 kuukauden kohdalla Pienimmän neliösumman keskiarvo (95 %:n luottamusväli) | 1,36 (0,61; 2,11) | -0,19 (-1,22; 0,84) |
Ero lumehoitoon nähden Estimaatti (95 %:n luottamusväli) p-arvo2 | 1,55 (0,30; 2,81) 0,0156 | |
| Toissijaiset päätetapahtumat: | ||
| Niiden potilaiden osuus, joilla MFM32-kokonaispisteiden muutos lähtötilanteesta1 on 3 tai enemmän 12 kuukauden kohdalla (95 %:n luottamusväli)1 | 38,3 % (28,9; 47,6) | 23,7 % (12,0; 35,4) |
Kokonaisvasteen vetokertoimien suhde (odds ratio) (95 %:n luottamusväli) Vakioitu (vakioimaton) p-arvo3,4 | 2,35 (1,01; 5,44) 0,0469 (0,0469) | |
RULM-kokonaispisteiden muutos lähtötilanteesta5 12 kuukauden kohdalla Pienimmän neliösumman keskiarvo (95 %:n luottamusväli) | 1,61 (1,00; 2,22) | 0,02 (-0,83; 0,87) |
Ero lumehoidon estimaattiin nähden (95 %:n luottamusväli) Vakioitu (vakioimaton) p-arvo2,4 | 1,59 (0,55; 2,62) 0,0469 (0,0028) | |
- Perustuu MFM32-pisteiden osalta puuttuvien tietojen sääntöön, sillä 6 potilasta jätettiin pois analyysista (risdiplaami n = 115; lumehoitoa saanut verrokkiryhmä n = 59).
- Tiedot analysoitiin käyttämällä toistettujen mittausten sekamallia, jossa muuttujia olivat lähtötilanteen kokonaispisteet, hoito, käynti, ikäryhmä, hoito käynneittäin ja lähtötilanne käynneittäin.
- Tiedot analysoitiin käyttämällä logistista regressiota, jossa muuttujat olivat lähtötilanteen kokonaispisteet, hoito ja ikäryhmä.
- Hierarkkiseen testaukseen otettiin mukaan päätetapahtumille saatu vakioitu p-arvo, joka perustui päätetapahtumista hierarkkisessa järjestyksessä senhetkiseen päätetapahtumaan saakka saatuihin kaikkiin p-arvoihin.
- Perustuu RULM-pisteiden osalta puuttuvien tietojen sääntöön, sillä 3 potilasta jätettiin pois analyysista (risdiplaami n = 119; lumehoitoa saanut verrokkiryhmä n = 58).
12 hoitokuukauden päätyttyä 117 potilasta jatkoi hoitoa risdiplaamilla. 24 kuukauden analyysin ajankohtana näiden risdiplaamihoitoa 24 kuukauden ajan saaneiden potilaiden motorinen toiminta parani edelleen kuukausien 12 ja 24 välillä. MFM32-mittarilla todettu keskimääräinen muutos lähtötilanteesta oli 1,83 (95 %:n luottamusväli: 0,74; 2,92) ja RULM-mittarilla se oli 2,79 (95 %:n luottamusväli: 1,94; 3,64).
Kuva 3. MFM32-kokonaispisteiden keskimääräinen muutos lähtötilanteesta 12 kuukauden aikana SUNFISH-tutkimuksen osassa 21
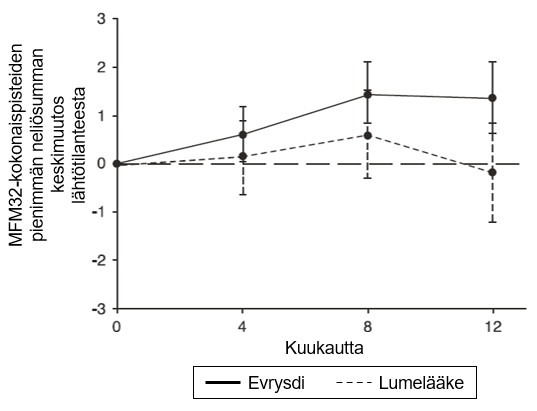
1Pienimmän neliösumman keskiarvon ero muutoksessa lähtötilanteen MFM32-pisteistä [95 %:n luottamusväli]
Kuva 4.RULM-kokonaispisteiden keskimääräinen muutos lähtötilanteesta 12 kuukauden aikana SUNFISH-tutkimuksen osassa 21
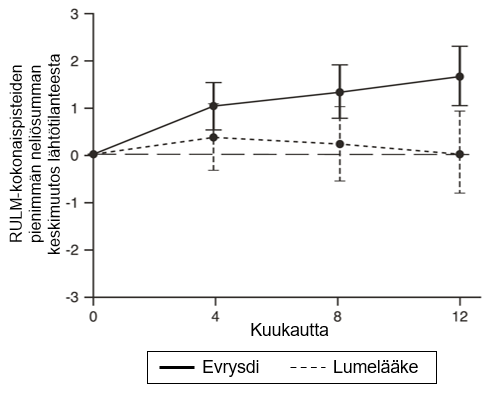
1Pienimmän neliösumman keskiarvon ero muutoksessa lähtötilanteen RULM-pisteistä [95 %:n luottamusväli]
SUNFISH-tutkimuksen osa 1
SUNFISH-tutkimuksen osan 1 (annoshakuosa) tulokset myös tukivat tehoa myöhemmin alkavaa spinaalista lihasatrofiaa sairastaville potilaille. Osaan 1 otettiin mukaan 51 tyyppien 2 ja 3 spinaalista lihasatrofiaa sairastavaa potilasta (mukaan lukien 7 kävelykykyistä potilasta), jotka olivat iältään 2–25-vuotiaita. Yhden hoitovuoden jälkeen motorisessa toiminnassa todettiin MFM32-mittarilla mitattuna kliinisesti merkittävää paranemista, ja keskimääräinen muutos lähtötilanteesta oli 2,7 pistettä (95 %:n luottamusväli: 1,5; 3,8). Parantuneet MFM32-pisteet säilyivät hoidon aikana 2 vuoteen saakka (keskimääräinen muutos 2,7 pistettä [95 %:n luottamusväli: 1,2; 4,2]).
