
BLINCYTO kuiva-aine välikonsentraatiksi ja liuos infuusionestettä varten, liuos 38,5 mikrog
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Hoitaja
Lisätietoja siitä, miten Blincyto-valmisteen käyttöön liittyvät neurologiset tapahtumat, mukaan lukien immuuniefektorisoluihin liittyvä neurotoksinen oireyhtymä (ICANS), voidaan minimoida tai ehkäistä. Sisältää linkin ja QR-koodin verkkopohjaiseen vuorovaikutteiseen versioon.
Potilas
painettuja kortteja voi tilata Amgenilta (yhteystiedot esim. potilaan ja hänestä huolehtivan henkilön oppaassa)
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo kuiva-ainetta sisältää 38,5 mikrogrammaa blinatumomabia.
Kun kuiva-aine liuotetaan injektionesteisiin käytettävään veteen, lopullinen blinatumomabipitoisuus on 12,5 mikrogrammaa/ml.
Blinatumomabi on tuotettu yhdistelmä‑DNA‑tekniikalla kiinanhamsterin munasarjasoluissa.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi ja liuos infuusionestettä varten, liuos.
Kliiniset tiedot
Käyttöaiheet
Blincyto on tarkoitettu monoterapiaksi aikuisille CD19‑positiivisen, uusiutuneen tai refraktaarisen B‑solulinjan akuutin lymfaattisen leukemian (pre-B-ALL) hoitoon. Jos potilaan pre‑B‑ALL on Philadelphia‑kromosomipositiivinen, edellytetään, että hoito vähintään kahdella tyrosiinikinaasin estäjällä (TKI) on epäonnistunut, eikä muita hoitovaihtoehtoja ole.
Blincyto on tarkoitettu monoterapiaksi aikuisille Philadelphia‑kromosominegatiivisen, CD19‑positiivisen, ensimmäisessä tai toisessa täydellisessä remissiossa olevan B-solulinjan akuutin lymfaattisen leukemian (pre‑B‑ALL) jäännöstaudin (MRD) hoitoon, kun sen määrä on suurempi tai yhtä suuri kuin 0,1 %.
Blincyto on tarkoitettu monoterapiaksi vähintään yhden kuukauden ikäisille lapsille Philadelphia‑kromosominegatiivisen, CD19‑positiivisen, B‑solulinjan akuutin lymfaattisen leukemian (pre‑B‑ALL) hoitoon, kun tauti on refraktaarinen tai uusiutunut vähintään kahden aiemman hoidon jälkeen tai uusiutunut aiemman allogeenisen hematopoieettisen kantasolusiirron (HSCT) jälkeen.
Blincyto on tarkoitettu monoterapiaksi vähintään yhden kuukauden ikäisille lapsille ensimmäistä kertaa uusiutuneen korkean riskin Philadelphia‑kromosominegatiivisen, CD19‑positiivisen, B‑solulinjan akuutin lymfaattisen leukemian (pre‑B‑ALL) hoitoon osana vakautushoitoa (ks. kohta Annostus ja antotapa).
Blincyto on tarkoitettu monoterapiaksi osaksi vakautushoitoa sellaisten aikuispotilaiden hoidossa, joilla on juuri diagnosoitu Philadelphia‑kromosominegatiivinen, CD19‑positiivinen, B‑solulinjan akuutti lymfaattinen leukemia (pre‑B‑ALL).
Ehto
Valmiste tulee antaa pahanlaatuisten veritautien solunsalpaajahoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito on aloitettava ja toteutettava pahanlaatuisten veritautien hoitoon perehtyneen lääkärin johdolla ja valvonnassa. Blincyto-hoitoa saaville potilaille on annettava potilaan ja hänestä huolehtivan henkilön opas sekä potilaskortti.
Uusiutuneen tai refraktaarisen pre-B‑ALL:n hoitoa aloitettaessa suositellaan sairaalahoitoa vähintään ensimmäisen hoitojakson 9 ensimmäisen päivän ajan ja toisen hoitojakson 2 ensimmäisen päivän ajan.
Philadelphia‑kromosominegatiivisen, MRD‑positiivisen pre-B‑ALL:n hoidossa suositellaan sairaalahoitoa vähintään ensimmäisen hoitojakson 3 ensimmäisen päivän ajan ja seuraavien hoitojaksojen 2 ensimmäisen päivän ajan.
Pre‑B‑ALL:n vakautushoitovaiheessa sairaalahoitoa suositellaan ensimmäisen hoitojakson 3 ensimmäisen päivän ajan ja toisen hoitojakson 2 ensimmäisen päivän ajan.
Jos potilaalla on tai on aikaisemmin ollut kliinisesti merkittäviä keskushermoston sairauksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), sairaalahoitoa suositellaan ensimmäisen hoitojakson yhteydessä vähintään 14 ensimmäisen päivän ajan. Toisen hoitojakson yhteydessä sairaalahoitoa suositellaan vähintään 2 päivän ajan, ja kliiniset päätökset tehdään sen perusteella, kuinka hyvin potilas sieti ensimmäisen Blincyto-hoitojakson. Varovaisuutta on noudatettava, sillä joissakin tapauksissa ensimmäiset neurologiset tapahtumat ovat ilmaantuneet viivästyneesti.
Kaikkien myöhempien hoitojaksojen aloituksen ja uudelleenaloittamisen (esim. jos hoito on keskeytetty vähintään 4 tunnin ajaksi) tulisi tapahtua terveydenhoitohenkilökunnan valvonnassa tai sairaalassa.
Annostus
Uusiutunut tai refraktaarinen pre-B‑ALL
Potilaat, joilla on uusiutunut tai refraktaarinen pre‑B‑ALL, voivat saada 2 hoitojaksoa. Yksi hoitojakso on 28 vuorokauden (4 viikon) jatkuva infuusio. Hoitojaksojen välissä on 14 vuorokauden (2 viikon) hoitotauko.
Potilaat, jotka saavuttavat täydellisen remission (CR/CRh*) 2 hoitojakson jälkeen, voivat saada vielä jopa 3 Blincyto-hoitojaksoa vakautushoitona yksilöllisen hyöty-riskiarvion perusteella.
Suositeltu vuorokausiannos määritetään kehonpainon perusteella (ks. taulukko 1). Jos potilas painaa vähintään 45 kg, hänelle annetaan kiinteä vuorokausiannos. Alle 45 kg painaville potilaille annos lasketaan kehon pinta‑alan (BSA) perusteella.
Taulukko 1. Suositeltu Blincyto-annostus uusiutuneen tai refraktaarisen pre‑B‑ALL:n hoitoon
| Kehonpaino | 1. hoitojakso | Myöhemmät hoitojaksot | |||
| Päivät 1–7 | Päivät 8–28 | Päivät 29–42 | Päivät 1–28 | Päivät 29–42 | |
| Vähintään 45 kg (kiinteä annos) | 9 mikrog/vrk jatkuvana infuusiona | 28 mikrog/vrk jatkuvana infuusiona | 14 vrk:n hoitotauko | 28 mikrog/vrk jatkuvana infuusiona | 14 vrk:n hoitotauko |
| Alle 45 kg (annos lasketaan kehon pinta-alan mukaan) | 5 mikrog/m2/ vrk jatkuvana infuusiona (enintään 9 mikrog/vrk) | 15 mikrog/m2/vrk jatkuvana infuusiona (enintään 28 mikrog/vrk) | 15 mikrog/m2/vrk jatkuvana infuusiona (enintään 28 mikrog/vrk) | ||
Esilääkitystä ja lisälääkitystä koskevat suositukset
Aikuispotilaille annetaan deksametasonia 20 mg laskimoon tuntia ennen jokaisen Blincyto-hoitojakson aloittamista.
Lapsipotilaille annetaan deksametasonia 10 mg/m2 (enintään 20 mg) suun kautta tai laskimoon 6–12 tuntia ennen Blincyto-hoidon aloittamista (1. hoitojakso, päivä 1). Tämän jälkeen annetaan deksametasonia 5 mg/m2 suun kautta tai laskimoon 30 minuutin kuluessa ennen Blincyto-hoidon aloittamista (1. hoitojakso, päivä 1).
Kuumelääkkeitä (esim. parasetamolia) suositellaan kuumeen alentamiseen jokaisen hoitojakson 48 ensimmäisen tunnin aikana.
Profylaktista intratekaalista solunsalpaajahoitoa suositellaan ennen Blincyto-hoitoa ja sen aikana ALL:n keskushermostorelapsin ehkäisemiseksi.
Edeltävä (pre-phase) hoito potilaille, joilla on suuri kasvaintaakka
Jos leukeemisten blastien osuus luuytimen soluista on ≥ 50 % tai leukeemisten blastien määrä perifeerisessä veressä on > 15 000/mikrolitra, annetaan deksametasonia (enintään 24 mg/vrk).
MRD‑positiivinen pre-B‑ALL
Kun Blincyton käyttöä harkitaan Philadelphia‑kromosominegatiivisen, MRD‑positiivisen pre-B‑ALL:n hoitoon, mitattava MRD on varmistettava validoidulla määritysmenetelmällä, jonka herkkyys on vähintään 10-4 (ks. kohta Farmakodynamiikka). Käytettävästä tekniikasta riippumatta MRD:n kliininen testaus on tehtävä kyseiseen tekniikkaan perehtyneessä laboratoriossa vakiintuneiden teknisten käytäntöjen mukaisesti.
Potilaat voivat saada 1 hoitojakson mittaisen induktiohoidon ja sen jälkeen vielä vakautushoitona enintään 3 hoitojaksoa Blincytoa. Yksi hoitojakso Blincytoa induktio- tai vakautushoitona on 28 vuorokauden (4 viikon) jatkuva infuusio laskimoon, jonka jälkeen on 14 vuorokauden (2 viikon) hoitotauko (yhteensä 42 vuorokautta). Valtaosa potilaista, joilla blinatumomabi tehoaa, saavuttaa hoitovasteen 1 hoitojakson jälkeen (ks. kohta Farmakodynamiikka). Tämän vuoksi hoitavan lääkärin on arvioitava hoidon jatkamisesta mahdollisesti saatavaa hyötyä ja hoidon riskejä potilailla, joiden hematologinen ja/tai kliininen tila ei kohennu 1 hoitojakson jälkeen. Katso suositeltu vuorokausiannos taulukosta 2.
Taulukko 2. Suositeltu Blincyto-annostus aikuispotilaille, joilla on MRD-positiivinen pre‑B‑ALL
| Kehonpaino | Hoitojakso(t) | |
| Päivät 1–28 | Päivät 29–42 | |
Vähintään 45 kg (kiinteä annos) | 28 mikrog/vrk | 14 vrk:n hoitotauko |
Alle 45 kg (annos lasketaan kehon pinta-alan mukaan) | 15 mikrog/m2/vrk (enintään 28 mikrog/vrk) | 14 vrk:n hoitotauko |
Esilääkitystä ja lisälääkitystä koskevat suositukset
Tuntia ennen jokaisen Blincyto-hoitojakson aloittamista annetaan laskimoon 100 mg prednisonia tai vastaavaa (esim. 16 mg deksametasonia).
Kuumelääkkeitä (esim. parasetamolia) suositellaan kuumeen alentamiseen jokaisen hoitojakson 48 ensimmäisen tunnin aikana.
Profylaktista intratekaalista solunsalpaajahoitoa suositellaan ennen Blincyto-hoitoa ja sen aikana ALL:n keskushermostorelapsin ehkäisemiseksi.
Pre‑B‑ALL vakautushoitovaiheessa
Blincytoa annetaan jatkuvana infuusiona laskimoon vakionopeudella infuusiopumppua käyttäen. Yksi hoitojakso on 28 vuorokauden (4 viikon) jatkuva infuusio, jonka jälkeen on 14 vuorokauden (2 viikon) hoitotauko. Potilaat voivat saada enintään 4 Blincyto-hoitojaksoa vakautushoitona.
Taulukossa 3 esitetään kehonpainon mukaan määritellyt suositellut vuorokausiannokset aikuisille. Jos potilas painaa vähintään 45 kg, hänelle annetaan kiinteä vuorokausiannos. Alle 45 kg painaville potilaille annos lasketaan kehon pinta‑alan (BSA) perusteella.
Taulukko 3. Suositeltu Blincyto-annostus aikuisille pre-B-ALL:n vakautushoitovaiheessa
| Kehonpaino | Vakautushoitojaksot (jaksot 1–4) | |
| Päivät 1–28 | Päivät 29–42 | |
Vähintään 45 kg (kiinteä annos) | 28 mikrog/vrk | 14 vrk:n hoitotauko |
Alle 45 kg (annos lasketaan kehon pinta-alan mukaan) | 15 mikrog/m2/vrk (enintään 28 mikrog/vrk) | 14 vrk:n hoitotauko |
Esilääkitystä ja lisälääkitystä koskevat suositukset
Aikuispotilaille annetaan deksametasonia 20 mg laskimoon enintään tuntia ennen jokaisen Blincyto-hoitojakson aloittamista.
Profylaktista intratekaalista solunsalpaajahoitoa suositellaan ennen Blincyto-hoitoa ja sen aikana ALL:n keskushermostorelapsin ehkäisemiseksi.
Ensimmäistä kertaa uusiutunut korkean riskin pre‑B‑ALL
Lapsipotilaat, joilla on ensimmäistä kertaa uusiutunut korkean riskin pre‑B‑ALL, voivat saada 1 Blincyto-hoitojakson induktiohoidon ja vakautushoitona annetun 2 solunsalpaajahoitoblokin jälkeen. Yksi hoitojakso on 28 vuorokauden (4 viikon) jatkuva infuusio. Taulukossa 4 on lapsipotilaan kehonpainon perusteella määritellyt suositellut vuorokausiannokset.
Taulukko 4. Suositeltu Blincyto-annostus lapsipotilaiden ensimmäistä kertaa uusiutuneen korkean riskin pre‑B‑ALL:n hoitoon induktiohoidon ja sen jälkeisen solunsalpaajahoidon jälkeen
| Yksi vakautushoitojakso | Kehonpaino vähintään 45 kg (kiinteä annos) | Kehonpaino alle 45 kg (annos lasketaan kehon pinta-alan mukaan) |
| Päivät 1–28 | 28 mikrog/vrk | 15 mikrog/m2/vrk (enintään 28 mikrog/vrk) |
Esilääkitystä ja lisälääkitystä koskevat suositukset
Lapsipotilaille annetaan deksametasonia 5 mg/m2 (enintään 20 mg) ennen ensimmäisen hoitojakson ensimmäistä Blincyto-annosta ja aloitettaessa infuusiota uudelleen vähintään 4 tunnin keskeytyksen jälkeen ensimmäisen jakson aikana.
Profylaktista intratekaalista solunsalpaajahoitoa suositellaan ennen Blincyto-hoitoa ja sen aikana ALL:n keskushermostorelapsin ehkäisemiseksi.
Annoksen säätäminen kaikissa käyttöaiheissa
Blincyto-hoidon keskeyttämistä tai lopettamista on harkittava tarpeen mukaan, jos havaitaan seuraavia vaikeita (3. asteen) tai hengenvaarallisia (4. asteen) haittavaikutuksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet): sytokiinioireyhtymä, tuumorilyysioireyhtymä, neurologiset haittavaikutukset, maksaentsyymiarvojen kohoaminen ja muut kliinisesti merkittävät haittavaikutukset.
Jos hoito on keskeytettynä haittavaikutuksen jälkeen enintään 7 vuorokautta, jatketaan samaa hoitojaksoa kunnes infuusion kesto on yhteensä 28 vuorokautta. Tähän sisältyvät sekä keskeytystä edeltäneet että sitä seuraavat päivät saman hoitojakson aikana. Jos haittavaikutuksesta johtuva keskeytys kestää yli 7 vuorokautta, aloitetaan uusi hoitojakso. Jos haittatapahtuman korjautuminen kestää yli 14 vuorokautta, Blincyto lopetetaan pysyvästi, paitsi jos seuraavan taulukon 5 ohjeet poikkeavat tästä.
Taulukko 5. Haittavaikutusten hoitosuositukset (pois lukien ICANS-oireyhtymä)
| Haittavaikutus | Vaikeusaste* | Toimenpide, kun potilas painaa vähintään 45 kg | Toimenpide, kun potilas painaa alle 45 kg |
| Sytokiinioireyhtymä, tuumorilyysioireyhtymä | 3. aste | Blincyto keskeytetään, kunnes tila korjautuu, sen jälkeen Blincyto aloitetaan uudelleen annoksella 9 mikrog/vrk. Annos nostetaan tasolle 28 mikrog/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. | Blincyto keskeytetään, kunnes tila korjautuu, sen jälkeen Blincyto aloitetaan uudelleen annoksella 5 mikrog/m2/vrk. Annos nostetaan tasolle 15 mikrog/m2/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. |
| 4. aste | Blincyto lopetetaan pysyvästi. | Blincyto lopetetaan pysyvästi. | |
| Neurologinen haittavaikutus (pois lukien ICANS) | 3. aste | Blincyto keskeytetään, kunnes vaikeusaste on enintään 1 (lievä), ja vähintään 3 vuorokauden ajaksi, sen jälkeen Blincyto aloitetaan uudelleen annoksella 9 mikrog/vrk. Annos nostetaan tasolle 28 mikrog/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. Ennen uudelleenaloitusta annetaan esilääkityksenä 24 mg:n annos deksametasonia. Sitten deksametasoniannosta pienennetään asteittain 4 vuorokauden aikana. Jos haittavaikutus ilmaantui annoksen ollessa 9 mikrog/vrk tai jos sen korjautuminen kestää yli 7 vuorokautta, Blincyto lopetetaan pysyvästi. | Blincyto keskeytetään, kunnes vaikeusaste on enintään 1 (lievä), ja vähintään 3 vuorokauden ajaksi, sen jälkeen Blincyto aloitetaan uudelleen annoksella 5 mikrog/m2/vrk. Annos nostetaan tasolle 15 mikrog/m2/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. Jos haittavaikutus ilmaantui annoksen ollessa 5 mikrog/m2/vrk tai jos sen korjautuminen kestää yli 7 vuorokautta, Blincyto lopetetaan pysyvästi. |
| 4. aste | Blincyto lopetetaan pysyvästi. | Blincyto lopetetaan pysyvästi. | |
| Maksaentsyymiarvojen kohoaminen | 3. aste | Jos kliinisesti merkittävä, Blincyto keskeytetään, kunnes vaikeusaste on enintään 1 (lievä), sen jälkeen Blincyto aloitetaan uudelleen annoksella 9 mikrog/vrk. Annos nostetaan tasolle 28 mikrog/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. | Jos kliinisesti merkittävä, Blincyto keskeytetään, kunnes vaikeusaste on enintään 1 (lievä), sen jälkeen Blincyto aloitetaan uudelleen annoksella 5 mikrog/m2/vrk. Annos nostetaan tasolle 15 mikrog/m2/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. |
| 4. aste | Harkitaan Blincyton lopettamista pysyvästi. | Harkitaan Blincyton lopettamista pysyvästi. | |
| Muut (hoitavan lääkärin arvion mukaan) kliinisesti merkittävät haittavaikutukset | 3. aste | Blincyto keskeytetään, kunnes vaikeusaste on enintään 1 (lievä), sen jälkeen Blincyto aloitetaan uudelleen annoksella 9 mikrog/vrk. Annos nostetaan tasolle 28 mikrog/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. | Blincyto keskeytetään, kunnes vaikeusaste on enintään 1 (lievä), sen jälkeen Blincyto aloitetaan uudelleen annoksella 5 mikrog/m2/vrk. Annos nostetaan tasolle 15 mikrog/m2/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. |
| 4. aste | Harkitaan Blincyton lopettamista pysyvästi. | Harkitaan Blincyton lopettamista pysyvästi. | |
| * Perustuu NCI CTCAE-luokitukseen (National Cancer Institute Common Terminology Criteria for Adverse Events, versio 4.0). 3. asteen haittavaikutus on vaikea ja 4. asteen haittavaikutus on hengenvaarallinen. | |||
Taulukko 6. Immuuniefektorisoluihin liittyvän neurotoksisen oireyhtymän (ICANS) hoitosuositukset
| Vaikeusastea | Ilmenneet oireetb | Toimenpiteet |
| 1. aste | ICE-pisteet 7–9c CAPD-pisteet 1–8* tai alentunut tajunnantasod: herää spontaanisti. | Blincyto keskeytetään, kunnes ICANS-oireyhtymä häviää. Seuraa neurologisia oireita ja harkitse neurologin konsultaatiota lisäarviointia ja hoitoa varten. Harkitse ei-sedatoivia kouristuslääkkeitä (esim. levetirasetaamia) kouristuskohtausten ehkäisemiseksi. Toimenpide, kun potilas painaa ≥ 45 kg: Harkitse deksametasonihoitoa, jossa annetaan enintään 8 mg:n kerta-annos ja enintään 3 annosta 24 tunnin aikana. Hoidon uudelleenaloittamiseksi annetaan esilääkityksenä enintään 20 mg deksametasonia 1–3 tuntia ennen Blincyto-hoitoa. Toimenpide, kun potilas painaa < 45 kg: Harkitse deksametasonihoitoa, jossa kokonaisvuorokausiannos on enintään 0,2–0,4 mg/kg/vrk (enintään 24 mg/vrk). Hoidon uudelleenaloittamiseksi annetaan esilääkityksenä 5 mg/m2 deksametasonia (enintään 20 mg:n annos) 1–3 tuntia ennen Blincyto-hoitoa. |
| 2. aste | ICE-pisteet 3–6c CAPD-pisteet 1–8* tai alentunut tajunnantasod: herää puhuteltaessa. | Blincyto keskeytetään. Anna deksametasonia: Potilaille, jotka painavat ≥ 45 kg: Anna deksametasonia 8 mg:n kerta-annos ja enintään 3 annosta/vrk (enintään 24 mg/vrk) joko enintään 2 vuorokauden ajan tai kunnes tapahtuma korjautuu (sen mukaan, kumpi tapahtuu ensin). Potilaille, jotka painavat < 45 kg: Anna deksametasonihoitoa, jossa kokonaisvuorokausiannos on vähintään 0,2–0,4 mg/kg/vrk (enintään 24 mg/vrk) ja joka annetaan 3 annoksena joko enintään 2 vuorokauden ajan tai kunnes tapahtuma korjautuu (sen mukaan, kumpi tapahtuu ensin). Seuraa neurologisia oireita ja harkitse neurologin ja muiden erikoislääkäreiden konsultaatiota lisäarviointia ja hoitoa varten. Harkitse ei-sedatoivia kouristuslääkkeitä (esim. levetirasetaamia) kouristuskohtausten ehkäisemiseksi. Toimenpide, kun potilas painaa ≥ 45 kg: Blincyto keskeytetään, kunnes ICANS-oireyhtymä häviää, sen jälkeen Blincyto aloitetaan uudelleen annoksella 9 mikrog/vrk. Annos nostetaan tasolle 28 mikrog/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. Hoidon uudelleenaloittamiseksi annetaan esilääkityksenä enintään 20 mg deksametasonia 1–3 tuntia ennen Blincyto-hoitoa. Toimenpide, kun potilas painaa < 45 kg: Blincyto keskeytetään, kunnes ICANS-oireyhtymä häviää, sen jälkeen Blincyto aloitetaan uudelleen annoksella 5 mikrog/m2/vrk. Annos nostetaan tasolle 15 mikrog/m2/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. Hoidon uudelleenaloittamiseksi annetaan esilääkityksenä 5 mg/m2 deksametasonia (enintään 20 mg:n annos) 1–3 tuntia ennen Blincyto-hoitoa. |
| 3. aste | ICE-pisteet 0–2c CAPD ≥ 9 tai alentunut tajunnantasod:
tai kouristuskohtauksiad, joko
tai kohonnut kallonsisäinen paine:
| Blincyto keskeytetään. Anna deksametasonia: Potilaille, jotka painavat ≥ 45 kg: Anna deksametasonia 8 mg:n kerta-annos 3 annosta/vrk (enintään 24 mg/vrk), kunnes vaikeusaste lievenee 1. asteelle tai lievemmäksi, ja lopeta hoito sitten annosta vähitellen pienentämällä kliinisen tarpeen mukaisesti. Potilaille, jotka painavat < 45 kg: Anna deksametasonihoitoa, jossa kokonaisvuorokausiannos on vähintään 0,2–0,4 mg/kg/vrk (enintään 24 mg/vrk), kunnes vaikeusaste lievenee 1. asteelle tai lievemmäksi, ja lopeta hoito sitten annosta vähitellen pienentämällä kliinisen tarpeen mukaisesti. Seuraa neurologisia oireita ja harkitse neurologin ja muiden erikoislääkäreiden konsultaatiota lisäarviointia ja hoitoa varten. Harkitse ei-sedatoivia kouristuslääkkeitä (esim. levetirasetaamia) kouristuskohtausten ehkäisemiseksi. Anna tukihoitoa, johon voi kuulua myös tehohoitoa. Toimenpide, kun potilas painaa ≥ 45 kg: Blincyto keskeytetään, kunnes ICANS-oireyhtymä häviää, sen jälkeen Blincyto aloitetaan uudelleen annoksella 9 mikrog/vrk. Annos nostetaan tasolle 28 mikrog/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. Hoidon uudelleenaloittamiseksi annetaan esilääkityksenä enintään 20 mg deksametasonia 1–3 tuntia ennen Blincyto-hoitoa. Jos haittavaikutus ilmaantui annoksen ollessa 9 mikrog/vrk tai jos sen korjautuminen kestää yli 7 vuorokautta, Blincyto lopetetaan pysyvästi. Toimenpide, kun potilas painaa < 45 kg: Blincyto keskeytetään, kunnes ICANS-oireyhtymä häviää, sen jälkeen Blincyto aloitetaan uudelleen annoksella 5 mikrog/m2/vrk. Annos nostetaan tasolle 15 mikrog/m2/vrk 7 vuorokauden kuluttua, ellei haittavaikutus uusiudu. Hoidon uudelleenaloittamiseksi annetaan esilääkityksenä 5 mg/m2 deksametasonia (enintään 20 mg:n annos) 1–3 tuntia ennen Blincyto-hoitoa. Jos haittavaikutus ilmaantui annoksen ollessa 5 mikrog/m2/vrk tai jos sen korjautuminen kestää yli 7 vuorokautta, Blincyto lopetetaan pysyvästi. Toimenpide kaikille potilaille: Lopeta Blincyto pysyvästi, jos kohtauksia on enemmän kuin yksi. |
| 4. aste | ICE-pisteet 0c CAPD-arviointia ei voitu tehdä* tai alentunut tajunnantasod, joko
tai kouristuskohtauksiad, joko
tai motorisia löydöksiäd:
tai kohonnut kallonsisäinen paine / aivoturvotusd, johon liittyy esim. seuraavia oireita/löydöksiä:
| Blincyto lopetetaan pysyvästi. Anna deksametasonia: Potilaille, jotka painavat ≥ 45 kg: Anna deksametasonia 8 mg:n kerta-annos 3 annosta/vrk, kunnes vaikeusaste lievenee 1. asteelle tai lievemmäksi, ja lopeta hoito sitten annosta vähitellen pienentämällä kliinisen tarpeen mukaisesti. Vaihtoehtoisesti harkitse metyyliprednisolonin antamista laskimoon 1 000 mg/vrk 3 vuorokauden ajan. Lopeta hoito sitten annosta vähitellen pienentämällä kliinisen tarpeen mukaisesti. Potilaille, jotka painavat < 45 kg: Anna deksametasonihoitoa, jossa kokonaisvuorokausiannos on vähintään 0,2–0,4 mg/kg/vrk, kunnes vaikeusaste lievenee 1. asteelle tai lievemmäksi, ja lopeta hoito sitten annosta vähitellen pienentämällä kliinisen tarpeen mukaisesti. Vaihtoehtoisesti harkitse metyyliprednisolonin antamista laskimoon 30 mg/kg/vrk (enintään 1 000 mg/vrk) jaettuina annoksina 3 vuorokauden ajan. Lopeta hoito sitten annosta vähitellen pienentämällä kliinisen tarpeen mukaisesti. Seuraa neurologisia oireita ja harkitse neurologin ja muiden erikoislääkäreiden konsultaatiota lisäarviointia ja hoitoa varten. Harkitse ei-sedatoivia kouristuslääkkeitä (esim. levetirasetaamia) kouristuskohtausten ehkäisemiseksi. Anna tukihoitoa, johon voi kuulua myös tehohoitoa. |
Käytä vähintään 12-vuotiaille potilaille ICE-pisteytystä (immuuniefektorisoluihin liittyvä enkefalopatia-pisteytys). Käytä alle 12-vuotiaille potilaille CAPD (Cornell Assessment of Paediatric Delirium) -arviointityökalua. Lisätietoja CAPD-arvioinnista, ks. Lee et al, 2019. a Perustuu American Society for Transplantation and Cellular Therapy (ASTCT) -yhdistyksen ICANS-luokitukseen (2019). b Hoito määritetään sellaisen vaikeimman tapahtuman mukaan, jolle ei ole muuta syytä. c Jos potilas on heräteltävissä ja ICE-arviointi voidaan tehdä, arvioi: orientaatio (tietää vuoden, kuukauden, paikkakunnan, sairaalan = 4 pistettä); nimeäminen (nimeä 3 esinettä, esim. osoita kelloa, kynää, nappia = 3 pistettä); kehotusten noudattaminen (esim. "näytä minulle 2 sormea" tai "sulje silmäsi ja näytä kieltä" = 1 piste); kirjoittaminen (kykenee kirjoittamaan tavanomaisen virkkeen = 1 piste); ja tarkkaavaisuus (luettele numeroita takaperin 100:sta kymmenen numeron välein = 1 piste). Jos potilas ei ole heräteltävissä eikä ICE-arviointia voida tehdä (4. asteen ICANS) = 0 pistettä. d Ei ole muuta syytä. e Kaikki viittaukset deksametasonin antoon tarkoittavat deksametasonia tai vastaavia lääkevalmisteita. * Pisteet 1–8 saattavat tarkoittaa ei heikentymistä tai 1. tai 2. asteen ICANS-oireyhtymä, joten tällaiseen tulokseen on yhdistettävä kliininen arviointi. | ||
Erityisryhmät
Iäkkäät potilaat
Iäkkäiden (≥ 65‑vuotiaiden) potilaiden annosta ei tarvitse muuttaa, ks. kohta Farmakodynamiikka. Blincytosta on vain vähän kokemuksia ≥ 75‑vuotiaiden potilaiden hoidossa.
