
SKYRIZI infuusiokonsentraatti, liuosta varten 600 mg
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 600 mg risankitsumabia 10,0 millilitrassa liuosta.
Risankitsumabi on humanisoitu immunoglobuliini G1 -luokan (IgG1) monoklonaalinen vasta-aine, joka valmistetaan kiinanhamsterin munasarjasoluissa yhdistelmä-DNA-tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti)
Kliiniset tiedot
Käyttöaiheet
Crohnin tauti
Skyrizi on tarkoitettu keskivaikean tai vaikean aktiivisen Crohnin taudin hoitoon aikuispotilaille, jotka eivät ole saaneet riittävää vastetta tavanomaisille hoidoille tai biologiselle hoidolle, joilla vaste on menetetty tai jotka eivät ole sietäneet tällaisia hoitoja.
Haavainen paksusuolitulehdus
Skyrizi on tarkoitettu keskivaikean tai vaikean aktiivisen haavaisen paksusuolitulehduksen hoitoon aikuispotilaille, jotka eivät ole saaneet riittävää vastetta tavanomaisille hoidoille tai biologiselle hoidolle, joilla vaste on menetetty tai jotka eivät ole sietäneet tällaisia hoitoja.
Ehto
Valmiste on tarkoitettu käytettäväksi käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus ja antotapa
Tämä lääkevalmiste on tarkoitettu käytettäväksi kohdassa Käyttöaiheet mainittujen käyttöaiheiden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja valvonnassa.
Annostus
Crohnin tauti
Suositeltu annos on 600 mg infuusiona laskimoon viikolla 0, viikolla 4 ja viikolla 8, minkä jälkeen 360 mg injektiona ihon alle viikolla 12 ja tämän jälkeen 8 viikon välein. Jos 24 hoitoviikon jälkeen ei todeta näyttöä terapeuttisesta hyödystä, on harkittava hoidon lopettamista.
Katso ihon alle annettavan ylläpitohoidon annostus Skyrizi 360 mg injektioneste, liuos, sylinteriampulli -valmisteyhteenvedon kohdasta Annostus ja antotapa.
Haavainen paksusuolitulehdus
Suositeltu induktioannos on 1 200 mg infuusiona laskimoon viikolla 0, viikolla 4 ja viikolla 8. Viikosta 12 alkaen 8 viikon välein annettava suositeltu ylläpitohoitoannos perustuu potilaan yksilölliseen vasteeseen:
- Injektiona ihon alle annettavaa 180 mg:n annosta suositellaan potilaille, joilla taudin aktiivisuuden paranema on riittävä induktion jälkeen
- Injektiona ihon alle annettavaa 360 mg:n annosta suositellaan potilaille, joilla taudin aktiivisuuden paranema on riittämätön induktion jälkeen
Jos 24 hoitoviikon jälkeen ei todeta näyttöä terapeuttisesta hyödystä, on harkittava hoidon lopettamista.
Katso ihon alle annettavan ylläpitohoidon annostus Skyrizi 180 mg ja 360 mg injektioneste, liuos, sylinteriampulli -valmisteyhteenvedon kohdasta Annostus ja antotapa.
Annoksen unohtuminen
Jos annos unohtuu, se on annettava mahdollisimman pian. Tämän jälkeen lääkkeen annostelua jatketaan tavanomaisella aikataululla.
Erityisryhmät
Iäkkäät
Annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
65 vuotta täyttäneiden potilaiden hoidosta on rajallisesti tietoa.
Munuaisten tai maksan vajaatoiminta
Maksan tai munuaisten vajaatoiminnan vaikutusta Skyrizi-valmisteen farmakokinetiikkaan ei ole tutkittu spesifisissä tutkimuksissa. Näiden tilojen ei yleisesti odoteta vaikuttavan merkitsevästi monoklonaalisten vasta-aineiden farmakokinetiikkaan, eikä annosmuutoksia pidetä tarpeellisina (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Skyrizi-valmisteen turvallisuutta ja tehoa 0‑17 vuoden ikäisten lasten Crohnin taudin ja haavaisen paksusuolitulehduksen hoidossa ei ole vielä varmistettu. Saatavilla olevat tiedot kuvataan kohdissa Farmakodynamiikka ja Farmakokinetiikka, mutta annostussuositusta ei voida antaa.
Ylipainoiset potilaat
Annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Antotapa
Annetaan laskimoon.
Skyrizi infuusiokonsentraatti, liuosta varten, on tarkoitettu annettavaksi vain laskimoon. 600 mg:n annos on annettava vähintään yhden tunnin kestoisena infuusiona ja 1 200 mg:n annos vähintään kahden tunnin kestoisena infuusiona. Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Kliinisesti merkittävä aktiivinen infektio (esim. aktiivinen tuberkuloosi, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Risankitsumabi voi suurentaa infektion riskiä.
Jos potilaalla on krooninen infektio, toistuvia infektioita tai tiedossa olevia infektion riskitekijöitä, risankitsumabia on käytettävä varoen. Jos potilaalla on mikä tahansa kliinisesti merkittävä aktiivinen infektio, risankitsumabihoitoa ei saa aloittaa ennen kuin infektio on parantunut tai hoidettu asianmukaisesti.
Risankitsumabihoitoa saaneita potilaita on kehotettava kääntymään lääkärin puoleen, jos heillä on kliinisesti merkittävän kroonisen tai akuutin infektion oireita tai merkkejä. Jos potilaalle kehittyy tällainen infektio tai infektion standardihoito ei tuota vastetta, potilaan tilannetta on seurattava tarkoin eikä risankitsumabia saa antaa ennen kuin infektio on parantunut.
Tuberkuloosi
Ennen risankitsumabihoidon aloittamista potilaat on arvioitava tuberkuloosi-infektion varalta. Risankitsumabia saavien potilaiden vointia on seurattava aktiivisen tuberkuloosin oireiden ja löydösten varalta. Tuberkuloosilääkityksen käyttöä on harkittava ennen risankitsumabihoidon aloittamista, jos potilaalla on anamneesissa latentti tai aktiivinen tuberkuloosi eikä asianmukaisen hoitojakson toteutumista pystytä vahvistamaan.
Immunisaatiot
Ennen risankitsumabihoidon aloittamista on harkittava kaikkien asianmukaisten rokotusten antamista ajankohtaisten rokotussuositusten mukaisesti. Jos potilas on saanut eläviä rokotteita (virus- tai bakteerirokotteita), on suositeltavaa odottaa vähintään 4 viikkoa ennen risankitsumabihoidon aloittamista. Risankitsumabihoitoa saaville potilaille ei pidä antaa eläviä rokotteita hoidon aikana eikä ainakaan 21 viikkoon hoidon jälkeen (ks. kohta Farmakokinetiikka).
Yliherkkyys
Vakavia yliherkkyysreaktioita, mukaan lukien anafylaksiaa, on raportoitu risankitsumabin käytön yhteydessä (ks. kohta Haittavaikutukset). Jos potilaalle kehittyy vakava yliherkkyysreaktio, risankitsumabin anto on lopetettava heti ja asianmukainen hoito on aloitettava.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per injektiopullo eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Risankitsumabi ei oletettavasti metaboloidu maksaentsyymien välityksellä eikä eliminoidu munuaisteitse. Risankitsumabilla ei odoteta olevan interaktioita lääkevalmisteiden metaboliaan osallistuvien entsyymien estäjien, indusorien tai substraattien kanssa, eikä annosta tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Samanaikainen immunosuppressiivinen hoito
Risankitsumabin ja immunosuppressanttien (myös biologisten lääkkeiden) yhdistelmän turvallisuutta ja tehoa ei ole arvioitu.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisymenetelmää hoidon aikana ja vähintään 21 viikon ajan sen jälkeen.
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja (alle 300 raskaudesta) risankitsumabin käytöstä raskaana oleville naisille. Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia. Varmuuden vuoksi risankitsumabin käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö risankitsumabi ihmisen rintamaitoon. Ihmisen IgG:tä tiedetään erittyvän rintamaitoon muutaman päivän ajan synnytyksen jälkeen, minkä jälkeen pitoisuudet pienevät nopeasti matalalle tasolle. Rintaruokittavaan imeväiseen kohdistuvia riskejä ei siis voida poissulkea tämän lyhyen jakson aikana. On päätettävä, lopetetaanko risankitsumabihoito tai pidättäydytäänkö risankitsumabihoidosta, ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja risankitsumabihoidosta koituvat hyödyt äidille.
Hedelmällisyys
Risankitsumabin vaikutusta ihmisen hedelmällisyyteen ei ole arvioitu. Eläinkokeissa ei ole havaittu suoria tai epäsuoria hedelmällisyyteen kohdistuvia haittavaikutuksia.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Risankitsumabilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitettuja haittavaikutuksia olivat ylähengitystieinfektiot (15,6 % Crohnin tautia sairastavilla potilailla ja 26,2 % haavaista paksusuolitulehdusta sairastavilla potilailla).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa risankitsumabin käytön yhteydessä todetut haittavaikutukset (taulukko 1) luetellaan MedDRA-elinjärjestelmäluokittain seuraavan käytännön mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on kussakin yleisyysluokassa esitetty vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Haittavaikutukset
| Elinjärjestelmä | Esiintymistiheys | Haittavaikutukset |
| Infektiot | Hyvin yleinen | Ylähengitystieinfektioa |
| Yleinen | Silsainfektiob | |
| Melko harvinainen | Karvatupen tulehdus | |
| Hermosto | Yleinen | Päänsärkyc |
| Iho ja ihonalainen kudos | Yleinen | Kutina Ihottuma Ekseema |
| Melko harvinainen | Nokkosihottuma | |
| Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Uupumusd Pistoskohdan reaktiote |
| Immuunijärjestelmä | Harvinainen | Anafylaktiset reaktiot |
a Mukana: hengitystieinfektio (virusperäinen, bakteeriperäinen tai määrittelemätön), sinuiitti (myös akuutti), riniitti, nenänielutulehdus, nielutulehdus (myös virusperäinen), tonsilliitti, laryngiitti, trakeiitti b Mukana: jalkasilsa, nivussilsa, vartalosilsa, tinea versicolor, käsisilsa, kynsisilsa, ihon sieni-infektio c Mukana: päänsärky, jännityspäänsärky, sivuontelopäänsärky d Mukana: uupumus, voimattomuus, huonovointisuus e Mukana: pistoskohdan mustelma, punoitus, hematooma, verenvuoto, ärsytys, kipu, kutina, reaktio, turvotus, kovettuma, yliherkkyys, kyhmy, ihottuma, nokkosihottuma, nesterakkulat, kuumotus; infuusiokohdan punoitus, ekstravasaatio, reaktio, turvotus | ||
Valikoitujen haittavaikutusten kuvaus
Psoriaasi
Infektiot
Infektioiden esiintyvyys koko kliinisessä psoriaasiohjelmassa, jossa tutkittavat altistuivat risankitsumabille myös pitkäaikaisesti, oli 75,5 tapahtumaa / 100 potilasvuotta. Valtaosa tapauksista oli ei-vakavia ja vaikeusasteeltaan lieviä tai keskivaikeita, eikä valtaosa tapauksista johtanut risankitsumabihoidon lopettamiseen. Vakavien infektioiden esiintyvyys oli 1,7 tapahtumaa / 100 potilasvuotta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Crohnin tauti
Risankitsumabihoitoa saaneilla Crohnin tautia sairastavilla potilailla todettu turvallisuusprofiili vastasi yleisesti ottaen muiden käyttöaiheiden potilailla todettua turvallisuusprofiilia.
