
RIASTAP injektio/infuusiokuiva-aine, liuosta varten 1 g
Vaikuttavat aineet ja niiden määrät
Riastap on injektio-/infuusiokuiva-aine, liuosta varten, joka sisältää 1 g:n ihmisen fibrinogeeniä injektiopulloa kohden.
50 ml:aan injektionesteisiin käytettävää vettä käyttövalmiiksi saatettu valmiste sisältää noin 20 mg/ml ihmisen fibrinogeeniä.
Hyytymiskykyisen fibrinogeenin sisältö määräytyy ihmisen fibrinogeeniä koskevan Euroopan farmakopean monografian mukaan.
Apuaineet, joiden vaikutus tunnetaan:
Natriumia enintään 164 mg (7,1 mmol) per injektiopullo.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektio-/infuusiokuiva-aine, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Verenvuodon hoito potilailla, joilla on synnynnäiseen hypo- tai afibrinogenemiaan liittyvä vuototaipumus.
Ehto
Hoito tulee aloittaa hyytymishäiriöihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito on aloitettava hyytymishäiriöiden hoitoon perehtyneen lääkärin valvonnassa.
Annostus
Korvaushoidon annostus ja kesto riippuvat sairauden vaikeusasteesta, verenvuodon sijainnista ja laajuudesta sekä potilaan kliinisestä tilasta.
(Toiminnallisen) fibrinogeenin pitoisuus on määritettävä, jotta yksilöllinen annostus voidaan laskea, ja annettava määrä ja antotiheys on määritettävä potilaalle yksilöllisesti plasman fibrinogeenipitoisuuden mittauksen ja potilaan kliinisen tilan jatkuvan seurannan perusteella sekä potilaan käyttämien muiden korvaushoitojen perusteella.
Plasman normaali fibrinogeenipitoisuus on 1,5−4,5 g/l. Kriittinen plasman fibrinogeenipitoisuus on noin 0,5−1,0 g/l, jota pienempien pitoisuuksien yhteydessä saattaa esiintyä verenvuotoja. Jos potilaalle tehdään suuri leikkaus, korvaushoidon tarkka seuranta veren hyytymistestien avulla on välttämätöntä.
Aloitusannos
Jos potilaan fibrinogeenipitoisuutta ei tiedetä, suositeltu annos on 70 mg painokiloa kohden (mg/kg) laskimoon.
Seuraavat annokset
Vähäisten verenvuotojen (esim. nenäverenvuoto, lihaksensisäinen verenvuoto tai menorragia) yhteydessä tavoitepitoisuus (1 g/l) on ylläpidettävä vähintään kolmen päivän ajan. Suurten verenvuotojen (esim. pään vamma tai kallonsisäinen verenvuoto) yhteydessä tavoitepitoisuus (1,5 g/l) on ylläpidettävä vähintään seitsemän päivän ajan.
| Fibrinogeeniannos (mg painokiloa kohden [mg/kg]) | = | [tavoitepitoisuus (g/l) - mitattu pitoisuus (g/l)] 0,017 (g/l per mg/kg ruumiinpainoa kohden) |
Vastasyntyneiden, imeväisikäisten ja lasten annostus
Kliinisistä tutkimuksista on saatu vähän tietoja Riastap-annostuksesta lapsille. Näiden tutkimusten sekä fibrinogeenivalmisteista saadun pitkän kliinisen käyttökokemuksen perusteella annostussuositukset lapsille ovat samat kuin aikuisille.
Antotapa
Infuusiona tai injektiona laskimoon.
Riastap on saatettava käyttövalmiiksi kohdassa Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet annettujen ohjeiden mukaisesti. Käyttövalmiiksi saatettu liuos on lämmitettävä huoneen- tai ruumiinlämpöiseksi ennen antoa, minkä jälkeen se annetaan hitaana injektiona tai infuusiona nopeudella, jonka potilas kokee miellyttäväksi. Injektio- tai infuusionopeus saa olla enintään noin 5 ml minuutissa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Kun ihmisen fibrinogeenikonsentraatilla hoidetaan potilaita, joilla on synnynnäinen puutos, hoitoon liittyy tromboosiriski, etenkin jos valmistetta annetaan suurina annoksina tai toistuvasti. Kun potilaalle on annettu ihmisen fibrinogeenikonsentraattia, hänen tilaansa on seurattava tarkoin tromboosiin viittaavien oireiden tai löydösten havaitsemiseksi.
