
VENCLYXTO tabletti, kalvopäällysteinen 10 mg, 50 mg, 100 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Potilaskortti / Patientkort / Patient card
Vaikuttavat aineet ja niiden määrät
Venclyxto 10 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 10 mg venetoklaksia.
Venclyxto 50 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 50 mg venetoklaksia.
Venclyxto 100 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 100 mg venetoklaksia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti).
Kliiniset tiedot
Käyttöaiheet
Venclyxto on tarkoitettu käytettäväksi kroonisen lymfaattisen leukemian (KLL) hoitoon aikuispotilailla, jotka eivät ole saaneet aiempaa hoitoa:
- yhdessä akalabrutinibin kanssa joko yhdistettynä obinututsumabiin tai ilman obinututsumabia
- yhdessä obinututsumabin kanssa (ks. kohta Farmakodynamiikka)
- yhdessä ibrutinibin kanssa.
Venclyxto on tarkoitettu käytettäväksi yhdessä rituksimabin kanssa KLL:n hoitoon aikuispotilailla, jotka ovat saaneet aiemmin vähintään yhtä hoitoa.
Venclyxto on tarkoitettu käytettäväksi ainoana lääkkeenä KLL:n hoitoon
- aikuispotilailla, joilla on 17p-deleetio tai TP53-mutaatio ja joille B-solureseptorireitin estäjähoito ei sovellu tai joilla se on epäonnistunut; tai
- aikuispotilailla, joilla ei ole 17p-deleetiota eikä TP53-mutaatiota ja joilla sekä kemoimmunoterapia että B-solureseptorireitin estäjähoito ovat epäonnistuneet.
Venclyxto on tarkoitettu käytettäväksi yhdessä hypometyloivan lääkkeen kanssa äskettäin diagnosoidun akuutin myelooisen leukemian (AML) hoitoon aikuispotilailla, joille intensiivinen solunsalpaajahoito ei sovi.
Ehto
Valmistetta tulee käyttää vain syövän hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Venetoklaksihoidon aloittaa ja sitä valvoo syövän lääkehoitoon perehtynyt lääkäri. Venetoklaksihoitoa saaville potilaille voi kehittyä tuumorilyysioireyhtymä. Tässä kohdassa kuvattuja tietoja, mukaan lukien riskinarviointi, ehkäisytoimet, annostitrausaikataulu, laboratorioseuranta ja lääkeaineiden yhteisvaikutukset, on noudatettava tuumorilyysioireyhtymän ehkäisemiseksi ja sen riskin pienentämiseksi.
Annostus
Krooninen lymfaattinen leukemia
Annostitrausaikataulu
Venetoklaksihoidon aloitusannos on 20 mg kerran vuorokaudessa 7 päivän ajan. Annosta on suurennettava vähitellen 5 viikon kuluessa 400 milligrammaan vuorokaudessa taulukossa 1 esitettävällä tavalla.
Taulukko 1: Annoksen suurentamisaikataulu KLL-potilailla
Viikko | Venetoklaksivuorokausiannos |
1 | 20 mg |
2 | 50 mg |
3 | 100 mg |
4 | 200 mg |
5 | 400 mg |
Viisiviikkoinen annostitrausaikataulu on suunniteltu pienentämään kasvainkuormaa vähitellen (debulking) ja pienentämään tuumorilyysioireyhtymän riskiä.
Venetoklaksin ja akalabrutinibin yhdistelmähoito obinututsumabin kanssa tai ilman obinututsumabia
Venetoklaksin ja akalabrutinibin yhdistelmähoitoa obinututsumabin kanssa tai ilman obinututsumabia jatketaan, kunnes tauti etenee, todetaan sietämätöntä toksisuutta tai 14 hoitojaksoa on annettu. Kunkin hoitojakson pituus on 28 päivää.
Akalabrutinibia annetaan 100 mg:n annos suun kautta hoitojakson 1 päivänä 1 noin 12 tunnin välein yhteensä 14 hoitojakson ajan. Kunkin hoitojakson pituus on 28 päivää.
Venetoklaksin 5 viikon pituinen annostitrausaikataulu (ks. taulukko 1) aloitetaan hoitojakson 3 päivänä 1. Annostitrausaikataulun päätyttyä venetoklaksin suositusannos on 400 mg kerran vuorokaudessa aina hoitojakson 14 viimeiseen päivään asti.
Jos venetoklaksia annetaan yhdessä akalabrutinibin ja obinututsumabin kanssa, obinututsumabia annetaan 100 mg:n annos hoitojakson 2 päivänä 1, minkä jälkeen annetaan 900 mg:n annos, joka voidaan antaa päivänä 1 tai päivänä 2. Tämän jälkeen annetaan 1 000 mg obinututsumabia hoitojakson 2 päivinä 8 ja 15 ja hoitojaksojen 3–7 päivänä 1. Obinututsumabia annetaan yhteensä 6 hoitojakson ajan.
Venetoklaksin ja obinututsumabin yhdistelmähoito
Venetoklaksia annetaan yhteensä 12 hoitojakson ajan. Kunkin hoitojakson pituus on 28 päivää. Venetoklaksia annetaan 6 hoitojakson ajan yhdessä obinututsumabin kanssa. Tämän jälkeen annetaan 6 hoitojakson ajan pelkkää venetoklaksia.
Obinututsumabia annetaan 100 mg:n annos hoitojakson 1 päivänä 1, minkä jälkeen annetaan 900 mg:n annos päivänä 1 tai päivänä 2. Tämän jälkeen annetaan 1 000 mg obinututsumabia hoitojakson 1 päivinä 8 ja 15 ja kunkin myöhemmän 28-päiväisen hoitojakson päivänä 1 yhteensä 6 hoitojakson ajan.
Venetoklaksin 5 viikon pituinen annostitrausaikataulu (ks. taulukko 1) aloitetaan hoitojakson 1 päivänä 22 ja sitä jatketaan hoitojakson 2 päivään 28 asti.
Annostitrausaikataulun päätyttyä venetoklaksin suositusannos on 400 mg kerran vuorokaudessa obinututsumabin hoitojakson 3 päivästä 1 alkaen aina hoitojakson 12 viimeiseen päivään asti.
Venetoklaksin ja ibrutinibin yhdistelmähoito
Hoito aloitetaan antamalla pelkkää ibrutinibia (420 mg kerran vuorokaudessa) 3 hoitojakson ajan (1 hoitojakson pituus on 28 päivää), minkä jälkeen venetoklaksia annetaan yhdessä ibrutinibin kanssa 12 hoitojakson ajan. Hoitojakson 4 päivästä 1 alkaen venetoklaksia annetaan annostitrausaikataulun mukaan (ks. taulukko 1). Annostitrausaikataulun päätyttyä potilaiden hoitoa jatketaan antamalla 400 mg venetoklaksia kerran vuorokaudessa ja 420 mg ibrutinibia suun kautta kerran vuorokaudessa hoitojakson 15 viimeiseen päivään asti.
Katso lisätietoja ibrutinibin valmisteyhteenvedosta.
Annostitrauksen jälkeinen annos venetoklaksin ja rituksimabin yhdistelmähoitoa käytettäessä
Suositeltu venetoklaksiannos yhdessä rituksimabin kanssa on 400 mg kerran vuorokaudessa (yhdistelmähoidon tiedot, ks. kohta Farmakodynamiikka).
Rituksimabi annetaan, kun potilas on suorittanut annostitrausaikataulun loppuun ja käyttänyt venetoklaksia suositusannoksina (400 mg/vrk) 7 päivän ajan.
Venetoklaksia käytetään 24 kk ajan ensimmäisen rituksimabihoitojakson ensimmäisestä päivästä laskettuna (ks. kohta Farmakodynamiikka).
Annostitrauksen jälkeinen annos, kun venetoklaksia käytetään ainoana lääkkeenä
Suositeltu venetoklaksiannos on 400 mg kerran vuorokaudessa. Hoitoa jatketaan, kunnes tauti etenee tai potilas ei enää siedä hoitoa.
Akuutti myelooinen leukemia
Venetoklaksin suositeltu annostusaikataulu (annostitraus mukaan lukien) esitetään taulukossa 2.
Taulukko 2: Annoksen suurentamisaikataulu AML-potilailla
Päivä | Venetoklaksivuorokausiannos |
1 | 100 mg |
2 | 200 mg |
3 ja siitä eteenpäin | 400 mg |
Atsasitidiinia annetaan 75 mg/m2 kehon pinta-alaa kohden joko laskimoon tai ihon alle kunkin 28 päivän hoitojakson päivinä 1–7 hoitojakson 1 päivästä 1 alkaen.
Desitabiinia annetaan 20 mg/m2 kehon pinta-alaa kohden laskimoon kunkin 28 päivän hoitojakson päivinä 1–5 hoitojakson 1 päivästä 1 alkaen.
Venetoklaksin anto voidaan tarvittaessa tauottaa hematologisen toksisuuden hoitamiseksi ja verisolujen määrän palautumiseksi (ks. taulukko 6).
Venetoklaksin ja hypometyloivan lääkkeen yhdistelmähoitoa jatketaan, kunnes tauti etenee tai todetaan sietämätöntä toksisuutta.
Tuumorilyysioireyhtymän ehkäisy
Venetoklaksihoitoa saaville potilaille saattaa kehittyä tuumorilyysioireyhtymä. Yksityiskohtaiset tiedot hoidosta ovat jäljempänä asianmukaisessa kohdassa käyttöaiheen mukaisesti.
Krooninen lymfaattinen leukemia
Venetoklaksi voi pienentää kasvaimen kokoa nopeasti, joten sen käyttöön liittyy tuumorilyysioireyhtymän riski hoidon alussa pidettävän 5 viikon annostitrausvaiheen aikana kaikilla KLL-potilailla kasvainkuormasta ja potilaan muista ominaisuuksista riippumatta. Tuumorilyysioireyhtymään viittaavia elektrolyyttiarvojen muutoksia, jotka edellyttävät nopeaa hoitoa, voi ilmetä jo 6–8 tunnin kuluessa ensimmäisestä venetoklaksiannoksesta ja aina annoksen suurentamisen yhteydessä. Potilaskohtaiset tekijät on arvioitava tuumorilyysioireyhtymän riskitason suhteen ja ehkäisevää nesteytystä ja hyperurikemialääkkeitä on annettava potilaille ennen ensimmäistä venetoklaksiannosta tuumorilyysioireyhtymän riskin pienentämiseksi.
Tuumorilyysioireyhtymän riski on jatkumo ja riippuu useista tekijöistä, mm. oheissairauksista, erityisesti heikentyneestä munuaistoiminnasta (kreatiniinipuhdistuma [CrCl] < 80 ml/min) ja kasvainkuormasta. Splenomegalia voi myötävaikuttaa tuumorilyysioireyhtymän kokonaisriskiin. Riski voi pienentyä, kun kasvainkuorma pienenee venetoklaksihoidon myötä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ennen venetoklaksihoidon aloittamista kaikille potilaille on tehtävä kasvainkuorman arviointi, johon kuuluu radiologinen arviointi (esim. TT-kuvaus). Veren kemiallinen koostumus (kalium, virtsahappo, fosfori, kalsium ja kreatiniini) on arvioitava ja mahdolliset poikkeavuudet korjattava.
Taulukossa 3 kuvataan suositeltu tuumorilyysioireyhtymän ehkäisy ja seuranta venetoklaksihoidon aikana, perustuen kasvainkuorman määrittämiseen kliinisten tutkimusten tiedoista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Lisäksi kaikki potilaan oheissairaudet on otettava huomioon riskin mukaista ehkäisyä ja seurantaa harkittaessa, joko avohoidossa tai sairaalassa.
Taulukko 3: Suositellut tuumorilyysioireyhtymän ehkäisytoimenpiteet, jotka perustuvat KLL-potilaan kasvainkuormaan.
Kasvainkuorma | Ehkäisytoimenpide | Veren kemiallisten arvojen seurantac,d | ||
Nesteytysa | Hyperurikemialääkkeetb | Arviointien järjestäminen ja tiheys | ||
Alhainen | Kaikki LN < 5 cm JA ALC < 25 x109/l | Suun kautta (1,5–2 l) | Allopurinoli | Avohoitopotilas
|
Keskisuuri | Mikä tahansa LN 5 cm – < 10 cm TAI ALC ≥ 25 x109/l | Suun kautta (1,5–2 l) ja harkitse lisäksi laskimoannostelua | Allopurinoli | Avohoitopotilas
|
Suuri | Mikä tahansa LN ≥ 10 cm TAI ALC ≥ 25 x109/l JA mikä tahansa LN ≥ 5 cm | Suun kautta (1,5–2 l) ja laskimoon (150–200 ml/h sietokyvyn mukaan) | Allopurinoli; harkitse rasburikaasia, jos lähtötilanteessa virtsahappo on koholla | Sairaalassa
Avohoitopotilas
|
ALC = absoluuttinen lymfosyyttimäärä; CrCl = kreatiniinipuhdistuma; LN = imusolmuke.aKehota potilaita juomaan vettä päivittäin, alkaen 2 päivää ennen annosta ja koko annostitrausvaiheen ajan, erityisesti ennen annosta aloitusvaiheessa ja ennen kunkin myöhemmän annoksen suurentamisen päivää, sekä kyseisinä päivinä. Anna nesteytys laskimoon potilaalle, joka ei siedä suun kautta annettavaa nesteytystä. bAloita allopurinoli tai ksantiinioksidaasin estäjä 2–3 päivää ennen venetoklaksin aloittamista.cArvioi veren kemialliset arvot (kalium, virtsahappo, fosfori, kalsium ja kreatiniini); arvioi reaaliaikaisesti. dMyöhemmin tehtävissä annoksen suurentamisissa seuraa veren kemiallisia arvoja 6–8 tunnin ja 24 tunnin kohdalla potilailla, joilla on edelleen tuumorilyysioireyhtymän riski. | ||||
Annosmuutokset tuumorilyysioireyhtymän ja muiden toksisuuksien vuoksi
Krooninen lymfaattinen leukemia
Lääkityksen tauotus ja/tai annoksen pienentäminen voi olla tarpeen toksisuuksien vuoksi. Katso taulukosta 4 ja 5 suositellut annosmuutokset venetoklaksiin liittyvissä toksisuustapauksissa.
Katso lisätietoja toksisuuksien hoidosta venetoklaksin kanssa yhdistelmähoitona käytettävien lääkevalmisteiden valmisteyhteenvedoista.
Taulukko 4: Suositellut venetoklaksin annosmuutokset toksisuuksien vuoksia KLL:ssä
Tapahtuma | Ilmeneminen | Toimintaohje |
Tuumorilyysioireyhtymä | ||
Tuumorilyysioireyhtymään viittaavat veren kemiallisen koostumuksen muutokset tai oireet | Milloin tahansa | Älä anna seuraavan päivän annosta. Jos muutokset/oireet korjautuvat 24–48 tunnin sisällä viimeisimmästä annoksesta, aloita uudelleen samalla annoksella. |
Jos veren kemiallisen koostumuksen muutosten korjautuminen vie yli 48 tuntia, aloita uudelleen aiempaa pienemmällä annoksella (ks. taulukko 5). | ||
Kaikkien kliinisten tuumorilyysioireyhtymätapahtumienb tapauksessa aloita uudelleen aiempaa pienemmällä annoksella korjautumisen jälkeen (ks. taulukko 5). | ||
Ei-hematologiset toksisuudet | ||
Asteen 3 tai 4 ei-hematologiset toksisuudet | Ensimmäinen ilmenemiskerta | Keskeytä venetoklaksin käyttö. Kun toksisuus on korjautunut asteen 1 tasolle tai lähtötasolle, venetoklaksihoito voidaan aloittaa uudelleen samalla annoksella. Annoksen muuttaminen ei ole tarpeen. |
Toinen tai myöhempi ilmenemiskerta | Keskeytä venetoklaksin käyttö. Noudata taulukossa 5 esitettäviä annoksen pienentämisohjeita, kun venetoklaksihoito aloitetaan uudelleen toksisuuden korjauduttua. Annosta voidaan pienentää enemmänkin lääkärin harkinnan mukaan. | |
Hematologiset toksisuudet | ||
Asteen 3 neutropenia, johon liittyy infektio tai kuume; tai asteen 4 hematologiset toksisuudet (paitsi lymfopenia) | Ensimmäinen ilmenemiskerta | Keskeytä venetoklaksin käyttö. Neutropeniaan liittyvien infektioriskien pienentämiseksi granulosyyttiryhmiä stimuloivaa kasvutekijää (G-CSF) voidaan antaa venetoklaksin kanssa, jos tämä on kliinisesti aiheellista. Kun toksisuus on korjautunut asteen 1 tasolle tai lähtötasolle, venetoklaksihoito voidaan aloittaa uudelleen samalla annoksella. |
Toinen tai myöhempi ilmenemiskerta | Keskeytä venetoklaksin käyttö. Harkitse G-CSF:n käyttöä, jos se on kliinisesti aiheellista. Noudata taulukossa 5 esitettäviä annoksen pienentämisohjeita, kun venetoklaksihoito aloitetaan uudelleen toksisuuden korjauduttua. Annosta voidaan pienentää enemmänkin lääkärin harkinnan mukaan. | |
Harkitse venetoklaksihoidon lopettamista, jos potilaan annos on pienennettävä alle 100 milligrammaan yli 2 viikon ajaksi.aHaittavaikutukset luokiteltiin käyttäen NCI CTCAE -versiota 4.0. bKliininen tuumorilyysioireyhtymä määriteltiin laboratorioarvoista todetuksi tuumorilyysioireyhtymäksi, joka on aiheuttanut kliinisiä seuraamuksia, kuten akuuttia munuaisten vajaatoimintaa, sydämen rytmihäiriöitä, kouristuskohtauksia ja/tai äkkikuoleman (ks. kohta Haittavaikutukset). | ||
Taulukko 5: Annosmuutokset tuumorilyysioireyhtymän tai muun toksisuuden vuoksi KLL-potilailla
Annos tauotushetkellä (mg) | Annos hoidon uudelleen aloittamisen yhteydessä (mga) |
400 | 300 |
300 | 200 |
200 | 100 |
100 | 50 |
50 | 20 |
20 | 10 |
aMuutettua annosta käytetään 1 viikon ajan, minkä jälkeen annosta suurennetaan. | |
Jos potilaan lääkitys on tauotettu yli 1 viikon ajaksi alkuvaiheen 5 viikon annostitrausvaiheen aikana tai yli 2 viikon ajaksi annostitrausvaiheen päättymisen jälkeen, on aiheellista arvioida tuumorilyysioireyhtymän riski uudelleen ja selvittää, tuleeko hoito aloittaa uudelleen pienemmällä annoksella (esim. kaikki tai jotkin annostitraustasot, ks. taulukko 5).
Akuutti myelooinen leukemia
Venetoklaksivuorokausiannoksen titraus on 3 päivää atsasitidiinin tai desitabiinin käytön yhteydessä (ks. taulukko 2).
Jäljempänä lueteltavat profylaktiset toimet on toteutettava:
Kaikkien potilaiden veren valkosolujen määrän on oltava < 25 × 109/l ennen venetoklaksihoidon aloittamista ja sytoreduktio ennen hoitoa saattaa olla tarpeen.
Kaikkien potilaiden nesteytyksen on oltava riittävä ja heidän on saatava hyperurikemialääkkeitä ennen ensimmäisen venetoklaksiannoksen antamista ja annostitrausvaiheen aikana.
Veren kemiallinen koostumus (kalium-, virtsahappo-, fosfori-, kalsium- ja kreatiniiniarvot) on arvioitava ja aiemmat poikkeavuudet on korjattava ennen venetoklaksihoidon aloittamista.
Veren kemiallista koostumusta on seurattava tuumorilyysioireyhtymän varalta ennen lääkkeen antoa, 6–8 tunnin kuluttua kunkin uuden annoksen jälkeen titrauksen aikana ja 24 tunnin kuluttua suositellun ylläpitoannoksen saavuttamisesta.
Potilaille, joilla on tuumorilyysioireyhtymän riskitekijöitä (esim. kiertäviä blasteja, luuytimen suuri leukemiakuorma, kohonneet veren laktaattidehydrogenaasiarvot [LDH] ennen hoitoa tai heikentynyt munuaistoiminta), on harkittava lisätoimia, kuten laboratorioseurannan tehostamista ja venetoklaksin aloitusannoksen pienentämistä.
Verisolumääriä on seurattava tiheästi sytopenioiden korjautumisen aikana. Annostuksen muuttaminen ja lääkkeen tauottaminen sytopenioiden vuoksi riippuu remissiotilasta. Venetoklaksin annostusmuutokset haittavaikutusten vuoksi on esitetty taulukossa 6.
Taulukko 6: Suositellut annostusmuutokset haittavaikutusten vuoksi AML-potilailla
Haittavaikutus | Esiintyminen | Annostusmuutos |
Hematologiset haittavaikutukset | ||
Asteen 4 neutropenia (ANC < 500/mikrolitra), johon voi liittyä kuume tai infektio, tai asteen 4 trombosytopenia (trombosyyttiarvo < 25 × 103/mikrolitra) | Esiintyminen ennen remission saavuttamistaa | Useimmissa tapauksissa älä tauota venetoklaksin ja atsasitidiinin tai desitabiinin yhdistelmähoitoa sytopenioiden takia ennen remission saavuttamista. |
Esiintyminen ensimmäisen kerran remission saavuttamisen jälkeen ja kesto vähintään 7 päivää | Viivytä seuraavaa venetoklaksin ja atsasitidiinin tai desitabiinin yhdistelmähoitojaksoa ja seuraa veriarvoja. Anna granulosyyttejä stimuloivaa kasvutekijää (G-CSF), jos se on kliinisesti tarpeellista neutropenian vuoksi. Kun neutropenia tai trombosytopenia on korjautunut asteen 1 tai 2 tasolle, aloita venetoklaksin anto uudestaan samalla annoksella yhdessä atsasitidiinin tai desitabiinin kanssa. | |
Seuraavat esiintymiset remission saavuttamisen jälkeen ja kesto vähintään 7 päivää | Viivytä seuraavaa venetoklaksin ja atsasitidiinin tai desitabiinin yhdistelmähoitojaksoa ja seuraa veriarvoja. Anna G-CSF:ää, jos se on kliinisesti tarpeellista neutropenian vuoksi. Kun neutropenia tai trombosytopenia on korjautunut asteen 1 tai 2 tasolle, aloita venetoklaksin anto uudestaan samalla annoksella yhdessä atsasitidiinin tai desitabiinin kanssa ja lyhennä venetoklaksin annon kestoa 7 päivällä kunkin seuraavan hoitojakson aikana, esimerkiksi 21 päivää 28 päivän sijaan. Lisätietoja on atsasitidiinin valmisteyhteenvedossa. | |
Ei-hematologiset haittavaikutukset | ||
Asteen 3 ja 4 ei-hematologiset toksisuudet | Esiintyminen milloin tahansa | Tauota venetoklaksin anto, jos toksisuus ei korjaudu tukihoidolla. Kun toksisuus on korjautunut asteen 1 tai lähtötilanteen tasolle, aloita venetoklaksin anto uudestaan samalla annoksella. |
aLuuydintutkimusta on harkittava. | ||
Annosmuutokset CYP3A:n estäjien samanaikaisen käytön yhteydessä
Venetoklaksin käyttö samanaikaisesti vahvojen tai keskivahvojen CYP3A:n estäjien kanssa suurentaa venetoklaksialtistusta (Cmax- ja AUC-arvoa) ja voi suurentaa tuumorilyysioireyhtymän riskiä hoidon alussa ja annostitrausvaiheen aikana sekä muun toksisuuden riskiä (ks. kohta Yhteisvaikutukset).