Infektiot
Infektioiden esiintyvyys 12 viikkoa kestäneiden induktiotutkimusten yhdistetyissä tiedoissa oli 83,3 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat 600 mg risankitsumabia laskimoon, ja 117,7 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat lumelääkettä. Vakavien infektioiden esiintyvyys oli 3,4 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat 600 mg risankitsumabia laskimoon, ja 16,7 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat lumelääkettä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Infektioiden esiintyvyys 52 viikkoa kestäneessä ylläpitohoitotutkimuksessa oli 57,7 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat 360 mg risankitsumabia ihon alle risankitsumabi-induktiohoidon jälkeen, ja 76,0 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat lumelääkettä risankitsumabi-induktiohoidon jälkeen. Vakavien infektioiden esiintyvyys oli 6,0 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat 360 mg risankitsumabia ihon alle risankitsumabi-induktiohoidon jälkeen, ja 5,0 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat lumelääkettä risankitsumabi-induktiohoidon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haavainen paksusuolitulehdus
Risankitsumabihoitoa saaneilla haavaista paksusuolitulehdusta sairastavilla potilailla todettu turvallisuusprofiili vastasi yleisesti ottaen muiden käyttöaiheiden potilailla todettua turvallisuusprofiilia.
Infektiot
Infektioiden esiintyvyys 12 viikkoa kestäneen induktiotutkimuksen yhdistetyissä tiedoissa oli 78,3 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat 1 200 mg risankitsumabia laskimoon, verrattuna 74,2 tapahtumaan / 100 potilasvuotta tutkittavilla, jotka saivat lumelääkettä. Vakavien infektioiden esiintyvyys oli 3,0 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat 1 200 mg risankitsumabia laskimoon, verrattuna 5,4 tapahtumaan / 100 potilasvuotta tutkittavilla, jotka saivat lumelääkettä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Infektioiden esiintyvyys 52 viikkoa kestäneessä ylläpitohoitotutkimuksessa oli 67,4 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat 180 mg risankitsumabia ihon alle, ja 56,5 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat 360 mg risankitsumabia ihon alle risankitsumabi-aloitushoidon jälkeen, verrattuna 64,6 tapahtumaan / 100 potilasvuotta tutkittavilla, jotka saivat lumelääkettä risankitsumabi-induktiohoidon jälkeen. Vakavien infektioiden esiintyvyys oli 1,1 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat 180 mg risankitsumabia ihon alle, ja 0,6 tapahtumaa / 100 potilasvuotta tutkittavilla, jotka saivat 360 mg risankitsumabia ihon alle risankitsumabi-induktiohoidon jälkeen, verrattuna 2,3 tapahtumaan / 100 potilasvuotta tutkittavilla, jotka saivat lumelääkettä risankitsumabi-induktiohoidon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Immunogeenisuus
Kun risankitsumabia annettiin Crohnin tautia sairastaville tutkittaville Crohnin tautia koskeneissa kliinisissä tutkimuksissa suositeltuina laskimoon annettavina induktioannoksina ja ihon alle annettavina ylläpitoannoksina enintään 64 viikon ajan, hoidon aikana kehittyneitä vasta-aineita lääkettä kohtaan todettiin 3,4 %:lla arvioiduista tutkittavista (2/58) ja neutraloivia vasta-aineita 0 %:lla arvioiduista tutkittavista (0/58).
Kun risankitsumabia annettiin haavaista paksusuolitulehdusta sairastaville tutkittaville haavaista paksusuolitulehdusta koskeneissa kliinisissä tutkimuksissa suositeltuina laskimoon annettavina induktioannoksina ja ihon alle annettavina ylläpitoannoksina (180 mg tai 360 mg) enintään 64 viikon ajan, hoidon aikana kehittyneitä lääkevasta-aineita todettiin 8,9 %:lla (8/90) ja neutraloivia vasta-aineita 6,7 %:lla (6/90) arvioiduista tutkittavista, jotka saivat 180 mg:n annoksia ihon alle, ja vastaavasti 4,4 %:lla (4/91) ja 2,2 %:lla (2/91) arvioiduista tutkittavista, jotka saivat 360 mg:n annoksia ihon alle.
Risankitsumabia kohtaan muodostuneisiin vasta-aineisiin ei liittynyt kliinisen vasteen eikä turvallisuuden muutoksia.
Iäkkäät
65 vuotta täyttäneiden potilaiden hoidosta on niukasti turvallisuustietoa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksissa on suositeltavaa seurata potilaan vointia haittavaikutusten oireiden ja löydösten varalta ja aloittaa viipymättä asianmukainen oireenmukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, interleukiinin estäjät, ATC-koodi: L04AC18
Vaikutusmekanismi
Risankitsumabi on humanisoitu immunoglobuliini G1 -luokan (IgG1) monoklonaalinen vasta-aine, joka sitoutuu selektiivisesti ja suurella affiniteetilla ihmisen interleukiini-23-sytokiinin (IL-23) p19-alayksikköön sitoutumatta IL-12-sytokiiniin ja estää IL-23:n vuorovaikutusta IL-23-reseptorikompleksin kanssa. IL-23 on inflammaatioon ja immuunivasteisiin osallistuva sytokiini. Estämällä IL-23:n sitoutumisen reseptoriinsa risankitsumabi estää IL-23-riippuvaista solujen signalointia ja inflammaatiota edistävien sytokiinien vapautumista.
Farmakodynaamiset vaikutukset
Psoriaasia sairastaneilla tutkittavilla tehdyissä tutkimuksissa todettiin, että IL-23/IL-17-akseliin liittyvien geenien ilmentyminen ihossa väheni risankitsumabikerta-annoksen jälkeen. Psoriaasimuutoksissa todettiin myös epidermiksen ohenemista, tulehdussoluinfiltraation vähenemistä ja psoriaasin tautimarkkerien ilmentymisen vähenemistä.
Crohnin tautia sairastaneilla tutkittavilla tehdyssä vaiheen 2 tutkimuksessa todettiin, että IL-23/Th17-akseliin liittyvien geenien ilmentyminen suoliston kudoksissa väheni useiden risankitsumabiannosten jälkeen. Myös ulosteen kalprotektiinin (FCP), seerumin C-reaktiivisen proteiinin (CRP) ja IL-22:n pitoisuuksien todettiin pienentyneen useiden annosten jälkeen Crohnin tautia sairastaneilla potilailla tehdyissä vaiheen 3 induktiotutkimuksissa. FCP- ja CRP-arvojen ja seerumin IL-22-arvojen vähenemä säilyi ylläpitohoitotutkimuksen viikolle 52 asti.
Haavaista paksusuolitulehdusta sairastaneilla tutkittavilla tehdyssä vaiheen 2b/3 tutkimuksessa induktiotutkimuksen viikolla 12 havaittiin tilastollisesti merkitsevää ja kliinisesti merkittävää FCP- ja CRP-tulehdusmerkkiainepitoisuuksien ja IL-23-reittiin liittyvän merkkiaineen, seerumin IL-22:n, pitoisuuden laskua lähtötilanteesta. FCP- ja CRP- ja seerumin IL-22-arvojen vähenemä säilyi ylläpitohoitotutkimuksen viikolle 52 asti.
Kliininen teho ja turvallisuus
Crohnin tauti
Risankitsumabin tehoa ja turvallisuutta arvioitiin 1 419 tutkittavalla, joilla oli keskivaikea tai vaikea aktiivinen Crohnin tauti, kolmessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa kliinisessä monikeskustutkimuksessa. Tutkittavat olivat 16 vuotta täyttäneitä potilaita, joiden Crohnin taudin aktiivisuutta kuvaavat pisteet (Crohn’s Disease Activity Index, CDAI) olivat 220–450, joiden päivittäisten ulostuskertojen määrä (stool frequency, SF) oli ≥ 4 ja/tai joiden keskimääräiset päivittäiset vatsakipupisteet (abdominal pain score, APS) olivat ≥ 2, ja joiden Crohnin taudin yksinkertaiset endoskooppiset pisteet (Simple Endoscopic Score for CD (SES-CD)) olivat ≥ 6 tai ≥ 4 mikäli kyseessä oli keskitetysti vahvistettu ainoastaan ileumin alueella esiintyvä tauti ilman ahtaumia.
Kahteen 12 viikkoa kestäneeseen, laskimoon annettavaa induktiohoitoa koskeneeseen tutkimukseen (ADVANCE ja MOTIVATE) sisältyi 12 viikon pituinen jatkovaihe tutkittaville, jotka eivät olleet saavuttaneet kliinistä SF/APS-vastetta viikolla 12 (≥ 30 %:n vähenemä SF-pisteissä ja/tai ≥ 30 %:n vähenemä APS-pisteissä, eikä kumpikaan pistearvo saanut olla lähtöarvoa huonompi). ADVANCE- ja MOTIVATE-tutkimuksia seurasi 52 viikkoa kestänyt satunnaistettu ihonalaisinjektioilla toteutettu ylläpitohoitotutkimus (FORTIFY), jossa osalta induktion saaneista potilaista risankitsumabihoito lopetettiin satunnaistamalla heidät lumelääkkeelle. Ylläpitohoitotutkimukseen otettiin laskimoon annetulla induktiohoidolla kliinisen SF/APS-vasteen saavuttaneita tutkittavia, jotka saivat hoitoa vähintään 64 viikon ajan.
ADVANCE ja MOTIVATE
ADVANCE- ja MOTIVATE-tutkimuksissa tutkittavat satunnaistettiin saamaan joko 600 mg (suositeltu annos) tai 1 200 mg risankitsumabia tai lumelääkettä viikolla 0, viikolla 4 ja viikolla 8.
