
UPTRAVI tabletti, kalvopäällysteinen 200 mikrog, 400 mikrog, 600 mikrog, 800 mikrog, 1000 mikrog, 1200 mikrog, 1400 mikrog, 1600 mikrog
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Terveydenhuollon ammattilainen
Vaikuttavat aineet ja niiden määrät
Uptravi 100 mikrogrammaa kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 100 mikrogrammaa seleksipagia.
Uptravi 200 mikrogrammaa kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 200 mikrogrammaa seleksipagia.
Uptravi 400 mikrogrammaa kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 400 mikrogrammaa seleksipagia.
Uptravi 600 mikrogrammaa kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 600 mikrogrammaa seleksipagia.
Uptravi 800 mikrogrammaa kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 800 mikrogrammaa seleksipagia.
Uptravi 1 000 mikrogrammaa kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 1 000 mikrogrammaa seleksipagia.
Uptravi 1 200 mikrogrammaa kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 1 200 mikrogrammaa seleksipagia.
Uptravi 1 400 mikrogrammaa kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 1 400 mikrogrammaa seleksipagia.
Uptravi 1 600 mikrogrammaa kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 1 600 mikrogrammaa seleksipagia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti
Kliiniset tiedot
Käyttöaiheet
Uptravi on tarkoitettu WHO:n toimintakykyluokkiin II-III kuuluvien aikuispotilaiden keuhkovaltimoiden verenpainetaudin (pulmonaaliarteriahypertensio, PAH) pitkäaikaishoitoon joko yhdistelmähoidossa potilaille, joiden tauti ei ole riittävän hyvin hallinnassa endoteliinireseptoriantagonisti- ja/tai fosfodiesteraasi 5:n (PDE-5:n) estäjä -hoidolla, tai monoterapiana potilaille, joille nämä hoidot eivät sovellu.
Teho on osoitettu PAH-populaatiossa, jonka potilailla oli idiopaattinen tai perinnöllinen PAH, sidekudossairauksiin liittyvä PAH tai korjattuun yksinkertaiseen synnynnäiseen sydänvikaan liittyvä PAH (ks. kohta Farmakodynamiikka).
Ehto
Hoito tulee aloittaa vain pulmonaalihypertension hoitoon perehtyneen lääkärin määräyksestä ja toteuttaa hänen valvonnassaan.
Annostus ja antotapa
Hoito on aloitettava keuhkovaltimoiden verenpainetaudin hoitoon perehtyneen lääkärin määräyksestä ja valvonnassa.
Annostus
Yksilöllinen annostitraus
Kunkin potilaan annos titrataan suurimpaan yksilöllisesti siedettyyn annokseen, joka vaihtelee 200 mikrogrammasta kahdesti vuorokaudessa 1 600 mikrogrammaan kahdesti vuorokaudessa (yksilöllinen ylläpitoannos).
Suositeltava aloitusannos on 200 mikrogrammaa (mikrog.) kahdesti vuorokaudessa (x 2) noin 12 tunnin välein. Annosta suurennetaan 200 mikrog. x 2 kerrallaan, yleensä viikon välein. Hoidon alussa ja kunkin annoksen suurentamisvaiheen alussa on suositeltavaa ottaa ensimmäinen annos illalla. Annostitrauksen aikana voi esiintyä tiettyjä haittavaikutuksia, jotka liittyvät seleksipagin vaikutustapaan (esim. päänsärkyä, ripulia, pahoinvointia ja oksentelua, leukakipua, lihaskipua, ääreisosien kipua, nivelkipua ja kuumia aaltoja). Ne ovat yleensä ohimeneviä tai hoidettavissa oireenmukaisesti (ks. kohta Haittavaikutukset). Jos potilas ei siedä saavutettua annostasoa, annosta on pienennettävä edelliselle annostasolle.
Jos annoksen suurentamista rajoitti jokin muu tekijä kuin seleksipagin vaikutustapaan liittyvä haittavaikutus, voidaan harkita uutta yritystä jatkaa annoksen suurentamista suurimpaan yksilöllisesti siedettyyn annokseen (enimmäisannos on 1 600 mikrog. x 2).
Yksilöllinen ylläpitoannos
Hoitoa ylläpidetään suurimmalla siedetyllä annoksella, joka on saavutettu annostitrauksen aikana. Jos tietyllä annoksella toteutetun hoidon siedettävyys huononee ajan mittaan, on harkittava oireenmukaista hoitoa ja/tai annoksen pienentämistä yhden annostason verran.
Hoidon keskeytyminen ja lopettaminen
Jos annos jää väliin, se on otettava mahdollisimman pian. Väliin jäänyttä annosta ei saa ottaa, jos seuraavan annoksen ottoajankohta on noin 6 tunnin kuluessa.
Jos hoito keskeytyy vähintään 3 päivän ajaksi, Uptravi-valmisteen käyttö on aloitettava uudelleen pienemmällä annoksella, minkä jälkeen annos titrataan suuremmaksi.
Seleksipagin äkillisestä lopettamisesta keuhkovaltimoiden verenpainetautia sairastavilla potilailla on niukasti kokemusta. Näyttöä akuutista rebound-ilmiöstä ei ole havaittu.
Jos Uptravi-hoito kuitenkin päätetään lopettaa, hoidon lopetuksen on tapahduttava vähitellen samalla, kun toinen hoitovaihtoehto otetaan käyttöön.
Annoksen muuttaminen annettaessa samanaikaisesti keskivahvojen CYP2C8:n estäjien kanssa
Annettaessa Uptravi-valmistetta samanaikaisesti keskivahvojen CYP2C8:n estäjien kanssa (esim. klopidogreeli, deferasiroksi ja teriflunomidi) Uptravi-valmisteen kokonaisvuorokausiannos pitää pienentää puoleen antamalla puolikas kerta-annos kaksi kertaa vuorokaudessa. Vaihtoehtoisesti, puolikkaan vuorokausiannoksen saavuttamiseksi, Uptravi-hoitoa voidaan jatkaa antotiheydellä kerran vuorokaudessa, jos potilas on sillä jo hyvässä hoitotasapainossa, tai potilaille, joille ei ole saatavissa sopivaa annosvahvuutta, joka tukisi puolikkaan annoksen annostelua kaksi kertaa vuorokaudessa. Jos hoitoa ei siedetä tietyllä annoksella, on harkittava oireenmukaista hoitoa ja/tai annoksen pienentämistä seuraavaksi pienempään annokseen. Kun keskivahvan CYP2C8:n estäjän samanaikainen käyttö loppuu, Uptravi-valmisteen kokonaisvuorokausiannosta pitää suurentaa tarpeen mukaan. Maksimiannosta 1 600 mikrogrammaa kahdesti vuorokaudessa ei saa ylittää (ks. kohta Yhteisvaikutukset).
Erityiset potilasryhmät
Iäkkäät potilaat (≥ 65 v)
Annosta ei tarvitse muuttaa iäkkäille (ks. kohta Farmakokinetiikka). Yli 75-vuotiaiden potilaiden hoidosta on niukasti kliinistä kokemusta, joten tässä potilasryhmässä Uptravi-valmisteen käytössä on noudatettava varovaisuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksan vajaatoiminta
Uptravi-valmistetta ei saa antaa potilaille, joilla on vaikea maksan vajaatoiminta (Child‑Pugh‑luokka C; ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos potilaalla on keskivaikea maksan vajaatoiminta (Child‑Pugh‑luokka B), hoidon aloitusannoksen on oltava 100 mikrog. x 2 vuorokaudessa, ja annosta on suurennettava viikon välein 100 mikrog. x 2 vuorokaudessa, kunnes ilmenee seleksipagin vaikutustapaan liittyviä haittavaikutuksia, jotka ovat sietämättömiä tai joita ei pystytä hoitamaan lääketieteellisesti. Näille potilaille maksimiannos on 800 mikrog. x 2 vuorokaudessa. Vaihtoehtoisesti, puolikkaan vuorokausiannoksen saavuttamiseksi, Uptravi-hoitoa voidaan jatkaa antotiheydellä kerran vuorokaudessa, jos potilas on sillä jo hyvässä hoitotasapainossa, tai potilaille, joille ei ole saatavissa sopivaa annosvahvuutta, joka tukisi puolikkaan annoksen annostelua kaksi kertaa vuorokaudessa. Annosta ei tarvitse muuttaa, jos potilaalla on lievä maksan vajaatoiminta (Child‑Pugh‑luokka A).
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta. Aloitusannosta ei tarvitse muuttaa, jos potilaalla on vaikea munuaisten vajaatoiminta (arvioitu glomerulusten suodatusnopeus [eGFR] < 30 ml/min/1,73 m2); näiden potilaiden kohdalla annostitrauksessa on noudatettava varovaisuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Seleksipagin turvallisuutta ja tehoa 2 – < 18 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Saatavissa olevan väliaikatiedon perusteella, joka on kuvattu kohdissa Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta. Seleksipagin antoa pediatrisille potilaille ei suositella.
Seleksipagin turvallisuutta ja tehoa alle 2 vuoden ikäisten lasten hoidossa ei ole tutkittu, sillä eläintutkimukset viittaavat lisääntyneeseen suolentuppeuman riskiin. Näiden löydösten kliinistä merkitystä ei tunneta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Antotapa
Suun kautta.
Kalvopäällysteiset tabletit otetaan suun kautta aamuisin ja iltaisin. Siedettävyyden parantamiseksi on suositeltavaa, että Uptravi otetaan ruoan kanssa ja että annosta suurennettaessa ensimmäinen suurennettu annos otetaan aina illalla.
Kalvopäällysteiset tabletit on tarkoitettu nieltäväksi veden kera. Tabletteja ei saa jakaa eikä murskata, koska tabletin kalvopäällyste suojaa vaikuttavaa ainetta valolta.