Munuaisten vajaatoiminta
Farmakokineettisten analyysien perusteella lievä tai kohtalainen munuaisten vajaatoiminta ei vaadi annoksen sovittamista (ks. kohta Farmakokinetiikka). Blincyton turvallisuutta ja tehoa ei ole tutkittu vaikean munuaisten vajaatoiminnan yhteydessä.
Maksan vajaatoiminta
Farmakokineettisten analyysien perusteella maksan toiminta lähtötilanteessa ei todennäköisesti vaikuta blinatumomabialtistukseen eikä aloitusannosta tarvitse muuttaa (ks. kohta Farmakokinetiikka). Blincyton turvallisuutta ja tehoa ei ole tutkittu vaikean maksan vajaatoiminnan yhteydessä.
Pediatriset potilaat
Blincyton käytöstä alle 1‑vuotiaiden lapsipotilaiden hoidossa on vain rajallisesti kokemusta. Saatavissa olevat, lapsipotilaita koskevat tiedot on kuvattu kohdissa Haittavaikutukset ja Farmakodynamiikka.
Antotapa
Blincyto annetaan laskimoon.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen käsittelystä ja saattamisesta käyttökuntoon ennen lääkkeen antoa.
Anna Blincyto jatkuvana infuusiona laskimoon vakionopeudella infuusiopumppua käyttäen enintään 96 tunnin aikana. Pumpun on oltava ohjelmoitava ja lukittava, se ei saa olla elastomeerinen ja siinä on oltava hälytin.
Aloitustilavuus (270 ml) on suurempi kuin potilaalle annettava tilavuus (240 ml), jotta varmistetaan että infuusioletkusto saadaan täytettyä ja potilas saa täyden Blincyto-annoksen.
Noudata käyttövalmiin Blincyto-infuusioliuoksen antamisessa käyttövalmiiseen pussiin kiinnitettyjä apteekin ohjeita ja annostele infuusio jollakin seuraavista vakionopeuksista:
- Infuusionopeus 10 ml/h ja infuusion kesto 24 tuntia
- Infuusionopeus 5 ml/h ja infuusion kesto 48 tuntia
- Infuusionopeus 3,3 ml/h ja infuusion kesto 72 tuntia
- Infuusionopeus 2,5 ml/h ja infuusion kesto 96 tuntia
Anna käyttövalmis Blincyto-infuusioliuos käyttäen infuusioletkustoa, jossa on steriili, pyrogeeniton, heikosti proteiinia sitova, 0,2 mikrometrin kiinteä (in-line) suodatin.
Tärkeää: Älä huuhtele Blincyto-infuusioletkua, varsinkaan kun vaihdat infuusiopussin. Jos pussinvaihdon yhteydessä tai infuusion päättyessä suoritetaan huuhtelu, potilaan annos saattaa ylittyä, mikä voi johtaa komplikaatioihin. Käytettäessä moniluumenista laskimokatetria Blincyto on infusoitava oman luumeninsa kautta.
Hoitava lääkäri valitsee infuusion keston ottaen huomioon infuusiopussien vaihtotiheyden ja potilaan painon. Aiottu potilaalle annettava Blincyto-hoitoannos ei muutu.
Infuusiopussin vaihtaminen
Steriiliyden säilyttämiseksi terveydenhoitohenkilökunnan on vaihdettava infuusiopussi vähintään 96 tunnin välein.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Imettäminen (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Neurologiset tapahtumat, mukaan lukien ICANS
Neurologisia tapahtumia, myös kuolemaan johtaneita, on havaittu. Vähintään 3. asteen (vaikeita tai hengenvaarallisia) (CTCAE versio 4.0) neurologisia tapahtumia (mukaan lukien ICANS), joita on havaittu blinatumomabihoidon aloittamisen jälkeen, ovat olleet enkefalopatia, kouristuskohtaukset, puhehäiriöt, tajunnan häiriöt, sekavuus ja ajan ja paikan tajun hämärtyminen sekä koordinaatio- ja tasapainohäiriöt. Potilailla, joilla neurologisia tapahtumia esiintyi, ensimmäinen tapahtuma ilmaantui keskimäärin 2 ensimmäisen hoitoviikon aikana (mediaaniaika). Suurin osa tapahtumista korjautui hoidon keskeyttämisen jälkeen, ja ne johtivat vain harvoin Blincyto-hoidon lopettamiseen.
Iäkkäät potilaat saattavat olla alttiimpia vakaville neurologisille tapahtumille, kuten kognitiivisille häiriöille, enkefalopatialle ja sekavuudelle.
Neurologiset tapahtumat (kuten vapina, huimaus, sekavuustila, enkefalopatia ja ataksia) olivat yleisempiä potilailla, joilla oli aikaisemmin esiintynyt neurologisia muutoksia tai oireita (kuten huimausta, tuntoaistin heikkenemistä, heijasteiden heikkoutta, vapinaa, tuntohäiriöitä, parestesioita tai muistin heikkenemistä). Näillä potilailla ensimmäinen neurologinen tapahtuma ilmaantui keskimäärin ensimmäisen hoitojakson aikana (mediaaniaika).
Potilaat, joilla on tai on aikaisemmin ollut kliinisesti merkittäviä keskushermoston sairauksia (esim. epilepsia, kouristuskohtauksia, pareesi, afasia, aivohalvaus, vaikeita aivovammoja, dementia, Parkinsonin tauti, pikkuaivosairaus, elimellinen aivo-oireyhtymä tai psykoosi), suljettiin pois kliinisistä tutkimuksista, joten näistä potilasryhmistä on vain vähän kokemuksia. Neurologisten tapahtumien riski saattaa olla tavanomaista suurempi tässä potilasjoukossa. Hoidon mahdollisia hyötyjä on punnittava huolellisesti neurologisten tapahtumien riskiä vastaan, ja on noudatettava erityisen suurta varovaisuutta, jos Blincyto-hoitoa annetaan näille potilaille.
Blinatumomabista on hyvin vähän kokemuksia sellaisten potilaiden hoidossa, joilla on dokumentoitu aktiivinen ALL keskushermostossa tai aivo-selkäydinnesteessä. Kliinisissä tutkimuksissa potilaille on kuitenkin annettu blinatumomabihoitoa sen jälkeen, kun blastisolut on hävitetty aivo-selkäydinnesteestä keskushermostoon kohdennetulla hoidolla (kuten intratekaalisella solunsalpaajahoidolla). Blincyto-hoito voidaan siis aloittaa, kun aivo-selkäydinneste on puhdistettu.
Potilailla, joilla on Downin syndrooma, saattaa olla suurempi kouristuskohtausten riski Blincyto-hoidon aikana. Harkitse näille potilaille kouristuskohtausten estohoitoa ennen Blincyto-hoidon aloittamista.
Suositellaan, että ennen Blincyto-hoidon aloittamista tehdään neurologinen tutkimus ja potilaita seurataan kliinisesti (esim. kirjoitustestin avulla; tämä voi olla osa kattavaa neurologista tutkimusta) neurologisiin tapahtumiin (mukaan lukien ICANS) viittaavien muutosten ja oireiden havaitsemiseksi. Näiden muutosten ja oireiden saaminen hallintaan voi vaatia Blincyto-hoidon keskeyttämistä tai lopettamista pysyvästi ja/tai kortikosteroidihoitoa (ks. kohta Annostus ja antotapa). Kouristuskohtausten sekundaariseksi estohoidoksi suositellaan sopivia epilepsialääkkeitä (esim. levetirasetaamia).
Infektiot
Blinatumomabia saavilla potilailla on esiintynyt vakavia infektioita, kuten sepsistä, keuhkokuumetta, bakteremiaa, opportunistisia infektioita ja katetri-infektioita, jotka ovat joissakin tapauksissa olleet hengenvaarallisia tai johtaneet kuolemaan. Vakavia infektioita esiintyi enemmän aikuispotilailla, joiden ECOG (Eastern Cooperative Oncology Group) ‑toimintakykyluokka lähtötilanteessa oli 2, kuin potilailla, joiden ECOG‑toimintakykyluokka oli alle 2. Blincyton käytöstä on hyvin vähän kokemuksia sellaisten potilaiden hoidossa, joilla on aktiivinen, huonosti hallinnassa oleva infektio.
Potilaiden tilaa on seurattava kliinisesti Blincyto-hoidon aikana, jotta infektioon viittaavat muutokset ja oireet havaitaan ajoissa ja voidaan hoitaa asianmukaisesti. Infektioiden hoito voi vaatia Blincyton keskeyttämistä tai lopettamista pysyvästi (ks. kohta Annostus ja antotapa).
Sytokiinioireyhtymä ja infuusioreaktiot
Blincyto-hoitoa saavilla potilailla on raportoitu sytokiinioireyhtymää, joka voi olla hengenvaarallinen tai johtaa kuolemaan (vaikeusasteluokka ≥ 4) (ks. kohta Haittavaikutukset).
Vakavia haittavaikutuksia, jotka voivat liittyä sytokiinioireyhtymään, olivat kuume, voimattomuus, päänsärky, verenpaineen lasku, kohonnut kokonaisbilirubiiniarvo ja pahoinvointi. Nämä tapahtumat vaativat melko harvoin Blincyto-hoidon lopettamista. Mediaaniaika sytokiinioireyhtymään liittyvän tapahtuman ilmaantumiseen oli 2 vuorokautta. Potilaiden tilaa on seurattava tarkoin sytokiinioireyhtymään viittaavien muutosten ja oireiden havaitsemiseksi.
Sytokiinioireyhtymän yhteydessä on esiintynyt yleisesti disseminoitunutta intravaskulaarista koagulaatiota ja kapillaarivuoto-oireyhtymää (esim. verenpaineen laskua, hypoalbuminemiaa, turvotusta ja hemokonsentraatiota) (ks. kohta Haittavaikutukset). Kapillaarivuoto-oireyhtymä vaatii pikaista hoitoa.
Hemofagosyyttistä lymfohistiosytoosia (HLH) / makrofagiaktivaatio-oireyhtymää (MAS) on raportoitu melko harvoin sytokiinioireyhtymän yhteydessä.
Infuusioreaktioita voi olla mahdotonta erottaa kliinisesti sytokiinioireyhtymän ilmenemismuodoista (ks. kohta Haittavaikutukset). Infuusioreaktiot olivat yleensä nopeita ja ilmaantuivat 48 tunnin kuluessa infuusion aloittamisesta. Joillakin potilailla infuusioreaktiot ilmaantuivat kuitenkin viivästyneesti tai myöhempien hoitojaksojen yhteydessä. Infuusioreaktioiden mahdollista ilmaantumista on seurattava tarkoin, varsinkin ensimmäistä ja toista hoitojaksoa aloitettaessa, ja tila on hoidettava asianmukaisesti. Kuumelääkkeitä (esim. parasetamolia) suositellaan kuumereaktion lievittämiseksi jokaisen hoitojakson 48 ensimmäisen tunnin aikana. Sytokiinioireyhtymän riskin pienentämiseksi on tärkeää, että Blincyto-hoito aloitetaan (1. hoitojakso, päivät 1–7) kohdassa Annostus ja antotapa suositellulla aloitusannoksella.
Näiden tapahtumien hoito saattaa vaatia Blincyton keskeyttämistä tai lopettamista pysyvästi (ks. kohta Annostus ja antotapa).
Hemofagosyyttinen lymfohistiosytoosi / immuuniefektorisoluihin liittyvä hemofagosyyttisen lymfohistiosytoosin kaltainen oireyhtymä (IEC‑HS)
Blincyto-hoitoa saavilla potilailla on havaittu hemofagosyyttistä lymfohistiosytoosia (HLH) / immuuniefektorisoluihin liittyvää hemofagosyyttisen lymfohistiosytoosin kaltaista oireyhtymää (IEC‑HS). Hemofagosyyttinen lymfohistiosytoosi on hengenvaarallinen oireyhtymä, jonka oireita ovat kuume, hyperferritinemia, hepato- ja/tai splenomegalia, sytopeniat, hyytymishäiriöön liittyvä hypofibrinogenemia, hemofagosytoosi ja hypertransaminasemia. Immuuniefektorisoluihin liittyvällä hemofagosyyttisen lymfohistiosytoosin kaltaisella oireyhtymällä tarkoitetaan immuuniefektorisoluhoitoihin liittyvien hemofagosyyttista lymfohistiosytoosia muistuttavien toksisuuksien ilmenemistä. Niille tyypillistä on immuunivasteen liiallinen aktivoituminen ja sytokiinituotannon säätelyhäiriö, mikä johtaa monisysteemiseen inflammaatioon ja elinhäiriöihin. Hemofagosyyttisen lymfohistiosytoosin / immuuniefektorisoluihin liittyvän hemofagosyyttisen lymfohistiosytoosin kaltaisen oireyhtymän mahdollisuus on huomioitava, jos sytokiinioireyhtymän oireet ovat epätavalliset tai pitkittyneet. Potilaiden tilaa on seurattava hemofagosyyttiseen lymfohistiosytoosiin / immuuniefektorisoluihin liittyvään hemofagosyyttisen lymfohistiosytoosin kaltaiseen oireyhtymään viittaavien kliinisten muutosten ja oireiden havaitsemiseksi. Jos epäillään hemofagosyyttistä lymfohistiosytoosia / immuuniefektorisoluihin liittyvää hemofagosyyttisen lymfohistiosytoosin kaltaista oireyhtymää, Blincyto‑hoito on keskeytettävä diagnostista tutkimusta varten. Potilas on arvioitava myös hemofagosyyttista lymfohistiosytoosia aiheuttavien muiden vaihtoehtoisten syiden, kuten infektioiden, maligniteettien ja autoimmuunisairauksien, suhteen. Hemofagosyyttisen lymfohistiosytoosin / immuuniefektorisoluihin liittyvän hemofagosyyttisen lymfohistiosytoosin kaltaisen oireyhtymän hoito (esim. kortikosteroidit, immunosuppressiivinen tai sytokiineihin kohdistuva hoito sekä elintoimintoja tukeva hoito) on aloitettava nopeasti laitoskohtaisten tai julkaistujen hoitosuositusten mukaan.
Tuumorilyysioireyhtymä
Blincyto-hoitoa saavilla potilailla on esiintynyt tuumorilyysioireyhtymää, joka voi olla hengenvaarallinen tai johtaa kuolemaan (vaikeusasteluokka ≥ 4).
Tuumorilyysioireyhtymän ehkäisystä ja hoidosta, joihin kuuluvat tehokas nesteytys ja hyperurikemian lääkehoito (esim. allopurinoli tai rasburikaasi), on huolehdittava Blincyto-hoidon aikana, varsinkin jos potilaalla on huomattava leukosytoosi tai suuri kasvaintaakka. Tuumorilyysioireyhtymään viittaavia muutoksia tai oireita, myös munuaisten toimintaa ja nestetasapainoa, on tarkkailtava 48 tunnin ajan ensimmäisen infuusion jälkeen. Kliinisissä tutkimuksissa tuumorilyysioireyhtymää esiintyi useammin potilailla, joilla oli kohtalainen munuaisten vajaatoiminta, kuin potilailla, joiden munuaisten toiminta oli normaali tai lievästi heikentynyt. Näiden tapahtumien hoito voi vaatia Blincyton keskeyttämistä tai lopettamista pysyvästi (ks. kohta Annostus ja antotapa).
Neutropenia ja kuumeinen neutropenia
Blincyto-hoitoa saavilla potilailla on havaittu neutropeniaa ja kuumeista neutropeniaa, myös hengenvaarallisia tapauksia. Laboratorioarvoja (muun muassa valkosolujen määrää ja absoluuttista neutrofiilimäärää) on seurattava rutiininomaisesti Blincyto-infuusion aikana, erityisesti ensimmäisen hoitojakson 9 ensimmäisen vuorokauden aikana, ja häiriöt on hoidettava asianmukaisesti.
Maksaentsyymiarvojen kohoaminen
Blincyto-hoitoon on liittynyt ohimenevää maksaentsyymiarvojen kohoamista. Suurin osa näistä tapahtumista havaittiin ensimmäisen viikon aikana hoidon aloittamisen jälkeen. Ne eivät vaatineet Blincyto-hoidon keskeyttämistä tai lopettamista (ks. kohta Haittavaikutukset).
Alaniiniaminotransferaasi- (ALAT), aspartaattiaminotransferaasi- (ASAT) ja gammaglutamyylitransferaasiarvoa (GGT) sekä veren kokonaisbilirubiiniarvoa on seurattava ennen Blincyto-hoidon aloittamista ja hoidon aikana, varsinkin kahden ensimmäisen hoitojakson 48 ensimmäisen tunnin aikana. Näiden tapahtumien hoito voi vaatia Blincyton keskeyttämistä tai lopettamista pysyvästi (ks. kohta Annostus ja antotapa).
Haimatulehdus
Hengenvaarallisia tai kuolemaan johtaneita haimatulehduksia on raportoitu potilailla, jotka ovat saaneet Blincyto-hoitoa kliinisissä tutkimuksissa tai markkinoille tulon jälkeen. Joissakin tapauksissa suuriannoksinen steroidihoito on voinut vaikuttaa haimatulehduksen kehittymiseen.
Potilaiden tilaa on seurattava tarkoin haimatulehdukseen viittaavien muutosten ja oireiden havaitsemiseksi. Tilaa voidaan arvioida fysikaalisen tutkimuksen, seerumin amylaasi- ja lipaasipitoisuuksien määritysten sekä vatsan kuvantamistutkimusten, kuten kaikukuvauksen, ja muiden asianmukaisten diagnostisten toimenpiteiden avulla. Haimatulehduksen hoito voi vaatia Blincyton keskeyttämistä tai lopettamista pysyvästi (ks. kohta Annostus ja antotapa).