ADVANCE-tutkimuksessa 58 %:lla (491/850) tutkittavista vähintään yksi biologinen hoito oli epäonnistunut tai potilas ei ollut sietänyt sitä (aiempi biologisen hoidon epäonnistuminen) ja 42 %:lla (359/850) tutkittavista tavanomaiset hoidot, mutta ei biologiset hoidot, olivat epäonnistuneet tai potilas ei ollut sietänyt niitä (ei aiempaa biologisen hoidon epäonnistumista). ADVANCE-tutkimuksessa 87 % (314/359) niistä tutkittavista, joilla ei ollut todettu aiempaa biologisen hoidon epäonnistumista, ei ollut saanut aiemmin mitään biologisia hoitoja, ja loput 13 % oli saanut jotakin biologista hoitoa, mutta hoito ei ollut epäonnistunut eikä aiheuttanut siedettävyysongelmia. Kaikilla MOTIVATE-tutkimukseen osallistuneilla potilailla oli todettu aiempi biologisen hoidon epäonnistuminen.
Molemmissa tutkimuksissa suurempi osa risankitsumabihoitoa saaneista tutkittavista saavutti rinnakkaiset ensisijaiset päätetapahtumat eli kliinisen remission viikolla 12 ja endoskooppisen vasteen viikolla 12 lumeryhmään verrattuna. Risankitsumabia saaneilla tutkittavilla parantunut kliininen SF/APS-vaste ja kliininen remissio olivat merkitseviä jo viikolla 4 ja paranivat entisestään viikolle 12 saakka (taulukko 2).
Taulukko 2. ADVANCE- ja MOTIVATE-tutkimusten tehotulokset
| ADVANCE | MOTIVATE | |||||
Lumelääke laskimoon (N = 175) % | Risankitsumabi 600 mg laskimoon (N = 336) % | Hoitojen välinen erod (95 %:n lv) | Lumelääke laskimoon (N = 187) % | Risankitsumabi 600 mg laskimoon (N = 191) % | Hoitojen välinen erod (95 %:n lv) | |
| Rinnakkaiset ensisijaiset päätetapahtumat | ||||||
| Kliininen remissio viikolla 12e | 22 % | 43 % | 22 % [14 %, 30 %]a | 19 % | 35 % | 15 % [6 %, 24 %]b |
| Endoskooppinen vaste viikolla 12f | 12 % | 40 % | 28 % [21 %, 35 %]a | 11 % | 29 % | 18 % [10 %, 25 %]a |
| Muut päätetapahtumat | ||||||
| Parantunut kliininen SF/APS-vaste viikolla 4g | 31 % | 46 % | 15 % [6 %, 23 %]b | 32 % | 45 % | 14 % [4 %; 23 %]c |
| Parantunut kliininen SF/APS-vaste viikolla 12g | 42 % | 63 % | 21 % [12 %, 30 %]a | 39 % | 62 % | 23 % [13 %, 33 %]a |
| CDAI < 150 viikolla 4 | 10 % | 18 % | 8 % [1 %; 14 %]c | 11 % | 21 % | 10 % [2 %; 17 %]c |
| CDAI < 150 viikolla 12 | 25 % | 45 % | 21 % [12 %, 29 %]a | 20 % | 42 % | 22 % [13 %, 31 %]a |
| Limakalvon paraneminen viikolla 12h | (N = 173) 8 % | (N = 336) 21 % | 14 % [8 %, 19 %]a | (N = 186) 4 % | (N = 190) 14 % | 9 % [4 %, 15 %]b |
| Endoskooppinen remissio viikolla 12i | 9 % | 24 % | 15 % [9 %, 21 %]a | 4 % | 19 % | 15 % [9 %, 21 %]a |
a Tilastollisesti merkitsevä multiplisiteetin suhteen kontrolloituna, risankitsumabi vs. lume (p < 0,001). b Tilastollisesti merkitsevä multiplisiteetin suhteen kontrolloituna, risankitsumabi vs. lume (p ≤ 0,01). c Nimellinen p-arvo ≤ 0,05, risankitsumabi vs. lume. d Mukautettu hoitojen välinen ero. e SF/APS-pisteisiin perustuva kliininen remissio: keskimääräinen päivittäinen SF ≤ 2,8 eikä lähtöarvoa huonompi ja keskimääräinen päivittäinen APS ≤ 1 eikä lähtöarvoa huonompi. f Endoskooppinen vaste: yli 50 %:n vähenemä SES‑CD-pisteissä lähtötilanteeseen nähden tai vähintään 2 pisteen vähenemä tutkittavilla, joiden pistearvo oli lähtötilanteessa 4 ja joilla oli ainoastaan ileumin alueella esiintyvä tauti. g Parantunut kliininen SF/APS-vaste: ≥ 60 %:n vähenemä keskimääräisissä päivittäisissä SF-pisteissä ja/tai ≥ 35 %:n vähenemä keskimääräisissä päivittäisissä APS-pisteissä, eikä kumpikaan lähtöarvoa huonompi, ja/tai kliininen remissio. h Limakalvon paraneminen: SES-CD-luokituksessa haavaumien peittämän limakalvon pinta-alaa kuvaavan osion pistearvo 0 tutkittavilla, joilla lähtöarvo oli ≥ 1. i Endoskooppinen remissio: SES-CD ≤ 4 ja vähintään 2 pisteen vähenemä lähtöarvosta, eikä yhdenkään osion pistearvo yli 1 minkään yksittäisen muuttujan suhteen. | ||||||
Viikolla 12 suurempi osa risankitsumabihoitoa saaneista tutkittavista saavutti vähintään 100 pisteen vähenemän lähtötilanteen CDAI-pistearvossa lumeryhmään verrattuna (ADVANCE, risankitsumabi = 60 %, lume = 37 %, p < 0,001; MOTIVATE, risankitsumabi = 60 %, lume = 30 %, p < 0,001).
Viikolla 12 suurempi osa risankitsumabihoitoa saaneista tutkittavista saavutti sekä parantuneen kliinisen SF/APS-vasteen että endoskooppisen vasteen lumeryhmään verrattuna (ADVANCE, risankitsumabi = 31 %, lume = 8 %, p < 0,001; MOTIVATE, risankitsumabi = 21 %, lume = 7 %, p < 0,001).
Rinnakkaisten ensisijaisten päätetapahtumien tulokset alaryhmien (ilman multiplisiteetin sallimista) tutkittavilla, joilla oli todettu aiempi biologisen hoidon epäonnistuminen tai joilla ei ollut todettu aiempaa biologisen hoidon epäonnistumista, esitetään taulukossa 3.
Taulukko 3. ADVANCE-tutkimuksen tehotulokset viikolla 12 alaryhmien tutkittavilla, joilla oli todettu aiempi biologisen hoidon epäonnistuminen, ja tutkittavilla, joilla ei ollut todettu aiempaa biologisen hoidon epäonnistumista
| ADVANCE | |||
| Lumelääke laskimoon | Risankitsumabi 600 mg | Hoitojen välinen ero (95 %:n lv) | |
| Kliininen remissio SF/APS-pisteiden perusteella | |||
| Aiempi biologisen hoidon epäonnistuminen | 23 % (N = 97) | 41 % (N = 195) | 18 % [7 %, 29 %] |
| Ei aiempaa biologisen hoidon epäonnistumista | 21 % (N = 78) | 48 % (N = 141) | 27 % [15 %, 39 %] |
| Endoskooppinen vaste | |||
| Aiempi biologisen hoidon epäonnistuminen | 11 % (N = 97) | 33 % (N = 195) | 21 % [12 %, 31 %] |
| Ei aiempaa biologisen hoidon epäonnistumista | 13 % (N = 78) | 50 % (N = 141) | 38 % [27 %, 49 %] |
ADVANCE-tutkimuksessa suurempi osa risankitsumabihoitoa saaneista tutkittavista, joilla oli todettu aiempi biologisen hoidon epäonnistuminen tai joilla ei ollut todettu aiemman biologisen hoidon epäonnistumista, saavutti CDAI-pistearvon < 150 lumeryhmään verrattuna (aiempi biologisen hoidon epäonnistuminen, risankitsumabi = 42 %, lume = 26 %; ei aiempaa biologisen hoidon epäonnistumista, risankitsumabi = 49 %, lume = 23 %).
Crohnin tautiin liittyvät sairaalajaksot
Risankitsumabihoitoa saaneilla tutkittavilla esiintyi vähemmän Crohnin tautiin liittyviä sairaalajaksoja viikolle 12 asti kuin lumelääkettä saaneilla tutkittavilla (ADVANCE, risankitsumabi = 3 %, lume = 12 %, p < 0,001; MOTIVATE, risankitsumabi = 3 %, lume = 11 %, p ≤ 0,01).
FORTIFY
FORTIFY-ylläpitohoitotutkimuksessa arvioitiin 462 tutkittavaa, jotka olivat saavuttaneet kliinisen SF/APS-vasteen ADVANCE- ja MOTIVATE-tutkimuksissa laskimoon annetun, 12 viikkoa kestäneen risankitsumabi-induktiohoidon jälkeen. Tutkittavat satunnaistettiin jatkamaan risankitsumabihoitoa ylläpitoannoksella 360 mg ihon alle (suositeltu annos) tai 180 mg ihon alle 8 viikon välein tai lopettamaan risankitsumabi-induktiohoidon ja saamaan lumelääkettä ihon alle 8 viikon välein enintään 52 viikon ajan.
Rinnakkaiset ensisijaiset päätetapahtumat olivat kliininen remissio viikolla 52 ja endoskooppinen vaste viikolla 52. Rinnakkaiset ensisijaiset päätetapahtumat mitattiin myös tutkittavilta, joilla oli todettu aiempi biologisen hoidon epäonnistuminen, ja tutkittavilta, joilla ei ollut todettu aiempaa biologisen hoidon epäonnistumista (ks. taulukko 4).
