
BIMZELX injektioneste, liuos, esitäytetty kynä 160 mg, 320 mg, injektioneste, liuos, esitäytetty ruisku 160 mg
Vaikuttavat aineet ja niiden määrät
Bimzelx 160 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 160 mg bimekitsumabia 1 ml:ssa liuosta.
Bimzelx 320 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 320 mg bimekitsumabia 2 ml:ssa liuosta.
Bimzelx 160 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kynä sisältää 160 mg bimekitsumabia 1 ml:ssa liuosta.
Bimzelx 320 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kynä sisältää 320 mg bimekitsumabia 2 ml:ssa liuosta.
Bimekitsumabi on humanisoitu monoklonaalinen IgG1-vasta-aine, joka tuotetaan geneettisesti muunnellussa kiinanhamsterin munasarjasolulinjassa (CHO) yhdistelmä-DNA-tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste).
Kliiniset tiedot
Käyttöaiheet
Läiskäpsoriaasi
Bimzelx on tarkoitettu keskivaikean tai vaikean läiskäpsoriaasin hoitoon aikuispotilaille, joille harkitaan systeemistä hoitoa.
Nivelpsoriaasi
Bimzelx on tarkoitettu yksinään tai yhdistelmänä metotreksaatin kanssa aktiivisen nivelpsoriaasin hoitoon aikuispotilaille, joilla on ollut riittämätön vaste yhteen tai useampaan perinteiseen reumalääkkeeseen (DMARD) tai jotka eivät ole sietäneet niitä.
Aksiaalinen spondylartriitti
Aksiaalinen spondylartriitti ilman radiografista näyttöä selkärankareumasta (röntgennegatiivinen aksiaalinen spondylartriitti)
Bimzelx on tarkoitettu aktiivisen röntgennegatiivisen aksiaalisen spondylartriitin hoitoon aikuispotilaille, joilla on objektiivisia tulehduksen merkkejä, kuten kohonnut C-reaktiivisen proteiinin (CRP) pitoisuus ja/tai magneettikuvauksessa (MRI) todettuja löydöksiä, silloin kun tulehduskipulääkkeillä (NSAID) ei ole saatu riittävää vastetta tai kun potilas ei siedä niitä.
Selkärankareuma (röntgenpositiivinen aksiaalinen spondylartriitti)
Bimzelx on tarkoitettu aktiivisen selkärankareuman hoitoon aikuispotilaille, jotka eivät ole saaneet riittävää vastetta tavanomaisella hoidolla tai jotka eivät ole sietäneet tavanomaista hoitoa.
Hidradenitis suppurativa (HS-tauti)
Bimzelx on tarkoitettu aktiivisen keskivaikean tai vaikean hidradenitis suppurativan (märkivän hikirauhastulehduksen, taiveaknen) hoitoon aikuispotilaille, jotka eivät ole saaneet riittävää vastetta tavanomaiseen systeemiseen HS-hoitoon (ks. kohta Farmakodynamiikka).
Ehto
Valmiste on tarkoitettu käytettäväksi käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus ja antotapa
Tämä lääkevalmiste on tarkoitettu käytettäväksi sen käyttöaiheina olevien sairauksien diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja valvonnassa.
Annostus
Läiskäpsoriaasi
Suositeltu annos läiskäpsoriaasia sairastaville aikuispotilaille on 320 mg (annetaan kahtena 160 mg:n injektiona ihon alle tai yhtenä 320 mg:n injektiona ihon alle) viikoilla 0, 4, 8, 12, 16 ja sen jälkeen joka 8. viikko.
Nivelpsoriaasi
Suositeltu annos aktiivista nivelpsoriaasia sairastaville aikuispotilaille on 160 mg (annetaan yhtenä 160 mg:n injektiona ihon alle) joka 4. viikko.
Niille nivelpsoriaasia sairastaville potilaille, joilla on samanaikainen keskivaikea tai vaikea läiskäpsoriaasi, suositeltu annos on sama kuin läiskäpsoriaasin hoidossa (320 mg [kahtena 160 mg:n injektiona ihon alle tai yhtenä 320 mg:n injektiona ihon alle] viikoilla 0, 4, 8, 12, 16 ja sen jälkeen joka 8. viikko). 16 viikon jälkeen suositellaan säännöllistä hoidon tehon arviointia, ja jos riittävää nivelten kliinistä hoitovastetta ei pystytä ylläpitämään, voidaan harkita vaihtamista 160 mg:n annokseen joka 4. viikko.
Aksiaalinen spondylartriitti (röntgennegatiivinen ja röntgenpositiivinen)
Suositeltu annos aksiaalista spondylartriittia sairastaville aikuispotilaille on 160 mg (annetaan yhtenä 160 mg:n injektiona ihon alle) 4 viikon välein.
Hidradenitis suppurativa (HS-tauti)
Suositeltu annos HS-tautia sairastaville aikuispotilaille on 320 mg (annetaan kahtena 160 mg:n injektiona ihon alle tai yhtenä 320 mg:n injektiona ihon alle) 2 viikon välein viikolle 16 saakka ja sen jälkeen 4 viikon välein.
Edellä olevien käyttöaiheiden osalta on harkittava hoidon lopettamista niillä potilailla, joilla ei ilmene ollenkaan oireiden lievittymistä hoitoviikkoon 16 mennessä.
Erityispotilasryhmät
Ylipainoiset läiskäpsoriaasia sairastavat potilaat
Joillekin läiskäpsoriaasia (mukaan lukien nivelpsoriaasi, johon liittyy samanaikaisesti keskivaikea tai vaikea psoriaasi) sairastaville ja ≥ 120 kg:n painoisille potilaille, joiden iho ei ollut viikolla 16 täysin oireeton, 320 mg joka 4. viikko voi edelleen parantaa hoitovastetta viikon 16 jälkeen (ks. kohta Farmakodynamiikka).
Iäkkäät (≥ 65-vuotiaat)
Annoksen muuttaminen ei ole tarpeen (ks. kohta Farmakokinetiikka).
Munuaisten tai maksan vajaatoiminta
Bimekitsumabia ei ole tutkittu näissä potilasryhmissä. Farmakokinetiikan perusteella annoksen muuttamista ei katsota tarpeelliseksi (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Bimekitsumabin turvallisuutta ja tehoa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Tämä lääkevalmiste annetaan injektiona ihon alle. 320 mg:n annos voidaan antaa kahtena 160 mg:n injektiona ihon alle tai yhtenä 320 mg:n injektiona ihon alle.
Sopivia injektioalueita ovat reidet, vatsa ja olkavarsi. Injektiokohtia pitää vaihdella eikä injektioita pidä antaa psoriaasiläiskiin tai alueille, joiden iho on arka, mustelmilla, punoittava tai kovettunut. Valmisteen saa antaa olkavarteen vain terveydenhuollon ammattilainen tai potilasta hoitava henkilö.
Esitäytettyä ruiskua tai esitäytettyä kynää ei saa ravistaa.
Kun potilaat ovat saaneet asianmukaisen ihonalaista injektiotekniikkaa koskevan koulutuksen, he voivat injisoida Bimzelx-valmisteen itse esitäytetyllä ruiskulla tai esitäytetyllä kynällä, jos lääkäri katsoo sen sopivaksi ja lääketieteellinen seuranta voidaan toteuttaa tarpeen mukaan. Potilaita pitää neuvoa injisoimaan täysi määrä Bimzelx-valmistetta pakkausselosteessa olevien käyttöohjeiden mukaisesti.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Kliinisesti merkittävä aktiivinen infektio (esim. aktiivinen tuberkuloosi, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Bimekitsumabi voi lisätä infektioiden, kuten ylähengitysteiden infektioiden ja suun kandidiaasi-infektioiden, riskiä (ks. kohta Haittavaikutukset).
Bimekitsumabin käyttöä pitää harkita tarkoin, jos potilaalla on krooninen infektio tai anamneesissa uusiutuvia infektioita. Jos potilaalla on jokin kliinisesti merkittävä aktiivinen infektio, bimekitsumabihoitoa ei saa aloittaa ennen kuin infektio on parantunut tai hoidettu asianmukaisesti (ks. kohta Vasta-aiheet).
Bimekitsumabihoitoa saaneita potilaita on kehotettava kääntymään lääkärin puoleen, jos heillä on infektioon viittaavia oireita tai löydöksiä. Jos potilaalle kehittyy infektio, potilaan tilannetta pitää seurata huolellisesti. Jos infektio muuttuu vakavaksi tai se ei reagoi tavanomaiseen hoitoon, hoito on keskeytettävä siihen saakka, kun infektio on parantunut.
Tuberkuloosin tutkiminen ennen hoitoa
Ennen bimekitsumabihoidon aloittamista potilaat on tutkittava tuberkuloosi-infektion varalta. Bimekitsumabia ei pidä antaa potilaille, joilla on aktiivinen tuberkuloosi (ks. kohta Vasta-aiheet). Bimekitsumabia saavien potilaiden vointia on seurattava aktiivisen tuberkuloosin oireiden ja löydösten varalta. Jos potilaalla on aiemmin ollut piilevä tai aktiivinen tuberkuloosi eikä asianmukaisen hoitojakson toteutumista pystytä varmistamaan, ennen bimekitsumabihoidon aloittamista pitää harkita tuberkuloosilääkitystä.
Tulehduksellinen suolistosairaus
Bimekitsumabin käytön yhteydessä on raportoitu tulehduksellisen suolistosairauden puhkeamista tai pahenemista (ks. kohta Haittavaikutukset). Bimekitsumabia ei suositella potilaille, joilla on tulehduksellinen suolistosairaus. Jos potilaalle kehittyy tulehduksellisen suolistosairauden oireita ja löydöksiä tai jo ennestään sairastettu tulehduksellinen suolistosairaus pahenee, bimekitsumabihoito pitää lopettaa ja aloittaa asianmukainen hoito.
Yliherkkyys
IL-17:n estäjien käytön yhteydessä on esiintynyt vakavia yliherkkyysreaktioita, mukaan lukien anafylaktisia reaktioita. Jos potilaalle ilmaantuu vakava yliherkkyysreaktio, bimekitsumabin anto pitää lopettaa heti ja aloittaa asianmukainen hoito.
Rokotukset
Ennen bimekitsumabihoidon aloittamista kaikkien asianmukaisten rokotusten antamista pitää harkita voimassa olevien rokotussuositusten mukaisesti.
Bimekitsumabihoitoa saavalle potilaalle ei pidä antaa eläviä taudinaiheuttajia sisältäviä rokotteita.
Bimekitsumabilla hoidetut potilaat voivat saada inaktivoituja eli tapettuja taudinaiheuttajia sisältäviä rokotteita. Terveillä henkilöillä, jotka saivat yhden 320 mg:n annoksen bimekitsumabia kaksi viikkoa ennen inaktivoitua kausi-influenssarokotetta, oli samankaltainen vasta-ainevaste kuin henkilöillä, jotka eivät saaneet bimekitsumabia ennen rokotusta.
Apuaineet, joiden vaikutus tunnetaan
Polysorbaatti 80
Tämä lääkevalmiste sisältää 0,4 mg polysorbaatti 80:tä per 1 ml liuosta. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
IL-17A:n tai IL-17F:n osuudesta CYP450-entsyymien ilmentymisessä ei ole suoraa näyttöä. Joidenkin CYP450-entsyymien muodostuminen vähenee kroonisen tulehduksen aikana kohonneiden sytokiinipitoisuuksien seurauksena. Siksi anti-inflammatoriset lääkkeet, kuten IL-17A:n ja IL-17F:n estäjä bimekitsumabi, voivat johtaa CYP450-tasojen normalisoitumiseen ja siten pienempään altistukseen CYP450:n metaboloimille lääkevalmisteille. Näin ollen kliinisesti oleellista vaikutusta CYP450:n substraatteihin, joiden terapeuttinen indeksi on kapea ja joiden annosta säädetään yksilöllisesti (esim. varfariini), ei voida sulkea pois. Jos potilasta hoidetaan tämäntyyppisillä lääkevalmisteilla, hoidon seurantaa pitää harkita bimekitsumabihoidon aloittamisen yhteydessä.
Populaatiofarmakokineettiset analyysit viittaavat siihen, että samanaikainen perinteisten reumalääkkeiden (cDMARD), mukaan lukien metotreksaatti, antaminen tai aiempi altistus biologisille lääkkeille ei vaikuta kliinisesti merkittävällä tavalla bimekitsumabin puhdistumaan.
Eläviä taudinaiheuttajia sisältäviä rokotteita ei pidä antaa samanaikaisesti bimekitsumabin kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, pitää käyttää tehokasta ehkäisymenetelmää hoidon aikana ja vähintään 17 viikon ajan hoidon jälkeen.
Raskaus
On vain vähän tietoja bimekitsumabin käytöstä raskaana oleville naisille. Eläinkokeissa ei ole havaittu suoria tai epäsuoria haitallisia vaikutuksia tiineyteen, alkion/sikiön kehitykseen, synnytykseen tai syntymän jälkeiseen kehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi Bimzelx-valmisteen käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö bimekitsumabi ihmisen rintamaitoon. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko Bimzelx-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Bimekitsumabin vaikutusta ihmisen hedelmällisyyteen ei ole selvitetty. Eläinkokeissa ei ole havaittu suoria tai epäsuoria hedelmällisyyteen kohdistuvia haittavaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Bimzelx-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin raportoituja haittavaikutuksia olivat ylähengitysteiden infektiot (14,5 % läiskäpsoriaasia sairastavilla, 14,6 % nivelpsoriaasia sairastavilla, 16,3 % aksiaalista spondylartriittia sairastavilla ja 8,8 % HS-tautia sairastavilla) ja suun kandidiaasi-infektio (7,3 % läiskäpsoriaasia sairastavilla, 2,3 % nivelpsoriaasia sairastavilla, 3,7 % aksiaalista spondylartriittia sairastavilla ja 5,6 % HS-tautia sairastavilla).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa ja markkinoille tulon jälkeisissä ilmoituksissa (taulukko 1) havaitut haittavaikutukset on luokiteltu MedDRA-elinjärjestelmäluokan ja esiintymistiheyden mukaan seuraavan esitystavan mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Yhteensä 5 862:ta potilasta on hoidettu bimekitsumabilla läiskäpsoriaasin, nivelpsoriaasin, aksiaalisen spondylartriitin (röntgennegatiivisen ja röntgenpositiivisen) ja HS-taudin sokkoutetuissa ja avoimissa kliinisissä tutkimuksissa. Nämä potilaat edustavat 11 468,6 potilasvuoden altistusta. Näistä potilaista yli 4 660 altistui bimekitsumabille vähintään yhden vuoden ajan. Bimekitsumabin turvallisuusprofiili on kokonaisuutena yhdenmukainen kaikissa sen käyttöaiheissa.
Taulukko 1: Luettelo haittavaikutuksista
| Elinjärjestelmä | Esiintymistiheys | Haittavaikutus |
| Infektiot | Hyvin yleinen | Ylähengitysteiden infektio |
| Yleinen | Suun kandidiaasi-infektio Silsainfektio Korvatulehdukset Herpes simplex -infektiot Suun ja nielun kandidiaasi-infektio Maha-suolitulehdus Karvatuppitulehdus Vulvovaginaalinen sieni-infektio (mukaan lukien vulvovaginaalinen kandidiaasi) | |
| Melko harvinainen | Limakalvojen ja ihon kandidiaasi-infektio (mukaan lukien ruokatorven kandidiaasi-infektio) Sidekalvotulehdus | |
| Veri ja imukudos | Melko harvinainen | Neutropenia |
| Hermosto | Yleinen | Päänsärky |
| Ruoansulatuselimistö | Melko harvinainen | Tulehduksellinen suolistosairaus |
| Iho ja ihonalainen kudos | Yleinen | Ihottuma, dermatiitti ja ekseema Akne |
| Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Injektiokohdan reaktiota Uupumus |
| a) Sisältää: injektiokohdan punoituksen, reaktion, ödeeman, kivun, turvotuksen ja hematooman. | ||
Valikoitujen haittavaikutusten kuvaukset
Infektiot
Vaiheen III kliinisten läiskäpsoriaasitutkimusten lumekontrolloidussa vaiheessa infektioita raportoitiin 36,0 %:lla potilaista, joita oli hoidettu bimekitsumabilla enintään 16 viikon ajan, verrattuna 22,5 %:iin lumelääkettä saaneista potilaista. Vakavia infektioita esiintyi 0,3 %:lla bimekitsumabihoitoa saaneista potilaista ja 0 %:lla lumelääkettä saaneista potilaista.
Suurin osa infektioista oli ei-vakavia, lieviä tai keskivaikeita ylähengitysteiden infektioita, kuten nenänielun tulehduksia. Bimekitsumabihoitoa saaneilla potilailla esiintyi yleisemmin suun ja suunielun kandidiaasi-infektioita (suun kandidiaasi-infektioita 7,3 %:lla ja suunielun kandidiaasi-infektioita 1,2 %:lla verrattuna lumelääkkeellä hoidettuihin potilaisiin, joilla luku oli 0 %). Yli 98 % tapauksista oli ei-vakavia, vaikeusasteeltaan lieviä tai keskivaikeita, eivätkä ne vaatineet hoidon keskeyttämistä. Suun kandidiaasi-infektioiden ilmaantuvuuden raportoitiin olleen < 70 kg painavilla potilailla hieman suurempi (8,5 % verrattuna 7,0 %:iin potilaista, jotka painoivat ≥ 70 kg).