Jos potilaalla on aiemmin ollut sepelvaltimotauti tai sydäninfarkti tai potilaalla on maksasairaus samoin kuin peri- ja postoperatiivisilla potilailla, vastasyntyneillä tai jos potilaalla on tromboembolisten tapahtumien tai yleistyneen suonensisäisen hyytymisen vaara (disseminated intravascular coagulation, DIC), ihmisen plasman fibrinogeenikonsentraattihoidosta saatavaa mahdollista hyötyä on punnittava tromboembolisten komplikaatioiden riskiin nähden. Hoidossa on oltava varovainen ja potilasta on seurattava tarkoin.
Jos potilaalle ilmaantuu allergisia tai anafylaksian kaltaisia reaktioita, injektion/infuusion antaminen on lopetettava heti. Anafylaktisen sokin yhteydessä on annettava tähän vakiintunutta hoitoa.
Vasta-ainereaktioita on havaittu, kun potilas on saanut hyytymistekijöitä sisältävää korvaushoitoa muiden synnynnäisten puutosten hoitoon, mutta fibrinogeenia koskevia tietoja ei tällä hetkellä ole.
Riastap sisältää natriumia enintään 164 mg (7,1 mmol) per injektiopullo. Tämä vastaa 11,5 mg:aa (0,5 mmol) natriumia painokiloa kohden (mg/kg), jos potilaalle annetaan suositeltu aloitusannos 70 mg painokiloa kohden (mg/kg). Potilaiden, joilla on ruokavalion natriumrajoitus, tulee ottaa tämä huomioon.
Virusturvallisuus
Vakiintuneita toimenpiteitä ihmisen verestä tai plasmasta valmistetuista lääkevalmisteista aiheutuvien infektioiden estämiseksi ovat verenluovuttajien valinta, erityisten infektiomerkkiaineiden seulominen luovutetusta verestä ja plasmapooleista sekä valmistuksenaikaiset tehokkaat toimenpiteet virusten inaktivoimiseksi/poistamiseksi. Näistä toimenpiteistä huolimatta ei ihmisverestä tai plasmasta valmistettujen lääkevalmisteiden antoon liittyvää infektion mahdollisuutta voida sulkea kokonaan pois. Tämä koskee myös kaikkia tuntemattomia tai nykyisin tuntemattomia uusia viruksia ja muita patogeenejä.
Käytössä olevien toimenpiteiden katsotaan tehoavan vaipallisiin viruksiin, kuten HI-virukseen, hepatiitti B- ja C-viruksiin sekä vaipattomiin viruksiin, kuten hepatiitti A -virukseen ja parvovirus B19:ään.
Ihmisen plasmasta valmistettuja valmisteita säännöllisesti/toistuvasti saavien potilaiden on tavallisesti harkittava asianmukaisten rokotusten (hepatiitti A:ta ja hepatiitti B:tä vastaan) ottamista.
Valmisteen nimi ja eränumero kehotetaan kirjaamaan Riastap-valmisteen jokaisen antokerran yhteydessä, jotta potilas ja valmiste-erä voidaan yhdistää.
Yhteisvaikutukset
Ihmisen plasman fibrinogeenikonsentraatin ja muiden lääkevalmisteiden välisiä yhteisvaikutuksia ei tunneta.
Raskaus ja imetys
Raskaus
Riastap-valmisteella ei ole tehty lisääntymistä koskevia eläinkokeita (ks. kohta Prekliiniset tiedot turvallisuudesta). Koska vaikuttava aine on ihmisperäinen, se kataboloituu samalla tavoin kuin potilaan oma proteiini. Näiden ihmisen veren fysiologisten aineosien ei odoteta aiheuttavan lisääntymiseen tai sikiöön kohdistuvia haittavaikutuksia.
