
YERVOY infuusiokonsentraatti, liuosta varten 5 mg/ml
Vaikuttavat aineet ja niiden määrät
Yksi millilitra konsentraattia sisältää 5 mg ipilimumabia.
Yksi 10 ml:n injektiopullo sisältää 50 mg ipilimumabia.
Yksi 40 ml:n injektiopullo sisältää 200 mg ipilimumabia.
Ipilimumabi on täysin ihmisperäinen CTLA-4:n monoklonaalinen vasta-aine (IgG1κ), joka on tuotettu kiinanhamsterin munasarjasoluissa yhdistelmä-DNA-tekniikalla.
Apuaine, jonka vaikutus tunnetaan:
Yksi millilitra konsentraattia sisältää 0,1 mmol (= 2,30 mg) natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Melanooma
YERVOY monoterapiana tai yhdistelmähoitona nivolumabin kanssa on tarkoitettu aikuisten sekä 12-vuotiaiden ja sitä vanhempien nuorten (ks. lisätietoja kohdassa Varoitukset ja käyttöön liittyvät varotoimet) edenneen melanooman hoitoon (jota ei voida kirurgisesti poistaa tai joka on metastasoinut).
Nivolumabi–ipilimumabi-yhdistelmähoito on osoittanut nivolumabi-monoterapiaan verrattuna etenemisvapaan elinajan ja kokonaiselinajan kasvua ainoastaan potilailla, joilla on vähäinen kasvaimen PD-L1-ilmentymä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet sekä Farmakodynamiikka).
Munuaiskarsinooma (RCC)
YERVOY yhdistelmähoitona nivolumabin kanssa on tarkoitettu ensilinjan hoidoksi aikuisille, joilla on kohtalaisen/huonon ennusteen edennyt munuaiskarsinooma (ks. kohta Farmakodynamiikka).
Ei-pienisoluinen keuhkosyöpä (NSCLC)
YERVOY yhdistelmähoitona nivolumabin ja kahden platinapohjaisen kemoterapiajakson kanssa on tarkoitettu etäpesäkkeisen ei-pienisoluisen keuhkosyövän ensilinjan hoitoon aikuisille, joiden kasvaimissa ei ole herkistävää EGFR-mutaatiota tai ALK-translokaatiota.
Keuhkopussin pahanlaatuinen mesoteliooma (MPM)
YERVOY yhdistelmähoitona nivolumabin kanssa on tarkoitettu ensilinjan hoitoon aikuisille, joilla on leikkaukseen soveltumaton keuhkopussin pahanlaatuinen mesoteliooma.
Kolorektaalisyöpä (CRC), johon liittyy DNA:n kopiovirheenkorjaus- eli MMR-järjestelmän geenien puutteellinen toiminta (dMMR) tai mikrosatelliitti-instabiliteetti (MSI-H)
YERVOY yhdistelmähoitona nivolumabin kanssa on tarkoitettu sellaisen aikuisten kolorektaalisyövän hoitoon, johon liittyy DNA:n kopiovirheenkorjaus- eli MMR-järjestelmän geenien puutteellinen toiminta tai mikrosatelliitti-instabiliteetti seuraavissa tapauksissa:
-
leikkaukseen soveltumattoman tai etäpesäkkeisen kolorektaalisyövän ensilinjan hoito
-
etäpesäkkeisen kolorektaalisyövän hoito aiemman fluoropyrimidiinipohjaisen yhdistelmäsolunsalpaajahoidon jälkeen (ks. kohta Farmakodynamiikka).
Ruokatorven levyepiteelikarsinooma (OSCC)
YERVOY yhdistelmähoitona nivolumabin kanssa on tarkoitettu ensilinjan hoitoon aikuisille, joilla on leikkaukseen soveltumaton, edennyt, uusiutunut tai etäpesäkkeinen ruokatorven levyepiteelikarsinooma, jonka kasvainsolujen PD-L1‑ilmentymä on ≥ 1 %.
Hepatosellulaarinen karsinooma (HCC)
YERVOY yhdistelmähoitona nivolumabin kanssa on tarkoitettu ensilinjan hoitoon aikuisille, joilla on leikkaukseen soveltumaton tai edennyt hepatosellulaarinen karsinooma.
Ehto
Valmistetta tulee käyttää vain syövän hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoidon aloittavan ja sitä valvovan lääkärin on oltava syövän hoitoon perehtynyt erikoislääkäri.
PD-L1‑määritys
Jos kyseisessä käyttöaiheessa näin mainitaan, potilaat valitaan saamaan YERVOY‑valmistetta sen perusteella, todetaanko heillä validoidulla testillä varmistettu PD-L1-ligandin ilmentyminen (ks. kohdat Käyttöaiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
MSI-/MMR-testaus
Jos käyttöaiheessa mainitaan, potilaan lääkehoidon Yervoylla on perustuttava potilaan kasvaimen MSI‑H-/dMMR‑statukseen. Kasvaimen statuksen määritys on tehtävä CE-merkityllä (diagnostiikan ja lääkehoidon yhdistävällä), tähän käyttötarkoitukseen tarkoitetulla, lääkinnällisellä laitteella. Mikäli tällaista ei ole saatavilla, on käytettävä vaihtoehtoista, lainsäädännön vaatimukset täyttävää lääkinnällistä laitetta (ks. kohdat Käyttöaiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Annostus
YERVOY monoterapiana
Melanooma
Aikuiset sekä 12‑vuotiaat ja sitä vanhemmat nuoret
Suositeltu aloitushoito koostuu 4:stä kolmen viikon välein annettavasta 3 mg/kg:n YERVOY-annoksesta, jotka annetaan laskimoon 30 minuutin infuusiona. Aloitushoito on annettava kokonaisuudessaan (4 annosta) jos potilas tämän sietää, riippumatta uusien leesioiden kehittymisestä tai jo olemassa olevien leesioiden kasvamisesta. Hoitovaste tulisi arvioida vasta koko aloitushoidon jälkeen.
YERVOY yhdistelmähoitona nivolumabin kanssa
Melanooma
Aikuisten sekä 12‑vuotiaiden ja sitä vanhempien vähintään 50 kg painavien nuorten hoidossa suositeltu annos on 3 mg/kg ipilimumabia laskimoon yhdessä nivolumabin kanssa, jota annostellaan 1 mg/kg laskimoon. Yhdistelmähoitoa annetaan 3 viikon välein 4 ensimmäistä annosta. Tämän jälkeen seuraa toinen vaihe, jossa nivolumabia annostellaan monoterapiana laskimoon joko 240 mg 2 viikon välein tai 480 mg 4 viikon välein (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka) taulukon 1 mukaisesti. Monoterapiavaiheen alkaessa ensimmäinen nivolumabiannos annetaan
-
3 viikkoa viimeisimmän nivolumabi–ipilimumabi‑yhdistelmähoitoannoksen jälkeen, jos annostelu on 240 mg 2 viikon välein tai
-
6 viikkoa viimeisimmän nivolumabi–ipilimumabi‑yhdistelmähoitoannoksen jälkeen, jos annostelu on 480 mg 4 viikon välein.
12‑vuotiaiden ja sitä vanhempien alle 50 kg painavien nuorten hoidossa suositeltu annos on 3 mg/kg ipilimumabia laskimoon yhdessä nivolumabin kanssa, jota annostellaan 1 mg/kg laskimoon. Yhdistelmähoitoa annetaan 3 viikon välein 4 ensimmäistä annosta. Tämän jälkeen seuraa toinen vaihe, jossa nivolumabia annostellaan monoterapiana laskimoon joko 3 mg/kg 2 viikon välein tai 6 mg/kg 4 viikon välein (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka) taulukon 1 mukaisesti. Monoterapiavaiheen alkaessa ensimmäinen nivolumabiannos annetaan
-
3 viikkoa viimeisimmän nivolumabi–ipilimumabi-yhdistelmähoitoannoksen jälkeen, jos annostelu on 3 mg/kg 2 viikon välein tai
-
6 viikkoa viimeisimmän nivolumabi–ipilimumabi-yhdistelmähoitoannoksen jälkeen, jos annostelu on 6 mg/kg 4 viikon välein.
Taulukko 1: Laskimoon yhdistelmähoitona annettavien ipilimumabin ja nivolumabin suositellut annokset ja infuusioajat
| Yhdistelmähoitovaihe, 3 viikon välein 4 annosta | Monoterapiavaihe |
|---|---|---|
Nivolumabi | Aikuiset sekä 12‑vuotiaat ja sitä vanhemmat nuoret: | Aikuiset ja nuoret (12‑vuotiaat ja sitä vanhemmat, jotka painavat vähintään 50 kg):
Nuoret (12‑vuotiaat ja sitä vanhemmat, jotka painavat alle 50 kg): |
Ipilimumabi | Aikuiset sekä 12‑vuotiaat ja sitä vanhemmat nuoret: | - |
Munuaiskarsinooma
Suositeltu annos on 1 mg/kg ipilimumabia laskimoon yhdessä nivolumabin kanssa, jota annostellaan 3 mg/kg laskimoon. Yhdistelmähoitoa annetaan 3 viikon välein 4 ensimmäistä annosta. Tämän jälkeen seuraa toinen vaihe, jossa nivolumabia annostellaan monoterapiana laskimoon joko 240 mg 2 viikon välein tai 480 mg 4 viikon välein taulukon 2 mukaisesti.
Monoterapiavaiheen alkaessa ensimmäinen nivolumabiannos annetaan
-
3 viikkoa viimeisimmän ipilimumabi–nivolumabi‑yhdistelmähoitoannoksen jälkeen, jos annostelu on 240 mg 2 viikon välein tai
-
6 viikkoa viimeisimmän ipilimumabi–nivolumabi‑yhdistelmähoitoannoksen jälkeen, jos annostelu on 480 mg 4 viikon välein.
Taulukko 2: Laskimoon yhdistelmähoitona annettavien ipilimumabin ja nivolumabin suositellut annokset ja infuusioajat munuaiskarsinooman hoidossa
| Yhdistelmähoitovaihe, 3 viikon välein 4 annosta | Monoterapiavaihe |
Nivolumabi | 3 mg/kg 30 minuutin kuluessa | 240 mg 2 viikon välein 30 minuutin kuluessa tai |
Ipilimumabi | 1 mg/kg 30 minuutin kuluessa | - |
dMMR‑ tai MSI‑H-kolorektaalisyöpä
Suositeltu annos dMMR- tai MSI‑H-kolorektaalisyövän ensilinjan hoidossa on 1 mg/kg ipilimumabia ja 240 mg nivolumabia laskimoon. Yhdistelmähoitoa annetaan 3 viikon välein enintään 4 annosta. Tämän jälkeen nivolumabia annostellaan monoterapiana laskimoon joko 240 mg 2 viikon välein tai 480 mg 4 viikon välein taulukon 3 mukaisesti. Monoterapiavaiheessa ensimmäinen nivolumabiannos annetaan 3 viikon kuluttua viimeisestä nivolumabi- ja ipilimumabiyhdistelmän annoksesta. Nivolumabihoitoa suositellaan jatkettavan, kunnes tauti etenee, ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan potilaille, joiden tauti ei etene.
Suositeltu annos potilaille, jotka ovat saaneet aiempaa fluoropyrimidiinipohjaista yhdistelmäsolunsalpaajahoitoa dMMR- tai MSI‑H-kolorektaalisyövän hoitoon, on 1 mg/kg ipilimumabia ja 3 mg/kg nivolumabia laskimoon. Yhdistelmähoitoa annetaan 3 viikon välein 4 ensimmäistä annosta. Tämän jälkeen nivolumabia annostellaan monoterapiana laskimoon 240 mg 2 viikon välein taulukon 3 mukaisesti. Monoterapiavaiheessa ensimmäinen nivolumabiannos annetaan 3 viikon kuluttua viimeisestä nivolumabi- ja ipilimumabiyhdistelmän annoksesta.
Taulukko 3: Laskimoon yhdistelmähoitona annettavien nivolumabin ja ipilimumabin suositellut annokset ja infuusioajat dMMR- tai MSI‑H‑-kolorektaalisyövän hoidossa
| Yhdistelmähoitovaihe, 3 viikon välein 4 annosta | Monoterapiavaihe | |
|---|---|---|---|
Nivolumabi | Ensilinjan hoito | 240 mg 30 minuutin kuluessa | 240 mg 2 viikon välein 30 minuutin kuluessa tai |
Aiemman fluoropyrimidiinipohjaisen yhdistelmäsolunsalpaajahoidon jälkeen | 3 mg/kg 30 minuutin kuluessa | 240 mg 2 viikon välein 30 minuutin kuluessa | |
Ipilimumabi | 1 mg/kg 30 minuutin kuluessa | - | |
Keuhkopussin pahanlaatuinen mesoteliooma
Suositeltu annos on 1 mg/kg ipilimumabia laskimoon 30 minuutin kuluessa kuuden viikon välein yhdistelmähoitona nivolumabin kanssa, jota annostellaan 360 mg laskimoon 30 minuutin kuluessa kolmen viikon välein. Hoitoa jatketaan enintään 24 kuukauden ajan potilailla, joiden tauti ei etene.
Ruokatorven levyepiteelikarsinooma
Suositeltu annos on 1 mg/kg ipilimumabia laskimoon 30 minuutin kuluessa kuuden viikon välein yhdistelmähoitona nivolumabin kanssa, jota annostellaan joko 3 mg/kg kahden viikon välein tai 360 mg kolmen viikon välein laskimoon 30 minuutin kuluessa. Hoitoa suositellaan, kunnes tauti etenee, ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan potilaille, joiden tauti ei etene.
Hepatosellulaarinen karsinooma
Suositeltu annos on 3 mg/kg ipilimumabia laskimoon yhdessä nivolumabin kanssa, jota annostellaan 1 mg/kg laskimoon. Yhdistelmähoitoa annetaan 3 viikon välein enintään 4 annosta. Tämän jälkeen seuraa toinen vaihe, jossa nivolumabia annostellaan monoterapiana laskimoon joko 240 mg 2 viikon välein tai 480 mg 4 viikon välein (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka) taulukon 4 mukaisesti. Hoitoa on suositeltavaa jatkaa, kunnes tauti etenee tai kunnes ilmenee ei-hyväksyttävää toksisuutta tai enintään 24 kuukauden ajan. Monoterapiavaiheen alkaessa ensimmäinen nivolumabiannos annetaan:
-
3 viikkoa viimeisimmän nivolumabi–ipilimumabi-yhdistelmähoitoannoksen jälkeen, jos annostelu on 240 mg 2 viikon välein tai 480 mg 4 viikon välein.
Taulukko 4: Laskimoon yhdistelmähoitona annettavien ipilimumabin ja nivolumabin suositellut annokset ja infuusioajat hepatosellulaarisen karsinooman hoidossa
| Yhdistelmähoitovaihe, 3 viikon välein 4 annosta | Monoterapiavaihe |
|---|---|---|
Nivolumabi | 1 mg/kg 30 minuutin kuluessa | 240 mg 2 viikon välein 30 minuutin kuluessa tai |
Ipilimumabi | 3 mg/kg 30 minuutin kuluessa | - |
YERVOY yhdistelmähoitona nivolumabin ja kemoterapian kanssa
Ei-pienisoluinen keuhkosyöpä
Suositusannos on 1 mg/kg ipilimumabia laskimoon 30 minuutin kuluessa joka kuudes viikko yhdessä nivolumabin kanssa, jota annetaan 360 mg laskimoon 30 minuutin kuluessa joka kolmas viikko, sekä yhdessä platinapohjaisen kemoterapian kanssa, jota annetaan joka kolmas viikko. Kun kaksi kemoterapiajaksoa on saatu päätökseen, hoitoa jatketaan ipilimumabilla annoksella 1 mg/kg joka kuudes viikko sekä nivolumabilla annoksella 360 mg, jota annetaan laskimoon joka kolmas viikko. Hoitoa suositellaan, kunnes tauti etenee, ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan potilaille, joiden tauti ei etene.
Hoidon kesto
YERVOY–nivolumabi-yhdistelmähoitoa jatketaan niin kauan kuin siitä todetaan olevan kliinistä hyötyä tai kunnes potilas ei enää siedä sitä (ja enintään hoidon enimmäiskeston ajan, jos indikaatiolle on sellainen määritetty).
Epätyypillisiä vasteita (esim. kasvain kasvaa aluksi tilapäisesti tai pieniä uusia leesioita kehittyy ensimmäisten kuukausien aikana kasvaimen pienenemisen jälkeen) on havaittu. Kliinisesti vakaiden potilaiden YERVOY–nivolumabi-yhdistelmähoitoa suositellaan jatkamaan taudin etenemisestä kertovien ensimmäisten merkkien ilmettyä, kunnes taudin eteneminen on vahvistettu.
Maksan ja kilpirauhasen toimintakokeet on suoritettava sekä lähtötilanteessa että ennen jokaista YERVOY-annosta. Lisäksi kaikki YERVOY-hoidon aikana mahdollisesti ilmenevät immuunivälitteisten haittavaikutusten merkit ja oireet, kuten ripuli ja koliitti, on tutkittava (ks. taulukot 5A, 5B ja kohta Varoitukset ja käyttöön liittyvät varotoimet).
Alle 12-vuotiaat lapset
Ipilimumabin turvallisuutta ja tehoa alle 12-vuotiaiden lasten hoidossa ei ole varmistettu.
Hoidon pysyvä lopettaminen tai annosten väliin jättäminen
Immuunivälitteisten haittavaikutusten hoitamiseksi YERVOY-annos voidaan joutua jättämään väliin tai YERVOY-hoito on ehkä lopetettava pysyvästi sekä on aloitettava systeeminen suuriannoksinen kortikosteroidihoito. Joissakin tapauksissa voidaan harkita lisäksi jotakin muuta immunosuppressiivista hoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annoksen suurentamista tai pienentämistä ei suositella. Annosten siirtäminen myöhemmäksi tai hoidon keskeytys voi olla tarpeen yksilöllisen turvallisuuden ja siedettävyyden vuoksi.
Ohjeet hoidon pysyvää lopettamista tai annosten väliin jättämistä koskien on kuvattu taulukoissa 5A ja 5B YERVOY-monoterapian osalta sekä taulukossa 4C YERVOY–nivolumabi-yhdistelmähoidon tai yhdistelmähoitoa seuraavan toisen hoitovaiheen (nivolumabi-monoterapia) osalta. Immuunivälitteisten haittavaikutusten yksityiskohtaiset hoito-ohjeet on kuvattu kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Taulukko 5A Milloin YERVOY-monoterapia on lopetettava pysyvästi
Lopeta YERVOY-hoito pysyvästi, jos potilaalla ilmenee seuraavia haittavaikutuksia. Haittavaikutusten hoitamiseksi voidaan myös tarvita systeemistä suuriannoksista kortikosteroidihoitoa, jos haittavaikutusten osoitetaan tai epäillään olevan immuunivälitteisiä (yksityiskohtaiset hoito-ohjeet, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). | |
Haittavaikutukset | Vaikeusaste (NCI-CTCAE v4a) |
Ruoansulatuselimistö: |
|
Maksa: |
|
Iho: |
|
Hermosto: |
|
Muut elinjärjestelmätb: |
|
a Toksisuuden vaikeusasteluokitus: National Cancer Institute Common Terminology Criteria for Adverse Events. Version 4.0 (NCI‑CTCAE v4).
b Kaikki muut haittavaikutukset, joiden osoitetaan tai epäillään olevan immuunivälitteisiä, on luokiteltava CTCAE:n mukaan. YERVOY-hoidon lopettamispäätöksen tulee perustua haittojen vaikeusasteeseen.
c Hoitoa voidaan jatkaa potilaille, joilla on vaikea (asteen 3 tai 4) umpierityshäiriö, joka pysyy hallinnassa hormonikorvaushoidolla.
Taulukko 5B Milloin YERVOY-monoterapia-annos on jätettävä antamatta
Jätä YERVOY-annosa väliin, jos potilaalla ilmenee taulukossa mainittuja immuunivälitteisiä haittavaikutuksia. Yksityiskohtaiset hoito-ohjeet, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet. | |
Haittavaikutukset | Toimi näin |
Ruoansulatuselimistö: |
|
Maksa: | |
Iho: | |
Umpieritys: Asteen 3 diabetes | |
Hermosto: | |
Muut keskivaikeat haittavaikutuksetc | |
a YERVOY-annoksen pienentämistä ei suositella.
b Toksisuuden vaikeusasteluokitus: National Cancer Institute Common Terminology Criteria for Adverse Events. Version 4.0 (NCI‑CTCAE v4).
c Kaikki muut elinjärjestelmien haittavaikutukset, joita pidetään immuunivälitteisinä, on luokiteltava CTCAE:n mukaan. YERVOY-annoksen antamatta jättämispäätöksen tulee perustua haittojen vaikeusasteeseen.
d Kunnes kaikki 4 annosta on annettu tai ensimmäisestä annoksesta on kulunut 16 viikkoa sen mukaan, kumpi on ensin.
Taulukko 5C: YERVOY–nivolumabi-yhdistelmähoidon tai yhdistelmähoitoa seuraavan toisen hoitovaiheen (nivolumabi-monoterapia) suositellut muutokset
Immuunivälitteinen haittavaikutus | Vaikeusaste | Hoidon muutos |
Immuunivälitteinen keuhkotulehdus | Asteen 2 keuhkotulehdus | Keskeytä hoito, kunnes oireet häviävät, radiologisesti havaittavat poikkeavuudet paranevat ja kortikosteroidihoito on toteutettu kokonaan. |
Asteen 3 tai 4 keuhkotulehdus | Lopeta hoito pysyvästi. | |
Immuunivälitteinen koliitti | Asteen 2 ripuli tai koliitti | Keskeytä hoito, kunnes oireet häviävät ja mahdollisesti tarvittava kortikosteroidihoito on toteutettu kokonaan. |
Asteen 3 tai 4 ripuli tai koliitti | Lopeta hoito pysyvästi. | |
Immuunivälitteinen maksatulehdus ilman hepatosellulaarista karsinoomaa | Asteen 2 nousu aspartaattiaminotransferaasi- (ASAT), alaniiniaminotransferaasi- (ALAT) tai kokonaisbilirubiiniarvossa | Keskeytä hoito, kunnes laboratorioarvot palaavat ennalleen ja mahdollisesti tarvittava kortikosteroidihoito on toteutettu kokonaan. |
Asteen 3 tai 4 nousu ASAT-, ALAT- tai kokonaisbilirubiiniarvossa | Lopeta hoito pysyvästi. | |
Immuunivälitteinen maksatulehdus ja hepatosellulaarinen karsinooma | Jos ASAT/ALAT on lähtötilanteessa normaalin rajoissa ja nousee tasolle > 3 ja ≤ 10 kertaa normaalin yläraja (ULN) | Keskeytä hoito, kunnes laboratorioarvot palaavat ennalleen ja mahdollisesti tarvittava kortikosteroidihoito on toteutettu kokonaan. |
ASAT/ALAT nousee tasolle > 10 kertaa ULN | Lopeta hoito pysyvästi | |
Immuunivälitteinen munuaistulehdus ja munuaisten toimintahäiriö | Asteen 2 tai 3 nousu kreatiniiniarvossa | Keskeytä hoito, kunnes kreatiniiniarvo palaa ennalleen ja kortikosteroidihoito on toteutettu kokonaan. |
Asteen 4 nousu kreatiniiniarvossa | Lopeta hoito pysyvästi. | |
Immuunivälitteiset umpierityshäiriöt | Oireiset asteen 2 tai 3 kilpirauhasen vajaatoiminta, kilpirauhasen liikatoiminta, hypofysiitti | Keskeytä hoito, kunnes oireet häviävät ja kortikosteroidihoito (jos sitä tarvitaan akuutin tulehduksen oireisiin) on toteutettu kokonaan. Hoitoa pitää jatkaa yhdessä hormonikorvaushoidona kanssa niin kauan kuin oireita on. |
Asteen 4 kilpirauhasen vajaatoiminta | Lopeta hoito pysyvästi. | |
Immuunivälitteiset ihohaitat | Asteen 3 ihottuma | Keskeytä hoito, kunnes oireet häviävät ja kortikosteroidihoito on toteutettu kokonaan. |
Asteen 4 ihottuma | Lopeta hoito pysyvästi. | |
Stevens–Johnsonin oireyhtymä (SJS) tai toksinen epidermaalinen nekrolyysi (TEN) | Lopeta hoito pysyvästi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). | |
Immuunivälitteinen myokardiitti | Asteen 2 myokardiitti | Keskeytä hoito, kunnes oireet häviävät ja kortikosteroidihoito on toteutettu kokonaan.b |
Asteen 3 tai 4 myokardiitti | Lopeta hoito pysyvästi. | |
Muut immuunivälitteiset haittavaikutukset | Asteen 3 (ensimmäistä kertaa) | Keskeytä hoito. |
Asteen 4 tai uusiutunut asteen 3; jatkuva asteen 2 tai 3 huolimatta hoidon muutoksista; kortikosteroidin vuorokausiannosta ei pystytä vähentämään 10 mg:aan prednisonia tai vastaavaa | Lopeta hoito pysyvästi. | |
Myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymäc | Asteen 2 myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymä | Keskeytä hoito, kunnes oireet häviävät ja kortikosteroidihoito on toteutettu kokonaan. |
Asteen 3 tai 4 myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymä | Lopeta hoito pysyvästi. |
Huom.: Toksisuus on luokiteltu NCI‑CTCAE-haittavaikutusluokituksen (National Cancer Institute Common Terminology Criteria for Adverse Events) version 4.0 mukaan.
a Suositukset hormonikorvaushoidon käytöstä on annettu kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
b Nivolumabihoidon tai nivolumabi–ipilimumabi-yhdistelmähoidon uudelleen aloittamisen turvallisuutta potilaille, joilla on aikaisemmin ollut immuunivälitteinen myokardiitti, ei tunneta.
c Ilmenee näistä joko kahden tai kaikkien kolmen sairauden limittymisenä. Hoitoon suositeltavan muutoksen arvioinnissa on huomioitava yksittäisten tapahtumien vaikein CTCAE-aste.
YERVOY–nivolumabi-yhdistelmähoito on lopetettava pysyvästi, jos potilaalla on
-
asteen 4 tai uusiutunut asteen 3 haittavaikutus
-
jatkuva asteen 2 tai 3 haittavaikutus sen hoidosta huolimatta.
Jos yhden lääkeaineen antaminen keskeytetään annettaessa YERVOY-valmistetta nivolumabin kanssa yhdistelmähoitona, pitää myös toisen lääkeaineen antaminen keskeyttää. Jos hoitoa jatketaan, sitä voidaan potilaan arvioinnin pohjalta jatkaa joko yhdistelmähoitona tai nivolumabi-monoterapiana.
Erityisryhmät
Pediatriset potilaat
YERVOY-valmisteen turvallisuutta ja tehoa monoterapiana alle 12-vuotiaiden lasten hoidossa ei ole varmistettu. Tietoja on vain vähän. YERVOY-valmistetta ei pidä käyttää alle 12-vuotiaille lapsille.
Lukuun ottamatta melanoomaa sairastavien 12‑vuotiaiden ja sitä vanhempien nuorten hoitoa, YERVOY-valmisteen turvallisuutta ja tehoa yhdistelmähoitona nivolumabin kanssa alle 18‑vuotiaiden lasten hoidossa ei ole varmistettu. Tämänhetkiset tiedot kuvataan kohdissa Annostus ja antotapa, Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka.
Iäkkäät
Iäkkäiden (≥ 65 v) ja nuorempien (< 65 v) potilaiden välillä ei ole raportoitu eroja turvallisuus- tai tehoprofiilin suhteen. Yli 75-vuotiaista ja sitä vanhemmista potilaista, jotka ovat saaneet ipilimumabia munuaiskarsinooman ensilinjan hoitona, on niin vähän tutkimustietoa, ettei ryhmää koskevia johtopäätöksiä voida tehdä (ks. kohta Farmakodynamiikka). Annosta ei ole tarpeen muuttaa tässä potilasjoukossa (ks. kohta Farmakodynamiikka).
Munuaisten vajaatoiminta
YERVOY-valmisteen turvallisuutta ja tehoa ei ole tutkittu potilailla, joiden munuaisten toiminta on heikentynyt. Populaatiofarmakokineettisten tutkimusten tulosten perusteella annosta ei tarvitse muuttaa potilaille, joilla on lievä tai keskivaikea munuaisen toimintahäiriö (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
YERVOY-valmisteen turvallisuutta ja tehoa ei ole tutkittu potilaissa, joiden maksan toiminta on heikentynyt. Populaatiofarmakokineettisten tutkimusten tulosten perusteella annosta ei tarvitse muuttaa potilaille, joilla on lievä maksan vajaatoiminta (ks. kohta Farmakokinetiikka).Varovaisuutta on noudatettava YERVOY-valmisteen annossa potilaalle, jonka transaminaasien lähtöarvo on ≥ 5 x ULN tai bilirubiinin lähtöarvo on > 3 x ULN (ks. kohta Farmakodynamiikka).
Antotapa
YERVOY on tarkoitettu laskimonsisäiseen käyttöön. Infuusion suositeltu kesto on 30 minuuttia.
YERVOY-valmisteen voi infusoida laskimoon laimentamattomana tai laimennettuna pitoisuuteen 1–4 mg/ml. YERVOY laimennetaan joko 9 mg/ml (0,9 %) natriumkloridi-injektionesteeseen tai 50 mg/ml (5 %) glukoosi-injektionesteeseen.
YERVOY-valmistetta ei saa antaa nopeana injektiona tai boluksena laskimoon.
YERVOY–nivolumabi-yhdistelmähoidossa tai Yervoy‑valmisteen, nivolumabin ja kemoterapian yhdistelmähoidossa annetaan ensin nivolumabi, sen jälkeen samana päivänä Yervoy ja sitten kemoterapia (jos annetaan). Käytä kullekin infuusiolle erillistä infuusiopussia ja suodatinta.
Ks. kohdasta Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja käsittelyyn ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
PD-L1-statuksen arviointi
Kasvaimen PD-L1-statuksen määritykseen on tärkeää valita hyvin validoitu ja luotettava menetelmä.
MSI-/MMR-statuksen määritys
Kasvaimen MSI‑H- ja dMMR-statuksen määritykseen on tärkeää valita hyvin validoitu ja luotettava menetelmä.
Ipilimumabi yhdistelmähoitona nivolumabin kanssa
Kun ipilimumabia annetaan yhdistelmähoitona, lue yhdistelmähoidon muiden lääkeaineiden valmisteyhteenvedot ennen hoidon aloittamista. Lue nivolumabin valmisteyhteenvedosta lisätietoa nivolumabihoitoon liittyvistä varoituksista ja varotoimista. Useimmat immuunivälitteiset haittavaikutukset paranivat tai hävisivät asianmukaisella hoidolla, johon kuului kortikosteroidihoito ja muutos hoidossa (ks. kohta Annostus ja antotapa). Immuunivälitteisiä haittavaikutuksia on havaittu enemmän nivolumabi–ipilimumabi-yhdistelmähoidon kuin nivolumabi-monoterapian yhteydessä.
Yhdistelmähoidossa on raportoitu myös sydämeen ja keuhkoihin liittyviä haittavaikutuksia, kuten keuhkoemboliaa. Potilaita on seurattava sydämeen ja keuhkoihin liittyvien haittavaikutusten varalta jatkuvasti sekä elektrolyyttihäiriön ja kuivumisen kliinisten merkkien ja oireiden sekä laboratorioarvojen huononemisen varalta ennen hoidon aloittamista ja säännöllisesti sen aikana. Ipilimumabi–nivolumabi-yhdistelmähoito pitää keskeyttää, jos hengenvaarallisia tai uusiutuvia vakavia sydämeen ja keuhkoihin liittyviä haittavaikutuksia ilmenee (ks. kohta Annostus ja antotapa.).
Potilaita on seurattava jatkuvasti (vähintään 5 kuukautta viimeisen annoksen jälkeen), sillä haittavaikutus voi tulla missä vaiheessa ipilimumabi–nivolumabi-yhdistelmähoitoa hyvänsä tai vasta sen päätyttyä.
Immuunivälitteiset reaktiot
Ipilimumabin käyttöön liittyy immuunijärjestelmän kiihtyneen tai liiallisen toiminnan aiheuttamia tulehduksellisia (immuunivälitteisiä) haittavaikutuksia, jotka ovat todennäköisesti yhteydessä ipilimumabin vaikutusmekanismiin. Immuunivälitteiset haittavaikutukset, jotka saattavat olla vakavia tai henkeä uhkaavia, voivat kohdistua maha-suolikanavaan, maksaan, ihoon, hermostoon, umpieritysrauhasiin tai johonkin muuhun elinjärjestelmään. Vaikka useimmat immuunivälitteiset haittavaikutukset ilmenivät hoitojakson aikana, niiden ilmaantumista on myös raportoitu kuukausienkin kuluttua viimeisestä ipilimumabiannoksesta. Ripulia, ulostamiskertojen tihenemistä, veriulosteita, maksan toimintakoearvojen suurenemista, ihottumaa ja umpierityshäiriöitä on pidettävä inflammatorisina ja ipilimumabihoitoon liittyvinä, jollei näille ole tunnistettu jotain muuta syytä. Haittavaikutusten varhainen diagnosointi ja asianmukainen hoito ovat keskeisiä hengenvaarallisten komplikaatioiden minimoimiseksi.
Vaikeiden immuunivälitteisten haittavaikutusten hoitamiseksi voidaan tarvita systeemistä suuriannoksista kortikosteroidihoitoa, johon voidaan yhdistää myös muuta immunosuppressiivista hoitoa.
Ipilimumabi-monoterapian sekä ipilimumabi–nivolumabi-yhdistelmähoidon immuunivälitteisten haittavaikutusten hoito-ohjeet on kuvattu alla.
Immuunivälitteistä haittavaikutusta epäiltäessä potilaan tila on arvioitava riittävän tarkasti, jotta pystytään vahvistamaan haittavaikutuksen etiologia tai sulkemaan pois muut syyt. Haittavaikutuksen vakavuuden perusteella ipilimumabi- tai ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä ja annettava kortikosteroideja. Jos yhdistelmähoidon seurauksena ilmenevän haittavaikutuksen hoitoon käytetään kortikosteroideilla aikaansaatua immunosuppressiota, kortikosteroidiannoksen vähentäminen on aloitettava tilan alkaessa parantua, ja siihen on käytettävä vähintään kuukausi. Kortikosteroidihoidon liian nopea lopetus voi pahentaa haittavaikutusta tai aiheuttaa haittavaikutuksen uusiutumisen. Muu immunosuppressiivinen lääke on syytä lisätä hoito-ohjelmaan, jos haittavaikutus pahenee tai ei parane kortikosteroidien käytöstä huolimatta.
Ipilimumabi–nivolumabi-yhdistelmähoitoa ei saa aloittaa uudelleen potilaan saadessa immunosuppressiivisia annoksia kortikosteroideja tai muuta immunosuppressiivista lääkettä. Immunosuppressiivista hoitoa saaville potilaille on annettava profylaktista antibioottihoitoa opportunististen infektioiden ehkäisemiseksi.
Ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi, jos yksikään vakava immuunivälitteinen haittavaikutus uusiutuu tai on hengenvaarallinen.
Maha-suolikanavan immuunivälitteiset reaktiot
Ipilimumabi monoterapiana
Ipilimumabiin liittyy maha-suolikanavan vakavia immuunivälitteisiä reaktioita. Kliinisissä tutkimuksissa on raportoitu maha-suolikanavan perforaatiosta johtuneita kuolemantapauksia (ks. kohta Haittavaikutukset).
Pitkälle edennyttä melanoomaa (jota ei voitu kirurgisesti poistaa tai joka oli metastasoinut) sairastaneet potilaat, jotka saivat ipilimumabia 3 mg/kg monoterapiana faasin III tutkimuksessa (MDX010-20, ks. kohta Farmakodynamiikka), maha-suolikanavan vaikeiden tai kuolemaan johtaneiden (vaikeusaste 3–5) immuunivälitteisten reaktioiden mediaani-ilmaantumisaika oli 8 viikkoa (vaihteluväli 5–13 vk) hoidon aloittamisesta. Kun reaktiot hoidettiin tutkimusprotokollan hoito-ohjeiden mukaisesti, ne lievittyivät (vaikeusaste ≤ aste 1, tai potilaan tila korjaantui lähtötasolle) useimmissa tapauksissa (90 %:lla) 4 viikossa (mediaani; vaihteluväli 0,6–22 vk) ilmenemisestä.
Potilasta on seurattava sellaisten maha-suolikanavan merkkien ja oireiden varalta, jotka voivat viitata immuunivälitteiseen koliittiin tai maha-suolikanavan perforaatioon. Nämä voivat ilmetä kliinisesti mm. ripulina, ulostamiskertojen lisääntymisenä, vatsakipuna tai veriulosteena, johon voi liittyä kuumetta. Kliinisissä tutkimuksissa immuunivälitteinen koliitti assosioitui näyttöön limakalvotulehduksesta, joka saattoi olla myös haavainen ja sisältää lymfosyytti- ja neutrofiili-infiltraatteja. Markkinoille tulon jälkeen on ilmoitettu tapauksia, joissa sytomegaloviruksen aiheuttama infektio on kehittynyt tai aktivoitunut uudelleen niillä potilailla, joilla on kortikosteroidihoitoon reagoimaton immuunivälitteinen koliitti. Ripulin tai koliitin ilmaantumisen yhteydessä on tehtävä ulosteen viljelytutkimukset, jotta voidaan sulkea pois infektioperäiset tai muut vaihtoehtoiset etiologiat.