KLL-potilailla venetoklaksin ja vahvojen CYP3A:n estäjien samanaikainen käyttö on vasta-aiheista hoidon alussa ja annostitrausvaiheen aikana (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Jos CYP3A:n estäjiä on käytettävä, lääkeaineiden yhteisvaikutusten hallintaa koskevia suosituksia on noudatettava kaikilla potilailla (yhteenveto taulukossa 7). Potilaita on seurattava tavallista tiiviimmin toksisuuden merkkien varalta ja annosta on mahdollisesti muutettava edelleen. Venetoklaksiannos, jota potilas käytti ennen CYP3A:n estäjän käyttöönottoa, otetaan jälleen käyttöön 2–3 päivän kuluttua CYP3A:n estäjän käytön päättymisestä (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Taulukko 7: Venetoklaksin ja CYP3A:n estäjien mahdollisten yhteisvaikutusten hallinta
Estäjä | Vaihe | KLL | AML |
Vahva CYP3A:n estäjä | Hoidon aloitus ja annostitrausvaihe | Vasta-aiheinen | Päivä 1 – 10 mg Päivä 2 – 20 mg Päivä 3 – 50 mg Päivä 4 – 100 mg tai alle |
Vakaa vuorokausiannos (annostitrausvaiheen jälkeen) | Pienennä venetoklaksin -annos 100 mg:aan tai alle (tai vähintään 75 %:lla, jos annosta on jo muutettu muista syistä) | ||
Keskivahva CYP3A:n estäjäa | Kaikki | Pienennä venetoklaksin annosta vähintään 50 % | |
aVenetoklaksin ja keskivahvojen CYP3A:n estäjien samanaikaista käyttöä on vältettävä hoidon alussa ja annostitrausvaiheen aikana KLL-potilailla. Harkitse muita lääkevalmisteita tai pienennä venetoklaksin annosta tässä taulukossa kuvattuun tapaan. | |||
Väliin jäänyt annos
Jos venetoklaksiannos myöhästyy enintään 8 tuntia tavanomaisesta lääkkeenottoajankohdasta, potilaan on otettava väliin jäänyt annos mahdollisimman pian samana päivänä. Jos annos myöhästyy yli 8 tuntia, se jätetään ottamatta ja potilas palaa tavanomaiseen lääkkeenottoaikatauluun seuraavana päivänä.
Jos potilas oksentaa lääkkeen ottamisen jälkeen, kyseisenä päivänä ei saa ottaa uutta annosta. Seuraava lääkemääräyksen mukainen annos otetaan seuraavana päivänä tavanomaiseen aikaan.
Erityisryhmät
Iäkkäät
Iäkkäillä (≥ 65-vuotiailla) potilailla ei tarvita erityisiä annosmuutoksia (ks. kohta Farmakodynamiikka).
Munuaisten vajaatoiminta
Jos potilaan munuaistoiminta on heikentynyt (kreatiniinipuhdistuma < 80 ml/min), tehokkaampi profylaksi ja seuranta voi olla tarpeen tuumorilyysioireyhtymän riskin pienentämiseksi hoidon alussa ja annostitrausvaiheen aikana (ks. kohta ”Tuumorilyysioireyhtymän ehkäisy” edellä). Venetoklaksia saa antaa vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma ≥ 15 ml/min, mutta < 30 ml/min) tai dialyysihoitoa vaativaa loppuvaiheen munuaistautia (end-stage renal disease, ESRD) (kreatiniinipuhdistuma < 15 ml/min) sairastaville potilaille vain, jos hyödyt ylittävät riskit; lisäksi potilaita on seurattava tarkoin toksisuuden varalta, sillä tuumorilyysioireyhtymän riski on suurentunut (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annosta ei tarvitse muuttaa, jos potilaalla on lievä, keskivaikea, vaikea munuaisten vajaatoiminta tai dialyysihoitoa vaativa loppuvaiheen munuaistauti (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annosmuutoksia ei suositella potilaille, joilla on lievä tai keskivaikea maksan vajaatoiminta. Potilaita, joilla on keskivaikea maksan vajaatoiminta, on seurattava tavanomaista tiiviimmin toksisuuden merkkien varalta hoidon alussa ja annostitrausvaiheen aikana (ks. kohta Haittavaikutukset).
Vaikeaa maksan vajaatoimintaa sairastaville potilaille suositellaan annoksen pienentämistä vähintään 50 % koko hoidon ajan (ks. kohta Farmakokinetiikka). Näitä potilaita on seurattava tarkemmin toksisuuden merkkien varalta (ks. kohta Haittavaikutukset).
Pediatriset potilaat
Venclyxto-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tällä hetkellä saatavilla olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta.
Antotapa
Kalvopäällysteiset Venclyxto-tabletit otetaan suun kautta. Potilaita kehotetaan nielemään tabletit kokonaisina veden kera suunnilleen samaan aikaan joka päivä. Tabletit on otettava aterian kanssa, jotta lääkkeen tehottomuuden riskiltä vältytään (ks. kohta Farmakokinetiikka). Tabletteja ei saa pureskella, murskata eikä pilkkoa ennen nielemistä.
Annostitrausvaiheessa venetoklaksi otetaan aamuisin laboratorioseurannan helpottamiseksi.
Greippituotteita, pomeranssia ja karambolaa on vältettävä venetoklaksihoidon aikana (ks. kohta Yhteisvaikutukset).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Vahvojen CYP3A:n estäjien samanaikainen käyttö hoidon alussa ja annostitrausvaiheen aikana KLL-potilailla (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Mäkikuismaa sisältävien valmisteiden samanaikainen käyttö kaikilla potilailla (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Tuumorilyysioireyhtymä
Potilailla on ilmennyt venetoklaksihoidon aikana tuumorilyysioireyhtymää, myös kuolemaan johtaneita tapahtumia ja dialyysiä edellyttävää munuaisten vajaatoimintaa (ks. kohta Haittavaikutukset).
Venetoklaksi voi pienentää kasvaimen kokoa nopeasti, joten sen käyttöön liittyy tuumorilyysioireyhtymän riski hoidon alussa ja annostitrausvaiheen aikana. Tuumorilyysioireyhtymään viittaavia elektrolyyttiarvojen muutoksia, jotka edellyttävät nopeaa hoitoa, voi ilmetä jo 6–8 tunnin kuluessa ensimmäisestä venetoklaksiannoksesta ja aina annoksen suurentamisen yhteydessä. Markkinoilletulon jälkeisen haittaseurannan aikana tuumorilyysioireyhtymää, mukaan lukien kuolemaan johtaneita tapauksia, on raportoitu yksittäisen 20 mg:n venetoklaksiannoksen jälkeen. Kohdassa Annostus ja antotapa kuvattuja ohjeita, mukaan lukien riskinarviointi, ehkäisytoimet, annostitraus ja -muutokset, laboratorioseuranta ja lääkeaineiden yhteisvaikutukset, on noudatettava tuumorilyysioireyhtymän ehkäisemiseksi ja sen riskin pienentämiseksi.
Tuumorilyysioireyhtymän riski on jatkumo ja riippuu useista tekijöistä, mm. oheissairauksista (erityisesti heikentyneestä munuaistoiminnasta), kasvainkuormasta ja splenomegaliasta KLL:ssa.
Kaikki potilaat on arvioitava tuumorilyysioireyhtymän riskin varalta, ja asianmukaisiin tuumorilyysioireyhtymän ehkäisytoimiin on ryhdyttävä (mm. nesteytys ja hyperurikemialääkkeet). Veren kemiallista koostumusta on seurattava ja poikkeavuudet on hoidettava välittömästi. Kokonaisriskin suurentuessa on ryhdyttävä tehokkaampiin toimiin (nesteytys laskimoon, tiheä seuranta, sairaalahoito). Valmisteen anto on tauotettava tarvittaessa; kun venetoklaksin käyttö aloitetaan uudelleen, annosmuutoksia koskevia ohjeita on noudatettava (ks. taulukko 4 ja 5). Kohdassa ”Tuumorilyysioireyhtymän ehkäisy” esitettäviä ohjeita on noudatettava (ks. kohta Annostus ja antotapa).
Tämän lääkevalmisteen käyttö samanaikaisesti vahvojen tai keskivahvojen CYP3A:n estäjien kanssa suurentaa venetoklaksialtistusta ja voi suurentaa tuumorilyysioireyhtymän riskiä hoidon alussa ja annostitrausvaiheen aikana (ks. kohdat Annostus ja antotapa ja Vasta-aiheet). Myös P-gp:n tai BCRP:n estäjien käyttö voi suurentaa venetoklaksialtistusta (ks. kohta Yhteisvaikutukset).
Neutropenia ja infektiot
Venetoklaksihoitoa saaneilla KLL-potilailla on ilmoitettu asteen 3 tai 4 neutropeniaa yhdistelmähoito- ja monoterapiatutkimuksissa (ks. kohta Haittavaikutukset).
Asteen 3 ja 4 neutropenia on yleinen AML-potilailla ennen hoidon aloittamista. Neutrofiiliarvot voivat huonontua venetoklaksin ja hypometyloivan lääkkeen yhdistelmähoidon yhteydessä. Neutropenia voi uusiutua myöhemmissä hoitojaksoissa.
Täydellistä verenkuvaa on seurattava koko hoitovaiheen ajan. Jos potilaalla on vaikea neutropenia, suositellaan lääkkeen tauottamista tai annoksen pienentämistä (ks. kohta Annostus ja antotapa).
Vakavia infektioita (mukaan lukien kuolemaan johtaneita sepsistapauksia) on ilmoitettu (ks. kohta Haittavaikutukset). Potilaiden vointia on seurattava infektion oireiden ja löydösten varalta. Infektioepäilyn yhteydessä potilaille on annettava ripeää hoitoa, mm. mikrobilääkkeitä, venetoklaksin anto on tauotettava tai sen annosta on pienennettävä ja kasvutekijöitä (esim. G-CSF) on käytettävä asianmukaisesti (ks. kohta Annostus ja antotapa).
Immunisaatio
Eläviä, heikennettyjä taudinaiheuttajia sisältävillä rokotteilla toteutetun immunisaation turvallisuutta ja tehoa ei ole arvioitu venetoklaksihoidon aikana eikä sen jälkeen. Eläviä rokotteita ei pidä antaa hoidon aikana eikä sen jälkeen ennen kuin B-solut ovat toipuneet.
CYP3A:n indusoijat
CYP3A4:n indusoijien samanaikainen anto voi johtaa venetoklaksialtistuksen pienenemiseen ja aiheuttaa siten hoidon tehottomuuden riskin. Vahvojen tai keskivahvojen CYP3A4:n indusoijien samanaikaista käyttöä on vältettävä (ks. kohdat Vasta-aiheet ja Yhteisvaikutukset).
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisymenetelmää venetoklaksihoidon aikana (ks. kohta Raskaus ja imetys).
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) tablettia kohden eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Venetoklaksi metaboloituu lähinnä CYP3A-välitteisesti.
Aineet, jotka saattavat muuttaa plasman venetoklaksipitoisuuksia
CYP3A:n estäjät
Ketokonatsolin (vahva CYP3A:n, P-gp:n ja BCRP:n estäjä) samanaikainen anto 400 mg annoksina kerran vuorokaudessa 7 päivän ajan 11 potilaalle suurensi venetoklaksin Cmax-arvon 2,3-kertaiseksi ja AUC∞-arvon 6,4-kertaiseksi. Ritonaviirin (vahva CYP3A:n ja P-gp:n estäjä) samanaikainen anto 50 mg annoksina kerran vuorokaudessa 14 päivän ajan 6 terveelle henkilölle suurensi venetoklaksin Cmax‑arvon 2,4-kertaiseksi ja AUC-arvon 7,9-kertaiseksi. Verrattuna venetoklaksin antoon 400 mg annoksina ainoana lääkkeenä, posakonatsolin (vahva CYP3A:n ja P-gp:n estäjä) samanaikainen anto 300 mg annoksina 7 päivän ajan 12 potilaalle yhdessä venetoklaksin kanssa suurensi venetoklaksin Cmax-arvon 1,6-kertaiseksi ja AUC-arvon 1,9-kertaiseksi, kun venetoklaksiannos oli 50 mg, ja Cmax-arvon 1,9-kertaiseksi ja AUC-arvon 2,4-kertaiseksi, kun venetoklaksiannos oli 100 mg. Venetoklaksin anto samanaikaisesti muiden vahvojen CYP3A4:n estäjien kanssa oletetaan suurentavan venetoklaksin AUC-arvon keskimäärin 5,8–7,8-kertaiseksi.
Potilaille, jotka tarvitsevat venetoklaksin samanaikaista käyttöä yhdessä vahvojen CYP3A:n estäjien (esim. itrakonatsoli, ketokonatsoli, posakonatsoli, vorikonatsoli, klaritromysiini, ritonaviiri) kanssa tai keskivahvojen CYP3A:n estäjien (esim. siprofloksasiini, diltiatseemi, erytromysiini, flukonatsoli, verapamiili) kanssa, venetoklaksiannos on annettava taulukon 7 mukaisesti. Potilaiden vointia on seurattava tavallista tiiviimmin toksisuuden merkkien varalta, ja annosta on mahdollisesti muutettava edelleen. Venetoklaksiannos, jota potilas käytti ennen CYP3A:n estäjän käyttöönottoa, on otettava jälleen käyttöön 2–3 päivän kuluttua CYP3A:n estäjän käytön päättymisestä (ks. kohta Annostus ja antotapa).
Greippituotteita, pomeranssia ja karambolaa on vältettävä venetoklaksihoidon aikana, sillä ne sisältävät CYP3A:n estäjiä.
P-gp:n ja BCRP:n estäjät
Venetoklaksi on P-gp:n ja BCRP:n substraatti. Rifampisiinin (P-gp:n estäjä) samanaikainen anto 600 mg kerta-annoksena 11 terveelle henkilölle suurensi venetoklaksin Cmax-arvoa 106 % ja sen AUC-arvoa 78 %. Venetoklaksin käyttöä samanaikaisesti P-gp:n tai BCRP:n estäjien kanssa on vältettävä hoidon aloittamisen yhteydessä ja annostitrausvaiheessa; mikäli P-gp:n tai BCRP:n estäjän käyttö on välttämätöntä, potilaita on seurattava tiiviisti toksisuuden merkkien varalta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ibrutinibi
Ibrutinibin (420 mg) ja venetoklaksin (400 mg) yhdistelmähoitoa koskeneissa tutkimuksissa KLL-potilailla venetoklaksin altistus suureni noin 1,8‑kertaiseksi (AUC-arvon perusteella) verrattuna venetoklaksin monoterapiahoitoon.
CYP3A:n indusoijat
Kun 10 terveelle henkilölle annettiin 13 päivän ajan samanaikaisesti 600 mg rifampisiinia (vahva CYP3A:n indusoija) kerran vuorokaudessa, venetoklaksin Cmax pieneni 42 % ja sen AUC taas 71 %. Venetoklaksin käyttöä samanaikaisesti vahvojen CYP3A:n indusoijien (esim. karbamatsepiini, fenytoiini, rifampisiini) tai keskivahvojen CYP3A:n indusoijien (esim. bosentaani, efavirentsi, etraviriini, modafiniili, nafsilliini) kanssa on vältettävä. Muita hoitovaihtoehtoja, joihin liittyy vähemmän CYP3A:n induktiota, on harkittava. Mäkikuismaa sisältävät valmisteet ovat vasta-aiheisia venetoklaksihoidon aikana, sillä venetoklaksin teho voi heikentyä (ks. kohta Vasta-aiheet).
Atsitromysiini
12 terveellä henkilöllä toteutetussa lääkeinteraktiotutkimuksessa venetoklaksin samanaikainen anto atsitromysiinin kanssa (500 mg atsitromysiiniä ensimmäisenä päivänä ja tämän jälkeen 250 mg atsitromysiiniä kerran päivässä 4 päivän ajan) pienensi venetoklaksin Cmax‑arvoa 25 % ja AUC‑arvoa 35 %. Annosmuutosta ei tarvita, mikäli atsitromysiiniä käytetään lyhyen aikaa samanaikaisesti venetoklaksin kanssa.
Mahahappoa vähentävät valmisteet
Populaatiofarmakokineettisen analyysin perusteella mahahappoa vähentävät valmisteet (esim. protonipumpun estäjät, H2-reseptorin salpaajat, antasidit) eivät vaikuta venetoklaksin biologiseen hyötyosuuteen.
Sappihappoja sitovat lääkeaineet
Sappihappoja sitovien lääkeaineiden anto samanaikaisesti venetoklaksin kanssa ei ole suositeltavaa, sillä venetoklaksin imeytyminen voi heikentyä. Jos sappihappoja sitovaa lääkeainetta aiotaan antaa yhdessä venetoklaksin kanssa, sappihappoja sitovan lääkkeen valmisteyhteenvedon ohjeita on noudatettava yhteisvaikutusriskin pienentämiseksi ja venetoklaksi on annettava aikaisintaan 4–6 tuntia sappihappoja sitovan lääkkeen jälkeen.
Aineet, joiden pitoisuuksiin plasmassa venetoklaksi voi vaikuttaa
Varfariini
Kolmella terveellä vapaaehtoisella toteutetussa lääkeaineinteraktiotutkimuksessa 400 mg venetoklaksikerta-annoksen anto 5 mg varfariiniannoksen kanssa suurensi R-varfariinin ja S-varfariinin Cmax- ja AUC-arvoja 18–28 %. Venetoklaksin antoa ei jatkettu vakaan tilan saavuttamiseen saakka. Näin ollen on suositeltavaa, että varfariinihoitoa saavien potilaiden INR-arvoa (International Normalized Ratio) seurataan tarkoin.
P-gp:n, BCRP:n ja OATP1B1:n substraatit
Venetoklaksi on P-gp:n, BCRP:n ja OATP1B1:n estäjä in vitro. Lääkeinteraktiotutkimuksessa 100 mg:n kerta-annos venetoklaksia yhdessä 0,5 mg:n digoksiiniannoksen kanssa (P-gp:n substraatti) suurensi digoksiinin Cmax-arvoa 35 % ja AUC-arvoa 9 %. Kapean terapeuttisen indeksin omaavien P-gp:n tai BCRP:n substraattien (esim. digoksiini, dabigatraani, everolimuusi, sirolimuusi) antoa samanaikaisesti venetoklaksin kanssa on vältettävä.
Jos kapean terapeuttisen indeksin omaavan P-gp:n tai BCRP:n substraatin käyttö on välttämätöntä, käytössä on noudatettava varovaisuutta. Jos kyseessä on suun kautta annettava P-gp:n tai BCRP:n substraatti, joka on herkkä ruoansulatuskanavassa tapahtuvalle estovaikutukselle (esim. dabigatraanieteksilaatti), kyseisen lääkkeen ja venetoklaksin antoajankohtien välillä on pidettävä mahdollisimman pitkä väli mahdollisten yhteisvaikutusten minimoimiseksi.
Jos statiinia (OATP:n substraatti) käytetään samanaikaisesti venetoklaksin kanssa, statiiniin liittyvän toksisuuden tarkka seuranta on suositeltavaa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Raskauden ehkäisy
Naisten on vältettävä raskaaksi tulemista Venclyxto-hoidon aikana ja vähintään 30 päivän ajan hoidon päätyttyä. Naisten, jotka voivat tulla raskaaksi, onkin käytettävä erittäin tehokasta ehkäisyä venetoklaksihoidon aikana ja 30 päivän ajan sen päättymisen jälkeen. Toistaiseksi ei tiedetä, heikentääkö venetoklaksi hormonaalisten ehkäisyvalmisteiden tehoa. Näin ollen hormonaalista ehkäisyä käyttävien naisten on käytettävä lisäksi jotakin estemenetelmää.
Raskaus
Eläimillä tehtyjen alkio- ja sikiötoksisuustutkimusten perusteella (ks. kohta Prekliiniset tiedot turvallisuudesta) venetoklaksin anto raskaana olevalle naiselle voi aiheuttaa sikiöhaittoja.
Ei ole olemassa tarkkoja, hyvin kontrolloituja tietoja venetoklaksin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Venetoklaksin käyttöä ei suositella raskauden aikana eikä naisille, jotka voivat tulla raskaaksi eivätkä käytä erittäin tehokasta ehkäisyä.
Imetys
Ei tiedetä, erittyvätkö venetoklaksi tai sen metaboliitit ihmisen rintamaitoon.
Imetettävään lapseen kohdistuvia riskejä ei voida poissulkea.
Imetys on lopetettava Venclyxto-hoidon ajaksi.
Hedelmällisyys
Venetoklaksin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Koirilla on todettu kivestoksisuutta kliinisesti relevanteilla altistuksilla, joten venetoklaksihoito saattaa heikentää miehen hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta). Joidenkin miespotilaiden kohdalla voidaan harkita siittiöiden tallettamista koskevaa neuvontaa ennen hoidon aloittamista.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Venclyxto-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Joillakin venetoklaksihoitoa saaneilla potilailla on ilmoitettu uupumusta ja huimausta. Asia on otettava huomioon potilaan ajokykyä ja koneidenkäyttökykyä arvioitaessa.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Krooninen lymfaattinen leukemia
Venclyxton kokonaisturvallisuusprofiili perustuu tietoihin 1 187 KLL-potilaasta, jotka saivat venetoklaksihoitoa kliinisissä tutkimuksissa yhdessä obinututsumabin, ibrutinibin tai rituksimabin kanssa tai ainoana lääkkeenä. Turvallisuusanalyysiin otettiin potilaita kolmesta vaiheen 3 tutkimuksesta (CLL14, GLOW ja MURANO), kolmesta vaiheen 2 tutkimuksesta (CAPTIVATE, M13-982 ja M14-032) ja yhdestä vaiheen 1 tutkimuksesta (M12-175). CLL14 oli satunnaistettu, kontrolloitu tutkimus, jossa 212 potilasta, joilla oli aiemmin hoitamaton KLL ja liitännäissairauksia, sai venetoklaksia yhdessä obinututsumabin kanssa. GLOW oli avoin, satunnaistettu tutkimus, jossa 106 potilasta, joilla oli aiemmin hoitamaton KLL, sai venetoklaksia yhdessä ibrutinibin kanssa. MURANO oli satunnaistettu, kontrolloitu tutkimus, jossa 194 aiemmin hoidettua KLL-potilasta sai venetoklaksia yhdessä rituksimabin kanssa. CAPTIVATE oli 2 kohortilla toteutettu monikeskustutkimus, jossa 323 potilasta, joilla oli aiemmin hoitamaton KLL, sai venetoklaksia yhdessä ibrutinibin kanssa. M13-982-, M14-032- ja M12‑175-tutkimuksiin otettiin 352 potilasta, jotka olivat saaneet aiempia hoitoja KLL:n hoitoon. Heistä 212 potilaalla oli 17p-deleetio ja 146 potilaan kohdalla B-solureseptorireitin estäjähoito oli epäonnistunut. Potilaat saivat venetoklaksia ainoana hoitona (ks. kohta Farmakodynamiikka).
Yhdistelmähoitotutkimuksissa (obinututsumabin, ibrutinibin tai rituksimabin kanssa) yleisimpiä venetoklaksihoitoa saaneilla potilailla ilmenneitä haittavaikutuksia (≥ 20 %; kaikki asteet) olivat ripuli, neutropenia, pahoinvointi, ylähengitystieinfektio, uupumus ja oksentelu. Monoterapiatutkimuksissa yleisimpiä haittavaikutuksia olivat neutropenia/neutrofiilimäärän pieneneminen, ripuli, pahoinvointi, anemia, uupumus ja ylähengitystieinfektio.
Yleisimmin ilmoitettuja vakavia haittavaikutuksia (≥ 2 %), kun venetoklaksia annettiin yhdessä obinututsumabin, ibrutinibin tai rituksimabin kanssa, olivat keuhkokuume, kuumeinen neutropenia, sepsis, neutropenia, anemia, ripuli ja tuumorilyysioireyhtymä. Tutkimuksissa, joissa venetoklaksia annettiin ainoana lääkkeenä, yleisimmin ilmoitettuja vakavia haittavaikutuksia (≥ 2 %) olivat keuhkokuume ja kuumeinen neutropenia.