Vaiheen III läiskäpsoriaasitutkimusten koko hoitojakson aikana infektioita raportoitiin 63,2 %:lla bimekitsumabihoitoa saaneista potilaista (120,4 potilasta 100 potilasvuotta kohti). Vakavia infektioita raportoitiin 1,5 %:lla bimekitsumabihoitoa saaneista potilaista (1,6 potilasta 100 potilasvuotta kohti) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Nivelpsoriaasia ja aksiaalista spondylartriittia (röntgennegatiivista ja röntgenpositiivista) sairastavilla potilailla vaiheen III kliinisissä tutkimuksissa havaittujen infektioiden esiintyvyys oli muuten yhtä yleistä kuin läiskäpsoriaasia sairastavilla, mutta suun ja suunielun kandidiaasia esiintyi näillä potilailla bimekitsumabihoitoa saaneiden ryhmässä vähemmän kuin bimekitsumabiryhmässä läiskäpsoriaasia sairastavilla (2,3 % ja 0 % nivelpsoriaasia sairastavilla ja 3,7 % ja 0,3 % aksiaalista spondylartriittia sairastavilla verrattuna lumelääkkeellä hoidettuihin potilaisiin, joilla luku oli 0 %).
Hidradenitis suppurativan (HS-taudin) vaiheen III kliinisissä tutkimuksissa havaitut infektioiden määrät olivat samanlaisia kuin muissa käyttöaiheissa havaitut määrät. Lumelääkekontrolloidun jakson aikana suun kandidiaasia esiintyi 7,1 %:lla ja suun ja nielun kandidiaasia 0 %:lla bimekitsumabihoitoa saaneista potilaista verrattuna lumelääkkeellä hoidettuihin potilaisiin, joilla suun kandidiaasia sekä suun ja nielun kandidiaasia esiintyi 0 %:lla.
Neutropenia
Vaiheen III kliinisissä läiskäpsoriaasin tutkimuksissa havaittiin bimekitsumabihoidon yhteydessä neutropeniaa. Vaiheen III tutkimusten koko hoitojakson aikana 1 %:lla bimekitsumabihoitoa saaneista potilaista havaittiin asteen 3/4 neutropeniaa.
Neutropeniaa esiintyi yhtä yleisesti nivelpsoriaasin, aksiaalisen spondylartriitin (röntgennegatiivisen ja röntgenpositiivisen) ja HS-taudin kliinisissä tutkimuksissa kuin läiskäpsoriaasin tutkimuksissa.
Yli 80 % tapauksista oli ohimeneviä eivätkä vaatineet hoidon lopettamista. Neutropeniaan ei liittynyt vakavia infektioita.
Yliherkkyys
IL-17:n estäjien käytön yhteydessä on havaittu vakavia yliherkkyysreaktioita, mukaan lukien anafylaktisia reaktioita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Immunogeenisuus
Läiskäpsoriaasi
Läiskäpsoriaasipotilaista, jotka saivat bimekitsumabihoitoa enintään 56 viikon ajan suositeltuna annostuksena (320 mg joka neljäs viikko viikkoon 16 asti ja sen jälkeen 320 mg joka kahdeksas viikko), noin 45 %:lle kehittyi lääkevasta-aineita. Niistä potilaista, jolle lääkevasta-aineita kehittyi, noin 34 %:lla (16 % kaikista bimekitsumabihoitoa saaneista potilaista) oli neutraloiviksi luokiteltuja vasta-aineita.
Nivelpsoriaasi
Noin 31 %:lla bimekitsumabihoitoa suositellulla annostuksella (160 mg joka neljäs viikko) saaneista nivelpsoriaasipotilaista esiintyi lääkevasta-aineita 16 viikon hoidon jälkeen. Näistä potilaista noin 33 %:lla (10 %:lla kaikista bimekitsumabihoitoa saaneista) oli vasta-aineita, jotka luokiteltiin neutraloiviksi. Viikkoon 52 mennessä biologisille reumalääkkeille aikaisemmin altistumattomista nivelpsoriaasipotilaista, jotka olivat mukana BE OPTIMAL ‑tutkimuksessa ja saivat bimekitsumabihoitoa suositellulla annostuksella (160 mg joka neljäs viikko), noin 47 %:lla oli lääkevasta-aineita. Näistä potilaista noin 38 %:lla (18 %:lla kaikista BE OPTIMAL ‑tutkimuksessa bimekitsumabihoitoa saaneista potilaista) oli vasta-aineita, jotka luokiteltiin neutraloiviksi.
Aksiaalinen spondylartriitti (röntgennegatiivinen ja röntgenpositiivinen)
Noin 57 %:lla bimekitsumabihoitoa suositellulla annostuksella (160 mg joka neljäs viikko) enintään 52 viikon ajan saaneista röntgennegatiivista aksiaalista spondylartriittia sairastavista potilaista esiintyi lääkevasta-aineita. Näistä potilaista noin 44 %:lla (25 %:lla kaikista bimekitsumabihoitoa saaneista potilaista) oli lääkevasta-aineita, jotka luokiteltiin neutraloiviksi.
Noin 44 %:lla bimekitsumabihoitoa suositellulla annostuksella (160 mg joka neljäs viikko) enintään 52 viikon ajan saaneista röntgenpositiivista aksiaalista spondylartriittia sairastavista potilaista esiintyi lääkevasta-aineita. Näistä potilaista noin 44 %:lla (20 %:lla kaikista bimekitsumabihoitoa saaneista potilaista) oli lääkevasta-aineita, jotka luokiteltiin neutraloiviksi.
Hidradenitis suppurativa (HS-tauti)
Noin 59 %:lle HS-tautia sairastavista potilaista, jotka saivat bimekitsumabihoitoa suositellulla annostuksella (320 mg joka toinen viikko viikolle 16 saakka ja sen jälkeen 320 mg joka neljäs viikko) enintään 48 viikon ajan, kehittyi lääkevasta-aineita. Näistä potilaista noin 63 %:lla (37 %:lla kaikista bimekitsumabihoitoa saaneista potilaista) oli lääkevasta-aineita, jotka luokiteltiin neutraloiviksi.
Bimekitsumabin vasta-aineiden kehittymiseen ei liittynyt kliinisesti merkittävää vaikutusta kliiniseen vasteeseen missään käyttöaiheissa, eikä yhteyttä immunogeenisuuden ja hoidosta aiheutuvien haittavaikutusten välillä ole selkeästi osoitettu.
Iäkkäät potilaat (≥ 65-vuotiaat)
Iäkkäiden potilaiden altistuksesta on vain vähän tietoa.
Iäkkäille potilaille voi ilmetä bimekitsumabin käytön aikana todennäköisemmin tiettyjä haittavaikutuksia, kuten suun kandidiaasi-infektioita, dermatiittia ja ekseemaa.
Vaiheen III kliinisten läiskäpsoriaasitutkimusten lumelääkekontrolloidussa jaksossa suun kandidiaasi-infektioita havaittiin 18,2 %:lla ≥ 65-vuotiaista potilaista verrattuna 6,3 %:iin < 65-vuotiaista, dermatiittia ja ekseemaa havaittiin 7,3 %:lla ≥ 65-vuotiaista potilaista verrattuna 2,8 %:iin < 65-vuotiaista.
Vaiheen III kliinisten nivelpsoriaasitutkimusten lumelääkekontrolloidussa jaksossa suun kandidiaasi-infektioita havaittiin 7,0 %:lla ≥ 65-vuotiaista potilaista verrattuna 1,6 %:iin < 65-vuotiaista, dermatiittia ja ekseemaa havaittiin 1,2 %:lla ≥ 65‑vuotiaista potilaista verrattuna 2,0 %:iin < 65-vuotiaista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa on annettu 640 mg:n kerta-annoksia laskimoon tai 640 mg ihon alle, minkä jälkeen on annettu viisi 320 mg:n annosta ihon alle kahden viikon välein ilman annosta rajoittavaa toksisuutta. Yliannostapauksissa on suositeltavaa seurata potilaan vointia haittavaikutusten oireiden ja löydösten varalta ja aloittaa viipymättä asianmukainen oireenmukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, interleukiinin estäjät, ATC-koodi: L04AC21
Vaikutusmekanismi
Bimekitsumabi on humanisoitu monoklonaalinen IgG1/κ-vasta-aine, joka sitoutuu selektiivisesti suurella affiniteetilla IL-17A-, IL-17F- ja IL-17AF-sytokiineihin ja estää siten niiden vuorovaikutuksen IL-17RA/IL-17RC-reseptorikompleksin kanssa. Kohonneet IL-17A- ja IL-17F-pitoisuudet on yhdistetty useiden immuunivälitteisten tulehdussairauksien, kuten läiskäpsoriaasin, nivelpsoriaasin, aksiaalisen spondylartriitin ja HS-taudin patogeneesiin. IL-17A ja IL-17F toimivat yhdessä ja/tai vaikuttavat synergisesti muiden tulehdusreaktioita välittävien sytokiinien kanssa tulehduksen aikaansaamiseksi. Luontaisen immuniteetin solut tuottavat merkittäviä määriä IL-17F-sytokiiniä. Tämä tuotanto voi olla IL-23:sta riippumatonta. Bimekitsumabi estää tulehdusreaktiota välittävien sytokiinien toimintaa, mikä johtaa ihotulehduksen normalisoitumiseen ja merkittävään paikallisen ja systeemisen tulehduksen vähenemiseen, sekä tämän seurauksena psoriaasiin, nivelpsoriaasiin, aksiaaliseen spondylartriittiin ja HS-tautiin liittyvien kliinisten löydösten ja oireiden paranemiseen. In vitro ‑malleissa on osoitettu, että bimekitsumabi estää psoriaasiin liittyvää geeniekspressiota, sytokiinituotantoa, tulehdussolujen migraatiota ja patologista osteogeneesiä enemmän kuin pelkkä IL-17A:n esto.
Kliininen teho ja turvallisuus
Läiskäpsoriaasi
Bimekitsumabin turvallisuutta ja tehoa arvioitiin kolmessa vaiheen III satunnaistetussa, lumelääkkeellä ja/tai aktiivisella vertailuvalmisteella kontrolloidussa monikeskustutkimuksessa 1 480 potilaalla, joilla oli keskivaikea tai vaikea läiskäpsoriaasi. Potilaat olivat vähintään 18-vuotiaita, heidän Psoriasis Area and Severity Index (PASI) ‑pisteensä olivat ≥ 12, psoriaasi-ihottuma peitti vähintään 10 % kehon pinta-alasta (Body Surface Area, BSA), IGA (Investigators Global Assessment) -pistemäärä oli ≥ 3 viisiportaisella asteikolla ja heille harkittiin systeemistä psoriaasihoitoa ja/tai valohoitoa. Bimekitsumabin tehoa ja turvallisuutta arvioitiin verrattuna lumelääkkeeseen ja ustekinumabiin (BE VIVID – PS0009), lumelääkkeeseen (BE READY – PS0013) ja adalimumabiin (BE SURE – PS0008).
BE VIVID -tutkimuksessa 567:ää potilasta seurattiin 52 viikon ajan. Potilaat satunnaistettiin saamaan joko 320 mg bimekitsumabia 4 viikon välein, ustekinumabia (potilaan painon mukaan 45 mg tai 90 mg lähtötilanteessa ja viikolla 4 ja sen jälkeen 12 viikon välein) tai lumelääkettä ensimmäisten 16 viikon ajan, minkä jälkeen he saivat 320 mg bimekitsumabia 4 viikon välein.
BE READY -tutkimuksessa seurattiin 435:ttä potilasta 56 viikon ajan. Potilaat satunnaistettiin saamaan 320 mg bimekitsumabia 4 viikon välein tai lumelääkettä. Viikolla 16 potilaat, jotka saavuttivat PASI 90 -vasteen, siirtyivät 40 viikkoa kestävään satunnaistettuun hoidon lopettamista tutkivaan jaksoon. Potilaat, jotka satunnaistettiin alun perin saamaan 320 mg bimekitsumabia 4 viikon välein, satunnaistettiin uudelleen saamaan joko 320 mg bimekitsumabia 4 viikon välein, 320 mg bimekitsumabia 8 viikon välein tai lumelääkettä (ts. bimekitsumabin käyttö lopetettiin). Potilaat, jotka satunnaistettiin alun perin saamaan lumelääkettä, jatkoivat lumelääkkeen saamista, jos he saavuttivat PASI 90 -vasteen. Potilaat, jotka eivät saavuttaneet PASI 90 -vastetta viikolla 16, siirtyivät avoimeen (ei-sokkoutettuun) vaihtoehtoiseen hoitoon, joka oli 320 mg bimekitsumabia 4 viikon välein 12 viikon ajan. Myös ne potilaat, joiden oireet uusiutuivat (eivät saavuttaneet PASI 75 -vastetta) satunnaistetun hoidon lopetusjakson aikana, siirtyivät samaan 12 viikon vaihtoehtoiseen hoitoon.
BE SURE -tutkimuksessa seurattiin 478:aa potilasta 56 viikon ajan. Potilaat satunnaistettiin saamaan joko 320 mg bimekitsumabia 4 viikon välein viikkoon 56 asti, 320 mg bimekitsumabia 4 viikon välein viikkoon 16 asti, jonka jälkeen he saivat 320 mg bimekitsumabia 8 viikon välein viikkoon 56 asti, tai adalimumabia valmisteyhteenvedon suosituksen mukaisesti viikkoon 24 asti, jonka jälkeen he saivat 320 mg bimekitsumabia 4 viikon välein viikkoon 56 asti.
Potilaiden ominaisuudet lähtötilanteessa olivat yhdenmukaiset kaikissa kolmessa tutkimuksessa: potilaat olivat enimmäkseen miehiä (70,7 %) ja valkoihoisia (84,1 %), ikä oli keskimäärin 45,2 vuotta (18–83 vuotta), ja 8,9 % oli ≥ 65-vuotiaita. BSA-arvon mediaani lähtötilanteessa oli 20 %, PASI-pisteiden mediaani lähtötilanteessa oli 18 ja IGA-pistemäärä lähtötilanteessa vastasi vaikeaa sairautta 33 %:lla potilaista. Potilaan oirepäiväkirjassa (PSD) kipua, kutinaa ja hilseilyä kuvastavien asteikolla 0–10 arvioitujen osioiden pisteiden mediaani oli lähtötilanteessa 6–7 ja Dermatology Life Quality Index (DLQI) -kokonaispistemäärän mediaani oli lähtötilanteessa 9.
Kaikissa kolmessa tutkimuksessa 38 % potilaista oli saanut aiempaa biologista hoitoa: 23 % oli saanut vähintään yhtä IL-17:n estäjää (jos aiempi hoito IL-17:n estäjällä oli päättynyt primaarin hoitovasteen puuttumiseen, potilas suljettiin pois tutkimuksesta) ja 13 % oli saanut vähintään yhtä TNF:n estäjää. Potilaista 22 % ei ollut aiemmin saanut mitään systeemistä hoitoa (ei-biologista eikä biologista), ja 39 % potilaista oli saanut aiemmin valohoitoa tai fotokemoterapiaa.
Bimekitsumabin tehoa arvioitiin ihotaudin osalta yleisesti ja spesifisillä kehon alueilla (hiuspohja, kynnet, kämmenet ja jalkapohjat) sekä potilaan raportoimien oireiden ja elämänlaatuun liittyvän vaikutuksen kannalta. Kaikissa kolmessa tutkimuksessa yhdistetty ensisijainen päätetapahtuma oli niiden potilaiden osuus, joilla 1) PASI 90 -vaste ja 2) IGA-vaste ”oireeton tai lähes oireeton” (IGA 0/1 ja vähintään kahden pisteen paraneminen lähtötilanteesta) viikolla 16. PASI 100- ja IGA 0 ‑vaste viikolla 16 sekä PASI 75 ‑vaste viikolla 4 olivat kaikissa kolmessa tutkimuksessa toissijaisia päätetapahtumia.
Ihotaudin kokonaistilanne
Bimekitsumabihoito johti merkitsevään paranemiseen tehon päätetapahtumissa lumelääkkeeseen, ustekinumabiin tai adalimumabiin verrattuna viikolla 16. Pääasialliset tehotulokset esitetään taulukossa 2.
Taulukko 2: Yhteenveto kliinisistä vasteista tutkimuksissa BE VIVID, BE READY ja BE SURE
| BE VIVID | BE READY | BE SURE | |||||
Lumelääke (N = 83) n (%) | Bimekitsumabi 320 mg Q4W (N = 321) n (%) | Ustekinumabi (N = 163) n (%) | Lumelääke (N = 86) n (%) | Bimekitsumabi 320 mg Q4W (N = 349) n (%) | Bimekitsumabi 320 mg Q4W (N = 319) n (%) | Adalimumabi (N = 159) n (%) | |
PASI 100 Viikko 16 |
0 (0,0) |
188 (58,6)a |
34 (20,9) |
1 (1,2) |
238 (68,2)a |
194 (60,8)a |
38 (23,9) |
PASI 90 Viikko 16 |
4 (4,8) |
273 (85,0)a, b |
81 (49,7) |
1 (1,2) |
317 (90,8)a |
275 (86,2)a |
75 (47,2) |
PASI 75 Viikko 4 Viikko 16 |
2 (2,4) 6 (7,2) |
247 (76,9)a, b 296 (92,2) |
25 (15,3) 119 (73,0) |
1 (1,2) 2 (2,3) |
265 (75,9)a 333 (95,4) |
244 (76,5)a 295 (92,5) |
50 (31,4) 110 (69,2) |
IGA 0 Viikko 16 |
0 (0,0) |
188 (58,6)a |
36 (22,1) |
1 (1,2) |
243 (69,6)a |
197 (61,8) |
39 (24,5) |
IGA 0/1 Viikko 16 |
4 (4,8) |
270 (84,1)a, b |
87 (53,4) |
1 (1,2) |
323 (92,6)a |
272 (85,3)a |
91 (57,2) |
Absoluut-tinen PASI-arvo ≤ 2 Viikko 16 |
3 (3,6) |
273 (85,0) |
84 (51,5) |
1 (1,2) |
315 (90,3) |
280 (87,8) |
86 (54,1) |
PSD:n kuvaama kivun vähenemä ≥ 4 (N) Viikko 16 | (N = 48) 5 (10,4) | (N = 190) 140 (73,7) | (N=90) 54 (60,0) | (N = 49) 0 (0,0) | (N = 209) 148 (70,8) | (N = 222) 143 (64,4) | (N = 92) 43 (46,7) |
PSD:n kuvaama kutinan vähenemä ≥ 4 (N) Viikko 16 | (N = 53) 6 (11,3) | (N = 222) 151 (68,0) | (N = 104) 57 (54,8) | (N = 60) 0 (0,0) | (N = 244) 161 (66,0) | (N = 248) 153 (61,7) | (N = 107) 42 (39,3) |
PSD:n kuvaama hilseilyn vähenemä ≥ 4 (N) Viikko 16 | (N = 56) 6 (10,7) | (N = 225) 171 (76,0) | (N = 104) 59 (56,7) | (N = 65) 1 (1,5) | (N = 262) 198 (75,6) | (N = 251) 170 (67,7) | (N = 109) 42 (38,5) |
Bimekitsumabi 320 mg Q4W = bimekitsumabi 4 viikon välein. Tulokset perustuvat non-responder imputaatioon (NRI) eli vastetiedon puuttuminen on tulkittu niin, että vastetta ei saavutettu.
