
SPEVIGO infuusiokonsentraatti, liuosta varten 450 mg
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 450 mg spesolimabia 7,5 ml:ssa.
Yksi millilitra infuusiokonsentraattia, liuosta varten, sisältää 60 mg spesolimabia.
Laimennuksen jälkeen yksi millilitra liuosta sisältää 9 mg spesolimabia (ks. kohta Käyttö- ja käsittelyohjeet).
Spesolimabi valmistetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa.
Apuaineet, joiden vaikutus tunnetaan
Yksi injektiopullo sisältää 3 mg polysorbaatti 20:tä (E432).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti)
Kliiniset tiedot
Käyttöaiheet
Spevigo on tarkoitettu yleistyneen pustulaarisen psoriaasin (GPP) pahenemisvaiheiden hoitoon monoterapiana aikuispotilaille ja vähintään 12 vuoden ikäisille nuorille.
Ehto
Valmisteen käyttöaiheissa mainittujen sairauksien hoitoon perehtyneen lääkärin on aloitettava hoito ja valvottava sitä.
Annostus ja antotapa
Hoidon aloittaa ja sitä valvoo lääkäri, jolla on kokemusta tulehduksellisten ihosairauksien hoidosta.
Hoito voidaan aloittaa esitäytetyillä ruiskuilla eli ihonalaisena injektiona GPP:n pahenemisvaiheiden ehkäisemiseksi (ks. Spevigo 150 mg ja 300 mg injektioneste, liuos, esitäytetty ruisku -valmisteyhteenveto) tai laskimoon annettavalla spesolimabiannoksella GPP:n pahenemisvaiheen hoitamiseksi.
Annostus
Suositeltu annos GPP:n pahenemisvaiheen hoitoon aikuisille ja vähintään 12 vuoden ikäisille nuorille, joiden paino on vähintään 40 kg, on 900 mg:n kerta-annos (kaksi 450 mg:n injektiopulloa) infuusiona laskimoon. Jos pahenemisvaiheen oireet jatkuvat, voidaan antaa toinen 900 mg:n annos 1 viikon kuluttua ensimmäisestä annoksesta.
Spevigo-valmistetta ei ole tutkittu alle 40 kg painavilla potilailla. Farmakokineettisen mallinnuksen ja simulaation perusteella suositeltu annos vähintään 12 vuoden ikäisille, ≥ 30 ja – <40 kg painaville nuorille on 450 mg:n kerta-annos (yksi 450 mg:n injektiopullo) infuusiona laskimoon (ks. kohta Farmakokinetiikka). Jos pahenemisvaiheen oireet jatkuvat, voidaan antaa toinen 450 mg:n annos (yksi 450 mg:n injektiopullo) 1 viikon kuluttua ensimmäisestä annoksesta.
Kliinisiä tietoja seuraavien pahenemisvaiheiden hoidosta on hyvin vähän (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kliinisiä tietoja spesolimabin käytöstä samanaikaisesti muiden GPP-hoitojen kanssa on vain vähän. Spesolimabia ei pidä käyttää pahenemisvaiheen hoitoon yhdistelmänä muiden GPP-hoitojen, kuten systeemisten immunosuppressanttien, kanssa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Erityisryhmät
Iäkkäät
Annoksen muuttaminen ei ole tarpeen.
Munuaisten tai maksan vajaatoiminta
Spesolimabia ei ole tutkittu virallisesti näissä potilasryhmissä. Näiden tilojen ei yleisesti odoteta vaikuttavan kliinisesti merkittävästi monoklonaalisten vasta-aineiden farmakokinetiikkaan, eikä annosmuutosta pidetä tarpeellisena.
Pediatriset potilaat
Spesolimabin turvallisuutta ja tehoa alle 12 vuoden ikäisten lasten hoidossa ei ole varmistettu.
Tietoja ei ole saatavilla.
Antotapa
Tämä lääkevalmiste on tarkoitettu annettavaksi vain infuusiona laskimoon. Sitä ei saa antaa nopeana injektiona eikä bolusinjektiona laskimoon.
Kun valmiste on laimennettu 9 mg/ml (0,9 %) natriumkloridi-injektionesteellä, se annetaan jatkuvana laskimoinfuusiona steriilillä, pyrogeenittomalla, niukasti proteiinia sitovalla letkunsisäisellä suodattimella (huokoskoko 0,2 mikronia) varustetun infuusioletkun kautta 90 minuutin aikana. Saman laskimoyhteyden kautta ei saa antaa samanaikaisesti muita infuusioita.
Jos infuusionopeutta hidastetaan tai infuusio keskeytetään tilapäisesti, infuusion kokonaiskesto (mukaan lukien keskeytykseen kulunut aika) ei saa olla yli 180 minuuttia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Vaikea tai henkeä uhkaava yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kliinisesti merkittävät aktiiviset infektiot (esim. aktiivinen tuberkuloosi, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Spesolimabi saattaa suurentaa infektioriskiä (ks. kohta Haittavaikutukset).
Jos potilaalla on krooninen infektio tai hänellä on aiemmin ollut toistuvia infektioita, hoidon mahdollisia riskejä ja odotettavissa olevia kliinisiä hyötyjä on punnittava ennen spesolimabin määräämistä. Spesolimabihoitoa ei pidä aloittaa potilaille, joilla on jokin kliinisesti merkittävä aktiivinen infektio, ennen kuin infektio on parantunut tai tyydyttävästi hoidettu. Potilaita tulee neuvoa hakeutumaan lääkärin hoitoon, jos heillä ilmenee kliinisesti merkittävän infektion merkkejä tai oireita spesolimabihoidon jälkeen.
Tuberkuloosin arviointi ennen hoitoa
Potilaat on arvioitava tuberkuloosi-infektion varalta ennen spesolimabihoidon aloittamista. Spesolimabi on vasta-aiheista potilaille, joilla on aktiivinen tuberkuloosi-infektio (ks. kohta Vasta-aiheet).