Leukoenkefalopatia ja etenevä multifokaalinen leukoenkefalopatia
Blincyto-hoitoa saavilla potilailla on havaittu aivojen magneettikuvauksessa leukoenkefalopatiaan viittaavia muutoksia. Niitä on esiintynyt varsinkin potilailla, jotka ovat aikaisemmin saaneet aivojen sädehoitoa ja solunsalpaajia leukemian hoitoon (kuten systeemistä suuriannoksista metotreksaattihoitoa tai intratekaalista sytarabiinihoitoa). Näiden muutosten kliinistä merkitystä ei tunneta.
Koska etenevän multifokaalisen leukoenkefalopatian (PML) kehittyminen on mahdollista, siihen viittaavia muutoksia ja oireita on seurattava. Epäilyttävissä tapauksissa on harkittava neurologin konsultointia, aivojen magneettikuvausta ja selkäydinnestetutkimusta, ks. kohta Haittavaikutukset.
CD19-negatiivinen uusiutuminen
CD19-negatiivista pre‑B‑ALL:ää on raportoitu Blincyto-hoitoa saavilla potilailla, joiden tauti on uusiutunut. CD19-ilmentymän arviointiin on kiinnitettävä erityistä huomiota luuydinnäytteen analyysissa.
Solulinjan vaihtuminen ALL:stä akuutiksi myelooiseksi leukemiaksi (AML)
Solulinjan vaihtumista ALL:stä AML:ksi on raportoitu harvoin Blincyto-hoitoa saaneilla potilailla, joiden tauti on uusiutunut, mukaan lukien ne potilaat, joilla alkuperäisessä diagnoosissa ei todettu immunofenotyyppisiä ja/tai sytogeneettisiä poikkeavuuksia. Kaikkia potilaita, joilla tauti on uusiutunut, on tarkkailtava AML:n esiintymisen varalta.
Rokotukset
Eläviä viruksia sisältävien rokotteiden turvallisuutta Blincyto-hoidon aikana tai sen jälkeen ei ole tutkittu. Eläviä viruksia sisältävien rokotteiden antamista ei suositella 2 viikon aikana ennen Blincyto-hoidon aloittamista, hoidon aikana eikä ennen kuin B‑lymfosyyttien määrä on palautunut normaalialueelle viimeisen hoitojakson jälkeen.
Vastasyntyneillä, jotka ovat sikiöaikana altistuneet blinatumomabille, voi esiintyä B‑solujen niukkuutta. Siksi vastasyntyneiden B‑solumäärää on seurattava, ja rokotukset eläviä viruksia sisältävillä rokotteilla voidaan aloittaa vasta, kun lapsen B‑solumäärä on korjautunut (ks. kohta Raskaus ja imetys).
Raskauden ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä Blincyto-hoidon aikana ja vähintään 48 tuntia hoidon päättymisen jälkeen (ks. kohta Raskaus ja imetys).
Lääkitysvirheet
Lääkitysvirheitä on havaittu Blincyto-hoidon aikana. On erittäin tärkeää, että Blincyton valmistamista infuusiota varten (liuottamista ja laimentamista) ja infuusion antamista koskevia ohjeita noudatetaan erityisen huolellisesti lääkitysvirheiden (yli- ja aliannostelun) välttämiseksi (ks. kohta Annostus ja antotapa).
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) 24 tunnin infuusiota kohti eli sen voidaan sanoa olevan "natriumiton".
Yhteisvaikutukset
Varsinaisia yhteisvaikutustutkimuksia ei ole tehty. Ihmisen maksasoluissa tehdyn in vitro ‑tutkimuksen tulokset viittaavat siihen, ettei blinatumomabi vaikuta CYP450‑entsyymien toimintaan.
Blincyto-hoidon aloittaminen aiheuttaa ensimmäisinä hoitopäivinä ohimenevää sytokiinien vapautumista, joka saattaa estää CYP450‑entsyymien toimintaa. Jos potilas saa lääkkeitä, jotka ovat CYP450‑entsyymien ja kuljettajaproteiinien substraatteja ja joilla on kapea terapeuttinen indeksi, haittavaikutusten ilmaantumista (esim. varfariini) tai lääkeainepitoisuuksia (esim. siklosporiini) on seurattava näiden päivien aikana. Näiden lääkkeiden annostusta on muutettava tarvittaessa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Raskauden ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä blinatumomabihoidon aikana ja vähintään 48 tuntia hoidon päättymisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus
Blinatumomabilla ei ole tehty lisääntymistoksisuustutkimuksia. Hiirillä tehdyssä alkio- ja sikiötoksisuustutkimuksessa hiiren korvikemolekyyli läpäisi istukan eikä aiheuttanut alkiotoksisuutta eikä teratogeenisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Odotettua B‑ ja T‑solujen vähenemistä havaittiin tiineillä hiirillä mutta sikiöihin kohdistuneita hematologisia vaikutuksia ei arvioitu.
Ei ole olemassa tietoja blinatumomabin käytöstä raskaana oleville naisille.
Blinatumomabia ei pidä käyttää raskauden aikana, paitsi jos mahdollinen hyöty on suurempi kuin sikiölle mahdollisesti aiheutuva vaara.
Valmisteen farmakologisten ominaisuuksien vuoksi sikiöaikainen altistuminen voi aiheuttaa vastasyntyneille B‑solujen niukkuutta. Siksi vastasyntyneiden B‑solumäärää on seurattava, ja rokotukset eläviä viruksia sisältävillä rokotteilla voidaan aloittaa vasta, kun lapsen B‑solumäärä on korjautunut (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Imetys
Ei tiedetä, erittyykö blinatumomabi tai erittyvätkö sen metaboliitit ihmisen rintamaitoon. Valmisteen farmakologisten ominaisuuksien perusteella imeväiseen kohdistuvia riskejä ei voida poissulkea. Siksi on varmuuden vuoksi pidättäydyttävä imettämisestä blinatumomabihoidon aikana ja vähintään 48 tuntia hoidon jälkeen.
Hedelmällisyys
Tutkimuksia blinatumomabin vaikutuksista hedelmällisyyteen ei ole tehty. Uros- tai naarashiirten lisääntymiselimiin kohdistuneita haittavaikutuksia ei havaittu hiiren korvikemolekyylillä tehdyissä 13 viikon toksisuustutkimuksissa (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Blinatumomabilla on huomattava vaikutus ajokykyyn ja koneidenkäyttökykyyn. Sekavuutta ja ajan ja paikan tajun hämärtymistä, koordinaatio- ja tasapainohäiriöitä, kouristuskohtauksia ja tajunnan häiriöitä voi esiintyä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Koska neurologiset tapahtumat ovat mahdollisia, blinatumomabia saavien potilaiden on pidättäydyttävä ajamasta ja tekemästä riskialttiita työtehtäviä ja toimintoja, kuten raskaiden tai vaarallisten koneiden ajamista tai käyttöä, sinä aikana, kun heille annetaan blinatumomabia. Potilaille on kerrottava, että heillä voi esiintyä neurologisia oireita.
Haittavaikutukset
Tiivistelmä turvallisuustiedoista
Tässä kohdassa kuvattuja haittavaikutuksia on esiintynyt kliinisissä tutkimuksissa potilailla, joilla oli pre-B‑ALL (N = 1 045).
Vakavimpia haittavaikutuksia, joita voi esiintyä blinatumomabihoidon aikana, ovat: infektiot (22,6 %), neurologiset tapahtumat (12,2 %), neutropenia / kuumeinen neutropenia (9,1 %), sytokiinioireyhtymä (2,7 %) ja tuumorilyysioireyhtymä (0,8 %).
Yleisimmät haittavaikutukset olivat: kuume (70,8 %), määrittämättömän patogeenin aiheuttamat infektiot (41,4 %), infuusioon liittyvät reaktiot (33,4 %), päänsärky (32,7 %), pahoinvointi (23,9 %), anemia (23,3 %), trombosytopenia (21,6 %), edeema (21,4 %), neutropenia (20,8 %), kuumeinen neutropenia (20,4 %), ripuli (19,7 %), oksentaminen (19,0 %), ihottuma (18,0 %), kohonneet maksaentsyymiarvot (17,2 %), yskä (15,0 %), bakteeri-infektiot (14,1 %), vapina (14,1 %), sytokiinioireyhtymä (13,8 %), leukopenia (13,8 %), ummetus (13,5 %), alentuneet immunoglobuliiniarvot (13,4 %), virusinfektiot (13,3 %), hypotensio (13,0 %), selkäkipu (12,5 %), vilunväristykset (11,7 %), vatsakipu (10,6 %), takykardia (10,6 %), unettomuus (10,4 %), raajakipu (10,1 %) ja sieni-infektiot (9,6 %).
Haittavaikutustaulukko
Haittavaikutukset luetellaan alla elinjärjestelmäluokan ja yleisyyden mukaan. Yleisyysluokat määritettiin niiden haittavaikutusten vakioimattomien ilmaantuvuuslukujen perusteella, joita raportoitiin kliinisissä tutkimuksissa potilailla, joilla oli pre-B‑ALL (N = 1 045). Haittavaikutukset on esitetty kussakin yleisyysluokassa vakavuuden mukaan alenevassa järjestyksessä.
| Elinjärjestelmä (MedDRA) | Hyvin yleinen (≥ 1/10) | Yleinen (≥ 1/100, < 1/10) | Melko harvinainen (≥ 1/1 000, < 1/100) |
| Infektiot | Bakteeri-infektiota, b Virusinfektiota, b Infektiot – patogeenia ei määriteltya, b | Sepsis Keuhkokuume Sieni-infektiota, b | |
| Veri ja imukudos | Kuumeinen neutropenia Anemia1 Neutropenia2 Trombosytopenia3 Leukopenia4 | Leukosytoosi5 Lymfopenia6 | Lymfadenopatia Hemofagosyyttinen lymfohistiosytoosi (HLH) |
| Immuunijärjestelmä | Sytokiinioireyhtymäa | Yliherkkyys | Sytokiinimyrsky |
| Aineenvaihdunta ja ravitsemus | Tuumorilyysioireyhtymä | ||
| Psyykkiset häiriöta | Unettomuus18 | Sekavuustila18 Ajan ja paikan tajun hämärtyminen18 | |
| Hermostoa | Päänsärky18 Vapina18 | Enkefalopatia18 Afasia18 Parestesiat18 Kouristuskohtaus18 Kognitiivinen häiriö18 Muistin heikkeneminen Huimaus18 Uneliaisuus18 Heikentynyt tuntoaisti18 Aivohermosairausb Ataksia18 Immuuniefektorisoluihin liittyvä neurotoksinen oireyhtymä (ICANS) | Puhehäiriö18 |
| Sydän | Takykardia7 | ||
| Verisuonisto | Hypotensio8 Hypertensio9 | Kasvojen ja kaulan punoitus | Kapillaarivuoto-oireyhtymä |
| Hengityselimet, rintakehä ja välikarsina | Yskä | Hengenahdistus Limaa tuottava yskä Hengitysvajaus Hengityksen vinkuminen | Rasitushengenahdistus Äkillinen hengitysvajaus |
| Ruoansulatuselimistö | Pahoinvointi Ripuli Oksentelu Ummetus Vatsakipu | Haimatulehdusa | |
| Maksa ja sappi | Hyperbilirubinemiaa, 10 | ||
| Iho ja ihonalainen kudos | Ihottuma11 | ||
| Luusto, lihakset ja sidekudos | Selkäkipu Raajakipu | Luukipu | |
| Yleisoireet ja antopaikassa todettavat haitat | Kuume12 Vilunväristykset Turvotus (edeema)13 | Rintakipu14 Kipu | |
| Tutkimukset | Kohonneet maksaentsyymiarvota, 15 Alentuneet immunoglobuliiniarvot16 | Painon nousu Kohonnut veren alkalinen fosfataasiarvo | |
| Vammat ja myrkytykset | Infuusioon liittyvät reaktiot17 | ||
a Lisätietoja on kohdassa Tärkeimpien haittavaikutusten kuvaus. b MedDRA‑järjestelmän ylemmän tason ryhmätermit (high level group terms) (MedDRA versio 23.0). Samaa lääketieteellistä käsitettä tai tilaa kuvaavat haittatapahtumatermit on yhdistetty ja raportoitu yhtenä haittavaikutuksena yllä olevassa taulukossa. Kuhunkin haittavaikutukseen sisältyvät termit luetellaan seuraavassa: 1 Anemia kattaa termit anemia ja pienentynyt hemoglobiinipitoisuus. 2 Neutropenia kattaa termit neutropenia ja pienentynyt neutrofiilimäärä. 3 Trombosytopenia kattaa termit pienentynyt verihiutalemäärä ja trombosytopenia. 4 Leukopenia kattaa termit leukopenia ja pienentynyt veren valkosolumäärä. 5 Leukosytoosi kattaa termit leukosytoosi ja suurentunut veren valkosolumäärä. 6 Lymfopenia kattaa termit pienentynyt lymfosyyttimäärä ja lymfopenia. 7 Takykardia kattaa termit sinustakykardia, supraventrikulaarinen takykardia, takykardia, eteistakykardia ja kammiotakykardia. 8 Hypotensio kattaa termit alentunut verenpaine ja hypotensio. 9 Hypertensio kattaa termit kohonnut verenpaine ja hypertensio. 10 Hyperbilirubinemia kattaa termit kohonnut veren bilirubiiniarvo ja hyperbilirubinemia. 11 Ihottuma kattaa termit ihon punoitus, ihottuma, punoittava ihottuma, yleistynyt ihottuma, makulaarinen ihottuma, makulopapulaarinen ihottuma, kutiava ihottuma, katetrikohdan ihottuma, pustulaarinen ihottuma, sukupuolielinten ihottuma, papulaarinen ihottuma ja rakkulainen ihottuma. 12 Kuume kattaa termit kohonnut ruumiinlämpö ja kuume. 13 Turvotus (edeema) kattaa termit luuydinturvotus, periorbitaalinen turvotus, silmäluomiturvotus, silmien turvotus, huulten turvotus, kasvojen turvotus, paikallinen turvotus, yleistynyt turvotus, turvotus/edeema, perifeerinen turvotus/edeema, infuusiokohdan turvotus, turvonnut munuainen, kivespussiturvotus, sukupuolielinten turvotus, keuhkopöhö, kurkunpään turvotus, angioedeema, suunympäryksen turvotus ja imunesteturvotus. 14 Rintakipu kattaa termit epämiellyttävä tuntemus rintakehässä, rintakipu, luu- ja lihasperäinen rintakipu ja muu kuin sydänperäinen rintakipu. 15 Kohonneet maksaentsyymiarvot kattaa termit kohonnut alaniiniaminotransferaasiarvo, kohonnut aspartaattiaminotransferaasiarvo, kohonnut gamma-glutamyylitransferaasiarvo, kohonnut maksaentsyymiarvo, kohonneet maksan toimintakokeen arvot ja kohonneet aminotransferaasiarvot. 16 Alentuneet immunoglobuliiniarvot kattaa termit immunoglobuliini G:n alentunut pitoisuus, immunoglobuliini A:n alentunut pitoisuus, immunoglobuliini M:n alentunut pitoisuus, globuliinien alentuneet pitoisuudet, hypogammaglobulinemia, hypoglobulinemia ja immunoglobuliinien alentuneet pitoisuudet veressä. 17 Infuusioon liittyvät reaktiot on yhteistermi, joka kattaa termin infuusioon liittyvä reaktio ja seuraavat tapahtumat, jotka ilmaantuvat infuusion ensimmäisten 48 tunnin aikana, ja tapahtuma kesti enintään 2 vuorokautta: kuume, sytokiinioireyhtymä, hypotensio, lihaskipu, akuutti munuaisvaurio, hypertensio, ihottuma, tiheä hengitys (takypnea), kasvojen turvotus, kasvoedeema ja punoittava ihottuma. 18 Tapahtumat saattavat johtua ICANS-oireyhtymästä. | |||
Tärkeimpien haittavaikutusten kuvaus
Neurologiset tapahtumat, mukaan lukien ICANS
Kolmannen vaiheen satunnaistetussa kliinisessä tutkimuksessa (N = 267) ja toisen vaiheen yhden hoitohaaran kliinisessä tutkimuksessa (N = 189) 66,0 prosentilla Blincyto-hoitoa saaneista potilaista, joilla oli Philadelphia‑kromosominegatiivinen uusiutunut tai refraktaarinen pre‑B‑ALL, esiintyi yksi tai useampia neurologisia haittavaikutuksia (psyykkiset häiriöt mukaan lukien), pääasiassa keskushermostovaikutuksia. Vakavia neurologisia haittavaikutuksia esiintyi 11,6 prosentilla ja ≥ 3. asteen neurologisia haittavaikutuksia 12,1 prosentilla potilaista, ja näistä yleisimpiä vakavia haittavaikutuksia olivat enkefalopatia, vapina, afasia ja sekavuustila. Suurin osa (80,5 %) neurologisista tapahtumista oli kliinisesti korjautuvia, ja ne hävisivät, kun Blincyto-hoito keskeytettiin. Ensimmäinen tapahtuma ilmaantui keskimäärin 2 ensimmäisen hoitoviikon aikana (mediaaniaika). Aikaisemmassa toisen vaiheen yhden hoitohaaran kliinisessä tutkimuksessa on raportoitu yksi kuolemaan johtanut enkefalopatiatapaus.
Neurologisia tapahtumia raportoitiin 62,2 prosentilla aikuispotilaista, joilla oli Philadelphia‑kromosomipositiivinen uusiutunut tai refraktaarinen pre‑B‑ALL (N = 45). Sekä vakavia että ≥ 3. asteen neurologisia tapahtumia raportoitiin 13,3 prosentilla aikuispotilaista, joilla oli Philadelphia‑kromosomipositiivinen uusiutunut tai refraktaarinen pre‑B‑ALL.
Neurologisia tapahtumia raportoitiin 71,5 prosentilla aikuispotilaista, joilla oli MRD‑positiivinen pre-B‑ALL (N = 137), ja 22,6 prosentilla potilaista ilmeni vakavia tapahtumia. ≥ 3. asteen tapahtumia raportoitiin 16,1 prosentilla ja ≥ 4. asteen tapahtumia 2,2 prosentilla aikuispotilaista, joilla oli MRD‑positiivinen pre-B‑ALL.
Neurologisia tapahtumia raportoitiin 61,2 prosentilla aikuispotilaista, joilla oli CD19‑positiivinen pre‑B‑ALL ja jotka olivat vakautushoitovaiheessa, jossa Blincytoa annettiin vuorotellen solunsalpaajahoidon kanssa (N = 147). ≥ 3. asteen tapahtumia raportoitiin 28,6 prosentilla ja ≥ 4. asteen tapahtumia 2,0 prosentilla aikuispotilaista, joilla oli CD19‑positiivinen pre‑B‑ALL ja jotka olivat vakautushoitovaiheessa.
ICANS-tapauksia, myös vähintään 3. asteen ICANS-tapauksia, raportoitiin kliinisissä tutkimuksissa ja markkinoille tulon jälkeen. ICANS-oireyhtymän yleisimmät kliiniset ilmenemismuodot olivat sekavuustila, afasia, ajan ja paikan tajun hämärtyminen, tajunnantilan muutos, dysartria, enkefalopatia, kouristuskohtaus, mielentilan muutokset, uneliaisuus ja dysgrafia.
Havaintojen mukaan ICANS-oireyhtymä ilmaantui 0–299 vuorokaudessa; suurin osa ICANS-tapauksista ilmeni ensimmäisten kolmen viikon aikana.
Lisätietoja neurologisten tapahtumien ja ICANS-oireyhtymän kliinisestä hoidosta on kohdassa Annostus ja antotapa.
Infektiot
Hengenvaarallisia tai kuolemaan johtaneita (vaikeusasteluokka ≥ 4) virus-, bakteeri- ja sieni-infektioita on raportoitu Blincyto-hoitoa saavilla potilailla. Lisäksi toisen vaiheen kliinisessä tutkimuksessa on havaittu virusinfektioiden (esim. polyoomaviruksen (BK)) uudelleenaktivoitumista Philadelphia‑kromosominegatiivista, uusiutunutta tai refraktaarista pre-B‑ALL:ää sairastavilla aikuisilla. Vakavia infektioita esiintyi enemmän niillä Philadelphia‑kromosominegatiivista, uusiutunutta tai refraktaarista pre-B‑ALL:ää sairastavilla potilailla, joiden ECOG‑toimintakykyluokka lähtötilanteessa oli 2, kuin potilailla, joiden ECOG‑toimintakykyluokka oli < 2.
Infektioita raportoitiin 34,7 prosentilla aikuispotilaista, joilla oli CD19‑positiivinen pre‑B‑ALL ja jotka olivat vakautushoitovaiheessa, jossa Blincytoa annettiin vuorotellen solunsalpaajahoidon kanssa (N = 147). ≥ 3. asteen tapahtumia raportoitiin 28,6 prosentilla ja ≥ 4. asteen tapahtumia 10,2 prosentilla aikuispotilaista, joilla oli CD19‑positiivinen pre-B‑ALL ja jotka olivat vakautushoitovaiheessa.
Lisätietoja infektioiden kliinisestä hoidosta on kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Sytokiinioireyhtymä
Sytokiinioireyhtymää ilmeni 14,7 prosentilla Philadelphia‑kromosominegatiivista uusiutunutta tai refraktaarista pre‑B‑ALL:ää sairastavista potilaista, jotka saivat Blincyto-hoitoa kolmannen vaiheen satunnaistetussa kliinisessä tutkimuksessa (N = 267) ja toisen vaiheen yhden hoitohaaran kliinisessä tutkimuksessa (N = 189). Vakavia sytokiinioireyhtymään liittyviä reaktioita raportoitiin 2,4 prosentilla, ja mediaaniaika niiden ilmaantumiseen oli 2 vuorokautta.
Sytokiinioireyhtymää raportoitiin 8,9 prosentilla aikuispotilaista, joilla oli Philadelphia‑kromosomipositiivinen uusiutunut tai refraktaarinen pre‑B‑ALL (N = 45), ja 2,2 prosentilla potilaista ilmeni vakavia tapahtumia. ≥ 3. tai ≥ 4. asteen tapahtumia ei raportoitu.
Sytokiinioireyhtymää raportoitiin 2,9 prosentilla aikuispotilaista, joilla oli MRD‑positiivinen pre-B‑ALL (N = 137). Sekä 3. asteen tapahtumia että vakavia tapahtumia raportoitiin 1,5 prosentilla aikuispotilaista, joilla oli MRD‑positiivinen pre-B‑ALL; ≥ 4. asteen tapahtumia ei raportoitu.
Sytokiinioireyhtymää raportoitiin 15,6 prosentilla aikuispotilaista, joilla oli CD19‑positiivinen pre‑B‑ALL ja jotka olivat vakautushoitovaiheessa, jossa Blincytoa annettiin vuorotellen solunsalpaajahoidon kanssa (N = 147). ≥ 3. asteen tapahtumia raportoitiin 4,1 prosentilla ja ≥ 4. asteen tapahtumia 0,7 prosentilla aikuispotilaista, joilla oli CD19‑positiivinen pre‑B‑ALL ja jotka olivat vakautushoitovaiheessa.
Kapillaarivuoto‑oireyhtymä todettiin yhdellä potilaalla toisen vaiheen kliinisessä tutkimuksessa aikuispotilailla, joilla oli Philadelphia‑kromosominegatiivinen uusiutunut tai refraktaarinen pre‑B‑ALL, sekä yhdellä potilaalla toisen vaiheen kliinisessä tutkimuksessa aikuispotilailla, joilla oli MRD‑positiivinen pre‑B‑ALL. Kapillaarivuoto‑oireyhtymää ei todettu lainkaan toisen vaiheen kliinisessä tutkimuksessa aikuispotilailla, joilla oli Philadelphia‑kromosomipositiivinen uusiutunut tai refraktaarinen pre‑B‑ALL. Kapillaarivuoto-oireyhtymä raportoitiin yhdellä potilaalla (0,7 %), jolla oli CD19‑positiivinen pre‑B‑ALL ja joka oli vakautushoitovaiheessa, jossa Blincytoa annettiin vuorotellen solunsalpaajahoidon kanssa (N = 147). Kyseessä oli asteen 3 tapahtuma.