Käyttö potilaille, jotka ovat aiemmin saaneet muita spinaalisen lihasatrofian hoitoon käytettäviä lääkkeitä (JEWELFISH)
BP39054-tutkimus (JEWELFISH, n = 174) on yhden hoitohaaran avoin tutkimus, jossa selvitetään risdiplaamin turvallisuutta, siedettävyyttä, farmakokinetiikkaa ja farmakodynamiikkaa potilailla, joilla on imeväisiässä tai myöhemmin alkava spinaalinen lihasatrofia (iän mediaani 14 vuotta [vaihteluväli 1–60 vuotta]) ja jotka olivat aiemmin saaneet hoitoa muilla myyntiluvallisilla (nusinerseeni n = 76, onasemnogeeniabeparvoveekki n = 14) tai tutkimusvaiheessa olevilla spinaalisen lihasatrofian hoitoon käytettävillä lääkkeillä. Lähtötilanteessa 168:sta iältään 2–60-vuotiaasta potilaasta 83 %:lla oli skolioosi ja 63 %:lla HFMSE-pisteet (Hammersmith Functional Motor Scale Expanded) olivat < 10 pistettä.
Hoidon 24 kuukauden aikapisteessä tehdyssä analyysissä 2–60‑vuotiailla potilailla todettiin yleisesti MFM-32- ja RULM-pisteillä mitattuna motoristen toimintojen stabiloitumista (MFM-32-pisteet n = 137 ja RULM-pisteet n = 133). Iältään alle 2-vuotiaiden potilaiden (n = 6) motorinen kehitystaso, kuten pään hallinta, kääntyminen ja istuminen tuetta, oli säilynyt tai parantunut. Kaikkien kävelykykyisten potilaiden (ikä 5–46 vuotta, n = 15) kävelykyky oli säilynyt.
Oireeton spinaalinen lihasatrofia (RAINBOWFISH)
BN40703-tutkimus (RAINBOWFISH) on avoin, yhden haaran kliininen monikeskustutkimus, jossa selvitetään risdiplaamin tehoa, turvallisuutta, farmakokinetiikkaa ja farmakodynamiikkaa imeväisikäisillä syntymästä 6 viikon ikään saakka (ensimmäisen annoksen ajankohta), kun imeväisikäisellä on geneettisesti todettu spinaalinen lihasatrofia, mutta oireita ei ole vielä ilmennyt.
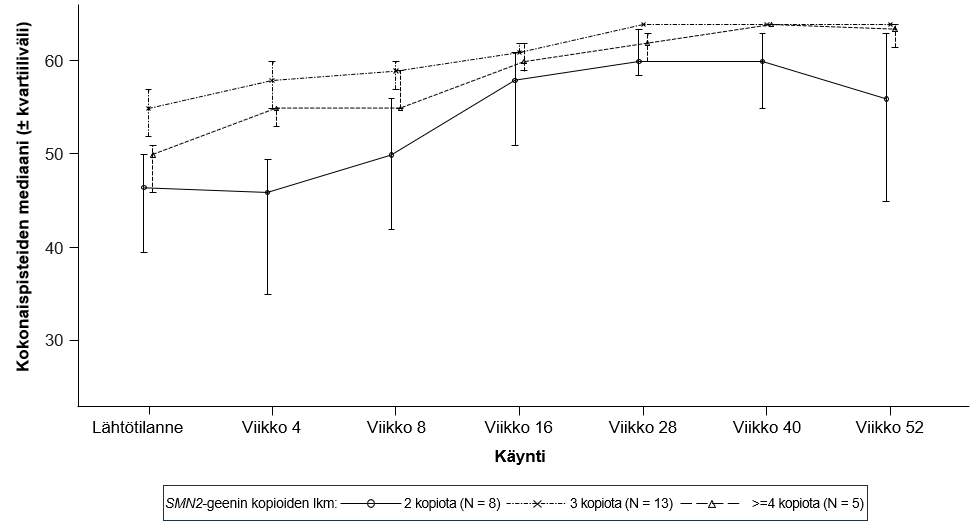
Teho vielä oireetonta spinaalista lihasatrofiaa sairastavilla potilailla arvioitiin 12 kuukauden aikapisteessä 26:lla risdiplaamihoitoa saaneella potilaalla (hoitoaikeen mukainen [ITT] potilasjoukko): kahdeksalla potilaalla oli kaksi SMN2-geenin kopiota, kolmellatoista potilaalla oli kolme SMN2-geenin kopiota ja viidellä potilaalla oli vähintään neljä SMN2-geenin kopiota. Näiden potilaiden iän mediaani ensimmäisen annoksen ajankohtana oli 25 vuorokautta (vaihteluväli: 16−41 vuorokautta), 62 % oli tyttöjä, 85 % oli valkoihoisia. Lähtötilanteessa CHOP-INTEND-pisteiden mediaani oli 51,5 (vaihteluväli: 35,0–62,0), HINE-2-pisteiden mediaani oli 2,5 (vaihteluväli: 0–6,0) ja ulnaarisen CMAP-mittauksen (ulnar nerve compound muscle action potential) amplitudin mediaani oli 3,6 mV (vaihteluväli: 0,5–6,7 mV).