Heikkonäköisiä ja sokeita potilaita on kehotettava pyytämään toisen henkilön apua Uptravi-valmisteen annostitrauksen aikana.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Vaikea sepelvaltimotauti tai epästabiili angina pectoris.
- Sydäninfarkti edeltävien 6 kuukauden aikana.
- Kompensoitumaton sydämen vajaatoiminta, ellei potilas ole lääkärin tiiviissä seurannassa.
- Vaikeat rytmihäiriöt.
- Aivoverisuonitapahtumat (esim. ohimenevä aivoverenkiertohäiriö, aivohalvaus) edeltävien 3 kuukauden aikana.
- Synnynnäiset tai hankinnaiset läppäviat, joihin liittyy kliinisesti merkittävä sydänlihaksen toimintahäiriö, jolla ei ole yhteyttä keuhkovaltimoiden verenpainetautiin.
- Vahvojen CYP2C8:n estäjien samanaikainen käyttö (esim. gemfibrotsiili; ks. kohta Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Hypotensio
Seleksipagi laajentaa verisuonia, mikä voi johtaa verenpaineen laskuun. Ennen Uptravi-valmisteen määräämistä lääkärin on pohdittava tarkoin, voiko verisuonia laajentavasta vaikutuksesta olla haittaa potilaille, joilla on tiettyjä perussairauksia (esim. potilaat, joilla on verenpainelääkitys, hypotensiota levossa, hypovolemiaa, vaikea vasemman kammion ulosvirtauseste tai autonomisen hermoston toimintahäiriö) (ks. kohta Haittavaikutukset).
Hypertyreoosi
Uptravi-valmisteen käytön yhteydessä on havaittu hypertyreoosia. On suositeltavaa tehdä kilpirauhasen toimintakokeita kliinisen tarpeen mukaan, mikäli potilaalla on hypertyreoosin oireita tai löydöksiä (ks. kohta Haittavaikutukset).
Veno-okklusiivinen keuhkosairaus
Vasodilataattorien (lähinnä prostasykliinien) käytön yhteydessä on raportoitu keuhkopöhöä potilailla, joilla on veno-okklusiivinen keuhkosairaus. Tästä syystä veno‑okklusiivisen keuhkosairauden mahdollisuus on otettava huomioon, jos PAH-potilaalla on keuhkopöhön merkkejä Uptravi-hoidon aikana. Jos sairaus varmistuu, hoito on lopetettava.
Iäkkäät potilaat (≥ 65 v)
Seleksipagin käytöstä yli 75-vuotiaiden potilaiden hoidossa on niukasti kliinistä kokemusta, joten Uptravi-valmisteen käytössä on noudatettava varovaisuutta tässä potilasryhmässä (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Seleksipagin käytöstä vaikeaa maksan vajaatoimintaa sairastavilla (Child-Pugh-luokka C) ei ole kliinistä kokemusta, joten hoitoa ei saa antaa näille potilaille. Keskivaikeaa maksan vajaatoimintaa sairastavilla (Child-Pugh-luokka B) altistus seleksipagille ja sen aktiiviselle metaboliitille suurenee (ks. Kohta Farmakokinetiikka). Keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla Uptravi-valmisteen kokonaisvuorokausiannosta on pienennettävä (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Vaikeaa munuaisten vajaatoimintaa sairastavilla (eGFR < 30 ml/min/1,73 m2) annostitrauksessa on noudatettava varovaisuutta. Uptravi-valmisteen käytöstä dialyysihoitoa saavilla potilailla ei ole kokemusta (ks. kohta Farmakokinetiikka), joten valmistetta ei pidä käyttää tässä potilasryhmässä.
Käyttö naisille, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä seleksipagihoidon aikana (ks. kohta Hedelmällisyys, raskaus ja imetys).
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset seleksipagiin
Seleksipagi hydrolysoituu aktiiviseksi metaboliitikseen karboksyyliesteraasien vaikutuksesta (ks. kohta Farmakokinetiikka). Seleksipagi ja sen aktiivinen metaboliitti läpikäyvät lähinnä CYP2C8-välitteisen ja vähäisemmässä määrin CYP3A4-välitteisen oksidatiivisen metabolian. Aktiivisen metaboliitin glukuronidaatiota katalysoivat UGT1A3 ja UGT2B7. Seleksipagi ja sen aktiivinen metaboliitti ovat OATP1B1:n ja OATP1B3:n substraatteja. Seleksipagi on P‑gp-ulosvirtauspumpun heikko substraatti. Aktiivinen metaboliitti on rintasyöpäresistenssiproteiini BCRP:n (breast cancer resistance protein) heikko substraatti.
Varfariini ei vaikuta seleksipagin eikä sen aktiivisen metaboliitin farmakokinetiikkaan.
CYP2C8:n estäjät
Kun gemfibrotsiilia (vahva CYP2C8:n estäjä) annettiin 600 mg x 2, seleksipagialtistus suureni noin 2‑kertaiseksi ja altistus aktiiviselle metaboliitille, jolla on merkittävä vaikutus hoidon tehoon, suureni noin 11-kertaiseksi. Uptravi-valmisteen anto samanaikaisesti vahvojen CYP2C8:n estäjien (esim. gemfibrotsiilin) kanssa on vasta-aiheista (ks. kohta Vasta-aiheet).
Uptravi-valmisteen annolla samanaikaisesti klopidogreelin kanssa (300 mg:n latausannos tai ylläpitoannos 75 mg kerran vuorokaudessa), joka on CYP2C8:n keskivahva estäjä, ei ollut oleellista vaikutusta seleksipagialtistukseen, mutta se lisäsi altistusta aktiiviselle metaboliitille noin 2,2‑kertaisesti latausannoksen jälkeen ja 2,7-kertaisesti ylläpitoannoksen jälkeen. Uptravi-valmisteen kokonaisvuorokausiannosta on vähennettävä pienentämällä jokainen annos puoleen annettaessa Uptravi-valmistetta samanaikaisesti keskivahvojen CYP2C8:n estäjien kanssa (esim. klopidogreeli, deferasiroksi, teriflunomidi). Vaihtoehtoisesti, puolikkaan vuorokausiannoksen saavuttamiseksi, Uptravi-hoitoa voidaan jatkaa antotiheydellä kerran vuorokaudessa, jos potilas on sillä jo hyvässä hoitotasapainossa, tai potilaille, joille ei ole saatavissa sopivaa annosvahvuutta, joka tukisi puolikkaan annoksen annostelua kaksi kertaa vuorokaudessa. Kun keskivahvan CYP2C8:n estäjän samanaikainen käyttö loppuu, Uptravi-valmisteen kokonaisvuorokausiannosta suurennetaan tarpeen mukaan. Maksimiannosta 1 600 mikrog. x 2 vuorokaudessa ei saa ylittää (ks. kohta Annostus ja antotapa).
CYP2C8:n induktorit
Kun rifampisiinia, joka on CYP2C8:n (ja UGT-entsyymien) induktori, annettiin 600 mg x 1, seleksipagialtistus ei muuttunut mutta altistus aktiiviselle metaboliitille pieneni puoleen. Seleksipagiannosta on mahdollisesti muutettava, jos samanaikaisesti annetaan CYP2C8:n induktoreja (esim. rifampisiini, karbamatsepiini, fenytoiini).
UGT1A3:n ja UGT2B7:n estäjät
Vahvojen UGT1A3:n ja UGT2B7:n estäjien (valproiinihappo, probenesidi ja flukonatsoli) vaikutusta altistukseen seleksipagille ja sen aktiiviselle metaboliitille ei ole tutkittu. Varovaisuus on tarpeen, jos näitä lääkevalmisteita annetaan samanaikaisesti Uptravi-valmisteen kanssa. Mahdollista farmakokineettistä yhteisvaikutusta UGT1A3:n ja UGT2B7:n vahvojen estäjien kanssa ei voida sulkea pois.
CYP3A4:n estäjät ja induktorit
Kun lopinaviiria/ritonaviiria (vahva CYP3A4:n estäjä) annettiin 400 mg/100 mg x 2, seleksipagialtistus suureni noin kaksinkertaiseksi, kun taas altistus seleksipagin aktiiviselle metaboliitille ei muuttunut. Vaikutuksella ei ole kliinistä merkitystä, kun otetaan huomioon, että aktiivisen metaboliitin teho on noin 37-kertainen. Vahva CYP3A4:n estäjä ei vaikuttanut aktiivisen metaboliitin farmakokinetiikkaan, mikä viittaa siihen, että CYP3A4-reitti ei osallistu merkittävästi aktiivisen metaboliitin eliminaatioon. Voidaan siis olettaa, että CYP3A4:n induktorit eivät vaikuta aktiivisen metaboliitin farmakokinetiikkaan.
Keuhkovaltimoiden verenpainetaudin spesifiset hoidot
PAH-potilailla toteutetussa vaiheen 3 lumekontrolloidussa tutkimuksessa seleksipagin käyttö yhdessä sekä endoteliinireseptoriantagonistin että PDE-5:n estäjän kanssa pienensi altistusta aktiiviselle metaboliitille 30 %.
Kuljettajaproteiinien estäjät (lopinaviiri/ritonaviiri)
Kun lopinaviiria/ritonaviiria (vahva OATP:n [OATP1B1 ja OATP1B3] ja P-gp:n estäjä) annettiin 400 mg/100 mg x 2, seleksipagialtistus suureni noin kaksinkertaiseksi, kun taas altistus seleksipagin aktiiviselle metaboliitille ei muuttunut. Tällä vaikutuksella ei ole kliinistä merkitystä, sillä farmakologinen vaikutus johtuu pääasiallisesti aktiivisesta metaboliitista.
Seleksipagin vaikutus muihin lääkevalmisteisiin
Seleksipagi ja sen aktiivinen metaboliitti eivät estä eivätkä indusoi sytokromi P450 -entsyymien eivätkä kuljettajaproteiinien toimintaa kliinisesti relevantteina pitoisuuksina.