IGA 0/1 ‑vasteen määritelmä oli oireeton (0) tai lähes oireeton (1) ja vähintään 2 portaan paranema lähtötasoon verrattuna viikolla 16. IGA 0 -vasteen määritelmä oli oireeton (0) ja vähintään 2 portaan paranema lähtötasoon verrattuna viikolla 16.
PSD (Patient Symptoms Diary) tarkoittaa potilaan oirepäiväkirjaa, joka tunnetaan myös nimellä P-SIM (Psoriasis Symptoms and Impacts Measure). Sillä mitataan psoriaasioireiden vaikeusastetta asteikolla 0 (ei oireita) – 10 (hyvin vaikeat oireet). Vasteeksi määritellään asteikolla 0–10 vähenemä ≥ 4 kivun, kutinan ja hilseilyn osalta lähtötilanteesta viikkoon 16 mennessä.
a) p < 0,001 vs. lumelääke (BE VIVID ja BE READY), vs. adalimumabi (BE SURE), korjattu kerrannaisuuden suhteen.
b) p < 0,001 vs. ustekinumabi (BE VIVID), korjattu kerrannaisuuden suhteen.
Bimekitsumabiin liittyi tehon nopea ilmeneminen. BE VIVID ‑tutkimuksessa PASI 90 -vasteosuudet viikolla 2 ja viikolla 4 olivat bimekitsumabihoitoa saaneilla potilailla merkittävästi suuremmat (12,1 % viikolla 2 ja 43,6 % viikolla 4) lumelääkkeeseen (1,2 % viikolla 2 ja 2,4 % viikolla 4) ja ustekinumabiin (1,2 % viikolla 2 ja 3,1 % viikolla 4) verrattuna.
BE VIVID -tutkimuksessa bimekitsumabihoitoa (neljän viikon välein) saaneet potilaat saavuttivat viikolla 52 merkitsevästi suuremmat vasteosuudet kuin ustekinumabihoitoa saaneet potilaat, kun päätetapahtumana oli PASI 90 (bimekitsumabi 81,9 % vs. ustekinumabi 55,8 %, p < 0,001), IGA 0/1 (bimekitsumabi 78,2 % vs. ustekinumabi 60,7 %, p < 0,001) ja PASI 100 (bimekitsumabi 64,5 % vs. ustekinumabi 38,0 %).
Kuva 1: BE VIVID -tutkimuksen PASI 90 -vasteosuudet aikajanalla

BKZ 320 mg Q4W = bimekitsumabi 4 viikon välein; Uste = ustekinumabi. Käytössä on NRI-imputointi (Non-Responder Imputation)
BE SURE -tutkimuksessa merkitsevästi suurempi prosentuaalinen osuus bimekitsumabihoitoa saaneista potilaista (Q4W/Q4W ja Q4W/Q8W [Q8W = bimekitsumabi kahdeksan viikon välein], yhdistetyt annostushaarat) saavutti viikolla 24 PASI 90- ja IGA 0/1 ‑vasteet verrattuna adalimumabiin (bimekitsumabihoidossa PASI 90-vaste 85,6 % ja IGA 0/1 ‑vaste 86,5 % vs. adalimumabihoidossa PASI 90 -vaste 51,6 % ja IGA 0/1 ‑vaste 57,9 %, p < 0,001). 70,2 % bimekitsumabihoitoa kahdeksan viikon välein saaneista potilaista saavutti viikolla 56 PASI 100 -vasteen. Niistä 65 potilaasta, joilla ei ollut vastetta adalimumabihoitoon viikolla 24 (< PASI 90), 78,5 % saavutti PASI 90 -vasteen 16 viikon bimekitsumabihoidon jälkeen. Turvallisuusprofiilin havaittiin olevan samankaltainen potilailla, jotka vaihtoivat adalimumabista bimekitsumabiin ilman lääkkeetöntä hoitotaukoa, ja potilailla, jotka aloittivat bimekitsumabin aiempien systeemisten hoitojen jälkeisen lääkkeettömän hoitotauon jälkeen.
Kuva 2: BE SURE -tutkimuksen PASI 90 -vasteosuudet aikajanalla
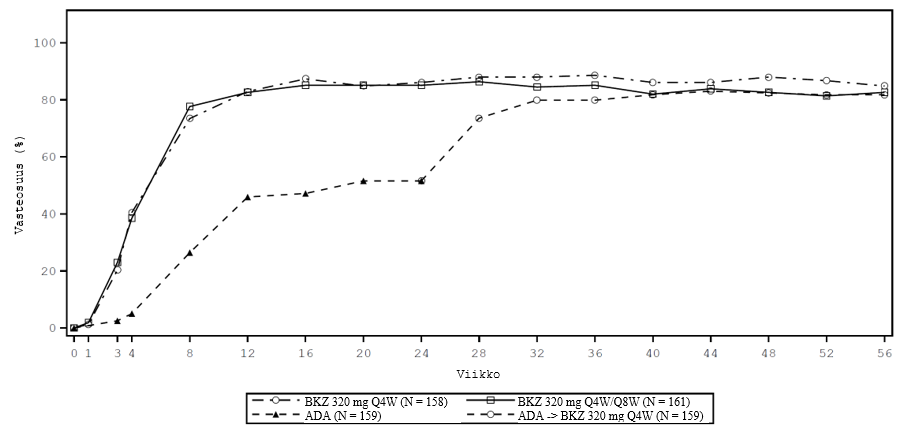
BKZ 320 mg Q4W = bimekitsumabi 4 viikon välein; BKZ 320 mg Q8W = bimekitsumabi 8 viikon välein; ADA = adalimumabi.
BKZ Q4W/Q8W -ryhmän potilaat vaihtoivat Q4W-annostelusta Q8W-annosteluun viikolla 16. ADA/BKZ 320 mg Q4W -ryhmän potilaat vaihtoivat ADA-annostelusta BKZ Q4W-annosteluun viikolla 24. Käytössä on NRI-imputointi (Non-Responder Imputation).
Bimekitsumabin teho osoitettiin riippumatta iästä, sukupuolesta, etnisestä taustasta, sairauden kestosta, painosta, sairauden vaikeusasteesta lähtötilanteessa PASI-pisteiden perusteella ja aiemmasta biologisesta hoidosta. Bimekitsumabi oli tehokas biologista hoitoa aiemmin saaneilla potilailla, mukaan lukien TNF:n ja IL-17:n estäjillä hoitoa saaneet potilaat, sekä potilailla, jotka eivät olleet aiemmin saaneet systeemistä hoitoa. Tehoa ei ole tutkittu potilailla, joilla aiempi hoito IL-17:n estäjällä on päättynyt primaarin hoitovasteen puuttumiseen.
Populaatiofarmakokineettisen/-farmakodynaamisen analyysin perusteella, jota kliiniset tiedot tukivat, painavammat potilaat (≥ 120 kg), jotka eivät saavuttaneet täysin oireetonta ihoa viikolla 16, hyötyivät ensimmäisten 16 hoitoviikon jälkeen jatkuvasta bimekitsumabihoidosta 320 mg:n annoksina neljän viikon välein. BE SURE -tutkimuksessa potilaat saivat 320 mg bimekitsumabia neljän viikon välein viikkoon 16 asti ja sen jälkeen joko neljän tai kahdeksan viikon välein viikkoon 56 asti riippumatta vastestatuksesta viikolla 16. Potilailla, jotka saivat hoitoa neljän viikon välein ja painoivat ≥ 120 kg (N = 37), PASI 100 -vaste yleistyi enemmän viikkojen 16 (23,5 %) ja 56 (70,6 %) välillä verrattuna kahdeksan viikon välein hoitoa saaneisiin potilaisiin (45,0 % viikolla 16 vs. 60,0 % viikolla 56).
Bimekitsumabihoitoa saaneiden potilaiden psoriaasin havaittiin viikolla 16 vähentyneen hiuspohjassa, kynsissä, kämmenissä ja jalkapohjissa (ks. taulukko 3).
Taulukko 3: Hiuspohjan, kämmenten, jalkapohjien ja kynsien vasteet BE VIVID-, BE READY- ja BE SURE -tutkimuksissa viikolla 16
| BE VIVID | BE READY | BE SURE | |||||
| Lumelääke | Bimekitsumabi 320 mg Q4W | Ustekinumabi | Lumelääke | Bimekitsumabi 320 mg Q4W | Bimekitsumabi 320 mg Q4W | Adalimumabi | |
Hius-pohjan IGA (N)a Hiuspohjan IGA 0/1, n (%) | (72) 11 (15,3) | (285) 240 (84,2)b | (146) 103 (70,5) | (74) 5 (6,8) | (310) 286 (92,3)b | (296) 256 (86,5) | (138) 93 (67,4) |
Kämmenten ja jalka-pohjien IGA (N)a pp-IGA 0/1, n (%) | (29) 7 (24,1) | (105) 85 (81,0) | (47) 39 (83,0) | (31) 10 (32,3) | (97) 91 (93,8) | (90) 75 (83,3) | (34) 24 (70,6) |
mNAPSI 100 (N)a mNAPSI 100, n (%) | (51) 4 (7,8) | (194) 57 (29,4) | (109) 15 (13,8) | (50) 3 (6,0) | (210) 73 (34,8) | (181) 54 (29,8) | (95) 21 (22,1) |
Bimekitsumabi 320 mg Q4W = bimekitsumabi 4 viikon välein. Käytössä on NRI-imputointi (Non-Responder Imputation).
Hiuspohjan IGA 0/1- ja kämmenten ja jalkapohjien IGA 0/1 ‑vasteiden määritelmä oli oireeton (0) tai lähes oireeton (1) ja vähintään 2 portaan paranema lähtötilanteeseen verrattuna.
a) Mukana vain potilaat, joilla tutkijan yleisarvio hiuspohjasta (IGA) sekä kämmenistä ja jalkapohjista (pp-IGA) on lähtötilanteessa vähintään 2 ja kynsipsoriaasin modifioitu vaikeusasteindeksi (modified Nail Psoriasis Severity Index; mNAPSI) > 0.
b) p < 0,001 vs. lumelääke, korjattu kerrannaisuuden suhteen
Hiuspohjan IGA-vasteet ja kämmenten ja jalkapohjien IGA-vasteet säilyivät bimekitsumabihoitoa saaneilla potilailla viikkoihin 52/56 asti. Kynsipsoriaasi lieveni edelleen viikon 16 jälkeen. BE VIVID -tutkimuksessa 60,3 %:lla bimekitsumabihoitoa 320 mg:n annoksina 4 viikon välein saaneista potilaista oli viikolla 52 täysin oireettomat kynnet (mNAPSI 100). BE READY -tutkimuksessa niistä potilaista, joilla oli viikolla 16 PASI 90 ˗vaste, kynnet olivat viikolla 56 täysin oireettomat 67,7 %:lla bimekitsumabia 320 mg:n annoksina 8 viikon välein saaneista ja 69,8 %:lla bimekitsumabia 320 mg:n annoksina 4 viikon välein saaneista.
Vasteen säilyminen
Taulukko 4: Vasteiden säilyminen bimekitsumabilla viikolla 52 PASI 100 -, PASI 90 -, IGA 0/1 - ja absoluuttisen PASI-vasteen ≤ 2 saaneilla viikolla 16*
| PASI 100 | PASI 90 | IGA 0/1 | Absoluuttinen PASI-arvo ≤ 2 | ||||
320 mg Q4W (N = 355) n (%) | 320 mg Q8W (N = 182) n (%) | 320 mg Q4W (N = 516) n (%) | 320 mg Q8W (N = 237) n (%) | 320 mg Q4W (N = 511) n (%) | 320 mg Q8W (N = 234) n (%) | 320 mg Q4W (N = 511) n (%) | 320 mg Q8W (N = 238) n (%) |
| 295 (83,1) | 161 (88,5) | 464 (89,9) | 214 (90,3) | 447 (87,5) | 214 (91,5) | 460 (90,0) | 215 (90,3) |
* Yhdistetty analyysi BE VIVID-, BE READY- ja BE SURE -tutkimuksista. NRI.
320 mg Q4W: 320 mg bimekitsumabia joka neljäs viikko ja sen jälkeen 320 mg bimekitsumabia joka neljäs viikko viikosta 16 alkaen.
320 mg Q8W: 320 mg bimekitsumabia joka neljäs viikko ja sen jälkeen 320 mg bimekitsumabia joka kahdeksas viikko viikosta 16 alkaen.
Vasteen kesto (bimekitsumabin käytön lopettamisen jälkeen)
Kuva 3: PASI 90 -vasteosuudet aikajanalla PASI 90 -vasteen viikolla 16 saaneilla – BE READY -tutkimuksen satunnaistettu hoidon lopetusjakso
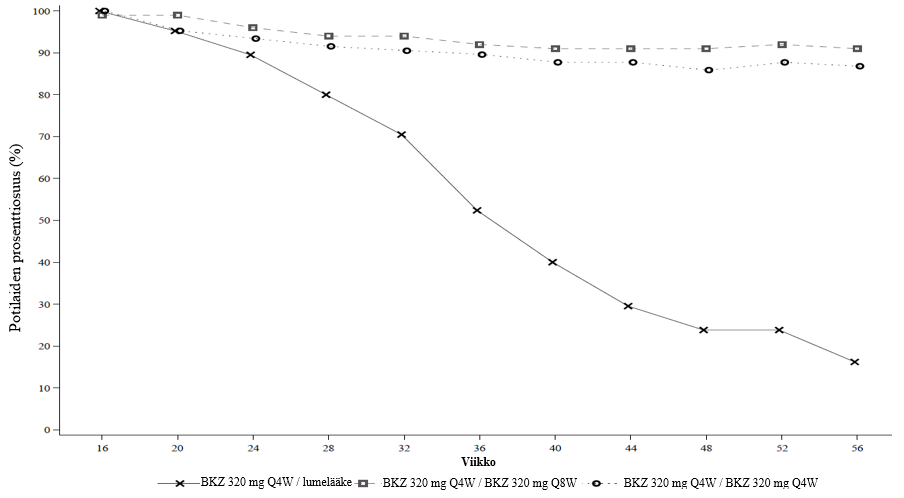
NRI.
Viikolla 16 hoidon satunnaistetun lopetusvaiheen aloitti 105 potilasta bimekitsumabi 320 mg Q4W/lumelääke ˗ryhmässä, 100 potilasta bimekitsumabi 320 mg Q4W/Q8W ˗ryhmässä ja 106 potilasta bimekitsumabi 320 mg Q4W/Q4W ˗ryhmässä.
BE READY -tutkimuksessa viikolla 16 PASI 90 -vasteen saaneilla, jotka satunnaistettiin uudelleen saamaan lumelääkettä ja lopettamaan bimekitsumabihoidon, mediaaniaika PASI 75 -vasteen häviämisenä määriteltyyn oireiden uusiutumiseen oli noin 28 viikkoa (32 viikkoa viimeisen bimekitsumabiannoksen jälkeen). Näistä potilaista 88,1 % sai PASI 90 ‑vasteen uudelleen 12 viikon kuluessa bimekitsumabihoidon aloittamisesta annoksina 320 mg 4 viikon välein.
Terveyteen liittyvä elämänlaatu / potilaan raportoimat hoitotulokset
Kaikissa kolmessa tutkimuksessa suuremmalla osalla bimekitsumabihoitoa saaneista potilaista psoriaasi ei vaikuttanut viikolla 16 Dermatology Life Quality Index (DLQI) -indeksillä mitattuun elämänlaatuun verrattuna lumelääkkeellä tai aktiivisella vertailuvalmisteella hoitoa saaneisiin potilaisiin (taulukko 5).
Taulukko 5: Elämänlaatu BE VIVID-, BE READY- ja BE SURE -tutkimuksissa
| BE VIVID | BE READY | BE SURE | |||||
Lumelääke (N = 83) n (%) | Bimekitsumabi 320 mg Q4W (N = 321) n (%) | Ustekinumabi (N = 163) n (%) | Lumelääke (N = 86) n (%) | Bimekitsumabi 320 mg Q4W (N = 349) n (%) | Bimekitsumabi 320 mg Q4W (N = 319) n (%) | Adalimumabi (N = 159) n (%) | |
| DLQI 0/1aLähtötaso | 3 (3,6) | 16 (5,0) | 5 (3,1) | 4 (4,7) | 11 (3,2) | 10 (3,1) | 13 (8,2) |
DLQI 0/1a Viikko 16 |
10 (12,0) |
216 (67,3) |
69 (42,3) |
5 (5,8) |
264 (75,6) |
201 (63,0) |
74 (46,5) |
a) DLQI:n absoluuttinen pistemäärä 0 tai 1 tarkoittaa, että sairaus ei vaikuta terveyteen liittyvään elämänlaatuun. NRI.