Riastap-valmisteen turvallisuutta ihmisen raskauden aikana ei ole tutkittu kontrolloiduissa kliinisissä tutkimuksissa.
Obstetristen komplikaatioiden hoitoon käytetystä fibrinogeenikonsentraatista saatu kliininen kokemus viittaa siihen, ettei haitallisia vaikutuksia raskauden kulkuun tai sikiön tai vastasyntyneen terveyteen ole odotettavissa.
Imetys
Ei tiedetä, erittyykö Riastap ihmisen rintamaitoon. Riastap-valmisteen käyttöä imettäville naisille ei ole tutkittu kliinisissä tutkimuksissa.
Imeväiseen kohdistuvia riskejä ei voida poissulkea. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko Riastap-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Hedelmällisyyttä koskevia tietoja ei ole.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Riastap-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneiden käyttökykyyn.
Haittavaikutukset
Haittavaikutustaulukko
Taulukkoon on yhdistetty kliinisissä tutkimuksissa ja markkinoilletulon jälkeen tunnistetut haittavaikutukset. Taulukossa esitetyt esiintymistiheydet perustuvat poolattuihin analyyseihin kahdesta yhtiön toimeksiantamasta lumekontrolloidusta kliinisestä tutkimuksesta, jotka toteutettiin aorttaleikkauksen ja siihen mahdollisesti liittyvien muiden kirurgisten toimenpiteiden yhteydessä [BI3023-2002 (N = 61) ja BI3023_3002 (N = 152)]. Esiintymistiheydet on luokiteltu seuraavasti: hyvin yleiset (≥ 1/10); yleiset (≥ 1/100, < 1/10); melko harvinaiset (≥ 1/1 000, < 1/100); harvinaiset (≥ 1/10 000, < 1/1 000); hyvin harvinaiset (< 1/10 000). Jos haittavaikutus on raportoitu spontaanisti markkinoilletulon jälkeen, ilmoitetuksi esiintymistiheydeksi on merkitty ”tuntematon”. Kun otetaan huomioon, että tutkimukset toteutettiin vain suppeassa populaatiossa aorttaleikkauksen yhteydessä, tutkimuksissa havaitut haittavaikutusprosentit eivät välttämättä vastaa kliinisessä käytännössä havaittavia prosentteja, eikä niitä tunneta muussa kuin tutkittuun käyttöaiheeseen liittyvässä kliinisessä käytössä.
MedDRA-elinjärjestelmäluokka | Haittavaikutus | Esiintymistiheys (aorttaleikkauksessa, johon saattoi liittyä muita kirurgisia toimenpiteitä) |
Yleisoireet ja antopaikassa todettavat haitat | Kuume | Hyvin yleiset
|
Immuunijärjestelmä | Anafylaktiset reaktiot (mm. anafylaktinen sokki) | Melko harvinaiset
|
| Allergiset reaktiot (mm. yleistynyt nokkosihottuma, ihottuma, hengenahdistus, takykardia, pahoinvointi, oksentelu, vilunväristykset, kuume, rintakipu, yskä, verenpaineen lasku) | Tuntematon |
Verisuonisto | Tromboemboliset tapahtumat* (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Yleiset** |
* Yksittäistapauksissa tapahtuma on johtanut kuolemaan.
** Kahden kliinisen tutkimuksen tulosten perusteella (aorttaleikkauksen ja siihen mahdollisesti liittyvien muiden kirurgisten toimenpiteiden yhteydessä) tromboembolisten tapahtumien poolattu ilmaantuvuusprosentti oli pienempi fibrinogeenihoitoa saaneilla tutkittavilla (N = 8, 7,4 %) lumehoitoon verrattuna (N = 11, 10,4 %).
Turvallisuus tarttuvien taudinaiheuttajien suhteen, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty−haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Plasman fibrinogeenipitoisuutta on seurattava säännöllisesti hoidon aikana, jotta yliannostus voidaan välttää (ks. Annostus ja antotapa).
Yliannostustapauksessa tromboembolisten komplikaatioiden kehittymisen vaara on lisääntynyt.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: hemostaatit, fibrinogeeni, ihmisen, ATC-koodi: B02BB01
Ihmisen fibrinogeeni (hyytymistekijä I) muutetaan trombiinin, aktivoidun hyytymistekijä XIII:n (F XIIIa) ja kalsiumionien läsnäollessa stabiiliksi ja joustavaksi kolmiulotteiseksi hemostaattiseksifibriinihyytymäksi.
Ihmisen fibrinogeenikonsentraatin antaminen suurentaa plasman fibrinogeenipitoisuutta ja saattaa korjata tilapäisesti potilaan fibrinogeenipuutoksesta johtuvaa hyytymishäiriötä.
Vaiheen II pivotaalitutkimuksessa arvioitiin kerta-annoksen farmakokinetiikkaa (ks. kohta Farmakokinetiikka) ja saatiin tietoa tehosta käyttäen korvattua päätetapahtumaa maksimaalinen hyytymän lujuus (MCF; maximum clot firmness) sekä tietoa turvallisuudesta.
MCF määritettiin kaikilta potilailta ennen Riastap-valmisteen kerta-annoksen (70 mg/kg) antoa (lähtötaso) ja tunti sen jälkeen. Riastap-valmisteen todettiin parantavan hyytymän lujuutta potilailla, joilla oli synnynnäinen fibrinogeenipuutos (afibrinogenemia) tromboelastometrialla mitattuna. Hemostaattinen teho akuutissa vuodossa ja sen yhteys maksimaaliseen hyytymän lujuuteen (MCF) varmistetaan markkinoille tulon jälkeisessä tutkimuksessa.
Farmakokinetiikka
Ihmisen fibrinogeeni on ihmisen plasman normaali osa ja toimii kuten endogeeninen fibrinogeeni. Fibrinogeenin biologinen puoliintumisaika plasmassa on 3–4 päivää. Riastap käyttäytyy hajoamisen suhteen samoin kuin endogeeninen fibrinogeeni.
Valmiste annetaan laskimoon, joten plasmassa on heti annettua annosta vastaava pitoisuus.
Farmakokineettisessä tutkimuksessa arvioitiin kerta-annoksen farmakokinetiikkaa ennen ihmisen fibrinogeenikonsentraatin antamista potilaille, joilla on afibrinogenemia, sekä sen jälkeen. Tässä prospektiivisessa, avoimessa, kontrolloimattomassa monikeskustutkimuksessa oli mukana 5 naispotilasta ja 10 miespotilasta, joiden ikä vaihteli 8–61 vuoteen (2 lasta, 3 nuorta ja 10 aikuista). Annoksen mediaani oli 77,0 mg painokiloa kohden (mg/kg) (vaihteluväli 76,6–77,4 mg/kg).
15 potilaalta otettiin verikoe (joista 14 oli analysoitavissa) fibrinogeeniaktiivisuuden määrittämiseksi lähtötilanteessa ja enintään 14 päivän kuluttua infuusion antamisesta. Tämän lisäksi määritettiin 4 tuntiin saakka infuusion jälkeen lisäsaanto (in vivo recovery, IVR), joka määriteltiin plasman fibrinogeenipitoisuuden maksimilisäykseksi painokiloa kohden. Lisäsaannon mediaani oli 1,7 (vaihteluväli 1,30–2,73) mg/dl painokiloa kohden. Seuraavassa taulukossa esitetään farmakokineettiset tulokset.