Lisätietoja sytokiinioireyhtymän kliinisestä hoidosta on kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Maksaentsyymiarvojen kohoaminen
Kohonneita maksaentsyymiarvoja ja niihin liittyviä löydöksiä/oireita raportoitiin 22,4 prosentilla Philadelphia‑kromosominegatiivista uusiutunutta tai refraktaarista pre‑B‑ALL:ää sairastavista potilaista, jotka saivat Blincyto-hoitoa kolmannen vaiheen satunnaistetussa kliinisessä tutkimuksessa (N = 267) ja toisen vaiheen yhden hoitohaaran kliinisessä tutkimuksessa (N = 189). Potilaista 1,5 prosentilla esiintyi vakavia ja 13,6 prosentilla ≥ 3. asteen haittavaikutuksia (kuten kohonneita ALAT‑arvoja, kohonneita ASAT‑arvoja ja kohonneita veren bilirubiiniarvoja). Mediaaniaika ensimmäisen tapahtuman ilmaantumiseen oli 4 vuorokautta Blincyto-hoidon aloittamisesta.
Maksaentsyymiarvojen kohoamista raportoitiin 17,8 prosentilla aikuispotilaista, joilla oli Philadelphia‑kromosomipositiivinen uusiutunut tai refraktaarinen pre‑B‑ALL (N = 45), ja 2,2 prosentilla potilaista ilmeni vakavia tapahtumia. ≥ 3. asteen tapahtumia raportoitiin 13,3 prosentilla ja ≥ 4. asteen tapahtumia 6,7 prosentilla aikuispotilaista, joilla oli Philadelphia‑kromosomipositiivinen uusiutunut tai refraktaarinen pre‑B‑ALL.
Maksaentsyymiarvojen kohoamista raportoitiin 12,4 prosentilla aikuispotilaista, joilla oli MRD‑positiivinen pre-B‑ALL (N = 137). ≥ 3. asteen tapahtumia raportoitiin 8,0 prosentilla ja ≥ 4. asteen tapahtumia 4,4 prosentilla aikuispotilaista, joilla oli MRD‑positiivinen pre-B‑ALL.
Maksaentsyymiarvojen kohoamista raportoitiin 15,6 prosentilla aikuispotilaista, joilla oli CD19 ‑positiivinen pre‑B‑ALL ja jotka olivat vakautushoitovaiheessa, jossa Blincytoa annettiin vuorotellen solunsalpaajahoidon kanssa (N = 147). ≥ 3. asteen tapahtumia raportoitiin 8,8 prosentilla ja ≥ 4. asteen tapahtumia 2,7 prosentilla aikuispotilaista, joilla oli CD19‑positiivinen pre‑B‑ALL ja jotka olivat vakautushoitovaiheessa.
Maksavaikutukset ovat yleensä olleet lyhytkestoisia, ja ne ovat korjautuneet usein nopeasti, vaikka Blincyto-hoitoa on jatkettu keskeytyksettä.
Lisätietoja kohonneiden maksaentsyymiarvojen kliinisestä hoidosta on kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Haimatulehdus
Hengenvaarallisia tai kuolemaan johtaneita haimatulehduksia on raportoitu potilailla, jotka ovat saaneet Blincyto-hoitoa kliinisissä tutkimuksissa tai markkinoille tulon jälkeen. Mediaaniaika haimatulehduksen ilmaantumiseen oli 7,5 vuorokautta. Lisätietoja haimatulehduksen kliinisestä hoidosta on kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Leukoenkefalopatia ja etenevä multifokaalinen leukoenkefalopatia
Leukoenkefalopatiaa on raportoitu. Potilailla, joiden aivojen magneettikuvaus- tai tietokonetomografialöydökset viittasivat leukoenkefalopatiaan, esiintyi samanaikaisesti vakavia haittavaikutuksia, joita olivat sekavuustila, vapina, kognitiivinen häiriö, enkefalopatia ja kouristukset. Vaikka etenevän multifokaalisen leukoenkefalopatian (PML) kehittyminen on mahdollista, kliinisissä tutkimuksissa ei ole raportoitu yhtään varmistettua PML-tapausta.
Pediatriset potilaat
Blincyton turvallisuutta ja tehoa on arvioitu kahdessa avoimessa tutkimuksessa lapsipotilailla, joilla oli Philadelphia‑kromosominegatiivinen uusiutunut tai refraktaarinen pre‑B‑ALL: yksihaaraisessa vaiheen I/II tutkimuksessa (MT103‑205) ja satunnaistetussa, kontrolloidussa vaiheen III tutkimuksessa (20120215).
Tutkimus MT103‑205 oli annosmääritys/-arviointitutkimus lapsipotilailla, joilla oli uusiutunut tai refraktaarinen pre‑B‑ALL. Tutkimus oli vaiheen I/II yhden hoitohaaran annosmääritys-/arviointitutkimus (MT103‑205), johon osallistui 70 potilasta (iältään 7 kk–17 vuotta), jotka saivat suositellun Blincyto-annostuksen.
Yleisimpiä vakavia haittavaikutuksia olivat kuume (11,4 %), kuumeinen neutropenia (11,4 %), sytokiinioireyhtymä (5,7 %), sepsis (4,3 %), laitteeseen liittyvä infektio (4,3 %), yliannostus (4,3 %), kouristukset (2,9 %), hengitysvajaus (2,9 %), hypoksia (2,9 %), keuhkokuume (2,9 %) ja monielinhäiriö (2,9 %).
Blincyto-hoidon haittavaikutukset olivat lapsipotilailla samantyyppisiä kuin on havaittu aikuispotilailla. Aikuisiin verrattuna lapsipotilailla havaittiin enemmän seuraavia haittavaikutuksia (ero ≥ 10 %): anemia, trombosytopenia, leukopenia, kuume, infuusioon liittyvät reaktiot, painon nousu ja hypertensio.
Haittavaikutukset ja niiden yleisyys olivat samantyyppisiä kaikissa lapsipotilasryhmissä (sukupuoli, ikä ja maantieteellinen alue).
Tutkimuksessa MT103‑205 sattui yksi sydämen vajaatoiminnan aiheuttama kuolemantapaus hengenvaarallisen sytokiinioireyhtymän ja tuumorilyysioireyhtymän yhteydessä, kun annos oli suurempi kuin suositeltu annos, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Blincytoa on myös arvioitu vaiheen III satunnaistetussa ja kontrolloidussa avoimessa tutkimuksessa (20120215) lapsipotilaiden ensimmäistä kertaa uusiutuneen korkean riskin pre‑B‑ALL:n hoidossa. Tutkimukseen osallistui 54 potilasta (iältään 1–18 vuotta), jotka saivat ensimmäistä kertaa uusiutuneen korkean riskin pre‑B‑ALL:n hoitoon suositellun Blincyto-annostuksen. Tutkimuksessa 20120215 Blincyton turvallisuusprofiili oli vastaava kuin tutkimuksessa lapsipotilailla, joilla oli uusiutunut tai refraktaarinen pre‑B‑ALL.
Muut erityisryhmät
Blincytosta on vain vähän kokemuksia ≥ 75‑vuotiaiden potilaiden hoidossa. Blincyton turvallisuus oli iäkkäillä (≥ 65‑vuotiailla) potilailla yleisesti samanlainen kuin alle 65‑vuotiailla potilailla. Iäkkäät potilaat saattavat kuitenkin olla alttiimpia vakaville neurologisille tapahtumille, kuten kognitiiviselle häiriölle, enkefalopatialle ja sekavuudelle.
Blincyto-hoitoa saavilla iäkkäillä potilailla, joilla on MRD‑positiivinen ALL, hypogammaglobulinemian riski saattaa olla suurentunut nuorempiin potilaisiin verrattuna. Suositellaan, että iäkkäiden potilaiden immunoglobuliiniarvoja seurataan Blincyto-hoidon aikana.
Blincyton turvallisuutta ei ole tutkittu vaikean munuaisten vajaatoiminnan yhteydessä.
Immunogeenisuus
Blincyto-hoitoa saaneiden aikuisten ALL‑potilaiden kliinisissä tutkimuksissa alle 2 prosentilla potilaista todettiin blinatumomabin vasta-aineita. Niistä potilaista, joille kehittyi blinatumomabin vasta-aineita, suurimmalla osalla esiintyi neutraloivaa aktiivisuutta in vitro osoittavia vasta-aineita. Kliinisissä tutkimuksissa ei todettu blinatumomabin vasta‑aineita lapsipotilailla, joilla oli uusiutunut tai refraktaarinen ALL ja joita hoidettiin blinatumomabilla.
Vasta-aineiden muodostuminen blinatumomabille saattaa vaikuttaa Blincyton farmakokinetiikkaan.
Kaiken kaikkiaan kliininen kokonaisnäyttö tukee löydöstä, että blinatumomabin vasta-aineilla ei näyttäisi olevan kliinistä vaikutusta Blincyton turvallisuuteen tai tehoon.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannoksia on havaittu, ja yksi potilas sai Blincyton suositellun hoitoannoksen 133‑kertaisena lyhyen ajan kuluessa. Yliannosten aiheuttamat haittavaikutukset vastasivat suositeltujen hoitoannosten yhteydessä havaittuja reaktioita, ja niitä olivat kuume, vapinat ja päänsärky. Yliannostapauksessa infuusio on keskeytettävä ja potilaan tilaa on seurattava. Blincyton uudelleenaloittamista oikealla hoitoannoksella on harkittava, kun kaikki haittavaikutukset ovat hävinneet, kuitenkin aikaisintaan 12 tunnin kuluttua infuusion keskeyttämisestä (ks. kohta Annostus ja antotapa).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, muut monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, ATC-koodi: L01FX07.
Vaikutusmekanismi
Blinatumomabi on kaksoisspesifinen T‑solun kytkevä molekyyli, joka sitoutuu spesifisesti B‑solulinjan solujen pinnalla ilmentyvään CD19‑antigeeniin ja T‑solujen pinnalla ilmentyvään CD3‑antigeeniin. Se aktivoi endogeeniset T‑solut kytkemällä T‑solureseptorikompleksin (TCR) CD3‑antigeenin ja hyvänlaatuisten ja pahanlaatuisten B‑solujen CD19‑antigeenin toisiinsa. Blinatumomabi-immunoterapian syöpäsoluja tuhoava vaikutus ei ole riippuvainen tiettyä T‑solureseptoria ilmentävistä T‑soluista eikä syöpäsolujen esittelemistä peptidiantigeeneista, vaan se on luonteeltaan polyklonaalinen ja riippumaton kohdesolun pinnan HLA‑molekyyleistä. Blinatumomabin vaikutuksesta T‑solun ja kasvainsolun väliin muodostuu sytolyyttinen liitos, ja tämän seurauksena vapautuu proteolyyttisiä entsyymejä, jotka tuhoavat sekä proliferoivat että lepäävät kohdesolut. Blinatumomabi aiheuttaa ohimenevää solun adheesiomolekyylien lisääntymistä, sytolyyttisten proteiinien muodostumista, inflammatoristen sytokiinien vapautumista ja T‑solujen proliferaatiota ja johtaa CD19‑positiivisten solujen tuhoutumiseen.
Farmakodynaamiset vaikutukset
Tutkituilla potilailla havaittiin yhdenmukainen immunofarmakodynaaminen vaste. Neljä viikkoa kestäneen jatkuvan infuusion aikana farmakodynaamisen vasteen tyypillisiä piirteitä olivat T‑solujen aktivaatio ja alkuvaiheen uudelleenjakautuminen, perifeeristen B‑solujen nopea väheneminen ja sytokiinien ohimenevä lisääntyminen.
Perifeeristen T‑solujen uudelleenjakautumista (eli T‑solujen kiinnittymistä verisuonten endoteeliin ja/tai siirtymistä kudoksiin) esiintyi blinatumomabi-infuusion aloittamisen tai annoksen suurentamisen jälkeen. T‑solumäärät pienenivät aluksi 1–2 vuorokauden aikana ja palautuivat sitten lähtötasolle 7–14 vuorokauden kuluessa suurimmalla osalla potilaista. Muutamilla potilailla T‑solumäärä nousi lähtötason yläpuolelle (T‑solujen monistuminen).
Suurimmalla osalla potilaista perifeeristen B‑solujen määrä pieneni nopeasti havaitsemisrajan alapuolelle hoidon aikana, kun annos oli ≥ 5 mikrog/m2/vrk tai ≥ 9 mikrog/vrk. Perifeerisen veren B‑solumäärän ei havaittu palautuvan 2 viikon hoitotauon aikana hoitojaksojen välillä. Epätäydellistä B‑solujen vähenemistä on esiintynyt annostasoilla 0,5 mikrog/m2/vrk ja 1,5 mikrog/m2/vrk ja muutamilla suurempia annoksia saaneilla potilailla, jotka eivät saavuttaneet hoitovastetta.
Perifeerisiä lymfosyyttejä ei mitattu lapsipotilaista.
Sytokiinien IL‑2, IL‑4, IL‑6, IL‑8, IL‑10, IL‑12, TNF‑α ja IFN‑γ pitoisuudet määritettiin, ja eniten koholla olivat IL‑6, IL‑10 ja IFN‑γ. Ohimenevää sytokiinipitoisuuksien nousua havaittiin 2 ensimmäisen vuorokauden aikana blinatumomabi-infuusion aloittamisen jälkeen. Kohonneet sytokiinipitoisuudet palautuivat lähtötasolle 24–48 tunnin kuluessa infuusion aikana. Myöhempien hoitojaksojen yhteydessä sytokiinipitoisuuksien nousu oli harvinaisempaa ja lievempää kuin ensimmäisen hoitojakson 48 ensimmäisen tunnin aikana.
Kliininen teho ja turvallisuus
Philadelphia‑kromosominegatiivinen, uusiutunut tai refraktaarinen pre-B‑ALL
Jäljempänä kuvatuissa toisen ja kolmannen vaiheen kliinisissä tutkimuksissa Blincyto‑hoitoa annettiin yhteensä 456:lle vähintään 18‑vuotiaalle potilaalle, joilla oli uusiutunut tai refraktaarinen pre-B‑ALL.
Kolmannen vaiheen satunnaistetussa, avoimessa monikeskustutkimuksessa (TOWER) arvioitiin Blincyton turvallisuutta ja tehoa tavanomaiseen solunsalpaajahoitoon verrattuna. Tutkimukseen otettiin ≥ 18‑vuotiaita potilaita, joiden ECOG‑toimintakykyluokka oli ≤ 2 ja joilla oli uusiutunut tai refraktaarinen pre-B‑ALL (> 5 % blastisoluja luuytimessä ja joko uusiutuminen milloin tahansa allogeenisen kantasolusiirron jälkeen, hoitamaton ensimmäinen uusiutuminen, kun ensimmäisen remission kesto < 12 kuukautta tai tehoton viimeinen hoito).
Potilaat jaettiin satunnaistetusti suhteessa 2:1 ryhmiin, jotka saivat Blincyto‑hoitoa tai jotakin neljästä etukäteen määritellystä, tutkijan valitsemasta tavanomaisesta solunsalpaajapohjaisesta hoito-ohjelmasta. Satunnaistaminen ositettiin iän (< 35 vuotta tai ≥ 35 vuotta), aikaisemman salvage-hoidon (kyllä tai ei) ja aikaisemman allogeenisen kantasolusiirron (kyllä tai ei) mukaan suostumuksen ajankohtana arvioituna. Potilaiden demografiset ja muut ominaisuudet lähtötilanteessa olivat samankaltaiset molemmissa hoitohaaroissa (ks. taulukko 7).
Taulukko 7. Potilaiden demografiset ja muut ominaisuudet lähtötilanteessa kolmannen vaiheen tutkimuksessa (TOWER)
| Ominaisuus | Blincyto (N = 271) | Tavanomainen solunsalpaajahoito (N = 134) |
| Ikä | ||
| Mediaani, vuotta (min, max) | 37 (18, 80) | 37 (18, 78) |
| Keskiarvo, vuotta (SD) | 40,8 (17,1) | 41,1 (17,3) |
| ≥ 65 vuotta, n (%) | 33 (12,2) | 15 (11,2) |
| Aikaisempi salvage-hoito | 164 (60,5) | 80 (59,7) |
| 0 | 114 (42,1) | 65 (48,5) |
| 1 | 91 (33,6) | 43 (32,1) |
| ≥ 2 | 66 (24,3) | 26 (19,4) |
| Aikaisempi allogeeninen kantasolusiirto | 94 (34,7) | 46 (34,3) |
| ECOG‑toimintakykyluokka, n (%) | ||
| 0 | 96 (35,4) | 52 (38,8) |
| 1 | 134 (49,4) | 61 (45,5) |
| 2 | 41 (15,1) | 20 (14,9) |
| Hoito tehoton, n (%) | ||
| Ensisijainen hoito tehoton | 46 (17,0) | 27 (20,1) |
| Salvage-hoito tehoton | 87 (32,1) | 34 (25,4) |
| Keskuslaboratoriossa / paikallisesti määritettyjen luuytimen blastien maksimimäärä, n (%) | ||
| ≥ 50 % | 201 (74,2) | 104 (77,6) |
Blincyto annettiin jatkuvana infuusiona laskimoon. Ensimmäisessä hoitojaksossa aloitusannos oli 9 mikrog/vrk ensimmäisen viikon ajan, jonka jälkeen annos nostettiin 28 mikrog:aan/vrk kolmen viikon ajaksi. Toisen ja myöhempien hoitojaksojen aikana tavoiteannosta 28 mikrog/vrk annettiin jokaisen jakson ensimmäisestä päivästä alkaen. Annoksen säätäminen oli mahdollista, jos haittavaikutuksia ilmaantui. Blincyto‑hoitoa saaneilla 267 potilaalla kokonaisten hoitojaksojen lukumäärän keskiarvo oli 2,0, ja tavanomaista solunsalpaajahoitoa saaneilla 109 potilaalla hoitojaksojen lukumäärän keskiarvo oli 1,3.
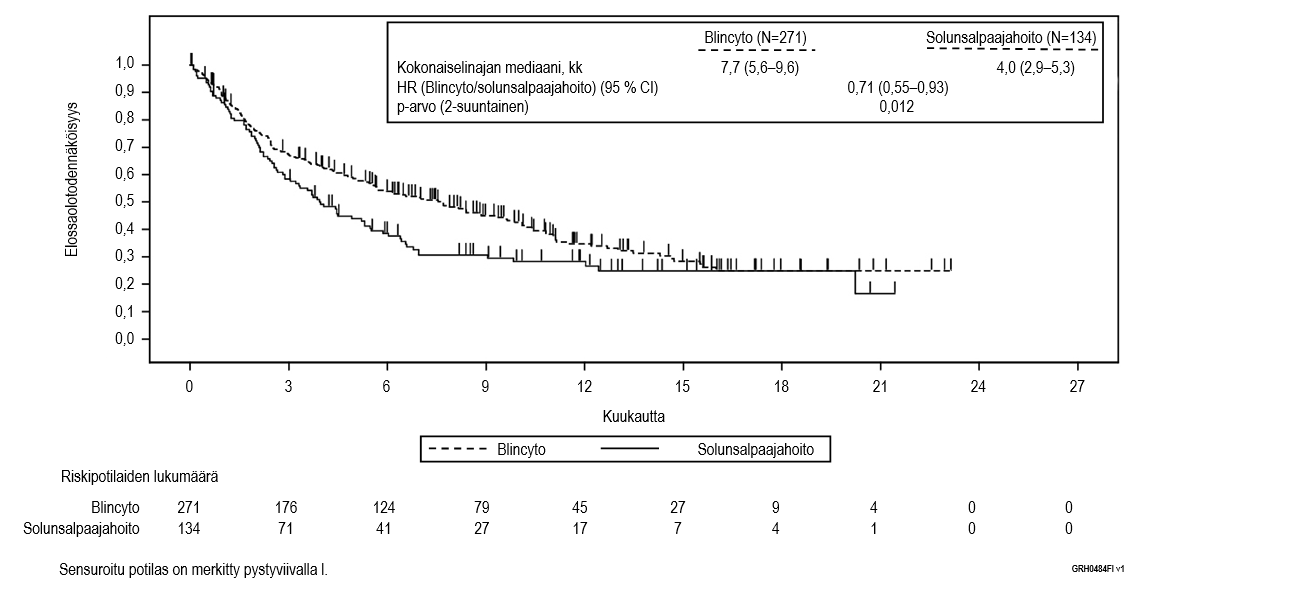
Ensisijainen päätetapahtuma oli kokonaiselinaika (OS). Kokonaiselinajan mediaani oli 4,0 kuukautta (95 % CI: 2,9–5,3) tavanomaista solunsalpaajahoitoa saaneessa haarassa ja 7,7 kuukautta (95 % CI: 5,6–9,6) Blincytoa saaneessa haarassa. Hoitohaarojen välinen vaarasuhde (HR) (95 % CI) oli 0,71 (0,55–0,93) Blincyton hyväksi, mikä osoitti, että riski oli pienentynyt 29 % Blincytoa saaneessa haarassa (p‑arvo = 0,012 (ositettu logrank-testi)), ks. kuva 1. Kokonaiselinaikatulokset olivat yhdenmukaiset osituksessa käytettyjen tekijöiden mukaisissa alaryhmissä.
Tulokset olivat yhdenmukaiset, kun kantasolusiirron ajankohtaa käytettiin sensurointiin: kokonaiselinajan mediaani sensuroituna kantasolusiirron ajankohtana oli 6,9 kuukautta (95 % CI: 5,3–8,8) Blincyto‑ryhmässä ja 3,9 kuukautta (95 % CI: 2,8–4,9) tavanomaista hoitoa saaneessa ryhmässä (HR 0,66; 95 % CI: 0,50−0,88; p‑arvo = 0,004). Kuolleisuus oli allogeenisen kantasolusiirron jälkeen kaikkien niiden hoitovasteen saaneiden potilaiden ryhmässä, jotka eivät saaneet leukemiahoitoa, Blincyto‑ryhmässä 10/38 (26,3 %; 95 % CI: 13,4–43,1) ja tavanomaista hoitoa saaneessa ryhmässä 3/12 (25 %; 95 % CI: 5,5–57,2). Vastaavat kuolleisuusluvut 100 vuorokauden kuluttua allogeenisesta kantasolusiirrosta olivat Blincyto‑ryhmässä 4/38 (12,4 %; 95 % CI: 4,8–29,9 %) ja tavanomaista hoitoa saaneessa ryhmässä 0/12 (0 %; 95 % CI ei arvioitavissa). Taulukossa 8 on tiivistelmä tutkimuksen muihin tärkeisiin päätetapahtumiin perustuvista tehoa kuvaavista tuloksista.