Venetoklaksin ja akalabrutinibin yhdistelmähoidon (yhdessä obinututsumabin kanssa tai ilman obinututsumabia) turvallisuutta arvioitiin satunnaistetussa ja kontrolloidussa AMPLIFY-tutkimuksessa 575 potilaalla, joilla oli aiemmin hoitamaton KLL ilman del (17p)- tai TP53-mutaatiota. Yleisimpiä venetoklaksia yhdessä akalabrutinibin kanssa saaneilla 291 potilaalla ilmenneitä haittavaikutuksia (≥ 20 %; kaikki asteet) olivat infektiot, neutropenia, päänsärky, mustelma, ripuli ja tuki- ja liikuntaelimistön kipu. Yleisimmin ilmoitettu asteen ≥ 3 haittavaikutus (≥ 5 %) oli neutropenia. Yleisimpiä venetoklaksia yhdessä akalabrutinibin ja obinututsumabin kanssa saaneilla 284 potilaalla ilmenneitä haittavaikutuksia (≥ 20 %; kaikki asteet) olivat infektiot, neutropenia, päänsärky, mustelma, ripuli, pahoinvointi ja tuki- ja liikuntaelimistön kipu. Yleisimmin ilmoitettuja asteen ≥ 3 haittavaikutuksia (≥ 5 %) olivat neutropenia ja trombosytopenia.
Akuutti myelooinen leukemia
Venclyxton kokonaisturvallisuusprofiili perustuu tietoihin 314 potilaasta, joilla oli äskettäin diagnosoitu akuutti myelooinen leukemia (AML) ja jotka saivat kliinisissä tutkimuksissa venetoklaksin ja hypometyloivan lääkkeen (atsasitidiinin tai desitabiinin) yhdistelmähoitoa (faasin 3 satunnaistettu tutkimus VIALE‑A ja faasin 1 satunnaistamaton tutkimus M14‑358).
VIALE‑A-tutkimuksessa yleisimpiä venetoklaksin ja atsasitidiinin yhdistelmähoitoa saaneilla potilailla ilmenneitä haittavaikutuksia (≥ 20 %; kaikki asteet) olivat trombosytopenia, neutropenia, kuumeinen neutropenia, pahoinvointi, ripuli, oksentelu, anemia, uupumus, keuhkokuume, hypokalemia ja ruokahalun heikkeneminen.
Yleisimmin ilmoitettuja vakavia haittavaikutuksia (≥ 5 %), kun venetoklaksia annettiin yhdessä atsasitidiinin kanssa, olivat kuumeinen neutropenia, keuhkokuume, sepsis ja verenvuoto.
M14‑358-tutkimuksessa yleisimpiä venetoklaksin ja desitabiinin yhdistelmähoitoa saaneilla potilailla ilmenneitä haittavaikutuksia (≥ 20 %; kaikki asteet) olivat trombosytopenia, kuumeinen neutropenia, pahoinvointi, verenvuoto, keuhkokuume, ripuli, uupumus, huimaus/pyörtyminen, oksentelu, neutropenia, hypotensio, hypokalemia, ruokahalun heikkeneminen, päänsärky, vatsakipu ja anemia. Yleisimmin ilmoitettuja vakavia haittavaikutuksia (≥ 5 %) olivat kuumeinen neutropenia, keuhkokuume, bakteremia ja sepsis.
30 päivän kuolleisuus oli VIALE‑A-tutkimuksessa 7,4 % (21/283), kun venetoklaksia annettiin yhdessä atsasitidiinin kanssa, ja 6,3 % (9/144) tutkimushaarassa, jossa annettiin lumelääkettä yhdessä atsasitidiinin kanssa.
M14‑358-tutkimuksessa, jossa venetoklaksia annettiin yhdessä desitabiinin kanssa, 30 päivän kuolleisuus oli 6,5 % (2/31).
Haittavaikutustaulukko
Haittavaikutukset luetellaan alla MedDRAn elinjärjestelmä- ja esiintymistiheysluokituksen mukaisesti esitettyinä. Esiintymistiheydet määritellään seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000), yleisyys tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Krooninen lymfaattinen leukemia
Venclyxto-hoidon yhteydessä (kun Venclyxtoa käytettiin joko yhdistelmähoitona obinututsumabin, ibrutinibin tai rituksimabin kanssa tai ainoana lääkkeenä KLL-potilaille) ilmoitettujen haittavaikutusten esiintymistiheydet esitetään yhteenvetomuodossa taulukossa 8.
Taulukko 8: Venetoklaksihoitoa saaneilla KLL-potilailla ilmoitetut haittavaikutukset
Elinjärjestelmä | Yleisyys | Kaikki asteeta | Aste ≥ 3a |
Infektiot | Hyvin yleinen | Keuhkokuume Ylähengitystieinfektio Virtsatieinfektio |
|
Yleinen | Sepsis | Sepsis Keuhkokuume Virtsatieinfektio Ylähengitystieinfektio | |
Veri ja imukudos | Hyvin yleinen | Neutropenia Anemia Lymfopenia | Neutropenia Anemia |
Yleinen | Kuumeinen neutropenia | Kuumeinen neutropenia Lymfopenia | |
Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Hyperkalemia Hyperfosfatemia Hypokalsemia |
|
Yleinen | Tuumorilyysioireyhtymä Hyperurikemia | Tuumorilyysioireyhtymä Hyperkalemia Hyperfosfatemia Hypokalsemia Hyperurikemia | |
Ruoansulatuselimistö | Hyvin yleinen | Ripuli Oksentelu Pahoinvointi Ummetus | Ripuli |
Yleinen |
| Oksentelu Pahoinvointi | |
Melko harvinainen |
| Ummetus | |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Uupumus |
|
Yleinen |
| Uupumus | |
Tutkimukset | Yleinen | Veren kreatiniinipitoisuuden suureneminen |
|
Melko harvinainen |
| Veren kreatiniinipitoisuuden suureneminen | |
a Vain suurin tutkimuksissa havaittu yleisyysluokka ilmoitetaan (CLL14-, GLOW-, CAPTIVATE-, MURANO-, M13-982-, M14-032- ja M12-175-tutkimusten pohjalta). | |||
AMPLIFY
Kun venetoklaksia annetaan yhdistelmähoitona akalabrutinibin kanssa joko yhdistettynä obinututsumabiin tai ilman obinututsumabia, katso haittavaikutusten kuvaus akalabrutinibin valmisteyhteenvedosta ennen hoidon aloittamista.
Akuutti myelooinen leukemia
Venclyxto-hoidon yhteydessä (kun Venclyxtoa käytettiin yhdistelmähoitona hypometyloivan lääkkeen kanssa AML-potilaille) ilmoitettujen haittavaikutusten esiintymistiheydet esitetään yhteenvetomuodossa taulukossa 9.
Taulukko 9: Venetoklaksihoitoa saaneilla AML-potilailla ilmoitetut haittavaikutukset
Elinjärjestelmä | Yleisyys | Kaikki asteeta | Aste ≥ 3a |
Infektiot | Hyvin yleinen | Keuhkokuumeb Sepsisb Virtsatieinfektio | Keuhkokuumeb Sepsisb |
Yleinen |
| Virtsatieinfektio | |
Veri ja imukudos | Hyvin yleinen | Neutropeniab Kuumeinen neutropenia Anemiab Trombosytopeniab | Neutropeniab Kuumeinen neutropenia Anemiab Trombosytopeniab |
Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Hypokalemia Ruokahalun heikkeneminen | Hypokalemia |
Yleinen | Tuumorilyysioireyhtymä | Ruokahalun heikkeneminen | |
Melko harvinainen |
| Tuumorilyysioireyhtymä | |
Hermosto | Hyvin yleinen | Huimaus/pyörtyminenb Päänsärky |
|
Yleinen |
| Huimaus/pyörtyminenb | |
Melko harvinainen |
| Päänsärky | |
Verisuonisto | Hyvin yleinen | Hypotensio Verenvuotob | Verenvuotob |
Yleinen |
| Hypotensio | |
Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Hengenahdistus |
|
Yleinen |
| Hengenahdistus | |
Ruoansulatuselimistö | Hyvin yleinen | Pahoinvointi Ripuli Oksentelu Stomatiitti Vatsakipu |
|
Yleinen |
| Pahoinvointi Ripuli Oksentelu | |
Melko harvinainen |
| Stomatiitti | |
Maksa ja sappi | Yleinen | Kolekystiitti/sappikivetb | Kolekystiitti/sappikivetb |
Luusto, lihakset ja sidekudos | Hyvin yleinen | Nivelkipu |
|
Melko harvinainen |
| Nivelkipu | |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Uupumus Astenia |
|
Yleinen |
| Uupumus Astenia | |
Tutkimukset | Hyvin yleinen | Painon lasku Veren bilirubiiniarvon suureneminen |
|
Yleinen |
| Painon lasku Veren bilirubiiniarvon suureneminen | |
aVain suurin tutkimuksissa havaittu yleisyysluokka ilmoitetaan (VIALE-A ja M14-358-tutkimusten pohjalta). bSisältää useita haittavaikutustermejä. | |||
Hoidon lopettaminen tai annoksen pienentäminen haittavaikutusten vuoksi.
Krooninen lymfaattinen leukemia
AMPLIFY-tutkimuksessa 8 % venetoklaksin ja akalabrutinibin yhdistelmähoitoa saaneista potilaista ja 20 % venetoklaksin, akalabrutinibin ja obinututsumabin yhdistelmähoitoa saaneista potilaista lopetti hoidon haittavaikutusten vuoksi.
CLL14-tutkimuksessa 16 % venetoklaksin ja obinututsumabin yhdistelmähoitoa saaneista potilaista ja MURANO-tutkimuksessa 16 % venetoklaksin ja rituksimabin yhdistelmähoitoa saaneista potilaista sekä GLOW-tutkimuksessa 21 % ja CAPTIVATE-tutkimuksessa 7 % venetoklaksin ja ibrutinibin yhdistelmähoitoa saaneista potilaista lopetti hoidon haittavaikutusten vuoksi. Venetoklaksin monoterapiatutkimuksissa 11 % potilaista lopetti hoidon haittavaikutusten vuoksi.
Annostusta pienennettiin haittavaikutusten vuoksi 14 %:lla AMPLIFY-tutkimuksen potilaista, jotka saivat venetoklaksin ja akalabrutinibin yhdistelmähoitoa ja 21 %:lla potilaista, jotka saivat venetoklaksin, akalabrutinibin ja obinututsumabin yhdistelmähoitoa.
Annostusta pienennettiin haittavaikutusten vuoksi 21 %:lla CLL14-tutkimuksen potilaista, jotka saivat venetoklaksin ja obinututsumabin yhdistelmähoitoa, 26 %:lla GLOW-tutkimuksen ja 20 %:lla CAPTIVATE-tutkimuksen potilaista, jotka saivat venetoklaksin ja ibrutinibin yhdistelmähoitoa, 15 %:lla MURANO-tutkimuksen potilaista, jotka saivat venetoklaksin ja rituksimabin yhdistelmähoitoa, ja 14 %:lla venetoklaksin monoterapiatutkimusten potilaista.
Hoito tauotettiin haittavaikutusten vuoksi 50 %:lla venetoklaksin ja akalabrutinibin yhdistelmähoitoa saaneista potilaista ja 65 %:lla venetoklaksin, akalabrutinibin ja obinututsumabin yhdistelmähoitoa saaneista potilaista AMPLIFY-tutkimuksessa. Yleisin venetoklaksin tauottamiseen johtanut haittavaikutus AMPLIFY-tutkimuksessa oli neutropenia (33 % obinututsumabia saaneilla potilailla ja 26 % potilailla, jotka eivät saaneet obinututsumabia).
Hoito tauotettiin haittavaikutusten vuoksi 74 %:lla venetoklaksin ja obinututsumabin yhdistelmää saaneista potilaista CLL14-tutkimuksessa, 67 %:lla venetoklaksin ja ibrutinibin yhdistelmää saaneista potilaista GLOW-tutkimuksessa ja 71 %:lla venetoklaksin ja rituksimabin yhdistelmää saaneista potilaista MURANO-tutkimuksessa. Yleisin venetoklaksin tauottamiseen johtanut haittavaikutus oli neutropenia (41 % CLL-tutkimuksessa, 19 % GLOW-tutkimuksessa ja 43 % MURANO-tutkimuksessa). Tutkimuksissa, joissa venetoklaksia annettiin ainoana lääkkeenä, hoito tauotettiin haittavaikutusten vuoksi 40 %:lla potilaista. Yleisin venetoklaksin tauottamiseen johtanut haittavaikutus oli neutropenia (5 %).
Akuutti myelooinen leukemia
VIALE‑A-tutkimuksessa 24 % venetoklaksin ja atsasitidiinin yhdistelmähoitoa saaneista potilaista lopetti hoidon haittavaikutusten vuoksi. Venetoclax-valmisteen annosta pienennettiin haittavaikutusten vuoksi 2 %:lla potilaista. Venetoclax-hoito tauotettiin haittavaikutusten vuoksi 72 %:lla potilaista. Potilaista, joiden luuytimessä ei enää ollut leukemiasoluja, 53 %:lla hoito tauotettiin ANC-arvon < 500/mikrolitra vuoksi. Yleisimmät venetoklaksin tauottamiseen johtaneet haittavaikutukset (> 10 %) olivat kuumeinen neutropenia, neutropenia, keuhkokuume ja trombosytopenia.
M14‑358-tutkimuksessa 26 % venetoklaksin ja desitabiinin yhdistelmähoitoa saaneista potilaista lopetti hoidon haittavaikutusten vuoksi. Annostusta pienennettiin haittavaikutusten vuoksi 6 %:lla potilaista. Hoito tauotettiin haittavaikutusten vuoksi 65 %:lla potilaista; yleisimmät venetoklaksin tauottamiseen johtaneet haittavaikutukset (≥ 5 %) olivat kuumeinen neutropenia, neutropenia / neutrofiiliarvon pieneneminen, keuhkokuume, trombosyyttiarvon pieneneminen ja veren valkosoluarvon pieneneminen.
Tiettyjen haittavaikutusten kuvaus
Tuumorilyysioireyhtymä
Tuumorilyysioireyhtymä on tärkeä, tiedossa oleva venetoklaksihoidon aloitukseen liittyvä riski.
Krooninen lymfaattinen leukemia
Ensimmäisissä vaiheen 1 annosmääritystutkimuksissa, joissa titrausvaihe oli lyhyempi (2–3 viikkoa) ja aloitusannos suurempi, tuumorilyysioireyhtymän ilmaantuvuus oli 13 % (10/77; 5 laboratorioarvoista todettua ja 5 kliinisesti todettua tuumorilyysioireyhtymätapausta). Mukana oli 2 kuolemaan johtanutta tapahtumaa, 3 akuuttiin munuaisten vajaatoimintaan johtanutta tapahtumaa ja yksi dialyysihoitoa vaatinut tapahtuma.
Tuumorilyysioireyhtymän riski pieneni, kun lääkkeen antoprotokollaa korjattiin ja profylaksi- ja seurantatoimia muutettiin. Kliinisissä venetoklaksitutkimuksissa potilaat, joilla oli mikä tahansa mitattavissa oleva ≥ 10 cm imusolmuke tai sekä ALC ≥ 25 x 109/l että mikä tahansa mitattavissa oleva ≥ 5 cm imusolmuke, otettiin titrausvaiheessa sairaalahoitoon, jotta nesteytys ja seuranta pystyttiin toteuttamaan tehokkaammin 20 mg ja 50 mg annosten ensimmäisenä antopäivänä (ks. kohta Annostus ja antotapa).
Kun 168 KLL-potilasta sai M13-982- ja M14-032-tutkimuksissa aluksi 20 mg vuorokausiannoksia ja heidän lääkeannoksensa suurennettiin 5 viikon kuluessa 400 mg vuorokausiannokseen, tuumorilyysioireyhtymän esiintymistiheys oli 2 %. Kaikki tapahtumat olivat laboratorioarvojen perusteella todettuja tuumorilyysioireyhtymätapauksia (laboratorioarvojen poikkeavuuksia, joissa ≥ 2 seuraavista kriteereistä täyttyi 24 tunnin kuluessa toisistaan: kalium > 6 mmol/l, virtsahappo > 476 mikromol/l, kalsium < 1,75 mmol/l tai fosfori > 1,5 mmol/l; tai tuumorilyysioireyhtymätapahtumaksi ilmoitettu tapahtuma), ja ne ilmenivät potilailla, joilla oli vähintään yksi ≥ 5 cm kokoinen imusolmuke tai joiden ALC-arvo oli ≥ 25 x 109/l. Näillä potilailla ei todettu yhtään tuumorilyysioireyhtymätapausta, joka olisi aiheuttanut kliinisiä seurauksia kuten akuuttia munuaisten vajaatoimintaa, sydämen rytmihäiriöitä, äkkikuoleman ja/tai kouristuskohtauksia. Kaikkien potilaiden kreatiniinipuhdistuma oli ≥ 50 ml/min.
Avoimessa, satunnaistetussa vaiheen 3 tutkimuksessa (MURANO) tuumorilyysioireyhtymän ilmaantuvuus oli 3 % (6/194) venetoklaksin ja rituksimabin yhdistelmähoitoa saaneilla. Kun tutkimukseen oli rekrytoitu 77 potilasta 389:stä, tutkimussuunnitelmaan lisättiin nykyiset tuumorilyysioireyhtymän esto- ja seurantatoimet, jotka kuvataan kohdassa “Annostus” (ks. kohta Annostus ja antotapa). Kaikki tuumorilyysioireyhtymätapahtumat ilmenivät venetoklaksin annostitrausvaiheen aikana ja korjautuivat kahden vuorokauden kuluessa. Kaikki kuusi potilasta suorittivat annostitrauksen loppuun ja saavuttivat suositellun venetoklaksiannoksen 400 mg/vrk. Kliinistä tuumorilyysioireyhtymää ei todettu potilailla, jotka noudattivat nykyistä 5 viikon annostitrausaikataulua ja nykyisiä tuumorilyysioireyhtymän esto- ja seurantatoimia (ks. kohta Annostus ja antotapa). Tuumorilyysioireyhtymän kannalta merkittävien asteen ≥ 3 laboratorioarvojen poikkeavuuksien ilmaantuvuudet olivat seuraavat: hyperkalemia 1 %, hyperfosfatemia 1 % ja hyperurikemia 1 %.
Avoimessa, satunnaistetussa vaiheen 3 tutkimuksessa (CLL14) tuumorilyysioireyhtymän ilmaantuvuus oli 1,4 % (3/212) venetoklaksin ja obinututsumabin yhdistelmähoitoa saaneilla. Kaikki kolme tuumorilyysioireyhtymätapahtumaa korjautuivat, eikä mikään niistä johtanut tutkimuksesta vetäytymiseen. Kahdessa tapauksessa obinututsumabin anto viivästyi tuumorilyysioireyhtymätapahtumien vuoksi.
Avoimessa, satunnaistetussa vaiheen 3 tutkimuksessa (AMPLIFY) tuumorilyysioireyhtymän ilmaantuvuus oli 0,3 % (1/291) venetoklaksin ja akalabrutinibin yhdistelmähoitoa saaneilla ja 0,4 % (1/284) venetoklaksin, akalabrutinibin ja obinututsumabin yhdistelmähoitoa saaneilla potilailla. Obinututsumabin anto viivästyi tuumorilyysioireyhtymätapahtuman vuoksi. Molemmissa tapauksissa kyse oli laboratorioarvoista todetusta tuumorilyysioireyhtymästä, joka korjautui eikä johtanut tutkimuksesta vetäytymiseen.
Tuumorilyysioireyhtymää ei todettu haittatapahtumana satunnaistetussa vaiheen 3 GLOW-tutkimuksessa.
Laboratorioarvoista todetun tuumorilyysioireyhtymän ilmaantuvuus oli 0,3 % (1/323) yhdellä hoitoryhmällä toteutetussa vaiheen 2 CAPTIVATE-tutkimuksessa, ja sitä raportoitiin yhdellä potilaalla MRD-ohjatussa kohortissa.
Markkinoilletulon jälkeisen haittaseurannan aikana tuumorilyysioireyhtymää, mukaan lukien kuolemaan johtaneita tapauksia, on raportoitu yksittäisen 20 mg:n venetoklaksiannoksen jälkeen (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Akuutti myelooinen leukemia
Satunnaistetussa, faasin 3 tutkimuksessa (VIALE‑A), jossa venetoklaksia annettiin yhdessä atsasitidiinin kanssa, tuumorilyysioireyhtymän ilmaantuvuus oli 1,1 % (3/283; 1 kliinisesti todettu tuumorilyysioireyhtymätapaus). Tutkimuksessa edellytettiin veren valkosoluarvon pieneneminen arvoon < 25 x 109/l ennen venetoklaksihoidon aloittamista ja annostitrausaikataulu tavanomaisten estohoito- ja seurantatoimien lisäksi (ks. kohta Annostus ja antotapa). Kaikki tuumorilyysioireyhtymätapaukset ilmenivät annostitrauksen aikana.
M14‑358-tutkimuksessa ei ilmoitettu yhtään laboratorioarvoista todettua tai kliinisesti todettua tuumorilyysioireyhtymätapahtumaa venetoklaksin ja desitabiinin yhdistelmähoidon käytön yhteydessä.
Neutropenia ja infektiot
Neutropenia on tiedossa oleva Venclyxto-hoitoon liittyvä riski.
Krooninen lymfaattinen leukemia
AMPLIFY-tutkimuksessa neutropeniaa / pienentynyttä neutrofiilimäärää / kuumeista neutropeniaa (kaikki asteet) ilmoitettiin 37 %:lla venetoklaksin ja akalabrutinibin yhdistelmähoitoa saaneista potilaista. Hoito tauotettiin 26 %:lla potilaista ja 0,7 % lopetti venetoklaksin käytön neutropenian / pienentyneen neutrofiilimäärän / kuumeisen neutropenian vuoksi. Asteen ≥ 3 neutropeniaa / pienentynyttä neutrofiilimäärää / kuumeista neutropeniaa ilmoitettiin 32 %:lla potilaista. Asteen ≥ 3 infektioita ilmoitettiin 12 %:lla ja vakavia infektioita 12 %:lla potilaista. Kuolemaan johtaneita infektioita ilmeni 3,1 %:lla potilaista (useimmiten ilmoitettuja olivat COVID‑19 tai COVID‑19-pneumonia).
AMPLIFY-tutkimuksessa neutropeniaa / pienentynyttä neutrofiilimäärää / kuumeista neutropeniaa (kaikki asteet) ilmoitettiin 50 %:lla venetoklaksin, akalabrutinibin ja obinututsumabin yhdistelmähoitoa saaneista potilaista. Hoito tauotettiin 33 %:lla potilaista ja 1 % lopetti venetoklaksin käytön neutropenian / pienentyneen neutrofiilimäärän / kuumeisen neutropenian vuoksi. Asteen ≥ 3 neutropeniaa / pienentynyttä neutrofiilimäärää / kuumeista neutropeniaa ilmoitettiin 46 %:lla potilaista. Asteen ≥ 3 infektioita ilmoitettiin 24 %:lla ja vakavia infektioita 24 %:lla potilaista. Kuolemaan johtaneita infektioita ilmeni 6 %:lla potilaista (useimmiten ilmoitettuja olivat COVID‑19 tai COVID‑19-pneumonia).
CLL14-tutkimuksessa neutropeniaa (kaikki asteet) ilmoitettiin 58 %:lla venetoklaksin ja obinututsumabin yhdistelmähoitoa saaneista. Hoito tauotettiin 41 %:lla venetoklaksin ja obinututsumabin yhdistelmähoitoa saaneista ja 2 % lopetti venetoklaksin käytön neutropenian vuoksi. Asteen 3 neutropeniaa ilmoitettiin 25 %:lla potilaista ja asteen 4 neutropeniaa 28 %:lla potilaista. Asteen 3 tai 4 neutropenian mediaanikesto oli 22 vrk (vaihteluväli 2–363 vrk). Kuumeista neutropeniaa ilmoitettiin 6 %:lla potilaista, asteen ≥ 3 infektioita 19 %:lla ja vakavia infektioita 19 %:lla potilaista. Infektioista johtuvia kuolemantapauksia todettiin 1,9 %:lla potilaista hoidon aikana ja 1,9 %:lla potilaista hoidon päätyttyä.
