
SPRAVATO nenäsumute, liuos 28 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Spravato-nenäsumute (esketamiini): Mitkä ovat hoidon riskit?
Terveydenhuollon ammattilainen
Terveydenhuollon ammattilaisten tarkistuslista potilaan kotiuttamisen arviointiin
Spravato-nenäsumutetta saavia potilaita koskevat riskienminimointitoimenpiteet
Vaikuttavat aineet ja niiden määrät
Yksi nenäsumutin sisältää esketamiinihydrokloridia määrän, joka vastaa 28 mg:aa esketamiinia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet
Lääkemuoto
Nenäsumute, liuos.
Kliiniset tiedot
Käyttöaiheet
Spravato on tarkoitettu yhdistelmänä jonkin serotoniinin takaisinoton estäjän (SSRI) tai serotoniinin ja noradrenaliinin takaisinoton estäjän (SNRI) kanssa aikuisille hoitoresistentin masennustilan hoitoon, kun potilaan parhaillaan sairastamaan keskivaikeaan tai vaikeaan masennusjaksoon ei ole saatu vastetta vähintään kahdella eri masennuslääkehoidolla.
Spravato on tarkoitettu yhdessä suun kautta otettavan masennuslääkehoidon kanssa akuutiksi lyhytaikaiseksi hoidoksi masennusoireiden nopeaan vähentämiseen kliiniseen arvioon perustuvassa psykiatrisessa hätätilanteessa aikuispotilailla, joilla on keskivaikea tai vaikea masennustila.
Ks. kohdasta Farmakodynamiikka kuvaus tutkituista potilasjoukoista.
Ehto
Päätöksen tämän valmisteen määräämisestä tekee psykiatri tai hänen valvonnassaan oleva lääkäri. Valmistetta saa toimittaa edellyttäen että potilas on allekirjoittanut apteekkisopimuksen. Apteekki toimittaa valmisteen hoitopaikkaan, jossa annostelu tapahtuu. Lääkettä ei saa luovuttaa apteekista suoraan potilaalle. Valmiste on tarkoitettu potilaan itse annosteltavaksi terveydenhuollon ammattilaisen suorassa valvonnassa. Hoitokertaan kuuluu nenäsumutteen annostelu ja annostelun jälkeinen tarkkailu. Tämän valmisteen sekä annostelun että annostelun jälkeisen tarkkailun pitää tapahtua asianmukaisessa hoitopaikassa.
Annostus ja antotapa
Päätöksen tämän lääkevalmisteen määräämisestä tekee psykiatri.
Se on tarkoitettu potilaan itse annosteltavaksi terveydenhuollon ammattilaisen suorassa valvonnassa.
Hoitokertaan kuuluu nenäsumutteen annostelu ja annostelun jälkeinen tarkkailu. Sekä annostelun että annostelun jälkeisen tarkkailun pitää tapahtua asianmukaisessa hoitopaikassa.
Potilaan tutkiminen ennen hoitoa
Verenpaine pitää mitata ennen Spravato‑nenäsumutteen annostelua.
Jos verenpaine on lähtötilanteessa koholla, verenpaineen lyhytaikaisen nousun riskit ja hoidon hyödyt pitää arvioida (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Lääkevalmistetta ei saa antaa potilaalle, jos verenpaineen tai kallonsisäisen paineen noususta aiheutuu vakava riski (ks. kohta Vasta-aiheet).
Jos potilaalla on kliinisesti merkittävä tai epästabiili sydän- ja verisuonitauti tai hengityselinsairaus, lisävarotoimet ovat tarpeen. Lääkevalmistetta saa tällöin antaa vain hoitopaikassa, jossa on asianmukaiset elvytysvälineet saatavissa ja jossa on paikalla painelu-puhalluselvytykseen koulutuksen saanut terveydenhuollon ammattilainen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Seuranta lääkkeen annostelun jälkeen
Potilaan verenpaine pitää mitata uudelleen noin 40 minuutin kuluttua Spravato‑nenäsumutteen annostelusta, ja tämän jälkeen kliinisen tarpeen mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilaan pitää mahdollisen sedaation, dissosiaatio-oireiden ja verenpaineen nousun vuoksi olla terveydenhuollon ammattilaisen seurannassa niin pitkään, kunnes potilaan tilan katsotaan olevan kliinisesti stabiili ja hänet voidaan kotiuttaa hoitopaikasta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Hoitoresistentti masennustila
Annossuositukset hoitoresistentin masennustilan hoitoon esitetään taulukossa 1 ja taulukossa 2 (≥ 65-vuotiaat aikuiset). Ylläpitohoidossa suositellaan jatkamaan sen annoksen käyttöä, jonka potilas saa hoidon aloitusvaiheen lopussa. Annosmuutosten pitää perustua edellisen annoksen tehoon ja siedettävyyteen. Ylläpitohoidossa annostukseen pitää valita yksilöllisesti pisin antoväli, jolla remissio/vaste säilyy.
| Taulukko 1. Spravato‑suositusannos hoitoresistentin masennustilan hoitoon < 65-vuotiaille aikuisille | |
| Hoidon aloitusvaihe | Ylläpitohoito |
Viikot 1‑4: Aloitusannos päivänä 1: 56 mg Seuraavat annokset: 56 mg tai 84 mg kaksi kertaa viikossa | Viikot 5‑8: 56 mg tai 84 mg kerran viikossa Viikosta 9 eteenpäin: 56 mg tai 84 mg joka toinen viikko tai kerran viikossa |
| Hoidon hyödyt pitää arvioida aloitusvaiheen lopussa ja tehdä päätös hoidon jatkamisen tarpeesta. | Hoidon jatkamisen tarve pitää arvioida uudelleen säännöllisin väliajoin. |
| Taulukko 2. Spravato‑suositusannos hoitoresistentin masennustilan hoitoon ≥ 65-vuotiaille aikuisille | |
| Hoidon aloitusvaihe | Ylläpitohoito |
Viikot 1‑4: Aloitusannos päivänä 1: 28 mg Seuraavat annokset: 28 mg, 56 mg tai 84 mg kaksi kertaa viikossa; kaikki annosmuutokset on tehtävä 28 mg:n lisäyksinä | Viikot 5‑8: 28 mg, 56 mg tai 84 mg kerran viikossa; kaikki annosmuutokset on tehtävä 28 mg:n lisäyksinä Viikosta 9 eteenpäin: 28 mg, 56 mg tai 84 mg joka toinen viikko tai kerran viikossa; kaikki annosmuutokset on tehtävä 28 mg:n lisäyksinä |
| Hoidon hyödyt pitää arvioida aloitusvaiheen lopussa ja tehdä päätös hoidon jatkamisen tarpeesta. | Hoidon jatkamisen tarve pitää arvioida uudelleen säännöllisin väliajoin. |
Hoitoa suositellaan jatkamaan vähintään 6 kuukauden ajan masennusoireiden vähenemisen jälkeen.
Masennustilasta aiheutuvan psykiatrisen hätätilanteen akuutti lyhytaikainen hoito
Annossuositus aikuispotilaille (< 65-vuotiaille) on 84 mg kaksi kertaa viikossa 4 viikon ajan. Annos pienennetään 56 mg:aan siedettävyyden perusteella. Neljän viikon Spravato-hoidon jälkeen jatketaan suun kautta otettavaa masennuslääkehoitoa kliiniseen arvioon perustuen.
Näillä potilailla Spravato-hoidon pitää olla osa kattavaa kliinistä hoitosuunnitelmaa.
Ruokailua ja juomista koskevat suositukset ennen lääkkeen antoa
Joillekin potilaille voi ilmaantua pahoinvointia ja oksentelua lääkevalmisteen käytön jälkeen, joten potilaita pitää kehottaa olemaan syömättä vähintään 2 tuntia ennen lääkkeen antoa ja olemaan juomatta nestettä vähintään 30 minuuttia ennen lääkkeen annostelua (ks. kohta Haittavaikutukset).
Nenään annettavat kortikosteroidit ja nenän tukkoisuutta vähentävät valmisteet
Jos potilas tarvitsee nenään annettavaa kortikosteroidia tai nenän tukkoisuutta vähentävää valmistetta lääkkeenantopäivänä, potilasta pitää neuvoa olla ottamatta tällaisia lääkkeitä vähintään 1 tunti ennen esketamiinin annostelua.
Hoitokertojen jääminen väliin
Jos potilaan hoitokerta/hoitokertoja on jäänyt väliin ensimmäisten neljän hoitoviikon aikana, potilaan pitää jatkaa senhetkisen hoitoaikataulun noudattamista.
Jos hoitoresistenttiä masennustilaa sairastavan potilaan hoitokerta/hoitokertoja jää väliin ylläpitojakson aikana ja masennusoireet pahenevat, harkitse kliiniseen arvioon perustuen aiempaan hoitoaikatauluun palaamista (ks. taulukot 1 ja 2).
Erityispotilasryhmät
Iäkkäät (65‑vuotiaat ja vanhemmat)
Hoitoresistenttiä masennustilaa sairastavien iäkkäiden potilaiden Spravato‑aloitusannos on 28 mg esketamiinia (aloitusannos päivänä 1, ks. taulukko 2 edellä). Seuraavia annoksia pitää suurentaa tehon ja siedettävyyden perusteella 28 mg:n lisäyksinä 56 mg:aan tai 84 mg:aan saakka.
Spravato-nenäsumutetta ei ole tutkittu iäkkäillä potilailla masennustilasta aiheutuvan psykiatrisen hätätilanteen akuuttina lyhytaikaisena hoitona.
Maksan vajaatoiminta
Lievää (Child–Pugh‑luokka A) tai keskivaikeaa (Child–Pugh‑luokka B) maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa. Keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden hoidossa 84 mg:n maksimiannosta pitää kuitenkin käyttää varoen.
Spravato‑valmistetta ei ole tutkittu vaikeaa maksan vajaatoimintaa (Child–Pugh‑luokka C) sairastavilla potilailla. Käyttöä tälle potilasryhmälle ei suositella (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa. Dialyysipotilaita ei tutkittu.
Pediatriset potilaat
Spravato‑valmisteen turvallisuutta ja tehoa 17 vuoden ikäisten ja nuorempien pediatristen potilaiden hoidossa ei ole varmistettu. Ei ole asianmukaista käyttää Spravato‑valmistetta alle 7 vuoden ikäisille lapsille.
Antotapa
Tämä lääkevalmiste on tarkoitettu annettavaksi vain nenään. Nenäsumutin on kertakäyttöinen laite, joka vapauttaa yhteensä 28 mg esketamiinia kahtena suihkeena (yksi suihke kumpaankin sieraimeen). Laitetta ei pidä koesuihkuttaa, jotta lääkevalmistetta ei mene hukkaan. Nenäsumutin on tarkoitettu potilaan käyttöön terveydenhuollon ammattilaisen valvonnassa. Potilas käyttää 1 nenäsumuttimen (28 mg:n annokseen), 2 nenäsumutinta (56 mg:n annokseen) tai 3 nenäsumutinta (84 mg:n annokseen), ja pitää kunkin nenäsumuttimen käytön välillä 5 minuutin tauon.
Aivastaminen lääkkeen annon jälkeen
Jos potilas aivastaa heti lääkkeen annostelun jälkeen, uutta nenäsumutinta ei pidä ottaa käyttöön.
Kahden suihkeen antaminen peräkkäin samaan sieraimeen
Jos lääke annetaan epähuomiossa samaan sieraimeen, uutta nenäsumutinta ei pidä ottaa käyttöön.
Hoidon lopettaminen ei vaadi annoksen pienentämistä vähitellen, sillä kliinisistä tutkimuksista saatujen tietojen perusteella vieroitusoireiden riski on vähäinen.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle, ketamiinille tai kohdassa Apuaineet mainituille apuaineille.
-
Verenpaineen tai kallonsisäisen paineen noususta potilaalle aiheutuva vakava riski (ks. kohta Haittavaikutukset)
- aneurysmaattinen verisuonitauti (mukaan lukien kallonsisäisissä, rintakehän tai vatsa‑aortan valtimoissa tai ääreisvaltimoissa)
- aiempi aivojensisäinen verenvuoto
- äskettäinen (edeltävien 6 viikon aikana) sydän- ja verisuonitapahtuma, mukaan lukien sydäninfarkti.
Varoitukset ja käyttöön liittyvät varotoimet
Itsemurha-ajatukset tai itsetuhoiset ajatukset tai tilan kliininen paheneminen
Esketamiinin tehoa itsemurhan estämiseen tai itsemurha-ajatusten tai itsetuhoisen käyttäytymisen vähentämiseen ei ole osoitettu (ks. kohta Farmakodynamiikka). Esketamiinin käyttö ei sulje pois sairaalahoidon tarvetta, jos se on kliinisesti aiheellista, vaikka potilas kokisikin vointinsa parantuneen ensimmäisen esketamiiniannoksen jälkeen.
Etenkin hoidon alkuvaiheessa ja annosmuutosten jälkeen pitää tarkkailla erityisesti suuren riskin potilaita. Potilaille (ja potilasta hoitaville henkilöille) pitää kertoa, että voinnin heikentymistä, itsemurhakäyttäytymistä tai itsemurha-ajatuksia sekä käyttäytymisen epätavallisia muutoksia pitää seurata ja että tällaisten oireiden ilmetessä pitää hakeutua heti lääkäriin.
Masennukseen liittyy lisääntynyt itsemurha-ajatusten, itsetuhoisuuden ja itsemurhan riski. Koska tämä riski on koholla siihen saakka kunnes kliinisesti merkittävä remissio saavutetaan, pitää potilaita seurata tarkasti. Yleisen kliinisen kokemuksen perusteella itsemurhariski saattaa lisääntyä toipumisen alkuvaiheessa.