Ripulin ja koliitin hoitosuositukset perustuvat oireiden vaikeusasteeseen (NCI-CTCAE v4 -luokituksen mukaan). Jos ripuli on lievää tai keskivaikeaa (aste 1 tai 2; enintään 6 ulostamiskertaa/vrk) tai epäilty koliitti on lievä tai keskivaikea (esim. vatsakipua tai verta ulosteessa), ipilimumabihoitoa voidaan jatkaa. Potilaalle tulee antaa oireenmukaista hoitoa (esim. loperamidi, nesteytys), ja häntä tulee seurata tarkoin. Jos lievät tai keskivaikeat oireet uusiutuvat tai jatkuvat 5–7 päivää, on aloitettava kortikosteroidihoito (esim. 1 mg/kg prednisonia suun kautta kerran vuorokaudessa tai vastaavaa) ja hoito-ohjelman mukainen ipilimumabihoito jätetään väliin. Jos oireet lievittyvät asteelle 0 tai 1 tai potilaan tila korjaantuu lähtötasolle ipilimumabihoito voidaan aloittaa uudestaan (ks. kohta Annostus ja antotapa).
Ipilimumabihoito on lopetettava pysyvästi ja aloitettava heti systeeminen suuriannoksinen kortikosteroidihoito laskimoon, jos ripuli tai koliitti on vaikeaa (aste 3 tai 4) (ks. kohta Annostus ja antotapa). (Kliinisissä tutkimuksissa on käytetty 2 mg/kg/vrk metyyliprednisolonia.) Kun ripuli ja muut oireet on saatu hallintaan, aloitetaan kortikosteroidiannoksen asteittainen pienentäminen kliinisen harkinnan mukaan. Kliinisissä tutkimuksissa annoksen pienentäminen nopeasti (< 1 kuukauden aikana) johti joillakin potilailla ripulin tai koliitin uusiutumiseen. Potilas on tutkittava maha-suolikanavan perforaation tai vatsakalvontulehduksen varalta.
Kliinisistä tutkimuksista on saatu vain vähän kokemusta kortikosteroidiin vastaamattoman ripulin tai koliitin hoidosta. Kortikosteroidihoidon täydentämistä vaihtoehtoisella immunosuppressantilla on harkittava kortikosteroidihoitoon vastaamattoman immuunivälitteisen koliitin yhteydessä, jos muut syyt on suljettu pois (mukaan lukien sytomegaloviruksen aiheuttama infektio / sen uudelleenaktivoituminen, kun se on todettu biopsiasta tehdyllä viruksen PCR-tutkimuksella, ja muut virus-, bakteeri- ja loiseläinperäiset etiologiset syyt). Kliinisissä tutkimuksissa hoitoon lisättiin yksi 5 mg/kg:n infliksimabiannos, jollei tämä ollut vasta-aiheinen. Infliksimabia ei saa käyttää, jos potilaalla epäillään maha-suolikanavan perforaatiota tai sepsistä (ks. infliksimabivalmisteen valmisteyhteenveto).
Immuunivälitteinen koliitti
Ipilimumabi yhdistelmähoitona nivolumabin kanssa
Ipilimumabi–nivolumabi-yhdistelmähoidon yhteydessä on todettu vaikeaa ripulia ja koliittia (ks. kohta Haittavaikutukset). Potilaita on seurattava ripulin ja koliitin oireiden varalta, joita ovat esimerkiksi vatsakipu ja veriset tai limaiset ulosteet. Infektiot ja sairauksiin liittyvät syyt on suljettava pois.
Jos potilaalla on asteen 4 ripuli tai koliitti, ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 3 ripuli tai koliitti ipilimumabi–nivolumabi-yhdistelmähoidon yhteydessä, hoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 2 ripuli tai koliitti, ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä. Jos ripuli tai koliitti jatkuu, se on hoidettava antamalla kortikosteroideja annoksina, jotka vastaavat 0,5–1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua ipilimumabi–nivolumabi-yhdistelmähoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannosta on suurennettava tasolle, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia, ja ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi.
Immuunivälitteinen keuhkotulehdus
Ipilimumabi yhdistelmähoitona nivolumabin kanssa
Ipilimumabi–nivolumabi-yhdistelmähoidon yhteydessä on todettu vaikeaa keuhkotulehdusta tai interstitiaalista keuhkosairautta, joka on joissain tapauksissa johtanut kuolemaan (ks. kohta Haittavaikutukset). Potilaita on seurattava keuhkotulehduksen löydösten ja oireiden varalta. Näitä ovat esimerkiksi radiologiset muutokset (kuten fokaaliset mattalasimuutokset, läiskikkäiset infiltraatit), dyspnea ja hypoksia. Infektiot ja sairauksiin liittyvät syyt on suljettava pois.
Jos potilaalla on asteen 3 tai 4 keuhkotulehdus, ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 2–4 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 2 (oireinen) keuhkotulehdus, ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua ipilimumabi–nivolumabi-yhdistelmähoitoa voi jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannosta on suurennettava tasolle, joka vastaa 2–4 mg/kg/vrk metyyliprednisolonia, ja ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi.
Immuunivälitteinen maksatoksisuus
Ipilimumabi monoterapiana
Ipilimumabiin liittyy vakavaa immuunivälitteistä maksatoksisuutta. Kliinisissä tutkimuksissa on raportoitu kuolemaan johtanutta maksan vajaatoimintaa (ks. kohta Haittavaikutukset).
MDX010-20-tutkimuksessa keskivaikeaa, vaikeaa tai kuolemaan johtavaa (aste 2–5) immuunivälitteistä maksatoksisuutta ilmeni ipilimumabia 3 mg/kg monoterapiana saaneilla potilailla 3–9 viikon kuluttua hoidon aloittamisesta. Kun reaktiot hoidettiin tutkimusprotokollan hoito-ohjeiden mukaisesti, ne lievittyivät 0,7–2 viikossa.
Maksan transaminaasi- ja bilirubiiniarvot on määritettävä ennen jokaista ipilimumabiannosta, koska laboratorioarvojen varhaiset muutokset voivat viitata alkavaan immuunivälitteiseen maksatulehdukseen (ks. kohta Annostus ja antotapa). Maksan toimintakoearvot voivat suurentua ilman kliinisiä oireita. Jos ASAT- ja ALAT- tai kokonaisbilirubiiniarvot suurenevat, potilas on tutkittava maksavaurion muiden syiden, kuten infektioiden, kasvaimen etenemisen tai samanaikaisen lääkityksen aiheuttamien reaktioiden, poissulkemiseksi, ja potilaan tilaa on seurattava, kunnes oireet lievittyvät. Immuunivälitteistä maksatoksisuutta kokeneiden potilaiden maksakudosnäytteissä oli merkkejä akuutista tulehduksesta (neutrofiilit, lymfosyytit ja makrofagit).
Jos transaminaasi‑ tai kokonaisbilirubiiniarvossa on asteen 2 suureneminen, hoito-ohjelman mukainen ipilimumabiannos on jätettävä väliin ja maksan toimintakoearvoja on seurattava, kunnes arvot korjaantuvat. Kun arvot ovat korjautuneet, ipilimumabihoito voidaan aloittaa uudestaan (ks. kohta Annostus ja antotapa).
Jos transaminaasi‑ tai kokonaisbilirubiiniarvossa on asteen 3 tai 4 suureneminen, hoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa) ja on aloitettava heti systeeminen suuriannoksinen kortikosteroidihoito laskimoon (esim. 2 mg/kg/vrk metyyliprednisolonia tai vastaavaa). Tällaisten potilaiden maksan toimintakoearvoja on seurattava normalisoitumiseen saakka. Kun oireet ovat hävinneet ja maksan toimintakoearvot ovat kestävästi parantuneet tai korjaantuneet lähtötasolle, aloitetaan kortikosteroidiannoksen asteittainen pienentäminen kliinisen harkinnan mukaan. Annosta tulee pienentää asteittain vähintään 1 kuukauden aikana. Jos maksan toimintakoearvot suurenevat kortikosteroidiannoksen asteittaisen pienentämisen aikana, kortikosteroidiannosta voidaan suurentaa uudelleen ja sitten pienentää edelliskertaa hitaammin.
Jos maksan toimintakoearvot suurenevat merkitsevästi ja reagoivat huonosti kortikosteroidihoitoon, voidaan harkita kortikosteroidihoidon täydentämistä vaihtoehtoisella immunosuppressiivisella hoidolla. Kliinisissä tutkimuksissa käytettiin mykofenolaattimofetiilia potilaille, joille ei saatu vastetta kortikosteroidihoitoon tai joiden maksan toimintakoearvot suurenivat kortikosteroidiannoksen asteittaisen pienentämisen aikana eikä vastetta kortikosteroidiannoksen suurentamisella enää saatu (ks. mykofenolaattimofetiilivalmisteen valmisteyhteenveto).
Ipilimumabi yhdistelmähoitona nivolumabin kanssa
Ipilimumabi–nivolumabi-yhdistelmähoidon yhteydessä on todettu vaikeaa maksatulehdusta (ks. kohta Haittavaikutukset). Potilaita on seurattava maksatulehduksen merkkien ja oireiden, kuten transaminaasi- ja kokonaisbilirubiiniarvon nousun, varalta. Infektiot ja sairauksiin liittyvät syyt on suljettava pois.
Jos potilaalla on asteen 3 tai 4 transaminaasi- tai kokonaisbilirubiiniarvon nousu, ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 2 transaminaasi- tai kokonaisbilirubiiniarvon nousu, ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä. Jos nämä laboratorioarvot ovat jatkuvasti koholla, potilaalle on annettava kortikosteroideja annoksina, jotka vastaavat 0,5–1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua ipilimumabi–nivolumabi-yhdistelmähoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannosta on suurennettava tasolle, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia, ja ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi.
Immuunivälitteiset ihohaitat
Varovaisuutta on noudatettava, kun harkitaan ipilimumabin tai ipilimumabi–nivolumabi-yhdistelmähoidon käyttöä potilaalle, jolla on aikaisemmin ollut vaikea tai henkeäuhkaava ihohaitta aiemman immuunijärjestelmää stimuloivan syöpälääkehoidon aikana.
Ipilimumabi monoterapiana
Ipilimumabiin liittyy ihon vakavia, mahdollisesti immuunivälitteisiä haittavaikutuksia. Harvinaisena haittavaikutuksena on havaittu toksista epidermaalista nekrolyysiä (TEN) (mukaan lukien Stevens–Johnsonin oireyhtymä), joka on joissain tapauksissa johtanut kuolemaan. Kliinisissä tutkimuksissa ja myyntiluvan jälkeisessä käytössä on myös harvinaisina tapauksina raportoitu lääkkeeseen liittyviä yleisoireisia eosinofiilisiä reaktioita (Drug Reaction with Eosinophilia and Systemic Symptoms, DRESS) (ks. kohta Haittavaikutukset).
Iho- ja systeemioireinen lääkereaktio (DRESS) ilmaantuu eosinofiilisena ihottumana, johon liittyy yksi tai useampi seuraavista oireista: kuume, lymfadenopatia, kasvojen turvotus ja sisäelinten (maksan, munuaisten, keuhkojen) oireet. Pitkä oireettomuus (kahdesta kahdeksaan viikkoon) lääkkeelle altistumisesta taudin alkamiseen voi liittyä DRESS:iin.
Ipilimumabin aiheuttamat ihottuma ja kutina olivat pääasiassa lieviä tai keskivaikeita (aste 1 tai 2) ja reagoivat hyvin oireenmukaiseen hoitoon. MDX010-20-tutkimuksessa keskivaikeita, vaikeita, hyvin vaikeita tai kuolemaan johtavia (aste 2–5) ihon haittavaikutuksia ilmeni ipilimumabia 3 mg/kg monoterapiana saaneilla 3 viikon (mediaani; vaihteluväli 0,9–16 vk) kuluttua hoidon aloittamisesta. Kun haittavaikutukset hoidettiin tutkimusprotokollan hoito-ohjeiden mukaisesti, ne lievittyivät useimmissa tapauksissa (87 %:lla) 5 viikossa (mediaani; vaihteluväli 0,6–29 vk).
Ipilimumabin aiheuttamat ihottuma ja kutina on hoidettava vaikeusasteen mukaan. Jos ihottuma on lievä tai keskivaikea (aste 1 tai 2), voidaan jatkaa ipilimumabihoitoa ja täydentää sitä oireenmukaisella hoidolla (esim. antihistamiinilla). Jos lievä tai keskivaikea ihottuma tai lievä kutina jatkuu 1–2 viikkoa eikä lievity paikallisilla kortikosteroideilla, on aloitettava suun kautta annettava kortikosteroidihoito (esim. 1 mg/kg prednisonia kerran vuorokaudessa tai vastaavaa).
Jos ihottuma on vaikea (aste 3), hoito-ohjelman mukainen ipilimumabiannos on jätettävä väliin. Jos oireet lieventyvät (aste 1) tai häviävät, ipilimumabihoidon voi aloittaa uudestaan (ks. kohta Annostus ja antotapa).
Ipilimumabihoito on lopetettava pysyvästi, jos ihottuma on hyvin vaikea (aste 4) tai kutina on vaikeaa (aste 3) (ks. kohta Annostus ja antotapa). Tällöin on aloitettava heti systeeminen suuriannoksinen kortikosteroidihoito laskimoon (esim. 2 mg/kg/vrk metyyliprednisolonia). Kun ihottuma tai kutina on saatu hallintaan, aloitetaan kortikosteroidiannoksen asteittainen pienentäminen kliinisen harkinnan mukaan. Annosta pienennetään asteittain vähintään 1 kuukauden aikana.
Ipilimumabi yhdistelmähoitona nivolumabin kanssa
Ipilimumabi–nivolumabi-yhdistelmähoidon yhteydessä on todettu vaikeaa ihottumaa (ks. kohta Haittavaikutukset). Ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä asteen 3 ihottumassa ja lopetettava asteen 4 ihottumassa. Vaikeaa ihottumaa on hoidettava suurella kortikosteroidiannoksella, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia.
Stevens–Johnsonin oireyhtymää (SJS) ja toksista epidermaalista nekrolyysiä (TEN) on havaittu harvoin. Joissain tapauksissa tila on johtanut kuolemaan. Jos SJS:n tai TEN:n oireita tai merkkejä ilmenee, ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä ja potilas on ohjattava niihin erikoistuneeseen yksikköön arviointia ja hoitoa varten. Jos potilaalle kehittyy SJS tai TEN ipilimumabi–nivolumabi-yhdistelmähoidon aikana, suositellaan hoidon lopettamista pysyvästi (ks. kohta Annostus ja antotapa).
Hermoston immuunivälitteiset reaktiot
Ipilimumabi monoterapiana
Ipilimumabiin liittyy hermoston vakavia immuunivälitteisiä haittavaikutuksia. Kliinisissä tutkimuksissa on raportoitu kuolemaan johtanutta Guillain-Barrén oireyhtymää. Myös myasthenia gravis -tyyppisiä oireita on raportoitu (ks. kohta Haittavaikutukset). Potilaalla voi ilmetä lihasheikkoutta. Lisäksi voi ilmetä sensorista neuropatiaa.
Selittämätön motorinen neuropatia, lihasheikkous tai yli 4 päivää kestävä sensorinen neuropatia on arvioitava, ja muut kuin tulehdukselliset syyt, kuten taudin eteneminen, infektiot, metabolinen oireyhtymä ja samanaikainen lääkitys, on poissuljettava. Jos keskivaikea (asteen 2) neuropatia (motorinen neuropatia, johon voi liittyä myös sensorinen neuropatia) on todennäköisesti yhteydessä ipilimumabihoitoon, hoito-ohjelman mukainen annos on jätettävä väliin. Jos neurologiset oireet lievittyvät lähtötasolle, ipilimumabihoidon voi aloittaa uudestaan (ks. kohta Annostus ja antotapa).
Jos vaikean (asteen 3 tai 4) sensorisen neuropatian epäillään johtuvan ipilimumabista, ipilimumabihoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa). Potilasta on hoidettava sensorista neuropatiaa koskevien vakiintuneiden hoitokäytäntöjen mukaisesti ja aloitettava heti laskimonsisäinen kortikosteroidihoito (esim. 2 mg/kg/vrk metyyliprednisolonia).
Motorisen neuropatian eteneviä merkkejä on pidettävä immuunivälitteisinä ja hoidettava sen mukaisesti. Jos potilaalle kehittyy vaikea (asteen 3 tai 4) motorinen neuropatia, ipilimumabihoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa) syy-yhteydestä riippumatta.
Immuunivälitteinen munuaistulehdus ja munuaisten toimintahäiriö
Ipilimumabi yhdistelmähoitona nivolumabin kanssa
Ipilimumabi–nivolumabi-yhdistelmähoidon yhteydessä on todettu vaikeaa munuaistulehdusta ja munuaisten toimintahäiriöitä (ks. kohta Haittavaikutukset). Potilaita on seurattava munuaistulehduksen tai munuaisten toimintahäiriön merkkien ja oireiden varalta. Useimpien potilaiden seerumin kreatiniiniarvo suurenee ilman oireita. Sairauksiin liittyvät syyt on suljettava pois.
Jos potilaalla on asteen 4 seerumin kreatiniinipitoisuuden nousu, ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 2 tai 3 seerumin kreatiniinipitoisuuden nousu, ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 0,5–1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua ipilimumabi–nivolumabi-yhdistelmähoitoa voi jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannosta on suurennettava tasolle, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia, ja ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi.
Immuunivälitteinen umpierityshäiriö
Ipilimumabi monoterapiana
Ipilimumabi voi aiheuttaa umpieritysjärjestelmän elinten tulehduksia, jotka ilmenevät hypofysiittinä, hypopituitarismina, lisämunuaisten vajaatoimintana, kilpirauhasen vajaatoimintana, tyypin 1 diabetes mellituksena ja diabeettisena ketoasidoosina (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Potilailla voi esiintyä epäspesifisiä oireita, jotka saattavat muistuttaa oireita, jotka johtuvat perussairaudesta, kuten aivojen etäpesäkkeistä. Yleisimmät kliiniset ilmenemismuodot ovat päänsärky ja uupumus. Oireita voivat olla myös näkökenttäpuutokset, käytösmuutokset, elektrolyyttihäiriöt ja matala verenpaine. Addisonin kriisi oireiden aiheuttajana on poissuljettava. Kliininen kokemus ipilimumabiin liittyvästä umpierityshäiriöstä on vähäistä.
MDX010-20-tutkimuksessa keskivaikea, vaikea tai hyvin vaikea (asteen 2–4) immuunivälitteinen umpierityshäiriö ilmeni ipilimumabia 3 mg/kg monoterapiana saaneilla 7 viikosta lähemmäs 20 viikon kuluttua hoidon aloittamisesta. Kliinisissä tutkimuksissa todettu immuunivälitteinen umpierityshäiriö saatiin yleensä hallintaan immunosuppressiivisella hoidolla ja hormonikorvaushoidolla.
Jos potilaalla ilmenee Addisonin kriisin oireita, kuten elimistön vaikeaa kuivumista, matalaa verenpainetta tai sokki, suositellaan välitöntä laskimonsisäistä hoitoa kortikosteroidilla, jolla on mineralokortikoidista aktiivisuutta. Sepsiksen tai infektion mahdollisuus tulee arvioida. Jos on merkkejä lisämunuaisten vajaatoiminnasta, mutta potilaalla ei ole Addisonin kriisiä, on harkittava lisätutkimuksia, mukaan lukien laboratorio- ja kuvantamistutkimukset. Ennen kortikosteroidihoidon aloittamista voidaan arvioida potilaan umpieritystoiminta laboratoriovastausten perusteella. Jos aivolisäkkeen kuvantamistulokset tai umpieritystoiminnan laboratoriotulokset ovat poikkeavia, suositellaan lyhyttä suuriannoksista kortikosteroidihoitoa (esim. 4 mg deksametasonia 6 tunnin välein tai vastaavaa) rauhastulehduksen rauhoittamiseksi, ja hoito-ohjelman mukainen ipilimumabiannos on jätettävä väliin (ks. kohta Annostus ja antotapa). Toistaiseksi ei tiedetä, palautuuko umpirauhasen toimintahäiriö kortikosteroidihoidolla. Asianmukainen hormonihoito on aloitettava. Pitkäaikainen hormonikorvaushoito saattaa olla välttämätön.
Jos potilaalla on diabeteksen oireita, ipilimumabihoito on keskeytettävä ja insuliinikorvaushoito aloitettava tarpeen mukaan. Verensokeripitoisuuksia on seurattava edelleen, jotta pystytään varmistamaan, että insuliinikorvaushoito on asianmukaisella tasolla. Ipilimumabihoito on lopetettava pysyvästi, jos potilaalla on henkeäuhkaava diabetes.
Kun oireet tai laboratorioarvojen poikkeavuudet on saatu hallintaan ja potilaan vointi on selkeästi kohentunut ipilimumabihoito voidaan aloittaa uudestaan ja kortikosteroidiannoksen asteittainen pienentäminen aloitetaan kliinisen harkinnan mukaan. Annosta pienennetään asteittain vähintään 1 kuukauden aikana.
Ipilimumabi yhdistelmähoitona nivolumabin kanssa
Ipilimumabi–nivolumabi-yhdistelmähoidon yhteydessä on todettu vaikeita umpierityshäiriöitä, kuten kilpirauhasen vajaa- ja liikatoimintaa, lisämunuaisten vajaatoimintaa (mukaan lukien lisämunuaiskuoren sekundaarista vajaatoimintaa), hypofysiittiä (mukaan lukien hypopituitarismia), diabetesta ja diabeettista ketoasidoosia (ks. kohta Haittavaikutukset).
Potilaita on tarkkailtava umpierityshäiriöiden kliinisten merkkien ja oireiden ja hyperglykemian sekä kilpirauhasen toiminnan muutosten varalta (hoidon alussa, jaksoittain hoidon aikana ja kliinisen arvioinnin pohjalta). Potilailla saattaa esiintyä uupumusta, päänsärkyä, mielialamuutoksia, vatsakipuja, epätavallista vatsantoimintaa ja matalaa verenpainetta tai epäspesifisiä oireita, jotka voivat muistuttaa muita syitä, kuten aivojen etäpesäke tai taustasairaus. Jos vaihtoehtoisia syitä ei ole tunnistettu, umpierityshäiriöiden oireet ja löydökset on katsottava immuunivälitteiseksi.
Jos potilaalla on kilpirauhasen vajaatoiminnan oireita, ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä ja korvaushoito kilpirauhashormonilla aloitettava tarpeen mukaan. Jos potilaalla on kilpirauhasen liikatoiminnan oireita, ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä ja kilpirauhasen toimintaa estävä lääkitys on aloitettava tarpeen mukaan. Kortikosteroidien käyttöä 1–2 mg/kg/vrk metyyliprednisolonia vastaavina annoksina on harkittava myös, jos epäillään akuuttia kilpirauhastulehdusta. Tilan parannuttua ipilimumabi–nivolumabi-yhdistelmähoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Kilpirauhasen toimintaa on seurattava edelleen, jotta pystytään varmistamaan, että hormonikorvaushoito on asianmukaisella tasolla. Ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi, jos potilaalla on henkeäuhkaava kilpirauhasen liika- tai vajaatoiminta.
Jos potilaalla on asteen 2 lisämunuaisen vajaatoiminnan oireita, ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä ja fysiologinen kortikosteroidikorvaushoito aloitettava tarpeen mukaan. Ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi, jos potilaalla on vakava (asteen 3) tai henkeäuhkaava (asteen 4) lisämunuaisen vajaatoiminta. Lisämunuaisten toimintaa ja hormonipitoisuuksia on seurattava edelleen, jotta pystytään varmistamaan, että kortikosteroidikorvaushoito on asianmukaisella tasolla.
Jos potilaalla on asteen 2 tai 3 hypofysiitin oireita, ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä ja hormonikorvaushoito aloitettava tarpeen mukaan. Kortikosteroidien käyttöä 1–2 mg/kg/vrk metyyliprednisolonia vastaavina annoksina on harkittava myös, jos epäillään akuuttia aivolisäkkeen tulehdusta. Tilan parannuttua ipilimumabi–nivolumabi-yhdistelmähoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi, jos potilaalla on henkeäuhkaava (asteen 4) hypofysiitti. Aivolisäkkeen toimintaa ja hormonipitoisuuksia on seurattava edelleen, jotta pystytään varmistamaan, että hormonikorvaushoito on asianmukaisella tasolla.
Jos potilaalla on diabeteksen oireita, ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä ja insuliinikorvaushoito aloitettava tarpeen mukaan. Verensokeripitoisuuksia on seurattava edelleen, jotta pystytään varmistamaan, että insuliinikorvaushoito on asianmukaisella tasolla. Ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi, jos potilaalla on henkeäuhkaava diabetes.
Infuusioreaktio
Ipilimumabi monoterapiana tai yhdistelmähoitona nivolumabin kanssa
Kliinisissä tutkimuksissa on raportoitu vaikeita infuusioreaktioita ipilimumabi-monoterapian tai ipilimumabi–nivolumabi-yhdistelmähoidon yhteydessä (ks. kohta Haittavaikutukset). Jos infuusioreaktio on vaikea tai henkeäuhkaava, ipilimumabi- tai ipilimumabi–nivolumabi-infuusio on lopetettava ja aloitettava asianmukainen lääketieteellinen hoito. Jos infuusioreaktio on lievä tai keskivaikea, ipilimumabia tai ipilimumabi–nivolumabi-yhdistelmähoitoa voidaan antaa tarkassa valvonnassa ja käyttämällä infuusioreaktioita estävää esilääkitystä paikallisten hoitosuositusten mukaisesti.
Muut immuunivälitteiset haittavaikutukset
Ipilimumabi monoterapiana
MDX010-20-tutkimuksessa ipilimumabia 3 mg/kg monoterapiana saaneilla potilailla on raportoitu seuraavia haittavaikutuksia, joita epäillään immuunivälitteisiksi: uveiitti, eosinofilia, lipaasiarvojen suureneminen ja glomerulonefriitti. Lisäksi potilailla, jotka saivat MDX010-20-tutkimuksessa ipilimumabin (3 mg/kg) ja gp100-peptidirokotteen yhdistelmää, on raportoitu iriittiä, hemolyyttistä anemiaa, amylaasiarvon suurenemista, monielinhäiriötä ja pneumoniittia. Vogt-Koyanagi-Haradan oireyhtymätapauksia, verkkokalvon nesteistä irtaumaa ja ei-infektiivistä virtsarakkotulehdusta on raportoitu markkinoilletulon jälkeen (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Jos nämä oireet ovat vaikeita (aste 3 tai 4), voidaan tarvita heti systeemisiä suuriannoksista kortikosteroidihoitoa ja ipilimumabihoito on mahdollisesti lopetettava (ks. kohta Annostus ja antotapa). Ipilimumabihoitoon liittyvään uveiittiin, iriittiin, verkkokalvon nesteiseen irtaumaan tai episkleriittiin on harkittava kortikosteroidi-silmätippoja lääketieteellisen arvion mukaisesti. Ohimenevää näön menetystä on raportoitu potilailla, joilla on ipilimumabihoitoon liittyviä silmätulehduksia.
Kiinteän elimen siirron jälkeisiä hyljintäreaktioita on raportoitu valmisteen markkinoilletulon jälkeen potilailla, joita on hoidettu ipilimumabilla. Ipilimumabihoito saattaa suurentaa hyljintäreaktion riskiä kiinteän elimen siirron saajilla. Ipilimumabihoidon hyöty ja mahdollisen hyljintäreaktion riski on otettava huomioon näiden potilaiden kohdalla.
Ipilimumabi monoterapiana tai yhdistelmähoitona PD‑1:n tai PD‑L1:n estäjää sisältävän hoidon kanssa
Ipilimumabi-monoterapian sekä ipilimumabin ja PD‑1:n tai PD‑L1:n estäjää (mukaan lukien nivolumabia) sisältävän yhdistelmähoidon yhteydessä on todettu hemofagosyyttistä lymfohistiosytoosia (HLH). Varovaisuutta tulee noudattaa, kun ipilimumabia annetaan monoterapiana taiyhdistelmähoitona PD‑1:n tai PD‑L1:n estäjän kanssa. Jos hemofagosyyttinen lymfohistiosytoosi vahvistetaan, ipilimumabin tai ipilimumabin ja PD‑1:n tai PD‑L1:n estäjän yhdistelmähoito on lopetettava ja hemofagosyyttisen lymfohistiosytoosin hoito aloitettava.
Ipilimumabi yhdistelmähoitona nivolumabin kanssa
Seuraavia immuunivälitteisiä haittavaikutuksia on raportoitu alle 1 %:lla ipilimumabi–nivolumabi-yhdistelmähoitoa saaneista potilaista kliinisissä tutkimuksissa eri annoksilla ja kasvaintyypeillä: pankreatiitti, uveiitti, demyelinaatio, autoimmuunineuropatia (mukaan lukien kasvo- ja loitontajahermon halvaus), Guillain–Barrén oireyhtymä, myasthenia gravis, myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymä, myasteeninen oireyhtymä, aseptinen aivokalvotulehdus, aivotulehdus, gastriitti, sarkoidoosi, duodeniitti, myosiitti, myokardiitti, rabdomyolyysi ja myeliitti. Vogt–Koyanagi–Haradan oireyhtymätapauksia, verkkokalvon nesteistä irtaumaa ja ei‑infektiivistä virtsarakkotulehdusta on raportoitu markkinoilletulon jälkeen (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Ohimenevää näön menetystä on raportoitu potilailla, joilla on ipilimumabihoitoon liittyviä silmätulehduksia.
Immuunivälitteistä haittavaikutusta epäiltäessä potilaan tila on arvioitava riittävän tarkasti, jotta pystytään vahvistamaan haittavaikutuksen etiologia tai sulkemaan pois muut syyt. Haittavaikutuksen vakavuuden perusteella ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä ja annettava kortikosteroideja. Tilan parannuttua ipilimumabi–nivolumabi-yhdistelmähoitoa voi jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi, jos yksikään vakava immuunivälitteinen haittavaikutus uusiutuu tai on hengenvaarallinen.
Myotoksisuutta (myosiitti, myokardiitti ja rabdomyolyysi) on raportoitu ipilimumabi–nivolumabi-yhdistelmähoidon yhteydessä. Joissain tapauksissa tila on johtanut kuolemaan. Jos potilaalle kehittyy myotoksisuuden merkkejä tai oireita, potilasta on seurattava huolellisesti ja hänet on välittömästi ohjattava asiantuntijalle arviointia ja hoitoa varten. Myotoksisuuden vakavuuden perusteella ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä tai lopetettava (ks. kohta Annostus ja antotapa) ja asianmukainen hoito on aloitettava.
Myokardiitin diagnosointi vaatii vahvan epäilyn. Potilaat, joilla on sydän- tai sydän- ja keuhko-oireita, on syytä arvioida mahdollisen myokardiitin varalta. Myokardiittia epäiltäessä on aloitettava ripeästi hoito suurella steroidiannoksella (prednisoni 1–2 mg/kg/päivä tai metyyliprednisoloni 1–2 mg/kg/päivä) ja konsultoitava viipymättä kardiologian asiantuntijaa diagnoosin varmistamista varten nykyisten hoitosuositusten mukaisesti. Myokardiittidiagnoosin varmistuttua nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä tai lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymän (ilmenee näistä joko kahden tai kaikkien kolmen sairauden limittymisenä) tapauksia, jotka ovat joskus johtaneet kuolemaan, on raportoitu ipilimumabi–nivolumabi-yhdistelmähoidossa. Oireyhtymän varhainen tunnistaminen ja aggressiivinen hoito ovat oleellisia siihen liittyvän sairastuvuuden ja kuolleisuusriskin vuoksi.
Jos potilaalla todetaan asteen 3 tai 4 myokardiitti-myosiitti-myasthenia gravis overlap -oireyhtymä, ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa). Kortikosteroidien käyttö on aloitettava kliinisen tarpeen mukaan.
Jos potilaalla todetaan asteen 2 myokardiitti-myosiitti-myasthenia gravis overlap -oireyhtymä, ipilimumabi–nivolumabi-yhdistelmähoito on keskeytettävä ja kortikosteroidien käyttö aloitettava kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa). Tilan parannuttua ipilimumabi–nivolumabihoitoa voidaan harkita kortikosteroidiannoksen asteittaisen pienentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidien käytön aloittamisesta huolimatta, kortikosteroidiannosta on muutettava kliinisen tarpeen mukaan ja ipilimumabi–nivolumabi-yhdistelmähoito on lopetettava pysyvästi.
Sairauskohtaiset varotoimet
Melanooma
MDX010‑20‑tutkimukseen ei otettu mukaan potilaita, joilla oli silmämelanooma, primaarinen keskushermoston melanooma tai aktiivisia aivojen etäpesäkkeitä (ks. kohta Farmakodynamiikka).
CA184‑169‑tutkimukseen ei otettu mukaan potilaita, joilla oli silmämelanooma. Tutkimukseen otettiin kuitenkin mukaan potilaita, joilla oli aivojen etäpesäkkeitä, jos potilailla ei ollut metastasoineisiin aivoleesioihin liittyviä neurologisia oireita ja jos heitä ei ollut tarvetta hoitaa tai heitä ei hoidettu systemisillä kortikosteroideilla 10 päivää ennen ipilimumabihoidon alkamista (ks. kohta Farmakodynamiikka).
Lapsilla tehtyyn CA184070‑tutkimukseen ei otettu mukaan aiempaa ipilimumabihoitoa saaneita potilaita sekä potilaita, joilla oli silmämelanooma tai aktiivisia aivojen etäpesäkkeitä (ks. kohta Farmakodynamiikka).
Lapsilla tehtyyn CA184178‑tutkimukseen ei otettu mukaan potilaita, joilla oli silmämelanooma, aktiivisia aivojen etäpesäkkeitä ja jotka olivat saaneet aiempaa hoitoa CTLA-4:ään, PD-1:een, PD-L1:een, tai CD137:ään kohdistuvilla valmisteilla (ks. kohta Farmakodynamiikka).
Potilaat, joiden lähtötason suorituskykyluokka oli ≥ 2, joilla oli aktiivinen aivometastaasi tai autoimmuunisairaus tai jotka olivat saaneet systeemistä immunosuppressiivista hoitoa ennen tutkimukseen ottamista, suljettiin pois kliinisistä tutkimuksista, jotka koskivat ipilimumabi–nivolumabi-yhdistelmähoitoa. Melanoomatutkimuksista suljettiin pois potilaat, joilla oli silmän/uvean melanooma. Tutkimustiedon puuttuessa nivolumabia on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty–riski-suhde on arvioitava tarkasti kunkin potilaan kohdalla.
Ipilimumabi–nivolumabi-yhdistelmähoito on osoittanut nivolumabi-monoterapiaan verrattuna etenemisvapaan elinajan kasvavan ainoastaan potilailla, joilla on vähäinen kasvaimen PD-L1-ilmentymä. Kokonaiselinajan parantuminen oli samaa luokkaa ipilimumabi–nivolumabi-yhdistelmähoitoa ja nivolumabi-monoterapiaa saaneilla potilailla, joilla oli suuri kasvaimen PD-L1-ilmentymä (PD-L1 ≥ 1 %). Ennen yhdistelmähoidon aloittamista lääkärin tulee arvioida tarkkaan potilas ja kasvaimen ominaisuudet sekä huomioida yhdistelmähoidon havaitut edut ja toksisuus nivolumabi-monoterapiaan verrattuna (ks. kohdat Haittavaikutukset ja Farmakodynamiikka).
Ipilimumabi–nivolumabi-yhdistelmähoito potilaille, joilla on nopeasti etenevä melanooma.
Lääkärien tulee huomioida ipilimumabi–nivolumabi-yhdistelmähoidon viivästynyt vaikutuksen alkaminen ennen hoidon aloittamista potilaille, joilla on nopeasti etenevä sairaus (ks. kohta Farmakodynamiikka).
Munuaiskarsinooma
Ipilimumabi–nivolumabi-yhdistelmähoitoa koskevista kliinisistä tutkimuksista suljettiin pois ne potilaat joilla oli, tai joilla oli ollut, aivometastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa ipilimumabi–nivolumabi‑yhdistelmähoitoa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty–riski-suhde on arvioitava tarkasti kunkin potilaan kohdalla.
Ei-pienisoluinen keuhkosyöpä
Ei-pienisoluisen keuhkosyövän ensilinjan hoitoa koskevasta kliinisestä avaintutkimuksesta suljettiin pois ne potilaat, joilla oli aktiivinen autoimmuunisairaus, oireinen interstitiaalinen keuhkosairaus, systeemistä immunosuppressiota vaativa tila, aktiivinen (hoitamaton) aivojen etäpesäke, tai jotka olivat saaneet aiempaa systeemistä hoitoa edenneeseen tautiin tai joilla oli herkistäviä EGFR-mutaatioita tai ALK:n translokaatioita (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tietoja käytöstä iäkkäille potilaille (≥ 75‑vuotiaille) on vain vähän (ks. kohta Farmakodynamiikka). Näiden potilaiden osalta ipilimumabin annossa yhdessä nivolumabin ja kemoterapian kanssa on noudatettava varovaisuutta; tällöin hoidon mahdollisia hyötyjä ja riskejä on arvioitava huolellisesti potilaskohtaisesti.
Keuhkopussin pahanlaatuinen mesoteliooma
Keuhkopussin pahanlaatuisen mesoteliooman ensilinjan hoitoa koskevasta pivotaalitutkimuksesta suljettiin pois potilaat, joilla oli primitiivinen vatsakalvon, sydänpussin, kiveksen tai tuppikalvon mesoteliooma, interstitiaalinen keuhkosairaus, aktiivinen autoimmuunisairaus, systeemistä immunosuppressiota vaativa tila tai aivojen etäpesäke (ellei etäpesäkettä ollut poistettu kirurgisesti tai hoidettu stereotaktisella sädehoidolla siten, ettei etäpesäke ollut kasvanut kolmeen kuukauteen ennen tutkimukseen ottamista) (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa ipilimumabi–nivolumabi‑yhdistelmähoitoa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
dMMR- tai MSI-H-tyyppinen kolorektaalisyöpä
Potilaat, joiden lähtötason suorituskykyluokka oli ≥ 2, joilla oli aktiivisia aivometastaaseja tai leptomeningeaalisia metastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila, suljettiin pois dMMR- ja MSI-H-tyyppistä metastaattista kolorektaalisyöpää koskevista kliinisistä tutkimuksista (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa ipilimumabi–nivolumabi‑yhdistelmähoitoa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Ruokatorven levyepiteelikarsinooma
Potilaat, joiden lähtötason suorituskykyluokka oli ≥ 2, joilla oli tai oli ollut aivometastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila tai joilla oli suuri riski verenvuodolle tai fistelille kasvaimen havaittavan invaasion ruokatorven kasvainta lähellä oleviin elimiin aiheuttamana, suljettiin pois ruokatorven levyepiteelikarsinoomaa koskevasta kliinisestä tutkimuksesta (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa ipilimumabi–nivolumabi‑yhdistelmähoitoa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Ruokatorven levyepiteelikarsinooman ensilinjan hoidon tutkimuksessa ipilimumabi–nivolumabi‑yhdistelmähoitoa saaneista potilaista kuoli 4 kuukauden aikana useampi kuin solunsalpaajahoitoa saaneista. Lääkärien tulee huomioida ipilimumabi–nivolumabi‑yhdistelmähoidon viivästynyt vaikutuksen alkaminen ennen hoidon aloittamista potilaille, joilla on huonompi ennuste ja/tai aggressiivinen sairaus (ks. kohta Farmakodynamiikka).
Hepatosellulaarinen karsinooma
Hepatosellulaarista karsinoomaa koskevasta kliinisestä tutkimuksesta suljettiin pois potilaat, joiden lähtötason ECOG-toimintakykyluokka oli ≥ 2, joille oli tehty aiemmin maksansiirto tai joilla oli Child‑Pugh-luokan C maksasairaus, aiemmin todettuja samanaikaisia aivometastaaseja, aiempi maksaperäinen enkefalopatia (12 kuukauden sisällä satunnaistamisesta), kliinisesti merkittävä askites, HIV-infektio tai aktiivinen samanaikainen hepatiitti B -virusinfektio (HBV) ja hepatiitti C-virusinfektio (HCV) tai HBV ja hepatiitti D -virusinfektio (HDV), aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiivista hoitoa vaativa tila (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Hepatosellulaarista karsinoomaa sairastavista potilaista, joilla on Child‑Pugh-luokan B maksasairaus, on saatavilla vain vähän tietoa. Tutkimustiedon puuttuessa ipilimumabi–nivolumabi-yhdistelmähoitoa ja sen jälkeen annettavaa nivolumabihoitoa on käytettävä varoen näille ryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Ipilimumabi–nivolumabi-yhdistelmähoitoa saaneista hepatosellulaarista karsinoomaa sairastavista potilaista kuoli 6 kuukauden aikana useampi kuin lenvatinibia tai sorafenibia saaneista. Suurempi kuolemanriski voi olla yhteydessä huonoon ennusteeseen. Lääkäreiden tulee huomioida tämä riski ennen ipilimumabi–nivolumabi-yhdistelmähoidon aloittamista potilaille, joiden ennuste on huono.
Potilaat, joilla on autoimmuunisairaus
Kliinisissä tutkimuksissa ei tutkittu potilaita, joilla oli joskus aiemmin ollut autoimmuunisairaus (muu kuin vitiligo tai riittävästi hallinnassa oleva umpierityksen vajaustila, kuten kilpirauhasen vajaatoiminta), eikä myöskään niitä, jotka tarvitsivat systeemistä immunosuppressiivista hoitoa heillä jo entuudestaan olevaan aktiiviseen autoimmuunisairauteen tai elinsiirteen vuoksi. Ipilimumabi on T-solujen potentoija ja mahdollistaa immuunivasteen (ks. kohta Farmakodynamiikka). Ipilimumabi saattaa haitata immunosuppressiivista hoitoa, jolloin perussairaus pahenee tai elinsiirteen hyljintäriski suurenee. Ipilimumabia ei tulisi antaa potilaalle, jolla on vaikea aktiivinen autoimmuunisairaus, jossa immuunijärjestelmän lisäaktivaatio voisi aiheuttaa välittömän hengenvaaran. Ipilimumabin annossa on noudatettava varovaisuutta myös silloin, jos potilaalla on joskus aiemmin ollut autoimmuunisairaus; tällöin ipilimumabihoidon mahdollisia riskejä ja hyötyjä on arvioitava potilaskohtaisesti.
Potilaat, joilla on vähäsuolainen ruokavalio
Tämä lääkevalmiste sisältää 23 mg natriumia per 10 ml:n injektiopullo ja 92 mg natriumia per 40 ml:n injektiopullo, jotka vastaavat 1,15 %:a ja 4,60 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille. Ipilimumabin natriumsisältö tulee ottaa huomioon, jos hoidettavalla potilaalla on vähäsuolainen ruokavalio.
Samanaikainen vemurafenibin anto
Faasin I tutkimuksessa raportoitiin transaminaasiarvojen (ALAT/ASAT > 5 x ULN) ja bilirubiinin (kokonaisbilirubiini > 3 x ULN) oireetonta, asteen 3 suurenemista ipilimumabin (3 mg/kg) ja vemurafenibin (960 mg x 2/vrk tai 720 mg x 2/vrk) samanaikaisen annon yhteydessä. Näiden alustavien tietojen perusteella ipilimumabin ja vemurafenibin samanaikaista antoa ei suositella.
Sekventiaalinen anto vemurafenibin kanssa
Faasin II tutkimuksessa potilailla, joilla oli BRAF‑mutatoitunut metastaattinen melanooma ja jotka saivat sekventiaalista hoitoa ensin vemurafenibilla, minkä jälkeen ipilimumabilla annoksella 10 mg/kg, havaittiin enemmän asteen 3+ ihohaittoja kuin potilailla, joita hoidettiin vain ipilimumabilla. Varovaisuutta on noudatettava, kun ipilimumabia annetaan vemurafenibihoidon jälkeen.
Pediatriset potilaat
Ipilimumabin käytöstä 12-vuotiaille ja sitä vanhemmille nuorille on saatavilla rajoitetusti turvallisuustietoja, eivätkä ne koske pitkäaikaiskäyttöä.
Tietoja käytöstä alle 12-vuotiaille lapsille on vain vähän. Ipilimumabia ei pidä sen vuoksi käyttää alle 12-vuotiaille lapsille.
Ennen kuin ipilimumabi-monoterapia aloitetaan 12-vuotiaalle tai sitä vanhemmalle nuorelle, lääkäriä kehotetaan arvioimaan jokainen potilas huolellisesti ja huomioimaan saatavilla olevien tietojen rajoitettu määrä, havaitut hyödyt ja ipilimumabi-monoterapian toksisuus pediatrisilla potilailla (ks. kohdat Haittavaikutukset ja Farmakodynamiikka).
Yhteisvaikutukset
Ipilimumabi on ihmisperäinen monoklonaalinen vasta-aine, joka ei metaboloidu sytokromi P450 ‑entsyymien, eikä muiden lääkeaineita metaboloivien entsyymien välityksellä.
Lääkeyhteisvaikutusta CYP-isotsyymien (erityisesti CYP1A2, CYP2E1, CYP2C8 ja CYP3A4) kanssa on selvitetty yhdessä aikuisilla tehdyssä tutkimuksessa edennyttä melanoomaa sairastaneilla potilailla, jotka eivät olleet saaneet aiempaa hoitoa: potilaille annettiin joko pelkkää ipilimumabia tai ipilimumabia yhdessä kemoterapian (dakarbatsiini tai paklitakseli/karboplatiini) kanssa. Ipilimumabin ja paklitakselin/karboplatiinin, dakarbatsiinin tai sen metaboliitin (5- aminoimidatsoli-4-karboksamidi, AIC) välillä ei todettu kliinisesti merkityksellisiä farmakokineettisiä lääke-lääkeyhteisvaikutuksia.
Muut yhteisvaikutukset
Kortikosteroidit
Lähtötilanteessa, ennen ipilimumabihoidon aloittamista, olisi vältettävä systeemisten kortikosteroidien käyttöä, koska ne saattavat haitata ipilimumabin farmakodynaamista aktiivisuutta ja tehoa. Systeemisiä kortikosteroideja tai muita immunosuppressantteja voi kuitenkin käyttää ipilimumabihoidon aloittamisen jälkeen immuunivälitteisten haittavaikutusten hoitoon. Systeemisten kortikosteroidien käyttö ipilimumabihoidon aloittamisen jälkeen ei tunnu heikentävän ipilimumabin tehoa.
Antikoagulantit
Antikoagulanttien käyttö suurentaa tunnetusti maha-suolikanavan verenvuodon riskiä. Koska maha-suolikanavan verenvuoto on ipilimumabin haittavaikutus (ks. kohta Haittavaikutukset), samanaikaista antikoagulanttihoitoa tarvitsevaa potilasta on seurattava tarkoin.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja ipilimumabin käytöstä raskaana oleville naisille. Eläimillä tehdyissä lisääntymistutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta prekliiniset tiedot turvallisuudesta). Ihmisen IgG1 läpäisee istukan. Hoidon mahdollisesti aiheuttamaa riskiä kehittyvälle sikiölle ei tunneta. YERVOY-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten hedelmällisessä iässä olevien naisten hoitoon, jotka eivät käytä ehkäisyä, jollei kliininen hyöty ole mahdollista riskiä suurempi
Imetys
Tiineysaikana ipilimumabihoitoa saaneiden cynomolgus-apinoiden rintamaidossa on todettu ipilimumabia vain hyvin pieninä pitoisuuksina. Ei tiedetä, erittyykö ipilimumabi ihmisen rintamaitoon. IgG erittyy ihmisen rintamaitoon yleensä vain vähäisessä määrin, ja suun kautta saadun IgG:n biologinen hyötyosuus on pieni. Imeväiseen kohdistuva systeeminen altistus ei odotettavasti ole merkitsevä, eikä rintaruokittavaan vastasyntyneeseen/imeväiseen oletettavasti kohdistu lääkkeen vaikutuksia. Rintaruokittuun imeväiseen kohdistuvan haittavaikutusriskin vuoksi on päätettävä joko imetyksen tai YERVOY-hoidon lopettamisesta ottaen huomioon rintaruokinnasta lapselle koituva hyöty ja YERVOY-hoidosta naiselle koituva hyöty.
Hedelmällisyys
Ipilimumabin vaikutusta hedelmällisyyteen ei ole selvitetty tutkimuksin. Näin ollen ei tiedetä, miten ipilimumabi vaikuttaa miehen ja naisen hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
YERVOY-valmisteella on vähäinen vaikutus ajokykyyn ja koneiden käyttökykyyn.
Koska hoito voi aiheuttaa haittavaikutuksia, kuten uupumusta (ks. kohta Haittavaikutukset), potilasta on kehotettava olemaan varovainen autoa ajaessaan tai koneita käyttäessään, kunnes on varmaa, ettei ipilimumabi vaikuta näihin toimiin haitallisesti.
Haittavaikutukset
Ipilimumabi monoterapiana (ks. kohta Annostus ja antotapa)
a. Tiivistelmä turvallisuusprofiilista
Ipilimumabia on annettu yli noin 10 000 potilaalle kliinisessä tutkimusohjelmassa, jossa arvioitiin ipilimumabia eri annoksilla eri syöpätyypeissä. Jollei toisin mainita, seuraavat tiedot koskevat altistusta ipilimumabiannokselle 3 mg/kg kliinisissä melanoomatutkimuksissa. Faasin III MDX010-20-tutkimuksessa (ks. kohta Farmakodynamiikka) potilaat saivat 4 annosta (mediaani; vaihteluväli 1–4).
Ipilimumabiin liittyy eniten haittavaikutuksia, jotka johtuvat immuunijärjestelmän kiihtyneestä tai liiallisesta toiminnasta. Useimmat tällaisista haittavaikutuksista, myös vaikeista reaktioista, korjaantuivat antamalla potilaalle asianmukaista lääketieteellistä hoitoa tai lopettamalla ipilimumabihoito (immuunivälitteisten haittavaikutusten hoito, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
MDX010-20-tutkimuksessa yleisimmin ilmoitetut haittavaikutukset (≥ 10 %:lla potilaista) ipilimumabia 3 mg/kg monoterapiana saaneilla olivat ripuli, ihottuma, kutina, uupumus, pahoinvointi, oksentelu, ruokahalun heikkeneminen ja vatsakipu. Suurin osa haittavaikutuksista oli lieviä tai keskivaikeita (aste 1 tai 2). Ipilimumabihoito lopetettiin haittavaikutusten vuoksi 10 %:lla potilaista.
b. Haittavaikutustaulukko
Taulukossa 6 on haittavaikutukset, joita raportoitiin markkinoilletulon jälkeisessä seurannassa sekä kliinisissä tutkimuksissa pitkälle edennyttä melanoomaa sairastaneilla potilailla, jotka saivat ipilimumabia 3 mg/kg (n = 767).
Haittavaikutukset on esitetty elinjärjestelmittäin ja yleisyysluokittain. Yleisyysluokkien määritelmät ovat: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000; < 1/1 000); hyvin harvinainen (< 1/10 000) tuntematon (ei voida arvioida markkinoilletulon jälkeen saatujen tietojen pohjalta). Haittavaikutukset on esitetty kussakin yleisyysluokassa vakavimmasta lievimpään. Immuunivälitteisiä haittavaikutuksia ilmeni ipilimumabia MDX010-20-tutkimuksessa saaneilla HLA-A2*0201-positiivisilla potilailla saman verran kuin koko kliinisessä tutkimusohjelmassa.
Kun tarkastellaan yhdistettyjä tietoja faasien II ja III kliinisten tutkimusten potilaista, jotka eivät olleet aiemmin saaneet kemoterapiaa (n = 75), kahden retrospektiivisen havaintotutkimuksen aiemmin hoitamattomista potilaista (n = 273 ja n = 157) sekä CA184‑169‑tutkimuksen potilaista (n = 362), ipilimumabin turvallisuusprofiili oli annoksella 3 mg/kg samankaltainen kuin edennyttä melanoomaa sairastaneilla, jotka olivat saaneet aiempaa hoitoa.
Niiden potilaiden turvallisuustiedot, joilla oli melanooma (jota ei voitu kirurgisesti poistaa tai joka oli metastasoitunut), joita hoidettiin ipilimumabilla (3 mg/kg, seuranta‑aika vähintään 3 vuotta) ja jotka oli otettu kansainväliseen, prospektiiviseen, CA184143‑havaintotutkimukseen (N= 1151), olivat samankaltaiset kuin ilmoitetut turvallisuustiedot kliinisistä tutkimuksista, joissa tutkittiin ipilimumabia edenneen melanooman hoidossa.
Taulukko 6: Haittavaikutukset edennyttä melanoomaa sairastaneilla, joiden ipilimumabiannos oli 3 mg/kga
Infektiot | |
Yleinen | sepsisb, virtsatieinfektio, hengitystieinfektio |
Melko harvinainen | septinen sokkib, keuhkokuume |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | |
Yleinen | kasvaimen aiheuttama kipu |
Melko harvinainen | paraneoplastinen oireyhtymä |
Veri ja imukudos | |
Yleinen | anemia, lymfosytopenia, trombosytopenia, neutropenia |
Melko harvinainen | hemolyyttinen anemiab, eosinofilia |
Tuntematon | hemofagosyyttinen lymfohistiosytoosie |
Immuunijärjestelmä | |
Melko harvinainen | yliherkkyys |
Hyvin harvinainen | anafylaktinen reaktio |
Tuntematon | hyljintäreaktio kiinteän elimen siirron jälkeene |
Umpieritys | |
Yleinen | hypopituitarismi (myös hypofysiitti)c, kilpirauhasen vajaatoimintac |
Melko harvinainen | lisämunuaisten vajaatoimintac, sekundaarinen lisämunuaiskuoren vajaatoimintad, kilpirauhasen liikatoimintac, hypogonadismi |
Harvinainen | autoimmuunityreoidiittid, kilpirauhastulehdusd |
Aineenvaihdunta ja ravitsemus | |
Hyvin yleinen | ruokahalun heikkeneminen |
Yleinen | elimistön kuivuminen, hypokalemia, painonlasku, hyponatremia |
Melko harvinainen | alkaloosi, hypofosfatemia, tuumorilyysioireyhtymä, hypokalsemiad |
Harvinainen | tyypin 1 diabetes mellitus (myös diabeettinen ketoasidoosi)h |
Psyykkiset häiriöt | |
Yleinen | sekavuustila, masennus |
Melko harvinainen | mielentilan muutokset, sukupuolivietin heikkeneminen |
Hermosto | |
Yleinen | perifeerinen sensorinen neuropatia, heitehuimaus, päänsärky, letargia, kraniaalinen neuropatia, aivojen turvotus, perifeerinen neuropatia |
Melko harvinainen | Guillain–Barrén oireyhtymäb,c, (aseptinen) aivokalvotulehdus, sentraalinen autoimmuunineuropatia (aivotulehdus)d, pyörtyminen, ataksia, vapina, myoklonus, dysartria |
Harvinainen | myasthenia gravisd |
Tuntematon | myeliitti |
Silmät | |
Yleinen | näön sumeneminen, silmäkipu |
Melko harvinainen | uveiittic, lasiaisen verenvuoto, iriittic, silmän turvotusd, silmäluomitulehdusd, näöntarkkuuden heikkeneminen, vierasesinetuntemus silmissä, sidekalvotulehdus |
Harvinainen | Vogt–Koyanagi–Haradan oireyhtymäe, verkkokalvon nesteinen irtauma |
Sydän | |
Yleinen | rytmihäiriö, eteisvärinä |
Verisuonisto | |
Yleinen | hypotensio, kasvojen ja kaulan punehtuminen, kuumat aallot |
Melko harvinainen | verisuonitulehdus, verisuonisairausb, perifeerinen iskemia, ortostaattinen hypotensio |
Harvinainen | ohimovaltimotulehdusd |
Hengityselimet, rintakehä ja välikarsina | |
Yleinen | hengenahdistus, yskä, allerginen nuha |
Melko harvinainen | hengityksen vajaatoiminta, akuutti hengitysvaikeusoireyhtymäb, keuhkoinfiltraatti, keuhkopöhö, pneumoniitti |
Ruoansulatuselimistö | |
Hyvin yleinen | ripulic, oksentelu, pahoinvointi, ummetus, vatsakipu |
Yleinen | maha-suolikanavan verenvuoto, koliittib,c, gastroesofageaalinen refluksitauti, limakalvotulehdusd, gastroenteriitti, suutulehdus |
Melko harvinainen | maha-suolikanavan puhkeaminenb,c, paksusuolen puhkeaminenb,c, suolen puhkeaminenb,c, vatsakalvontulehdusb, divertikuliitti, pankreatiitti, enterokoliitti, mahahaava, paksusuolen haavauma, ruokatorvitulehdus, ileusd, peräsuolitulehdusd |
Harviainen | haiman eksokriininen vajaatoiminta, keliakia |
Maksa ja sappi | |
Yleinen | maksan epänormaali toiminta |
Melko harvinainen | maksan vajaatoimintab,c, maksatulehdus, hepatomegalia, keltaisuus |
Iho ja ihonalainen kudos | |
Hyvin yleinen | ihottumac, kutinac |
Yleinen | ihotulehdus, ihon punoitus, vitiligo, nokkosihottuma, ekseemad, hiustenlähtö, yöhikoilu, kuiva iho |
Melko harvinainen | toksinen epidermaalinen nekrolyysib,c, leukosytoklastinen verisuonitulehdus, ihon kesiminen, hiusten värimuutoksetd |
Harvinainen | erythema multiformed, psoriaasid, lääkkeeseen liittyvä yleisoireinen eosinofiilinen reaktio (Drug Reaction with Eosinophilia and Systemic Symptoms, DRESS)d |
Tuntematon | pemfigoidi |
Luusto, lihakset ja sidekudos | |
Hyvin yleinen | muskuloskeletaalinen kipuf |
Yleinen | artralgia, lihaskipu, lihaskouristukset, niveltulehdus |
Melko harvinainen | polymyalgia rheumatica, myosiittid, lihasheikkousd |
Harvinainen | polymyosiittid |
Munuaiset ja virtsatiet | |
Yleinen | munuaisten vajaatoimintab |
Melko harvinainen | glomerulonefriittic, autoimmuuninefriittid, munuaisperäinen asidoosi, hematuriad, ei‑infektiivinen virtsarakkotulehdusg, proteinuriad |
Sukupuolielimet ja rinnat | |
Melko harvinainen | kuukautisten poisjäänti |
Yleisoireet ja antopaikassa todettavat haitat | |
Hyvin yleinen | uupumus, pistokohdan reaktio, kuume, turvotus, kipu |
Yleinen | vilunväristykset, voimattomuus, influenssankaltainen sairausd |
Melko harvinainen | monielinhäiriöb,c, yleistynyt tulehdusvasteoireyhtymä (SIRS)d, infuusioreaktio |
Tutkimukset | |
Yleinen | alaniiniaminotransferaasin suureneminenc, aspartaattiaminotransferaasin suureneminenc, veren alkalinen fosfataasi -pitoisuuden suureneminend, veren bilirubiinin suureneminen, lisääntynyt lipaasic |
Melko harvinainen | lisääntynyt gammaglutamyylitransferaasid, veren lisääntynyt kreatiniini, veren kilpirauhasta stimuloivan hormonin lisääntyminen, veren kortisolin pieneneminen, veren kortikotropiinin pieneneminen, veren lisääntynyt amylaasic, positiiviset tumavasta-aineetd, veren testosteronin pieneneminen |
Harvinainen | veren tyreotropiinipitoisuuden pieneneminend, tyroksiinipitoisuuden pieneneminend, epänormaali veren prolaktiinipitoisuusd |
Taulukossa 6 esitetyt yleisyysluokat eivät välttämättä liity täysin pelkästään ipilimumabiin, vaan niihin voi vaikuttaa myös perussairaus.
a esiintymistiheydet perustuvat yhdistettyihin tuloksiin 9 kliinisestä tutkimuksesta, joissa arvioitiin 3 mg/kg:n ipilimumabiannosta melanooman hoidossa.
b aiheuttanut myös kuolemia.
c lisätietoa näistä mahdollisesti tulehduksellisista haittavaikutuksista on alakohdassa ”Valikoitujen haittavaikutusten kuvaus” ja kohdassa Varoitukset ja käyttöön liittyvät varotoimet. Näiden kohtien tiedot perustuvat ensisijaisesti kokemuksiin faasin III MDX010‑20-tutkimuksessa.
d esiintymistiheyden määrityksissä huomioitiin myös tiedot, jotka saatiin muualta kuin 9:stä loppuun saatetusta kliinisestä melanoomatutkimuksesta.
e Markkinoilletulon jälkeinen tapahtuma (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet).
f Tuki- ja liikuntaelinten kipu on termi, joka kattaa selkäkivun, luukivun, muskuloskeletaalisen rintakivun, muskuloskeletaaliset vaivat, lihaskivun, niskakivun, raajakivun ja selkärankakivun.
g Ilmoitettu kliinisissä tutkimuksissa ja valmisteen markkinoilletulon jälkeen.
h Tyypin 1 diabetes mellitus, johon saattaa liittyä diabeettinen ketoasidoosi.
Kliinisissä melanoomatutkimuksissa on raportoitu muitakin kuin taulukossa 6 lueteltuja haittavaikutuksia potilailla, jotka saivat jotakin muuta ipilimumabiannosta (joko < tai > 3 mg/kg). Näitä muita reaktioita ilmeni alle 1 %:lla, jollei muuta ole ilmoitettu: meningismi, myokardiitti, perikardiaalinen effuusio, sydänlihassairaus, autoimmuunihepatiitti, erythema nodosum, autoimmuunipankreatiitti, hyperpituitarismi, lisäkilpirauhasten vajaatoiminta, infektioperitoniitti, episkleriitti, skleriitti, Raynaud’n oireyhtymä, käsi jalkaoireyhtymä, sytokiinien vapautumisesta aiheutuva oireyhtymä, sarkoidoosi, veren gonadotropiiniarvon pieneneminen, leukopenia, polysytemia, lymfosytoosi, silmälihastulehdus ja sensorineuraalinen huonokuuloisuus.
CA184‑169‑tutkimuksessa (n = 362) ipilimumabin yleinen turvallisuusprofiili oli annoksella 3 mg/kg samankaltainen kuin ipilimumabia saaneiden, edennyttä melanoomaa sairastaneiden potilaiden tavanomainen turvallisuusprofiili.
Ipilimumabi yhdistelmähoitona nivolumabin kanssa (kemoterapian kanssa tai ilman) (ks. kohta Annostus ja antotapa)
a. Tiivistelmä turvallisuusprofiilista
Kun ipilimumabia annetaan yhdistelmähoitona, lue muiden lääkeaineiden valmisteyhteenvedot ennen hoidon aloittamista. Kun ipilimumabia annetaan yhdistelmähoitona muiden lääkeaineiden kanssa, lue muiden lääkeaineiden valmisteyhteenvedosta lisätietoa turvallisuusprofiilista.
Yhdistetyissä tutkimustiedoissa (n = 2626) eri kasvaintyyppien hoidossa käytetyn ipilimumabi–nivolumabi-yhdistelmähoidon (kemoterapian kanssa tai ilman) yleisimmät haittavaikutukset (≥ 10 %) olivat uupumus (47 %), ripuli (35 %), ihottuma (37 %), pahoinvointi (27 %), kutina (29 %), muskuloskeletaalinen kipu (26 %), kuume (23 %), ruokahalun heikkeneminen (22 %), yskä (21 %), vatsakipu (18 %), oksentelu (18 %), ummetus (18 %), artralgia (18 %), dyspnea (17 %), kilpirauhasen vajaatoiminta (16 %), päänsärky (15 %), ylähengitystieinfektio (13 %), turvotus (13 %) ja heitehuimaus (10 %), kun seuranta-aika oli vähintään 6–47 kuukautta. Asteen 3–5 haittavaikutusten ilmaantuvuus oli 66 %, kun ipilimumabia käytettiin yhdistelmähoitona nivolumabin kanssa (kemoterapian kanssa tai ilman). Tutkimuslääkkeestä johtuvien, kuolemaan johtaneiden haittavaikutusten ilmaantuvuus oli 1,0 %. Seuraavien haittavaikutusten ilmaantuvuus oli ≥ 10 % suurempi potilailla, jotka saivat yhdistelmähoitona ipilimumabia 3 mg/kg ja nivolumabia 1 mg/kg melanooman hoitoon, kuin ilmaantuvuus yhdistetyissä tutkimustiedoissa, jotka koskivat ipilimumabi–nivolumabi-yhdistelmähoitoa (kemoterapian kanssa tai ilman): uupumus (62 %), ihottuma (57 %), ripuli (52 %), pahoinvointi (42 %), kutina (40 %), kuume (36 %) ja päänsärky (26 %). Seuraavien haittavaikutusten ilmaantuvuus oli ≥ 10 % suurempi potilailla, jotka saivat yhdistelmähoitona ipilimumabia 1 mg/kg ja nivolumabia 360 mg sekä kemoterapiaa ei-pienisoluisen keuhkosyövän hoitoon, kuin ilmaantuvuus yhdistetyissä tutkimustiedoissa, jotka koskivat ipilimumabi–nivolumabi-yhdistelmähoitoa (kemoterapian kanssa tai ilman): anemia (32 %) ja neutropenia (15 %).
b. Haittavaikutustaulukko
Taulukossa 7 on esitetty yhdistetyistä tutkimustiedoista kootut haittavaikutukset potilailla, jotka saivat ipilimumabin ja nivolumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) (n = 2626), ja markkinoilletulon jälkeen ilmoitetut haittavaikutukset. Haittavaikutukset on esitetty elinjärjestelmittäin ja yleisyysluokittain. Yleisyysluokkien määritelmät ovat: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (ei voida arvioida markkinoilletulon jälkeen saatujen tietojen pohjalta). Haittavaikutukset on esitetty kussakin yleisyysluokassa vakavimmasta lievimpään.
Taulukko 7: Ipilimumabin ja muiden lääkeaineiden yhdistelmähoitoon liittyvät haittavaikutukset
| Yhdistelmähoito nivolumabin kanssa (kemoterapian kanssa tai ilman) | ||
|---|---|---|---|
Infektiot | |||
Hyvin yleinen | ylähengitystieinfektio | ||
Yleinen | keuhkokuume, keuhkoputkentulehdus, konjunktiviitti | ||
Harvinainen | aseptinen aivokalvotulehdus | ||
Veri ja imukudos | |||
Hyvin yleinen | anemiab,i, trombosytopeniab, leukopeniab, lymfosytopeniab, neutropeniab | ||
Yleinen | eosinofilia | ||
Melko harvinainen | kuumeinen neutropenia | ||
Tuntematon | hemofagosyyttinen lymfohistiosytoosi | ||
Immuunijärjestelmä | |||
Yleinen | infuusioreaktio (mukaan lukien sytokiinioireyhtymä), yliherkkyys | ||
Harvinainen | sarkoidoosi | ||
Tuntematon | hyljintäreaktio kiinteän elimen siirron jälkeenf | ||
Umpieritys | |||
Hyvin yleinen | kilpirauhasen vajaatoiminta | ||
Yleinen | kilpirauhasen liikatoiminta, kilpirauhastulehdus, lisämunuaisten vajaatoiminta, hypofysiitti, hypopituitarismi, diabetes mellitus | ||
Melko harvinainen | diabeettinen ketoasidoosi | ||
Harvinainen | lisäkilpirauhasten vajaatoiminta | ||
Aineenvaihdunta ja ravitsemus | |||
Hyvin yleinen | ruokahalun heikkeneminen, hyperglykemiab, hypoglykemiab | ||
Yleinen | elimistön kuivuminen, hypoalbuminemia, hypofosfatemia, painonlasku | ||
Melko harvinainen | metabolinen asidoosi | ||
Tuntematon | tuumorilyysioireyhtymäg | ||
Hermosto | |||
Hyvin yleinen | päänsärky | ||
Yleinen | huimaus, perifeerinen neuropatia | ||
Melko harvinainen | polyneuropatia, peroneuspareesi, autoimmuunineuropatia (mukaan lukien kasvo- ja loitontajahermon halvaus), aivotulehdus, myasthenia gravis, myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymäk | ||
Harvinainen | Guillain–Barrén oireyhtymä, neuriitti, myeliitti (mukaan lukien transversaalinen myeliitti) | ||
Silmät | |||
Yleinen | sumentunut näkö, kuivat silmät | ||
Melko harvinainen | uveiitti, episkleriitti | ||
Harvinainen | Vogt–Koyanagi–Haradan oireyhtymä, verkkokalvon nesteinen irtauma | ||
Sydän | |||
Yleinen | sydämen tiheälyöntisyys, eteisvärinä | ||
Melko harvinainen | myokardiittia, rytmihäiriö (mukaan lukien kammioperäinen rytmihäiriö)a, sydämen harvalyöntisyys | ||
Tuntematon | perikardiaaliset häiriöth | ||
Verisuonisto | |||
Yleinen | kohonnut verenpaine | ||
Hengityselimet, rintakehä ja välikarsina | |||
Hyvin yleinen | yskä, dyspnea | ||
Yleinen | pneumoniittia, keuhkoemboliaa, keuhkopussin nestekertymä | ||
Ruoansulatuselimistö | |||
Hyvin yleinen | ripuli, oksentelu, pahoinvointi, vatsakipu, ummetus | ||
Yleinen | koliittia, pankreatiitti, suutulehdus, gastriitti, suun kuivuminen | ||
Melko harvinainen | duodeniitti | ||
Harvinainen | suolen puhkeaminena, haiman eksokriininen vajaatoiminta, keliakia | ||
Maksa ja sappi | |||
Yleinen | maksatulehdus | ||
Iho ja ihonalainen kudos | |||
Hyvin yleinen | ihottumac, kutina | ||
Yleinen | alopesia, vitiligo, nokkosihottuma, kuiva iho, ihon punoitus | ||
Melko harvinainen | Stevens–Johnsonin oireyhtymä, erythema multiforme, psoriaasi, muut jäkälätauditj | ||
Harvinainen | toksinen epidermaalinen nekrolyysia,d, valkojäkälä | ||
Luusto, lihakset ja sidekudos | |||
Hyvin yleinen | muskuloskeletaalinen kipue, artralgia | ||
Yleinen | lihaskouristukset, lihasheikkous, artriitti | ||
Melko harvinainen | polymyalgia rheumatica, myopatia, myosiitti (mukaan lukien polymyosiitti)a | ||
Harvinainen | spondylartropatia, Sjögrenin oireyhtymä, rabdomyolyysia | ||
Munuaiset ja virtsatiet | |||
Yleinen | munuaisten vajaatoiminta (mukaan lukien akuutti munuaisvaurio)a | ||
Melko harvinainen | tubulointerstitiaalinen nefriitti, nefriitti | ||
Harvinainen | ei‑infektiivinen virtsarakkotulehdus | ||
Yleisoireet ja antopaikassa todettavat haitat | |||
Hyvin yleinen | uupumus, kuume, turvotus (mukaan lukien perifeerinen ödeema) | ||
Yleinen | rintakipu, kipu, vilunväristykset | ||
Tutkimukset | |||
Hyvin yleinen | lisääntynyt alkalinen fosfataasib, lisääntynyt ASATb, lisääntynyt ALATb, lisääntynyt kokonaisbilirubiinib, lisääntynyt kreatiniinib, lisääntynyt amylaasib, lisääntynyt lipaasib, hyponatremiab, hyperkalemiab, hypokalemiab, hyperkalsemiab, hypokalsemiab | ||
Yleinen | hypernatremiab, hypermagnesemiab, kilpirauhasta stimuloivan hormonin lisääntyminen, lisääntynyt gammaglutamyylitransferaasi | ||
Taulukossa 7 esitetyt yleisyysluokat eivät välttämättä liity täysin pelkästään ipilimumabiin tai ipilimumabin ja muiden lääkeaineiden yhdistelmähoitoihin, vaan niihin voivat vaikuttaa myös perussairaudet tai muut yhdistelmänä käytetyt lääkevalmisteet.
a Kuolemaan johtaneita tapauksia on ilmoitettu päättyneissä ja jatkuvissa kliinisissä tutkimuksissa.
b Yleisyys laskettiin sen mukaan, kuinka suurella osalla potilaista laboratorioarvo huononi lähtötasolta. Katso jäljempää ”Valikoitujen haittavaikutusten kuvaus, Poikkeavat laboratorioarvot”.
c Ihottuma on yhdistelmätermi, joka sisältää makulopapulaarisen ihottuman, punoittavan ihottuman, kutiavan ihottuman, follikulaarisen ihottuman, makulaarisen ihottuman, tuhkarokkomaisen ihottuman, näppyläisen ihottuman, märkärakkulaihottuman, näppyläisen ja suomuilevan ihottuman, vesirakkulaisen ihottuman, laajalle levinneen ihottuman, kesivän ihottuman, ihotulehduksen, aknea muistuttavan ihottuman, allergisen ihottuman, atooppisen ihottuman, rakkulaisen ihottuman, kesivän ihotulehduksen, psoriaasia muistuttavan ihottuman, lääkeihottuman, nodulaarisen ihottuman ja pemfigoidin.
d Ilmoitettu myös yhdistetyn aineiston ulkopuolisissa tutkimuksissa. Yleisyys perustuu kehitysohjelman laajuiseen altistukseen.
e Tuki- ja liikuntaelinten kipu on termi, joka kattaa selkäkivun, luukivun, muskuloskeletaalisen rintakivun, muskuloskeletaaliset vaivat, lihaskivun, kylkivälilihasten kivun, niskakivun, raajakivun ja selkärankakivun.
f Markkinoilletulon jälkeen (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet).
g Ilmoitettu kliinisissä tutkimuksissa ja valmisteen markkinoilletulon jälkeen.
h Perikardiaaliset häiriöt on termi, joka kattaa perikardiitin, perikardiaalisen effuusion, perikardiumin tamponaation ja Dresslerin oireyhtymän.
i Anemia on yhdistelmätermi, joka sisältää muiden syiden lisäksi hemolyyttisen anemian ja autoimmuunianemian, alhaisen hemoglobiinin, raudanpuuteanemian ja pienentyneen punasolumäärän.
j Jäkälätaudit on termi, joka kattaa jäkälän aiheuttaman keratoosin ja punajäkälän.
k Myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymän (ilmenee näistä joko kahden tai kaikkien kolmen sairauden limittymisenä) tapauksia, jotka ovat joskus johtaneet kuolemaan, on raportoitu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Valikoitujen haittavaikutusten kuvaus
Ellei toisin mainita, ipilimumabi-monoterapiaa koskevat tiedot perustuvat potilaisiin, jotka saivat joko ipilimumabia 3 mg/kg monoterapiana (n = 131) tai ipilimumabin (3 mg/kg) ja gp100-peptidirokotteen yhdistelmää (n = 380) faasin III tutkimuksessa, pitkälle edennyttä melanoomaa (jota ei voitu kirurgisesti poistaa tai joka oli metastasoitunut) sairastaneilla potilailla (MDX010-20, ks. kohta Farmakodynamiikka).