Ensisijaisessa tehoa koskevassa potilasjoukossa (N = 5) oli mukana potilaita, joilla oli kaksi SMN2-geenin kopiota ja joiden CMAP-mittauksen amplitudi oli lähtötilanteessa ≥ 1,5 mV. Lähtötilanteessa näiden potilaiden CHOP-INTEND-pisteiden mediaani oli 48,0 (vaihteluväli: 36,0–52,0), HINE-2-pisteiden mediaani oli 2,0 (vaihteluväli: 1,0–3,0) ja CMAP-mittauksen amplitudin mediaani oli 2,6 mV (vaihteluväli: 1,6–3,8 mV).
Ensisijainen päätetapahtuma oli niiden potilaiden osuus ensisijaisessa tehon potilasjoukossa, jotka kykenivät 12 kuukauden aikapisteessä istumaan tuetta vähintään 5 sekuntia (BSID-III-asteikon karkeamotoriikkaa arvioiva osio, kohta 22 [Item 22]); tämän kehitystason saavuttaneiden potilaiden osuus oli tilastollisesti merkitsevä ja kliinisesti merkittävä verrattuna ennalta määriteltyyn 5 %:n toimintakykykriteeriin.
Risdiplaamihoitoa saaneiden potilaiden keskeiset tehon päätetapahtumat esitetään taulukoissa 5 ja 6 sekä kuvassa 5.
Taulukko 5. Vielä oireettomien potilaiden BSID-III-asteikon kohdalla 22 määritelty kyky istua 12 kuukauden aikapisteessä
| Tehon päätetapahtuma | Potilasjoukko | ||
| Ensisijainen teho (N = 5) | Potilaita, joilla kaksi SMN2-geenin kopiotaa (N = 8) | Hoitoaikeen mukainen (ITT) potilasjoukko (N = 26) | |
| Niiden potilaiden osuus, jotka kykenivät istumaan tuetta vähintään 5 sekunnin ajan (BSID-III-asteikon kohta 22); (90 %:n luottamusväli) | 80 % (34,3 %; 99,0 %) p < 0,0001b | 87,5 % (52,9 %; 99,4 %) | 96,2 % (83,0 %; 99,8 %) |
Lyhenteet: BSID-III = Bayley Scales of Infant and Toddler Development – Third Edition.
a Potilailla, joilla oli kaksi SMN2-geenin kopiota, CMAP-mittauksen amplitudin mediaani lähtötilanteessa oli 2,0 (vaihteluväli: 0,5–3,8).
b p-arvo perustuu yksitahoiseen eksaktiin binomitestiin. Tulosta verrataan 5 %:n raja-arvoon.
Lisäksi 80 % (4/5) ensisijaisesta tehon potilasjoukosta, 87,5 % (7/8) potilaista, joilla oli kaksi SMN2-geenin kopiota, ja 80,8 % (21/26) hoitoaikeen mukaisen (ITT) potilasjoukon potilaista kykeni istumaan tuetta 30 sekuntia (BSID-III-asteikon kohta 26).