Potilailla, joilla on aiemmin ollut itsetuhoisuutta tai itsemurhayrityksiä tai joilla on huomattavan voimakkaita itsemurha-ajatuksia ennen hoidon aloittamista, tiedetään olevan tavanomaista suurempi itsemurha-ajatusten tai itsemurhayritysten riski, joten heitä pitää seurata tarkoin hoidon aikana.
Neuropsykiatristen ja motoristen toimintojen heikkeneminen
Esketamiinin on kliinisissä tutkimuksissa raportoitu aiheuttaneen uneliaisuutta, sedaatiota, dissosiatiivisia oireita, aistivääristymiä, huimausta, kiertohuimausta ja ahdistuneisuutta (ks. kohta Haittavaikutukset). Tällaiset vaikutukset saattavat heikentää keskittymiskykyä, arviointikykyä, ajattelua, reaktionopeutta ja motorisia taitoja. Potilasta pitää tarkkailla jokaisella hoitokerralla terveydenhuollon ammattilaisen valvonnassa sen arvioimiseksi, milloin potilaan tila on kliinisen arvion perusteella vakaa (ks. kohta Vaikutus ajokykyyn ja koneidenkäyttökykyyn).
Hengityslama
Suurten esketamiini- ja ketamiiniannosten antamisesta anestesiaa varten nopeana injektiona laskimoon voi aiheutua hengityslamaa. Syvää sedaatiota on raportoitu harvinaisina tapauksina. Esketamiinin ja keskushermostoa lamaavien aineiden samanaikainen käyttö voi lisätä sedaation riskiä (ks. kohta Yhteisvaikutukset). Hengityslamaa on havaittu valmisteen markkinoille tulon jälkeen harvinaisina tapauksina. Valtaosa näistä tapauksista on raportoitu käytettäessä samanaikaisesti keskushermostoa lamaavia lääkeaineita ja/tai potilailla, joilla on muita samanaikaisia sairauksia, kuten lihavuutta, ahdistuneisuutta, sydän- ja verisuonisairauksia sekä hengityselinsairauksia. Nämä tapahtumat olivat luonteeltaan ohimeneviä ja hävisivät puhuteltaessa/kosketettaessa potilasta tai annettaessa lisähappea. Potilasta on seurattava tarkoin sedaation ja hengityslaman havaitsemiseksi.
Vaikutus verenpaineeseen
Esketamiini voi nostaa systolista ja/tai diastolista verenpainetta tilapäisesti. Verenpaineen nousu on korkeimmillaan noin 40 minuuttia lääkevalmisteen annon jälkeen ja se pysyy koholla noin 1–2 tuntia (ks. kohta Haittavaikutukset). Merkittävää verenpaineen nousua voi esiintyä minkä tahansa hoitokerran jälkeen. Esketamiini on vasta-aiheista potilaille, joille verenpaineen tai kallonsisäisen paineen noususta aiheutuu vakava riski (ks. kohta Vasta-aiheet). Muita sydän‑ ja verisuonitauteja ja aivoverisuonisairauksia sairastavat potilaat on tutkittava tarkoin ennen esketamiinin määräämistä sen selvittämiseksi, ovatko esketamiinin mahdolliset hyödyt sen riskejä suuremmat.
Jos potilaan verenpaineen katsotaan olevan koholla ennen annoksen antamista (yleisohje: < 65‑vuotiailla potilailla > 140/90 mmHg ja ≥ 65‑vuotiailla potilailla > 150/90 mmHg), elintapoja ja/tai lääkehoitoa on muutettava verenpaineen alentamiseksi ennen esketamiinihoidon aloittamista. Jos verenpaine on koholla ennen esketamiiniannoksen antamista, esketamiinihoidon siirtämisestä myöhemmäksi on päätettävä potilaan yksilöllisen hyöty‑riskiarvion perusteella.
Verenpainetta pitää seurata annoksen antamisen jälkeen. Verenpaine pitää mitata noin 40 minuuttia annoksen antamisen jälkeen ja sen jälkeen kliinisen tarpeen mukaan, kunnes verenpaine alenee. Jos verenpaine pysyy koholla pidemmän aikaa, on viipymättä konsultoitava lääkäriä, jolla on kokemusta verenpaineen hoidosta. Jos potilaalle ilmaantuu hypertensiivisen kriisin oireita, potilas on heti lähetettävä päivystykselliseen hoitoon.
Potilaat, joilla on kliinisesti merkittävä tai epästabiili sydän- ja verisuonitauti tai hengityselinsairaus
Jos potilaalla on kliinisesti merkittävä tai epästabiili sydän- ja verisuonitauti tai hengityselinsairaus, aloita esketamiinihoito vain, jos hyödyt ovat riskejä suuremmat. Esketamiinihoito pitää tällöin antaa hoitopaikassa, jossa on asianmukaiset elvytysvälineet saatavissa ja jossa on paikalla painelu-puhalluselvytykseen koulutuksen saanut terveydenhuollon ammattilainen. Esimerkkejä tällaisista sairauksista ovat mm.:
- merkittävä keuhkojen vajaatoiminta, mukaan lukien keuhkoahtaumatauti
- uniapnea, johon liittyy sairaalloinen lihavuus (painoindeksi [BMI] ≥ 35)
- huonossa hoitotasapainossa oleva brady- tai takyarytmia, josta aiheutuu hemodynaamista epävakautta
- aiemmin sairastettu sydäninfarkti; potilaan pitää tällöin olla ennen lääkkeen annostelua kliinisesti stabiili eikä sydänoireita saa olla
- hemodynaamisesti merkittävä sydämen läppäsairaus tai sydämen vajaatoiminta (NYHA-luokka III‑IV).
Lääkkeiden ja huumausaineiden väärinkäyttö, riippuvuus, vieroitusoireet
Henkilöillä, joilla on aiempaa lääkkeiden tai huumeiden väärinkäyttö- tai riippuvuustaustaa, saattaa olla suurempi riski esketamiinin väärälle tai virheelliselle käytölle. Jokaisen potilaan riski käyttää esketamiinia väärin tai virheellisesti pitää arvioida ennen lääkkeen määräämistä, ja esketamiinia saavia potilaita pitää seurata hoidon aikana väärinkäyttöön tai virheelliseen käyttöön liittyvän käyttäytymisen tai sairauksien, myös päihdehakuisen käyttäytymisen, havaitsemiseksi.
Ketamiinin pitkäaikaiskäytössä on raportoitu riippuvuutta ja toleranssia. Ketamiiniriippuvaisilla henkilöillä on havaittu ketamiinin käytön lopettamisen yhteydessä vieroitusoireina ketamiininhimoa, ahdistuneisuutta, vapinaa, hikoilua ja sydämentykytystä.
Ketamiini on R-ketamiinin ja S-ketamiinin (esketamiini) raseeminen seos. Se on lääkevalmiste, jonka käyttöön on raportoitu liittyneen väärinkäyttöä. Esketamiinin väärinkäytön, virheellisen käytön ja luvattoman käytön mahdollisuus on minimoitu, sillä lääkkeen käyttö tapahtuu terveydenhuollon ammattilaisen suorassa valvonnassa. Spravato sisältää esketamiinia, joten on mahdollista, että sitä voidaan käyttää väärin ja hyväksytystä käyttöaiheesta poikkeavasti.
Muut riskiryhmät
Seuraavia sairauksia sairastavien potilaiden Spravato‑hoidossa pitää olla varovainen. Nämä potilaat on tutkittava tarkoin ennen Spravato‑nenäsumutteen määräämistä, ja hoidon saa aloittaa vain, jos hyödyt ovat riskejä suuremmat:
- psykoosi (parhaillaan tai aiemmin sairastettu)
- mania tai kaksisuuntainen mielialahäiriö (parhaillaan tai aiemmin todettu)
- riittämättömästi hoidettu hypertyreoosi
- aiemmin todettu aivovamma, hypertensiivinen enkefalopatia, selkäydinnesteeseen annettu hoito aivokammiosuntilla tai jokin muu sairaus, johon liittyy kallonsisäisen paineen nousua.
Iäkkäät (65-vuotiaat ja vanhemmat)
Spravato-hoitoa saavilla iäkkäillä potilailla saattaa olla jalkeille pääsyn jälkeen tavanomaista suurempi kaatumisten riski. Näitä potilaita pitää siksi seurata tarkoin.
Vaikea maksan vajaatoiminta
Spravato-nenäsumutteen käyttöä ei suositella potilaille, joilla on Child–Pugh-luokan C (vaikea-asteinen) maksan vajaatoiminta, sillä altistus oletettavasti suurenee, eikä tästä potilasryhmästä ole kliinistä kokemusta.
Ketamiinin pitkäaikaiskäyttöön on raportoitu liittyvän maksatoksisuutta, joten tällaisen vaikutuksen mahdollisuutta ei voida sulkea pois Spravato-nenäsumutteen pitkäaikaiskäytössä. Pitkäkestoisessa kliinisessä tutkimuksessa, joissa potilaat saivat hoitoa keskimäärin yhteensä 42,9 kuukauden altistuksen ajan (enintään 79 kuukautta), ei havaittu viitteitä maksatoksisuudesta.
Virtsatieoireet
Spravato-nenäsumutteen käytön yhteydessä on raportoitu virtsatie- ja virtsarakko-oireita (ks. kohta Haittavaikutukset). On suositeltavaa tarkkailla virtsatie- ja virtsarakko-oireita hoidon aikana ja lähettää potilas asianmukaisen terveydenhuollon ammattilaisen hoitoon, jos oireet jatkuvat.
Yhteisvaikutukset
Spravato-nenäsumutteen samanaikainen käyttö keskushermostoa lamaavien aineiden kanssa (esim. bentsodiatsepiinit, opioidit, alkoholi) saattaa voimistaa sedaatiota, joten sitä pitää tarkkailla.
Verenpainetta pitää seurata tarkoin, jos Spravato-nenäsumutteen kanssa käytetään samanaikaisesti psykostimulantteja (esim. amfetamiinit, metyylifenidaatti, modafiniili, R-modafiniili) tai muita verenpainetta nostavia lääkevalmisteita (esim. ksantiinijohdokset, ergometriini, kilpirauhashormonit, vasopressiini tai MAO:n estäjät, kuten tranyylisypromiini, selegiliini, feneltsiini).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Spravato-nenäsumutetta ei suositella raskauden aikana eikä naisille, jotka voivat tulla raskaaksi eivätkä käytä ehkäisyä.
Raskaus
Esketamiinin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja. Eläinkokeissa ketamiinin (R- ja S-ketamiinin raseeminen seos) on havaittu olevan neurotoksinen kehittyvälle sikiölle (ks. kohta Prekliiniset tiedot turvallisuudesta). Esketamiinin osalta samankaltaista riskiä ei voida sulkea pois.
Jos nainen tulee raskaaksi Spravato‑hoidon aikana, hoito pitää lopettaa, ja potilaalle pitää mahdollisimman pian kertoa sikiölle mahdollisesti aiheutuneesta riskistä sekä hoitovaihtoehdoista.
Imetys
Ei tiedetä, erittyykö esketamiini ihmisillä äidinmaitoon. Tiedot koe-eläimistä ovat osoittaneet esketamiinin erittyvän äidinmaitoon. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. On päätettävä, lopetetaanko imetys vai pidättäydytäänkö Spravato-hoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Eläinkokeissa on osoitettu, ettei esketamiini vaikuta haitallisesti hedelmällisyyteen ja lisääntymiskykyyn.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Spravato‑valmisteella on huomattava vaikutus ajokykyyn ja koneidenkäyttökykyyn. Spravato-nenäsumutteen on kliinisissä tutkimuksissa raportoitu aiheuttaneen uneliaisuutta, sedaatiota, dissosiatiivisia oireita, aistivääristymiä, huimausta, kiertohuimausta ja ahdistuneisuutta (ks. kohta Haittavaikutukset). Potilaita pitää neuvoa ennen Spravato-nenäsumutteen annostelua, ettei täydellistä vireystilaa ja motorista koordinaatiokykyä edellyttävää toimintaa, kuten ajoneuvon ajamista tai koneiden käyttöä, joihin liittyy vaaratilanteen riski, pidä tehdä ennen kuin seuraavana päivänä rauhallisen yöunen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Spravato‑nenäsumutetta saaneiden potilaiden yleisimmät haittavaikutukset olivat huimaus (31 %), dissosiaatio-oireet (27 %), pahoinvointi (27 %), päänsärky (23 %), uneliaisuus (18 %), makuaistin häiriö (18 %), kiertohuimaus (16 %), hypestesia (11 %), oksentelu (11 %) ja verenpaineen nousu (10 %).