Ipilimumabi-yhdistelmähoitoon liittyy immuunivälitteisiä haittavaikutuksia. Immuunivälitteiset haittavaikutukset hävisivät useimmissa tapauksissa oikeanlaisella lääkehoidolla. Hoito piti yleensä lopettaa pysyvästi ipilimumabi–nivolumabi‑yhdistelmähoitoa saaneiden kohdalla useammin kuin pelkkää nivolumabi-monoterapiaa saaneiden kohdalla. Taulukossa 8 esitetään niiden potilaiden prosenttiosuus, joilla oli immuunivälitteisiä haittavaikutuksia ja joiden hoito lopetettiin pysyvästi. Lisäksi sellaisten potilaiden osalta, joilla oli haittatapahtuma, taulukossa 8 esitetään niiden potilaiden prosenttiosuus, jotka tarvitsivat suuriannoksista kortikosteroidihoitoa (joka vastasi vähintään 40 mg:aa prednisonia vuorokaudessa). Näiden haittavaikutusten hoito-ohjeet on kuvattu kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Taulukko 8: Immuunivälitteiset haittavaikutukset, joiden takia hoito lopetettiin pysyvästi tai potilas tarvitsi suuriannoksista kortikosteroidihoitoa
| Ipilimumabi yhdessä nivolumabin kanssa (kemoterapian kanssa tai ilman) | ||||
|---|---|---|---|---|---|
Immuunivälitteiset haittavaikutukset, joiden takia hoito lopetettiin pysyvästi | |||||
Pneumoniitti | 2,1 | ||||
Koliitti | 6 | ||||
Maksatulehdus | 5 | ||||
Munuaistulehdus ja munuaisten toimintahäiriö | 1,1 | ||||
Umpierityshäiriöt | 2,2 | ||||
Iho | 1,0 | ||||
Yliherkkyys-/infuusio- reaktio | 0,3 | ||||
Immuunivälitteiset haittavaikutukset, joiden takia potilas tarvitsi suuriannoksista kortikosteroidihoitoaa,b | |||||
Pneumoniitti | 59 | ||||
Koliitti | 32 | ||||
Maksatulehdus | 39 | ||||
Munuaistulehdus ja munuaisten toimintahäiriö | 27 | ||||
Umpierityshäiriöt | 18 | ||||
Iho | 8 | ||||
Yliherkkyys-/infuusioreaktio | 18 | ||||
a Hoito vastasi vähintään 40 mg:aa prednisonia vuorokaudessa.
b Yleisyys perustuu niiden potilaiden lukumäärään, jotka saivat immuunivälitteisen haittavaikutuksen.
Maha-suolikanavan immuunivälitteiset reaktiot
Ipilimumabiin liittyy maha-suolikanavan vakavia immuunivälitteisiä reaktioita. Maha-suolikanavan perforaatiosta johtuneita kuolemia raportoitiin alle 1 %:lla potilaista, jotka saivat ipilimumabin (3 mg/kg) ja gp100-peptidirokotteen yhdistelmää.
Ipilimumabia 3 mg/kg monoterapiana saaneiden ryhmässä raportoitiin vaikeudeltaan eriasteista ripulia 27 %:lla ja koliittia 8 %:lla potilaista. Sekä vaikeaa (asteen 3 tai 4) ripulia että vaikeaa (asteen 3 tai 4) koliittia ilmeni 5 %:lla potilaista. Mediaaniaika maha-suolikanavan vaikeiden tai kuolemaan johtaneiden (asteen 3–5) immuunivälitteisten reaktioiden ilmenemiselle oli 8 viikkoa (vaihteluväli 5–13 vk) hoidon aloittamisesta. Tutkimusprotokollassa määriteltyjen ohjeiden mukaisella hoidolla oireet lievittyivät (vaikeusaste ≤ 1 tai potilaan tila korjaantui lähtötasolle) useimmissa tapauksissa (90 %:lla) 4 viikossa (mediaani; vaihteluväli 0,6–22 vk) niiden alkamisesta. Kliinisissä tutkimuksissa immuunivälitteinen koliitti assosioitui näyttöön limakalvotulehduksesta, joka saattoi olla myös haavainen ja sisältää lymfosyytti- ja neutrofiili-infiltraatteja.
Immuunivälitteinen koliitti
Ipilimumabia ja nivolumabia yhdistelmähoitona (kemoterapian kanssa tai ilman) saaneilla potilailla ripulin tai koliitin ilmaantuvuus oli 26,0 % (682/2626). Potilaista 8,1 prosentilla (212/2626) ilmoitettiin vaikeusasteen 2 tapauksia, 6,4 prosentilla (167/2626) vaikeusasteen 3 tapauksia ja 0,2 prosentilla (4/2626) vaikeusasteen 4 tapauksia. Kahden potilaan (< 0,1 %) tapaus johti kuolemaan. Haittavaikutusten ilmenemisajan mediaani oli 1,4 kuukautta (vaihteluväli 0,0–48,9). Haittavaikutus korjaantui 618 potilaalla (91 %). Korjaantumisajan mediaani oli 2,9 viikkoa (vaihteluväli 0,1–170,0+). Ipilimumabin (3 mg/kg) ja nivolumabin (1 mg/kg) yhdistelmähoitoa melanooman hoitoon saaneilla potilailla ripulin tai koliitin ilmaantuvuus oli 46,7 %, johon sisältyi asteen 2 tapauksia (13,6 %), asteen 3 tapauksia (15,8 %) ja asteen 4 tapauksia (0,4 %).
Immuunivälitteinen keuhkotulehdus
Ipilimumabin ja nivolumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) saaneilla potilailla keuhkotulehduksen, mukaan lukien interstitiaalinen keuhkosairaus, ilmaantuvuus oli 6,0 % (157/2626). Potilaista 3,0 prosentilla (78/2626) ilmoitettiin vaikeusasteen 2 tapauksia, 1,0 prosentilla (27/2626) vaikeusasteen 3 tapauksia ja 0,3 prosentilla (8/2626) vaikeusasteen 4 tapauksia. Neljän potilaan (0,2 %) tapaus johti kuolemaan. Haittavaikutusten ilmenemisajan mediaani oli 2,7 kuukautta (vaihteluväli 0,1–56,8). Haittavaikutus korjaantui 129 potilaalla (82,2 %). Korjaantumisajan mediaani oli 6,1 viikkoa (vaihteluväli 0,1+–149,3+).
Immuunivälitteinen maksatoksisuus
Ipilimumabiin liittyy vakavaa immuunivälitteistä maksatoksisuutta. Kuolemaan johtanutta maksan vajaatoimintaa raportoitiin alle 1 %:lla potilaista, jotka saivat ipilimumabia monoterapiana annoksella 3 mg/kg.
ASAT-arvon eriasteista suurenemista raportoitiin 1 %:lla potilaista ja ALAT-arvon suurenemista 2 %:lla potilaista. Yhdelläkään potilaalla ei raportoitu ASAT- eikä ALAT-arvon vaikea-asteista (asteen 3 tai 4) suurenemista. Keskivaikea, vaikea tai kuolemaan johtava (asteen 2–5) immuunivälitteinen maksatoksisuus ilmeni 3–9 viikon kuluttua hoidon aloittamisesta. Tutkimusprotokollassa määriteltyjen ohjeiden mukaisella hoidolla oireet korjaantuivat 0,7–2 viikossa. Kliinisissä tutkimuksissa immuunivälitteistä maksatoksisuutta saaneiden potilaiden maksanäytteissä havaittiin merkkejä akuutista tulehduksesta (neutrofiilit, lymfosyytit ja makrofagit).
Ipilimumabiin liittyvää immuunivälitteistä maksatoksisuutta ilmeni useammin potilailla, jotka olivat saaneet ipilimumabia suositeltua suuremman annoksen ja dakarbatsiinia kuin pelkkää ipilimumabia 3 mg/kg saaneilla potilailla.
Ipilimumabin ja nivolumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) saaneilla potilailla maksan toimintakokeiden poikkeavien tulosten ilmaantuvuus oli 21,2 % (556/2626). Potilaista 5,0 prosentilla (132/2626) ilmoitettiin vaikeusasteen 2 tapauksia, 8,3 prosentilla (218/2626) vaikeusasteen 3 tapauksia ja 1,3 prosentilla (34/2626) vaikeusasteen 4 tapauksia. Seitsemän potilaan (0,3 %) tapaus johti kuolemaan. Haittavaikutusten ilmenemisajan mediaani oli 1,5 kuukautta (vaihteluväli 0,0–36,6). Haittavaikutus korjaantui 482 potilaalla (87,0 %). Korjaantumisajan mediaani oli 5,9 viikkoa (vaihteluväli 0,1–175,9+). Ipilimumabin (3 mg/kg) ja nivolumabin (1 mg/kg) yhdistelmähoitoa melanooman hoitoon saaneilla potilailla maksan toimintakokeiden poikkeavien tulosten ilmaantuvuus oli 30,1 %. Näistä asteen 2 tapauksia oli 6,9 %, asteen 3 tapauksia 15,8 % ja asteen 4 tapauksia 1,8 %. Ipilimumabin (3 mg/kg) ja nivolumabin (1 mg/kg) yhdistelmähoitoa hepatosellulaarisen karsinooman hoitoon saaneilla potilailla maksan toimintakokeiden poikkeavien tulosten ilmaantuvuus oli 34,3 %. Näistä asteen 2 tapauksia oli 8,4 %, asteen 3 tapauksia 14,2 % ja asteen 4 tapauksia 2,7 %.
Immuunivälitteiset ihohaitat
Ipilimumabiin liittyy ihon vakavia, mahdollisesti immuunivälitteisiä haittavaikutuksia. Kuolemaan johtavaa toksista epidermaalista nekrolyysiä (mukaan lukien Stevens–Johnsonin oireyhtymä) raportoitiin alle 1 %:lla potilaista, jotka saivat ipilimumabin ja gp100-peptidirokotteen yhdistelmää (ks. kohta Farmakodynamiikka). Kliinisissä tutkimuksissa ja myyntiluvan jälkeisessä käytössä on ipilimumabilla harvoin raportoitu yleisoireista eosinofiilistä reaktiota (Drug Reaction with Eosinophilia and Systemic Symptoms, DRESS). Markkinoilletulon jälkeisessä käytössä on ilmoitettu satunnaisia pemfigoiditapauksia.
Ipilimumabia 3 mg/kg monoterapiana saaneiden ryhmässä raportoitiin eriasteista ihottumaa ja kutinaa kumpaakin 26 %:lla potilaista. Ipilimumabin aiheuttamat ihottuma ja kutina olivat pääasiassa lieviä (aste 1) tai keskivaikeita (aste 2) ja reagoivat hyvin oireenmukaiseen hoitoon. Mediaaniaika ihon keskivaikeiden, vaikeiden tai kuolemaan johtavien haittavaikutusten (asteen 2–5) ilmenemiseen oli 3 viikkoa (vaihteluväli 0,9–16 vk) hoidon aloittamisesta. Tutkimusprotokollassa määriteltyjen ohjeiden mukaisella hoidolla oireet korjaantuivat useimmissa tapauksissa (87 %:lla) mediaaniajan korjautumiseen ollessa viisi viikkoa (vaihteluväli 0,6–29 vk).
Ipilimumabin ja nivolumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) saaneilla potilailla ihottuman ilmaantuvuus oli 46,1 % (1210/2626). Potilaista 14,3 prosentilla (375/2626) ilmoitettiin vaikeusasteen 2 tapauksia, 4,6 prosentilla (120/2626) vaikeusasteen 3 tapauksia ja 0,1 prosentilla (3/2626) vaikeusasteen 4 tapauksia. Haittavaikutusten ilmenemisajan mediaani oli 0,7 kuukautta (vaihteluväli 0,0–33,8). Haittavaikutus korjaantui 843 potilaalla (70 %). Korjaantumisajan mediaani oli 12,1 viikkoa (vaihteluväli 0,1–268,7+). Ipilimumabin (3 mg/kg) ja nivolumabin (1 mg/kg) yhdistelmähoitoa melanooman hoitoon saaneilla potilailla ihottuman ilmaantuvuus oli 65,2 %, johon sisältyi vaikeusasteen 2 tapauksia (20,3 %) ja vaikeusasteen 3 tapauksia (7,8 %).
Hermoston immuunivälitteiset reaktiot
Ipilimumabiin liittyy hermoston vakavia immuunivälitteisiä reaktioita. Kuolemaan johtanutta Guillain-Barrén oireyhtymää raportoitiin alle 1 %:lla potilaista, jotka saivat ipilimumabin (3 mg/kg) ja gp100-peptidirokotteen yhdistelmää. Kliinisissä tutkimuksissaraportoitiin myasthenia gravis -tyyppisiä oireita alle 1 %:lla potilaista, jotka saivat tutkimuksissa tätä suurempia ipilimumabiannoksia.
Immuunivälitteinen munuaistulehdus ja munuaisten toimintahäiriö
Ipilimumabin ja nivolumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) saaneilla potilailla munuaistulehduksen tai munuaisten toimintahäiriön ilmaantuvuus oli 5,4 % (141/2626). Potilaista 2,0 prosentilla (52/2626) ilmoitettiin vaikeusasteen 2 tapauksia, 0,8 prosentilla (21/2626) vaikeusasteen 3 tapauksia ja 0,4 prosentilla (11/2626) vaikeusasteen 4 tapauksia. Kahden potilaan (< 0,1 %) tapaus johti kuolemaan. Haittavaikutusten ilmenemisajan mediaani oli 2,6 kuukautta (vaihteluväli 0,0–34,8). Haittavaikutus korjaantui 110 potilaalla (78,0 %). Korjaantumisajan mediaani oli 5,9 viikkoa (vaihteluväli 0,1–172,1+).
Immuunivälitteiset umpierityshäiriöt
Ipilimumabia 3 mg/kg monoterapiana saaneiden ryhmässä raportoitiin eriasteista hypopituitarismia 4 %:lla potilaista. Potilaista 2 %:lla raportoitiin eriasteista lisämunuaisten vajaatoimintaa, kilpirauhasen liikatoimintaa ja kilpirauhasen vajaatoimintaa. Vaikeaa (asteen 3 tai 4) hypopituitarismia raportoitiin 3 %:lla potilaista. Keskivaikeat, vaikeat ja hyvin vaikeat (asteen 2–4) immuunivälitteiset umpierityshäiriöt ilmenivät 7–20 viikon kuluessa hoidon aloittamisesta. Kliinisissä tutkimuksissa havaitut immuunivälitteiset umpierityshäiriöt saatiin yleensä hallintaan hormonikorvaushoidolla.
Ipilimumabin ja nivolumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) saaneilla potilailla kilpirauhasen toimintahäiriöiden ilmaantuvuus oli 23,2 % (608/2626). Potilaista 12,7 prosentilla (333/2626) ilmoitettiin vaikeusasteen 2 kilpirauhasen toimintahäiriöitä ja 1,0 prosentilla (27/2626) vaikeusasteen 3 kilpirauhasen toimintahäiriöitä. Potilaista 1,9 prosentilla (49/2626) ilmoitettiin vaikeusasteen 2 hypofysiittitapauksia (mukaan lukien lymfosyyttista hypofysiittia) ja 1,5 prosentilla (40/2626) vaikeusasteen 3 hypofysiittitapauksia. Vaikeusasteen 2 hypopituitarismia ilmeni 0,6 prosentilla potilaista (16/2626) ja vaikeusasteen 3 hypopituitarismia 0,5 prosentilla (13/2626) potilaista. Vaikeusasteen 2 lisämunuaisten vajaatoimintaa (mukaan lukien lisämunuaiskuoren sekundaarista vajaatoimintaa, akuuttia lisämunuaiskuoren vajaatoimintaa, pienentynyttä veren kortikotropiinipitoisuutta ja immuunivälitteistä lisämunuaisten vajaatoimintaa) ilmoitettiin 2,7 prosentilla (72/2626) potilaista, vaikeusasteen 3 vajaatoimintaa ilmoitettiin 1,6 prosentilla (43/2626) ja vaikeusasteen 4 vajaatoimintaa ilmoitettiin 0,2 prosentilla (4/2626) potilaista. Asteen 1 diabetes mellitusta (mukaan lukien tyypin 1 diabetes mellitus ja diabeettinen ketoasidoosi) esiintyi < 0,1 prosentilla (1/2626), asteen 2 diabetes mellitusta 0,3 prosentilla (8/2626), asteen 3 diabetes mellitusta 0,3 prosentilla (7/2626) ja asteen 4 diabetes mellitusta 0,2 prosentilla (6/2626) potilaista. Näiden umpierityshäiriöiden ilmenemisajan mediaani oli 2,1 kuukautta (vaihteluväli 0,0–28,1). Haittavaikutus korjaantui 297 potilaalla (40,0 %). Korjaantumisajan vaihteluväli oli 0,3–257,1+ viikkoa.
Infuusioreaktiot
Ipilimumabin ja nivolumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) saaneilla potilailla yliherkkyys-/infuusioreaktioiden ilmaantuvuus oli 4,5 % (118/2626). Potilaista 1,9 prosentilla (49/2626) ilmoitettiin vaikeusasteen 1 tapauksia; 2,4 prosentilla (62/2626) vaikeusasteen 2 tapauksia, 0,2 prosentilla (6/2626) vaikeusasteen 3 tapauksia ja < 0,1 prosentilla (1/2626) vaikeusasteen 4 tapauksia. Ipilimumabia (1 mg/kg) ja nivolumabia (3 mg/kg) yhdistelmähoitona keuhkopussin pahanlaatuisen mesoteliooman (MPM) hoitoon saaneilla potilailla yliherkkyys-/infuusioreaktioiden ilmaantuvuus oli 12 %.
Immunogeenisuus
Faasien II ja III kliinisissä tutkimuksissa, joissa ipilimumabia saivat pitkälle edennyttä melanoomaa sairastaneet potilaat, vasta-aineita ipilimumabille kehittyi alle 2 %:lle potilaista. Yhdelläkään ei ilmennyt infuusioon liittyviä tai infuusiota edeltäviä tai sen jälkeisiä yliherkkyysreaktioita tai anafylaktisia reaktioita. Ipilimumabia neutraloivia vasta-aineita ei havaittu. Vasta-ainemuodostuksen ja haittavaikutusten välillä ei kaiken kaikkiaan havaittu ilmeistä yhteyttä.
Niistä potilaista, jotka saivat ipilimumabi–nivolumabi-yhdistelmähoitoa ja joilta voitiin arvioida ipilimumabin vasta-aineiden esiintyvyys, 6,3–13,7 %:lle kehittyi ipilimumabin vasta-aineita. 0–0,4 %:lle kehittyi ipilimumabia neutraloivia vasta-aineita. Niistä potilaista, jotka saivat ipilimumabin, nivolumabin ja kemoterapian yhdistelmähoitoa ja joilta voitiin arvioida ipilimumabin vasta-aineiden tai ipilimumabia neutraloivien vasta-aineiden esiintyvyys, 7,5 %:lle kehittyi ipilimumabin vasta-aineita ja 1,6 %:lle kehittyi ipilimumabia neutraloivia vasta-aineita. Potilaista, joilta voitiin arvioida nivolumabin vasta-aineiden esiintyvyys, nivolumabia (3 mg/kg) ja ipilimumabia (1 mg/kg) yhdistelmähoitona joka kolmas viikko saaneista 26,0 %:lle kehittyi nivolumabin vasta-aineita. Nivolumabia (3 mg/kg) joka toinen viikko ja ipilimumabia (1 mg/kg) joka kuudes viikko saaneista 24,9 %:lle kehittyi nivolumabin vasta-aineita. Nivolumabia (1 mg/kg) ja ipilimumabia (3 mg/kg) yhdistelmähoitona joka kolmas viikko saaneista 37,8 %:lle kehittyi nivolumabin vasta-aineita. Nivolumabia (360 mg joka kolmas viikko), ipilimumabia (1 mg/kg joka kuudes viikko) ja kemoterapiaa yhdistelmähoitona saaneista 33,8 %:lle kehittyi nivolumabin vasta-aineita. Nivolumabia (3 mg/kg) ja ipilimumabia (1 mg/kg) yhdistelmähoitona joka kolmas viikko saaneista 0,8 %:lle kehittyi nivolumabia neutraloivia vasta-aineita. Nivolumabia (3 mg/kg) joka toinen viikko ja ipilimumabia (1 mg/kg) joka kuudes viikko yhdistelmähoitona saaneista 1,5 %:lle kehittyi nivolumabia neutraloivia vasta-aineita. Nivolumabia (1 mg/kg) ja ipilimumabia (3 mg/kg) yhdistelmähoitona joka kolmas viikko saaneista 4,6 %:lle kehittyi nivolumabia neutraloivia vasta-aineita. Nivolumabia (360 mg joka kolmas viikko), ipilimumabia (1 mg/kg joka kuudes viikko) ja kemoterapiaa yhdistelmähoitona saaneista 2,6 %:lle kehittyi nivolumabia neutraloivia vasta-aineita.
Yhdistelmähoidossa nivolumabin kanssa ipilimumabin puhdistuma pysyi ipilimumabin vasta-aineiden läsnäollessa samana, eikä toksisuusprofiilin muutoksista ollut merkkejä.
Poikkeavat laboratorioarvot
Ipilimumabia ja nivolumabia yhdistelmähoitona (kemoterapian kanssa tai ilman) saaneista potilaista niiden potilaiden osuudet, joilla laboratorioarvo muuttui lähtötasolta asteen 3 tai 4 poikkeavuudeksi, olivat seuraavat: anemia 4,8 %, trombosytopenia 1,8 %, leukopenia 2,2 %, lymfosytopenia 6,9 %, neutropenia 3,3 %, lisääntynyt alkalinen fosfataasi 2,7 %, lisääntynyt ASAT 9,8 %, lisääntynyt ALAT 9,3 %, lisääntynyt kokonaisbilirubiini 2,3 %, lisääntynyt kreatiniini 1,8 %, hypoalbuminemia 1,4 %, hyperglykemia 7,1 %, hypoglykemia 0,7 %, lisääntynyt amylaasi 7,8 %, lisääntynyt lipaasi 16,3 %, hypokalsemia 0,8 %, hypernatremia 0,2 %, hyperkalsemia 0,8 %, hyperkalemia 2,0 %, hypermagnesemia 0,8 %, hypomagnesemia 0,4 %, hypokalemia 3,0 % ja hyponatremia 8,7 %.
Ipilimumabin (3 mg/kg) ja nivolumabin (1 mg/kg) yhdistelmähoitoa melanooman hoitoon saaneilla potilailla lisääntynyt ALAT-arvo muuttui suuremmalla osalla lähtötasosta asteeseen 3 tai 4 (15,3 %).
Pediatriset potilaat
Ipilimumabi monoterapiana
Uusia haittavaikutuksia ei raportoitu 12-vuotiailla ja sitä vanhemmilla nuorilla.
CA184070‑tutkimuksessa asteen 3 tai vakavampia immuunivälitteisiä haittavaikutuksia ei raportoitu ainoallakaan 12-vuotiaalla tai sitä vanhemmalla potilaalla, joka sai ipilimumabia 3 mg/kg. Asteen 3 tai 4 haittatapahtumia raportoitiin kahdella (25,0 %) kahdeksasta potilaasta, jotka saivat 5 mg/kg:n annosta, ja yhdellä (11,1 %) yhdeksästä potilaasta, jotka saivat 10 mg/kg:n annosta. Yksikään haittatapahtumista ei johtanut kuolemaan. Immuunivälitteiset haittavaikutukset olivat yhteneväisiä aikuisista saatujen tietojen kanssa. Kaikissa ryhmissä useimmin raportoidut immuunivälitteiset haittavaikutukset olivat maha-suolikanavan haittavaikutukset (0 [3 mg/kg], 62,5 % [5 mg/kg], ja 44,4 % [10 mg/kg]), maksan toimintaan liittyvät haittavaikutukset (0 [3 mg/kg], 75,0 % [5 mg/kg], 33,3 % [10 mg/kg]) ja ihoon liittyvät haittavaikutukset (0 [3 mg/kg], 25,0 % [5 mg/kg], 33,3 % [10 mg/kg]). Tutkimuksessa ei havaittu uusia tai odottamattomia immuunivälitteisiä haittavaikutuksia. Raportoiduissa immuunivälitteisissä haittavaikutuksissa ei ollut selviä eroja aikuisten ja pediatristen potilaiden välillä.
CA184178‑tutkimuksessa ei havaittu uusia tai odottamattomia immuunivälitteisiä haittavaikutuksia. Havaittujen haittavaikutusten esiintymistiheys, vakavuusaste ja esiintymispaikka olivat samankaltaisia kuin aikuisilla tehdyissä tutkimuksissa. 10 mg/kg saaneessa ryhmässä kaksi potilasta sai tutkimuksen aikana asteen 1 ja asteen 3 umpieritykseen liittyvän immuunivälitteisen haittavaikutuksen, joka oli hyperglykemia. Muita umpieritykseen liittyviä poikkeavuuksia ei raportoitu.
Tiivistelmä 12-vuotiaiden ja sitä vanhempien nuorten sekä aikuisten haittavaikutuksista on esitetty taulukossa 9.
Taulukko 9: Tiivistelmä haittavaikutuksista korkeintaan neljän annoksen (3, 5 ja 10 mg/kg) jälkeen, kaikki hoidetut potilaat
| Potilaiden määrä (%) | ||||||||
Ikä ≥ 12–21 vuotta | Ikä 12 – alle 18 vuotta | Aikuiset | |||||||
Edennyt melanooma ja ei‑melanoottiset kiinteät kasvaimet | Edennyt melanooma | Edennyt melanooma | |||||||
CA184070 | CA184178 | CA184004/ | CA184004/007 | ||||||
3 mg/kg | 5 mg/kg | 10 mg/kg | 3 mg/kg | 10 mg/kg | 3 mg/kg | 10 mg/kg | |||
n = 1 | n = 8 | n = 9 | n = 4 | n = 8 | n = 111 | n = 325 | |||
Kaikki kuolemat, n (%) | 1 (100,0) | 4 (50,0) | 2 (22,2) | 2 (50,0) | 3 (37,5) | 26 (23,4) | 71 (21,8) | ||
Hoitoon liittyvät kuolemat, n (%) | 0 | 0 | 0 | 0 | 0 | 2 (1,8) | 6 (1,8) | ||
Vakavat haittatapahtumat, n (%) | 1 (100,0) | 7 (87,5) | 4 (44,4) | 1 (25,0) | 6 (75,0) | 50 (45,0) | 168 (51,7) | ||
Vakavat lääkkeeseen liittyvät haittatapahtumat, n (%) | 1 (100,0) | 5 (62,5) | 4 (44,4) | 1 (25,0) | 5 (62,5) | 19 (17,1) | 95 (29,2) | ||
Haittatapahtumat, jotka johtivat tutkimuslääkkeen käytön keskeyttämiseen, n (%) | 0 | 3 (37,5) | 2 (22,2) | 1 (25,0) | 5 (62,5) | 12 (10,8) | 88 (27,1) | ||
Lääkkeeseen liittyvät haittatapahtumat, jotka johtivat tutkimuslääkkeen käytön keskeyttämiseen, n (%) | 0 | 3 (37,5) | 2 (22,2) | 1 (25,0) | 5 (62,5) | 9 (8,1) | 61 (18,8) | ||
Immuunivälitteiset haittatapahtumat, n (%) | 1 (100,0) | 7 (87,5) | 7 (77,8) | 2 (50,0) | 4 (50,0) | 68 (61,3) | 234 (72,0) | ||
Haittatapahtumat, n (%) | 1 (100,0) | 8 (100,0) | 9 (100,0) | 4 (100,0) | 8 (100,0) | 108 (97,3) | 315 (96,9) | ||
Lääkkeeseen liittyvät haittatapahtumat, n (%) | 1 (100,0) | 7 (87,5) | 9 (100,0) | 2 (50,0) | 7 (87,5) | 88 (79,3) | 274 (84,3) | ||
CA184070-tutkimuksessa MedDRAn versio 17.0, CA184178-tutkimuksessa versio 19.0 ja aikuisten yhdistetyissä turvallisuustiedoissa versio 12.1. NA = not assessed, ei arvioitu
Aikuisilla raportoidut kuolemantapaukset tapahtuivat 70 päivän kuluessa viimeisen annoksen saamisesta riippumatta siitä, liittyikö kuolema tutkimukseen. Pediatristen potilaiden kuolemantapaukset ovat tutkimuksen aikana raportoituja tapahtumia, jotka esiintyivät 30 päivän kuluessa viimeisen annoksen saamisesta, lukuun ottamatta riviä ”Kaikki kuolemat”, johon liittyvät kuolemantapaukset tapahtuivat yli 30 päivää viimeisen annoksen saamisen jälkeen.
CA184178-tutkimuksessa kuolemantapaukset raportoitiin vähintään 90 päivän ajalta viimeisen annoksen jälkeen.
CA184178-tutkimuksessa ja aikuisten yhdistetyissä turvallisuustiedoissa ipilimumabin vaikutuksen raportoitiin olevan mahdollinen, todennäköinen, varma tai puuttuva ja CA184070-tutkimuksessa kuolemaan liittyvä tai puuttuva.
Ipilimumabi yhdistelmähoitona nivolumabin kanssa
Ipilimumabin turvallisuutta (1 mg/kg 3 viikon välein) yhdistelmähoitona nivolumabin kanssa (1 mg/kg tai 3 mg/kg 4 ensimmäistä annosta, minkä jälkeen nivolumabia 3 mg/kg monoterapiana 2 viikon välein) arvioitiin kliinisessä CA209070-tutkimuksessa 33 pediatrisella potilaalla, jotka olivat ≥ 1–< 18 vuoden ikäisiä (mukaan lukien 20 potilasta, jotka olivat 12–< 18 vuoden ikäisiä) ja joilla oli uusiutunut tai hoitoon vastaamaton kiinteä tai hematologinen kasvain, edennyt melanooma mukaan lukien. Pediatristen potilaiden turvallisuusprofiili oli yleisesti ottaen samankaltainen kuin ipilimumabia yhdistelmähoitona nivolumabin kanssa saaneilla aikuisilla. Uusia turvallisuussignaaleja ei havaittu.
Ipilimumabi–nivolumabi-yhdistelmähoidon yleisimmät haittavaikutukset (ilmoitettiin vähintään 20 %:lla pediatrisista potilaista) olivat uupumus (33,3 %) ja makulopapulaarinen ihottuma (21,2 %). Suurin osa ipilimumabi–nivolumabi-yhdistelmähoidon haittavaikutuksista oli vaikeusastetta 1 tai 2. Kymmenellä potilaalla (30 %) oli yksi tai useampi vaikeusasteen 3 tai 4 haittavaikutus.
Kliinisessä CA209908-tutkimuksessa, johon osallistui 74 korkean asteen primaarista keskushermoston syöpää sairastavaa pediatrista potilasta, ei havaittu uusia turvallisuussignaaleja (ks. kohta Farmakodynamiikka) verrattuna aikuispotilaista saatuihin tietoihin kaikista indikaatioista.
Iäkkäät
Keuhkopussin pahanlaatuista mesotelioomaa sairastavista potilaista 75-vuotiailla ja sitä vanhemmilla oli enemmän vakavia haittavaikutuksia (68 %) ja lääkitys lopetettiin useammin haittavaikutusten vuoksi (35 %) verrattuna kaikkiin nivolumabi–ipilimumabi-yhdistelmähoitoa saaneisiin potilaisiin, joista vakavia haittavaikutuksia oli 54 %:lla ja joista lääkitys lopetettiin haittavaikutusten vuoksi 28 %:lla. 75-vuotiaista ja sitä vanhemmista dMMR- tai MSI-H-tyyppistä kolorektaalisyöpää sairastavista potilaista on vain vähän tietoa (ks. kohta Farmakodynamiikka). Hepatosellulaarista karsinoomaa sairastavista potilaista 75‑vuotiailla ja sitä vanhemmilla oli enemmän vakavia haittavaikutuksia (67 %) ja lääkitys lopetettiin useammin haittavaikutusten vuoksi (35 %) kuin yhteensä kaikilla ipilimumabi–nivolumabi-yhdistelmähoitoa saaneilla potilailla, joista 53 %:lla oli vakavia haittavaikutuksia ja lääkitys lopetettiin haittavaikutusten vuoksi 27 %:lla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Ipilimumabille ei ole määritetty suurinta siedettyä annosta. Kliinisissä tutkimuksissa annettu enimmäisannos 20 mg/kg ei aiheuttanut ilmeisiä toksisia vaikutuksia.
Yliannostustapauksessa potilasta on seurattava tarkasti haittavaikutusten merkkien tai oireiden varalta ja aloitettava sopiva oireemukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut syöpälääkkeet, monoklonaaliset vasta-aineet
ATC-koodi: L01XC11
Vaikutusmekanismi
Sytotoksinen T‑soluantigeeni‑4 (CTLA‑4) on keskeinen T‑solujen aktiivisuuden säätelijä. Ipilimumabi on CTLA‑4-immuunijärjestelmän tarkistuspisteen estäjä, joka salpaa CTLA‑4-reitin indusoiman T‑solujen inhibitorisen signaalin. Tämä lisää reaktiivisten T‑efektorisolujen määrää, mikä käynnistää välittömän T‑solujen immuunipuolustuksen nousun syöpäsoluja vastaan. CTLA‑4-salpaus voi myös vähentää säätelijä‑T‑solujen toimintaa, mikä voi olla osallisena kasvaimen kasvua ehkäisevään immuunivasteeseen. Ipilimumabi voi selektiivisesti kuluttaa säätelijä‑T‑solut loppuun kasvaimen kohdalla. Tämä aiheuttaa kohonneen kasvaimensisäisen T‑efektorisolu/säätelijä‑Tsolu suhteen, mikä johtaa syöpäsolun tuhoutumiseen.
Farmakodynaamiset vaikutukset
Ipilimumabia saaneilla melanoomapotilailla ääreisverenkierron lymfosyyttien absoluuttisen määrän keskiarvo kohosi koko hoitojakson ajan. Faasin II tutkimuksissa lymfosyyttien absoluuttisen määrän keskiarvo ääreisverenkierrossa suureni annoksesta riippuvaisesti. MDX010-20-tutkimuksessa (ks. kohta Farmakodunamiikka) ipilimumabiannos 3 mg/kg, joko monoterapiana tai yhdessä gp100-peptidirokotteen kanssa, suurensi lymfosyyttien absoluuttisen määrän keskiarvoa ääreisverenkierrossa koko hoitojakson ajan. Sen sijaan verrokkiryhmässä, joka sai tutkimusvaiheessa olevaa gp100-peptidirokotetta monoterapiana, ei todettu merkitsevää muutosta ääreisverenkierron lymfosyyttien absoluuttisen määrän keskiarvossa.
Melanoomapotilaiden ääreisverenkierrossa aktivoituneiden HLA-DR+-, CD4-positiivisten ja CD8-positiivisten T-solujen osuus (%) suureni keskimäärin ipilimumabihoidon jälkeen, mikä on yhdenmukaista ipilimumabin vaikutusmekanismin kanssa. Ipilimumabihoidon lopettamisen jälkeen huomattiin myös sentraalisiin T-muistisoluihin (CCR7+-, CD45RA-) lukeutuvien CD4-positiivisten ja CD8-positiivisten T-solujen prosenttiosuuden keskimäärin lisääntyneen ja efektorimuistisoluihin lukeutuvien (CCR7-, CD45RA-) CD8-positiivisten T-solujen lisääntyneen (%) keskimäärin vähemmän mutta kuitenkin tilastollisesti merkitsevästi.
Kliininen teho ja turvallisuus
Ipilimumabi yhdistelmähoitona nivolumabin kanssa
Katso nivolumabin valmisteyhteenvedosta lisätietoa kliinisestä tehosta ja turvallisuudesta liittyen nivolumabin suositusannoksiin, kun nivolumabia annetaan monoterapiana nivolumabi–ipilimumabi-yhdistelmähoidon jälkeen.
Annoksen/altistuksen tehon ja turvallisuuden suhteiden mallintamisen perusteella nivolumabiannosten 240 mg 2 viikon välein tai 3 mg/kg 2 viikon välein välillä ei ole kliinisesti merkittäviä eroja tehossa ja turvallisuudessa. Lisäksi näiden suhteiden perusteella nivolumabiannosten 480 mg 4 viikon välein tai 3 mg/kg 2 viikon välein välillä ei ole kliinisesti merkittäviä eroja hoidettaessa edennyttä melanoomaa ja munuaiskarsinoomaa.