Taulukko 4. FORTIFY-tutkimuksen tehotulokset viikolla 52 (64 viikkoa induktiohoidon aloittamisesta)
| FORTIFY | |||
| Risankitsumabi-induktiohoito laskimoon / lumelääke ihon allef (N = 164) % | Risankitsumabi-induktiohoito laskimoon / risankitsumabi 360 mg ihon alle (N = 141) % | Hoitojen välinen ero (95 %:n lv) | |
| Rinnakkaiset ensisijaiset päätetapahtumat | |||
| Kliininen remissio | 40 % | 52 % | 15 % [5 %, 25 %]a,g |
| Aiempi biologisen hoidon epäonnistuminen | 34 % (N = 123) | 48 % (N = 102) | 14 % [1 %, 27 %] |
| Ei aiempaa biologisen hoidon epäonnistumista | 56 % (N = 41) | 62 % (N = 39) | 5 % [-16 %, 27 %] |
| Endoskooppinen vaste | 22 % | 47 % | 28 % [19 %, 37 %]b,g |
| Aiempi biologisen hoidon epäonnistuminen | 20 % (N = 123) | 44 % (N = 102) | 23 % [11 %, 35 %] |
| Ei aiempaa biologisen hoidon epäonnistumista | 27 % (N = 41) | 54 % (N = 39) | 27 % [6 %, 48 %] |
| Muut päätetapahtumat | |||
| Parantunut kliininen SF/APS-vaste | 49 % | 59 % | 13 % [2 %, 23 %]e,g |
| Kliinisen remission säilyminenh | (N = 91) 51 % | (N = 72) 69 % | 21 % [6 %, 35 %]d,g |
| Endoskooppinen remissio | 13 % | 39 % | 28 % [20 %, 37 %]c,g |
| Limakalvon paraneminen | (N = 162) 10 % | (N = 141) 31 % | 22 % [14 %, 30 %]c,g |
a Tilastollisesti merkitsevä multiplisiteetin suhteen kontrolloituna, risankitsumabi vs. lume (p ≤ 0,01). b Tilastollisesti merkitsevä multiplisiteetin suhteen kontrolloituna, risankitsumabi vs. lume (p < 0,001). c Nimellinen p-arvo < 0,001 risankitsumabi vs. lume vertailu ilman yleistä tyypin I virheiden kontrollointia. d Nimellinen p-arvo ≤ 0,01 risankitsumabi vs. lume vertailu ilman yleistä tyypin I virheiden kontrollointia. e Nimellinen p-arvo ≤ 0,05 risankitsumabi vs. lume vertailu ilman yleistä tyypin I virheiden kontrollointia. f Vain induktio -ryhmä koostui tutkittavista, jotka saavuttivat kliinisen vasteen risankitsumabi-induktiohoidolle ja jotka satunnaistettiin saamaan lumelääkettä ylläpitohoitotutkimuksessa (FORTIFY). g Mukautettu hoitojen välinen ero. h Kliinisen remission säilyminen: kliininen remissio viikolla 52 tutkittavilla, jotka olivat kliinisessä remissiossa viikolla 0. | |||
Syvä remissio (kliininen remissio ja endoskooppinen remissio) todettiin viikolla 52 useammin risankitsumabia laskimoon / risankitsumabia ihon alle saaneilla tutkittavilla kuin tutkittavilla, jotka olivat saaneet risankitsumabia laskimoon / lumelääkettä ihon alle (28 % vs. 10 %, nimellinen p-arvo < 0,001).
Viikolla 52 suurempi osa risankitsumabia laskimoon / risankitsumabia ihon alle saaneista tutkittavista saavutti CDAI-pistearvon < 150 verrattuna tutkittaviin, jotka olivat saaneet risankitsumabia laskimoon / lumelääkettä ihon alle (52 % vs. 41 %, nimellinen p-arvo ≤ 0,01). Suurempi osa risankitsumabia laskimoon / risankitsumabia ihon alle saaneista tutkittavista saavutti vähintään 100 pisteen vähenemän CDAI-pistearvossa verrattuna tutkittaviin, jotka olivat saaneet risankitsumabia laskimoon / lumelääkettä ihon alle (62 % vs. 48 %, nimellinen p-arvo ≤ 0,01).
91 tutkittavaa, joilla ei todettu kliinistä SF/APS-vastetta ADVANCE- ja MOTIVATE-tutkimuksissa 12 viikkoa risankitsumabi-induktiohoidon jälkeen, sai ihon alle 360 mg:n annoksen risankitsumabia viikolla 12 ja viikolla 20. Näistä tutkittavista 64 % (58/91) saavutti kliinisen SF/APS-vasteen viikolla 24. Kliinisen SF/APS-vasteen saavuttaneista tutkittavista 33 osallistui FORTIFY-tutkimukseen ja jatkoi ihon alle annettavaa risankitsumabihoitoa annoksella 360 mg 8 viikon välein enintään 52 viikon ajan. Näistä tutkittavista 55 % (18/33) saavutti kliinisen remission ja 45 % (15/33) endoskooppisen vasteen viikolla 52.
FORTIFY-tutkimuksessa 30 tutkittavaa menetti vasteensa risankitsumabihoidolle ihon alle annettavalla 360 mg:n annoksella ja sai pelastushoitona risankitsumabia (1 200 mg kerta-annoksena laskimoon, minkä jälkeen 360 mg ihon alle 8 viikon välein). Näistä tutkittavista 57 % (17/30) saavutti kliinisen SF/APS-vasteen viikolla 52. Lisäksi 20 % (6/30) tutkittavista saavutti kliinisen remission ja 34 % (10/29) endoskooppisen vasteen viikolla 52.
Terveyteen ja elämänlaatuun liittyvät tulokset
Terveyteen liittyvää elämänlaatua arvioitiin tulehduksellisia suolistosairauksia koskevalla kyselyllä (Inflammatory Bowel Disease Questionnaire, IBDQ) ja lyhyellä 36-kohtaisella terveyskyselyllä (36-Item Short Form Health Survey, SF‑36). Väsymyksen lievittymistä arvioitiin kroonisen sairauden hoidon toiminnallisen arvioinnin väsymysasteikolla (Functional Assessment of Chronic Illness Therapy‑Fatigue, FACIT‑Fatigue). Työn tuottavuutta arvioitiin Work Productivity and Activity Impairment CD (WPAI-CD) -kyselyllä.
Viikolla 12 ADVANCE- ja MOTIVATE-tutkimuksissa risankitsumabihoitoa saaneet tutkittavat saavuttivat kliinisesti merkitsevästi suuremman paraneman lähtötasosta IBDQ-kokonaispisteissä, kaikkien IBDQ-kyselyn osioiden pisteissä (suolisto-oireet, systeeminen toimintakyky, emotionaalinen toimintakyky ja sosiaalinen toimintakyky), SF‑36-kyselyn fyysisen ja psyykkisen osion yhteenvetopistemäärässä, FACIT-Fatigue-pisteissä ja WPAI-CD-pisteissä lumelääkettä saaneisiin tutkittaviin verrattuna. WPAI-CD-kyselyllä arvioituna työkyvyn heikkenemisessä, työkyvyn yleisessä heikkenemisessä ja aktiivisuuden heikkenemisessä osoitettiin suurempia vähenemiä ADVANCE-tutkimuksessa ja aktiivisuuden heikkenemisessä osoitettiin suurempi vähenemä MOTIVATE-tutkimuksessa. Nämä paranemat säilyivät FORTIFY-tutkimuksessa risankitsumabia laskimoon / risankitsumabia ihon alle saaneilla tutkittavilla viikolle 52 asti.
Haavainen paksusuolitulehdus
Risankitsumabin tehoa ja turvallisuutta keskivaikeaa tai vaikeaa aktiivista haavaista paksusuolitulehdusta sairastavilla tutkittavilla arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa kliinisessä monikeskustutkimuksessa. Tutkimukseen otetut tutkittavat olivat iältään 18 – ≤ 80-vuotiaita, ja heidän keskitetysti vahvistetut mukautetut Mayo-pistemääränsä (adapted Mayo Score, aMS) olivat 5–9 (Mayo-pisteytysjärjestelmä lukuun ottamatta lääkärin yleisarviota [Physician’s Global Assessment]), joista endoskooppisen osion pistemäärät (ES) 2 tai 3 seulonnassa tehdyn endoskopian perusteella.
12 viikon pituiseen laskimonsisäisen hoidon induktiotutkimukseen (INSPIRE) sisältyi 12 viikon pituinen jatkovaihe niille tutkittaville, jotka eivät olleet saavuttaneet kliinistä vastetta viikolla 12 (vasteen määritelmä oli aMS-pisteiden lasku ≥ 2 pisteellä ja ≥ 30 %:lla lähtötilanteesta sekä peräsuolen verenvuotoa koskevan osion pistemäärän (RBS) lasku ≥ 1 pisteellä tai absoluuttinen RBS ≤ 1). INSPIRE-tutkimusta seurasi 52 viikon pituinen satunnaistettu ihonalaisen ylläpitohoidon lopettamista koskeva tutkimus (COMMAND), johon osallistuvat tutkittavat olivat saavuttaneet kliinisen vasteen 12 viikkoa kestäneelle laskimoon annetulle risankitsumabi-induktiohoidolle ja joiden hoidon kokonaispituudeksi tuli vähintään 64 viikkoa.
INSPIRE
INSPIRE-tutkimuksessa 975 tutkittavaa satunnaistettiin saamaan joko 1 200 mg risankitsumabia tai lumelääkettä viikolla 0, viikolla 4 ja viikolla 8.
INSPIRE-tutkimuksen tutkittavista 52 %:lla (503/975) aikaisempi hoito yhdellä tai useammalla biologisella hoidolla, JAK-estäjällä ja/tai S1P-reseptorin modulaattorilla oli epäonnistunut (riittämättömän vasteen tai sietokyvyttömyyden vuoksi). Näistä 503 tutkittavasta biologinen hoito oli epäonnistunut 488 tutkittavalla (97 %) ja JAK-estäjähoito 90 tutkittavalla (18 %).
Tutkittavien sallittiin käyttää suun kautta otettavia kortikosteroideja (enintään 20 mg/vrk prednisonia tai ekvivalentti annos jotain toista kortikosteroidia), immuunivasteen muuntajia ja aminosalisylaatteja vakaalla annoksella. INSPIRE-tutkimuksen lähtötilanteessa 36 % tutkittavista sai kortikosteroideja, 17 % immuunivasteen muuntajia ja 73 % aminosalisylaatteja. Taudin aktiivisuus oli keskivaikea (aMS ≤ 7) 58 %:lla tutkittavista ja vaikea (aMS > 7) 42 %:lla tutkittavista.
INSPIRE-tutkimuksessa merkitsevästi suurempi osa risankitsumabia saaneista tutkittavista saavutti ensisijaisen päätetapahtuman eli kliinisen remission aMS-pistemäärän perusteella (määritelmänä ulostustiheyttä koskevan osion pistemäärä [SFS] ≤ 1 eikä lähtötilanteen pistemäärää korkeampi, RBS = 0 sekä ES ≤ 1 ilman näyttöä kosketusverenvuodosta) viikolla 12 verrattuna lumelääkettä saaneisiin tutkittaviin (taulukko 5). Ensisijaista päätetapahtumaa ja tärkeimpiä toissijaisia päätetapahtumia koskevat tulokset on esitetty taulukossa 5.