Hoitoaikeen mukaisen (ITT) potilasjoukon potilaat saavuttivat myös motorisia kehitystasoja, mikä mitattiin 12 kuukauden aikapisteessä HINE-2-pisteillä (N = 25). Tässä potilasjoukossa 96,0 % potilaista kykeni istumaan (yksi potilas [yksi potilas kahdeksasta, joilla oli kaksi SMN2-geenin kopiota] kykeni istumaan tukevasti ja 23 potilasta [kuusi potilasta kahdeksasta, joilla oli kaksi SMN2-geenin kopiota, 13 potilasta 13:sta, joilla oli kolme SMN2-geenin kopiota, ja neljä potilasta neljästä, joilla oli vähintään neljä SMN2-geenin kopiota] kykeni kääntymään istuessaan / pyörähtämään). Lisäksi 84 % potilaista kykeni seisomaan: 32 % (N = 8) potilaista kykeni seisomaan tuettuna (kolme potilasta kahdeksasta, joilla oli kaksi SMN2-geenin kopiota, kolme potilasta 13:sta, joilla oli kolme SMN2-geenin kopiota, ja kaksi potilasta neljästä, joilla oli vähintään neljä SMN2-geenin kopiota) ja 52 % (N = 13) potilaista kykeni seisomaan tuetta (yksi potilas kahdeksasta, joilla oli kaksi SMN2-geenin kopiota, kymmenen potilasta 13:sta, joilla oli kolme SMN2-geenin kopiota, ja kaksi potilasta neljästä, joilla oli vähintään neljä SMN2-geenin kopiota). Lisäksi 72 % potilaista kykeni pomppimaan, nousemaan tukea vasten seisomaan tai kävelemään; 8 % (N = 2) potilaista kykeni pomppimaan (kaksi potilasta kahdeksasta, joilla oli kaksi SMN2-geenin kopiota), 16 % (N = 4) kykeni nousemaan tukea vasten seisomaan (kolme potilasta 13:sta, joilla oli kolme SMN2-geenin kopiota, ja yksi potilas neljästä potilaasta, joilla oli vähintään neljä SMN2-geenin kopiota) ja 48 % (N = 12) kykeni kävelemään itsenäisesti (yksi potilas kahdeksasta, joilla oli kaksi SMN2-geenin kopiota, yhdeksän potilasta 13:sta, joilla oli kolme SMN2-geenin kopiota, ja kaksi potilasta neljästä, joilla oli vähintään neljä SMN2-geenin kopiota). Seitsemällä potilaalla ei testattu kävelykykyä 12 kuukauden aikapisteessä.
Taulukko 6. Yhteenveto keskeisistä tehon päätetapahtumista vielä oireettomilla potilailla 12 kuukauden aikapisteessä
| Tehon päätetapahtumat | Hoitoaikeen mukainen (ITT) potilasjoukko (N = 26) |
| Motorinen toimintakyky | |
| Niiden potilaiden osuus, jotka saavuttavat CHOP-INTEND-kokonaispisteet 50 tai enemmän (90 %:n luottamusväli) | 92 %a (76,9 %; 98,6 %) |
| Niiden potilaiden osuus, jotka saavuttavat CHOP-INTEND-kokonaispisteet 60 tai enemmän (90 %:n luottamusväli) | 80 %a (62,5 %; 91,8 %) |
| Nieleminen ja syöminen | |
| Niiden potilaiden osuus, jotka kykenevät syömään (90 %:n luottamusväli) | 96,2 %b (83,0 %; 99,8 %) |
| Terveydenhuollon palveluiden käyttö | |
| Niiden potilaiden osuus, joilla ei sairaalahoitojaksojac (90 %:n luottamusväli) | 92,3 % (77,7 %; 98,6 %) |
| Elossaoloaika ilman tapahtumiad | |
| Niiden potilaiden osuus, jotka ovat elossa ilman tapahtumia (90 %:n luottamusväli) | 100 % (100 %; 100 %) |
| Lyhenteet: CHOP‑INTEND = Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders a Peruste: N = 25 b Yhtä potilasta ei arvioitu. c Sairaalahoitojaksot käsittävät kaikki vähintään kaksi päivää kestävät sairaalahoitojaksot, jotka eivät liity tutkimuksen vaatimuksiin. d Tapahtuma viittaa kuolemaan tai pysyvään ventilaatioon; pysyväksi ventilaatioksi määritellään trakeostomia tai ≥ 16 tunnin ei-invasiivinen ventilaatio päivässä tai intubaatio > 21 peräkkäisenä päivänä ilman akuuttia korjautuvaa tapahtumaa tai sellaisen häviämisen jälkeen. | |
Kuva 5. CHOP-INTEND-kokonaispisteiden mediaani käynneittäin ja SMN2-geenin kopioiden lukumäärän mukaan (hoitoaikeen mukainen [ITT] potilasjoukko)
Lyhenteet: SMN2 = Survival of Motor Neuron 2.
Farmakokinetiikka
Farmakokineettisiä parametreja on tutkittu terveillä aikuisilla tutkittavilla ja spinaalista lihasatrofiaa sairastavilla potilailla.
Kun hoito annettiin oraaliliuoksena, risdiplaamin farmakokinetiikka oli lähes lineaarinen annosvälillä 0,6–18 mg. Risdiplaamin farmakodynamiikkaa kuvasti parhaiten populaatiofarmakokineettinen malli, jossa oli kolmen läpikulkutilan imeytyminen, kaksitila-altistus ja ensimmäisen asteen eliminaatio. Painon ja iän havaittiin vaikuttavan farmakokinetiikkaan merkittävästi.