Kliiniset ipilimumabi-monoterapiatutkimukset
Melanooma
Kokonaiselinaikahyöty (overall survival, OS) ipilimumabin suositellulla annoksella (3 mg/kg) on osoitettu faasin III tutkimuksessa (MDX010-20) pitkälle edenneessä melanoomassa (jota ei voitu kirurgisesti poistaa tai joka oli metastasoitunut), johon oli annettu jo muuta aiempaa hoitoa. MDX010‑20‑tutkimukseen ei otettu mukaan potilaita, joilla oli silmämelanooma, primaarinen keskushermoston melanooma, aktiivisia aivojen etäpesäkkeitä, HI-virus tai hepatiitti B tai C. Kliinisiin tutkimuksiin ei otettu mukaan potilaita, joiden ECOG-suorituskykyluokka oli > 1 tai joilla oli limakalvon melanooma. Kliinisiin tutkimuksiin ei myöskään otettu potilaita joiden ASAT-lähtöarvo oli > 2,5 x ULN ilman etäpesäkkeitä maksassa; tai potilaita, joilla oli etäpesäkkeitä maksassa ja joiden ASAT-lähtöarvo oli > 5 x ULN; eikä potilaita, joiden kokonaisbilirubiinin lähtöarvo oli ≥ 3 x ULN.
Potilaat, joilla on aiemmin ollut jokin autoimmuunisairaus, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
MDX010-20
Faasin III kaksoissokkotutkimukseen otettiin edennyttä melanoomaa (jota ei voitu kirurgisesti poistaa tai joka oli metastasoitunut) sairastaneita potilaita, jotka olivat aiemmin saaneet vähintään yhtä seuraavista hoidoista: IL-2, dakarbatsiini, temotsolomidi, fotemustiini tai karboplatiini. Potilaat satunnaistettiin suhteessa 3:1:1 saamaan joko ipilimumabia (3 mg/kg) ja tutkimusvaiheessa olevaa gp100-peptidirokotetta yhdistelmähoitona, ipilimumabia 3 mg/kg monoterapiana tai gp100-peptidirokotetta monoterapiana. Kaikki potilaat olivat HLA-A2*0201-kudostyyppiä. Tämä HLA-tyyppi tukee gp100:n immuunipresentaatiota. Potilaan BRAF-mutaatiostatus lähtötilanteessa ei vaikuttanut tutkimukseen mukaan ottamiseen. Potilaat saivat ipilimumabia 3 viikon välein yhteensä 4 annosta, jos sietivät hoidon (aloitushoito). Ne potilaat, joiden kasvainkuorma kasvoi selkeästi ennen hoitojakson päättymistä, jatkoivat hoitoa, jos sietivät sen ja jos heidän toimintakykynsä oli riittävä. Hoitovaste ipilimumabihoitoon arvioitiin hoitojakson jälkeen noin viikolla 12.
Ipilimumabin lisähoitoa (uusintahoitoa) tarjottiin potilaille, joiden sairaus eteni alkuperäisen kliinisen vasteen (osittainen tai täydellinen vaste) tai stabiilin sairausvaiheen jälkeen (modifioitujen WHO-kriteerien perusteella) > 3 kuukauden kuluttua ensimmäisestä hoidon vastearviosta. Ensisijainen päätetapahtuma oli kokonaiselinaika ipilimumabin ja gp100-peptidirokotteen yhdistelmää saaneessa ryhmässä verrattuna gp100-peptidirokotetta monoterapiana saaneeseen ryhmään. Keskeiset toissijaiset päätetapahtumat olivat kokonaiselinaika ipilimumabin ja gp100-peptidirokotteen yhdistelmää saaneessa ryhmässä verrattuna ipilimumabia monoterapiana saaneeseen ryhmään sekä kokonaiselinaika ipilimumabia monoterapiana saaneessa ryhmässä verrattuna gp100-peptidirokotetta monoterapiana saaneeseen ryhmään.
Tutkimuksessa satunnaistettiin yhteensä 676 potilasta: 137 ipilimumabiryhmään, 403 ipilimumabin ja gp100-peptidirokotteen yhdistelmää saaneeseen ryhmään ja 136 gp100-peptidirokoteryhmään. Suurin osa potilaista oli saanut aloitushoidossa kaikki 4 annosta. Potilaista 32 sai uusintahoidon: 8 ipilimumabiryhmässä, 23 ipilimumabin ja gp100-peptidirokotteen yhdistelmää saaneessa ryhmässä ja 1 gp100-peptidirokoteryhmässä. Seuranta–aika oli enintään 55 kuukautta. Lähtötilanteessa ryhmät olivat keskenään vertailukelpoisia. Mediaani-ikä oli 57 vuotta. Suurimmalla osalla (71–73 %) oli M1c-vaiheen sairaus ja laktaattidehydrogenaasin (LDH) lähtöarvo oli suurentunut 37–40 %:lla tutkittavista. Yhteensä 77 potilasta oli saanut aiempaa hoitoa aivojen etäpesäkkeisiin.
Kokonaiselinaika oli tilastollisesti merkitsevästi parempi ipilimumabiryhmissä verrattuna gp100-verrokkiryhmään. Vertailtaessa kokonaiselinaikaa pelkän ipilimumabin ja gp100-peptidirokotteen välillä riskitiheyssuhde (hazard ratio, HR) oli 0,66 (95 %:n luottamusväli; 0,51–0,87; p = 0,0026).
Alaryhmäanalyysissa kokonaiselinajan suhteen havaittu hyöty oli yhdenmukainen suurimmassa osassa alaryhmien potilaita (M [metastaasit]-vaihe, aiempi interleukiini-2-hoito, LDH-lähtöarvo, ikä, sukupuoli ja aiemman hoidon tyyppi ja hoitokerrat). Tiedot ipilimumabihoidon kokonaiselinaikaan kohdistuvasta hyödystä olivat kuitenkin yli 50-vuotiaiden naisten osalta vähäisiä. Alaryhmäanalyysissa oli mukana vain vähän potilaita, joten sen tiedoista ei voida tehdä luotettavia johtopäätöksiä.
Elossa olleiden osuus (mediaani ja arvioitu 1 ja 2 vuoden jälkeen) on esitetty taulukossa 10.
Taulukko 10: MDX010-20: Elossa olleiden osuus
| Ipilimumabi 3 mg/kg | gp100 a |
|---|---|---|
Mediaani kk (95 %:n luottamusväli) | 10 kk (8,0,13,8) | 6 kk (5,5, 8,7) |
Elossa olleiden osuus, 1 v % (95 %:n luottamusväli) | 46 % (37,0, 54,1) | 25 % (18,1, 32,9) |
Elossa olleiden osuus, 2 v % (95 %:n luottamusväli) | 24 % (16,0, 31,5) | 14 % (8,0, 20,0) |
a gp100-peptidirokote on tutkimusvaiheessa oleva vertailuhoito.
Ipilimumabia 3 mg/kg monoterapiana saaneessa ryhmässä kokonaiselinajan mediaani oli 22 kk stabiilin sairausvaiheen (SD) saavuttaneilla potilailla ja 8 kk potilailla, joiden sairaus eteni hoidosta huolimatta (PD). Analyysihetkellä mediaaniarvoja ei ollut saavutettu täydellisen (CR) tai osittaisen vasteen (PR) saaneilla potilailla.
Niistä potilaista, jotka tarvitsivat uusintahoidon, hoitovaste (BORR) saavutettiin 38 %:lla (3/8 potilasta) ipilimumabiryhmässä ja 0 %:lla gp100-peptidirokoteryhmässä. Sairaus saatiin hallintaan (disease control rate, DCR. Määritelmä: CR + PR + SD) 75 %:lla (6/8 potilasta) ipilimumabiryhmässä ja 0 %:lla gp100-peptidirokoteryhmässä. Koska näissä alaryhmäanalyyseissa oli mukana vain vähän potilaita, ipilimumabihoidon uusintahoidon tehosta ei voida tehdä luotettavia johtopäätöksiä.
Kliinisen aktiivisuuden kehittyminen tai säilyminen ipilimumabihoidon jälkeen oli potilailla samankaltaista riippumatta siitä, käyttivätkö he systeemisiä kortikosteroideja vai eivät.
CA184-169
Faasin III kaksoissokkoutettuun tutkimukseen otettiin aiemmin hoidettuja ja hoitamattomia potilaita, joilla oli asteen III tai IV melanooma, jota ei voitu kirurgisesti poistaa. Yhteensä 727 potilasta satunnaistettiin: 362 sai ipilimumabia 3 mg/kg annoksena ja 365 sai ipilimumabia 10 mg/kg annoksena joka kolmas viikko yhteensä neljä annosta. Ipilimumabia 10 mg/kg annoksena saaneessa ryhmässä kokonaiselinajan mediaani (95 %:n luottamusväli) oli 16 kuukautta (11,63, 17,84) ja ipilimumabia 3 mg/kg annoksena saaneessa ryhmässä kokonaiselinajan mediaani (95 %:n luottamusväli) oli 12 kuukautta (9,86, 13,27). Verrattaessa kokonaiselinaikaa ipilimumabia 10 mg:n/kg annoksena ja 3 mg:n/kg annoksena saaneiden ryhmien välillä, riskitiheyssuhde oli 0,84 (95 %:n luottamusväli: 0,70, 0,99; p‑arvo = 0,04). Etenemisvapaassa elinajassa (progression-free survival, PFS) ei havaittu tilastollisesti merkittävää eroa 10 mg/kg annosta ja 3 mg/kg annosta saaneiden ryhmien välillä (riskitiheyssuhde 0,89 [95 %:n luottamusväli: 0,76, 1,04 ja log‑rank‑testin p‑arvo = 0,1548]). Hoitovaste (BORR) oli samankaltainen 10 mg/kg annosta ja 3 mg/kg annosta saaneissa ryhmissä. 10 mg/kg annosta saaneessa ryhmässä hoitovaste (BORR) oli 15,3 % (95 %:n luottamusväli: 11,8, 19,5) ja 3 mg/kg annosta saaneessa ryhmässä 12,2 % (95 %:n luottamusväli: 9,0, 16,0). Ipilimumabia 10 mg/kg annoksena saaneessa ryhmässä esiintyi enemmän haittavaikutuksia kuin 3 mg/kg annosta saaneessa ryhmässä. Vakavia haittavaikutuksia esiintyi 37 %:lla 10 mg/kg annosta saaneista ja 18 %:lla 3 mg/kg annosta saaneista. Kolme yleisintä vakavaa haittavaikutusta olivat ripuli, jota esiintyi 10,7 %:lla 10 mg/kg annosta saaneista ja 5,5 %:lla 3 mg/kg annosta saaneista; koliitti, jota esiintyi 8,0 %:lla 10 mg/kg annosta saaneista ja 3,0 %:lla 3 mg/kg annosta saaneista sekä hypofysiitti, jota esiintyi 4,4 %:lla 10 mg/kg annosta saaneista ja 1,9 %:lla 3 mg/kg annosta saaneista. Hoito lopetettiin haittavaikutusten vuoksi 31 %:lla 10 mg/kg annosta saaneista potilaista ja 19 %:lla 3 mg/kg annosta saaneista potilaista. Kuolemaan johtaneita haittavaikutuksia oli neljällä 10 mg/kg annosta saaneista potilaista ja kahdella 3 mg/kg annosta saaneista potilaista.
Suositeltua 3 mg/kg annosta käytettäessä kokonaiselinajan mediaani oli yli 50‑vuotiaiden naisten alaryhmässä samankaltainen kuin koko potilasjoukossa: (11,40 kuukautta vs. 11,53 kuukautta). Alaryhmässä, jonka potilailla oli aivojen etäpesäkkeitä, kokonaiselinajan mediaani oli lähtötasolla 5,67 kuukautta, kun käytettiin suositeltua 3 mg/kg:n annosta.
Muut ipilimumabi-monoterapiatutkimukset
Melanooma
CA184332 ja CA184338
Kun tarkastellaan yhdistettyjä tietoja faasien II ja III kliinisten tutkimusten potilaista, jotka eivät olleet aiemmin saaneet kemoterapiaa (n = 78, satunnaistettuja), ja kahden retrospektiivisen havaintotutkimuksen aiemmin hoitamattomista potilaista (n = 273 ja n = 157), ipilimumabilla saavutettu kokonaiselinaika oli 3 mg/kg monoterapiassa yleisesti ottaen yhteneväinen. Näissä kahdessa havaintotutkimuksessa todettiin edenneen melanooman diagnosointihetkellä etäpesäkkeitä aivoissa 12,1 %:lla potilaista toisessa tutkimuksessa ja 33,1 %:lla toisessa. Kokonaiselinajan mediaani sekä arivoidut ensimmäisen, toisen, kolmannen ja neljännen vuoden elossaolo-osuudet esitetään taulukossa 11. Faasien II ja III kliinisten tutkimusten yhdistetyt tulokset potilaista, jotka eivät olleet aiemmin saaneet kemoterapiaa (n = 78), osoittivat, että ensimmäisen vuoden elossa olleiden osuus oli arviolta 54,1 % (95 %:n luottamusväli: 42,5–65,6), toisen vuoden arviolta 31,6 % (95 %:n luottamusväli: 20,7–42,9) ja kolmannen vuoden arviolta 23,7 % (95 %:n luottamusväli: 14,3–34,4).
Taulukko 11: Elossa olleiden osuus havaintotutkimuksissa
| CA184338 | CA184332 |
|---|---|---|
Kokonaiselinajan mediaani | 14 kk (12,8, 18,7) | 10 kk (7,0, 12,8) |
Elossa olleiden osuus, 1 v % | 59 % (52,5, 64,3) | 44 % (35,5, 51,4) |
Elossa olleiden osuus, 2 v % | 39 % (33,1, 44,8) | 26 % (18,9, 33,3) |
Elossa olleiden osuus, 3 v % | 31 % (25,5, 36,7) | 22 % (15,5, 29,2) |
Elossa olleiden osuus, 4 v % | 26 % (20,4, 31,3) | 22 % (15,5, 29,2) |
CA184332‑tutkimuksessa potilailla, joilla oli aivojen etäpesäkkeitä, kokonaiselinajan mediaani oli 7 kuukautta (95 %:n luottamusväli: 5,06, 12,81) ja potilailla, joilla ei ollut aivojen etäpesäkkeitä, kokonaiselinajan mediaani oli 14,1 kuukautta (95 %:n luottamusväli: 9,96, ei arvioitu).
CA184338‑tutkimuksessa potilailla, joilla oli aivojen etäpesäkkeitä, kokonaiselinajan mediaani oli 6,3 kuukautta (95 %:n luottamusväli: 3,2, 12,0) ja potilailla, joilla ei ollut aivojen etäpesäkkeitä, kokonaiselinajan mediaani oli 17,7 kuukautta (95 %:n luottamusväli: 13,6, 12,1).
Ipilimumabihoidon (3 mg/kg) pitkäaikainen elinaikahyöty on osoitettu aiemmin hoidetuilla ja hoitamattomilla edennyttä melanoomaa sairastavilla potilailla (N=965) tehtyjen kliinisten tutkimusten yhdistettyjen kokonaiselinaikatuloksien analyysissa. Kaplan-Meierin kokonaiselinaikakäyrässä näkyy suunnilleen kolmannen vuoden aikana (elossa olleiden osuus [OS] = 21 % [95 %:n luottamusväli: 17–24]) alkava tasannevaihe, joka jatkui joillain potilailla jopa 10 vuoteen asti (ks. kuva 1).
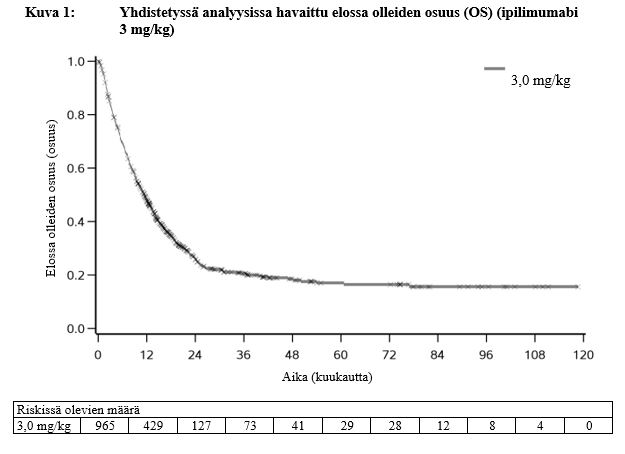
Kliiniset ipilimumabi–nivolumabi-yhdistelmähoidon tutkimukset
Melanooma
Satunnaistettu faasin III tutkimus, ipilimumabi–nivolumabi-yhdistelmähoito tai nivolumabi-monoterapia vs. ipilimumabi-monoterapia (CA209067)
Ipilimumabin (3 mg/kg) ja nivolumabin (1 mg/kg) yhdistelmähoidon tai nivolumabi-monoterapian (3 mg/kg) vs. ipilimumabi-monoterapian (3 mg/kg) turvallisuutta ja tehoa edenneen melanooman (jota ei voida kirurgisesti poistaa tai joka on metastasoinut) hoidossa arvioitiin satunnaistetussa faasin III kaksoissokkoutetussa tutkimuksessa (CA209067). Kahden nivolumabia saaneen ryhmän eroja arvioitiin deskriptiivisesti. Tutkimukseen otettiin mukaan diagnosoituja, aiemmin hoitamattomia aikuisia potilaita, joilla oli III tai IV asteen melanooma. Potilaiden ECOG-suorituskykyluokan tuli olla 0 tai 1. Tutkimukseen otettiin mukaan potilaita, jotka eivät olleet aikaisemmin saaneet systemaattista syöpähoitoa melanoomaan, jota ei voitu kirurgisesti poistaa tai joka oli metastasoinut. Aikaisempi liitännäishoito tai esiliitännäishoito sallittiin, jos se oli päättynyt vähintään 6 viikkoa ennen satunnaistamista. Potilaita, joilla oli aktiivinen autoimmuunisairaus, silmän/uvean melanooma tai aktiivisia aivometastaaseja tai leptomeningeaalisia metastaaseja, ei otettu mukaan tutkimukseen.
Kaikkiaan 945 potilaista satunnaistettiin saamaan ipilimumabi–nivolumabi-yhdistelmähoitoa (n = 314), nivolumabi-monoterapiaa (n = 316) tai ipilimumabi-monoterapiaa (n = 315). Yhdistelmähoitohaarassa potilaat saivat ensimmäiset neljä annosta nivolumabia 1 mg/kg 60 minuutin kuluessa ja ipilimumabia 3 mg/kg 90 minuutin kuluessa laskimonsisäisesti kolmen viikon välein, jonka jälkeen heille annettiin nivolumabia 3 mg/kg monoterapiana joka toinen viikko. Nivolumabi-monoterapiahaarassa potilaille annettiin nivolumabia 3 mg/kg joka toinen viikko. Vertailuhaaran potilaat saivat neljä ensimmäistä annosta ipilimumabia 3 mg/kg ja nivolumabin lumelääkettä laskimonsisäisesti joka kolmas viikko, jonka jälkeen heille annettiin lumelääkettä joka toinen viikko. Satunnaistaminen stratifioitiin PD-L1-statuksen (≥ 5 %:ssa vs. < 5 %:ssa kasvainsolun membraanissa ilmentyvä), BRAF-statuksen ja American Joint Committee on Cancer (AJCC) ‑järjestelmän määrittelemän M-vaiheen mukaan. Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt hoitoa. Kasvaimet arvioitiin 12 viikkoa satunnaistamisen jälkeen, sen jälkeen ensimmäisen vuoden ajan joka kuudes viikko ja sen jälkeen joka kahdestoista viikko. Primaariset muuttujat olivat etenemisvapaa elinaika ja kokonaiselinaika. Myös objektiivisen vasteen saavuttaneiden osuus (ORR) ja vasteen kesto arvioitiin.
Ryhmien lähtötason ominaisuudet olivat kussakin ryhmässä tasapainossa. Iän mediaani oli 61 vuotta (vaihteluväli 18–90 vuotta), 65 % oli miehiä ja 97 % valkoihoisia. ECOG-suorituskykyluokka oli 0 (73 %) tai 1 (27 %). Useimmilla potilailla oli AJCC-järjestelmän mukaisesti asteen IV sairaus (93 %), 58 %:lla potilaista sairaus oli tutkimukseen otettaessa luokkaa M1c. 22 % potilaista oli saanut aiemmin liitännäishoitoa. 32 %:lla potilaista oli BRAF-mutaatiopositiivinen melanooma; 26,5 %:lla oli PD-L1 ≥ 5 %:ssa kasvainsolun membraanissa ilmentyvänä. 4 %:lla potilaista oli aiemmin ollut aivometastaasi ja 36 %:lla LDH-arvo oli tutkimukseen otettaessa normaalia suurempi. Potilaat, joilla oli määritettävissä oleva PD-L1:n ilmentyminen, jaettiin tasaisesti kolmeen hoitoryhmään. Kasvaimen PD-L1-ilmentyminen määriteltiin PD-L1 IHC 28–8 pharmDx ‑testausmenetelmän avulla.
Primaarianalyysissa (seuranta‑aika vähintään 9 kuukautta) etenemisvapaan elinajan mediaani oli 6,9 kuukautta nivolumabiryhmässä, kun taas ipilimumabiryhmässä se oli 2,9 kuukautta (riskitiheyssuhde = 0,57, 99,5 %:n luottamusväli: 0,43, 0,76; p < 0,0001). Etenemisvapaan elinajan mediaani oli 11,5 kuukautta ipilimumabin ja nivolumabin yhdistelmää saaneiden ryhmässä, kun taas ipilimumabiryhmässä se oli 2,9 kuukautta (riskitiheyssuhde = 0,42, 99,5 %:n luottamusväli: 0,31, 0,57; p < 0,0001).
Deskriptiivisen analyysin mukaiset etenemisvapaan elinajan tulokset (seuranta-ajan ollessa vähintään 90 kuukautta) esitetään kuvassa 2 (kaikki satunnaistetut populaatiot), kuvassa 3 (PD-L1-ilmentymän raja-arvolla 5 %) ja kuvassa 4 (PD-L1-ilmentymän raja-arvolla 1 %).
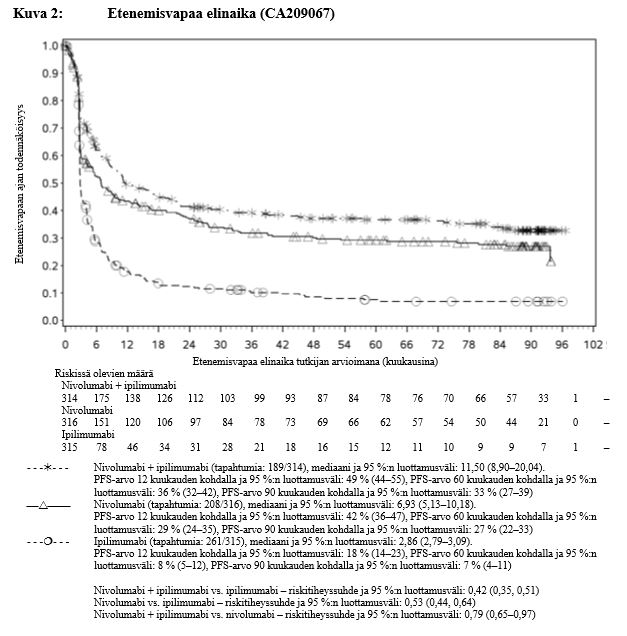
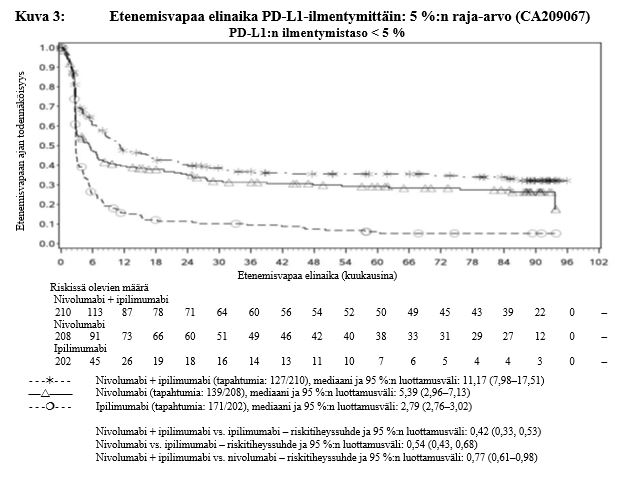
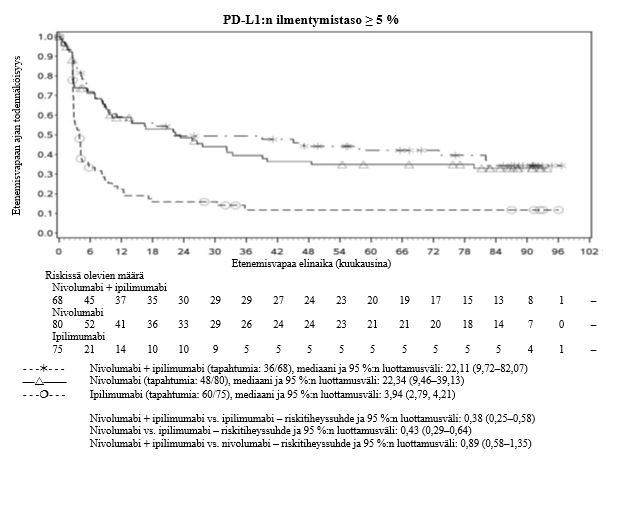
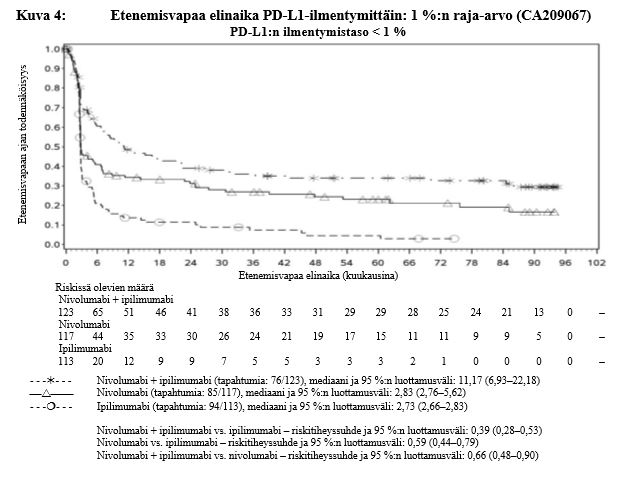
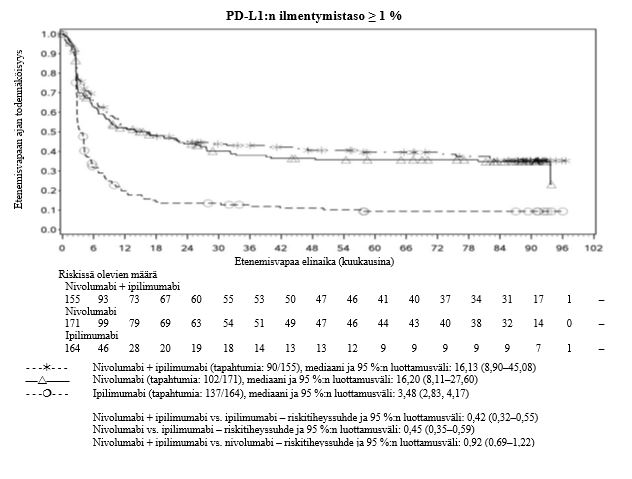
Lopullinen kokonaiselinajan (primaari)analyysi tehtiin, kun kaikkia potilaita oli seurattu vähintään 28 kuukauden ajan. Kokonaiselinajan mediaania ei saavutettu 28 kuukauden kohdalla nivolumabia saaneiden ryhmässä, kun taas ipilimumabia saaneiden ryhmässä kokonaiselinajan mediaani oli 19,98 kuukautta (riskitiheyssuhde = 0,63, 98 %:n luottamusväli: 0,48, 0,81; p‑arvo: < 0,0001). Kokonaiselinajan mediaania ei saavutettu ipilimumabin ja nivolumabin yhdistelmähoitoa saaneiden ryhmässä ipilimumabia saaneiden ryhmään verrattuna (riskitiheyssuhde = 0,55, 98 %:n luottamusväli: 0,42, 0,72; p‑arvo: < 0,0001).
Vähintään 90 kuukauden seuranta-ajan jälkeen tehdyn ylimääräisen deskriptiivisen analyysin mukaiset kokonaiselinajan tulokset olivat yhteneviä alkuperäisen primaarianalyysin kanssa. Tämän seuranta-analyysin mukaiset kokonaiselinajan tulokset esitetään kuvassa 5 (kaikki satunnaistetut), kuvissa 6 ja 7 (kasvaimen PD‑L1:n ilmentymän raja-arvoilla 5 % ja 1 %).
Kokonaiselinaika-analyysissa ei huomioitu tutkimuslääkkeen jälkeistä lääkehoitoa. Potilaat saivat myöhempää systeemistä hoitoa seuraavasti: 36 % yhdistelmähoitoa saaneista, 49,1 % nivolumabi-monoterapiaa saaneista ja 66,3 % ipilimumabia saaneista. Potilaat saivat myöhempää immuunihoitoa (mukaan lukien anti-PD-1-hoitoa, anti-CTLA-4-hoitoa tai muuta immuunihoitoa) seuraavasti: 19,1 % yhdistelmähoitoa saaneista, 34,2 % nivolumabi-monoterapiaa saaneista ja 48,3 % ipilimumabia saaneista.
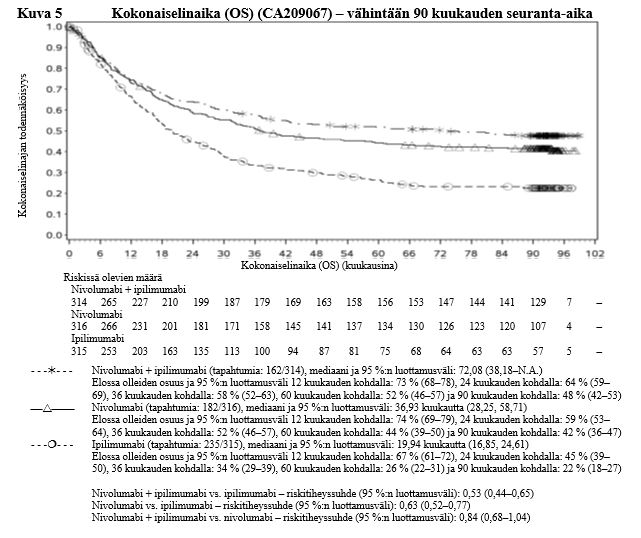
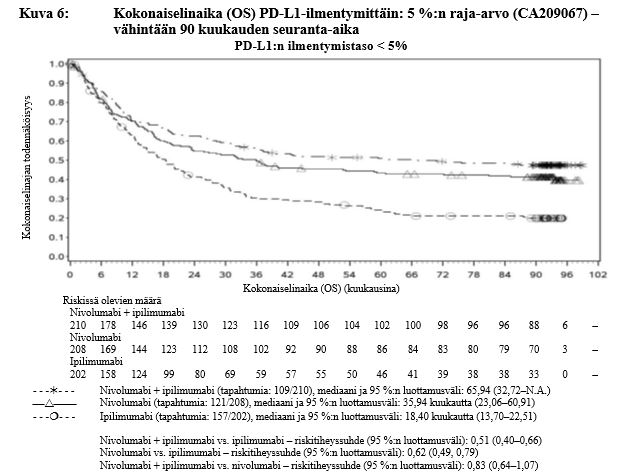
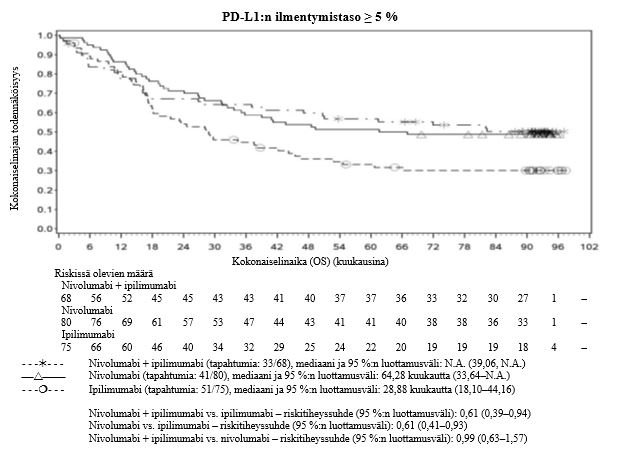
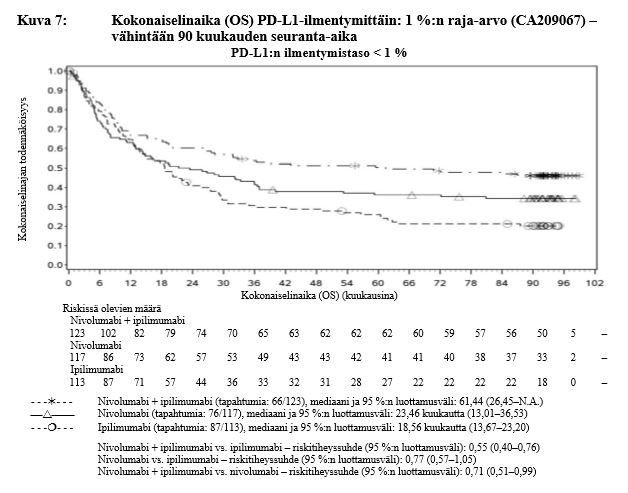
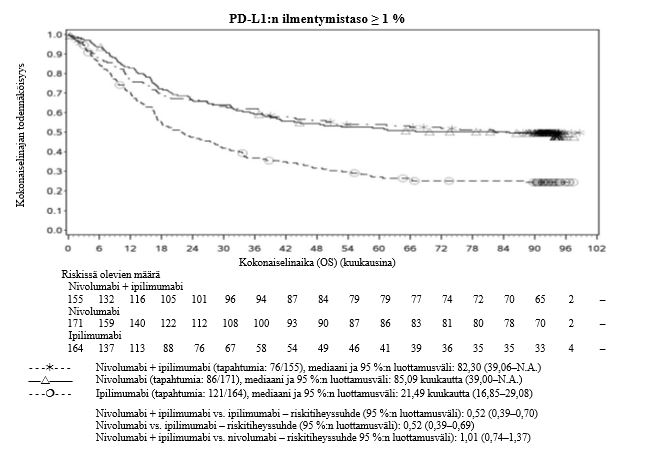
Objektiivisen vasteen saavuttaneiden osuuden analyysin seuranta-aika oli vähintään 90 kuukautta. Vasteiden yhteenveto on taulukossa 12.
Taulukko 12: Objektiivinen vaste (CA209067)
| nivolumabi + | nivolumabi | ipilimumabi |
|---|---|---|---|
Objektiivinen vaste | 183 (58 %) | 142 (45 %) | 60 (19 %) |
(95 %:n luottamusväli) | (52,6, 63,8) | (39,4–50,6) | (14,9, 23,8) |
Ristitulosuhde (vs. ipilimumabi) | 6,35 | 3,5 |
|
(95 %:n luottamusväli) | (4,38, 9,22) | (2,49–5,16) |
|
Täydellinen vaste (CR) | 71 (23 %) | 59 (19 %) | 19 (6 %) |
Osittainen vaste (PR) | 112 (36 %) | 83 (26 %) | 41 (13 %) |
Stabiili tauti (SD) | 38 (12 %) | 29 (9 %) | 69 (22 %) |
Vasteen kesto |
|
|
|
Mediaani (vaihteluväli), kuukausina | N.A. (69,1–N.A.) | 90,8 (45,7–N.A.) | 19,3 (8,8–47,4) |
Osuus ≥ 12 kuukauden keston kohdalla | 68 % | 73 % | 44 % |
Osuus ≥ 24 kuukauden keston kohdalla | 58 % | 63 % | 30 % |
Objektiivisen vasteen saavuttaneiden osuus (95 %:n luottamusväli) kasvaimen PD‑L1-ilmentymittäin |
|
| |
< 5 % | 56 % (48,7, 62,5) n = 210 | 43 % (36, 49,8) n = 208 | 18 % (12,8, 23,8) n = 202 |
≥ 5 % | 72 % (59,9, 82,3) n = 68 | 59 % (47,2–69,6) | 21 % (12,7, 32,3) n = 75 |
< 1 % | 54 % (44,4, 62,7) n = 123 | 36 % (27,2, 45,3) n = 117 | 18 % (11,2, 26,0) n = 113 |
≥ 1 % | 65 % (56,4, 72) n = 155 | 55 % (47,2–62,6) | 20 % (13,7, 26,4) n = 164 |
Kummatkin nivolumabihoitoa sisältäneet tutkimushaarat osoittivat pelkkään ipilimumabihoitoon verrattuna merkitsevää etua etenemisvapaan elinajan ja kokonaiselinajan sekä objektiivisen vasteen saavuttaneiden osuuden suhteen. Havaitut etenemisvapaan elinajan tulokset osoitettiin 18 kuukauden kohdalla ja objektiivisen vasteen saavuttaneiden osuuden sekä kokonaiselinajan tulokset 28 kuukauden kohdalla tehdyssä seurannassa johdonmukaisesti kaikissa alaryhmissä, joihin sisältyi ECOG-suorituskykyluokka, BRAF-status, M‑vaihe, ikä, aiempi aivometastaasi ja LDH lähtötilanteessa. Tämä havainto pysyi muuttumattomana kokonaiselinajan tulosten osalta seuranta-ajan ollessa vähintään 90 kuukautta.
Haittavaikutuksen vuoksi yhdistelmähoidon 28 kuukauden seuranta-ajan jälkeen keskeyttäneiden 131 potilaan objektiivisen vasteen saavuttaneiden osuus oli 71 % (93/131), josta 20 % (26/131) saavutti täydellisen vasteen, eikä kokonaiselinajan mediaania saavutettu.
Kummallakin nivolumabihoitoa sisältäneellä tutkimushaaralla oli pelkkään ipilimumabihoitoon verrattuna parempi objektiivinen vaste PD‑L1-ilmentymistasosta riippumatta. Objektiivisen vasteen saavuttaneiden osuudet olivat kaikilla PD‑L1-ilmentymistasoilla korkeammat nivolumabi–ipilimumabi-yhdistelmähoitoa saaneessa ryhmässä kuin nivolumabi-monoterapiaa saaneessa ryhmässä (taulukko 12) 90 kuukauden seuranta-ajan jälkeen parhaan saavutetun kokonaisvasteen, täydellisen vasteen, korreloidessa paremman eloonjäämisasteen kanssa.
Potilailla, joilla oli PD‑L1 ≥ 5 % ‑ilmentymistason kasvain, vasteen mediaanikesto oli 78,19 kuukautta 90 kuukauden seuranta-ajan jälkeen yhdistelmähoitoa saaneessa ryhmässä (vaihteluväli 18,07–N.A.), 77,21 kuukautta nivolumabi-monoterapiaa saaneessa ryhmässä (vaihteluväli 26,25–N.A.) ja 31,28 kuukautta (vaihteluväli 6,08–N.A.) ipilimumabia saaneessa ryhmässä. Potilailla, joilla oli PD‑L1 < 5 % ‑ilmentymistason kasvain, vasteen mediaanikestoa ei saavutettu yhdistelmähoitoa saaneessa ryhmässä (vaihteluväli 61,93–N.A.). Vasteen mediaanikesto oli 90,84 kuukautta nivolumabi-monoterapiaa saaneessa ryhmässä (vaihteluväli 50,43–N.A.) ja 19,25 kuukautta (vaihteluväli 5,32–47,44) ipilimumabi-monoterapiaa saaneessa ryhmässä.
Kasvaimen vasteita ja etenemisvapaata elinaikaa sekä kokonaiselinaikaa koskevat merkitykselliset päätetapahtumat huomioon ottaen selkeää raja-arvoa PD‑L1-ilmentymiselle ei voida luotettavasti määritellä. Tämän havainnollistavan usean satunnaismuuttujan analyysin tulosten perusteella voitiin tunnistaa potilaan ja kasvaimen erityispiirteitä (ECOG-suorituskykyluokka, M‑vaihe, LDH lähtötilanteessa, BRAF-mutaatiostatus, PD‑L1-status ja sukupuoli), joilla voi olla merkitystä eloonjäämisen kannalta.
Teho BRAF-statuksen mukaan:
Ipilimumabi–nivolumabi-yhdistelmähoidolle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla PFS-mediaani oli 90 kuukauden seurannan jälkeen 16,76 kuukautta (95 %:n luottamusväli 8,28–32,0) ja potilailla, joilla oli BRAF-villityypin kasvain, PFS-mediaani oli 11,17 kuukautta (95 %:n luottamusväli 7,0–19,32). Nivolumabi-monoterapialle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla PFS-mediaani oli 5,62 kuukautta (95 %:n luottamusväli 2,79–9,46) ja potilailla, joilla oli BRAF-villityypin kasvain, etenemisvapaan elinajan mediaani oli 8,18 kuukautta (95 %:n luottamusväli 5,13–19,55). Ipilimumabi-monoterapialle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla etenemisvapaan elinajan mediaani oli 3,09 kuukautta (95 %:n luottamusväli 2,79–5,19) ja potilailla, joilla oli BRAF-villityypin kasvain, etenemisvapaan elinajan mediaani oli 2,83 kuukautta (95 %:n luottamusväli: 2,76–3,06).
Ipilimumabi–nivolumabi-yhdistelmähoidolle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla objektiivisen vasteen saavuttaneiden osuus oli 90 kuukauden seurannan jälkeen 67,0 % (95 %:n luottamusväli 57,0–75,9; n = 103), ja potilailla, joilla oli BRAF-villityypin kasvain, objektiivisen vasteen saavuttaneiden osuus oli 54,0 % (95 %:n luottamusväli 47,1–60,9; n = 211). Nivolumabi-monoterapialle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla objektiivisen vasteen saavuttaneiden osuus oli 37,87 % (95 %:n luottamusväli 28,2–48,1; n = 98), ja potilailla, joilla oli BRAF-villityypin kasvain, objektiivisen vasteen saavuttaneiden osuus oli 48,2 % (95 %:n luottamusväli 41,4–55,0; n = 218). Ipilimumabi-monoterapialle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla objektiivisen vasteen saavuttaneiden osuus oli 23,0 % (95 %:n luottamusväli 15,2, 32,5; n = 100) ja potilailla, joilla oli BRAF-villityypin kasvain, objektiivisen vasteen saavuttaneiden osuus oli 17,2 % (95 %:n luottamusväli 12,4, 22,9; n = 215).
Kokonaiselinajan mediaania ei saavutettu BRAF V600 ‑mutaatiopositiivisilla potilailla 90 kuukauden seuranta-ajan jälkeen yhdistelmähoitoa saaneessa ryhmässä, ja nivolumabi-monoterapiaryhmässä kokonaiselinajan mediaani oli 45,5 kuukautta. Kokonaiselinajan mediaani oli BRAF V600 ‑mutaatiopositiivisilla potilailla ipilimumabi-monoterapiaryhmässä 24,6 kuukautta. Kokonaiselinajan mediaani potilailla, joilla oli BRAF-villityypin kasvain, oli yhdistelmähoitoa saaneessa ryhmässä 39,06 kuukautta, nivolumabi-monoterapiaryhmässä 34,37 kuukautta ja ipilimumabi-monoterapiaryhmässä 18,5 kuukautta. Kokonaiselinajan riskitiheyssuhteet olivat verrattaessa ipilimumabi–nivolumabi-yhdistelmähoitoa nivolumabi-monoterapiaan BRAF V600 ‑mutaatiopositiivisilla potilailla 0,66 (95 %:n luottamusväli 0,44–0,98) ja BRAF-villityypin potilailla 0,95 (95 %:n luottamusväli0,74–1,22).
Satunnaistettu faasin II tutkimus, ipilimumabi–nivolumabi ja ipilimumabi (CA209069)
CA209069 oli satunnaistettu, faasin II kaksoissokkoutettu tutkimus, jossa verrattiin nivolumabi–ipilimumabi-yhdistelmähoitoa ipilimumabi-monoterapiaan. Tutkimukseen osallistui 142 potilasta, joilla oli edennyt melanooma (jota ei voitu kirurgisesti poistaa tai joka oli metastasoinut). Sisäänottokriteerit olivat samanlaiset kuin tutkimuksessa CA209067 ja primaarianalyysi potilaiden BRAF-villityypin melanoomasta oli samanlainen (77 %:lla potilaista). Tutkijan arvioima objektiivisen vasteen saavuttaneiden osuus oli 61 % (95 %:n luottamusväli 48,9, 72,4) yhdistelmähoitoryhmässä (n = 72), kun se ipilimumabi-monoterapiaryhmässä (n = 37) oli 11 % (95 %:n luottamusväli 3,0, 25,4). Arvioidut 2 ja 3 vuoden kokonaiselinaikaluvut olivat yhdistelmähoitoryhmässä (n = 73) 68 % (95 %:n luottamusväli 56, 78) ja 61 % (95 %:n luottamusväli 49, 71) ja ipilimumabi-monoterapiaryhmässä (n = 37) 53 % (95 %:n luottamusväli 36, 68) ja 44 % (95 %:n luottamusväli 28, 60).
Munuaiskarsinooma (RCC)
Satunnaistettu faasin III tutkimus, ipilimumabi–nivolumabi-yhdistelmähoito vs. sunitinibi (CA209214)
Ipilimumabin (1 mg/kg) ja nivolumabin (3 mg/kg) yhdistelmähoidon turvallisuutta ja tehoa edenneen/metastasoituneen munuaiskarsinooman hoidossa arvioitiin satunnaistetussa faasin III avoimessa tutkimuksessa (CA209214). Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joilla oli aiemmin hoitamaton, edennyt tai metastasoitunut kirkassoluinen munuaiskarsinooma. Tehon arviointiin ensisijaisesti käytettävässä potilasjoukossa oli kohtalaisen/huonon ennusteen potilaita, joilla oli vähintään 1 tai useampi International Metastatic RCC Database Consortiumin (IMDC) kriteerien mukaisista kuudesta prognostisesta riskitekijästä (alle vuosi ensimmäisestä munuaiskarsinoomadiagnoosista satunnaistamiseen, Karnofskyn asteikolla pisteitä < 80 %, viitearvon alarajaa matalampi hemoglobiini, korjattu kalsiumpitoisuus yli 10 mg/dl, viitearvon ylärajaa korkeampi trombosyyttien määrä ja viitearvon ylärajaa korkeampi neutrofiilien määrä). Tutkimukseen otettiin potilaita riippumatta kasvaimen PD‑L1-statuksesta. Tutkimuksesta suljettiin pois ne potilaat, joilla oli Karnofskyn asteikolla pisteitä < 70 %, sekä potilaat joilla oli tai joilla oli ollut aivometastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila. Potilaat stratifioitiin IMDC:n prognostisen pistemäärän ja esiintymispaikan mukaan.
Tutkimukseen satunnaistettiin yhteensä 1 096 potilasta, joista 847 potilaalla oli kohtalaisen/huonon ennusteen munuaiskarsinooma. Potilaille annettiin joko 1 mg/kg ipilimumabia (n = 425) laskimoon 30 minuutin kuluessa sekä 3 mg/kg nivolumabia laskimoon 60 minuutin kuluessa joka kolmas viikko yhteensä 4 annosta, minkä jälkeen annettiin nivolumabi-monoterapiaa 3 mg/kg:n annoksena joka toinen viikko tai 50 mg/vrk sunitinibia (n = 422) suun kautta neljän viikon ajan, minkä jälkeen pidettiin aina kahden viikon tauko. Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt hoitoa. Kasvaimet arvioitiin ensimmäisen kerran 12 viikkoa satunnaistamisen jälkeen, sen jälkeen ensimmäisen vuoden ajan joka kuudes viikko ja sen jälkeen joka kahdestoista viikko taudin etenemiseen tai hoidon lopettamiseen saakka, kumpi näistä tapahtui jälkimmäisenä. Hoito taudin etenemisen jälkeen sallittiin tutkijan suorittaman ensimmäisen kiinteiden kasvainten vasteenarviointikriteerien (RECIST) version 1.1 mukaisesti tekemän arvioinnin jälkeen, jos tutkija määritteli, että potilaalle on kliinistä hyötyä tutkimuslääkkeestä ja hän sietää hoitoa. Ensisijaiset tehokkuusmuuttujat olivat kokonaiselinaika, objektiivisen vasteen saavuttaneiden osuus ja etenemisvapaa elinaika riippumattoman keskitetyn arvioijatahon (Blinded Independent Central Review, BICR) arvioimana kohtalaisen/huonon ennusteen potilailla.
Ryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Iän mediaani oli 61 vuotta (vaihteluväli 21–85). 38 % potilaista oli ≥ 65 vuotta ja 8 % oli ≥ 75 vuotta. Suurin osa potilaista oli miehiä (73 %) ja valkoihoisia (87 %). 31 %:lla potilaista lähtötason Karnofskyn suorituskykypisteet (KPS) olivat 70–80 % ja 69 %:lla potilaista 90–100 %. Ajan mediaani ensimmäisestä diagnoosista satunnaistamiseen oli 0,4 vuotta sekä ryhmässä, joka sai 1 mg/kg ipilimumabia ja 3 mg/kg nivolumabia, että ryhmässä, joka sai sunitinibia. Ipilimumabia ja nivolumabia saaneiden potilaiden hoidon keston mediaani oli 7,9 kuukautta (vaihteluväli 1 päivä – 21,4+ kuukautta) ja sunitinibia saaneiden 7,8 kuukautta (vaihteluväli 1 päivä – 20,2+ kuukautta). Ipilimumabi- ja nivolumabihoitoa jatkettiin taudin etenemisen jälkeen 29 %:lla potilaista.
Kohtalaisen/huonon ennusteen potilaiden tehoon liittyvät tulokset esitetään taulukossa 13 (primaarianalyysi, kun seuranta-aika oli vähintään 17,5 kuukautta ja vähintään 60 kuukautta) sekä kuvassa 8 (seuranta-aika vähintään 60 kuukautta).
Vähintään 60 kuukauden seuranta‑ajan jälkeen tehdyn ylimääräisen deskriptiivisen analyysin mukaiset kokonaiselinajan tulokset olivat yhteneviä alkuperäisen primaarianalyysin kanssa.
Taulukko 13: Kohtalaisen/huonon ennusteen potilaiden tehoon liittyvät tulokset (CA209214)
| nivolumabi + ipilimumabi | sunitinibi |
|---|---|---|
| Primaarianalyysi | |
Kokonaiselinaika (OS) | ||
Tapahtumat | 140 (33 %) | 188 (45 %) |
Riskitiheyssuhdea | 0,63 | |
(99,8 %:n luottamusväli) | (0,44, 0,89) | |
p‑arvob, c | < 0,0001 | |
| ||
Mediaani (95 %:n luottamusväli) | NE (28,2, NE) | 25,9 (22,1, NE) |
Osuus (95 %:n luottamusväli) |
|
|
6 kuukauden kohdalla | 89,5 (86,1, 92,1) | 86,2 (82,4, 89,1) |
12 kuukauden kohdalla | 80,1 (75,9, 83,6) | 72,1 (67,4, 76,2) |
Etenemisvapaa elinaika | ||
Tapahtumat | 228 (53,6 %) | 228 (54,0 %) |
Riskitiheyssuhdea | 0,82 | |
(99,1 %:n luottamusväli) | (0,64, 1,05) | |
p‑arvob,h | 0,0331 | |
Mediaani (95 %:n luottamusväli) | 11,6 (8,71, 15,51) | 8,4 (7,03, 10,81) |
Vahvistettu objektiivinen vaste (BICR) | 177 (41,6 %) | 112 (26,5 %) |
(95 %:n luottamusväli) | (36,9, 46,5) | (22,4, 31,0) |
Ero objektiivisen vasteen saavuttaneiden osuudessa (95 %:n luottamusväli)d | 16,0 (9,8, 22,2) | |
p‑arvoe,f | < 0,0001 | |
| ||
Täydellinen vaste (CR) | 40 (9,4 %) | 5 (1,2 %) |
Osittainen vaste (PR) | 137 (32,2 %) | 107 (25,4 %) |
Stabiili tauti (SD) | 133 (31,3 %) | 188 (44,5 %) |
| ||
Vasteen keston mediaanig | ||
Kuukausina (vaihteluväli) | NE (1,4+–25,5+) | 18,17 (1,3+–23,6+) |
| ||
Vasteen saavuttamisajan mediaani | ||
Kuukausina (vaihteluväli) | 2,8 (0,9–11,3) | 3,0 (0,6–15,0) |
| Päivitetty analyysi* Seuranta-aika vähintään: 60 kuukautta | |
Kokonaiselinaika (OS) | ||
Tapahtumat | 242 (57 %) | 282 (67 %) |
Riskitiheyssuhdea | 0,68 | |
95 %:n luottamusväli | (0,58, 0,81) | |
| ||
Mediaani (95 %:n luottamusväli) | 46,95 (35,35, 57,43) | 26,64 (22,08, 33,54) |
Osuus (95 %:n luottamusväli) |
|
|
24 kuukauden kohdalla | 66,3 (61,5, 70,6) | 52,4 (47,4, 57,1) |
36 kuukauden kohdalla | 54,6 (49,7, 59,3) | 43,7 (38,7, 48,5) |
48 kuukauden kohdalla | 49,9 (44,9, 54,6) | 35,8 (31,1, 40,5) |
60 kuukauden kohdalla | 43,0 (38,1, 47,7) | 31,3 (26,8, 35,9) |
Etenemisvapaa elinaika | ||
Tapahtumat | 245 (57,6 %) | 253 (60,0 %) |
Riskitiheyssuhdea | 0,73 | |
95 %:n luottamusväli | (0,61, 0,87) | |
Mediaani (95 %:n luottamusväli) | 11,6 (8,44, 16,63) | 8,3 (7,03, 10,41) |
Vahvistettu objektiivinen vaste (BICR) | 179 (42,1 %) | 113 (26,8 %) |
(95 %:n luottamusväli) | (37,4, 47,0) | (22,6, 31,3) |
Ero objektiivisen vasteen saavuttaneiden osuudessa (95 %:n luottamusväli)d,e | 16,2 (10,0, 22,5) | |
| ||
Täydellinen vaste (CR) | 48 (11,3 %) | 9 (2,1 %) |
Osittainen vaste (PR) | 131 (30,8 %) | 104 (24,6 %) |
Stabiili tauti (SD) | 131 (30,8 %) | 187 (44,3 %) |
| ||
Vasteen keston mediaanig | ||
Kuukausina (vaihteluväli) | NE (50,89–NE) | 19,38 (15,38–25,10) |
| ||
Vasteen saavuttamisajan mediaani | ||
Kuukausina (vaihteluväli) | 2,8 (0,9–35,0) | 3,1 (0,6–23,6) |
a Perustuen ositettuun verrannollisten riskitiheyksien malliin.
b Perustuen ositettuun log-rank-merkitsevyystestiin.
c Tilastollisen merkitsevyyden saavuttamiseksi p-arvoa on verrattu α 0,002:een.
d Osajoukkojen suhteen korjattu ero.
e Perustuen ositettuun DerSimonian–Lairdin testiin.
f Tilastollisen merkitsevyyden saavuttamiseksi p-arvoa on verrattu α 0,001:een.
g Laskettu Kaplan–Meierin menetelmällä.
h Tilastollisen merkitsevyyden saavuttamiseksi p-arvoa on verrattu α 0,009:een.
”+” tarkoittaa sensuroitunutta havaintoa.
NE = non‑estimable, ei arvioitavissa
* Deskriptiivinen analyysi perustuu tietojen keruun katkaisuajankohtaan 26.2.2021.
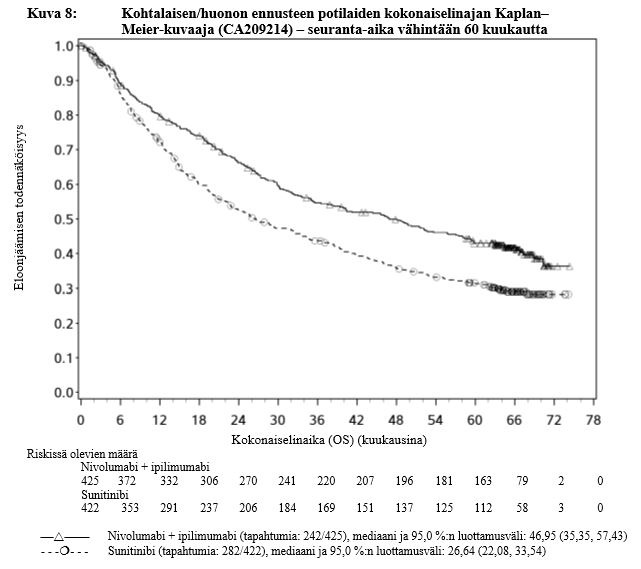
Kokonaiselinajan päivitetty deskriptiivinen analyysi tehtiin, kun kaikki potilaat olivat olleet seurannassa vähintään 24 kuukautta. Analyysihetkellä riskitiheyssuhde oli 0,66 (99,8 %:n luottamusväli: 0,48–0,91), ja tapahtumia oli yhdistelmähoitohaarassa 166/425 ja sunitinibihaarassa 209/422. Ipilimumabi–nivolumabi‑yhdistelmähoitoa saaneilla kohtalaisen/huonon ennusteen potilailla kokonaiselinajan hyöty oli sunitinibia saaneita parempi riippumatta kasvainten PD‑L1:n ilmentymisasteesta. PD‑L1:n ilmentymistason ollessa ≥ 1 % ipilimumabi–nivolumabi‑yhdistelmähoitoa saaneiden ryhmässä kokonaiselinajan mediaania ei saavutettu, ja sunitinibiryhmässä se oli 19,61 kuukautta (riskitiheyssuhde = 0,52; 95 %:n luottamusväli: 0,34, 0,78). PD‑L1:n ilmentymistason ollessa < 1 % ipilimumabi–nivolumabi‑yhdistelmähoitoa saaneiden ryhmässä kokonaiselinajan mediaani oli 34,7 kuukautta ja sunitinibiryhmässä 32,2 kuukautta (riskitiheyssuhde = 0,70; 95 %:n luottamusväli: 0,54, 0,92).
CA209214‑tutkimuksessa satunnaistettiin IMDC:n kriteerien mukaisesti lisäksi 249 hyvän ennusteen potilasta saamaan joko ipilimumabia ja nivolumabia (n = 125) tai sunitinibia (n = 124). Näitä potilaita ei arvioitu osana tehon arviointiin ensisijaisesti käytettyä potilasjoukkoa. Kun seuranta-aika oli vähintään 24 kuukautta, kokonaiselinajan riskitiheyssuhde oli ipilimumabia ja nivolumabia saaneilla hyvän ennusteen potilailla sunitinibiin verrattuna 1,13 (95 %:n luottamusväli: 0,64, 1,99; p = 0,6710). Kun seuranta-aika oli vähintään 60 kuukautta, kokonaiselinajan riskitiheyssuhde oli 0,94 (95 %:n luottamusväli: 0,65, 1,37).
Ipilimumabin yhteiskäytöstä nivolumabin kanssa ei ole tietoa munuaiskarsinooman ensilinjan hoidossa potilailla, joilla on ainoastaan ei-kirkassoluinen histologinen kasvaintyyppi.
CA209214-tutkimuksessa 8 % kaikista kohtalaisen/huonon ennusteen potilaista oli ≥ 75-vuotiaita, ja koko potilasjoukkoon verrattuna tässä alaryhmässä ipilimumabi–nivolumabi-hoito vaikutti lukumääräisesti vähemmän kokonaiselinaikaan (riskitiheyssuhde 0,97, 95 %:n luottamusväli: 0,48, 1,95), kun seuranta-aika oli vähintään 17,5 kuukautta. Koska alaryhmä oli pieni, sen tiedoista ei voida tehdä luotettavia johtopäätöksiä
Ei-pienisoluisen keuhkosyövän ensilinjan hoito
Satunnaistettu faasin 3 tutkimus ipilimumabista yhdistelmähoitona nivolumabin ja kahden platinapohjaisen kemoterapiajakson kanssa vs. neljä platinapohjaista kemoterapiajaksoa (CA2099LA)
Kuuden viikon välein annetun ipilimumabin (1 mg/kg) ja kolmen viikon välein annetun nivolumabin (360 mg) sekä kahden platinapohjaisen kemoterapiajakson yhdistelmähoidon turvallisuutta ja tehoa arvioitiin satunnaistetussa faasin 3 avoimessa tutkimuksessa (CA2099LA). Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joilla oli histologisesti vahvistettu ei-levyepiteeliperäinen tai levyepiteeliperäinen IV asteen tai uusiutunut ei-pienisoluinen keuhkosyöpä (International Association for the Study of Lung Cancer ‑yhdistyksen 7:nnen luokittelun mukaisesti), joiden ECOG‑suorituskykyluokka oli 0 tai 1 ja jotka eivät olleen saaneet aiempaa syöpähoitoa (mukaan lukien EGFR:n ja ALK:n estäjät). Potilaan kasvaimen PD-L1-status ei vaikuttanut tutkimukseen mukaan ottamiseen.
Tutkimuksesta suljettiin pois ne potilaat, joilla oli herkistäviä EGFR-mutaatioita tai ALK:n translokaatioita, aktiivinen (hoitamaton) aivojen etäpesäke, karsinomatoottinen aivokalvotulehdus, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila. Ne potilaat, jotka olivat saaneet aiempaa hoitoa aivojen etäpesäkkeisiin, voitiin ottaa mukaan tutkimukseen, jos heidän neurologinen tilansa palautui lähtötasolle vähintään 2 viikkoa ennen tutkimukseen ottamista ja jos he eivät joko saaneet lainkaan kortikosteroideja tai saivat vakaata tai pienentyvää annosta, joka vastasi < 10 mg:aa prednisonia vuorokaudessa. Satunnaistaminen stratifioitiin histologian (levyepiteeliperäinen vs. ei-levyepiteeliperäinen), kasvaimen PD-L1‑ilmentymistason (≥ 1 % vs. < 1 %) ja sukupuolen (mies vs. nainen) mukaan.
Kaikkiaan 719 potilaista satunnaistettiin saamaan joko ipilimumabin, nivolumabin ja platinapohjaisen kemoterapian yhdistelmähoitoa (n = 361) tai platinapohjaista kemoterapiaa (n = 358). Ipilimumabin, nivolumabin ja platinapohjaisen kemoterapian yhdistelmähoitoa saaneen ryhmän potilaat saivat ipilimumabia 1 mg/kg laskimoon annosteltuna 30 minuutin kuluessa joka kuudes viikko yhdessä nivolumabin kanssa, jota he saivat 360 mg laskimoon annosteltuna 30 minuutin kuluessa joka kolmas viikko, sekä platinapohjaisen kemoterapian kanssa, jota annettiin joka kolmas viikko kahden hoitojakson ajan. Kemoterapiaryhmän potilaat saivat platinapohjaista kemoterapiaa, jota annettiin kolmen viikon välein neljän hoitojakson ajan; ne potilaat, joiden syöpä oli ei-levyepiteeliperäinen, saattoivat saada valinnaista pemetreksedi-ylläpitohoitoa.
Platinapohjainen kemoterapia koostui karboplatiinista (AUC 5 tai 6) ja pemetreksedistä (500 mg/m2); tai sisplatiinista (75 mg/m2) ja pemetreksedistä (500 mg/m2) ei-levyepiteeliperäisen ei-pienisoluisen keuhkosyövän hoitoon; tai karboplatiinista (AUC 6) ja paklitakselista (200 mg/m2) levyepiteeliperäisen ei-pienisoluisen keuhkosyövän hoitoon.
Hoitoa jatkettiin, kunnes tauti eteni, ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä, tai enintään 24 kuukauden ajan. Hoitoa voitiin jatkaa taudin etenemisen jälkeenkin, jos potilas oli kliinisesti stabiili ja jos tutkija arvioi, että potilas sai hoidosta kliinistä hyötyä. Potilaat, jotka eivät jatkaneet yhdistelmähoitoa ipilimumabin aiheuttaman haittavaikutuksen takia, saivat jatkaa nivolumabi-monoterapiaa. Kasvaimet arvioitiin kuuden viikon välein ensimmäisen tutkimushoidon saamisen jälkeen ensimmäisten 12 kuukauden ajan, minkä jälkeen 12 viikon välein, kunnes tauti eteni tai tutkimushoito lopetettiin.
CA2099LA‑tutkimuksen kaikkien ryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Iän mediaani oli 65 vuotta (vaihteluväli 26–86). 51 % potilaista oli ≥ 65‑vuotiaita ja 10 % oli ≥ 75‑vuotiaita. Suurin osa potilasta oli valkoihoisia (89 %) ja miehiä (70 %). Lähtötason ECOG‑suorituskykyluokka oli 0 (31 %) tai 1 (68 %). 57 %:lla potilaista PD-L1 oli ≥ 1 % ja 37 %:lla PD-L1 oli < 1 %. 31 %:lla histologia oli levyepiteeliperäinen ja 69 %:lla ei-levyepiteeliperäinen. 17 %:lla oli aivojen etäpesäkkeitä, ja 86 % oli entisiä/nykyisiä tupakoitsijoita. Kukaan potilaista ei ollut saanut aiempaa immunoterapiaa.
CA2099LA‑tutkimuksen ensisijainen tehokkuusmuuttuja oli kokonaiselinaika. Tehon muita päätetapahtumia olivat etenemisvapaa elinaika (PFS), objektiivisen vasteen saavuttaneiden osuus (ORR) ja vasteen kesto BICR:n arvioimana.
Tutkimuksen ennalta määritetty välianalyysi, joka tehtiin, kun 351 tapahtumaa oli todettu (87 % lopullisen analyysin tapahtumien suunnitellusta määrästä), osoitti tilastollisesti merkitsevää etua objektiivisen vasteen saavuttaneiden osuudessa, etenemisvapaassa elinajassa ja kokonaisvasteessa potilailla, jotka saivat ipilimumabin, nivolumabin ja platinapohjaisen kemoterapian yhdistelmähoitoa, verrattuna potilaisiin, jotka saivat vain platinapohjaista kemoterapiaa. Kokonaiselinajan seuranta-aika oli vähintään 8,1 kuukautta.
Tehon tulokset esitetään kuvassa 9 (päivitetty kokonaiselinajan analyysi, kun seuranta-aika oli vähintään 12,7 kuukautta) ja taulukossa 14 (primaarianalyysi, kun seuranta-aika oli vähintään 8,1 kuukautta).
Tehon päivitetty analyysi tehtiin, kun kaikki potilaat olivat olleet seurannassa vähintään 12,7 kuukautta (ks. kuva 9). Analyysihetkellä kokonaiselinajan riskitiheyssuhde oli 0,66 (95 %:n luottamusväli: 0,55, 0,80) ja etenemisvapaan elinajan riskitiheyssuhde oli 0,68 (95 %:n luottamusväli: 0,57, 0,82).
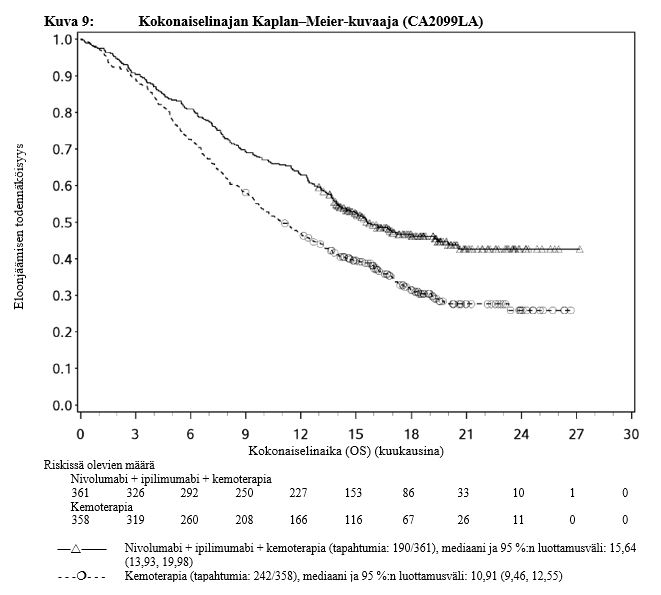
Taulukko 14: Tehoon liittyvät tulokset (CA2099LA)
| ipilimumabi + nivolumabi + kemoterapia | kemoterapia |
|---|---|---|
Kokonaiselinaika | ||
Tapahtumat | 156 (43,2 %) | 195 (54,5 %) |
Riskitiheyssuhde (96,71 %:n luottamusväli)a | 0,69 (0,55, 0,87) | |
Stratifioitu log-rank-testin p‑arvob | 0,0006 | |
Mediaani (kuukautta) (95 %:n luottamusväli) | 14,1 | 10,7 |
Osuus (95 %:n luottamusväli) 6 kuukauden kohdalla | 80,9 (76,4, 84,6) | 72,3 (67,4, 76,7) |
|
|
|
Etenemisvapaa elinaika | ||
Tapahtumat | 232 (64,3 %) | 249 (69,6 %) |
Riskitiheyssuhde (97,48 %:n luottamusväli)a | 0,70 (0,57, 0,86) | |
Stratifioitu log-rank-testin p‑arvoc | 0,0001 | |
Mediaani (kuukautta)d | 6,83 | 4,96 |
Osuus (95 %:n luottamusväli) 6 kuukauden kohdalla | 51,7 (46,2, 56,8) | 35,9 (30,5, 41,3) |
|
|
|
Kokonaishoitovastee | 136 (37,7 %) | 90 (25,1 %) |
(95 %:n luottamusväli) | (32,7, 42,9) | (20,7, 30,0) |
Stratifioitu CMH-testin p-arvof | 0,0003 | |
Täydellinen vaste (CR) | 7 (1,9 %) | 3 (0,8 %) |
Osittainen vaste (PR) | 129 (35,7 %) | 87 (24,3 %) |
|
|
|
Vasteen kesto | ||
Mediaani (kuukautta) | 10,02 | 5,09 |
%, kun kesto oli ≥ 6 kuukauttag | 74 | 41 |
a Perustuu ositettuun Coxin suhteellisen vaaran malliin.
b p‑arvoa on verrattu allokoituun α 0,0329:een tätä välianalyysia varten.
c p‑arvoa on verrattu allokoituun α 0,0252:een tätä välianalyysia varten.
d Kaplan–Meier-arvio.
e Osuus täydellisellä tai osittaisella vasteella; luottamusväli perustuu Clopper–Pearsonin menetelmään.
f p‑arvoa on verrattu allokoituun α 0,0025:een tätä välianalyysia varten.
g Perustuu Kaplan–Meierin arvioihin vasteen kestosta.
CMH = Cochran–Mantel–Haenszel
Potilaat saivat myöhempää systeemistä hoitoa seuraavasti: 28,8 % yhdistelmähoitoa saaneista ja 41,1 % kemoterapiaa saaneista. Potilaat saivat myöhempää immuunihoitoa (mukaan lukien anti-PD-1-hoitoa, anti-PD-L1-hoitoa ja anti-CTLA-4-hoitoa) seuraavasti: 3,9 % yhdistelmähoitoa saaneista ja 27,9 % kemoterapiaa saaneista.
CA2099LA‑tutkimuksessa kemoterapiaan liittyvä alaryhmien deskriptiivinen analyysi osoitti kokonaiselinaikahyötyä potilailla, joita hoidettiin ipilimumabin, nivolumabin ja kemoterapian yhdistelmähoidolla ja joilla histologia oli levyepiteeliperäinen (riskitiheyssuhde [95 %:n luottamusväli] 0,65 [0,46, 0,93], n = 227) ja joilla histologia oli ei-levyepiteeliperäinen (riskitiheyssuhde [95 %:n luottamusväli] 0,72 [0,55, 0,93], n = 492).
Taulukossa 15 on yhteenveto tehoon liittyvistä ennalta määritettyjen alaryhmien analyysien tuloksista mitattuna objektiivisen vasteen saavuttaneiden osuudella, etenemisvapaalla elinajalla ja kokonaisvasteella kasvaimen PD-L1-ilmentymistason mukaan.
Taulukko 15: Tehoon liittyvät tulokset kasvaimen PD-L1-ilmentymistason mukaan (CA2099LA)
| ipilimumabi + nivolumabi + kemoterapia | kemo-terapia | ipilimumabi + nivolumabi + kemoterapia | kemo-terapia | ipilimumabi + nivolumabi + kemoterapia | kemo-terapia | ipilimumabi + nivolumabi + kemoterapia | kemo-terapia |
| PD-L1:n ilmentymistaso < 1 % (n = 264) | PD-L1:n ilmentymistaso ≥ 1 % (n = 406) | PD-L1:n ilmentymistaso ≥ 1 % – 49 % (n = 233) | PD-L1:n ilmentymistaso ≥ 50 % (n = 173) | ||||
Kokonais-elinajan riskitiheys-suhde (95 %:n luottamus-väli)a | 0,65 | 0,67 | 0,69 | 0,64 | ||||
Etenemis-vapaan elinajan riskitiheys-suhde (95 %:n luottamus-väli)a | 0,77 | 0,67 | 0,71 | 0,59 | ||||
Objektiivisen vasteen saavuttaneiden osuus, % | 31,1 | 20,9 | 41,9 | 27,6 | 37,8 | 24,5 | 48,7 | 30,9 |
a Riskitiheyssuhde perustuu ei‑stratifioituun Coxin suhteellisen vaaran malliin
Yhteensä 70 ei-pienisoluista keuhkosyöpää sairastavaa ≥ 75‑vuotiasta potilasta otettiin mukaan CA2099LA‑tutkimukseen (37 potilasta ipilimumabin, nivolumabin ja kemoterapian yhdistelmähoitoa saaneiden ryhmässä ja 33 potilasta kemoterapiaa saaneiden ryhmässä). Tutkimuksen tässä alaryhmässä todettiin ipilimumabin, nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla kokonaiselinajan riskitiheyssuhde 1,36 (95 %:n luottamusväli: 0,74, 2,52) ja etenemisvapaan elinajan riskitiheyssuhde 1,12 (95 %:n luottamusväli: 0,64, 1,96) verrattuna kemoterapiaa saaneisiin. Objektiivisen vasteen saavuttaneiden osuus oli 27,0 % ipilimumabin, nivolumabin ja kemoterapian yhdistelmähoitoa saaneiden ryhmässä ja 15,2 % kemoterapiaa saaneiden ryhmässä. 43 %:lla potilaista, jotka olivat ≥ 75‑vuoden ikäisiä, ipilimumabin, nivolumabin ja kemoterapian yhdistelmähoito lopetettiin. Tietoa ipilimumabin, nivolumabin ja kemoterapian yhdistelmähoidon tehosta ja turvallisuudesta tässä potilasjoukossa on vain vähän.
Alaryhmäanalyysissa todettiin pienentynyt elinaikahyöty ipilimumabin, nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla verrattuna kemoterapiaa saaneisiin potilailla, jotka eivät olleet koskaan tupakoineet. Pienen potilasmäärän vuoksi näistä tiedoista ei voida kuitenkaan tehdä luotettavia johtopäätöksiä.
Keuhkopussin pahanlaatuinen mesoteliooma
Satunnaistettu vaiheen 3 tutkimus: ipilimumabi–nivolumabi-yhdistelmähoito vs. kemoterapia (CA209743)
Ipilimumabi–nivolumabi-yhdistelmähoidon (ipilimumabia 1 mg/kg kuuden viikon välein ja nivolumabia 3 mg/kg kahden viikon välein) turvallisuutta ja tehoa arvioitiin vaiheen 3 satunnaistetussa avoimessa tutkimuksessa (CA209743). Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joilla oli histologisesti vahvistettu ja aiemmin hoitamaton keuhkopussin pahanlaatuinen mesoteliooma, jonka histologia oli epiteloidinen tai ei-epiteloidinen; joiden ECOG-suorituskykyluokka oli 0 tai 1 ja jotka eivät olleet saaneet palliatiivista sädehoitoa 14 vuorokauden sisällä ensimmäisen tutkimushoidon saannista. Tutkimukseen otettiin potilaita riippumatta kasvaimen PD-L1-statuksesta.
Tutkimuksen ulkopuolelle suljettiin potilaat, joilla oli primitiivinen vatsakalvon, sydänpussin, kiveksen tai tuppikalvon mesoteliooma, interstitiaalinen keuhkosairaus, aktiivinen autoimmuunisairaus, systeemistä immunosuppressiota vaativa tila tai aivojen etäpesäke (ellei etäpesäkettä ollut poistettu kirurgisesti tai hoidettu stereotaktisella sädehoidolla siten, ettei etäpesäke ollut kasvanut kolmeen kuukauteen ennen tutkimukseen ottamista). Satunnaistaminen stratifioitiin histologian (epiteloidinen vs. sarkomatoidinen tai sekamuotoinen histologinen alatyyppi) ja sukupuolen (miehet vs. naiset) mukaan.
Yhteensä 605 potilasta satunnaistettiin saamaan joko ipilimumabi–nivolumabi-yhdistelmähoitoa (n = 303) tai kemoterapiaa (n = 302).Ipilimumabi–nivolumabi-yhdistelmähoitoryhmän potilaat saivat 1 mg/kg ipilimumabia laskimoon annosteltuna 30 minuutien kuluessa joka kuudes viikko ja 3 mg/kg nivolumabia laskimoon annosteltuna 30 minuutin kuluessa joka toinen viikko enintään kahden vuoden ajan. Kemoterapiaryhmän potilaat saivat enintään 6 hoitojaksoa kemoterapiaa (yksi hoitojakso oli 21 päivää). Kemoterapia koostui sisplatiinista (75 mg/m2) ja pemetreksedistä (500 mg/m2) tai karboplatiinista (5 AUC) ja pemetreksedistä (500 mg/m2).
Hoitoa jatkettiin, kunnes tauti eteni tai kunnes ilmeni ei-hyväksyttävää toksisuutta tai enintään 24 kuukauden ajan. Hoitoa voitiin jatkaa taudin etenemisen jälkeenkin, jos potilas oli kliinisesti stabiili ja jos tutkija arvioi, että potilas sai hoidosta kliinistä hyötyä. Potilaat, jotka eivät jatkaneet yhdistelmähoitoa ipilimumabin aiheuttaman haittavaikutuksen takia, saivat jatkaa nivolumabi-monoterapiaa. Kasvaimet arvioitiin kuuden viikon välein ensimmäisen tutkimushoidon saamisen jälkeen ensimmäisten 12 kuukauden ajan, minkä jälkeen 12 viikon välein, kunnes tauti eteni tai tutkimushoito lopetettiin.
CA209743-tutkimuksen kaikkien ryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Iän mediaani oli 69 vuotta (vaihteluväli 25–89). 72 prosenttia potilaista oli ≥ 65-vuotiaita ja 26 prosenttia ≥ 75-vuotiaita. Suurin osa potilaista oli valkoihoisia (85 %) ja miehiä (77 %). Lähtötason ECOG-suorituskykyluokka oli 0 (40 %) tai 1 (60 %). 80 prosentilla potilaista PD-L1 oli ≥ 1 % ja 20 prosentilla PD-L1 oli < 1 %. 75 prosentilla histologia oli epiteloidinen ja 25 prosentilla ei-epiteloidinen.
CA209743-tutkimuksen ensisijainen tehokkuusmuuttuja oli kokonaiselinaika. Keskeisiä toissijaisia päätetapahtumia olivat etenemisvapaa elinaika (PFS), objektiivisen vasteen saavuttaneiden osuus (ORR) ja riippumattoman keskitetyn arvioijatahon (Blinded Independent Central Review, BICR) arvioima vasteen kesto, jonka arvioinnissa hyödynnettiin keuhkopussin mesotelioomaa varten muokattuja kiinteiden kasvainten vasteenarviointikriteerejä (RECIST). Toissijaisten päätetapahtumien deskriptiiviset analyysit esitetään taulukossa 16.
Kemoterapiaan verrattuna tutkimuksessa todettiin tilastollisesti merkitsevää hyötyä kokonaiselinajassa potilailla, jotka oli satunnaistettu saamaan ipilimumabi–nivolumabi-yhdistelmähoitoa. Tulos perustui ennaltamääriteltyyn välianalyysiin, jossa arvioitiin 419 tapahtumaa (89 % suunnitellusta loppuanalyysin tapahtumien määrästä). Kokonaiselinajan seuranta-aika oli vähintään 22 kuukautta.
Tulokset tehosta esitetään kuvassa 10 ja taulukossa 16.
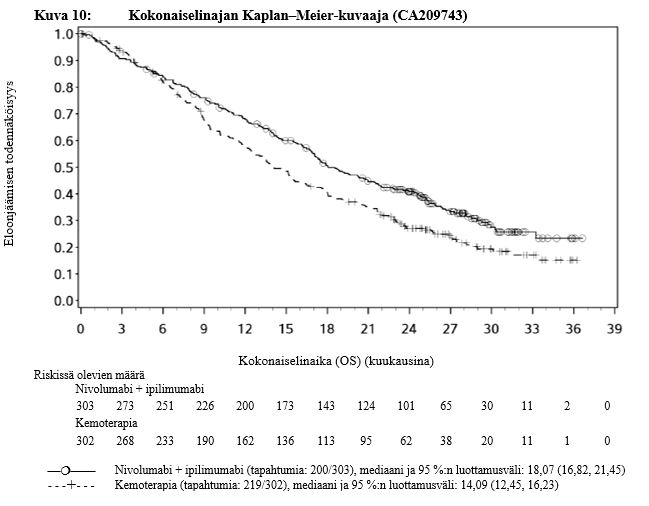
Taulukko 16: Tehoon liittyvät tulokset (CA209743)
| ipilimumabi + nivolumabi | kemoterapia |
|---|---|---|
Kokonaiselinaika (OS) | ||
Tapahtumat | 200 (66 %) | 219 (73 %) |
Riskitiheyssuhde (96,6 %:n luottamusväli)a | 0,74 (0,60, 0,91) | |
Stratifioitu log-rank-testin p‑arvob | 0,002 | |
Mediaani (kuukautta)c (95 %:n luottamusväli) | 18,1 | 14,1 |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdallac | 41 % (35,1, 46,5) | 27 % (21,9, 32,4) |
|
|
|
Etenemisvapaa elinaika | ||
Tapahtumat | 218 (72 %) | 209 (69 %) |
Riskitiheyssuhde (95 %:n luottamusväli)a | 1,0 (0,82, 1,21) | |
Mediaani (kuukautta)c | 6,8 | 7,2 |
|
|
|
Kokonaishoitovaste | 40 % | 43 % |
(95 %:n luottamusväli) | (34,1, 45,4) | (37,1, 48,5) |
Täydellinen vaste (CR) | 1,7 % | 0 |
Osittainen vaste (PR) | 38 % | 43 % |
|
|
|
Vasteen kesto |
|
|
Mediaani (kuukautta)c | 11,0 | 6,7 |
a Stratifioitu Coxin suhteellisen vaaran malli.
b p‑arvoa on verrattu allokoituun α 0,0345:een tätä välianalyysiä varten.
c Kaplan–Meier-arvio.
Potilaat saivat myöhempää systeemistä hoitoa seuraavasti: 44,2 prosenttia yhdistelmähoitoa saaneista ja 40,7 prosenttia kemoterapiaa saaneista. Potilaat saivat myöhempää immuunihoitoa (mukaan lukien anti-PD-1-hoitoa, anti-PD-L1-hoitoa ja anti-CTLA-4-hoitoa) seuraavasti: 3,3 prosenttia yhdistelmähoitoa saaneista ja 20,2 prosenttia kemoterapiaa saaneista.
Taulukossa 17 on yhteenveto tehoon liittyvistä ennalta määritettyjen alaryhmien analyysien tuloksista histologian mukaan ja mitattuna kokonaiselinajalla, etenemisvapaalla elinajalla ja objektiivisen vasteen saavuttaneiden osuudella.
Taulukko 17: Tulokset tehosta histologian mukaan (CA209743)
| Epiteloidinen | Ei-epiteloidinen | ||
| ipilimumabi | kemoterapia | ipilimumabi | kemoterapia |
Kokonaiselinaika (OS) | ||||
Tapahtumat | 157 | 164 | 43 | 55 |
Riskitiheyssuhde (95 %:n luottamusväli)a | 0,85 | 0,46 | ||
Mediaani (kuukautta) | 18,73 (17,05, 21,72) | 16,23 (14,09, 19,15) | 16,89 (11,83, 25,20) | 8,80 (7,62, 11,76) |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 41,2 (34,7, 47,6) | 31,8 (25,7, 38,1) | 39,5 (27,5, 51,2) | 9,7 (3,8, 18,9) |
| ||||
Etenemisvapaa elinaika | ||||
Riskitiheyssuhde | 1,14 (0,92, 1,41) | 0,58 (0,38, 0,90) | ||
Mediaani (kuukautta) | 6,18 (5,49, 7,03) | 7,66 (7,03, 8,31) | 8,31 (3,84, 11,01) | 5,59 (5,13, 7,16) |
|
|
|
|
|
Kokonaishoitovaste | 38,6 % | 47,2 % | 43,3 % | 26,9 % |
(95 %:n luottamusväli)b | (32,3, 45,1) | (40,7, 53,8) | (31,2, 56,0) | (16,8, 39,1) |
| ||||
Vasteen kesto | 8,44 | 6,83 | 24,02 | 4,21 |
Mediaani (kuukautta) | (7,16, 14,59) | (5,59, 7,13) | (8,31, N.A.) | (2,79, 7,03) |
a Riskitiheyssuhde perustuu ei-stratifioituun Coxin suhteellisen vaaran malliin.
b Luottamusväli perustuu Clopper–Pearsonin menetelmään.
c Mediaani laskettu Kaplan–Meierin menetelmän avulla.
Taulukossa 18 on yhteenveto tehoon liittyvistä ennalta määritettyjen alaryhmien analyysien tuloksista mitattuna kokonaiselinajalla, etenemisvapaalla elinajalla ja objektiivisen vasteen saavuttaneiden osuudella kasvaimen lähtötason PD-L1-ilmentymistason mukaan.
Taulukko 18: Tehoon liittyvät tulokset kasvaimen PD-L1-ilmentymistason mukaan (CA209743)
| PD‑L1:n ilmentymistaso < 1 % | PD‑L1:n ilmentymistaso ≥ 1 % | ||
|---|---|---|---|---|
| ipilimumabi | kemoterapia | ipilimumabi | kemoterapia |
Kokonaiselinaika (OS) | ||||
Tapahtumat | 40 | 58 | 150 | 157 |
Riskitiheyssuhde | 0,94 (0,62, 1,40) | 0,69 (0,55, 0,87) | ||
Mediaani (kuukautta) | 17,3 (10,1, 24,3) | 16,5 (13,4, 20,5) | 18,0 (16,8, 21,5) | 13,3 (11,6, 15,4) |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 38,7 (25,9, 51,3) | 24,6 (15,5, 35,0) | 40,8 (34,3, 47,2) | 28,3 (22,1, 34,7) |
| ||||
Etenemisvapaa elinaika | ||||
Riskitiheyssuhde | 1,79 (1,21, 2,64) | 0,81 (0,64, 1,01) | ||
Mediaani (kuukautta) | 4,1 (2,7, 5,6) | 8,3 (7,0, 11,1) | 7,0 (5,8, 8,5) | 7,1 (6,2, 7,6) |
|
|
|
|
|
Kokonaishoitovaste | 21,1 % | 38,5 % | 43,5 % | 44,3 % |
(95 %:n luottamusväli)c | (11,4, 33,9) | (27,7, 50,2) | (37,1, 50,2) | (37,6, 51,1) |
a Riskitiheyssuhde perustuu ei-stratifioituun Coxin suhteellisen vaaran malliin.
b Mediaani laskettu Kaplan–Meierin menetelmän avulla.
c Luottamusväli perustuu Clopper–Pearsonin menetelmään.
CA209743-tutkimukseen osallistui yhteensä 157 keuhkopussin pahanlaatuista mesotelioomaa sairastavaa ≥ 75-vuotiasta potilasta (78 ipilimumabi–nivolumabi-yhdistelmähoitoa saaneessa ryhmässä ja 79 kemoterapiaryhmässä). Tässä alaryhmässä kokonaiselinajan riskitiheyssuhde ipilimumabi–nivolumabi-yhdistelmähoitoryhmässä oli 1,02 (95 %:n luottamusväli: 0,70, 1,48) kemoterapiaryhmään verrattuna. Tutkimuksessa todettiin, että 75-vuotiailla ja tätä vanhemmilla ilmeni enemmän vakavia haittavaikutuksia kuin kaikilla ipilimumabi–nivolumabi-yhdistelmähoitoa saaneilla ja että 75-vuotiaat ja tätä vanhemmat keskeyttivät hoidon haittavaikusten takia useammin kuin kaikki ipilimumabi–nivolumabi-yhdistelmähoitoa saaneet potilaat (ks. kohta Haittavaikutukset). Alaryhmäanalyysin havainnollistavan luonteen vuoksi luotettavia johtopäätöksiä ei kuitenkaan voida tehdä.
dMMR- tai MSI-H-tyyppinen kolorektaalisyöpä
Avoin tutkimus nivolumabin ja ipilimumabin yhdistelmähoidosta verrattuna solunsalpaajahoitoon dMMR- tai MSI‑H-tyyppisen kolorektaalisyövän hoidossa potilailla, jotka eivät olleet saaneet aiempaa hoitoa etäpesäkkeiseen tautiin
Ipilimumabin (1 mg/kg) ja nivolumabin (240 mg/kg) yhdistelmän, jota annettiin 3 viikon välein enintään 4 annosta ja jonka jälkeen nivolumabia annettiin monoterapiana 480 mg 4 viikon välein, turvallisuutta ja tehoa leikkaukseen soveltumattoman tai etäpesäkkeisen kolorektaalisyövän (kasvaimen MSI‑H- tai dMMR-status oli tiedossa) ensilinjan hoidossa arvioitiin satunnaistetussa, useilla hoitoryhmillä tehdyssä, vaiheen 3 avoimessa tutkimuksessa (CA2098HW). Tutkimuksen hoitoryhmiä olivat nivolumabi monoterapiana, nivolumabin ja ipilimumabin yhdistelmä tai tutkijan valitsema solunsalpaajahoito. Kasvaimen MSI‑H- tai dMMR-status määritettiin paikallisen käytännön mukaan PCR-, NGS- tai IHC-menetelmillä. MSI‑H-status määritettiin keskitetysti PCR-testillä (Idylla MSI) ja dMMR-status IHC-testillä (Omnis MMR) retrospektiivisesti potilaiden kasvainnäytteistä, joita käytettiin MSI‑H-/dMMR-statuksen määrittämiseen paikallisesti. Ensisijainen tehopopulaatio koostui potilaista, joiden MSI‑H-/dMMR-status vahvistettiin kummalla tahansa keskitetyllä testillä. Tutkimuksesta suljettiin pois potilaat, joilla oli oireita aiheuttavia aivometastaaseja tai aktiivinen autoimmuunisairaus tai jotka käyttivät systeemisiä kortikosteroideja tai immunosuppressantteja tai joita oli hoidettu tarkistuspisteen estäjillä. Satunnaistaminen ositettiin kasvaimen sijainnin (oikea vs. vasen) perusteella. Solunsalpaajaryhmään satunnaistetuille potilaille voitiin antaa nivolumabin ja ipilimumabin yhdistelmää BICR:n arvioiman taudin etenemisen jälkeen.
Tutkimukseen satunnaistettiin yhteensä 303 aiemmin hoitamatonta potilasta, joilla oli etäpesäkkeinen tauti. 202 potilasta satunnaistettiin saamaan nivolumabin ja ipilimumabin yhdistelmää ja 101 satunnaistettiin saamaan solunsalpaajahoitoa. Näistä potilaista 255:n MSI‑H-/dMMR-status oli vahvistettu keskitetysti. Heistä 171 oli nivolumabin ja ipilimumabin yhdistelmää saavassa ryhmässä ja 84 solunsalpaajahoitoa saavassa ryhmässä. Nivolumabin ja ipilimumabin yhdistelmää saavassa ryhmässä potilaat saivat ipilimumabia 1 mg/kg ja nivolumabia 240 mg 3 viikon välein enintään 4 annosta. Tämän jälkeen nivolumabia annettiin monoterapiana 480 mg 4 viikon välein. Solunsalpaajaryhmän potilaat saivat hoitoa seuraavasti: mFOLFOX6-hoito (oksaliplatiinia, leukovoriinia ja fluorourasiilia) joko bevasitsumabin tai setuksimabin kanssa tai ilman niitä: oksaliplatiinia 85 mg/m2, leukovoriinia 400 mg/m2 ja fluorourasiilia 400 mg/m2 boluksena, minkä jälkeen fluorourasiilia 2400 mg/m2 46 tunnin aikana 2 viikon välein. Bevasitsumabia 5 mg/kg tai setuksimabia 500 mg/m2 annettiin ennen mFOLFOX6-hoitoa 2 viikon välein; tai FOLFIRI-hoitoa (irinotekaania, leukovoriinia ja fluorourasiilia) joko bevasitsumabin tai setuksimabin kanssa tai ilman niitä: irinotekaania 180 mg/m2, leukovoriinia 400 mg/m2 ja fluorourasiilia 400 mg/m2 boluksena, minkä jälkeen fluorourasiilia 2400 mg/m2 46 tunnin aikana 2 viikon välein. Bevasitsumabia 5 mg/kg tai setuksimabia 500 mg/m2 annettiin ennen FOLFIRI-hoitoa 2 viikon välein. Hoitoa jatkettiin, kunnes tauti eteni tai kunnes ilmeni ei-hyväksyttävää toksisuutta tai nivolumabin ja ipilimumabin yhdistelmän kohdalla enintään 24 kuukauden ajan. Potilaat, jotka eivät jatkaneet yhdistelmähoitoa ipilimumabin aiheuttaman haittavaikutuksen takia, saivat jatkaa pelkän nivolumabin käyttöä. RECIST v1.1:n mukaiset kasvainten arvioinnit tehtiin 6 viikon välein ensimmäisten 24 viikon ajan, sen jälkeen 8 viikon välein viikolle 96 asti, sen jälkeen 16 viikon välein viikolle 146 asti ja sen jälkeen 24 viikon välein.
Lähtötason ominaisuudet kaikilla satunnaistetuilla potilailla, jotka eivät olleet saaneet aiemmin hoitoa etäpesäkkeiseen tautiin: iän mediaani oli 63 vuotta (vaihteluväli 21–87) ja 46 % oli ≥ 65‑vuotiaita ja 18 % ≥ 75‑vuotiaita; 46 % oli miehiä ja 86 % oli valkoihoisia. Lähtötason ECOG-toimintakykyluokka oli 0 (54 %) ja ≥ 1 (46 %), kasvain sijaitsi oikealla puolella 68 %:lla potilaista ja vasemmalla puolella 32 %:lla potilaista, ja 39 potilaalla niistä 223 potilaista, joiden status tunnettiin, oli vahvistettu Lynchin oireyhtymä. Lähtötason ominaisuudet potilailla, jotka eivät olleet saaneet aiemmin hoitoa etäpesäkkeiseen tautiin ja joiden MSI‑H-/dMMR-status oli keskitetysti vahvistettu, olivat yhdenmukaiset kaikkien niiden satunnaistettujen potilaiden kanssa, jotka eivät olleet saaneet aiemmin hoitoa. Solunsalpaajahoitoon satunnaistetuista 101 potilaasta 88 sai tutkimussuunnitelman mukaista solunsalpaajahoitoa, mukaan lukien oksaliplatiinia sisältäviä hoitoja (58 %) ja irinotekaania sisältäviä hoitoja (42 %). Tämän lisäksi 66 potilasta sai täsmälääkkeenä joko bevasitsumabia (64 %) tai setuksimabia (11 %).
Tutkimuksen ensisijainen tehokkuusmuuttuja oli BICR:n arvioima RECIST 1.1 -kriteerien mukainen etenemisvapaa elinaika (PFS). Muita tehokkuusmuuttujia olivat BICR:n arvioima objektiivisen vasteen saavuttaneiden osuus (ORR), kokonaiselinaika (OS) ja vasteen kesto.
Tutkimuksen ensisijainen päätetapahtuma saavutettiin suunnitellussa välianalyysissa, jossa BICR:n arvioiman etenemisvapaan elinajan osoitettiin parantuneen tilastollisesti merkitsevästi nivolumabin ja ipilimumabin yhdistelmää saaneilla potilailla, joiden MSI‑H-/dMMR-status oli keskitetysti vahvistettu, solunsalpaajahoitoa saaneeseen ryhmään verrattuna. BICR:n arvioiman etenemisvapaan elinajan tulokset on esitetty taulukossa 19 ja kuvassa 11. Välianalyysin ajankohtana muita päätetapahtumia (mukaan lukien tiedot pelkkää nivolumabia saaneesta ryhmästä) ei testattu testaushierarkian takia.
Taulukko 19: Tulokset tehosta keskitetysti vahvistetun MSI‑H-/dMMR-tyyppisen kolorektaalisyövän ensilinjan hoidossa (CA2098HW)a
| ipilimumabi + nivolumabi | kemoterapia | |
|---|---|---|---|
Etenemisavapaa elinaika |
|
| |
Tapahtumia | 48 (28 %) | 52 (62 %) | |
Riskitiheyssuhde |
| 0,21 |
|
95 %:n luottamusväli |
| (0,14–0,32) |
|
p‑arvob |
| < 0,0001 |
|
| |||
Mediaani (95 %:n luottamusväli) (kuukautta) | NR (38,4–NR) | 5,9 (4,4–7,8) | |
a Seurannan mediaani 31,5 kuukautta (vaihteluväli 6,1–48,4 kuukautta).
b Ositetun 2‑puolisen log‑rank-testin perusteella
Kuva 11: Etenemisvapaan elinajan Kaplan‑Meier-kuvaaja keskitetysti vahvistetun MSI‑H-/dMMR-tyyppisen kolorektaalisyövän ensilinjan hoidossa (CA2098HW)

Avoin tutkimus nivolumabin ja ipilimumabin yhdistelmähoidosta dMMR- tai MSI‑H-tyyppisen kolorektaalisyövän hoidossa potilailla, jotka olivat saaneet aiempaa fluoropyrimidiinipohjaista yhdistelmäsolunsalpaajahoitoa
Ipilimumabi 1 mg/kg- ja nivolumabi 3 mg/kg -yhdistelmähoidon turvallisuutta ja tehoa dMMR- ja MSI-H-tyyppisen metastaattisen kolorektaalisyövän hoidossa arvioitiin yksihaaraisessa avoimessa vaiheen 2 monikeskustutkimuksessa (CA209142).
Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joilla oli paikallisesti määritetty dMMR- tai MSI-H-status ja joiden tauti oli edennyt aiemman fluoropyrimidiini- ja oksaliplatiinihoidon tai irinotekaanihoidon aikana tai sen jälkeen tai jotka eivät olleet sietäneet fluoropyrimidiini- ja oksaliplatiinihoitoa tai irinotekaanihoitoa. Niillä potilailla, joiden viimeisin aiempi hoito oli liitännäishoito, taudin oli pitänyt edetä hoidon aikana tai kuuden kuukauden sisällä liitännäishoitona annetusta kemoterapiasta. Potilaiden ECOG-suorituskykyluokka oli 0 tai 1, ja heidät otettiin mukaan tutkimukseen kasvaimen PD-L1-ilmentymistasosta riippumatta. Potilaat, joilla oli aktiivisia aivometastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila, suljettiin pois tutkimuksesta.
Yhteensä 119 potilasta sai joka kolmas viikko neljän annoksen ajan yhdistelmähoitoa, jossa ipilimumabia 1 mg/kg annosteltiin laskimoon 90 minuutin ajan ja nivolumabia 3 mg/kg annosteltiin laskimoon 60 minuutin ajan. Tämän jälkeen potilaille annettiin nivolumabia monoterapiana 3 mg/kg joka toinen viikko. Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt hoitoa. Kasvaimet arvioitiin kiinteiden kasvainten vasteenarviointikriteerien (RECIST) version 1.1 mukaisesti ensimmäisen 24 viikon ajan joka 6. viikko ja tämän jälkeen joka 12. viikko. Ensisijainen muuttuja oli tutkijan arvioima objektiivisen vasteen saavuttaneiden osuus (ORR). Toissijaisia muuttujia olivat riippumattoman keskitetyn arvioijatahon (Blinded Independent Central Review, BICR) arvioima objektiivisen vasteen saavuttaneiden osuus ja niiden potilaiden määrä, joilla sairaus saatiin hallintaan. Vasteen kesto ja vasteen saavuttamisaika sisältyivät objektiivisen vasteen saavuttaneiden osuuden analyysiin. Havainnollistavia (eksploratiivisia) muuttujia olivat etenemisvapaa elinaika (PFS) ja kokonaiselinaika (OR).
Potilaiden mediaani-ikä oli 58 vuotta (vaihteluväli 21–88). Potilaista 32 % oli iältään ≥65-vuotiaita ja 9 % oli iältään ≥75-vuotiaita. Potilaista 59 % oli miehiä ja 92 % valkoihoisia. Lähtötason ECOG-suorituskykyluokka oli 0 (45 %) tai 1 (55 %). Potilaista 25 %:lla oli BRAF-mutaatioita, 37 %:lla oli KRAS-mutaatioita ja 12 %:lla mutaatiostatus oli tuntematon. 119:stä hoidetusta potilaasta 109 oli saanut aiempaa fluoropyrimidiinipohjaista kemoterapiaa metastaattiseen tautiin ja 9 liitännäishoitona. Ennen tutkimukseen osallistumista kaikista 119:sta hoitoa saaneesta potilaasta 118 potilasta (99 %) oli saanut fluorourasiilia, 111 potilasta (93 %) oli saanut oksaliplatiinia, 87 potilasta (73 %) oli saanut irinotekaania osana aiempia hoitoja ja 82 potilasta (69 %) oli saanut aiempaa fluoropyrimidiini-, oksaliplatiini- ja irinotekaanihoitoa. 23 % potilaista oli saanut aiemmin yhtä hoitoa, 36 % kahta hoitoa, 24 % kolmea hoitoa ja 16 % neljää tai useampaa hoitoa. 29 % potilaista oli saanut EGFR:n estäjää.
Tulokset tehosta (seuranta-aika vähintään 46,9 kuukautta; seuranta-ajan mediaani 51,1 kuukautta) esitetään taulukossa 20.
Taulukko 20: Tulokset tehosta (CA209142) MSI-H- tai dMMR-tyyppistä kolorektaalisyöpää sairastavilla potilailla*
| ipilimumabi + nivolumabi |
Vahvistettu objektiivinen vaste, n (%) | 77 (64,7) |
(95 %:n luottamusväli) | (55,4, 73,2) |
Täydellinen vaste (CR), n (%) | 15 (12,6) |
Osittainen vaste (PR), n (%) | 62 (52,1) |
Stabiili tauti (SD), n (%) | 25 (21,0) |
Vasteen kesto |
|
Mediaani (vaihteluväli) kuukausina | NR (1,4, 58,0+) |
Vasteen saavuttamisajan mediaani |
|
Kuukausina (vaihteluväli) | 2,8 (1,1, 37,1) |
* tutkijan arvion mukaan
”+” tarkoittaa sensuroitunutta havaintoa
NR = ei saavutettu
BICR:n arvioima objektiivisen vasteen saavuttaneiden osuus oli 61,3 % (95 %:n luottamusväli: 52,0, 70,1), mukaan lukien täydellisen vasteen osuus 20,2 % (95 %:n luottamusväli: 13,4, 28,5), osittaisen vasteen osuus 41,2 % (95 %:n luottamusväli: 32,2, 50,6) ja stabiilin taudin raportoitu osuus 22,7 %. BICR:n arviot olivat yleisesti ottaen yhteneviä tutkijan arvion kanssa. Vahvistettuja vasteita havaittiin kasvaimen BRAF- tai KRAS-mutaatiostatuksesta ja PD-L1-ilmentymistasosta riippumatta.
119 potilaasta 11 (9,2 %) oli ≥ 75-vuotiaita. Tutkijan arvioima objektiivisen vasteen saavuttaneiden osuus ≥ 75-vuotiailla potilailla oli 45,5 % (95 %:n luottamusväli: 16,7, 76,6).
Ruokatorven levyepiteelikarsinooma
Satunnaistettu faasin 3 tutkimus ipilimumabista yhdistelmähoitona nivolumabin kanssa vs. kemoterapia ensilinjan hoitona (CA209648)
Ipilimumabin ja nivolumabin yhdistelmähoidon turvallisuutta ja tehoa arvioitiin satunnaistetussa, aktiivisella vertailuvalmisteella kontrolloidussa avoimessa faasin 3 tutkimuksessa (CA209648). Tutkimukseen osallistui aikuisia potilaita (18‑vuotiaita tai tätä vanhempia), joilla oli aiemmin hoitamaton, leikkaukseen soveltumaton, edennyt, uusiutunut tai etäpesäkkeinen ruokatorven levyepiteelikarsinooma (OSCC). Tutkimukseen otettiin potilaita riippumatta kasvainsolujen PD-L1‑statuksesta, ja kasvainsolujen PD-L1‑ilmentyminen määritettiin PD‑L1 IHC 28‑8 pharmDx ‑ testausmenetelmän avulla. Potilailla piti olla ruokatorven levyepiteelikarsinooma tai adenoskvamoosi karsinooma, jota ei voitu hoitaa kemosädehoidolla ja/tai leikkauksella. Aikaisempi liitännäishoitona, esiliitännäishoitona tai definitiivisenä hoitona annettu kemoterapia, sädehoito tai kemosädehoito sallittiin, jos se oli annettu osana parantavassa tarkoituksessa annettua hoitoa ennen tutkimukseen osallistumista. Potilaat, joiden lähtötason suorituskykyluokka oli ≥ 2, joilla oli oireisia aivometastaaseja tai aktiivinen autoimmuunisairaus, jotka käyttivät systeemisiä kortikosteroideja tai immunosuppressiivisia lääkkeitä tai joilla oli suuri riski verenvuodolle tai fistelille kasvaimen havaittavan invaasion ruokatorven kasvainta lähellä oleviin elimiin aiheuttamana, suljettiin pois tutkimuksesta. Satunnaistaminen stratifioitiin kasvainsolujen PD-L1‑statuksen (≥ 1 % vs. < 1 % tai määrittämätön), alueen (Itä-Aasia vs. muu Aasia vs. muu maailma), ECOG‑suorituskykyluokan (0 vs. 1) ja metastaaseja sisältävien elinten lukumäärän (≤ 1 vs. ≥ 2) mukaan.
Yhteensä 649 potilasta satunnaistettiin saamaan joko ipilimumabin ja nivolumabin yhdistelmähoitoa (n = 325) tai kemoterapiaa (n = 324). Näistä potilaista 315 potilaalla kasvainsolujen PD-L1‑ilmentymä oli ≥ 1 %; 158 potilaalla ipilimumabi–nivolumabi-yhdistelmähoidon ryhmässä ja 157 potilaalla kemoterapiaryhmässä. Ipilimumabi–nivolumabi-yhdistelmähoitoryhmässä olleet potilaat saivat ipilimumabia 1 mg/kg kuuden viikon välein ja nivolumabia 3 mg/kg kahden viikon välein. Kemoterapiaryhmässä olleet potilaat saivat fluorourasiilia 800 mg/m2/vrk laskimoon päivästä 1 päivään 5 (5 päivän ajan) ja sisplatiinia 80 mg/m2 laskimoon päivänä 1 (4 viikon jaksosta). Hoitoa jatkettiin, kunnes tauti eteni tai kunnes ilmeni ei-hyväksyttävää toksisuutta tai enintään 24 kuukauden ajan. Potilaat, jotka eivät jatkaneet yhdistelmähoitoa ipilimumabin aiheuttaman haittavaikutuksen takia, saivat jatkaa nivolumabi-monoterapiaa.
Hoitoryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Potilasryhmässä, jossa kasvainsolujen PD-L1‑ilmentymä oli ≥ 1 %, iän mediaani oli 63 vuotta (vaihteluväli: 26–85), 8,2 % oli ≥ 75‑vuotiaita, 81,8 % oli miehiä, 73,1 % oli aasialaisia ja 23,3 % oli valkoihoisia. Potilaiden ruokatorven levyepiteelikarsinooma (98,9 %) tai adenoskvamoosi karsinooma (1,1 %) oli histologisesti vahvistettu. Lähtötason ECOG‑suorituskykyluokka oli 0 (45,2 %) tai 1 (54,8 %).
Ensisijaiset tehokkuusmuuttujat olivat etenemisvapaa elinaika (BICR:n arvioimana) ja kokonaiselinaika, jotka arvioitiin potilailla, joiden kasvainsolujen PD-L1‑ilmentymä oli ≥ 1 %. Toissijaiset päätetapahtumat ennalta määritellyn hierarkkisen testausjärjestyksen mukaan olivat muun muassa kokonaiselinaika, etenemisvapaa elinaika (BICR:n arvioimana) ja objektiivisen vasteen saavuttaneiden osuus (BICR:n arvioimana) kaikilla satunnaistetuilla potilailla. Kasvaimet arvioitiin RECIST v1.1 ‑kriteerien mukaan joka 6. viikko viikkoon 48 asti, viikko 48 mukaan lukien, ja sen jälkeen joka 12. viikko.
Kokonaiselinajan osoitettiin primaarisessa ennalta määritetyssä analyysissä, kun seuranta‑aika oli vähintään 13,1 kuukautta, olevan tilastollisesti merkitsevästi parempi potilailla, joiden kasvainsolujen PD-L1‑ilmentymä oli ≥ 1 %. Tulokset tehosta esitetään taulukossa 21.
Taulukko 21: Tulokset tehosta potilailla, joiden kasvainsolujen PD-L1 oli ≥ 1 % (CA209648)
| ipilimumabi + nivolumabi | kemoterapiaa | |
Kokonaiselinaika |
|
| |
Tapahtumat | 106 (67,1 %) | 121 (77,1 %) | |
Riskitiheyssuhde (98,6 %:n luottamusväli)b | 0,64 (0,46, 0,90) | ||
p-arvoc | 0,0010 | ||
Mediaani (95 %:n luottamusväli) (kuukausina)d | 13,70 (11,24, 17,02) | 9,07 (7,69, 9,95) | |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdallad | 57,1 (49,0, 64,4) | 37,1 (29,2, 44,9) | |
Etenemisvapaa elinaikae |
|
| |
Tapahtumat | 123 (77,8 %) | 100 (63,7 %) | |
Riskitiheyssuhde (98,5 %:n luottamusväli)b | 1,02 (0,73, 1,43) | ||
p-arvoc | 0,8958 | ||
Mediaani (95 %:n luottamusväli) (kuukausina)d | 4,04 (2,40, 4,93) | 4,44 (2,89, 5,82) | |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdallad | 26,4 (19,5, 33,9) | 10,5 (4,7, 18,8) | |
Kokonaisvaste, n (%)e | 56 (35,4) | 31 (19,7) | |
(95 %:n luottamusväli) | (28,0, 43,4) | (13,8, 26,8) | |
Täydellinen vaste | 28 (17,7) | 8 (5,1) | |
Osittainen vaste | 28 (17,7) | 23 (14,6) | |
Vasteen kestoe |
|
| |
Mediaani (95 %:n luottamusväli) (kuukausina)d | 11,83 (7,10, 27,43) | 5,68 (4,40, 8,67) | |
Vaihteluväli | 1,4+, 34,5+ | 1,4+, 31,8+ | |
a Fluorourasiili ja sisplatiini.
b Perustuu stratifioituun Coxin suhteellisen vaaran malliin.
c Perustuu stratifioituun kaksipuoliseen log‑rank-merkitsevyystestiin.
d Perustuu Kaplan–Meierin arvioihin.
e BICR:n arvioimana.
Vähintään 20 kuukauden seuranta‑ajan jälkeen tehdyn päivitetyn deskriptiivisen analyysin mukaiset kokonaiselinajan tulokset olivat yhteneviä primaarianalyysin kanssa. Kokonaiselinajan mediaani oli 13,70 kuukautta (95 %:n luottamusväli: 11,24, 17,41) ipilimumabin ja nivolumabin yhdistelmähoitoa saaneilla vs. 9,07 kuukautta (95 %:n luottamusväli: 7,69, 10,02) kemoterapiaa saaneilla (riskistiheysuhde = 0,63; 95 %:n luottamusväli: 0,49, 0,82). Etenemisvapaan elinajan mediaani oli 4,04 kuukautta (95 %:n luottamusväli: 2,40, 4,93) ipilimumabin ja nivolumabin yhdistelmähoitoa saaneilla vs. 4,44 kuukautta (95 %:n luottamusväli: 2,89, 5,82) kemoterapiaa saaneilla (riskitiheyssuhde = 1,02; 95 %:n luottamusväli: 0,77, 1,34). Objektiivisen vasteen saavuttaneiden osuus oli 35,4 % (95 %:n luottamusväli: 28,0, 43,4) ipilimumabin ja nivolumabin yhdistelmähoitoa saaneilla vs. 19,7 % (95 %:n luottamusväli: 13,8, 26,8) kemoterapiaa saaneilla.
Kuvassa 12 esitetään Kaplan–Meier-kuvaajat kokonaiselinajasta vähintään 20 kuukauden seuranta-ajalla.
Kuva 12: Kaplan–Meier-kuvaajat kokonaiselinajasta potilailla, joiden kasvainsolujen PD‑L1 oli ≥ 1 % (CA209648)

Hepatosellulaarinen karsinooma
Ipilimumabin (3 mg/kg) ja nivolumabin (1 mg/kg) yhdistelmän, jota annettiin 3 viikon välein enintään 4 annosta ja jonka jälkeen nivolumabia annettiin monoterapiana 480 mg 4 viikon välein, turvallisuutta ja tehoa leikkaukseen soveltumattoman tai edenneen hepatosellulaarisen karsinooman (HCC) ensilinjan hoidossa arvioitiin satunnaistetussa, aktiivikontrolloidussa, vaiheen 3 avoimessa tutkimuksessa (CA2099DW). Tutkimuksessa oli mukana aikuispotilaita (vähintään 18‑vuotiaita), joilla oli histologisesti vahvistettu hepatosellulaarinen karsinooma, Child‑Pugh-luokan A maksasairaus ja ECOG-toimintakykyluokka 0 tai 1 ja jotka eivät olleet saaneet aiemmin systeemistä hoitoa edenneeseen tautiin. Esofagogastroduodenoskopia ei ollut pakollinen ennen tutkimukseen ottamista. Tutkimukseen otettiin aikuisia, joiden tauti ei soveltunut hoidettavaksi leikkauksella ja/tai lokoregionaalisella hoidolla tai oli edennyt näiden hoitojen jälkeen. Aiemmat systeemiset neoadjuvantti- tai adjuvanttihoidot olivat sallittuja. Tutkimuksesta suljettiin pois potilaat, joille oli tehty aiemmin maksansiirto tai joilla oli aktiivinen autoimmuunisairaus, aivometastaaseja tai leptomeningeaalisia metastaaseja, aiempi maksaperäinen enkefalopatia (12 kuukauden sisällä satunnaistamisesta), kliinisesti merkittävä askites, systeemistä immunosuppressiivista hoitoa vaativa tila, HIV-infektio tai aktiivinen samanaikainen hepatiitti B -virusinfektio (HBV) ja hepatiitti C -virusinfektio (HCV) tai HBV ja hepatiitti D -virusinfektio (HDV). Satunnaistaminen ositettiin etiologian (HBV vs. HCV vs. ei-virusperäinen), makrovaskulaarisen invaasion ja/tai maksan ulkopuolelle leviämisen (kyllä tai ei) sekä alfafetoproteiiniarvojen (≥ 400 tai < 400 ng/ml) perusteella.
Yhteensä 668 potilasta satunnaistettiin saamaan joko ipilimumabi–nivolumabi-yhdistelmähoitoa (n = 335) tai tutkijan valitsemaa (n = 333) lenvatinibi- tai sorafenibihoitoa. Tutkijan valitsemaa hoitoa saaneessa ryhmässä 85 % potilaista sai lenvatinibia ja 15 % sorafenibia. Ipilimumabin ja nivolumabin yhdistelmää saavassa ryhmässä potilaat saivat ipilimumabia 3 mg/kg ja nivolumabia 1 mg/kg 3 viikon välein enintään 4 annosta. Tämän jälkeen nivolumabia annettiin monoterapiana 480 mg 4 viikon välein. Tutkijan valitsemaa hoitoa saaneessa ryhmässä potilaat saivat joko lenvatinibia 8 mg vuorokaudessa suun kautta (jos paino oli < 60 kg) tai 12 mg vuorokaudessa suun kautta (jos paino oli ≥ 60 kg) tai sorafenibia 400 mg kahdesti vuorokaudessa suun kautta. Hoitoa jatkettiin, kunnes tauti eteni tai kunnes ilmeni ei-hyväksyttävää toksisuutta tai enintään 24 kuukauden ajan. Potilaat, jotka eivät jatkaneet yhdistelmähoitoa ipilimumabin aiheuttaman haittavaikutuksen takia, saivat jatkaa pelkän nivolumabin käyttöä. Kasvaimet arvioitiin lähtötasolla, satunnaistamisen jälkeen viikolla 9 ja viikolla 16, sen jälkeen 8 viikon välein viikolle 48 saakka ja sen jälkeen 12 viikon välein taudin etenemiseen, hoidon lopettamiseen tai seuraavan hoidon aloittamiseen saakka.
Hoitoryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Iän mediaani oli 66 vuotta (vaihteluväli: 20–89). Potilaista 53 % oli ≥ 65‑vuotiaita ja 16 % oli ≥ 75‑vuotiaita. Potilaista 53 % oli valkoihoisia, 44 % aasialaisia, 2,2 % tummaihoisia ja 82 % miehiä. Lähtötason ECOG-toimintakykyluokka oli 0 (71 %) tai 1 (29 %). Potilaista 34 %:lla oli HBV-infektio, 28 %:lla oli HCV-infektio ja 36 %:lla ei ollut näyttöä HBV- tai HCV-infektiosta. Potilaista 19 %:lla oli alkoholin aiheuttama maksasairaus ja 11 %:lla alkoholiin liittymätön rasvamaksa. Suurimmalla osalla potilaista oli lähtötilanteessa BCLC-luokituksen mukainen asteen C tauti (73 %), 19 %:lla oli asteen B tauti ja 6 %:lla asteen A tauti. Child‑Pugh-pistearvo oli 77 %:lla potilaista 5, 20 %:lla potilaista 6 ja 3 %:lla potilaista ≥ 7. Kaikkiaan 54 %:lla potilaista tauti oli levinnyt maksan ulkopuolelle, 25 %:lla esiintyi makrovaskulaarista invaasiota ja 33 %:lla AFP-arvo oli ≥ 400 µg/l.
Tutkimus osoitti tilastollisesti merkitsevää hyötyä kokonaiselinajassa ja kokonaisvasteessa potilailla, jotka oli satunnaistettu saamaan ipilimumabia yhdistelmähoitona nivolumabin kanssa, verrattuna potilaisiin, jotka oli satunnaistettu saamaan tutkijan valitsemaa lenvatinibi- tai sorafenibihoitoa. Tulokset tehosta esitetään taulukossa 22 ja kuvassa 13.
Taulukko 22: Tulokset tehosta hepatosellulaarisen karsinooman ensilinjan hoidossa (CA2099DW)a
| ipilimumabi + nivolumabi | lenvatinibi tai sorafenibi |
|---|---|---|
Kokonaiselinaika |
|
|
Tapahtumia | 194 (58 %) | 228 (68 %) |
Mediaani (kuukautta) (95 %:n luottamusväli) | 23,7 (18,8–29,4) | 20,6 (17,5–22,5) |
Riskitiheyssuhde (95 %:n luottamusväli)b | 0,79 (0,65–0,96) | |
p‑arvoc | 0,0180 | |
Kokonaisvaste, n (%)d | 121 (36,1) | 44 (13,2) |
(95 %:n luottamusväli) | (31,0–41,5) | (9,8–17,3) |
p‑arvoe | < 0,0001 | |
Täydellinen vaste (%) | 23 (6,9) | 6 (1,8) |
Osittainen vaste (%) | 98 (29,3) | 38 (11,4) |
Vasteen kesto (kuukausina)d |
|
|
Mediaani (95 %:n luottamusväli) | 30,4 (21,2–N.A.) | 12,9 (10,2–31,2) |
a Seuranta-aika vähintään 26,8 kuukautta. Seurannan mediaani 35,2 kuukautta.
b Perustuu stratifioituun Coxin suhteellisen vaaran malliin.
c Perustuu kaksipuoliseen ositettuun log‑rank‑merkitsevyystestiin. Tilastollisen merkitsevyyden p‑arvon raja ≤ 0,0257.
d BICR:n arvioima ja RECIST 1.1 -kriteerien mukainen.
e Perustuu kaksipuoliseen stratifioituun Cochran-Mantel-Haenszelin testiin. Tilastollisen merkitsevyyden p‑arvon raja ≤ 0,025.
Kuva 13: Kokonaiselinajan Kaplan‑Meier-kuvaaja hepatosellulaarisen karsinooman ensilinjan hoidossa (CA2099DW)
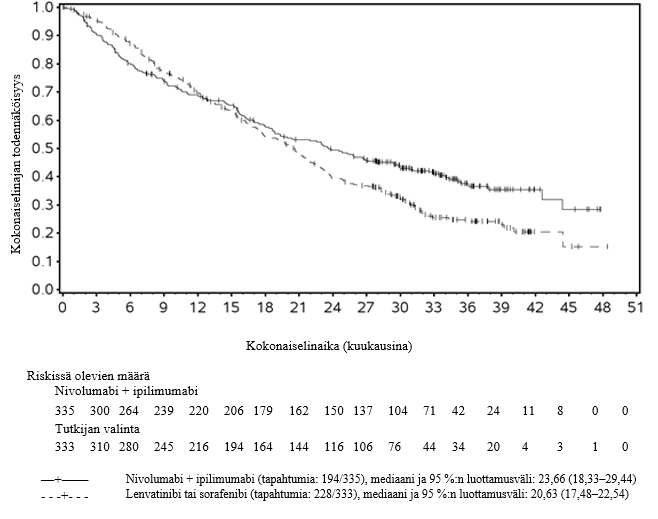
Pediatriset potilaat
Ipilimumabi monoterapiana
CA184070‑tutkimus oli faasin I avoin, suurenevin ipilimumabiannoksin tehty monikeskustutkimus, jossa tutkittiin yli 1-vuotiaita – alle 21-vuotiaita pediatrisia potilaita, joilla oli mitattavissa/arvioitavissa oleva hoitamaton, uusiutunut tai hoitoon vastaamaton kiinteä, pahanlaatuinen kasvain, jota ei voida parantaa tavanomaisin hoidoin. Tutkimukseen otettiin mukaan 13 alle 12-vuotiasta potilasta ja 20 12‑vuotiasta tai sitä vanhempaa potilasta. Ipilimumabia annettiin joka kolmas viikko yhteensä neljän annoksen verran. Sen jälkeen ipilimumabia annettiin vielä 12 viikon ajan, jos annosta rajoittavaa toksisuutta ei havaittu eikä tauti edennyt. Ensisijaiset päätetapahtumat olivat turvallisuus ja farmakokinetiikka. Kolmelle 12‑vuotiaalle tai sitä vanhemmalle potilaalle, joilla oli edennyt melanooma, annettiin ipilimumabia 5 mg/kg:n annoksena ja kahdelle 10 mg/kg:n annoksena. Stabiilin sairausvaiheen saavutti kaksi potilasta, jotka saivat ipilimumabia 5 mg/kg:n annoksena, ja heistä toisella kesto oli > 22 kuukautta.
CA184178‑tutkimus oli faasin II satunnaistamaton, avoin monikeskustutkimus, johon osallistui 12 – alle 18-vuotiaita aiemmin hoidettuja tai hoitamattomia potilaita, joilla oli pahanlaatuinen, leikkaukseen soveltumaton asteen III tai IV melanooma. Ipilimumabia annettiin joka kolmas viikko yhteensä 4 annosta. Tehon ensisijainen päätetapahtuma oli ensimmäisen vuoden kokonaisalinaika (OS). Tehon toissijaiset päätetapahtumat hoitovaste (BORR), stabiili tauti (SD), sairauden hallinnan aste (DCR) ja etenemisvapaa elinaika (PFS) perustuivat modifioituihin WHO‑kriteereihin, ja ne määriteltiin tutkijan arvion mukaan. Myös kokonaiselinaika (OS) arvioitiin. Kasvain arvioitiin viikolla 12. Kaikkia potilaita seurattiin vähintään vuoden ajan. Ipilimumabia annettiin 3 mg/ml:n annoksena neljälle potilaalle ja 10 mg/kg:n annoksena kahdeksalle potilaalle. Suurin osa potilaista oli miespuolisia (58 %) ja valkoihoisia (92 %). Mediaani-ikä oli 15 vuotta. Yksi ipilimumabia 3 mg/kg:n annoksena saanut potilas saavutti stabiilin sairausvaiheen 260 päivän ajaksi ja yksi 10 mg/kg:n annosta saanut potilas saavutti stabiilin sairausvaiheen noin 14 kuukauden ajaksi. Kaksi ipilimumabia 10 mg/kg:n annoksena saanutta potilasta saavutti osittaisen vasteen, ja heistä toisella se säilyi yli vuoden ajan. Muut tehoon liittyvät tulokset esitetään taulukossa 23.
Taulukko 23: Tehoon liittyvät tulokset CA184178‑tutkimuksessa
| Ipilimumabi 3 mg/kg | Ipilimumabi 10 mg/kg |
|---|---|---|
OS, 1 v (%) (95 %:n luottamusväli) | 75 % (12,8, 96,1) | 62,5 % (22,9, 86,1) |
BORR (%) (95 %:n luottamusväli) | 0 % (0, 60,2) | 25 % (3,2, 65,1) |
SD (n/N)a | 1/4 | 1/8 |
DCR (%) (95 %:n luottamusväli) | 25 % (0,6, 80,6) | 37,5 % (8,5, 75,5) |
Etenemisvapaan elinajan mediaani (kuukautta) (95 %:n luottamusväli) | 2,6 (2,3, 8,5) | 2,9 (0,7, NEa) |
OS-mediaani (kuukautta) (95 %:n luottamusväli) | 18,2 (8,9, 18,2) | Ei saavutettu (5,2, NE) |
a NE = not estimable, ei arvioitavissa
Ipilimumabi yhdistelmähoitona nivolumabin kanssa
CA209070-tutkimus oli avoin faasin 1/2 yksihaarainen annoksen varmistus- ja laajennustutkimus, jossa nivolumabia tutkittiin ainoana lääkkeenä ja yhdistelmähoitona ipilimumabin kanssa pediatrisilla potilailla ja nuorilla aikuispotilailla, joilla oli uusiutunut tai hoitoon vastaamaton kiinteä tai hematologinen kasvain, mukaan lukien neuroblastooma, osteosarkooma, rabdomyosarkooma, Ewingin sarkooma, edennyt melanooma, klassinen Hodgkinin lymfooma ja non-Hodgkin-lymfooma (NHL). Hoidetuista 126 potilaasta 97 oli pediatrisia potilaita (12 kuukauden ikäisistä < 18‑vuotiaisiin). Näistä 97 pediatrisesta potilaasta 64 sai nivolumabi-monoterapiaa (3 mg/kg annosteltuna 60 minuutin kuluessa laskimoon joka toinen viikko) ja 33 sai ipilimumabi–nivolumabi-yhdistelmähoitoa (nivolumabia 1 mg/kg tai 3 mg/kg annosteltuna 60 minuutin kuluessa laskimoon yhdessä ipilimumabiannoksen 1 mg/kg kanssa, joka annosteltiin 90 minuutin kuluessa laskimoon, joka kolmas viikko ensimmäiset neljä annosta, minkä jälkeen nivolumabi-monoterapiaa 3 mg/kg joka toinen viikko). Potilaat saivat joko nivolumabi-monoterapiaa kaksi annosta (mediaani, vaihteluväli: 1–89) tai ipilimumabi–nivolumabi-yhdistelmähoitoa kaksi annosta (mediaani, vaihteluväli: 1–24). Tärkeimmät ensisijaiset muuttujat olivat turvallisuus, siedettävyys ja kasvaimeen kohdistuva estovaikutus arvioituna deskriptiivisillä objektiivisen vasteen saavuttaneiden osuudella ja kokonaiselinajalla.
Nivolumabi-monoterapiaa saaneista 64 pediatrisesta potilaasta 60 potilaan vaste oli arvioitavissa (melanooma n = 1, kiinteitä kasvaimia n = 47 ja hematologisia kasvaimia n = 12). Niillä 48 pediatrisella potilaalla, joilla oli melanooma tai kiinteitä kasvaimia ja joiden vaste oli arvioitavissa, ei havaittu objektiivisia vasteita. Niillä 12 pediatrisella potilaalla, joilla oli hematologisia kasvaimia ja joiden vaste oli arvioitavissa, objektiivisen vasteen saavuttaneiden osuus oli 25,0 % (95 %:n luottamusväli: 5,5, 57,2), mukaan lukien yksi täydellinen vaste klassista Hodgkinin lymfoomaa sairastaneella ja kaksi osittaista vastetta, joista toinen klassista Hodgkinin lymfoomaa sairastaneella ja toinen non-Hodgkin-lymfoomaa sairastaneella. Näiden nivolumabi-monoterapiaa saaneiden 64 pediatrisen potilaan deskriptiivisissä analyyseissä kokonaiselinajan mediaani oli 6,67 kuukautta (95 %:n luottamusväli: 5,98, NA), 6,14 kuukautta (95 %:n luottamusväli: 5,39, 24,67) potilailla, joilla oli melanooma tai kiinteitä kasvaimia, eikä sitä saavutettu potilailla, joilla oli hematologisia kasvaimia.
Niillä 30 pediatrisella potilaalla, jotka saivat ipilimumabi–nivolumabi-yhdistelmähoitoa (vain muita kiinteitä kasvaimia kuin melanooma) ja joiden vaste oli arvioitavissa, ei havaittu objektiivisia vasteita. Ipilimumabi–nivolumabi-yhdistelmähoitoa saaneilla 33 pediatrisella potilaalla kokonaiselinajan mediaani oli 8,25 kuukautta (95 %:n luottamusväli: 5,45, 16,95) deskriptiivisessä analyysissä.
CA209908‑tutkimus oli avoin, sekventiaalisten hoitoryhminen, vaiheen 1b/2 kliininen tutkimus nivolumabi-monoterapiasta ja ipilimumabin ja nivolumabin yhdistelmähoidosta pediatrisilla potilailla ja nuorilla aikuispotilailla, joilla oli korkean asteen primaarinen keskushermoston syöpä, mukaan lukien diffuusi sisäinen aivosillan gliooma (DIPG), korkean asteen gliooma, medulloblastooma, ependymooma ja muut korkean asteen keskushermoston syövän uusiutuneet alatyypit (esim. pineoblastooma, epätyypillinen teratoidi-rhabdoidikasvain ja keskushermoston embryonaaliset kasvaimet). Tutkimukseen otetuista 151 pediatrisesta potilaasta (≥ 6 kuukauden ikäisistä < 18‑vuotiaisiin) 77:ää hoidettiin nivolumabi-monoterapialla (3 mg/kg joka toinen viikko) ja 74:ää hoidettiin ipilimumabin ja nivolumabin yhdistelmähoidolla (ipilimumabia 1 mg/kg ja nivolumabia 3 mg/kg, joka kolmas viikko 4 annoksen ajan, minkä jälkeen nivolumabi‑monoterapiaa 3 mg/kg joka toinen viikko). Ensisijaiset tehokkuusmuuttujat olivat kokonaiselinaika DIPG‑kohortissa ja etenemisvapaa elinaika tutkijan arvioimana RANO‑kriteerien mukaan kaikissa muissa kasvaintyypeissä. Kokonaiselinajan mediaani DIPG‑kohortissa oli 10,97 kuukautta (80 %:n luottamusväli: 9,92, 12,16) nivolumabi‑monoterapialla hoidetuilla potilailla ja 10,50 kuukautta (80 %:n luottamusväli: 9,10, 12,32) ipilimumabin ja nivolumabin yhdistelmähoidolla hoidetuilla potilailla. Kaikkien muiden tutkimuksessa tutkittujen pediatristen keskushermoston kasvaintyyppien osalta etenemisvapaan elinajan mediaani vaihteli 1,23 kuukaudesta 2,35 kuukauteen nivolumabi‑monoterapialla hoidetuilla potilailla ja 1,45 kuukaudesta 3,09 kuukauteen ipilimumabin ja nivolumabin yhdistelmähoidolla hoidetuilla potilailla. Tutkimuksessa ei havaittu objektiivisia vasteita, paitsi yhdellä ependymoomaa sairastavalla, nivolumabi-monoterapialla hoidetulla potilaalla, joka sai osittaisen vasteen. CA209908‑tutkimuksessa havaitut tulokset kokonaiselinajassa, etenemisvapaassa elinajassa ja objektiivisen vasteen saavuttaneiden osuudessa eivät näytä antavan viitteitä kliinisesti merkittävästä edusta verrattuna siihen, mitä tällaisilta potilaspopulaatioilta voidaan muuten odottaa.
Farmakokinetiikka
Ipilimumabin farmakokinetiikkaa on tutkittu 785:llä pitkälle edennyttä melanoomaa sairastaneella potilaalla, jotka saivat aloitushoidon, 4 infuusiota kolmen viikon välein annoksella 0,3–10 mg/kg. Ipilimumabin Cmax , Cmin ja AUC-arvojen todettiin olevan suhteessa annokseen tutkitulla annosvälillä. Kolmen viikon välein toistuvassa annostelussa ipilimumabin puhdistuman ei todettu muuttuvan ajan myötä. Ipilimumabin todettiin kertyvän elimistöön hyvin vähäisessä määrin, sillä kertymisindeksi oli enintään 1,5 kertainen. Vakaa tila ipilimumabilla saavutettiin kolmanteen annokseen mennessä. Populaatiofarmakokineettisen tutkimuksen perusteella ipilimumabin keskimääräiset (variaatiokerroinprosentti, CV %) farmakokineettiset parametrit olivat: terminaalinen puoliintumisaika 15,4 vrk (34,4 %); systeeminen puhdistuma 16,8 ml/h (38,1 %) ja vakaan tilan jakaantumistilavuus 7,47 l (10,1 %). Ipilimumabin keskimääräinen (variaatiokerroinprosentti, CV %) vakaan tilan Cmin oli 3 mg/kg aloitushoidon yhteydessä 19,4 mikrog/ml (74,6 %).
Ipilimumabin puhdistuma suureni potilaan lähtöpainon ja LDH-lähtöarvon suurentuessa. Mg/kg perusteista annosta ei kuitenkaan tarvitse muuttaa koholla olevan LDH-arvon tai potilaan painon mukaan. Seuraavat tekijät eivät vaikuttaneet puhdistumaan: ikä (vaihteluväli 23–88 v), sukupuoli, samanaikainen budesonidin tai dakarbatsiinin käyttö, toimintakyky, kudostyyppi HLA-A2*0201, lievä maksan vajaatoiminta, munuaisten vajaatoiminta, immunogeenisuus ja aiempi syöpähoito. Rodun vaikutusta ei tutkittu, koska tiedot muista kuin valkoihoisista potilaista olivat riittämättömiä. Ipilimumabin farmakokinetiikkaa lapsissa tai maksan tai munuaisten vajaatoimintapotilaissa ei ole selvitetty kontrolloiduissa tutkimuksissa.
Altistus-vasteanalyysissa, joka tehtiin 497:lle edennyttä melanoomaa sairastaneelle, aiempi systeeminen syöpähoito ei vaikuttanut kokonaiselinaikaan vaan tämä piteni sen mukaan, mitä suurempi ipilimumabin Cmin-ss pitoisuus plasmassa oli.
YERVOY yhdistelmähoitona nivolumabin kanssa: Kun ipilimumabia annetiin 1 mg/kg yhdistelmähoitona nivolumabin 3 mg/kg kanssa, ipilimumabin puhdistuma väheni 1,5 %:lla ja nivolumabin puhdistuma kasvoi 1 %:lla. Näitä muutoksia ei pidetä kliinisesti merkityksellisinä. Kun ipilimumabia annettiin 3 mg/kg yhdistelmähoitona nivolumabin 1 mg/kg kanssa, ipilimumabin puhdistuma kasvoi 9 %:lla ja nivolumabin puhdistuma kasvoi 29 %:lla, mitä ei pidetä kliinisesti merkityksellisenä.
Yhdistelmähoidossa nivolumabin kanssa ipilimumabin puhdistuma kasvoi 5,7 %:lla ipilimumabin vasta-aineiden läsnäollessa ja nivolumabin puhdistuma kasvoi 20 %:lla nivolumabin vasta-aineiden läsnäollessa. Näitä muutoksia ei pidetä kliinisesti merkityksellisinä.
YERVOY yhdistelmähoitona nivolumabin ja kemoterapian kanssa: Kun ipilimumabia (1 mg/kg joka kuudes viikko) annettiin yhdistelmähoitona nivolumabin (360 mg joka kolmas viikko) ja kahden kemoterapiajakson kanssa, ipilimumabin puhdistuma kasvoi noin 22 %:lla ja nivolumabin puhdistuma vähentyi noin 10 %:lla. Näitä muutoksia ei pidetty kliinisesti merkityksellisinä.
Munuaisten vajaatoiminta
Populaatiofarmakokineettisessa analyysissa arvioitiin metastasoinutta melanoomaa sairastaneiden kliinisiä tutkimustuloksia: potilailla entuudestaan ollut lievä tai keskivaikea munuaisten vajaatoiminta ei vaikuttanut ipilimumabin puhdistumaan. Kliiniset ja farmakokineettiset tiedot potilaista, joilla on entuudestaan vaikea munuaisten vajaatoiminta, ovat vähäisiä, joten mahdollista tarvetta muuttaa annosta ei voida määrittää.
Maksan vajaatoiminta
Farmakokineettisessa populaatioanalyysissa arvioitiin kliinisten tutkimusten tuloksia metastasoinutta melanoomaa sairastaneilla: potilailla entuudestaan ollut lievä maksan vajaatoiminta ei vaikuttanut ipilimumabin puhdistumaan. Kliiniset ja farmakokineettiset tiedot potilaista, joilla on entuudestaan keskivaikea maksan vajaatoiminta, ovat vähäisiä, joten mahdollista tarvetta muuttaa annosta ei voida määrittää. Kliinisissä tutkimuksissa ei tunnistettu yhtään potilasta, jolla olisi ollut entuudestaan vaikea maksan vajaatoiminta.
Pediatriset potilaat
Populaatiofarmakokineettisen analyysin perusteella ipilimumabi-monoterapiassa ipilimumabin puhdistuma suureni potilaan lähtöpainon suurentuessa. Analyysissa käytettiin saatavilla olevia yhdistettyjä tietoja neljästä aikuisilla tehdystä faasin II tutkimuksesta, joihin osallistui 565 potilasta (N = 521), ja kahdesta pediatrisilla potilailla tehdystä tutkimuksesta (N = 44). Iällä (2–87 vuotta) ei ollut kliinisesti merkittävää vaikutusta ipilimumabin puhdistumaan. Arvioitu keskimääräinen geometrinen puhdistuma (CL) on 12‑vuotiailla ja sitä vanhemmilla – alle 18‑vuotiailla nuorilla 8,72 ml/h. Nuorten potilaiden altistuminen on verrattavissa samaa annosta (mg/kg) saavien aikuisten altistumiseen. Aikuisilla ja pediatrisilla potilailla tehdyn simulaation perusteella vertailukelpoinen altistus saavutetaan aikuisille ja pediatrisille potilaille suositellulla 3 mg/kg:n annoksella, joka annetaan kolmen viikon välein.
Ipilimumabi–nivolumabi-yhdistelmähoidossa 12‑vuotiaiden ja sitä vanhempien pediatristen potilaiden ipilimumabi- ja nivolumabialtistusten oletetaan vastaavan aikuispotilaiden vastaavia suositellulla annoksella.
Prekliiniset tiedot turvallisuudesta
Apinoilla tehdyssä toistuvan laskimonsisäisen annon toksisuustutkimuksessa ipilimumabi oli yleensä hyvin siedetty. Immuunivälitteisiä haittavaikutuksia havaittiin harvoin (~3 %:lla), ja niitä olivat koliitti (aiheutti yhden yksilön kuoleman), ihotulehdus ja infuusioreaktio (saattoi johtua nopean injisoinnin aiheuttamasta sytokiinin äkillisestä vapautumisesta). Yhdessä tutkimuksessa todettiin kilpirauhasen ja kivesten painon pienenemistä, mutta tähän ei liittynyt histopatologisia löydöksiä. Tämän löydöksen kliinistä merkitystä ei tiedetä.
Ipilimumabin vaikutuksia sikiöaikaiseen ja syntymänjälkeiseen kehitykseen on selvitetty yhdessä tutkimuksessa cynomolgus-apinoilla. Tiineet apinat saivat ipilimumabia 3 viikon välein ensimmäisellä raskauskolmanneksella käynnistyvästä organogeneesistä synnytykseen saakka. Apinat altistuivat pitoisuuksille (AUC), jotka olivat joko samaa luokkaa tai suuremmat kuin mitä ipilimumabin hoitoannos 3 mg/kg tuottaa. Ensimmäisen ja toisen raskauskolmanneksen aikana ei havaittu hoidosta johtuvia lisääntymiseen kohdistuvia haittavaikutuksia. Viimeisen raskauskolmanneksen alusta lähtien keskenmenojen, kuolleena syntyneiden poikasten ja ennenaikaisten synnytysten (ja niihin liittyvä pieni syntymäpaino) ja poikaskuolleisuuden ilmaantuvuudet lisääntyivät molemmissa ipilimumabiryhmissä suhteessa verrokkieläimiin; nämä löydökset olivat annoksesta riippuvaisia. Lisäksi kahdella sikiöaikanaan ipilimumabille altistuneella imeväisellä todettiin ulkoisten tai sisäisten virtsa- ja sukupuolielinten kehityshäiriöitä: yhdeltä naaraspoikaselta puuttui synnynnäisesti vasen munuainen ja virtsanjohdin, ja yhdellä koiraspoikasella todettiin virtsaputken synnynnäinen umpeuma, johon liittyi virtsaamisen estyminen ja kivespussien ihonalainen turvotus. Näiden epämuodostumien yhteys hoitoon on epäselvä.
Ipilimumabin mutageenisuutta ja karsinogeenisuutta ei ole arvioitu tutkimuksissa. Hedelmällisyystutkimuksia ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Trishydrokloridi (2-amino-2-hydroksimetyyli-1,3-propaanidiolihydrokloridi), natriumkloridi, mannitoli (E421), dietyleenitriamiinipentaetikkahappo, polysorbaatti 80, natriumhydroksidi (pH:n säätöön), kloorivetyhappo (pH:n säätöön), injektionesteisiin käytettävä vesi.
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamaton injektiopullo
3 vuotta.
Avaamisen jälkeen
Mikrobiologisista syistä lääkevalmiste on infusoitava tai laimennettava ja infusoitava heti injektiopullon avaamisen jälkeen. Laimentamattoman tai laimennetun (1-4 mg/ml) konsentraatin on osoitettu pysyvän kemiallisesti ja fysikaalisesti stabiilina 24 tuntia +25 °C:ssa ja 2–8 °C:ssa. Jos (laimentamatonta tai laimennettua) infuusioliuosta ei käytetä heti, sitä voi säilyttää enintään 24 tuntia joko jääkaapissa (2–8 °C) tai huoneenlämmössä (20–25 °C).
Säilytys
Säilytä jääkaapissa (2–8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Avatun tai laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
YERVOY infuusiokonsentraatti, liuosta varten
5 mg/ml (L:ei) 10 ml (5120,44 €), 40 ml (19939,94 €)
PF-selosteen tieto
10 ml konsentraatti injektiopullossa (tyypin I lasia), jossa on tulppa (pinnoitettua butyylikumia) ja repäisykorkki (alumiinia). Pakkauskoko: 1 injektiopullo.
40 ml konsentraatti injektiopullossa (tyypin I lasia), jossa on tulppa (pinnoitettua butyylikumia) ja repäisykorkki (alumiinia). Pakkauskoko: 1 injektiopullo.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai hieman samea ja väritön tai vaaleankeltainen liuos, jossa voi olla muutamia hiukkasia. Liuoksen pH on 7,0 ja osmolaarisuus 260–300 mOsm/kg.
Käyttö- ja käsittelyohjeet
Terveydenhuollon ammattihenkilöstön on saatettava valmiste käyttökuntoon aseptista tekniikkaa noudattaen.
Annoksen määritys:
Ipilimumabi-monoterapia tai ipilimumabi–nivolumabi-yhdistelmähoito:
Annos määritetään mg/kg-perusteisesti. Laske potilaalle annettava kokonaisannos hänelle määrätyn annoksen perusteella. Yhteen kokonaisannokseen voidaan tarvita useampi kuin yksi injektiopullo YERVOY-konsentraattia.
-
Yhdestä 10 ml:n YERVOY-injektiopullosta saadaan 50 mg ipilimumabia ja yhdestä 40 ml:n YERVOY-injektiopullosta saadaan 200 mg ipilimumabia.
-
Ipilimumabin kokonaisannos (mg) = potilaan paino (kg) x määrätty annos (mg/kg).
-
Annokseen tarvittava YERVOY-konsentraatin määrä (ml) = kokonaisannos (mg) jaettuna 5:llä (YERVOY-konsentraatin vahvuus on 5 mg/ml).
Infuusion valmistelu:
Noudata aseptista tekniikkaa infuusion valmistelussa.
YERVOY voi antaa laskimoon joko:
-
laimentamattomana, kun infuusioliuos on ensin siirretty infuusiopulloon tai -pussiin asianmukaisella steriilillä ruiskulla;
tai -
laimennettuna enintään viisinkertaiseksi lähtötilavuudestaan (enintään 4 osaa injektionestettä 1 osaan konsentraattia). Loppupitoisuuden tulee olla 1–4 mg/ml. YERVOY-konsentraatin voi laimentaa joko
-
9 mg/ml (0,9 %) natriumkloridi-injektionesteeseen; tai
-
50 mg/ml (5 %) glukoosi-injektionesteeseen.
VAIHE 1
-
Ota tarvittava määrä YERVOY-injektiopulloja huoneenlämpöön noin 5 minuutiksi.
-
Tarkasta, ettei YERVOY-konsentraatissa ole hiukkasia ja ettei se ole värjäytynyt. YERVOY-konsentraatti on kirkas tai hieman samea ja väritön tai vaaleankeltainen liuos. Liuoksessa voi olla muutamia hiukkasia. Älä käytä, jos liuoksessa on epätavallisen paljon hiukkasia tai merkkejä värjäytymisestä.
-
Vedä injektiopullosta tarvittava määrä YERVOY-konsentraattia asianmukaisella steriilillä ruiskulla.
VAIHE 2
-
Siirrä konsentraatti steriiliin, vakuumitekniikalla tyhjennettyyn lasipulloon tai infuusiopussiin (PVC tai muu materiaali).
-
Jos tarpeen, laimenna tarvittavaan määrään 9 mg/ml (0,9 %) natriumkloridi-injektionestettä tai 50 mg/ml (5 %) glukoosi-injektionestettä. Käyttökuntoon saattamisen helpottamiseksi konsentraatti voidaan siirtää suoraan esitäytettyyn pussiin, jossa on sopiva määrä 9 mg/ml (0,9 %) natriumkloridi-injektionestettä tai 50 mg/ml (5 %) glukoosi-injektionestettä. Sekoita infuusioliuos varovasti kääntelemällä infuusiopussia/-pulloa käsin.
Anto:
YERVOY-infuusiota ei saa antaa boluksena laskimoon.
Anna YERVOY-infuusio laskimoon 30 minuutin aikana.
YERVOY-infuusioliuosta ja muita lääkeaineita ei saa infusoida yhtä aikaa saman laskimolinjan kautta. Käytä YERVOY-infuusioon erillistä laskimolinjaa.
Käytä infuusiolaitteistoa ja steriiliä niukasti proteiinia sitovaa ja pyrogeenitonta in-line-suodatinta (huokoskoko 0,2–1,2 mikrom).
YERVOY-infuusioliuos on yhteensopiva:
-
PVC-infuusiovälineiden kanssa
-
polyeetterisulfonista (0,2–1,2 mikrom) ja polyamidista (0,2 mikrom) valmistettujen in-line-suodatinten kanssa.
Huuhtele laskimolinja 9 mg/ml (0,9 %) natriumkloridi-injektionesteellä tai 50 mg/ml (5 %) glukoosi-injektionesteellä infuusion jälkeen.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
YERVOY infuusiokonsentraatti, liuosta varten
5 mg/ml 10 ml, 40 ml
- Ei korvausta.
ATC-koodi
L01FX04
Valmisteyhteenvedon muuttamispäivämäärä
07.05.2026
Yhteystiedot
09 2512 1244
www.bms.com/fi
medinfo.finland@bms.com