Haittavaikutustaulukko
Esketamiinin käytön yhteydessä raportoidut haittavaikutukset luetellaan taulukossa 3. Kunkin elinjärjestelmän haittavaikutukset luetellaan esiintyvyyttä osoittavan otsikon alla seuraavan esitystavan mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
| Taulukko 3. Luettelo haittavaikutuksista | ||||
| Elinjärjestelmä | Haittavaikutus | |||
| Esiintyvyys | ||||
| Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | |
| Psyykkiset häiriöt | dissosiaatio | ahdistuneisuus, euforinen mieliala, sekavuustila, derealisaatio, ärtyisyys, aistiharhat, mukaan lukien näköharhat, agitaatio, havaintovirheet, paniikkikohtaus, muuttunut ajan hahmottaminen | psykomotorinen hidastuneisuus, tunneperäinen kuormitus, dysforia | |
| Hermosto | huimaus, päänsärky, uneliaisuus, makuaistin häiriö, hypestesia | parestesiat, sedaatio, vapina, henkisen suorituskyvyn heikkeneminen, letargia, dysartria, keskittymishäiriöt | silmävärve, psykomotorinen ylivilkkaus | kouristuskohtaukset |
| Silmät | näön sumeneminen | |||
| Kuulo ja tasapainoelin | kiertohuimaus | tinnitus, hyperakusia | ||
| Sydän | takykardia | bradykardia | ||
| Verisuonisto | hypertensio | hypotensio | ||
| Hengityselimet, rintakehä ja välikarsina | epämukavat tuntemukset nenässä, nielun ärsytys, suunielun kipu, nenän kuivuminen, mukaan lukien karstanmuodostus nenässä, nenän kutina | hengityslama | ||
| Ruoansulatuselimistö | pahoinvointi, oksentelu | suun hypestesia, suun kuivuminen | syljen liikaeritys | |
| Iho ja ihonalainen kudos | hyperhidroosi | kylmänhiki | ||
| Munuaiset ja virtsatiet | pollakisuria, dysuria, virtsaamispakko | |||
| Yleisoireet ja antopaikassa todettavat haitat | poikkeava olo, päihtyneisyyden tunne, voimattomuus, itkuisuus, tunne kehon lämpötilan muuttumisesta | kävelyn häiriöt | ||
| Tutkimukset | verenpaineen nousu | |||
Pitkäaikainen turvallisuus
Pitkäaikaista turvallisuutta arvioitiin monikeskustutkimuksena tehdyssä vaiheen 3 avoimessa jatkotutkimuksessa (TRD3008) 1 148 aikuisella potilaalla, joilla oli hoitoresistentti masennustila ja jotka käsittivät 3 777 potilasvuoden altistuksen. Potilaat saivat esketamiinihoitoa keskimäärin yhteensä 42,9 kuukauden altistuksen ajan (enintään 79 kuukautta), ja 63 % potilaista sai hoitoa vähintään 3 vuoden ajan ja 28 % potilaista sai hoitoa vähintään 5 vuoden ajan. Esketamiinin turvallisuusprofiili oli yhdenmukainen keskeisissä kliinisissä tutkimuksissa havaitun tunnetun turvallisuusprofiilin kanssa. Uusia turvallisuushuolia ei tunnistettu.
Valikoitujen haittavaikutusten kuvaus
Dissosiaatio
Yksi yleisimmistä esketamiinin psykologisista haittavaikutuksista oli dissosiaatio (27 %). Muita tähän liittyviä termejä olivat derealisaatio (2,2 %), depersonalisaatio (2,2 %), kuvitelmat (1,3 %) ja aikakäsityksen vääristyminen (1,2 %). Näiden haittavaikutusten raportoitiin olleen ohimeneviä ja hävinneen itsestään, ja niitä esiintyi lääkkeenottopäivänä. Vaikea-asteisen dissosiaation ilmaantuvuus oli alle 4 % kaikissa tutkimuksissa. Dissosiaatio‑oireet hävisivät tyypillisesti 1,5 tunnin kuluessa annoksen jälkeen, ja niiden vaikeusaste näytti vähenevän ajan mittaan toistetussa annostelussa.
Sedaatio/uneliaisuus/hengityslama
Kliinisissä tutkimuksissa haittavaikutuksina esiintyneet sedaatio (9,3 %) ja uneliaisuus (18,2 %) olivat vaikeusasteeltaan pääasiassa lieviä tai keskivaikeita, niitä esiintyi lääkkeenottopäivänä, ja ne hävisivät itsestään samana päivänä. Sedatiiviset vaikutukset hävisivät tyypillisesti 1,5 tunnin kuluessa annoksen jälkeen. Uneliaisuuden esiintyvyys oli pitkäaikaishoidossa ajan mittaan suhteellisen tasaista. Sedaatioon ei liittynyt hengitysvaikeuksia, ja hemodynaamiset parametrit (mukaan lukien vitaalimerkit ja happisaturaatio) pysyivät normaaliarvoissa. Valmisteen markkinoille tulon jälkeisessä käytössä on havaittu harvinaisina tapauksina hengityslamaa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Verenpaineen muutokset
Spravato‑nenäsumutetta ja suun kautta otettavaa masennuslääkettä yhdistelmänä hoitoresistenttiä masennustilaa koskeneissa kliinisissä tutkimuksissa saaneilla potilailla todettiin systolisen ja diastolisen verenpaineen nousua. Nousu oli noin 7–9 mmHg (systolinen verenpaine) ja 4–6 mmHg (diastolinen verenpaine) 40 minuuttia annoksen jälkeen, ja 2–5 mmHg (systolinen verenpaine) ja 1–3 mmHg (diastolinen verenpaine) 1,5 tuntia annoksen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Esketamiinin ja suun kautta otettavan masennuslääkkeen yhdistelmää saaneilla potilailla systolisen verenpaineen merkittävän kohoamisen (≥ 40 mmHg:n nousu) esiintyvyys oli 8 %:sta (< 65-vuotiaat) 17 %:iin (≥ 65-vuotiaat), ja diastolisen verenpaineen merkittävän kohoamisen (≥ 25 mmHg:n nousu) esiintyvyys oli 13 %:sta (< 65-vuotiaat) 14 %:iin (≥ 65-vuotiaat). Systolisen verenpaineen nousua (≥ 180 mmHg) todettiin 3 %:lla ja diastolisen verenpaineen nousua (≥ 110 mmHg) todettiin 4 %:lla potilaista.
Kognition ja muistin heikkeneminen
Ketamiinin pitkäaikaiskäytössä tai väärinkäytössä on raportoitu kognition ja muistin heikkenemistä. Nämä vaikutukset eivät lisääntyneet ajan mittaan, ja ne korjautuivat ketamiinin käytön lopettamisen jälkeen. Kognitiivisten toimintojen havaittiin pysyvän vakaina, kun niitä arvioitiin ajan mittaan esketamiininenäsumutteen pitkäkestoisissa kliinisissä tutkimuksissa, mukaan lukien kliinisessä tutkimuksessa, jossa potilaat saivat hoitoa keskimäärin yhteensä 42,9 kuukauden altistuksen ajan (enintään 79 kuukautta).
Virtsatieoireet
Suurten ketamiiniannosten päivittäisessä ja pitkäaikaisessa käytössä on raportoitu interstitiaalista kystiittiä. Esketamiinilla tehdyissä kliinisissä tutkimuksissa ei todettu interstitiaalista kystiittia, mutta alempien virtsateiden oireiden (pollakisuria, dysuria, virtsaamispakko, nykturia ja kystiitti) havaittiin lisääntyneen esketamiinihoitoa saaneilla potilailla lumehoitoa saaneisiin potilaisiin verrattuna. Pitkäkestoisessa kliinisessä tutkimuksessa, jossa potilaat saivat hoitoa keskimäärin yhteensä 42,9 kuukauden altistuksen ajan (enintään 79 kuukautta), ei havaittu interstitiaalista kystiittiä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Potilaan ottaman Spravato-yliannoksen mahdollisuus on minimoitu valmisteen suunnittelulla sekä sillä, että lääkkeen käyttö tapahtuu terveydenhuollon ammattilaisen valvonnassa (ks. kohta Annostus ja antotapa).
Oireet
Terveillä vapaaehtoisilla tutkittu esketamiininenäsumutteen suurin kerta-annos oli 112 mg, jonka ei havaittu olevan toksinen eikä vaikuttavan haitallisesti kliiniseen lopputulokseen. Suositusannoksiin verrattuna esketamiininenäsumutteen 112 mg:n annokseen liittyi kuitenkin yleisemmin haittavaikutuksia, kuten huimausta, liikahikoilua, uneliaisuutta, tuntoaistin heikentymistä, poikkeavaa oloa, pahoinvointia ja oksentelua.
Ketamiinin käyttökokemusten perusteella henkeä uhkaavia oireita on odotettavissa annoksilla, jotka ovat 25‑kertaisia tavanomaiseen ketamiinin anestesia‑annokseen nähden. Kliinisinä oireina on kuvattu kouristuksia, sydämen rytmihäiriöitä ja hengityspysähdystä. Vastaavien hoitoannoksia suurempien esketamiiniannosten antaminen nenään ei todennäköisesti ole mahdollista.
Hoito
Esketamiiniyliannostukseen ei ole spesifistä vasta‑ainetta. Yliannostustilanteessa on otettava huomioon usean eri lääkkeen yhteisvaikutuksen mahdollisuus. Spravato‑yliannostuksen hoitoon pitää kuulua kliinisten oireiden hoito ja asiaankuuluva seuranta. Potilasta on tarkkailtava ja seurattava toipumiseen saakka.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Masennuslääkkeet ja keskushermostoa stimuloivat lääkkeet, muut masennuslääkkeet. ATC‑koodi: N06AX27.
Vaikutusmekanismi
Esketamiini on raseemisen ketamiinin S‑enantiomeeri. Se on ionotrooppisen N‑metyyli‑D‑aspartaatti (NMDA) -glutamaattireseptorin epäselektiivinen, kilpailematon estäjä. NMDA‑reseptoria estämällä esketamiini lisää ohimenevästi glutamaatin vapautumista, mikä puolestaan lisää α‑amino‑3‑hydroksi‑5‑metyyli‑4‑isoksatsolipropionihapporeseptorin (AMPA‑reseptorin) stimulaatiota. Tämän seurauksena hermokasvutekijäsignalointi lisääntyy, mikä saattaa osaltaan palauttaa synapsien toiminnan mielialaa ja emotionaalista käyttäytymistä säätelevillä aivoalueilla. Esketamiinin nopeaan vasteeseen voivat liittyä dopaminergisen hermovälityksen korjautuminen mielihyvään ja motivaatioon liittyvillä aivoalueilla sekä stimulaation väheneminen anhedoniaa säätelevillä aivoalueilla.
Farmakodynaamiset vaikutukset
Väärinkäytön mahdollisuus
Lääkkeen väärinkäytön mahdollisuutta selvitettiin tutkimuksessa, jossa oli mukana huumeiden satunnaisia sekakäyttäjiä (n = 41). Esketamiininenäsumutteen kerta‑annoksiin (84 mg ja 112 mg) ja positiivisena vertailuvalmisteena käytettyyn laskimoon annettavaan ketamiiniin (0,5 mg/kg 40 minuutin kestoisena infuusiona) liittyvät pisteet ”lääkkeestä saatavasta mielihyvästä” ja muista lääkkeen vaikutusten subjektiivisista mittareista olivat merkittävästi korkeammat kuin lumevalmisteella.
Kliininen teho ja turvallisuus
Esketamiininenäsumutteen tehoa ja turvallisuutta hoitoresistentin masennuksen hoitoon selvitettiin viidessä vaiheen 3 kliinisessä tutkimuksessa (TRD3001, TRD3002, TRD3003, TRD3004 ja TRD3005) aikuispotilailla (18–86‑vuotiailla), jotka täyttivät vaikea‑asteisen masennuksen DSM‑5‑kriteerit ja jotka eivät olleet saaneet vastetta parhaillaan sairastettuun vaikea‑asteiseen masennusjaksoon vähintään kahdella suun kautta otettavalla masennuslääkkeellä riittävin annoksin ja riittävän pituisena hoitona. Tutkimuksiin otettiin mukaan 1 833 aikuispotilasta, joista 1 601 potilasta sai esketamiinia. Lisäksi Japanissa tehdyssä vaiheen 2 tutkimuksessa (TRD2005) satunnaistettiin 202 potilasta (122 potilasta sai esketamiinia), pääasiassa Kiinassa tehdyssä vaiheen 3 tutkimuksessa (TRD3006) satunnaistettiin 252 potilasta (126 potilasta sai esketamiinia) ja vaiheen 3 tutkimuksessa (TRD3013) satunnaistettiin 676 potilasta (334 potilasta sai esketamiinia).
Esketamiininenäsumutteen tehoa ja turvallisuutta selvitettiin kahdessa vaiheen 3 kliinisessä tutkimuksessa aikuispotilailla (18–64-vuotiaita), joilla oli keskivaikea tai vaikea masennustila (MADRS-kokonaispisteet > 28) ja joilla oli tämän varmistavia vastauksia MINI (Mini International Neuropsychiatric Interview)-kysymyksiin B3 (Ajatteletko [edes hetkittäin] vahingoittavasi tai satuttavasi itseäsi vähintään jossakin määrin aikomuksena tai tietoisena, että saatat sen vuoksi kuolla, tai ajatteletko itsemurhaa [eli itsesi tappamista]?) ja B10 (Onko sinulla ollut kuluneiden 24 tunnin aikana aikomuksia toteuttaa itsemurha-ajatuksesi?). Tutkimukseen otettiin mukaan 456 aikuista potilasta, ja näistä 227 potilasta sai Spravato-nenäsumutetta.