Antikoagulantit ja trombosyyttiaggregaation estäjät
Seleksipagi estää trombosyyttiaggregaatiota in vitro. PAH-potilailla toteutetussa vaiheen 3 lumekontrolloidussa tutkimuksessa seleksipagihoitoon ei todettu liittyvän suurentunutta vuotoriskiä verrattuna lumehoitoon, ei myöskään tilanteissa, joissa seleksipagia annettiin yhdessä antikoagulanttien (esim. hepariini, kumariinityyppiset antikoagulantit) tai trombosyyttiaggregaation estäjien kanssa. Terveillä henkilöillä tehdyssä tutkimuksessa seleksipagi (400 mikrog. x 2) ei vaikuttanut altistukseen S-varfariinille (CYP2C9:n substraatti) eikä altistukseen R-varfariinille (CYP3A4:n substraatti) 20 mg:n varfariinikerta-annoksen jälkeen. Seleksipagi ei muuttanut varfariinin farmakodynaamista vaikutusta INR-arvoon.
Midatsolaami
Kun seleksipagiannos oli titrattu tasolle 1 600 mikrogrammaa x 2, vakaassa tilassa ei todettu kliinisesti merkittäviä muutoksia altistuksessa midatsolaamille (joka on herkkä CYP3A4:n substraatti suolistossa ja maksassa) eikä altistuksessa midatsolaamin metaboliitille 1-hydroksimidatsolaamille. Seleksipagin anto samanaikaisesti CYP3A4:n substraattien kanssa ei edellytä annosmuutoksia.
Hormonaalinen ehkäisy
Hormonaalisia ehkäisyvalmisteita koskevia spesifisiä lääkeaineinteraktiotutkimuksia ei ole tehty. Seleksipagi ei vaikuttanut altistukseen midatsolaamille eikä R-varfariinille (CYP3A4:n substraatteja) eikä altistukseen S-varfariinille (CYP2C9:n substraatti), joten sen ei odoteta vähentävän hormonaalisten ehkäisyvalmisteiden tehoa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä seleksipagihoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus
Ei ole olemassa tietoja seleksipagin käytöstä raskaana oleville naisille. Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia. Seleksipagin ja sen päämetaboliitin teho prostasykliinireseptoriin (IP) in vitro oli lisääntymistoksisuuskokeissa käytetyillä eläinlajeilla 20-80 kertaa pienempi kuin ihmisellä. Mahdollisten IP-reseptorin kautta välittyvien, lisääntymiseen kohdistuvien vaikutusten turvallisuusmarginaalit ovat tämän mukaisesti pienemmät kuin IP-reseptoriin liittymättömien vaikutusten turvallisuusmarginaalit (ks. kohta Prekliiniset tiedot turvallisuudesta).
Uptravi-valmisteen käyttöä ei suositella raskauden aikana, eikä hedelmällisessä iässä oleville naisille, jotka eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyvätkö seleksipagi tai sen metaboliitit ihmisen rintamaitoon. Rotalla seleksipagi tai sen metaboliitit erittyvät maitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Imeväiseen kohdistuvia riskejä ei voida poissulkea. Uptravi-valmistetta ei pidä käyttää rintaruokinnan aikana.
Hedelmällisyys
Kliinisiä tietoja ei ole saatavilla. Rottatutkimuksissa suuret seleksipagiannokset aiheuttivat ohimeneviä kiimakierron häiriöitä, jotka eivät vaikuttaneet hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta). Ilmiön merkitystä ihmisille ei tunneta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Uptravi-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaan kliininen tila ja seleksipagin haittavaikutusprofiili (kuten päänsärky, hypotensio, ks. kohta Haittavaikutukset) on otettava huomioon potilaan ajokyvyn ja koneidenkäyttökyvyn arvioinnissa.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitettuja haittavaikutuksia ovat päänsärky, ripuli, pahoinvointi ja oksentelu, leukakipu, lihaskipu, raajakipu, nivelkipu ja punastuminen. Näitä reaktioita esiintyy yleisemmin annoksen suurentamisvaiheessa. Valtaosa niistä on lieviä tai keskivaikeita.
Seleksipagin turvallisuutta arvioitiin lumekontrolloidussa vaiheen 3 pitkäaikaistutkimuksessa 1 156 aikuisella potilaalla, joilla oli oireinen keuhkovaltimoiden verenpainetauti (GRIPHON-tutkimus). Hoidon keston keskiarvo oli seleksipagiryhmän potilailla 76,4 viikkoa (mediaani 70,7 viikkoa) ja lumeryhmän potilailla 71,2 viikkoa (mediaani 63,7 viikkoa). Seleksipagialtistus kesti enimmillään 4,2 vuotta.
Haittavaikutustaulukko
Kliinisessä avaintutkimuksessa (GRIPHON) ja valmisteen markkinoille tulon jälkeisessä valvonnassa todetut haittavaikutukset esitetään seuraavassa taulukossa. Haittavaikutukset on luokiteltu elinjärjestelmäluokittain esiintyvyyden mukaisesti ja esitetään vakavuuden mukaan alenevassa järjestyksessä. Esiintyvyydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000).
| Elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen |
| Veri ja imukudos | Anemia* Hemoglobiinipitoisuuden pieneneminen* | ||
| Umpieritys | Hypertyreoosi* Tyreotropiinipitoisuuden pieneneminen | ||
| Aineenvaihdunta ja ravitsemus | Ruokahalun heikentyminen Painon lasku | ||
| Hermosto | Päänsärky* | ||
| Sydän | Sinustakykardia* | ||
| Verisuonisto | Punastuminen* | Hypotensio* | |
| Hengityselimet, rintakehä ja välikarsina | Nenänielutulehdus (muu kuin infektioperäinen) | Nenän tukkoisuus | |
| Ruoansulatuselimistö | Ripuli* Oksentelu* Pahoinvointi* | Vatsakipu Dyspepsia* | |
| Iho ja ihonalainen kudos | Ihottuma Angioedeema† | ||
| Luusto, lihakset ja sidekudos | Leukakipu* Lihaskipu* Nivelkipu* Raajakipu* | ||
| Yleisoireet ja antopaikassa todettavat haitat | Kipu |
* Ks. kohta Valikoitujen haittavaikutusten kuvaus.
† Valmisteen markkinoille tulon jälkeen on raportoitu angioedeematapauksia, joiden latenssiaika voi olla yli 30 hoitopäivää.
Valikoitujen haittavaikutusten kuvaus
Titraukseen ja ylläpitohoitoon liittyvät farmakologiset vaikutukset
Seleksipagin vaikutustapaan liittyviä haittavaikutuksia on todettu usein, etenkin yksilöllisen annostitrausvaiheen aikana. Ne esitetään taulukossa alla:
| Prostasykliinityyppiset liittyvät haittavaikutukset | Titraus | Ylläpito | ||
| Seleksipagi | Lume | Seleksipagi | Lume | |
| Päänsärky | 64 % | 28 % | 40 % | 20 % |
| Ripuli | 36 % | 12 % | 30 % | 13 % |
| Pahoinvointi | 29 % | 13 % | 20 % | 10 % |
| Leukakipu | 26 % | 4 % | 21 % | 4 % |
| Lihaskipu | 15 % | 5 % | 9 % | 3 % |
| Raajakipu | 14 % | 5 % | 13 % | 6 % |
| Oksentelu | 14 % | 4 % | 8 % | 6 % |
| Punastuminen | 11 % | 4 % | 10 % | 3 % |
| Nivelkipu | 7 % | 5 % | 9 % | 5 % |
Nämä vaikutukset ovat yleensä ohimeneviä tai hoidettavissa oireenmukaisesti. 7,5 % seleksipagia saaneista potilaista lopetti hoidon näiden haittavaikutusten vuoksi. Vakavia haittavaikutuksia esiintyi seleksipagiryhmässä noin 2,3 %:lla ja lumeryhmässä noin 0,5 %:lla. Kliinisessä työssä ruoansulatuskanavan tapahtumien on todettu reagoivan ripuli-, oksentelu- ja pahoinvointilääkkeisiin ja/tai toiminnallisten vatsavaivojen lääkkeisiin.
Kipuun liittyviä tapahtumia on usein hoidettu kipulääkkeillä (esim. parasetamolilla).
Hemoglobiinipitoisuuden pieneneminen
PAH-potilailla toteutetussa vaiheen 3 lumekontrolloidussa tutkimuksessa hemoglobiinipitoisuuksien absoluuttisten muutoksien keskiarvot vaihtelivat seurantakäyntien mittauksissa verrattuna lähtötilanteeseen seleksipagiryhmässä ‑0,34:stä ‑0,02 g/dl:aan verrattuna lumeryhmän ‑0,05:sta 0,25 g/dl:aan. Hemoglobiinipitoisuus laski lähtötilanteesta alle arvon 10 g/dl 8,6 %:lla seleksipagiryhmän potilaista ja 5,0 %:lla lumeryhmän potilaista.
PAH-diagnoosin äskettäin saaneilla potilailla toteutetussa vaiheen 3 lumekontrolloidussa tutkimuksessa hemoglobiinipitoisuuksien absoluuttisten muutoksien keskiarvot vaihtelivat seurantakäyntien mittauksissa verrattuna lähtötilanteeseen kolmoishoitoryhmässä (seleksipagi, masitentaani, tadalafiili) ‑1,77:stä ‑1,26 g/dl:aan verrattuna kaksoishoitoryhmän (lumelääke, masitentaani ja tadalafiili) ‑1,61:stä ‑1,28 g/dl:aan. Hemoglobiinipitoisuuden lasku lähtötilanteesta alle arvon 10 g/dl raportoitiin 19,0 %:lla kolmoishoitoryhmän potilaista ja 14,5 %:lla kaksoishoitoryhmän potilaista. Anemiaa raportoitiin kolmoishoitoryhmässä hyvin yleisesti (13,4 %) ja kaksoishoitoryhmässä yleisesti (8,3 %).
Kilpirauhasen toimintakokeet
PAH-potilailla toteutetussa vaiheen 3 lumekontrolloidussa tutkimuksessa hypertyreoosia ilmoitettiin 1,6 %:lla seleksipagiryhmän potilaista, kun taas lumeryhmässä ei ilmoitettu yhtään tapausta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Seleksipagiryhmässä todettiin tyreotropiinin (TSH) mediaanipitoisuuden pienentymistä (enintään ‑0,3 mU/l, lähtötilanteen mediaaniarvo 2,5 mU/l) useimmilla seurantakäynneillä. Lumeryhmässä mediaanipitoisuuksissa ei havaittu juurikaan muutosta. Trijodityroniini- ja tyroksiinipitoisuuksien keskiarvot eivät muuttuneet kummassakaan ryhmässä.
Sykkeen nopeutuminen
PAH-potilailla toteutetussa vaiheen 3 lumekontrolloidussa tutkimuksessa todettiin syketiheyden keskiarvon suurentuvan ohimenevästi 3–4 lyöntiä/min 2–4 tunnin kuluttua annoksesta. EKG-tutkimuksissa todettiin sinustakykardiaa 11,3 %:lla seleksipagiryhmän potilaista ja 8,8 %:lla lumeryhmän potilaista (ks. kohta Farmakodynamiikka).
Hypotensio
PAH-potilailla toteutetussa vaiheen 3 lumekontrolloidussa tutkimuksessa hypotensiota ilmoitettiin 5,8 %:lla seleksipagiryhmän potilaista ja 3,8 %:lla lumeryhmän potilaista. Systolisen verenpaineen keskimääräinen absoluuttinen muutos lähtötilanteesta oli säännöllisillä käynneillä seleksipagiryhmässä ‑2,0 – ‑1,5 mmHg ja lumeryhmässä ‑1,3 – 0,0 mmHg, ja diastolisen verenpaineen osalta vastaava muutos oli seleksipagiryhmässä ‑1,6 – ‑0,1 mmHg ja lumeryhmässä ‑1,1 – 0,3 mmHg. Systolisen verenpaineen todettiin laskeneen alle 90 mmHg 9,7 %:lla seleksipagiryhmän potilaista ja 6,7 %:lla lumeryhmän potilaista.
Dyspepsia
PAH-diagnoosin äskettäin saaneilla potilailla tehdyssä vaiheen 3 lumekontrolloidussa tutkimuksessa dyspepsiaa raportoitiin kolmoishoitoa (seleksipagi, masitentaani, tadalafiili) saaneilla potilailla hyvin yleisesti (16,8 %) ja kaksoishoitoa (lumelääke, masitentaani ja tadalafiili) saaneilla potilailla yleisesti (8,3 %).
Pitkäaikaisturvallisuus
Avaintutkimukseen osallistuneista 1 156 potilaasta 709 potilasta tuli mukaan pitkäkestoiseen avoimeen jatkotutkimukseen (330 seleksipagin käyttöä jatkanutta potilasta GRIPHON-tutkimuksesta ja 379 lumehoitoa GRIPHON-tutkimuksessa saanutta potilasta, jotka siirtyivät seleksipagihoitoon). Pitkäaikaisseurannassa seleksipagihoidon keston mediaani oli 30,5 kuukautta ja enintään 103 kuukautta, ja turvallisuusprofiilin osoitettiin olevan samankaltainen kuin edellä kuvatussa kliinisessä avaintutkimuksessa havaittu turvallisuusprofiili.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yksittäisiä yliannostustapauksia (enintään 3 200 mikrog.) on ilmoitettu aikuisilla. Ainoa ilmoitettu seuraus oli lievä, ohimenevä pahoinvointi. Yliannostustapauksessa on ryhdyttävä tarpeellisiin tukitoimiin. Dialyysi ei todennäköisesti tehoa, sillä seleksipagi ja sen aktiivinen metaboliitti sitoutuvat suuressa määrin proteiineihin.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antitromboottiset lääkeaineet, aggregaation estäjät lukuun ottamatta hepariinia, ATC-koodi: B01AC27
Vaikutusmekanismi
Seleksipagi on selektiivinen IP-reseptorin agonisti, joka eroaa prostasykliinistä ja sen analogeista. Seleksipagi hydrolysoituu karboksyyliesteraasien välityksellä aktiiviseksi metaboliitiksi, joka on noin 37 kertaa tehokkaampi kuin seleksipagi. Seleksipagi ja sen aktiivinen metaboliitti ovat vahva-affiniteettisia IP-reseptorin agonisteja, jotka sitoutuvat hyvin selektiivisesti IP-reseptoriin ja heikommin muihin prostanoidireseptoreihin (EP1-EP4, DP, FP ja TP). Selektiivisyys EP1-, EP3-, FP- ja TP-reseptoreja vastaan on tärkeää, sillä näitä tarkoin kuvattuja supistavia reseptoreja esiintyy ruoansulatuskanavassa ja verisuonissa. Selektiivisyys EP2-, EP4- ja DP1-reseptoreja vastaan on tärkeää, sillä nämä reseptorit välittävät immuunijärjestelmän toimintaa lamaavia vaikutuksia.
Seleksipagin ja sen aktiivisen metaboliitin aiheuttama IP-reseptoristimulaatio johtaa vasodilatoiviin, proliferaatiota estäviin ja antifibroottisiin vaikutuksiin. Seleksipagi estää sydämen ja keuhkojen uudelleenmuotoutumista rotan PAH-mallissa ja pienentää keuhko- ja ääreisverisuonten painetta suhteessa annokseen. Tämä viittaa siihen, että ääreisverisuonten vasodilataatio kuvaa valmisteen farmakodynaamista tehoa keuhkoissa. Seleksipagi ei aiheuta IP-reseptorien desensitisaatiota in vitro eikä johda takyfylaksiin rottamallissa.
Farmakodynaamiset vaikutukset
Sydämen elektrofysiologia
Terveillä aikuisilla henkilöillä tehdyssä perusteellisessa QT-tutkimuksessa toistuvat 800 mikrog. ja 1 600 mikrog. x 2 seleksipagiannokset eivät vaikuttaneet sydämen repolarisaatioon (QTc-aika) eivätkä johtumiseen (PR- ja QRS-ajat) ja nopeuttivat lievästi syketiheyttä (lumekorjattu, lähtötasoarvon suhteen korjattu sykkeen nopeutuminen 6-7 lyöntiä/min 1,5-3 h kuluttua 800 mikrog. seleksipagiannoksesta ja 9-10 lyöntiä/min vastaavan ajan kuluttua 1 600 mikrog. seleksipagiannoksesta).
Hyytymistekijät
Vaiheiden 1 ja 2 tutkimuksissa todettiin, että von Willebrand -tekijän (vWF) pitoisuudet plasmassa pienenivät hieman seleksipagia saaneilla; vWF-pitoisuudet pysyivät normaalialueen alarajan yläpuolella.
Keuhkojen hemodynamiikka
Vaiheen 2 kaksoissokkoutetussa, lumekontrolloidussa kliinisessä tutkimuksessa arvioitiin hemodynamiikan muuttujia 17 hoitoviikon jälkeen aikuisilla PAH-potilailla, joiden WHO:n toimintakykyluokka oli II–III ja jotka saivat samanaikaisesti endoteliinireseptoriantagonistihoitoa ja/tai PDE-5:n estäjiä. Kun seleksipagiannos titrattiin yksilöllisesti siedettyyn annokseen (annosta suurennettiin 200 mikrog. x 2 kerrallaan enimmäisannokseen 800 mikrog. x 2; N = 33), keuhkojen verisuonivastuksen keskiarvo pieneni tilastollisesti merkitsevästi 30,3 % (95 %:n luottamusväli [lv] ‑44,7 %, ‑12,2 %; p = 0,0045) ja sydänindeksi (cardiac index) suureni (hoitovaikutuksen keskiarvo) 0,48 l/min/m2 (95 %:n lv 0,13, 0,83) verrattuna lumehoitoon (N = 10).
Kliininen teho ja turvallisuus
Teho aikuisilla PAH-potilailla (GRIPHON)
Seleksipagin vaikutus keuhkovaltimoiden verenpainetaudin etenemiseen todettiin pitkäkestoisessa (altistuksen enimmäiskesto noin 4,2 v), kaksoissokkoutetussa, lumekontrolloidussa, rinnakkaisryhmissä toteutetussa, tapahtumien määrään perustuvassa vaiheen 3 monikeskustutkimuksessa (GRIPHON), johon osallistuneilla 1 156 potilaalla oli oireinen (WHO:n toimintakykyluokat I–IV) keuhkovaltimoiden verenpainetauti.
Potilaat satunnaistettiin saamaan joko lumetta (N = 582) tai seleksipagia (N = 574) kahdesti vuorokaudessa. Annosta suurennettiin viikon välein 200 mikrog. x 2 kerrallaan yksilöllisen ylläpitoannoksen määrittämiseksi (200–1 600 mikrog. x 2).
Tutkimuksen ensisijainen päätetapahtuma oli aika ensimmäiseen sairastavuus- tai kuolleisuustapahtumaan hoidon loppuun mennessä; tämä määriteltiin seuraavien yhdistelmäksi: kuolema (kaikki syyt), tai PAH:sta johtuva sairaalahoito, tai PAH:n etenemisestä johtuva keuhkonsiirron tai pallokatetrilla tehtävän eteisseptostomian tarve, tai parenteraalisen prostanoidihoidon tai pitkäkestoisen happihoidon aloitus, tai muu taudin etenemistapahtuma, jonka vahvistettiin (potilailla, joiden WHO:n toimintakykyluokka lähtötilanteessa II tai III) 6 minuutin kävelytestin (6MWD) tuloksen huononemisella lähtötilanteesta (≥ 15 %) sekä WHO:n toimintakykyluokan huononemisella tai (potilailla, joiden WHO:n toimintakykyluokka lähtötilanteessa III tai IV) 6MWD-kokeen tuloksen huononemisella lähtötilanteesta (≥ 15 %) sekä tarpeella ottaa käyttöön lisää PAH:n spesifisiä hoitoja.