Imeväisiässä alkavaa spinaalista lihasatrofiaa sairastavilla potilailla (ikä tutkimukseen mukaan tullessa 2–7 kuukautta) arvioitu altistus (keskimääräinen AUC0-24h) hoitoannoksilla 0,2 mg/kg kerran päivässä oli 1930 ng.h/ml. RAINBOWFISH-tutkimuksen vielä oireettomilla imeväisikäisillä (ikä 16 vuorokaudesta < 2 kuukauteen) arvioitu keskimääräinen altistus, kun annoksia 0,15 mg/kg oli annettu kerran päivässä kahden viikon ajan, oli 2020 ng.h/ml. SUNFISH-tutkimuksen (osa 2) myöhemmin alkavaa spinaalista lihasatrofiaa sairastavilla potilailla (ikä tutkimukseen mukaan tullessa 2–25 vuotta) arvioitu altistus hoitoannoksilla (< 20 kg:n painoisille potilaille 0,25 mg/kg kerran päivässä; ≥ 20 kg:n painoisille potilaille 5 mg kerran päivässä) oli 2070 ng.h/ml yhden hoitovuoden jälkeen ja 1940 ng.h/ml viiden hoitovuoden jälkeen. Aiemmin hoitoa saaneille potilaille (tutkimukseen mukaan tullessa ikä 1–60 vuotta) spinaalisen lihasatrofian hoidosta aiheutunut arvioitu altistus (keskimääräinen AUC0‑24h) käytettäessä hoitoannosta 0,25 mg/kg tai 5 mg oli 1700 ng.h/ml. Havaittu maksimipitoisuus (keskimääräinen Cmax) FIREFISH-tutkimuksessa käytetyillä annoksilla 0,2 mg/kg oli 194 ng/ml, SUNFISH-tutkimuksen osassa 2 se oli 140 ng/ml ja JEWELFISH-tutkimuksessa se oli 129 ng/ml. Arvioitu maksimipitoisuus RAINBOWFISH-tutkimuksessa käytetyllä annoksella 0,15 mg/kg oli 111 ng/ml.
Imeytyminen
Risdiplaami imeytyi paastotilassa nopeasti, ja käyttökuntoon saatetun jauheen oraaliliuosta varten annon jälkeen plasman tmax-arvo oli 1–5 tuntia. 47:ää tervettä tutkittavaa koskevien tietojen perusteella ruoalla (runsasrasvainen, runsasenergiainen aamiainen) ei ollut oleellista vaikutusta risdiplaamialtistukseen. Risdiplaami annettiin kliinisissä tutkimuksissa aamuaterian yhteydessä tai imetyksen jälkeen.
Jakautuminen
Risdiplaami jakautuu tasaisesti kaikkialle elimistöön, mukaan lukien keskushermostoon läpäisemällä veri-aivoesteen, minkä seurauksena SMN-proteiinin määrä lisääntyy keskushermostossa ja kaikkialla elimistössä. Risdiplaamin pitoisuus plasmassa ja SMN-proteiinin pitoisuus veressä kuvastavat sen jakautumista ja farmakodynaamisia vaikutuksia kudoksissa, kuten aivo- ja lihaskudoksissa.
Populaatiofarmakokineettisten parametrien estimaatit olivat: laskennallinen jakautumistilavuus keskustilassa 98 l, perifeerisessä tilassa 93 l ja tilojen välinen puhdistuma 0,68 l/h.
Risdiplaami sitoutuu pääasiassa seerumin albumiinin, mutta se ei sitoudu lainkaan happamaan alfa-1-glykoproteiiniin, ja sen vapaa fraktio on 11 %.
Biotransformaatio
Risdiplaami metaboloituu pääasiassa FMO1:n ja FMO3:n sekä CYP-entsyymien 1A1, 2J2, 3A4 ja 3A7 välityksellä.
Annettaessa 200 mg itrakonatsolia (voimakas CYP3A:n estäjä) kaksi kertaa päivässä yhdessä suun kautta annetun 6 mg:n risdiplaamikerta-annoksen kanssa ei todettu kliinisesti oleellista vaikutusta risdiplaamin farmakokinetiikkaan (AUC-arvo suureni 11 %, Cmax-arvo pieneni 9 %).
Eliminaatio
Populaatiofarmakokineettisistä analyyseistä saatu risdiplaamin laskennallisen puhdistuman (CL/F) estimaatti on 2,6 l/h.
Risdiplaamin efektiivinen puoliintumisaika spinaalista lihasatrofiaa sairastavilla potilailla oli noin 50 tuntia.
Risdiplaami ei ole ihmisen monilääkeresistenssiproteiinin 1 (MDR1) substraatti.
Annoksesta erittyi ulosteisiin noin 53 % (14 % muuttumatonta risdiplaamia) ja virtsaan 28 % (8 % muuttumatonta risdiplaamia). Kanta-aine oli pääasiallinen plasmassa havaittu komponentti, joka käsitti 83 % verenkierrossa olleesta lääkkeeseen liittyneestä aineesta. Farmakologisesti inaktiivinen metaboliitti M1 tunnistettiin pääasialliseksi verenkierrossa olevaksi metaboliitiksi.
Farmakokinetiikka erityispotilasjoukoissa
Pediatriset potilaat
Populaatiofarmakokineettisessä analyysissa paino ja ikä tunnistettiin kovariaateiksi. Tällaisen mallinnuksen mukaisesti annosta muutetaan iän (alle ja yli 2 kuukautta ja 2 vuotta) ja painon (20 kg:aan saakka) perusteella, jotta eri-ikäisille ja -painoisille potilaille saadaan samankaltainen altistus. Alle 20 vuorokauden ikäisistä potilaista on vähän farmakokineettisiä tietoja saatavilla, koska kliinisissä tutkimuksissa vain yksi 16 vuorokauden ikäinen vastasyntynyt sai risdiplaamia pienempänä annoksena (0,04 mg/kg).
