
PADCEV kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 20 mg, 30 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Yleinen
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Padcev 20 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo kuiva-ainetta välikonsentraatiksi infuusionestettä, liuosta varten sisältää 20 mg enfortumabivedotiinia.
Padcev 30 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo kuiva-ainetta välikonsentraatiksi infuusionestettä, liuosta varten sisältää 30 mg enfortumabivedotiinia.
Käyttökuntoon saattamisen jälkeen yksi millilitra liuosta sisältää 10 mg enfortumabivedotiinia.
Enfortumabivedotiini koostuu täysin ihmisperäisestä IgG1 kappa‑vasta-aineesta, joka on konjugoitu mikrotubulustoimintaan vaikuttavaan aineeseen, monometyyliauristatiini E:hen (MMAE), proteaasilla pilkkoutuvalla maleimidokaproyyli-valiini-sitrulliinilinkkerillä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos.
Kliiniset tiedot
Käyttöaiheet
Lihasinvasiivinen virtsarakkosyöpä
Padcev yhdessä pembrolitsumabin kanssa, neoadjuvanttihoitona ja radikaalin kystektomian jälkeen adjuvanttihoitona, on tarkoitettu hoidoksi sellaisille aikuispotilaille, joilla on leikkaukseen soveltuva lihasinvasiivinen virtsarakkosyöpä ja joille sisplatiinipohjainen kemoterapia ei sovellu.
Leikkaukseen soveltumaton tai metastaattinen uroteelisyöpä
Padcev yhdessä pembrolitsumabin kanssa on tarkoitettu ensilinjan hoidoksi sellaisille aikuispotilaille, joilla on leikkaukseen soveltumaton tai metastaattinen uroteelisyöpä ja joille platinapohjainen kemoterapia soveltuu.
Paikallisesti edennyt tai metastaattinen uroteelisyöpä
Padcev on monoterapiana tarkoitettu sellaisten aikuispotilaiden hoitoon, joilla on paikallisesti edennyt tai metastaattinen uroteelisyöpä ja jotka ovat aikaisemmin saaneet platinaa sisältävää kemoterapiaa ja ohjelmoituneen solukuoleman reseptori 1 tai ohjelmoituneen solukuoleman ligandi 1 ‑estäjää (ks. kohta Farmakodynamiikka).
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Padcev-hoidon saa aloittaa ja sitä saa valvoa vain lääkäri, jolla on kokemusta syöpähoidoista. Hyvä laskimoyhteys on varmistettava ennen hoidon aloittamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Taulukko 1. Yhdessä pembrolitsumabin kanssa käytetyn enfortumabivedotiinin suositeltu annos* | ||
Käyttöaihe | Suositeltu enfortumabivedotiinin annos | Hoidon kesto |
Lihasinvasiivisen virtsarakkosyövän neoadjuvantti- ja adjuvanttihoito | Enfortumabivedotiini 1,25 mg/kg (enintään 125 mg potilaille, jotka painavat ≥ 100 kg) 21 päivän syklin päivinä 1 ja 8. | Potilaat, joille sisplatiini ei sovellu Neoadjuvanttihoito: 3 sykliä tai kunnes sairaus etenee ja estää parantavaan tarkoitukseen tähtäävän radikaalin kystektomian tai ei‑hyväksyttävään toksisuuteen asti. Adjuvanttihoito: 6 sykliä tai kunnes sairaus uusiutuu tai ei‑hyväksyttävään toksisuuteen asti. |
Leikkaukseen soveltumaton tai metastaattinen uroteelisyöpä | Enfortumabivedotiini 1,25 mg/kg (enintään 125 mg potilaille, jotka painavat ≥ 100 kg) 21 päivän syklin päivinä 1 ja 8. | Kunnes sairaus etenee tai ei‑hyväksyttävään toksisuuteen asti. |
*Jos potilaille annetaan pembrolitsumabia samana päivänä, se tulee antaa enfortumabivedotiinin jälkeen. Katso pembrolitsumabin valmisteyhteenvedosta lisätiedot pembrolitsumabin antamisesta. | ||
Taulukko 2. Monoterapiana käytetyn enfortumabivedotiinin suositeltu annos | ||
Käyttöaihe | Suositeltu enfortumabivedotiinin annos | Hoidon kesto |
Paikallisesti edennyt tai metastaattinen uroteelisyöpä | Enfortumabivedotiini 1,25 mg/kg (enintään 125 mg potilaille, jotka painavat ≥ 100 kg) 28 päivän syklin päivinä 1, 8 ja 15. | Kunnes sairaus etenee tai ei‑hyväksyttävään toksisuuteen asti. |
Taulukko 3. Suositeltu enfortumabivedotiinin annoksenpienennys haittavaikutusten vuoksi | |
Annoksen pienennyksen suunnitelma | Annostaso |
Aloitusannos | 1,25 mg/kg tai enintään 125 mg |
Ensimmäinen annoksen pienennys | 1,0 mg/kg tai enintään 100 mg |
Toinen annoksen pienennys | 0,75 mg/kg tai enintään 75 mg |
Kolmas annoksen pienennys | 0,5 mg/kg tai enintään 50 mg |
Annoksen muutokset
Taulukko 4. Enfortumabivedotiinin antamisen keskeytys, annoksen pienennys ja antamisen lopettaminen | ||
Haittavaikutus | Vaikeusaste* | Annoksen muutos* |
Ihoreaktiot | Epäilty Stevens‑Johnsonin oireyhtymä (SJS) tai toksinen epidermaalinen nekrolyysi (TEN) tai rakkulaiset ihovauriot | Keskeytä hoito välittömästi ja lähetä potilas erikoissairaanhoitoon. |
Vahvistettu SJS tai TEN; aste 4 tai toistuvasti aste 3 | Hoito lopetettava pysyvästi. | |
Asteen 2 huononeminen Kuumeinen aste 2 Aste 3 |
| |
Hyperglykemia | Veren glukoosi > 13,9 mmol/l (> 250 mg/dl) |
|
Pneumoniitti/ interstitiaalinen keuhkosairaus (ILD) | Aste 2 |
|
Aste ≥ 3 |
| |
Perifeerinen neuropatia | Aste 2 |
|
Aste ≥ 3 | Hoito lopetettava pysyvästi. | |
*Toksisuus luokiteltiin National Cancer Institue (Kansallinen syöpäinstituutti) Common Terminology Criteria for Adverse Events Version 5.0 (NCI-CTCAE v5.0) (Yleiset terminologiakriteerit haittavaikutuksille, versio 5.0) ‑luokituksen mukaan, missä aste 1 on lievä, aste 2 on keskivaikea, aste 3 on vaikea ja aste 4 on henkeä uhkaava. | ||
Erityisryhmät
Iäkkäät
Annosta ei tarvitse muuttaa ≥ 65-vuotiaille potilaille (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa potilaille, joilla on lievä (kreatiniinipuhdistuma [CrCL] > 60–90 ml/min), keskivaikea (CrCL 30–60 ml/min) tai vaikea (CrCL 15–< 30 ml/min) munuaisten vajaatoiminta. Enfortumabivedotiinia ei ole arvioitu potilailla, joilla on loppuvaiheen munuaistauti (CrCL < 15 ml/min) (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa potilaille, joilla on lievä maksan vajaatoiminta (kokonaisbilirubiini 1–1,5 × normaalin yläraja [ULN] ja ASAT mikä tahansa tai kokonaisbilirubiini ≤ ULN ja ASAT > ULN). Enfortumabivedotiinia on arvioitu vain pienellä määrällä potilaita, jolla oli keskivaikea tai vaikea maksan vajaatoiminta. Maksan vajaatoiminnan odotetaan lisäävän systeemistä MMAE-altistusta (sytotoksinen lääke), ja siksi potilaita on seurattava tiiviisti haittatapahtumien varalta. Keskivaikeaa ja vaikeaa maksan vajaatoimintaa sairastavista potilaista on vain niukasti tietoja, joten tarkkoja annossuosituksia ei voida antaa (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Enfortumabivedotiinilla ei ole asianmukaista käyttöaihetta pediatrisille potilaille paikallisesti edenneen tai metastaattisen uroteelisyövän ja lihasinvasiivisen virtsarakkosyövän hoitoon.
Antotapa
Padcev annetaan laskimoon. Suositeltu annos on annettava infuusiona laskimoon 30 minuutin ajan. Enfortumabivedotiinia ei saa antaa pikaboluksena tai bolusinjektiona.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Ihoreaktiot
Enfortumabivedotiiniin liittyy ihoreaktioita enfortumabivedotiinin iholla ekspressoituvaan nektiini-4:ään sitoutumisen vuoksi. Kuume tai flunssan kaltaiset oireet voivat olla ensimmäinen merkki vakavasta ihoreaktiosta. Potilaita tulee tarkkailla, jos tällaisia oireita ilmenee.
Lieviä tai keskivaikeita ihoreaktioita, ensisijaisesti makulopapulaarista ihottumaa, on raportoitu enfortumabivedotiinin käytön yhteydessä. Ihoreaktioiden ilmaantuvuus oli suurempi, kun enfortumabivedotiinia annettiin yhdessä pembrolitsumabin kanssa, verrattuna enfortumabivedotiinin antoon monoterapiana (ks. kohta Haittavaikutukset). Myös kuolemaan johtaneita vaikeita ihoon liittyviä haittavaikutuksia, kuten SJS:ää ja TEN:ä, on esiintynyt enfortumabivedotiinilla hoidetuilla potilailla, pääasiassa ensimmäisen hoitojakson aikana.
Potilaita on seurattava ihoreaktioiden varalta ensimmäisestä hoitojaksosta lähtien sekä koko hoidon keston ajan. Asianmukaista hoitoa, kuten paikallisesti käytettäviä kortikosteroideja ja antihistamiineja, voidaan harkita lieviin ja keskivaikeisiin ihoreaktioihin. Epäillyssä SJS:ssä tai TEN:ssä, tai jos potilaalle ilmaantuu rakkulaisia ihovaurioita, hoito on keskeytettävä välittömästi ja potilas lähetettävä erikoishoitoon. Histologinen varmistus, mukaan lukien useammat biopsiat, on ratkaisevan tärkeää ihoreaktion varhaisessa tunnistamisessa, sillä diagnoosi ja toimenpiteet voivat parantaa ennustetta. Padcev-hoito on lopetettava pysyvästi, jos SJS tai TEN varmistuu tai asteen 4 ihoreaktioita tai toistuvia asteen 3 ihoreaktioita ilmenee. Asteen 2 huonontuessa, kuumeisessa asteessa 2 tai asteen 3 ihoreaktioissa, hoito on keskeytettävä, kunnes reaktiot ovat parantuneet asteeseen ≤ 1. Potilaan lähettämistä erikoishoitoon on harkittava. Hoito voidaan aloittaa uudelleen samalla annostasolla tai annoksen pienentämistä yhdellä annostasolla voidaan harkita (ks. kohta Annostus ja antotapa).
Pneumoniitti/ILD
Enfortumabivedotiinilla hoidetuilla potilailla on ilmennyt vaikeaa, henkeä uhkaavaa tai kuolemaan johtanutta pneumoniittia/ILD:tä. Pneumoniitin/ILD:n, myös vaikeiden tapausten, ilmaantuvuus oli suurempi, kun enfortumabivedotiinia annettiin yhdessä pembrolitsumabin kanssa, verrattuna enfortumabivedotiinin antoon monoterapiana (ks. kohta Haittavaikutukset).
Potilaita on seurattava pneumoniittiin/ILD:hen viittaavien merkkien ja oireiden, kuten hypoksian, yskän, hengenahdistuksen tai röntgentutkimuksissa havaittavien interstitiaalisten infiltraattien varalta. Kortikosteroideja on annettava asteen ≥ 2 tapahtumiin (esim. aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaavaa, jonka jälkeen annosta vähennetään asteittain). Keskeytä Padcev-hoito, jos potilaalla ilmenee asteen 2 pneumoniitti/ILD, ja harkitse annoksen pienentämistä. Padcev-hoito on lopetettava pysyvästi, jos potilaalla ilmenee asteen ≥ 3 pneumoniitti/ILD (ks. kohta Annostus ja antotapa).
Hyperglykemia
Enfortumabivedotiinilla hoidetuilla potilailla, joilla oli tai ei ollut aiempaa diabetes mellitusta, on ilmennyt hyperglykemiaa ja diabeettista ketoasidoosia (DKA), myös kuolemaan johtavana (ks. kohta Haittavaikutukset). Hyperglykemiaa ilmeni useammin potilailla, joilla oli aiemmin ollut hyperglykemiaa tai korkea painoindeksi (≥ 30 kg/m2). Kliinisiin tutkimuksiin ei otettu potilaita, joiden HbA1c oli lähtötilanteessa ≥ 8 %. Veren glukoositasoja on seurattava ennen valmisteen antamista ja säännöllisesti hoidon aikana kliinisen tarpeen mukaan potilailla, joilla on diabetes mellituksen tai hyperglykemian riski. Jos veren glukoositaso nousee arvoon > 13,9 mmol/l (> 250 mg/dl), Padcev-hoito on keskeytettävä, kunnes veren glukoosi on laskenut arvoon ≤ 13,9 mmol/l (≤ 250 mg/dl), ja potilaalle on annettava asianmukaista hoitoa (ks. kohta Annostus ja antotapa).
Vakavat infektiot
Vakavia infektioita, kuten sepsistä tai pneumoniaa (mukaan lukien kuolemaan johtaneet), on raportoitu potilailla, joita on hoidettu Padcev-valmisteella. Potilaita tulee seurata huolellisesti hoidon aikana mahdollisten vakavien infektioiden ilmaantumisen varalta.
Perifeerinen neuropatia
Perifeeristä neuropatiaa, pääasiassa perifeeristä sensorista neuropatiaa, on ilmennyt enfortumabivedotiinihoidon yhteydessä, mukaan lukien asteen ≥ 3 reaktiot (ks. kohta Haittavaikutukset). Kliinisiin tutkimuksiin ei otettu potilaita, joilla oli ennestään asteen ≥ 2 perifeeristä neuropatiaa. Potilaita on seurattava uuden tai pahenevan perifeerisen neuropatian oireiden varalta, koska näiden potilaiden enfortumabivedotiinihoitoa on ehkä lykättävä, annosta pienennettävä tai hoito keskeytettävä (ks. taulukko 3). Padcev-hoito on lopetettava pysyvästi, jos potilaalla ilmenee asteen ≥ 3 perifeeristä neuropatiaa (ks. kohta Annostus ja antotapa).
Silmät
Silmiin liittyviä haittavaikutuksia, pääasiassa silmien kuivumista, on esiintynyt enfortumabivedotiinihoitoa saaneilla potilailla (ks. kohta Haittavaikutukset). Potilaita on seurattava silmiin liittyvien haittavaikutusten varalta. Jos silmäoireet eivät häviä tai pahenevat, on harkittava keinokyyneleitä silmien kuivumisen ennaltaehkäisyyn ja lähetettä silmälääkärin arviointiin.
Infuusiokohdan ekstravasaatio
Enfortumabivedotiinin antamisen jälkeen on havaittu ekstravasaatiosta johtuneita ihon ja pehmytkudoksen vaurioita (ks. kohta Haittavaikutukset). Ennen Padcev-hoidon aloittamista on tarkistettava, että laskimoyhteys on hyvä, ja antamisen aikana infuusion antokohtaa on tarkkailtava mahdollisen ekstravasaation varalta. Mikäli ekstravasaatiota ilmenee, infuusio on lopetettava ja potilasta seurattava haittavaikutusten varalta.
Alkio- ja sikiötoksisuus ja ehkäisy
Raskaana oleville naisille on kerrottava mahdollisesta sikiöön kohdistuvasta riskistä (ks. kohdat Raskaus ja imetys ja Prekliiniset tiedot turvallisuudesta). Hedelmällisessä iässä olevia naisia on neuvottava tekemään raskaustesti 7 päivän sisällä ennen enfortumabivedotiinihoidon aloittamista sekä käyttämään tehokasta ehkäisyä hoidon aikana ja vähintään 6 kuukauden ajan hoidon päättymisen jälkeen. Enfortumabivedotiinilla hoidettavia miehiä kehotetaan olemaan hankkimatta lapsia hoidon aikana ja vähintään 4 kuukauden ajan viimeisen Padcev-annoksen jälkeen.
Potilaan tietopaketti
Lääkkeen määrääjän on keskusteltava potilaan kanssa Padcev-hoidon riskeistä, mukaan lukien riskit yhdistelmähoidossa pembrolitsumabin kanssa. Potilaalle on annettava potilastiedote ja potilaskortti jokaisen lääkemääräyksen yhteydessä.
Yhteisvaikutukset
Enfortumabivedotiinilla ei ole tehty virallisia lääkkeiden välisiä yhteisvaikutustutkimuksia. Samanaikaisella enfortumabivedotiinin ja CYP3A4-substraattien kautta metaboloituvien lääkevalmisteiden käytöllä ei ollut kliinisesti relevanttia riskiä aiheuttaa farmakokineettisiä yhteisvaikutuksia (ks. kohta Farmakokinetiikka).
Muiden lääkevalmisteiden vaikutukset enfortumabivedotiiniin
CYP3A4:n estäjät, substraatit tai indusoijat
Fysiologiaan perustuvan farmakokineettisen (PBPK) mallinnuksen perusteella enfortumabivedotiinin ja ketokonatsolin (yhdistetty P‑gp:n ja vahva CYP3A:n estäjä) samanaikaisen käytön ennustetaan suurentavan konjugoitumattoman MMAE:n Cmax- ja AUC-arvoja vähäisesti ilman muutoksia vasta-aine-lääkekonjugaatin (ADC)-altistukseen. Varovaisuutta on noudatettava annettaessa samanaikaisesti CYP3A4-estäjiä. Samanaikaisesti vahvoja CYP3A4-estäjiä (esim. bosepreviiri, klaritromysiini, kobisistaatti, indinaviiri, itrakonatsoli, nefatsodoni, nelfinaviiri, posakonatsoli, ritonaviiri, sakinaviiri, telapreviiri, telitromysiini, vorikonatsoli) saavia potilaita on seurattava tarkemmin toksisuuden merkkien varalta.
Konjugoitumattoman MMAE:n ei ennusteta muuttavan samanaikaisesti käytettävien CYP3A4-subtraattivalmisteiden (esim. midatsolaami) AUC-arvoa.
Vahvat CYP3A4-indusoijat (esim. rifampisiini, karbamatsepiini, fenobarbitaali, fenytoiini, mäkikuisma [Hypericum perforatum]) voivat vähentää konjugoitumattoman MMAE:n altistusta kohtalaisesti (ks. kohta Farmakokinetiikka).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / miesten ja naisten ehkäisy
Raskaustestin tekemistä suositellaan naisille, jotka voivat tulla raskaaksi, 7 päivän sisällä ennen hoidon aloittamista. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 6 kuukautta hoidon lopettamisen jälkeen. Enfortumabivedotiinilla hoidettavia miehiä kehotetaan olemaan hankkimatta lapsia hoidon aikana ja vähintään 4 kuukauden ajan viimeisen Padcev-annoksen jälkeen.
Raskaus
Eläinkokeiden löydösten perusteella Padcev voi aiheuttaa haittaa sikiölle, kun sitä annetaan raskaana oleville naisille. Naarasrotilla tehdyt alkio- ja sikiökehitystutkimukset ovat osoittaneet, että enfortumabivedotiinin antaminen laskimoon aiheutti elinkelpoisten sikiöiden määrän vähenemistä, poikuekoon pienenemistä ja lisääntynyttä varhaista resorboitumista (ks. kohta Prekliiniset tiedot turvallisuudesta). Padcev-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä tehokasta ehkäisyä.
Imetys
Ei tiedetä, erittyykö enfortumabivedotiini ihmisen rintamaitoon. Imeväiseen kohdistuvia riskejä ei voida poissulkea. Imetys on keskeytettävä Padcev-hoidon ajaksi ja vähintään 6 kuukaudeksi viimeisen Padcev-annoksen jälkeen.
Hedelmällisyys
Rotilla havaittiin kivestoksisuutta toistuvassa annostelussa, mikä voi muuttaa miesten hedelmällisyyttä. MMAE:lla on osoitettu olevan aneugeenisiä ominaisuuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Sen vuoksi tällä lääkevalmisteella hoidettavia miehiä neuvotaan pakastuttamaan siemennestenäytteitä ja asettamaan ne säilytykseen ennen hoitoa. Padcev-valmisteen vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Padcev-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Enfortumabivedotiini yhdessä pembrolitsumabin kanssa
Jos enfortumabivedotiinia käytetään yhdessä pembrolitsumabin kanssa, tutustu pembrolitsumabin valmisteyhteenvetoon ennen hoidon aloittamista.
Lihasinvasiivinen virtsarakkosyöpä
Enfortumabivedotiinin turvallisuutta yhdessä pembrolitsumabin kanssa käytettynä arvioitiin 167 potilaalla, jotka saivat vähintään yhden 1,25 mg/kg annoksen enfortumabivedotiinia yhdessä pembrolitsumabin kanssa vaiheen 3 tutkimuksessa (EV-303) (ks. taulukko 5). Potilaat saivat enfortumabivedotiinia yhdessä pembrolitsumabin kanssa mediaaniltaan 6,3 kuukautta kestäneen hoitojakson ajan (vaihteluväli: 0,03–19,7 kuukautta). Pelkän enfortumabivedotiinialtistuksen mediaani oli 5,5 kuukautta (vaihteluväli: 0,03–14,1 kuukautta).
Yleisimpiä haittavaikutuksia enfortumabivedotiini- ja pembrolitsumabihoidon yhteydessä olivat kutina (47,3 %), hiustenlähtö (34,7 %), ripuli (34,1 %), väsymys (32,3 %), anemia (30,5 %), ruokahaluttomuus (28,1 %), makuhäiriöt (28,1 %), pahoinvointi (25,7 %), ihottuma (25,1 %), aspartaattiaminotransferaasiarvon nousu (24 %), painon lasku (19,8 %), alaniiniaminotransferaasiarvon nousu (19,2 %), makulopapulaarinen ihottuma (16,2 %), ihon kuivuminen (15 %), hypotyreoosi (14,4 %), perifeerinen sensorinen neuropatia (13,8 %), hyperglykemia (12,6 %) ja perifeerinen neuropatia (10,2 %).
Yleisin vakava haittavaikutus (≥ 2 %) oli ripuli (2,4 %). 41 % potilaista lopetti enfortumabivedotiinihoidon pysyvästi haittavaikutusten vuoksi; yleisin hoidon lopetukseen johtanut haittavaikutus (≥ 2 %) oli perifeerinen sensorinen neuropatia (2,4 %).
Hoidon keskeytykseen johtaneita haittavaikutuksia esiintyi 44 %:lla potilaista. Yleisimmät hoidon keskeytykseen johtaneet haittavaikutukset (≥ 2 %) olivat ripuli (4,2 %), ihottuma (4,2 %), neutropenia (3,6 %), väsymys (3 %), hyperglykemia (3 %) ja kutina (2,4 %).
Enfortumabivedotiiniannoksen pienennykseen johtaneita haittavaikutuksia esiintyi 17 %:lla potilaista. Yleisimmät annoksen pienennykseen johtaneet haittavaikutukset (≥ 2 %) olivat ihottuma (2,4 %) ja painon lasku (2,4 %).
Leikkaukseen soveltumaton tai metastaattinen uroteelisyöpä
Enfortumabivedotiinin turvallisuutta yhdessä pembrolitsumabin kanssa käytettynä arvioitiin 564 potilaalla, jotka saivat vähintään yhden 1,25 mg/kg annoksen enfortumabivedotiinia yhdessä pembrolitsumabin kanssa, yhdessä vaiheen 2 tutkimuksessa (EV-103) ja yhdessä vaiheen 3 tutkimuksessa (EV-302) (ks. taulukko 5). Potilaat saivat enfortumabivedotiinia yhdessä pembrolitsumabin kanssa mediaaniltaan 9,4 kuukautta kestäneen hoitojakson ajan (vaihteluväli: 0,3–34,4 kuukautta).
Yleisimpiä haittavaikutuksia enfortumabivedotiini- ja pembrolitsumabihoidon yhteydessä olivat perifeerinen sensorinen neuropatia (53,4 %), kutina (41,1 %), väsymys (40,4 %), ripuli (39,2 %), hiustenlähtö (38,5 %), makulopapulaarinen ihottuma (36 %), painon lasku (36 %), ruokahaluttomuus (33,9 %), pahoinvointi (28,4 %), anemia (25,7 %), makuhäiriöt (24,3 %), ihon kuivuminen (18,1 %), alaniiniaminotransferaasiarvon nousu (16,8 %), hyperglykemia (16,7 %), aspartaattiaminotransferaasiarvon nousu (15,4 %), silmien kuivuminen (14,4 %), oksentelu (13,3 %), makulaarinen ihottuma (11,3 %), hypotyreoosi (10,5 %) ja neutropenia (10,1 %).
Yleisimmät vakavat haittavaikutukset (≥ 2 %) olivat ripuli (3 %) ja pneumoniitti (2,3 %). 36 % potilaista lopetti enfortumabivedotiinihoidon pysyvästi haittavaikutusten vuoksi; yleisimmät hoidon lopetukseen johtaneet haittavaikutukset (≥ 2 %) olivat perifeerinen sensorinen neuropatia (12,2 %) ja makulopapulaarinen ihottuma (2 %).
Hoidon keskeytykseen johtaneita haittavaikutuksia esiintyi 72 %:lla potilaista. Yleisimmät hoidon keskeytykseen johtaneet haittavaikutukset (≥ 2 %) olivat perifeerinen sensorinen neuropatia (17 %), makulopapulaarinen ihottuma (6,9 %), ripuli (4,8 %), väsymys (3,7 %), pneumoniitti (3,7 %), hyperglykemia (3,4 %), neutropenia (3,2 %), alaniiniaminotransferaasiarvon nousu (3 %), kutina (2,3 %) ja anemia (2 %).
Enfortumabivedotiiniannoksen pienennykseen johtaneita haittavaikutuksia esiintyi 42,4 %:lla potilaista. Yleisimmät annoksen pienennykseen johtaneet haittavaikutukset (≥ 2 %) olivat perifeerinen sensorinen neuropatia (9,9 %), makulopapulaarinen ihottuma (6,4 %), väsymys (3,2 %), ripuli (2,3 %) ja neutropenia (2,1 %).
Enfortumabivedotiini monoterapiana
Enfortumabivedotiinin turvallisuutta monoterapiana arvioitiin 793 potilaalla, jotka saivat vähintään yhden 1,25 mg/kg annoksen enfortumabivedotiinia kahdessa vaiheen 1 tutkimuksessa (EV-101 ja EV‑102), kolmessa vaiheen 2 tutkimuksessa (EV-103, EV-201 ja EV-203) sekä yhdessä vaiheen 3 tutkimuksessa (EV-301) (ks. taulukko 5). Potilaiden saaman enfortumabivedotiinin hoitojakson mediaani oli 4,7 kuukautta (vaihteluväli: 0,3–55,7 kuukautta).
Yleisimpiä haittavaikutuksia enfortumabivedotiinihoidon yhteydessä olivat hiustenlähtö (47,7 %), ruokahaluttomuus (47,2 %), väsymys (46,8 %), ripuli (39,1 %), perifeerinen sensorinen neuropatia (38,5 %), pahoinvointi (37,8 %), kutina (33,4 %), makuhäiriöt (30,4 %), anemia (29,1 %), painon lasku (25,2 %), makulopapulaarinen ihottuma (23,6 %), ihon kuivuminen (21,8 %), oksentelu (18,7 %), aspartaattiaminotransferaasiarvon nousu (17 %), hyperglykemia (14,9 %), silmien kuivuminen (12,7 %), alaniiniaminotransferaasiarvon nousu (12,7 %) ja ihottuma (11,6 %).
Yleisimmät vakavat haittavaikutukset (≥ 2 %) olivat ripuli (2,1 %) ja hyperglykemia (2,1 %). 21 % potilaista lopetti enfortumabivedotiinihoidon pysyvästi haittavaikutusten vuoksi; yleisimmät hoidon lopetukseen johtaneet haittavaikutukset (≥ 2 %) olivat perifeerinen sensorinen neuropatia (4,8 %). Hoidon keskeytykseen johtaneita haittavaikutuksia esiintyi 62 %:lla potilaista; yleisimmät hoidon keskeytykseen johtaneet haittavaikutukset (≥ 2 %) olivat perifeerinen sensorinen neuropatia (14,8 %), väsymys (7,4 %), makulopapulaarinen ihottuma (4 %), aspartaattiaminotransferaasiarvon nousu (3,4 %), alaniiniaminotransferaasiarvon nousu (3,2 %), anemia (3,2 %), hyperglykemia (3,2 %), neutrofiiliarvon lasku (3 %), ripuli (2,8 %), ihottuma (2,4 %) ja perifeerinen motorinen neuropatia (2,1 %). 38 % potilaista edellytti annoksen pienennystä haittavaikutuksen vuoksi; yleisimmät annoksen pienennykseen johtaneet haittavaikutukset (≥ 2 %) olivat perifeerinen sensorinen neuropatia (10,3 %), väsymys (5,3 %), makulopapulaarinen ihottuma (4,2 %) ja ruokahaluttomuus (2,1 %).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa, joissa enfortumabivedotiinia käytettiin monoterapiana tai yhdessä pembrolitsumabin kanssa, sekä enfortumabivedotiinin markkinoille tulon jälkeisen käytön yhteydessä havaitut haittavaikutukset on esitetty tässä kohdassa yleisyysluokittain. Yleisyysluokat määritellään seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Kussakin esiintymistiheysryhmässä haittavaikutukset on esitetty laskevassa vakavuusjärjestyksessä.
Taulukko 5. Enfortumabivedotiinilla hoidetuilla potilailla todetut haittavaikutukset | ||
Yleisyys | Monoterapia | Yhdessä pembrolitsumabin kanssa |
Infektiot |
| |
Yleinen | Sepsis, pneumonia | Sepsis, pneumonia |
Veri ja imukudos |
| |
Hyvin yleinen | Anemia | Anemia |
Yleinen | Trombosytopenia | Trombosytopenia |
Tuntematon1 | Neutropenia, kuumeinen neutropenia, neutrofiiliarvon lasku | Neutropenia, kuumeinen neutropenia, neutrofiiliarvon lasku |
Umpieritys | ||
Hyvin yleinen |
| Hypotyreoosi |
Aineenvaihdunta ja ravitsemus |
| |
Hyvin yleinen | Hyperglykemia, ruokahaluttomuus | Hyperglykemia, ruokahaluttomuus |
Tuntematon1 | Diabeettinen ketoasidoosi | Diabeettinen ketoasidoosi |
Hermosto |
| |
Hyvin yleinen | Perifeerinen sensorinen neuropatia, makuhäiriöt | Perifeerinen sensorinen neuropatia, makuhäiriöt |
Yleinen | Perifeerinen neuropatia, perifeerinen motorinen neuropatia, perifeerinen sensorimotorinen neuropatia, parestesia, hypoestesia, kävelyhäiriö, lihasheikkous | Perifeerinen neuropatia, perifeerinen motorinen neuropatia, perifeerinen sensorimotorinen neuropatia, parestesia, hypoestesia, kävelyhäiriö, lihasheikkous, neurotoksisuus |
Melko harvinainen | Demyelinoiva polyneuropatia, polyneuropatia, neurotoksisuus, motorinen toimintahäiriö, dysestesia, lihasatrofia, neuralgia, peroneushermon halvaus, tuntohäiriö, polttelu, ihon kirvely | Dysestesia, polyneuropatia, myasthenia gravis, neuralgia, peroneushermon halvaus, ihon kirvely |
Silmät |
| |
Hyvin yleinen | Silmien kuivuminen | Silmien kuivuminen |
Hengityselimet, rintakehä ja välikarsina |
| |
Yleinen | Pneumoniitti/ILD2 | Pneumoniitti/ILD2 |
Ruoansulatuselimistö |
| |
Hyvin yleinen | Ripuli, oksentelu, pahoinvointi | Ripuli, oksentelu, pahoinvointi |
Iho ja ihonalainen kudos |
| |
Hyvin yleinen | Hiustenlähtö, kutina, ihottuma, makulopapulaarinen ihottuma, ihon kuivuminen | Hiustenlähtö, kutina, makulopapulaarinen ihottuma, ihon kuivuminen |
Yleinen | Lääkkeen aiheuttama ihoreaktio, ihon kuoriutuminen, sidekalvotulehdus, rakkulaihottuma, rakkula, stomatiitti, palmoplantaarinen erytrodysestesiaoireyhtymä, ekseema, punoitus, punoittava ihottuma, makulaarinen ihottuma, papulaarinen ihottuma, kutiava ihottuma, rakkulaihottuma | Ihottuma, ihon kuoriutuminen, sidekalvotulehdus, rakkulaihottuma, rakkula, stomatiitti, palmoplantaarinen erytrodysestesiaoireyhtymä, ekseema, punoitus, punoittava ihottuma, makulaarinen ihottuma, papulaarinen ihottuma, kutiava ihottuma, rakkulaihottuma, ihotulehdus |
Melko harvinainen | Yleistynyt eksfoliatiivinen ihotulehdus, erythema multiforme, eksfoliatiivinen ihottuma, pemfigoidi, makulovesikulaarinen ihottuma, ihottuma, allerginen ihottuma, kosketusihottuma, hiertymä, ihoärsytys, staasi-ihottuma, verirakkula | Lääkkeen aiheuttama ihoreaktio, yleistynyt eksfoliatiivinen ihotulehdus, erythema multiforme, eksfoliatiivinen ihottuma, pemfigoidi, allerginen ihottuma, kosketusihottuma, hiertymä, ihoärsytys, staasi-ihottuma |
Tuntematon1 | Toksinen epidermaalinen nekrolyysi, ihon hyperpigmentaatio, ihon värjäytyminen, pigmentaatiohäiriö, Stevens-Johnsonin oireyhtymä, epidermaalinen nekroosi, symmetrinen lääkkeeseen liittyvä ihopoimu- ja taive-eksanteema | Toksinen epidermaalinen nekrolyysi, ihon hyperpigmentaatio, ihon värjäytyminen, pigmentaatiohäiriö, Stevens-Johnsonin oireyhtymä, epidermaalinen nekroosi, symmetrinen lääkkeeseen liittyvä ihopoimu- ja taive-eksanteema |
Luusto, lihakset ja sidekudos | ||
Melko harvinainen |
| Myosiitti |
Yleisoireet ja antopaikassa todettavat haitat |
| |
Hyvin yleinen | Väsymys | Väsymys |
Yleinen | Infuusiokohdan ekstravasaatio | Infuusiokohdan ekstravasaatio |
Tutkimukset |
| |
Hyvin yleinen | Alaniiniaminotransferaasiarvon nousu, aspartaattiaminotransferaasiarvon nousu, painon lasku | Alaniiniaminotransferaasiarvon nousu, aspartaattiaminotransferaasiarvon nousu, painon lasku |
Yleinen |
| Suurentunut lipaasiarvo |
Vammat, myrkytykset ja hoitokomplikaatiot | ||
Yleinen | Infuusioon liittyvä reaktio | Infuusioon liittyvä reaktio |
1Maailmanlaajuisen markkinoille tulon jälkeisen kokemuksen perusteella. 2Sisältää: akuutti hengitysvajausoireyhtymä, autoimmuuni keuhkosairaus, immuunivälitteinen keuhkosairaus, interstitiaalinen keuhkosairaus, keuhkojen sameus, organisoituva keuhkokuume, pneumoniitti, keuhkofibroosi, keuhkotoksisuus, keuhkosarkoidoosi ja sarkoidoosi. | ||
Valikoitujen haittavaikutusten kuvaus
Immunogeenisuus
Yhteensä 159 potilasta testattiin enfortumabivedotiini-immunogeenisuuden varalta enfortumabivedotiinin ja pembrolitsumabin annon jälkeen lihasinvasiivisen virtsarakkosyövän hoidossa; 3 potilasta vahvistettiin lähtötilanteessa positiivisiksi lääkevasta-aineiden (ADA) osalta, ja lähtötilanteessa negatiivisista potilaista (n = 156) yhteensä 2 (1,3 %) oli positiivisia lähtötilanteen jälkeen.
Yhteensä 490 potilasta testattiin enfortumabivedotiini-immunogeenisuuden varalta enfortumabivedotiinin ja pembrolitsumabin annon jälkeen leikkaukseen soveltumattoman tai metastaattisen uroteelisyövän hoidossa; 24 potilasta vahvistettiin lähtötilanteessa positiivisiksi lääkevasta-aineiden (ADA) osalta ja lähtötilanteessa negatiivisista potilaista (n = 466) yhteensä 14 (3 %) oli positiivisia lähtötilanteen jälkeen.
Yhteensä 697 potilasta testattiin immunogeenisuuden varalta enfortumabivedotiinimonoterapialla 1,25 mg/kg; 16 potilasta vahvistettiin lähtötilanteessa positiivisiksi lääkevasta-aineiden (ADA) osalta, ja lähtötilanteessa negatiivisista potilaista (n = 681) yhteensä 24 (3,5 %) oli positiivisia lähtötilanteen jälkeen.
Hoidon aikana muodostuneiden enfortumabivedotiinivasta-aineiden ilmaantuvuus oli johdonmukaista, kun sitä arvioitiin enfortumabivedotiinimonoterapian ja enfortumabivedotiinin ja pembrolitsumabin yhdistelmän käytön yhteydessä.
Koska sellaisia potilaita, joilla oli vasta-aineita Padcev-valmistetta vastaan, oli niukasti, immunogeenisuuden mahdollisesta vaikutuksesta tehoon, turvallisuuteen tai farmakokinetiikkaan ei voida tehdä johtopäätöksiä.
Ihoreaktiot
Kliinisissä tutkimuksissa, joissa enfortumabivedotiinia käytettiin yhdessä pembrolitsumabin kanssa, ihoreaktioita ilmeni 89 %:lla (648) 731 potilaasta ja useimmat näistä reaktioista olivat makulopapulaarista ihottumaa, makulaarista ihottumaa, ihottumaa ja papulaarista ihottumaa. Vaikeita (asteen 3 tai 4) ihoreaktioita ilmeni 19 %:lla (135) potilaista (aste 3: 17 %, aste 4: 2 %). Kuolemaan johtava toksinen epidermaalinekrolyysitapahtuma ilmeni yhdellä potilaalla. Mediaaniaika vaikeiden ihoreaktioiden alkamiseen oli 1,6 kuukautta (vaihteluväli: 0,1–17,2 kuukautta). Ihoreaktioita saaneista potilaista, joilta oli saatavilla tietoja reaktioiden paranemisesta (n = 496), 76 % parani täysin, 16 % parani osittain ja 8 % ei ollut parantunut lainkaan viimeisen arvioinnin ajankohtana. Niistä 24 %:sta potilaita, joilla oli edelleen ihoreaktioita viimeisen arvioinnin ajankohtana, 25 %:lla oli asteen ≥ 2 tapahtumia.
Kliinisissä tutkimuksissa, joissa enfortumabivedotiinia käytettiin monoterapiana, ihoreaktioita ilmeni 57 %:lla (452) 793 potilaasta, jotka saivat enfortumabivedotiinia 1,25 mg/kg. Vaikeita (asteen 3 tai 4) ihoreaktioita ilmeni 14 %:lla (108) potilaista ja useimmat näistä reaktioista olivat makulopapulaarista ihottumaa, stomatiittia, punoittavaa ihottumaa, ihottumaa tai lääkkeen aiheuttamia ihoreaktioita. Mediaaniaika vaikeiden ihoreaktioiden alkamiseen oli 0,7 kuukautta (vaihteluväli: 0,1–8,2 kuukautta). Vakavia ihoreaktioita esiintyi 4,3 %:lla (34) potilaista. Ihoreaktioita saaneista potilaista, joilta oli saatavilla tietoja reaktioiden paranemisesta (n = 366), 61 % oli parantunut täysin, 24 % oli parantunut osittain ja 15 % ei ollut parantunut lainkaan viimeisen arvioinnin ajankohtana. Niistä 39 %:sta potilaita, joilla oli edelleen ihoreaktioita viimeisen arvioinnin ajankohtana, 38 %:lla oli asteen ≥ 2 tapahtumia.
Pneumoniitti/ILD
Kliinisissä tutkimuksissa, joissa enfortumabivedotiinia käytettiin yhdessä pembrolitsumabin kanssa, pneumoniittia/ILD:tä ilmeni 9,4 %:lla (69) 731 potilaasta. Vaikeaa (asteen 3 tai 4) pneumoniittia/ILD:tä ilmeni 20 potilaalla (aste 3: 2,3 %, aste 4: 0,4 %). Pneumoniitti/ILD johti enfortumabivedotiinihoidon keskeyttämiseen 2,1 %:lla potilaista. Kolmella potilaalla ilmeni kuolemaan johtanut pneumoniitti/ILD-tapahtuma. Mediaaniaika minkä tahansa asteisen pneumoniitin/ILD:n puhkeamiseen oli 4,1 kuukautta (vaihteluväli: 0,3–26,2 kuukautta).
Kliinisissä tutkimuksissa, joissa enfortumabivedotiinia käytettiin monoterapiana, pneumoniittia/ILD:tä ilmeni 3,3 %:lla (26) 793 potilaasta, jotka saivat enfortumabivedotiinia 1,25 mg/kg. Vaikeaa (asteen 3 tai 4) pneumoniittia/ILD:tä (aste 3: 0,5 %, aste 4: 0,3 %) ilmeni alle 1 %:lla potilaista. Pneumoniitti/ILD johti enfortumabivedotiinihoidon keskeyttämiseen 0,5 %:lla potilaista. Pneumoniitista/ILD:stä johtuvia kuolemantapauksia ei ollut. Mediaaniaika minkä tahansa asteisen pneumoniitin/ILD:n puhkeamiseen oli 2,7 kuukautta (vaihteluväli: 0,6–6,0 kuukautta) ja pneumoniitin/ILD:n mediaanikesto oli 1,6 kuukautta (vaihteluväli: 0,1–43,0 kuukautta). Niistä 26 potilaasta, joilla esiintyi keuhkotulehdusta/ILD:tä, oireet hävisivät 8:lla (30,8 %).
Hyperglykemia
Kliinisissä tutkimuksissa, joissa enfortumabivedotiinia käytettiin monoterapiana, hyperglykemiaa (veren glukoosi > 13,9 mmol/l) ilmeni 17 %:lla (133) 793 potilaasta, jotka saivat enfortumabivedotiinia 1,25 mg/kg. Vakavia hyperglykemiatapahtumia esiintyi 2,5 %:lla potilaista, 7 %:lle potilaista kehittyi vaikea (asteen 3 tai 4) hyperglykemia ja 0,3 %:lla potilaista ilmeni kuolemaan johtanut haittavaikutus, toisella hyperglykemia ja toisella diabeettinen ketoasidoosi. Asteen 3–4 hyperglykemian ilmaantuvuus lisääntyi tasaisesti potilailla, joilla oli korkeampi painoindeksi, ja potilailla, joilla oli korkeampi hemoglobiini A1C (HbA1c) -arvo lähtötilanteessa. Mediaaniaika hyperglykemian alkamiseen oli 0,5 kuukautta (vaihteluväli: 0–20,3). Potilaista, joilla esiintyi hyperglykemiaa ja joilta oli saatavilla tietoja oireiden paranemisesta (n = 106), 66 % oli parantunut täysin, 19 % oli parantunut osittain ja 15 % ei ollut parantunut lainkaan viimeisen arvioinnin ajankohtana. Niistä 34 %:sta potilaita, joilla oli edelleen hyperglykemiaa viimeisen arvioinnin ajankohtana, 64 %:lla oli asteen ≥ 2 tapahtumia.
Perifeerinen neuropatia
Kliinisissä tutkimuksissa, joissa enfortumabivedotiinia käytettiin monoterapiana, perifeeristä neuropatiaa ilmeni 53 %:lla (422) 793 potilaasta, jotka saivat enfortumabivedotiinia 1,25 mg/kg. Viidellä prosentilla potilaista ilmeni vaikeaa (asteen 3 tai 4) perifeeristä neuropatiaa, mukaan luettuna sensorisia ja motorisia vaikutuksia. Mediaaniaika asteen ≥ 2 perifeerisen neuropatian alkamiseen oli 5 kuukautta (vaihteluväli: 0,1–20,2 kuukautta). Potilaista, joilla esiintyi neuropatiaa ja joilta oli saatavilla tietoja oireiden paranemisesta (n = 340), 14 % oli parantunut täysin, 46 % oli parantunut osittain ja 41 % ei ollut parantunut lainkaan viimeisen arvioinnin ajankohtana. Niistä 86 %:sta potilaita, joilla oli edelleen neuropatiaa viimeisen arvioinnin ajankohtana, 51 %:lla oli asteen ≥ 2 tapahtumia.
Silmät
Kliinisissä tutkimuksissa, joissa enfortumabivedotiinia käytettiin monoterapiana, 30 % potilaista koki silmien kuivuutta saadessaan hoitoa enfortumabivedotiiniannoksella 1,25 mg/kg. Potilaista 1,5 %:n hoito keskeytettiin ja 0,1 % potilaista lopetti hoidon pysyvästi silmien kuivuuden vuoksi. Vaikeaa (asteen 3) silmien kuivuutta esiintyi vain kolmella potilaalla (0,4 %). Mediaaniaika silmien kuivuuden alkamiseen oli 1,7 kuukautta (vaihteluväli: 0–30,6 kuukautta.
Erityisryhmät
Iäkkäät
Yhdessä pembrolitsumabin kanssa käytettyä enfortumabivedotiinia on tutkittu 202 potilaalla, joiden ikä oli < 65 vuotta, ja 529 potilaalla, joiden ikä oli ≥ 65 vuotta. Yleisesti ottaen haittatapahtumien esiintyvyys oli suurempi ≥ 65-vuotiailla potilailla verrattuna < 65-vuotiaisiin. Tämä koski erityisesti vakavia haittatapahtumia (58,8 % ja vastaavasti 40,1 %) sekä asteen ≥ 3 tapahtumia (79,8 % ja vastaavasti 65,3 %). Nämä vastasivat kemoterapiavertailuvalmisteella tehtyjä havaintoja.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Enfortumabivedotiinin yliannostukseen ei ole tunnettua vasta-ainetta. Yliannostustapauksessa potilasta on seurattava tarkasti haittavaikutusten varalta ja tukihoitoa annettava tarpeen mukaan ottaen huomioon 3,6 vuorokauden (ADC) ja 2,6 vuorokauden (MMAE) puoliintumisaika.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, muut antineoplastiset lääkeaineet, monoklonaaliset vasta-aineet, ATC-koodi: L01FX13
Vaikutusmekanismi
Enfortumabivedotiini on vasta-aineen ja lääkkeen konjugaatti (ADC), joka kohdistuu nektiini-4:ään, uroteelisyöpäsolujen pinnalla olevaan adheesioproteiiniin. Se koostuu täysin ihmisperäisestä IgG1 kappa-vasta-aineesta, joka on konjugoitunut mikrotubulustoimintaan vaikuttavaan aineeseen, MMAE:hen, proteaasia pilkkovalla maleimidokaproyyli-valiini-sitrulliinilinkkerillä. Prekliinisten tietojen mukaan enfortumabivedotiinin syöpää torjuva vaikutus johtuu ADC:n sitoutumisesta nektiini-4:ää ilmentäviin soluihin, minkä jälkeen ADC:n ja nektiini-4:n muodostama kompleksi otetaan soluun sisään ja MMAE:ta vapautuu proteolyyttisen pilkkoutumisen kautta. MMAE:n vapautuminen häiritsee solun sisäistä mikrotubulusverkostoa, mikä aiheuttaa solusyklin pysähtymisen, apoptoosin ja immunogeenisen solukuoleman. Enfortumabivedotiinin kohdesoluista vapautuva MMAE voi diffusoitua läheisiin nektiini-4:ää heikosti ilmentäviin soluihin, mikä aiheuttaa sytotoksisen solukuoleman. Enfortumabivedotiinin yhdistäminen PD-1-estäjiin tehostaa kasvaimia tuhoavaa vaikutusta, mikä on yhdenmukaista toisiaan täydentävien mekanismien eli MMAE:n aiheuttaman sytotoksisuuden ja immunogeenisen solukuoleman sekä PD-1:n eston aiheuttaman immuunijärjestelmän toiminnan tehostumisen kanssa.
Sydämen elektrofysiologia
Edennyttä uroteelisyöpää sairastavien potilaiden EKG-tietojen tutkimuksen perusteella suositellulla 1,25 mg/kg annoksella enfortumabivedotiini ei pidentänyt keskimääräistä QTc-väliä kliinisesti merkittävästi.
Kliininen teho ja turvallisuus
Enfortumabivedotiini yhdessä pembrolitsumabin kanssa
Neoadjuvanttihoito ja adjuvanttihoito sisplatiinihoitoon soveltumattomille potilaille, joilla on lihasinvasiivinen virtsarakkosyöpä
EV-303 (KEYNOTE-905)
Padcev-valmisteen tehoa yhdessä pembrolitsumabin kanssa neoadjuvanttihoitona ja radikaalin kystektomian jälkeen jatkettuna adjuvanttihoitona arvioitiin tutkimuksessa EV-303 (KEYNOTE-905), joka oli avoin, satunnaistettu, vaiheen 3 monikeskustutkimus, johon otettiin mukaan potilaita, jotka saivat enfortumabivedotiinia ja pembrolitsumabia neoadjuvanttihoitona ja adjuvanttihoitona verrattuna radikaaliin kystektomiaan ja lantion alueen imusolmukedissektioon. Tärkeimmät soveltuvuuskriteerit olivat aiemmin hoitamaton lihasinvasiivinen cT2-T4aN0M0- tai cT1-T4aN1M0-virtsarakkosyöpä potilailla, jotka eivät soveltuneet saamaan sisplatiinipohjaista kemoterapiaa tai jotka kieltäytyivät siitä ja olivat soveltuvia radikaaliin kystektomiaan ja lantion alueen imusolmukedissektioon huolimatta PD‑L1-statuksesta. Potilaat, joilla oli aktiivinen autoimmuunisairaus, joka edellytti systeemistä hoitoa 2 vuoden sisällä hoidosta, tai sairaus, joka edellytti immunosuppressiota, eivät soveltuneet tutkimukseen.
Potilaat satunnaistettiin suhteessa 1:1:1 saamaan joko neoadjuvantti- ja adjuvanttienfortumabivedotiinia yhdessä pembrolitsumabin kanssa, neoadjuvantti- ja adjuvanttipembrolitsumabimonoterapiaa tai pelkästään radikaali kystektomia ja lantion alueen imusolmukedissektio. Hypoteesin testausta päivitettiin priorisoimaan yhdessä pembrolitsumabin kanssa annetun enfortumabivedotiinihoitohaaran vertailu pelkän radikaalin kystektomian ja lantion alueen imusolmukedissektion hoitohaaraan päätetapahtumattoman elinajan (EFS) osalta; sen vuoksi satunnaistaminen pembrolitsumabimonoterapiahoitohaaraan lopetettiin aikaisin. Tehon arviointi perustui seuraavien hoitohaarojen vertailuun:
-
Enfortumabivedotiini yhdessä pembrolitsumabin kanssa (n = 170): neoadjuvanttienfortumabivedotiini 1,25 mg/kg infuusiona laskimoon 30 minuutin ajan jokaisen 3 viikon (21 päivää) syklin päivinä 1 ja 8 3 syklin ajan ja neoadjuvanttipembrolitsumabi 200 mg 30 minuutin ajan jokaisen 3 viikon (21 päivää) syklin päivänä 1 3 syklin ajan. Radikaalin kystektomian ja lantion alueen imusolmukedissektion jälkeen adjuvanttienfortumabivedotiini 1,25 mg/kg 30 minuutin ajan jokaisen 3 viikon (21 päivää) syklin päivinä 1 ja 8 6 syklin ajan ja adjuvanttipembrolitsumabi 200 mg 30 minuutin ajan jokaisen 3 viikon (21 päivää) syklin päivänä 1 14 syklin ajan.
-
Pelkkä radikaali kystektomia ja lantion alueen imusolmukedissektio (n = 174).
Hoitoa jatkettiin, kunnes se oli suoritettu loppuun, kunnes sairaus eteni ja esti radikaalin kystektomian ja lantion alueen imusolmukedissektion, radikaalia kystektomiaa ja lantion alueen imusolmukedissektiota ei tehty tai potilas kieltäytyi siitä, sairaus uusiutui adjuvanttivaiheessa tai ei‑hyväksyttävään toksisuuteen asti. Kasvaimen statuksen arviointi suoritettiin lähtötilanteessa, 5 viikon sisällä ennen radikaalia kystektomiaa ja lantion alueen imusolmukedissektiota ja 6 viikkoa leikkauksen jälkeen. Radikaalin kystektomian ja lantion alueen imusolmukedissektion jälkeen kasvaimen statuksen arviointi suoritettiin 12 viikon välein 2 vuoteen asti ja 24 viikon välein sen jälkeen. Satunnaistaminen stratifioitiin kasvaimen asteen (T2N0 vs. T3/T4aN0 vs. T1-T4aN1), sisplatiinin soveltuvuuden (sisplatiini ei sovellu vs. sisplatiini soveltuu, mutta potilas kieltäytyi) ja maantieteellisen alueen (Yhdysvallat vs. Euroopan unioni vs. muu maailma) perusteella.
Mediaani-ikä oli 73 vuotta (vaihteluväli: 46–87 vuotta); 78 % oli miehiä ja 78 % valkoihoisia. Potilaiden ECOG-toimintakyky oli lähtötilanteessa 0–1 (86 %) tai 2 (14 %). 47 %:lla CPS-arvo oli ≥ 10 ja 52 %:lla CPS-arvo oli < 10. 18 %:lla oli T2N0, 77 %:lla T3/T4aN0 ja 5 %:lla T1-T4aN1. 41 %:lla potilaista lähtötilanteen kreatiniinipuhdistuma oli ≥ 60 ml/min, 59 %:lla < 60 ml/min mutta ≥ 30 ml/min ja 0,3 %:lla < 30 ml/min. 91 %:lla potilaista oli histologisesti uroteelikarsinooma; 4 %:lla oli uroteelikarsinooma, jossa oli mukana levyepiteelidifferentaatiota, 3 %:lla oli uroteelikarsinooma, jossa oli mukana glandulaarista differentaatiota, ja 2 %:lla oli uroteelikarsinooma, jossa oli mukana muita histologisia variantteja.
Kokonaisväestöstä 149 (88 %) potilaalle yhdessä pembrolitsumabin kanssa annetusta enfortumabivedotiinihoitohaarasta ja 156 (90 %) potilaalle pelkästä radikaalin kystektomian ja lantion alueen imusolmukedissektion hoitohaarasta tehtiin radikaali kystektomia ja lantion alueen imusolmukedissektio. Potilaista, joita hoidettiin enfortumabivedotiinilla yhdessä pembrolitsumabin kanssa, 100 potilasta aloitti adjuvanttihoidon ja 43 potilasta suorittivat kaikki suunnitellut syklit.
Ensisijainen tehon tulosmuuttuja oli päätetapahtumaton elinaika (EFS), jonka arvioi sokkoutettu riippumaton keskitetty arvioijataho (BICR). Toissijaisia tehon tulosmuuttujia olivat kokonaiselinaika (OS) ja patologinen täydellinen hoitovaste (pCR), jotka arvioi sokkoutettu riippumaton patologia-arvioijataho (BIPR).
Tutkimuksessa osoitettiin tilastollisesti merkitseviä parannuksia päätetapahtumattomassa elinajassa (EFS), kokonaiselinajassa (OS) ja patologisesti täydellisessä hoitovasteessa (pCR) potilailla, joita hoidettiin neoadjuvantti- ja adjuvanttienfortumabivedotiinilla yhdessä pembrolitsumabin kanssa verrattuna pelkkään radikaaliin kystektomiaan ja lantion alueen imusolmukedissektioon. Tämän tutkimuksen seuranta-ajan mediaani oli 18,5 kuukautta (vaihteluväli: 0,6–53,7 kuukautta).
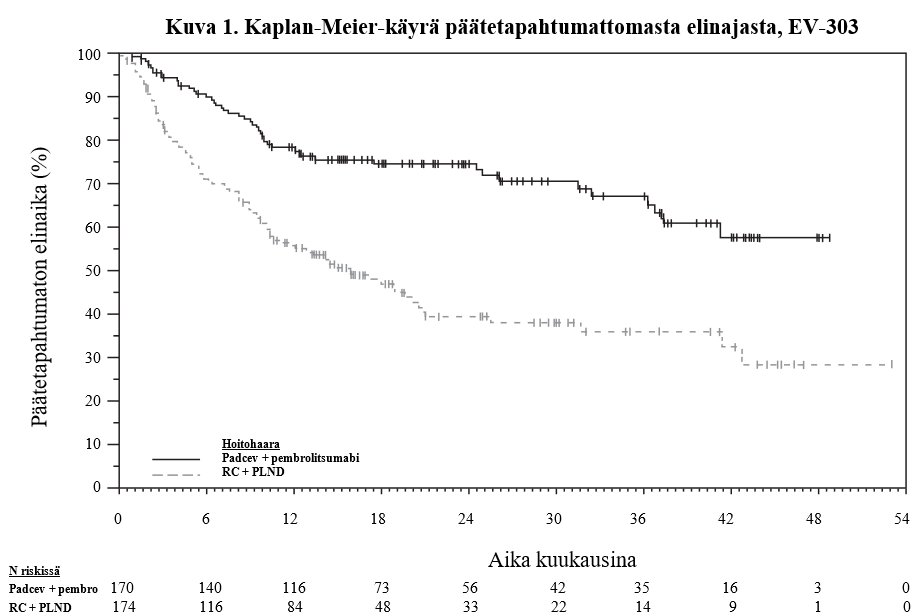
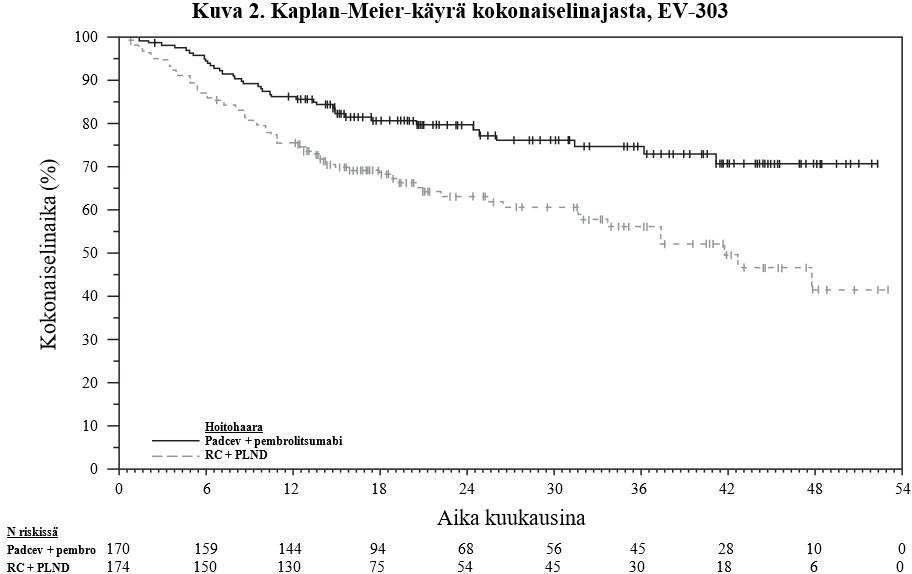
Taulukossa 6 ja kuvissa 1 ja 2 on esitetty yhteenveto EV-303-tutkimuksen tehotuloksista.
Taulukko 6. EV-303-tutkimuksen tehotulokset
Päätetapahtuma | Padcev + pembrolitsumabi | Pelkkä radikaali kystektomia (RC) ja lantion alueen imusolmukedissektio (PLND) |
Päätetapahtumaton elinaikaa | ||
Potilaiden määrä (%), joilla esiintyi tapahtumia | 48 (28,2) | 95 (54,6) |
Mediaani kuukausina (95 %:n CI)b | NR (37,3; NR) | 15,7 (10,3; 20,5) |
Riskisuhdec (95 %:n CI) | 0,40 (0,28; 0,57) | |
1-puolinen p-arvod | < 0,0001 | |
Kokonaiselinaika | ||
Potilaiden määrä (%), joilla esiintyi tapahtumia | 38 (22,4) | 68 (39,1) |
Mediaani kuukausina (95 %:n CI)b | NR (NR; NR) | 41,7 (31,8; NR) |
Riskisuhdec (95 %:n CI) | 0,50 (0,33; 0,74) | |
1-puolinen p-arvod | 0,0002 | |
Patologinen täydellinen hoitovaste | ||
Potilaiden määrä, joilla oli patologinen täydellinen hoitovaste | 97 | 15 |
pCR-arvo (%) (95 %:n CI) | 57,1 (49,3; 64,6) | 8,6 (4,9; 13,8) |
Hoitoeron arvio (%) (95 %:n CI)e | 48,3 (39,5; 56,5) | |
1-puolinen p-arvoe | < 0,000001 | |
NR = Not reached (ei saavutettu)
a. Päätetapahtumaton elinaika määritellään aikana satunnaistamisesta mihin tahansa seuraavista tapahtumista: radiografinen sairauden eteneminen, joka estää paranemiseen tähtäävän leikkauksen sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) arvioimana ennen radikaalia kystektomiaa ja lantion alueen imusolmukedissektiota; leikkauksen suorittamisen epäonnistuminen osallistujille, jolla on lihasinvasiivisen sairauden jäännös ja radiografinen sairaus; suuri jäännössairauden määrä leikkauksen aikaan (kirurgi ei pysty suorittamaan paranemiseen tähtäävää leikkausta leikkaukseen soveltumattoman kasvaimen tai vasta havaitun metastaattisen sairauden vuoksi); sairauden paikallinen tai etäuusiutuminen radikaalin kystektomian ja lantion alueen imusolmukedissektion jälkeen sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) TT- tai MK-kuvauksella arvioimana ja/tai biopsian perusteella – jos biopsia ei ole mahdollinen osallistujan turvallisuuden vuoksi, TT/MK yksinään riittää; kuolema mistä tahansa syystä.
b. Perustuu Kaplan-Meierin estimaatteihin.
c. Perustuu stratifioituun Coxin regressiomalliin Efronin sidoskäsittelymenetelmällä hoidon ollessa kovariaatti.
d. Perustuu stratifioituun log-rank-testiin.
e. Perustuu stratifioituun Miettisen ja Nurmisen menetelmään.
Aiemmin hoitamaton, paikallisesti edennyt tai metastaattinen uroteelisyöpä
EV-302 (KEYNOTE-A39)
Yhdessä pembrolitsumabin kanssa käytetyn Padcev-valmisteen tehoa arvioitiin tutkimuksessa EV-302 (KEYNOTE-A39), avoimessa, satunnaistetussa, vaiheen 3 monikeskustutkimuksessa, johon otetuilla 886 potilaalla oli leikkaukseen soveltumaton tai metastaattinen uroteelisyöpä ja jotka eivät olleet saaneet aiemmin mitään systeemistä hoitoa paikallisesti edenneeseen tai metastaattiseen sairauteen. Tutkimukseen otettiin potilaita, jotka olivat saaneet neoadjuvanttikemoterapiaa tai jotka olivat saaneet adjuvanttikemoterapiaa kystektomian jälkeen, mikäli sairaus oli uusiutunut > 12 kuukautta hoidon päättymisen jälkeen. Potilaiden katsottiin olevan sisplatiinihoitoon soveltumattomia, mikäli he täyttivät vähintään yhden seuraavista kriteereistä: glomerulusten suodatusnopeus (GFR) 30–59 ml/min, ECOG -toimintakyky ≥ 2, asteen ≥ 2 kuulonmenetys tai New York Heart Association (NYHA) -luokan III sydämen vajaatoiminta.
Potilaat satunnaistettiin suhteessa 1:1 saamaan joko enfortumabivedotiinia yhdessä pembrolitsumabin kanssa (hoitohaara A) tai gemsitabiinia ja platinapohjaista kemoterapiaa (sisplatiinia tai karboplatiinia) (hoitohaara B). Hoitohaaran A potilaat saivat 1,25 mg/kg enfortumabivedotiinia infuusiona laskimoon 30 minuutin ajan jokaisen 21 päivän syklin päivinä 1 ja 8 ja sitten 200 mg pembrolitsumabia jokaisen 21 päivän syklin päivänä 1 noin 30 minuuttia enfortumabivedotiinin jälkeen. Hoitohaaran B potilaat saivat 1 000 mg/m2 gemsitabiinia jokaisen 21 päivän syklin päivinä 1 ja 8 sekä 70 mg/m2 sisplatiinia tai karboplatiinia (AUC = 4,5 tai 5 mg/ml/min paikallisten ohjeiden mukaan) jokaisen 21 päivän syklin päivänä 1. Hoitoa jatkettiin, kunnes sairaus eteni, ei-hyväksyttävään toksisuuteen asti tai kunnes suurin sallittu määrä hoitosyklejä oli suoritettu loppuun (kemoterapia, 6 sykliä; pembrolitsumabi, 35 sykliä; enfortumabivedotiini, ei enimmäismäärää).
Potilailla, jotka oli satunnaistettu gemsitabiinia ja platinapohjaista kemoterapiaa saavaan hoitohaaraan, voitiin antaa ylläpitohoitona immunoterapiaa (esim. avelumabia). Satunnaistaminen stratifioitiin sisplatiinin soveltuvuuden (soveltuu vs. ei sovellu), PD-L1:n ilmentymisen (CPS ≥ 10 vs. CPS < 10) ja maksametastaasien (on vs. ei ole) perusteella. PD-L1:n ilmentyminen perustui PD-L1 IHC 22C3 pharmDx -määritykseen.
Potilaat suljettiin pois tutkimuksesta, jos heillä oli aktiivisia keskushermoston metastaaseja, jatkuvaa asteen ≥ 2 sensorista tai motorista neuropatiaa, huonossa hoitotasapainossa oleva diabetes, jonka määritelmänä oli hemoglobiini A1C (HbA1c) ≥ 8 % tai HbA1c ≥ 7 % ja liittyviä diabetesoireita, autoimmuunisairaus tai immunosuppressiota vaativa sairaus, pneumoniitti tai muun tyyppinen interstitiaalinen keuhkosairaus.
Mediaani-ikä oli 69 vuotta (vaihteluväli: 22–91 vuotta); 77 % oli miehiä ja useimmat potilaat olivat valkoihoisia (67 %) tai aasialaisia (22 %). Potilaiden ECOG-toimintakyky oli lähtötilanteessa 0 (49 %), 1 (47 %) tai 2 (3 %). 47 %:lla potilaista lähtötilanteen HbA1c oli dokumentoidusti < 5,7 %. Lähtötilanteessa 95 %:lla potilaista oli metastaattinen uroteelisyöpä ja 5 %:lla oli leikkaukseen soveltumaton uroteelisyöpä. 72 %:lla potilaista oli lähtötilanteessa sisäelinmetastaaseja, mukaan lukien 22 %:lla maksametastaaseja. 85 %:lla potilaista oli histologisesti uroteelikarsinooma, 6 %:lla oli uroteelikarsinooma, jossa oli mukana levyepiteelidifferentiaatiota, ja 2 %:lla oli muita uroteelikarsinooman sekamuotoisia histologisia variantteja. 46 %:lle potilaista ei voitu antaa sisplatiinia ja 54 % potilaalle voitiin antaa sisplatiinia satunnaistamisvaiheessa. Niistä 877 testatusta potilaasta, joiden kudoksesta voitiin arvioida PD-L1:n ilmentyminen, 58 %:lla oli PD-L1:tä ilmentäviä kasvaimia, joiden CPS-arvo oli ≥ 10, ja 42 %:lla oli PD-L1:tä ilmentäviä kasvaimia, joiden CPS-arvo oli < 10. Seuranta-ajan mediaani oli 17,3 kuukautta (vaihteluväli: 0,3–37,2 kuukautta).
Ensisijaisia tehon tulosmuuttujia olivat kokonaiselinaika (OS) ja elinaika ilman taudin etenemistä (PFS), jotka arvioi sokkoutettu riippumaton keskitetty arvioijataho (BICR) RECIST v1.1 -kriteerien mukaisesti. Toissijaisia tehon tulosmuuttujia olivat objektiivinen vasteprosentti (ORR), jonka arvioi sokkoutettu riippumaton keskitetty arvioijataho RECIST v1.1 -kriteerien mukaisesti.
Tutkimuksessa potilailla, jotka oli satunnaistettu saamaan enfortumabivedotiinia yhdessä pembrolitsumabin kanssa, osoitettiin tilastollisesti merkitseviä OS:n, PFS:n ja ORR:n parannuksia verrattuna gemsitabiiniin ja platinapohjaiseen kemoterapiaan.
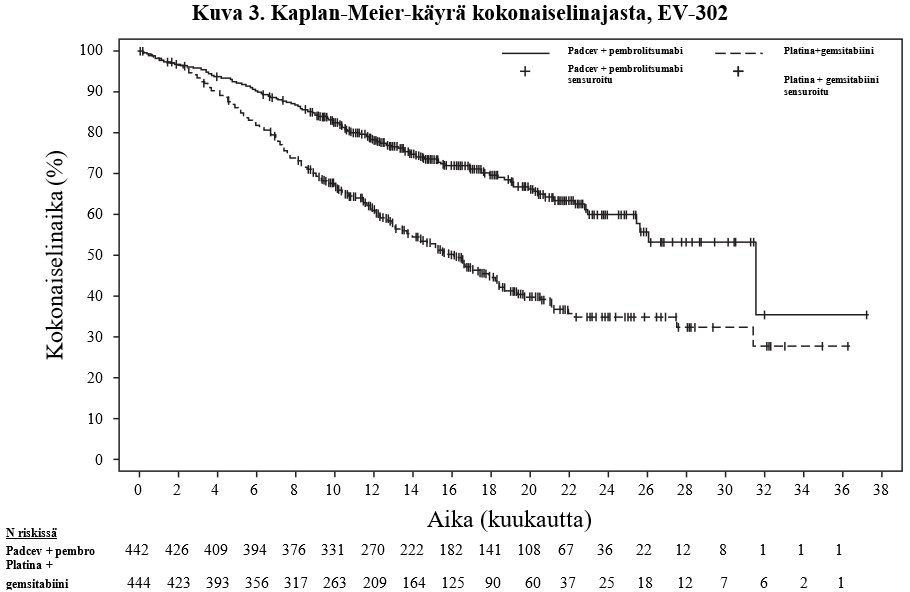
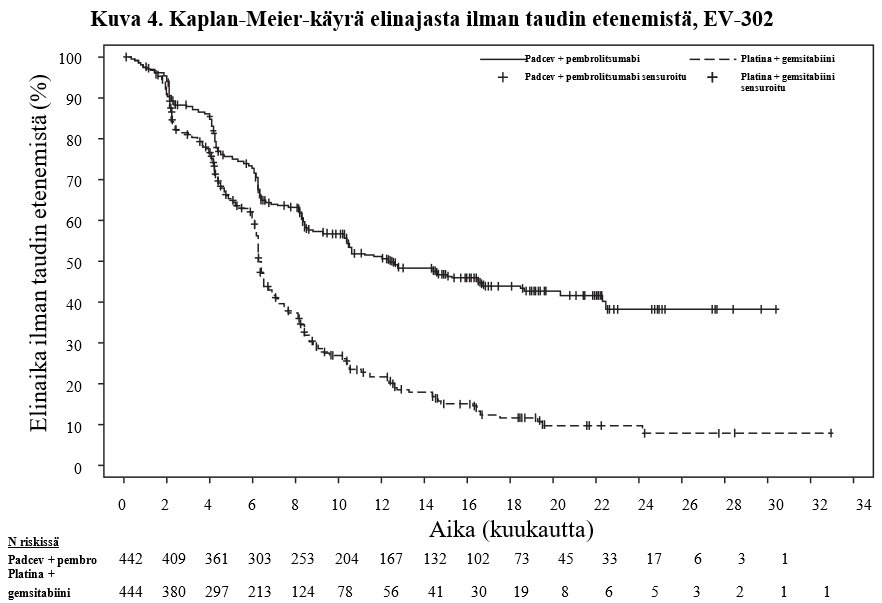
Taulukossa 7 ja kuvissa 3 ja 4 on esitetty yhteenveto EV-302-tutkimuksen tehotuloksista.
Taulukko 7. EV-302-tutkimuksen tehotulokset
Päätetapahtuma | Padcev + pembrolitsumabi n = 442 | Gemsitabiini + platina n = 444 |
|---|---|---|
Kokonaiselinaika | ||
Potilaiden määrä (%), joilla esiintyi tapahtumia | 133 (30,1) | 226 (50,9) |
Mediaani kuukausina (95 %:n CI)a | 31,5 (25,4; -) | 16,1 (13,9; 18,3) |
Riskisuhdeb (95 %:n CI) | 0,468 (0,376; 0,582) | |
2-puolinen p-arvoc | < 0,00001 | |
Elinaika ilman taudin etenemistäd | ||
Potilaiden määrä (%), joilla esiintyi tapahtumia | 223 (50,5) | 307 (69,1) |
Mediaani kuukausina (95 %:n CI)a | 12,5 (10,4; 16,6) | 6,3 (6,2; 6,5) |
Riskisuhdeb (95 %:n CI) | 0,450 (0,377; 0,538) | |
2-puolinen p-arvoc | < 0,00001 | |
Objektiivinen vasteprosentti (CR + PR)d,f | ||
Vahvistettu ORR (%) (95 %:n CI)e | 67,7 (63,1; 72,1) | 44,4 (39,7; 49,2) |
2-puolinen p-arvog | < 0,00001 | |
Vasteen kestod,f | ||
Mediaani kuukausina (95 %:n CI)a | NR (20,2; -) | 7,0 (6,2; 10,2) |
NR = Not reached (ei saavutettu). | ||
Enfortumabivedotiini monoterapiana
Aiemmin hoidettu, paikallisesti edennyt tai metastaattinen uroteelisyöpä
EV‑301
Monoterapiana käytetyn Padcev-valmisteen tehoa arvioitiin tutkimuksessa EV‑301, avoimessa, satunnaistetussa, vaiheen 3 monikeskustutkimuksessa, johon otetuilla 608 potilaalla oli paikallisesti edennyt tai metastaattinen uroteelisyöpä ja jotka olivat aiemmin saaneet platinaa sisältävää kemoterapiaa ja ohjelmoituneen solukuoleman reseptori 1 (PD-1)- tai ohjelmoituneen solukuoleman ligandi 1 (PD-L1) ‑inhibiittoria. Tutkimuksen ensisijainen päätetapahtuma oli kokonaiselinaika (OS), ja toissijaisiin päätetapahtumiin kuuluivat elinaika ilman taudin etenemistä (PFS) ja objektiivinen vaste (ORR) [PFS ja ORR arvioitiin tutkijan arvion perusteella RECIST v1.1 -menetelmää käyttäen]. Potilaat satunnaistettiin suhteessa 1:1 saamaan joko enfortumabivedotiinia 1,25 mg/kg jokaisen 28 päivän jakson päivinä 1, 8 ja 15 tai yhtä seuraavista kemoterapioista tutkijan päätöksen mukaan: dosetakseli 75 mg/m2 (38 %), paklitakseli 175 mg/m2 (36 %) tai vinfluniini 320 mg/m2 (25 %) 21 päivän jakson päivänä 1.
Potilaat suljettiin pois tutkimuksesta, jos heillä oli aktiivisia keskushermoston metastaaseja, jatkuvaa asteen ≥ 2 sensorista tai motorista neuropatiaa, tunnetusti ihmisen immuunikatovirus (HIV) ‑infektio (HIV 1 tai 2), aktiivinen hepatiitti B tai C tai huonossa hoitotasapainossa oleva diabetes, kuten HbA1c ≥ 8 % tai HbA1c ≥ 7 % ja liittyviä diabetesoireita.
Mediaani-ikä oli 68 vuotta (vaihteluväli: 30–88 vuotta), 77 % oli miehiä ja useimmat potilaat olivat valkoihoisia (52 %) tai aasialaisia (33 %). Kaikilla potilailla ECOG‑toimintakyky oli lähtötilanteessa 0 (40 %) tai 1 (60 %). 95 %:lla potilaista oli metastaattinen sairaus ja 5 %:lla oli paikallisesti edennyt sairaus. 80 %:lla potilaista oli sisäelinmetastaaseja, mukaan lukien 31 %:lla maksametastaaseja. 76 %:lla potilaista oli histologisesti uroteelikarsinooma / transitiosellulaarinen karsinooma (TCC), 14 %:lla oli sekamuotoinen uroteelikarsinooma ja noin 10 %:lla oli muita histologisia variantteja. Yhteensä 76 (13 %) potilasta oli aiemmin saanut ≥ 3 linjan systeemistä hoitoa. 52 % (314) potilaista oli saanut aiempaa PD-1-estäjää, 47 % (284) aiempaa PD-L1-estäjää ja lisäksi 1 % (9) potilaista oli saanut sekä PD-1- että PD-L1-estäjähoitoa. Vain 18 %:lla (111) potilaista oli saavutettu vaste aiempaan PD-1- tai PD-L1-estäjähoitoon. 63 % (383) potilaista oli saanut aiempaa sisplatiinipohjaista hoitoa, 26 % (159) karboplatiinipohjaista hoitoa ja lisäksi 11 % (65) oli saanut sekä sisplatiini- että karboplatiinipohjaista hoitoa.
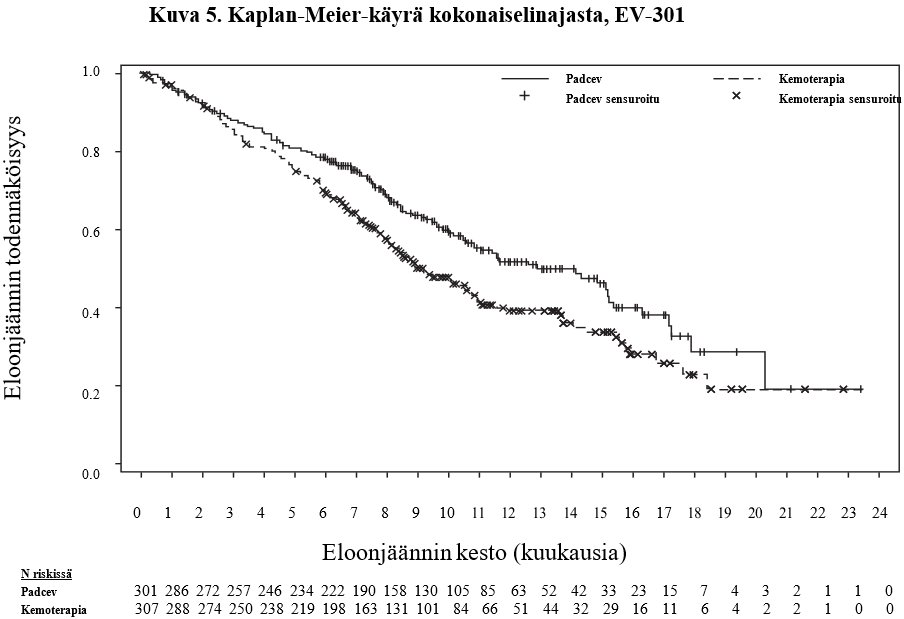
Taulukossa 8 on esitetty yhteenveto EV-301 -tutkimuksen tehotuloksista, kun seuranta-ajan mediaani oli 11,1 kuukautta (95 % CI: 10,6–11,6).
Taulukko 8. EV‑301 -tutkimuksen tehotulokset |
| |
Päätetapahtuma | Padcev n=301 | Kemoterapia n=307 |
Kokonaiselinaika (OS) | ||
Potilaiden määrä (%), joilla esiintyi tapahtumia | 134 (44,5) | 167 (54,4) |
Mediaani kuukausina (95 %:n CI) | 12,9 (10,6; 15,2) | 9,0 (8,1; 10,7) |
Riskisuhde (95 %:n CI) | 0,702 (0,556, 0,886) | |
1‑puolinen p‑arvo | 0,00142a | |
Elinaika ilman taudin etenemistä (PFS)b | ||
Potilaiden määrä (%), joilla esiintyi tapahtumia | 201 (66,8) | 231 (75,2) |
Mediaani kuukausina (95 %:n CI) | 5,6 (5,3; 5,8) | 3,7 (3,5; 3,9) |
Riskisuhde (95 %:n CI) | 0,615 (0,505; 0,748) | |
1‑puolinen p‑arvo | < 0,00001c | |
Objektiivinen vasteprosentti (CR + PR)b | ||
ORR (%) (95 %:n CI) | 40,6 (35,0; 46,5) | 17,9 (13,7; 22,8) |
1‑puolinen p‑arvo | < 0,001d | |
Täydellinen vaste (%) | 4,9 | 2,7 |
Osittainen vaste (%) | 35,8 | 15,2 |
Vasteen kesto niillä, joilla esiintyi vaste | ||
Mediaani kuukausina (95 %:n CI) | 7,4 (5,6; 9,5) | 8,1 (5,7; 9,6) |
a Etukäteen määritetty tehon raja = 0,00679, 1‑puolinen (mukautettuna havaituilla 301 kuolemalla). b Tutkijan arvioima RECIST v1.1 ‑kriteerien mukaan. c Etukäteen määritetty tehon raja = 0,02189, 1‑puolinen (mukautettuna havaituilla 432 PFS1-tapahtumalla). d Etukäteen määritetty tehon raja = 0,025, 1‑puolinen (mukautettuna 100 %:n informaatiofraktiolla). | ||
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset enfortumabivedotiinin käytöstä uroteelisyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Jakautuminen
Keskimääräinen arvio ADC:n vakaan tilan jakautumistilavuudesta oli 12,8 l, kun annettu enfortumabivedotiinia 1,25 mg/kg. Konjugoitumattoman MMAE:n sitoutuminen ihmisen plasmaproteiineihin in vitro vaihteli välillä 68 – 82 %. Konjugoitumaton MMAE ei todennäköisesti syrjäytä voimakkaasti proteiiniin sitoutuvia lääkevalmisteita tai joudu niiden syrjäyttämäksi. In vitro ‑tutkimukset osoittavat, että konjugoitumaton MMAE on P-glykoproteiinin substraatti.
Biotransformaatio
Enfortumabivedotiinista vapautunut pieni konjugoitumaton MMAE-fraktio metaboloituu. In vitro ‑tiedot osoittavat, että konjugoitumattoman MMAE:n metabolia tapahtuu ensisijaisesti CYP3A4:n oksidaation kautta.
Eliminaatio
ADC:n ja konjugoitumattoman MMAE:n keskimääräinen puhdistuma potilailla oli 0,11 l/h ja 2,11 l/h. ADC:n eliminaatiossa todettiin multieksponentiaalinen lasku puoliintumisajan ollessa 3,6 vuorokautta.
Konjugoitumattoman MMAE:n eliminoitumista vaikutti rajoittavan sen vapautumisnopeus enfortumabivedotiinista. Konjugoitumattoman MMAE:n eliminaatiossa todettiin multieksponentiaalinen lasku puoliintumisajan ollessa 2,6 vuorokautta.
Erittyminen
Konjugoitumaton MMAE erittyy pääasiassa ulosteeseen ja vähäisemmässä määrin virtsaan. Toisen konjugoitumatonta MMAE:ta sisältävän ADC:n kerta-annoksen jälkeen noin 24 % annetusta konjugoitumattoman MMAE:n kokonaismäärästä erittyi ulosteeseen ja virtsaan muuttumattomana konjugoitumattomana MMAE:na 1 viikon kuluessa. Suurin osa todetusta konjugoitumattomasta MMAE:sta erittyi ulosteeseen (72 %). Konjugoitumattoman MMAE:n erittymisprofiilin odotetaan olevan samankaltainen enfortumabivedotiinin antamisen jälkeen.
Erityisryhmät
Iäkkäät
Populaatiofarmakokineettinen analyysi osoittaa, että iällä (vaihteluväli: 24 – 90 vuotta; 60 % (450/748) > 65 vuotta, 19 % (143/748) > 75 vuotta) ei ole kliinisesti merkittävää vaikutusta enfortumabivedotiinin farmakokinetiikkaan.
Rotu ja sukupuoli
Populaatiofarmakokineettisen analyysin perusteella rodulla (69 % [519/748] valkoihoisia, 21 % [158/748] aasialaisia, 1 % [10/748] mustaihoisia ja 8 % [61/748] muita tai tuntemattomia) ja sukupuolella (73 % [544/748] miehiä) ei ole kliinisesti merkittävää vaikutusta enfortumabivedotiinin farmakokinetiikkaan.
Munuaisten vajaatoiminta
ADC:n ja konjugoitumattoman MMAE:n farmakokinetiikkaa arvioitiin, kun lievää (CrCL > 60 – 90 ml/min), keskivaikeaa (CrCL 30 – 60 ml/min) ja vaikeaa (CrCL 15 – 30 ml/min) munuaisten vajaatoimintaa sairastaneille potilaille oli annettu 1,25 mg/kg enfortumabivedotiinia. ADC:n tai konjugoitumattoman MMAE:n AUC-altistuksessa ei havaittu merkittäviä eroja potilailla, joilla oli lievä, keskivaikea tai vaikea munuaisten vajaatoiminta, verrattuna potilaisiin, joiden munuaiset toimivat normaalisti. Enfortumabivedotiinia ei ole arvioitu potilailla, joilla on loppuvaiheen munuaistauti (CrCL < 15 ml/min).
Maksan vajaatoiminta
Kun käytettiin tietoja kliinisistä tutkimuksista, joihin osallistuneilla potilailla oli metastaattinen uroteelisyöpä, populaatiofarmakokineettisen analyysin perusteella ADC:lle altistumisessa ei ollut merkittäviä eroja ja konjugoitumattoman MMAE:n keskimääräisissä pitoisuuksissa havaittiin 37 %:n nousu aiemmin hoidettua, paikallisesti edennyttä tai metastaattista uroteelisyöpää sairastavilla potilailla ja 16 %:n nousu aiemmin hoitamatonta, paikallisesti edennyttä tai metastaattista uroteelisyöpää sairastavilla potilailla, joilla oli lievä maksan vajaatoiminta (kokonaisbilirubiini 1–1,5 × ULN ja ASAT mikä tahansa tai kokonaisbilirubiini ≤ ULN ja ASAT > ULN) verrattuna potilaisiin, joiden maksa toimi normaalisti. Enfortumabivedotiinia on tutkittu vain pienellä määrällä potilaita, joilla oli keskivaikea maksan vajaatoiminta (n = 5) tai vaikea maksan vajaatoiminta (n = 1). Keskivaikean tai vaikean maksan vajaatoiminnan (kokonaisbilirubiini > 1,5 x ULN ja ASAT mikä tahansa) tai maksansiirron vaikutusta ADC:n tai konjugoitumattoman MMAE:n farmakokinetiikkaan ei tunneta.
Fysiologiaan perustuvan farmakokineettisen mallinnuksen ennusteet
Enfortumabivedotiinin ja ketokonatsolin (yhdistetty P‑gp:n estäjä ja vahva CYP3A:n estäjä) samanaikaisen käytön ennustetaan suurentavan konjugoitumattoman MMAE:n Cmax- ja AUC-altistusta vähäisesti ilman muutoksia ADC-altistukseen.
Enfortumabivedotiinin ja rifampisiinin (yhdistetty P‑gp:n indusoija ja vahva CYP3A:n indusoija) samanaikaisen käytön ennustetaan pienentävän konjugoitumattoman MMAE:n Cmax- ja AUC-altistusta kohtalaisesti ilman muutoksia ADC:n altistukseen. olla todellista pienempi. Fysiologiaan perustuva farmakokineettinen malli voi aliarvioida rifampisiinin täyttä vaikutusta konjugoitumattoman MMAE:n Cmax-arvoon.
Samanaikaisen enfortumabivedotiinin käytön ei ennusteta vaikuttavan midatsolaamialtistukseen (herkkä CYP3A:n substraatti). In vitro ‑tutkimukset ihmisen maksan mikrosomeilla osoittavat, että konjugoitumaton MMAE estää CYP3A4/5:ä, mutta ei muita CYP450-isoformeja. Konjugoitumaton MMAE ei indusoinut merkittäviä CYP450-entsyymejä ihmisen maksasoluissa.
In vitro ‑tutkimukset
In vitro ‑tutkimukset osoittavat, että konjugoitumaton MMAE on ulosvirtaustransportterin P‑glykoproteiinin (P‑gp) substraatti, ei estäjä. In vitro ‑tutkimuksissa on todettu, että konjugoitumaton MMAE ei ollut rintasyövän resistenssiproteiinin (BCRP), monilääkeresistenssiin liittyvän proteiinin 2 (MRP2), orgaanisia anioneja kuljettavan polypeptidi 1B1:n tai 1B3:n (OATP1B1 tai OATP1B3), orgaanisten kationien transportterin 2 (OCT2) tai orgaanisen anionitransportterin 1 tai 3 (OAT1 tai OAT3) substraatti. Konjugoitumaton MMAE ei ollut sappisuolojen poistopumpun (BSEP), P‑gp:n, BCRP:n, MRP2:n, OCT1:n, OCT2:n, OAT1:n, OAT3:n, OATP1B1:n tai OATP1B3:n estäjä kliinisesti relevantteina pitoisuuksina.
Prekliiniset tiedot turvallisuudesta
Genotoksisuustutkimukset osoittivat, että MMAE:lla ei ollut havaittavaa genotoksisuuspotentiaalia bakteerien käänteismutaatiotestissä (Amesin testi) tai L5178Y TK+/‑ hiiren lymfoomamutaatiomäärityksessä. MMAE aiheutti kromosomipoikkeavuuksia rotille mikrotumatestissä, mikä on yhdenmukaista mikrotubuluksiin vaikuttavien aineiden farmakologisen toiminnan kanssa.
Iholeesioita havaittiin toistuvan annoksen tutkimuksissa rotilla (4- ja 13-viikkoiset) ja apinoilla (4-viikkoiset). Ihomuutokset hävisivät täysin 6 viikon toipumisjakson loppuun mennessä.
Kliinisissä tutkimuksissa raportoitua hyperglykemiaa ei ilmennyt rotilla ja apinoilla tehdyissä toksisuustutkimuksissa, eikä kummankaan lajin haimoissa ollut histopatologisia löydöksiä.
Sikiötoksisuutta (pienempi poikueen koko tai täydellinen poikueen menetys) havaittiin ja poikuekoon pieneneminen ilmeni varhaisten resorptioiden lisääntymisenä. Sikiön keskimääräinen ruumiinpaino annostasolla 2 mg/kg henkiin jääneillä sikiöillä oli pienempi kontrolliryhmään verrattuna.
Enfortumabivedotiiniin liittyvät sikiön luuston poikkeamat katsottiin kehitysviivästymisiksi. 2 mg/kg annos (suurin piirtein sama altistus kuin ihmisille suositellulla annoksella) aiheutti emoon kohdistuvia toksisia vaikutuksia, alkio- ja sikiökuolemia ja rakenteellisia epämuodostumia, kuten vatsahalkioita, takaraajojen kiertymishäiriöitä, etukäpälän puuttumista, sisäelinten virheasentoja ja kaularangan nikamien fuusioitumista. Lisäksi havaittiin luuston poikkeavuuksia (rintalastan osien epäsymmetrisyys, fuusioituminen, puutteellinen luutuminen ja epämuodostumat, kaularangan nikamien epämuodostumat ja rintakehän keskustan toispuoleinen luutuminen) ja sikiöiden painon pienenemistä.
Kivestoksisuutta havaittiin vain rotilla ja se korjaantui osittain 24 viikon toipumisjakson loppuun mennessä.
Enfortumabivedotiinin ja pembrolitsumabin yhdistelmällä ei ole tehty nimenomaisia prekliinisiä turvallisuustutkimuksia.
Farmaseuttiset tiedot
Apuaineet
Histidiini
Histidiinihydrokloridimonohydraatti
Trehaloosidihydraatti
Polysorbaatti 20
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamaton injektiopullo
4 vuotta
Käyttökuntoon saatettu liuos injektiopullossa
Mikrobiologiselta kannalta käyttökuntoon saattamisen jälkeen injektiopullojen liuos on lisättävä välittömästi infuusiopussiin. Jos valmistetta ei käytetä välittömästi, käyttökuntoon saatettujen injektiopullojen säilytysajat ja käyttöä edeltävät olosuhteet ovat käyttäjän vastuulla eivätkä normaalisti ole yli 24 tuntia jääkaapissa lämpötilassa 2 °C – 8 °C). Ei saa jäätyä.
Laimennettu liuos infuusiopussissa
Mikrobiologiselta kannalta infuusiopussiin laimennettu liuos on annettava potilaalle välittömästi laimentamisen jälkeen. Jos valmistetta ei käytetä välittömästi, laimennetun liuoksen säilytysajat ja käyttöä edeltävät olosuhteet ovat käyttäjän vastuulla eivätkä normaalisti ole yli 16 tuntia jääkaapissa lämpötilassa 2 °C – 8 °C infuusioaika mukaan luettuna. Ei saa jäätyä.
Säilytys
Avaamattomat injektiopullot
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
PADCEV kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
20 mg (L:ei) 1 kpl (816,66 €)
30 mg (L:ei) 1 kpl (1202,78 €)
PF-selosteen tieto
Padcev 20 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos, injektiopullossa
10 ml:n tyypin I lasinen injektiopullo, jossa on harmaa bromobutyylikumisuljin, 20 mm:n alumiinitiiviste ja vihreä rengas ja vihreä korkki. Yksi pahvikotelo sisältää yhden injektiopullon.
Padcev 30 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos, injektiopullossa
10 ml:n tyypin I lasinen injektiopullo, jossa on harmaa bromobutyylikumisuljin, 20 mm:n alumiinitiiviste ja hopeanvärinen rengas ja keltainen korkki. Yksi pahvikotelo sisältää yhden injektiopullon.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen kylmäkuivattu kuiva-aine.
Käyttö- ja käsittelyohjeet
Valmistelu- ja anto-ohjeet
Käyttökuntoon saattaminen kerta-annosinjektiopullossa
1. Noudata syöpälääkevalmisteita koskevia asianmukaisia käsittely- ja hävitystoimenpiteitä.
2. Käytä käyttökuntoon saattamiseen ja annostusliuosten valmistelemiseen asianmukaista aseptista tekniikkaa.
3. Laske suositeltu annos potilaan painon perusteella, jotta voit määrittää tarvittavien injektiopullojen määrän ja vahvuuden (20 mg tai 30 mg).
4. Saata jokainen injektiopullo käyttökuntoon seuraavasti ja mikäli mahdollista ohjaa steriili injektionesteisiin käytettävä vesi injektiopullon seinämiä pitkin, ei suoraan kylmäkuivattuun kuiva-aineeseen:
a. 20 mg:n injektiopullo: Lisää 2,3 ml steriiliä injektionesteisiin käytettävää vettä, mikä tuottaa enfortumabivedotiinin vahvuudeksi 10 mg/ml.
b. 30 mg:n injektiopullo: Lisää 3,3 ml steriiliä injektionesteisiin käytettävää vettä, mikä tuottaa enfortumabivedotiinin vahvuudeksi 10 mg/ml.
5. Pyöritä jokaista injektiopulloa hitaasti, kunnes sisältö on liuennut kokonaan. Anna käyttökuntoon saatetun injektiopullon (saatettujen injektiopullojen) seistä vähintään 1 minuutin ajan, kunnes kuplat ovat hävinneet. Injektiopulloa ei saa ravistaa. Ei saa altistaa suoralle auringonvalolle.
6. Liuos on tarkastettava visuaalisesti hiukkasten ja värimuutosten varalta. Käyttökuntoon saatetun liuoksen pitäisi olla kirkasta tai hieman opalisoivaa, väritöntä tai vaaleankeltaista eikä siinä saa olla näkyviä hiukkasia. Hävitä kaikki injektiopullot, joissa on näkyviä hiukkasia tai värimuutoksia.
Laimennus infuusiopussissa
7. Ota laskettu annosmäärä käyttökuntoon saatettua liuosta injektiopullo(i)sta ja siirrä se infuusiopussiin.
8. Laimenna enfortumabivedotiinia joko dekstroosilla 50 mg/ml (5 %), natriumkloridilla 9 mg/ml (0,9 %) tai Ringerin laktaattiliuoksella. Infuusiopussiin on mahduttava tarpeeksi liuotinta, jotta lopullinen pitoisuus on 0,3 – 4 mg/ml enfortumabivedotiinia.
Laimennettu enfortumabivedotiiniliuos on yhteensopiva polyvinyylikloridista (PVC), etyylivinyyliasetaatista tai polypropeenin (PP) kaltaisesta polyolefiinista valmistettujen infuusiopussien kanssa, polyeteenistä (PE) tai glykolimuokatusta polyeteenitereftalaatista valmistettujen infuusiopullojen kanssa, sellaisten PVC:stä valmistettujen infuusioletkustojen kanssa, joissa on käytetty pehmentimenä (bis(2-etyyliheksyyli)ftalaattia (DEHP) tai tris(2-etyyliheksyyli) trimellitaattia (TOTM)) ja polyeteeniä, sekä polyeetterisulfonista, polyvinylideenidifluoridista tai sekoitetuista selluloosaestereistä valmistettujen suodatinkalvojen (huokoskoko: 0,2–1,2 μm) kanssa.
9. Sekoita laimennettu liuos kääntelemällä varovasti. Pussia ei saa ravistaa. Ei saa altistaa suoralle auringonvalolle.
10. Infuusiopussi on tarkastettava visuaalisesti hiukkasten ja värimuutosten varalta ennen käyttöä. Käyttökuntoon saatetun liuoksen pitäisi olla kirkasta tai hieman opalisoivaa, väritöntä tai vaaleankeltaista eikä siinä saa olla näkyviä hiukkasia. Infuusiopussia ei saa käyttää, jos siinä näkyy hiukkasia tai värimuutoksia.
11. Kerta-annosinjektiopulloihin jäänyt käyttämätön osuus on hävitettävä.
Antaminen
12. Anna infuusio 30 minuutin kuluessa laskimoletkulla. Valmistetta ei saa antaa pikaboluksena tai boluksena laskimoon.
Yhteensopimattomuuksia ei ole havaittu akryylinitriilibutadieenistyreenistä (ABS), akryylistä, aktiivihiilestä, etyleenipropyleenidieenimonomeeristä, metakrylaatti-ABS:stä, polykarbonaatista, polyisopreenistä, polyoksimetyleenistä, PP:stä, silikonista, ruostumattomasta teräksestä ja lämpömuovautuvasta elastomeeristä koostuvien suljetun järjestelmän siirtolaitteiden ja käyttökuntoon saatetun liuoksen välillä.
13. Ei saa antaa samanaikaisesti muiden lääkevalmisteiden kanssa samalla infuusioletkulla.
14. Antamisen aikana on suositeltavaa käyttää letkusuodattimia tai ruiskusuodattimia (huokoskoko: 0,2–1,2 µm, suositellut materiaalit: polyeetterisulfoni, polyvinyylideenidifluoridi, sekoitetut selluloosaesterit).
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Lisätietoa tästä lääkevalmisteesta on Euroopan lääkeviraston verkkosivulla http://www.ema.europa.eu.
Korvattavuus
PADCEV kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
20 mg 1 kpl
30 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FX13
Valmisteyhteenvedon muuttamispäivämäärä
19.06.2026
Yhteystiedot
Hatsinanpuisto 8
02600 Espoo
09 8560 6000
www.astellas.fi