Tuberkuloosihoitoa on harkittava ennen spesolimabihoidon aloittamista, jos potilaalla on piilevä tuberkuloosi, jos hänellä on aiemmin ollut tuberkuloosi, tai jos hän on saattanut aiemmin altistua aktiivista tuberkuloosia sairastaville henkilöille, eikä riittävän hoitokuurin saamista voida varmistaa. Spesolimabihoidon jälkeen potilaita tulee seurata aktiivisen tuberkuloosin merkkien ja oireiden varalta.
Yliherkkyys ja infuusioon liittyvät reaktiot
Spesolimabin kaltaisten monoklonaalisten vasta-aineiden käytön yhteydessä voi esiintyä yliherkkyyttä ja infuusioon liittyviä reaktioita. Yliherkkyysreaktiot voivat olla välittömiä reaktioita, kuten anafylaksia, sekä viiveellä ilmeneviä reaktioita, kuten yleisoireinen eosinofiilinen oireyhtymä (DRESS).
Välittömiä yliherkkyysreaktioita, mukaan lukien anafylaktisia reaktioita, on raportoitu spesolimabihoitoa saaneilla potilailla (ks. kohta Haittavaikutukset).
Jos potilaalle kehittyy anafylaksian tai muun vakavan yliherkkyysreaktion merkkejä, spesolimabihoito on lopetettava välittömästi ja asianmukainen hoito on aloitettava (ks. kohta Vasta-aiheet).
Jos potilaalle kehittyy laskimoinfuusion aikana lieviä tai keskivaikeita yliherkkyysreaktioita tai muita infuusioon liittyviä reaktioita, hoito on lopetettava ja asianmukaisen lääkehoidon (esim. systeemisten antihistamiinien ja/tai kortikosteroidien) antamista on harkittava. Reaktion korjaannuttua infuusio voidaan aloittaa uudelleen hitaammalla nopeudella, jota lisätään vähitellen, kunnes infuusio on annettu (ks. kohta Annostus ja antotapa).
Käyttö potilaille, joilla on välitön, henkeä uhkaava GPP:n pahenemisvaihe
Spesolimabin käytöstä ei ole kokemusta potilaille, joilla on välitön, henkeä uhkaava GPP:n pahenemisvaihe tai tehohoitoa edellyttävä pahenemisvaihe.
Samanaikainen käyttö muiden GPP-hoitojen kanssa
Spesolimabin turvallisuutta ja tehoa yhteiskäytössä immunosuppressanttien kanssa, biologiset lääkkeet mukaan lukien, ei ole systemaattisesti arvioitu (ks. kohta Yhteisvaikutukset). GPP:n pahenemisvaiheen hoitoa koskevassa kliinisessä tutkimuksessa useimmat muut hoidot lopetettiin lääkityskatkon (washout) jälkeen (biologiset lääkkeet, muut systeemiset immuunivastetta muuntavat hoidot), kun taas osa hoidoista lopetettiin ennen spesolimabihoidon aloittamista ilman lääkityskatkoa (metotreksaatti, siklosporiini, retinoidit, paikallishoidot) (ks. kohta Farmakodynamiikka).
Spesolimabin käyttöä samanaikaisesti muiden immunosuppressanttien kanssa ei suositella. Kun spesolimabihoito aloitetaan, muut GPP-hoidot on lopetettava, ja pahenemisvaiheissa ei pidä käyttää samanaikaisesti muita hoitoja (esim. systeemisiä immunosuppressantteja).
Uusintahoito
Spesolimabi-uusintahoidon tehosta ja turvallisuudesta uusien pahenemisvaiheiden hoidossa on saatavilla hyvin vähän tietoa. Effisayil 1 -tutkimuksessa viisi potilasta sai uusintahoitoa myöhempään uuteen pahenemisvaiheeseen, ja heidän vointiaan seurattiin vähintään 8 viikon ajan.
Rokotukset
Ei tiedetä, vaikuttaako spesolimabi rokotteiden tehoon.
Tietoja ei ole saatavilla elävien rokotteiden seurauksena mahdollisesti tulleista infektioista spesolimabia saavilla potilailla (ks. kohta Yhteisvaikutukset). Elävien rokotteiden antamisen ja spesolimabihoidon aloittamisen välillä on oltava vähintään 4 viikon tauko. Eläviä rokotteita ei saa antaa ainakaan 16 viikkoon spesolimabihoidon jälkeen.
Lisätietoa rokotuksista ennen GPP:n pahenemisvaiheiden ehkäisyyn annettavaa hoitoa, ks. Spevigo 150 mg injektioneste, liuos, esitäytetty ruisku valmisteyhteenveto.
Perifeerinen neuropatia
Ei tiedetä, voiko spesolimabi aiheuttaa perifeeristä neuropatiaa. Spesolimabilla tehdyissä kliinisissä tutkimuksissa on raportoitu perifeerisiä neuropatiatapauksia. Lääkärien on tarkkailtava potilaita ensimmäistä kertaa ilmenevään perifeeriseen neuropatiaan viittaavien oireiden varalta.
Apuaineet, joiden vaikutus tunnetaan
Polysorbaatit
Tämä lääkevalmiste sisältää 3 mg polysorbaatti 20:tä per 7,5 ml:n injektiopullo. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. GPP-potilailla spesolimabin ei odoteta aiheuttavan sytokiinivälitteisiä CYP-yhteisvaikutuksia.
Eläviä rokotteita ei saa antaa samanaikaisesti spesolimabin kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Spesolimabin ja immunosuppressanttien samanaikaisesta käytöstä GPP-potilaille on vain vähän kokemusta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus ja imetys
Raskaus
Spesolimabin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja. Ei-kliinisissä tutkimuksissa, joissa spesolimabin sijaan käytettiin hiirispesifistä IL-36R:n monoklonaalista vasta-ainetta, ei havaittu suoria eikä epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Ihmisen immunoglobuliinin (IgG) tiedetään läpäisevän istukan. Varmuuden vuoksi spesolimabin käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö spesolimabi ihmisillä äidinmaitoon. Ihmisillä IgG-vasta-aineita erittyy maitoon muutamien päivien ajan synnytyksen jälkeen, ja pitoisuudet laskevat pieniksi pian sen jälkeen. Näin ollen IgG-vasta-aineiden kulkeutumista vastasyntyneeseen äidinmaidon kautta voi tapahtua näiden ensimmäisten päivien aikana. Tämän lyhyen jakson aikana imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. Sen jälkeen spesolimabia voi käyttää imetyksen aikana, jos se on kliinisesti tarpeen. Jos hoito on keskeytetty ennen raskauden viimeistä kolmannesta, imetys voidaan aloittaa heti lapsen syntymän jälkeen.