Iäkkäät potilaat
Farmakokinetiikkaa ei ole tutkittu erityisesti yli 60-vuotiailla spinaalista lihasatrofiaa sairastavilla potilailla. Kliinisissä farmakokineettisissä tutkimuksissa oli mukana spinaalista lihasatrofiaa sairastamattomia tutkittavia 69 ikävuoteen saakka, ja ne osoittavat, ettei enintään 69-vuotiaiden potilaiden annosta tarvitse muuttaa.
Munuaisten vajaatoiminta
Risdiplaamin farmakokinetiikan tutkimiseksi munuaisten vajaatoimintaa sairastavilla potilailla ei ole tehty tutkimuksia. Risdiplaamin eliminaatio erittymällä muuttumattomana aineena munuaisten kautta on vähäistä (8 %).
Maksan vajaatoiminta
Lievä tai keskivaikea maksan vajaatoiminta ei vaikuttanut merkittävästi risdiplaamin farmakokinetiikkaan. Cmax- ja AUC-arvojen keskimääräinen suhde suun kautta annetun 5 mg:n risdiplaamikerta-annoksen jälkeen oli lievää (n = 8) maksan vajaatoimintaa sairastavilla 0,95 (Cmax) ja 0,80 (AUC) ja keskivaikeaa (n = 8) maksan vajaatoimintaa sairastavilla 1,20 (Cmax) ja 1,08 (AUC) kaltaistettuihin terveisiin verrokkeihin (n = 10) verrattuna. Turvallisuutta ja farmakokinetiikkaa ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla.
Etninen tausta
Risdiplaamin farmakokinetiikassa ei ole eroja japanilaisten ja valkoihoisten tutkittavien välillä.
Prekliiniset tiedot turvallisuudesta
Hedelmällisyyden heikkeneminen
Risdiplaamihoitoon systeemisillä altistuksilla haittavaikutuksia aiheuttamattomalla annostasolla (NOAEL, no observed adverse effect level) ilman turvallisuusmarginaaleja liittyi rotilla ja apinoilla urosten itusolujen muodostumisen lakkaamista. Tällaiset vaikutukset johtivat spermatosyyttien rappeutumiseen, siementiehyiden epiteelin rappeutumiseen/nekroosiin ja oligospermiaan/aspermiaan lisäkiveksissä. Risdiplaamin vaikutukset siittiösoluihin liittyvät todennäköisesti risdiplaamin jakautuvien solujen solusykliä häiritsevään vaikutukseen, ja ne ovat vaihespesifisiä ja oletettavasti korjautuvia. Rotilla ja apinoilla ei havaittu risdiplaamihoidon jälkeen vaikutuksia naaraiden lisääntymiselimissä.
Hedelmällisyyttä ja alkion varhaisvaiheen kehitystä koskevia tutkimuksia ei tehty risdiplaamin samanaikaisessa käytössä, sillä siittiösolujen muodostumisen lakkaaminen ja alkiotoksisuus hoidon aikana oli tunnistettu rottien ja apinoiden hoidossa jo muissa toksisuustutkimuksissa. Kahdessa tutkimuksessa, joissa rotat parittelivat, ei havaittu urosten eikä naaraiden hedelmällisyyden heikkenemistä. Sitä ei havaittu vieroitusajankohtana aloitetun 13 viikon hoitojakson päättymisen jälkeen eikä 8 viikkoa 4 päivän iässä aloitetun 4 viikon hoitojakson päättymisen jälkeen.
Vaikutus verkkokalvon rakenteeseen
Apinoiden pitkäaikaisessa risdiplaamihoidossa havaittiin vaikutus verkkokalvoon, joka ilmeni valoaistinsolujen rappeumana verkkokalvon reuna-alueelta alkaen. Retinografiassa havaittavat vaikutukset korjautuivat osittain hoidon lopettamisen jälkeen, mutta valoaistinsolujen rappeuma ei korjautunut. Vaikutuksia seurattiin silmän valokerroskuvauksella (OCT) ja elektroretinografiatutkimuksella (ERG). Vaikutuksia havaittiin systeemisillä altistuksilla NOAEL-tasolla ilman turvallisuusmarginaaleja, kun altistukset olivat yli kaksinkertaisia verrattuna ihmisen altistukseen hoitoannoksilla. Tällaisia vaikutuksia ei havaittu albiinorotilla tai pigmentoiduilla rotilla, kun niille annettiin jatkuvasti risdiplaamia siten, että altistus oli suurempi kuin apinoilla. Tällaisia vaikutuksia ei ole havaittu spinaalista lihasatrofiaa sairastavilla potilailla tehdyissä kliinisissä tutkimuksissa, kun potilaat ovat olleet säännöllisessä oftalmologisessa seurannassa (mukaan lukien spektrialueen silmän valokerroskuvaus [spectral domain optical coherence tomography, SD-OCT] ja näkökyvyn arviointi).
Vaikutus epiteelikudoksiin
Risdiplaamihoitoa saaneilla rotilla ja apinoilla vaikutukset ihon, kurkunpään ja silmäluomien histologiaan sekä maha-suolikanavaan olivat ilmeisiä. Muutoksia alkoi ilmaantua, kun hoito suurilla annoksilla oli kestänyt 2 viikkoa ja pidempään. Pitkäaikaista hoitoa 39 viikon ajan saaneilla apinoilla NOAEL-taso oli yli kaksinkertainen altistus verrattuna ihmisen keskimääräiseen altistukseen hoitoannoksilla.
Vaikutus hematologisiin parametreihin
Rotilla tehdyssä akuutissa luuytimen mikrotumatestissä havaittiin yli 50 %:n vähenemä polykromaattisten (varhaisvaiheen) ja normokromaattisten (kypsien) erytrosyyttien suhteessa, mikä viittaa merkittävään luuydintoksisuuteen. Tämä havainto tehtiin suurilla annoksilla, joiden käytössä altistus oli yli 15-kertainen verrattuna ihmisen keskimääräiseen altistukseen hoitoannoksia käytettäessä. Rottien saatua hoitoa 26 viikon ajan altistuksen marginaali NOAEL-tasoon oli noin 4‑kertainen verrattuna ihmisen keskimääräiseen altistukseen hoitoannoksia käytettäessä.
Genotoksisuus
Risdiplaami ei ole mutageeninen bakteereilla tehtävässä käänteisessä mutaatiotestissä. Nisäkässoluissa in vitro ja rotan luuytimessä risdiplaami lisää mikrotumaisten solujen esiintyvyyttä. Useissa rotilla tehdyissä toksisuustutkimuksissa havaittiin mikrotumainduktiota luuytimessä (aikuisilla ja nuorilla eläimillä). Tutkimuksissa NOAEL-tasoon liittyi noin 1,5-kertainen altistus verrattuna ihmisen altistukseen hoitoannoksilla. Tiedot osoittivat, että tämä vaikutus on epäsuora ja aiheutuu risdiplaamin jakautuvien solujen solusykliä häiritsevästä vaikutuksesta. Risdiplaami ei vaurioita DNA:ta suoraan.
Lisääntymistoksisuus
Risdiplaamihoitoa saaneilla tiineillä rotilla tehdyissä tutkimuksissa todettiin alkio- ja sikiötoksisuutta, johon liittyi tavanomaista pienempi sikiön paino ja kehityksen viivästymistä. Tämän vaikutuksen NOAEL-taso oli noin kaksi kertaa suurempi kuin potilaiden altistus risdiplaamin hoitoannoksilla. Tiineillä kaniineilla tehdyissä tutkimuksissa havaittiin dysmorfogeenisia vaikutuksia altistuksilla, joihin liittyi myös emoon kohdistuvaa toksisuutta. Tällaisia olivat neljän poikueen (22 %) neljä sikiötä (4 %), joilla oli hydrokefalus. Tämän vaikutuksen NOAEL-taso oli noin nelinkertainen verrattuna potilaiden altistukseen risdiplaamin hoitoannoksilla. Pre- ja postnataalista kehitystä koskeneessa tutkimuksessa, jossa rotat saivat risdiplaamihoitoa päivittäin, risdiplaami pidensi hieman gestaation kestoaikaa. Tiineillä ja imettävillä rotilla tehdyt tutkimukset osoittivat, että risdiplaami läpäisee istukkaesteen ja erittyy maitoon.
Karsinogeenisuus
Risdiplaami ei osoittanut karsinogeenisuutta siirtogeenisillä rasH2-hiirillä 6 kuukauden aikana eikä rotilla tehdyssä kahden vuoden tutkimuksessa altistuksilla, jotka vastasivat ihmisellä todettua altistusta ihmisille suositellulla enimmäisannoksella (MRHD). Urosrottien esinahkarauhasen ja naarasrottien häpykielirauhasen kasvaimet lisääntyivät merkittävästi, kun annos oli neljä kertaa suurempi kuin ihmiselle suositeltu enimmäisannos (MRHD). Koska nämä molemmat ovat jyrsijöille spesifisiä elimiä, löydöksellä ei ole merkitystä ihmiselle.
Nuorilla eläimillä tehdyt eläinkokeet
Nuorista eläimistä saadut tiedot eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
mannitoli (E421)
isomalti (E953)
mansikka-aromi: luontainen (luontaiset) aromiaine(et), aromivalmiste(et), maissimaltodekstriini, muunnettu vahamaissitärkkelys (E1450)
viinihappo (E334)
natriumbentsoaatti (E211)
makrogoli 6000 (E1521)
sukraloosi
askorbiinihappo (E300)
dinatriumedetaattidihydraatti
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Jauhe oraaliliuosta varten
2 vuotta
Käyttökuntoon saatettu oraaliliuos
64 päivää jääkaapissa säilytettynä (2–8 ºC).