GLOW-tutkimuksessa 42 %:lla venetoklaksin ja ibrutinibin yhdistelmähoitoa saaneista potilaista ilmoitettiin minkä tahansa asteista neutropeniaa / pienentynyttä neutrofiilimäärää, mukaan lukien asteen 3 tai 4 tapahtumia 35 %:lla potilaista. Hoito tauotettiin 19 %:lla potilaista ja 8 %:lla venetoklaksiannosta pienennettiin neutropenian / pienentyneen neutrofiilimäärän vuoksi. Seuraavia ilmoitettiin venetoklaksin ja ibrutinibin yhdistelmähoitoa saaneilla potilailla vs. obinututsumabin ja klorambusiilin yhdistelmähoitoa saaneilla potilailla: kuumeinen neutropenia 2 % vs. 3 %, asteen ≥ 3 infektiot 17 % vs. 11 % ja vakavat infektiot 12 % vs. 9 %.
CAPTIVATE-tutkimuksessa 47 %:lla venetoklaksin ja ibrutinibin yhdistelmähoitoa saaneista potilaista ilmoitettiin minkä tahansa asteista neutropeniaa / pienentynyttä neutrofiilimäärää, mukaan lukien asteen 3 tai 4 tapahtumia 37 %:lla potilaista. Hoito tauotettiin 14 %:lla potilaista, annosta pienennettiin 4 %:lla potilaista ja yksi potilas (0,3 %) lopetti venetoklaksihoidon neutropenian / pienentyneen neutrofiilimäärän vuoksi. Kuumeista neutropeniaa ilmoitettiin 1 %:lla, asteen ≥ 3 infektioita 8 %:lla ja vakavia infektioita 8 %:lla potilaista.
MURANO-tutkimuksessa neutropeniaa (kaikki asteet) ilmoitettiin 61 %:lla venetoklaksin ja rituksimabin yhdistelmähoitoryhmän potilaista. Hoito tauotettiin 43 %:lla venetoklaksin ja rituksimabin yhdistelmähoitoa saaneista ja 3 % lopetti venetoklaksin käytön neutropenian vuoksi. Asteen 3 neutropeniaa ilmoitettiin 32 %:lla potilaista ja asteen 4 neutropeniaa 26 %:lla potilaista. Asteen 3 tai 4 neutropenian mediaanikesto oli 8 vrk (vaihteluväli 1–712 vrk). Venetoklaksin ja rituksimabin yhdistelmähoidon yhteydessä kuumeista neutropeniaa ilmoitettiin 4 %:lla potilaista, asteen ≥ 3 infektioita 18 %:lla ja vakavia infektioita 21 %:lla potilaista.
Akuutti myelooinen leukemia
VIALE‑A-tutkimuksessa neutropeniaa (aste ≥ 3) ilmoitettiin 45 %:lla potilaista. Myös seuraavista ilmoitettiin venetoklaksin ja atsasitidiinin yhdistelmähoitoryhmässä: kuumeinen neutropenia 42 %:lla, infektiot (aste ≥ 3) 64 %:lla ja vakavat infektiot 57 %:lla, kun taas vastaavat luvut lumelääkkeen ja atsasitidiinin yhdistelmähoitoryhmässä olivat 19 %, 51 % ja 44 %.
M14‑358-tutkimuksessa neutropeniaa ilmoitettiin 35 %:lla (kaikki asteet), ja 35 %:lla ilmoitettiin asteen 3 tai 4 neutropeniaa venetoklaksin ja desitabiinin yhdistelmähoitoryhmän potilaista.
Pediatriset potilaat
Venetoklaksin turvallisuusprofiili pediatrisilla potilailla perustuu tietoihin avoimesta vaiheen 1 tutkimuksesta (M13-833), johon osallistuneilla 140 pediatrisella ja nuorella aikuisella potilaalla oli uusiutuneita tai huonosti hoitoon vastaavia pahanlaatuisia kasvaimia (ks. kohta Farmakodynamiikka). Tutkimuksessa ei tunnistettu mitään uusia riskejä tai turvallisuushuolia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta.
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 Fimea
Yliannostus
Venetoklaksille ei ole spesifistä vastalääkettä. Jos potilas saa yliannoksen, hänen vointiaan on seurattava tarkoin ja asianmukaista tukihoitoa on tarjottava. Annostitrausvaiheessa hoito on tauotettava ja potilaan vointia on seurattava tarkoin tuumorilyysioireyhtymän oireiden ja löydösten varalta (kuume, vilunväristykset, pahoinvointi, oksentelu, sekavuus, hengenahdistus, kouristuskohtaukset, epäsäännöllinen syke, tumma tai samea virtsa, poikkeava väsymys, lihas- tai nivelkipu, vatsakipu ja vatsan pullotus) ja muun toksisuuden varalta (ks. kohta Annostus ja antotapa). Dialyysi ei poista venetoklaksia.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, muut antineoplastiset lääkeaineet, ATC-koodi: L01XX52
Vaikutusmekanismi
Venetoklaksi on voimakas, selektiivinen B-solulymfooma 2 ‑proteiinin (B-cell lymphoma-2, BCL-2) estäjä. BCL-2 on apoptoosia estävä proteiini. BCL-2:n on todettu yli-ilmentyvän KLL- ja AML-soluissa, joissa se välittää kasvainsolujen eloonjäämistä ja on ollut yhteydessä resistenssiin kemoterapialääkkeille. Venetoklaksi sitoutuu suoraan BCL-2:n BH3-sitoutumisuurteeseen, syrjäyttää BH3-motiivin sisältäviä proapoptoottisia proteiineja kuten BIM-proteiinia ja käynnistää mitokondrion ulkokalvon läpäisevyyden lisääntymisen (MOMP, mitochondrial outer membrane permeabilization), kaspaasi- aktivaation ja ohjelmoituneen solukuoleman. Ei-kliinisissä tutkimuksissa on todettu, että venetoklaksilla on sytotoksinen vaikutus BCL-2-proteiinia yli-ilmentäviin kasvainsoluihin.
Farmakodynaamiset vaikutukset
Sydämen elektrofysiologia
Toistuvien, kerran vuorokaudessa annettujen enintään 1200 mg venetoklaksiannosten vaikutusta QTc-aikaan arvioitiin avoimessa, yksiryhmäisessä tutkimuksessa 176 potilaalla. Venetoklaksi ei vaikuttanut QTc-aikaan, eikä venetoklaksialtistuksella ollut yhteyttä QTc-ajan muutokseen.
Kliininen teho ja turvallisuus
Krooninen lymfaattinen leukemia
Venetoklaksin ja akalabrutinibin yhdistelmähoito yhdessä obinututsumabin kanssa tai ilman obitutsumabia potilailla, joilla oli aiemmin hoitamaton KLL – ACE-CL-311-tutkimus (AMPLIFY)
Satunnaistetussa (1:1:1), avoimessa vaiheen 3 monikeskustutkimuksessa 867 potilaalla arvioitiin venetoklaksin ja akalabrutinibin yhdistelmän turvallisuutta ja tehoa verrattuna venetoklaksin, akalabrutinibin ja obinututsumabin yhdistelmään ja verrattuna tutkijan valitsemaan kemoimmunoterapiaan eli joko FCR:ään (fludarabiini, syklofosfamidi ja rituksimabi) tai BR:ään (bendamustiini ja rituksimabi) potilailla, joilla oli aiemmin hoitamaton KLL. AMPLIFY-tutkimukseen otettiin vähintään 18‑vuotiaita potilaita, jotka eivät olleet saaneet aikaisemmin KLL-hoitoja ja joilla ei ollut del(17p)- tai TP53-mutaatiota. Antitromboottiset lääkkeet varfariinia ja muita K‑vitamiiniantagonisteja lukuun ottamatta olivat tutkimuksessa sallittuja.
Potilaat satunnaistettiin suhteessa 1:1:1 kolmeen ryhmään, ja he saivat jotakin seuraavista:
-
Venetoklaksi + akalabrutinibi: akalabrutinibia annettiin 100 mg kahdesti vuorokaudessa hoitojakson 1 päivästä 1 alkaen yhteensä 14 hoitojakson ajan tai kunnes tauti eteni tai todettiin sietämätöntä toksisuutta. Hoitojakson 3 päivänä 1 potilailla alkoi venetoklaksin 5 viikon pituinen annostitrausaikataulu, jossa annos oli aluksi 20 mg ja sitä suurennettiin viikoittain 50 mg:aan, 100 mg:aan, 200 mg:aan ja lopulta 400 mg:aan kerran vuorokaudessa. Venetoklaksia annettiin yhteensä 12 hoitojakson ajan. Kunkin hoitojakson pituus oli 28 päivää.
-
Venetoklaksi + akalabrutinibi + obinututsumabi: akalabrutinibia annettiin 100 mg kahdesti vuorokaudessa hoitojakson 1 päivästä 1 alkaen yhteensä 14 hoitojakson ajan tai kunnes tauti eteni tai todettiin sietämätöntä toksisuutta. Hoitojakson 3 päivänä 1 potilailla alkoi venetoklaksin 5 viikon pituinen annostitrausaikataulu, jossa annos oli aluksi 20 mg ja sitä suurennettiin viikoittain 50 mg:aan, 100 mg:aan, 200 mg:aan ja lopulta 400 mg:aan kerran vuorokaudessa. Venetoklaksia annettiin yhteensä 12 hoitojakson ajan. Obinututsumabia annettiin 1 000 mg:n annos hoitojakson 2 päivänä 1 tai päivinä 1 ja 2 (100 mg päivänä 1 ja 900 mg päivinä 1 tai 2), 8 ja 15, minkä jälkeen annettiin 1 000 mg:n annos hoitojaksojen 3–7 päivänä 1. Kunkin hoitojakson pituus oli 28 päivää.
-
Tutkijan valitsema kemoimmunoterapia (FCR/BR):
Fludarabiini, syklofosfamidi ja rituksimabi (FCR): fludarabiinia (25 mg/m2) ja syklofosfamidia (250 mg/m2) annettiin enintään 6 hoitojakson ajan päivinä 1–3. Rituksimabia annettiin 375 mg/m2 hoitojakson 1 päivänä 1 ja 500 mg/m2 hoitojaksojen 2–6 päivänä 1. Kunkin hoitojakson pituus oli 28 päivää.
Bendamustiini ja rituksimabi (BR): bendamustiinia annettiin 90 mg/m2 enintään 6 hoitojakson ajan päivinä 1 ja 2. Rituksimabia annettiin 375 mg/m2 hoitojakson 1 päivänä 1 ja 500 mg/m2 hoitojaksojen 2–6 päivänä 1. Kunkin hoitojakson pituus oli 28 päivää.
Potilaat stratifioitiin iän (> 65 vuotta tai ≤ 65 vuotta), IGHV-mutaatiostatuksen (mutatoitunut vs. mutatoitumaton), Rai-asteen (korkea riski [≥ 3] vs. ei korkeaa riskiä) ja maantieteellisen alueen (Pohjois-Amerikka vs. Länsi-Eurooppa vs. muu) perusteella. Taulukossa 10 esitetään yhteenveto tutkimuspopulaation lähtötilanteen demografisista tiedoista ja taudin piirteistä.
Taulukko 10: Lähtötilanteen ominaisuudet (AMPLIFY) potilailla, joilla oli aiemmin hoitamaton KLL
Ominaisuus | Venetoklaksi + akalabrutinibi N = 291 | Venetoklaksi + akalabrutinibi + obinututsumabi N = 286 | FCR/BR N = 290 |
Ikä, vuotta; mediaani (vaihteluväli) | 61 (31–84) | 61 (29–81) | 61 (26–86) |
Miehiä; % | 61,2 | 69,2 | 63,1 |
Valkoihoisia; % | 91,1 | 86,7 | 86,9 |
ECOG-toimintakykyluokka 0–1; % | 90,0 | 95,1 | 90,3 |
Mediaaniaika diagnoosista satunnaistamiseen (kuukautta) | 28,5 | 26,1 | 29,6 |
Suurikokoisia imusolmukkeita ≥ 5 cm; % | 38,8 | 35,0 | 42,8 |
Sytogeneettinen/FISH-luokka; % |
|
|
|
11q-deleetio | 17,5 | 19,6 | 15,9 |
Kompleksinen karyotyyppi (≥ 3 poikkeavuutta) | 15,5 | 16,1 | 14,5 |
Mutatoitumaton IGHV; % | 57,4 | 59,1 | 59,3 |
Rai-aste; % |
|
|
|
0 | 1,0 | 0,3 | 1,4 |
I | 16,2 | 21,3 | 21,4 |
II | 35,7 | 37,8 | 33,4 |
III | 23,7 | 17,8 | 20,3 |
IV | 23,4 | 22,7 | 23,4 |
Ensisijainen päätetapahtuma oli riippumattoman arviointitoimikunnan arvioima etenemisvapaa elossaolo (PFS) venetoklaksin ja akalabrutinibin yhdistelmää saaneessa ryhmässä verrattuna tutkijan valitsemaa kemoimmunoterapiaa (FCR/BR) saaneeseen ryhmään IWCLL 2018 -kriteerien mukaisesti. Muita tehon päätetapahtumia olivat riippumattoman arviointitoimikunnan arvioima etenemisvapaa elossaolo venetoklaksin, akalabrutinibin ja obinututsumabin yhdistelmää saaneessa ryhmässä verrattuna tutkijan valitsemaa hoitoa (FCR/BR) saaneeseen ryhmään sekä kokonaiselossaolo (OS) venetoklaksin ja akalabrutinibin yhdistelmää saaneessa ryhmässä verrattuna tutkijan valitsemaa hoitoa (FCR/BR) saaneeseen ryhmään ja venetoklaksin, akalabrutinibin ja obinututsumabin yhdistelmää saaneessa ryhmässä verrattuna tutkijan valitsemaa hoitoa (FCR/BR) saaneeseen ryhmään.
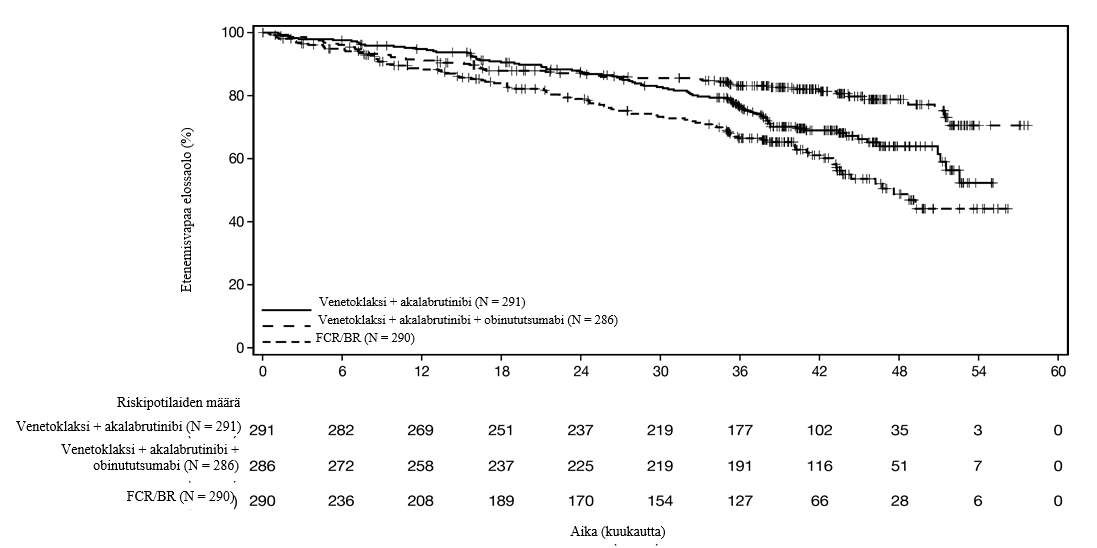
Tehotulokset esitetään taulukossa 11. Riippumattoman arviointitoimikunnan arvioiman etenemisvapaan elossaolon Kaplan-Meier-käyrä esitetään kuvassa 1.
Taulukko 11: Tehotulokset (AMPLIFY) potilailla, joilla oli aiemmin hoitamaton KLL
| Venetoklaksi + akalabrutinibi | Venetoklaksi + akalabrutinibi + obinututsumabi N = 286 | FCR/BRa N = 290 |
Etenemisvapaa elossaolo* | |||
Tapahtumia (%) | 89 (30,6) | 56 (19,6) | 95 (32,8) |
PD, n (%) | 77 (26,5) | 23 (8,0) | 66 (22,8) |
Kuolemia (%) | 12 (4,1) | 33 (11,5) | 29 (10,0) |
Mediaani (95 % lv), kk | EA (51,1; EA) | EA (EA, EA) | 47,6 (43,3; EA) |
Riskitiheyksien suhde† (95 % lv) | 0,65 (0,49; 0,87) | 0,42 (0,30; 0,59) | - |
P-arvo | 0,0038 | ˂ 0,0001 | - |
Kokonaiselossaolob | |||
Kuolemia (%) | 23 (7,9) | 37 (12,9) | 44 (15,2) |
Riskitiheyksien suhde† (95 % lv) | 0,42 (0,25; 0,70)c | 0,75 (0,48; 1,16) | - |
lv = luottamusväli; EA = ei arvioitavissa; PD = etenevä tauti. *Riippumattoman arviointitoimikunnan arvion mukaan. †Perustuu stratifioituun Coxin suhteellisen vaaran malliin. aTutkijan valinnan mukaan 143 potilaan oli tarkoitus saada FCR-hoitoa ja 147 potilaan BR-hoitoa. bKokonaiselossaolotiedot 6 kuukauden lisäseurannassa etenemisvapaan elossaolon välianalyysista. cP-arvo ei ole merkitsevä multiplisiteettikorjauksen jälkeen. | |||
Kuva 1: Riippumattoman arviointitoimikunnan arvioiman etenemisvapaan elossaolon Kaplan-Meier-käyrä (lähtöryhmien mukainen populaatio) AMPLIFY-tutkimuksessa
Venetoklaksi yhdessä obinututsumabin kanssa potilailla, joilla oli aiemmin hoitamaton KLL – BO25323-tutkimus (CLL14)
Satunnaistetussa (1:1), avoimessa vaiheen 3 monikeskustutkimuksessa arvioitiin venetoklaksin ja obinututsumabin yhdistelmän tehoa ja turvallisuutta verrattuna obinututsumabin ja klorambusiilin yhdistelmään potilailla, joilla oli aiemmin hoitamaton KLL ja liitännäissairauksia (Cumulative Illness Rating Scale ‑asteikon [CIRS] kokonaispisteet > 6 tai kreatiniinipuhdistuma < 70 ml/min). Tutkittavat arvioitiin tuumorilyysioireyhtymän riskin suhteen, ja heille annettiin tämän mukaista profylaktista hoitoa ennen obinututsumabin antoa. Kaikki tutkittavat saivat obinututsumabia 100 mg:n annoksen hoitojakson 1 päivänä 1 ja tämän jälkeen 900 mg:n annoksen joko päivänä 1 tai päivänä 2. Tämän jälkeen annettiin 1 000 mg:n annoksia hoitojakson 1 päivänä 8 ja 15 ja kunkin myöhemmän hoitojakson päivänä 1 yhteensä 6 hoitojakson ajan. Hoitojakson 1 päivänä 22 venetoklaksin ja obinututsumabin yhdistelmäryhmän potilaat aloittivat 5 viikon pituisen venetoklaksin annostitrausaikataulun, joka jatkui hoitojakson 2 päivään 28 asti. Annostitrausaikataulun päätyttyä tutkittavat jatkoivat venetoklaksin käyttöä annoksella 400 mg kerran vuorokaudessa hoitojakson 3 päivästä 1 alkaen aina hoitojakson 12 viimeiseen päivään asti. Kukin hoitojakso kesti 28 vuorokautta. Obinututsumabin ja klorambusiilin yhdistelmäryhmään satunnaistetut tutkittavat saivat 0,5 mg/kg klorambusiilia suun kautta hoitojaksojen 1–12 päivinä 1 ja 15. Hoidon päätyttyä tutkittavien vointia seurattiin taudin etenemisen varalta ja kokonaiselossaolon suhteen.
Lähtötilanteen demografiset tiedot ja taudin piirteet olivat samankaltaiset molemmissa tutkimusryhmissä. Tutkittavien mediaani-ikä oli 72 v (vaihteluväli 41–89 v), 89 % oli valkoihoisia ja 67 % oli miehiä. Tauti oli 36 %:lla Binet’n astetta B ja 43 %:lla Binet’n astetta C. CIRS-pisteiden mediaani oli 8,0 (vaihteluväli 0–28) ja 58 %:lla tutkittavista kreatiniinipuhdistuma oli < 70 ml/min. 17p-deleetio todettiin 8 %:lla potilaista, TP53-mutaatio 10 %:lla, 11q-deleetio 19 %:lla ja mutatoitumaton IgVH-geeni 57 %:lla. Ensisijaisen analyysin ajankohtana seurannan mediaanikesto oli 28 kk (vaihteluväli 0–36 kk).
Lymfosyyttiarvon mediaani oli lähtötilanteessa 55 x 109 solua/l molemmissa tutkimusryhmissä. Hoitojakson 1 päivänä 15 mediaaniarvo oli pienentynyt 1,03 x 109 soluun/l (vaihteluväli 0,2–43,4 x 109 solua/l) obinututsumabin ja klorambusiilin yhdistelmäryhmässä ja 1,27 x 109 soluun/l (vaihteluväli 0,2–83,7 x 109 solua/l) venetoklaksin ja obinututsumabin yhdistelmäryhmässä.
Tutkijat arvioivat etenemisvapaan elossaolon (PFS) IWCLL:n (International Workshop for Chronic Lymphocytic Leukemia) vuonna 2008 päivittämien NCI-WG-suositusten (National Cancer Institute-sponsored Working Group) pohjalta.
Ensisijaisen analyysin aikana (tiedonkeruun katkaisuajankohta 17.8.2018) 14 %:lla (30/216) venetoklaksin ja obinututsumabin yhdistelmäryhmän potilaista oli ollut tutkijan arvioima etenemisvapaan elossaolon tapahtuma (taudin etenemistapahtuma tai kuolemantapaus), kun osuus obinututsumabin ja klorambusiilin yhdistelmäryhmässä oli 36 % (77/216) (riskitiheyksien suhde [HR] 0,35 [95 % luottamusväli [lv] 0,23; 0,53]; p < 0,0001, stratifioitu log-rank-testi). Etenemisvapaan elossaoloajan mediaania ei saavutettu kummassakaan tutkimusryhmässä.
Myös riippumaton arviointitoimikunta (IRC) arvioi etenemisvapaan elossaolon, ja tulokset vastasivat tutkijan arvioimaa etenemisvapaata elossaoloa.
Tutkijan arvioima kokonaisvasteosuus (ORR) oli venetoklaksin ja obinututsumabin yhdistelmäryhmässä 85 % (95 % lv 79,2; 89,2) ja obinututsumabin ja klorambusiilin yhdistelmäryhmässä 71 % (95 % lv 64,8; 77,2) (p = 0,0007, Cochran–Mantel–Haenszelin testi). Tutkijan arvioiman täydellisen remission + täydellisen remission ilman täydellistä luuydintoiminnan korjautumista (CR + CRi) osuus oli venetoklaksin ja obinututsumabin yhdistelmäryhmässä 50 % ja obinututsumabin ja klorambusiilin yhdistelmäryhmässä 23 % (p < 0,0001, Cochran–Mantel–Haenszelin testi).
Minimaalinen jäännöstauti (MRD) hoidon lopussa arvioitiin alleelispesifisten oligonukleotidien polymeraasiketjureaktiotestillä (ASO-PCR). MRD-negatiivisuuden raja-arvo oli alle 1 KLL-solu 104 leukosyyttiä kohti. Ääreisverestä tutkittuna MRD-negatiivisten osuus oli venetoklaksin ja obinututsumabin yhdistelmäryhmässä 76 % (95 % lv 69,2; 81,1) ja obinututsumabin ja klorambusiilin yhdistelmäryhmässä 35 % (95 % lv 28,8; 42,0) (p < 0,0001). Tutkimussuunnitelman mukaisesti luuytimen MRD arvioitiin vain, jos potilas saavutti vasteen (CR/CRi ja osittainen remissio [PR]). Luuytimestä tutkittuna MRD-negatiivisten osuus oli venetoklaksin ja obinututsumabin yhdistelmäryhmässä 57 % (95 % lv 50,1; 63,6) ja obinututsumabin ja klorambusiilin yhdistelmäryhmässä 17 % (95 % lv 12,4; 22,8) (p < 0,0001).