Riippumaton arviointitoimikunta vahvisti kaikki tapahtumat, hoitoryhmän suhteen sokkoutetusti.
Potilaat olivat keskimäärin 48,1 vuotiaita (vaihteluväli 18-80 vuotta). Valtaosa tutkittavista oli valkoihoisia (65,0 %) ja naisia (79,8 %). 17,9 % potilaista oli ≥ 65-vuotiaita ja 1,1 % ≥ 75-vuotiaita. Lähtötilanteessa noin 1 % kuului WHO:n toimintakykyluokkaan I, 46 % luokkaan II, 53 % luokkaan III ja 1 % luokkaan IV.
Yleisin etiologia tutkimuspopulaatiossa oli idiopaattinen tai perinnöllinen PAH (58 %). Seuraavaksi yleisimpiä olivat sidekudostaudista johtuva PAH (29 %), yksinkertaiseen korjattuun synnynnäiseen sydänvikaan liittyvä PAH (10 %) ja muihin syihin liittyvä PAH (lääkkeet, huumeet ja toksiinit [2 %] ja HIV [1 %]).
Valtaosa mukaan otetuista potilaista (80 %) sai lähtötilanteessa vakaa-annoksista PAH-spesifistä hoitoa: joko endoteliinireseptoriantagonistia (15 %) tai PDE-5:n estäjää (32 %) tai sekä endoteliinireseptoriantagonistia että PDE-5:n estäjää (33 %).
Kaksoissokkoutetun hoidon kokonaiskeston mediaani oli lumeryhmässä 63,7 viikkoa ja seleksipagiryhmässä 70,7 viikkoa. 23 % seleksipagia saaneista potilaista saavutti 200-400 mikrog. ylläpitoannokset, 31 % saavutti 600-1 000 mikrog. ylläpitoannokset, ja 43 % saavutti 1 200-1 600 mikrog. ylläpitoannokset.
Seleksipagihoito annoksilla 200–1 600 mikrog. x 2 pienensi sairastavuus- ja kuolleisuustapahtumien riskiä 40 % (riskisuhde [HR] 0,60; 99 %:n lv 0,46, 0,78; 1-suuntainen log-rank p-arvo < 0,0001) 7 päivään asti viimeisen annoksen jälkeen verrattuna lumeeseen (kuva 1). Seleksipagin hyöty johtui lähinnä keuhkovaltimoiden verenpainetaudista johtuvien sairaalahoitojen vähenemisestä ja muiden taudin etenemistapahtumien vähenemisestä (taulukko 1).
Kuva 1 Kaplan-Meier-estimaatit ensimmäisestä sairastavuus-kuolleisuustapahtumasta
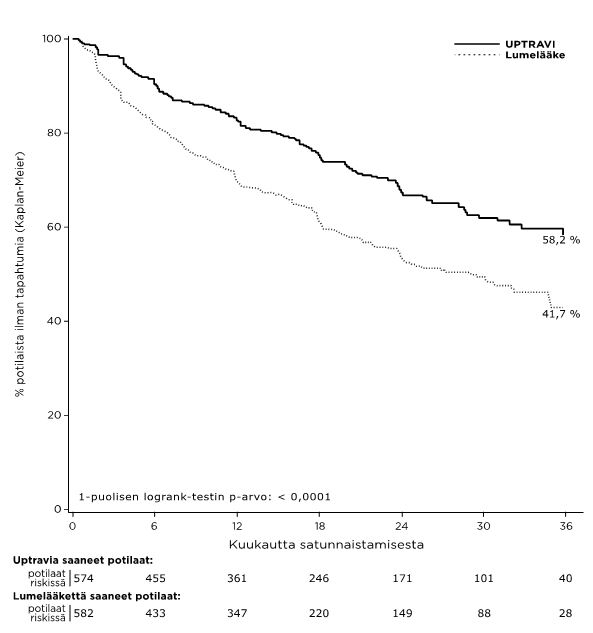
Taulukko 1 Päätetapahtumien yhteenveto
| Päätetapahtumat ja tilastot | Potilaita, joilla tapahtuma | Hoitojen vertailu: seleksipagi vs. lume | ||||
| Lume (N = 582) | Seleksipagi (N = 574) | Absoluuttisen riskin pienentyminen | Suhteellisen riskin pienentyminen (99 %:n lv) | riskisuhde (99 %:n lv) | p-arvo | |
| Sairastavuus-kuolleisuustapahtumaa | 58,3 % | 41,8 % | 16,5 % | 40 % (22 %; 54 %) | 0,60 (0,46; 0,78) | < 0,0001 |
| PAH:sta johtuva sairaalahoitob n (%) | 109 (18,7 %) | 78 (13,6 %) | 5,1 % | 33 % (2 %; 54 %) | 0,67 (0,46; 0,98) | 0,04 |
| Taudin eteneminenb n (%) | 100 (17,2 %) | 38 (6,6 %) | 10,6 % | 64 % (41 %; 78 %) | 0,36 (0,22; 0,59) | < 0,0001 |
| I.v./s.c. prostanoidihoidon aloitus tai happihoitob,c n (%) | 15 (2,6 %) | 11 (1,9 %) | 0,7 % | 32 % (-90 %; 76 %) | 0,68 (0,24; 1,90) | 0,53 |
| Kuolema viimeistään 7 päivän kuluttua hoidon päättymisestäd n (%) | 37 (6,4 %) | 46 (8,0 %) | -1,7 % | -17 % (-107 %; 34 %) | 1,17 (0,66; 2,07) | 0,77 |
Kuolema tutkimuksen loppuun mennessäd n (%) | 105 (18,0 %) | 100 (17,4 %) | 0,6 % | 3 % (-39 %; 32 %) | 0,97 (0,68; 1,39) | 0,42 |
lv = luottamusväli; HR = riskisuhde; i.v. = laskimoon; PAH = keuhkovaltimoiden verenpainetauti, pulmonaaliarteriahypertensio; s.c. = ihon alle.
a Potilaita, joilla tapahtuma 36 kuukauteen mennessä (%) = 100 × (1 – Kaplan–Meier-estimaatti); riskisuhde estimoitu Coxin suhteellisen riskin mallilla; stratifioimaton, yksisuuntainen log-rank p-arvo
b Potilaita, joilla ensisijaiseen päätetapahtumaan kuuluva tapahtuma viimeistään 7 päivän kuluttua hoidon päättymisestä (%); riskisuhde estimoitu Aalen–Johansenin menetelmällä; 2-suuntainen p-arvo Grayn testillä
c Mukana ”Keuhkonsiirron tai eteisseptostomian tarve” (1 potilas seleksipagiryhmässä ja 2 lumeryhmässä)
d Potilaita, joilla tapahtuma viimeistään 7 päivän kuluttua hoidon päättymisestä tai tutkimuksen loppuun mennessä (%); riskisuhde estimoitu Coxin suhteellisen riskin mallilla; stratifioimaton, yksisuuntainen log-rank p-arvo
Matemaattisen mallinnuksen avulla selvitettiin tarkemmin, miksi kuolemien määrä oli numeerisesti lisääntynyt hoidon päättyessä ja 7 päivää sen jälkeen, kun taas tutkimuksen loppuessa kuolemien määrä ei ollut lisääntynyt. Kyseisen mallinnuksen mukaan kuolemantapausten määrän epäjohdonmukaisuus vastaa oletusta hoidon neutraalista vaikutuksesta PAH-kuolleisuuteen ja ei-fataaleja tapahtumia vähentävästä vaikutuksesta.
Seleksipagin havaittu vaikutus ensisijaiseen päätetapahtumaan verrattuna lumeeseen oli johdonmukainen erilaisilla yksilöllisillä ylläpitoannoksilla, osoitettuna riskisuhteilla kolmessa ennalta määritellyssä ryhmässä (0,60 annosryhmässä 200-400 mikrog. x 2; 0,53 annosryhmässä 600-1 000 mikrog. x 2; ja 0,64 annosryhmässä 1 200-1 600 mikrog. x 2), joka oli linjassa hoidon kokonaisvaikutuksen kanssa (0,60).
Seleksipagin teho ensisijaiseen päätetapahtumaan oli johdonmukainen eri alaryhmissä (ikä, sukupuoli, etninen tausta, etiologia, maantieteellinen alue, WHO:n toimintakykyluokka) ja sekä monoterapiana käytettäessä että yhdessä joko endoteliinireseptoriantagonistin tai PDE-5:n estäjän käytettäessä että kolmoishoitona endoteliinireseptoriantagonistin ja PDE-5:n estäjän kanssa käytettäessä.
Toissijaisena päätetapahtumana oli aika PAH:iin liittyvään kuolemaan tai sairaalahoitoon. Tähän päätetapahtumaan kuuluvan tapahtuman riski pieneni seleksipagia saaneilla 30 % verrattuna lumeryhmään (HR 0,70, 99 %:n lv 0,50, 0,98; yksisuuntainen log-rank p-arvo = 0,0031). Seleksipagiryhmässä 28,9 %:lla potilaista oli tapahtuma 36 kuukauteen mennessä; lumeryhmässä vastaava osuus oli 41,3 %, ja absoluuttinen riski pieneni 12,4 %.
Niiden potilaiden määrä, joiden ensimmäinen tapahtuma oli PAH:ista johtuva kuolema tai sairaalahoito hoidon päättymiseen mennessä, oli seleksipagiryhmässä 102 (17,8 %) ja lumeryhmässä 137 (23,5 %). Seleksipagiryhmässä 16 potilasta (2,8 %) kuoli PAH:n vuoksi (päätetapahtuman komponenttina); lumeryhmässä vastaava määrä oli 14 (2,4 %). PAH:ista johtuvia sairaalahoitoja todettiin seleksipagiryhmässä 86 potilaalla (15,0 %) ja lumeryhmässä 123 potilaalla (21,1 %). Seleksipagi pienensi PAH-sta johtuvan sairaalahoidon riskiä ensimmäisenä päätetapahtumana verrattuna lumeeseen (HR 0,67, 99 %:n lv 0,46: 0,98; yksisuuntainen log-rank p-arvo = 0,04).
Kuolemantapausten kokonaismäärä (kaikki syyt) tutkimuksen loppuun mennessä oli seleksipagiryhmässä 100 (17,4 %) ja lumeryhmässä 105 (18,0 %) (HR 0,97, 99 %:n lv 0,68, 1,39). PAH:ista johtuvien kuolemantapausten määrä tutkimuksen loppuun mennessä oli seleksipagiryhmässä 70 (12,2 %) ja lumeryhmässä 83 (14,3 %).
Oireenmukaiset päätetapahtumat
Suorituskyky oli toissijainen päätetapahtuma. 6MWD:n mediaani oli lähtötilanteessa seleksipagiryhmässä 376 m (vaihteluväli: 90-482 m) ja lumeryhmässä 369 m (vaihteluväli: 50-515 m). Seleksipagihoidon lumekorjatun vaikutuksen mediaani 6MWD:ssä oli 12 m (99 %:n lv 1, 24 m; yksisuuntainen p-arvo = 0,0027) jäännöspitoisuuksien (”at trough”; eli noin 12 tunnin kuluttua annoksesta) kohdalla, viikolla 26 mitattuina. Potilailla, joilla ei ollut muuta samanaikaista PAH-spesifistä hoitoa, jäännöspitoisuuksien kohdalla mitattu, lumekorjattu hoitovaikutus oli 34 m (99 %:n lv 10, 63 m).
Elämänlaatua arvioitiin GRIPHON-tutkimuksen potilaspopulaation osajoukossa CAMPHOR-kyselyllä (Cambridge Pulmonary Hypertension Outcome Review). Viikkoon 26 mennessä ei todettu merkitsevää hoitovaikutusta verrattuna lähtötilanteeseen.
PAH:n pitkäaikaistiedot
Avaintutkimukseen (GRIPHON) mukaan otetut potilaat soveltuivat osallistumaan pitkäkestoiseen avoimeen jatkotutkimukseen. GRIPHON-tutkimuksessa yhteensä 574 potilasta sai seleksipagihoitoa, ja näistä 330 potilasta jatkoi seleksipagihoitoa avoimessa jatkotutkimuksessa. Seurannan keston mediaani oli 4,5 vuotta ja seleksipagialtistuksen keston mediaani oli 3 vuotta. Seurannan aikana 28,4 %:lle potilaista lisättiin seleksipagihoitoon vähintään yksi muu PAH:n hoitoon käytettävä lääkevalmiste. Kaikilla 574 potilaalla suurin osa hoidolle altistumisesta (86,3 %) kumuloitui ilman uuden lääkevalmisteen lisäämistä PAH:n hoitoon. Näiden 574 potilaan Kaplan–Meierin estimaatit elossaolosta GRIPHON-tutkimuksessa ja jatkotutkimuksessa olivat 1 vuoden kohdalla 92 %, 2 vuoden kohdalla 85 %, 5 vuoden kohdalla 71 % ja 7 vuoden kohdalla 63 %. Avaintutkimuksen lähtötilanteessa 273 potilaan WHO:n toimintakykyluokka oli II, ja näiden potilaiden elossaolo oli 1 vuoden kohdalla 97 %, 2 vuoden kohdalla 91 %, 5 vuoden kohdalla 80 % ja 7 vuoden kohdalla 70 %; lähtötilanteessa 294 potilaan WHO:n toimintakykyluokka oli III, ja näiden potilaiden elossaolo oli 1 vuoden kohdalla 88 %, 2 vuoden kohdalla 80 %, 5 vuoden kohdalla 62 % ja 7 vuoden kohdalla 56 %. Koska lisälääkitys PAH:n hoitoon aloitettiin pienelle osalle potilaista eikä jatkotutkimuksessa ollut vertailuryhmää, seleksipagin hyötyä elossaolon suhteen ei voida varmistaa näiden tietojen perusteella.
Alkuvaiheen kolmoisyhdistelmähoito seleksipagilla, masitentaanilla ja tadalafiililla PAH-diagnoosin äskettäin saaneilla potilailla
Kaksoissokkoutetussa lumekontrolloidussa tutkimuksessa yhteensä 247 PAH-diagnoosin äskettäin saanutta potilasta satunnaistettiin, jotta alkuvaiheen kolmoishoidon (seleksipagi, masitentaani ja tadalafiili) (N = 123) ja alkuvaiheen kaksoishoidon (lumelääke, masitentaani ja tadalafiili) (N = 124) hoitovaikutusta voitiin verrata keskenään.
Ensisijaisessa päätetapahtumassa, joka oli keuhkovastuksen muutos lähtötilanteesta viikon 26 aikapisteessä, ei todettu tilastollisesti merkitsevää eroa ryhmien välillä, mutta kummassakin hoitoryhmässä todettiin paranemista lähtötilanteesta (suhteellinen vähenemä 54 % alkuvaiheen kolmoishoidon ryhmässä vs 52 % alkuvaiheen kaksoishoidon ryhmässä).
Kahden vuoden (mediaani) seurannassa kolmoishoidon ryhmässä kuoli 4 potilasta (3,4 %) ja kaksoishoidon ryhmässä kuoli 12 potilasta (9,4 %).
Pediatriset potilaat
Väliaikatiedot tehosta ja turvallisuudesta pediatrisilla PAH-potilailla (SALTO)
Tehoa ja turvallisuutta iältään ≥ 2 – < 18-vuotiailla pediatrisilla PAH-potilailla arvioitiin deskriptiivisesti satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa, rinnakkaisryhmillä tehdyssä vaiheen 3 monikeskustutkimuksessa (SALTO). Yhteensä 138 potilasta satunnaistettiin suhteessa 1:1 saamaan joko seleksipagia (N = 69) tai lumehoitoa (N = 69) kahdesti vuorokaudessa. Seleksipagiannokset 100, 150 tai 200 mikrogrammaa suurennettiin titraamalla 800, 1 200 tai 1 600 mikrogrammaan kahdesti vuorokaudessa painoluokan ja siedettävyyden perusteella (ks. kohta Farmakokinetiikka). Välianalyysi tehtiin, kun 92 potilasta oli saanut hoitoa 24 viikkoa.
Tutkimuksen ensisijainen päätetapahtuma oli ensimmäiseen CEC-toimikunnan (Clinical Events Committee) enintään 7 päivää viimeisen tutkimuslääkeannoksen jälkeen vahvistamaan sairauden etenemiseen kulunut aika. Toissijaisia ja eksploratiivisia päätetapahtumia olivat turvallisuus ja siedettävyys, muutos 6MWD-kokeessa, WHO:n toimintakykyluokassa ja B‑tyypin natriureettista N‑terminaalista propeptidiä (NT-proBNP) koskevissa tiedoissa, kaikukardiografiassa, fyysisessä aktiivisuudessa ja elämänlaatua koskevissa mittareissa.
Hoidon keston mediaani oli 50 viikkoa, ja noin 50 % potilaista sai hoitoa 12 kuukauden ajan. Valtaosalla potilaista oli idiopaattinen PAH (55,8 %), runkohoitona yhdistelmähoito (74,6 %) ja WHO:n toimintakykyluokka II (76,8 %). Keskimääräinen ikä oli 11,8 vuotta (vaihteluväli 3–18 vuotta).
CEC‑toimikunnan vahvistamia sairauden etenemistä koskevia tapahtumia raportoitiin 16 (23,2 %) potilaalla seleksipagiryhmässä ja 11 (15,9 %) potilaalla lumeryhmässä.
Raportoitujen haittatapahtumien luonne oli yhdenmukainen seleksipagin tunnetun turvallisuusprofiilin kanssa (tyypillisiä pääasiassa prostasykliinityyppiset liittyvät haittavaikutukset, ks. kohta Haittavaikutukset) ja PAH-potilaspopulaatiolla oletettujen tapahtumien, mukaan lukien PAH:n etenemiseen liittyvien haittatapahtumien, kanssa. Titrausjakson aikana oksentelua raportoitiin haittavaikutuksena yleisemmin (19:llä [27,5 %] seleksipagiryhmässä ja 5:llä [7,2 %] lumeryhmässä) kuin aikuisilla (ks. kohta Haittavaikutukset). PAH:n eteneminen oli yleisimmin raportoitu vakava haittatapahtuma (8 [11,6 %] potilaalla seleksipagiryhmässä verrattuna 4:ään [5,8 %] lumeryhmässä). Mistä tahansa syystä aiheutuneiden kuolemien kokonaislukumäärä oli 7 (10,1 %) seleksipagiryhmässä ja 5 (7,2 %) lumeryhmässä. Näistä 5 (7,2 %) tapahtui seleksipagihoidon aikana ja 3 (4,3 %) lumehoidon aikana. Yhtä lukuun ottamatta kaikki kuolemat liittyivät PAH:iin.
Farmakokinetiikka
Seleksipagin ja sen aktiivisen metaboliitin farmakokinetiikkaa on arvioitu pääasiassa terveillä henkilöillä. Seleksipagin ja sen aktiivisen metaboliitin farmakokinetiikka oli sekä kerta-annoksen että toistuvien annosten jälkeen suhteessa annokseen, kun arvioinnissa käytettiin enintään 800 mikrog kerta-annoksia ja enintään 1 800 mikrog. x 2 toistuvia annoksia. Toistuvan annostelun jälkeen seleksipagin ja sen aktiivisen metaboliitin vakaa tila saavutettiin 3 päivässä. Toistuvan annostelun jälkeen kanta-aine ja aktiivinen metaboliitti eivät kumuloituneet plasmaan.