DLQI 0/1 ‑vasteiden osuus suureni viikosta 16 eteenpäin edelleen ja säilyi viikkoon 52/56 asti. BE VIVID -tutkimuksen viikolla 52 DLQI 0/1 ‑vasteosuus bimekitsumabihoitoa 320 mg:n annoksina neljän viikon välein saaneilla potilailla oli 74,8 %. BE SURE -tutkimuksen viikolla 56 DLQI 0/1 ‑vasteosuus 320 mg bimekitsumabia kahdeksan viikon välein saaneilla potilailla oli 78,9 % ja 320 mg bimekitsumabia neljän viikon välein saaneilla potilailla 74,1 %.
Vaiheen 3 avoin jatkotutkimus
Potilaat, jotka olivat loppuun asti mukana yhdessä alkuperäisistä vaiheen 3 tutkimuksista, saivat mahdollisuuden osallistua 144 viikkoa kestäneeseen avoimeen jatkotutkimukseen (PS0014), jossa arvioitiin bimekitsumabin pitkäkestoista turvallisuutta ja tehoa.
344 potilasta, jotka saivat bimekitsumabihoitoa 320 mg kahdeksan viikon välein (BKZ 320 mg Q8W) tai neljän viikon välein (BKZ 320 mg Q4W) alkuperäisessä tutkimuksessa ja jotka olivat saavuttaneet PASI 90 ‑vasteen alkuperäisen tutkimuksen päättyessä, saivat bimekitsumabia 320 mg kahdeksan viikon välein koko PS0014-tutkimuksen ajan. Näistä potilaista 293 (85,2 %) sai bimekitsumabia 320 mg kahdeksan viikon välein 144 viikkoa kestäneen hoidon loppuun asti. Hoitojakson aikana 48 potilasta (14,0 %) keskeytti tutkimuksen. Heistä 21 (6,1 %) keskeytti haittatapahtuman ja 4 (1,2 %) tehon puutteen takia.
Alkuperäisissä tutkimuksissa bimekitsumabilla saavutetut parannukset tehon päätetapahtumiksi määritetyissä PASI 90- ja IGA 0/1 ‑vasteissa säilyivät tutkimuksessa pysyneillä potilailla koko 144 viikkoa kestäneen avoimen jatkohoitojakson ajan.
Vaiheen 3b suora vertaileva tutkimus sekukinumabin kanssa
Bimekitsumabin tehoa ja turvallisuutta arvioitiin myös kaksoissokkoutetussa tutkimuksessa (BE RADIANT - PS0015), jossa sitä verrattiin IL-17A:n estäjään sekukinumabiin. Potilaat satunnaistettiin saamaan bimekitsumabia (N = 373, 320 mg viikoilla 0, 4, 8, 12 ja 16 (Q4W) ja sen jälkeen 320 mg 4 viikon välein (Q4W/Q4W) tai 320 mg 8 viikon välein (Q4W/Q8W)) tai sekukinumabia (N = 370, 300 mg viikoilla 0, 1, 2, 3 ja 4 ja sen jälkeen 300 mg 4 viikon välein). Potilaiden ominaisuudet lähtötilanteessa olivat yhdenmukaiset keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastavien potilaiden kanssa. Tutkittavien potilaiden BSA-arvon mediaani oli 19 % ja PASI-pisteiden mediaani 18.
Bimekitsumabihoitoa saaneet potilaat saavuttivat merkittävästi suurempia vasteosuuksia kuin sekukinumabia saaneet potilaat, kun ensisijaisena päätetapahtumana oli PASI 100 ‑vaste (täysin oireeton iho) viikolla 16. Bimekitsumabilla saavutettiin merkittävästi suurempia vasteosuuksia myös toissijaisessa päätetapahtumassa, jossa mitattiin PASI 100 ‑vastetta viikolla 48 (sekä Q4W/Q4W-annosteluvälillä että Q4W/Q8W-annosteluvälillä). PASI-vasteosuuksien vertailutiedot esitetään taulukossa 6.
Erot vasteosuuksissa bimekitsumabilla ja sekukinumabilla hoidettujen potilaiden välillä huomattiin niinkin varhain kuin viikolla 1 PASI 75 ‑vasteen kohdalla (7,2 % vs. 1,4 %) ja niinkin varhain kuin viikolla 2 PASI 90 ‑vasteen kohdalla (7,5 % vs. 2,4 %).
Taulukko 6: PASI-vasteosuudet BE RADIANT ‑tutkimuksessa: bimekitsumabi vs. sekukinumabi
| Viikko 4 | Viikko 16 | Viikko 48a) | |||||
Bimekitsumabi 320 mg Q4W | Sekukinumabi | Bimekitsumabi 320 mg Q4W | Sekukinumabi | Bimekitsumabi 320 mg Q4W/Q4W | Bimekitsumabi 320 mg Q4W/Q8W | Sekukinumabi | |
(N = 373) n (%) | (N = 370) n (%) | (N = 373) n (%) | (N = 370) n (%) | (N = 147) n (%) | (N = 215) n (%) | (N = 354) n (%) | |
| PASI 100 | 52 (13,9) | 23 (6,2) | 230 (61,7)* | 181 (48,9) | 108 (73,5)* | 142 (66,0)* | 171 (48,3) |
| PASI 90 | 134 (35,9) | 65 (17,6) | 319 (85,5) | 275 (74,3) | 126 (85,7) | 186 (86,5) | 261 (73,7) |
| PASI 75 | 265 (71,0)* | 175 (47,3) | 348 (93,3) | 337 (91,1) | 134 (91,2) | 196 (91,2) | 301 (85,0) |
| Absoluuttinen PASI < 2 | 151 (40,5) | 75 (20,3) | 318 (85,3) | 283 (76,5) | 127 (86,4) | 186 (86,5) | 269 (76,0) |
a) Tiedot ovat ylläpitojoukosta, joka koostuu potilaista, jotka saivat ainakin yhden annoksen tutkimuslääkettä viikolla 16 tai myöhemmin.
* p < 0,001 vs. sekukinumabi, korjattu kerrannaisuuden suhteen. NRI.
Bimekitsumabin ja sekukinumabin PASI 100 ‑vasteosuudet viikkoon 48 asti esitetään kuvassa 4.
Kuva 4: BE RADIANT ‑tutkimuksen PASI 100 ‑vasteosuudet aikajanalla
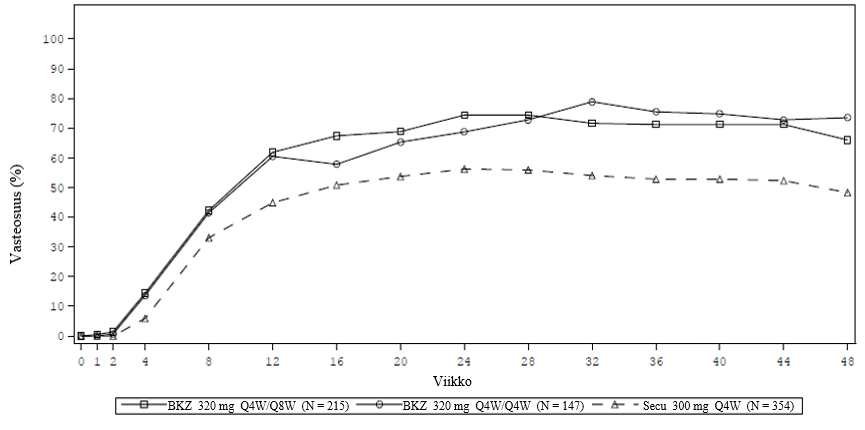
NRI. Ylläpitojoukko koostuu potilaista, jotka saivat ainakin yhden annoksen tutkimuslääkettä viikolla 16 tai myöhemmin.
Bimekitsumabin teho BE RADIANT ‑tutkimuksessa oli yhdenmukainen BE VIVID-, BE READY- ja BE SURE ‑tutkimusten kanssa.
Vaiheen 3b avoin jatkotutkimusjakso
Viikolla 48 potilailla oli mahdollisuus jatkaa 96 viikkoa kestävään avoimeen jatkotutkimusjaksoon. He joko aloittivat bimekitsumabihoidon tai jatkoivat sitä 320 mg:n annoksina neljän viikon välein (Q4W) tai 320 mg:n annoksina kahdeksan viikon välein (Q8W) riippuen siitä, olivatko he saavuttaneet PASI 90 -vasteen viikkoon 48 mennessä vai eivät. Tutkittavat, jotka saivat avoimen jatkotutkimusjakson aikana aluksi bimekitsumabia 320 mg neljän viikon välein (Q4W), siirtyivät bimekitsumabihoitoon 320 mg:n annoksina kahdeksan viikon välein (QW8) viikolla 72 tai myöhemmin.
231 potilasta, jotka olivat saaneet bimekitsumabia 320 mg kahdeksan tai neljän viikon välein ja jotka olivat saavuttaneet PASI 90 -vasteen viikkoon 48 mennessä, saivat bimekitsumabia 320 mg kahdeksan viikon välein koko avoimen jatkotutkimusjakson ajan. Näistä potilaista 31 (13,4 %) -keskeytti tutkimuksen avoimen jatkotutkimusjakson aikana. Heistä 10 (4,3 %) keskeytti haittatapahtuman takia ja 1 (0,4 %) tehon puutteen takia.
116 potilasta, jotka olivat saaneet sekukinumabihoitoa ja saavuttaneet PASI 90 -vasteen viikkoon 48 mennessä, saivat bimekitsumabia 320 mg kahdeksan viikon välein koko avoimen jatkotutkimusjakson ajan. Näistä potilaista 16 (13,8 %) -keskeytti tutkimuksen avoimen jatkotutkimusjakson aikana. Heistä 6 (5,2 %) keskeytti haittatapahtuman takia ja 1 (0,9 %) tehon puutteen takia.
Bimekitsumabilla tai sekukinumabilla viikkoon 48 mennessä saavutetut parannukset tehon päätetapahtumiksi määritetyissä PASI 100-, PASI 90-, PASI 75- ja PASI ≤ 2 ‑vasteissa säilyivät tutkimuksessa pysyneillä potilailla, jotka saivat avoimessa jatkotutkimusvaiheessa bimekitsumabia 320 mg kahdeksan viikon välein, koko 96 viikkoa kestäneen avoimen jatkotutkimusjakson ajan.
Bimekitsumabin turvallisuusprofiili oli 144 viikon aikana yhdenmukainen viikkoon 48 mennessä havaitun turvallisuusprofiilin kanssa.
Nivelpsoriaasi
Bimekitsumabin turvallisuutta ja tehoa arvioitiin 1 112:lla aktiivista nivelpsoriaasia sairastavalla aikuispotilaalla (vähintään 18-vuotiaalla) kahdessa satunnaistetussa, kaksoissokkoutetussa, lumelääkkeellä kontrolloidussa monikeskustutkimuksessa (PA0010 - BE OPTIMAL ja PA0011 - BE COMPLETE). BE OPTIMAL ‑tutkimus sisälsi aktiivisen vertailuhoidon haaran (adalimumabi) (N = 140).
Molemmissa tutkimuksissa potilailla oli ollut Classification Criteria for Psoriatic Arthritis (CASPAR) ‑luokituskriteerien mukainen aktiivinen nivelpsoriaasi vähintään 6 kuukauden ajan, ja heillä oli aktiivinen tauti, jossa arkojen nivelten määrä (TJC) oli ≥ 3 ja turvonneiden nivelten määrä (SJC) ≥ 3. Potilaiden nivelpsoriaasin toteamisesta kuluneen ajan mediaani oli 3,6 vuotta BE OPTIMAL ‑tutkimuksessa ja 6,8 vuotta BE COMPLETE ‑tutkimuksessa. Näihin tutkimuksiin otettiin mukaan potilaita siten, että edustettuina olivat kaikki nivelpsoriaasin alatyypit, mukaan lukien polyartikulaarinen symmetrinen artriitti, oligoartikulaarinen asymmetrinen artriitti, distaalisten interfalangeaalisten nivelten suhteen vallitseva, spondyliitin suhteen vallitseva ja mutiloiva artriitti. Lähtötilanteessa 55,9 %:lla potilaista oli aktiivinen läiskäpsoriaasi ≥ 3 %:ssa kehon pinta-alasta. 10,4 %:lla potilaista oli keskivaikea tai vaikea läiskäpsoriaasi. 31,9 %:lla oli entesiitti ja 12,3 %:lla daktyliitti lähtötilanteessa. Ensisijaisena päätetapahtumana molemmissa tutkimuksissa oli American College of Rheumatology (ACR) 50 ‑vaste viikon 16 kohdalla.
BE OPTIMAL ‑tutkimuksessa arvioitiin 852:ta potilasta, jotka eivät olleet aiemmin altistuneet millekään nivelpsoriaasin tai psoriaasin hoitoon käytettävälle biologiselle reumalääkkeelle. Potilaat satunnaistettiin (3:2:1) saamaan bimekitsumabia 160 mg joka neljäs viikko viikkoon 52 saakka tai lumelääkettä viikkoon 16 saakka, jonka jälkeen bimekitsumabia 160 mg joka neljäs viikko viikkoon 52 saakka, tai aktiiviseen vertailuhoitohaaraan (adalimumabi 40 mg joka toinen viikko) viikkoon 52 saakka. Tässä tutkimuksessa 78,3 % potilaista oli saanut aiemmin hoitoa vähintään yhdellä perinteisellä reumalääkkeellä ja 21,7 % potilaista ei ollut saanut aiemmin hoitoa millään perinteisellä reumalääkkeellä. Lähtötilanteessa 58,2 % potilaista sai samanaikaisesti metotreksaattia, 11,3 % sai samanaikaisesti muita perinteisiä reumalääkkeitä kuin metotreksaattia ja 30,5 % ei saanut mitään perinteistä reumalääkettä.
BE COMPLETE ‑tutkimuksessa arvioitiin 400:aa potilasta, joilla ei ollut nivelpsoriaasin tai psoriaasin hoidossa saavutettu riittävää vastetta (tehoa) yhteen tai kahteen tuumorinekroositekijä alfan (TNFα) estäjään tai jotka eivät sietäneet niitä. Potilaat satunnaistettiin (2:1) saamaan bimekitsumabia 160 mg joka neljäs viikko tai lumelääkettä enintään viikolle 16 saakka. Lähtötilanteessa 42,5 % potilaista sai samanaikaisesti metotreksaattia, 8,0 % sai samanaikaisesti muita perinteisiä reumalääkkeitä kuin metotreksaattia ja 49,5 % ei saanut mitään perinteistä reumalääkettä. Tässä tutkimuksessa 76,5 % tutkittavista ei ollut saanut riittävää vastetta yhteen TNFα:n estäjään, 11,3 % ei ollut saanut riittävää vastetta kahteen TNFα:n estäjään ja 12,3 % ei sietänyt TNFα:n estäjähoitoa.
Löydökset ja oireet
Potilailla, joita ei ollut aiemmin hoidettu biologisilla reumalääkkeillä (BE OPTIMAL), ja potilailla, jotka eivät olleet saaneet riittävää vastetta TNFα:n estäjiin tai eivät sietäneet niitä (BE COMPLETE), bimekitsumabihoidolla saavutettiin merkittävää parannusta taudin löydöksissä ja oireissa sekä tautiaktiivisuudessa lumelääkkeeseen verrattuna viikon 16 kohdalla. Vasteosuuksien määrät olivat samanlaisia molemmissa potilaspopulaatioissa (ks. taulukko 7). Kliiniset vasteet säilyivät viikolle 52 saakka BE OPTIMAL ‑tutkimuksessa ACR 20-, ACR 50-, ACR 70-, MDA-, PASI 90-, PASI 100- ja ACR 50 / PASI 100 ‑vastemittareilla arvioituna.