65 kuukauden seuranta
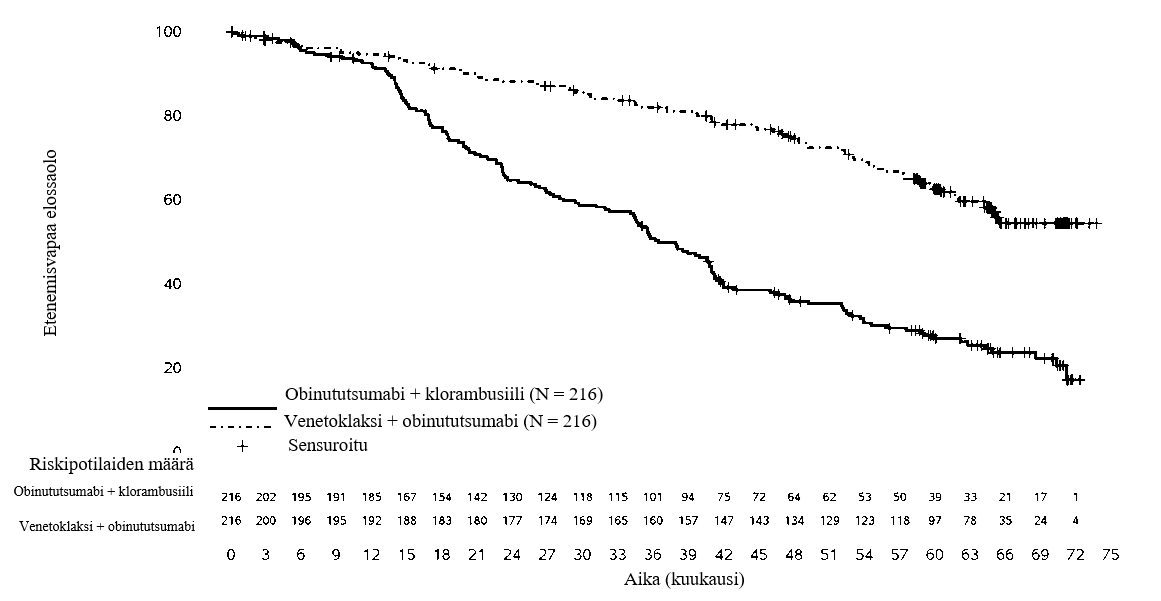
Teho arvioitiin 65 kuukauden mediaaniseuranta-ajan jälkeen (tiedonkeruun katkaisuajankohta 8.11.2021). CLL14-tutkimuksen 65 kuukauden seurannan tehotulokset esitetään taulukossa 12. Kaplan-Meier-käyrä tutkijan arvioimasta etenemisvapaasta elossaolosta esitetään kuvassa 2.
Taulukko 12: Tutkijan arvioimat tehotulokset CLL14-tutkimuksessa (65 kuukauden seuranta)
Päätetapahtuma | Venetoklaksi + obinututsumabi (N = 216) | Obinututsumabi + klorambusiili (N = 216) |
Etenemisvapaa elossaolo | ||
Tapahtumia (%) | 80 (37) | 150 (69) |
Mediaani, kk (95 % lv) | ES (64,8; EA) | 36,4 (34,1; 41,0) |
Riskitiheyksien suhde, stratifioitu (95 % lv) | 0,35 (0,26; 0,46) | |
Kokonaiselossaolo | ||
Tapahtumia (%) | 40 (19) | 57 (26) |
Riskitiheyksien suhde, stratifioitu (95 % lv) | 0,72 (0,48; 1,09) | |
lv = luottamusväli; EA = ei arvioitavissa; ES = ei saavutettu | ||
Kuva 2: Tutkijan arvioiman etenemisvapaan elossaolon Kaplan-Meier-käyrä (lähtöryhmien mukainen populaatio) CLL14-tutkimuksessa 65 kk:n seurannan kohdalla
Venetoklaksin ja obinututsumabin yhdistelmän todettiin parantaneen etenemättömyysaikaa verrattuna obinututsumabin ja klorambusiilin yhdistelmään kaikissa tutkittujen potilaiden alaryhmissä, mukaan lukien korkean riskin potilaat, joilla oli 17p-deleetio ja/tai TP53-mutaatio ja/tai mutatoitumaton IgVH-geeni.
Venetoklaksi yhdessä ibrutinibin kanssa potilailla, joilla oli aiemmin hoitamaton KLL – CLL3011-tutkimus (GLOW)
GLOW oli satunnaistettu, avoin vaiheen 3 tutkimus, jossa venetoklaksin ja ibrutinibin yhdistelmää verrattiin klorambusiilin ja obinututsumabin yhdistelmään potilailla, joilla oli aiemmin hoitamaton aktiivinen KLL ja jotka olivat vähintään 65‑vuotiaita, sekä < 65‑vuotiailla aikuispotilailla, joiden CIRS-pistearvo oli > 6 tai CrCL-arvo ≥ 30 – < 70 ml/min. Tutkimuksesta suljettiin pois potilaat, joilla oli 17p-deleetio tai tiedossa olevia TP53-mutaatioita. Potilaat (n = 211) satunnaistettiin suhteessa 1:1 saamaan joko venetoklaksin ja ibrutinibin yhdistelmää tai klorambusiilin ja obinututsumabin yhdistelmää.
Venetoklaksin ja ibrutinibin yhdistelmää saaneen ryhmän potilaat saivat pelkkää ibrutinibia 3 hoitojakson ajan ja sen jälkeen venetoklaksin ja ibrutinibin yhdistelmää 12 hoitojakson ajan (mukaan lukien 5 viikon pituinen venetoklaksiannoksen titraus). Kunkin hoitojakson pituus oli 28 päivää. Ibrutinibia annettiin 420 mg vuorokaudessa. Venetoklaksia annettiin 5 viikon annostitrausaikataulun mukaisesti ja sen jälkeen suositeltuna annoksena 400 mg vuorokaudessa (ks. kohta Annostus ja antotapa).
Klorambusiilin ja obinututsumabin yhdistelmään satunnaistetut potilaat saivat hoitoa 6 hoitojakson ajan. Obinututsumabia annettiin 1 000 mg:n annos hoitojakson 1 päivinä 1 (tai 100 mg päivänä 1 ja 900 mg päivänä 2), 8 ja 15. Hoitojaksojen 2–6 aikana annettiin 1 000 mg obinututsumabia päivänä 1. Klorambusiilia annettiin 0,5 mg/painokilo hoitojaksojen 1–6 päivinä 1 ja 15. Potilaita, joiden taudin vahvistettiin edenneen IWCLL-kriteerien mukaisesti kumman tahansa määräaikaisen hoito-ohjelman päättymisen jälkeen, voitiin hoitaa pelkällä ibrutinibilla.
Mediaani-ikä oli 71 vuotta (vaihteluväli: 47–93 vuotta), 58 % oli miehiä ja 96 % oli valkoihoisia. Kaikkien potilaiden ECOG-toimintakykyluokka oli lähtötilanteessa 0 (35 %), 1 (53 %) tai 2 (12 %). Lähtötilanteessa 18 %:lla potilaista oli 11q-deleetio ja 52 %:lla mutatoitumaton IGHV. Lähtötilanteessa tehdyssä tuumorilyysioireyhtymän riskin arvioinnissa 25 %:lla potilaista oli suuri kasvaintaakka. Kolmen pelkällä ibrutinibilla toteutetun aloitushoitojakson jälkeen 2 %:lla potilaista oli suuri kasvaintaakka. Suuri kasvaintaakka määriteltiin seuraavasti: mikä tahansa imusolmuke ≥ 10 cm; tai mikä tahansa imusolmuke ≥ 5 cm ja absoluuttinen lymfosyyttimäärä ≥ 25 × 109/l.
Riippumattoman arviointitoimikunnan IWCLL 2008 -kriteerien mukaan arvioimat GLOW-tutkimuksen tehotulokset (mediaaniseuranta-aika tutkimuksessa 28 kuukautta) esitetään taulukossa 13, etenemisvapaan elossaolon Kaplan–Meier-käyrä esitetään kuvassa 3 ja MRD-negatiivisuuden osuudet esitetään taulukossa 14.
Taulukko 13: CLL3011-tutkimuksen (GLOW) tehotulokset potilailla, joilla oli aiemmin hoitamaton KLL
Päätetapahtumaa | Venetoklaksi + ibrutinibi N = 106 | Klorambusiili + obinututsumabi N = 105 |
Etenemisvapaa elossaolo |
|
|
Tapahtumia (%) | 22 (21) | 67 (64) |
Mediaani, kk (95 % lv) | EA (31,2; EA) | 21 (16,6; 24,7) |
HR (95 % lv) | 0,22 (0,13; 0,36) | |
p-arvob | < 0,0001 | |
Täydellinen vasteprosentti (%)c | 39 | 11 |
95 % lv | (29,4; 48,0) | (5,3; 17,5) |
p-arvod | < 0,0001 | |
Kokonaisvasteprosentti (%)e | 87 | 85 |
95 % lv | (80,3; 93,2) | (77,9; 91,6) |
lv = luottamusväli; CR = täydellinen vaste; HR = riskitiheyksien suhde; IRC = riippumaton arviointitoimikunta; EA = ei arvioitavissa; nPR = osittainen nodulaarinen vaste; PR = osittainen vaste. aPerustuu riippumattoman arviointitoimikunnan arvioon. bStratifioitu log-rank-testi. cKattaa kolme venetoklaksin ja ibrutinibin yhdistelmää saanutta potilasta, jotka saavuttivat täydellisen vasteen ilman täydellistä luuytimen toiminnan korjautumista (CRi). dCochran-Mantel-Haenszelin khiin neliötesti. eKokonaisvaste = CR + CRi + nPR + PR. | ||
Kuva 3: Etenemisvapaan elossaolon Kaplan-Meier-käyrä (lähtöryhmien mukainen populaatio) potilailla, joilla oli aiemmin hoitamaton KLL CLL3011-tutkimuksessa (GLOW)
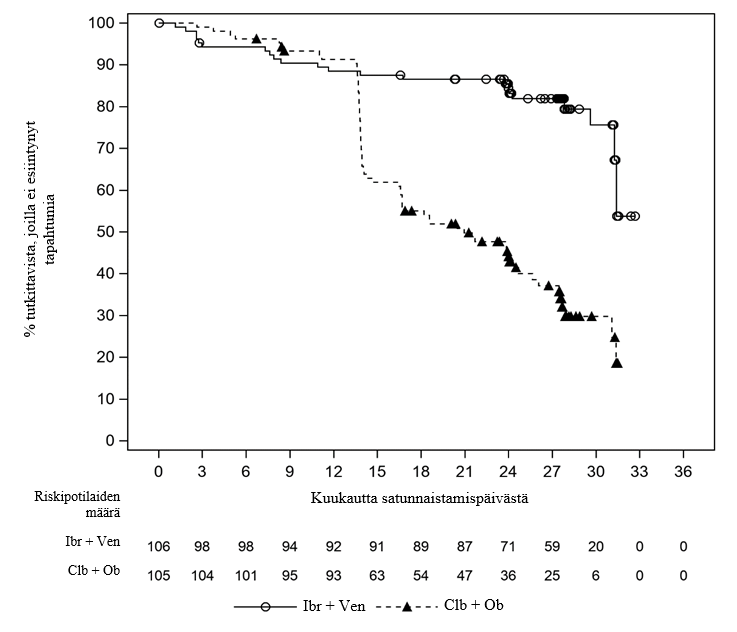
Venetoklaksin ja ibrutinibin yhdistelmän hoitovaikutus etenemisvapaaseen elossaoloon verrattuna klorambusiilin ja obinututsumabin yhdistelmän vaikutukseen oli johdonmukainen ennalta määritetyissä alaryhmissä ja myös suuren riskin populaatiossa (TP53-mutaatio, 11q-deleetio tai mutatoitumaton IGHV); etenemisvapaan elossaolon riskitiheyksien suhde oli 0,23 (95 % lv [0,13; 0,41]).
Kun seuranta-ajan mediaani oli 28 kuukautta, kokonaiselossaoloa koskevat tiedot eivät olleet valmiita ja kuolemia oli esiintynyt 23: 11 (10 %) venetoklaksin ja ibrutinibin yhdistelmää saaneessa ryhmässä ja 12 (11 %) klorambusiilin ja obinututsumabin yhdistelmää saaneessa ryhmässä.
Taulukko 14: Negatiivisuus minimaalisen jäännöstaudin suhteen potilailla, joilla oli aiemmin hoitamaton KLL CLL3011-tutkimuksessa (GLOW)
| NGS-määritysa | Virtaussytometriab | ||
| Venetoklaksi + ibrutinibi N = 106 | Klorambusiili + obinututsumabi N = 105 | Venetoklaksi + ibrutinibi N = 106 | Klorambusiili + obinututsumabi N = 105 |
MRD-negatiivisten osuus | ||||
Luuydin, n (%) | 59 (56) | 22 (21) | 72 (68) | 24 (23) |
95 % lv | (46,2; 65,1) | (13,2; 28,7) | (59,0; 76,8) | (14,8; 30,9) |
p-arvo | < 0,0001 |
| ||
Ääreisveri, n (%) | 63 (59) | 42 (40) | 85 (80) | 49 (47) |
95 % lv | (50,1, 68,8) | (30,6; 49,4) | (72,6; 87,8) | (37,1; 56,2) |
MRD-negatiivisten osuus 3 kuukautta hoidon päättymisen jälkeen | ||||
Luuydin, n (%) | 55 (51,9) | 18 (17,1) | 60 (56,6) | 17 (16,2) |
95 % lv | (42,4; 61,4) | (9,9; 24,4) | (47,2; 66,0) | (9,1; 23,3) |
Ääreisveri, n (%) | 58 (54,7) | 41 (39,0) | 65 (61,3) | 43 (41,0) |
95 % lv | (45,2; 64,2) | (29,7; 48,4) | (52,0; 70,6) | (31,5; 50,4) |
lv = luottamusväli; NGS = seuraavan sukupolven sekvensointi. p-arvot on saatu Cochran-Mantel-Haenszelin khiin neliötestillä. Lukuun ottamatta NGS-määrityksellä mitatun luuytimen MRD-negatiivisuuden p-arvoa, joka on ensisijainen MRD-analyysi ja GLOW-tutkimuksen ensimmäinen keskeinen toissijainen päätetapahtuma, kaikki muut p-arvot ovat nimellisiä. aPerustuu raja-arvoon 10-4 seuraavan sukupolven sekvensointimääritystä (clonoSEQ) käytettäessä. bMRD arvioitiin ääreisverestä tai luuytimestä virtaussytometrialla keskuslaboratoriossa. Negatiivisen statuksen raja oli < 1 KLL-solu 104 leukosyyttiä kohti. | ||||
Kun hoidon päättymisestä oli kulunut kaksitoista kuukautta, ääreisveren MRD-negatiivisuuden osuudet olivat venetoklaksin ja ibrutinibin yhdistelmää saaneilla potilailla 49 % (52/106) NGS-määrityksen mukaan ja 55 % (58/106) virtaussytometrian mukaan ja klorambusiilin ja obinututsumabin yhdistelmää saaneilla potilailla samassa aikapisteessä 12 % (13/105) NGS-määrityksen mukaan ja 16 % (17/105) virtaussytometrian mukaan.
Tuumorilyysioireyhtymää ilmoitettiin kuudella klorambusiilin ja obinututsumabin yhdistelmää saaneella potilaalla eikä yhdelläkään venetoklaksin ja ibrutinibin yhdistelmää saaneella potilaalla.
64 kuukauden seuranta
Teho arvioitiin, kun seuranta-ajan mediaani GLOW-tutkimuksessa oli 64,0 kuukautta (tiedonkeruun katkaisuajankohta 24. helmikuuta 2024). Tutkijan arvioima etenemisvapaan elossaolon riskitiheyksien suhde oli 0,27 [95 % lv (0,18; 0,39), nimellinen p < 0,0001, tyypin 1 virhettä ei kontrolloitu]. Etenemisvapaan elossaolon mediaani oli 65 kuukautta [95 % lv (58,7; EA)] venetoklaksin ja ibrutinibin yhdistelmää saaneessa ryhmässä ja 23 kuukautta [95 % lv (16,9; 31,2)] klorambusiilin ja obinututsumabin yhdistelmää saaneessa ryhmässä.
Kun seuranta-ajan mediaani tutkimuksessa oli 64 kuukautta, venetoklaksin ja ibrutinibin yhdistelmää saaneessa ryhmässä oli todettu 20 (19 %) kuolemantapausta ja klorambusiilin ja obinututsumabin yhdistelmää saaneessa ryhmässä 40 (38 %) kuolemantapausta. Kokonaiselossaolon riskitiheyksien suhde oli 0,462 (95 % lv: 0,269; 0,791, nimellinen p = 0,0039, tyypin 1 virhettä ei kontrolloitu).
Kokonaiselossaolon Kaplan-Meier-käyrä esitetään kuvassa 4.
Kuva 4: Kokonaiselossaolon Kaplan-Meier-käyrä (lähtöryhmien mukainen populaatio) potilailla, joilla oli aiemmin hoitamaton KLL CLL3011-tutkimuksessa (GLOW) (64 kuukauden seuranta)
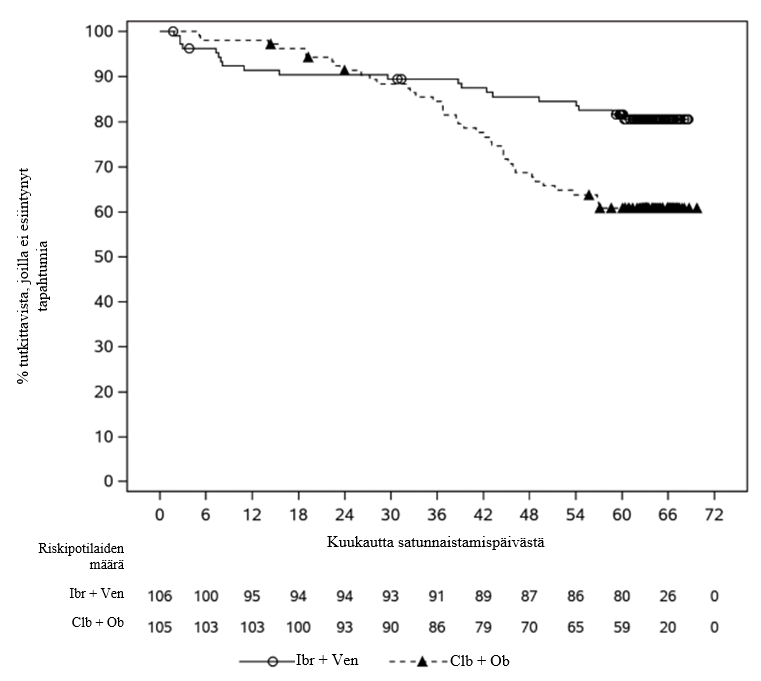
Venetoklaksi yhdessä ibrutinibin kanssa potilailla, joilla oli aiemmin hoitamaton KLL – PCYC-1142-CA-tutkimus (CAPTIVATE)
CAPTIVATE oli kahdella kohortilla toteutettu vaiheen 2 monikeskustutkimus, jossa arvioitiin sekä MRD-ohjattua hoidon lopettamista ja määräaikaista hoitoa venetoklaksin ja ibrutinibin yhdistelmällä. Tutkimus suoritettiin enintään 70-vuotiailla aikuispotilailla, joilla oli aiemmin hoitamaton aktiivinen KLL. Tutkimukseen otettiin 323 potilasta. Näistä potilaista 159 sai määräaikaista hoitoa, joka koostui pelkästä ibrutinibista 3 hoitojakson ajan ja sen jälkeen venetoklaksin ja ibrutinibin yhdistelmästä 12 hoitojakson ajan (mukaan lukien 5 viikon pituinen annoksen titraus). Kunkin hoitojakson pituus oli 28 päivää. Ibrutinibia annettiin 420 mg vuorokaudessa. Venetoklaksia annettiin 5 viikon annostitrausaikataulun mukaisesti ja sen jälkeen suositeltuna annoksena 400 mg vuorokaudessa (ks. kohta Annostus ja antotapa).
Potilaita, joiden taudin vahvistettiin edenneen IWCLL-kriteerien mukaisesti määräaikaisen hoito-ohjelman päättymisen jälkeen, voitiin hoitaa pelkällä ibrutinibilla.
Määräaikaista hoitoa saaneessa kohortissa mediaani-ikä oli 60 vuotta (vaihteluväli: 33–71 vuotta), 67 % oli miehiä ja 92 % oli valkoihoisia. Kaikkien potilaiden ECOG-toimintakykyluokka oli lähtötilanteessa 0 (69 %) tai 1 (31 %). Lähtötilanteessa 13 %:lla potilaista oli 17p-deleetio, 18 %:lla 11q-deleetio, 17 %:lla 17p-deleetio tai TP53-mutaatio, 56 %:lla mutatoitumaton IGHV ja 19 %:lla kompleksinen karyotyyppi. Lähtötilanteessa tehdyssä tuumorilyysioireyhtymän riskin arvioinnissa 21 %:lla potilaista oli suuri kasvaintaakka. Kolmen pelkällä ibrutinibilla toteutetun aloitushoitojakson jälkeen 1 %:lla potilaista oli suuri kasvaintaakka. Suuri kasvaintaakka määriteltiin seuraavasti: mikä tahansa imusolmuke ≥ 10 cm; tai mikä tahansa imusolmuke ≥ 5 cm ja absoluuttinen lymfosyyttimäärä ≥ 25 × 109/l.
Riippumattoman arviointitoimikunnan IWCLL 2008 -kriteerien mukaan arvioimat CAPTIVATE-tutkimuksen tehotulokset (mediaaniseuranta-aika tutkimuksessa 28 kuukautta) esitetään taulukossa 15 ja MRD-negatiivisuuden osuudet esitetään taulukossa 16.
Taulukko 15: PCYC-1142-CA-tutkimuksen (CAPTIVATE) tehotulokset potilailla, joilla oli aiemmin hoitamaton KLL, määräaikaisen hoidon kohorttia
Päätetapahtumaa | Venetoklaksi + ibrutinibi | |
| Ei 17p-deleetiota (N = 136) | Kaikki (N = 159) |
Kokonaisvasteprosentti, n (%)b | 130 (96) | 153 (96) |
95 % lv (%) | (92,1; 99,0) | (93,3; 99,2) |
Täydellinen vasteprosentti, n (%)c | 83 (61,0) | 95 (59,7) |
95 % lv (%) | (52,8; 69,2) | (52,1; 67,4) |
CR:n mediaanikesto, kk (vaihteluväli)d | EA (0,03+; 24,9+) | EA (0,03+; 24,9+) |
lv = luottamusväli; CR = täydellinen vaste; CRi = täydellinen vaste ilman täydellistä luuytimen toiminnan korjautumista; nPR = osittainen nodulaarinen vaste; PR = osittainen vaste; EA = ei arvioitavissa. aPerustuu riippumattoman arviointitoimikunnan arvioon. bKokonaisvaste = CR + CRi + nPR + PR. cKattaa 3 potilasta, jotka saavuttivat täydellisen vasteen ilman täydellistä luuytimen toiminnan korjautumista (CRi). dPlusmerkki tarkoittaa sensuroitua havaintoa. | ||
Taulukko 16: Negatiivisuus minimaalisen jäännöstaudin suhteen potilailla, joilla oli aiemmin hoitamaton KLL PCYC-1142-CA-tutkimuksessa (CAPTIVATE); määräaikaista hoitoa saanut kohortti
Päätetapahtuma | Venetoklaksi + ibrutinibi | |
| Ei 17p-deleetiota (N = 136) | Kaikki (N = 159) |
MRD-negatiivisten osuus | ||
Luuydin, n (%) | 84 (62) | 95 (60) |
95 % lv | (53,6; 69,9) | (52,1; 67,4) |
Ääreisveri, n (%) | 104 (77) | 122 (77) |
95 % lv | (69,3; 83,6) | (70,2; 83,3) |
MRD-negatiivisten osuus 3 kuukautta hoidon päättymisen jälkeen | ||
Luuydin, n (%) | 74 (54,4) | 83 (52,2) |
95 % lv | (46,0; 62.8) | (44,4; 60,0) |
Ääreisveri, n (%) | 78 (57,4) | 90 (56,6) |
95 % lv | (49,0; 65,7) | (48,9; 64,3) |
lv = luottamusväli. MRD arvioitiin ääreisverestä tai luuytimestä virtaussytometrialla keskuslaboratoriossa. Negatiivisen statuksen raja oli < 1 KLL-solu 104 leukosyyttiä kohti. | ||
Määräaikaista hoitoa saaneessa kohortissa yhdelläkään venetoklaksin ja ibrutinibin yhdistelmää saaneella potilaalla ei ilmoitettu tuumorilyysioireyhtymää.