Terveillä henkilöillä tutkittavien välinen vaihtelu altistuksessa (area under the curve (AUC)-ala annosvälin aikana) seleksipagilla vaihteli vakaassa tilassa 43 % ja vastaava altistus aktiiviselle metaboliitille 39 %. Saman tutkittavan altistusarvojen vaihtelu oli seleksipagin kohdalla 24 % ja aktiivisen metaboliitin kohdalla 19 %.
Vakaan tilan altistus seleksipagille ja aktiiviselle metaboliitille oli samaa luokkaa sekä PAH-potilailla että terveillä henkilöillä. Seleksipagin ja aktiivisen metaboliitin farmakokinetiikka PAH-potilailla ei riippunut taudin vaikeusasteesta eikä muuttunut ajan mittaan.
Imeytyminen
Seleksipagi imeytyy nopeasti ja hydrolysoituu karboksyyliesteraasien välityksellä aktiiviseksi metaboliitikseen.
Peroraalisen annon jälkeen suurimmat havaitut plasman seleksipagipitoisuudet saavutetaan 1-3 tunnissa ja suurimmat havaitut aktiivisen metaboliitin pitoisuudet plasmassa 3-4 tunnissa.
Seleksipagin absoluuttinen biologinen hyötyosuus on ihmisellä noin 49 %. Tämä johtuu todennäköisimmin seleksipagin ensikierron metaboliasta, sillä valmisteen anto suun kautta tuottaa samankaltaiset aktiivisen metaboliitin pitoisuudet plasmassa kuin saman annoksen anto laskimoon.
Ruoan yhteydessä 400 mikrog. seleksipagin kerta-annoksen tuottama altistus suureni 10 % valkoihoisilla ja pieneni 15 % japanilaisilla, kun taas altistus aktiiviselle metaboliitille pieneni 27 % (valkoihoiset) ja 12 % (japanilaiset). Lääkkeen ottaminen tyhjään mahaan suurensi haittatapahtumista ilmoittaneiden tutkittavien määrää verrattuna tilanteeseen, jossa lääke otettiin ruoan kanssa.
Jakautuminen
Seleksipagi ja sen aktiivinen metaboliitti sitoutuvat voimakkaasti plasman proteiineihin (noin 99 % kokonaismäärästä; yhtä runsaasti albumiiniin ja happamaan alfa-1-glykoproteiiniin). Seleksipagin vakaan tilan jakautumistilavuus on 11,7 l.
Biotransformaatio
Seleksipagi hydrolysoituu aktiiviseksi metaboliitikseen maksassa ja suolessa karboksyyliesteraasien välityksellä. Lähinnä CYP2C8:n ja vähäisemmässä määrin myös CYP3A4:n katalysoima oksidatiivinen metabolia johtaa hydroksylaatio- ja dealkylaatiotuotteiden muodostukseen. UGT1A3 ja UGT2B7 osallistuvat aktiivisen metaboliitin glukuronidaatioon. Aktiivista metaboliittia lukuun ottamatta minkään ihmisplasmassa kiertävän metaboliitin osuus kaikesta lääkemateriaalista ei ylitä 3 prosenttia. Suun kautta annettaessa altistus aktiiviselle metaboliitille on vakaassa tilassa noin 3–4 kertaa suurempaa kuin altistus kanta-aineelle sekä terveillä henkilöillä että PAH-potilailla.
Eliminaatio
Seleksipagi eliminoituu pääasiassa metaboloitumalla, ja sen terminaalisen puoliintumisajan keskiarvo on 0,8-2,5 h. Aktiivisen metaboliitin puoliintumisaika on 6,2-13,5 h. Seleksipagin kokonaispuhdistuma elimistöstä on 17,9 l/h. Terveillä henkilöillä lääke eliminoitui täysin 5 päivän kuluessa annosta, lähinnä ulosteeseen (93 % annetusta annoksesta); virtsaan erittyi 12 %.
Erityisryhmät
Sukupuolen, etnisen taustan, iän ja painon ei ole todettu vaikuttavan kliinisesti merkittävällä tavalla seleksipagin eikä sen aktiivisen metaboliitin farmakokinetiikkaan terveillä henkilöillä eikä PAH-potilailla.
Pediatriset potilaat
Seleksipagin farmakokinetiikkaa iältään ≥ 2 – < 18-vuotiailla pediatrisilla PAH-potilailla tutkittiin yhden hoitohaaran avoimessa vaiheen 2 tutkimuksessa (AC-065A203 [N = 62]) ja SALTO-tutkimuksessa (N = 36) (ks. kohta Farmakodynamiikka).
Pediatrisille potilaille annettiin seleksipagia aloitusannoksena 100 mikrogrammaa kahdesti vuorokaudessa (paino ≥ 9 kg – < 25 kg), 150 mikrogrammaa kahdesti vuorokaudessa (paino ≥ 25 kg – < 50 kg) ja 200 mikrogrammaa kahdesti vuorokaudessa (paino ≥ 50 kg). Annos suurennettiin titraamalla suurimpaan yksilöllisesti siedettyyn annokseen enintään 800 mikrogrammaan kahdesti vuorokaudessa (paino ≥ 9 kg – < 25 kg), 1 200 mikrogrammaan kahdesti vuorokaudessa (paino ≥ 25 kg – < 50 kg) ja 1 600 mikrogrammaan kahdesti vuorokaudessa (paino ≥ 50 kg). Käytetyistä painonmukaisista hoito-ohjelmista saatu yhdistetty altistus seleksipagille ja sen aktiiviselle metaboliitille oli verrannollinen aikuisilla havaitun yhdistetyn altistuksen kanssa.
Munuaisten vajaatoiminta
Altistuksen seleksipagille ja sen aktiiviselle metaboliitille (maksimipitoisuus plasmassa ja pitoisuus-aikakäyrän alle jäävä AUC-pinta-ala) todettiin suurentuneen 1,4-1,7-kertaiseksi vaikeaa munuaisten vajaatoimintaa sairastavilla (eGFR < 30 ml/min/1,73 m2).
Maksan vajaatoiminta
Lievää (Child-Pugh-luokka A) tai keskivaikeaa (Child-Pugh-luokka B) maksan vajaatoimintaa sairastavilla seleksipagialtistus oli 2 kertaa suurempi (luokka A) ja 4 kertaa suurempi (luokka B) kuin terveillä henkilöillä. Altistus aktiiviselle metaboliitille ei muuttunut juurikaan lievää maksan vajaatoimintaa sairastavilla ja kaksinkertaistui keskivaikeaa maksan vajaatoimintaa sairastavilla. Vain kahdelle vaikeaa (Child-Pugh-luokka C) maksan vajaatoimintaa sairastavalle henkilölle annettiin seleksipagia. Näillä kahdella henkilöllä altistus seleksipagille ja sen aktiiviselle metaboliitille oli samaa luokkaa kuin henkilöillä, joilla oli keskivaikea (Child-Pugh-luokka B) maksan vajaatoiminta.
Maksan vajaatoimintaa sairastavilla tehdyn tutkimuksen perusteella tehdyn mallinnuksen ja simulaatiotietojen perusteella arvioidaan, että vakaan tilan seleksipagialtistus on keskivaikeaa maksan vajaatoimintaa (Child-Pugh-luokka B) sairastavilla kerran vuorokaudessa tapahtuvan annostelun jälkeen noin 2 kertaa suurempi kuin terveillä henkilöillä kahdesti vuorokaudessa tapahtuvan annostelun jälkeen. Vakaan tilan altistus aktiiviselle metaboliitille arvioidaan olevan näillä potilailla lääkettä kerran vuorokaudessa annosteltaessa samaa luokkaa kuin terveillä henkilöillä kahdesti vuorokaudessa tapahtuvan annostelun jälkeen. Vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavilla todettiin, että ennustettu vakaan tilan altistus oli samaa luokkaa kuin keskivaikeaa maksan vajaatoimintaa sairastavilla, kun lääke annosteltiin kerran vuorokaudessa.
Prekliiniset tiedot turvallisuudesta
Jyrsijöillä tehdyissä toistuvaisannosten toksisuustutkimuksissa korostuneen farmakologisen vaikutuksen aiheuttama voimakas verenpaineen lasku aiheutti ohimeneviä kliinisiä löydöksiä ja vähensi syömistä ja painon nousua. Aikuisilla ja nuorilla koirilla suoli ja luu/luuydin todettiin seleksipagihoidon jälkeen tärkeimmiksi toksisuuden kohde-elimiksi. Nuorilla koirilla todettiin reisiluun ja/tai tibian epifyseaalisen kasvulevyn sulkeutumisen hidastumista. NOAEL-annostasoa eli annostasoa, joka ei aiheuttanut havaittuja haittavaikutuksia, ei määritetty. Nuorilla koirilla havaittiin sporadisesti suolentuppeumaa, joka johtui prostasykliinin kaltaisista vaikutuksista suolen motiliteettiin. IP-reseptoriin kohdistuvan tehon suhteen korjatut aktiivisen metaboliitin turvallisuusmarginaalit olivat 2-kertaiset (kokonaisaltistuksen perusteella) verrattuna ihmisen altistukseen hoitoannoksilla. Kyseistä löydöstä ei todettu hiirellä eikä rotalla tehdyissä toksisuustutkimuksissa. Koska koirien taipumus suolentuppeumaan on lajispesifinen, löydöksellä ei katsota olevan merkitystä aikuisille ihmisille.
Koiratutkimuksissa todettiin luun luutumisen lisääntymistä ja tähän liittyviä luuydinmuutoksia, minkä katsotaan johtuvan EP4-reseptorien aktivaatiosta koirilla. Seleksipagi ja sen aktiivinen metaboliitti eivät aktivoi ihmisen EP4-reseptoreja, joten vaikutus on lajispesifinen eikä sillä siis ole merkitystä ihmiselle.
Seleksipagi ja sen aktiivinen metaboliitti eivät ole genotoksisia perustuen genotoksisuustutkimusten tuottamaan kokonaisnäyttöön.
Kaksivuotisissa karsinogeenisuustutkimuksissa seleksipagi suurensi kilpirauhasadenoomien ilmaantuvuutta hiirellä ja leydiginsoluadenoomien ilmaantuvuutta rotalla. Mekanismit ovat jyrsijöille spesifisiä. Kahden hoitovuoden jälkeen todettiin verkkokalvon arteriolien kiemuraisuutta vain rotalla. Mekanistisesti ajatellen vaikutuksen katsotaan johtuvan elinikäisestä vasodilataatiosta ja tämän aiheuttamista muutoksista silmän hemodynamiikassa. Muita seleksipagin aiheuttamia histopatologisia löydöksiä on todettu vain silloin, kun on käytetty altistusta, joka ylittää suurimman ihmisille käytettävän annostuksen niin huomattavasti, että asialla on ihmiselle vain vähäinen merkitys.
Rotalla tehdyssä hedelmällisyystutkimuksessa todettiin kiimakierron pitenemistä ja paritteluun kuluvan ajan (päiviä) pitenemistä, kun altistus oli 173-kertaisesti terapeuttista altistusta suurempi (kokonaisaltistusten perusteella); annostaso, jolla ei todettu vaikutuksia, oli 30 kertaa terapeuttista altistusta suurempi. Hedelmällisyysparametreissa ei tapahtunut muita muutoksia.
Seleksipagi ei ollut teratogeeninen rotalla eikä kanilla (altistus seleksipagille 13-kertainen ja altistus aktiiviselle metaboliitille 43-kertainen verrattuna terapeuttiseen altistukseen; luvut perustuvat kokonaisaltistukseen). Mahdollisten IP-reseptoriin liittyvien lisääntymiseen kohdistuvien vaikutusten turvallisuusmarginaali hedelmällisyyden suhteen oli 20 ja turvallisuusmarginaali alkion- ja sikiönkehityksen suhteen rotalla 5 ja kanilla 1 (vapaan altistuksen perusteella), kun tiedot korjattiin reseptoriin kohdistuvan tehon erojen suhteen. Rotalla tehdyssä pre- ja postnataalista kehitystä koskeneessa tutkimuksessa seleksipagi ei aiheuttanut emon eikä poikasten lisääntymistoimintoihin kohdistuneita muutoksia.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mannitoli (E421)
Maissitärkkelys
Matalasubstituutioasteinen hydroksipropyyliselluloosa
Hydroksipropyyliselluloosa
Magnesiumstearaatti
Kalvopäällyste
Uptravi 100 mikrogrammaa kalvopäällysteinen tabletti
Hypromelloosi (E464)
Propyleeniglykoli (E1520)
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Karnaubavaha
Talkki
Uptravi 200 mikrogrammaa kalvopäällysteinen tabletti
Hypromelloosi (E464)
Propyleeniglykoli (E1520)
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Karnaubavaha
Uptravi 400 mikrogrammaa kalvopäällysteinen tabletti
Hypromelloosi (E464)
Propyleeniglykoli (E1520)
Titaanidioksidi (E171)
Punainen rautaoksidi (E172)
Karnaubavaha
Uptravi 600 mikrogrammaa kalvopäällysteinen tabletti
Hypromelloosi (E464)
Propyleeniglykoli (E1520)
Titaanidioksidi (E171)
Punainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Karnaubavaha
Uptravi 800 mikrogrammaa kalvopäällysteinen tabletti
Hypromelloosi (E464)
Propyleeniglykoli (E1520)
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Karnaubavaha
Uptravi 1 000 mikrogrammaa kalvopäällysteinen tabletti
Hypromelloosi (E464)
Propyleeniglykoli (E1520)
Titaanidioksidi (E171)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Karnaubavaha
Uptravi 1 200 mikrogrammaa kalvopäällysteinen tabletti
Hypromelloosi (E464)
Propyleeniglykoli (E1520)
Titaanidioksidi (E171)
Musta rautaoksidi (E172)
Punainen rautaoksidi (E172)
Karnaubavaha
Uptravi 1 400 mikrogrammaa kalvopäällysteinen tabletti
Hypromelloosi (E464)
Propyleeniglykoli (E1520)
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Karnaubavaha
Uptravi 1 600 mikrogrammaa kalvopäällysteinen tabletti
Hypromelloosi (E464)
Propyleeniglykoli (E1520)
Titaanidioksidi (E171)
Musta rautaoksidi (E172)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Karnaubavaha
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Läpipainopakkaus: 4 vuotta.
Avaamaton purkki: 2 vuotta. Avattu purkki: 5 kuukautta tai viimeiseen käyttöpäivämäärään saakka (sen mukaan, kumpi näistä määräajoista umpeutuu ensin).
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
UPTRAVI tabletti, kalvopäällysteinen
200 mikrog (L:ei) 60 fol (3934,82 €), 140 fol (8940,46 €)
400 mikrog (L:ei) 60 fol (3934,82 €)
600 mikrog (L:ei) 60 fol (3934,82 €)
800 mikrog (L:ei) 60 fol (3934,82 €)
1000 mikrog (L:ei) 60 fol (3934,82 €)
1200 mikrog (L:ei) 60 fol (3934,82 €)
1400 mikrog (L:ei) 60 fol (3934,82 €)
1600 mikrog (L:ei) 60 fol (3934,82 €)
PF-selosteen tieto
Uptravi 100 mikrogrammaa kalvopäällysteinen tabletti
Suurtiheyspolyeteenipurkki (HDPE); purkissa on kierreturvasuljin, jonka korkissa on kuivatusaineena 1 g:n silikakapseli, sekä kuumasaumattu sinetti.
Kotelo, jossa 60 kalvopäällysteistä tablettia (purkit).
Kotelo, jossa 140 kalvopäällysteistä tablettia (titrauspakkaus, purkit).
Uptravi 200 mikrogrammaa kalvopäällysteinen tabletti
Polyamidi / alumiini / HDPE / PE, johon upotettu kuivatusainetta / HDPE-läpipainopakkaus, sinetöity alumiinifoliolla.
Yksi läpipainoliuska sisältää 10 kalvopäällysteistä tablettia.
Kotelo, jossa 10 tai 60 kalvopäällysteistä tablettia (1 tai 6 läpipainoliuskaa).
Kotelo, jossa 60 tai 140 kalvopäällysteistä tablettia (titrauspakkaukset, 6 tai 14 läpipainoliuskaa).
Uptravi 400 mikrogrammaa, 600 mikrogrammaa, 800 mikrogrammaa, 1 000 mikrogrammaa, 1 200 mikrogrammaa, 1 400 mikrogrammaa ja 1 600 mikrogrammaa, kalvopäällysteinen tabletti
Polyamidi / alumiini / HDPE / PE, johon upotettu kuivatusainetta / HDPE-läpipainopakkaus, sinetöity alumiinifoliolla.
Yksi läpipainoliuska sisältää 10 kalvopäällysteistä tablettia.
Kotelo, jossa 60 kalvopäällysteistä tablettia (6 läpipainoliuskaa).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Uptravi 100 mikrogrammaa kalvopäällysteinen tabletti
Pyöreä, 3,0 mm:n läpimittainen, vaaleankeltainen, kalvopäällysteinen tabletti, jonka toisella puolella on kaiverrus ”1”.
Uptravi 200 mikrogrammaa kalvopäällysteinen tabletti
Pyöreä, 7,3 mm:n läpimittainen, vaaleankeltainen, kalvopäällysteinen tabletti, jonka toisella puolella on kaiverrus ”2”.
Uptravi 400 mikrogrammaa kalvopäällysteinen tabletti
Pyöreä, 7,3 mm:n läpimittainen, punainen, kalvopäällysteinen tabletti, jonka toisella puolella on kaiverrus ”4”.
Uptravi 600 mikrogrammaa kalvopäällysteinen tabletti
Pyöreä, 7,3 mm:n läpimittainen, vaaleanvioletti, kalvopäällysteinen tabletti, jonka toisella puolella on kaiverrus ”6”.
Uptravi 800 mikrogrammaa kalvopäällysteinen tabletti
Pyöreä, 7,3 mm:n läpimittainen, vihreä, kalvopäällysteinen tabletti, jonka toisella puolella on kaiverrus ”8”.
Uptravi 1 000 mikrogrammaa kalvopäällysteinen tabletti
Pyöreä, 7,3 mm:n läpimittainen, oranssi, kalvopäällysteinen tabletti, jonka toisella puolella on kaiverrus ”10”.
Uptravi 1 200 mikrogrammaa kalvopäällysteinen tabletti
Pyöreä, 7,3 mm:n läpimittainen, tummanvioletti, kalvopäällysteinen tabletti, jonka toisella puolella on kaiverrus ”12”.
Uptravi 1 400 mikrogrammaa kalvopäällysteinen tabletti
Pyöreä, 7,3 mm:n läpimittainen, tummankeltainen, kalvopäällysteinen tabletti, jonka toisella puolella on kaiverrus ”14”.
Uptravi 1 600 mikrogrammaa kalvopäällysteinen tabletti
Pyöreä, 7,3 mm:n läpimittainen, ruskea, kalvopäällysteinen tabletti, jonka toisella puolella on kaiverrus ”16”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
UPTRAVI tabletti, kalvopäällysteinen
200 mikrog 60 fol, 140 fol
400 mikrog 60 fol
600 mikrog 60 fol
800 mikrog 60 fol
1000 mikrog 60 fol
1200 mikrog 60 fol
1400 mikrog 60 fol
1600 mikrog 60 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Seleksipagi: Vaikeahoitoisen pulmonaalihypertension hoito erityisin edellytyksin (383).
ATC-koodi
B01AC27
Valmisteyhteenvedon muuttamispäivämäärä
04.02.2026
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com