Taulukko 5. INSPIRE-tutkimuksen tehotulokset viikolla 12
| Päätetapahtuma | Lumelääke laskimoon (N = 325) % | Risankitsumabi 1 200 mg laskimoon (N = 650) % | Hoitojen välinen ero (95 %:n lv) |
| Taudin aktiivisuus ja haavaisen paksusuolitulehduksen oireet | |||
| Kliininen remissioab | 6 % | 20 % | 14 %f (10 %, 18 %) |
| Aiempi biologisen ja/tai JAK-estäjähoidon epäonnistuminen | 4 % (N = 170) | 11 % (N = 333) | 7 % (3 %, 12 %) |
| Ei aiempaa biologisen ja/tai JAK-estäjähoidon epäonnistumista | 8 % (N = 155) | 30 % (N = 317) | 21 % (15 %, 28 %) |
| Kliininen vastec | 36 % | 64 % | 29 %f (22 %, 35 %) |
| Aiempi biologisen ja/tai JAK-estäjähoidon epäonnistuminen | 31 % (N = 170) | 55 % (N = 333) | 24 % (15 %, 33 %) |
| Ei aiempaa biologisen ja/tai JAK-estäjähoidon epäonnistumista | 41 % (N = 155) | 74 % (N = 317) | 33 % (24 %, 42 %) |
| Endoskooppinen ja histologinen arviointi | |||
| Limakalvon paraneminend | 12 % | 37 % | 24 %f (19 %, 29 %) |
| Aiempi biologisen ja/tai JAK-estäjähoidon epäonnistuminen | 10 % (N = 170) | 26 % (N = 333) | 16 % (9 %, 22 %) |
| Ei aiempaa biologisen ja/tai JAK-estäjähoidon epäonnistumista | 14 % (N = 155) | 48 % (N = 317) | 33 % (26 %, 41 %) |
| Histologis-endoskooppinen limakalvon paraneminene | 8 % | 24 % | 17 %f (12 %, 21 %) |
| Aiempi biologisen ja/tai JAK-estäjähoidon epäonnistuminen | 7 % (N = 170) | 16 % (N = 333) | 9 % (3 %, 14 %) |
| Ei aiempaa biologisen ja/tai JAK-estäjähoidon epäonnistumista | 8 % (N = 155) | 33 % (N = 317) | 25 % (18 %, 32 %) |
a Ensisijainen päätetapahtuma b Kliininen remissio aMS-pistemäärän perusteella: SFS ≤ 1 eikä lähtötilanteen pistemäärää korkeampi, RBS = 0 sekä ES ≤ 1 ilman näyttöä kosketusverenvuodosta c Kliininen vaste aMS-pistemäärän perusteella: pistemäärän ≥ 2 pisteen ja ≥ 30 %:n lasku lähtötilanteesta sekä RBS-pistemäärän ≥ 1 pisteen lasku tai absoluuttinen RBS ≤ 1 d ES ≤ 1 ilman näyttöä kosketusverenvuodosta e ES ≤ 1 ilman näyttöä kosketusverenvuodosta ja Geboes-pistemäärä ≤ 3,1 (jolloin neutrofiili-infiltraatiota on < 5 %:ssa kryptoista, kryptojen tuhoutumista ei ole tapahtunut, eikä eroosioita, haavaumia tai granulaatiokudosta esiinny) f p < 0,00001, korjattu hoitojen välinen ero (95 %:n luottamusväli) | |||
Kliininen taudin aktiivisuus ja oireet
Osittainen mukautettu Mayo-pistemäärä (partial adapted Mayo score, paMS) koostuu SFS- ja RBS-pistemääristä. Kliininen vaste paMS-pistemäärän perusteella määriteltiin paMS-pistemäärän ≥ 1 pisteen ja ≥ 30 %:n laskuna lähtötilanteesta sekä RBS-pistemäärän ≥ 1 pisteen laskuna tai absoluuttisena RBS-pistemääränä ≤ 1. paMS-pistemäärään perustuvaa kliinistä vastetta koskevat tulokset ajan myötä INSPIRE-tutkimuksessa on esitetty kuvassa 1. Teho ilmeni nopeasti, ja suurempi osa risankitsumabia saaneista tutkittavista saavutti kliinisen vasteen aikaisimmillaan viikolla 4 verrattuna lumelääkettä saaneisiin tutkittaviin (52 % vs. 31 %, p < 0,00001).
Kuva 1. paMS-pistemäärään perustuvan kliinisen vasteen saavuttaneiden tutkittavien osuus ajan myötä INSPIRE-induktiotutkimuksessa
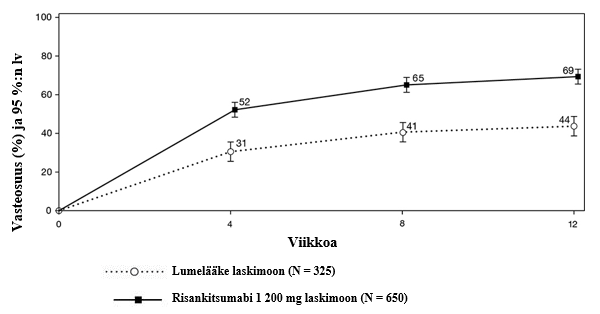
Merkitsevästi suuremmalla osalla risankitsumabia saaneista tutkittavista verrattuna lumelääkettä saaneisiin tutkittaviin ei esiintynyt vatsakipua (36 % vs. 26 %, p < 0,01) ja ulostamispakkoa (44 % vs. 28 %, p < 0,00001) viikolla 12.
Muut haavaisen paksusuolitulehduksen oireet
Ulosteinkontinenssiepisodien viikoittainen lukumäärä oli vähentynyt merkitsevästi enemmän risankitsumabia saaneilla tutkittavilla verrattuna lumelääkettä saaneisiin tutkittaviin viikolla 12 (muutos lähtötilanteesta: risankitsumabi = -3,8, lumelääke = -2,2, p = 0,00003).
Merkitsevästi suuremmalla osalla risankitsumabia saaneista tutkittavista verrattuna lumelääkettä saaneisiin tutkittaviin ei esiintynyt yöaikaista ulostamista viikolla 12 (67 % vs. 43 %, p < 0,00001).
Merkitsevästi suuremmalla osalla risankitsumabia saaneista tutkittavista verrattuna lumelääkettä saaneisiin tutkittaviin ei esiintynyt tenesmusta viikolla 12 (49 % vs. 30 %, p < 0,00001).
Niiden päivien viikoittainen lukumäärä, jolloin tutkittavilla esiintyi haavaisen paksusuolitulehduksen oireiden aiheuttamaa unen keskeytymistä, oli vähentynyt merkitsevästi enemmän risankitsumabia saaneilla tutkittavilla verrattuna lumelääkettä saaneisiin tutkittaviin viikolla 12 (muutos lähtötilanteesta: risankitsumabi = ‑2,5, lumelääke = -1,5, p < 0,00001).
Haavaiseen paksusuolitulehdukseen liittyvät sairaalajaksot
Risankitsumabia saaneilla tutkittavilla esiintyi merkitsevästi vähemmän haavaiseen paksusuolitulehdukseen liittyviä sairaalajaksoja viikkoon 12 mennessä verrattuna lumelääkettä saaneisiin tutkittaviin (1 % vs. 6 %, p < 0,00001).
Pidennetty hoito tutkittavilla, jotka eivät olleet saavuttaneet vastetta viikolla 12
Yhteensä 141 tutkittavaa, jotka eivät olleet saavuttaneet kliinistä vastetta risankitsumabille INSPIRE-induktiotutkimuksen viikolla 12, saivat joko 180 mg tai 360 mg risankitsumabia ihon alle viikolla 12 ja viikolla 20. Risankitsumabia 180 mg ihon alle saaneista 71 tutkittavasta 56 % ja 360 mg ihon alle saaneista 70 tutkittavasta 57 % saavutti kliinisen vasteen viikolla 24.
COMMAND
COMMAND-ylläpitohoitotutkimuksessa arvioitiin 548 tutkittavaa, jotka olivat saavuttaneet kliinisen vasteen risankitsumabille INSPIRE-tutkimuksen 12 viikon pituisen laskimoon annetun induktiohoidon aikana. Tutkittavat satunnaistettiin saamaan ihon alle annettavaa risankitsumabiylläpitohoitoa 180 mg:n tai 360 mg:n annoksella 8 viikon välein tai lopettamaan risankitsumabi-induktiohoidon ja saamaan ihon alle annettavaa lumelääkettä 8 viikon välein enintään 52 viikon ajan.
COMMAND-tutkimuksen tutkittavista 75 %:lla (411/548) aikaisempi hoito yhdellä tai useammalla biologisella hoidolla, JAK-estäjällä ja/tai S1P-reseptorin modulaattorilla oli epäonnistunut (riittämättömän vasteen tai sietokyvyttömyyden vuoksi) ennen induktiovaiheen lähtötilannetta. Näistä 411 tutkittavasta biologinen hoito oli epäonnistunut 407 tutkittavalla (99 %) ja JAK-estäjähoito 78 tutkittavalla (19 %).
COMMAND-tutkimuksessa merkitsevästi suurempi osa edellä mainituista 548:sta risankitsumabia ihon alle 180 mg:n tai 360 mg:n annoksella saaneesta tutkittavasta saavutti ensisijaisen päätetapahtuman eli kliinisen remission aMS-pistemäärän perusteella viikolla 52 verrattuna lumelääkettä saaneisiin tutkittaviin (ks. taulukko 6). Ensisijaista päätetapahtumaa ja tärkeimpiä toissijaisia päätetapahtumia koskevat tulokset on esitetty taulukossa 6.