KLL ja del 17p-/TP53-mutaatio PCYC-1142-CA-tutkimuksessa (CAPTIVATE)
Potilailla, joilla oli del 17p-/TP53 -mutaatio (n = 27) riippumattoman arviointitoimikunnan arvioon perustuva kokonaisvasteprosentti oli 96,3 %. Täydellisen vasteen saavuttaneiden prosenttiosuus oli 55,6 %, eikä täydellisen vasteen mediaanikestoa saavutettu (vaihteluväli: 4,3–22,6 kuukautta). MRD-negatiivisten osuus potilailla, joilla oli del 17p-/TP53-mutaatio, oli 3 kuukautta hoidon päättymisen jälkeen luuytimen osalta 40,7 % ja ääreisveren osalta 59,3 %.
Venetoklaksi yhdessä rituksimabin kanssa vähintään yhtä aiempaa hoitoa saaneiden KLL-potilaiden hoidossa – GO28667-tutkimus (MURANO)
Satunnaistetussa (1:1), avoimessa vaiheen 3 monikeskustutkimuksessa venetoklaksin ja rituksimabin yhdistelmän tehoa ja turvallisuutta verrattiin bendamustiini + rituksimabi -hoitoon potilailla, joilla oli aiemmin hoidettu KLL. Venetoklaksin ja rituksimabin yhdistelmähoitoryhmän potilaat suorittivat Venclyxton 5-viikkoisen annostitrausaikataulun kokonaan ja saivat tämän jälkeen 400 mg kerran vuorokaudessa 24 kk ajan ensimmäisen rituksimabihoitojakson ensimmäisestä päivästä laskettuna, mikäli tauti ei edennyt eikä kehittynyt sietämätöntä toksisuutta. Rituksimabin käyttö aloitettiin 5-viikkoisen annostitrausaikataulun jälkeen. Rituksimabiannos oli 375 mg/m2 ensimmäisellä hoitojaksolla ja 500 mg/m2 hoitojaksoilla 2–6. Kukin hoitojakso kesti 28 vrk. Bendamustiini + rituksimabi -hoitoon satunnaistetut potilaat saivat bendamustiinia (70 mg/m2 päivinä 1 ja 2) 6 hoitojakson ajan ja rituksimabia edellä kuvattuun tapaan.
Iän mediaani oli 65 vuotta (vaihteluväli 22–85), 74 % oli miehiä ja 97 % valkoihoisia. Mediaaniaika diagnoosista oli 6,7 v (vaihteluväli 0,3–29,5). Aiempien hoitolinjojen mediaanimäärä oli 1 (vaihteluväli 1–5), ja niihin kuului alkyloivia lääkkeitä (94 %), CD20-vasta-aineita (77 %), B-solureseptorireitin estäjiä (2 %) ja aiempia puriinianalogihoitoja (81 %; 55 % oli saanut fludarabiini + syklofosfamidi + rituksimabi (FCR) -hoitoa). Lähtötilanteessa 47 %:lla potilaista oli vähintään yksi ≥ 5 cm:n kokoinen imusolmuke, ja 68 %:n ALC-arvo oli ≥ 25 x 109/l. 17p-deleetio todettiin 27 %:lla potilaista, TP53-mutaatio 26 %:lla, 11q-deleetio 37 %:lla ja mutatoitumaton IgVH-geeni 68 %:lla. Ensisijaisen analyysin ajankohtana seurannan mediaanikesto oli 23,8 kk (vaihteluväli 0,0–37,4 kk).
Tutkijat arvioivat etenemisvapaan elossaolon IWCLL:n (International Workshop for Chronic Lymphocytic Leukaemia) vuonna 2008 päivittämien NCI-WG-suositusten (National Cancer Institute-sponsored Working Group) pohjalta.
Ensisijaisen analyysin aikana (tiedonkeruun katkaisuajankohta 8.5.2017) 16 %:lla (32/194) venetoklaksi- ja rituksimabiryhmän potilaista oli ollut etenemisvapaan elossaolon tapahtuma, kun osuus bendamustiini- ja rituksimabiryhmässä oli 58 % (114/195) (riskitiheyksien suhde 0,17 [95 % lv 0,11; 0,25]; p < 0,0001, stratifioitu log-rank-testi). Venetoklaksi- ja rituksimabiryhmässä etenemisvapaan elossaolon tapahtumista 21 oli taudin etenemistapahtumia ja 11 kuolemantapauksia, ja bendamustiini- ja rituksimabiryhmässä taudin etenemistapahtumia oli 98 ja kuolemantapauksia 16. Etenemisvapaan elossaoloajan mediaania ei saavutettu venetoklaksi- ja rituksimabiryhmässä, bendamustiini- ja rituksimabiryhmässä se oli 17,0 kuukautta (95 % lv 15,5; 21,6).
12 kuukauden etenemisvapaan elossaolon estimaatti oli venetoklaksi- ja rituksimabiryhmässä 93 % (95 % lv 89,1; 96,4) ja 24 kuukauden etenemisvapaan elossaolon estimaatti 85 % (95 % lv 79,1; 90,6). Bendamustiini- ja rituksimabiryhmässä vastaavat luvut olivat 73 % (95 % lv 65,9; 79,1) ja 36 % (95 % lv 28,5; 44,0).
Myös riippumaton arviointitoimikunta arvioi ensisijaisen analyysin tehotulokset. Tässä arvioinnissa todettiin, että venetoklaksi- ja rituksimabihoitoa saaneilla oli tilastollisesti merkitsevästi 81 % pienempi taudin etenemisen tai kuoleman riski (riskitiheyksien suhde 0,19 [95 % lv 0,13; 0,28]; p < 0,0001).
Tutkijan arvioima ORR venetoklaksia ja rituksimabia saaneille potilaille oli 93 % (95 % lv 88,8; 96,4), jossa CR + CRi ‑osuus oli 27 %, nodulaarinen osittainen remissio (nPR) -osuus 3 % ja PR-osuus 63 %. Bendamustiinia ja rituksimabia saaneiden potilaiden kokonaisvasteosuus (ORR) oli 68 % (95 % lv 60,6; 74,2), jossa CR + CRi ‑osuus oli 8 %, nPR-osuus 6 % ja PR-osuus 53 %. Vasteen keston mediaania ei saavutettu, kun seurannan mediaanikesto oli noin 23,8 kk. Riippumattoman arviointitoimikunnan arvioima kokonaisvasteosuus (ORR) venetoklaksia ja rituksimabia saaneille potilaille oli 92 % (95 % lv 87,6; 95,6), jossa CR + CRi ‑osuus oli 8 %, nPR-osuus 2 % ja PR-osuus 82 %. Bendamustiinia ja rituksimabia saaneille potilaille riippumattoman arviointitoimikunnan arvioima kokonaisvasteosuus (ORR) oli 72 % (95 % lv 65,5; 78,5), jossa CR + CRi ‑osuus oli 4 %, nPR-osuus 1 % ja PR-osuus 68 %. Ero riippumattoman arviointitoimikunnan arvioimien ja tutkijan arvioimien CR-osuuksien välillä johtuu TT-kuvissa todetun residuaalisen adenopatian tulkinnasta. 18 potilaalla venetoklaksi- ja rituksimabiryhmässä ja kolmella potilaalla bendamustiini- ja rituksimabiryhmässä oli negatiivinen luuydintulos ja imusolmukkeiden koko < 2 cm.
MRD yhdistelmähoidon lopussa arvioitiin ASO-PCR:llä ja/tai virtaussytometrialla. MRD-negatiivisuuden raja-arvo oli alle 1 KLL-solu 104 leukosyyttiä kohti. Ääreisverestä tutkittuna MRD-negatiivisten osuus oli venetoklaksi- ja rituksimabiryhmässä 62 % (95 % lv 55,2; 69,2) ja 13 % (95 % lv 8,9; 18,9) bendamustiini- ja rituksimabiryhmässä. Potilaista, joilta oli saatavilla ääreisveren MRD-testitulokset, 72 % (121/167) venetoklaksi- ja rituksimabiryhmän potilaista ja 20 % (26/128) bendamustiini- ja rituksimabiryhmän potilaista todettiin MRD-negatiivisiksi. Luuytimestä tutkittuna MRD-negatiivisten osuus oli venetoklaksi- ja rituksimabiryhmässä 16 % (95 % lv 10,7; 21,3) ja 1 % (95 % lv 0,1; 3,7) bendamustiini- ja rituksimabiryhmässä. Potilaista, joilta oli saatavilla luuytimen MRD-testitulokset, 77 % (30/39) venetoklaksi- ja rituksimabiryhmän potilaista ja 7 % (2/30) bendamustiini- ja rituksimabiryhmän potilaista todettiin MRD-negatiivisiksi.
Kokonaiselossaoloajan mediaania ei saavutettu kummassakaan hoitoryhmässä. Kuolemantapauksia todettiin 8 %:lla (15/194) venetoklaksilla ja rituksimabilla hoidetuista potilaista ja 14 %:lla (27/195) bendamustiinilla ja rituksimabilla hoidetuista potilaista (riskitiheyksien suhde 0,48 [95 % lv 0,25; 0,90]).
Tiedonkeruun katkaisuajankohtaan mennessä 12 % (23/194) venetoklaksi- ja rituksimabiryhmän potilaista ja 43 % (83/195) bendamustiini- ja rituksimabiryhmän potilaista oli aloittanut uuden leukemiahoidon tai kuollut (stratifioitu riskitiheyksien suhde 0,19 [95 % lv 0,12; 0,31]). Mediaaniaikaa uuden leukemiahoidon aloittamiseen tai kuolemaan ei saavutettu venetoklaksi- ja rituksimabiryhmässä, ja bendamustiini- ja rituksimabiryhmässä se oli 26,4 kuukautta.
59 kuukauden seuranta
Teho arvioitiin 59 kuukauden mediaaniseuranta-ajan jälkeen (tiedonkeruun katkaisuajankohta 8.5.2020). MURANO-tutkimuksen 59 kuukauden seurannan tehotulokset esitetään taulukossa 17.
Taulukko 17: Tutkijan arvioimat tehotulokset MURANO-tutkimuksessa (59 kuukauden seuranta)
Päätetapahtuma | Venetoklaksi + rituksimabi N = 194 | Bendamustiini + rituksimabi N = 195 |
Etenemisvapaa elossaolo | ||
Tapahtumia (%)a | 101 (52) | 167 (86) |
Mediaani, kk (95 % lv) | 54 (48,4; 57,0) | 17 (15,5; 21,7) |
Riskitiheyksien suhde, stratifioitu (95 % lv) | 0,19 (0,15; 0,26) | |
Kokonaiselossaolo | ||
Tapahtumia (%) | 32 (16) | 64 (33) |
Riskitiheyksien suhde (95 % lv) | 0,40 (0,26; 0,62) | |
60 kk:n estimaatti, % (95 % lv) | 82 (76,4; 87,8) | 62 (54,8; 69,6) |
Aika seuraavan leukemiahoidon aloittamiseen | ||
Tapahtumia (%)b | 89 (46) | 149 (76) |
Mediaani, kk (95 % lv) | 58 (55,1; EA) | 24 (20,7; 29,5) |
Riskitiheyksien suhde, stratifioitu (95 % lv) | 0,26 (0,20; 0,35) | |
MRD-negatiivisetc | ||
Ääreisveri, hoidon lopussa, n (%)d | 83 (64) | EOf |
Etenemisvapaa elossaolo, 3 vuoden estimaatti hoidon lopusta, % (95 % lv)e | 61 (47,3; 75,2) | EOf |
Kokonaiselossaolo, 3 vuoden estimaatti hoidon lopusta, % (95 % lv)e | 95 (90,0; 100,0) | EOf |
lv = luottamusväli; MRD = minimaalinen jäännöstauti; EA = ei arvioitavissa; OS = kokonaiselossaolo; PFS = etenemisvapaa elossaolo; EO = ei oleellinen. aVenetoklaksi- ja rituksimabiryhmässä tapahtumista 87 oli taudin etenemistapahtumia ja 14 kuolemantapauksia, ja bendamustiini- ja rituksimabiryhmässä taudin etenemistapahtumia oli 148 ja kuolemantapauksia 19. bVenetoklaksi- ja rituksimabiryhmässä tapahtumista 68 johtui uuden leukemiahoidon aloittamisesta ja 21 oli kuolemantapauksia, ja bendamustiini- ja rituksimabiryhmässä tapahtumista 123 johtui uuden leukemiahoidon aloittamisesta ja 26 oli kuolemantapauksia. cMinimaalinen jäännöstauti arvioitiin alleelispesifisten oligonukleotidien polymeraasiketjureaktiotestillä (ASO-PCR) ja/tai virtaussytometrialla. Negatiivisuuden raja-arvo oli yksi KLL-solu 104 leukosyyttiä kohti. dPotilaat, jotka suorittivat venetoklaksihoidon loppuun ilman taudin etenemistä (130 potilasta). ePotilaat, jotka suorittivat venetoklaksihoidon loppuun ilman taudin etenemistä ja olivat MRD-negatiivisia (83 potilasta). fEi vastaavaa hoidon loppukäyntiä bendamustiini- ja rituksimabiryhmässä. | ||
Venetoklaksi- ja rituksimabiryhmässä yhteensä 130 potilasta suoritti 2 vuoden venetoklaksihoidon loppuun ilman taudin etenemistä. Arvioitu etenemisvapaa elossaolo-osuus 3 vuoden kuluttua hoidon päättymisestä oli näillä potilailla 51 % (95 % lv 40,2; 61,9).
Kaplan-Meier-käyrä tutkijan arvioimasta etenemisvapaasta elossaolosta esitetään kuvassa 5.
Kuva 5: Tutkijan arvioiman etenemisvapaan elossaolon Kaplan–Meier-käyrä (lähtöryhmien mukainen populaatio) MURANO-tutkimuksessa (tiedonkeruun katkaisuajankohta 8.5.2020), 59 kuukauden seuranta
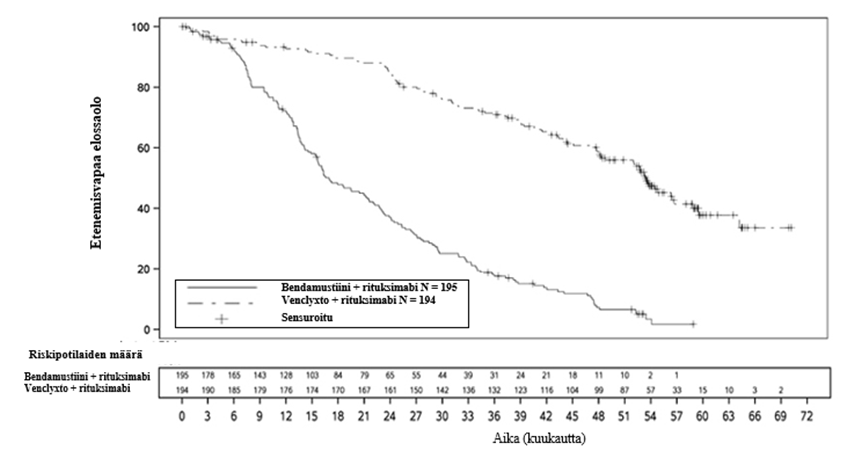
Alaryhmäanalyysien tulokset
Venetoklaksin ja rituksimabin yhdistelmän todettiin parantaneen taudin etenemisvapaata aikaa bendamustiinin ja rituksimabin yhdistelmään verrattuna johdonmukaisesti kaikissa tutkittujen potilaiden alaryhmissä, mukaan lukien korkean riskin potilaat, joilla 17p-deleetio/TP53-mutaatio ja/tai mutatoitumaton IgVH-geeni (kuva 6).
Kuva 6: Tutkijan arvioiman etenemisvapaan elossaolon metsikkökuvio (forest plot) MURANO-tutkimuksen eri alaryhmissä (tiedonkeruun katkaisuajankohta 8.5.2020), 59 kuukauden seuranta
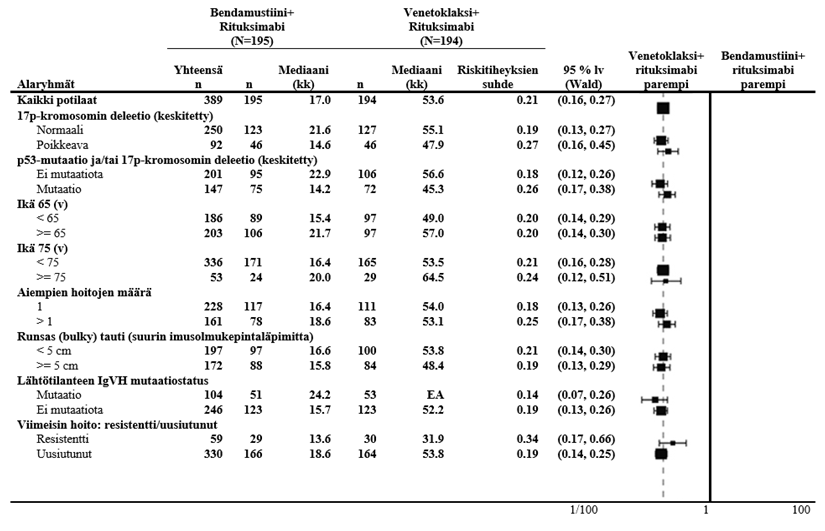
17p-deleetiostatus määritettiin keskuslaboratorion testitulosten pohjalta.
Stratifioimaton riskitiheyksien suhde esitetään X-akselilla logaritmista asteikkoa käyttäen.
EA = ei arvioitavissa.
Kokonaiselossaolon loppuanalyysi (86 kuukauden seuranta)
Kokonaiselossaolon loppuanalyysin ajankohtana (tiedonkeruun katkaisuajankohta 3.8.2022) yhteensä 144 satunnaistettua potilasta oli kuollut; 60/194 potilasta (31%) venetoklaksin ja rituksimabin yhdistelmähoitoryhmässä ja 84/195 potilasta (43%) bendamustiinin ja rituksimabin yhdistelmähoitoryhmässä. Kokonaiselossaolon mediaania ei saavutettu venetoklaksin ja rituksimabin yhdistelmähoitoryhmässä, ja se oli 88 kuukautta bendamustiinin ja rituksimabin yhdistelmähoitoryhmässä. Arvioitu kuoleman riski pieneni 47% potilailla, joita hoidettiin venetoklaksilla ja rituksimabilla (stratifioitu riskitiheyksien suhde = 0,53; 95% lv 0,37; 0,74). Kokonaiselossaolon loppuanalyysi ei ollut tyypin I virhekontrolloitu. Kaplan-Meier-käyrä kokonaiselossaolosta esitetään kuvassa 7.
Kuva 7: Kokonaiselossaolon Kaplan-Meier käyrä (lähtöryhmien mukainen populaatio) MURANO-tutkimuksessa (tiedonkeruun katkaisuajankohta 3.8.2022), 86 kuukauden seuranta
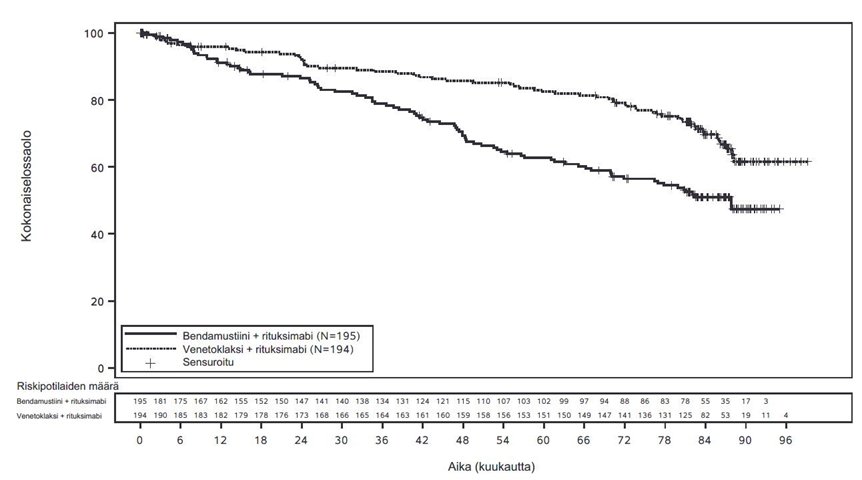
Venetoklaksi ainoana hoitona KLL-potilailla, joilla on 17p-deleetio tai TP53-mutaatio – M13-982-tutkimus
Venetoklaksin turvallisuutta ja tehoa arvioitiin yksiryhmäisessä, avoimessa monikeskustutkimuksessa (M13‑982) 107:llä aiemmin hoidetulla KLL-potilaalla, joilla oli 17p-deleetio. Potilaat noudattivat 4–5 viikon annostitrausaikataulua, jossa 20 mg aloitusannoksesta siirryttiin ottamaan 50 mg, 100 mg, 200 mg ja lopulta 400 mg annoksia kerran vuorokaudessa. Potilaat jatkoivat venetoklaksihoitoa annostuksella 400 mg x 1, kunnes tauti eteni tai todettiin sietämätöntä toksisuutta. Potilaiden mediaani-ikä oli 67 v (vaihteluväli 37–85 v), 65 % oli miehiä ja 97 % oli valkoihoisia. Mediaaniaika diagnoosista oli 6,8 v (vaihteluväli 0,1–32 v, N = 106). Aiempien KLL-hoitojen mediaanimäärä oli 2 (vaihteluväli 1–10 aiempaa hoitoa); 49,5 % oli saanut aiempaa nukleosidianalogihoitoa, 38 % aiempaa rituksimabihoitoa ja 94 % aiempaa hoitoa alkyloivalla lääkkeellä (33 % oli saanut aiemmin bendamustiinia). Lähtötilanteessa 53 %:lla potilaista oli vähintään yksi ≥ 5 cm kokoinen imusolmuke, ja 51 %:n ALC-arvo oli ≥ 25 x 109/l. Potilaista 37 % (34/91) oli resistenttejä fludarabiinille, 81 %:lla (30/37) oli mutatoitumaton IgVH-geeni ja 72 %:lla (60/83) oli TP53-mutaatio. Hoidon mediaanikesto oli arviointihetkellä 12 kk (vaihteluväli 0–22 kk).
Ensisijainen tehon päätetapahtuma oli riippumattoman arviointitoimikunnan arvioima kokonaisvasteosuus (ORR), joka arvioitiin IWCLL:n vuonna 2008 päivittämien NCI‑WG-suositusten mukaisesti. Tehotulokset esitetään taulukossa 18. Esitettävät tehotiedot perustuvat 107 potilaan tietoihin, joiden tiedonkeruun katkaisupäivä oli 30.4.2015. Lisäksi 51 potilasta otettiin turvallisuustietojen ekspansiokohorttiin. Tutkijan arvioimat tehotiedot esitetään 158 potilaasta. Näiden tietojen tiedonkeruu katkaistiin myöhemmin, 10.6.2016. Hoidon mediaanikesto oli näiden 158 potilaan ryhmässä 17 kk (vaihteluväli 0–34 kk).