Hoitoresistentin masennuksen lyhytkestoiset tutkimukset
Esketamiinia arvioitiin hoitoresistenttiä masennusta sairastavilla potilailla kolmessa vaiheen 3 lyhytkestoisessa (4 viikkoa) satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa tutkimuksessa. Tutkimukset TRANSFORM‑1 (TRD3001) ja TRANSFORM‑2 (TRD3002) tehtiin 18‑ – < 65‑vuotiailla aikuisilla, ja tutkimus TRANSFORM‑3 (TRD3005) tehtiin ≥ 65‑vuotiailla aikuisilla. Tutkimuksissa TRD3001 ja TRD3002 mukana olleet potilaat saivat ensimmäisenä hoitopäivänä 56 mg:n annoksen esketamiinia ja uuden (uutena aloitettu) päivittäin suun kautta otettavan masennuslääkkeen tai uuden päivittäin suun kautta otettavan masennuslääkkeen ja lumenenäsumutteen. Esketamiiniannoksia jatkettiin sen jälkeen kaksi kertaa viikossa 4 viikon kaksoissokkoutetun aloitusvaiheen 56 mg:n annoksella tai annos nostettiin 84 mg:aan tai käytettiin vastaava annos lumenenäsumutetta. TRD3001-tutkimuksessa potilaiden esketamiiniannos oli vakio: joko 56 mg tai 84 mg. TRD3002-tutkimuksessa annos oli vaihteleva. TRD3005-tutkimuksessa potilaat (≥ 65-vuotiaat) saivat ensimmäisenä hoitopäivänä 28 mg:n esketamiiniannoksen ja uuden päivittäin suun kautta otettavan masennuslääkkeen tai uuden päivittäin suun kautta otettavan masennuslääkkeen ja lumenenäsumutteen. Esketamiiniannos nostettiin 56 mg:aan tai 84 mg:aan tai vastaavaan määrään lumenenäsumutetta ja annosteltiin kaksi kertaa viikossa 4 viikon kaksoissokkoutetun aloitusvaiheen ajan. Vaihtelevan annoksen TRD3002- ja TRD3005-tutkimuksissa, esketamiiniannosta nostettiin kliinisen arvion perusteella, ja annosta voitiin myös laskea siedettävyyden perusteella. Kaikissa tutkimuksissa aloitettiin ensimmäisenä päivänä avoimena hoitona uusi suun kautta otettava masennuslääke (SNRI-lääke: duloksetiini, pitkävaikutteinen venlafaksiini; SSRI-lääke: essitalopraami, sertraliini). Tutkija valitsi uuden suun kautta otettavan masennuslääkkeen perustuen potilaan hoitohistoriaan. Kaikissa lyhytkestoisissa tutkimuksissa tehon ensisijainen päätetapahtuma oli MADRS-kokonaispisteiden muutos lähtötilanteesta päivään 28.
Taulukossa 4 esitetään TRD3002-, TRD3001- ja TRD3005-tutkimuksissa mukana olleiden potilaiden lähtötilanteen demografiset ja sairauden ominaisuuksia koskevat tiedot.
| Taulukko 4. Demografiset ominaisuudet TRD3002-, TRD3001- ja TRD3005-tutkimusten lähtötilanteessa (koko analyysiaineisto) | |||
TRD3002-tutkimus (N = 223) | TRD3001-tutkimus (N = 342) | TRD3005-tutkimus (N = 137) | |
| Ikä, vuotta | |||
| Mediaani (vaihteluväli) | 47,0 (19; 64) | 47,0 (18; 64) | 69,0 (65; 86) |
| Sukupuoli, n (%) | |||
| Mies | 85 (38,1 %) | 101 (29,5 %) | 52 (38,0 %) |
| Nainen | 138 (61,9 %) | 241 (70,5 %) | 85 (62,0 %) |
| Etninen tausta, n (%) | |||
| Valkoihoinen | 208 (93,3 %) | 262 (76,6 %) | 130 (94,9 %) |
| Mustaihoinen tai afroamerikkalainen | 11 (4,9 %) | 19 (5,6 %) | ‑‑ |
| Aiempia suun kautta otettuja masennuslääkehoitoja ilman vastetta (eli masennuslääkehoito epäonnistunut) | |||
| Spesifisten masennuslääkkeiden lkm, n (%) | |||
| 2 | 136 (61,0 %) | 167 (48,8 %) | 68 (49,6 %) |
| 3 tai enemmän | 82 (36,8 %) | 167 (48,8 %) | 58 (42,3 %) |
| Uusi suun kautta otettava masennuslääkehoito, joka aloitettiin satunnaistamisen yhteydessä, n (%) | |||
| SNRI‑lääke | 152 (68,2 %) | 196 (57,3 %) | 61 (44,5 %) |
| SSRI‑lääke | 71 (31,8 %) | 146 (42,7 %) | 76 (55,5 %) |
| Keskeyttänyt tutkimuksen (mistä tahansa syystä), n/N (%) | 30/227 (13,2 %) | 31/346 (9,0 %) | 16/138 (11,6 %) |
Vaihtelevan annoksen TRD3002-tutkimuksessa 28. päivänä esketamiinihoitoon satunnaistetuista potilaista 67 % käytti annosta 84 mg. TRD3002-tutkimuksessa havaittiin esketamiinin ja uuden suun kautta otettavan masennuslääkehoidon yhdistelmän olevan kliinisesti ja tilastollisesti merkitsevästi parempi verrattuna uuden suun kautta otettavan masennuslääkkeen (SNRI‑lääke: duloksetiini, pitkävaikutteinen venlafaksiini; SSRI‑lääke: essitalopraami, sertraliini) ja lumenenäsumutteen yhdistelmään (taulukko 5). Lisäksi oireiden havaittiin vähentyneen jo 24 tunnin kuluttua annostelusta.
TRD3001-tutkimuksessa havaittiin kliinisesti merkittävä hoitovaste MADRS‑kokonaispisteiden muutoksena lähtötilanteeseen verrattuna esketamiinin ja uuden suun kautta otettavan masennuslääkkeen yhdistelmän eduksi verrattuna uuden suun kautta otettavan masennuslääkkeen (SNRI‑lääke: duloksetiini, pitkävaikutteinen venlafaksiini; SSRI‑lääke: essitalopraami, sertraliini) ja lumenenäsumutteen yhdistelmään (taulukko 5). TRD3001-tutkimuksessa esketamiinia 84 mg:n annoksina ja suun kautta otettavaa masennuslääkettä saaneessa ryhmässä hoidon teho ei ollut tilastollisesti merkitsevä verrattaessa ryhmään, joka sai suun kautta otettavaa masennuslääkettä ja lumenenäsumutetta.
TRD3005-tutkimuksen 28. päivänä esketamiinihoitoon satunnaistetuista potilaista 64 % käytti annosta 84 mg, 25 % käytti annosta 56 mg ja 10 % käytti annosta 28 mg. TRD3005-tutkimuksessa havaittiin 4 viikon aloitusvaiheen lopussa kliinisesti merkittävä, mutta ei tilastollisesti merkitsevä, hoitovaste MADRS‑kokonaispisteiden muutoksena lähtötilanteeseen verrattuna esketamiinin ja uuden suun kautta otettavan masennuslääkkeen yhdistelmän eduksi verrattuna uuden suun kautta otettavan masennuslääkkeen (SNRI‑lääke: duloksetiini, pitkävaikutteinen venlafaksiini; SSRI‑lääke: essitalopraami, sertraliini) ja lumenenäsumutteen yhdistelmään (taulukko 5). Alaryhmäanalyysit viittaavat siihen, että yli 75-vuotiailla potilailla teho on rajallinen.
| Taulukko 5. Ensisijaiset tehoa koskevat tulokset MADRS‑kokonaispisteiden muutoksena 4 viikon pituisissa kliinisissä tutkimuksissa (ANCOVA BOCF*) | |||||
| Tutkimuksen nro | Hoitoryhmä§ | Potilaiden lkm | Lähtötilanteen pisteiden keskiarvo (keskihajonta) | Pienimmän neliösumman keskiarvon muutos lähtötilanteesta viikon 4 loppuun (keskivirhe) | Pienimmän neliösumman keskiarvojen ero (95 %:n luottamusväli)† |
| TRD3001 | Spravato 56 mg + suun kautta otettava masennuslääke | 115 | 37,4 (4,8) | ‑18,9 (1,3) | ‑4,3 (‑7,8; ‑0,8)# |
| Spravato 84 mg + suun kautta otettava masennuslääke | 114 | 37,8 (5,6) | ‑16,2 (1,3) | ‑1,2 (‑4,7; 2,3)# | |
| Suun kautta otettava masennuslääke + lumenenäsumute | 113 | 37,5 (6,2) | ‑14,7 (1,3) | ||
| TRD3002 | Spravato (56 mg tai 84 mg) + suun kautta otettava masennuslääke | 114 | 37,0 (5,7) | ‑17,7 (1,3) | ‑3,5 (‑6,7; ‑0,3)‡ |
| Suun kautta otettava masennuslääke + lumenenäsumute | 109 | 37,3 (5,7) | ‑14,3 (1,3) | ||
| TRD3005 (≥ 65‑vuotiaat) | Spravato (28 mg, 56 mg tai 84 mg) + suun kautta otettava masennuslääke | 72 | 35,5 (5,9) | ‑10,1 (1,7) | ‑2,9 (‑6,5; 0,6)# |
| Suun kautta otettava masennuslääke + lumenenäsumute | 65 | 34,8 (6,4) | ‑6,8 (1,7) | ||
* ANCOVA-analyysi tehtiin BOCF-menetelmällä (Baseline Observation Carried Forward), mikä tarkoittaa, että hoidon keskeyttäneen potilaan masennuksen vaikeusasteen oletetaan palaavan lähtötilanteen tasolle (eli masennuksen vaikeusaste on sama kuin ennen hoidon aloittamista). § Nenään annosteltava esketamiini tai lumelääke; suun kautta otettava masennuslääke = uusi (uutena aloitettu) masennuslääke (SNRI‑lääke: duloksetiini, pitkävaikutteinen venlafaksiini; SSRI‑lääke: essitalopraami, sertraliini) † Ero (Spravaton ja suun kautta otettavan masennuslääkkeen yhdistelmän ja suun kautta otettavan masennuslääkkeen ja lumenenäsumutteen yhdistelmän erotus) ilmoitettuna pienimmän neliösumman keskiarvon muutoksena lähtötilanteesta ‡ Hoitoryhmä, joka oli tilastollisesti merkitsevästi parempi kuin suun kautta otettavan masennuslääkkeen ja lumenenäsumutteen yhdistelmä # Mediaani-harhaton estimaatti (eli suun kautta otettavan masennuslääkkeen ja lumenenäsumutteen yhdistelmästä saadun eron pienimmän neliösumman keskiarvojen painotettu yhdistelmä) ja 95 %:n joustava luottamusväli | |||||
Vaste‑ ja remissioluvut
Vasteeksi määriteltiin MADRS‑kokonaispisteiden väheneminen ≥ 50 % aloitusvaiheen lähtötilanteesta. Lähtötilanteen MADRS‑kokonaispisteiden vähenemän perusteella hoitovasteen saaneita potilaita oli enemmän esketamiinia ja suun kautta otettavaa masennuslääkettä saaneiden ryhmässä verrattuna masennuslääkkeen ja lumenenäsumutteen yhdistelmää saaneisiin. Tämä ero oli havaittavissa koko 4 viikon kaksoissokkoutetun aloitusvaiheen ajan (taulukko 6) TRD3001-, TRD3002- ja TRD3005-tutkimuksissa.
Remissioksi määriteltiin MADRS‑kokonaispisteet ≤ 12. Kaikissa kolmessa tutkimuksessa 4 viikon pituisen kaksoissokkoutetun aloitusvaiheen lopussa remissiossa oli suurempi osa esketamiinia ja suun kautta otettavan masennuslääkkeen yhdistelmää saaneista potilaista kuin suun kautta otettavan masennuslääkkeen ja lumenenäsumutteen yhdistelmää saaneista potilaista (taulukko 6).