Taulukko 6. COMMAND-tutkimuksen tehotulokset viikolla 52 (64 viikon jälkeen induktiohoidon aloittamisesta)
| Päätetapahtuma | Risankitsumabi induktio laskimoon / lumelääke ihon alle+ (N = 183) % | Risankitsumabi induktio laskimoon / risankitsumabi 180 mg ihon alle (N = 179) % | Risankitsumabi induktio laskimoon / risankitsumabi 360 mg ihon alle (N=186) % | Hoitojen välinen ero (97,5 %:n lv)++ | |
| Risankitsumabi induktio laskimoon / risankitsumabi 180 mg ihon alle | Risankitsumabi induktio laskimoon / risankitsumabi 360 mg ihon alle | ||||
| Taudin aktiivisuus ja haavaisen paksusuolitulehduksen oireet | |||||
| Kliininen remissioab | 25 % | 40 % | 38 % | 16 %h (6 %, 27 %) | 14 %h (4 %, 24 %) |
| Aiempi biologisen ja/tai JAK-estäjähoidon epäonnistuminen | 23 % (N = 138) | 37 % (N = 134) | 29 % (N = 139) | 13 % (1 %, 26 %) | 6 % (-6 %, 18 %) |
| Ei aiempaa biologisen ja/tai JAK-estäjähoidon epäonnistumista | 31 % (N = 45) | 51 % (N = 45) | 62 % (N = 47) | 20 % (-3 %, 43 %) | 31 % (8 %, 53 %) |
| Kliinisen remission säilyminenc | 40 % (N = 53) | 70 % (N = 44) | 50 % (N = 40) | 29 %h (7 %, 51 %) | 13 %k (-11 %, 36 %) |
| Aiempi biologisen ja/tai JAK-estäjähoidon epäonnistuminen | 37 % (N = 35) | 65 % (N = 26) | 44 % (N = 25) | 28 % (0 %, 56 %) | 7 % (-22 %, 36 %) |
| Ei aiempaa biologisen ja/tai JAK-estäjähoidon epäonnistumista | 44 % (N = 18) | 77 % (N = 18) | 60 % (N = 15) | 33 % (-2 %, 67 %) | 16 % (-23 %, 54 %) |
| Kliininen remissio ilman kortikosteroidejad | 25 % | 40 % | 37 % | 16 % h (6 %, 26 %) | 14 % h (3 %, 24 %) |
| Aiempi biologisen ja/tai JAK-estäjähoidon epäonnistuminen | 23 % (N = 138) | 36 % (N = 134) | 29 % (N = 139) | 13 % (0 %, 25 %) | 6 % (-6 %, 18 %) |
| Ei aiempaa biologisen ja/tai JAK-estäjähoidon epäonnistumista | 31% (N = 45) | 51 % (N = 45) | 60 % (N = 47) | 20 % (-3 %, 43 %) | 28 % (6 %, 51 %) |
| Kliininen vastee | 52 % | 68 % | 62 % | 17 %i (6 %, 28 %) | 11 %j (0 %, 23 %) |
| Aiempi biologisen ja/tai JAK-estäjähoidon epäonnistuminen | 46 % (N = 138) | 63 % (N = 134) | 57 % (N = 139) | 18 % (4 %, 31 %) | 11 % (-2 %, 25 %) |
| Ei aiempaa biologisen ja/tai JAK-estäjähoidon epäonnistumista | 71 % (N = 45) | 82 % (N = 45) | 79 % (N = 47) | 11 % (-9 %, 31 %) | 8 % (-13 %, 28 %) |
| Endoskooppinen ja histologinen arviointi | |||||
| Limakalvon paraneminenf | 32 % | 51 % | 48% | 20 % h (9 %, 31 %) | 17 % h (7 %, 28 %) |
| Aiempi biologisen ja/tai JAK-estäjähoidon epäonnistuminen | 30 % (N = 138) | 48 % (N = 134) | 39 % (N = 139) | 17 % (4 %, 30 %) | 8 % (-4 %, 21 %) |
| Ei aiempaa biologisen ja/tai JAK-estäjähoidon epäonnistumista | 36 % (N = 45) | 60 % (N = 45) | 76 % (N = 47) | 24 % (1 %, 47 %) | 41 % (19 %, 62 %) |
| Histologis-endoskooppinen limakalvon paranemineng | 23 % | 43 % | 42 % | 20 %h (10 %, 31 %) | 20 % h (10 %, 30 %) |
| Aiempi biologisen ja/tai JAK-estäjähoidon epäonnistuminen | 22 % (N = 138) | 39 % (N = 134) | 33 % (N = 139) | 17 % (5 %, 29 %) | 11 % (-1 %, 23 %) |
| Ei aiempaa biologisen ja/tai JAK-estäjähoidon epäonnistumista | 29 % (N = 45) | 55 % (N = 45) | 69 % (N = 47) | 26 % (3 %, 49 %) | 40 % (19 %, 62 %) |
+ Vain induktiohoitoa saaneiden ryhmä koostui tutkittavista, jotka saavuttivat kliinisen vasteen risankitsumabi-induktiohoidolle ja jotka satunnaistettiin saamaan lumelääkettä ylläpitohoitotutkimuksessa (COMMAND). ++ Mukautettu hoitoero. a Ensisijainen päätetapahtuma b Kliininen remissio aMS-pistemäärän perusteella: SFS ≤ 1 eikä lähtötilanteen pistemäärää korkeampi, RBS = 0 sekä ES ≤ 1 ilman näyttöä kosketusverenvuodosta c Kliininen remissio aMS-pistemäärän perusteella viikolla 52 niillä tutkittavilla, jotka olivat saavuttaneet kliinisen remission induktiohoidon päättyessä d Kliininen remissio aMS-pistemäärän perusteella viikolla 52 niillä tutkittavilla, jotka olivat olleet ilman kortikosteroideja ≥ 90 päivän ajan e Kliininen vaste aMS-pistemäärän perusteella: pistemäärän ≥ 2 pisteen ja ≥ 30 %:n lasku lähtötilanteesta sekä RBS-pistemäärän ≥ 1 pisteen lasku tai absoluuttinen RBS ≤ 1 f ES ≤ 1 ilman näyttöä kosketusverenvuodosta g ES ≤ 1 ilman näyttöä kosketusverenvuodosta ja Geboes-pistemäärä ≤ 3,1 (jolloin neutrofiili-infiltraatiota on < 5 %:ssa kryptoista, kryptojen tuhoutumista ei ole tapahtunut, eikä eroosioita, haavaumia tai granulaatiokudosta esiinny) h Tilastollisesti merkitsevä multiplisiteetin suhteen kontrolloituna, risankitsumabi vs. lumelääke (p ≤ 0,01). i Nimellinen p ≤ 0,01, risankitsumabi vs. lumelääke j Nimellinen p ≤ 0,05, risankitsumabi vs. lumelääke k p = 0,2234 | |||||
Kliininen taudin aktiivisuus ja oireet
Merkitsevästi suuremmalla osalla risankitsumabia laskimoon ja 180 mg risankitsumabia ihon alle saaneista tutkittavista verrattuna risankitsumabia laskimoon ja lumelääkettä saaneisiin tutkittaviin ei esiintynyt vatsakipua (47 % vs. 30 %, p < 0,001) ja ulostamispakkoa (54 % vs. 31 %, p < 0,00001) viikolla 52. Suuremmalla osalla risankitsumabia laskimoon ja 360 mg risankitsumabia ihon alle saaneista tutkittavista verrattuna risankitsumabia laskimoon ja lumelääkettä saaneisiin tutkittaviin ei esiintynyt ulostamispakkoa (49 % vs. 31 %, p < 0,001) viikolla 52, ja numeerisesti suuremmalla osalla näistä tutkittavista verrattuna risankitsumabia laskimoon ja lumelääkettä saaneisiin tutkittaviin ei esiintynyt vatsakipua (38 % vs. 30 %, p < 0,0895) viikolla 52.
Muut haavaisen paksusuolitulehduksen oireet
Suuremmalla osalla risankitsumabia laskimoon ja 180 mg risankitsumabia ihon alle saaneista tutkittavista sekä risankitsumabia laskimoon ja 360 mg risankitsumabia ihon alle saaneista tutkittavista verrattuna risankitsumabia laskimoon ja lumelääkettä saaneisiin tutkittaviin ei esiintynyt yöaikaista ulostamista viikolla 52 (42 % ja 43 % vs. 30 %, p < 0,01 ja p < 0,001).
Suuremmalla osalla risankitsumabia laskimoon ja 180 mg risankitsumabia ihon alle saaneista tutkittavista sekä risankitsumabia laskimoon ja 360 mg risankitsumabia ihon alle saaneista tutkittavista verrattuna risankitsumabia laskimoon ja lumelääkettä saaneisiin tutkittaviin ei esiintynyt tenesmusta viikolla 52 (37 % ja 37 % vs. 23 %, p < 0,01).
Haavaiseen paksusuolitulehdukseen liittyvät sairaalajaksot
Risankitsumabia laskimoon ja 180 mg risankitsumabia ihon alle saaneilla tutkittavilla sekä risankitsumabia laskimoon ja 360 mg risankitsumabia ihon alle saaneilla tutkittavilla esiintyi numeerisesti vähemmän haavaiseen paksusuolitulehdukseen liittyviä sairaalajaksoja viikkoon 52 mennessä verrattuna risankitsumabia laskimoon ja lumelääkettä saaneisiin tutkittaviin (0,6 / 100 potilasvuotta ja 1,2 / 100 potilasvuotta vs. 3,1 / 100 potilasvuotta, p = 0,0949 ja p = 0,2531).
Endoskooppinen ja histologinen arviointi
Endoskooppinen remissio (limakalvon endoskooppisen ulkonäön normalisoituminen) määriteltiin ES-pistemääränä 0. INSPIRE-tutkimuksen viikolla 12 merkitsevästi suurempi osa risankitsumabia saaneista tutkittavista verrattuna lumelääkettä saaneisiin tutkittaviin saavutti endoskooppisen remission (11 % vs. 3 %, p < 0,00001). COMMAND-tutkimuksen viikolla 52 merkitsevästi suurempi osa risankitsumabia laskimoon ja 180 mg risankitsumabia ihon alle saaneista tutkittavista sekä risankitsumabia laskimoon ja 360 mg risankitsumabia ihon alle saaneista tutkittavista verrattuna risankitsumabia laskimoon ja lumelääkettä saaneisiin tutkittaviin saavutti endoskooppisen remission (23 % ja 24 % vs. 15 %, p < 0,05).
Limakalvon syvä paraneminen määriteltiin ES-pistemääränä 0 ja Geboes-pistemääränä ≤ 2,0 (jolloin neutrofiili-infiltraatiota kryptoissa tai limakalvon tukikerroksessa ei ole, eosinofiilien määrä ei ole kasvanut, kryptojen tuhoutumista ei ole tapahtunut, eikä eroosioita, haavaumia tai granulaatiokudosta esiinny). INSPIRE-tutkimuksen viikolla 12 merkitsevästi suurempi osa risankitsumabia saaneista tutkittavista verrattuna lumelääkettä saaneisiin tutkittaviin saavutti limakalvon syvän paranemisen (6 % vs. 1 %, p < 0,00001). COMMAND-tutkimuksen viikolla 52 numeerisesti suurempi osa risankitsumabia laskimoon ja 180 mg risankitsumabia ihon alle saaneista tutkittavista sekä risankitsumabia laskimoon ja 360 mg risankitsumabia ihon alle saaneista tutkittavista verrattuna risankitsumabia laskimoon ja lumelääkettä saaneisiin tutkittaviin saavutti limakalvon syvän paranemisen (13 % ja 16 % vs. 10 %, p = 0,2062 ja p = 0,0618).