Taulukko 7: Kliininen vaste BE OPTIMAL- ja BE COMPLETE -tutkimuksissa
| BE OPTIMAL (potilaita ei ollut aiemmin hoidettu biologisilla reumalääkkeillä) | BE COMPLETE (potilaat eivät olleet saaneet riittävää vastetta TNFα:n estäjiin tai eivät sietäneet niitä) | ||||||
Lumelääke (N = 281) n (%) | Bimekitsumabi 160 mg Q4W (N = 431) n (%) | Ero lumelääkkeeseen verrattuna (95 %:n luottamusväli)(d) | Vertailuryhmä(e) (adalimumabi) (N = 140) n (%) | Lumelääke (N = 133) n (%) | Bimekitsumabi 160 mg Q4W (N = 267) n (%) | Ero lumelääkkeeseen verrattuna (95 %:n luottamusväli)(d) | |
ACR 20 Viikko 16 Viikko 24 Viikko 52 |
67 (23,8) - |
268 (62,2) 282 (65,4) 307 (71,2) | 38,3 (31,4; 45,3) |
96 (68,6) 99 (70,7) 102 (72,9) | 21 (15,8) | 179 (67,0) | 51,2 (42,1; 60,4) |
ACR 50 Viikko 16 Viikko 24 Viikko 52 |
28 (10,0) - |
189 (43,9)* 196 (45,5) 235 (54,5) | 33,9 (27,4; 40,4) |
64 (45,7) 66 (47,1) 70 (50,0) | 9 (6,8) | 116 (43,4)* | 36,7 (27,7; 45,7) |
ACR 70 Viikko 16 Viikko 24 Viikko 52 |
12 (4,3) - |
105 (24,4) 126 (29,2) 169 (39,2) | 20,1 (14,7; 25,5) |
39 (27,9) 42 (30,0) 53 (37,9) | 1 (0,8) | 71 (26,6) | 25,8 (18,2; 33,5) |
MDA(a) Viikko 16 Viikko 24 Viikko 52 |
37 (13,2) - |
194 (45,0)* 209 (48,5) 237 (55,0) | 31,8 (25,2; 38,5) |
63 (45,0) 67 (47,9) 74 (52,9) | 8 (6,0) | 118 (44,2)* | 38,2 (29,2; 47,2) |
| Potilaat, joiden kehon pinta-alasta ≥ 3 % läiskäpsoriaasia | (N = 140) | (N = 217) | (N = 68) | (N = 88) | (N = 176) | ||
PASI 90 Viikko 16 Viikko 24 Viikko 52 |
4 (2,9) - |
133 (61,3)* 158 (72,8) 155 (71,4) | 58,4 (49,9; 66,9) |
28 (41,2) 32 (47,1) 41 (60,3) | 6 (6,8) | 121 (68,8)* | 61,9 (51,5; 72,4) |
PASI 100 Viikko 16 Viikko 24 Viikko 52 |
3 (2,1) - |
103 (47,5) 122 (56,2) 132 (60,8) | 45,3 (36,7; 54,0) |
14 (20,6) 26 (38,2) 33 (48,5) | 4 (4,5) | 103 (58,5) | 54,0 (43,1; 64,8) |
ACR 50 / PASI 100 Viikko 16 Viikko 24 Viikko 52 |
0 - |
60 (27,6) 68 (31,3) 102 (47,0) | NC (NC, NC) |
11 (16,2) 17 (25,0) 24 (35,3) | 1 (1,1) | 59 (33,5) | 32,4 (22,3; 42,5) |
| Potilaat, joiden LDI > 0(b) | (N = 47) | (N = 90) | |||||
Ei daktyliittiä(b) Viikko 16 |
24 (51,1) |
68 (75,6)*** |
24,5 (8,4; 40,6) | ||||
| Potilaat, joiden LEI > 0(c) | (N = 106) | (N = 249) | |||||
Ei entesiittiä(c) Viikko 16 |
37 (34,9) |
124 (49,8)** |
14,9 (3,7; 26,1) | ||||
ACR 50 / PASI 100 = yhdistetty ACR 50- ja PASI 100 ‑vaste. BKZ 160 mg Q4W = 160 mg bimekitsumabia 4 viikon välein. CI = Luottamusväli. NC = Ei laskettavissa.
(a) Potilaan luokiteltiin saavuttaneen minimaalisen taudin aktiivisuuden (Minimal Disease Activity, MDA), kun hän täytti 5 seuraavista 7 kriteeristä: arkojen nivelten määrä ≤ 1; turvonneiden nivelten määrä ≤ 1; Psoriasis Activity and Severity Index -indeksi ≤ 1 tai kehon pinta-alasta läiskäpsoriaasia ≤ 3 %; potilaan kipu visuaalisella analogisella asteikolla (VAS) ≤ 15; potilaan yleisvointi VAS-asteikolla ≤ 20; Health Assessment Questionnaire Disability Index ‑indeksi ≤ 0,5; entesiittikipupisteitä ≤ 1.
(b) Perustuu yhdistettyihin tietoihin BE OPTIMAL- ja BE COMPLETE ‑tutkimuksista potilailta, joiden lähtötilanteen Leeds Dactylitis Index (LDI) ‑indeksi > 0. Potilaalla ei ollut daktyliittiä, kun LDI = 0.
c) Perustuu yhdistettyihin tietoihin BE OPTIMAL- ja BE COMPLETE ‑tutkimuksista potilailta, joiden lähtötilanteen Leeds Enthesitis Index (LEI) ‑indeksi > 0. Potilaalla ei ollut entesiittiä, kun LEI = 0.
(d) Esitetty korjaamattomina eroina.
(e) Tilastollista vertailua bimekitsumabiin tai lumelääkkeeseen ei ole tehty.
* p < 0,001 lumelääkkeeseen verrattuna, korjattu kerrannaisuuden suhteen. ** p = 0,008 lumelääkkeeseen verrattuna, korjattu kerrannaisuuden suhteen. *** p = 0,002 lumelääkkeeseen verrattuna, korjattu kerrannaisuuden suhteen. NRI:tä on käytetty. Muut viikon 16 päätepisteet ja kaikki viikkojen 24 ja 52 päätepisteet eivät ole osa sekvenssin mukaista testihierarkiaa, joten kaikki vertailut ovat nimellisiä.
BE OPTIMAL ‑tutkimuksessa bimekitsumabihoitoa käytettäessä havaittiin parannusta lähtötilanteeseen nähden kaikissa yksittäisissä ACR-mittarin komponenteissa viikon 16 kohdalla, ja nämä parannukset säilyivät viikolle 52 saakka.
Bimekitsumabihoitoa käytettäessä hoitovasteet olivat merkitsevästi parempia kuin lumelääkkeellä niinkin varhain kuin viikolla 2 ACR 20 ‑vasteella mitattuna (BE OPTIMAL, 27,1 % vs. 7,8 %, nimellinen p < 0,001) ja viikolla 4 ACR 50 ‑vasteella mitattuna (BE OPTIMAL, 17,6 % vs. 3,2 %, nimellinen p < 0,001 ja BE COMPLETE, 16,1 % vs. 1,5 %, nimellinen p < 0,001).
Kuva 5: BE OPTIMAL ‑tutkimuksen ACR 50 ‑vasteosuudet viikolle 52 saakka (NRI)
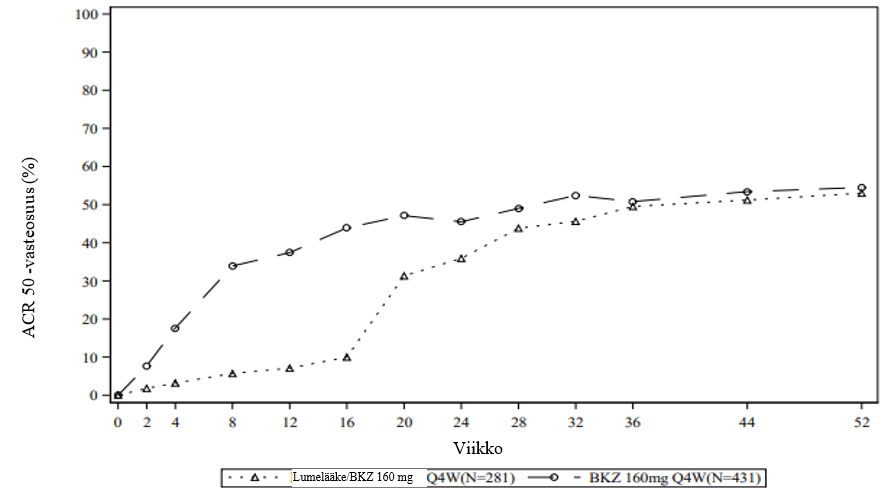
Lumelääkeryhmän potilaat vaihtoivat saamaan bimekitsumabia (BKZ) 160 mg 4 viikon välein (Q4W) viikolla 16.
Kuva 6: BE COMPLETE ‑tutkimuksen ACR 50 ‑vasteosuudet tutkimusviikolle 16 saakka (NRI)
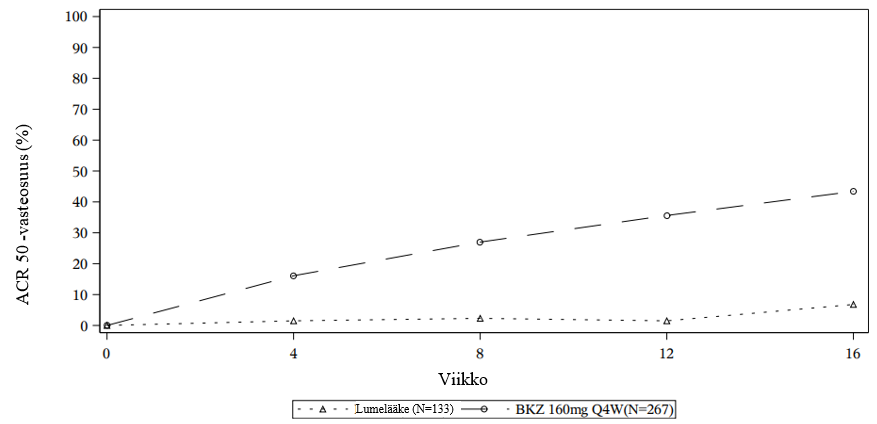
Bimekitsumabihoitoa saaneista potilaista, jotka saavuttivat ACR 50 ‑vasteen viikon 16 kohdalla BE OPTIMAL ‑tutkimuksessa, 87,2 %:lla tämä vaste säilyi viikolle 52 saakka.
Bimekitsumabin teho ja turvallisuus osoitettiin iästä, sukupuolesta, etnisestä taustasta, lähtötilanteen painosta, lähtötilanteen psoriaasin tilanteesta, lähtötilanteen CRP-arvosta, taudin kestosta ja aiemmasta perinteisten reumalääkkeiden käytöstä riippumatta. Molemmissa tutkimuksissa havaittiin samanlaiset vasteet bimekitsumabiin riippumatta siitä, saivatko potilaat samanaikaisesti perinteisiä reumalääkkeitä (mukaan lukien metotreksaatti) vai eivät.
Muokatut Psoriatic Arthritis Response Criteria (PsARC) ‑kriteerit muodostavat yhdessä erityisen vastetta kuvaavan mittarin, johon sisältyy arkojen nivelten määrä, turvonneiden nivelten määrä sekä potilaan ja lääkärin yleisarviointi. Muokatut PsARC‑kriteerit viikolla 16 saavuttaneiden potilaiden osuus oli suurempi bimekitsumabihoitoa saaneilla potilailla kuin lumelääkettä saaneilla (80,3 % vs. 40,2 % BE OPTIMAL ‑tutkimuksessa ja 85,4 % vs. 30,8 % BE COMPLETE ‑tutkimuksessa). PsARC-vaste säilyi viikkoon 52 saakka BE OPTIMAL ‑tutkimuksessa.
Radiografinen vaste
BE OPTIMAL ‑tutkimuksessa rakenteellisten vaurioiden etenemisen estoa arvioitiin radiografisesti, ja se ilmaistiin muutoksena lähtötilanteesta Van der Heijde modified total Sharp Score (vdHmTSS) ‑kokonaispistemäärässä ja tämän mittarin komponenttien eli eroosiopisteiden (Erosion Score, ES) ja nivelraon kaventumispisteiden määrässä (Joint Space Narrowing score, JSN) viikon 16 kohdalla (ks. taulukko 8).
Taulukko 8: vdHmTSS-kokonaispistemäärän muutos BE OPTIMAL ‑tutkimuksessa viikon 16 kohdalla
| Lumelääke | Bimekitsumabi 160 mg Q4W | Ero lumelääkkeeseen verrattuna (95 %:n luottamusväli)a) | |
| Potilasjoukko, jolla oli kohonnut hs-CRP-arvo ja/tai vähintään yksi luueroosio lähtötilanteessa | (N = 227) | (N = 361) | |
| Keskimääräinen muutos lähtötilanteesta (keskivirhe) | 0,36 (0,10) | 0,04 (0,05)* | –0,32 (–0,35; –0,30) |
| Koko potilasjoukko | (N = 269) | (N = 420) | |
| Keskimääräinen muutos lähtötilanteesta (keskivirhe) | 0,32 (0,09) | 0,04 (0,04)* | –0,26 (–0,29; –0,23) |
*p = 0,001 lumelääkkeeseen verrattuna. p-arvot perustuvat viitepohjaiseen imputaatioon, jossa käytetään pienimpien neliösummien keskiarvon eroa ANCOVA-mallin avulla, jossa hoito, luueroosio lähtötilanteessa ja alue ovat kiinteitä vaikutuksia ja lähtötilanteen pistemäärä toimii kovariaattina.
Viikon 16 yhteenvetotiedot perustuvat ensisijaisen analyysin ensimmäiseen tulkittujen tietojen joukkoon.
a) Korjaamattomat erot on esitetty.
Lumelääkkeeseen verrattuna bimekitsumabin todettiin viikon 16 kohdalla hidastaneen merkittävästi nivelvaurioiden etenemistä sekä potilasjoukossa, jossa potilailla oli koholla oleva hs-CRP-arvo ja/tai vähintään yksi luueroosio lähtötilanteessa, että koko potilasjoukossa. Samalla kun viitepohjainen imputaatio määritettiin puuttuvien tietojen käsittelymenetelmäksi tilastollisessa testimenettelyssä, jolla vertailtiin bimekitsumabia ja lumelääkettä, muutokset lähtötilanteesta laskettiin myös käyttäen tavanomaista moni-imputaatiota sekä potilasjoukossa, jossa potilailla oli koholla oleva hs-CRP-arvo ja/tai vähintään yksi luueroosio lähtötilanteessa, että koko potilasjoukossa viikon 16 kohdalla bimekitsumabihaarassa (keskimääräinen muutos lähtötilanteesta edempänä mainitussa potilasjoukossa 0,01 ja vastaavasti koko potilasjoukossa 0,01) ja adalimumabihaarassa (keskimääräinen muutos lähtötilanteesta edempänä mainitussa potilasjoukossa –0,05 ja vastaavasti koko potilasjoukossa -0,03). Nivelvaurioiden etenemistä estävää vaikutusta pystyttiin ylläpitämään sekä potilasjoukossa, jossa potilailla oli koholla oleva hs-CRP-arvo ja/tai vähintään yksi luueroosio lähtötilanteessa, että koko potilasjoukossa viikkoon 52 saakka bimekitsumabihaarassa (keskimääräinen muutos lähtötilanteesta edempänä mainitussa potilasjoukossa 0,10 ja vastaavasti koko potilasjoukossa 0,10) ja adalimumabihaarassa (keskimääräinen muutos lähtötilanteesta edempänä mainitussa potilasjoukossa –0,17 ja vastaavasti koko potilasjoukossa –0,12).
Niiden potilaiden havaittu prosenttiosuus, joilla ei ollut radiografista näyttöä nivelvaurion etenemisestä (määritelmänä ≤ 0,5 pisteen muutos lähtötilanteesta mTSS-mittarilla mitattuna) satunnaistamisesta viikolle 52, oli bimekitsumabilla hoidetuista potilaista 87,9 % (N = 276/314) ja lumelääkkeellä hoidetuista bimekitsumabiin vaihtaneista tutkittavista 84,8 % (N = 168/198) ja adalimumabilla hoidetuista potilaista 94,1 % (N = 96/102). Tällä potilasjoukolla oli havaittu koholla oleva hs-CRP-arvo ja/tai vähintään yksi luueroosio. Vastaavanlaisia määriä havaittiin koko potilasjoukossa (89,3 %:lla [N = 326/365] bimekitsumabihoitoa saaneista ja 87,3 %:lla [N = 207/237] lumelääkettä saaneista bimekitsumabiin vaihtaneista tutkittavista ja 94,1 %:lla [N = 111/118] adalimumabia saaneista).
Fyysinen toimintakyky ja muut terveyteen liittyvät tulokset
Sekä potilailla, joita ei ollut aiemmin hoidettu biologisilla reumalääkkeillä (BE OPTIMAL), että potilailla, jotka eivät olleet saaneet riittävää vastetta TNFα:n estäjiin tai eivät sietäneet niitä (BE COMPLETE), fyysisen toimintakyvyn todettiin viikon 16 kohdalla parantuneen merkittävästi lähtötilanteesta bimekitsumabihoitoa saaneiden ryhmässä verrattuna lumelääkettä saaneisiin potilaisiin (p < 0,001). Fyysisen toimintakyvyn muutosta arvioitiin HAQ-DI-mittarilla (pienimpien neliösummien keskiarvon muutos lähtötilanteesta: –0,3 vs. –0,1 BE OPTIMAL ‑tutkimuksessa ja –0,3 vs. 0 BE COMPLETE ‑tutkimuksessa). Molemmissa tutkimuksissa viikon 16 kohdalla kliinisesti merkittävän HAQ-DI-pistemäärän vähenemisen lähtötilanteeseen verrattuna (vähintään 0,35) saavuttaneiden potilaiden osuus oli suurempi bimekitsumabiryhmässä kuin lumelääkeryhmässä.
Bimekitsumabihoitoa saaneiden potilaiden raportoidut Short Form 36-Item Health Survey Physical Component Summary (SF-36 PCS) ‑pisteet olivat viikon 16 kohdalla parantuneet merkittävästi lähtötilanteesta verrattuna lumelääkettä saaneisiin potilaisiin (pienimpien neliösummien keskiarvon muutos lähtötilanteesta: 6,3 vs. 1.9, p < 0,001 BE OPTIMAL ‑tutkimuksessa ja 6.2 vs. 0,1 p < 0,001 BE COMPLETE ‑tutkimuksessa).
Molemmissa tutkimuksissa bimekitsumabihoitoa saaneet potilaat raportoivat uupumuksen vähentyneen merkittävästi lähtötilanteesta verrattuna lumelääkettä saaneisiin potilaisiin. Uupumusta mitattiin Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue ‑pisteillä viikon 16 kohdalla. Myös Psoriatic Arthritis Impact of Disease-12 (PsAID-12) ‑pisteiden todettiin viikon 16 kohdalla parantuneen merkittävästi lähtötilanteesta bimekitsumabihoitoa saaneessa ryhmässä verrattuna lumelääkeryhmään.
Noin 74 %:lla potilaista oli aksiaalisia oireita lähtötilanteessa (määritelmänä Bath Ankylosing Spondylitis Disease Activity Index [BASDAI] ‑pistemäärä ≥ 4), ja näillä potilailla BASDAI-pistemäärä oli parantunut enemmän lähtötilanteesta kuin lumelääkettä saaneilla viikon 16 kohdalla.