Taulukko 18: Tehotulokset aiemmin hoidetuilla KLL-potilailla, joilla oli 17p-deleetio (M13-982-tutkimus)
Päätetapahtuma | Riippumattoman arviointitoimikunnan arvio (N = 107)a | Tutkijan arvio (N = 158)b |
Tiedonkeruun katkaisupäivä | 30.4.2015 | 10.6.2016 |
Kokonaisvasteosuus, % (95 % lv) | 79 (70,5; 86,6) | 77 (69,9; 83,5) |
CR + CRi, % | 7 | 18 |
nPR, % | 3 | 6 |
PR, % | 69 | 53 |
Vasteen kesto, kk, mediaani (95 % lv) | ei saavutettu | 27,5 (26,5; ei saavutettu) |
Etenemisvapaa elossaolo, % (95 % lv) 12 kk:n estimaatti 24 kk:n estimaatti | 72 (61,8; 79,8) ei saatavilla | 77 (69,1; 82,6) 52 (43; 61) |
Etenemisvapaa elossaolo, kk, mediaani (95 % lv) | ei saavutettu | 27,2 (21,9; ei saavutettu) |
Ensimmäiseen vasteeseen kulunut aika, kk, mediaani (vaihteluväli) | 0,8 (0,1–8,1) | 1,0 (0,5–4,4) |
aYhdellä potilaalla ei ollut 17p-deleetiota. bMukana 51 lisäpotilasta turvallisuustietojen ekspansiokohortista. CR = täydellinen remissio; CRi = täydellinen remissio ilman täydellistä luuydintoiminnan korjautumista; lv = luottamusväli; nPR = nodulaarinen PR; PR = osittainen remissio. | ||
Minimaalista jäännöstautia (MRD) arvioitiin virtaussytometrialla 93:lla 158 potilaasta, jotka saavuttivat venetoklaksihoidolla täydellisen remission (CR), täydellisen remission ilman täydellistä luuydintoiminnan korjautumista (CRi) tai osittaisen remission (PR), jossa jäännöstauti oli rajallinen. MRD-negatiivisuus määriteltiin tilanteeksi, jossa tulos oli alle 0,0001 (näytteessä < 1 KLL-solu 104 leukosyyttiä kohti). 27 % potilaista (42/158) oli ääreisveren tutkimuksissa MRD-negatiivisia, ja heistä 16 potilaalla myös luuydin oli MRD-negatiivinen.
Venetoklaksi ainoana hoitona KLL-potilailla, joilla B-solureseptorireitin estäjähoito oli epäonnistunut – M14-032-tutkimus
Venetoklaksin tehoa ja turvallisuutta arvioitiin avoimessa, satunnaistamattomassa vaiheen 2 monikeskustutkimuksessa (M14-032) KLL-potilailla, joilla aiempi ibrutinibi- tai idelalisibihoito oli epäonnistunut. Potilaat saivat venetoklaksihoitoa suositellun annostitrausaikataulun mukaisesti. Potilaat jatkoivat venetoklaksihoitoa annostuksella 400 mg x 1, kunnes tauti eteni tai todettiin sietämätöntä toksisuutta.
Tiedonkeruun katkaisuajankohtana (26.7.2017) 127 potilasta oli mukana tutkimuksessa ja sai venetoklaksihoitoa. Heistä 91 potilasta oli saanut aiemmin ibrutinibihoitoa (ryhmä A) ja 36 oli saanut aiemmin idelalisibihoitoa (ryhmä B). Potilaiden mediaani-ikä oli 66 v (vaihteluväli 28–85 v), 70 % oli miehiä ja 92 % oli valkoihoisia. Mediaaniaika diagnoosista oli 8,3 v (vaihteluväli 0,3–18,5 v; N = 96). Kromosomipoikkeavuuksia olivat 11q-deleetio (34 %, 43/127), 17p-deleetio (40 %, 50/126), TP53-mutaatio (38 %, 26/68) ja mutatoitumaton IgVH (78 %, 72/92). Lähtötilanteessa 41 %:lla potilaista oli vähintään yksi ≥ 5 cm kokoinen imusolmuke, ja 31 %:n ALC-arvo oli ≥ 25 x 109/l. Aiempien syöpähoitojen mediaanimäärä oli ibrutinibihoitoa saaneilla 4 (vaihteluväli 1–15) ja idelalisibia saaneilla 3 (vaihteluväli 1–11). Yhteensä 65 % potilaista oli saanut aiemmin nukleosidianalogihoitoa, 86 % rituksimabia, 39 % muuta monoklonaalista vasta-ainetta ja 72 % alkyloivaa ainetta (41 % oli saanut bendamustiinia). Arviointiajankohtana venetoklaksihoidon mediaanikesto oli 14,3 kk (vaihteluväli 0,1–31,4 kk).
Ensisijainen tehon päätetapahtuma oli kokonaisvasteosuus IWCLL:n päivittämien NCI-WG-suositusten mukaisesti arvioituna. Vaste arvioitiin 8 viikon ja 24 viikon kohdalla ja 12 viikon välein tämän jälkeen.
Taulukko 19: Tehoa koskevat tulokset tutkijan arvion mukaan potilailla, joilla B-solureseptorireitin estäjähoito oli epäonnistunut (M14‑032-tutkimus)
Päätetapahtuma | Ryhmä A (ibrutinibihoito epäonnistunut) (N = 91) | Ryhmä B (idelalisibihoito epäonnistunut) (N = 36) | Yhteensä (N = 127) |
Kokonaisvasteosuus, % (95 % lv) | 65 (54,1; 74,6) | 67 (49,0; 81,4) | 65 (56,4; 73,6) |
CR + CRi, % | 10 | 11 | 10 |
nPR, % | 3 | 0 | 2 |
PR, % | 52 | 56 | 53 |
Etenemisvapaa elossaolo, % (95 % lv) 12 kk:n estimaatti 24 kk:n estimaatti | 75 (64,7; 83,2) 51 (36,3; 63,9) | 80 (63,1; 90,1) 61 (39,6; 77,4) | 77 (68,1; 83,4) 54 (41,8; 64,6) |
Etenemisvapaa elossaolo, kk, mediaani (95 % lv) | 25 (19,2; ES) | ES (16,4; ES) | 25 (19,6; ES) |
Kokonaiselossaolo, % (95 % lv) 12 kk:n estimaatti | 91 (82,8; 95,4) | 94,2 (78,6; 98,5) | 92 (85,6; 95,6) |
Ensimmäiseen vasteeseen kulunut aika, kk, mediaani (vaihteluväli) | 2,5 (1,6–14,9) | 2,5 (1,6–8,1) | 2,5 (1,6–14,9) |
17p-deleetio- ja/tai TP53-mutaatiostatus Kokonaisvasteosuus, % (95 % lv) | |||
Kyllä | (n = 28) 61 (45,4; 74,9) | (n = 7) 58 (27,7; 84,8) | (n = 35) 60 (46,6; 73,0) |
Ei | (n = 31) 69 (53,4; 81,8) | (n = 17) 71 (48,9; 87,4) | (n = 48) 70 (57,3; 80,1) |
CR = täydellinen remissio; CRi = täydellinen remissio ilman täydellistä luuydintoiminnan korjautumista; ES = ei saavutettu; lv = luottamusväli; nPR = nodulaarinen PR; PR = osittainen remissio. | |||
Tehotietoja arvioi myös riippumaton arviointitoimikunta, ja ne osoittivat, että kokonaisvasteosuus oli yhteensä 70 % (ryhmä A: 70 %; ryhmä B: 69 %). Yhdellä potilaalla (jolla ibrutinibihoito oli epäonnistunut) saavutettiin täydellinen remissio ilman täydellistä luuydintoiminnan korjautumista. Potilailla, joilla oli 17p-deleetio ja/tai TP53-mutaatio, kokonaisvasteosuus oli ryhmässä A 72 % (33/46) (95 % lv 56,5; 84,0) ja ryhmässä B 67 % (8/12) (95 % lv 34,9; 90,1). Potilailla, joilla ei ollut 17p-deleetiota eikä TP53-mutaatiota, kokonaisvasteosuus oli ryhmässä A 69 % (31/45) (95 % lv 53,4; 81,8) ja ryhmässä B 71 % (17/24) (95 % lv 48,9; 87,4).
Kokonaiselossaoloajan ja vasteen keston mediaaneja ei saavutettu, kun seurannan mediaanikesto oli ryhmässä A noin 14,3 kk ja ryhmässä B noin 14,7 kk.
Ääreisveri oli MRD-negatiivinen 25 %:lla potilaista (32/127). Heistä 8 potilaalla myös luuydin oli MRD-negatiivinen.
Akuutti myelooinen leukemia
Venetoklaksia tutkittiin aikuispotilailla, jotka olivat ≥ 75-vuotiaita tai joilla oli oheissairauksia, jotka estivät intensiivisen solunsalpaajahoidon käytön vähintään yhden seuraavan kriteerin takia: Eastern Cooperative Oncology Group (ECOG) -toimintakykyluokka 2–3, vaikea samanaikainen sydän- tai keuhkosairaus, keskivaikea maksan vajaatoiminta, kreatiniinipuhdistuma < 45 ml/min tai muu oheissairaus.
Venetoklaksi yhdessä atsasitidiinin kanssa potilailla, joilla oli äskettäin diagnosoitu AML – M15‑656-tutkimus (VIALE-A)
VIALE‑A oli satunnaistettu (2:1), kaksoissokkoutettu, lumekontrolloitu faasin 3 tutkimus, jossa arvioitiin venetoklaksin ja atsasitidiinin yhdistelmähoidon tehoa ja turvallisuutta potilailla, joilla oli äskettäin diagnosoitu AML ja joille intensiivinen solunsalpaajahoito ei sopinut.
VIALE‑A-tutkimuksen potilaat kävivät läpi 3 päivän asteittaisen annostitrauksen lopulliseen kerran päivässä annettuun 400 mg annokseen ensimmäisen 28 päivän hoitojakson aikana (ks. kohta Annostus ja antotapa) ja saivat venetoklaksia 400 mg suun kautta kerran päivässä seuraavien hoitojaksojen aikana. Atsasitidiinia 75 mg/m2 oli annosteltu joko laskimoon tai ihon alle kunkin 28 päivän hoitojakson päivinä 1–7 hoitojakson 1 päivästä 1 alkaen. Titrauksen aikana potilaat saivat tuumorilyysioireyhtymän estohoitoa ja olivat sairaalassa seurattavina. Kun remissio oli varmistettu luuydintutkimuksella (määritelmänä leukemiablasteja alle 5 %) ja potilaalla oli asteen 4 sytopenia hoitojakson 1 jälkeen, venetoklaksi tai lumelääke tauotettiin enintään 14 päiväksi tai kunnes ANC oli ≥ 500/mikrolitra ja trombosyyttiarvo ≥ 50 × 103/mikrolitra. Potilaille, joilla oli resistentti tauti hoitojakson 1 lopussa, tehtiin luuydintutkimus hoitojakson 2 tai 3 jälkeen ja kliinisen tarpeen mukaan. Atsasitidiinihoito aloitettiin uudelleen samana päivänä venetoklaksin tai lumelääkkeen kanssa tauotuksen jälkeen (ks. kohta Annostus ja antotapa). Atsasitidiinin annosta pienennettiin kliinisessä tutkimuksessa hematologisen toksisuuden hoitamiseksi (ks. atsasitidiinin valmisteyhteenveto). Potilaat jatkoivat hoitojaksoja, kunnes tauti eteni tai todettiin sietämätöntä toksisuutta.
Kaikkiaan 431 potilasta satunnaistettiin: 286 venetoklaksin ja atsasitidiinin yhdistelmähoitoryhmään ja 145 lumelääkkeen ja atsasitidiinin yhdistelmähoitoryhmään. Lähtötilanteen demografiset tiedot ja taudin piirteet olivat samankaltaiset venetoklaksin ja atsasitidiinin sekä lumelääkkeen ja atsasitidiinin yhdistelmähoitoryhmissä. Iän mediaani oli 76 vuotta (vaihteluväli: 49–91 vuotta), 76 % oli valkoihoisia ja 60 % oli miehiä. Lähtötilanteen ECOG-toimintakykyluokka oli 55 %:lla potilaista 0 tai 1, 40 %:lla potilaista 2 ja 5 %:lla potilaista 3. 75 %:lla potilaista oli de novo AML ja 25 %:lla sekundaarinen AML. Lähtötilanteessa 29 %:lla potilaista luuytimen blastimäärä oli < 30 %, 22 %:lla potilaista se oli ≥ 30 % – < 50 % ja 49 %:lla ≥ 50 %. Sytogeneettinen riski oli keskisuuri 63 %:lla potilaista ja korkea 37 %:lla potilaista. Seuraavat mutaatiot tunnistettiin: TP53-mutaatioita 21 %:lla (52/249), IDH1- ja/tai IDH2-mutaatio 24 %:lla (89/372) [9 %:lla (34/372) IDH1-mutaatio ja 16 %:lla (58/372) IDH2-mutaatio], 16 %:lla (51/314) FLT3-mutaatio ja 18 %:lla (44/249) NPM1-mutaatio.
Tutkimuksen ensisijaiset tehon päätetapahtumat olivat kokonaiselossaolo (OS), mitattuna satunnaistamispäivästä mistä tahansa syystä johtuvaan kuolemaan, täydellisen remission (CR) yhdistelmä (täydellinen remissio + täydellinen remissio ilman täydellistä luuydintoiminnan korjautumista [CR + CRi]). Seurannan keston mediaani oli analyysihetkellä 20,5 kk (vaihteluväli: < 0,1–30,7 kk).
Venetoklaksin ja atsasitidiinin yhdistelmähoito pienensi kuoleman riskiä 34 %:lla verrattuna lumelääkkeen ja atsasitidiinin yhdistelmähoitoon (p < 0,001). Tulokset esitetään taulukossa 20.
Taulukko 20: VIALE‑A-tutkimuksen tehotulokset
Päätetapahtuma | Venetoklaksi + atsasitidiini | Lumelääke + atsasitidiini |
Kokonaiselossaoloa | (N = 286) | (N = 145) |
Tapahtumia n (%) | 161 (56) | 109 (75) |
Elossaolon mediaani, kk (95 % lv) | 14,7 (11,9; 18,7) | 9,6 (7,4; 12,7) |
Riskisuhdeb (95 % lv) | 0,66 (0,52; 0,85) | |
p-arvob | < 0,001 | |
CR + CRi -osuusc | (N = 147) | (N = 79) |
n (%) | 96 (65) | 20 (25) |
(95 % lv) | (57; 73) | (16; 36) |
p-arvod | < 0,001 | |
lv = luottamusväli; CR = täydellinen remissio, jonka määritelmänä oli absoluuttinen neutrofiiliarvo > 1 000/mikrolitra, trombosyytit > 100 000/mikrolitra, riippumattomuus punasolusiirroista ja < 5 % blasteja luuytimessä. Kiertävien blastien ja Auerin sauvojen puuttuminen; ekstramedullaarisen taudin puuttuminen; CRi = täydellinen remissio ilman täydellistä luuydintoiminnan korjautumista. aKaplan–Meierin estimaatti toisessa välianalyysissa (tiedonkeruun katkaisuajankohta 4. tammikuuta 2020). bRiskisuhteen estimaatti (venetoklaksi + atsasitidiini vs. lumelääke + atsasitidiini) perustuu Coxin malliin sytogenetiikan (keskisuuri riski, korkea riski) ja iän (18 – < 75, ≥ 75) mukaan stratifioituina satunnaistamishetkellä määriteltynä; p-arvo perustuu log-rank-testiin samoilla tekijöillä stratifioituina. cCR + CRi -osuus on suunnitellusta välianalyysista, joka tehtiin ensimmäisestä 226 satunnaistetusta potilaasta, joita oli seurattu 6 kuukauden ajan ensimmäisessä välianalyysissa (tiedonkeruun katkaisuajankohta 1. lokakuuta 2018). dP-arvo on Cochran–Mantel–Haenszelin testistä iän (18 – < 75, ≥ 75) ja sytogeneettisen riskin (keskisuuri riski, korkea riski) mukaan stratifioituina satunnaistamishetkellä määriteltynä. | ||
Kuva 8: Kokonaiselossaolon Kaplan-Meier-käyrä VIALE‑A-tutkimuksessa
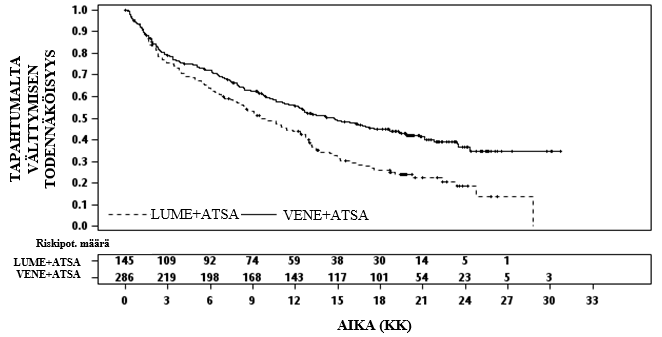
Tärkeimmät sekundaariset tehon päätetapahtumat esitetään taulukossa 21:
Taulukko 21: VIALE‑A-tutkimuksen muita tehon päätetapahtumia
Päätetapahtuma | Venetoklaksi + atsasitidiini N = 286 | Lumelääke + atsasitidiini N = 145 |
CR-osuus n (%) (95 % lv) p-arvoa Vasteen keston mediaanib, kk (95 % lv) | 105 (37) (31; 43) | 26 (18) (12; 25) |
< 0,001 | ||
17,5 (15,3; –) | 13,3 (8,5; 17,6) | |
CR + CRi -osuus n(%) (95 % lv) Vasteen keston mediaanib, kk (95 % lv) | 190 (66) (61; 72) 17,5 (13,6; –) | 41(28) (21; 36) 13,4 (5,8; 15,5) |
CR + CRi -osuus hoitojakson 2 alussa, n (%) (95 % lv) | 124 (43) (38; 49) | 11 (8) (4; 13) |
p-arvoa | < 0,001 | |
Riippumattomuus verensiirroista, trombosyytit n (%) (95 % lv) | 196 (69) (63; 74) | 72 (50) (41; 58) |
p-arvoa | < 0,001 | |
Riippumattomuus verensiirroista, punasolut n (%) (95 % lv) | 171 (60) (54; 66) | 51 (35) (27; 44) |
p-arvoa | < 0,001 | |
CR + CRi MRD -vasted n (%) (95 % lv) | 67 (23) (19; 29) | 11 (8) (4; 13) |
p-arvoa | < 0,001 | |
Elossaolo ilman tapahtumaa (EFS) Tapahtumia, n (%) EFS:n mediaanie, kk (95 % lv) | 191 (67) 9,8 (8,4; 11,8) | 122 (84) 7,0 (5,6; 9,5) |
Riskisuhde (95 % lv)c | 0,63 (0,50; 0,80) | |
p-arvoc | < 0,001 | |
lv = luottamusväli; CR = täydellinen remissio; CRi = täydellinen remissio ilman täydellistä luuydintoiminnan korjautumista; MRD = minimaalinen / mitattavissa oleva jäännöstauti; n = vasteiden määrä tai tapahtumien määrä; – = ei saavutettu. CR:n (täydellinen remissio) määritelmänä oli absoluuttinen neutrofiiliarvo > 1 000/mikrolitra, trombosyytit > 100 000/mikrolitra, riippumattomuus punasolusiirroista ja < 5 % blasteja luuytimessä. Kiertävien blastien ja Auerin sauvojen puuttuminen; ekstramedullaarisen taudin puuttuminen. Verensiirroista riippumattomuuden määritelmänä oli vähintään 56 peräkkäisen päivän (≥ 56 päivän) jakso ilman verensiirtoa ensimmäisen tutkimuslääkeannoksen jälkeen ja viimeisen tutkimuslääkeannoksen antopäivänä tai sitä ennen + 30 päivää tai ennen relapsia tai taudin etenemistä tai ennen hoidon jälkeisen hoidon aloittamista, sen mukaan mikä niistä on aiemmin. aP-arvo on Cochran–Mantel–Haenszelin testistä iän (18 – < 75, ≥ 75) ja sytogeneettisen riskin (keskisuuri riski, korkea riski) mukaan stratifioituina satunnaistamishetkellä määritettynä. bVasteen keston määritelmänä oli aika ensimmäisestä CR-vasteesta CR:n vasteen keston osalta tai aika ensimmäisestä CR- tai CRi-vasteesta CR + CRi:n vasteen keston osalta varmistetun morfologisen relapsin tai varmistetun taudin etenemisen ensimmäiseen esiintymispäivään tai taudin etenemisestä johtuvaan kuolemaan, sen mukaan mikä niistä ilmeni aiemmin. Vasteen keston mediaani on Kaplan–Meierin estimaatista. cRiskisuhteen estimaatti (venetoklaksi + atsasitidiini vs. lumelääke + atsasitidiini) perustuu Coxin malliin iän (18 – < 75, ≥ 75) ja sytogenetiikan (keskisuuri riski, korkea riski) mukaan stratifioituina satunnaistamishetkellä määriteltynä; p-arvo perustuu log-rank-testiin samoilla tekijöillä stratifioituina. d CR + CRi MRD -vasteprosentin määritelmänä on niiden potilaiden prosenttimäärä, jotka saavuttivat CR:n tai CRi:n ja joiden MRD-vaste oli < 10-3 blastia luuytimessä tavanomaisella keskitetyllä monivärisellä virtaussytometriatutkimuksella määritettynä. eKaplan–Meierin estimaatti. | ||
Potilailla, joilla oli FLT3-mutaatio, CR + CRi -osuudet olivat 72 % (21/29; [95 % lv 53; 87]) venetoklaksin ja atsasitidiinin yhdistelmähoitoryhmässä ja 36 % (8/22; [95 % lv 17; 59]) lumelääkkeen ja atsasitidiinin yhdistelmähoitoryhmässä (p = 0,021).
Potilailla, joilla oli IDH1/IDH2-mutaatioita, CR + CRi -osuudet olivat 75 % (46/61; [95 % lv 63; 86]) venetoklaksin ja atsasitidiinin yhdistelmähoitoryhmässä ja 11 % (3/28; [95 % lv 2; 28]) lumelääkkeen ja atsasitidiinin yhdistelmähoitoryhmässä (p < 0,001).
Potilaista, jotka olivat lähtötilanteessa punasolusiirroista riippuvaisia ja jotka saivat venetoklaksin ja atsasitidiinin yhdistelmähoitoa, 49 % (71/144) tuli verensiirroista riippumattomiksi. Potilaista, jotka olivat lähtötilanteessa trombosyyttisiirroista riippuvaisia ja jotka saivat venetoklaksin ja atsasitidiinin yhdistelmähoitoa, 50 % (34/68) tuli verensiirroista riippumattomiksi.
Mediaaniaika ensimmäiseen CR- tai CRi-vasteeseen oli 1,3 kuukautta (vaihteluväli: 0,6–9,9 kuukautta) venetoklaksin ja atsasitidiinin yhdistelmähoitoa saaneilla. Mediaaniaika parhaaseen CR- tai CRi-vasteeseen oli 2,3 kuukautta (vaihteluväli: 0,6–24,5 kuukautta).
Kuva 9: Kokonaiselossaolon metsikkökuvio alaryhmittäin VIALE‑A-tutkimuksesta
– = ei saavutettu.
Ennalta määritetty sekundaarinen päätetapahtuma OS IDH1/2-mutaatioalaryhmässä, p <0,0001 (stratifioimaton log-rank-testi).
Stratifioimaton riskisuhde (HR) esitetään X-akselilla logaritmista asteikkoa käyttäen.
Venetoklaksi yhdessä atsasitidiinin tai desitabiinin kanssa potilailla, joilla oli äskettäin diagnosoitu AML – M14‑358-tutkimus
M14‑358-tutkimus oli satunnaistamaton, faasin 1/2 kliininen tutkimus, jossa tutkittiin venetoklaksia yhdessä atsasitidiinin (n = 84) tai desitabiinin (n = 31) kanssa potilailla, joilla oli äskettäin diagnosoitu AML ja joille intensiivinen solunsalpaajahoito ei sopinut. Potilaat saivat venetoklaksia päivittäisen titrauksen kautta lopullisen annoksen 400 mg kerran vuorokaudessa. Atsasitidiinia annettiin M14‑358-tutkimuksessa samalla tavoin kuin satunnaistetussa VIALE‑A-tutkimuksessa. Desitabiinia annettiin 20 mg/m2 laskimoon kunkin 28 päivän hoitojakson päivinä 1–5 hoitojakson 1 päivästä 1 alkaen.
Seurannan mediaani oli 40,4 kuukautta (vaihteluväli: 0,7–42,7 kuukautta) venetoklaksin ja desitabiinin yhdistelmähoitoryhmässä.
Venetoklaksin ja desitabiinin yhdistelmähoitoa saaneiden potilaiden iän mediaani oli 72 vuotta (vaihteluväli: 65–86 vuotta), 87 % oli valkoihoisia, 48 % oli miehiä ja 87 %:lla ECOG-toimintakykyluokka oli 0 tai 1. CR + CRi -vasteen saavutti 74 % (95 % lv 55; 88).
Iäkkäät potilaat
194 potilaasta, joilla oli aiemmin hoidettu KLL ja jotka saivat venetoklaksia yhdessä rituksimabin kanssa, 50 % oli täyttänyt 65 vuotta.
M13-982-tutkimuksen tehoarvioinnin 107 potilaasta 57 % oli täyttänyt 65 vuotta.
M14-032-tutkimuksen tehoarvioinnin 127 potilaasta 58 % oli täyttänyt 65 vuotta.
Kolmessa avoimessa monoterapiatutkimuksessa arvioitiin turvallisuutta 352 potilaalla, joista 57 % oli täyttänyt 65 vuotta.
Niistä 283:sta äskettäin diagnosoidusta AML-potilaasta, joita hoidettiin kliinisessä VIALE‑A-tutkimuksessa (venetoklaksin ja atsasitidiinin yhdistelmähoitoryhmä), 96 % oli ≥ 65-vuotiaita ja 60 % oli ≥ 75-vuotiaita.
Niistä 31:stä potilaasta, jotka saivat venetoklaksin ja desitabiinin yhdistelmähoitoa kliinisessä M14-358-tutkimuksessa, 100 % oli ≥ 65-vuotiaita ja 26 % oli ≥ 75-vuotiaita.
Turvallisuudessa ja tehossa ei todettu kliinisesti merkittäviä eroja iäkkäämpien ja nuorempien potilaiden välillä yhdistelmähoitotutkimuksissa eikä monoterapiatutkimuksissa.
Pediatriset potilaat
Venetoklaksin turvallisuutta, tehoa ja farmakokinetiikkaa arvioitiin kaksiosaisessa, avoimessa vaiheen 1 monikeskustutkimuksessa (M13‑833), jossa venetoklaksia annettiin monoterapiana tai yhdessä kemoterapian kanssa 140 pediatriselle ja nuorelle aikuiselle potilaalle, joilla oli uusiutuneita tai huonosti hoitoon vastaavia pahanlaatuisia kasvaimia. Potilaat saivat pelkkää venetoklaksia tai venetoklaksin ja kemoterapian yhdistelmää iän tai painon mukaan sovitettuna annoksena, joka vastasi aikuisten tavoitevuorokausiannosta 400 mg tai 800 mg jatkuvasti tai jaksottaisesti (päivinä 1–10) 21 vuorokauden sykleinä.
Osassa 1 oli mukana 22 potilasta annoksenhakukohortissa (AML (n = 10), akuutti lymfoblastinen leukemia [ALL] (n = 5), neuroblastooma (n = 3) ja kiinteät kasvaimet (n = 4)) ja 18 potilasta annoksen suurennus-/pienennyskohortissa (neuroblastooma (n = 7) ja kiinteät kasvaimet (n = 11).
Tutkimuksen osassa 2 oli mukana 100 potilasta, joilla oli seuraavia sairauksia: AML (n = 27), ALL (n = 26), non-Hodgkin-lymfooma [NHL] (n = 2), neuroblastooma (n = 26) ja eksploratiivinen kohortti, johon sisältyivät muut BCL‑2:ta ilmentävät kasvaimet tai transkriptiotekijä 3-maksan leukemiatekijä-ALL (n = 19; kiinteät kasvaimet n = 8 ja muut kasvaimet n = 11). Yleisesti ottaen osissa 1 ja 2 potilaiden mediaani-ikä oli AML-potilailla 6 vuotta (vaihteluväli: 0–17 vuotta), ALL-potilailla 9 vuotta (vaihteluväli: 0–25 vuotta), NHL-potilailla 12 vuotta (vaihteluväli: 3–21 vuotta), neuroblastoomapotilailla 8 vuotta (vaihteluväli: 1–17 vuotta), kiinteitä kasvaimia sairastavilla potilailla 16 vuotta (vaihteluväli: 3–24 vuotta) ja muita kasvaimia sairastavilla potilailla 10 vuotta (vaihteluväli: 5–19 vuotta).
Tehon analyysiin otettiin potilaita osasta 1 ja osasta 2 (n = 129), ja siitä suljettiin pois muiden kasvainten eksploratiivinen kohortti. AML-kohortissa kokonaisvasteosuus (ORR) oli 24 %, täydellisen vasteen (CR) osuus 16 % ja vasteen arvioitu mediaanikesto 2,6 kuukautta (95 % lv: 0,5; 7,9). ALL-kohortissa ORR oli 42 % (kaikki täydellisiä vasteita) ja vasteen arvioitu mediaanikesto 10,2 kuukautta (95 % lv: 2,8; 14,2). Toinen NHL-kohortin potilaista saavutti osittaisen vasteen, jonka kesto oli 1,4 kuukautta. Vasteen mediaanikestoa ei voitu arvioida, ja merkityksellisiä johtopäätöksiä voitiin tehdä vain vähän pienen otoskoon vuoksi. Neuroblastoomakohortissa ORR oli 31 %, CR-osuus 22 % ja vasteen arvioitu mediaanikesto 9,3 kuukautta (95 % lv: 3,9; ei arvioitavissa). Kiinteiden kasvainten kohortissa ORR oli 22 %, CR-osuus 4 % ja vasteen arvioitu mediaanikesto 11,1 kuukautta (95 % lv: 3,1; ei arvioitavissa).
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Venclyxto-valmisteen käytöstä hematopoieettisen kudoksen ja imukudoksen pahanlaatuisten kasvainten hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Kun venetoklaksia annettiin toistuvasti suun kautta, maksimipitoisuudet plasmassa saavutettiin 5‑8 tunnin kuluttua annoksesta. Venetoklaksin vakaan tilan AUC suureni suhteessa annokseen 150‑800 mg:n annosalueella. Kun 400 mg venetoklaksia otettiin kerran vuorokaudessa vähärasvaisen aterian yhteydessä, venetoklaksin vakaan tilan Cmax-arvon keskiarvo (± keskihajonta) oli 2,1 ± 1,1 mikrogramma/ml ja AUC24-arvon keskiarvo (± keskihajonta) oli 32,8 ± 16,9 mikrogramma•h/ml.
Ruoan vaikutus
Venetoklaksin anto vähärasvaisen aterian kanssa suurensi venetoklaksialtistusta noin 3,4-kertaiseksi ja sen anto runsasrasvaisen aterian kanssa taas 5,1–5,3-kertaiseksi verrattuna lääkkeen ottamiseen tyhjään mahaan. On suositeltavaa ottaa venetoklaksi aterian yhteydessä (ks. kohta Annostus ja antotapa).
Jakautuminen
Venetoklaksi sitoutuu laajalti ihmisen plasman proteiineihin, ja sitoutumattoman lääkkeen fraktio plasmassa on < 0,01, kun lääkepitoisuus on 1–30 mikromol/l (0,87‑26 mikrogramma/ml). Veren ja plasman lääkepitoisuuksien suhteen keskiarvo oli 0,57. Venetoklaksin näennäisen jakautumistilavuuden (Vdss/F) populaatioestimaatti oli potilailla 256‑321 l.
Biotransformaatio
In vitro ‑tutkimuksissa todettiin, että venetoklaksi metaboloituu lähinnä sytokromi P450 ‑entsyymi CYP3A4:n kautta. Tärkeäksi plasmassa esiintyväksi metaboliitiksi todettiin M27, jonka BCL-2-toimintaa estävä vaikutus on vähintään 58-kertaisesti venetoklaksin vastaavaa vaikutusta pienempi in vitro.
In vitro ‑yhteisvaikutustutkimukset
CYP:n ja UGT:n substraattien samanaikainen käyttö
In vitro ‑tutkimukset viittasivat siihen, että kliinisesti merkittävät venetoklaksipitoisuudet eivät estä eivätkä indusoi CYP1A2-, CYP2B6-, CYP2C19-, CYP2D6- eivätkä CYP3A4-toimintaa. Venetoklaksi on heikko CYP2C8:n, CYP2C9:n ja UGT1A1:n estäjä in vitro, mutta sen ei uskota estävän niiden toimintaa kliinisesti merkittävästi. Venetoklaksi ei estä UGT1A4-, UGT1A6-, UGT1A9- eikä UGT2B7-toimintaa.
Kuljettajaproteiinien substraattien/estäjien samanaikainen käyttö
Venetoklaksi on P-gp:n ja BCRP:n substraatti, P-gp:n ja BCRP:n estäjä ja heikko OATP1B1:n estäjä in vitro (ks. kohta Yhteisvaikutukset). Kliinisesti merkittävät venetoklaksipitoisuudet eivät oletettavasti estä OATP1B3-, OCT1-, OCT2-, OAT1-, OAT3-, MATE1- eivätkä MATE2K-toimintaa.
Eliminaatio
Venetoklaksin terminaalivaiheen eliminaation puoliintumisajan populaatioestimaatti oli noin 26 tuntia. Venetoklaksin kertyminen on minimaalista ja sen kumuloitumissuhde on 1,30–1,44. Kun terveille henkilöille annettiin suun kautta 200 mg kerta-annos radioaktiivisesti leimattua [14C]-venetoklaksia, > 99,9 % annoksesta erittyi ulosteeseen ja < 0,1 % annoksesta erittyi virtsaan 9 päivän kuluessa. 20,8 % annetusta radioaktiivisuudesta erittyi muuttumattomana venetoklaksina ulosteeseen. Venetoklaksin farmakokinetiikka ei muutu ajan mittaan.
Erityisryhmät
Pediatriset potilaat
Uusiutuneita / huonosti hoitoon vastaavia pahanlaatuisia kasvaimia sairastavilla pediatrisilla potilailla tehdyn farmakokineettisen analyysin perusteella painoon perustuva annostus vähintään 2‑vuotiailla potilailla saa aikaan plasmassa venetoklaksialtistuksen, joka on vertailukelpoinen kaikissa pediatristen potilaiden painoon perustuvissa alaryhmissä ja verrattavissa 400 mg:n venetoklaksiannosta saavilla aikuispotilailla todettuun altistukseen taulukossa 22 esitetyllä tavalla.
Taulukko 22: Venetoklaksialtistukset pediatrisissa painoryhmissä vähintään 2‑vuotiailla potilailla aikuisten 400 mg:n annostusta vastaavalla annoksella
Pediatrinen alaryhmä (n) | 10 – ≤ 20 kg (5) | 20 – ≤ 30 kg (4) | 30 – ≤ 45 kg (6) | ≥ 45 kg (13) | Aikuiset |
AUC24* (mikrog•h/ml) | 22,4 ± 13,1 | 27,5 ± 27,5 | 38,3 ± 36,9 | 26,0 ± 24,3 | 32,8 ± 16,9 |
*Keskiarvo ± keskihajonta
Munuaisten vajaatoiminta
Populaatiofarmakokinetiikan analyysissa, johon osallistui 321 lievää munuaisten vajaatoimintaa sairastavaa henkilöä (kreatiniinipuhdistuma ≥ 60, mutta < 90 ml/min), 219 keskivaikeaa munuaisten vajaatoimintaa sairastavaa (kreatiniinipuhdistuma ≥ 30, mutta < 60 ml/min), 5 vaikeaa munuaisten vajaatoimintaa sairastavaa (kreatiniinipuhdistuma ≥ 15, mutta < 30 ml/min) ja 224 henkilöä, joiden munuaistoiminta oli normaali (kreatiniinipuhdistuma ≥ 90 ml/min), venetoklaksialtistus oli lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavilla samankaltainen kuin munuaistoiminnaltaan normaaleilla henkilöillä. Venetoklaksin farmakokinetiikkaa on tutkittu kuudella dialyysihoitoa vaativaa loppuvaiheen munuaistautia (end-stage renal disease, ESRD) sairastavalla potilaalla. Tutkittavilla sitoutumattoman venetoklaksin Cmax ja AUC 100 mg:n venetoklaksikerta-annoksen jälkeen olivat verrattavissa munuaistoiminnaltaan normaaleihin henkilöihin päivänä, jolloin tutkittavat eivät saaneet dialyysihoitoa. Sitoutumattoman venetoklaksin Cmax ja AUC olivat dialyysipäivänä noin 1,8–1,9-kertaisia verrattuna altistukseen päivänä, jolloin tutkittavat eivät saaneet dialyysihoitoa. Kuitenkin yksilöllinen vaihteluväli kokonais- ja sitoutumattoman venetoklaksin altistuksessa dialyysipäivänä oli yleisesti ottaen verrattavissa vastaavaan vaihteluväliin munuaistoiminnaltaan normaaleilla henkilöillä. Lisäksi venetoklaksipitoisuudet plasmassa olivat dialyysin aikana valtimo- ja laskimonäytteissä keskenään vertailukelpoisia, mikä osoittaa, että dialyysillä ei ole vaikutusta venetoklaksin puhdistumaan (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Populaatiofarmakokinetiikan analyysissa, johon osallistui 74 lievää maksan vajaatoimintaa sairastavaa henkilöä, 7 keskivaikeaa maksan vajaatoimintaa sairastavaa ja 442 henkilöä, joiden maksan toiminta oli normaali, venetoklaksialtistus oli lievää tai keskivaikeaa maksan vajaatoimintaa sairastavilla samankaltainen kuin henkilöillä, joiden maksa toimi normaalisti. Lievä maksan vajaatoiminta määriteltiin tilanteeksi, jossa kokonaisbilirubiini oli normaali ja ASAT-arvo (aspartaattiaminotransferaasi) > viitealueen yläraja (ULN) tai kokonaisbilirubiini > 1,0 – 1,5 x ULN. Keskivaikea maksan vajaatoiminta määriteltiin tilanteeksi, jossa kokonaisbilirubiini oli > 1,5 – 3,0 x ULN, ja vaikea maksan vajaatoiminta tilanteeksi, jossa kokonaisbilirubiini oli > 3,0 x ULN.
Maksan vajaatoimintaa selvittäneessä tutkimuksessa venetoklaksin Cmax ja AUC 50 mg:n kerta-annoksen jälkeen oli samankaltainen henkilöillä, joilla oli lievä (Child-Pugh A; n = 6) tai kohtalainen (Child-Pugh B; n = 6) maksan vajaatoiminta, verrattuna henkilöihin, joilla oli normaali maksan toiminta. Henkilöillä, joilla oli vaikea (Child-Pugh C; n = 5) maksan vajaatoiminta, keskimääräinen venetoklaksin Cmax oli samankaltainen kuin henkilöillä, joilla oli normaali maksan toiminta, mutta venetoklaksin AUCinf oli keskimäärin 2,7-kertainen (vaihteluväli: ei muutoksia - 5-kertainen) verrattuna venetoklaksin AUCinf -arvoihin henkilöillä, joilla oli normaali maksan toiminta (ks. kohta Annostus ja antotapa).
Iän, sukupuolen, painon ja rodun vaikutus
Populaatiofarmakokineettisten analyysien perusteella ikä, sukupuoli ja paino eivät vaikuta venetoklaksin puhdistumaan. Altistus on 67 % suurempi aasialaisilla tutkittavilla verrattuna ei-aasialaisiin tutkittaviin. Eron ei katsota olevan kliinisesti merkittävä.
Prekliiniset tiedot turvallisuudesta
Venetoklaksin eläintutkimuksissa todettuja toksisuuksia olivat mm. lymfosyyttimäärien ja veren punasolujen massan annosriippuvainen väheneminen. Molemmat löydökset korjautuivat, kun venetoklaksin anto lopetettiin. Lymfosyyttiarvot korjautuivat 18 viikon kuluttua hoidon päättymisestä. Lääke vaikutti sekä B- että T-soluihin, mutta B-solumäärät vähenivät merkittävimmin.
Venetoklaksi aiheutti myös yksittäisten solujen nekroosia eri kudoksissa, mm. sappirakossa ja eksokriinisessa haimassa. Kudosten rakenteen häiriytymistä tai elinten toimintahäiriötä ei todettu. Löydökset olivat minimaalisia tai lieviä.
Kun venetoklaksia annettiin päivittäin koirille noin 3 kuukauden ajan, se aiheutti etenevää turkin värin muuttumista (turkin valkaistumista), joka johtui karvojen melaniinipigmentin häviämisestä.
Karsinogeenisuus/genotoksisuus
Venetoklaksi ja sen tärkein ihmisellä todettava metaboliitti M27 eivät olleet karsinogeenisia 6 kuukauden pituisessa karsinogeenisuustutkimuksessa siirtogeenisillä hiirillä (Tg.rasH2), joille annettiin suun kautta enintään 400 mg/kg/vrk venetoklaksia, eivätkä tilanteessa, jossa M27-metaboliitin kerta-annostaso oli 250 mg/kg/vrk. Altistusmarginaalit (AUC) verrattuna kliinisessä käytössä annoksella 400 mg/vrk saavutettavaan AUC-arvoon olivat venetoklaksin kohdalla noin 2-kertaiset ja M27-metaboliitin kohdalla 5,8-kertaiset.
Venetoklaksi ei ollut genotoksinen bakteereilla tehdyssä mutageenisuuskokeessa, in vitro ‑kromosomipoikkeavuuskokeessa eikä hiiren in vivo ‑mikrotumakokeessa. M27-metaboliitti ei ollut genotoksinen bakteereilla tehdyssä mutageenisuuskokeessa eikä kromosomipoikkeavuuskokeessa.
Lisääntymistoksisuus
Hedelmällisyyttä ja varhaisvaiheen alkionkehitystä koskevissa tutkimuksissa uros- ja naarashiirillä ei todettu hedelmällisyyteen kohdistuvia vaikutuksia. Koirilla tehdyissä yleistä toksisuutta koskeneissa tutkimuksissa todettiin kivestoksisuutta (itusolukatoa), kun altistus oli 0,5–18-kertainen verrattuna ihmisen AUC-altistukseen 400 mg:n annoksilla. Ilmiön korjautumista ei ole osoitettu.
Hiirillä tehdyissä alkion- ja sikiönkehitystutkimuksissa venetoklaksi lisäsi implantoituneiden alkioiden kuolemia ja pienensi sikiöiden painoa altistuksilla, jotka olivat 1,1-kertaisia verrattuna ihmisen AUC-altistukseen 400 mg:n annoksilla. M27-metaboliitilla, joka on tärkein ihmisellä todettava metaboliitti, oli yhteys implantoituneiden alkioiden kuolemiin ja resorptioihin, kun altistus oli noin 9-kertainen verrattuna ihmisen M27-altistuksen AUC-arvoon 400 mg:n venetoklaksiannoksia käytettäessä. Kaneilla venetoklaksi oli toksinen emolle mutta ei sikiölle, kun altistus oli 0,1-kertainen verrattuna ihmisen AUC-altistukseen 400 mg:n annoksilla.
Farmaseuttiset tiedot
Apuaineet
Venclyxto 10 mg kalvopäällysteiset tabletit
Tabletin ydin
Kopovidoni (K 28)
Vedetön kolloidinen piidioksidi (E551)
Polysorbaatti 80 (E433)
Natriumstearyylifumaraatti
Vedetön kalsiumvetyfosfaatti (E341 (ii))
Kalvopäällyste
Keltainen rautaoksidi (E172)
Polyvinyylialkoholi (E1203)
Titaanidioksidi (E171)
Makrogoli 3350 (E1521)
Talkki (E553b)
Venclyxto 50 mg kalvopäällysteiset tabletit
Tabletin ydin
Kopovidoni (K 28)
Vedetön kolloidinen piidioksidi (E551)
Polysorbaatti 80 (E433)
Natriumstearyylifumaraatti
Vedetön kalsiumvetyfosfaatti (E341 (ii))
Kalvopäällyste
Keltainen rautaoksidi (E172)
Punainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Polyvinyylialkoholi (E1203)
Titaanidioksidi (E171)
Makrogoli 3350 (E1521)
Talkki (E553b)
Venclyxto100 mg kalvopäällysteiset tabletit
Tabletin ydin
Kopovidoni (K 28)
Vedetön kolloidinen piidioksidi (E551)
Polysorbaatti 80 (E433)
Natriumstearyylifumaraatti
Vedetön kalsiumvetyfosfaatti (E341 (ii))
Kalvopäällyste
Keltainen rautaoksidi (E172)
Polyvinyylialkoholi (E1203)
Titaanidioksidi (E171)
Makrogoli 3350 (E1521)
Talkki (E553b)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Venclyxto 10 mg kalvopäällysteiset tabletit
2 vuotta.
Venclyxto 50 mg kalvopäällysteiset tabletit
2 vuotta.
Venclyxto 100 mg kalvopäällysteiset tabletit
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VENCLYXTO tabletti, kalvopäällysteinen
10 mg (L:ei) 14 fol (91,90 €)
50 mg (L:ei) 7 fol (217,09 €)
100 mg (L:ei) 7 fol (418,18 €), 112 fol (5874,78 €)
PF-selosteen tieto
Venclyxto kalvopäällysteiset tabletit on pakattu PVC/PE/PCTFE-alumiinifolioläpipainopakkauksiin, joissa on joko 1, 2 tai 4 kalvopäällysteistä tablettia.
Venclyxto 10 mg kalvopäällysteiset tabletit
Kalvopäällysteiset tabletit toimitetaan pahvipakkauksissa, joissa on joko 10 tai 14 tablettia (kahden tabletin läpipainopakkauksissa).
Venclyxto 50 mg kalvopäällysteiset tabletit
Kalvopäällysteiset tabletit toimitetaan pahvipakkauksissa, joissa on joko 5 tai 7 tablettia (yhden tabletin läpipainopakkauksissa).
Venclyxto 100 mg kalvopäällysteiset tabletit
Kalvopäällysteiset tabletit toimitetaan pahvipakkauksissa, joissa on 7 (yhden tabletin läpipainopakkauksissa) tai 14 tablettia (kahden tabletin läpipainopakkauksissa), tai monipakkauksessa, jossa on 112 tablettia (4 x 28 tablettia (neljän tabletin läpipainopakkauksissa)).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Venclyxto 10 mg kalvopäällysteiset tabletit
Vaaleankeltainen, pyöreä, kaksoiskupera tabletti, jonka läpimitta on 6 mm ja jonka toisella puolella on kaiverrus V ja toisella puolella kaiverrus 10.
Venclyxto 50 kalvopäällysteiset tabletit
Beige, pitkänomainen, kaksoiskupera tabletti, jonka pituus on 14 mm ja leveys 8 mm ja jonka toisella puolella on kaiverrus V ja toisella puolella kaiverrus 50.
Venclyxto 100 mg kalvopäällysteiset tabletit
Vaaleankeltainen, pitkänomainen, kaksoiskupera tabletti, jonka pituus on 17,2 mm ja leveys 9,5 mm ja jonka toisella puolella on kaiverrus V ja toisella puolella kaiverrus 100.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VENCLYXTO tabletti, kalvopäällysteinen
10 mg 14 fol
50 mg 7 fol
100 mg 7 fol, 112 fol
- Ylempi erityiskorvaus (100 %). Venetoklaksi: Aikuisten kroonisen lymfaattisen leukemian ja akuutin myelooisen leukemian hoito erityisin edellytyksin (1518).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Venetoklaksi: Kroonisen lymfaattisen leukemian ja akuutin myelooisen leukemian hoito erityisin edellytyksin (399).
ATC-koodi
L01XX52
Valmisteyhteenvedon muuttamispäivämäärä
26.05.2026
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi