
NUBEQA tabletti, kalvopäällysteinen 300 mg
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää 300 mg darolutamidia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 186 mg laktoosimonohydraattia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kliiniset tiedot
Käyttöaiheet
NUBEQA on tarkoitettu aikuisille miehille
- etäpesäkkeettömän kastraatioresistentin eturauhassyövän hoitoon, kun potilaalla on korkea etäpesäkkeisen taudin kehittymisen riski (ks. kohta Farmakodynamiikka).
- etäpesäkkeisen hormonisensitiivisen eturauhassyövän hoitoon yhdistelmänä androgeenideprivaatiohoidon kanssa (ks. kohta Farmakodynamiikka).
- etäpesäkkeisen hormonisensitiivisen eturauhassyövän hoitoon yhdistelmänä dosetakselin ja androgeenideprivaatiohoidon kanssa (ks. kohta Farmakodynamiikka).
Ehto
Hoito on aloitettava ja toteutettava eturauhassyövän lääkehoitoon perehtyneen erikoislääkärin valvonnassa.
Annostus ja antotapa
Hoito on aloitettava ja toteutettava eturauhassyövän lääkehoitoon perehtyneen erikoislääkärin valvonnassa.
Annostus
Suositeltu annos on 600 mg darolutamidia (kaksi 300 mg:n tablettia) kahdesti päivässä, mikä vastaa 1 200 mg:n kokonaisannosta vuorokaudessa (ks. kohta Farmakokinetiikka).
Darolutamidihoitoa jatketaan, kunnes tauti etenee tai potilas ei enää siedä hoitoa.
Lääkkeellistä kastraatiota lutenisoivan hormonin vapauttajahormonin (LHRH) agonistilla tai antagonistilla (GnRH-analogilla) on jatkettava hoidon aikana, jos potilaalle ei ole tehty kirurgista kastraatiota.
Etäpesäkkeinen hormonisensitiivinen eturauhassyöpä – darolutamidihoito yhdistelmänä dosetakselin kanssa
Etäpesäkkeistä hormonisensitiivistä eturauhassyöpää sairastavien potilaiden darolutamidihoito on aloitettava yhdistelmänä dosetakselin kanssa (ks. kohta Farmakokinetiikka). Ensimmäinen kuudesta dosetakselisyklistä on annettava 6 viikon sisällä darolutamidihoidon aloittamisesta. Dosetakselin valmistetiedoissa annettuja suosituksia on noudatettava. Darolutamidihoitoa jatketaan, kunnes tauti etenee tai potilas ei enää siedä hoitoa. Darolutamidihoitoa tulee jatkaa siitä huolimatta, että dosetakselisykli viivästyisi, se jouduttaisiin keskeyttämään tai lopettamaan.
Unohtunut annos
Jos annos unohtuu, potilaan pitää ottaa se heti muistaessaan ennen seuraavan annoksen ottoaikaa. Unohtuneen annoksen korvaamiseksi ei pidä ottaa kahta annosta yhdessä.
Annoksen muuttaminen
Jos potilaalla on darolutamidiin liittyvää ≥ asteen 3 toksisuutta tai sietämättömiä haittavaikutuksia (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset), hoito on keskeytettävä tai sitä on pienennettävä annokseen 300 mg kahdesti päivässä, kunnes oireet lieventyvät. Sen jälkeen hoitoa voidaan jatkaa ottamalla 600 mg:n annos kahdesti päivässä.
Ei ole suositeltavaa pienentää annosta alle 300 mg kahdesti päivässä, koska tehoa ei ole varmistettu.
Erityisryhmät
Iäkkäät
Iäkkäiden potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa. Vaikeaa munuaisten vajaatoimintaa sairastaville potilaille (eGFR 15-29 ml/min/1,73 m2), jotka eivät saa hemodialyysihoitoa, suositeltu aloitusannos on 300 mg kahdesti päivässä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa.
Tietoa darolutamidin farmakokinetiikasta keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla on saatavilla rajoitetusti. Darolutamidia ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla.
Keskivaikeaa ja vaikeaa maksan vajaatoimintaa sairastaville potilaille (Child-Pugh-luokat B ja C) suositeltu aloitusannos on 300 mg kahdesti päivässä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka.).
Pediatriset potilaat
Ei ole asianmukaista käyttää darolutamidia pediatrisille potilaille.
Antotapa
NUBEQA otetaan suun kautta.
Tabletit otetaan kokonaisina ruoan kanssa (ks. kohta Farmakokinetiikka).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille. Naiset, jotka ovat raskaana tai voivat tulla raskaaksi (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Munuaisten vajaatoiminta
Tietoa tämän valmisteen käytöstä potilaille, joilla on vaikea munuaisten vajaatoiminta, on saatavilla rajoitetusti.
Koska altistus saattaa olla suurentunut, näitä potilaita on seurattava tarkasti haittavaikutusten varalta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Maksan vajaatoiminta
Tietoa tämän valmisteen käytöstä keskivaikeaa maksan vajaatoimintaa sairastaville potilaille on rajoitetusti, eikä darolutamidia ole tutkittu potilailla, joilla on vaikea maksan vajaatoiminta.
Koska altistus saattaa olla suurentunut, näitä potilaita on seurattava tarkasti haittavaikutusten varalta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Äskettäin todettu sydän- tai verisuonitauti
Kliinisistä tutkimuksista suljettiin pois potilaat, joilla oli edellisten 6 kuukauden aikana ollut kliinisesti merkittävä sydän- tai verisuonitauti, mukaan lukien aivohalvaus, sydäninfarkti, vaikea-asteinen tai epästabiili angina pectoris, sepelvaltimon tai ääreisverenkierron ohitussiirre tai oireinen kongestiivinen sydämen vajaatoiminta. Darolutamidin turvallisuutta näille potilasryhmille ei ole siksi varmistettu.
Jos NUBEQA-valmistetta määrätään, pitää potilaan kliinisesti merkittävää sydän- tai verisuonitautia hoitaa vakiintuneiden hoito-ohjeiden mukaan.
Maksatoksisuus
Jos potilaalla ilmenee maksa-arvojen poikkeavuuksian jotka viittaavat idiosynkraattiseen, lääkkeen aiheuttamaan maksavaurioon, on darolutamidihoito lopetettava pysyvästi (ks. kohta Haittavaikutukset).
Muiden lääkevalmisteiden samanaikainen käyttö
Voimakkaiden CYP3A4:n ja P-gp:n indusoijien käyttö darolutamidihoidon aikana voi pienentää darolutamidin pitoisuutta plasmassa eikä ole suositeltavaa, jos jokin muu hoitovaihtoehto on olemassa. On suositeltavaa valita sellainen vaihtoehtoinen, samanaikaisesti käytettävä lääkevalmiste, jonka indusoiva vaikutus CYP3A4-entsyymiin tai P-gp-kuljetusproteiiniin on heikompi (ks. kohta Yhteisvaikutukset).
Potilaita on tarkkailtava BCRP:n, OATP1B1:n ja OATP1B3:n substraattien haittavaikutusten varalta, sillä niiden samanaikainen käyttö darolutamidin kanssa voi lisätä näiden substraattien pitoisuutta plasmassa.
Samanaikaista antoa rosuvastatiinin kanssa on vältettävä, jos jokin muu hoitovaihtoehto on olemassa (ks. kohta Yhteisvaikutukset).
Androgeenideprivaatiohoito voi pidentää QT-aikaa
Jos potilaalla on aiemmin todettu QT-ajan pidentymisen riskitekijöitä ja potilas käyttää samanaikaisesti QT-aikaa mahdollisesti pidentäviä lääkevalmisteita (ks. kohta Yhteisvaikutukset), lääkärin on ennen NUBEQA-hoidon aloittamista arvioitava hoidon hyöty-riskisuhde, mukaan lukien kääntyvien kärkien takykardian mahdollisuus.
Tietoa apuaineista
NUBEQA sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset darolutamidiin
CYP3A4:n ja P-gp:n induktorit
Darolutamidi on CYP3A4:n ja P-glykoproteiinin (P-gp) substraatti.
Voimakkaiden ja kohtalaisten CYP3A4:n induktorien ja P-gp:n indusoijien (esim. karbamatsepiini, fenobarbitaali, mäkikuisma, fenytoiini ja rifampisiini) käyttö darolutamidihoidon aikana ei ole suositeltavaa, jos jokin muu hoitovaihtoehto on olemassa. On suositeltavaa valita sellainen vaihtoehtoinen, samanaikaisesti käytettävä lääkevalmiste, joka ei indusoi CYP3A4-entsyymiä tai P-gp-kuljetusproteiinia tai indusoi niitä vain heikosti.
Rifampisiinin (600 mg), joka on voimakas CYP3A4:n ja P-gp:n indusoija, toistuva anto aterian yhteydessä otetun darolutamidin kerta-annoksen (600 mg) kanssa pienensi keskimääräistä altistusta 72 % (AUC0-72) ja darolutamidin Cmax-arvoa 52 %.
CYP3A4:n, P-gp:n ja BCRP:n estäjät
Darolutamidi on CYP3A4:n, P-gp:n ja rintasyöpäresistenssiproteiinin (BCRP) substraatti.
CYP3A4:n, P-gp:n tai BCRP:n estäjää annettaessa ei ole odotettavissa lääkeaineiden välisiä kliinisesti merkittäviä yhteisvaikutuksia. Darolutamidia voidaan antaa samanaikaisesti CYP3A4:n P-gp:n tai BCRP:n estäjien kanssa. Darolutamidin samanaikainen käyttö yhdistetyn P-gp:n ja voimakkaan CYP3A4:n estäjän kanssa lisää darolutamidialtistusta, mikä voi lisätä darolutamidin haittavaikutusten riskiä. On suositeltavaa tarkkailla potilaita useammin darolutamidin haittavaikutusten varalta ja tarvittaessa muuttaa darolutamidiannosta.
Voimakkaan CYP3A4:n, P-gp:n ja BCRP:n estäjän itrakonatsolin anto (200 mg kahdesti päivässä päivänä 1 ja kerran päivässä seuraavien 7 päivän ajan) darolutamidin kerta-annoksen (600 mg päivänä 5 aterian yhteydessä) kanssa johti keskimääräisen altistuksen 1,7-kertaiseen suurenemiseen (AUC0-72) ja darolutamidin Cmax-arvon 1,4-kertaiseen suurenemiseen.
UGT1A9:n estäjät
Darolutamidi on UGT1A9:n substraatti.
UGT1A9:n estäjää annettaessa ei ole odotettavissa lääkeaineiden välisiä kliinisesti merkittäviä yhteisvaikutuksia.
Darolutamidia voidaan antaa samanaikaisesti UGT1A9:n estäjien kanssa. Populaatiofarmakokineettinen analyysi osoitti, että UGT1A9:n estäjien ja darolutamidin samanaikainen anto johti darolutamidin altistuksen (AUC0-72) lisääntymiseen 1,2-kertaiseksi.
Dosetakseli
Darolutamidin antaminen yhdistelmänä dosetakselin kanssa ei aiheuttanut kliinisesti merkittäviä muutoksia darolutamidin farmakokinetiikkaan etäpesäkkeistä hormonisensitiivistä eturauhassyöpää sairastavilla potilailla (ks. kohta Farmakodynamiikka).
Darolutamidin vaikutukset muihin lääkevalmisteisiin
BCRP:n, OATP1B1:n ja OATP1B3:n substraatit
Darolutamidi on rintasyöpäresistenssiproteiinin (BCRP) ja orgaanisten anionien kuljettajapolypeptidien (OATP) 1B1 ja 1B3 estäjä.
Samanaikaista antoa rosuvastatiinin kanssa on vältettävä, jos jokin muu hoitovaihtoehto on olemassa. On suositeltavaa valita sellainen vaihtoehtoinen, samanaikaisesti käytettävä lääkevalmiste, jonka estävä vaikutus BCRP-, OATP1B1- ja OATP1B3-proteiineihin on heikompi.
Darolutamidin anto (600 mg kahdesti päivässä 5 päivän ajan) ennen samanaikaista rosuvastatiinin kerta-annoksen antamista (5 mg) aterian yhteydessä johti rosuvastatiinin keskimääräisen altistuksen (AUC) ja Cmax-arvon noin 5-kertaiseen suurenemiseen.
Darolutamidin samanaikaista antoa muiden BCRP:n substraattien kanssa on vältettävä, jos mahdollista.
Darolutamidin samanaikainen anto voi lisätä muiden samanaikaisesti käytettyjen BCRP:n, OATP1B1:n ja OATP1B3:n substraattien (esim. metotreksaatti, sulfasalatsiini, fluvastatiini, atorvastatiini, pitavastatiini) pitoisuutta plasmassa. Sen vuoksi on suositeltavaa tarkkailla potilaita BCRP:n, OATP1B1:n ja OATPB3:n substraattien haittavaikutusten varalta. Lisäksi on noudatettava näiden substraattien valmistetietoihin sisältyvää asiaan liittyvää suositusta, kun niitä annetaan yhdessä darolutamidin kanssa.
P-gp:n substraatit
P-gp:n substraattia annettaessa ei ole odotettavissa lääkeaineiden välisiä kliinisesti merkityksellisiä yhteisvaikutuksia. Darolutamidia voidaan antaa samanaikaisesti P-gp:n substraattien (esim. digoksiini, verapamiili tai nifedipiini) kanssa. Kun darolutamidia annettiin samanaikaisesti yhdessä herkän P-gp:n substraatin dabigatraanieteksilaatin kanssa, dabigatraanin altistuksessa (AUC ja Cmax) ei havaittu mitään suurenemista.
CYP3A4:n substraatit
Darolutamidi on CYP3A4:n heikko indusoija.
CYP-entsyymien substraattia annettaessa ei ole odotettavissa lääkeaineiden välisiä kliinisesti merkityksellisiä yhteisvaikutuksia. Darolutamidia voidaan antaa samanaikaisesti CYP-entsyymien substraattien (esim. varfariini, levotyroksiini, omepratsoli) kanssa.
Darolutamidin anto (600 mg kahdesti päivässä 9 päivän ajan) ennen samanaikaista herkän CYP3A4:n substraatin midatsolaamin kerta-annoksen (1 mg) antamista aterian yhteydessä pienensi midatsolaamin keskimääräistä altistusta (AUC) 29 % ja Cmax-arvoa 32 %.
Kliinisesti merkityksellisinä pitoisuuksina annettuina darolutamidi ei estänyt valikoitujen CYP- entsyymien substraattien aineenvaihduntaa in vitro.
Dosetakseli
Darolutamidin antaminen yhdistelmänä dosetakselin kanssa ei aiheuttanut kliinisesti merkittäviä muutoksia dosetakselin farmakokinetiikkaan etäpesäkkeistä hormonisensitiivistä eturauhassyöpää sairastavilla potilailla (ks. kohta Farmakodynamiikka).
Lääkevalmisteet, jotka pidentävät QT-aikaa
Androgeenideprivaatiohoito voi pidentää QT-aikaa, joten NUBEQA-valmisteen ja QT-aikaa tunnetusti pidentävien lääkevalmisteiden tai kääntyvien kärkien takykardiaa mahdollisesti aiheuttavien lääkevalmisteiden samanaikaista käyttöä pitää arvioida tarkoin. Näitä lääkevalmisteita ovat muun muassa IA-ryhmän (esim. kinidiini, disopyramidi) tai III-ryhmän (esim. amiodaroni, sotaloli, dofetilidi, ibutilidi) rytmihäiriölääkevalmisteet, metadoni, moksifloksasiini ja psykoosilääkkeet (esim. haloperidoli).
Raskaus ja imetys
Tätä lääkevalmistetta ei ole tarkoitettu naisille, jotka voivat tulla raskaaksi. Sitä ei pidä käyttää naisille, jotka ovat tai saattavat olla raskaana tai jotka imettävät tai saattavat imettää (ks. kohdat Käyttöaiheet ja Vasta-aiheet).
Naiset, jotka voivat tulla raskaaksi / ehkäisy miehille ja naisille
Ei tiedetä, erittyykö darolutamidia tai sen metaboliitteja siemennesteeseen. Jos potilas on sukupuoliyhteydessä naisen kanssa, joka voi tulla raskaaksi, potilaan on käytettävä raskauden ehkäisemiseksi jotakin erittäin luotettavaa (ehkäisyn pettäminen < 1 % / vuosi) ehkäisymenetelmää NUBEQA-hoidon aikana ja 1 viikon ajan hoidon päättymisen jälkeen.
Raskaus
Vaikutusmekanismistaan johtuen darolutamidi saattaa aiheuttaa haittaa sikiölle. Prekliinisiä lisääntymistoksisuustutkimuksia ei ole tehty (ks. kohta Prekliiniset tiedot turvallisuudesta).
Ei tiedetä, erittyykö darolutamidia tai sen metaboliitteja siemennesteeseen. Jos potilas on sukupuoliyhteydessä raskaana olevan naisen kanssa, potilaan on käytettävä kondomia NUBEQA- hoidon aikana ja 1 viikon ajan hoidon päättymisen jälkeen. Sikiön altistumista androgeenireseptorin estäjälle raskaana olevaan naiseen siirtyneen siemennesteen kautta on vältettävä, sillä tämä saattaa vaikuttaa sikiön kehitykseen.
Imetys
Ei tiedetä, erittyvätkö darolutamidi tai sen metaboliitit ihmisen rintamaitoon. Eläinkokeita, joissa olisi tutkittu darolutamidin tai sen metaboliitin erittymistä maitoon, ei ole tehty (ks. kohta Prekliiniset tiedot turvallisuudesta).
Rintaruokittuun lapseen kohdistuvia riskejä ei voida poissulkea.
Hedelmällisyys
Ei ole olemassa tietoja darolutamidin vaikutuksesta ihmisten hedelmällisyyteen. Eläinkokeet ovat osoittaneet, että NUBEQA saattaa heikentää lisääntymiskykyisten miesten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
NUBEQA-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Useimmin havaittuja haittavaikutuksia
- potilailla, joilla on etäpesäkkeetön kastraatioresistentti eturauhassyöpä tai etäpesäkkeinen hormonisensitiivinen eturauhassyöpä ja jotka saavat darolutamidia, ovat uupumus / asteeniset tilat (13,7 %)
- potilailla, joilla on etäpesäkkeinen hormonisensitiivinen eturauhassyöpä ja jotka saavat darolutamidia yhdistelmänä dosetakselin kanssa, ovat ihottuma (16,6 %) ja hypertensio (13,8 %).
Lisätietoja turvallisuudesta käytettäessä darolutamidia yhdistelmänä muiden lääkevalmisteiden kanssa on kyseisten lääkevalmisteiden valmistetiedoissa.
Haittavaikutustaulukko
Käytön yhteydessä etäpesäkkeetöntä kastraatioresistenttiä eturauhassyöpää tai etäpesäkkeistä hormonisensitiivistä eturauhassyöpää sairastavilla, darolutamidihoitoa saavilla potilailla havaitut haittavaikutukset on lueteltu taulukossa 1. Käytön yhteydessä etäpesäkkeistä hormonisensitiivistä eturauhassyöpää sairastavilla, darolutamidin ja dosetakselin yhdistelmää saavilla potilailla havaitut haittavaikutukset on lueteltu taulukossa 2.
Haittavaikutukset on luokiteltu elinjärjestelmittäin. Ne on ryhmitelty yleisyyden mukaan. Yleisyysluokat ovat seuraavat: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Haittavaikutukset, joita raportoitiin etäpesäkkeetöntä kastraatioresistenttiä eturauhassyöpää ja etäpesäkkeistä hormonisensitiivistä eturauhassyöpää sairastavilla, darolutamidihoitoa saavilla potilailla ARAMIS- ja ARANOTE-tutkimuksissaa
| Elinjärjestelmä (MedDRA) | Hyvin yleinen | Yleinen |
| Sydän | Iskeeminen sydänsairausb Sydämen vajaatoimintac | |
| Iho ja ihonalainen kudos | Ihottumad | |
| Luusto, lihakset ja sidekudos | Raajakipu Murtumat | |
| Yleisoireet ja antopaikassa todettavat haitat | Uupumus/asteeniset tilate | |
| Tutkimuksetf | Neutrofiilimäärän lasku Veren bilirubiiniarvon nousu ALAT-arvon nousu ASAT-arvon nousu |
a Altistuksen keston mediaani ARAMIS- ja ARANOTE-tutkimuksissa oli 18,2 kuukautta (vaihteluväli: 0,0–44,3 kuukautta) darolutamidihoitoa saaneilla potilailla ja 11,6 kuukautta (vaihteluväli: 0,0–40,5 kuukautta) lumelääkettä saaneilla potilailla.
b Tähän sisältyy sepelvaltimokovettuma, sepelvaltimotauti, sepelvaltimotukos, sepelvaltimon ahtauma, äkillinen sepelvaltimo-oireyhtymä, äkillinen sydäninfarkti, angina pectoris, epävakaa angina pectoris, sydäninfarkti, sydänlihaksen hapenpuute.
c Tähän sisältyy sydämen vajaatoiminta, äkillinen sydämen vajaatoiminta, krooninen sydämen vajaatoiminta, kongestiivinen sydämen vajaatoiminta, kardiogeeninen sokki ja sydämen vajaatoiminta, jossa ejektiofraktio on säilynyt.
d Tähän sisältyy ihottuma, makulaarinen ihottuma, makulopapulaarinen ihottuma, papulaarinen ihottuma, pustulaarinen ihottuma, eryteema ja dermatiitti.
e Tähän sisältyy uupumus ja astenia, letargia ja pahoinvointi.
f CTCAE -luokitus (Common Terminology Criteria for Adverse Events), versio 5.0. Ilmaantuvuus perustuu laboratorioarvojen poikkeavuuksina raportoituihin arvoihin.
Taulukko 2: Haittavaikutukset, joita raportoitiin ARASENS-tutkimuksessa etäpesäkkeistä hormonisensitiivistä eturauhassyöpää sairastavilla, darolutamidin ja dosetakselin yhdistelmää saaneilla potilaillaa, b
Elinjärjestelmä (MedDRA) | Hyvin yleinen | Yleinen |
|---|---|---|
| Verisuonisto | Hypertensioc | |
| Iho ja ihonalainen kudos | Ihottumad,e | |
| Luusto, lihakset ja sidekudos | Murtumat | |
| Sukupuolielimet ja rinnat | Gynekomastia | |
| Tutkimuksetf | Neutrofiilimäärän lasku Veren bilirubiiniarvon nousu ALAT-arvon nousu ASAT-arvon nousu |
a Altistuksen keston mediaani oli 41,0 kuukautta (vaihteluväli: 0,1–56,5 kuukautta) darolutamidin ja dosetakselin yhdistelmää saaneilla potilailla ja 16,7 kuukautta (vaihteluväli: 0,3–55,8 kuukautta) lumelääkkeen ja dosetakselin yhdistelmää saaneilla potilailla.
b Haittavaikutusten ilmaantuvuudet eivät välttämättä liity pelkästään darolutamidiin, sillä myös muut samanaikaisesti käytetyt lääkevalmisteet saattavat vaikuttaa niihin.
c Tähän sisältyy hypertensio, verenpaineen nousu ja hypertensiivinen hätätilanne.
d Tähän sisältyy ihottuma, lääkeaineihottuma, erytematoottinen ihottuma, follikulaarinen ihottuma, makulaarinen ihottuma, makulopapulaarinen ihottuma, papulaarinen ihottuma, kutiseva ihottuma, pustulaarinen ihottuma, vesikulaarinen ihottuma, eryteema ja dermatiitti.
e Ilmaantuvuus oli suurimmillaan 6 ensimmäisen hoitokuukauden aikana.
f CTCAE -luokitus (Common Terminology Criteria for Adverse Events), versio 4.03. Ilmaantuvuus perustuu laboratorioarvojen poikkeavuuksina raportoituihin arvoihin.
Valikoitujen haittavaikutusten kuvaus
Maksan toimintakokeet
Darolutamidilla hoidetuilla potilailla on raportoitu idiosynkraattisia, lääkkeen aiheuttamia maksavaurioita, joihin liittyi asteen 3 ja 4 alaniiniaminotransferaasiarvon (ALAT) ja/tai aspartaattiaminotransferaasiarvon (ASAT) nousu ≥ 5-kertaiseksi ja ≥ 20-kertaiseksi normaaliarvojen ylärajaan (ULN) nähden mukaan lukien transaminaasiarvojen nousua, johon on liittynyt samanaikainen kokonaisbilirubiiniarvon nousu vähintään kaksinkertaiseksi normaaliarvojen ylärajaan nähden. Näiden vaikutusten alkamiseen kulunut aika vaihteli 1 kuukaudesta 12 kuukauteen darolutamidihoidon aloittamisesta. Monissa tapauksissa ALAT- ja ASAT-arvojen nousut korjaantuivat, kun darolutamidihoito lopetettiin. Katso tarkat suositukset kohdasta Haittavaikutukset.
Etäpesäkkeetön kastraatioresistentti eturauhassyöpä ja etäpesäkkeinen hormonisensitiivinen eturauhassyöpä – darolutamidihoito
Uupumus
Uupumusta/asteenisia tiloja raportoitiin 13,7 %:lla darolutamidihoitoa saaneista potilaista ja 11,7 %:lla lumelääkettä saaneista potilaista. Pahimman eli 3. asteen tapahtumia raportoitiin 0,4 %:lla darolutamidihoitoa saaneista potilaista ja 0,9 %:lla lumelääkettä saaneista potilaista. Uupumusta (kun mukaan ei luettu asteniaa, letargiaa ja pahoinvointia) esiintyi suurimmalla osalla potilaista (10,0 %:lla darolutamidihoitoa saaneista potilaista ja 8,5 %:lla lumelääkettä saaneista potilaista).
Murtumat
Murtumia esiintyi 4,1 %:lla darolutamidihoitoa saaneista potilaista ja 3,2 %:lla lumelääkettä saaneista potilaista.
Iskeeminen sydänsairaus ja sydämen vajaatoiminta
Iskeemistä sydänsairautta esiintyi 3,4 %:lla darolutamidihoitoa saaneista potilaista ja 2,2 %:lla lumelääkettä saaneista potilaista. Asteen 5 tapahtumia esiintyi 0,4 %:lla darolutamidihoitoa saaneista potilaista ja 0,4 %:lla lumelääkettä saaneista potilaista. Sydämen vajaatoimintaa esiintyi 1,6 %:lla darolutamidihoitoa saaneista potilaista ja 0,9 %:lla lumelääkettä saaneista potilaista.
Neutrofiilimäärän lasku
Neutrofiilimäärän laskua raportoitiin laboratorioarvojen poikkeavuutena 17,3 %:lla darolutamidihoitoa saaneista potilaista ja 7,4 %:lla lumelääkettä saaneista potilaista. Mediaaniaika nadiiriin oli 225 vuorokautta. Laboratorioarvojen poikkeavuudet ilmenivät pääasiassa voimakkuudeltaan asteen 1 tai 2 tapahtumina. Neutrofiilimäärän asteen 3 ja 4 laskua raportoitiin 2,6 %:lla ja vastaavasti 0,3 %:lla potilaista. Vain yksi potilas lopetti darolutamidihoidon pysyvästi neutropenian vuoksi. Neutropenia oli joko ohimenevä tai korjautuva (83 %:lla potilaista) eikä se liittynyt mihinkään kliinisesti merkityksellisiin löydöksiin tai oireisiin.
Veren bilirubiiniarvon nousu
Bilirubiiniarvon nousua raportoitiin laboratorioarvojen poikkeavuutena 16,1 %:lla darolutamidihoitoa saaneista potilaista ja 6,1 %:lla lumelääkettä saaneista potilaista. Nämä tapahtumat olivat enimmäkseen voimakkuudeltaan asteen 1 tai 2 tapahtumia, eivät liittyneet mihinkään kliinisesti merkityksellisiin löydöksiin tai oireisiin ja korjautuivat darolutamidihoidon lopettamisen jälkeen.
Asteen 3 ja 4 bilirubiiniarvon nousua raportoitiin 0,2 %:lla darolutamidihoitoa saaneista potilaista ja 0 %:lla lumelääkettä saaneista potilaista. Darolutamidia saaneiden potilaiden tutkimushaarassa keskimääräinen aika bilirubiiniarvon nousun alkamiseen oli 187 päivää ja ensimmäisen tapahtuman keskimääräinen kesto oli 172 päivää. Yhden potilaan hoitoa lopetettiin bilirubiiniarvon nousun vuoksi.
ALAT- ja ASAT-arvon nousu
ALAT-arvon nousua raportoitiin laboratorioarvojen poikkeavuutena 13,3 %:lla darolutamidihoitoa saaneista potilaista ja 9,7 %:lla lumelääkettä saaneista potilaista. ASAT-arvon nousua raportoitiin laboratorioarvojen poikkeavuutena 22,0 %:lla darolutamidihoitoa saaneista potilaista ja 13,4 %:lla lumelääkettä saaneista potilaista. Nämä tapahtumat olivat enimmäkseen voimakkuudeltaan asteen 1 tai 2 tapahtumia, eivät liittyneet mihinkään kliinisesti merkityksellisiin löydöksiin tai oireisiin ja korjautuivat darolutamidihoidon lopettamisen jälkeen. Asteen 3 ja 4 ALAT-arvon nousua raportoitiin 0,9 %:lla darolutamidihoitoa saaneista potilaista ja 0,3 %:lla lumelääkettä saaneista potilaista. Asteen 3 ja 4 ASAT-arvon nousua raportoitiin 1,2 %:lla darolutamidihoitoa saaneista potilaista ja 0,3 %:lla lumelääkettä saaneista potilaista. Darolutamidia saaneiden potilaiden tutkimushaarassa keskimääräinen aika ALAT-arvon nousun alkamiseen oli 253 päivää ja ASAT-arvon nousun alkamiseen 257 päivää. Ensimmäisen tapahtuman keskimääräinen kesto oli ALAT-arvon nousun osalta 122 päivää ja ASAT-arvon nousun osalta 121 päivää. Kahden potilaan hoito lopetettiin ALAT-arvon nousun ja 3 potilaan hoito ASAT-arvon nousun vuoksi.
Etäpesäkkeinen hormonisensitiivinen eturauhassyöpä – darolutamidihoito yhdistelmänä dosetakselin kanssa
Hypertensio
ARASENS-tutkimuksessa hypertensiota raportoitiin 13,8 %:lla darolutamidin ja dosetakselin yhdistelmää saaneista potilaista ja 9,4 %:lla lumelääkkeen ja dosetakselin yhdistelmää saaneista potilaista.
Asteen 3 hypertensiota raportoitiin 6,4 %:lla darolutamidin ja dosetakselin yhdistelmää saaneista potilaista ja 3,5 %:lla lumelääkkeen ja dosetakselin yhdistelmää saaneista potilaista. Molemmissa hoitoryhmissä yhdellä potilaalla esiintyi asteen 4 hypertensiota.
Yksi darolutamidin ja dosetakselin yhdistelmää saaneessa ryhmässä esiintynyt tapaus raportoitiin asteen 5 hypertensiona, johon liittyi asteen 5 arterioskleroosi. Kyseinen potilas oli tupakoinut ja sairastanut hypertensiota jo kauan, ja tapaus ilmeni yli 3 vuotta darolutamidihoidon aloittamisen jälkeen. Molemmissa hoitoryhmissä potilailla, joilla ei ollut anamneesissa hypertensiota, raportoitiin yleisemmin hypertensiotapahtumia.
Murtumat
Murtumia esiintyi 7,5 %:lla darolutamidin ja dosetakselin yhdistelmää saaneista potilaista ja 5,1 %:lla lumelääkkeen ja dosetakselin yhdistelmää saaneista potilaista.
Neutrofiilimäärän lasku
Neutrofiilimäärän laskua raportoitiin laboratorioarvojen poikkeavuutena 50,6 %:lla darolutamidin ja dosetakselin yhdistelmää saaneista potilaista ja 45,5 %:lla lumelääkkeen ja dosetakselin yhdistelmää saaneista potilaista. Neutrofiilimäärän asteen 3 ja 4 laskua raportoitiin 34,4 %:lla darolutamidin ja dosetakselin yhdistelmää saaneista potilaista ja 31,4 %:lla lumelääkkeen ja dosetakselin yhdistelmää saaneista potilaista. Molemmissa hoitoryhmissä neutrofiilimäärän laskun ja neutropenian esiintyvyys oli suurin hoidon ensimmäisten kuukausien aikana, minkä jälkeen tapahtumien ilmaantuvuus ja vakavuus vähenivät.
Veren bilirubiiniarvon nousu
Bilirubiiniarvon nousua raportoitiin laboratorioarvojen poikkeavuutena 19,6 %:lla darolutamidin ja dosetakselin yhdistelmää saaneista potilaista ja 10 %:lla lumelääkkeen ja dosetakselin yhdistelmää saaneista potilaista. Nämä tapahtumat olivat enimmäkseen voimakkuudeltaan asteen 1 tai 2 tapahtumia. Asteen 3 ja 4 bilirubiiniarvon nousua raportoitiin 0,5 %:lla darolutamidin ja dosetakselin yhdistelmää saaneista potilaista ja 0,3 %:lla lumelääkkeen ja dosetakselin yhdistelmää saaneista potilaista.
ALAT- ja ASAT-arvojen nousu
Alaniiniaminotransferaasiarvon (ALAT) nousua raportoitiin laboratorioarvojen poikkeavuuksina 42,3 %:lla darolutamidin ja dosetakselin yhdistelmää saaneista potilaista ja 38,0 %:lla lumelääkkeen ja dosetakselin yhdistelmää saaneista potilaista. Aspartaattiaminotransferaasiarvon (ASAT) nousuja raportoitiin laboratorioarvojen poikkeavuuksina 43,9 %:lla darolutamidin ja dosetakselin yhdistelmää saaneista potilaista ja 39,3 %:lla lumelääkkeen ja dosetakselin yhdistelmää saaneista potilaista. ALAT- ja ASAT-arvojen nousut olivat enimmäkseen voimakkuudeltaan asteen 1 tapahtumia. Asteen 3 ja 4 ALAT-arvojen nousua raportoitiin 3,7 %:lla darolutamidin ja dosetakselin yhdistelmää saaneista potilaista ja 3,0 %:lla lumelääkkeen ja dosetakselin yhdistelmää saaneista potilaista. Asteen 3 ja 4 ASAT-arvojen nousua raportoitiin 3,6 %:lla darolutamidin ja dosetakselin yhdistelmää saaneista potilaista ja 2,3 %:lla lumelääkkeen ja dosetakselin yhdistelmää saaneista potilaista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri PL 55
00034 FIMEA.
Yliannostus
Kliinisissä tutkimuksissa tutkittu suurin darolutamidiannos oli 900 mg kahdesti päivässä, joka vastaa 1 800 mg:n päivittäistä kokonaisannosta. Annosta rajoittavaa toksisuutta ei havaittu tällä annoksella.
Kun otetaan huomioon kyllästyvä absorptio (ks. kohta Farmakokinetiikka) ja se, että näyttöä akuutista toksisuudesta ei ole, suositeltua annosta suuremman darolutamidiannoksen saannin ei odoteta johtavan toksisuuteen.
Jos potilas on ottanut suositeltua suuremman annoksen, darolutamidihoitoa voidaan jatkaa ottamalla seuraava annos normaalin aikataulun mukaan.
Darolutamidiyliannostukseen ei tunneta spesifistä vastalääkettä eikä yliannostuksen oireita ole määritetty.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: endokrinologiset lääkeaineet, anti-androgeenit; ATC-koodi: L02BB06
Vaikutusmekanismi
Darolutamidi on androgeenireseptorin estäjä. Sen joustava polaarisesti korvattu pyratsolirakenne sitoutuu suurella voimakkuudella suoraan reseptorin ligandia sitovaan osaan.
Darolutamidi estää kilpailevasti androgeenin sitoutumisen, androgeenireseptorin nukleaarisen translokaation ja androgeenireseptorivälitteisen transkription. Darolutamidi ja sen pääasiallinen metaboliitti, ketodarolutamidi, olivat in vitro -aktiivisuudeltaan vastaavat. Darolutamidihoito vähentää eturauhassyövän kasvainsolujen proliferaatiota, mikä johtaa voimakkaaseen antituumorivaikutukseen.
Farmakodynaamiset vaikutukset
Keskimääräisessä QTcF-ajassa ei havaittu pidentymistä (ts. suurempaa kuin 10 ms) verrattuna lumelääkkeeseen sen jälkeen, kun darolutamidia oli annettu suun kautta 600 mg kahdesti päivässä.
Kliininen teho ja turvallisuus
Teho ja turvallisuus varmistettiin kolmessa satunnaistetussa, lumekontrolloidussa, faasin III monikeskustutkimuksessa potilailla, joilla oli etäpesäkkeetön kastraatioresistentti eturauhassyöpä (ARAMIS) ja etäpesäkkeinen hormonisensitiivinen eturauhassyöpä (ARANOTE ja ARASENS). Kaikki potilaat saivat samanaikaisesti luteinisoivan hormonin vapauttajahormonin (LHRH) agonistia tai antagonistia tai heille oli tehty bilateraalinen orkiektomia.
Etäpesäkkeetön kastraatioresistentti eturauhassyöpä - darolutamidihoito
Darolutamidin kliininen teho ja turvallisuus arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa faasin III monikeskustutkimuksessa (ARAMIS) potilailla, joilla oli etäpesäkkeetön (arvioituna tavanomaisella CT-kuvauksella, luustokartoituksella, MRI:llä) kastraatioresistentti eturauhassyöpä ja joilla prostataspesifisen antigeenin kahdentumisaika (PSADT) oli ≤ 10 kuukautta.
Potilaat otettiin mukaan tutkimukseen seuraavin ehdoin: heiltä oli saatu 3 suurenevaa prostataspesifisen antigeenin (PSA) pitoisuutta nadiirin jälkeen vähintään 1 viikon välein androgeenideprivaatiohoidon aikana, PSA ≥ 2 ng/ml seulonnassa ja seerumin testosteronin kastraattipitoisuus < 1,7 nmol/l.
Tutkimukseen hyväksyttiin mukaan potilaat, joiden esitiedoissa oli kouristuskohtauksia. Darolutamidihaaraan otettiin 12 potilasta (0,21 % potilaista), joilla oli aiemmin ollut kouristuskohtauksia.
Tutkimuksesta suljettiin pois potilaat, joilla oli huonossa hoitotasapainossa oleva korkea verenpaine tai hiljattain (viimeisten 6 kuukauden aikana) esiintynyt aivohalvaus, sydäninfarkti, vaikea/epästabiili angina pectoris, sepelvaltimon- tai ääreisvaltimon ohitusleikkaus, NYHA (New York Heart Association) III-IV -luokan sydämen vajaatoiminta.
Tutkimuksesta suljettiin pois potilaat, joita oli aiemmin hoidettu toisen sukupolven androgeenireseptorien estäjillä kuten entsalutamidi, apalutamidi ja darolutamidi tai CYP17‑entsyymin estäjillä kuten abirateroniasetaatti samoin kuin potilaat, joita oli hoidettu 28 vuorokauden sisällä ennen satunnaistamista systeemisellä kortikosteroidilla, jonka annos oli suurempi kuin 10 mg prednisonia/vrk.
Kaikkiaan 1 509 potilasta satunnaistettiin suhteessa 2:1 saamaan joko 600 mg darolutamidia suun kautta kahdesti päivässä (n = 955) tai lumelääkettä (n = 554).
Potilaat, joilla oli lyhyen akselin näkymässä aortan haarautumiskohdan alapuolella lantion imusolmukkeita, joiden koko oli < 2 cm, hyväksyttiin mukaan tutkimukseen. Metastaasin esiintyminen tai esiintymättömyys arvioitiin riippumattomassa keskitetyssä radiologisessa arviossa. Jälkeenpäin tarkasteltuna 89 potilaalla tunnistettiin olleen metastaasi lähtötilanteessa. Satunnaistamisessa käytettiin PSADT-arvoa (≤ 6 kuukautta tai > 6 kuukautta) ja osteoklasteihin vaikuttavan hoidon käyttöä tutkimukseen osallistumishetkellä (kyllä tai ei).
Potilaiden seuraavat demografiset ja sairautta koskevat lähtötilanteen ominaisuudet olivat tasapainossa hoitoryhmien kesken. Iän mediaani oli 74 vuotta (vaihteluväli 48‑95 vuotta), ja 9 % tutkittavista oli vähintään 85‑vuotiaita. Potilaat jakautuivat etnisen taustan mukaan seuraavasti: 79 % oli valkoihoisia, 13 % aasialaisia ja 3 % mustaihoisia. Valtaosalla (73 %) potilaista Gleason-pisteet olivat diagnoosihetkellä vähintään 7. Mediaani PSADT oli 4,5 kuukautta. Yhdeksälle prosentille (9 %) potilaista oli aiemmin tehty orkiektomia, 25 %:lle potilaista oli aiemmin tehty prostatektomia ja 50 % potilaista oli saanut aiemmin ainakin kerran sädehoitoa. Seitsemänkymmentäkuusi prosenttia (76 %) potilaista oli aiemmin saanut enemmän kuin yhden antihormonaalisen hoitokuurin. Tutkimukseen osallistumishetkellä potilaiden ECOG-toimintakykyluokka (Eastern Cooperative Oncology Group Performance Status) oli 0 (69 %:lla) tai 1 (31 %:lla).
Darolutamidihoitoa jatkettiin, kunnes taudin eteneminen vahvistettiin sokkoutetulla keskitetyllä radiologisella arviolla käyttäen tavanomaisia kuvantamismenetelmiä (CT, luustokartta, MRI) tai kunnes potilas ei enää sietänyt hoitoa, tai kunnes potilas vetäytyi tutkimuksesta.
Tehon ensisijainen päätetapahtuma oli metastaasivapaa elossaoloaika (MFS). Toissijaisia päätetapahtumia olivat kokonaiselossaoloaika (OS), aika kivun etenemiseen, aika ensimmäiseen eturauhassyövän hoitoon käytetyn sytotoksisen kemoterapian aloittamiseen ja aika oireileviin luustotapahtumiin (ulkoinen sädehoito kivun lievittämiseksi, uusi oireileva patologinen luunmurtuma, selkäytimen kompressio tai kasvaimeen liittyvä kirurginen toimenpide).
