
LORVIQUA tabletti, kalvopäällysteinen 25 mg, 100 mg
Vaikuttavat aineet ja niiden määrät
Lorviqua 25 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 25 mg lorlatinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 1,58 mg laktoosimonohydraattia.
Lorviqua 100 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 100 mg lorlatinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 4,20 mg laktoosimonohydraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti).
Kliiniset tiedot
Käyttöaiheet
Lorviqua on tarkoitettu monoterapiana anaplastinen lymfoomakinaasi (ALK) ‑positiivisen edenneen ei-pienisoluisen keuhkosyövän (NSCLC) hoitoon aikuispotilaille, joita ei ole aikaisemmin hoidettu ALK-estäjällä.
Lorviqua on tarkoitettu monoterapiana ALK‑positiivisen edenneen ei-pienisoluisen keuhkosyövän hoitoon aikuispotilaille, joiden sairaus on edennyt
- ensimmäisenä ALK-tyrosiinikinaasin estäjänä (TKI) annetun alektinibi‑ tai seritinibihoidon jälkeen tai
- kritsotinibilla ja vähintään yhdellä muulla ALK-tyrosiinikinaasin estäjällä annetun hoidon jälkeen.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Lorlatinibihoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Ei-pienisoluisen keuhkosyövän ALK-positiivisuuden toteaminen on edellytys lorlatinibihoitoon soveltuvien potilaiden valitsemiseksi, koska hyöty on osoitettu vain näillä potilailla. Ei-pienisoluista keuhkosyöpää sairastavien potilaiden ALK-positiivisuus on määritettävä laboratorioissa, joiden osaaminen käytetyssä erityistekniikassa on osoitettu. Virheellisesti tehty määritys voi johtaa epäluotettaviin testituloksiin.
Annostus
Suositeltu annostus on 100 mg lorlatinibia suun kautta kerran vuorokaudessa.
Hoidon kesto
Lorlatinibihoitoa tulisi jatkaa, kunnes tauti etenee, tai ilmenee toksisuutta, joka ei ole hyväksyttävissä.
Annoksen unohtaminen tai ottaminen tavanomaista myöhemmin
Jos Lorviqua-annos jää ottamatta, potilaan tulisi ottaa se heti, kun hän huomaa annoksen unohtuneen.
Potilaan ei kuitenkaan tule ottaa unohtunutta annosta, jos seuraavan annoksen ottamisajankohtaan on alle 4 tuntia. Kahta annosta ei tule ottaa samanaikaisesti yhden unohtuneen annoksen korvaamiseksi.
Annosmuutokset
Yksilöllinen turvallisuus ja siedettävyys saattavat edellyttää annostelun keskeyttämistä tai annoksen pienentämistä. Ohjeet lorlatinibiannoksen pienentämiseen ovat seuraavat:
- Ensimmäinen annoslasku: 75 mg suun kautta kerran vuorokaudessa.
- Toinen annoslasku: 50 mg suun kautta kerran vuorokaudessa.
Lorlatinibihoito tulee lopettaa pysyvästi, jos potilas ei siedä 50 mg:n annosta suun kautta kerran vuorokaudessa.
Taulukossa 1 on esitetty suositellut annosmuutokset toksisuuksien vuoksi ja tilanteisiin, joissa potilaille kehittyy eteis-kammiokatkos (AV-katkos).
Taulukko 1. Suositellut lorlatinibiannoksen muutokset haittavaikutusten vuoksi
Haittavaikutusa | Lorlatinibiannostus |
Hyperkolesterolemia tai hypertriglyseridemia | |
Lievä hyperkolesterolemia (kolesteroli normaalin viitevälin yläraja [ULN] – 300 mg/dl tai ULN – 7,75 mmol/l) TAI Kohtalainen hyperkolesterolemia (kolesteroli 301–400 mg/dl tai 7,76–10,34 mmol/l) TAI Lievä hypertriglyseridemia (triglyseridit 150–300 mg/dl tai 1,71–3,42 mmol/l) TAI Kohtalainen hypertriglyseridemia (triglyseridit 301–500 mg/dl tai 3,43–5,7 mmol/l) | Aloita lipidilääkitysb tai muuta lipidilääkitystä käytetyn lääkkeen valmisteyhteenvedon mukaisesti; jatka lorlatinibihoitoa samalla annoksella. |
Vaikea hyperkolesterolemia (kolesteroli 401–500 mg/dl tai 10,35–12,92 mmol/l) TAI Vaikea hypertriglyseridemia (triglyseridit 501–1 000 mg/dl tai 5,71–11,4 mmol/l) | Aloita lipidilääkitysb; jos potilas saa jo lipidilääkitystä, suurenna kyseisen lääkkeenb annosta valmisteyhteenvedon mukaisesti tai vaihda toiseen lipidilääkitykseenb. Jatka lorlatinibihoitoa samalla annoksella keskeytyksettä. |
Henkeä uhkaava hyperkolesterolemia (kolesteroli yli 500 mg/dl tai yli 12,92 mmol/l) TAI Henkeä uhkaava hypertriglyseridemia (triglyseridit yli 1 000 mg/dl tai yli 11,4 mmol/l) | Aloita lipidilääkitysb tai suurenna lipidilääkityksenb annosta valmisteyhteenvedon mukaisesti tai vaihda toiseen lipidilääkitykseenb. Keskeytä lorlatinibihoito, kunnes hyperkolesterolemia ja/tai hypertriglyseridemia on lievittynyt vaikeusasteeltaan kohtalaiseksi tai lieväksi. Aloita lorlatinibihoito uudestaan samalla annoksella ja samanaikaisesti maksimoimalla lipidilääkitystäb kyseisen lääkkeen valmisteyhteenvedon mukaisesti. Jos vaikea hyperkolesterolemia ja/tai hypertriglyseridemia uusiutuu huolimatta valmisteyhteenvedon mukaisesta maksimaalisesta lipidilääkityksestäb, pienennä lorlatinibiannosta yhdellä annostasolla. |
Keskushermostovaikutukset (sisältäen psykoottiset vaikutukset ja kognition, mielialan, mielentilan tai puheen muutokset) | |
Aste 2: Kohtalainen TAI Aste 3: Vaikea | Keskeytä hoito, kunnes toksisuus on lievittynyt vähintään asteelle 1. Aloita lorlatinibihoito sitten uudestaan yhtä annostasoa pienemmällä annoksella. |
Aste 4: Henkeä uhkaava/kiireellinen hoito tarpeen | Lopeta lorlatinibihoito pysyvästi. |
Lipaasi‑/amylaasipitoisuuden kohoaminen | |
Aste 3: Vaikea TAI Aste 4: Henkeä uhkaava/kiireellinen hoito tarpeen | Keskeytä lorlatinibihoito, kunnes lipaasi‑ tai amylaasipitoisuus korjaantuu lähtötasolle. Aloita lorlatinibihoito sitten uudestaan yhtä annostasoa pienemmällä annoksella. |
Interstitiaalinen keuhkosairaus (ILD)/keuhkotulehdus | |
Aste 1: Lievä TAI Aste 2: Kohtalainen | Keskeytä lorlatinibihoito, kunnes oireet ovat lievittyneet lähtötasolle, ja harkitse kortikosteroidihoidon aloittamista. Aloita lorlatinibihoito uudestaan yhtä annostasoa pienemmällä annoksella. Lopeta lorlatinibihoito pysyvästi, jos ILD/keuhkotulehdus uusiutuu tai se ei parane 6 viikon lorlatinibihoidon keskeytyksestä ja steroidihoidosta huolimatta. |
Aste 3: Vaikea TAI Aste 4: Henkeä uhkaava/kiireellinen hoito tarpeen | Lopeta lorlatinibihoito pysyvästi. |
PR-ajan piteneminen/eteis-kammiokatkos (AV-katkos) | |
Ensimmäisen asteen eteis-kammiokatkos: Oireeton | Jatka lorlatinibihoitoa samalla annoksella keskeytyksettä. Arvioi samanaikaisten lääkevalmisteiden vaikutuksia sekä määritä elektrolyyttiarvot ja korjaa PR-aikaa mahdollisesti pidentävät elektrolyyttihäiriöt. Seuraa tarkoin EKG:tä/eteis-kammiokatkokseen mahdollisesti liittyviä oireita. |
Ensimmäisen asteen eteis-kammiokatkos: Oireinen | Keskeytä lorlatinibihoito. Arvioi samanaikaisten lääkevalmisteiden vaikutuksia sekä määritä elektrolyyttiarvot ja korjaa PR-aikaa mahdollisesti pidentävät elektrolyyttihäiriöt. Seuraa tarkoin EKG:tä/eteis-kammiokatkokseen mahdollisesti liittyviä oireita. Jos oireet häviävät, aloita lorlatinibihoito uudestaan yhtä annostasoa pienemmällä annoksella. |
Toisen asteen eteis-kammiokatkos: Oireeton | Keskeytä lorlatinibihoito. Arvioi samanaikaisten lääkevalmisteiden vaikutuksia sekä määritä elektrolyyttiarvot ja korjaa PR-aikaa mahdollisesti pidentävät elektrolyyttihäiriöt. Seuraa tarkoin EKG:tä/eteis-kammiokatkokseen mahdollisesti liittyviä oireita. Jos seuraavassa EKG:ssä ei todeta toisen asteen eteis-kammiokatkosta, aloita lorlatinibihoito uudestaan yhtä annostasoa pienemmällä annoksella. |
Toisen asteen eteis-kammiokatkos: Oireinen | Keskeytä lorlatinibihoito. Arvioi samanaikaisten lääkevalmisteiden vaikutuksia sekä määritä elektrolyyttiarvot ja korjaa PR-aikaa mahdollisesti pidentävät elektrolyyttihäiriöt. Lähetä potilas sydänvalvontaan ja ‑seurantaan. Harkitse tahdistimen asentamista, jos oireinen eteis-kammiokatkos jatkuu. Jos oireet ja toisen asteen eteis-kammiokatkos häviävät tai jos potilaan tila korjaantuu oireettomaksi ensimmäisen asteen eteis-kammiokatkokseksi, aloita lorlatinibihoito uudestaan yhtä annostasoa pienemmällä annoksella. |
Täydellinen eteis-kammiokatkos | Keskeytä lorlatinibihoito. Arvioi samanaikaisten lääkevalmisteiden vaikutuksia sekä määritä elektrolyyttiarvot ja korjaa PR-aikaa mahdollisesti pidentävät elektrolyyttihäiriöt. Lähetä potilas sydänvalvontaan ja ‑seurantaan. Tahdistimen asentaminen voi olla tarpeen eteis-kammiokatkokseen liittyvissä vaikeissa oireissa. Jos eteis-kammiokatkos ei korjaannu, voidaan harkita pysyvän tahdistimen asentamista. Jos tahdistin asennetaan, aloita lorlatinibihoito uudestaan enimmäisannoksella. Jos tahdistinta ei asenneta, aloita lorlatinibihoito uudestaan yhtä annostasoa pienemmällä annoksella vasta sen jälkeen, kun oireet ovat hävinneet ja PR-aika on alle 200 ms. |
Hypertensio | |
Aste 3 (Systolinen verenpaine ≥ 160 mmHg tai diastolinen verenpaine ≥ 100 mmHg; lääketieteellinen hoito tarpeen; useamman kuin yhden verenpainelääkkeen tai aikaisempaa tehokkaamman hoidon käyttö tarpeen) | Keskeytä lorlatinibihoito, kunnes hypertensio on lievittynyt vähintään asteelle 1 (systolinen verenpaine alle 140 mmHg ja diastolinen verenpaine alle 90 mmHg). Aloita lorlatinibihoito sitten uudestaan samalla annoksella. Jos asteen 3 hypertensio uusiutuu, keskeytä lorlatinibihoito, kunnes hypertensio on lievittynyt vähintään asteelle 1. Aloita hoito sitten uudestaan pienemmällä annoksella. Lopeta lorlatinibihoito pysyvästi, jos hypertensiota ei saada riittävästi hallintaan optimaalisella lääkehoidolla. |
Aste 4 (Henkeä uhkaavia oireita, kiireellinen hoito tarpeen) | Keskeytä lorlatinibihoito, kunnes hypertensio on lievittynyt vähintään asteelle 1. Aloita lorlatinibihoito sitten uudestaan pienemmällä annoksella tai lopeta hoito pysyvästi. Lopeta lorlatinibihoito pysyvästi, jos asteen 4 hypertensio uusiutuu. |
Hyperglykemia | |
Aste 3 TAI Aste 4 (Optimaalisesta verensokeria alentavasta lääkityksestä huolimatta hyperglykemia > 250 mg/dl jatkuu) | Keskeytä lorlatinibihoito, kunnes hyperglykemia saadaan riittävästi hallintaan. Aloita lorlatinibihoito sitten uudestaan yhtä annostasoa pienemmällä annoksella. Lopeta lorlatinibihoito pysyvästi, jos hyperglykemiaa ei saada riittävästi hallintaan optimaalisella lääkehoidolla. |
Muut haittavaikutukset | |
Aste 1: Lievä TAI Aste 2: Kohtalainen | Harkitse kliinisen tarpeen mukaan joko annoksen pitämistä ennallaan tai pienentämistä yhdellä annostasolla. |
Aste 3 tai suurempi: Vaikea | Keskeytä lorlatinibihoito, kunnes oireet ovat lievittyneet korkeintaan vaikeusasteelle 2 tai lähtötasolle. Aloita lorlatinibihoito sitten uudestaan yhtä annostasoa pienemmällä annoksella. |
Lyhenteet: CTCAE = Common Terminology Criteria for Adverse Events (yhteiset terminologiakriteerit haittatapahtumille), EKG = elektrokardiogrammi, HMG CoA = 3‑hydroksi‑3‑metyyliglutaryylikoentsyymi A, NCI = National Cancer Institute (Yhdysvaltain kansallinen syöpäinstituutti), ULN = normaalin viitevälin yläraja. a Vaikeusasteluokat perustuvat NCI CTCAE ‑luokitukseen. b Lipidilääkitys voi sisältää seuraavia: HMG‑CoA‑reduktaasin estäjä, nikotiinihappo, fibriinihapon johdokset tai omega‑3-rasvahappojen etyyliesterit. | |
Voimakkaat sytokromi P450 (CYP) 3A4/5:n estäjät
Lorlatinibin samanaikainen käyttö voimakkaiden CYP3A4/5:n estäjien tai greippimehuvalmisteiden kanssa saattaa suurentaa plasman lorlatinibipitoisuutta. Jotakin vaihtoehtoista samanaikaista lääkevalmistetta, joka ei ole yhtä potentiaalinen CYP3A4/5:n estäjä, tulisi harkita (ks. kohta Yhteisvaikutukset). Jos potilas tarvitsee samanaikaista hoitoa voimakkaalla CYP3A4/5:n estäjällä, lorlatinibin aloitusannos 100 mg kerran vuorokaudessa tulee pienentää 75 mg:aan kerran vuorokaudessa (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka). Jos voimakkaan CYP3A4/5:n estäjän samanaikainen käyttö lopetetaan, lorlatinibihoitoa tulee jatkaa annoksella, jota potilas sai ennen voimakkaan CYP3A4/5:n estäjän aloitusta, mutta vasta puhdistumisjakson (voimakkaan CYP3A4/5:n estäjän 3–5 puoliintumisajan) jälkeen.
Erityisryhmät
Iäkkäät (≥ 65‑vuotiaat)
Annossuosituksia 65‑vuotiaille ja tätä vanhemmille potilaille ei voida antaa, koska tästä potilasjoukosta on vain vähän tietoja (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa potilaille, joilla on normaali munuaisten toiminta tai lievä tai kohtalainen munuaisten vajaatoiminta (absoluuttinen glomerulusten laskennallinen suodatusnopeus [eGFR] ≥ 30 ml/min). Potilaille, joilla on vaikea munuaisten vajaatoiminta (absoluuttinen eGFR < 30 ml/min), suositellaan pienennettyä lorlatinibiannosta, esim. aloitusannosta 75 mg suun kautta kerran vuorokaudessa (ks. kohta Farmakokinetiikka). Tietoja ei ole saatavilla munuaisdialyysihoitoa tarvitsevista potilaista.
Maksan vajaatoiminta
Annosmuutoksia ei suositella potilaille, joilla on lievä tai kohtalainen maksan vajaatoiminta. Potilaille, joilla on vaikea (Child-Pugh C) maksan vajaatoiminta, lorlatinibin aloitusannosta suositellaan pienentämään 100 mg:sta 50 mg:aan suun kautta kerran vuorokaudessa (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Lorlatinibihoidon turvallisuutta ja tehoa alle 18-vuotiaiden pediatristen potilaiden hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Lorviqua otetaan suun kautta.
Potilaita tulee kehottaa ottamaan lorlatinibiannos suurin piirtein samaan aikaan joka päivä joko aterian yhteydessä tai tyhjään mahaan (ks. kohta Farmakokinetiikka). Tabletit on nieltävä kokonaisina (niitä ei saa pureskella, murskata eikä puolittaa ennen nielemistä). Tablettia ei pidä ottaa, jos se on rikkoutunut, lohjennut tai muuten vahingoittunut.
Vasta-aiheet
Yliherkkyys lorlatinibille tai kohdassa Apuaineet mainituille apuaineille.
Voimakkaiden CYP3A4/5:n induktoreiden samanaikainen käyttö (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Hyperlipidemia
Lorlatinibin käyttöön on liitetty seerumin kolesteroli‑ ja triglyseridipitoisuuksien kohoamista (ks. kohta Haittavaikutukset). Mediaaniaika seerumin kolesteroliarvon vaikea-asteiseen kohoamiseen on 201 päivää (vaihteluväli: 29–729 päivää) ja triglyseridiarvon vaikea-asteiseen kohoamiseen 127 päivää (vaihteluväli: 15 – 1367 päivää). Seerumin kolesteroli‑ ja triglyseridipitoisuudet tulee määrittää ennen lorlatinibihoidon aloittamista, 2, 4 ja 8 viikkoa lorlatinibihoidon aloittamisen jälkeen ja sen jälkeen säännöllisesti. Potilaalle tulee aloittaa hoito lipidejä alentavilla lääkevalmisteilla tai näiden annosta tulee suurentaa, jos tämä on lääketieteellisesti aiheellista (ks. kohta Annostus ja antotapa).
Keskushermostovaikutukset
Lorlatinibia saavilla potilailla on havaittu keskushermostovaikutuksia, mukaan lukien psykoottisia vaikutuksia ja kognitiivisen toiminnan, mielialan, mielentilan tai puhekyvyn muutoksia (ks. kohta Haittavaikutukset). Jos keskushermostovaikutuksia ilmenee, annosta voidaan joutua muuttamaan tai hoito lopettamaan (ks. kohta Annostus ja antotapa).
Eteis-kammiokatkos
Lorlatinibin tutkimuspopulaatiosta oli poissuljettu potilaat, joilla oli toisen tai kolmannen asteen eteis-kammiokatkos (paitsi jos potilaalla oli tahdistin) tai mikä tahansa eteis-kammiokatkos, jossa PR‑aika oli > 220 ms. Lorlatinibia saavilla potilailla on raportoitu PR-ajan pitenemistä ja eteis-kammiokatkoksia (ks. kohta Farmakokinetiikka). Sydänsähkökäyrä (EKG) tulee rekisteröidä ennen lorlatinibihoidon aloittamista ja sen jälkeen kuukausittain, erityisesti potilailla, joilla on kliinisesti merkittävien sydäntapahtumien ilmaantumiselle altistavia sairauksia. Jos eteis-kammiokatkos ilmenee, annosta voidaan joutua muuttamaan (ks. kohta Annostus ja antotapa).
Vasemman kammion ejektiofraktion pieneneminen
Vasemman kammion ejektiofraktion (LVEF) pienenemistä on raportoitu lorlatinibia saavilla potilailla, joille tehtiin LVEF-mittaus lähtötilanteessa ja vähintään yhdellä seurantakäynnillä. Saatavilla olevien kliinisten tutkimustietojen perusteella ei ole mahdollista määrittää syy-yhteyttä sydämen supistumiskyvyn muutoksiin kohdistuvien vaikutusten ja lorlatinibin välillä. Jos potilaalla on sydämeen liittyviä riskitekijöitä tai vasemman kammion ejektiofraktioon mahdollisesti vaikuttava sairaus, sydäntutkimuksia, mukaan lukien LVEF-mittausta, tulee harkita lähtötilanteessa ja hoidon aikana. Jos potilaalle kehittyy merkityksellisiä sydämeen liittyviä merkkejä/oireita hoidon aikana, sydäntutkimuksia, mukaan lukien LVEF-mittausta, tulee harkita.
Lipaasi‑ ja amylaasipitoisuuksien kohoaminen
Lorlatinibia saavilla potilailla on ilmennyt lipaasi‑ ja/tai amylaasipitoisuuksien kohoamista (ks. kohta Haittavaikutukset). Mediaaniaika seerumin lipaasiarvon kohoamiseen on 169 päivää (vaihteluväli: 1 − 1755 päivää) ja seerumin amylaasiarvon kohoamiseen 158 päivää (vaihteluväli: 1 – 1932 päivää). Lorlatinibia saavilla potilailla tulee huomioida haimatulehduksen riski samanaikaisen hypertriglyseridemian ja/tai mahdollisen sisäsyntyisen mekanismin takia. Potilaiden lipaasi‑ ja amylaasipitoisuudet tulee määrittää kohonneiden pitoisuuksien varalta ennen lorlatinibihoidon aloittamista ja sen jälkeen säännöllisesti kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa).
Interstitiaalinen keuhkosairaus/keuhkotulehdus
Lorlatinibihoidon yhteydessä on ilmennyt interstitiaaliseen keuhkosairauteen/keuhkotulehdukseen sopivia vaikeita tai henkeä uhkaavia keuhkohaittavaikutuksia (ks. kohta Haittavaikutukset). Jos potilaalla ilmenee interstitiaaliseen keuhkosairauteen/keuhkotulehdukseen viittaavien hengitysoireiden (esim. hengenahdistus, yskä ja kuume) pahenemista, hänet on viipymättä arvioitava interstitiaalisen keuhkosairauden/keuhkotulehduksen varalta. Lorlatinibihoito on keskeytettävä ja/tai lopetettava pysyvästi oireiden vaikeusasteen perusteella (ks. kohta Annostus ja antotapa).
Hypertensio
Hypertensiota on raportoitu lorlatinibihoitoa saavilla potilailla (ks. kohta Haittavaikutukset). Verenpaine on tarkistettava ja sen on oltava hallinnassa ennen lorlatinibihoidon aloittamista. Verenpaine tulee mitata kahden viikon kuluttua hoidon aloittamisesta ja sen jälkeen vähintään kuukausittain lorlatinibihoidon aikana. Verenpaineen noustessa lorlatinibihoito on tarvittaessa keskeytettävä ja aloitettava uudestaan pienemmällä annoksella tai lopetettava pysyvästi hypertension vaikeusasteen perusteella (ks. kohta Annostus ja antotapa).
Hyperglykemia
Hyperglykemiaa on ilmennyt lorlatinibihoitoa saavilla potilailla (ks. kohta Haittavaikutukset). Seerumin paastoglukoosiarvo tulee määrittää ennen lorlatinibihoidon aloittamista ja tämän jälkeen sitä on seurattava säännöllisesti kansallisten hoitosuositusten mukaisesti. Verensokerin noustessa lorlatinibihoito on tarvittaessa keskeytettävä ja aloitettava uudestaan pienemmällä annoksella tai lopetettava pysyvästi hyperglykemian vaikeusasteen perusteella (ks. kohta Annostus ja antotapa).
Lääkkeiden yhteisvaikutukset
Terveille vapaaehtoisille tutkittaville tehdyssä tutkimuksessa lorlatinibin ja rifampisiinin (voimakas CYP3A4/5:n induktori) samanaikaiseen käyttöön liittyi alaniiniaminotransferaasi‑ (ALAT) ja aspartaattiaminotransferaasipitoisuuksien (ASAT) kohoamista; bilirubiini ja alkalinen fosfataasi eivät sen sijaan kohonneet (ks. kohta Yhteisvaikutukset). Voimakkaan CYP3A4/5:n induktorin samanaikainen käyttö on vasta-aiheista (ks. kohdat Vasta-aiheet ja Yhteisvaikutukset). Terveillä tutkittavilla ei havaittu kliinisesti merkittäviä muutoksia maksan toimintakokeissa sen jälkeen, kun he olivat saaneet lorlatinibia yhdessä kohtalaisen voimakkaan CYP3A4/5:n induktorin modafiniilin kanssa (ks. kohta Yhteisvaikutukset).
Lorlatinibin samanaikaista antoa kapean terapeuttisen leveyden omaavien CYP3A4/5:n substraattien kanssa tulee välttää, koska lorlatinibi voi pienentää tällaisten lääkevalmisteiden pitoisuutta. Tällaisia CYP3A4/5:n substraatteja ovat mm. alfentaniili, siklosporiini, dihydroergotamiini, ergotamiini, fentanyyli, hormonaaliset ehkäisyvalmisteet, pimotsidi, kinidiini, sirolimuusi ja takrolimuusi (ks. kohta Yhteisvaikutukset).
Hedelmällisyys ja raskaus
Miespotilaiden, joiden naiskumppani voi tulla raskaaksi, on käytettävä tehokasta ehkäisyä (mukaan lukien kondomia) lorlatinibihoidon aikana ja vähintään 14 viikon ajan viimeisestä annoksesta laskettuna. Miespotilaiden on käytettävä kondomia kumppanin raskausaikana (ks. kohta Raskaus ja imetys). Miehen hedelmällisyys voi vaarantua lorlatinibihoidon aikana (ks. kohta Prekliiniset tiedot turvallisuudesta). Miesten tulisi saada ennen hoitoa tietoa toimenpiteistä lisääntymiskyvyn säilyttämiseksi. Naisia, jotka voivat tulla raskaaksi, on neuvottava välttämään raskaaksi tuloa lorlatinibihoidon aikana. Naispotilaiden on käytettävä lorlatinibihoidon aikana hyvin tehokasta ei-hormonaalista ehkäisyä, koska lorlatinibi voi tehdä hormonaaliset ehkäisyvalmisteet tehottomiksi (ks. kohdat Yhteisvaikutukset ja Raskaus ja imetys). Jos hormonaalisen ehkäisymenetelmän käyttöä ei voida välttää, hormonaalisen menetelmän lisäksi on käytettävä kondomia. Tehokkaan ehkäisyn käyttöä on jatkettava vähintään 35 päivän ajan hoidon päättymisen jälkeen (ks. kohta Raskaus ja imetys). Lorlatinibin vaikutuksia naisen hedelmällisyyteen ei tunneta.
Laktoosi-intoleranssi
Tämä lääkevalmiste sisältää laktoosia apuaineena. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Natriumin saanti
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per 25 mg:n tai 100 mg:n tabletti. Vähäsuolaista ruokavaliota noudattaville potilaille tulisi kertoa, että tämän lääkevalmisteen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Farmakokineettiset yhteisvaikutukset
In vitro ‑tiedot viittaavat siihen, että lorlatinibi metaboloituu ensisijaisesti CYP3A4:n ja uridiinidifosfaatti‑glukuronosyylitransferaasi (UGT)1A4:n välityksellä ja vähäisessä määrin myös CYP2C8:n, CYP2C19:n, CYP3A5:n ja UGT1A3:n välityksellä.
Muiden lääkevalmisteiden vaikutus lorlatinibiin
CYP3A4/5:n induktorit
Kun terveille vapaaehtoisille tutkittaville annettiin 600 mg rifampisiinia (voimakas CYP3A4/5:n induktori) suun kautta kerran vuorokaudessa 12 päivän ajan, lorlatinibin suun kautta annetun 100 mg:n kerta-annoksen pitoisuus-aikakuvaajan alle jäävä keskimääräinen pinta-ala (AUCinf) pieneni 85 % ja Cmax 76 %; myös ASAT‑ ja ALAT‑arvojen kohoamista havaittiin. Lorlatinibin samanaikainen anto voimakkaiden CYP3A4/5:n induktoreiden (esim. rifampisiini, karbamatsepiini, entsalutamidi, mitotaani, fenytoiini ja mäkikuisma) kanssa saattaa pienentää plasman lorlatinibipitoisuutta. Voimakkaan CYP3A4/5:n induktorin käyttö lorlatinibin kanssa on vasta-aiheista (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet). Maksan toimintakokeiden tuloksissa ei havaittu kliinisesti merkittäviä muutoksia sen jälkeen, kun terveille tutkittaville annettiin 100 mg:n kerta-annos lorlatinibia suun kautta yhdessä kohtalaisen voimakkaan CYP3A4/5:n induktorin modafiniilin (400 mg kerran päivässä 19 päivän ajan) kanssa. Modafiniilin samanaikaisella käytöllä ei ollut kliinisesti merkittävää vaikutusta lorlatinibin farmakokinetiikkaan.
CYP3A4/5:n estäjät
Kun terveille vapaaehtoisille tutkittaville annettiin 200 mg itrakonatsolia (voimakas CYP3A4/5:n estäjä) suun kautta kerran vuorokaudessa 5 päivän ajan, suun kautta annetun lorlatinibin 100 mg:n kerta-annoksen keskimääräinen AUCinf suureni 42 % ja Cmax 24 %. Lorlatinibin samanaikainen anto voimakkaiden CYP3A4/5:n estäjien kanssa (esim. bosepreviiri, kobisistaatti, itrakonatsoli, ketokonatsoli, posakonatsoli, troleandomysiini, vorikonatsoli, ritonaviiri, paritapreviiri yhdessä ritonaviirin ja ombitasviirin ja/tai dasabuviirin kanssa ja ritonaviiri yhdessä joko elvitegraviirin, indinaviirin, lopinaviirin tai tipranaviirin kanssa) saattaa suurentaa plasman lorlatinibipitoisuutta. Myös greippimehuvalmisteet saattavat suurentaa plasman lorlatinibipitoisuutta, ja niitä on vältettävä. Jotakin vaihtoehtoista samanaikaista lääkevalmistetta, joka ei yhtä todennäköisesti estä CYP3A4/5-entsyymejä, tulee harkita. Jos potilaalle on annettava samanaikaisesti voimakasta CYP3A4/5:n estäjää, suositellaan lorlatinibiannoksen pienentämistä (ks. kohta Annostus ja antotapa).
Lorlatinibin vaikutus muihin lääkevalmisteisiin
CYP3A4/5:n substraatit
In vitro ‑tutkimukset osoittivat, että lorlatinibi sekä estää että indusoi CYP3A4/5-entsyymejä ajasta riippuvaisesti. 150 mg lorlatinibia suun kautta kerran vuorokaudessa 15 päivän ajan pienensi suun kautta otetun midatsolaamin (herkkä CYP3A:n substraatti) 2 mg:n kerta-annoksen AUCinf‑arvoa 61 % ja Cmax‑arvoa 50 %. Lorlatinibi on siten kohtalainen CYP3A:n induktori. Lorlatinibin samanaikaista antoa kapean terapeuttisen leveyden omaavien CYP3A4/5:n substraattien kanssa tulee välttää, koska lorlatinibi voi pienentää tällaisten lääkevalmisteiden pitoisuutta. Tällaisia CYP3A4/5:n substraatteja ovat mm. alfentaniili, siklosporiini, dihydroergotamiini, ergotamiini, fentanyyli, hormonaaliset ehkäisyvalmisteet, pimotsidi, kinidiini, sirolimuusi ja takrolimuusi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
CYP2B6:n substraatit
100 mg lorlatinibia kerran vuorokaudessa 15 päivän ajan pienensi suun kautta otetun bupropionin (yhdistetty CYP2B6:n ja CYP3A4:n substraatti) 100 mg:n kerta-annoksen AUCinf-arvoa 49,5 % ja Cmax-arvoa 53 %. Lorlatinibi on siten heikko CYP2B6:n induktori. Kun lorlatinibia käytetään yhdistelmänä pääasiassa CYP2B6:n välityksellä metaboloituvien lääkevalmisteiden kanssa, annosta ei ole tarpeen muuttaa.
CYP2C9:n substraatit
100 mg lorlatinibia kerran vuorokaudessa 15 päivän ajan pienensi suun kautta otetun tolbutamidin (herkkä CYP2C9:n substraatti) 500 mg:n kerta-annoksen AUCinf-arvoa 43 % ja Cmax-arvoa 15 %. Lorlatinibi on siten heikko CYP2C9:n induktori. Pääasiassa CYP2C9:n välityksellä metaboloituvien lääkevalmisteiden annosta ei ole tarpeen muuttaa. Potilaita tulee kuitenkin seurata, jos samanaikaisesti käytetään CYP2C9:n välityksellä metaboloituvia lääkevalmisteita, joiden terapeuttinen indeksi on kapea (esim. kumariiniantikoagulantit).
UGT:n substraatit
100 mg lorlatinibia kerran vuorokaudessa 15 päivän ajan pienensi suun kautta otetun parasetamolin (UGT:n, SULT:n sekä CYP1A2:n, 2A6:n, 2D6:n ja 3A4:n substraatti) 500 mg:n kerta-annoksen AUCinf-arvoa 45 % ja Cmax-arvoa 28 %. Lorlatinibi on siten heikko UGT:n induktori. Pääasiassa UGT:n välityksellä metaboloituvien lääkevalmisteiden annosta ei ole tarpeen muuttaa. Potilaita tulee kuitenkin seurata, jos samanaikaisesti käytetään UGT:n välityksellä metaboloituvia lääkevalmisteita, joiden terapeuttinen indeksi on kapea.
P-glykoproteiinin substraatit
100 mg lorlatinibia kerran vuorokaudessa 15 päivän ajan pienensi suun kautta otetun feksofenadiinin (herkkä P-glykoproteiinin [P-gp] substraatti) 60 mg:n kerta-annoksen AUCinf-arvoa 67 % ja Cmax-arvoa 63 %. Lorlatinibi on siten kohtalainen P‑gp:n induktori. Lääkevalmisteita, jotka ovat P‑gp:n substraatteja ja joiden terapeuttinen indeksi on kapea (esim. digoksiini, dabigatraanieteksilaatti), tulee käyttää varoen yhdistelmänä lorlatinibin kanssa, koska todennäköisesti näiden substraattien pitoisuus plasmassa pienenee.
In vitro ‑tutkimukset muiden CYP-entsyymien estoa ja induktiota koskien
In vitro -tutkimuksien mukaan on epätodennäköistä, että lorlatinibi aiheuttaisi lääkkeiden yhteisvaikutuksia CYP1A2:ta indusoimalla.
In vitro ‑tutkimukset muilla lääkeaineiden kuljettajaproteiineilla kuin P-gp:llä
In vitro ‑tutkimukset osoittivat, että lorlatinibi saattaa kliinisesti merkittävinä pitoisuuksina estää seuraavia: BCRP (maha-suolikanava), OATP1B1, OATP1B3, OCT1, MATE1 ja OAT3. Lorlatinibia tulee käyttää varoen yhdessä BCRP:n, OATP1B1:n, OATP1B3:n, OCT1:n, MATE1:n ja OAT3:n substraattien kanssa, koska kliinisesti merkittäviä muutoksia näiden substraattien plasma-altistuksessa ei voida sulkea pois.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi/raskaudenehkäisy naisilla ja miehillä
Naisia, jotka voivat tulla raskaaksi, on neuvottava välttämään raskaaksi tuloa lorlatinibihoidon aikana. Naispotilaiden on käytettävä lorlatinibihoidon aikana hyvin tehokasta ei-hormonaalista ehkäisyä, koska lorlatinibi voi tehdä hormonaaliset ehkäisyvalmisteet tehottomiksi (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset). Jos hormonaalisen ehkäisymenetelmän käyttöä ei voida välttää, hormonaalisen menetelmän lisäksi on käytettävä kondomia. Tehokkaan ehkäisyn käyttöä on jatkettava vähintään 35 päivän ajan hoidon päättymisen jälkeen.
Miespotilaiden, joiden naiskumppani voi tulla raskaaksi, on käytettävä tehokasta ehkäisyä (mukaan lukien kondomia) lorlatinibihoidon aikana ja vähintään 14 viikon ajan viimeisestä annoksesta laskettuna. Miespotilaiden on käytettävä kondomia kumppanin raskausaikana.
Raskaus
Eläinkokeissa on osoitettu alkio‑/sikiötoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Ei ole olemassa tietoja lorlatinibin käytöstä raskaana oleville naisille. Raskaana olevalle naiselle annettu lorlatinibi voi vahingoittaa sikiötä.
Lorlatinibin käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyvätkö lorlatinibi ja sen metaboliitit ihmisen rintamaitoon. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea.
Lorlatinibia ei pidä käyttää rintaruokinnan aikana. Rintaruokinta on lopetettava lorlatinibihoidon ja viimeistä annosta seuraavien 7 päivän ajaksi.
Hedelmällisyys
Prekliinisten turvallisuuslöydösten perusteella miehen hedelmällisyys voi vaarantua lorlatinibihoidon aikana (ks. kohta Prekliiniset tiedot turvallisuudesta). Lorlatinibin vaikutusta naisen hedelmällisyyteen ei tunneta. Miesten tulisi saada ennen hoitoa tietoa toimenpiteistä lisääntymiskyvyn säilyttämiseksi.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Lorlatinibilla on kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Autoa ajettaessa tai koneita käytettäessä on noudatettava varovaisuutta, koska keskushermostovaikutuksia saattaa ilmetä (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin raportoidut haittavaikutukset olivat hyperkolesterolemia (79,0 %), hypertriglyseridemia (67,5 %), edeema (55,4 %), perifeerinen neuropatia (44,2 %), väsymys (30,7 %), painonnousu (29,8 %), nivelkipu (27,8 %), kognitiiviset vaikutukset (27,4 %), ripuli (22,7 %) ja mielialavaikutukset (21,4 %).
Vakavia haittavaikutuksia raportoitiin 9,1 %:lla lorlatinibia saaneista potilaista. Yleisimmät vakavat haittavaikutukset olivat kognitiiviset vaikutukset ja keuhkotulehdus.
Haittavaikutusten vuoksi annosta oli pienennettävä 20,1 %:lla lorlatinibia saaneista potilaista. Yleisimmät annoksen pienentämiseen johtaneet haittavaikutukset olivat edeema, kognitiiviset vaikutukset ja perifeerinen neuropatia. Haittavaikutusten vuoksi hoito oli lopetettava pysyvästi 4,0 %:lla lorlatinibia saaneista potilaista. Yleisimmät hoidon pysyvään lopettamiseen johtaneet haittavaikutukset olivat kognitiiviset vaikutukset, perifeerinen neuropatia, keuhkotulehdus ja psykoottiset vaikutukset.
Haittavaikutustaulukko
Taulukossa 2 on esitetty haittavaikutukset edennyttä ei-pienisoluista keuhkosyöpää sairastavista aikuispotilaista, jotka saivat lorlatinibia 100 mg kerran vuorokaudessa (n = 547) tutkimuksessa A (n = 327), CROWN-tutkimuksessa (n = 149) ja tutkimuksessa B (n = 71).
Taulukossa 2 on lueteltu haittavaikutukset elinjärjestelmien ja esiintymistiheyksien mukaan. Esiintymistiheydet on määritelty seuraavan luokituksen mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2. Haittavaikutukset
Elinjärjestelmä ja haittavaikutus | Esiintymistiheys | Kaikki vaikeusasteet % | Vaikeusasteet 3–4 % |
Veri ja imukudos Anemia |
Hyvin yleinen | 19,6 | 4,4 |
Aineenvaihdunta ja ravitsemus Hyperkolesterolemiaa Hypertriglyseridemiab Hyperglykemia |
Hyvin yleinen Hyvin yleinen Yleinen | 79,0 67,5 9,7 | 19,2 20,3 3,7 |
Psyykkiset häiriöt Mielialavaikutuksetc Psykoottiset vaikutuksetd Mielentilan muutokset |
Hyvin yleinen Yleinen Yleinen | 21,4 6,9 1,1 | 1,3 0,9 0,9 |
Hermosto Kognitiiviset vaikutuksete Perifeerinen neuropatiaf Päänsärky Puhevaikeudetg |
Hyvin yleinen Hyvin yleinen Hyvin yleinen Yleinen | 27,4 44,2 18,6 8,2 | 3,5 2,6 0,7 0,7 |
Silmät Näköhäiriöh |
Hyvin yleinen | 16,1 | 0,2 |
Verisuonisto Hypertensio |
Hyvin yleinen | 14,8 | 6,0 |
Hengityselimet, rintakehä ja välikarsina Keuhkotulehdusi |
Yleinen | 2,4 | 0,7 |
Ruoansulatuselimistö Ripuli Pahoinvointi Ummetus |
Hyvin yleinen Hyvin yleinen Hyvin yleinen | 22,7 17,6 16,8 | 1,8 0,9 0,2 |
Maksa ja sappi Alaniiniaminotransferaasipitoisuuden (ALAT) kohoaminen Aspartaattiaminotransferaasipitoisuuden (ASAT) kohoaminen |
Hyvin yleinen
Hyvin yleinen | 14,8 13,9 | 2,0 1,5 |
Iho ja ihonalainen kudos Ihottumaj |
Hyvin yleinen | 14,6 | 0,2 |
Munuaiset ja virtsatiet Proteinuria |
Yleinen | 3,7 | 0,4 |
Luusto, lihakset ja sidekudos Nivelkipu Lihaskipuk |
Hyvin yleinen Hyvin yleinen | 27,8 15,0 | 0,7 0 |
Yleisoireet ja antopaikassa todettavat haitat Edeemal Väsymysm |
Hyvin yleinen Hyvin yleinen | 55,4 30,7 | 2,9 1,1 |
Tutkimukset Painonnousu Kohonnut lipaasi Kohonnut amylaasi PR-ajan piteneminen EKG:ssa |
Hyvin yleinen Hyvin yleinen Hyvin yleinen Melko harvinainen | 29,8 12,8 11,3 0,7 | 11 6,8 2,7 0 |
Samaa lääketieteellistä käsitettä tai sairaustilaa edustavat haittavaikutukset yhdistettiin ryhmiksi ja ne on raportoitu yksittäisenä haittavaikutuksena edellä olevassa taulukossa. Tutkimuksissa tosiasiallisesti raportoidut termit, jotka sisältyvät kyseiseen haittavaikutukseen, on lueteltu seuraavassa sulkeissa. a Hyperkolesterolemia (mukaan lukien kohonnut veren kolesterolipitoisuus, hyperkolesterolemia). b Hypertriglyseridemia (mukaan lukien kohonnut veren triglyseridipitoisuus, hypertriglyseridemia). c Mielialavaikutukset (mukaan lukien affektiivinen häiriö, affektilabiilius, aggressio, kiihtymys, vihaisuus, ahdistuneisuus, tyypin I kaksisuuntainen mielialahäiriö, masentuneisuus, masennus, masennusoire, euforinen mieliala, ärtyisyys, mania, mielialan muutos, mielialan vaihtelut, paniikkikohtaus, persoonallisuuden muutos, stressi). d Psykoottiset vaikutukset (mukaan lukien kuuloharhat, aistiharhat ja näköharhat). e Kognitiiviset vaikutukset (mukaan lukien ”Hermosto”-elinjärjestelmäluokan tapahtumat: muistinmenetys, kognitiivinen häiriö, dementia, tarkkaavuushäiriö, muistin heikkeneminen, henkisen suorituskyvyn heikkeneminen; sekä myös ”Psyykkiset häiriöt” ‑elinjärjestelmäluokan tapahtumat: tarkkaavuus‑ ja ylivilkkaushäiriö, sekavuustila, delirium, desorientaatio, lukemishäiriö). Näistä vaikutuksista useammin raportoitiin ”Hermosto”-elinjärjestelmäluokan termejä kuin ”Psyykkiset häiriöt” ‑elinjärjestelmäluokan termejä. f Perifeerinen neuropatia (mukaan lukien polttelun tunne, dysestesia, formikaatio, kävelyhäiriö, hypestesia, motorinen toimintahäiriö, lihasheikkous, hermokipu, perifeerinen neuropatia, neurotoksisuus, parestesia, perifeerinen motorinen neuropatia, perifeerinen sensorinen neuropatia, pohjehermohalvaus, tuntohäiriö). g Puhevaikeudet (dysartria, puheen hitaus, puhehäiriö). h Näköhäiriö (mukaan lukien kaksoiskuvat, valonarkuus, fotopsia, näön hämärtyminen, näöntarkkuuden heikkeneminen, näkökyvyn heikkeneminen, lasiaiskellujat). i Keuhkotulehdus (mukaan lukien interstitiaalinen keuhkosairaus, keuhkovarjostuma, keuhkotulehdus). j Ihottuma (mukaan lukien aknen kaltainen ihotulehdus, makulopapulaarinen ihottuma, kutiseva ihottuma, ihottuma). k Lihaskipu (mukaan lukien lihaksiin ja luihin liittyvä kipu, lihaskipu). l Edeema (mukaan lukien yleistynyt edeema, edeema, perifeerinen edeema, ääreisturvotus, turvotus). m Väsymys (mukaan lukien voimattomuus, väsymys). | |||
Valikoitujen haittavaikutusten kuvaus
Hyperkolesterolemia/hypertriglyseridemia
Seerumin kolesterolipitoisuuden kohoamista raportoitiin haittavaikutuksena 79,0 %:lla potilaista ja triglyseridipitoisuuden kohoamista 67,5 %:lla potilaista. Näistä potilaista 59,8 %:lla ilmeni hyperkolesterolemiaa ja 47,2 %:lla hypertriglyseridemiaa lievänä tai kohtalaisena haittavaikutuksena (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Mediaaniaika hyperkolesterolemian ilmenemiseen oli 15 päivää (vaihteluväli: 1 − 1921 päivää) ja hypertriglyseridemian ilmenemiseen 16 päivää (vaihteluväli: 1 – 1921 päivää). Hyperkolesterolemian mediaanikesto oli 526 päivää ja hypertriglyseridemian 519 päivää.
Keskushermostovaikutukset
Keskushermostoon kohdistuneet haittavaikutukset olivat pääasiassa kognitiivisia vaikutuksia (27,4 %), mielialavaikutuksia (21,4 %), puhevaikeuksia (8,2 %) ja psykoottisia vaikutuksia (6,9 %). Nämä olivat yleensä lieviä, ohimeneviä ja itsestään korjaantuvia, kun lääkkeen antoa siirrettiin ja/tai annosta pienennettiin (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Yleisin minkä tahansa vaikeusasteen kognitiivinen vaikutus oli muistin heikkeneminen (10,8 %), ja yleisimmät vaikeusasteen 3 tai 4 reaktiot olivat sekavuustila (1,6 %) ja kognitiivinen häiriö (0,7 %). Yleisin minkä tahansa vaikeusasteen mielialavaikutus oli ahdistuneisuus (7,3 %), ja yleisimmät vaikeusasteen 3 tai 4 reaktiot olivat ärtyisyys (0,7 %), masennus (0,4 %), ahdistuneisuus, agitaatio ja tyypin 1 kaksisuuntainen mielialahäiriö (kukin 0,2 %). Yleisin minkä tahansa vaikeusasteen puhevaikeus oli dysartria (3,8 %), ja vaikeusasteen 3 tai 4 reaktioita olivat dysartria (0,4 %), puheen hitaus ja puhehäiriö (kukin 0,2 %). Yleisin minkä tahansa vaikeusasteen psykoottinen vaikutus oli aistiharha (2,7 %), ja yleisimmät vaikeusasteen 3 tai 4 reaktiot olivat kuuloharhat ja näköharhat, harhaluulot, akuutti psykoosi ja skitsofreniaryhmän häiriö (kukin 0,2 %). Mediaaniaika kognitiivisen vaikutuksen ilmenemiseen oli 129 päivää, mielialavaikutuksen ilmenemiseen 57 päivää, puhevaikeuden ilmenemiseen 58 päivää ja psykoottisen vaikutuksen ilmenemiseen 27 päivää. Näiden häiriöiden mediaanikestot olivat seuraavat: kognitiivinen vaikutus 270 päivää, mielialavaikutus 145 päivää, puhevaikeus 147 päivää ja psykoottinen vaikutus 84 päivää.
Hypertensio
Hypertensiota raportoitiin haittavaikutuksena 14,8 %:lla potilaista tutkimuksessa A, CROWN (B7461006) -tutkimuksessa ja tutkimuksessa B (B7461027). Näistä potilaista 8,8 %:lla haittavaikutus ilmeni lievänä tai kohtalaisena (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Mediaaniaika hypertension alkamiseen oli 295 päivää (vaihteluväli: 1 − 1990 päivää). Hypertension mediaanikesto oli 505 päivää.
Hyperglykemia
Hyperglykemiaa raportoitiin haittavaikutuksena 9,7 %:lla potilaista tutkimuksessa A, CROWN (B7461006) -tutkimuksessa ja tutkimuksessa B (B7461027). Näistä potilaista 6,0 %:lla haittavaikutus ilmeni lievänä tai kohtalaisena (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Mediaaniaika hyperglykemian alkamiseen oli 148 päivää (vaihteluväli: 1 – 1637 päivää). Hyperglykemian mediaanikesto oli 118 päivää.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin.
Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tämän lääkevalmisteen yliannostuksen hoito käsittää yleiset elintoimintoja tukevat toimenpiteet. Koska lääkkeellä on annoksesta riippuvainen vaikutus PR‑aikaan, EKG-seurantaa suositellaan. Lorlatinibille ei ole vastalääkettä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut syöpälääkkeet, proteiinikinaasin estäjät, ATC-koodi: L01ED05
Vaikutusmekanismi
Lorlatinibi on selektiivinen, adenosiinitrifosfaatin (ATP) kanssa kilpaileva ALK‑ ja c-ros-syöpägeeni 1:n (ROS1) tyrosiinikinaasien estäjä.
Prekliinisissä tutkimuksissa lorlatinibi esti mutatoitumattoman ALK-kinaasin ja kliinisesti merkittävien mutatoituneiden ALK‑kinaasien katalyyttistä aktiivisuutta yhdistelmä-DNA-tekniikkaan perustuvissa entsyymimäärityksissä ja solupohjaisissa määrityksissä. Lorlatinibilla osoitettiin huomattava kasvainta estävä vaikutus hiirissä, joiden kasvainksenografti ilmensi EML4 (echinoderm microtubule‑associated protein‑like 4) ‑geenin fuusioita ALK‑variantti 1:n (v1) kanssa, mukaan lukien ALK‑mutaatiot L1196M, G1269A, G1202R ja I1171T. Kaksi näistä ALK‑mutanteista, G1202R ja I1171T, aiheuttavat tunnetusti resistenssiä alektinibille, brigatinibille, seritinibille ja kritsotinibille. Lorlatinibi kykeni myös läpäisemään veri-aivoesteen. Lorlatinibilla osoitettiin aktiivisuutta hiirissä, joille oli implantoitu ortotooppinen EML4‑ALK‑ tai EML4‑ALKL1196M‑aivokasvain.
Kliininen teho
Aikaisemmin hoitamaton, ALK-positiivinen edennyt NSCLC (CROWN-tutkimus)
Lorlatinibin teho hoidettaessa potilaita, joilla oli ALK-positiivinen NSCLC ja jotka eivät olleet saaneet aikaisempaa systeemistä hoitoa metastasoituneeseen tautiin, osoitettiin avoimessa, satunnaistetussa, vaikuttavalla aineella kontrolloidussa monikeskustutkimuksessa B7461006 (CROWN-tutkimus). Tutkimukseen mukaan otetuilla potilailta edellytettiin ECOG (Eastern Cooperative Oncology Group) -suorituskykyluokkaa 0–2 ja VENTANA ALK (D5F3) CDx -määrityksellä tunnistettua ALK‑positiivista ei-pienisoluista keuhkosyöpää. Tutkimukseen soveltuivat neurologiselta tilaltaan vakaat potilaat, joilla oli hoidettuja tai hoitamattomia keskushermoston etäpesäkkeitä, mukaan lukien leptomeningeaalisia etäpesäkkeitä. Edellytyksenä oli, että potilaan sädehoito, mukaan lukien stereotaktinen tai osittainen aivojen sädehoito, oli päättynyt vähintään 2 viikkoa ennen satunnaistamista ja koko aivoalueen sädehoito vähintään 4 viikkoa ennen satunnaistamista.
Potilaat satunnaistettiin suhteessa 1:1 saamaan 100 mg lorlatinibia suun kautta kerran päivässä tai 250 mg kritsotinibia suun kautta kahdesti päivässä. Satunnaistaminen ositettiin etnisen alkuperän (aasialainen vs. muu kuin aasialainen) ja lähtötilanteen keskushermoston etäpesäkkeiden mukaan (kyllä vs. ei). Molemmissa hoitohaaroissa hoitoa jatkettiin, kunnes tauti eteni, tai ilmeni toksisuutta, joka ei ollut hyväksyttävissä. Tärkein tehon päätetapahtuma oli taudin etenemisestä vapaa elinaika (progression‑free survival, PFS), jonka arvioi sokkoutettu riippumaton keskitetty arviointitaho (Blinded Independent Central Review, BICR) kiinteille kasvaimille tarkoitettujen vasteen arviointikriteerien (Response Evaluation Criteria in Solid Tumours, RECIST) version 1.1 perusteella. Muita tehon päätetapahtumia olivat kokonaiselinaika (overall survival, OS), tutkijan arvioima PFS, PFS2 ja BICR:n arvioimat kasvaimeen liittyvät tiedot, mukaan lukien objektiivisen vasteen saaneiden osuus (objective response rate, ORR), vasteen kesto (duration of response, DOR) ja aika taudin intrakraniaaliseen etenemiseen (time to intracranial progression, IC-TTP). Muita tehon päätetapahtumia potilailla, joilla oli keskushermoston etäpesäkkeitä lähtötilanteessa, olivat intrakraniaalisen objektiivisen vasteen saaneiden osuus (IC-ORR) ja intrakraniaalisen vasteen kesto (IC-DOR), molemmat BICR:n arvioimana.
Kaikkiaan 296 potilasta satunnaistettiin saamaan lorlatinibia (n = 149) tai kritsotinibia (n = 147). Koko tutkimuspotilasjoukon demografiset ominaisuudet olivat seuraavat: iän mediaani 59 vuotta (vaihteluväli: 26–90 vuotta), ikä ≥ 65 vuotta (35 %), 59 % naisia, 49 % valkoihoisia, 44 % aasialaisia ja 0,3 % mustaihoisia. Suurimmalla osalla potilaista oli adenokarsinooma (95 %) eivätkä he olleet koskaan tupakoineet (59 %). BICR:n neuroradiologien määrityksen perusteella keskushermoston etäpesäkkeitä oli 26 %:lla potilaista (n = 78) ja näistä 30 potilaalla oli mitattavissa olevia keskushermoston leesioita.
Yhteenveto CROWN-tutkimuksen tuloksista on esitetty taulukossa 3. Tietojen keruun katkaisuhetkellä OS- ja PFS2-tiedot eivät olleet kypsiä.
Taulukko 3. CROWN-tutkimuksen yleiset tehoa koskevat tulokset
Tehon mittari | Lorlatinibi n = 149 | Kritsotinibi n = 147 | |
Seurannan mediaanikesto, kuukausia (95 %:n luottamusväli)a | 18 (16–20) | 15 (13–18) | |
Taudin etenemisestä vapaa elinaika (BICR:n arvio) | |||
| Potilaat, joilla tapahtuma; n (%) | 41 (28 %) | 86 (59 %) | |
| Taudin eteneminen, n (%) | 32 (22 %) | 82 (56 %) | |
| Kuolema, n (%) | 9 (6 %) | 4 (3 %) | |
| Mediaani, kuukausia (95 %:n luottamusväli)a | NE (NE–NE) | 9 (8–11) | |
| Riskitiheyksien suhde (95 %:n luottamusväli)b | 0,28 (0,19–0,41) | ||
| p-arvo* | < 0,0001 | ||
| Kokonaiselinaika | |||
| Potilaat, joilla tapahtuma; n (%) | 23 (15 %) | 28 (19 %) | |
| Mediaani, kuukausia (95 %:n luottamusväli)a | NE (NE–NE) | NE (NE–NE) | |
| Riskitiheyksien suhde (95 %:n luottamusväli)b | 0,72 (0,41–1,25) | ||
Taudin etenemisestä vapaa elinaika (tutkijan arvio) | |||
| Potilaat, joilla tapahtuma; n (%) | 40 (27 %) | 104 (71 %) | |
| Taudin eteneminen, n (%) | 34 (23 %) | 99 (67 %) | |
| Kuolema, n (%) | 6 (4 %) | 5 (3 %) | |
| Mediaani, kuukausia (95 %:n luottamusväli)a | NE (NE–NE) | 9 (7–11) | |
| Riskitiheyksien suhde (95 %:n luottamusväli)b | 0,21 (0,14–0,31) | ||
| p-arvo* | < 0,0001 | ||
| Kokonaisvaste (BICR:n arvio) | |||
| Kokonaisvasteen saaneiden osuus, n (%) | 113 (76 %) | 85 (58 %) | |
| (95 %:n luottamusväli)c | (68–83) | (49–66) | |
| Aika taudin intrakraniaaliseen etenemiseen | |||
| Mediaani, kuukausia (95 %:n luottamusväli)a | NE (NE–NE) | 16,6 (11–NE) | |
| Riskitiheyksien suhde (95 %:n luottamusväli)b | 0,07 (0,03–0,17) | ||
| Vasteen kesto | |||
| Vasteen saaneet | 113 | 85 | |
| Mediaani, kuukausia (95 %:n luottamusväli)a | NE (NE–NE) | 11 (9–13) | |
| Intrakraniaalinen kokonaisvaste potilailla, joilla oli lähtötilanteessa mitattavissa olevia keskushermoston leesioita | n = 17 | n = 13 | |
| Intrakraniaalisen vasteen saaneiden osuus, n (%) | 14 (82 %) | 3 (23 %) | |
| (95 %:n luottamusväli)c | (57–96) | (5–54) | |
| Täydellisen vasteen saaneiden osuus | 71 % | 8 % | |
| Vasteen kesto | |||
| Vasteen saaneet | 14 | 3 | |
| Mediaani, kuukausia (95 %:n luottamusväli)a | NE (NE–NE) | 10 (9–11) | |
| Intrakraniaalinen kokonaisvaste potilailla, joilla oli lähtötilanteessa mitattavissa tai ei-mitattavissa olevia keskushermoston leesioita | n = 38 | n = 40 | |
| Intrakraniaalisen vasteen saaneiden osuus, n (%) | 25 (66 %) | 8 (20 %) | |
| (95 %:n luottamusväli)c | (49–80) | (9–36) | |
| Täydellisen vasteen saaneiden osuus | 61 % | 15 % | |
| Vasteen kesto | |||
| Vasteen saaneet | 25 | 8 | |
| Mediaani, kuukausia (95 %:n luottamusväli)a | NE (NE–NE) | 9 (6–11) | |
Lyhenteet: BICR = sokkoutettu riippumaton keskitetty arviointitaho, n = potilaiden lukumäärä, NE = ei arvioitavissa.
* p‑arvo perustuu 1-tahoiseen ositettuun log-rank-testiin.
a Perustuu Brookmeyer-Crowleyn menetelmään.
b Riskitiheyksien suhde perustuu Coxin suhteellisen vaaran regressiomalliin; suhteellisten riskien osalta riskitiheyksien suhde < 1 osoittaa riskitiheyksien suhteen alenemisen lorlatinibin eduksi.
c Käytettäessä binomijakaumaan perustuvaa eksaktia menetelmää.
Kuva 1. Taudin etenemisestä vapaata elinaikaa kuvaavat Kaplan-Meierin käyrätCROWN-tutkimuksessa (BICR:n arvio)
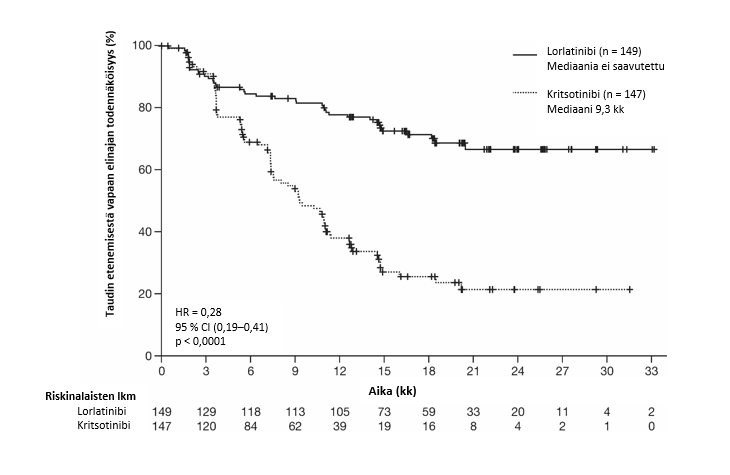
Lyhenteet: BICR = sokkoutettu riippumaton keskitetty arviointitaho, HR = riskitiheyksien suhde, CI = luottamusväli, n = potilaiden lukumäärä.
Lorlatinibihoidosta saatava hyöty oli samankaltainen eri alaryhmissä riippumatta potilaiden ja sairauden ominaisuuksista lähtötilanteessa, mukaan lukien potilaat, joilla oli lähtötilanteessa keskushermoston etäpesäkkeitä (n = 38, HR = 0,2, 95 %:n luottamusväli: 0,10–0,43) ja joilla ei ollut lähtötilanteessa keskushermoston etäpesäkkeitä (n = 111, HR = 0,32, 95 %:n luottamusväli: 0,20–0,49).
ALK-positiivinen edennyt NSCLC, jota on aikaisemmin hoidettu ALK-kinaasin estäjällä
Vaiheen 1/2 yksihaaraisessa monikeskustutkimuksessa A ja vaiheen 4 yksihaaraisessa monikeskustutkimuksessa B selvitettiin lorlatinibin käyttöä ALK‑positiivisessa edenneessä NSCLC:ssä, jota oli aiemmin hoidettu vähintään yhdellä toisen sukupolven ALK‑tyrosiinikinaasin estäjällä. Tutkimuksessa A vaiheeseen 2 otettiin mukaan yhteensä 139 potilasta, joilla oli ALK‑positiivinen edennyt NSCLC. Potilaiden tautia oli aiemmin hoidettu vähintään yhdellä toisen sukupolven ALK‑tyrosiinikinaasin estäjällä. Tutkimukseen B otettiin mukaan yhteensä 71 potilasta, joilla oli ALK-positiivinen edennyt NSCLC. Potilaiden tautia oli hoidettu aiemmin yhdellä ALK-tyrosiinikinaasin estäjällä (alektinibi tai seritinibi). Kummassakin tutkimuksessa potilaat saivat lorlatinibia jatkuvana hoitona suositusannoksella 100 mg suun kautta kerran vuorokaudessa.
Tutkimuksessa A tehon ensisijainen päätetapahtuma tutkimuksen vaiheessa 2 oli objektiivisen vasteen saaneiden osuus (ORR), mukaan lukien intrakraniaalinen ORR (IC-ORR). Vasteen arvioi riippumaton keskitetty arviointitaho (Independent Central Review, ICR) modifioitujen RECIST-kriteerien, versio 1.1, mukaisesti. Toissijaisia päätetapahtumia olivat vasteen kesto (DOR), intrakraniaalinen DOR (IC-DOR), aika kasvainvasteeseen (time‑to‑tumour response, TTR) ja taudin etenemisestä vapaa elinaika (PFS). Tutkimuksessa B tehon ensisijainen päätetapahtuma oli objektiivisen vasteen saaneiden osuus (ORR), jonka arvioi riippumaton keskitetty arviointitaho RECIST-kriteerien, versio 1.1, mukaisesti. Toissijaisia päätetapahtumia olivat intrakraniaalinen ORR (IC-ORR), vasteen kesto (DOR), intrakraniaalinen DOR (IC-DOR), aika kasvainvasteeseen (time‑to‑tumour response, TTR), aika taudin etenemiseen (time‑to‑tumour progression, TTP) ja taudin etenemisestä vapaa elinaika (PFS).
Seuraavassa on listattu demografisia tietoja 139 potilaasta, joilla oli ALK‑positiivinen edennyt NSCLC ja joita oli aiemmin hoidettu tutkimuksessa A vähintään yhdellä toisen sukupolven ALK‑tyrosiinikinaasin estäjällä: 56 % oli naisia, 48 % valkoihoisia, 38 % aasialaisia ja iän mediaani oli 53 vuotta (vaihteluväli: 29–83 vuotta), 16 % potilaista oli ≥ 65‑vuotiaita. Lähtötilanteen ECOG‑suorituskykyluokka oli 96 %:lla potilaista 0 tai 1. Aivojen etäpesäkkeitä oli lähtötilanteessa 67 %:lla potilaista. Tutkituista 139 potilaasta 20 % oli saanut aiemmin yhtä ALK‑tyrosiinikinaasin estäjää (pois lukien kritsotinibi), 47 % kahta ALK‑tyrosiinikinaasin estäjää ja 33 % vähintään kolmea ALK‑tyrosiinikinaasin estäjää.
Seuraavassa on listattu demografisia tietoja 71 potilaasta, jotka osallistuivat tutkimukseen B ja joilla oli ALK‑positiivinen edennyt NSCLC, joka oli edennyt yhden aiemman ALK‑tyrosiinikinaasin estäjällä (alektinibi tai seritinibi) yhdessä solunsalpaajahoidon kanssa tai ilman solunsalpaajahoitoa annetun hoidon jälkeen: 42 % oli naisia, 76 % valkoihoisia, 21 % aasialaisia ja iän mediaani oli 59 vuotta (vaihteluväli: 26–87 vuotta), 32 % potilaista oli ≥ 65‑vuotiaita. Lähtötilanteen ECOG‑suorituskykyluokka oli 52 %:lla potilaista 0 ja 48 %:lla potilaista 1. Aivojen etäpesäkkeitä oli lähtötilanteessa 42 %:lla potilaista. Tutkituista 71 potilaasta 85 % oli saanut aiempana ALK-tyrosiinikinaasin estäjänä alektinibia ja 15 % seritinibiä.
Taulukoissa 4 ja 5 on esitetty tutkimuksen A ja tutkimuksen B tehoa koskevat päätulokset.
Taulukko 4. Tutkimuksen A ja tutkimuksen B yleiset tehoa koskevat tulokset aiemman hoidon mukaan
| Tehon mittari | Yksi aiempi ALK-TKIa; aikaisempi solunsalpaajahoito oli mahdollinen (n = 99)b | Vähintään kaksi aiempaa ALK-TKI:ta; aikaisempi solunsalpaajahoito oli mahdollinen (n = 111)c |
Objektiivisen vasteen saaneiden osuusd (95 %:n luottamusväli) Täydellinen vaste, n Osittainen vaste, n | 42,4 % (32,5–52,8) 5 37 | 39,6 % (30,5–49,4) 2 42 |
Vasteen kesto Mediaani, kuukausia (95 %:n luottamusväli) | NE (7,8–NE) | 9,9 (5,7–24,4) |
Taudin etenemisestä vapaa elinaika Mediaani, kuukausia (95 %:n luottamusväli) | 8,3 (6,3–16,5) | 6,9 (5,4–9,5) |
Lyhenteet: ALK = anaplastinen lymfoomakinaasi, n = potilaiden lukumäärä, NE = ei arvioitavissa, TKI = tyrosiinikinaasin estäjä.
a Alektinibi, brigatinibi tai seritinibi.
b Tutkimusten A ja B yhdistetyt tehon tulokset
c Vain tutkimuksen A tehon tulokset
d Riippumattoman keskitetyn arviointitahon (ICR) mukaan.
Taulukko 5. Tutkimuksen A ja tutkimuksen B intrakraniaalista* tehoa koskevat tulokset aiemman hoidon mukaan
| Tehon mittari | Yksi aiempi ALK-TKIa; aikaisempi solunsalpaajahoito oli mahdollinen (n = 19)b | Vähintään kaksi aiempaa ALK-TKI:ta; aikaisempi solunsalpaajahoito oli mahdollinen (n = 48)c |
Objektiivisen vasteen saaneiden osuusd (95 %:n luottamusväli) Täydellinen vaste, n Osittainen vaste, n | 63,2 % (38,4–83,7) 4 8 | 52,1 % (37,2–66,7) 10 15 |
Intrakraniaalisen vasteen kesto Mediaani, kuukausia (95 %:n luottamusväli) | NE (4,2–NE) | 12,4 (6,0–NE) |
Lyhenteet: ALK = anaplastinen lymfoomakinaasi, n = potilaiden lukumäärä, NE = ei arvioitavissa, TKI = tyrosiinikinaasin estäjä.
* Potilaat, joilla oli lähtötilanteessa vähintään yksi mitattavissa oleva aivojen etäpesäke.
a Alektinibi, brigatinibi tai seritinibi.
b Tutkimusten A ja B yhdistetyt tehon tulokset
c Vain tutkimuksen A tehon tulokset
d Riippumattoman keskitetyn arviointitahon (ICR) mukaan.
Tehon arvioinnissa oli mukana yhteensä 210 potilasta. Niillä 86 potilaalla, joilla ICR vahvisti objektiivisen vasteen, TTR:n mediaani oli 1,4 kuukautta (vaihteluväli: 1,2–16,6 kuukautta). ORR oli aasialaisilla 48,5 % (95 %:n luottamusväli: 36,2–61,0) ja muilla kuin aasialaisilla 35,7 % (95 %:n luottamusväli: 27,4–44,6). Niillä 37 potilaalla, joilla ICR vahvisti objektiivisen intrakraniaalisen kasvainvasteen ja lähtötilanteessa vähintään yhden mitattavissa olevan aivojen etäpesäkkeen, IC-TTR:n mediaani oli 1,4 kuukautta (vaihteluväli: 1,2–16,2 kuukautta). IC-ORR oli aasialaisilla 58,3 % (95 %:n luottamusväli: 36,6–77,9) ja muilla kuin aasialaisilla 47,2 % (95 %:n luottamusväli: 30,4−64,5).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset lorlatinibin käytöstä keuhkosyövän (pienisoluisen ja ei-pienisoluisen keuhkosyövän) hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Lorlatinibin huippupitoisuus plasmassa saavutetaan nopeasti: Tmax‑arvon mediaani on 1,2 tuntia 100 mg:n kerta-annoksen jälkeen ja 2,0 tuntia annettaessa toistuvasti 100 mg kerran vuorokaudessa.
Suun kautta annettujen lorlatinibitablettien keskimääräinen absoluuttinen biologinen hyötyosuus on 80,8 % (90 %:n luottamusväli: 75,7–86,2) verrattuna laskimonsisäiseen antoon.
Lorlatinibin anto runsaasti rasvaa ja kaloreja sisältävän aterian yhteydessä suurensi altistusta 5 % verrattuna paastotilaan. Lorlatinibin voi ottaa joko ruokailun yhteydessä tai tyhjään mahaan.
Kun syöpäpotilaille annettiin 100 mg lorlatinibia kerran vuorokaudessa, plasman huippupitoisuuden geometrinen keskiarvo (% variaatiokerroin [CV]) oli 577 (42) ng/ml ja AUC24 oli 5 650 (39) ng·h/ml. Oraalisen puhdistuman geometrinen keskiarvo (% CV) oli 17,7 (39) l/h.
Jakautuminen
Lorlatinibi sitoutuu in vitro ihmisen plasman proteiineihin 66 %; kohtalainen määrä sitoutuu albumiiniin tai hapan alfa‑1‑glykoproteiiniin.
Biotransformaatio
Lorlatinibin ensisijaiset metaboliareitit ihmisessä ovat oksidaatio ja glukuronidaatio. In vitro ‑tiedot osoittavat, että lorlatinibi metaboloituu ensisijaisesti CYP3A4:n ja UGT1A4:n välityksellä ja vähäisessä määrin myös CYP2C8:n, CYP2C19:n, CYP3A5:n ja UGT1A3:n välityksellä.
Lorlatinibin päämetaboliitiksi plasmassa on todettu bentsoehappometaboliitti, jota syntyy lorlatinibin amidi‑ ja aromaattisten eetterisidosten oksidatiivisessa pilkkoutumisessa; tämä metaboliitti vastaa 21 % verenkierrosta mitatusta radioaktiivisuudesta. Tämä oksidatiivisesta pilkkoutumisesta muodostuva metaboliitti on farmakologisesti inaktiivinen.
Eliminaatio
Lorlatinibin puoliintumisaika plasmassa oli 23,6 tuntia 100 mg:n kerta-annoksen jälkeen. Lorlatinibin arvioitu efektiivinen puoliintumisaika plasmassa vakaassa tilassa autoinduktion päättymisen jälkeen oli 14,83 tuntia. Kun suun kautta annettiin radioaktiivisesti leimattu 100 mg:n lorlatinibiannos, keskimäärin 47,7 % radioaktiivisuudesta mitattiin virtsasta ja 40,9 % ulosteista; mitattu kokonaisradioaktiivisuus oli kaiken kaikkiaan keskimäärin 88,6 %.
Pääkomponentti ihmisen plasmassa ja ulosteissa oli muuttumaton lorlatinibi: sitä oli kokonaisradioaktiivisuudesta 44 % plasmassa ja 9,1 % ulosteissa. Alle 1 % muuttumattomasta lorlatinibista mitattiin virtsasta.
Lisäksi lorlatinibi on ihmisen pregnaani X ‑reseptorin (PXR) ja ihmisen konstitutiivisen androstaanireseptorin (CAR) induktori.
Lineaarisuus/ei-lineaarisuus
Kerta-annoksena annetun lorlatinibin systeeminen altistus (AUCinf ja Cmax) suureni annoksesta riippuvaisesti annosvälillä 10–200 mg. Vain vähän tietoa on saatavilla annosvälistä 10–200 mg; AUCinf‑ ja Cmax‑arvoissa ei kuitenkaan todettu poikkeamia lineaarisuudesta kerta-annoksen jälkeen.
Toistuvassa kerran päivässä annostelussa lorlatinibin Cmax-arvo suureni suhteessa annokseen ja myös AUCtau-arvo suureni, mutta suhteessa hieman vähemmän kuin annos, annosvälillä 10–200 mg kerran päivässä.
Lisäksi vakaan tilan lorlatinibialtistus plasmassa on pienempi kuin kerta-annoksen farmakokinetiikan perusteella olisi odotettavissa, mikä viittaa ajasta riippuvaiseen autoinduktion nettovaikutukseen.
Maksan vajaatoiminta
Koska lorlatinibi metaboloituu maksassa, maksan vajaatoiminta todennäköisesti suurentaa plasman lorlatinibipitoisuutta. Tehdyistä kliinisistä tutkimuksista suljettiin pois potilaat, joiden ASAT tai ALAT oli > 2,5 × ULN, tai jos syynä oli perussairautena oleva syöpäsairaus, > 5,0 × ULN, tai kokonaisbilirubiini > 1,5 × ULN. Populaatiofarmakokineettiset analyysit ovat osoittaneet, että lorlatinibialtistus ei muuttunut kliinisesti merkittävästi potilailla, joilla oli lievä maksan vajaatoiminta (n = 53). Maksan vajaatoimintaa koskeneessa tutkimuksessa suun kautta annetun 100 mg:n lorlatinibikerta-annoksen jälkeen lorlatinibin AUCinf-arvo suureni 15 % potilailla, joilla oli kohtalainen maksan vajaatoiminta (Child–Pugh B), ja 82 % potilailla, joilla oli vaikea maksan vajaatoiminta (Child–Pugh C), verrattuna tutkittaviin, joiden maksan toiminta oli normaali.
Annosmuutoksia ei suositella potilaille, joilla on lievä tai kohtalainen maksan vajaatoiminta. Potilaille, joilla on vaikea maksan vajaatoiminta, lorlatinibin aloitusannosta suositellaan pienentämään 50 mg:aan suun kautta kerran vuorokaudessa (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Alle 1 % annetusta annoksesta todetaan virtsasta muuttumattomana lorlatinibina. Populaatiofarmakokineettiset analyysit ovat osoittaneet, että plasman vakaan tilan lorlatinibialtistus ja Cmax-arvot suurenevat hieman, jos munuaisten toiminta lähtötilanteessa on heikentynyt. Munuaisten vajaatoimintaa koskevan tutkimuksen perusteella aloitusannoksen muutoksia ei suositella potilaille, joilla on lievä tai kohtalainen munuaisten vajaatoiminta (absoluuttinen eGFR ≥ 30 ml/min; MDRD [Modification of Diet in Renal Disease Study] -kaavalla laskettu eGFR [ml/min/1,73 m2] × mitattu kehon pinta-ala/1,73). Tässä tutkimuksessa lorlatinibin AUCinf -arvo suureni vaikeaa munuaisten vajaatoimintaa sairastavilla tutkittavilla (absoluuttinen eGFR < 30 ml/min) 41 % verrattuna tutkittaviin, joiden munuaisten toiminta oli normaali (absoluuttinen eGFR ≥ 90 ml/min). Potilaille, joilla on vaikea munuaisten vajaatoiminta, suositellaan pienennettyä lorlatinibiannosta, esim. aloitusannosta 75 mg suun kautta kerran vuorokaudessa (ks. kohta Annostus ja antotapa). Tietoja ei ole saatavilla munuaisdialyysihoitoa tarvitsevista potilaista.
Ikä, sukupuoli, etninen tausta, kehonpaino ja fenotyyppi
Populaatiofarmakokineettiset analyysit edennyttä NSCLC:tä sairastavista potilaista ja terveistä vapaaehtoisista tutkittavista osoittavat, että ikä, sukupuoli, etninen tausta, kehonpaino ja fenotyyppi CYP3A5:n ja CYP2C19:n suhteen eivät vaikuta kliinisesti merkittävästi.
Sydämen elektrofysiologia
Tutkimuksessa A Fridericia-kaavalla korjattu absoluuttinen QTc-aika (QTcF) oli 2 potilaalla (0,7 %) > 500 ms ja 5 potilaalla (1,8 %) QTcF muuttui lähtötilanteesta > 60 ms.
Lisäksi kaksisuuntaisessa vaihtovuoroisessa (crossover) tutkimuksessa, johon osallistui 16 tervettä vapaaehtoista tutkittavaa, arvioitiin suun kautta otetun lorlatinibin kerta-annoksen (50 mg, 75 mg ja 100 mg) vaikutusta sekä yhdessä kerran vuorokaudessa otetun 200 mg:n itrakonatsoliannoksen kanssa että ilman itrakonatsoliannosta. Tutkituilla keskimääräisillä lorlatinibipitoisuuksilla ei todettu keskimääräisen QTc-ajan pitenemistä.
Tutkimuksessa A rekisteröitiin EKG 295 potilaasta, jotka saivat lorlatinibin suositusannosta 100 mg kerran vuorokaudessa. Tutkimuspopulaatiosta oli suljettu pois potilaat, joiden QTc-aika oli > 470 ms. Tutkimuspopulaatiossa PR-ajan suurin keskimääräinen muutos lähtötilanteesta oli 16,4 ms (kaksisuuntaisen 90 %:n luottamusvälin yläraja 19,4 ms) (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Näistä potilaista 7:llä PR-aika oli lähtötilanteessa > 200 ms. Niistä 284 potilaasta, joiden PR-aika oli < 200 ms, 14 %:lla PR-aika piteni ≥ 200 ms:iin lorlatinibihoidon aloittamisen jälkeen. PR-ajan piteneminen tapahtui pitoisuudesta riippuvaisesti. Eteis-kammiokatkos ilmeni 1,0 %:lla potilaista.
Annosta voidaan joutua muuttamaan potilaille, joiden PR‑aika pitenee (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Toistuvan annon toksisuus
Havaitut merkittävimmät toksisuudet olivat tulehdus monissa kudoksissa (rotilla iho ja kohdunkaula ja koirilla keuhkot, henkitorvi, iho, imusolmukkeet ja/tai suuontelo, mukaan lukien alaleukaluu; tähän liittyi valkosolumäärän, fibrinogeeni‑ ja/tai globuliinipitoisuuksien kohoamista ja albumiinipitoisuuden pienenemistä) sekä muutokset haimassa (tähän liittyi amylaasi‑ ja lipaasipitoisuuksien kohoamista), hepatobiliaarisessa järjestelmässä (tähän liittyi maksaentsyymipitoisuuksien kohoamista), urosten lisääntymiselimissä, verenkiertojärjestelmässä, munuaisissa ja maha-suolikanavassa, ääreis‑ ja keskushermostossa (kognitiivisen toiminnan mahdollinen heikkeneminen) annoksella, joka vastaa ihmisen kliinistä altistusta suositusannostuksella. Eläimillä havaittiin myös verenpaineen ja syketiheyden sekä QRS‑kompleksin ja PR‑ajan muutoksia akuutin annostelun jälkeen (altistus Cmax‑arvon perusteella noin 2,6‑kertainen verrattuna kliiniseen altistukseen ihmisellä 100 mg:n kerta-annoksen jälkeen). Kaikki kohde-elinten löydökset korjaantuivat joko osittain tai täysin, lukuun ottamatta maksan sapenjohtimen hyperplasiaa.
Genotoksisuus
Lorlatinibi ei ole mutageeninen, mutta se on aneugeeninen in vitro ja in vivo. Korkein annostaso (NOAEL), joka ei aiheuta aneuploidiaa, on AUC‑arvon perusteella noin 16,5 kertaa suurempi kuin 100 mg:lla saavutettava kliininen altistus ihmisellä.
Karsinogeenisuus
Lorlatinibilla ei ole tehty karsinogeenisuustutkimuksia.
Lisääntymistoksisuus
Rotalla ja koiralla on havaittu siementiehyiden rappeutumista ja/tai kivesten surkastumista sekä lisäkivesten muutoksia (tulehdus ja/tai vakuolisaatio). Koiralla havaittiin eturauhasessa minimaalista tai lievää rauhassurkastumaa annoksella, joka vastaa ihmisen kliinistä altistusta suositusannostuksella. Urosten lisääntymiselimiin kohdistuneet vaikutukset korjaantuivat joko osittain tai täysin.
Rotilla ja kaniineilla tehdyissä alkio‑ ja sikiötoksisuutta selvittäneissä tutkimuksissa havaittiin lisääntynyttä alkiokuolleisuutta, sikiöpainon pienenemistä ja epämuodostumia. Sikiöiden morfologisia poikkeavuuksia olivat vääntyneet raajat, ylimääräiset varpaat, vatsahalkio, epämuodostuneet munuaiset, kupumainen pää, korkea suulaki ja aivokammioiden laajentuma. Pienin annos, jolle altistuminen aiheutti eläimille alkio‑ ja sikiövaikutuksia, vastasi AUC‑arvon perusteella 100 mg:lla saavutettavaa kliinistä altistusta ihmisellä.
Farmaseuttiset tiedot
Apuaineet
Tablettiydin
Mikrokiteinen selluloosa
Kalsiumvetyfosfaatti
Natriumtärkkelysglykolaatti
Magnesiumstearaatti
Kalvopäällyste
Hypromelloosi
Laktoosimonohydraatti
Makrogoli
Triasetiini
Titaanidioksidi (E171)
Musta rautaoksidi (E172)
Punainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LORVIQUA tabletti, kalvopäällysteinen
25 mg (L:kyllä) 90 fol (4258,60 €)
100 mg (L:kyllä) 30 fol (4972,25 €)
PF-selosteen tieto
OPA/Al/PVC-läpipainopakkaukset, joissa on alumiinifoliotausta. Läpipainolevyssä on 10 kalvopäällysteistä tablettia.
Lorviqua 25 mg kalvopäällysteiset tabletit
Yksi pakkaus sisältää 90 kalvopäällysteistä tablettia (9 läpipainolevyä).
Lorviqua 100 mg kalvopäällysteiset tabletit
Yksi pakkaus sisältää 30 kalvopäällysteistä tablettia (3 läpipainolevyä).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Lorviqua 25 mg kalvopäällysteiset tabletit
Pyöreä (8 mm), haalean vaaleanpunainen, välittömästi vapauttava kalvopäällysteinen tabletti, jonka toiselle puolelle on kaiverrettu ”Pfizer” ja toiselle puolelle ”25” ja ”LLN”.
Lorviqua 100 mg kalvopäällysteiset tabletit
Soikea (8,5 × 17 mm), vaaleanpunainen, välittömästi vapauttava kalvopäällysteinen tabletti, jonka toiselle puolelle on kaiverrettu ”Pfizer” ja toiselle puolelle ”LLN 100”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
LORVIQUA tabletti, kalvopäällysteinen
25 mg 90 fol
100 mg 30 fol
- Ylempi erityiskorvaus (100 %). Lorlatinibi: ALK-positiivisen ei-pienisoluisen keuhkosyövän hoito erityisin edellytyksin (1527).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Lorlatinibi: ALK-positiivisen ei-pienisoluisen keuhkosyövän hoito erityisin edellytyksin (3026).
ATC-koodi
L01ED05
Valmisteyhteenvedon muuttamispäivämäärä
12.06.2026
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com