| Taulukko 6. Vaste‑ ja remissioluvut 4 viikon kliinisten tutkimusten BOCF*‑tietojen perusteella | |||||||
| Tutkimuksen nro | Hoitoryhmä§ | Potilaiden lkm (%) | |||||
| Vasteluku† | Remissioluku‡ | ||||||
| 24 tuntia | Viikko 1 | Viikko 2 | Viikko 3 | Viikko 4 | Viikko 4 | ||
| TRD3001 | Spravato 56 mg + suun kautta otettava masennuslääke | 20 (17,4 %) | 21 (18,3 %) | 29 (25,2 %) | 52 (45,2 %) | 61 (53,0 %) | 40 (34,8 %) |
| Spravato 84 mg + suun kautta otettava masennuslääke | 17 (14,9 %)# | 16 (14,0 %) | 25 (21,9 %) | 33 (28,9 %) | 52 (45,6 %) | 38 (33,3 %) | |
| Suun kautta otettava masennuslääke + lumenenäsumute | 8 (7,1 %) | 5 (4,4 %) | 15 (13,3 %) | 25 (22,1 %) | 42 (37,2 %) | 33 (29,2 %) | |
| TRD3002 | Spravato 56 mg tai 84 mg + suun kautta otettava masennuslääke | 18 (15,8 %) | 15 (13,2 %) | 29 (25,4 %) | 54 (47,4 %) | 70 (61,4 %) | 53 (46,5 %) |
| Suun kautta otettava masennuslääke + lumenenäsumute | 11 (10,1 %) | 13 (11,9 %) | 23 (21,1 %) | 35 (32,1 %) | 52 (47,7 %) | 31 (28,4 %) | |
| TRD3005 (≥ 65‑vuotiaat) | Spravato 28 mg, 56 mg tai 84 mg + suun kautta otettava masennuslääke | NA | 4 (5,6 %) | 4 (5,6 %) | 9 (12,5 %) | 17 (23,6 %) | 11 (15,3 %) |
| Suun kautta otettava masennuslääke + lumenenäsumute | NA | 3 (4,6 %) | 8 (12,3 %) | 8 (12,3 %) | 8 (12,3 %) | 4 (6,2 %) | |
NA = ei saatavissa (not available) * BOCF-menetelmä (Baseline Observation Carried Forward), joka tarkoittaa, että hoidon keskeyttäneen potilaan masennuksen vaikeusasteen oletetaan palaavan lähtötilanteen tasolle (eli masennuksen vaikeusaste on sama kuin ennen hoidon aloittamista). § Nenään annettava Spravato tai lumelääke; suun kautta otettava masennuslääke = uusi (uutena aloitettu) aloitettu masennuslääke (SNRI‑lääke: duloksetiini, pitkävaikutteinen venlafaksiini; SSRI‑lääke: essitalopraami, sertraliini) † Vasteeksi määriteltiin MADRS‑kokonaispisteiden väheneminen ≥ 50 % lähtötilanteesta. ‡ Remissioksi määriteltiin MADRS‑kokonaispisteet ≤ 12. # Ensimmäinen annos oli Spravato-nenäsumutteen 56 mg:n annoksen ja suun kautta otettavan masennuslääkkeen yhdistelmä | |||||||
Hoitoresistentin masennuksen pitkäkestoiset tutkimukset
Relapsin estoa koskeva tutkimus
Relapsin estoa koskeneessa tutkimuksessa masennusta lievittävän tehon osoitettiin säilyvän. SUSTAIN‑1 (TRD3003) -tutkimus oli pitkäkestoinen, satunnaistettu, kaksoissokkoutettu, rinnakkaisryhmillä toteutettu, aktiivikontrolloitu relapsin estoa koskenut monikeskustutkimus. Masennuksen relapsin estoa, joka oli hoitotuloksen ensisijainen arvioitava mittari, mitattiin relapsiin kuluneella ajalla. Tutkimukseen otettiin mukaan yhteensä 705 potilasta, joista 437 potilasta otettiin mukaan suoraan, 150 potilasta siirtyi TRD3001-tutkimuksesta ja 118 TRD3002-tutkimuksesta. Tutkimukseen suoraan mukaan tulleille potilaille annettiin 4 viikon pituisen avoimen aloitusvaiheen aikana esketamiinia (56 mg tai 84 mg kaksi kertaa viikossa) ja suun kautta otettavaa masennuslääkettä. Avoimen aloitusvaiheen lopussa 52 % potilaista oli remissiossa (MADRS‑kokonaispisteet ≤ 12) ja 66 % potilaista oli saanut vasteen (MADRS‑pisteet parantuneet ≥ 50 %). Vasteen saaneet potilaat (455) jatkoivat 12 viikon optimointijakson aikana esketamiinin ja suun kautta otettavan masennuslääkkeen käyttöä. Aloitusvaiheen jälkeen potilaat saivat esketamiinia kerran viikossa 4 viikon ajan. Viikosta 8 alkaen antotiheys määritettiin MADRS‑pisteisiin perustuvalla algoritmilla. Remissiossa olevat potilaat (eli MADRS‑kokonaispisteet ≤ 12) saivat lääkettä joka toinen viikko, mutta jos MADRS‑kokonaispisteet kasvoivat > 12 pisteeseen, lääkkeen anto tihennettiin kerran viikossa tapahtuvaksi seuraavan 4 viikon ajaksi. Tavoitteena oli jatkaa potilaan hoitoa pisimmällä antovälillä, jolla vaste/remissio säilyi. 16 viikon hoitojakson lopussa stabiilissa remissiossa olevat potilaat (n = 176) tai pysyvän vasteen saaneet potilaat (n = 121) satunnaistettiin jatkamaan esketamiinihoitoa tai lopettamaan esketamiinihoito ja siirtymään lumenenäsumutteen käyttöön. Stabiiliksi remissioksi määriteltiin MADRS‑kokonaispisteet ≤ 12 vähintään kolmena optimointijakson neljästä viimeisestä viikosta. Pysyväksi vasteeksi määriteltiin MADRS‑kokonaispisteiden vähenemä ≥ 50 % lähtötilanteesta optimointijakson kahtena viimeisenä viikkona, silloin kun stabiilin remission määritelmä ei täyttynyt.
Stabiili remissio
Stabiilissa remissiossa olleilla esketamiinin ja suun kautta otettavan masennuslääkkeen käyttöä jatkaneilla potilailla masennusoireiden relapsiin kului tilastollisesti merkitsevästi pidempi aika kuin uutena aloitetun suun kautta otettavaa masennuslääkettä (SNRI‑lääke: duloksetiini, pitkävaikutteinen venlafaksiini; SSRI‑lääke: essitalopraami, sertraliini) ja lumenenäsumutetta saaneilla potilailla (kuva 1). Relapsiksi määriteltiin MADRS‑kokonaispisteet ≥ 22 kahdella peräkkäisellä viikolla tai sairaalahoito masennuksen pahenemisen vuoksi tai jokin muu kliinisesti oleellinen tapahtuma, joka osoitti relapsin. Mediaaniaika relapsiin oli uuden suun kautta otettavan masennuslääkkeen (SNRI‑lääke: duloksetiini, pitkävaikutteinen venlafaksiini; SSRI‑lääke: essitalopraami, sertraliini) ja lumenenäsumutteen yhdistelmää saaneessa ryhmässä 273 päivää, kun taas esketamiinia ja suun kautta otettavan masennuslääkettä saaneiden ryhmässä mediaania ei voitu määrittää, sillä tässä ryhmässä ei saavutettu 50 %:n relapsimäärää.
Kuva 1. Relapsiin kulunut aika stabiilissa remissiossa olleilla potilailla TRD3003-tutkimuksessa (koko analyysiaineisto)
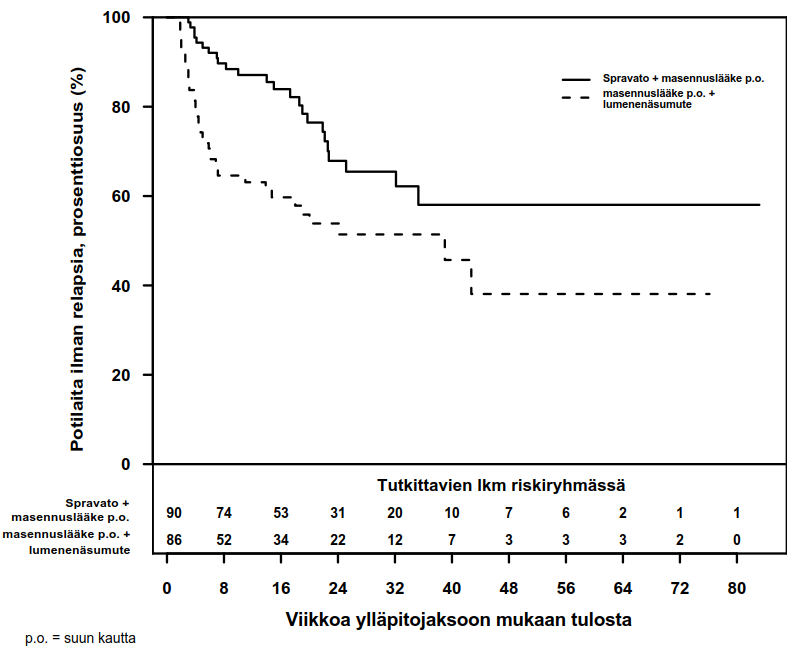
12 ja 24 viikon kaksoissokkoutetun seurantajakson aikana stabiilissa remissiossa olleiden potilaiden Kaplan–Meier ‑estimaattiin perustuva relapsien esiintymistiheys oli esketamiinia saaneiden ryhmässä 13 % (12 viikkoa) ja 32 % (24 viikkoa) ja lumenenäsumutetta käyttäneiden ryhmässä 37 % (12 viikkoa) ja 46 % (24 viikkoa).
Pysyvä vaste
Tehoa koskevat tulokset olivat yhdenmukaiset myös niillä pysyvän vasteen saaneilla potilailla, jotka jatkoivat esketamiinin ja suun kautta otettavan masennuslääkkeen käyttöä. Relapsiin kulunut aika oli tilastollisesti merkitsevästi pidempi kuin uutta suun kautta otettavaa masennuslääkettä (SNRI‑lääke: duloksetiini, pitkävaikutteinen venlafaksiini; SSRI‑lääke: essitalopraami, sertraliini) ja lumenenäsumutetta saaneilla potilailla (kuva 2). Mediaaniaika relapsiin oli uutta suun kautta otettavaa masennuslääkettä (SNRI‑lääke: duloksetiini, pitkävaikutteinen venlafaksiini; SSRI‑lääke: essitalopraami, sertraliini) ja lumenenäsumutetta saaneiden ryhmässä (88 päivää) lyhyempi kuin esketamiinia ja suun kautta otettavaa masennuslääkettä saaneiden ryhmässä (635 päivää).
Kuva 2. Relapsiin kulunut aika pysyvän vasteen saaneilla potilailla TRD3003-tutkimuksessa (koko analyysiaineisto)
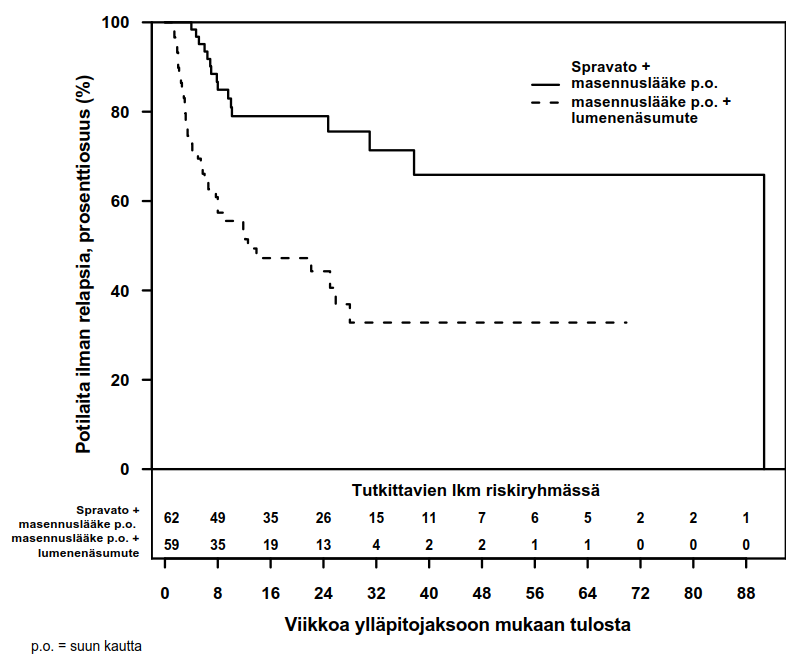
12 ja 24 viikon kaksoissokkoutetun seurantajakson aikana pysyvän vasteen saaneiden potilaiden Kaplan‑Meier ‑estimaatteihin perustuva relapsien määrä oli esketamiinia käyttäneiden ryhmässä 21 % (12 viikkoa) ja 21 % (24 viikkoa) ja lumenenäsumutetta käyttäneiden ryhmässä 47 % (12 viikkoa) ja 56 % (24 viikkoa).
Potilaita otettiin TRD3003-tutkimukseen mukaan porrastetusti noin 2 vuoden ajan. Ylläpitojakson kesto vaihteli, ja se jatkui kunnes potilaalle ilmeni masennuksen relapsi tai potilas keskeytti tutkimuksen jostain muusta syystä tai se päättyi, koska tutkimukseen vaadittava määrä relapseja oli saavutettu. Lääkehoidolle altistuneiden määrään vaikutti välianalyysiin perustuva ennaltamääritetty relapsien määrä, joka asetettiin tutkimuksen päättymiskriteeriksi. Stabiilissa remissiossa tai pysyvän vasteen saaneiden potilaiden mediaanihoitoaika esketamiinilla ja suun kautta otettavalla masennuslääkkeellä oli 4,2 kuukautta (vaihteluväli: 1 päivä – 21,2 kuukautta) 16 viikon aloitusvaiheen jälkeen. Tutkimuksen aikana potilaista 31,6 % sai esketamiinia pidempään kuin 6 kuukautta, ja 7,9 % potilaista sai esketamiinia pidempään kuin 1 vuoden.
Antotiheys
Taulukossa 7 esitetään antotiheys valtaosassa ylläpitojakson ajasta. Spravato‑hoitoon satunnaistetuista potilaista 60 % sai 84 mg:n annoksia ja 40 % sai 56 mg:n annoksia.
| Taulukko 7. Ylläpitojakson pääasiallinen antotiheys (TRD3003-tutkimus) | ||||
| Stabiili remissio | Pysyvän vasteen saaneet | |||
Spravato + suun kautta otettava masennuslääke (N = 90) | Suun kautta otettava masennuslääke + lumenenäsumute (N = 86) | Spravato + suun kautta otettava masennuslääke (N = 62) | Suun kautta otettava masennuslääke + lumenenäsumute (N = 59) | |
| Pääasiallinen antotiheys | ||||
| Kerran viikossa | 21 (23,3 %) | 27 (31,4 %) | 34 (54,8 %) | 36 (61,0 %) |
| Joka toinen viikko | 62 (68,9 %) | 48 (55,8 %) | 21 (33,9 %) | 19 (32,2 %) |
| Kerran viikossa tai joka toinen viikko | 7 (7,8 %) | 11 (12,8 %) | 7 (11,3 %) | 4 (6,8 %) |
TRD3013-tutkimus (ESCAPE-TRD)
Spravato-nenäsumutteen tehoa arvioitiin pitkäkestoisessa satunnaistetussa, avoimessa, arvioijasokkoutetussa, aktiivikontrolloidussa tutkimuksessa (TRD3013), jossa esketamiinia verrattiin pitkävaikutteiseen ketiapiiniin 676 aikuisella (18–74-vuotiaalla) hoitoresistenttiä masennusta sairastavalla potilaalla, jotka jatkoivat parhaillaan käyttämänsä suun kautta otettavan masennuslääkkeen (SSRI- tai SNRI-lääke) käyttöä. Potilaat saivat joustavasti annosteltuna esketamiinihoitoa (28, 56 tai 84 mg) tai pitkävaikutteista ketiapiinia tutkimuksen alkaessa käytössä olleen valmisteyhteenvedon annostussuositusten mukaisesti.
Ensisijainen tehon päätetapahtuma oli remissio (MADRS-kokonaispisteet ≤ 10) viikolla 8, ja keskeinen toissijainen päätetapahtuma oli säilyminen ilman relapseja viikon 8 remission jälkeen viikkoon 32 saakka. Relapsiksi määriteltiin MADRS-kokonaispisteet ≥ 22 kahtena peräkkäisenä viikkona tai sairaalahoito masennuksen pahenemisen vuoksi tai jokin muu kliinisesti oleellinen relapsin osoittava tapahtuma.
Potilaiden demografiset ja sairauden ominaisuudet esketamiinin ja suun kautta otettavan masennuslääkkeen yhdistelmää ja pitkävaikutteisen ketiapiinin ja suun kautta otettavan masennuslääkkeen yhdistelmää käyttäneillä ryhmillä olivat lähtötilanteessa samankaltaiset. Lähtötilanteen MADRS-kokonaispisteiden keskiarvo (keskihajonta) oli esketamiinin ja suun kautta otettavan masennuslääkkeen yhdistelmää käyttäneessä ryhmässä 31,4 (6,06) ja pitkävaikutteisen ketiapiinin ja suun kautta otettavan masennuslääkkeen yhdistelmää käyttäneessä ryhmässä 31,0 (5,83).
Esketamiinin ja suun kautta otettavan masennuslääkkeen yhdistelmän osoitettiin olevan sekä ensisijaisen (taulukko 8) että keskeisen toissijaisen (taulukko 9) tehon mittarin osalta kliinisesti ja tilastollisesti merkitsevästi parempi kuin pitkävaikutteisen ketiapiinin ja suun kautta otettavan masennuslääkkeen yhdistelmä.
| Taulukko 8. TRD3013-tutkimuksen ensisijaiset tehoa koskevat tulokseta | ||
| Hoitoryhmä | Spravato + suun kautta otettava masennuslääke | Pitkävaikutteinen ketiapiini + suun kautta otettava masennuslääke |
| Remissiossa viikolla 8 olleiden potilaiden lukumäärä | 91/336 (27,1 %) | 60/340 (17,6 %) |
| Vakioitu riskin prosentuaalinen ero (95 %:n luottamusväli) b | 9,5 (3,3; 15,8) | – |
| p-arvo c | p = 0,003 | – |
a Potilas, joka lopetti tutkimusintervention ennen viikkoa 8, katsottiin negatiiviseksi hoitotulokseksi (eli ei remissiota). Potilaisiin, joista ei ollut MADRS-tulosta saatavana viikon 8 käynnillä, mutta jotka eivät lopettaneet tutkimusinterventiota eivätkä vetäytyneet tutkimuksesta ennen viikkoa 8, sovellettiin viimeisimpiä MADRS-pisteitä (LOCF-menetelmä). b Käytettiin riskin eron Mantel–Haenszelin estimaattia, joka ositettiin ikäryhmittäin (18–64; ≥ 65) ja hoidon epäonnistumisten kokonaislukumäärän mukaan. Tämä estimoitu ero osoittaa esketamiinin paremmuuden. c Cochran–Mantel–Haenszelin (CMH) testi, joka on vakioitu ikäryhmän (18–64; ≥ 65) ja hoidon epäonnistumisten lukumäärän suhteen. | ||
| Taulukko 9. TRD3013-tutkimuksen keskeiset toissijaiset tehoa koskevat tulokseta | ||
| Hoitoryhmä | Spravato + suun kautta otettava masennuslääke | Pitkävaikutteinen ketiapiini + suun kautta otettava masennuslääke |
| Sekä remissiossa viikolla 8 että ilman relapseja viikolla 32 olleiden potilaiden lukumäärä | 73/336 (21,7 %) | 48/340 (14,1 %) |
| Vakioitu riskin prosentuaalinen ero (95 %:n luottamusväli)b | 7,7 (2,0; 13,5) | – |
| p-arvo c | p = 0,008 | – |
a Potilas, joka lopetti tutkimusintervention, katsottiin negatiiviseksi hoitotulokseksi. Potilaisiin, joista ei ollut MADRS-tulosta saatavana viikon 8 käynnillä, mutta jotka eivät lopettaneet tutkimusinterventiota eivätkä vetäytyneet tutkimuksesta ennen viikkoa 8, sovellettiin viimeisimpiä MADRS-pisteitä (LOCF-menetelmä). b Käytettiin riskin eron Mantel–Haenszelin estimaattia, joka ositettiin ikäryhmittäin (18–64; ≥ 65) ja hoidon epäonnistumisten kokonaislukumäärän mukaan. Tämä estimoitu ero osoittaa esketamiinin paremmuuden. c Cochran–Mantel–Haenszelin (CMH) testi, joka on vakioitu ikäryhmän (18–64; ≥ 65) ja hoidon epäonnistumisten lukumäärän suhteen. | ||
32 viikon hoitojakson aikana hoidon lopetti esketamiinin ja suun kautta otettavan masennuslääkkeen yhdistelmää käyttäneessä ryhmässä haittavaikutusten vuoksi 4,2 %, tehon puuttumisen vuoksi 8,3 % ja kaikkiaan 23,2 % potilaista ja pitkävaikutteisen ketiapiinin ja suun kautta otettavan masennuslääkkeen yhdistelmää käyttäneessä ryhmässä haittavaikutusten vuoksi 11,5 %, tehon puuttumisen vuoksi 15,0 % ja kaikkiaan 40,3 % potilaista.
Hoitoresistentin masennuksen lyhytkestoinen tutkimus japanilaisilla potilailla
Spravato-valmisteen tehoa on arvioitu myös lyhytkestoisessa (4 viikkoa) satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa tutkimuksessa (TRD2005) 202:lla aikuisella japanilaisella hoitoresistenttiä masennusta sairastavalla potilaalla. Potilaat saivat 4 viikon aloitushoidon esketamiinin vakioannoksella 28 mg, 56 mg tai 84 mg, tai lumenenäsumutetta. Samalla jatkettiin käytössä ollutta suun kautta otettavaa masennuslääkitystä. Ensisijainen tehon päätetapahtuma oli MADRS-kokonaispisteiden muutos lähtötilanteesta päivään 28. Potilaiden demografiset ja sairauden ominaisuudet esketamiinin ja masennuslääkkeen yhdistelmää ja lumenenäsumutteen ja masennuslääkkeen yhdistelmää käyttäneillä ryhmillä olivat lähtötilanteessa samankaltaiset.
TRD2005-tutkimuksessa MADRS-kokonaispisteiden muutoksessa lähtötilanteesta 4 viikon aloitusjakson loppuun ei havaittu tilastollista merkitsevyyttä minkään esketamiinin ja suun kautta otettavan masennuslääkkeen yhdistelmän annostuksen vertailussa lumenenäsumutteen ja suun kautta otettavan masennuslääkkeen yhdistelmään (taulukko 10).
| Taulukko 10. Ensisijaiset tehoa koskevat tulokset muutoksena MADRS-kokonaispisteissä 4 viikon TRD2005-tutkimuksessa japanilaisilla potilailla (MMRM) | ||||
| Hoitoryhmä | Potilaiden lkm | Lähtötilanteen pisteiden keskiarvo (keskihajonta) | Pienimmän neliösumman keskiarvon muutos lähtötilanteesta viikon 4 loppuun (keskivirhe) | Pienimmän neliösumman keskiarvojen ero (90 %:n luottamusväli)†,# |
| Spravato 28 mg + suun kautta otettava masennuslääke | 41 | 38,4 (6,1) | -15,6 (1,8) | -1,0 -5,77; 3,70 |
| Spravato 56 mg + suun kautta otettava masennuslääke | 40 | 37,9 (5,4) | -14,0 (1,9) | 0,6 -4,32; 5,47 |
| Spravato 84 mg + suun kautta otettava masennuslääke | 41 | 35,9 (5,3) | -15,5 (1,8) | -0,9 -5,66; 3,83 |
Suun kautta otettava masennuslääke + lume‑ nenäsumute | 80 | 37,7 (5,7) | -14,6 (1,3) | |
† Ero (Spravaton ja suun kautta otettavan masennuslääkkeen yhdistelmän ja suun kautta otettavan masennuslääkkeen ja lumenenäsumutteen yhdistelmän erotus) ilmoitettuna pienimmän neliösumman keskiarvon muutoksena lähtötilanteesta. # Luottamusväli perustuu Dunnettin korjaukseen. | ||||
Hoitoresistentin masennuksen lyhytkestoinen tutkimus kiinalaisilla potilailla
Spravato-valmisteen tehoa on arvioitu myös lyhytkestoisessa (4 viikkoa) satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa tutkimuksessa (TRD3006) 252:lla aikuisella hoitoresistenttiä masennusta sairastavalla potilaalla (224 kiinalaista potilasta, 28 muuta kuin kiinalaista potilasta).
Potilaat saivat 4 viikon aloitushoidon joustavalla esketamiiniannostuksella (56 mg tai 84 mg) tai lumenenäsumutetta. Samalla jatkettiin äskettäin käyttöön otettua suun kautta otettavaa masennuslääkitystä. Ensisijainen tehon päätetapahtuma oli MADRS-kokonaispisteiden muutos lähtötilanteesta päivään 28. Lähtötilanteen demografiset tiedot ja sairauden ominaisuudet olivat samankaltaiset esketamiinin ja masennuslääkkeen yhdistelmää ja lumenenäsumutteen ja masennuslääkkeen yhdistelmää käyttäneissä ryhmissä.
TRD3006-tutkimuksessa MADRS-kokonaispisteiden muutoksessa lähtötilanteesta 4 viikon aloitusjakson loppuun ei havaittu tilastollista merkitsevyyttä esketamiinin ja suun kautta otettavan masennuslääkkeen yhdistelmään, kun sitä verrattiin lumenenäsumutteen ja suun kautta otettavan masennuslääkkeen yhdistelmään (taulukko 11).
| Taulukko 11. Ensisijaiset tehoa koskevat tulokset muutoksena MADRS-kokonaispisteissä 4 viikon TRD3006-tutkimuksessa (MMRM) | ||||
| Hoitoryhmä | Potilaiden lkm# | Lähtötilanteen pisteiden keskiarvo (keskihajonta) | Pienimmän neliösumman keskiarvon muutos lähtötilanteesta viikon 4 loppuun (keskivirhe) | Pienimmän neliösumman keskiarvojen ero (95 %:n luottamusväli)† |
| Kaikki potilaat | ||||
| Spravato (56 mg tai 84 mg) + suun kautta otettava masennuslääke | 124 | 36,5 (5,21) | -11,7 (1,09) | -2,0 -4,64; 0,55 |
| Suun kautta otettava masennuslääke + lumenenäsumute | 126 | 35,9 (4,50) | -9,7 (1,09) | |
| Kiinalainen potilasjoukko | ||||
| Spravato (56 mg tai 84 mg) + suun kautta otettava masennuslääke | 110 | 36,2 (5,02) | -8,8 (0,95) | -0,7 -3,35; 1,94 |
| Suun kautta otettava masennuslääke + lumenenäsumute | 112 | 35,9 (4,49) | -8,1 (0,95) | |
# Kaksi potilasta ei saanut suun kautta otettavaa masennuslääkettä eikä heitä otettu mukaan tehon analyysiin. † Ero (Spravaton ja suun kautta otettavan masennuslääkkeen yhdistelmän ja suun kautta otettavan masennuslääkkeen ja lumenenäsumutteen yhdistelmän erotus) ilmoitettuna pienimmän neliösumman keskiarvon muutoksena lähtötilanteesta. | ||||
Masennustilasta aiheutuvan psykiatrisen hätätilanteen akuutti lyhytaikainen hoito
Spravato-valmistetta tutkittiin kahdessa identtisessä vaiheen 3 lyhytkestoisessa (4 viikkoa) satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa Aspire I (SUI3001) ja Aspire II (SUI3002). Niissä oli mukana aikuisia potilaita, joilla oli keskivaikea tai vaikea masennustila (MADRS-kokonaispisteet > 28) ja joilla oli tämän varmistavia vastauksia MINI-kysymyksiin B3 (Ajatteletko [edes hetkittäin] vahingoittavasi tai satuttavasi itseäsi vähintään jossakin määrin aikomuksena tai tietoisena, että saatat sen vuoksi kuolla, tai ajatteletko itsemurhaa [eli itsesi tappamista]?) ja B10 (Onko sinulla ollut kuluneiden 24 tunnin aikana aikomuksia toteuttaa itsemurha-ajatuksesi?). Potilaat saivat näissä tutkimuksissa hoitona 84 mg:aa esketamiinia tai lumenenäsumutetta kaksi kertaa viikossa 4 viikon ajan. Kaikki potilaat saivat tavanomaista kattavaa hoitoa, johon kuului alkuvaiheen sairaalahoito ja uutena aloitettu tai optimoitu suun kautta otettava masennuslääkehoito (masennuslääkehoito monoterapiana tai masennuslääkehoito plus lisälääkitys) tutkijan arvioon perustuen. Lääkärin arvion mukaan akuutti psykiatrinen sairaalahoito oli kliinisesti aiheellista tutkittavan välittömän itsemurhariskin vuoksi. Ensimmäisen annoksen jälkeen oli sallittua kerran pienentää esketamiiniannos 56 mg:aan, jos potilas ei sietänyt 84 mg:n annosta.
SUI3001- ja SUI3002-tutkimuksissa potilaiden demografiset ja sairauden ominaisuudet olivat lähtötilanteessa samankaltaiset ryhmien välillä, jotka saivat esketamiinin ja tavanomaisen hoidon yhdistelmää tai lumenenäsumutteen ja tavanomaisen hoidon yhdistelmää. Potilaiden iän mediaani oli 40 vuotta (vaihteluväli 18–64 vuotta), 61 % oli naisia, 73 % valkoihoisia ja 6 % mustaihoisia, ja 63 % potilaista oli yrittänyt itsemurhaa vähintään kerran. 92 % potilaista sai masennuslääkehoitoa jo ennen tutkimukseen mukaan tuloa. Osana tutkimuksenaikaista tavanomaista hoitoa 40 % potilaista sai masennuslääkehoitoa monoterapiana, 54 % potilaista sai masennuslääkehoitoa ja lisälääkehoitoa ja 6 % potilaista sai sekä masennuslääkehoitoa monoterapiana että masennuslääkehoitoa ja lisälääkehoitoa.
Ensisijainen tehon mittari oli vakavan masennustilan oireiden väheneminen, minkä osoitti 24 tuntia ensimmäisen annoksen jälkeen mitattu muutos lähtötilanteen MADRS-kokonaispisteistä (päivä 2).
SUI3001- ja SUI3002-tutkimuksissa osoitettiin, että Spravato-valmisteen ja tavanomaisen hoidon yhdistelmä on ensisijaisen tehon mittarin perusteella tilastollisesti parempi lumenenäsumutteen ja tavanomaisen hoidon yhdistelmään verrattuna (ks. taulukko 12).
| Taulukko 12. Ensisijaiset tehon tulokset MADRS-kokonaispisteissä havaitun muutoksen perusteella 24 tuntia ensimmäisen annoksen jälkeen (SUI3001- ja SUI3002-tutkimukset) (ANCOVA BOCF*) | |||||
| Tutkimusnro | Hoitoryhmä‡ | Potilaiden lkm | Lähtötilanteen pisteiden keskiarvo (keskihajonta) | Pienimmän neliösumman keskiarvon muutos 24 tunnin kuluttua lähtötilanteesta (keskivirhe) | Pienimmän neliösumman keskiarvojen ero (95 %:n luottamusväli)§ |
| Tutkimus 1 (SUI3001) | Spravato 84 mg + tavanomainen hoito | 112 | 41,2 (5,87) | -15,7 (1,05) | -3,7 (-6,41; -0,92)# p = 0,006 |
| Lumenenäsumute + tavanomainen hoito | 112 | 41,0 (6,29) | -12,1 (1,03) | – | |
| Tutkimus 2 (SUI3002) | Spravato 84 mg + tavanomainen hoito | 114 | 39,5 (5,19) | -15,9 (1,02) | -3,9 (-6,65; -1,12)# p =0,006 |
| Lumenenäsumute + tavanomainen hoito | 113 | 39,9 (5,76) | -12,0 (1,06) | – | |
| Yhdistetyt tutkimukset 1 ja 2 | Spravato 84 mg + tavanomainen hoito | 226 | 40,3 (5,60) | -15,8 (0,73) | -3,8 (-5,69; -1,82) |
| Lumenenäsumute + tavanomainen hoito | 225 | 40,4 (6,04) | -12,1 (0,73) | – | |
* ANCOVA-analyysi BOCF-menetelmällä (Baseline Observation Carried Forward): SUI3001-tutkimuksessa 2 tutkittavasta (1 tutkittava kummassakin ryhmässä) ei ollut päivän 2 (24 tuntia ensimmäisen annoksen jälkeen) MADRS‑kokonaispisteitä ja SUI3002-tutkimuksessa 6 tutkittavasta (4 esketamiinia saanutta tutkittavaa ja 2 lumevalmistetta saanutta tutkittavaa) ei ollut päivän 2 (24 tuntia ensimmäisen annoksen jälkeen) MADRS-kokonaispisteitä. Näiden tutkittavien masennuksen vaikeusasteen oletetaan palautuvan lähtötasolle (eli masennuksen vaikeusaste on sama kuin ennen hoidon aloittamista), joten analyysiin käytettiin lähtötilanteen MADRS-kokonaispisteitä. ‡ Nenään annettava esketamiini tai lumevalmiste § Lähtötilanteeseen verrattuna tapahtuneen muutoksen (Spravato-nenäsumutteen ja tavanomaisen hoidon yhdistelmän ja lumenenäsumutteen ja tavanomaisen hoidon yhdistelmän erotus) pienimmän neliösumman keskiarvo # Hoitoryhmät, jotka olivat tilastollisesti merkitsevästi parempia lumenenäsumutteen ja tavanomaisen hoidon yhdistelmään verrattuna. | |||||
Hoitojen välinen ero (95 %:n luottamusväli) MADRS-kokonaispisteiden muutoksessa lähtötilanteesta päivänä 2 (24 tuntia ensimmäisen annoksen jälkeen) verrattaessa esketamiinin ja tavanomaisen hoidon yhdistelmää lumenenäsumutteen ja tavanomaisen hoidon yhdistelmään oli ‑4,70 (‑7,16; ‑2,24) aiemman itsemurhayrityksen raportoineessa osajoukossa (N = 284) ja ‑2,34 (‑5,59; 0,91) osajoukossa, jossa ei raportoitu aiempaa itsemurhayritystä (N = 166).
Hoitovaste ajan kuluessa
Sekä SUI3001-tutkimuksessa että SUI3002-tutkimuksessa esketamiinihoidon ero lumevalmisteeseen verrattuna havaittiin 4 tunnin aikapisteestä lähtien. Hoitovaiheen lopussa, 4 tunnin ja päivän 25 välillä, tilanne sekä esketamiini- että lumeryhmässä parani edelleen, ja ryhmien välinen ero yleensä säilyi, mutta ei vaikuttanut suurenevan ajan mittaan päivään 25 mennessä. Kuva 3 kuvastaa ensisijaista tehon mittaria eli MADRS-kokonaispisteiden muutosta ajan kuluessa yhdistetyissä SUI3001- ja SUI3002-tutkimuksissa.
Kuva 3. Muutos MADRS-pisteissä pienimmän neliösumman keskiarvoina ajan suhteen SUI3001- ja SUI3002-tutkimuksissa* (yhdistetyt tiedot, turvallisuutta koskeva tietoaineisto) – ANCOVA BOCF
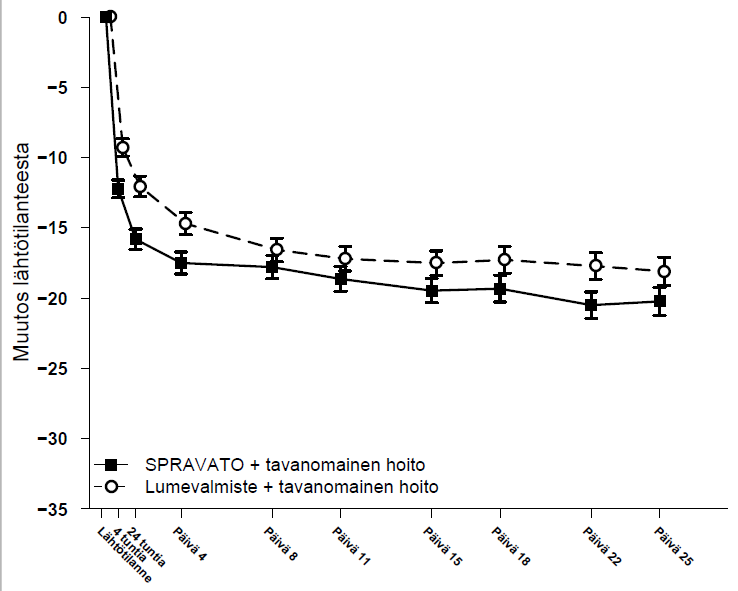
* Huom.: Näissä tutkimuksissa oli ensimmäisen annoksen jälkeen sallittua kerran pienentää Spravato-annos 56 mg:aan, jos potilas ei sietänyt 84 mg:n annosta. Noin 16 % potilaista pienensi Spravato-annoksen 84 mg annokseen 56 mg kaksi kertaa viikossa.
Remissioluvut
Remissioon päässeiden osuus (MADRS-kokonaispisteet ≤ 12 milloin tahansa tutkimuksen aikana), oli suurempi esketamiinin ja tavanomaisen hoidon yhdistelmää saaneessa ryhmässä kuin lumevalmisteen ja tavanomaisen hoidon yhdistelmää saaneessa ryhmässä kaikissa neljän viikon pituisen kaksoissokkoutetun hoitovaiheen aikapisteissä (taulukko 13) vaiheen 3 tutkimuksissa.
| Taulukko 13. Potilaat, joiden vakava masennustila remissiossa; kaksoissokkoutettu hoitojakso; koko tehoa koskeva tietoaineisto | ||||||
| SUI3001 | SUI3002 | Yhdistetyt tutkimukset (SUI3001 ja SUI3002) | ||||
| Lumevalmiste + tavanomainen hoito | Spravato + tavanomainen hoito | Lumevalmiste + tavanomainen hoito | Spravato + tavanomainen hoito | Lumevalmiste + tavanomainen hoito | Spravato + tavanomainen hoito | |
| 112 | 112 | 113 | 114 | 225 | 226 | |
| Päivä 1: 4 tuntia ensimmäisen annoksen jälkeen | ||||||
| Potilaat, joiden masennustila remissiossa | 9 (8,0 %) | 12 (10,7 %) | 4 (3,5 %) | 12 (10,5 %) | 13 (5,8 %) | 24 (10,6 %) |
| Päivä 2: 24 tuntia ensimmäisen annoksen jälkeen | ||||||
| Potilaat, joiden masennustila remissiossa | 10 (8,9 %) | 21 (18,8 %) | 12 (10,6 %) | 25 (21,9 %) | 22 (9,8 %) | 46 (20,4 %) |
| Päivä 25 (ennen annosta) | ||||||
| Potilaat, joiden masennustila remissiossa | 38 (33,9 %) | 46 (41,1 %) | 31 (27,4 %) | 49 (43,0 %) | 69 (30,7 %) | 95 (42,0 %) |
| Päivä 25 (4 tuntia annoksen jälkeen) | ||||||
| Potilaat, joiden masennustila remissiossa | 42 (37,5 %) | 60 (53,6 %) | 42 (37,2 %) | 54 (47,4 %) | 84 (37,3 %) | 114 (50,4 %) |
| Huom.: Remissio perustuu MADRS-kokonaispisteisiin ≤ 12. Tutkittavien, jotka eivät täyttäneet tätä kriteeriä tai jotka lopettivat hoidon mistä tahansa syystä ennen aikapistettä, ei katsottu olevan remissiossa. | ||||||
Vaikutukset itsemurha-alttiuteen
Kummankin hoitoryhmän potilaat kokivat yleisesti itsemurha-alttiutensa vaikeusasteen vähentyneen, mikä mitattiin CGI-SS-r-asteikolla (Clinical Global Impression – Severity of Suicidality - revised) 24 tunnin aikapisteessä, mutta hoitoryhmien välillä ei ollut tilastollisesti merkitsevää eroa.
Esketamiinin pitkäaikaistehoa itsemurhien estämisessä ei ole varmistettu.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Spravato-valmisteen käytöstä masennuksen hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Nenäsumutteena annetun 84 mg:n esketamiiniannoksen absoluuttisen biologisen hyötyosuuden keskiarvo on noin 48 %.
Nenään annettu esketamiini imeytyy nopeasti nenän limakalvon läpi ja on 28 mg:n annoksen jälkeen mitattavissa plasmasta 7 minuutin kuluessa. Huippupitoisuus plasmassa (tmax) saavutetaan tyypillisesti 20–40 minuutin kuluttua siitä, kun hoitokerran viimeinen nenäsumute on annosteltu (ks. kohta Annostus ja antotapa).
Esketamiininenäsumuteannokset 28 mg, 56 mg ja 84 mg suurentavat annosriippuvaisesti huippupitoisuutta plasmassa (Cmax) ja pitoisuus‑aikakuvaajan alle jäävää pinta‑alaa (AUC∞).
Esketamiinin farmakokineettinen profiili on samankaltainen kerta‑annoksen ja toistettujen annosten jälkeen, eikä kahdesti viikossa annettu esketamiini kumuloidu plasmaan.
Jakautuminen
Laskimoon annetun esketamiinin vakaan tilan jakautumistilavuuden keskiarvo on 709 l.
Ihmisen plasman proteiineihin sitoutuneen esketamiinin kokonaispitoisuuden osuus on keskimäärin 43–45 %. Esketamiinin sitoutumisaste plasman proteiineihin ei ole maksan tai munuaisten toiminnasta riippuvainen.
Esketamiini ei ole P‑glykoproteiini- (P‑gp; monilääkeresistenssiproteiini 1), BCRP:n (rintasyövän resistenssiproteiini) eikä OATP1B1- (OATP: orgaanisten anionien kuljettajaproteiini) tai OATP1B3- kuljettajaproteiinien substraatti. Esketamiini ei estä näitä kuljettajaproteiineja eikä MATE1:tä ja MATE2‑K:ta (MATE: multi‑drug and toxin extrusion) tai OCT2:ta (OCT: orgaanisten kationien kuljettajaproteiini), OAT1:tä eikä OAT3:a (OAT: orgaanisten anionien kuljettaja).
Biotransformaatio
Esketamiini metaboloituu laajasti maksassa. Esketamiinin pääasiallinen metaboliareitti ihmisen maksan mikrosomeissa on N‑demetylaatio noresketamiiniksi. Esketamiinin N‑demetylaatiosta pääasiallisesti vastaavat sytokromi P450 (CYP) ‑entsyymit ovat CYP2B6 ja CYP3A4. Muut CYP‑entsyymit, kuten CYP2C19 ja CYP2C9, osallistuvat siihen vähemmän. Noresketamiini metaboloituu sen jälkeen CYP‑riippuvaisten reittien kautta muiksi metaboliiteiksi, joista osa glukuronoituu.