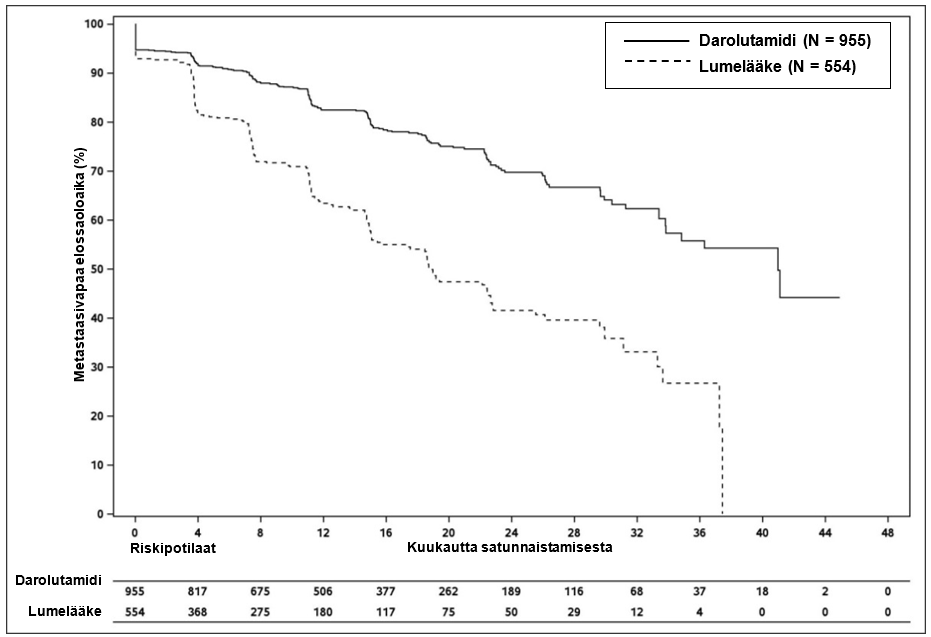
Darolutamidihoito pidensi metastaasivapaata elossaoloaikaa verrattuna lumelääkkeeseen (ks. taulukko 3 ja kuva 1).
MFS-tulokset olivat yhdenmukaisia kaikissa potilasryhmissä riippumatta PSADT-arvosta, luustoa vahvistavien lääkeaineiden aiemmasta käytöstä tai taudin paikallisesta levinneisyydestä. Muita alaryhmiä, joissa MFS-tulokset olivat yhdenmukaisia, olivat seuraavat: PSA lähtötilanteessa, Gleason-pisteet diagnoosihetkellä, ikä, maantieteellinen alue, ECOG-toimintakykyluokitus lähtötilanteessa, etninen tausta ja aiempien hormonihoitojen lukumäärä.
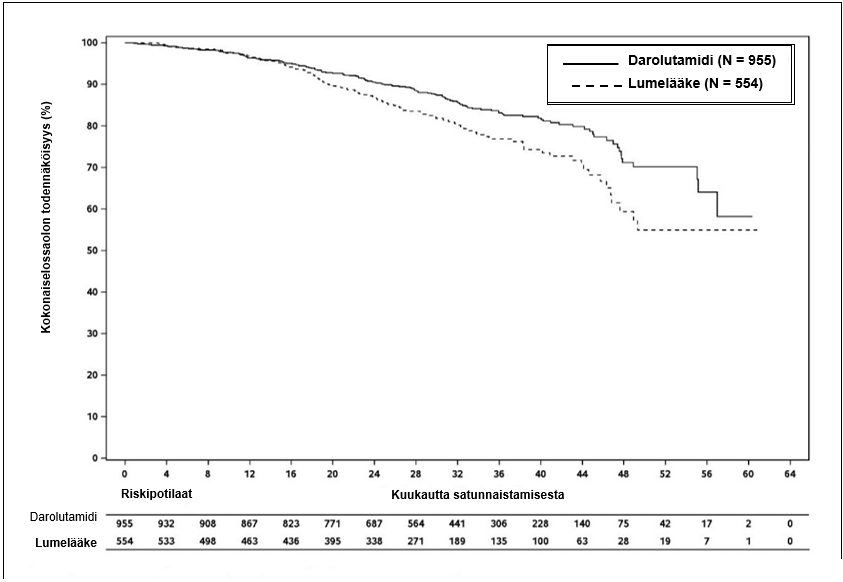
Ensisijaisen MFS-analyysin jälkeen, kun tutkimuksen sokkoutus oli purettu, lumelääkettä saaneille potilaille tarjottiin sokkouttamatonta hoitoa darolutamidilla (vaihtovuoroisen hoidon mahdollisuus). Lumelääkkeelle satunnaistetuista 554 potilaasta 170 potilasta (31 %) siirtyi saamaan darolutamidihoitoa. Kokonaiselossaoloajan analyysissä ei huomioitu vaihtovuoroisen hoidon aiheuttamia vaikutuksia.
Loppuanalyysin kohdalla lumelääkkeeseen verrattuna darolutamidihoito sai aikaan tilastollisesti merkitsevää pitenemistä kokonaiselinajassa (kummassakaan ryhmässä mediaania ei oltu saavutettu, ks. taulukko 3 ja kuva 2).
Darolutamidihoito sai aikaan myös tilastollisesti merkitsevän viiveen ajassa kivun etenemiseen, ajassa ensimmäisen sytotoksisen kemoterapian aloittamiseen ja ajassa ensimmäiseen oireilevaan luustotapahtumaan verrattuna lumelääkkeeseen (ks. taulukko 3).
Lopullisen analyysin ajankohtana hoidon mediaanikesto darolutamidihoitoa saaneilla potilailla oli 33,3 kuukautta (vaihteluväli 0,0–74,0 kuukautta) kaksoissokkoutetun ja avoimen vaiheen aikana yhteensä.
Kaikki analyysit suoritettiin täydellisessä analyysijoukossa.
Taulukko 3: Tehoa koskevat tulokset ARAMIS-tutkimuksesta
| Tehoa kuvaava muuttuja | Niiden potilaiden lukumäärä (%), joilla esiintyi tapahtumia | Mediaani (kuukautta) (95 %:n CI) | Riskisuhdeb (95 %:n luottamusväli [CI]) p‑arvo (2‑puolinen) | ||
Darolutamidi (N = 955) | Lumelääkea (N = 554) | Darolutamidi (N = 955) | Lumelääkea (N = 554) | ||
| Metastaasivapaa elossaoloaikac | 221 (23,1 %) | 216 (39,0 %) | 40,4 (34,3; NR) | 18,4 (15,5; 22,3) | 0,413 (0,341; 0,500) < 0,000001 |
| Kokonaiselossaoloaika | 148 (15,5 %) | 106 (19,1 %) | NR (56,1; NR) | NR (46,9; NR) | 0,685 (0,533; 0,881) 0,003048 |
| Aika kivun etenemiseenc, d | 251 (26,3 %) | 178 (32,1 %) | 40,3 (33,2; 41,2) | 25,4 (19,1; 29,6) | 0,647 (0,533; 0,785) 0,000008 |
| Aika 1. sytotoksisen kemoterapian aloittamiseen | 127 (13,3 %) | 98 (17,7 %) | NR (NR; NR) | NR (NR; NR) | 0,579 (0,444; 0,755) < 0,000044 |
| Aika 1. oireilevaan luustotapahtumaan | 29 (3.0 %) | 28 (5,1 %) | NR (NR; NR) | NR (NR; NR) | 0,484 (0,287; 0,815) 0,005294 |
a Mukaan lukien 170 potilasta, jotka siirtyivät avoimelle darolutamidille (vaihtovuoroinen asetelma)
b Riskisuhde < 1 suosii darolutamidia
c Metastaasivapaan elossaoloajan (MFS) ja aika kivun etenemiseen osalta analyysiä, joka suoritettu ensisijaisen analyysipisteen kohdalla, pidetään lopullisena analyysinä
d Potilaan ilmoittamat tulokset arvioituna kyselylomakkeen (Brief Pain Inventory-Short Form) avulla
NR Not reached (ei saavutettu)
Darolutamidihoito pidensi etenemisvapaata elossaoloaikaa (PFS, mediaani 36,8 vs. 14,8 kuukautta, HR = 0,380, nimellinen p < 0,000001) ja aikaa PSA:n etenemiseen (mediaani 29,5 vs. 7,2 kuukautta, HR = 0,164, nimellinen p < 0,000001). Vaikutus todettiin yhdenmukaiseksi kaikilla elossaoloajan mittareilla (MFS, OS ja PFS).
Kuva 1: Metastaasivapaan elossaoloajan Kaplan-Meier-käyrät (ARAMIS)
Kuva 2: Kokonaiselossaoloajan Kaplan-Meier-käyrät (ARAMIS)
ARAMIS-tutkimuksessa (kaksoissokkoutetussa vaiheessa) darolutamidia saaneiden potilaiden PSA-vasteprosentti oli merkitsevästi suurempi (määriteltiin ≥ 50 %:n vähenemäksi lähtötilanteesta) verrattuna lumelääkettä saaneisiin potilaisiin, vastaavasti 84,0 % vs. 7,9 % (ero = 76,1 %, p < 0,000001(nominaalinen p-arvo, vain tiedoksi)).
Etäpesäkkeinen hormonisensitiivinen eturauhassyöpä – darolutamidihoito
Darolutamidin tehoa ja turvallisuutta arvioitiin kaksoissokkoutetussa, lumekontrolloidussa faasin III monikeskustutkimuksessa (ARANOTE) potilailla, joilla oli etäpesäkkeinen hormonisensitiivinen eturauhassyöpä. Kaikkiaan 669 potilasta satunnaistettiin suhteessa 2:1 saamaan joko 600 mg darolutamidia suun kautta kahdesti päivässä (n = 446) tai lumelääkettä (n = 223). Darolutamidi- tai lumehoitoa jatkettiin taudin etenemiseen asti, tai kunnes potilaan syöpähoitoa muutettiin, potilas ei enää sietänyt hoitoa, potilas kuoli tai potilas vetäytyi tutkimuksesta.
Metastaasien esiintyminen arvioitiin riippumattomassa keskitetyssä radiologisessa arviossa. Potilaat, joiden tauti oli levinnyt vain alueellisiin imusolmukkeisiin (M0), suljettiin pois tutkimuksesta. Satunnaistaminen stratifioitiin viskeraalisten metastaasien ja aiemman paikallishoidon perusteella.
Tutkimuksesta suljettiin pois potilaat, joita oli aiemmin hoidettu toisen sukupolven androgeenireseptorien estäjillä kuten entsalutamidilla, apalutamidilla tai darolutamidilla, tai CYP17-entsyymin estäjillä kuten abirateroniasetaattilla, tai kemoterapialla, mukaan lukien dosetakseli.
Potilaiden demografiset ja sairautta koskevat seuraavat lähtötilanteen ominaisuudet olivat tasapainossa hoitoryhmien kesken. Iän mediaani oli 70 vuotta (vaihteluväli 43–93 vuotta), ja 4,2 % potilaista oli vähintään 85-vuotiaita. Potilaat jakautuivat etnisen taustan mukaan seuraavasti: 56,2 % oli valkoihoisia, 31,2 % aasialaisia, 9,7 % mustaihoisia tai afroamerikkalaisia ja 2,8 % muita. Valtaosalla (68,3 %) potilaista Gleason-pisteet olivat diagnoosihetkellä vähintään 8. Tutkimukseen osallistumishetkellä potilaiden ECOG-toimintakykyluokka oli 49,8 %:lla potilaista 0, 47,2 %:lla potilaista 1 ja 3,0 %:lla potilaista 2. Potilaista 72,5 %:lla oli de novo -tauti ja 21,7 %:lla uusiutunut tauti. Tutkimukseen osallistumishetkellä tautiluokitus oli 3,8 %:lla potilaista M1a (metastaaseja vain muissa kuin alueellisissa imusolmukkeissa), 77,1 %:lla M1b (luustometastaaseja joko imusolmukemetastaasien kanssa tai ilman) ja 19,1 %:lla M1c (viskeraalisia metastaaseja imusolmukemetastaasien kanssa tai ilman, tai luustometastaasien kanssa tai ilman). PSA-arvon mediaani oli lähtötilanteessa 21,3 mikrog/l. 70,6 %:lla potilaista oli korkean tautivolyymin sairaus ja 29,4 %:lla matalan tautivolyymin sairaus. Tauti luokiteltiin korkean volyymin sairaudeksi, kun potilaalla oli viskeraalisia metastaaseja tai 4 tai useampi leesio luustossa, joista vähintään yksi metastaasi oli selkärangan ja lantion luiden ulkopuolella. Tutkimukseen hyväksyttiin mukaan potilaat, joiden esitiedoissa oli kouristuskohtauksia, ja darolutamidiryhmään otettiin yksi tällainen potilas (0,2 %).
Tehon ensisijainen päätetapahtuma oli radiologinen etenemisvapaa elossaoloaika (rPFS).
Toissijaisia päätetapahtumia olivat kokonaiselossaoloaika (OS), aika seuraavan syöpähoidon aloittamiseen, aika kastraatioresistenttiin eturauhassyöpään, aika PSA-etenemiseen, aika havaitsemistason alapuolella olevaan PSA-arvoon ja aika kivun etenemiseen. Kivun etenemistä arvioitiin kyselylomakkeella (Brief Pain Inventory Short Form) potilaan ilmoittamien tulosten perusteella, ja se määriteltiin vähintään kahden pisteen huononemiseksi nadiirista tai ≥ 7 peräkkäistä päivää kestävän lyhyt- tai pitkävaikutteisten opioidikipulääkkeiden käytön aloittamiseksi syöpäsairauden takia. rPFS-analyysin käytettävien tietojen keruun päättymispäivämäärä oli 7. kesäkuuta 2024. rPFS:n ensisijaisen analyysin jälkeen, kun tutkimuksen sokkoutus oli purettu, lumelääkettä saaneille potilaille tarjottiin mahdollisuus siirtyä avoimeen darolutamidihaaraan. Niistä 63 potilaasta, jotka olivat edelleen lumelääkehoidossa ensisijaisen analyysin tietojen keruun päättymispäivänä, 60 potilasta (95 %) siirtyi saamaan avointa darolutamidihoitoa. Lopullisen kokonaiselossaoloaika-analyysin tietojen keruun päättymispäivä oli 10. tammikuuta 2025. Kokonaiselossaoloaika-analyysille ei tehty tilastollista korjausta avoimen darolutamidihaaran aiheuttamalle mahdolliselle vääristävälle vaikutukselle.
Taulukko 4: Tehoa koskevat tulokset ARANOTE-tutkimuksesta
| Tehoa kuvaava muuttuja | Niiden potilaiden lukumäärä (%), joilla esiintyi tapahtumia | Mediaani (kuukautta) (95 %:n CI) | Riskisuhdea (95 %:n luottamusväli [CI]) p arvo (1-puolinen)b | ||
| Darolutamidi (N = 446) | Lumelääke (N = 223) | Darolutamidi (N = 446) | Lumelääke (N = 223) | ||
| Radiologinen etenemisvapaa elossaoloaikac | 128 (28,7 %) | 94 (42,2 %) | A (A; A) | 25,0 (19,0; A) | 0,541 (0,413; 0,707) < 0,0001 |
| Kokonaiselossaoloaikad | 115 (25,8 %) | 70 (31,4 %) | A (A; A) | A (A; A) | 0,776 (0,577; 1,045) |
a Riskisuhde < 1 suosii darolutamidia
b Perustuu stratifioituun log rank -testiin
c rPFS-analyysin tietojen keruun päättymispäivä oli 7. kesäkuuta 2024.
d Kokonaiselossaoloajan p‑arvo ei ollut saavuttanut etukäteen määritettyä tilastollisen merkitsevyyden raja-arvoa kokonaiselossaoloajan lopullisen analyysin ajankohtana. Tietojen keruun päättymispäivä oli 10. tammikuuta 2025.
A: arvoa ei voida arvioida tietojen sensuroinnin vuoksi
Kuva 3: Kaplan Meier-käyrät radiologisesta etenemisvapaasta elossaoloajasta; etäpesäkkeistä hormonisensitiivistä eturauhassyöpää sairastava populaatio (ARANOTE)a
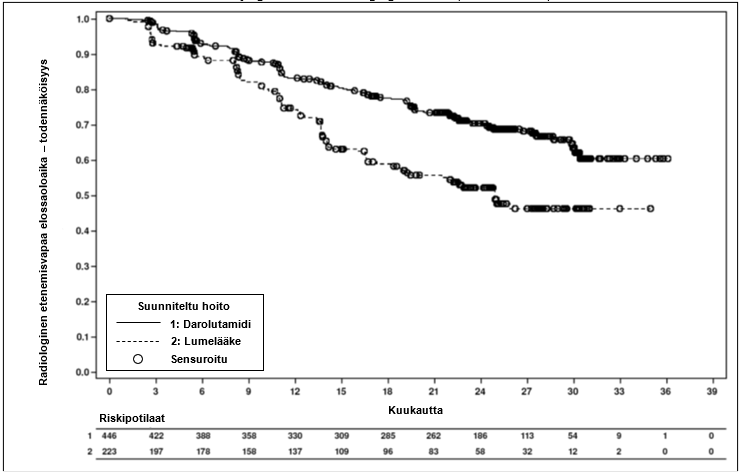
a rPFS:n prosenttiosuus 12 kuukauden kohdalla oli darolutamidiryhmässä 83,1 % (95 %:n luottamusväli 79,5-86,7 %) ja lumeryhmässä 74,1 % (95 %:n luottamusväli 68,0-80,2 %). rPFS:n prosenttiosuus 24 kuukauden kohdalla oli darolutamidiryhmässä 70,3 % (95 %:n luottamusväli 65,7-74,9 %) ja lumeryhmässä 52,1 % (95 %:n luottamusväli 44,7-59,5 %). Tietojen keruun päättymispäivä oli 7. kesäkuuta 2024.
Kuva 4: Kokonaiselossaoloajan Kaplan Meier-käyrät; etäpesäkkeistä hormonisensitiivistä eturauhassyöpää sairastava populaatio (ARANOTE)a
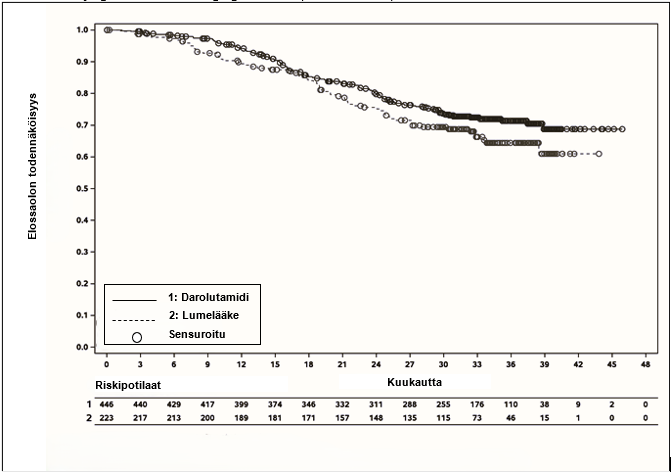
a OS:n prosenttiosuus 36 kuukauden kohdalla oli darolutamidiryhmässä 71,4 % (95 %:n luottamusväli 66,8-76,0 %) ja lumeryhmässä 64,4 % (95 %:n luottamusväli 57,3-71,4 %). Tietojen keruun päättymispäivä oli 10. tammikuuta 2025.
Etäpesäkkeinen hormonisensitiivinen eturauhassyöpä – darolutamidihoito yhdistelmänä dosetakselin kanssa
Darolutamidin ja dosetakselin yhdistelmän tehoa ja turvallisuutta arvioitiin kaksoissokkoutetussa, lumekontrolloidussa faasin III monikeskustutkimuksessa (ARASENS) potilailla, joilla oli etäpesäkkeinen hormonisensitiivinen eturauhassyöpä. Kaikkiaan 1 306 potilasta satunnaistettiin suhteessa 1:1 saamaan joko 600 mg darolutamidia suun kautta kahdesti päivässä (n = 651) tai lumelääkettä (n = 655) ja samanaikaisesti 75 mg/m2 dosetakselia 6 syklin ajan. Darolutamidi- tai lumehoitoa jatkettiin oireiseen taudin etenemiseen asti, tai kunnes potilaan syöpähoitoa muutettiin, potilas ei enää sietänyt hoitoa, potilas kuoli tai potilas vetäytyi tutkimuksesta.
Metastaasien esiintyminen arvioitiin riippumattomassa keskitetyssä radiologisessa arviossa. Potilaat, joiden tauti oli levinnyt vain alueellisiin imusolmukkeisiin (M0), suljettiin pois tutkimuksesta. Satunnaistaminen stratifioitiin taudin levinneisyyden (metastaaseja vain muissa kuin alueellisissa imusolmukkeissa [M1a], metastaaseja luustossa imusolmukemetastaasien kanssa tai ilman [M1b] tai metastaaseja sisäelimissä imusolmukemetastaasien tai luustometastaasien kanssa tai ilman [M1c]) ja alkalisen fosfataasiarvon (< tai ≥ normaaliarvojen yläraja) perusteella tutkimukseen osallistumishetkellä. Potilaat, joilla oli aivometastaaseja, saivat osallistua tutkimukseen. Yhtään tällaista potilasta ei kuitenkaan ollut tutkimuspotilaiden joukossa.
Potilaiden seuraavat demografiset ja sairautta koskevat ominaisuudet olivat tasapainossa hoitoryhmien kesken. Iän mediaani oli 67 vuotta (vaihteluväli 41–89 vuotta), ja 0,5 % tutkittavista oli vähintään 85‑vuotiaita. Potilaat jakautuivat etnisen taustan mukaan seuraavasti: 52 % oli valkoihoisia, 36 % aasialaisia ja 4 % mustaihoisia. Valtaosalla (78 %) potilaista Gleason-pisteet olivat diagnoosihetkellä vähintään 8. ECOG-toimintakykyluokka oli 71 %:lla potilaista 0 ja 29 %:lla potilaista 1. Potilaista 86,1 %:lla oli de novo -tauti ja 12,9 %:lla uusiutunut tauti. Tutkimukseen osallistumishetkellä tautiluokitus oli 3 %:lla potilaista M1a, 79,5 %:lla M1b ja 17,5 %:lla M1c. Alkalinen fosfataasi oli 44,5 %:lla potilaista < ULN ja 55,5 %:lla potilaista ≥ ULN. PSA-arvon mediaani oli lähtötilanteessa 30,3 mikrog/l darolutamidiryhmässä ja 24,2 mikrog/l lumeryhmässä. Tutkimukseen hyväksyttiin mukaan potilaat, joiden esitiedoissa oli kouristuskohtauksia, ja darolutamidin ja dosetakselin yhdistelmää saaneeseen ryhmään otettiin neljä tällaista potilasta (0,6 %).
77,0 %:lla potilaista oli korkean tautivolyymin sairaus ja 23,0 %:lla matalan tautivolyymin sairaus. Korkean volyymin sairaus määriteltiin, kun potilaalla oli viskeraalisia metastaaseja tai 4 tai useampi leesio luustossa, joista vähintään yksi metastaasi oli selkärangan ja lantion luiden ulkopuolella. Noin 25 % potilaista sai samanaikaista hoitoa bisfosfonaateilla tai denosumabilla.
Tehon ensisijainen päätetapahtuma oli kokonaiselossaoloaika (OS). Toissijaisia päätetapahtumia olivat aika kastraatioresistenttiin eturauhassyöpään, aika kivun etenemiseen, elossaoloaika ilman oireilevia luustotapahtumia (SSE-FS), aika ensimmäiseen oireilevaan luustotapahtumaan (SSE), aika seuraavan syöpähoidon aloittamiseen, aika sairauteen liittyvien fyysisten oireiden pahenemiseen ja aika ≥ 7 peräkkäistä päivää kestävän opioidien käytön aloittamiseen. Kivun etenemistä arvioitiin kyselylomakkeella (Brief Pain Inventory Short Form) potilaan ilmoittamien tulosten perusteella, ja se määriteltiin vähintään kahden pisteen huononemiseksi nadiirista tai ≥ 7 peräkkäistä päivää kestävän lyhyt- tai pitkävaikutteisten opioidikipulääkkeiden käytön aloittamiseksi.
Hoidon keston mediaani oli 41,0 kuukautta (vaihteluväli: 0,1–56,5 kuukautta) darolutamidin ja dosetakselin yhdistelmää saaneilla potilailla ja 16,7 kuukautta (vaihteluväli: 0,3–55,8 kuukautta) lumelääkkeen ja dosetakselin yhdistelmää saaneilla potilailla. Darolutamidin ja dosetakselin yhdistelmää saaneessa ryhmässä 87,6 % potilaista sai täydet 6 sykliä dosetakselia ja 1,5 % potilaista ei saanut dosetakselia. Lumelääkkeen ja dosetakselin yhdistelmää saaneessa ryhmässä 85,5 % potilaista sai täydet 6 sykliä dosetakselia ja 2,0 % potilaista ei saanut dosetakselia.
Taulukko 5: Tehoa koskevat tulokset ARASENS-tutkimuksesta
| Tehoa kuvaava muuttuja | Niiden potilaiden lukumäärä (%), joilla esiintyi tapahtumia | Mediaani (kuukautta) (95 %:n CI) | Riskisuhdeb (95 %:n luottamusväli [CI]) p‑arvo (1‑puolinen)c | ||
Darolutamidi + dosetakseli (N = 651) | Lumelääke + dosetakseli (N = 654)a | Darolutamidi + dosetakseli (N = 651) | Lumelääke + dosetakseli (N = 654)a | ||
| Kokonaiselossaoloaika d | 229 (35,2 %) | 304 (46,5 %) | NR (NR, NR) | 48,9 (44,4, NR) | 0,675 (0,568; 0,801) < 0,0001 |
a Yksi lumeryhmän potilas suljettiin pois kaikista analyyseista
b Riskisuhde < 1 suosii darolutamidia
c Perustuu stratifioituun log rank -testiin
d Kokonaiselossaoloajan tulokset olivat yhdenmukaisia potilaiden alaryhmissä, taudin levinneisyys ja alkalinen fosfataasiarvo mukaan lukien
NR: Not reached (ei saavutettu)
Seuraavat tehon toissijaiset päätetapahtumat osoittivat tilastollisesti merkitsevän edun darolutamidin ja dosetakselin yhdistelmää saaneiden potilaiden hyväksi verrattuna lumelääkettä ja dosetakselia saaneisiin potilaisiin: aika kastraatioresistenttiin eturauhassyöpään (mediaani NR vs. 19,1 kuukautta, HR = 0,357, p < 0,0001), aika ensimmäiseen oireilevaan luustotapahtumaan (mediaani NR vs. NR kuukautta, HR = 0,712, p < 0,0081), aika seuraavan syöpähoidon aloittamiseen (mediaani NR vs. 25,3 kuukautta, HR = 0,388, p < 0,0001), aika kivun etenemiseen (mediaani NR vs. 27,5 kuukautta, HR = 0,792, p < 0,0058), elossaoloaika ilman oireilevia luustotapahtumia (mediaani 51,2 vs. 39,7 kuukautta, HR = 0,609, p < 0,0001).
Kuva 5: Kokonaiselossaoloajan Kaplan‑Meier-kuvaajat (ARASENS)a
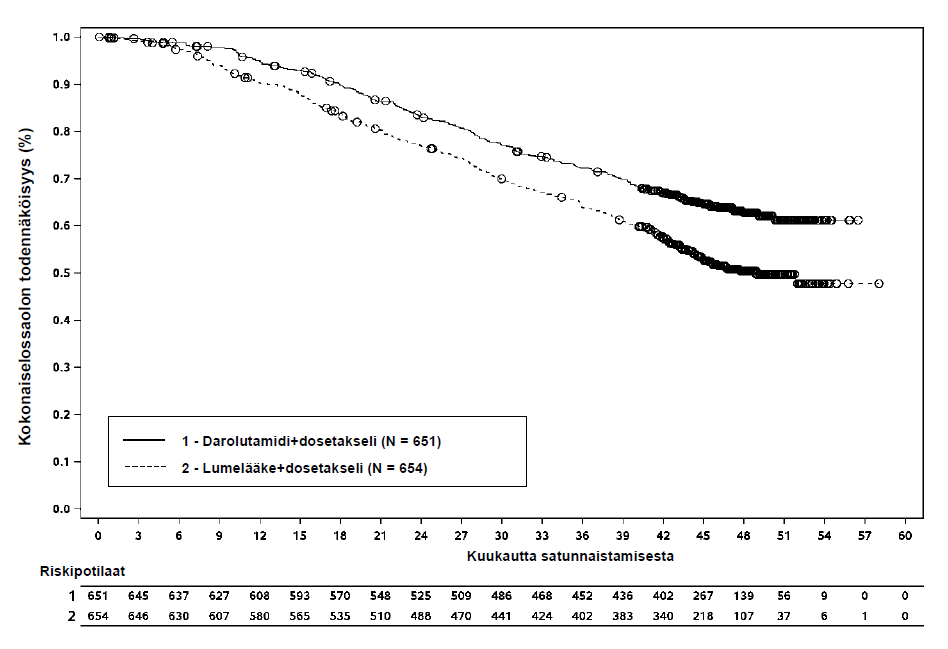
a Kokonaiselossaoloprosentti 36 kuukauden kohdalla oli 72,3 % (95 %:n luottamusväli 68,8–75,8 %) darolutamidin ja dosetakselin yhdistelmää saaneessa ryhmässä vs. 63,8 % (95 %:n luottamusväli 60,1‑67,6 %) lumelääkkeen ja dosetakselin yhdistelmää saaneessa ryhmässä.
Kokonaiselossaoloprosentti 48 kuukauden kohdalla oli 62,7 % (95 %:n luottamusväli 58,7–66,7 %) darolutamidin ja dosetakselin yhdistelmää saaneessa ryhmässä vs. 50,4 % (95 %:n luottamusväli 46,3‑54,6 % lumelääkkeen ja dosetakselin yhdistelmää saaneessa ryhmässä.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset darolutamidin käytöstä kaikissa pediatrisissa potilasryhmissä eturauhasen malignien neoplasmojen hoidossa (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Yleinen johdanto
Darolutamidi koostuu kahdesta diastereomeeristä [(S,R)darolutamidi ja (S,S)darolutamidi], jotka konvertoituvat keskenään tärkeimmän verenkierrossa esiintyvän metaboliitin, ketodarolutamidin, välityksellä. Kaikkien kolmen aineen farmakologinen aktiivisuus in vitro on samankaltainen.
Darolutamidi liukenee heikosti vesiliuottimiin laajalla pH-alueella. Yleisesti se liukenee paremmin orgaanisiin liuottimiin.
Imeytyminen
Suun kautta annetun 600 mg:n annoksen (kaksi 300 mg:n tablettia) kahdesti päivässä antamisen jälkeen darolutamidin huippupitoisuudet plasmassa vakaassa tilassa olivat 4,79 mg/l (variaatiokerroin: 30,9 %) etäpesäkkeetöntä kastraatioresistenttiä eturauhassyöpää sairastavilla potilailla ARAMIS-tutkimuksessa ja 3,84 mg/l (variaatiokerroin: 35,6 %) etäpesäkkeistä hormonisensitiivistä eturauhassyöpää sairastavilla potilailla ARASENS-tutkimuksessa. Mediaaniaika huippupitoisuuden saavuttamiseen plasmassa oli 3‑4 tuntia. Kahden diastereomeerin eli (S,R)darolutamidin ja (S,S)darolutamidin suhde muuttui tabletin sisältämästä 1:1-suhteesta niiden suhteeksi plasmassa, joka on noin 1:9 perustuen tietoihin AUC012 -arvosta vakaassa tilassa. Ruokailun yhteydessä suun kautta tapahtuneen annon jälkeen vakaan tilan pitoisuus saavutetaan 25 vuorokauden kuluttua, kun lääkettä on annettu toistuvasti kahdesti päivässä.
Absoluuttinen biologinen hyötyosuus vs. laskimoon annetun injektion hyötyosuus on noin 30 % tyhjään mahaan suun kautta otetun, 300 mg darolutamidia sisältävän NUBEQA-tabletin annon jälkeen. Darolutamidin biologinen hyötyosuus parani 2,0‑2,5‑kertaisesti, kun se annettiin ruoan kanssa. Samanlainen altistuksen lisääntyminen havaittiin lääkeaineen pääasiallisen metaboliitin eli ketodarolutamidin osalta.
Jakautuminen
Darolutamidin keskimääräinen laskennallinen jakautumistilavuus laskimoon annon jälkeen on 119 litraa, mikä osoittaa, että darolutamidi jakautuu laajasti koko elimistöön, sekä solunsisäisiin että solunulkoisiin nestetiloihin.
Darolutamidi sitoutuu kohtalaisesti (92 %) ihmisen plasman proteiineihin, eikä kahden eri diastereomeerin välillä ole eroa. Darolutamidin pääasiallinen metaboliitti ketodarolutamidi sitoutuu voimakkaasti (99,8 %) plasman proteiineihin.
Darolutamidin kulkeutumista veri-aivoesteen läpi ei ole tutkittu kliinisesti. Darolutamidin altistusprosentit aivoissa AUC0-24-arvon osalta ovat kuitenkin hyvin alhaisia: altistus plasmassa on 4,5 % rotille annetun kerta-annoksen jälkeen ja 1,9-3,9 % hiirille annetun toistuvan annoksen jälkeen. Tämä osoittaa, että darolutamidi kulkeutuu rotilla ja hiirillä ehjän veri-aivoesteen läpi vähäisessä määrin ja että todennäköisyys, että darolutamidi läpäisisi kliinisesti merkittävässä määrin ehjän veri-aivoesteen ihmisillä, on vähäinen.
Biotransformaatio
Diastereomeerit (S,R)-darolutamidi ja (S,S)-darolutamidi voivat konvertoitua keskenään ketodarolutamidi-metaboliitin kautta. Aineenvaihdunta suosii (S,S)-darolutamidia.
Sen jälkeen kun 300 mg 14 C-darolutamidia on annettu suun kautta kerta-annoksena oraaliliuoksen muodossa, ainoa pääasiallinen metaboliitti on ketodarolutamidi, jonka kokonaisaltistus plasmassa on noin 2 kertaa darolutamidin altistusta suurempi. Plasman 14 C-radioaktiivisuudesta 87,4 % johtui darolutamidin ja ketodarolutamidin yhteisestä vaikutuksesta, mikä osoittaa muiden metaboliittien vähäisen merkityksen.
Darolutamidi metaboloituu ensisijaisesti oksidatiivisen aineenvaihdunnan kautta pääasiassa CYP3A4.n välityksellä sekä suoran glukuronidaation kautta ensisijaisesti UGT1A9:n ja UGT1A1:n välityksellä. Lisäksi osoitettiin, että pääasiassa AKR1C-isoentsyymit katalysoivat ketodarolutamidin reduktiota diastereomeeriaineiksi.
Eliminaatio
Darolutamidin ja ketodarolutamidin efektiivinen puoliintumisaika potilaiden plasmassa on noin 18-20 tuntia. Kahdesta darolutamidia sisältävästä diastereomeerista (S,R)-darolutamidilla on lyhyempi efektiivinen puoliintumisaika (9 tuntia) verrattuna (S,S)-darolutamidiin, jonka puoliintumisaika on 22 tuntia. Darolutamidin puhdistuma laskimoon annon jälkeen oli 116 ml/min (CV: 39,7 %).
Lääkeaineeseen liittyvästä aineksesta noin 63,4 % erittyy virtsaan (noin 7 % muuttumattomana) ja 32,4 % erittyy ulosteeseen. Annoksesta yli 95 % poistui elimistöstä 7 päivän aikana annon jälkeen.
Lineaarisuus/ei-lineaarisuus
Annoksen ollessa välillä 100-700 mg (kerta-annoksen jälkeen ja vakaassa tilassa) molempien diastereomeerien ja pääasiallisen metaboliitin eli ketodarolutamidin altistus suurenee lineaarisesti lähes annosriippuvaisella tavalla. Darolutamidille altistumisessa ei havaittu lisää suurenemista, kun lääkeainetta annettiin 900 mg kahdesti päivässä. Tämä perustuu saturoituneeseen absorptioon.
Erityisryhmät
Iäkkäät
Darolutamidin farmakokinetiikassa ei havaittu kliinisesti merkityksellisiä eroja (65-95-vuotiaat).
Munuaisten vajaatoiminta
Kliinisessä farmakokineettisessä tutkimuksessa darolutamidin AUC oli 2,5 kertaa suurempi ja Cmax 1,6 kertaa suurempi vaikeaa munuaisten vajaatoimintaa (laskennallinen glomerulusten suodatusnopeus [eGFR] 15-29 ml/min/1,73 m2) sairastavilla potilailla verrattuna terveisiin vapaaehtoisiin.
Eräässä populaatiofarmakokineettisessä analyysissa osoitetaan, että potilailla, joilla on lievä, keskivaikea tai vaikea munuaisten vajaatoiminta (eGFR 15-89 ml/min/1,73 m2), darolutamidin altistus (AUC) on vastaavasti 1,1-kertaisesti, 1,3-kertaisesti ja noin 1,5-kertaisesti suurempi verrattuna potilaisiin, joilla munuaiset toimivat normaalisti.
Darolutamidin farmakokinetiikkaa ei ole tutkittu potilailla, joilla on loppuvaiheen munuaissairaus ja jotka saavat dialyysihoitoa (eGFR < 15 ml/min/1,73 m2).
Maksan vajaatoiminta
Kliinisessä farmakokineettisessä tutkimuksessa darolutamidin Cmax oli 1,5 kertaa suurempi ja AUC 1,9 kertaa suurempi potilailla, joilla oli keskivaikea maksan vajaatoiminta (Child-Pugh B) verrattuna terveisiin vapaaehtoisiin. Tietoja vaikeaa maksan vajaatoimintaa sairastavista potilaista (Child-Pugh C) ei ole.
Etnisten ryhmien väliset erot
Darolutamidin farmakokinetiikassa ei havaittu kliinisesti merkityksellisiä etnisyyteen perustuvia eroja (valkoinen, japanilainen, muu aasialainen kuin japanilainen, mustaihoinen tai afroamerikkalainen). Populaatiofarmakokineettinen analyysi osoitti altistuksen (AUC) geometrisen keskiarvon kohoavan jopa 1,56-kertaisesti (90 % luottamusväli: 1,43‑1,70) japanilaisilla potilailla verrattuna muiden alueiden potilaisiin sekä ARAMIS- että ARASENS-tutkimuksissa.
Prekliiniset tiedot turvallisuudesta
Systeeminen toksisuus
Rotilla ja koirilla tehdyissä toistuvan annoksen toksisuustutkimuksissa pääasialliset löydökset olivat muutokset urosten sukupuolielimissä (elinten painon pieneneminen, johon liittyi eturauhasen atrofiaa ja lisäkivesten tulehduksia). Nämä vaikutukset esiintyivät systeemisillä altistuksilla, jotka olivat samaa luokkaa kuin ihmisellä odotettavissa oleva altistus tai sen alle (AUC-vertailun perusteella). Muita lisääntymisjärjestelmän kudosten muutoksia olivat mm. rotilla hyvin pieni vakuolisaation lisääntyminen aivolisäkkeessä ja siemenrakkuloiden ja rintarauhasten atrofia ja eritystoiminnan väheneminen sekä koirilla kivesten hypospermia, siementiehyiden laajeneminen ja rappeuma. Urosten lisääntymiselinten muutokset olivat molemmilla eläinlajeilla yhdenmukaiset darolutamidin farmakologisen aktiivisuuden kanssa, ja muutokset palautuivat kokonaan tai osittain 4-8 viikon toipumisjakson jälkeen.
Sikiötoksisuus/teratogeenisuus
Kehitystoksikologisia tutkimuksia ei ole tehty.
Lisääntymistoksisuus
Lisääntymistoksikologisia tutkimuksia ei ole tehty. Rotilla ja koirilla tehtyjen toistuvan annoksen toksisuustutkimusten tulosten perusteella urosten hedelmällisyys kuitenkin todennäköisesti heikentyy, mikä on yhdenmukaista darolutamidin farmakologisen aktiivisuuden kanssa.
Genotoksisuus ja karsinogeenisuus
Darolutamidi ei aiheuttanut mutaatioita Amesin testissä (mikrobien mutageneesitesti). Suurina pitoisuuksina darolutamidi sai kylläkin aikaan rakenteellisia kromosomipoikkeavuuksia in vitro viljellyissä ihmisen lymfosyyteissä. Rotan maksassa ja pohjukaissuolessa in vivo tehdyissä yhdistetyissä luuytimen ja mikrotuman testeissä ja Comet- (SCGE-) -määrityksessä ei havaittu genotoksisuutta altistuksilla, jotka ylittivät ihmisen maksimialtistuksen.
Kun darolutamidia annettiin suun kautta rasH2-siirtogeenisille uroshiirille 6 kuukauden ajan, karsinogeenistä potentiaalia ei todettu enintään 1 000 mg/kg/vrk annoksilla, jotka vastaavat 0,9–1,3-kertaista kliinistä darolutamidialtistusta (AUC) ja 2,1–2,3-kertaista kliinistä ketodarolutamidialtistusta suositellulla kliinisellä vuorokausiannoksella 1 200 mg/vrk. Tämän tutkimuksen perusteella darolutamidin karsinogeenistä riskiä ei voida täysin poissulkea.
Turvallisuusfarmakologia
In vitro darolutamidi esti heikosti hERG-kaliumvirtaa ja L-tyypin kalsiumkanavaa. In vivo nukutetuilla koirilla darolutamidi lyhensi hieman QT-ajan kestoa, mutta tätä vaikutusta ei havaittu tajuissaan olleilla koirilla.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Kalsiumvetyfosfaatti (E341)
Kroskarmelloosinatrium
Laktoosimonohydraatti
Magnesiumstearaatti (E470b)
Povidoni (E1201)
Kalvopäällyste
Hypromelloosi
Laktoosimonohydraatti
Makrogoli (E1521)
Titaanioksidi (E171)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
NUBEQA tabletti, kalvopäällysteinen
300 mg (L:kyllä) 112 fol (3141,95 €)
PF-selosteen tieto
PVC/alumiinifolioläpipainopakkaukset, jotka sisältävät kukin 16 kalvopäällysteistä tablettia. Yksi pakkaus sisältää 112 kalvopäällysteistä tablettia.
Valmisteen kuvaus:
Tabletti, kalvopäällysteinen (tabletti).
Valkoinen tai luonnonvalkoinen, soikea tabletti, jonka pituus on 16 mm ja leveys 8 mm, ja jonka toisella puolella on merkintä ”300” ja toisella puolella merkintä ”BAYER”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
NUBEQA tabletti, kalvopäällysteinen
300 mg 112 fol
- Ylempi erityiskorvaus (100 %). Darolutamidi: Aikuisten etäpesäkkeisen hormonisensitiivisen ja etäpesäkkeettömän kastraatioresistentin eturauhassyövän hoito erityisin edellytyksin (1525).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Darolutamidi: Etäpesäkkeettömän kastraatioresistentin eturauhassyövän hoito erityisin edellytyksin (3036).
ATC-koodi
L02BB06
Valmisteyhteenvedon muuttamispäivämäärä
17.07.2025
Yhteystiedot
 BAYER OY
BAYER OY Tuulikuja 2, PL 73
02151 Espoo
020 785 21
www.bayer.fi