Potilas tai potilasta hoitava henkilö voi tarvittaessa säilyttää oraaliliuosta huoneenlämmössä (alle 40 °C) yhteenlaskettuna enintään 120 tunnin ajan (5 päivää). Oraaliliuos on palautettava jääkaappiin aina kun pullon säilytys huoneenlämmössä ei ole enää tarpeen. Seuraa yhteenlaskettua säilytysaikaa jääkaapin ulkopuolella (alle 40 °C).
Oraaliliuos on hävitettävä, jos pulloa on säilytetty huoneenlämmössä (alle 40 °C) yhteenlaskettuna pidempään kuin 120 tuntia (5 päivää), tai jos pulloa on säilytetty yli 40 °C lämpötilassa minkä tahansa aikajakson ajan.
Säilytys
Jauhe oraaliliuosta varten
Säilytä alle 25 °C.
Pidä pullo tiiviisti suljettuna. Herkkä kosteudelle.
Käyttökuntoon saatettu oraaliliuos
Säilytysolosuhteet lääkevalmisteen käyttökuntoon saattamisen jälkeen, ks. kohta Kestoaika.
Pidä oraaliliuos alkuperäisessä ruskeassa lasipullossa. Herkkä valolle. Pidä pullo aina pystysuorassa korkki tiiviisti suljettuna.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
EVRYSDI jauhe oraaliliuosta varten
0,75 mg/ml (L:ei) 1 pakkaus (80 ml, 1 sovitin, 2x1 ml+2x6 ml+1x12 ml mittaruisku annosasteikolla) (7989,40 €)
PF-selosteen tieto
Ruskea tyypin III lasipullo, jossa avaamattomuuden osoittava turvakierrekorkki.
Yksi kartonkikotelo sisältää yhden pullon, yhden pulloon kiinni painettavan sovittimen, kaksi useaan käyttökertaan tarkoitettua 1 ml:n ja kaksi useaan käyttökertaan tarkoitettua 6 ml:n ruskeaa mittaruiskua sekä yhden useaan käyttökertaan tarkoitetun 12 ml:n ruskean mittaruiskun. Mittaruiskuissa on annosasteikko.
Valmisteen kuvaus:
Vaaleankeltainen, keltainen, harmahtavan keltainen, vihertävän keltainen tai vaaleanvihreä jauhe.
Käyttö- ja käsittelyohjeet
Terveydenhuollon ammattilaisen (esim. apteekkihenkilökunnan) on ennen valmisteen potilaalle toimittamista sekoitettava risdiplaamijauhe käyttökuntoon oraaliliuokseksi.
Käyttökuntoon saattaminen
Evrysdi- jauheen oraaliliuosta varten käsittelyssä on oltava varovainen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Vältä valmisteen sisäänhengittämistä sekä ihon ja limakalvojen pääsyä suoraan kosketukseen kuivajauheen ja käyttökuntoon saatetun liuoksen kanssa.
Käytä kertakäyttökäsineitä käyttökuntoon saattamisessa sekä pyyhkiessäsi pullon/korkin ulkopintaa ja puhdistaessasi työskentelyalustaa valmisteen käyttökuntoon saattamisen jälkeen. Jos kontakti tapahtuu, pese kyseinen alue huolellisesti vedellä ja saippualla, huuhtele silmät vedellä.
Ohjeet käyttökuntoon saattamista varten:
- Napauta suljetun lasipullon pohjaa kevyesti, jotta jauhe irtoaa.
- Poista korkki. Älä heitä korkkia pois.
- Lisää risdiplaamipulloon varovasti 79 ml puhdistettua vettä tai injektionesteisiin käytettävää vettä, jotta saadaan 0,75 mg/ml oraaliliuos.
- Pitele toisella kädellä lääkepulloa pöydällä. Paina sovitin pullon suuaukkoon painamalla sovitinta toisella kädellä alaspäin. Varmista, että sovitin on painettu hyvin pullonsuun reunaa vasten.
- Laita korkki takaisin pulloon, ja sulje pullo tiiviisti. Varmista, että pullo on suljettu tiiviisti. Ravista 15 sekunnin ajan. Odota 10 minuuttia. Tuloksena pitäisi olla kirkas liuos. Ravista sen jälkeen hyvin vielä 15 sekunnin ajan.
- Kirjoita pullon etikettiin ja kartonkikoteloon liuoksen Hävitä viimeistään -päivämäärä. (Hävitä viimeistään -päivämääräksi lasketaan 64 päivää käyttökuntoon saattamisen jälkeen; käyttökuntoon saattamisen päivä on päivä 0.) Laita pullo takaisin alkuperäiseen kartonkikoteloon, joka sisältää ruiskut (suojapusseissa), pakkausselosteen ja käyttöohjekirjasen. Säilytä kartonkikotelo jääkaapissa (2–8 °C).
Hävitä käyttämättä jäävä liuos 64 päivää käyttökuntoon saattamisen jälkeen.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
EVRYSDI jauhe oraaliliuosta varten
0,75 mg/ml 1 pakkaus
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Risdiplaami: Spinaalisen lihasatrofian hoito erityisin edellytyksin (3082).
ATC-koodi
M09AX10
Valmisteyhteenvedon muuttamispäivämäärä
20.04.2026
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com