Kuva 1. Kaplan–Meier-kuvaaja kokonaiselinajasta
Taulukko 8. Tehoa mittaavat tulokset ≥ 18‑vuotiailla potilailla, joilla oli Philadelphia‑kromosominegatiivinen, uusiutunut tai refraktaarinen pre-B‑ALL (TOWER)
Blincyto (N = 271) | Tavanomainen solunsalpaajahoito (N = 134) | |
| Täydellinen remissio (CR) | ||
| CRa/CRh*b/CRic, n (%) [95 % CI] | 119 (43,9) (37,9–50,0) | 33 (24,6) (17,6–32,8) |
| Hoitojen välinen ero [95 % CI] | 19,3 (9,9–28,7) | |
| p‑arvo | < 0,001 | |
| CR, n (%) [95 % CI] | 91 (33,6) (28,0–39,5) | 21 (15,7) (10,0–23,0) |
| Hoitojen välinen ero [95 % CI] | 17,9 (9,6–26,2) | |
| p‑arvo | < 0,001 | |
| Elinaika ilman tapahtumiad | ||
| 6 kuukauden estimaatti, % [95 % CI] | 30,7 (25,0–36,5) | 12,5 (7,2–19,2) |
| 18 kuukauden estimaatti, % [95 % CI] | 9,5 (5,1–15,6) | 7,9 (3,7–14,2) |
| HR [95 % CI] | 0,55 (0,43–0,71) | |
Hematologisen vasteen kesto Mediaaniaika tapahtumaan [95 % CI] | ||
| CR | 8,3 (5,7–10,7) | 7,8 (2,2–19,0) |
| CR/CRh*/CRi | 7,3 (5,8–9,9) | 4,6 (1,8–19,0) |
| MRDe‑vaste CR/CRh*/CRi‑remissioryhmässä | ||
| Potilailla, joille MRD‑arviointi voitiin tehdä (%) [95% CI]f | 74/97 (76,3) (66,6–84,3) | 16/33 (48,5) (30,8–66,5) |
MRD‑vasteen kesto Mediaaniaika tapahtumaan [95 % CI] | 4,5 kuukautta (3,6–9,0) | 3,8 kuukautta (1,9–19,0) |
| Allogeeninen kantasolusiirto lähtötilanteen jälkeen, n (%) | ||
| Kaikki potilaat | 65 (24) | 32 (23,9) |
| Hematologisen vasteen saaneet (CR/CRh*/CRi) | 50 (42,0) | 18 (54,5) |
Aika allogeeniseen kantasolusiirtoon – kaikki siirron saaneet potilaat Mediaaniaika tapahtumaan (vaihteluväli) | 3,7 kuukautta (3,0–5,3) (N = 65) | 3,1 kuukautta (2,6–4,3) (N = 32) |
Aika allogeeniseen kantasolusiirtoon – CR/CRh*/CRi‑vasteen saaneet Mediaaniaika tapahtumaan [95 % CI] (KM‑estimaatti) | 11,3 kuukautta (5,2 – ei arvioitavissa) (N = 119) | 3,6 kuukautta (2,3–7,2) (N = 33) |
| Kuolleisuus 100 vuorokauden aikana allogeenisen kantasolusiirron jälkeen | ||
| n/N (%), [95 % CI] | 4/38, 12,4 % (4,8–29,9) | 0/12, 0,0 % (0,0–ei arvioitavissa) |
a Täydellisen remission (CR) kriteerit olivat: blastisolujen osuus luuytimen soluista ≤ 5 %, ei tautiin viittaavia löydöksiä ja perifeerisen veren solumäärien täydellinen palautuminen (trombosyytit > 100 000/mikrolitra ja absoluuttinen neutrofiilimäärä [ANC] > 1 000/mikrolitra). b CRh*‑remission (täydellinen remissio mutta vain osittainen hematologinen palautuminen) kriteerit olivat: blastisolujen osuus luuytimen soluista ≤ 5 %, ei tautiin viittaavia löydöksiä ja perifeerisen veren solumäärien osittainen palautuminen (trombosyytit > 50 000/mikrolitra ja ANC > 500/mikrolitra). c CRi‑remission (täydellinen remissio mutta epätäydellinen hematologinen palautuminen) kriteerit olivat: blastisolujen osuus luuytimen soluista ≤ 5 %, ei tautiin viittaavia löydöksiä ja perifeerisen veren solumäärien epätäydellinen palautuminen (trombosyytit > 100 000/mikrolitra tai ANC > 1 000/mikrolitra). d Elinaika ilman tapahtumia (EFS) laskettiin satunnaistamisajankohdasta siihen päivään, jolloin arvioinnissa todettiin taudin uusiutuminen CR/CRh*/CRi‑remission saavuttamisen jälkeen tai kuolema, sen mukaan, kumpi tulee ensiksi. Ellei potilas saavuta CR/CRh*/CRi‑remissiota 12 viikon kuluessa hoidon alkamisesta, hoidon katsotaan epäonnistuneen, ja elinajaksi ilman tapahtumia (EFS) merkitään 1 vuorokausi. e MRD‑vasteen (minimaalisen jäännöstaudin) kriteeri oli: polymeraasiketjureaktiolla (PCR) tai virtaussytometrialla määritetty MRD < 1 × 10−4. f Potilaat, jotka saavuttivat CR/CRh*/CRi‑remission ja joilla oli arvioitavissa oleva MRD‑määritystulos lähtötilanteen jälkeen. | ||
Terveyteen liittyvä elämänlaatu
Tässä avoimessa tutkimuksessa potilaiden raportoimaa terveyteen liittyvää elämänlaatua (Health related quality of life (HRQoL)) mitattiin EORTC QLQ‑C30-asteikolla (European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Core 30). Post hoc ‑herkkyysanalyysissä Blincyto viivästytti johdonmukaisesti terveyteen liittyvän elämänlaadun kliinisesti merkittävää huononemista (≥ 10 pisteen huononeminen lähtötasosta) tavanomaiseen hoitoon verrattuna yleisen terveydentilan osiossa [mediaani, Blincyto 8,1 kuukautta, tavanomainen hoito 1,0 kuukautta; HR = 0,60 (95 % CI = 0,42–0,85)], toiminta-asteikoissa, oireasteikoissa ja yksittäisissä kohdissa. Koska terveyteen liittyvän elämänlaadun tulokset perustuvat post hoc ‑herkkyysanalyysiin, tuloksia on tulkittava varoen.
Blincytoa arvioitiin myös toisen vaiheen avoimessa yhden hoitohaaran monikeskustutkimuksessa (MT103‑211) 189 potilaalla. Tutkimukseen otettiin ≥ 18‑vuotiaita potilaita, joilla oli Philadelphia‑kromosominegatiivinen uusiutunut tai refraktaarinen pre-B‑ALL (uusiutunut ≤ 12 kuukautta kestäneen ensimmäisen remission jälkeen ensimmäisen salvage-hoidon aikana tai uusiutunut tai refraktaarinen ensimmäisen salvage-hoidon jälkeen tai uusiutunut 12 kuukauden kuluessa allogeenisesta kantasolusiirrosta, ja blastisolujen osuus luuytimessä oli ≥ 10 %).
Esilääkitys, Blincyto‑annos hoitojaksoa kohti ja antoreitti olivat samat kuin kolmannen vaiheen tutkimuksessa. Potilaat saivat Blincyto‑hoidon aloittamista edeltävän viikon aikana esilääkityksenä pakollisen aivo-selkäydinnesteprofylaksin, joka koostui laitoskohtaisten tai kansallisten hoitosuositusten mukaisesta intratekaalisesta hoito-ohjelmasta. Blincyto annettiin jatkuvana infuusiona laskimoon. Ensimmäisessä hoitojaksossa aloitusannos oli 9 mikrog/vrk ensimmäisen viikon ajan, jonka jälkeen annos nostettiin 28 mikrog:aan/vrk kolmen viikon ajaksi. Tätä tavoiteannosta 28 mikrog/vrk annettiin toisen ja myöhempien hoitojaksojen aikana jokaisen jakson ensimmäisestä päivästä alkaen. Annosta voitiin säätää, jos haittavaikutuksia ilmaantui. Hoitoa saaneeseen potilasjoukkoon kuului 189 potilasta, jotka saivat vähintään yhden Blincyto‑infuusion. Hoitojaksojen lukumäärä potilasta kohti oli 1,6 (keskiarvo). Jos Blincyto‑hoidolla oli saavutettu hoitovaste mutta tauti oli myöhemmin uusiutunut, potilaalla oli mahdollisuus saada uusi Blincyto‑hoito. Hoitoa saaneiden potilaiden mediaani-ikä oli 39 vuotta (vaihteluväli: 18–79 vuotta; 25 potilasta oli ≥ 65‑vuotiaita), 189 potilaasta 64 (33,9 %) oli saanut kantasolusiirron ennen Blincyto‑hoitoa ja 189 potilaasta 32 (16,9 %) oli saanut aikaisemmin enemmän kuin kaksi salvage-hoitoa.
Ensisijainen päätetapahtuma oli täydellisen remission / täydellisen remission mutta vain osittaisen hematologisen palautumisen (CR/CRh*) saavuttaneiden potilaiden osuus kahden Blincyto‑hoitojakson kuluessa. CR/CRh*‑remission saavutti kahden ensimmäisen hoitojakson aikana 81 potilasta 189:stä (42,9 %), ja suurin osa hoitovasteista (64/81) saavutettiin yhden hoitojakson kuluessa. Iäkkäiden (≥ 65‑vuotiaiden) potilaiden ryhmässä 11 potilasta 25:stä (44,0 %) saavutti CR/CRh*‑remission kahden ensimmäisen hoitojakson kuluessa (ks. kohdasta Haittavaikutukset tietoa hoidon turvallisuudesta iäkkäiden potilaiden hoidossa). Neljä potilasta saavutti täydellisen remission (CR) vakautushoitojaksojen aikana, joten täydellisen remission saavuttaneiden kumulatiivinen osuus oli 35,4 % (67/189; 95 % CI: 28,6–42,7 %). Allogeeninen kantasolusiirto tehtiin 189 potilaasta 32:lle (17 %) Blincyto‑hoidolla saavutetun CR/CRh*‑remission aikana (ks. taulukko 9).
Taulukko 9. Tehoa mittaavat tulokset ≥ 18‑vuotiailla potilailla, joilla oli Philadelphia‑kromosominegatiivinen uusiutunut tai refraktaarinen pre-B‑ALL (MT103‑211)
n (%) n = 189 | 95 % CI | |
| Täydellinen remissio (CR)1 / Täydellinen remissio mutta vain osittainen hematologinen palautuminen (CRh*)2 | 81 (42,9 %) | [35,7–50,2 %] |
| CR | 63 (33,3 %) | [26,7–40,5 %] |
| CRh* | 18 (9,5 %) | [5,7–14,6 %] |
| Hypoplastinen tai aplastinen luuydin, ei blasteja3 | 17 (9,0 %) | [5,3–14,0 %] |
| Osittainen remissio4 | 5 (2,6 %) | [0,9–6,1 %] |
| CR/CRh*‑potilaiden elinaika ilman taudin uusiutumista5 (RFS) | 5,9 kuukautta | [4,8–8,3 kuukautta] |
| Kokonaiselinaika | 6,1 kuukautta | [4,2–7,5 kuukautta] |
1 Täydellisen remission (CR) kriteerit olivat: blastisolujen osuus luuytimen soluista ≤ 5 %, ei tautiin viittaavia löydöksiä ja perifeerisen veren solumäärien täydellinen palautuminen (trombosyytit > 100 000/mikrolitra ja absoluuttinen neutrofiilimäärä [ANC] > 1 000/mikrolitra). 2 CRh*:n kriteerit olivat: blastisolujen osuus luuytimen soluista ≤ 5 %, ei tautiin viittaavia löydöksiä ja perifeerisen veren solumäärien osittainen palautuminen (trombosyytit > 50 000/mikrolitra ja ANC > 500/mikrolitra). 3 Hypoplastinen tai aplastinen luuydin, ei blasteja: blastisolujen osuus luuytimen soluista ≤ 5 %, ei tautiin viittaavia löydöksiä, perifeerisen veren solumäärien riittämätön palautuminen: trombosyytit ≤ 50 000/mikrolitra ja/tai ANC ≤ 500/mikrolitra. 4 Osittaisen remission kriteerinä oli blastisolujen osuus luuytimen soluista 6–25 % ja osuus pienentynyt lähtötasosta vähintään 50 %. 5 Taudin uusiutumiseksi katsottiin hematologinen relapsi (blastisolujen osuus luuytimen soluista yli 5 % täydellisen remission jälkeen) tai ekstramedullaarinen relapsi. | ||
Etukäteen määritellyssä eksploratiivisessa analyysissä CR/CRh*‑remission saavuttaneista 73 potilaasta, joilla minimaalinen jäännöstauti (MRD) oli arvioitavissa, 60 potilaalla (82,2 %) todettiin vaste myös jäännöstaudin osalta (vasteen kriteerinä oli MRD < 1 × 10−4 polymeraasiketjureaktiolla (PCR) määritettynä).
Hoitovaste oli samanlainen aikaisemmin allogeenisen kantasolusiirron saaneilla potilailla kuin potilailla, jotka eivät olleet saaneet kantasolusiirtoa, ja iäkkäillä potilailla samanlainen kuin nuoremmilla potilailla, eikä aikaisempien salvage-hoitolinjojen lukumäärällä näyttänyt olevan selvää vaikutusta remissio-osuuksiin.
Potilailla, joilla oli seulontavaiheessa ekstramedullaarinen tauti (vähintään yksi ≥ 1,5 cm:n leesio) muualla kuin keskushermostossa tai kiveksissä (N = 8/189), kliininen vasteosuus oli pienempi (25 % [95 % CI: 3,2–65,1]) kuin potilailla, joilla ei ollut viitteitä ekstramedullaarisesta taudista (N = 181, 43,6 % [95 % CI: 36,3–51,2]) (ks. kuva 2).
Potilailla, joilla oli lähtötilanteessa suurin kasvaintaakka mitattuna blastisolujen osuudella luuytimessä (≥ 90 %), saavutettiin kuitenkin kliinisesti merkittävä vaste ja CR/CRh*‑osuus oli 21,6 % (95 % CI: 12,9–32,7) (ks. kuva 2). Potilailla, joilla oli pieni kasvaintaakka (< 50 %), saatiin paras vaste Blincyto‑hoidolle ja CR/CRh*‑osuus heillä oli 72,9 % (95 % CI: 59,7–83,6).
Kuva 2. CR/CRh*‑osuus 2 ensimmäisen hoitojakson aikana MT103‑211-tutkimuksessa (primaarinen analyysijoukko) – Forest plot ‑diagrammi
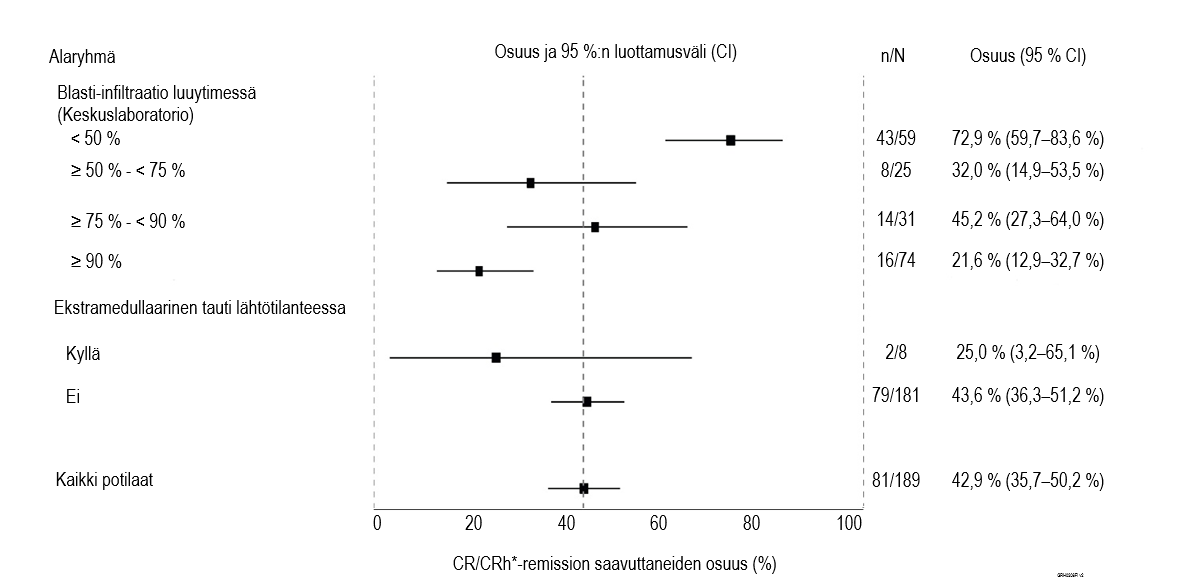
n = 2 ensimmäisen hoitojakson aikana CR‑ tai CRh*‑remission saavuttaneiden potilaiden lukumäärä kyseisessä alaryhmässä.
N = potilaiden kokonaismäärä kyseisessä alaryhmässä.
Tutkimustietoa on vain vähän potilaista, joilla pre-B‑ALL:n ensimmäinen uusiutuminen tapahtui myöhään eli yli 12 kuukautta ensimmäisen remission jälkeen tai yli 12 kuukautta ensimmäisen remission aikana annetun kantasolusiirron jälkeen. Toisen vaiheen kliinisissä tutkimuksissa niistä potilaista, joilla taudin ensimmäinen uusiutuminen tapahtui myöhään yksittäisten tutkimusten määritelmän mukaan, 88,9 % (8/9) saavutti CR/CRh*‑remission kahden ensimmäisen hoitojakson aikana ja 62,5 % (6/9) saavutti vasteen myös MRD:n osalta ja 37,5 prosentille (3/9) tehtiin allogeeninen kantasolusiirto Blincyto‑hoidon jälkeen. Kokonaiselinajan (OS) mediaani oli 17,7 kuukautta (95 % CI: 3,1 – ei arvioitavissa).
Kolmannen vaiheen satunnaistetussa, avoimessa monikeskustutkimuksessa (TOWER) siirron saaneista potilaista, joilla taudin ensimmäinen uusiutuminen tapahtui myöhään, 70 % (7/10) Blincyto‑hoitoa saaneista ja 20 % (1/5) tavanomaista solunsalpaajahoitoa saaneista saavutti CR/CRh*‑remission kahden ensimmäisen hoitojakson aikana. Vastaavasti 50 % (5/10) ja 0 % (0/5) saavutti MRD‑vasteen ja 20 prosentille (2/10) ja 40 prosentille (2/5) tehtiin hoidon jälkeen allogeeninen kantasolusiirto. Kokonaiselinajan mediaani oli Blincyto‑ryhmässä 15,6 kuukautta (95 % CI: 5,5 – ei arvioitavissa) ja tavanomaisen solunsalpaajahoidon ryhmässä 5,3 kuukautta (95 % CI: 1,1 – ei arvioitavissa).
Philadelphia‑kromosomipositiivinen uusiutunut tai refraktaarinen pre‑B‑ALL aikuispotilailla
Blincyton turvallisuutta ja tehoa arvioitiin toisen vaiheen avoimessa yhden hoitohaaran monikeskustutkimuksessa (ALCANTARA). Tutkimukseen otettiin ≥ 18-vuotiaita potilaita, joilla oli Philadelphia‑kromosomipositiivinen pre‑B‑ALL. Tauti oli uusiutunut tai ei reagoinut vähintään yhteen toisen sukupolven tai uudempaan tyrosiinikinaasin estäjään (TKI) tai vaihtoehtoisesti potilaat eivät sietäneet toisen sukupolven tyrosiinikinaasin estäjää ja eivät joko sietäneet imatinibimesylaattia tai se ei tehonnut.
Blincyto annettiin jatkuvana infuusiona laskimoon. Ensimmäisessä hoitojaksossa aloitusannos oli 9 mikrog/vrk ensimmäisen viikon ajan, jonka jälkeen annos nostettiin 28 mikrog:aan/vrk kolmen viikon ajaksi. Toisen ja myöhempien hoitojaksojen aikana annosta 28 mikrog/vrk annettiin jokaisen jakson ensimmäisestä päivästä alkaen. Annoksen säätäminen oli mahdollista, jos haittavaikutuksia ilmaantui. Hoitoa saaneeseen potilasjoukkoon kuului 45 potilasta, jotka saivat vähintään yhden Blincyto‑infuusion, ja hoitojaksojen lukumäärän keskiarvo oli 2,2. (Ks. potilaiden demografiset ja muut ominaisuudet lähtötilanteessa taulukosta 10.)
Taulukko 10. Potilaiden demografiset ja muut ominaisuudet lähtötilanteessa toisen vaiheen tutkimuksessa (ALCANTARA)
| Ominaisuus | Blincyto (N = 45) |
| Ikä | |
| Mediaani, vuotta (min, max) | 55 (23, 78) |
| Keskiarvo, vuotta (SD) | 52,8 (15) |
| ≥ 65 vuotta ja < 75 vuotta, n (%) | 10 (22,2) |
| ≥ 75 vuotta, n (%) | 2 (4,4) |
| Miehiä, n (%) | 24 (53,3) |
| Rotu, n (%) | |
| Aasialainen | 1 (2,2) |
| Musta (tai afroamerikkalainen) | 3 (6,7) |
| Muu | 2 (4,4) |
| Valkoinen | 39 (86,7) |
| Tautihistoria, n (%) | |
| Aikaisempi TKI-hoitoa | |
| 1 | 7 (15,6) |
| 2 | 21 (46,7) |
| ≥ 3 | 17 (37,8) |
| Aikaisempi salvage-hoito | 31 (61,9) |
| Aikaisempi allogeeninen kantasolusiirto | 20 (44,4) |
| Luuytimen blastien määräb, n (%) | |
| ≥ 50 % – < 75 % | 6 (13,3) |
| ≥ 75 % | 28 (62,2) |
a potilaat, joilla ponatinibihoito epäonnistui = 23 (51,1 %) b arvioitu keskuslaboratoriossa | |
Ensisijainen päätetapahtuma oli CR/CRh*‑osuus kahden Blincyto‑hoitojakson kuluessa. CR/CRh*‑remission saavutti kahden ensimmäisen hoitojakson aikana 16 potilasta 45:stä (35,6 %). Niistä 16 potilaasta, jotka saavuttivat CR/CRh*‑remission kahden ensimmäisen hoitojakson aikana, 12 potilasta 14:stä CR-remission saavuttaneesta potilaasta (85,7 %) ja molemmat (2) CRh*‑remission saavuttaneet potilaat (100 %) saavuttivat myös täydellisen MRD‑vasteen (ks. taulukko 11).
Seuraavien hoitojaksojen aikana kaksi potilasta saavutti täydellisen remission (CR), joten täydellisen remission saavuttaneiden kumulatiivinen osuus oli 35,6 % (16/45; 95 % CI: 21,9–51,2). Allogeeninen kantasolusiirto tehtiin 16 potilaasta viidelle (31,3 %) Blincyto‑hoidolla saavutetun CR/CRh*‑remission aikana.
Taulukko 11. Tehoa mittaavat tulokset ≥ 18‑vuotiailla potilailla, joilla oli Philadelphia‑kromosomipositiivinen uusiutunut tai refraktaarinen pre‑B‑ALL (ALCANTARA)
| N = 45 | |
| Täydellinen remissio (CR)a / Täydellinen remissio mutta vain osittainen hematologinen palautuminen (CRh*)b, n (%) [95 % CI] | 16 (35,6) [21,9–51,2] |
| CR | 14 (31,1) [18,2–46,6] |
| CRh* | 2 (4,4) [0,5–15,1] |
| CRic (ilman CRh*-remissiota), n (%) [95 % CI] | 2 (4,4) [0,5–15,1] |
| Hypoplastinen tai aplastinen luuydin, ei blasteja (ilman CRi-remissiota)d, n (%) [95 % CI] | 3 (6,7) [1,4–18,3] |
| Osittainen remissioe, n (%) [95 % CI] | 2 (4,4) [0,5–15,1] |
| Täydellinen MRD‑vastef, n (%), [95 % CI] | 18 (40,0) [25,7–55,7] |
| CR/CRh*‑potilaiden elinaika ilman taudin uusiutumista (RFS), mediaanig [95 % CI] | 6,7 kuukautta [4,4 – ei arvioitavissa] |
| Kokonaiselinajan mediaani [95 % CI] | 7,1 kuukautta [5,6 – ei arvioitavissa] |
a Täydellisen remission (CR) kriteerit olivat: blastisolujen osuus luuytimen soluista ≤ 5 %, ei tautiin viittaavia löydöksiä ja perifeerisen veren solumäärien täydellinen palautuminen (trombosyytit > 100 000/mikrolitra ja absoluuttinen neutrofiilimäärä [ANC] > 1 000/mikrolitra). b CRh*‑remission kriteerit olivat: blastisolujen osuus luuytimen soluista ≤ 5 %, ei tautiin viittaavia löydöksiä ja perifeerisen veren solumäärien osittainen palautuminen (trombosyytit > 50 000/mikrolitra ja ANC > 500/mikrolitra). c. CRi‑remission (täydellinen remissio mutta epätäydellinen hematologinen palautuminen) kriteerit olivat: blastisolujen osuus luuytimen soluista ≤ 5 %, ei tautiin viittaavia löydöksiä ja perifeerisen veren solumäärien epätäydellinen palautuminen (trombosyytit > 100 000/mikrolitra tai ANC > 1 000/mikrolitra). d Hypoplastinen tai aplastinen luuydin, ei blasteja: blastisolujen osuus luuytimen soluista ≤ 5 %, ei tautiin viittaavia löydöksiä, perifeerisen veren solumäärien riittämätön palautuminen: trombosyytit ≤ 50 000/mikrolitra ja/tai ANC ≤ 500/mikrolitra. e Osittaisen remission kriteerinä oli blastisolujen osuus luuytimen soluista 6–25 % ja osuus pienentynyt lähtötasosta vähintään 50 %. f Täydelliseksi MRD‑vasteeksi katsottiin havaittavan MRD:n puuttuminen vahvistettuna määritysmenetelmällä, jonka herkkyys on vähintään 10-4. g Taudin uusiutumiseksi katsottiin hematologinen relapsi (blastisolujen osuus luuytimen soluista yli 5 % täydellisen remission jälkeen) tai ekstramedullaarinen relapsi. | |
Potilailla, joilla oli lähtötilanteessa suurempi kasvaintaakka mitattuna blastisolujen osuudella luuytimessä (≥ 50 %), saavutettiin kuitenkin kliinisesti merkittävä vaste ja CR/CRh*‑osuus oli 26,5 % (95 % CI: 12,9–44,4). Potilailla, joilla oli pieni kasvaintaakka (< 50 %), saatiin paras vaste Blincyto‑hoidolle ja CR/CRh*‑osuus heillä oli 63,6 % (95 % CI: 30,8–89,1). Potilaista, joilla perifeerisen veren valkosolumäärät olivat suuria (≥ 3,0 × 109/l), 27,3 % saavutti vasteen (95 % CI: 10,7–50,2), kun taas niistä, joilla valkosolumäärät olivat pienempiä (< 3,0 × 109/l), vasteen saaneiden osuus oli 43,5 % (95 % CI: 23,2–65,5).