Edempänä mainittujen fyysisen toimintakyvyn mittareiden ja muiden terveyteen liittyvien tuloksien (HAQ-DI-, SF-36 PCS-, FACIT-Fatigue-, PsAID-12- ja BASDAI-pisteiden) parannukset, jotka oli saavutettu viikon 16 kohdalla, säilyivät viikolle 52 saakka BE OPTIMAL ‑tutkimuksessa.
BE OPTIMAL ‑tutkimuksen viikon 52 kohdalla 65,5 % bimekitsumabihoitoa saaneista potilaista saavutti täydellisen kynsien oireettomuuden (mNAPSI-paraneminen potilailla, joilla mNAPSI-arvo oli yli 0 lähtötilanteessa).
Aksiaalinen spondylartriitti (röntgennegatiivinen ja röntgenpositiivinen)
Bimekitsumabin tehoa ja turvallisuutta arvioitiin 586:lla aktiivista aksiaalista spondylartriittia sairastavalla aikuispotilaalla (vähintään 18-vuotiaalla) kahdessa satunnaistetussa, kaksoissokkoutetussa, lumelääkkeellä kontrolloidussa monikeskustutkimuksessa. Yksi tutkimus keskittyi röntgennegatiiviseen aksiaaliseen spondylartriittiin ja toinen selkärankareumaan, josta käytetään myös nimitystä röntgenpositiivinen aksiaalinen spondylartriitti. Ensisijainen päätetapahtuma molemmissa tutkimuksissa oli niiden potilaiden prosenttiosuus, jotka saavuttivat Assessment of SpondyloArthritis International Society (ASAS) 40 ‑vasteen viikkoon 16 mennessä. Molemmissa potilaspopulaatioissa havaittiin yhteneväisiä tuloksia.
BE MOBILE 1 ‑tutkimuksessa (AS0010) tutkittiin 254:ää aktiivista röntgennegatiivista aksiaalista spondylartriittia sairastavaa potilasta. Potilailla oli aksiaalinen spondylartriitti (ikä oireiden alkamisen aikaan korkeintaan 45 vuotta), joka vastasi ASAS-luokittelukriteereitä. Heillä oli aktiivinen tauti (Bath Ankylosing Spondylitis Disease Activity Index [BASDAI] ‑tautiaktiivisuusindeksi vähintään 4) ja selkärangan kipua, jonka voimakkuus oli numeroasteikolla (NRS, Numeric Rating Scale) 0–10 mitattuna vähintään 4 (BASDAI kohta: 2), eikä heillä ollut näyttöä risti-suoliluunivelen (SI-nivel) radiografisista muutoksista, jotka olisivat täyttäneet selkärankareuman muokatut New Yorkin kriteerit. Potilailla oli myös objektiivisia merkkejä tulehduksesta, kuten kohonnut C-reaktiivisen proteiinin (CRP) pitoisuus, ja/tai magneettikuvauksessa (MRI) todettu risti-suoliluunivelen tulehdus sekä aiempi riittämätön vaste kahteen eri tulehduskipulääkkeeseen (NSAID) tai intoleranssi tulehduskipulääkkeille tai vasta-aihe niiden käytölle. Potilaat satunnaistettiin (1:1) saamaan joko bimekitsumabia 160 mg neljän viikon välein viikkoon 52 asti tai lumelääkettä viikkoon 16 asti, jonka jälkeen lumelääkeryhmä sai bimekitsumabia 160 mg neljän viikon välein viikkoon 52 asti. Lähtötilanteessa potilailla oli ollut röntgennegatiivisen aksiaalisen spondylartriitin oireita keskimäärin 9 vuoden ajan (mediaani 5,5 vuotta). 10,6 %:a potilaista oli aiemmin hoidettu TNFα:n estäjällä.
BE MOBILE 2 ‑tutkimuksessa (AS0011) tutkittiin 332:ta potilasta, joilla oli aktiivinen selkärankareuma. Selkärankareuma määriteltiin dokumentoitujen radiologisten todisteiden (röntgenkuvien) avulla, joiden tuli täyttää selkärankareuman muokatut New Yorkin kriteerit. Potilailla oli aktiivinen tauti (BASDAI vähintään 4) ja selkärangan kipua, jonka voimakkuus oli numeroasteikolla 0–10 mitattuna vähintään 4 (BASDAI kohta: 2). Potilailta vaadittiin aiempaa riittämätöntä vastetta kahteen eri tulehduskipulääkkeeseen tai intoleranssia tulehduskipulääkkeille tai vasta-aihetta niiden käytölle. Potilaat satunnaistettiin (2:1) saamaan joko bimekitsumabia 160 mg neljän viikon välein viikkoon 52 asti tai lumelääkettä viikkoon 16 asti, jonka jälkeen lumelääkeryhmä sai bimekitsumabia 160 mg neljän viikon välein viikkoon 52 asti. Lähtötilanteessa potilailla oli ollut selkärankareuman oireita keskimäärin 13,5 vuoden ajan (mediaani 11 vuotta). 16,3 %:a potilaista oli aiemmin hoidettu TNFα:n estäjällä.
Kliininen vaste
Bimekitsumabihoidolla saavutettiin merkittävää parannusta röntgennegatiivista aksiaalista spondylartriittia ja selkärankareumaa sairastavien potilaiden oireissa ja löydöksissä sekä taudin aktiivisuudessa lumelääkehoitoon verrattuna 16 viikon hoidon jälkeen (katso taulukko 9). Kliiniset vasteet säilyivät viikkoon 52 asti molemmissa potilaspopulaatioissa, kuten taulukossa 9 esitetyt päätetapahtumat osoittavat.
Taulukko 9: Kliiniset vasteet BE MOBILE 1- ja BE MOBILE 2 ‑tutkimuksissa
| BE MOBILE 1 (röntgennegatiivinen aksiaalinen spondylartriitti) | BE MOBILE 2 (selkärankareuma) | |||||
Lumelääke (N = 126) n (%) | BKZ 160 mg Q4W (N = 128) n (%) | Ero lumelääkkeeseen verrattuna (95 %:n luottamusväli)a) | Lumelääke (N = 111) n (%) | BKZ 160 mg Q4W (N = 221) n (%) | Ero lumelääkkeeseen verrattuna (95 %:n luottamusväli)a) | |
ASAS 40 Viikko 16 Viikko 52 | 27 (21,4) |
61 (47,7)* 78 (60,9) | 26,2 (14,9; 37,5) | 25 (22,5) |
99 (44,8)* 129 (58,4) | 22,3 (11,5; 33,0) |
ASAS 40 potilailla, jotka eivät olleet saaneet TNFα:n estäjää Viikko 16 Viikko 52 |
(N = 109) 25 (22,9) |
(N = 118) 55 (46,6) 73 (61,9) |
24,8 (12,4; 37,1) |
(N = 94) 22 (23,4) |
(N = 184) 84 (45,7)* 108 (58,7) |
22,3 (10,5; 34,0) |
ASAS 20 Viikko 16 Viikko 52 | 48 (38,1) |
88 (68,8)* 94 (73,4) | 30,7 (19,0; 42,3) | 48 (43,2) |
146 (66,1)* 158 (71,5) | 22,8 (11,8; 33,8) |
ASAS – osittainen remissio Viikko 16 Viikko 52 | 9 (7,1) |
33 (25,8)* 38 (29,7) | 18,6 (9,7; 27,6) | 8 (7,2) |
53 (24,0)* 66 (29,9) | 16,8 (8,1; 25,5) |
ASDAS – merkittävä parannus Viikko 16 Viikko 52 | 9 (7,1) |
35 (27,3)* 47 (36,7) | 20,2 (11,2; 29,3) | 6 (5,4) |
57 (25,8)* 71 (32,1) | 20,4 (11,7; 29,1) |
BASDAI-50 Viikko 16 Viikko 52 | 27 (21,4) |
60 (46,9) 69 (53,9) | 25,3 (14,0; 36,6) | 29 (26,1) |
103 (46,6) 119 (53,8) | 20,5 (9,6; 31,4) |
BKZ 160 mg Q4W = bimekitsumabi 160 mg 4 viikon välein. ASDAS (Ankylosing Spondylitis Disease Activity Score) = Selkärankareuman aktiivisuuspisteet.
Käytössä on NRI (Non-Responder Imputation).
a) Esitetty korjaamattomina eroina.
*p < 0,001 lumelääkkeeseen verrattuna, korjattu kerrannaisuuden suhteen.
Vähintään alhaisen tautiaktiivisuuden (ASDAS < 2,1) viikolla 16 saavuttaneiden potilaiden (joilla tauti oli inaktiivinen tai tautiaktiivisuus oli alhainen) osuus BE MOBILE 1 ‑tutkimuksessa oli 46,1 % bimekitsumabiryhmässä verrattuna 21,1 %:iin lumelääkeryhmässä (moni-imputointi). Viikolla 52 bimekitsumabiryhmän potilaista 61,6 % saavutti vähintään alhaisen tautiaktiivisuuden (ASDAS < 2,1), ja näistä 25,2 %:lla tauti oli inaktiivinen (ASDAS < 1,3).
Vähintään alhaisen tautiaktiivisuuden (ASDAS < 2,1) viikolla 16 saavuttaneiden potilaiden (joilla tauti oli inaktiivinen tai tautiaktiivisuus oli alhainen) osuus BE MOBILE 2 ‑tutkimuksessa oli 44,8 % bimekitsumabiryhmässä verrattuna 17,4 %:iin lumelääkeryhmässä (moni-imputointi). Viikolla 52 bimekitsumabiryhmän potilaista 57,1 % saavutti vähintään alhaisen tautiaktiivisuuden (ASDAS < 2,1), ja näistä 23,4 %:lla tauti oli inaktiivinen (ASDAS < 1,3).
Kaikki neljä ASAS 40 ‑vasteen osa-aluetta (selkärangan kokonaiskipu, aamujäykkyys, Bath Ankylosing Spondylitis Functional Index [BASFI] ‑indeksi ja potilaan yleinen arvio sairauden aktiivisuudesta [PGADA]) paranivat bimekitsumabihoidon myötä ja vaikuttivat yleiseen ASAS 40 ‑vasteeseen viikolla 16. Nämä saavutetut parannukset säilyivät viikkoon 52 asti molemmissa potilaspopulaatioissa.
Muiden tehoa kuvaavien mittareiden parannukset on esitetty taulukossa 10.
Taulukko 10: Muut tehoa kuvaavat mittarit BE MOBILE 1- ja BE MOBILE 2 ‑tutkimuksissa
| BE MOBILE 1 (röntgennegatiivinen aksiaalinen spondylartriitti) | BE MOBILE 2 (selkärankareuma) | |||
Lumelääke (N = 126) | BKZ 160 mg Q4W (N = 128) | Lumelääke (N = 111) | BKZ 160 mg Q4W (N = 221) | |
Selkäkipu yöllä Lähtötilanne Keskimääräinen muutos lähtötilanteesta viikolla 16 Keskimääräinen muutos lähtötilanteesta viikolla 52 | 6,7 –1,7 |
6,9 –3,6* –4,3 | 6,8 –1,9 |
6,6 –3,3* –4,1 |
BASDAI Lähtötilanne Keskimääräinen muutos lähtötilanteesta viikolla 16 Keskimääräinen muutos lähtötilanteesta viikolla 52 | 6,7 –1,5 |
6,9 –3,1* –3,9 | 6,5 –1,9 |
6,5 –2,9* –3,6 |
BASMI Lähtötilanne Keskimääräinen muutos lähtötilanteesta viikolla 16 Keskimääräinen muutos lähtötilanteesta viikolla 52 | 3,0 –0,1 |
2,9 –0,4 –0,6 | 3,8 –0,2 |
3,9 –0,5** –0,7 |
hs-CRP (mg/l) Lähtötilanne (geometrinen keskiarvo) Suhde lähtötilanteeseen verrattuna viikolla 16 Suhde lähtötilanteeseen verrattuna viikolla 52 | 5,0 0,8 |
4,6 0,4 0,4 | 6,7 0,9 |
6,5 0,4 0,3 |
BASMI (Bath Ankylosing Spondylitis Metrology Index) = Selkärangan liikkuvuutta kuvaava indeksi. Hs-CRP = erittäin herkkä C-reaktiivinen proteiini.
Käytössä on moni-imputointi.
* p < 0,001 viitepohjainen imputointi lumelääkkeeseen verrattuna, korjattu kerrannaisuuden suhteen.
** p < 0,01 viitepohjainen imputointi lumelääkkeeseen verrattuna, korjattu kerrannaisuuden suhteen.
Bimekitsumabihoitoon liittyi nopea tehon ilmaantuminen sekä röntgennegatiivista aksiaalista spondylartriittia että selkärankareumaa sairastavassa potilaspopulaatiossa.
Bimekitsumabihoitoa saaneiden potilaiden ryhmässä ASAS 40 ‑hoitovasteen saavuttaneiden potilaiden prosenttiosuus oli suurempi kuin lumelääkehoitoa saaneiden ryhmässä jo viikolla 1 BE MOBILE 1 ‑tutkimuksessa (16,4 % vs. 1,6 %, nimellinen p < 0,001) ja viikolla 2 BE MOBILE 2 ‑tutkimuksessa (16,7 % vs. 7,2 %, nimellinen p < 0,019).
Bimekitsumabihoitoon liittyi myös nopea systeemisen tulehduksen väheneminen hs-CRP-pitoisuuksien mittausten perusteella jo viikolla 2 sekä röntgennegatiivista aksiaalista spondylartriittia että selkärankareumaa sairastavissa potilaspopulaatioissa, ja nimellinen p-arvo oli < 0,001 molemmissa tutkimuksissa.
Kuva 7: BE MOBILE 1 ‑tutkimuksen ASAS 40-vasteosuudet aikajanalla viikkoon 52 asti (NRI)

Lumelääkeryhmän potilaat vaihtoivat saamaan bimekitsumabia (BKZ) 160 mg 4 viikon välein (Q4W) viikolla 16
Kuva 8: BE MOBILE 2 ‑tutkimuksen ASAS 40 ‑vasteosuudet aikajanalla viikkoon 52 asti (NRI)
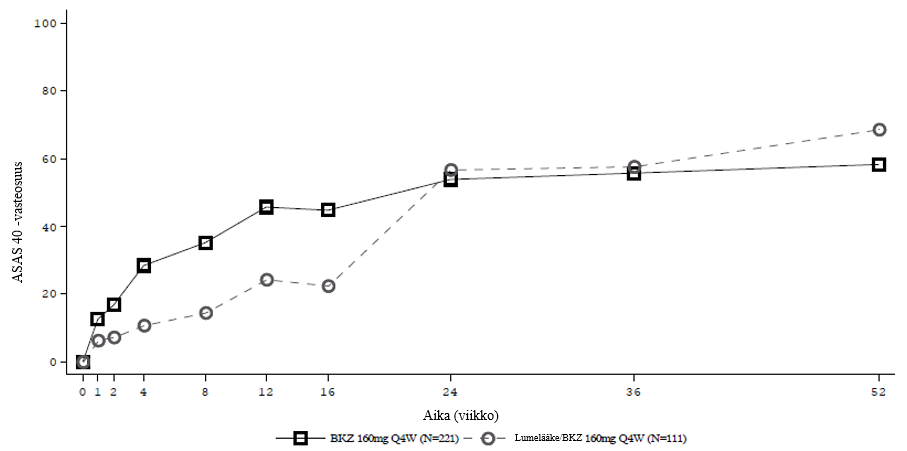
Lumelääkeryhmän potilaat vaihtoivat saamaan bimekitsumabia (BKZ) 160 mg 4 viikon välein (Q4W) viikolla 16
BE MOBILE 1- ja BE MOBILE 2 ‑tutkimusten integroidussa analyysissä todettiin, että bimekitsumabilla hoidetuista ASAS 40 ‑vasteen viikolla 16 saavuttaneista potilaista 82,1 %:lla vaste säilyi viikkoon 52 asti.
Bimekitsumabin teho osoitettiin iästä, sukupuolesta, etnisestä taustasta, sairauden kestosta, lähtötilanteen tulehdustilasta, lähtötilanteen tautiaktiivisuudesta (ASDAS-pisteytyksellä mitattuna) ja samanaikaisesti käytetyistä perinteisistä reumalääkkeistä riippumatta.
Samanlainen ASAS 40 ‑vaste todettiin potilailla aiemmasta TNFα:n estäjähoidosta riippumatta.
16 viikon hoidon jälkeen bimekitsumabilla hoidettujen (entesiitti todettu lähtötilanteessa) entesiittiin täydellisen hoitovasteen saaneiden potilaiden prosentuaalinen osuus (NRI) Maastricht Ankylosing Spondylitis Enthesitis (MASES) ‑indeksin perusteella arvioituna oli suurempi kuin lumelääkeryhmässä hoidettujen osuus. (BE MOBILE 1: 51,1 % vs. 23,9 % ja BE MOBILE 2: 51,5 % vs. 32,8 %). Bimekitsumabihoidolla saavutettu entesiitin täydellinen oireettomuus säilyi viikkoon 52 asti molemmissa tutkimuksissa (BE MOBILE 1: 54,3 % ja BE MOBILE 2: 50,8 %).
Tulehduksen vähentyminen
Bimekitsumabi vähensi tulehdusta hs-CRP-pitoisuudella mitattuna (katso taulukko 10) ja magneettikuvauksella arvioituna yhdessä kuvantamisosatutkimuksessa. Tulehduksen löydökset arvioitiin magneettikuvauksella lähtötilanteessa ja viikolla 16. Tulokset ilmoitettiin risti-suoliluunivelten osalta Spondyloarthritis Research Consortium of Canada (SPARCC) ‑pisteiden muutoksena ja selkärangan osalta Ankylosing Spondylitis spine Magnetic Resonance Imagine Activity (ASspiMRI-a-pisteytys, Berliinin muokkaus) –pisteiden muutoksena verrattuna lähtötilanteeseen. Tulehduslöydösten vähenemistä havaittiin sekä risti-suoliluunivelissä että selkärangassa bimekitsumabilla hoidetuilla potilailla verrattuna lumelääkkeellä hoidettuihin potilaisiin (katso taulukko 11). Bimekitsumabin tulehdusta hillitsevä vaikutus hs-CRP-pitoisuudella mitattuna ja magneettikuvauksen avulla arvioituna säilyi viikkoon 52 asti.
Taulukko 11: Tulehduksen vähentyminen magneettikuvauksella arvioituna BE MOBILE 1- ja BE MOBILE 2 ‑tutkimuksissa
| BE MOBILE 1 (röntgennegatiivinen aksiaalinen spondylartriitti) | BE MOBILE 2 (selkärankareuma) | |||
| Lumelääke | BKZ 160 mg Q4W | Lumelääke | BKZ 160 mg Q4W | |
SPARCC-pisteet Keskimääräinen muutos lähtötilanteestaa) viikolla 16 Keskimääräinen muutos lähtötilanteestaa) viikolla 52 | –1,56 (N = 62) |
–6,15 (N = 78) –7,57 (N = 67) | 0,59 (N = 46) |
–4,51 (N = 81) –4,67 (N = 78) |
ASspiMRI-a (Berliinin muokkaus) ‑pisteet Keskimääräinen muutos lähtötilanteestaa) viikolla 16 Keskimääräinen muutos lähtötilanteestaa) viikolla 52 | 0,03 (N = 60) |
–0,36 (N = 74) –0,70 (N = 65) | –0,34 (N = 46) |
–2,23 (N = 81) –2,38 (N = 77) |
a) Muutokset lähtötilanteen arvoista perustuvat havaittuihin tapauksiin viikolla 52 keskitetysti luettuina arvioidusta aineistosta.
Fyysinen toimintakyky ja muut terveyteen liittyvät tulokset
Bimekitsumabilla hoidettujen potilaiden fyysinen toimintakyky parani BASFI-pisteytyksellä arvioituna merkittävästi lähtötilanteesta lumelääkkeeseen verrattuna (pienimpien neliösummien keskiarvon muutos lähtötilanteesta viikolla 16 BE MOBILE 1‑tutkimuksessa: –2,4 vs. –0,9, p < 0,001, ja BE MOBILE 2 ‑tutkimuksessa: –2,0 vs. –1,0, p < 0,001). Bimekitsumabilla hoidettujen potilaiden raportoidut SF-36 PCS ‑pisteet paranivat merkittävästi lähtötilanteesta verrattuna lumelääkkeellä hoidettuihin potilaisiin (pienimpien neliösummien keskiarvon muutos lähtötilanteesta viikolla 16 BE MOBILE 1 ‑tutkimuksessa: 9,3 vs. 5,4, p < 0,001 ja BE MOBILE 2 ‑tutkimuksessa: 8,5 vs. 5,2, p < 0,001).
Bimekitsumabilla hoidettujen potilaiden terveyteen liittyvä elämänlaatu parani merkittävästi lähtötilanteesta verrattuna lumelääkkeellä hoidettuihin AS Quality of Life Questionnaire (ASQoL) ‑kyselyn perusteella (pienimpien neliösummien keskiarvon muutos lähtötilanteesta viikolla 16 BE MOBILE 1 ‑tutkimuksessa: –4,9 vs. –2,3, p < 0,001 ja BE MOBILE 2 ‑tutkimuksessa: –4,6 vs. –3,0, p < 0,001), ja heidän kokemansa väsymys väheni merkittävästi FACIT-väsymyspisteytyksellä arvioituna (keskimääräinen muutos lähtötasosta viikolla 16 BE MOBILE 1 ‑tutkimuksessa: 8,5 bimekitsumabilla hoidetut vs. 3,9 lumelääkkeellä hoidetut ja BE MOBILE 2 ‑tutkimuksessa: 8,4 bimekitsumabilla hoidetut vs. 5,0 lumelääkkeellä hoidetut).
Edempänä mainittujen fyysisen toimintakyvyn mittareiden ja muiden terveyteen liittyvien tuloksien (BASFI, SF-36 PCS, ASQoL ja FACIT-väsymyspisteet) parannukset, jotka oli saavutettu viikon 16 kohdalla, säilyivät molemmissa tutkimuksissa viikkoon 52 asti.
Nivelen ulkopuoliset taudin ilmenemismuodot
BE MOBILE 1 (röntgennegatiivinen aksiaalinen spondylartriitti) ‑tutkimuksesta ja BE MOBILE 2 (selkärankareuma) ‑tutkimuksesta viikon 16 kohdalla yhdistetyssä potilasaineistossa uveiittiin sairastuneiden potilaiden osuus oli pienempi bimekitsumabiryhmässä (0,6 %) kuin lumelääkeryhmässä (4,6 %). Uveiitin esiintyvyys pysyi alhaisena pitkäaikaisen bimekitsumabihoidon aikana (1,2 tapausta 100:aa potilasvuotta kohden vaiheen 2/3 tutkimuksista yhdistetyssä potilasaineistossa).
Hidradenitis suppurativa (HS-tauti)
Bimekitsumabin turvallisuutta ja tehoa arvioitiin 1 014:llä keskivaikeaa tai vaikeaa HS-tautia sairastavalla (vähintään 18-vuotiaalla) aikuispotilaalla kahdessa vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumelääkkeellä kontrolloidussa monikeskustutkimuksessa (HS0003 – BE HEARD I ja HS0004 – BE HEARD II). Potilailla oli vähintään kuusi kuukautta aikaisemmin todettu HS-tauti (Hurley-asteen II tai III tauti) ja ≥ 5 tulehduksellista leesiota (paiseiden ja tulehduksellisten kyhmyjen määrä yhteensä), ja he olivat aiemmin saaneet riittämättömän vasteen HS-taudin hoitoon käytettäviin systeemisiin antibiootteihin.
Molemmissa tutkimuksissa potilaat satunnaistettiin (suhteessa 2:2:2:1) saamaan bimekitsumabia 320 mg kahden viikon välein 48 viikon ajan tai bimekitsumabia 320 mg neljän viikon välein 48 viikon ajan tai bimekitsumabia 320 mg kahden viikon välein viikolle 16 saakka, jonka jälkeen bimekitsumabia 320 mg neljän viikon välein viikolle 48 saakka, tai lumelääkettä viikolle 16 saakka, jonka jälkeen bimekitsumabia 320 mg kahden viikon välein viikolle 48 saakka. Samanaikainen suun kautta otettavan antibiootin käyttö oli sallittua, jos potilas käytti vakaa-annoksista doksisykliiniä, minosykliiniä tai vastaavaa systeemistä tetrasykliiniä 28 vuorokauden ajan ennen lähtötilannetta.
Ensisijaisena päätetapahtumana molemmissa tutkimuksissa oli HiSCR50-vaste (Hidradenitis Suppurativa Clinical Response) viikolla 16, eli vähintään 50 %:n väheneminen paiseiden ja tulehduksellisten kyhmyjen kokonaismäärässä ilman paiseiden tai vuotavien fistelien määrän lisääntymistä lähtötilanteeseen nähden.
Lähtötilanteessa HS-tautia sairastavien potilaiden ominaisuudet olivat samanlaisia molemmissa tutkimuksissa ja vastasivat keskivaikeaa tai vaikeaa tautia. Potilaiden taudin keston mediaani oli 5,3 vuotta (keskiarvo 8,0 vuotta). Hurley-asteen II potilaita oli 55,7 % (50,3 % HS0003-tutkimuksessa ja 61,1 % HS0004-tutkimuksessa) ja Hurley-asteen III potilaita 44,3 % (49,7 % HS0003-tutkimuksessa ja 38,9 % HS0004-tutkimuksessa). Potilaista 8,5 % sai HS-tautiin samanaikaista antibioottihoitoa. Keskimääräinen lähtötilanteen DLQI (Dermatology Life Quality Index) ‑kokonaispistemäärä oli 11,4. 56,8 % potilaista oli naisia, ja kaikkien potilaiden keskimääräinen ikä oli 36,6 vuotta. Potilaista 79,7 % oli valkoihoisia ja 10,8 % mustaihoisia tai afroamerikkalaisia. Potilaista 45,6 % tupakoi tutkimushetkellä.
Kliininen vaste
Bimekitsumabihoito oli saanut aikaan kliinisesti merkittävää taudin aktiivisuuden lievittymistä lumelääkkeeseen verrattuna viikon 16 kohdalla. Keskeiset tehoa koskevat tulokset on esitetty taulukoissa 12 ja 13. Taulukon 12 tulokset kuvastavat ennalta määritettyä primäärianalyysiä, jossa mikä tahansa systeemisten antibioottien käyttö ennen viikkoa 16 johti vastetiedon puuttumisen tulkitsemiseen niin, että potilaan vastetieto katsottiin puuttuvaksi (non-responder imputation, NRI). Taulukossa 13 HS-taudin salvage-hoitoon vaadittu systeeminen antibioottihoito johti siihen, että potilaan vastetieto katsottiin puuttuvaksi.
Taulukko 12: Vaste BE HEARD I- ja BE HEARD II ‑tutkimuksissa viikon 16 kohdalla – primäärianalyysia
| BE HEARD I | BE HEARD II | |||||
Lumelääke (N = 72) | BKZ 320 mg Q4W (N = 144)
| BKZ 320 mg Q2W (N = 289)
| Lumelääke (N = 74)
| BKZ 320 mg Q4W (N = 144)
| BKZ 320 mg Q2W (N = 291)
| |
HiSCR50, % (95 %:n luottamusväli) | 28,7 (18,1; 39,3) | 45,3 (36,8; 53,8) | 47,8* (41,8; 53,7) | 32,2 (21,4; 42,9) | 53,8* (45,4; 62,1) | 52,0* (46,1; 57,8) |
HiSCR75, % (95 %:n luottamusväli) | 18,4 (9,3; 27,5) | 24,7 (17,3; 32,1) | 33,4* (27,8; 39,1) | 15,6 (7,2; 24,0) | 33,7* (25,7; 41,7) | 35,7* (30,1; 41,3) |
HSSDD Pahimpaan ihon kipuun liittyvä vasteb % (95 %:n luottamusväli) |
15,0 (3,6; 26,5) |
22,1 (12,7; 31,4) |
32,3 (25,1; 39,5) |
10,9 (1,7; 20,1) |
28,6 (19,5; 37,8) |
31,8 (25,1; 38,4) |
a) Potilaiden, jotka käyttivät systeemisiä antibiootteja mistä tahansa syystä tai jotka keskeyttivät hoidon haittatapahtuman tai tehon puutteen takia, katsotaan jääneen ilman vastetta kaikilla myöhemmillä käynneillä vastemuuttujien osalta (tai jatkuvien muuttujien tapauksessa heidän tiedoilleen tehdään moni-imputointi). Muut puuttuvat tiedot imputoitiin moni-imputoinnin avulla.
b) Ihon kipuun liittyvä vaste määritettiin potilaan kliinisesti merkittävän muutoksen kynnysarvon perusteella (määritelmänä vähintään kolmen pisteen väheneminen lähtötilanteeseen nähden HSSDD [Hidradenitis Suppurativa Symptom Daily Diary] ‑pisteissä) viikon 16 kohdalla niillä tutkittavilla, joiden lähtötilanteen pistemäärä oli ≥ 3. BE HEARD I ‑tutkimuksessa: lumelääke N = 46, bimekitsumabi 4 viikon välein N = 103 ja bimekitsumabi 2 viikon välein N = 190; BE HEARD II ‑tutkimuksessa: lumelääke N = 49, bimekitsumabi 4 viikon välein N = 108 ja bimekitsumabi 2 viikon välein N = 209.
*p < 0,025 lumelääkkeeseen verrattuna, korjattu kerrannaisuuden suhteen.
Taulukko 13: Vaste BE HEARD I- ja BE HEARD II ‑tutkimuksissa viikon 16 kohdalla – lisäanalyysia
| BE HEARD I | BE HEARD II | |||||
Lumelääke (N = 72) | BKZ 320 mg Q4W (N = 144)
| BKZ 320 mg Q2W (N = 289)
| Lumelääke (N = 74)
| BKZ 320 mg Q4W (N = 144)
| BKZ 320 mg Q2W (N = 291)
| |
HiSCR50, % (95 %:n luottamusväli) | 34,0 (23,0; 45,1) | 53,5 (45,0; 62,0) | 55,2 (49,2; 61,1) | 32,3 (21,5; 43,1) | 58,5 (50,2; 66,8) | 58,7 (53,0; 64,5) |
HiSCR75, % (95 %:n luottamusväli) | 18,3 (9,3; 27,3) | 31,4 (23,5; 39,4) | 38,7 (32,9; 44,5) | 15,7 (7,2; 24,1) | 36,4 (28,3; 44,5) | 39,7 (34,0; 45,5) |
HSSDD Pahimpaan ihon kipuun liittyvä vasteb % (95 %:n luottamusväli) |
16,1 (4,5; 27,8) |
25,3 (16,0; 34,7) |
36,7 (29,4; 44,1) |
11,1 (1,8; 20,4) |
32,9 (23,5; 42,4)´ |
36,7 (29,8; 43,6) |
a)Post-hoc‑analyysi (modified nonresponder imputation, mNRI): Potilaiden, jotka tutkijan määritelmän mukaan käyttivät systeemisiä antibiootteja HS-taudin salvage-hoitoon tai jotka keskeyttivät hoidon haittatapahtuman tai tehon puutteen takia, katsotaan jääneen ilman vastetta kaikilla myöhemmillä käynneillä vastemuuttujien osalta (tai jatkuvien muuttujien tapauksessa heidän tiedoilleen tehdään moni-imputointi). Muut puuttuvat tiedot imputoitiin moni-imputoinnin avulla.
b) Ihon kipuun liittyvä vaste määritettiin potilaan kliinisesti merkittävän muutoksen kynnysarvon perusteella (määritelmänä vähintään kolmen pisteen väheneminen lähtötilanteeseen nähden HSSDD [Hidradenitis Suppurativa Symptom Daily Diary] ‑pisteissä) viikon 16 kohdalla niillä tutkittavilla, joiden lähtötilanteen pistemäärä oli ≥ 3. BE HEARD I ‑tutkimuksessa: lumelääke N = 46, bimekitsumabi 4 viikon välein N = 103 ja bimekitsumabi 2 viikon välein N = 190; BE HEARD II ‑tutkimuksessa: lumelääke N = 49, bimekitsumabi 4 viikon välein N = 108 ja bimekitsumabi 2 viikon välein N = 209.
Molemmissa tutkimuksissa bimekitsumabin vaikutus alkoi jo viikolla 2.
Bimekitsumabin teho osoitettiin riippumatta aiemmasta biologisesta hoidosta ja systeemisten antibioottien käytöstä lähtötilanteessa.
Kliiniset vasteet säilyivät viikolle 48 saakka molemmissa tutkimuksissa (ks. taulukko 14).
Taulukko 14: Vaste BE HEARD I - ja BE HEARD II ‑tutkimuksissa viikon 48 kohdalla (mNRI*)
| BE HEARD I | BE HEARD II | |||||
BKZ 320 mg Q4W/Q4W (N = 144) | BKZ 320 mg Q2W/Q4W (N = 146) | BKZ 320 mg Q2W/Q2W (N = 143) | BKZ 320 mg Q4W/Q4W (N = 144) | BKZ 320 mg Q2W/Q4W (N = 146) | BKZ 320 mg Q2W/Q2W (N = 145) | |
| HiSCR50, % | 52,7 | 61,4 | 60,6 | 63,2 | 63,8 | 60,6 |
| HiSCR75, % | 40,5 | 44,7 | 47,6 | 53,9 | 48,8 | 47,3 |
* mNRI (modified nonresponder imputation, mNRI): Potilaiden, jotka tutkijan määritelmän mukaan käyttivät systeemisiä antibiootteja HS-taudin salvage-hoitona tai jotka keskeyttivät hoidon haittatapahtuman tai tehon puutteen takia, katsotaan jääneen ilman vastetta kaikilla myöhemmillä käynneillä vastemuuttujien osalta (tai jatkuvien muuttujien tapauksessa heidän tiedoilleen tehdään moni-imputointi). Muut puuttuvat tiedot imputoitiin moni-imputoinnin avulla. Tätä puuttuvien tietojen eksploratiivista käsittelytapaa käytettiin post hoc.
Terveyteen liittyvä elämänlaatu
Molemmissa tutkimuksissa bimekitsumabilla hoidettujen potilaiden terveyteen liittyvä elämänlaatu parani merkitsevästi lumelääkkeeseen verrattuna. Terveyteen liittyvää elämänlaatua mitattiin standardimuotoisella ihospesifisellä DLQI-kyselyllä (taulukko 15).
Taulukko 15: Terveyteen liittyvä elämänlaatu BE HEARD I- ja BE HEARD II ‑tutkimuksissa viikon 16 kohdalla
| BE HEARD I | BE HEARD II | |||||
Lumelääke (N = 72) | BKZ 320 mg Q4W (N = 144) | BKZ 320 mg Q2W (N = 289) | Lumelääke (N = 74) | BKZ 320 mg Q4W (N = 144) | BKZ 320 mg Q2W (N = 291) | |
DLQI-kokonaispistemäärä Keskimääräinen muutos lähtötilanteesta (keskivirhe) | –2,9 (0,8) | –5,4 (0,6) | –5,0 (0,4) | –3,2 (0,6) | –4,5 (0,5) | –4,6 (0,3) |
DLQI-kokonaispistemäärän vaihteluväli oli 0–30; korkeammat pistemäärät tarkoittavat huonompaa terveyteen liittyvää elämänlaatua.
Potilaisiin, jotka tutkijan määritelmän mukaan käyttivät systeemisiä antibiootteja HS-taudin salvage-hoitoon tai jotka keskeyttivät hoidon haittatapahtuman tai tehon puutteen takia, sovellettiin moni-imputointia. Muut puuttuvat tiedot imputoitiin moni-imputoinnin avulla.
Terveyteen liittyvän elämänlaadun mittareissa viikon 16 kohdalla bimekitsumabilla saavutettu elämänlaadun paraneminen säilyi viikolle 48 saakka.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Bimzelx-valmisteen käytöstä psoriaasin, kroonisen idiopaattisen artriitin ja hidradenitis suppurativan (HS-taudin) hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Bimekitsumabin farmakokineettiset ominaisuudet olivat samanlaisia läiskäpsoriaasia, nivelpsoriaasia ja aksiaalista spondylartriittia (röntgennegatiivista ja röntgenpositiivista) sairastavilla potilailla.
Populaatiofarmakokineettisten analyysien perusteella ja käyttäen viitepainona 90 kg:aa bimekitsumabin näennäinen puhdistuma oli 31 % suurempi ja jakautumistilavuus 18 % suurempi hidradenitis suppurativaa (HS-tautia) sairastavilla kuin edellä mainituissa käyttöaiheissa. HS-taudin osalta arvioitu puoliintumisaika oli 20 vuorokautta. Annoksella 320 mg neljän viikon välein vakaan tilan jäännöspitoisuuden mediaani oli noin 40 % pienempi HS-tautia sairastavilla potilailla kuin muissa käyttöaiheissa.
Imeytyminen
Populaatiofarmakokineettisen analyysin perusteella läiskäpsoriaasipotilaille ihon alle annetun 320 mg:n kerta-annoksen jälkeen 3–4 päivän kuluttua annoksen antamisesta plasmassa saavutettu bimekitsumabin huippupitoisuus oli 25 (12–50) mikrog/ml (mediaanin [2,5. ja 97,5. prosenttipiste]).
Populaatiofarmakokineettinen analyysi osoitti, että bimekitsumabi imeytyi siten, että sen absoluuttinen biologinen hyötyosuus terveillä vapaaehtoisilla oli keskimäärin 70,1 %.
Simuloitujen tietojen perusteella vakaan tilan huippu- ja jäännöspitoisuuksien mediaanit (2,5. ja 97,5. prosenttipiste) ihon alle 4 viikon välein annetun 320 mg:n annoksen jälkeen ovat 43 (20–91) mikrog/ml ja 20 (7–50) mikrog/ml; vakaa tila saavutetaan annoksia 4 viikon välein annettaessa noin 16 viikon jälkeen. Populaatiofarmakokineettinen analyysi osoitti, että huippupitoisuudet potilaiden plasmassa ja käyrän alle jäävä pinta-ala (AUC) suurenivat neljän viikon välein toistetun annon jälkeen 1,74-kertaisiksi kerta-annoksen jälkeiseen altistukseen verrattuna.
Vaihdettaessa 320 mg:n annoksen 4 viikon välein annosta 320 mg:n annoksen 8 viikon välein antoon viikolla 16 vakaa tila saavutetaan noin 16 viikkoa vaihdon jälkeen. Plasman huippupitoisuuden (2,5. ja 97,5. prosenttipiste) mediaani on 30 (14–60) mikrog/ml ja jäännöspitoisuuden mediaani on 5 (1−16) mikrog/ml.
Jakautuminen
Populaatiofarmakokineettisten analyysien perusteella jakautumistilavuuden (V/F) mediaani (variaatiokerroin, %) oli läiskäpsoriaasipotilailla vakaassa tilassa 11,2 (30,5 %) l.
Biotransformaatio
Bimekitsumabi on monoklonaalinen vasta-aine ja se oletettavasti hajoaa endogeenisten immunoglobuliinien tavoin katabolisten reittien kautta pieniksi peptideiksi ja aminohapoiksi.
Eliminaatio
Populaatiofarmakokineettisten analyysien perusteella bimekitsumabin laskennallisen puhdistuman (CL/F) mediaani (variaatiokerroin, %) oli 0,337 l/vrk (32,7 %) ja bimekitsumabin terminaalisen eliminaation puoliintumisajan keskiarvo läiskäpsoriaasipotilailla tehdyissä kliinisissä tutkimuksissa oli 23 vuorokautta.
Lineaarisuus/ei-lineaarisuus
Bimekitsumabin farmakokinetiikka oli läiskäpsoriaasipotilailla suhteessa annokseen, kun ihon alle annettiin useita 64–480 mg:n annoksia, ja laskennallinen puhdistuma (CL/F) oli annoksesta riippumaton.
Farmakokineettiset/farmakodynaamiset suhteet
Populaatiofarmakokineettinen/farmakodynaaminen malli kehitettiin käyttämällä kaikkia keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastavista potilaista saatavissa olevia tietoja. Analyysi osoitti, että suuremmat bimekitsumabipitoisuudet liittyvät parempiin PASI- ja IGA-vasteisiin. Valtaosalla keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastavista potilaista 320 mg:n annoksen neljän viikon välein osoitettiin olevan sopiva annos aloitushoitojaksolla, jonka jälkeen 320 mg:n annos kahdeksan viikon välein oli sopiva annos ylläpitojaksolla (ks. Erityispotilasryhmät, paino).
Erityispotilasryhmät
Paino
Populaatiofarmakokineettinen mallinnus osoitti, että altistus väheni painon noustessa. Aikuisilla ≥ 120 kg:n painoisilla potilailla keskimääräisen pitoisuuden plasmassa ennustettiin olevan ihon alle annetun 320 mg:n injektion jälkeen vähintään 30 % pienempi kuin aikuisilla potilailla, jotka painavat 90 kg. Joillakin potilailla annoksen muuttaminen voi olla tarpeen (ks. kohta Annostus ja antotapa).
Iäkkäät
Populaatiofarmakokineettisen analyysin perusteella, jossa oli mukana pieni joukko iäkkäitä potilaita (≥ 65-vuotiaiden n = 355 ja ≥ 75-vuotiaiden n = 47), laskennallinen puhdistuma (CL/F) oli iäkkäillä potilailla ja alle 65-vuotiailla potilailla samankaltainen. Annoksen muuttaminen ei ole tarpeen (ks. kohta Annostus ja antotapa).
Munuaisten tai maksan vajaatoiminta
Munuaisten tai maksan vajaatoiminnasta bimekitsumabin farmakokinetiikkaan aiheutuvan vaikutuksen selvittämiseksi ei ole tehty spesifisiä tutkimuksia. Muuttumattoman bimekitsumabin (monoklonaalinen IgG-vasta-aine) eliminaatio munuaisten kautta on oletettavasti vähäistä ja merkitykseltään vähäistä. IgG:t eliminoituvat vastaavasti pääasiassa solunsisäisen katabolian välityksellä, eikä maksan vajaatoiminta oletettavasti vaikuta bimekitsumabin puhdistumaan. Populaatiofarmakokineettisten analyysien perusteella maksan toiminnan markkereilla (ALAT/bilirubiini) ei ollut läiskäpsoriaasipotilailla vaikutusta bimekitsumabin puhdistumaan.
Etninen tausta
Kun japanilaisia ja kiinalaisia tutkittavia verrattiin kliinisessä farmakokineettisessä tutkimuksessa valkoihoisiin tutkittaviin, bimekitsumabialtistuksessa ei todettu kliinisesti merkittäviä eroja. Annoksen muuttaminen ei ole tarpeen.
Sukupuoli
Populaatiofarmakokineettinen mallinnus osoitti, että laskennallinen puhdistuma (CL/F) voi olla naisilla 10 % nopeampi kuin miehillä, mikä ei ole kliinisesti merkittävää. Annoksen muuttaminen ei ole tarpeen.
Prekliiniset tiedot turvallisuudesta
Kudosten ristireagointitestausten, toistuvan altistuksen aiheuttamaa toksisuutta koskevien tutkimusten (mukaan lukien turvallisuuteen liittyvät farmakologiset päätetapahtumat ja hedelmällisyyteen liittyvien päätetapahtumien arviointi) ja jaavanmakakeilla tutkitun synnytystä edeltävän ja sen jälkeisen kehityksen arvioinnin tulokset eivät viittaa erityiseen vaaraan ihmisille.
Jaavanmakakeilla bimekitsumabiin liittyvät vaikutukset rajoittuivat limakalvomuutoksiin, jotka olivat yhdenmukaisia kommensaalisen mikrobiston farmakologisen modulaation kanssa.
Bimekitsumabilla ei ole tehty mutageenisuus- tai karsinogeenisuustutkimuksia. Monoklonaaliset vasta-aineet eivät oletettavasti kuitenkaan vaurioita DNA:ta tai kromosomeja. 26 viikon pituisessa kroonista toksikologiaa koskeneessa tutkimuksessa jaavanmakakeilla ei havaittu preneoplastisia tai neoplastisia leesioita annoksella, josta aiheutuva altistus oli 109-kertainen ihmiselle 320 mg:n annoksesta 4 viikon välein aiheutuvaan altistukseen verrattuna.
Peri- ja postnataalista kehitystä koskevassa jaavanmakakeilla tehdyssä tutkimuksessa bimekitsumabilla ei todettu olevan vaikutuksia tiineyteen, synnytykseen, vastasyntyneiden poikasten eloonjäämiseen, sikiön ja synnytyksen jälkeiseen kehitykseen, kun bimekitsumabia annettiin organogeneesin ajan synnytykseen asti annoksina, joista aiheutuva altistus oli AUC-arvon perusteella 27-kertainen ihmiselle 320 mg:n bimekitsumabiannoksesta 4 viikon välein aiheutuvaan altistukseen verrattuna. Apinanpoikasten syntymähetkellä bimekitsumabipitoisuudet niiden seerumissa olivat verrannolliset emon seerumissa oleviin pitoisuuksiin nähden.
Farmaseuttiset tiedot
Apuaineet
Glysiini
Natriumasetaattitrihydraatti
Väkevä etikkahappo
Polysorbaatti 80
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Säilytys
Bimzelx 160 mg injektioneste, liuos, esitäytetty ruisku
Säilytä jääkaapissa (2–8 °C).
Ei saa jäätyä.
Pidä esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
Esitäytettyä ruiskua voidaan säilyttää huoneenlämmössä (enintään 25 °C) yhden enintään 25 päivän jakson ajan valolta suojattuna. Kun valmiste on otettu jääkaapista ja sitä on säilytetty näissä olosuhteissa, hävitä se 25 päivän kuluttua tai pakkaukseen merkittyyn viimeiseen käyttöpäivämäärään mennessä sen mukaan, kumpi on aiemmin. Ulkopakkauksessa on paikka päivämäärälle, jotta siihen merkitään se päivämäärä, jolloin valmiste on otettu pois jääkaapista.
Bimzelx 320 mg injektioneste, liuos, esitäytetty ruisku
Säilytä jääkaapissa (2–8 °C).
Ei saa jäätyä.
Pidä esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
Esitäytettyä ruiskua voidaan säilyttää huoneenlämmössä (enintään 25 °C) yhden enintään 25 päivän jakson ajan valolta suojattuna. Kun valmiste on otettu jääkaapista ja sitä on säilytetty näissä olosuhteissa, hävitä se 25 päivän kuluttua tai pakkaukseen merkittyyn viimeiseen käyttöpäivämäärään mennessä sen mukaan, kumpi on aiemmin. Ulkopakkauksessa on paikka päivämäärälle, jotta siihen merkitään se päivämäärä, jolloin valmiste on otettu pois jääkaapista.
Bimzelx 160 mg injektioneste, liuos, esitäytetty kynä
Säilytä jääkaapissa (2–8 °C).
Ei saa jäätyä.
Pidä esitäytetty kynä ulkopakkauksessa. Herkkä valolle.
Esitäytettyä kynää voidaan säilyttää huoneenlämmössä (enintään 25 °C) yhden enintään 25 päivän jakson ajan valolta suojattuna. Kun valmiste on otettu jääkaapista ja sitä on säilytetty näissä olosuhteissa, hävitä se 25 päivän kuluttua tai pakkaukseen merkittyyn viimeiseen käyttöpäivään mennessä sen mukaan, kumpi on aiemmin. Ulkopakkauksessa on paikka päivämäärälle, jotta siihen merkitään se päivämäärä, jolloin valmiste on otettu pois jääkaapista.
Bimzelx 320 mg injektioneste, liuos, esitäytetty kynä
Säilytä jääkaapissa (2–8 °C).
Ei saa jäätyä.
Pidä esitäytetty kynä ulkopakkauksessa. Herkkä valolle.
Esitäytettyä kynää voidaan säilyttää huoneenlämmössä (enintään 25 °C) yhden enintään 25 päivän jakson ajan valolta suojattuna. Kun valmiste on otettu jääkaapista ja sitä on säilytetty näissä olosuhteissa, hävitä se 25 päivän kuluttua tai pakkaukseen merkittyyn viimeiseen käyttöpäivään mennessä sen mukaan, kumpi on aiemmin. Ulkopakkauksessa on paikka päivämäärälle, jotta siihen merkitään se päivämäärä, jolloin valmiste on otettu pois jääkaapista.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BIMZELX injektioneste, liuos, esitäytetty kynä
160 mg (L:ei) 2 x 1 kpl (1 ml (160 mg/ml)) (1991,81 €)
320 mg (L:ei) 1 kpl (2 ml (160 mg/ml)) (1991,81 €)
PF-selosteen tieto
Bimzelx 160 mg injektioneste, liuos, esitäytetty ruisku
Yhden ml:n esitäytetty ruisku (tyypin I lasia), jossa on fluoripolymeerilaminoitu bromobutyylikumitulppa, kiinteä 27G:n, ½”:n ohutseinäinen neula ja jäykkä neulansuojus (joka koostuu termoplastisesta elastomeerista valmistetusta neulan turvamekanismista ja polypropeenista valmistetusta jäykästä suojuksesta), jotka ovat automaattisessa neulan turvalaitteessa.
Pakkauskoko 1 esitäytetty ruisku.
Pakkauskoko 2 esitäytettyä ruiskua.
Kerrannaispakkaus, joka sisältää 3 (3 x 1) esitäytettyä ruiskua.
Kerrannaispakkaus, joka sisältää 4 (2 x 2) esitäytettyä ruiskua.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Bimzelx 320 mg injektioneste, liuos, esitäytetty ruisku
Kahden ml:n esitäytetty ruisku (tyypin I lasia), jossa on fluoripolymeerilaminoitu bromobutyylikumitulppa, kiinteä 27G:n, ½”:n ohutseinäinen neula ja jäykkä neulansuojus (joka koostuu termoplastisesta elastomeerista valmistetusta neulan turvamekanismista ja polypropeenista valmistetusta jäykästä suojuksesta), jotka ovat automaattisessa neulan turvalaitteessa.
Pakkauskoko 1 esitäytetty ruisku.
Kerrannaispakkaus, joka sisältää 3 (3 x 1) esitäytettyä ruiskua.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Bimzelx 160 mg injektioneste, liuos, esitäytetty kynä
Yhden ml:n esitäytetty kynä, joka sisältää esitäytetyn ruiskun (tyypin I lasia), jossa on fluoripolymeerilaminoitu bromobutyylikumitulppa, kiinteä 27G:n, ½”:n ohutseinäinen neula ja jäykkä neulansuojus, joka koostuu termoplastisesta elastomeerista valmistetusta neulan turvamekanismista ja polypropeenista valmistetusta jäykästä suojuksesta.
Pakkauskoko 1 esitäytetty kynä.
Pakkauskoko 2 esitäytettyä kynää.
Kerrannaispakkaus, joka sisältää 3 (3 x 1) esitäytettyä kynää.
Kerrannaispakkaus, joka sisältää 4 (2 x 2) esitäytettyä kynää.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Bimzelx 320 mg injektioneste, liuos, esitäytetty kynä
Kahden ml:n esitäytetty kynä, joka sisältää esitäytetyn ruiskun (tyypin I lasia), jossa on fluoripolymeerilaminoitu bromobutyylikumitulppa, kiinteä 27G:n, ½”:n ohutseinäinen neula ja jäykkä neulansuojus, joka koostuu termoplastisesta elastomeerista valmistetusta neulan turvamekanismista ja polypropeenista valmistetusta jäykästä suojuksesta.
Pakkauskoko 1 esitäytetty kynä.
Kerrannaispakkaus, joka sisältää 3 (3 x 1) esitäytettyä kynää.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Liuos on kirkasta tai hieman opalisoivaa, ja se on väriltään väritöntä tai vaalean ruskehtavankeltaista.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BIMZELX injektioneste, liuos, esitäytetty kynä
160 mg 2 x 1 kpl
320 mg 1 kpl
BIMZELX injektioneste, liuos, esitäytetty ruisku
160 mg 2 kpl
- Alempi erityiskorvaus (65 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli ja tosilitsumabi (tulehdukselliset reumasairaudet): Nivelreuman, juveniilin polyartriitin, psoriaasiin liittyvän niveltulehduksen, selkärankareuman tai edellä mainittuja niveltulehduksia läheisesti muistuttavan niveltulehduksen hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman hoito erityisin edellytyksin (281).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli, tosilitsumabi ja ustekinumabi (tulehdukselliset reumasairaudet): Eräiden reumasairauksien hoito erityisin edellytyksin / Adalimumabi: Uveiitin hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman ja jättisoluarteriitin hoito erityisin edellytyksin (313), Adalimumabi, bimekitsumabi, brodalumabi, etanersepti, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sekukinumabi, sertolitsumabipegoli, tildrakitsumabi ja ustekinumabi (ihopsoriaasi): Vaikean kroonisen ihopsoriaasin hoito erityisin edellytyksin (319).
ATC-koodi
L04AC21
Valmisteyhteenvedon muuttamispäivämäärä
30.04.2026
Yhteystiedot
Bertel Jungin aukio 5, 6 krs.
02600 Espoo
+358 9 2514 4221
etunimi.sukunimi@ucb.com