Fibrinogeeniaktiivisuuden farmakokineettiset tulokset
| Parametri (n = 14) | Keskiarvo ± keskihajonta | Mediaani (vaihteluväli) |
| t1/2 [h] | 78,7 ± 18,13 | 77,1 (55,73–117,26) |
| Cmax [g/l] | 1,4 ± 0,27 | 1,3 (1,00–2,10) |
| Annoksen 70 mg/kg AUC-arvo [h•mg/ml] | 124,3 ± 24,16 | 126,8 (81,73–156,40) |
| AUC-arvon ekstrapoloitu osuus [%] | 8,4 ± 1,72 | 7,8 (6,13–12,14) |
| Cl [ml/h/kg] | 0,59 ± 0,13 | 0,55 (0,45–0,86) |
| MRT [h] | 92,8 ± 20,11 | 85,9 (66,14–126,44) |
| Vss [ml/kg] | 52,7 ± 7,48 | 52,7 (36,22–67,67) |
| IVR [mg/dl per mg/kg] | 1,8 ± 0,35 | 1,7 (1,30–2,73) |
t1/2 = eliminaation terminaalinen puoliintumisaika
h = tuntia
Cmax = suurin pitoisuus 4 tunnin kuluessa
AUC = pitoisuus−pinta-alakäyrän alle jäävä alue
Cl = puhdistuma
MRT = keskiviipymä
Vss = jakautumistilavuus vakaassa tilassa
SD = keskihajonta
IVR = saanto (in vivo recovery)
Prekliiniset tiedot turvallisuudesta
Kerta-altistuksen farmakologista turvallisuutta ja toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Toistetuilla annoksilla tehtäviä prekliinisiä tutkimuksia (krooninen toksisuus, karsinogeenisuus ja mutageenisuus) ei voi toteuttaa järkevästi tavanomaisilla eläinmalleilla heterologisten ihmisen proteiinien annon jälkeen kehittyvien vasta-aineiden vuoksi.
Farmaseuttiset tiedot
Apuaineet
Ihmisen albumiini, L-arginiinihydrokloridi, natriumhydroksidi (pH:n säätöön), natriumkloridi, natriumsitraatti.
Yhteensopimattomuudet
Lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden, liuottimien ja laimentimien kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet. Käyttövalmiiksi saatetun liuoksen antoon laskimoon huoneenlämmössä suositellaan normaalia infuusiovälineistöä.
Kestoaika
3 vuotta.
Käyttövalmiiksi saatetun valmisteen fysikaalisen ja kemiallisen säilyvyyden on osoitettu olevan 8 tuntia huoneenlämmössä (enintään 25 °C). Mikrobiologiselta kannalta valmiste tulisi käyttää heti käyttövalmiiksi saattamisen jälkeen. Jos käyttövalmiiksi saatettua valmistetta ei käytetä heti, sitä saa säilyttää enintään 8 tuntia huoneenlämpötilassa (enintään 25 °C). Käyttövalmiiksi saatettua valmistetta ei saa säilyttää jääkaapissa.
Säilytys
Säilytä alle 25 ºC. Ei saa jäätyä. Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
RIASTAP injektio/infuusiokuiva-aine, liuosta varten
1 g (L:ei) 1 kpl (663,43 €)
PF-selosteen tieto
Väritön tyypin II (Ph. Eur.) lasinen injektiopullo, joka on suljettu lateksittomalla tulpalla (bromobutyylikumia), alumiinikorkilla ja muovilevyllä.
1 g:n pakkaus (kuva 1)
1. Yksi injektiopullo, jossa 1 g ihmisen fibrinogeeniä
2. Suodatin: Pall®-ruiskusuodatin
3. Lääkkeenottokanyyli: Mini-Spike®-lääkkeenottokanyyli

Kuva 1
Valmisteen kuvaus:
Valkoinen jauhe.
Käyttö- ja käsittelyohjeet
Yleiset ohjeet
- Valmisteen käyttövalmiiksi saattaminen ja vetäminen ruiskuun on tehtävä aseptisissa olosuhteissa.
- Käyttövalmiiksi saatettu liuos on tarkistettava silmämääräisesti ennen antoa, ettei siinä ole havaittavissa hiukkasia eikä värimuutoksia.
- Liuoksen on oltava väritöntä tai kellertävää, kirkasta tai hieman opaalinhohtoista ja pH-arvoltaan neutraali. Älä käytä liuosta, jos se on sameaa tai siinä on sakkaa.
Käyttövalmiiksi saattaminen
- Lämmitä liuotin ja kuiva-aine avaamattomissa injektiopulloissa huoneen- tai kehonlämpöiseksi (ei yli 37 °C).
- Riastap on saatettava käyttövalmiiksi sekoittamalla se injektionesteisiin käytettävään veteen (50 ml, ei mukana pakkauksessa).
- Pese kädet ennen valmisteen käyttövalmiiksi sekoittamista tai käytä suojakäsineitä.
- Poista korkki Riastap-injektiopullosta, jolloin infuusiokorkin keskiosa tulee näkyviin.
- Pyyhi infuusiotulpan pinta antiseptisella liuoksella ja anna kuivua.
- Siirrä liuotin injektiopulloon asianmukaisen siirtolaitteen avulla. Varmista, että kuiva-aine kostuu kauttaaltaan.
- Pyörittele injektiopulloa varovasti, kunnes kuiva-aine on saatettu käyttövalmiiksi ja liuos on valmis käytettäväksi. Vältä ravistamista, koska se aiheuttaa vaahdon muodostumista. Kuiva-aine liukenee yleensä noin 5 minuutissa, ja sen täydelliseen liukenemiseen ei pitäisi kulua pidempään kuin 15 minuuttia.
- Avaa Riastap-pakkauksen sisältämän lääkkeenottokanyylin (Mini-Spike®-lääkkeenottokanyyli) muovinen läpipainopakkaus (kuva 2).

Kuva 2
- Ota pakkauksen sisältämä lääkkeenottokanyyli, ja työnnä se käyttövalmiiksi sekoitetun valmisteen sisältävän injektiopullon tulppaan (kuva 3).

Kuva 3
- Kun lääkkeenottokanyyli on työnnetty tulppaan, poista korkki. Kun korkki on poistettu, älä kosketa esillä olevaan pintaan.
- Avaa Riastap-pakkauksen sisältämän suodattimen (Pall®-ruiskusuodatin) läpipainopakkaus (kuva 4).
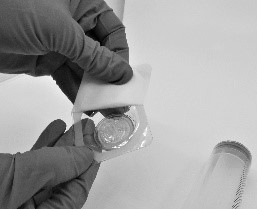
Kuva 4
- Kierrä ruisku kiinni suodattimeen (kuva 5).
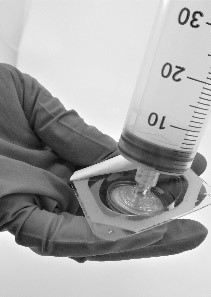
Kuva 5
- Kierrä ruisku ja siihen kiinnitetty suodatin kiinni lääkkeenottokanyyliin (kuva 6).

Kuva 6
- Vedä käyttövalmiiksi saatettu valmiste ruiskuun (kuva 7).
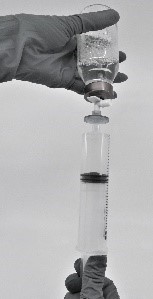
Kuva 7
- Kun tämä on tehty, irrota ruiskusta suodatin, lääkkeenottokanyyli ja tyhjä injektiopullo, ja hävitä nämä asianmukaisesti. Jatka lääkkeenantoa tavanomaiseen tapaan.
- Käyttövalmiiksi saatettu valmiste on annettava heti erillisen injektio-/infuusioantolaitteen avulla.
- Varmista, ettei valmisteen sisältävään ruiskuun pääse verta.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
RIASTAP injektio/infuusiokuiva-aine, liuosta varten
1 g 1 kpl
- Ei korvausta.
ATC-koodi
B02BB01
Valmisteyhteenvedon muuttamispäivämäärä
29.10.2024
Yhteystiedot
 CSL BEHRING AB
CSL BEHRING AB Box 712
182 17 Danderyd
Ruotsi
+46 (0) 8 544 966 70
www.cslbehring.fi
info@cslbehring.se