Hoidon vaikutukset arviointiin soveltuvissa alaryhmissä (esim. mutaatiostatus, aiempien TKI‑hoitojen määrä, mahdollinen aiempi kantasolusiirto ja uusiutuminen ilman aiempaa kantasolusiirtoa) olivat yhdenmukaisia koko potilasjoukon tulosten kanssa. Hoitovasteen saaneiden osuus niistä potilaista, joilla oli T315I‑mutaatio tai muita mutaatioita tai sytogeneettisiä poikkeavuuksia, oli samankaltainen kuin potilailla, joilla näitä mutaatioita tai poikkeavuuksia ei ollut.
MRD‑positiivinen pre-B‑ALL
Blincyton turvallisuutta ja tehoa MRD‑positiivista pre‑B‑ALL:ää sairastavilla aikuispotilailla arvioitiin avoimessa toisen vaiheen yhden hoitohaaran monikeskustutkimuksessa (BLAST). Tutkimukseen otettiin ≥ 18‑vuotiaita potilaita, jotka eivät olleet aiemmin saaneet kantasolusiirtoa, olivat saaneet vähintään 3 hoitoblokkia ALL:n tavanomaista induktiohoitoa, olivat täydellisessä hematologisessa remissiossa (kriteerit: blastisolujen osuus luuytimen soluista < 5 %, absoluuttinen neutrofiilimäärä ≥ 1 000/mikrolitra, trombosyytit ≥ 50 000/mikrolitra ja hemoglobiiniarvo ≥ 9 g/dl) ja joilla oli todettu molekulaarinen hoidon epäonnistuminen tai molekulaarinen relapsi (kriteeri: MRD ≥ 10-3), ks. taulukko 12. Minimaalinen jäännöstauti määritettiin seulonnan aikaan luuytimen aspiraationäytteestä virtaussytometrialla tai polymeraasiketjureaktiolla (PCR) paikallisessa laboratioriossa, jossa menetelmän herkkyys oli vähintään 10-4. Tämän jälkeen MRD:n aste vahvistettiin PCR‑menetelmällä keskuslaboratoriossa. MRD‑tulosten lopullisessa tulkinnassa noudatettiin EuroMRD Consortiumin ohjeistusta.
Taulukko 12. Potilaiden demografiset ja muut ominaisuudet lähtötilanteessa MRD‑tutkimuksessa (BLAST)
| Ominaisuus | Blincyto (N = 116) |
| Ikä | |
| Mediaani, vuotta (min, max) | 45 (18, 76) |
| Keskiarvo, vuotta (SD) | 44,6 (16,4) |
| ≥ 65 vuotta, n (%) | 15 (12,9) |
| Miehiä, n (%) | 68 (58,6) |
| Rotu, n (%) | |
| Aasialainen | 1 (0,9) |
| Muu (sekoittunut) | 1 (0,9) |
| Valkoinen | 102 (87,9) |
| Tuntematon | 12 (10,3) |
| Taudin uusiutumishistoria, n (%) | |
| Potilaat, jotka 1. CR:ssä | 75 (64,7) |
| Potilaat, jotka 2. CR:ssä | 39 (33,6) |
| Potilaat, jotka 3. CR:ssä | 2 (1,7) |
| MRD:n aste lähtötilanteessa*, n (%) | |
| ≥ 10-1 ja < 1 | 9 (7,8) |
| ≥ 10-2 ja < 10-1 | 45 (38,8) |
| ≥ 10-3 ja < 10-2 | 52 (44,8) |
| < 10-3 | 3 (2,6) |
| Alle mitattavan alarajan | 5 (4,3) |
| Tuntematon | 2 (1,7) |
| * Arvioitu keskuslaboratoriossa määritysmenetelmällä, jonka herkkyys on vähintään 10-4 | |
Blincyto annettiin jatkuvana infuusiona laskimoon. Potilaat saivat Blincyto‑hoitoa vakioannoksella 15 mikrog/m2/vrk (vastaa suositeltua annostusta 28 mikrog/vrk) kaikkien hoitojaksojen aikana. Potilaat saivat enintään 4 hoitojaksoa. Annoksen säätäminen oli mahdollista, jos haittavaikutuksia ilmaantui. Hoitoa saaneeseen potilasjoukkoon kuului 116 potilasta, jotka saivat vähintään yhden Blincyto‑infuusion. Kokonaisten hoitojaksojen lukumäärän keskiarvo oli 1,8 (vaihteluväli: 1–4).
Ensisijainen päätetapahtuma oli täydellisen MRD‑vasteen saavuttaneiden potilaiden osuus yhden Blincyto‑hoitojakson kuluessa. 88 potilasta 113:sta (77,9 %) arviointiin soveltuvasta potilaasta saavutti täydellisen MRD‑vasteen yhden hoitojakson jälkeen, ks. taulukko 13. Kaksi potilasta saavutti täydellisen MRD‑vasteen saatuaan yhden lisähoitojakson Blincytoa. MRD‑vasteosuudet iän ja lähtötilanteen MRD‑asteen mukaisissa alaryhmissä olivat yhdenmukaiset koko potilasjoukon tulosten kanssa. Philadelphia‑kromosominegatiivista pre-B-ALL:ää sairastavista elossa ilman taudin uusiutumista (RFS) oli 18 kuukauden kohdalla 54 % (33 %, 70 %), kun sensurointi tehtiin kantasolusiirron tai Blincyto‑hoidon jälkeen annetun solunsalpaajahoidon ajankohtana. Elossa ilman taudin uusiutumista (RFS) oli 18 kuukauden kohdalla 53 % (44 %, 62 %), kun sensurointia kantasolusiirron tai Blincyto‑hoidon jälkeen annetun solunsalpaajahoidon ajankohtana ei tehty.
Taulukko 13. Tehoa mittaavat tulokset ≥ 18‑vuotiailla potilailla, joilla oli MRD‑positiivinen pre‑B‑ALL (BLAST)
| Täydellinen MRD‑vastea, n/N (%), [95 % CI] | 88/113b (77,9) [69,1–85,1] |
| ≥ 65‑vuotiaat | 12/15 (80,0) [51,9–95,7] |
| Potilaat, jotka 1. CR:ssä | 60/73 (82,2) [71,5–90,2] |
| Potilaat, jotka 2. CR:ssä | 27/38 (71,1) [54,1–84,6] |
| Potilaat, jotka 3. CR:ssä | 1/2 (50,0) [1,3–98,7] |
| Täydellisen MRD‑vasteen kesto [95 % CI] | 17,3 kuukautta [12,6–23,3] |
a Täydelliseksi MRD‑vasteeksi katsottiin havaittavan MRD:n puuttuminen vahvistettuna määritysmenetelmällä, jonka herkkyys on vähintään 10-4. b Ensisijaisen päätetapahtuman koko analyysijoukkoon kuului 113 potilasta (97,4 %; 113/116). | |
Pre‑B‑ALL vakautushoitovaiheessa
Blincyton tehoa pre-B‑ALL:n vakautushoitovaiheessa aikuis- ja lapsipotilailla arvioitiin tutkimuksissa E1910 ja 20120215. Tutkimuksen E1910 tehoa koskevat tulokset on kuvattu alla, ja lapsia ja nuoria aikuisia koskevat tutkimukset on kuvattu Pediatriset potilaat -kohdassa.
Tutkimuksessa E1910 (20129152) Blincyton turvallisuutta ja tehoa arvioitiin vaiheen III satunnaistetussa kontrolloidussa tutkimuksessa aikuispotilailla, joilla oli juuri diagnosoitu Philadelphia‑kromosominegatiivinen pre-B‑ALL. Tutkimukseen soveltuvat potilaat saivat induktiohoitona solunsalpaajahoitoa. Induktion jälkeen potilaat, joilla oli hematologisesti täydellinen remissio (CR) tai CR, jossa perifeerisen veren solumäärät ovat epätäydellisesti palautuneet (CRi), jatkoivat tutkimusta ja saivat tehostusvaiheen solunsalpaajahoitoa. Tehostushoidon jälkeen 286 potilasta satunnaistettiin eli määrättiin saamaan Blincyto‑hoitoa vuorotellen vakauttavan solunsalpaajahoidon kanssa (n = 152) tai pelkkää tavanomaista (SOC) vakauttavaa solunsalpaajahoitoa (n = 134). Tutkimuksessa E1910 käytetyt solunsalpaajahoidon hoito-ohjelmat perustuivat tutkimussuunnitelmiin UKALL12/ECOG2993. Kumpikin hoitohaara sai ylläpitohoitoa, ja hoidon kokonaiskesto oli 2,5 vuotta tehostushoidon alkamisen jälkeen. Satunnaistaminen ositettiin MRD‑tilan (MRD‑negatiivisuudeksi määriteltiin < 1 × 10-4), iän (< 55 vuotta vs. ≥ 55 vuotta), CD20‑ilmentymisen, rituksimabin käytön ja suunnitellun allogeenisen kantasolusiirron perusteella.
Tutkimushoito Blincyto‑haarassa koostui 4 blinatumomabijaksosta ja 4 solunsalpaajahoitojaksosta seuraavassa järjestyksessä. Se sisälsi 2 Blincyto‑jaksoa (kukin jakso sisälsi 28 mikrogrammaa/vrk Blincytoa, jota annettiin jatkuvana laskimoinfuusiona 28 päivän ajan, ja jaksojen välillä oli 14 päivän hoitotauko), joita seurasivat 3 vakauttavaa solunsalpaajahoitojaksoa, kolmas Blincyto‑jakso ja sen jälkeen vielä yksi vakauttava solunsalpaajahoitojakso ja sitten neljäs Blincyto‑jakso. Jälkikäteisanalyysissä tutkittavilla, jotka eivät saaneet kantasolusiirtoa, havaittiin potilailla, jotka saivat 4 Blincyto‑jaksoa, numeerisesti suurempi OS kuin potilailla, jotka saivat 1–2 Blincyto‑jaksoa ensilinjan vakautushoitona. Potilaat saivat keskimäärin 3,04 Blincyto‑jaksoa. Jos potilas sai tämän jälkeen allogeenisen kantasolusiirron, blinatumomabia saamaan satunnaistetuille potilaille suositeltiin vahvasti kahden blinatumomabihoitojakson läpikäyntiä ennen allogeenisen kantasolusiirron saamista. Tutkimuksen SOC‑haara koostui neljästä vakauttavasta solunsalpaajahoitojaksosta. Kummankin hoitohaaran potilaat saivat saman määrän solunsalpaajien hoitojaksoja ja annoksia vakautushoidossa. Potilaat, jotka satunnaistettiin SOC‑haaraan, voivat saada suoraan allogeenisen kantasolusiirron tai jatkaa saamaan suoraan vakauttavaa solunsalpaajahoitoa.
Lähtötilanteen demografiset ja muut ominaisuudet olivat samanlaiset hoitohaarojen välillä. Demografiset ja muut ominaisuudet esitetään taulukossa 14.
Taulukko 14. Demografiset ja muut ominaisuudet (E1910)
| Ominaisuus | Blincyto‑haara (N = 152) | SOC-haaraa (N = 134) | ||
MRD-positiiviset (N = 40) | MRD-negatiiviset (N = 112) | MRD-positiiviset (N = 22) | MRD-negatiiviset (N = 112) | |
| Ikä | ||||
| Keskiarvo, vuotta (min, max) | 49,6 (30, 69) | 50,2 (30, 70) | ||
| Miehiä, n (%) | 69 (45,4) | 70 (52,2) | ||
| Rotu, n (%) | ||||
| Amerikan tai Alaskan alkuperäisasukas | 2 (1,3) | 1 (0,7) | ||
| Aasialainen | 4 (2,6) | 2 (1,5) | ||
| Mustaihoinen (tai afroamerikkalainen) | 12 (7,9) | 5 (3,7) | ||
| Latinalaisamerikkalainen (latino) | 21 (13,8) | 15 (11,2) | ||
| Havaijin tai muiden Tyynenmeren saarten alkuperäisasukas | 1 (0,7) | 0 (0,0) | ||
| Valkoihoinen | 117 (77,0) | 110 (82,1) | ||
| Saanut allogeenisen kantasolusiirron, n (%) | 37 (24,3) | 28 (20,9) | ||
| Allogeenisen kantasolusiirron saaneiden potilaiden Blincyto‑jaksojen keskimääräinen lukumäärä, n (jaksoa) | 15 (1,93) | 22 (1,95) | ||
| Niiden potilaiden Blincyto‑jaksojen keskimääräinen lukumäärä, jotka eivät saaneet allogeenistä kantasolusiirtoa, n (jaksoa) | 21 (2,90) | 89 (3,30) | ||
| a SOC = tavanomainen hoito. | ||||
Ensisijainen päätetapahtuma oli kokonaiselinaika (OS) potilailla, jotka olivat MRD‑negatiivisia. Toissijaisia päätetapahtumia olivat elinaika ilman taudin uusiutumista (RFS) potilailla, jotka olivat MRD-negatiivisia, sekä OS ja RFS potilailla, jotka olivat MRD-positiivisia.
Tutkimus osoitti, että sekä OS ja RFS paranivat. Taulukossa 15 on ilmoitettu ositetut vaarasuhteet (HR) sekä OS- ja RFS-Kaplan-Meier-estimaatit potilaille, jotka olivat MRD-negatiivisia, MRD-positiivisia, ja yhdessä kaikille potilaille MRD-tilasta riippumatta. Kaplan-Meierin kuvaaja OS:n suhteen potilaille, jotka olivat MRD-negatiivisia, esitetään kuvassa 3. Kaplan-Meierin kuvaaja OS:n suhteen yhdessä kaikille potilaille MRD-tilasta riippumatta esitetään kuvassa 4.
Kuva 3. Satunnaistamisen aikaan (ennen vakautushoidon aloittamista) MRD-negatiivisten potilaiden kokonaiselinajan Kaplan-Meier (E1910)
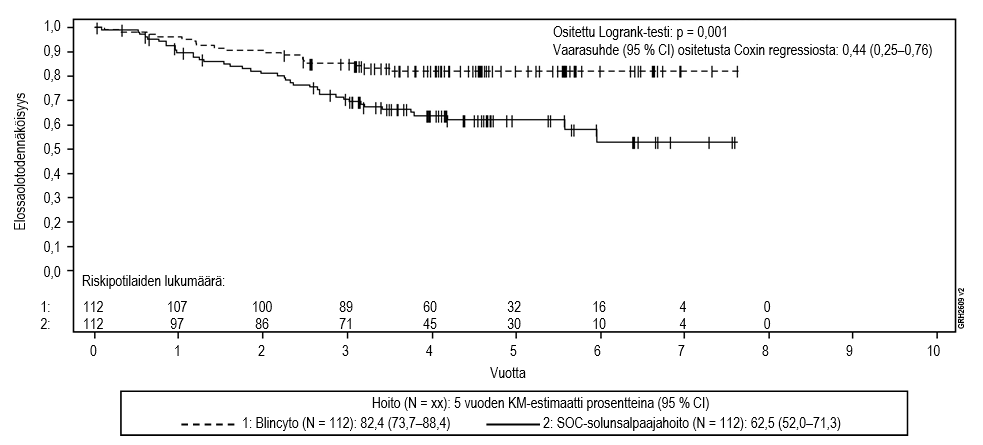
SOC = tavanomainen hoito, KM = Kaplan‑Meier, CI = luottamusväli, N = potilaiden määrä analyysijoukossa, sensurointi merkitty pystyviivalla.
Kuva 4. Kokonaiselinajan Kaplan-Meier, johon on yhdistetty satunnaistamisen aikaan (ennen vakautushoidon aloittamista) MRD-positiiviset potilaat ja MRD-negatiiviset potilaat (E1910)
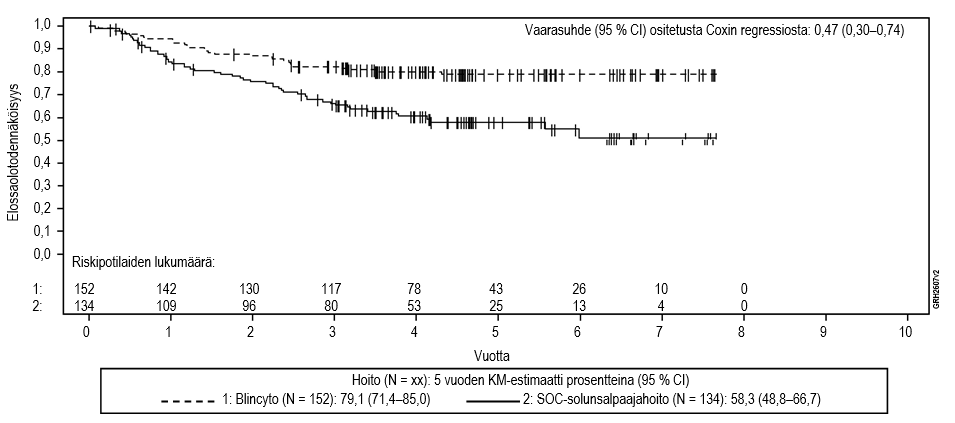
SOC = tavanomainen hoito, KM = Kaplan‑Meier, CI = luottamusväli, N = potilaiden määrä analyysijoukossa, sensurointi merkitty pystyviivalla.
Taulukko 15. Kokonaiselinaika ja elinaika ilman taudin uusiutumista MRD‑negatiivisilla ja MRD‑positiivisilla potilailla (E1910)
| Blincyto‑haara | SOC‑haara | |
| MRD‑negatiiviset | ||
| Potilaiden lukumäärä | 112 | 112 |
| Seurannan mediaanikesto (vuotta)a, b | 4,5 | 4,5 |
| Kokonaiselinaika | ||
| 5 vuoden Kaplan‑Meier‑estimaatti (%) [95 % CI] | 82,4 [73,7–88,4] | 62,5 [52,0–71,3] |
| Vaarasuhde [95 % CI]b | 0,44 [0,25–0,76] | |
| p‑arvo | 0,003 | |
| Elinaika ilman taudin uusiutumista | ||
| 5 vuoden Kaplan‑Meier‑estimaatti (%) [95 % CI] | 77,0 [67,8–83,8] | 60,5 [50,1–69,4] |
| Vaarasuhde [95 % CI]b | 0,53 [0,32–0,88] | |
| MRD‑positiiviset | ||
| Potilaiden lukumäärä | 40 | 22 |
| Seurannan mediaanikesto (vuotta)e, b | 4,6 | 5,0 |
| Kokonaiselinaika | ||
| 5 vuoden Kaplan‑Meier‑estimaatti (%) [95 % CI] | 70,1 [52,0–82,5] | 37,8 [17,8–57,7] |
| Vaarasuhde [95 % CI]f | 0,40 [0,14–1,12] | |
| Elinaika ilman taudin uusiutumista | ||
| 5 vuoden Kaplan‑Meier‑estimaatti (%) [95 % CI] | 71,8 [54,8–83,3] | 39,4 [19,3–59,0] |
| Vaarasuhde [95 % CI]g | 0,37 [0,13–1,03] | |
| MRD‑negatiiviset ja MRD‑positiiviset yhdessä | ||
| Potilaiden lukumäärä | 152 | 134 |
| Seurannan mediaanikesto (vuotta)a, b, e | 4,5 | 4,5 |
| Kokonaiselinaika | ||
| 5 vuoden Kaplan‑Meier‑estimaatti (%) [95 % CI] | 79,1 [71,4–85,0] | 58,3 [48,8–66,7] |
| Vaarasuhde [95 % CI]f | 0,47 [0,30–0,74] | |
| Elinaika ilman taudin uusiutumista | ||
| 5 vuoden Kaplan‑Meier‑estimaatti (%) [95 % CI] | 75,6 [67,8–81,8] | 57,2 [47,9–65,4] |
| Vaarasuhde [95 % CI]g | 0,53 [0,35–0,81] | |
Koko analyysijoukko sisältää kaikki satunnaistetut eli määrätyt potilaat, jotka on arvioitu keskitetysti MRD‑negatiivisiksi tai MRD‑positiivisiksi induktio- ja tehostamistarkoituksessa annetun solunsalpaajahoidon jälkeen. CI = luottamusväli. Elinaika ilman taudin uusiutumista (RFS) lasketaan satunnaistamis- tai rekisteröintihetkestä taudin uusiutumiseen tai mistä tahansa syystä johtuvaan kuolemaan. Kokonaiselinaika (OS) lasketaan satunnaistamis- tai rekisteröintihetkestä mistä tahansa syystä johtuvaan kuolemaan. MRD‑positiivinen määritellään MRD‑arvoksi ≥ 1 × 10-4 ja MRD‑negatiivinen MRD‑arvoksi < 1 × 10-4. a Vuodet lasketaan päivinä satunnaistamispäivästä tapahtumis-/sensurointipäivään jaettuna luvulla 365,25. b Sensurointiin kulunut aika mittaa seuranta-aikaa, joka on laskettu muuttamalla sensuroinnin ja tapahtuman tilaindikaattori käänteiseksi. c Vaarasuhteen arviot on saatu ositetun Coxin regressiomallin avulla. Vaarasuhde, joka on alle 1,0, tarkoittaa Blincyto‑haaran potilaiden alhaisempaa keskimääräistä kuolleisuutta ja pidempää elinaikaa verrattuna SOC‑haaran potilaisiin. d Vaarasuhteen arviot on saatu ositetun Coxin regressiomallin avulla. Vaarasuhde, joka on alle 1,0, tarkoittaa Blincyto‑haaran potilaiden alhaisempaa tapahtumien keskimääräistä esiintymisastetta ja pidempää elinaikaa ilman taudin uusiutumista verrattuna SOC‑haaran potilaisiin. e Vuodet lasketaan päivinä satunnaistamispäivästä tapahtumis-/sensurointipäivään jaettuna luvulla 365,25. f Vaarasuhteen arviot on saatu ositetun Coxin verrannollisten uhkien mallin avulla. Vaarasuhde, joka on alle 1,0, tarkoittaa Blincyto‑haaran potilaiden alhaisempaa keskimääräistä kuolleisuutta ja pidempää elinaikaa verrattuna SOC‑haaran potilaisiin. g Vaarasuhteen arviot on saatu ositetun Coxin verrannollisten uhkien mallin avulla. Vaarasuhde, joka on alle 1,0, tarkoittaa Blincyto‑haaran potilaiden alhaisempaa tapahtumien keskimääräistä esiintymisastetta ja pidempää elinaikaa ilman taudin uusiutumista verrattuna SOC‑haaran potilaisiin. | ||
Pediatriset potilaat
Blincyton turvallisuus ja teho on määritetty kahdessa avoimessa tutkimuksessa lapsipotilailla, joilla oli Philadelphia‑kromosominegatiivinen uusiutunut tai refraktaarinen pre‑B‑ALL: yksihaaraisessa vaiheen I/II tutkimuksessa (MT103-205) ja satunnaistetussa, kontrolloidussa vaiheen III tutkimuksessa (20120215).
Satunnaistetussa, kontrolloidussa, avoimessa monikeskustutkimuksessa (20120215) arvioitiin Blincyton turvallisuutta ja tehoa tavanomaiseen vakautushoitona käytettyyn solunsalpaajahoitoon verrattuna. Tutkimukseen soveltuivat 28 vrk–18-vuotiaat potilaat, joilla oli ensimmäistä kertaa uusiutunut korkean riskin Philadelphia-kromosominegatiivinen pre‑B‑ALL ja blastisolujen osuus luuytimen soluista oli < 25 %. Potilaiden korkean riskin määrittämiseen käytettiin IntReALL-kriteerejä. Tutkimuksesta suljettiin pois potilaat, joilla oli kliinisesti merkittävä ja hoitoa vaativa keskushermoston sairaus (esim. epilepsia, joka ei ole hoitotasapainossa) tai näyttöä ALL:n leviämisestä keskushermostoon. Tutkimukseen osallistuneet potilaat satunnaistettiin induktiohoidon ja vakautushoitona annetun 2 solunsalpaajahoitoblokin jälkeen.
Potilaat satunnaistettiin 1:1 saamaan joko Blincyto-hoitoa tai tavanomaisen vakautushoidon kolmannen solunsalpaajahoitoblokin (korkean riskin vakautushoito 3, HC3). Blincyto-hoitohaaran potilaat saivat yhden 4 viikon hoitojakson ajan Blincytoa 15 mikrog/m2/vrk jatkuvana infuusiona laskimoon (suurin sallittu vuorokausiannos oli 28 mikrog/vrk). Annoksen säätäminen oli mahdollista, jos haittavaikutuksia ilmaantui. Satunnaistaminen ositettiin iän (< 1 vuotta, 1–9 vuotta ja > 9 vuotta), sekä toisen vakautushoitona annetun solunsalpaajahoitoblokin lopuksi määritetyn luuytimen tilanteen ja induktiohoidon lopuksi määritetyn MRD-tilanteen (blastisoluja < 5 % ja MRD-aste < 10-3, blastisoluja < 5 % ja MRD-aste ≥ 10-3, ja blastisoluja ≥ 5 % mutta < 25 %) mukaan. Potilaiden demografiset ja muut ominaisuudet lähtötilanteessa olivat samankaltaiset molemmissa hoitohaaroissa (ks. taulukko 16). Kukaan potilaista ei ollut aiemmin saanut kantasolusiirtoa.
Taulukko 16. Potilaiden demografiset ja muut ominaisuudet lähtötilanteessa tutkimuksessa 20120215
| Ominaisuus | Blincyto (N = 54) | Tavanomainen solunsalpaajahoito (N = 54) |
| Ikä, n (%) | ||
| < 1 vuotta | 0 (0,0) | 0 (0,0) |
| 1–9 vuotta | 39 (72,2) | 38 (70,4) |
| ≥ 10–18 vuotta | 15 (27,8) | 16 (29,6) |
| Miehiä, n (%) | 30 (55,6) | 22 (40,7) |
| Rotu, n (%) | ||
| Amerikan tai Alaskan alkuperäisasukas | 0 (0,0) | 0 (0,0) |
| Aasialainen | 1 (1,9) | 3 (5,6) |
| Musta (tai afroamerikkalainen) | 0 (0,0) | 3 (5,6) |
| Havaijin tai muiden Tyynenmeren saarten alkuperäisasukas | 0 (0,0) | 0 (0,0) |
| Muu | 3 (5,6) | 5 (9,3) |
| Valkoinen | 50 (92,6) | 43 (79,6) |
| Mahdolliset geneettiset poikkeavuudet, n (%) | ||
| Ei | 34 (63,0) | 29 (53,7) |
| Kyllä | 20 (37,0) | 25 (46,3) |
| Hyperdiploidia | 6 (11,1) | 6 (11,1) |
| Hypodiploidia | 1 (1,9) | 0 (0,0) |
| t(v;11q23)/MLL:n uudelleenjärjestymä | 0 (0,0) | 4 (7,4) |
| t(12;21)(p13;q22)/TEL-AML1 | 2 (3,7) | 3 (5,6) |
| t(1;19)(q23;p13.3)/E2A-PBX1 | 2 (3,7) | 2 (3,7) |
| t(5;14)(q31;32)/IL3-IGH | 0 (0,0) | 0 (0,0) |
| Muu | 9 (16,7) | 10 (18,5) |
| Ekstramedullaarinen relapsi, n (%) | ||
| Ei | 44 (81,5) | 40 (74,1) |
| Kyllä | 10 (18,5) | 14 (25,9) |
| Solujen morfologia, n (%) | ||
| Blastisoluja < 5 % | 54 (100,0) | 51 (94,4) |
| Blastisoluja ≥ 5 % mutta < 25 % | 0 (0,0) | 2 (3,7) |
| Blastisoluja ≥ 25 % | 0 (0,0) | 0 (0,0) |
| Ei arvioitavissa | 0 (0,0) | 1 (1,9) |
| MRD PCR:llä, n (%) | ||
| ≥ 10-4 | 10 (18,5) | 13 (24,1) |
| < 10-4 | 20 (37,0) | 22 (40,7) |
| Aika uusiutumiseen ensimmäisestä diagnoosista (kk), n (%) | ||
| < 18 kuukautta | 19 (35,2) | 22 (40,7) |
| ≥ 18 kuukautta mutta ≤ 30 kuukautta | 32 (59,3) | 28 (51,9) |
| > 30 kuukautta | 3 (5,6) | 4 (7,4) |
| N = potilaiden lukumäärä analyysijoukossa; n = niiden potilaiden määrä, joilta on havaintoja; MRD = minimaalinen jäännöstauti; PCR = polymeraasiketjureaktio. | ||
Ensisijainen päätetapahtuma oli elinaika ilman tapahtumia (EFS). Tässä tutkimuksessa Blincyto-hoitoa saaneiden potilaiden elinaika ilman tapahtumia oli tilastollisesti merkitsevästi pidempi kuin tavanomaista solunsalpaajilla toteutettua vakautushoitoa saaneilla potilailla. Hoidon vaikutukset alaryhmissä (esim. ikä, kasvaintaakka / MRD:n aste, aika uusiutumiseen ensimmäisestä diagnoosista) olivat pääosin yhdenmukaisia koko potilasjoukon tulosten kanssa. Tutkimuksen 20120215 tehoa mittaavat tulokset primaarianalyysista on esitetty kuvassa 5 ja taulukossa 17.
Kuva 5. Kaplan–Meier‑kuvaaja elinajasta ilman tapahtumia
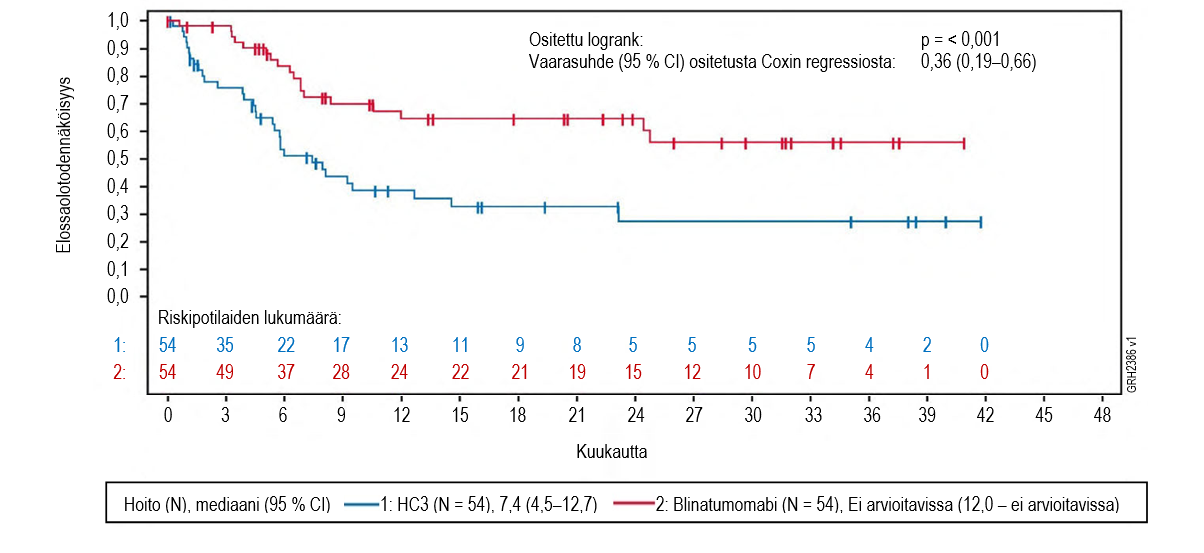
CI = luottamusväli, HC3 = korkean riskin vakautushoito 3, N = analyysijoukkoon kuuluvien potilaiden määrä.
Taulukko 17. Tehoa mittaavat tulokset lapsipotilailla, joilla oli ensimmäistä kertaa uusiutunut korkean riskin pre‑B‑ALL (20120215)
Blincyto (N = 54) | Tavanomainen solunsalpaajahoito (N = 54) | |
| Elinaika ilman tapahtumiaa | ||
| Tapahtumia (%) | 18 (33,3) | 31 (57,4) |
| Mediaani, kuukautta [95 % CI] | Ei arvioitavissa [12,0 – ei arvioitavissa] | 7,4 [4,5–12,7] |
| Vaarasuhde [95 % CI]b | 0,36 [0,19–0,66] | |
| p‑arvoc | < 0,001 | |
| Kokonaiselinaika | ||
| Kuolemien määrä (%) | 8 (14,8) | 16 (29,6) |
| 36 kuukauden estimaatti, % [95 % CI] | 81,1 [65,5–90,2] | 55,8 [36,9–71,0] |
| Vaarasuhde [95 % CI]b,c | 0,43 [0,18–1,01] | |
| p-arvod,e | 0,047 | |
| MRD-vastef | ||
| MRD-vasteen saaneiden määrä, n1/n2g (%) | 44/49 (89,8) | 26/48 (54,2) |
| [95 % CI] | [77,8–96,6] | [39,2–68,6] |
| p-arvoe,h | < 0,001 | |
Huomautus: Tehoa mittaavat tulokset saatu primaarianalyysista (mukana tiedot 17.7.2019 saakka). a Elinaika ilman tapahtumia (EFS) laskettiin satunnaistamisajankohdasta siihen päivään, jolloin todettiin relapsi tai kasvaintaakka oli ≥ 5 % mutta < 25 % blastisoluja täydellisen remission (CR) saavuttamisen jälkeen, täydellisen remission (CR) saavuttamatta jääminen hoidon päätyttyä, sekundaarinen maligniteetti tai kuolema sen syystä riippumatta, sen mukaan mikä tapahtui ensin. b Perustuu ositettuun Coxin malliin. c Kokonaiselinajan päivitetty vaarasuhde (sisältää tiedot 14.9.2020 saakka) oli 0,33 (95 % CI: 0,15–0,72). d p-arvo on johdettu ositetusta logrank-testistä. e Päätetapahtumaa ei ole testattu muodollisesti. Kerrannaisuutta ei ole huomioitu p-arvossa. f MRD-vasteen (minimaalisen jäännöstaudin) kriteeri oli: PCR:llä määritetty MRD < 1 x 10-4.g n1: MRD-vasteen saavuttaneiden potilaiden määrä, joilla lähtötilanteen MRD ≥ 10-4 tai < 10-4; n2: arvioitujen potilaiden määrä. h p‑arvo on johdettu Cochran-Mantel-Hänszelin testistä. | ||
Elinaikaa ilman tapahtumia (EFS) koskevan seuranta-ajan mediaani oli kaiken kaikkiaan 51,9 kuukautta (95 % CI: 47,2–62,1). Tavanomaista solunsalpaajilla toteutettua vakautushoitoa (HC3) saaneilla potilailla EFS:n Kaplan-Meierin estimaatti 5 vuoden kohdalla oli 27,6 % (95 % CI: 16,2–40,3) verrattuna 57,8 %:iin (95 % CI: 42,5–70,4) Blincyto-hoitoa saaneilla potilailla, ja vaarasuhde (95 % CI) oli 0,35 (0,20–0,61).
Kokonaiselinajan seuranta-ajan mediaani oli 55,2 kuukautta koko potilasjoukossa; tämä oli samaa luokkaa kummassakin hoitohaarassa. Kokonaiselinajan Kaplan-Meierin estimaatti 5 vuoden kohdalla oli 41,4 % (95 % CI: 26,3–55,9) solunsalpaajahoitoa saaneessa haarassa (HC3) ja 78,4 % (95 % CI: 64,2–87,4) Blincyto-hoitohaarassa, ja vaarasuhde (95 % CI) oli 0,33 (0,16–0,66). Mediaaniaika kantasolusiirtoon oli 1,7 kuukautta (vaihteluväli: 1–4 kuukautta) HC3-haarassa ja 1,9 kuukautta (vaihteluväli: 1–3 kuukautta) Blincyto-haarassa.
Allogeenisia kantasolusiirtoja tehtiin lähtötilanteen jälkeen lukumääräisesti enemmän Blincyto-haarassa kuin HC3-haarassa; 82,5 %:lla (47/57) potilaista HC3-haarassa ja 94,4 %:lla (51/54) potilaista Blincyto-haarassa. Täydellisessä remissiossa siirron sai HC3-haarassa 39 potilasta 57:stä (68,4 %) ja Blincyto-haarassa 51 potilasta 54:stä (94,4 %).
100 päivän kuluttua siirrosta kuolleisuus oli 3,9 % (95 % CI: 1,0–14,8) Blincyto-haarassa ja 5,1 % (95 % CI: 1,3–19,0) solunsalpaajaa saaneessa haarassa (HC3). Kaplan-Meierin estimaatin mukaan mediaaniaika kuolemaan oli HC3-haarassa 1 558,0 päivää (95 % CI: 431,0 päivää – ei arvioitavissa), ja sitä ei saavutettu blinatumomabihaarassa (95 % CI ei arvioitavissa – ei arvioitavissa).
Blincyton turvallisuutta ja tehoa arvioitiin myös avoimessa yhden hoitohaaran monikeskustutkimuksessa 93 lapsipotilaalla, joilla oli uusiutunut tai refraktaarinen pre‑B‑ALL (toinen tai myöhempi uusiutuminen luuytimessä, uusiutuminen luuytimessä allogeenisen kantasolusiirron jälkeen tai muu hoito tehotonta sekä > 25 % blastisoluja luuytimessä) (MT103‑205). Kaksiosaisessa tutkimuksessa määritettiin ensiksi soveltuva annostus, minkä jälkeen seurasi tutkimuksen yhden hoitohaaran osa, jossa tutkittiin kyseisen annostuksen tehoa.
Blincyto annettiin jatkuvana infuusiona laskimoon. Tutkimuksen annoksenmääritysosassa suurin arvioitu annos oli 30 mikrog/m2/vrk. Tutkimuksen farmakokinetiikan laajennusosaan ja tehoa arvioivaan osaan suositusannokseksi 1. hoitojaksossa määritettiin 5 mikrog/m2/vrk päivinä 1–7 ja 15 mikrog/m2/vrk päivinä 8–28 ja myöhemmissä hoitojaksoissa 15 mikrog/m2/vrk päivinä 1–28. Annosta voitiin säätää, jos haittavaikutuksia ilmaantui. Jos Blincyto‑hoidolla oli saavutettu hoitovaste, mutta tauti oli myöhemmin uusiutunut, potilaalla oli mahdollisuus saada uusi Blincyto‑hoito.
Hoitoa saaneeseen potilasjoukkoon kuului 70 potilasta (tutkimuksen annoksenmääritystä, farmakokinetiikan laajennusta ja tehoa arvioivat osat), jotka saivat vähintään yhden Blincyto-infuusion suositusannoksella. Hoitojaksojen lukumäärä potilasta kohti oli 1,5 (keskiarvo). Hoitoa saaneiden potilaiden mediaani-ikä oli 8 vuotta (vaihteluväli: 7 kk–17 vuotta), 70 potilaasta 40 (57,1 %) oli saanut allogeenisen kantasolusiirron ennen Blincyto-hoitoa ja 39 potilaalla 70 potilaasta (55,7 %) sairaus oli refraktaarinen. Useimmilla potilailla oli lähtötilanteessa suuri kasvaintaakka (leukeemisten blastien osuus luuytimen soluista ≥ 50 %) ja blastisolujen osuus luuytimen soluista oli 75,5 % (mediaani).
CR/CRh*‑remission saavutti kahden ensimmäisen hoitojakson aikana 20 potilasta 70 potilaasta (28,6 %), ja 20 potilaasta 17 (85 %) saavutti hoitovasteen ensimmäisen hoitojakson kuluessa. Neljällä potilaalla blastisolujen osuus luuytimen soluista oli ≤ 5 % (M1), mutta heillä perifeerisen veren solumäärien palautuminen ei riittänyt täyttämään CR‑ tai CRh*‑remission kriteereitä. Yksitoista CR/CRh*‑remission saavuttaneista 20 potilaasta (55 %) sai allogeenisen kantasolusiirron. Alle 2‑vuotiaista potilaista CR/CRh*‑remission saavutti 40,0 % (4/10), 2–6‑vuotiaista 30,0 % (6/20) ja 7–17‑vuotiaista 25,0 % (10/40). Kolmelle alle 1‑vuotiaalle potilaalle, joilla aikaisempi hoito oli ollut tehoton ja jotka eivät olleet saaneet allogeenista kantasolusiirtoa, annettiin Blincytoa yhden hoitojakson ajan 5–15 mikrog/m2/vrk. Kukaan kolmesta alle 1‑vuotiaasta potilaasta ei saavuttanut CR/CRh*‑remissiota: yhden potilaan sairaus oli etenevä (OS 2,3 kk) ja kahdella ei saatu hoitovastetta (OS 1,1 kk ja 8,7 kk). Haittavaikutukset imeväisikäisissä olivat samantyyppisiä kuin lapsipotilaissa yleisesti havaitut haittavaikutukset. Tehoa kuvaavat tulokset ovat taulukossa 18.
Taulukko 18. Tehoa mittaavat tulokset < 18‑vuotiailla potilailla, joilla oli uusiutunut tai refraktaarinen pre‑B‑ALL (MT103‑205)
| N = 70 | |
| CRa/CRh*b, n (%) [95 % CI] | 20 (28,6 %) [18,4–40,6 %] |
| CR, n (%) [95 % CI] | 11 (15,7 %) [8,1–26,4 %] |
| CRh*, n (%) [95 % CI] | 9 (12,9 %) [6,1–23,0 %] |
| Täydellinen MRD-vaste CR/CRh*-remissioryhmässäc, n1/n2d (%) [95 % CI] | 11/20 (55,0 %) [31,5–76,9] |
| CR, n1/n2d (%) [95 % CI] | 6/11 (54,5 %) [23,4–83,3] |
| CRh*, n1/n2d (%) [95 % CI] | 5/9 (55,6 %) [21,2–86,3] |
| CR/CRh*‑potilaiden elinaika ilman taudin uusiutumista (RFS), mediaanie [95 % CI] | 6,8 kk [2,2–12,0 kk] |
| Kokonaiselinajan mediaani [95 % CI] | 7,5 kk [4,0–11,8 kk] |
| Kuolleisuus 100 vuorokauden aikana allogeenisen kantasolusiirron jälkeenf | |
| n/N (%), [95 % CI] | 1/6 (16,7 %) [2,5 %–72,7 %] |
a Täydellisen remission (CR) kriteerit olivat blastisolujen osuus luuytimen soluista ≤ 5 % (M1), ei kiertäviin blastisoluihin tai ekstramedullaariseen tautiin viittaavia löydöksiä ja perifeerisen veren solumäärien täydellinen palautuminen (trombosyytit > 100 000/mikrolitra ja absoluuttinen neutrofiilimäärä [ANC] > 1 000/mikrolitra) eikä uusiutumista 28 vuorokauteen. b CRh*-remission kriteerit olivat blastisolujen osuus luuytimen soluista ≤ 5 % (M1), ei kiertäviin blastisoluihin tai ekstramedullaariseen tautiin viittaavia löydöksiä ja perifeerisen veren solumäärien osittainen palautuminen (trombosyytit > 50 000/mikrolitra ja absoluuttinen neutrofiilimäärä [ANC] > 500/mikrolitra) eikä uusiutumista 28 vuorokauteen. c Täydellinen MRD-vaste. polymeraasiketjureaktiolla (PCR) tai virtaussytometrialla ei havaittu merkkejä leukemiasoluista. d n1: MRD-vasteen saavuttaneiden potilaiden määrä kyseisessä remissioluokassa. n2: kyseisen remissioluokan saavuttaneiden potilaiden määrä. MRD‑tiedot puuttuivat yhdeltä CR/CRh*‑vasteen saavuttaneelta potilaalta; hänen ei tulkittu saavuttaneen MRD‑hoitovastetta. e Taudin uusiutumiseksi katsottiin hematologinen relapsi (blastisolujen osuus luuytimen soluista yli 25 % täydellisen remission jälkeen) tai ekstramedullaarinen relapsi. f Mukaan on luettu vain potilaat, joille on tehty kantasolusiirto (HSCT) CR/CRh*‑remission aikana (ei leukemialääkkeitä ennen kantasolusiirtoa). | |
Farmakokinetiikka
Blinatumomabin farmakokinetiikka näyttäisi olevan lineaarinen annosalueella 5–90 mikrog/m2/vrk (vastaa noin 9−162 mikrog/vrk) aikuisilla potilailla. Laskimoon annetun jatkuvan infuusion jälkeen vakaan tilan pitoisuus seerumissa (Css) saavutettiin vuorokauden kuluessa, ja pitoisuus pysyi stabiilina ajan myötä. Css-keskiarvot suurenivat suunnilleen suorassa suhteessa annokseen testatulla annosalueella. Uusiutuneen tai refraktaarisen ALL:n hoidossa käytettyjen kliinisten annosten aikaansaamat Css-keskiarvot (SD) olivat 228 (356) pg/ml, kun annos oli 9 mikrog/vrk, ja 616 (537) pg/ml, kun annos oli 28 mikrog/vrk. Blinatumomabin farmakokinetiikka potilailla, joilla oli MRD‑positiivinen pre-B‑ALL, oli samanlainen kuin potilailla, joilla oli uusiutunut tai refraktaarinen ALL. Blinatumomabin farmakokinetiikka vakautushoitovaiheessa oli samanlainen aikuisilla, joilla oli pre‑B‑ALL, mukaan lukien potilaat, joilla oli juuri diagnosoitu ALL ja ensimmäisen kerran uusiutunut ALL, ja aikuispotilailla, joilla oli uusiutunut tai refraktaarinen ALL.
Jakautuminen
Terminaalivaiheeseen (Vz) perustuva arvioitu jakautumistilavuuden keskiarvo (SD) oli 5,27 (4,37) litraa, kun blinatumomabia annettiin jatkuvana infuusiona laskimoon.
Biotransformaatio
Blinatumomabin metaboloitumistietä ei ole määritetty. Muiden proteiinilääkkeiden tavoin blinatumomabin oletetaan pilkkoutuvan pieniksi peptideiksi ja aminohapoiksi katabolisia reittejä pitkin.
Eliminaatio
Arvioidun systeemisen puhdistuman keskiarvo (SD) potilailla, jotka saivat kliinisissä tutkimuksissa blinatumomabia jatkuvana infuusiona laskimoon, oli 3,10 (2,94) l/h. Puoliintumisajan keskiarvo (SD) oli 2,20 (1,34) tuntia. Virtsaan erittyneet blinatumomabimäärät olivat merkityksettömiä testatuilla kliinisillä annostasoilla.
Erityispopulaatiot
Blinatumomabin farmakokinetiikassa ei havaittu kliinisesti merkityksellisiä eroja iän, sukupuolen, rodun, etnisen taustan, Philadelphia‑kromosomitilan tai lievän (kokonaisbilirubiini ≤ normaalin yläraja [ULN] ja ASAT > ULN tai kokonaisbilirubiini > 1–1,5 × ULN ja mikä tahansa ASAT) tai keskivaikean maksan vajaatoiminnan (kokonaisbilirubiini > 1,5–3 × ULN ja mikä tahansa ASAT) perusteella. Kehon pinta-ala (0,4–2,9 m2) vaikuttaa blinatumomabin farmakokinetiikkaan ja tukee kehon pinta-alaan pohjautuvaa annostusta alle 45 kg painavilla potilailla.
Munuaisten vajaatoiminta
Blinatumomabista ei ole tehty varsinaisia farmakokineettisiä tutkimuksia munuaisten vajaatoimintaa sairastavilla potilailla.
Farmakokineettiset analyysit osoittivat, että blinatumomabin puhdistuma-arvojen ero on noin kaksinkertainen kohtalaista munuaisten vajaatoimintaa sairastavien potilaiden ja niiden potilaiden välillä, joiden munuaisten toiminta on normaali. Koska potilaiden väliset erot olivat kuitenkin suuria (variaatiokerroin (CV%) enintään 98,4 %) ja puhdistuma-arvot olivat munuaisten vajaatoimintaa sairastavilla potilailla pääosin samalla alueella kuin potilailla, joiden munuaistoiminta oli normaali, munuaistoiminnan ei odoteta vaikuttavan kliinisesti merkittävässä määrin hoitotulokseen. Vaikean munuaisten vajaatoiminnan vaikutusta blinatumomabin farmakokinetiikkaan ei ole tutkittu.
Maksan vajaatoiminta
Blinatumomabin käytöstä ei ole tehty varsinaisia farmakokineettisiä tutkimuksia maksan vajaatoimintaa sairastavilla potilailla. Maksan vajaatoiminnan vaikutusta blinatumomabipuhdistumaan arvioitiin populaatiofarmakokineettisellä analyysillä potilailla, joilla oli lievä tai keskivaikea maksan vajaatoiminta ja joita verrattiin potilaisiin, joilla oli normaali maksan toiminta, National Cancer Institute ‑organisaation elinhäiriöitä käsittelevän työryhmän määrittelemien kriteerien mukaisesti. Blinatumomabipuhdistumassa ei havaittu kliinisesti merkityksellisiä eroja potilailla, joilla oli lievä tai keskivaikea maksan vajaatoiminta, ja potilailla, joilla maksan toiminta oli normaali. Vaikean maksan vajaatoiminnan vaikutusta blinatumomabin farmakokinetiikkaan ei ole tutkittu.
Pediatriset potilaat
Blinatumomabin farmakokinetiikka näyttäisi olevan lineaarinen annosalueella 5–30 mikrog/m2/vrk lapsipotilailla. Uusiutuneen tai refraktaarisen pre‑B‑ALL:n hoidossa käytetyillä suositelluilla annoksilla vakaan tilan pitoisuuden (Css) keskiarvo (SD) oli 162 (179) pg/ml, kun annos oli 5 mikrog/m2/vrk, ja 533 (392) pg/ml, kun annos oli 15 mikrog/m2/vrk. Arvioidut keskiarvot (SD) olivat: jakautumistilavuus (Vz) 4,14 (3,32) l/m2, puhdistuma (CL) 1,65 (1,62) l/h/m2 ja terminaalinen puoliintumisaika (t1/2,z) 2,14 (1,44) h.
Blinatumomabin farmakokinetiikka vakautushoitovaiheessa oli samanlainen lapsipotilailla, joilla oli pre‑B‑ALL, mukaan lukien potilaat, joilla oli ensimmäisen kerran uusiutunut pre‑B‑ALL, ja lapsipotilailla, joilla oli uusiutunut tai refraktaarinen pre‑B‑ALL.
Prekliiniset tiedot turvallisuudesta
Blinatumomabilla ja hiiren korvikemolekyylillä tehdyissä toistuvien annosten toksisuustutkimuksissa havaittiin odotettuja farmakologisia vaikutuksia (kuten sytokiinien vapautuminen, leukosyyttimäärien pieneneminen, B‑solujen häviäminen, T‑solujen väheneminen, solujen väheneminen imukudoksista). Nämä muutokset hävisivät, kun hoito lopetettiin.
Blinatumomabilla ei ole tehty lisääntymistoksisuustutkimuksia. Hiirillä tehdyssä alkio- ja sikiötoksisuustutkimuksessa korvikemolekyyli läpäisi istukan vain vähäisessä määrin (sikiön ja emon seerumista mitattujen pitoisuuksien suhde < 1 %) eikä se aiheuttanut alkio- eikä sikiötoksisuutta eikä epämuodostumia. Odotettua B‑ ja T‑solujen vähenemistä havaittiin tiineillä hiirillä mutta sikiöihin kohdistuneita hematologisia vaikutuksia ei arvioitu. Tutkimuksia hoidon aiheuttamista vaikutuksista hedelmällisyyteen ei ole tehty. Urosten tai naaraiden lisääntymiselimiin kohdistuneita vaikutuksia ei havaittu hiiren korvikemolekyylillä tehdyissä toksisuustutkimuksissa.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine
Sitruunahappomonohydraatti (E330)
Trehaloosidihydraatti
Lysiinihydrokloridi
Polysorbaatti 80 (E433)
Natriumhydroksidi (pH:n säätöön)
Liuos (stabilointiliuos)
Sitruunahappomonohydraatti (E330)
Lysiinihydrokloridi
Polysorbaatti 80 (E433)
Natriumhydroksidi (pH:n säätöön)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamattomat injektiopullot
5 vuotta
Käyttövalmis liuos
Kemiallisen ja fysikaalisen käytönaikaisen säilyvyyden on osoitettu olevan 24 tuntia 2–8 °C:ssa tai 4 tuntia enintään 27 °C:ssa.
Mikrobiologisista syistä käyttökuntoon saatettu liuos on laimennettava heti, paitsi jos käyttökuntoon saattamisessa on käytetty menetelmää, joka estää mikrobikontaminaation. Ellei sitä laimenneta heti, käytönaikaiset säilytysajat ja ‑olosuhteet ovat käyttäjän vastuulla.
Laimennettu liuos (käyttövalmis infuusiopussi)
Kemiallisen ja fysikaalisen käytönaikaisen säilyvyyden on osoitettu olevan 10 vuorokautta 2–8 °C:ssa tai 96 tuntia enintään 27 °C:ssa.
Mikrobiologisista syistä käyttövalmiit infuusiopussit on käytettävä heti. Ellei niitä käytetä heti, käytönaikaiset säilytysajat ja käyttöä edeltävät olosuhteet ovat käyttäjän vastuulla, eikä säilytysaika saa normaalisti ylittää 24 tuntia 2–8 °C:ssa, paitsi jos laimentaminen on tapahtunut valvotuissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä ja kuljeta kylmässä (2°C ‑ 8°C).
Ei saa jäätyä.
Säilytä injektiopulloja alkuperäispakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BLINCYTO kuiva-aine välikonsentraatiksi ja liuos infuusionestettä varten, liuos
38,5 mikrog (L:ei) 1 pakkaus (3749,92 €)
PF-selosteen tieto
Yksi Blincyto‑pakkaus sisältää 1 injektiopullon kuiva-ainetta välikonsentraatiksi infuusionestettä varten, liuos, ja 1 injektiopullon liuosta (stabilointiliuos):
- 38,5 mikrogrammaa blinatumomabikuiva-ainetta injektiopullossa (tyypin I lasia), jossa on tulppa (elastomeerikumia), suljin (alumiinia) ja suojakansi ja
- 10 ml liuosta injektiopullossa (tyypin I lasia), jossa on tulppa (elastomeerikumia), suljin (alumiinia) ja suojakansi.
Valmisteen kuvaus:
Blincyto kuiva-aine (kuiva-aine välikonsentraattia varten): Valkoinen tai luonnonvalkoinen jauhe.
Liuos (stabilointiliuos): Väritön tai kellertävä kirkas liuos, jonka pH on 7,0.
Käyttö- ja käsittelyohjeet
Aseptinen valmistaminen
Valmistamisessa infuusiota varten on noudatettava aseptista työskentelytapaa. Blincyton valmistaminen infuusiota varten:
- koulutetun henkilökunnan on valmistettava lääke infuusiota varten aseptisissa olosuhteissa noudattaen hyvää menettelytapaa ja erityisesti parenteraalisten lääkkeiden aseptista valmistamista koskevia ohjeita.
- valmistus tapahtuu laminaarivirtauskaapissa tai mikrobiologisessa suojakaapissa noudattaen laskimoon annettavien lääkkeiden turvallista käsittelyä koskevia standardeja varotoimenpiteitä.
On erittäin tärkeää, että tässä kohdassa kuvattuja ohjeita lääkkeen valmistamisesta ja antamisesta noudatetaan huolellisesti lääkitysvirheiden (kuten yli- ja aliannostelun) välttämiseksi.
Muut ohjeet
- Blincyto on yhteensopiva polyolefiinista, dietyyliheksyyliftalaattia (DEHP) sisältämättömästä PVC:stä ja etyylivinyyliasetaatista (EVA) valmistettujen infuusiopussien / infuusiopumpun lääkekasettien kanssa.
- Infuusion päätyttyä käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Infuusioliuoksen valmistaminen
Lisäksi tarvitaan seuraavat tarvikkeet, jotka eivät ole mukana pakkauksessa:
- Steriilejä kertakäyttöruiskuja
- 21–23 G:n neula (neuloja) (suositus)
- Injektionesteisiin käytettävää vettä
-
Infuusiopussi, jossa on 250 ml natriumkloridi 9 mg/ml (0,9 %) ‑infuusionestettä
- Aseptisten siirtojen vähentämiseksi käytä esitäytettyä 250 ml:n infuusiopussia. Blincyton annoslaskelmat perustuvat tavanomaisen ylitäytön sisältävään 265–275 ml:n natriumkloridi 9 mg/ml (0,9 %) ‑infuusionestetilavuuteen.
- Käytä ainoastaan polyolefiinista, dietyyliheksyyliftalaattia (DEHP) sisältämättömästä PVC:stä tai etyylivinyyliasetaatista (EVA) valmistettuja infuusiopusseja / infuusiopumpun lääkekasetteja.
-
Polyolefiinista, DEHP:ta sisältämättömästä PVC:stä tai EVA:sta valmistettu infuusioletkusto, jossa on steriili, pyrogeeniton, heikosti proteiinia sitova 0,2 mikrometrin kiinteä (in-line) suodatin
- Varmista, että letkusto on yhteensopiva infuusiopumpun kanssa.
Liuota Blincyto injektionesteisiin käytettävään veteen. Älä käytä liuosta (stabilointiliuosta) Blincyto-injektiopullojen sisällön liuottamiseen.
Esitäytä infuusioletku vain VALMISTA Blincyto-infuusioliuosta sisältävästä pussista. Älä esitäytä letkua natriumkloridi 9 mg/ml (0,9 %) ‑infuusionesteellä.
Blincyton liuottaminen käyttövalmiiksi
1. Määritä annoksen ja infuusion keston perusteella, montako Blincyto-injektiopulloa tarvitaan.
2. Liuota kunkin Blincyto kuiva-ainetta välikonsentraattia varten sisältävän injektiopullon sisältö käyttövalmiiksi lisäämällä pulloon injektioruiskua käyttäen 3 ml injektionesteisiin käytettävää vettä. Suuntaa vesi Blincyto-injektiopullon sisäseinämää kohti, älä suoraan kylmäkuivattuun kuiva-aineeseen.
- Älä käytä liuosta (stabilointiliuosta) Blincyto kuiva-aineen liuottamiseen välikonsentraattia varten.
- Kun injektionesteisiin käytettävä vesi on lisätty kuiva-aineeseen välikonsentraattia varten, näin saadun käyttövalmiin liuoksen kokonaismäärä on 3,08 ml ja lopullinen Blincyto-pitoisuus on 12,5 mikrogrammaa/ml.
3. Pyörittele injektiopullon sisältöä varovasti välttäen liiallista vaahdon muodostumista.
- Älä ravista.
4. Tarkista silmämääräisesti liuottamisen aikana ja ennen infuusiopussin valmistelua, ettei käyttövalmiissa liuoksessa ole hiukkasia eikä sen väri ole muuttunut. Käyttövalmiin liuoksen on oltava kirkasta tai hieman opalisoivaa ja väritöntä tai kellertävää.
- Älä käytä, jos liuos on sameaa tai siinä on sakkaa.
Blincyto-infuusioliuospussin valmisteleminen käyttövalmiiksi
Varmista potilaalle määrätty annos ja infuusion kesto erikseen jokaisen Blincyto-infuusioliuospussin kohdalla. Virheiden välttämiseksi noudata Blincyto‑infuusioliuospussin valmistelussa taulukoissa 19 ja 20 ilmoitettuja määriä.
- Taulukko 19, kun potilas painaa vähintään 45 kg
- Taulukko 20, kun potilas painaa alle 45 kg
1. Käytä 250 ml:n esitäytettyä infuusiopussia, jossa on natriumkloridi 9 mg/ml (0,9 %) ‑infuusionestettä. Tällaisen esitäytetyn pussin kokonaisnestemäärä on yleensä 265–275 ml.
2. Esikäsittele infuusiopussi siirtämällä aseptisesti injektioruiskua käyttäen 5,5 ml liuosta (stabilointiliuosta) infuusiopussiin. Sekoita pussin sisältö varovasti välttäen vaahdon muodostumista. Hävitä jäljelle jäänyt liuos (stabilointiliuos).
3. Siirrä aseptisesti injektioruiskua käyttäen tarvittava määrä käyttövalmista Blincyto‑liuosta infuusiopussiin, joka sisältää natriumkloridi 9 mg/ml (0,9 %) -infuusionestettä ja liuosta (stabilointiliuosta). Sekoita pussin sisältö varovasti välttäen vaahdon muodostumista.
- Käyttövalmiiksi liuotetun Blincyton tarkka määrä vähintään 45 kg painaville potilaille on taulukossa 19.
- Käyttövalmiiksi liuotetun Blincyton tarkka määrä alle 45 kg painaville potilaille (kehon pinta-alan mukaan laskettu annos) on taulukossa 20.
- Hävitä mahdollinen käyttämätöntä käyttövalmista Blincyto-liuosta sisältävä injektiopullo.
4. Poista ilma infuusiopussista. Tämä on erityisen tärkeää, jos käytetään kannettavaa infuusiopumppua.
5. Kiinnitä infuusiopussiin aseptisissa olosuhteissa infuusioletku, jossa on steriili 0,2 mikrometrin kiinteä (in-line) suodatin. Varmista, että infuusioletku on yhteensopiva infuusiopumpun kanssa.
6. Esitäytä infuusioletku vain VALMISTA Blincyto-infuusioliuosta sisältävästä pussista.
7. Säilytä kylmässä 2–8 °C:ssa, ellei käytetä heti.
Taulukko 19. Vähintään 45 kg painavat potilaat: natriumkloridi 9 mg/ml (0,9 %) ‑infuusionesteen ja infuusiopussiin lisättävien liuoksen (stabilointiliuoksen) ja käyttövalmiiksi liuotetun Blincyton tilavuudet
| Natriumkloridi 9 mg/ml (0,9 %) -infuusioneste (aloitustilavuus) | 250 ml (tavanomaisen ylitäytön sisältävä tilavuus 265–275 ml) | |||
| Liuos (stabilointiliuos) (kiinteä tilavuus 24, 48, 72 ja 96 tunnin mittaisia infuusioita varten) | 5,5 ml | |||
| Infuusion kesto | Annos | Infuusionopeus | Käyttövalmiiksi liuotettu Blincyto | |
| Tilavuus | Injektiopulloja | |||
| 24 tuntia | 9 mikrog/vrk | 10 ml/tunti | 0,83 ml | 1 |
| 28 mikrog/vrk | 10 ml/tunti | 2,6 ml | 1 | |
| 48 tuntia | 9 mikrog/vrk | 5 ml/tunti | 1,7 ml | 1 |
| 28 mikrog/vrk | 5 ml/tunti | 5,2 ml | 2 | |
| 72 tuntia | 9 mikrog/vrk | 3,3 ml/tunti | 2,5 ml | 1 |
| 28 mikrog/vrk | 3,3 ml/tunti | 8 ml | 3 | |
| 96 tuntia | 9 mikrog/vrk | 2,5 ml/tunti | 3,3 ml | 2 |
| 28 mikrog/vrk | 2,5 ml/tunti | 10,7 ml | 4 | |
Taulukko 20. Alle 45 kg painavat potilaat: natriumkloridi 9 mg/ml (0,9 %) ‑infuusionesteen ja infuusiopussiin lisättävien liuoksen (stabilointiliuoksen) ja käyttövalmiiksi liuotetun Blincyton tilavuudet
| Natriumkloridi 9 mg/ml (0,9 %) -infuusioneste (aloitustilavuus) | 250 ml (tavanomaisen ylitäytön sisältävä tilavuus 265–275 ml) | ||||
| Liuos (stabilointiliuos) (kiinteä tilavuus 24, 48, 72 ja 96 tunnin mittaisia infuusioita varten) | 5,5 ml | ||||
| Infuusion kesto | Annos | Infuusionopeus | BSA (m2)* | Käyttövalmiiksi liuotettu Blincyto | |
| Tilavuus | Injektiopulloja | ||||
| 24 tuntia | 5 mikrog/m2/vrk | 10 ml/tunti | 1,5–1,59 | 0,7 ml | 1 |
| 1,4–1,49 | 0,66 ml | 1 | |||
| 1,3–1,39 | 0,61 ml | 1 | |||
| 1,2–1,29 | 0,56 ml | 1 | |||
| 1,1–1,19 | 0,52 ml | 1 | |||
| 1–1,09 | 0,47 ml | 1 | |||
| 0,9–0,99 | 0,43 ml | 1 | |||
| 0,8–0,89 | 0,38 ml | 1 | |||
| 0,7–0,79 | 0,33 ml | 1 | |||
| 0,6–0,69 | 0,29 ml | 1 | |||
| 0,5–0,59 | 0,24 ml | 1 | |||
| 0,4–0,49 | 0,2 ml | 1 | |||
| 24 tuntia | 15 mikrog/m2/vrk | 10 ml/tunti | 1,5–1,59 | 2,1 ml | 1 |
| 1,4–1,49 | 2 ml | 1 | |||
| 1,3–1,39 | 1,8 ml | 1 | |||
| 1,2–1,29 | 1,7 ml | 1 | |||
| 1,1–1,19 | 1,6 ml | 1 | |||
| 1–1,09 | 1,4 ml | 1 | |||
| 0,9–0,99 | 1,3 ml | 1 | |||
| 0,8–0,89 | 1,1 ml | 1 | |||
| 0,7–0,79 | 1 ml | 1 | |||
| 0,6–0,69 | 0,86 ml | 1 | |||
| 0,5–0,59 | 0,72 ml | 1 | |||
| 0,4–0,49 | 0,59 ml | 1 | |||
| 48 tuntia | 5 mikrog/m2/vrk | 5 ml/tunti | 1,5–1,59 | 1,4 ml | 1 |
| 1,4–1,49 | 1,3 ml | 1 | |||
| 1,3–1,39 | 1,2 ml | 1 | |||
| 1,2–1,29 | 1,1 ml | 1 | |||
| 1,1–1,19 | 1 ml | 1 | |||
| 1–1,09 | 0,94 ml | 1 | |||
| 0,9–0,99 | 0,85 ml | 1 | |||
| 0,8–0,89 | 0,76 ml | 1 | |||
| 0,7–0,79 | 0,67 ml | 1 | |||
| 0,6–0,69 | 0,57 ml | 1 | |||
| 0,5–0,59 | 0,48 ml | 1 | |||
| 0,4–0,49 | 0,39 ml | 1 | |||
| 48 tuntia | 15 mikrog/m2/vrk | 5 ml/tunti | 1,5–1,59 | 4,2 ml | 2 |
| 1,4–1,49 | 3,9 ml | 2 | |||
| 1,3–1,39 | 3,7 ml | 2 | |||
| 1,2–1,29 | 3,4 ml | 2 | |||
| 1,1–1,19 | 3,1 ml | 2 | |||
| 1–1,09 | 2,8 ml | 1 | |||
| 0,9–0,99 | 2,6 ml | 1 | |||
| 0,8–0,89 | 2,3 ml | 1 | |||
| 0,7–0,79 | 2 ml | 1 | |||
| 0,6–0,69 | 1,7 ml | 1 | |||
| 0,5–0,59 | 1,4 ml | 1 | |||
| 0,4–0,49 | 1,2 ml | 1 | |||
| 72 tuntia | 5 mikrog/m2/vrk | 3,3 ml/tunti | 1,5–1,59 | 2,1 ml | 1 |
| 1,4–1,49 | 2 ml | 1 | |||
| 1,3–1,39 | 1,8 ml | 1 | |||
| 1,2–1,29 | 1,7 ml | 1 | |||
| 1,1–1,19 | 1,6 ml | 1 | |||
| 1–1,09 | 1,4 ml | 1 | |||
| 0,9–0,99 | 1,3 ml | 1 | |||
| 0,8–0,89 | 1,1 ml | 1 | |||
| 0,7–0,79 | 1 ml | 1 | |||
| 0,6–0,69 | 0,86 ml | 1 | |||
| 0,5–0,59 | 0,72 ml | 1 | |||
| 0,4–0,49 | 0,59 ml | 1 | |||
| 72 tuntia | 15 mikrog/m2/vrk | 3,3 ml/tunti | 1,5–1,59 | 6,3 ml | 3 |
| 1,4–1,49 | 5,9 ml | 3 | |||
| 1,3–1,39 | 5,5 ml | 2 | |||
| 1,2–1,29 | 5,1 ml | 2 | |||
| 1,1–1,19 | 4,7 ml | 2 | |||
| 1–1,09 | 4,2 ml | 2 | |||
| 0,9–0,99 | 3,8 ml | 2 | |||
| 0,8–0,89 | 3,4 ml | 2 | |||
| 0,7–0,79 | 3 ml | 2 | |||
| 0,6–0,69 | 2,6 ml | 1 | |||
| 0,5–0,59 | 2,2 ml | 1 | |||
| 0,4–0,49 | 1,8 ml | 1 | |||
| 96 tuntia | 5 mikrog/m2/vrk | 2,5 ml/tunti | 1,5–1,59 | 2,8 ml | 1 |
| 1,4–1,49 | 2,6 ml | 1 | |||
| 1,3–1,39 | 2,4 ml | 1 | |||
| 1,2–1,29 | 2,3 ml | 1 | |||
| 1,1–1,19 | 2,1 ml | 1 | |||
| 1–1,09 | 1,9 ml | 1 | |||
| 0,9–0,99 | 1,7 ml | 1 | |||
| 0,8–0,89 | 1,5 ml | 1 | |||
| 0,7–0,79 | 1,3 ml | 1 | |||
| 0,6–0,69 | 1,2 ml | 1 | |||
| 0,5–0,59 | 0,97 ml | 1 | |||
| 0,4–0,49 | 0,78 ml | 1 | |||
| 96 tuntia | 15 mikrog/m2/vrk | 2,5 ml/tunti | 1,5–1,59 | 8,4 ml | 3 |
| 1,4–1,49 | 7,9 ml | 3 | |||
| 1,3–1,39 | 7,3 ml | 3 | |||
| 1,2–1,29 | 6,8 ml | 3 | |||
| 1,1–1,19 | 6,2 ml | 3 | |||
| 1–1,09 | 5,7 ml | 3 | |||
| 0,9–0,99 | 5,1 ml | 2 | |||
| 0,8–0,89 | 4,6 ml | 2 | |||
| 0,7–0,79 | 4 ml | 2 | |||
| 0,6–0,69 | 3,4 ml | 2 | |||
| 0,5–0,59 | 2,9 ml | 2 | |||
| 0,4–0,49 | 2,3 ml | 1 | |||
BSA (Body Surface Area) = kehon pinta-ala * Blincyto-hoidon turvallisuutta ei ole määritetty, kun kehon pinta-ala on alle 0,4 m2. | |||||
Korvattavuus
BLINCYTO kuiva-aine välikonsentraatiksi ja liuos infuusionestettä varten, liuos
38,5 mikrog 1 pakkaus
- Ei korvausta.
ATC-koodi
L01FX07
Valmisteyhteenvedon muuttamispäivämäärä
27.11.2025
Yhteystiedot
Keilaranta 10, PL 86
02101 Espoo
09 5490 0500