COMMAND-tutkimuksen viikolla 52 induktiohoidon lopussa todetun limakalvon paranemisen havaittiin säilyneen (ES ≤ 1 ilman kosketusverenvuotoa) suuremmalla osalla risankitsumabia laskimoon ja 180 mg risankitsumabia ihon alle saaneista tutkittavista sekä risankitsumabia laskimoon ja 360 mg risankitsumabia ihon alle saaneista tutkittavista verrattuna risankitsumabia laskimoon ja lumelääkettä saaneisiin tutkittaviin (74 % ja 54 % vs. 47 %, p <0,01 ja p = 0,5629).
Pelastushoito
COMMAND-tutkimuksen aikana tutkittavat, jotka menettivät vasteensa ihon alle annetulle risankitsumabihoidolle, saivat pelastushoitoa risankitsumabilla (yksi induktioannos laskimoon, minkä jälkeen 360 mg ihon alle 8 viikon välein). Näistä tutkittavista 85 % (17/20) risankitsumabia 180 mg ihon alle saavien ryhmässä ja 74 % (26/35) risankitsumabia 360 mg ihon alle saavien ryhmässä saavutti kliinisen vasteen viikolla 52. Lisäksi 24 % (6/25) risankitsumabia 180 mg ihon alle saavien ryhmässä ja 35 % (13/37) risankitsumabia 360 mg ihon alle saavien ryhmässä saavutti kliinisen remission aMS-pistemäärän perusteella ja 38 % (10/26) risankitsumabia 180 mg ihon alle saavien ryhmässä ja 45 % (17/38) risankitsumabia 360 mg ihon alle saavien ryhmässä endoskooppisen paraneman viikolla 52.
Vasteen viikolla 24 saavuttaneet tutkittavat
COMMAND-tutkimuksessa 100 tutkittavaa ei ollut saavuttanut kliinistä vastetta 12 viikon pituisen induktiohoidon aikana, sai risankitsumabia ihon alle joko 180 mg:n annoksen (N = 56) tai 360 mg:n annoksen (N = 44) viikolla 12 ja viikolla 20, saavutti kliinisen vasteen viikolla 24 ja jatkoi 8 viikon välein annettavaa risankitsumabihoitoa 180 mg:n tai 360 mg:n annoksella ihon alle enintään 52 viikon ajan. Näistä tutkittavista 46 % risankitsumabia 180 mg ihon alle saavien ryhmässä ja 45 % risankitsumabia 360 mg ihon alle saavien ryhmässä saavutti kliinisen vasteen aMS-pistemäärän perusteella ja 18 % risankitsumabia 180 mg ihon alle saavien ryhmässä ja 23 % risankitsumabia 360 mg ihon alle saavien ryhmässä kliinisen remission aMS-pistemäärän perusteella viikolla 52.
Terveyteen ja elämänlaatuun liittyvät tulokset
Risankitsumabihoitoa saaneet tutkittavat saavuttivat kliinisesti merkittäviä paranemia lähtötilanteesta IBDQ (Inflammatory Bowel Disease Questionnaire) -kyselyn osioissa (suolisto-oireet, systeeminen toimintakyky, emotionaalinen toimintakyky ja sosiaalinen toimintakyky) verrattuna lumelääkettä saaneisiin tutkittaviin. IBDQ-kokonaispisteiden muutos lähtötilanteesta viikolla 12 oli risankitsumabia saaneilla 42,6 ja lumelääkettä saaneilla 24,3. IBDQ-kokonaispisteiden muutos lähtötilanteesta viikolla 52 oli risankitsumabia laskimoon ja risankitsumabia 180 mg ihon alle saaneilla tutkittavilla 52,6, risankitsumabia laskimoon ja risankitsumabia 360 mg ihon alle saaneilla tutkittavilla 50,3 ja risankitsumabia laskimoon ja lumelääkettä saaneilla tutkittavilla 35,0.
Risankitsumabia saaneilla tutkittavilla havaittiin merkitsevästi suurempaa paranemaa väsymyksessä lähtötilanteeseen nähden verrattuna lumelääkettä saaneisiin tutkittaviin viikolla 12 FACIT-Fatigue-kyselyn pisteillä mitattuna. FACIT-Fatigue-pisteiden muutos lähtötilanteesta viikolla 12 oli risankitsumabia saaneilla tutkittavilla 7,9 ja lumelääkettä saaneilla tutkittavilla 3,3. FACIT-Fatigue-pisteiden muutos lähtötilanteesta viikolla 52 oli risankitsumabia laskimoon ja risankitsumabia 180 mg ihon alle saaneilla tutkittavilla 10,9, risankitsumabia laskimoon ja risankitsumabia 360 mg ihon alle saaneilla tutkittavilla 10,3 ja risankitsumabia laskimoon ja lumelääkettä saaneilla tutkittavilla 7,0.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Skyrizi-valmisteen käytöstä Crohnin taudin ja haavaisen paksusuolitulehduksen hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Risankitsumabin farmakokinetiikka oli samankaltaista läiskäpsoriaasia sairastavien ja nivelpsoriaasia sairastavien sekä Crohnin tautia sairastavien ja haavaista paksusuolitulehdusta sairastavien potilaiden välillä.
Imeytyminen
Risankitsumabin farmakokinetiikka oli lineaarinen ja altistus suureni suhteessa annokseen, kun ihon alle annettu annos oli 18–360 mg ja 0,25–1 mg/kg ja laskimoon annettu annos oli 200–1 200 mg ja 0,01–5 mg/kg.
Kun risankitsumabia annettiin ihon alle, huippupitoisuudet plasmassa saavutettiin 3–14 päivän kuluttua annostelusta ja arvioitu absoluuttinen biologinen hyötyosuus oli 74–89 %. Kun valmistetta annettiin 150 mg:n annoksina viikolla 0, viikolla 4 ja tämän jälkeen 12 viikon välein, arvioidut vakaan tilan huippu- ja jäännöspitoisuudet olivat 12 µg/ml ja 2 µg/ml.
Crohnin tautia sairastavilla tutkittavilla, jotka saavat induktioannoksena 600 mg laskimoon viikoilla 0, 4 ja 8 ja sitten ylläpitoannoksena 360 mg ihon alle viikolla 12 ja tämän jälkeen 8 viikon välein, suurimpien huippu- ja jäännöspitoisuuksien mediaanit ovat induktiovaiheessa (viikoilla 8–12) arviolta 156 µg/ml ja 38,8 µg/ml ja vakaan tilan huippu- ja jäännöspitoisuuksien mediaanit ylläpitovaiheessa (viikoilla 40–48) arviolta 28,0 µg/ml ja 8,13 µg/ml.
Haavaista paksusuolitulehdusta sairastavilla tutkittavilla, jotka saavat induktioannoksena 1 200 mg laskimoon viikoilla 0, 4 ja 8 ja sitten ylläpitoannoksena 180 mg tai 360 mg ihon alle viikolla 12 ja tämän jälkeen 8 viikon välein, suurimpien huippu- ja jäännöspitoisuuksien mediaanit ovat induktiovaiheessa (viikoilla 8–12) arviolta 350 µg/ml ja 87,7 µg/ml ja vakaan tilan huippu- ja jäännöspitoisuuksien mediaanit ylläpitovaiheessa (viikoilla 40–48) arviolta 19,6 µg/ml ja 4,64 µg/ml annoksella 180 mg ihon alle ja 39,2 µg/ml ja 9,29 µg/ml annoksella 360 mg ihon alle.
Jakautuminen
Risankitsumabin vakaan tilan jakautumistilavuuden (Vss) keskiarvo (± keskihajonta) oli 11,4 (± 2,7) l vaiheen 3 tutkimuksissa psoriaasipotilailla. Tämä viittaa siihen, että risankitsumabin jakautuminen rajoittuu lähinnä vaskulaari- ja interstitiaalitiloihin. Tyypillisen Crohnin tautia sairastavan, 70 kg painavan tutkittavan Vss oli 7,68 l.
Biotransformaatio
Monoklonaaliset IgG-vasta-ainelääkkeet pilkkoutuvat tyypillisesti pieniksi peptideiksi ja aminohapoiksi kataboliareittien välityksellä samaan tapaan kuin endogeeniset IgG-molekyylit. Risankitsumabi ei oletettavasti metaboloidu sytokromi P450 -entsyymivälitteisesti.
Eliminaatio
Risankitsumabin systeemisen puhdistuman (CL) keskiarvo (± keskihajonta) oli 0,3 (± 0,1) l/vrk vaiheen 3 tutkimuksissa psoriaasipotilailla. Risankitsumabin terminaalisen eliminaation puoliintumisajan keskiarvo vaihteli 28 vuorokaudesta 29 vuorokauteen vaiheen 3 tutkimuksissa psoriaasipotilailla. Tyypillisen Crohnin tautia sairastavan, 70 kg painavan tutkittavan puhdistuma oli 0,30 l/vrk ja terminaalinen eliminaation puoliintumisaika 21 vuorokautta.
Risankitsumabi on IgG1-luokan monoklonaalinen vasta-aine, joten se ei oletettavasti suodatu munuaisissa glomerulussuodatuksen kautta eikä erity pilkkoutumattomana virtsaan.
Lineaarisuus/ei-lineaarisuus
Risankitsumabin farmakokinetiikka oli lineaarinen ja systeeminen altistus (Cmax ja AUC) suureni suunnilleen suhteessa annokseen arvioiduilla annosalueilla eli 18–360 mg:n annoksilla ja 0,25–1 mg/kg:n annoksilla ihon alle ja 200–1 800 mg:n annoksilla ja 0,01–5 mg/kg:n annoksilla laskimoon terveillä henkilöillä ja psoriaasia, Crohnin tautia tai haavaista paksusuolitulehdusta sairastavilla potilailla.
Interaktiot
Läiskäpsoriaasia, Crohnin tautia tai haavaista paksusuolitulehdusta sairastaneilla tutkittavilla toteutetuissa interaktiotutkimuksissa arvioitiin toistuvien risankitsumabiannosten vaikutusta sytokromi P450 (CYP) -toiminnan herkkien testisubstraattien farmakokinetiikkaan. Altistus kofeiinille (CYP1A2-substraatti), varfariinille (CYP2C9-substraatti), omepratsolille (CYP2C19-substraatti), metoprololille (CYP2D6-substraatti) ja midatsolaamille (CYP3A-substraatti) oli risankitsumabihoidon jälkeen verrattavissa tutkittavien altistukseen näille aineille ennen risankitsumabihoitoa. Tämä viittaa siihen, että näiden entsyymien kautta välittyviä kliinisesti merkittäviä interaktioita ei ole.