Hedelmällisyys
Ei ole olemassa tietoja spesolimabin vaikutuksesta ihmisen hedelmällisyyteen. Hiirillä tehdyissä tutkimuksissa, joissa spesolimabin sijaan käytettiin hiirispesifistä IL-36R:n monoklonaalista vasta-ainetta, ei ole havaittu IL-36R-salpauksesta johtuvia suoria tai epäsuoria vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Spevigo-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin raportoituja haittavaikutuksia olivat infektiot (17,1 %); yhdellä potilaalla raportoitiin vakava virtsatieinfektio (2,9 %) (ks. Valikoitujen haittavaikutusten kuvaus).
Haittavaikutustaulukko
Taulukossa 1 on luettelo kliinisissä tutkimuksissa sekä markkinoille saattamisen jälkeen ilmoitetuista haittavaikutuksista. Haittavaikutukset on luokiteltu MedDRA-elinjärjestelmäluokan ja esiintyvyyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1: Haittavaikutukset
Elinjärjestelmäluokka | Haittavaikutukset | Esiintyvyydet |
|---|---|---|
Infektiot | Infektioa) | Hyvin yleinen |
Immuunijärjestelmä | Yliherkkyysb) | Tuntematon |
Iho ja ihonalainen kudos | Kutina | Yleinen |
Yleisoireet ja antopaikassa todettavat haitat | Pistoskohdan reaktiot | Hyvin yleinenc) |
Väsymys | Yleinen |
a) Yleisimmin raportoituja infektioita olivat virtsatieinfektio (yleinen) ja ylähengitystieinfektio (hyvin yleinen)
b) Tiedot saatu avoimista jatkotutkimuksista ja markkinoille saattamisen jälkeisistä kokemuksista
c) Ei raportoitu Effisayil 1 -tutkimuksessa
Valikoitujen haittavaikutusten kuvaus
Infektiot
Effisayil 1 -tutkimuksessa viikon kestäneen lumekontrolloidun jakson aikana infektioita raportoitiin 17,1 %:lla spesolimabia saaneista potilaista ja 5,6 %:lla lumelääkettä saaneista potilaista. Effisayil 1 ‑tutkimuksenspesolimabiryhmässä yhdellä potilaalla (2,9 %) raportoitiin vakava infektio (virtsatieinfektio), kun taas lumeryhmässä ei raportoitu yhtään vakavaa infektiota. Effisayil 2 ‑tutkimuksen enintään 48 viikkoa kestäneen lumekontrolloidun jakson aikana infektioita raportoitiin 33,3 %:lla Spevigo-valmistetta saaneista potilaista ja 33,3 %:lla lumelääkettä saaneista potilaista. Effisayil 2 ‑tutkimuksen Spevigo-ryhmässä kolmella potilaalla (3,2 %) raportoitiin vakavia infektioita, kun taas lumeryhmässä ei raportoitu yhtään vakavaa infektiota. Spesolimabilla tehdyissä kliinisissä tutkimuksissa todetut infektiot olivat yleensä lieviä tai keskivaikeita, eikä patogeenien tai infektiotyyppien suhteen ollut nähtävissä mitään selkeää kaavaa.
Yliherkkyys
Yliherkkyys sisältää välittömät systeemiset yliherkkyysreaktiot, mukaan lukien anafylaktinen reaktio. Välittömiä systeemisiä yliherkkyysreaktioita on raportoitu avoimissa jatkotutkimuksissa ja markkinoille saattamisen jälkeen.
Pistoskohdan reaktiot
Pistoskohdan reaktioita ovat punoitus, turvotus, kipu, kovettuma, kuumotus, hilseily, papula, kutina, ihottuma ja nokkosihottuma pistoskohdassa. Pistoskohdan reaktiot olivat tyypillisesti lieviä tai keskivaikeita.
Pediatriset potilaat
Saatavilla olevat tiedot nuorista ovat vähäisiä. Effisayil 2 -tutkimukseen osallistui kahdeksan 14–17-vuotiasta nuorta, joilla oli GPP (ks. kohta Farmakodynamiikka). Yleisesti ottaen spesolimabihoitoa saaneiden nuorten (n = 6) turvallisuusprofiili vastasi aikuisten turvallisuusprofiilia, eikä uusia turvallisuutta koskevia huolenaiheita ole tunnistettu.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‐haitta‐tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Suurin kliinisissä tutkimuksissa annettu spesolimabiannos oli 1 200 mg laskimoon tai ihon alle. Haittavaikutukset, joita todettiin 1 200 mg kerta-annoksena tai toistuvasti saaneilla tutkittavilla, olivat yhdenmukaisia spesolimabin tunnetun turvallisuusprofiilin kanssa.
Yliannostustapauksessa suositellaan potilaan seurantaa haittavaikutusten merkkien tai oireiden varalta ja oireenmukaisen hoidon antamista tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: immunosuppressantit, interleukiinin estäjät, ATC-koodi: L04AC22
Vaikutusmekanismi
Spesolimabi on humanisoitu antagonistinen monoklonaalinen immunoglobuliini G1:n (IgG1) vasta-aine, joka estää interleukiini 36 -reseptorin (IL36R) signaalinvälitystä ihmisillä. Spesolimabin sitoutuminen IL36R:ään estää IL36R:n aktivaation sen ligandien (IL36 α, β ja γ) vaikutuksesta ja siten myös alavirran puoleisten proinflammatoristen reittien aktivaation.
Farmakodynaamiset vaikutukset
Laskimoon annetun spesolimabihoidon jälkeen GPP-potilaiden seerumissa ja ihossa todettiin viikolla 1 C‑reaktiivisen proteiinin (CRP), IL6:n, auttaja-T‑soluvälitteisten (Th1/Th17) sytokiinien, keratinosyyttivälitteisen tulehduksen merkkiaineiden, neutrofiilisten välittäjäaineiden ja proinflammatoristen sytokiinien määrän vähenemistä lähtötilanteesta sekä tähän liittyvää sairauden kliinisen vaikeusasteen lievittymistä. Näiden biomarkkerien väheneminen oli voimakkaampaa Effisayil 1 -tutkimuksen viimeisessä mittauksessa viikolla 8.
Kliininen teho ja turvallisuus
Effisayil 1 (1368‑0013)
Satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa (Effisayil 1) arvioitiin spesolimabin kliinistä tehoa ja turvallisuutta aikuispotilailla, joilla esiintyi European Rare And Severe Psoriasis Expert Network (ERASPEN) -asiantuntijaverkoston kriteerien mukaan diagnosoituja yleistyneen pustulaarisen psoriaasin (GPP) pahenemisvaiheita IL36RN-mutaatiostatuksesta riippumatta. Potilaat satunnaistettiin tutkimukseen, jos heillä oli yleistynyttä pustulaarista psoriaasia koskevan lääkärin yleisarvion (GPPGA) kokonaispisteiden (vaihteluväli: 0 [terve] – 4 [vaikea]) mukaan määritelty keskivaikea tai vaikea GPP:n pahenemisvaihe, jonka vaikeusaste oli vähintään 3 (keskivaikea) ja jos heillä oli myös tuoreita pusteleita (uusia pusteleita tai pustelien pahenemista), GPPGA:n pustulaatio-osion pistemäärä vähintään 2 (lievä) ja eryteemaa ja pusteleita vähintään 5 % kehon pinta-alasta. Potilaiden oli lopetettava GPP:n systeemiset hoidot ja paikallishoidot ennen satunnaistamista (ks. taulukko 2). Potilaita, joilla oli välittömästi henkeä uhkaava GPP:n pahenemisvaihe tai jotka tarvitsivat tehohoitoa, suljettiin pois tutkimuksesta.
Taulukko 2: GPP:n hoidossa rajoitettujen lääkkeiden lopettamisesta satunnaistamiseen kulunut vähimmäisaika (Effisayil 1)*
| Lääkityskatkon pituus | Lääkkeet tai lääkeluokat |
| 2 kuukautta | adalimumabi, alemtutsumabi, briakinumabi, brodalumabi, efalitsumabi, guselkumabi, infliksimabi, iksekitsumabi, natalitsumabi, risankitsumabi, rituksimabi, sekukinumabi, tildrakitsumabi, ustekinumabi, visilitsumabi, tutkimusvaiheessa olevat psoriaasilääkkeet (ei-biologiset) |
| 6 viikkoa | etanersepti |
| 30 vuorokautta | systeemiset immuunivastetta muuntavat hoidot (esim. kortikosteroidit**, siklofosfamidi), tofasitinibi, apremilasti; muut systeemiset psoriaasihoidot (esim. fumaraatit); mikä tahansa tutkimuslaite tai -valmiste (paitsi psoriaasivalmisteet); fotokemoterapia (esim. PUVA); granulosyytit ja monosyytit adsorptoiva afereesi |
| 7 vuorokautta | anakinra |
* Ei hoidon aloitusta, satunnaistamista edeltävän viikon aikana: valohoito (esim. UVA, UVB), psoriaasin tai muiden iho-ongelmien paikallishoito (esim. paikalliset kortikosteroidit, paikalliset D vitamiinianalogit, terva, antraliini, paikalliset retinoidit); ei hoidon aloitusta satunnaistamista edeltävien kahden viikon aikana, ei annoksen suurentamista satunnaistamista edeltävien kahden viikon aikana, ja hoito lopetettava ennen ensimmäisen annoksen antoa: metotreksaatti, siklosporiini, retinoidit.
** Ei rajoituksia astman hoidossa käytettäville inhaloitaville kortikosteroideille eikä silmään tai korvaan annosteltaville kortikosteroiditipoille.
Tutkimuksen ensisijainen päätetapahtuma oli niiden potilaiden osuus, joiden GPPGA:n pustulaatio-osion pistemäärä oli 0 (ei näkyviä pusteleita) viikolla 1 hoidon jälkeen. Tutkimuksen tärkein toissijainen päätetapahtuma oli niiden potilaiden osuus, joiden GPPGA-kokonaispistemäärä oli 0 tai 1 (terve tai lähes terve iho) viikolla 1. GPPGA:n pustulaatio-osion pistemäärään 0 ja GPPGA-kokonaispistemäärään 0/1 sovellettiin hoitoon vastaamattomien imputointia kohtauslääkkeen (tutkijan päätöksestä annettu hoito taudin pahentuessa) tai varalääkkeen (900 mg spesolimabia kerta-annoksena laskimoon) käytön ja puuttuvien tietojen huomioimiseksi.
Yhteensä 53 potilasta satunnaistettiin (2:1) saamaan 900 mg:n kerta-annos spesolimabia laskimoon (n = 35) tai lumelääkettä (n = 18). Kumpaan tahansa hoitoryhmään kuuluville potilaille, joilla oli pahenemisvaiheen oireita vielä viikon 1 kohdalla, voitiin antaa avoimesti 900 mg:n kerta-annos spesolimabia laskimoon. Tämän seurauksena 12 spesolimabiryhmän potilasta (34 %) sai toisen spesolimabiannoksen ja 15 lumeryhmän potilasta (83 %) sai yhden spesolimabiannoksen päivänä 8. Lisäksi 6 potilasta (4 spesolimabiryhmässä ja 2 lumeryhmässä) sai 900 mg:n kerta-annoksen spesolimabia laskimoon pahenemisvaiheen hoitona päivän 8 jälkeen uusiutuneen pahenemisvaiheen takia.
Tutkimuspopulaation potilaista 32 % oli miehiä ja 68 % naisia. Potilaiden keskimääräinen ikä oli 43 (vaihteluväli 21–69) vuotta; potilaista 55 % oli aasialaisia ja 45 % valkoihoisia. Useimpien tutkimukseen osallistuneiden potilaiden GPPGA:n pustulaatio-osion pistemäärä oli 3 (43 %) tai 4 (36 %) ja GPPGA-kokonaispistemäärä 3 (81 %) tai 4 (19 %). Potilaista 24,5 % oli saanut aiemmin biologista hoitoa GPP:hen.
Ensisijaiset ja tärkeimmät toissijaiset tehon päätetapahtumat
Viikolla 1 niiden potilaiden osuudessa, jotka saavuttivat GPPGA:n pustulaatio-osion pistemäärän 0 (ei näkyviä pusteleita) ja GPPGA-kokonaispistemäärän 0 tai 1 (terve tai lähes terve iho), oli tilastollisesti merkitsevä ero spesolimabiryhmän ja lumeryhmän välillä (ks. taulukko 3).
Taulukko 3: GPPGA:n pustulaatio-osion pistemäärä ja GPPGA-kokonaispistemäärä viikolla 1 (Effisayil 1)
| Lumelääke | Spesolimabi 900 mg i.v. | |
| Analysoitujen potilaiden lukumäärä | 18 | 35 |
| Potilaat, jotka saavuttivat GPPGA:n pustulaatio-osion pistemäärän 0, n (%) | 1 (5,6) | 19 (54,3) |
| p-arvo* | 0,0004 | |
| Potilaat, jotka saavuttivat GPPGA-kokonaispistemäärän 0 tai 1, n (%) | 2 (11,1) | 15 (42,9) |
| p-arvo* | 0,0118 | |
GPPGA = yleistynyttä pustulaarista psoriaasia koskeva lääkärin yleisarvio; i.v. = laskimoon
*Yksitahoinen p-arvo
Hoitovaikutus todettiin sekä ensisijaisen että tärkeimmän toissijaisen päätetapahtuman osalta kaikilla potilailla IL36RN-mutaatiostatuksesta riippumatta.
Effisayil 2 (1368-0027)
Satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen II b tutkimuksessa (Effisayil 2) arvioitiin ihon alle annettavan spesolimabin tehoa ja turvallisuutta aikuisilla ja nuorilla potilailla, joilla oli anamneesissa ERASPEN-kriteerien mukaan diagnosoitu yleistynyt pustulaarinen psoriaasi (GPP) IL36RN-mutaatiostatuksesta riippumatta ja joilla oli aikaisemmin esiintynyt vähintään kaksi keskivaikeaa tai vaikeaa GPP:n pahenemisvaihetta. Potilaat satunnaistettiin tutkimukseen, jos heidän GPPGA-kokonaispistearvonsa oli 0 tai 1 seulonta- ja satunnaistamisvaiheessa. Potilaiden oli lopetettava GPP:n systeemiset hoidot ja paikallishoidot ennen satunnaistamista. Näillä potilailla oli täytynyt olla aiemmin pahenemisvaihe samanaikaisen GPP-hoidon aikana tai näiden samanaikaisten lääkitysten annoksen pienentämisen tai lopettamisen yhteydessä.
Tutkimuksen ensisijainen päätetapahtuma oli ensimmäiseen GPP:n pahenemisvaiheeseen kulunut aika viikolle 48 asti (määriteltiin GPPGA:n pustulaatio-osion pistemääräksi ≥ 2 ja GPPGA-kokonaispistemäärän suurenemiseksi ≥ 2 pisteellä lähtötilanteesta). Tutkimuksen tärkein toissijainen päätetapahtuma oli vähintään yhden GPP:n pahenemisvaiheen esiintyminen viikkoon 48 mennessä. Muita toissijaisia päätetapahtumia viikolla 48 olivat psoriaasioireasteikon (PSS) ja ihotauteihin liittyvän elämänlaatuindeksin (DLQI) pistemäärien huononemiseen kulunut aika, joka määriteltiin 4 pisteen nousuksi kokonaispistemäärässä lähtötilanteeseen nähden.
Yhteensä 123 potilasta satunnaistettiin (1:1:1:1) saamaan jotakin neljästä hoidosta (ks. taulukko 4).
Taulukko 4: Effisayil 2 -tutkimuksen hoitoryhmät
| Latausannos | Myöhemmät annokset | |
| spesolimabi | 600 mg ihon alle | 300 mg ihon alle 4 viikon välein |
| spesolimabi | 600 mg ihon alle | 300 mg ihon alle 12 viikon välein |
| spesolimabi | 300 mg ihon alle | 150 mg ihon alle 12 viikon välein |
| Lumelääke | ihon alle annettava hoito | ihon alle annettava hoito 4 viikon välein |
Tutkimuspopulaation potilaista 38,2 % oli miehiä ja 61,8 % naisia. Potilaiden keskimääräinen ikä oli 40,4 (vaihteluväli 14–75) vuotta, ja mukana oli 8 (6,5 %) nuorta potilasta (2 kussakin hoitoryhmässä); potilaista 64,2 % oli aasialaisia ja 35,8 % valkoihoisia. Tutkimukseen osallistuneiden potilaiden GPPGA:n pustulaatio-osion pistemäärä oli 1 (28,5 %) tai 0 (71,5 %) ja GPPGA-kokonaispistemäärä 1 (86,2 %) tai 0 (13,8 %). Satunnaistamisvaiheessa 74,8 % potilaista sai systeemistä GPP-hoitoa, joka keskeytettiin satunnaistetun tutkimushoidon alkaessa.
Effisayil 2 -tutkimuksessa tutkittiin kolmea annostusta, mutta GPP:n pahenemisvaiheiden ehkäisyyn suositeltu annostus on 600 mg:n latausannos spesolimabia ihon alle ja sen jälkeen 300 mg ihon alle 4 viikon välein (ks. kohta Annostus ja antotapa). Tulokset, joiden yhteenveto on esitetty alla, koskevat suositeltua annostusta.
Potilaat, joilla esiintyi pahenemisvaihe, soveltuivat saamaan avoimesti enintään kaksi 900 mg:n annosta spesolimabia laskimoon (ks. kohta Annostus ja antotapa). Kaksi (6,7 %) potilasta suositeltua spesolimabiannosta saaneessa ryhmässä ja 15 (48,4 %) potilasta lumeryhmässä sai laskimonsisäistä hoitoa pahenemisvaiheeseen.
Hoito suositellulla spesolimabiannoksella sai aikaan tilastollisesti merkitseviä parannuksia ensisijaisessa ja tärkeimmässä toissijaisessa päätetapahtumassa lumelääkkeeseen verrattuna (ks. taulukko 5).
Taulukko 5: Ensimmäiseen GPP:n pahenemisvaiheeseen kulunut aika ja vähintään yhden GPP:n pahenemisvaiheen esiintyminen viikkoon 48 mennessä (Effisayil 2)
| Lumelääke | Suositeltu spesolimabiannos | |
| Analysoitujen potilaiden lukumäärä, N | 31 | 30 |
| Potilaat, joilla esiintyi GPP:n pahenemisvaiheita, N (%)* | 16 (51,6) | 3 (10,0) |
| Riskisuhde (HR)** ensimmäiseen pahenemisvaiheeseen kuluneelle ajalle vs. lumelääke (95 %:n luottamusväli) | 0,16 (0,05; 0,54) | |
| p-arvo*** | 0,0005 | |
| Ero GPP:n pahenemisvaiheen esiintymisriskissä vs. lumelääke (95 %:n luottamusväli) | -39,0 % (-62,1; -15,9) | |
| p-arvo**** | 0,0013 | |
* Laskimoon annettavan spesolimabihoidon tai tutkijan määräämän tavanomaisen hoidon käyttö GPP:n pahenemisen vuoksi katsottiin GPP:n pahenemisvaiheen alkamiseksi
** Coxin regressiomalli, joka stratifioitiin satunnaistamisvaiheessa käytettyjen systeemisten GPP-lääkitysten perusteella
*** Log-rank-testi, joka stratifioitiin satunnaistamisvaiheessa käytettyjen systeemisten GPP-lääkitysten perusteella, yksitahoinen p-arvo
**** Cochran-Mantel-Haenszelin testi moni-imputaation jälkeen, stratifioitiin satunnaistamisvaiheessa käytettyjen systeemisten GPP-lääkitysten perusteella, yksitahoinen p-arvo
Ihon alle annettavan suositellun spesolimabiannoksen teho lumelääkkeeseen verrattuna todettiin pian satunnaistamisen jälkeen, ja se säilyi viikolle 48 asti (ks. kuva 1).
Kuva 1: Ensimmäiseen GPP:n pahenemisvaiheeseen kulunut aika viikolle 48 asti (Effisayil 2)
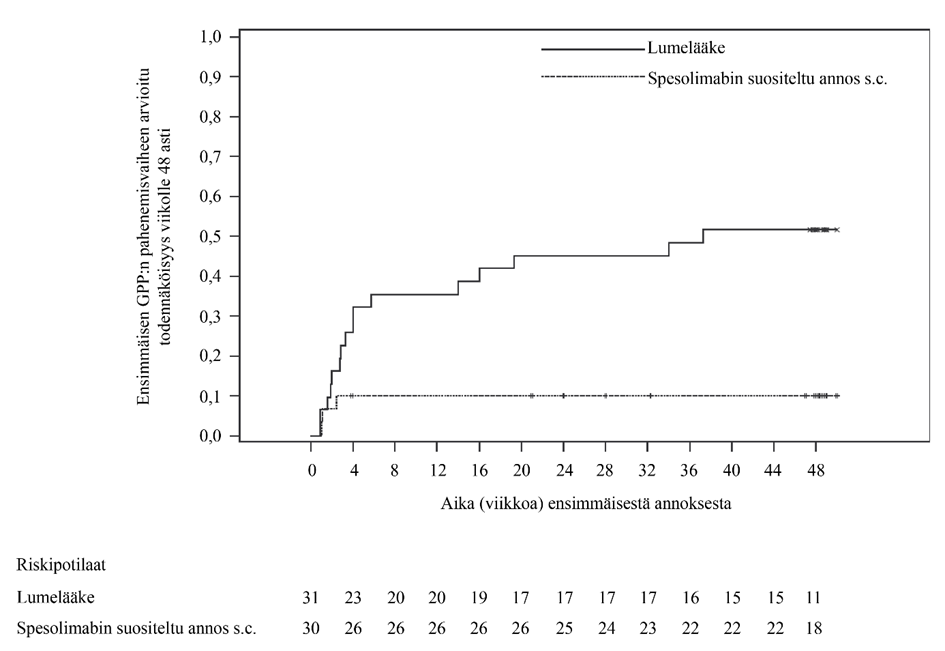
Sekä ensisijaisen että tärkeimmän toissijaisen päätetapahtuman kohdalla hoidon vaikutus oli todettavissa kaikilla potilailla IL36RN-mutaatiostatuksesta riippumatta.
Yksi lumeryhmän nuorista potilaista sai tutkijan määräämää tavanomaista hoitoa GPP:n pahenemisen vuoksi, ja tämä tulkittiin GPP:n pahenemisvaiheeksi. GPP:n pahenemisvaiheita ei esiintynyt yhdelläkään nuorella potilaalla suositeltua spesolimabiannosta saaneessa ryhmässä.
GPP:n pahenemisen estymistä todettiin myös PSS- ja DLQI-tuloksina mitattuna, sillä PSS-riskisuhde oli 0,42 (95 %:n luottamusväli 0,20; 0,91) ja DLQI-riskisuhde 0,26 (95 %:n luottamusväli 0,11; 0,62).
Immunogeenisuus
Effisayil 1 -tutkimuksessa laskimonsisäistä spesolimabihoitoa saaneilla GPP-potilailla lääkevasta-aineita kehittyi 46 %:lle potilaista. Suurimmalle osalle lääkevasta-ainepositiivisista potilaista kehittyi myös neutraloivia vasta-aineita. Effisayil 2 tutkimuksessa 41 %:lle potilaista kehittyi lääkevasta-aineita useiden ihon alle annettujen spesolimabiannosten jälkeen. Suurimmalle osalle lääkevasta-ainepositiivisista potilaista kehittyi myös neutraloivia vasta-aineita. Spesolimabin puhdistuma suureni ADA-titterien suurenemisen myötä.
Koska suurimmalla osalla potilaista ei esiintynyt myöhempiä uusia pahenemisvaiheita Effisayil 1 tutkimuksessa, uusintahoidosta potilailla, joilla oli lääkevasta-aineita (n = 4), on vain vähän tietoa. Tällä hetkellä ei tiedetä, onko spesolimabivasta-aineiden ja pahenemisvaiheiden hoidon tehon säilymisen välillä korrelaatiota. Kun spesolimabia annettiin ihon alle Effisayil 2 tutkimuksessa, lääkevasta-aineilla ei todettu ilmeistä vaikutusta tehoon tai turvallisuuteen.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Spevigo-valmisteen käytöstä yleistyneen pustulaarisen psoriaasin hoidossa alle 12 vuoden ikäisillä pediatrisilla potilailla (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Ehdollinen myyntilupa
Tämä lääkevalmiste on saanut ns. ehdollisen myyntiluvan. Se tarkoittaa, että lääkevalmisteesta odotetaan uutta tietoa. Euroopan lääkevirasto arvioi vähintään kerran vuodessa tätä lääkevalmistetta koskevat uudet tiedot, ja tarvittaessa tämä valmisteyhteenveto päivitetään.
Farmakokinetiikka
Terveistä henkilöistä, GPP-potilaista ja muita tauteja sairastavista potilaista kerättyjen tietojen perusteella laadittiin populaatiofarmakokineettinen malli. Laskimoon annetun 900 mg:n kerta-annoksen jälkeen populaatiofarmakokineettisellä mallilla arvioitu tyypillisen ADA-negatiivisen GPP-potilaan AUC0‑∞-arvo (95 %:n lv) oli 4 750 (4 510, 4 970) µg·vrk/ml ja Cmax-arvo (95 %:n lv) oli 238 (218, 256) µg/ml. Kun potilaille annettiin ihon alle 600 mg:n latausannos spesolimabia ja sen jälkeen ihon alle 300 mg spesolimabia 4 viikon välein, keskimääräinen (CV%) pienin pitoisuus vakaassa tilassa vaihteli välillä 33,4 µg/ml (37,6 %) ja 42,3 µg/ml (43,0 %).
Imeytyminen
Kun terveille vapaaehtoisille annettiin ihon alle kerta-annos spesolimabia, huippupitoisuudet plasmassa saavutettiin 5,5–7,0 vuorokauden kuluttua annoksen antamisesta. Kun lääke annettiin vatsan ihon alle, absoluuttinen biologinen hyötyosuus oli hieman korkeampi käytettäessä suurempia annoksia, arviolta 58 % annoksella 150 mg, 65 % annoksella 300 mg ja 72 % annoksella 600 mg. Vähäisten tietojen perusteella reiden ihon alle annetun spesolimabin absoluuttinen biologinen hyötyosuus oli noin 85 % annoksella 300 mg.
Jakautuminen
Populaatiofarmakokineettisen analyysin perusteella tyypillinen vakaan tilan jakautumistilavuus oli 6,4 l.
Biotransformaatio
Spesolimabin metaboliareittejä ei ole luonnehdittu. Spesolimabi on humanisoitu IgG1:n monoklonaalinen vasta-aine, joten sen oletetaan hajoavan pieniksi peptideiksi ja aminohapoiksi katabolisten reittien kautta samalla tavalla kuin endogeeninen IgG.
Eliminaatio
Populaatiofarmakokineettisen mallin perusteella lineaarisella annosalueella (0,3–20 mg/kg) spesolimabin puhdistuma (95 %:n lv) 70 kg painavalla tyypillisellä ADA-negatiivisella GPP-potilaalla oli 0,184 l/vrk. Terminaalinen puoliintumisaika oli 25,5 päivää.
Lineaarisuus/ei-lineaarisuus
Laskimoon annettuna spesolimabin farmakokinetiikan todettiin olevan lineaarista ja altistuksen suurenevan suhteessa annokseen kerta-annoksen ollessa 0,3–20 mg/kg. Sekä puhdistuma (CL) että terminaalinen puoliintumisaika olivat annoksesta riippumattomia. Ihon alle annetun kerta-annoksen jälkeen spesolimabialtistus suureni hieman enemmän kuin suhteessa annokseen annosalueella 150–600 mg. Tämä johtuu siitä, että biologinen hyötyosuus on hieman suurempi käytettäessä suurempia annoksia.
Paino
Spesolimabin pitoisuudet olivat pienempiä painavammilla tutkittavilla ja suurempia kevyemmillä tutkittavilla. Spesolimabia ei ole tutkittu yli 164 kg painavilla GPP-potilailla. Farmakokineettisen mallinnuksen ja simulaation perusteella 12 vuoden ikäisille, ≥ 30 ja < 40 kg painaville nuorille suositeltu annos on puolet aikuisille ja vähintään 12 vuoden ikäisille ja vähintään 40 kg painaville nuorille suositellusta annoksesta (ks. kohta Annostus ja antotapa). Pienempää annosta saavilla ≥ 30 ja < 40 kg painavilla potilailla altistuksen odotetaan olevan verrattavissa GPP-tutkimuksissa todettuihin altistuksiin.
Ikä/sukupuoli/rotu
Populaatiofarmakokineettisten analyysien perusteella ikä, sukupuoli ja rotu eivät vaikuta spesolimabin farmakokinetiikkaan kliinisesti merkittävästi.
Maksan ja munuaisten vajaatoiminta
Spesolimabi on monoklonaalinen vasta-aine, joten sen ei odoteta eliminoituvan maksan tai munuaisten kautta. Maksan tai munuaisten vajaatoiminnan vaikutuksesta spesolimabin farmakokinetiikkaan ei ole tehty muodollisia tutkimuksia.
Populaatiofarmakokineettisessä analyysissa lievän maksan vajaatoiminnan tai lievän tai keskivaikean munuaisten vajaatoiminnan ei todettu vaikuttavan systeemiseen spesolimabialtistukseen.
Pediatriset potilaat
Spesolimabin farmakokinetiikkaa ei ole vielä tutkittu alle 14 vuoden ikäisillä pediatrisilla potilailla.
Nuorilla todettu spesolimabin farmakokinetiikka plasmassa oli yhdenmukaista aikuisilla tehtyjen havaintojen kanssa.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Kehitys- ja lisääntymistoksisuus
Hiirillä tehdyissä ei-kliinisissä tutkimuksissa, joissa käytettiin hiiren IL-36R:ään kohdistuvaa monoklonaalista vasta-ainetta, ei ole havaittu suoria tai epäsuoria haitallisia vaikutuksia tiineyteen, alkion- tai sikiönkehitykseen tai hedelmällisyyteen.
Genotoksisuus
Spesolimabilla ei ole tehty genotoksisuustutkimuksia.
Karsinogeenisuus
Spesolimabilla ei ole tehty karsinogeenisuus- eikä mutageenisuustutkimuksia.
Farmaseuttiset tiedot
Apuaineet
Natriumasetaattitrihydraatti (E262)
Etikkahappo, väkevä (E260) (pH:n säätöön)
Sakkaroosi
Arginiinihydrokloridi
Polysorbaatti 20 (E432)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta.
Avaamisen jälkeen
Mikrobiologiselta kannalta lääkevalmiste tulisi laimentaa ja antaa infuusiona välittömästi avaamisen jälkeen.
Infuusion valmistelun jälkeen
Laimennetun liuoksen kemiallinen ja fysikaalinen käytönaikainen stabiliteetti on osoitettu 24 tunnin ajalta 2 °C – 30 °C:n lämpötilassa.
Mikrobiologiselta kannalta laimennettu infuusioliuos tulisi käyttää välittömästi. Jos sitä ei käytetä välittömästi, käytönaikaiset säilytysolosuhteet ovat käyttäjän vastuulla eivätkä normaalisti saa ylittää 24 tuntia 2 °C – 8 °C:n lämpötilassa, ellei laimennusta ole tehty kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa. Valmistelun ja infuusion aloittamisen välisenä aikana infuusioneste on suojattava valolta tavanomaisten paikallisten käytäntöjen mukaisesti.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Ennen käyttöä avaamatonta injektiopulloa voidaan säilyttää enintään 30 °C:n lämpötilassa alkuperäispakkauksessa valolta suojattuna enintään 24 tunnin ajan.
Avatun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SPEVIGO infuusiokonsentraatti, liuosta varten
450 mg (L:ei) 2 x 7,5 ml (60 mg/ml) (18340,60 €)
PF-selosteen tieto
7,5 ml konsentraattia värittömästä lasista (tyypin I lasista) valmistetussa 10 ml:n injektiopullossa, jossa on päällystetty kumitulppa ja alumiininen puristuskorkki, jossa on sininen muovipainike.
Pakkauskoko on 2 injektiopulloa.
Valmisteen kuvaus:
Kirkas tai hieman opalisoiva, väritön tai hieman rusehtavan keltainen liuos.
Käyttö- ja käsittelyohjeet
Tämä lääkevalmiste on yhteensopiva polyvinyylikloridista (PVC), polyeteenistä (PE), polypropeenista (PP), polybutadieenista ja polyuretaanista (PUR) valmistettujen infuusioletkujen ja polyeetterisulfonista (PES, neutraali ja positiivisesti varautunut) ja positiivisesti varautuneesta polyamidista (PA) valmistettujen letkunsisäisten suodatinkalvojen kanssa.
Käsittelyohjeet
- Injektiopullo on tarkistettava silmämääräisesti ennen käyttöä. Jos liuos on sameaa, siinä on värimuutoksia tai se sisältää suuria tai värillisiä hiukkasia, injektiopullo on hävitettävä.
- Spevigo on tarkoitettu vain yhtä käyttökertaa varten.
-
Infuusionesteen valmistelussa on käytettävä aseptista tekniikkaa.
o 900 mg:n suositeltu annos: poista ruiskulla 100 ml:n natriumkloridi 9 mg/ml (0,9 %) injektionestepakkauksesta 15 ml nestettä ja lisää tilalle 15 ml steriiliä spesolimabikonsentraattia (kaksi 450 mg / 7,5 ml:n injektiopulloa) hitaasti injektionestepakkaukseen ruiskuttamalla.
o 450 mg:n suositeltu annos: poista ruiskulla 100 ml:n natriumkloridi 9 mg/ml (0,9 %) injektionestepakkauksesta 7,5 ml nestettä ja lisää tilalle 7,5 ml steriiliä spesolimabikonsentraattia (yksi 450 mg / 7,5 ml:n injektiopullo) hitaasti injektionestepakkaukseen ruiskuttamalla.
o Sekoita varovasti ennen käyttöä. Laimennettu spesolimabi-infuusioneste on käytettävä välittömästi.
- Spevigo-valmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa. Aiemmin asetettua infuusioletkua voidaan käyttää laimennetun spesolimabi-infuusionesteen antamiseen, kunhan edellä mainitut yhteensopivuutta koskevat seikat otetaan huomioon. Infuusioletku on huuhdeltava 9 mg/ml (0,9 %) natriumkloridi-injektionesteellä ennen infuusiota ja infuusion lopussa. Saman laskimoyhteyden kautta ei saa antaa samanaikaisesti muita infuusioita.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SPEVIGO infuusiokonsentraatti, liuosta varten
450 mg 2 x 7,5 ml
- Ei korvausta.
ATC-koodi
L04AC22
Valmisteyhteenvedon muuttamispäivämäärä
22.01.2026
Yhteystiedot
Karhumäentie 3
01530 Vantaa
020 721 8440
www.leo-pharma.fi
info.fi@leo-pharma.com