Eliminaatio
Laskimoon annetun esketamiinin puhdistuman keskiarvo oli noin 89 l/tunti. Sen jälkeen kun nenään annetun esketamiinin huippupitoisuus oli saavutettu, esketamiinipitoisuus plasmassa pieneni nopeasti muutaman ensimmäisen tunnin ajan ja sen jälkeen hitaammin. Nenäsumutteen terminaalisen puoliintumisajan keskiarvo vaihteli yleensä 7 tunnista 12 tuntiin.
Kun radioaktiivisesti merkittyä esketamiinia annettiin laskimoon, noin 78 % radioaktiivisuudesta oli havaittavissa virtsassa ja noin 2 % ulosteissa. Kun radioaktiivisesti merkittyä esketamiinia annettiin suun kautta, noin 86 % radioaktiivisuudesta oli havaittavissa virtsassa ja noin 2 % ulosteissa. Havaittu radioaktiivisuus koostui pääasiassa esketamiinin metaboliiteista. Laskimoon ja suun kautta annettuna < 1 % annoksesta erittyi virtsaan muuttumattomana aineena.
Lineaarisuus/ei‑lineaarisuus
Esketamiinille altistuminen kasvaa siirryttäessä 28 mg:n annoksesta 84 mg:n annokseen. Annosten 28 mg ja 56 mg ja 84 mg välillä Cmax‑ ja AUC‑arvot nousivat vähemmän kuin annosriippuvaisesti. Annosten 56 mg ja 84 mg välinen arvojen nousu oli kuitenkin lähes annosriippuvuvainen.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutus esketamiiniin
Maksaentsyymien estäjät
Terveille tutkittaville esilääkityksenä suun kautta annettu tiklopidiini (250 mg kaksi kertaa vuorokaudessa 9 päivän ajan ennen esketamiinin annostelua sekä esketamiinin annostelupäivänä; tiklopidiini on maksan CYP2B6-entsyymin estäjä) ei vaikuttanut nenäsumutteena annetun esketamiinin Cmax-arvoon. Esketamiinin AUC∞-arvo suureni noin 29 %. Esilääkityksenä annettu tiklopidiini ei vaikuttanut esketamiinin terminaaliseen puoliintumisaikaan.
Esilääkityksenä suun kautta annettu klaritromysiini (500 mg kaksi kertaa vuorokaudessa 3 päivän ajan ennen esketamiinin annostelua sekä esketamiinin annostelupäivänä; klaritromysiini on maksan CYP3A4-entsyymin estäjä) suurentaa nenään annetun esketamiinin keskimääräistä Cmax-arvoa noin 11 % ja AUC∞-arvoa noin 4 %. Esilääkityksenä annettu klaritromysiini ei vaikuttanut esketamiinin terminaaliseen puoliintumisaikaan.
Maksaentsyymien indusoijat
Esilääkitys suun kautta otettavalla rifampisiinilla (600 mg/vrk esketamiinin annostelua edeltävinä 5 päivänä; rifampisiini on monien maksan CYP-entsyymien, esim. CYP3A4 ja CYP2B6, aktiivisuuden voimakas indusoija) pienensi nenäsumutteena annetun esketamiinin keskimääräistä Cmax-arvoa noin 17 % ja keskimääräistä AUC∞-arvoa noin 28 %.
Muut nenäsumutevalmisteet
Tutkittaville, joilla oli aiemmin ollut allergista nuhaa ja jotka oli altistettu heinien siitepölylle, annettiin esilääkityksenä oksimetatsoliinia nenäsumutteena (2 suihketta 0,05 %:n vahvuista liuosta 1 tunti ennen esketamiinin annostelua nenään), ja sen vaikutukset esketamiinin farmakokinetiikkaan olivat vähäisiä.
Terveille tutkittaville annettiin esilääkityksenä nenään mometasonifuroaattia (200 mikrog/vrk 2 viikon ajan, ja viimeinen mometasonifuroaattiannos 1 tunti ennen esketamiinin annostelua nenään), ja sen vaikutukset esketamiinin farmakokinetiikkaan olivat vähäisiä.
Esketamiinin vaikutus muihin lääkevalmisteisiin
Nenään annettu 84 mg:n esketamiiniannos kaksi kertaa viikossa 2 viikon ajan pienensi suun kautta otettavan midatsolaamin (6 mg:n kerta-annos; midatsolaami on maksan CYP3A4-entsyymin substraatti) keskimääräistä AUC∞-arvoa plasmassa noin 16 %.
Nenään annettu 84 mg:n esketamiiniannos kaksi kertaa viikossa 2 viikon ajan ei vaikuttanut suun kautta otettavan bupropionin (150 mg:n kerta-annos; bupropioni on maksan CYP2B6-entsyymin substraatti) keskimääräiseen AUC-arvoon plasmassa.
Erityispotilasryhmät
Iäkkäät (65‑vuotiaat ja vanhemmat)
Nenäsumutteena annetun esketamiinin farmakokinetiikkaa verrattiin terveiden iäkkäiden tutkittavien ja terveiden nuorempien aikuisten välillä. Esketamiinin Cmax‑ ja AUC∞‑arvojen keskiarvot 28 mg:n annoksen jälkeen olivat iäkkäillä tutkittavilla (65–81‑vuotiailla) 21 % (Cmax) ja 18 % (AUC∞) suuremmat nuorempiin (22–50‑vuotiaisiin) aikuisiin tutkittaviin verrattuna. Esketamiinin Cmax‑ ja AUC∞‑arvojen keskiarvot 84 mg:n annoksen jälkeen olivat iäkkäillä tutkittavilla (75–85‑vuotiailla) 67 % (Cmax) ja 38 % (AUC∞) suuremmat nuorempiin (24–54‑vuotiaisiin) aikuisiin tutkittaviin verrattuna. Esketamiinin terminaalinen puoliintumisaika oli iäkkäillä ja nuoremmilla aikuisilla tutkittavilla samankaltainen (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
28 mg:n esketamiininenäsumuteannoksen jälkeen esketamiinin Cmax oli lievää munuaisten vajaatoimintaa (kreatiniinipuhdistuma [CLCR] 58–77 ml/min), keskivaikeaa munuaisten vajaatoimintaa (CLCR 30–47 ml/min) tai vaikeaa munuaisten vajaatoimintaa (CLCR 5–28 ml/min, ei dialyysihoitoa) sairastavilla potilailla keskimäärin 20–26 % suurempi verrattuna tutkittaviin, joiden munuaisten toiminta oli normaali (CLCR 88–140 ml/min). AUC∞‑arvo oli lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavilla tutkittavilla 13–36 % suurempi.
Esketamiinin annosta nenäsumutteena dialyysihoitoa saaville potilaille ei ole kliinistä kokemusta.
Maksan vajaatoiminta
28 mg:n esketamiiniannoksen jälkeen Cmax‑ ja AUC∞‑arvot olivat samankaltaiset Child‑Pugh‑luokan A (lievä) maksan vajaatoimintaa sairastavillla ja terveillä tutkittavilla. Esketamiinin Cmax‑arvo oli 8 % suurempi ja AUC∞‑arvo oli 103 % suurempi tutkittavilla, joilla oli Child‑Pugh‑luokan B (keskivaikea) maksan vajaatoiminta, verrattuna terveisiin tutkittaviin.
Esketamiinin annosta nenäsumutteena Child‑Pugh‑luokan C (vaikea) maksan vajaatoimintaa sairastaville potilaille ei ole kliinistä kokemusta (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Etninen tausta
Esketamiininenäsumutteen farmakokinetiikkaa verrattiin terveiden aasialaisten ja valkoihoisten tutkittavien välillä. 56 mg:n esketamiinikerta‑annoksen jälkeen esketamiinin plasman Cmax‑ ja AUC∞‑arvojen keskiarvot olivat kiinalaisilla tutkittavilla noin 14 % (Cmax) ja 33 % (AUC∞) suuremmat kuin valkoihoisilla tutkittavilla. Korealaisilla tutkittavilla esketamiinin Cmax‑arvo oli keskimäärin 10 % pienempi ja AUC∞‑arvo keskimäärin 17 % suurempi kuin valkoihoisilla tutkittavilla. Populaatiofarmakokineettisessä analyysissa oli mukana terveiden japanilaisten tutkittavien lisäksi hoitoresistenttiä masennusta sairastavia japanilaisia potilaita. Tämän analyysin perusteella esketamiinin Cmax- ja AUC24h-arvot plasmassa olivat kunkin annoksen yhteydessä japanilaisilla tutkittavilla noin 20 % suuremmat kuin tutkittavilla, jotka eivät olleet aasialaista syntyperää. Esketamiinin terminaalinen puoliintumisaika plasmassa oli aasialaisilla tutkittavilla keskimäärin 7,1–8,9 tuntia, kun valkoihoisilla tutkittavilla se oli 6,8 tuntia.
Sukupuoli ja paino
Sukupuolella ja kokonaispainolla (> 39–170 kg) ei ollut merkittävää vaikutusta esketamiininenäsumutteen farmakokinetiikkaan populaatiofarmakokineettisen analyysin perusteella.
Allerginen nuha
Nenäsumutteena annostellun 56 mg:n esketamiinikerta‑annoksen farmakokinetiikka oli samankaltainen allergista nuhaa sairastavilla, jotka oli altistettu heinien siitepölylle, ja terveillä tutkittavilla.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, neurotoksisuutta, lisääntymistoksisuutta ja karsinogeenisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Ketamiinilla tehdyissä eläinkokeissa on todettu kehitykseen liittyvää neurotoksisuutta. Esketamiinista mahdollisesti kehittyvälle sikiölle aiheutuvia neurotoksisia vaikutuksia ei voida sulkea pois (ks. kohta Raskaus ja imetys).
Genotoksisuus
Esketamiini ei ollut Amesin testissä mutageeninen metabolisen aktivaation yhteydessä eikä ilman sitä. Esketamiinilla havaittiin genotoksisia vaikutuksia metabolisen aktivaation yhteydessä tehdyssä in vitro ‑mikrotumaseulontakokeessa. Laskimoon annetulla esketamiinilla ei kuitenkaan ollut genotoksisia vaikutuksia luuytimen in vivo ‑mikrotumakokeessa rotilla eikä rotan maksasoluilla tehdyssä Comet‑määrityksessä in vivo.
Lisääntymistoksisuus
Rotille nenään annetulla ketamiinilla tehdyssä alkion ja sikiön kehitystoksisuutta selvittäneessä tutkimuksessa emolle toksiset annokset, joista aiheutui AUC-arvon perusteella enimmillään kuusinkertainen altistus ihmisen altistukseen verrattuna, eivät vaikuttaneet jälkeläisiin haitallisesti. Nenään annettavalla ketamiinilla tehdyssä alkion ja sikiön kehitystoksisuutta kaniineilla selvittäneessä tutkimuksessa emolle toksisista annoksista havaittiin aiheutuneen sikiön luuston epämuodostumia ja painon laskua. Kaniinien altistus vastasi AUC-arvojen perusteella suunnilleen ihmisen altistusta.
Julkaistut eläimillä (myös kädellisillä) tehdyt tutkimukset, joissa annetuista annoksista aiheutui kevyt tai kohtalainen anestesia, osoittivat, että anestesia-aineiden käyttö aivojen nopean kasvun tai synaptogeneesin aikana aiheuttaa kehittyvissä aivoissa solukatoa ja että tähän voi liittyä pitkäaikaisia kognitiivisia puutoksia. Näiden prekliinisten havaintojen kliinistä merkitystä ei tiedetä.
Farmaseuttiset tiedot
Apuaineet
Sitruunahappomonohydraatti
Dinatriumedetaatti
Natriumhydroksidi (pH:n säätöön)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SPRAVATO nenäsumute, liuos
28 mg (L:ei) 1 kpl (243,42 €), 2 x 1 kpl (470,82 €), 3 x 1 kpl (696,40 €)
PF-selosteen tieto
Tyypin I lasista valmistettu injektiopullo, jossa on klooributyylikumitulppa. Täytetty ja tulpalla suljettu injektiopullo on liitetty osaksi käsikäyttöisesti aktivoitavaa nenäsumutinta. Nenäsumutin annostelee kaksi suihketta.
Jokaisessa pakkauksessa kukin laite on yksittäispakattuna sinetöityyn läpipainopakkaukseen.
Pakkauskoot: 1, 2, 3 tai 6 nenäsumutinta ja kerrannaispakkaukset, joissa 12 (4 x 3) tai 24 (8 x 3) nenäsumutinta.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas, väritön vesiliuos.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SPRAVATO nenäsumute, liuos
28 mg 1 kpl, 2 x 1 kpl, 3 x 1 kpl
- Ylempi erityiskorvaus (100 %). Esketamiini: Aikuisten hoitoresistentin masennuksen hoito yhdistelmänä serotoniinin takaisinoton estäjän (SSRI) tai serotoniinin ja noradrenaliinin takaisinoton estäjän (SNRI) kanssa erityisin edellytyksin (1539).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Esketamiini: Aikuisten hoitoresistentin masennuksen hoito yhdistelmänä serotoniinin takaisinoton estäjän (SSRI) tai serotoniinin ja noradrenaliinin takaisinoton estäjän (SNRI) kanssa erityisin edellytyksin (3062).
ATC-koodi
N06AX27
Valmisteyhteenvedon muuttamispäivämäärä
20.12.2024
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com