Populaatiofarmakokineettiset analyysit viittasivat siihen, että joidenkin kliinisiin tutkimuksiin osallistuneiden läiskäpsoriaasipotilaiden samanaikaisesti käyttämät lääkevalmisteet eivät vaikuttaneet risankitsumabialtistukseen. Samanaikaisten lääkevalmisteiden vaikutuksettomuus todettiin myös Crohnin tautia tai haavaista paksusuolitulehdusta koskeneissa populaatiofarmakokineettisissä analyyseissa.
Erityisryhmät
Pediatriset potilaat
Risankitsumabin farmakokinetiikkaa alle 16 vuoden ikäisillä pediatrisilla potilailla ei ole selvitetty. Risankitsumabille altistuneista Crohnin tautia sairastavasta 1 574 tutkittavasta 12 oli 16–17-vuotiaita. Crohnin tautia sairastavien 16–17-vuotiaiden tutkittavien risankitsumabialtistus oli samankaltainen kuin aikuisten. Iän ei havaittu vaikuttavan merkittävästi risankitsumabialtistukseen populaatiofarmakokineettisten analyysien perusteella.
Iäkkäät
Risankitsumabille altistuneista 2 234:stä läiskäpsoriaasia sairastavasta tutkittavasta 243 oli täyttänyt 65 vuotta ja 24 oli täyttänyt 75 vuotta. Risankitsumabille altistuneista 1 574:stä Crohnin tautia sairastavasta tutkittavasta 72 oli täyttänyt 65 vuotta ja 5 oli täyttänyt 75 vuotta. Risankitsumabille altistuneista 1 512:sta haavaista paksusuolitulehdusta sairastavasta tutkittavasta 103 oli täyttänyt 65 vuotta ja 8 oli täyttänyt 75 vuotta. Risankitsumabialtistuksessa ei todettu yleisesti eroja risankitsumabia saaneiden iäkkäiden tutkittavien ja nuorempien tutkittavien välillä.
Potilaat, joilla on munuaisten tai maksan vajaatoiminta
Munuaisten tai maksan vajaatoiminnan vaikutusta risankitsumabin farmakokinetiikkaan ei ole arvioitu spesifisissä tutkimuksissa. Populaatiofarmakokineettisten analyysien perusteella seerumin kreatiniinipitoisuus, kreatiniinipuhdistuma ja maksan toiminnan merkkiaineet (ALAT/ASAT/bilirubiini) eivät vaikuttaneet merkittävästi risankitsumabin puhdistumaan psoriaasia, Crohnin tautia tai haavaista paksusuolitulehdusta sairastavilla potilailla.
Risankitsumabi on IgG1-luokan monoklonaalinen vasta-aine, joten se eliminoituu lähinnä solunsisäisen katabolian kautta eikä todennäköisesti metaboloidu maksan sytokromi P450 -entsyymien kautta eikä eliminoidu munuaisteitse.
Paino
Risankitsumabin puhdistuma ja jakautumistilavuus suurenevat painon myötä, mikä voi johtaa tehon heikkenemiseen potilailla, joilla on merkittävä ylipaino (> 130 kg). Tämä havainto perustuu kuitenkin rajalliseen määrään läiskäpsoriaasipotilaita. Painolla ei ollut kliinisesti merkittävää vaikutusta risankitsumabialtistukseen tai risankitsumabin tehoon nivelpsoriaasia, Crohnin tautia tai haavaista paksusuolitulehdusta sairastavilla potilailla. Tämänhetkisen suosituksen mukaan annosta ei tarvitse muuttaa painon perusteella.
Sukupuoli tai etninen tausta
Sukupuoli ja etninen tausta eivät vaikuttaneet merkitsevästi risankitsumabin puhdistumaan aikuisilla läiskäpsoriaasia, Crohnin tautia tai haavaista paksusuolitulehdusta sairastavilla potilailla. Risankitsumabialtistuksessa ei todettu kliinisesti merkittäviä eroja, kun kiinalaisia ja japanilaisia tutkittavia verrattiin valkoihoisiin tutkittaviin terveillä vapaaehtoisilla tehdyissä kliinisissä farmakokinetiikan tutkimuksissa.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Mukana on farmakologisen turvallisuuden arviointeja sekä tehostettu pre- ja postnataalista kehitystoksisuutta selvittänyt tutkimus, jossa jaavanmakakiapinoille annetut annokset olivat enintään 50 mg/kg/viikko, jolloin altistus on noin 10-kertainen verrattuna induktiohoidon aikana saavutettavaan kliiniseen altistukseen annostuksella 600 mg laskimoon 4 viikon välein ja 39-kertainen verrattuna ylläpitohoidon aikana saavutettavaan kliiniseen altistukseen annostuksella 360 mg ihon alle 8 viikon välein Crohnin taudissa. Haavaisen paksusuolitulehduksen osalta tämä altistus on noin 5-kertainen verrattuna induktiohoidon aikana saavutettavaan kliiniseen altistukseen annostuksella 1 200 mg laskimoon 4 viikon välein ja 65- tai 32-kertainen verrattuna ylläpitohoidon aikana saavutettavaan kliiniseen altistukseen annostuksilla 180 mg ja 360 mg ihon alle 8 viikon välein.
Risankitsumabilla ei ole tehty mutageenisuus- eikä karsinogeenisuustutkimuksia. Jaavanmakakiapinoilla toteutetussa 26 viikon pituisessa pitkäaikaistoksisuuden tutkimuksessa, jossa annokset olivat enintään 50 mg/kg/viikko (jolloin altistus on 7-kertainen verrattuna induktiohoidon aikana saavutettavaan kliiniseen altistukseen annostuksella 600 mg laskimoon 4 viikon välein ja 28-kertainen verrattuna ylläpitohoidon aikana saavutettavaan kliiniseen altistukseen annostuksella 360 mg ihon alle 8 viikon välein Crohnin taudissa sekä 3-kertainen verrattuna induktiohoidon aikana saavutettavaan kliiniseen altistukseen annostuksella 1 200 mg laskimoon 4 viikon välein ja 45- tai 23-kertainen verrattuna ylläpitohoidon aikana saavutettavaan kliiniseen altistukseen annostuksilla 180 mg ja 360 mg ihon alle 8 viikon välein haavaisessa paksusuolitulehduksessa), ei todettu preneoplastisia tai neoplastisia muutoksia eikä immunotoksisia tai kardiovaskulaarisia haittavaikutuksia.
Farmaseuttiset tiedot
Apuaineet
Natriumasetaattitrihydraatti
Etikkahappo
Trehaloosidihydraatti
Polysorbaatti 20
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
2 vuotta
Laimennettu infuusioneste laskimoinfuusioon
Valmisteen on osoitettu säilyvän käytön aikana kemiallisesti ja fysikaalisesti stabiilina 20 tunnin ajan 2–8 °C:n lämpötilassa (valolta suojattuna) tai enintään 8 tunnin ajan huoneenlämmössä (auringonvalolta suojattuna). Säilytysaika huoneenlämmössä alkaa, kun laimennettu infuusioneste on valmistettu. Infuusio on annettava kokonaan 8 tunnin kuluessa infuusiopussiin laimentamisesta. Sisävalaistukselle altistuminen on hyväksyttävää huoneenlämmössä säilytyksen ja annon aikana.
Mikrobiologiselta kannalta käyttövalmis infuusioneste tulisi käyttää välittömästi. Jos sitä ei käytetä välittömästi, käytönaikainen säilytysaika ja olosuhteet ennen käyttöä ovat käyttäjän vastuulla, eikä säilytysaika saa ylittää 20 tuntia 2–8 °C:n lämpötilassa.
Ei saa jäätyä.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C). Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SKYRIZI infuusiokonsentraatti, liuosta varten
600 mg (L:ei) 1 kpl (10 ml (60 mg/ml)) (3415,69 €)
PF-selosteen tieto
10,0 ml infuusiokonsentraattia, liuosta varten, lasisessa injektiopullossa, jossa on pinnoitettu bromobutyylikumitulppa.
Skyrizi on saatavana 1 injektiopullon pakkauksissa.
Valmisteen kuvaus:
Liuos on väritön tai hieman kellertävä ja kirkas tai hieman opalisoiva.
Käyttö- ja käsittelyohjeet
Liuokset on tarkastettava silmämääräisesti hiukkasten ja värimuutosten varalta ennen potilaalle antoa. Liuoksen pitää olla väritöntä tai hieman kellertävää ja kirkasta tai hieman opalisoivaa. Nesteessä voi olla pienen pieniä valkoisia tai kirkkaita hiukkasia. Lääkevalmistetta ja sen laimennoksia ei saa käyttää, jos liuos on sameaa tai siinä on värimuutoksia tai vierashiukkasia.
Laimennusohjeet
Terveydenhuollon ammattilaisen on valmisteltava liuos aseptisella tekniikalla. Liuos on laimennettava ennen potilaalle antoa.
Infuusioneste valmistellaan laimentamalla konsentraatti infuusiopussiin tai lasipulloon, joka sisältää 5 % glukoosia vedessä (D5W) tai natriumkloridi 9 mg/ml (0,9 %) infuusionestettä. Lopullinen pitoisuus on noin 1,2–6 mg/ml. Katso potilaan käyttöaiheen mukaiset laimennusohjeet alla olevasta taulukosta.
Käyttöaihe | Laskimoon annettava induktioannos | 600 mg/ 10 ml injektiopullojen lukumäärä | 5 % glukoosi- tai natriumkloridi 9 mg/ml (0,9 %) infuusionesteen kokonaistilavuus |
Crohnin tauti | 600 mg | 1 | 100 ml tai 250 ml tai 500 ml |
Haavainen paksusuolitulehdus | 1 200 mg | 2 | 250 ml tai 500 ml |
Infuusiopussin tai lasipullon sisällön on oltava huoneenlämpöistä ennen laskimoinfuusion aloittamista.
Anna laimennettu liuos vähintään yhden tunnin kestoisena infuusiona, kun annos 600 mg, ja vähintään kahden tunnin kestoisena infuusiona, kun annos on 1 200 mg.
Injektiopullossa olevaa liuosta ja laimennoksia ei saa ravistaa.
Kukin injektiopullo on tarkoitettu vain yhtä käyttökertaa varten.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SKYRIZI infuusiokonsentraatti, liuosta varten
600 mg 1 kpl
- Ei korvausta.
ATC-koodi
L04AC18
Valmisteyhteenvedon muuttamispäivämäärä
19.06.2026
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi