
RYBREVANT infuusiokonsentraatti, liuosta varten 350 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi millilitra infuusiokonsentraattia sisältää 50 mg amivantamabia.
Yksi 7 ml:n injektiopullo sisältää 350 mg amivantamabia.
Amivantamabi on täysin humaani immunoglobuliini G1 (IgG1) -pohjainen bispesifinen vasta-aine, joka kohdentuu epidermaalisen kasvutekijän (EGF) ja mesenkymaalis-epidermaalisen siirtymän (MET) reseptoreihin. Se on tuotettu nisäkässolulinjalla (kiinanhamsterin munasarja [CHO]) rekombinantti-DNA-tekniikalla.
Apuaine, jonka vaikutus tunnetaan:
Yksi ml liuosta sisältää 0,6 mg polysorbaatti 80:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Rybrevant on tarkoitettu:
- yhdistelmänä latsertinibin kanssa edenneen ei-pienisoluisen keuhkosyövän (NSCLC) ensilinjan hoitoon aikuispotilailla, joilla on epidermaalisen kasvutekijäreseptorin (EGFR) eksonin 19 deleetioita tai eksonin 21 L858R‑substituutiomutaatioita
- yhdistelmänä karboplatiinin ja pemetreksedin kanssa edenneen ei-pienisoluisen keuhkosyövän (NSCLC) hoitoon aikuispotilailla, joilla on epidermaalisen kasvutekijäreseptorin (EGFR) eksonin 19 deleetioita tai eksonin 21 L858R-substituutiomutaatioita, aiemman EGFR:n tyrosiinikinaasin estäjää sisältäneen hoidon epäonnistumisen jälkeen
- yhdistelmänä karboplatiinin ja pemetreksedin kanssa edenneen ei-pienisoluisen keuhkosyövän (NSCLC) ensilinjan hoitoon aikuispotilailla, joilla on aktivoivia epidermaalisen kasvutekijäreseptorin (EGFR) eksonin 20 insertiomutaatioita
- monoterapiana edenneen ei-pienisoluisen keuhkosyövän hoitoon aikuispotilailla, joilla on aktivoivia EGFR:n eksonin 20 insertiomutaatioita, platinapohjaisen hoidon epäonnistumisen jälkeen.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Syöpälääkevalmisteiden käyttöön perehtyneen lääkärin on aloitettava Rybrevant-hoito ja valvottava sitä.
Rybrevant-valmiste on annettava terveydenhuollon ammattilaisen toimesta. Terveydenhuollon ammattilaisella pitää olla saatavilla asianmukainen lääketieteellinen tuki mahdollisten infuusioon liittyvien reaktioiden hoitoon.
Ennen Rybrevant-hoidon aloittamista EGFR:n mutaatiostatus täytyy määrittää validoidulla testimenetelmällä kasvainkudos- tai plasmanäytteistä. Jos plasmanäytteessä ei havaita mutaatiota, on mutaatiot testattava kasvainkudoksesta, jos sitä on saatavilla määrältään ja laadultaan riittävästi, sillä plasmatesteihin liittyy virheellisten negatiivisten tulosten mahdollisuus. Testaus voidaan tehdä milloin tahansa alkuvaiheen diagnoosin ja hoidon aloittamisen välillä; kun EGFR:n mutaatiostatus on varmistettu, testausta ei tarvitse tehdä uudelleen (ks. kohta Farmakodynamiikka).
Annostus
Esilääkitys on annettava Rybrevant-valmisteen käytön yhteydessä ilmenevien infuusioon liittyvien reaktioiden riskin vähentämiseksi (ks. jäljempänä kohdat "Annoksen muuttaminen" ja "Suositellut muut samanaikaisesti käytettävät lääkevalmisteet").
3 viikon välein
Suositellut Rybrevant-annostukset yhdistelmänä karboplatiinin ja pemetreksedin kanssa käytettäessä on esitetty taulukossa 1 (ks. jäljempänä kohta ”Infuusionopeudet” ja taulukko 5).
| Taulukko 1 Suositeltu Rybrevant-annostus 3 viikon välein | |||
| Paino lähtötilanteessaa | Rybrevant-annos | Hoitoaikataulu | Injektiopullojen lukumäärä |
| Alle 80 kg | 1 400 mg | Viikoittain (yhteensä 4 annosta) viikoilla 1–4
| 4 |
| 1 750 mg | 3 viikon välein viikosta 7 alkaen | 5 | |
| Vähintään 80 kg | 1 750 mg | Viikoittain (yhteensä 4 annosta) viikoilla 1–4
| 5 |
| 2 100 mg | 3 viikon välein viikosta 7 alkaen | 6 | |
| a Lähtötilanteen jälkeiset painon muutokset eivät edellytä annosmuutoksia. | |||
Yhdistelmänä karboplatiinin ja pemetreksedin kanssa käytettäessä Rybrevant-valmiste pitää antaa karboplatiinin ja pemetreksedin jälkeen seuraavassa järjestyksessä: pemetreksedi, karboplatiini ja sen jälkeen Rybrevant. Ks. kohta Farmakodynamiikka sekä karboplatiinin ja pemetreksedin annostusohjeet ko. lääkevalmisteiden valmistetiedoista.
2 viikon välein
Suositellut Rybrevant-monoterapian tai Rybrevant-valmisteen ja latsertinibin yhdistelmän annostukset on esitetty taulukossa 2 (ks. jäljempänä kohta "Infuusionopeudet” ja taulukko 6).
| Taulukko 2 Suositeltu Rybrevant-annostus 2 viikon välein | |||
| Paino lähtötilanteessaa | Rybrevant-annos | Hoitoaikataulu | 350 mg/7 ml Rybrevant-valmistetta sisältävien injektiopullojen lukumäärä |
| Alle 80 kg | 1 050 mg | Viikoittain (yhteensä 4 annosta) viikoilla 1–4
| 3 |
| 2 viikon välein viikosta 5 alkaen | |||
| Vähintään 80 kg | 1 400 mg | Viikoittain (yhteensä 4 annosta) viikoilla 1–4
| 4 |
| 2 viikon välein viikosta 5 alkaen | |||
| a Lähtötilanteen jälkeiset painon muutokset eivät edellytä annosmuutoksia. | |||
Yhdistelmänä latsertinibin kanssa käytettäessä Rybrevant-valmiste on suositeltavaa antaa milloin tahansa latsertinibin jälkeen, kun se annetaan samana päivänä. Ks. suositeltu latsertinibiannostus latsertinibin valmisteyhteenvedon kohdasta Annostus ja antotapa.
Hoidon kesto
On suositeltavaa, että potilaita hoidetaan Rybrevant-valmisteella, kunnes sairaus etenee tai ilmaantuu toksisuutta, joka ei ole hyväksyttävissä.
Annoksen väliin jääminen
Jos suunniteltu annos jää väliin, annos on annettava mahdollisimman pian. Annostusaikataulua on tällöin muutettava vastaavasti, säilyttäen hoitoväli.
Annoksen muuttaminen
Annostus on keskeytettävä vaikeusasteen 3 tai 4 tasoisten haittavaikutusten ilmetessä, kunnes haittavaikutus lievittyy ≤ asteeseen 1 tai lähtötasoon. Jos keskeytys on kestoltaan enintään 7 vuorokautta, hoito aloitetaan uudelleen nykyisellä annoksella. Jos keskeytys kestää pidempään kuin 7 vuorokautta, on suositeltavaa aloittaa hoito uudelleen pienemmällä annoksella taulukon 3 mukaisesti. Katso myös annosmuutokset tiettyjen haittavaikutusten osalta alla olevasta taulukosta 3.
Latsertinibin kanssa yhdistelmänä käytettäessä ks. tiedot annosmuutoksista latsertinibin valmisteyhteenvedon kohdasta Annostus ja antotapa.
| Taulukko 3 Suositellut annosmuutokset haittavaikutusten seurauksena | |||
| Annos, jota käytettäessä haittavaikutus ilmeni | Annos 1. keskeytyksen jälkeen haitta-vaikutuksen vuoksi | Annos 2. keskeytyksen jälkeen haitta-vaikutuksen vuoksi | Annos 3. keskeytyksen jälkeen haitta-vaikutuksen vuoksi |
| 1 050 mg | 700 mg | 350 mg | Rybrevant-hoito lopetetaan |
| 1 400 mg | 1 050 mg | 700 mg | |
| 1 750 mg | 1 400 mg | 1 050 mg | |
| 2 100 mg | 1 750 mg | 1 400 mg | |
Infuusioon liittyvät reaktiot
Infuusio pitää keskeyttää ensimmäisen infuusioon liittyvän reaktion merkin ilmetessä. Muita tukihoitona käytettäviä lääkevalmisteita (esim. ylimääräiset glukokortikoidit, antihistamiini, antipyreetit ja antiemeetit) on annettava kliinisen tarpeen mukaisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Vaikeusaste 1–3 (lievä–vaikea): Oireiden lievityttyä infuusiota jatketaan 50 %:n nopeudella aiempaan nopeuteen nähden. Jos muita oireita ei ilmene, nopeutta voidaan lisätä suositellun infuusionopeuden mukaisesti (ks. taulukot 5 ja 6). Muut samanaikaisesti annettavat lääkevalmisteet (mukaan lukien deksametasoni [20 mg] tai vastaava) on annettava seuraavan annoksen kohdalla (ks. taulukko 4).
- Toistuva vaikeusasteen 3 tasoinen tai vaikeusasteen 4 tasoinen (hengenvaarallinen): Rybrevant-hoito lopetetaan pysyvästi.
Laskimotromboemboliset tapahtumat käytettäessä samanaikaisesti latsertinibia
Potilaille, jotka saavat Rybrevant-valmistetta yhdistelmänä latsertinibin kanssa, pitää hoitoa aloitettaessa antaa estolääkityksenä antikoagulantteja laskimotromboembolisten tapahtumien ennaltaehkäisyyn. Kliinisten hoitosuositusten mukaisesti potilaille pitää antaa estolääkityksenä joko suun kautta otettavaa suoraa antikoagulanttia tai pienimolekyylistä hepariinia. K‑vitamiiniantagonistien käyttöä ei suositella.
Jos laskimotromboembolisiin tapahtumiin liittyy kliinistä epävakautta (esim. hengitysvajaus tai sydämen toimintahäiriö), hoitoa ei pidä antaa kummallakaan valmisteella ennen kuin potilaan tila on kliinisesti vakaa. Sen jälkeen kummankin lääkevalmisteen käyttöä voidaan jatkaa aiemmalla annoksella. Jos laskimotromboembolisia tapahtumia ilmenee uudelleen asianmukaisesta hyytymisenestohoidosta huolimatta, Rybrevant-hoito on lopetettava. Latsertinibihoitoa voidaan jatkaa aiemmalla annoksella.
Ihon ja kynsien reaktiot
Iho ja kynsireaktioiden riskin ja vaikeusasteen vähentämiseksi Rybrevant-hoitoa saavilla potilailla suositellaan estohoitoa suun kautta otettavilla ja paikallisesti käytettävillä antibiooteilla. Kasvoihin ja koko kehoon (päänahkaa lukuun ottamatta) suositellaan käyttämään ei-komedogeenista kosteusvoidetta (mieluiten keramidipohjainen tai muu ihoa pitkäkestoisesti kosteuttava koostumus, joka ei sisällä kuivattavia ainesosia), ja käsien ja jalkaterien pesemiseen suositellaan myös klooriheksidiiniliuosta. Potilaita on neuvottava rajoittamaan altistumista auringolle Rybrevant-hoidon aikana ja 2 kuukauden ajan sen jälkeen. Ks. kohdasta Varoitukset ja käyttöön liittyvät varotoimet lisätietoja ihon ja kynsien reaktioiden estohoidosta.
Jos potilaalle kehittyy vaikeusasteen 1–2 tasoinen ihon tai kynsien reaktio, tukihoito on aloitettava kliinisen tarpeen mukaan. Jos ilmenee pitkittynyt vaikeusasteen 2 ihottuma, joka ei lievity 2 viikossa, annoksen pienentämistä on harkittava (ks. taulukko 3). Jos potilaalle kehittyy vaikeusasteen 3 tasoinen ihon tai kynsien reaktio, tukihoito on aloitettava kliinisen tarpeen mukaan ja Rybrevant-hoidon keskeyttämistä on harkittava haittavaikutuksen lievittymiseen saakka. Kun ihon tai kynsien reaktio on lievittynyt ≤ vaikeusasteen 2 tasoiseksi, Rybrevant-hoitoa on jatkettava pienemmällä annoksella. Jos potilaalle kehittyy vaikeusasteen 4 tasoisia ihoreaktioita, Rybrevant-hoito on lopetettava pysyvästi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Interstitiaalinen keuhkosairaus
Interstitiaalista keuhkosairautta tai interstitiaalisen keuhkosairauden kaltaisia haittavaikutuksia (pneumoniittia) epäiltäessä Rybrevant-hoito pitää keskeyttää. Jos potilaalla varmistuu interstitiaalinen keuhkosairaus (ILD) tai interstitiaalisen keuhkosairauden kaltainen haittavaikutus (esim. pneumoniitti), Rybrevant-hoito on lopetettava pysyvästi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Suositellut muut samanaikaisesti käytettävät lääkevalmisteet
Kaksi päivää ennen ensimmäistä infuusiota:
Potilaan pitää ottaa ensimmäistä Rybrevant-infuusiota edeltävinä kahtena päivänä 8 mg deksametasonia suun kautta kaksi kertaa päivässä.
Infuusiopäivä:
Ensimmäisen infuusion antopäivänä (viikko 1, päivä 1) potilaan pitää ottaa 8 mg deksametasonia suun kautta tunti ennen infuusiota sen lisäksi, että hänelle annetaan deksametasonia laskimoon, infuusioon liittyvien reaktioiden riskin pienentämiseksi edelleen.
Ennen infuusiota (viikko 1, päivät 1 ja 2) on annettava antihistamiineja, antipyreettejä ja glukokortikoideja infuusioon liittyvien reaktioiden riskin vähentämiseksi (ks. taulukko 4). Myöhempiä annoksia varten on tarpeen antaa antihistamiineja ja antipyreettejä. Myös glukokortikoidien käyttö pitää aloittaa uudelleen hoidon oltua pitkään keskeytettynä. Antiemeettejä on annettava tarpeen mukaan.
| Taulukko 4 Esilääkitysten annostusaikataulu | |||
| Esilääkitys | Annos | Antoreitti | Suositeltu annostusikkuna ennen Rybrevant-valmisteen antoa |
| Antihistamiini* | Difenhydramiini (25–50 mg) tai vastaava | Laskimoon | 15–30 minuuttia |
| Suun kautta | 30–60 minuuttia | ||
| Antipyreetti* | Parasetamoli/asetaminofeeni (650–1 000 mg) | Laskimoon | 15–30 minuuttia |
| Suun kautta | 30–60 minuuttia | ||
| Glukokortikoidi‡ | Deksametasoni (8 mg) | Suun kautta | 60 minuuttia |
| Glukokortikoidi‡ | Deksametasoni (20 mg) tai vastaava | Laskimoon | 60–120 minuuttia |
| Glukokortikoidi+ | Deksametasoni (10 mg) tai vastaava | Laskimoon | 45–60 minuuttia |
| * Tarvitaan kaikilla annoksilla. ‡ Tarvitaan alkuannoksen yhteydessä (viikko 1, päivä 1) tai infuusioon liittyvän reaktion ilmetessä seuraavan annoksen yhteydessä. + Tarvitaan toisen annoksen yhteydessä (viikko 1, päivä 2); valinnainen myöhempien annosten yhteydessä. | |||
Erityiset potilasryhmät
Pediatriset potilaat
Ei ole asianmukaista käyttää amivantamabia pediatrisille potilaille ei-pienisoluisen keuhkosyövän hoitoon.
Iäkkäät potilaat
Annoksen muuttaminen ei ole tarpeen (ks. kohta Haittavaikutukset, kohta Farmakodynamiikka ja kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Amivantamabin käytöstä munuaisten vajaatoimintaa sairastaville potilaille ei ole tehty muodollisia tutkimuksia. Populaatiofarmakokineettisten analyysien perusteella annoksen muuttaminen ei ole tarpeen potilailla, joilla on lievä tai kohtalainen munuaisten vajaatoiminta. Varovaisuutta on noudatettava potilaiden kohdalla, joilla on vaikea munuaisten vajaatoiminta, koska amivantamabia ei ole tutkittu tässä potilasjoukossa (ks. kohta Farmakokinetiikka). Jos hoito aloitetaan, potilaita on seurattava haittavaikutusten varalta muuttaen annosta edellä esitettyjen suositusten mukaisesti.
Maksan vajaatoiminta
Amivantamabilla ei ole tehty muodollisia tutkimuksia maksan vajaatoimintaa sairastavilla potilailla. Populaatiofarmakokineettisten analyysien perusteella annoksen muuttaminen ei ole tarpeen potilailla, joilla on lievä maksan vajaatoiminta. Varovaisuutta on noudatettava, jos potilaalla on kohtalainen tai vaikea maksan vajaatoiminta, koska amivantamabia ei ole tutkittu tässä potilasjoukossa (ks. kohta Farmakokinetiikka). Jos hoito aloitetaan, potilasta on seurattava haittavaikutusten varalta muuttaen annosta edellä esitettyjen suositusten mukaisesti.
Antotapa
Rybrevant annetaan laskimoon. Se annetaan laskimoinfuusiona sen jälkeen, kun sitä on laimennettu steriilillä 5 % glukoosiliuoksella tai 9 mg/ml (0,9 %) natriumkloridiliuoksella injektiota varten. Rybrevant on annettava in-line-suodatuksen kanssa.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Infuusionopeudet
Laimentamisen jälkeen infuusio on annettava laskimoon alla olevassa taulukossa 5 tai 6 esitetyillä infuusionopeuksilla. Ensimmäisen annoksen kohdalla ilmenevien infuusioon liittyvien reaktioiden yleisyyden takia amivantamabi on infusoitava perifeerisen laskimon kautta viikolla 1 ja viikolla 2. Infuusio voidaan antaa keskuslaskimokatetrin kautta, kun infuusioon liittyvien reaktioiden riski on pienempi (ks. kohta Käyttö- ja käsittelyohjeet). On suositeltavaa, että ensimmäinen annos valmistetaan mahdollisimman lähellä infuusion antoa sen loppuun suorittamisen todennäköisyyden maksimoimiseksi siinä tapauksessa, että ilmenisi infuusioon liittyä reaktio.
| Taulukko 5 Infuusionopeudet 3 viikon välein annettavassa Rybrevant-hoidossa | |||
| Paino alle 80 kg | |||
| Viikko | Annos (per 250 ml:n pussi) | Alkuvaiheen infuusionopeus | Myöhempi infuusionopeus† |
| Viikko 1 (infuusioannos jaetaan kahdelle päivälle) | |||
| Viikko 1 päivä 1 | 350 mg | 50 ml/h | 75 ml/h |
| Viikko 1 päivä 2 | 1 050 mg | 33 ml/h | 50 ml/h |
| Viikko 2 | 1 400 mg | 65 ml/h | |
| Viikko 3 | 1 400 mg | 85 ml/h | |
| Viikko 4 | 1 400 mg | 125 ml/h | |
| Myöhemmät viikot* | 1 750 mg | 125 ml/h | |
| Paino vähintään 80 kg | |||
| Viikko | Annos (per 250 ml:n pussi) | Alkuvaiheen infuusionopeus | Myöhempi infuusionopeus† |
| Viikko 1 (infuusioannos jaetaan kahdelle päivälle) | |||
| Viikko 1 päivä 1 | 350 mg | 50 ml/h | 75 ml/h |
| Viikko 1 päivä 2 | 1 400 mg | 25 ml/h | 50 ml/h |
| Viikko 2 | 1 750 mg | 65 ml/h | |
| Viikko 3 | 1 750 mg | 85 ml/h | |
| Viikko 4 | 1 750 mg | 125 ml/h | |
| Myöhemmät viikot* | 2 100 mg | 125 ml/h | |
| * Viikosta 7 alkaen potilaille annetaan annos 3 viikon välein. † Alkuvaiheen infuusionopeutta lisätään myöhemmän infuusionopeuden tasoiseksi 2 tunnin jälkeen, mikäli infuusioon liittyviä reaktioita ei ilmene. | |||
| Taulukko 6 Infuusionopeudet 2 viikon välein annettavassa Rybrevant-hoidossa | |||
| Paino alle 80 kg | |||
| Viikko | Annos (per 250 ml:n pussi) | Alkuvaiheen infuusionopeus | Myöhempi infuusionopeus‡ |
| Viikko 1 (infuusioannos jaetaan kahdelle päivälle) | |||
| Viikko 1 päivä 1 | 350 mg | 50 ml/h | 75 ml/h |
| Viikko 1 päivä 2 | 700 mg | 50 ml/h | 75 ml/h |
| Viikko 2 | 1 050 mg | 85 ml/h | |
| Myöhemmät viikot* | 1 050 mg | 125 ml/h | |
| Paino vähintään 80 kg | |||
| Viikko | Annos (per 250 ml:n pussi) | Alkuvaiheen infuusionopeus | Myöhempi infuusionopeus‡ |
| Viikko 1 (infuusioannos jaetaan kahdelle päivälle) | |||
| Viikko 1 päivä 1 | 350 mg | 50 ml/h | 75 ml/h |
| Viikko 1 päivä 2 | 1 050 mg | 35 ml/h | 50 ml/h |
| Viikko 2 | 1 400 mg | 65 ml/h | |
| Viikko 3 | 1 400 mg | 85 ml/h | |
| Myöhemmät viikot* | 1 400 mg | 125 ml/h | |
| * Viikon 5 jälkeen potilaille annetaan annos 2 viikon välein. ‡ Alkuvaiheen infuusionopeutta lisätään myöhemmän infuusionopeuden tasoiseksi 2 tunnin jälkeen, mikäli infuusioon liittyviä reaktioita ei ilmene. | |||
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infuusioon liittyvät reaktiot
Infuusioon liittyviä reaktioita on ilmennyt usein amivantamabihoitoa saaneilla potilailla (ks. kohta Haittavaikutukset).
Ennen ensimmäistä infuusiota (viikko 1) on annettava antihistamiineja, antipyreettejä ja glukokortikoideja infuusioon liittyvien reaktioiden riskin vähentämiseksi. Myöhempiä annoksia varten on annettava antihistamiineja ja antipyreettejä. Viikolla 1 ensimmäinen infuusio on jaettava kahdelle antokerralle, jotka annetaan päivinä 1 ja 2.
Potilasta on hoidettava ympäristössä, jossa on asianmukainen lääketieteellinen tuki infuusioon liittyvien reaktioiden hoitamiseksi. Infuusiot on keskeytettävä ensimmäisen minkä tahansa asteisen infuusioon liittyvän reaktion merkin ilmetessä. Infuusion jälkeen annettavia lääkevalmisteita on annettava kliinisen tarpeen mukaisesti. Oireiden lievityttyä infuusiota tulee jatkaa 50 %:n nopeudella aiempaan nopeuteen nähden. Toistuvien vaikeusasteen 3 tasoisten tai vaikeusasteen 4 tasoisten infuusioon liittyvien reaktioiden ilmetessä Rybrevant-hoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Interstitiaalinen keuhkosairaus
Interstitiaalista keuhkosairautta tai interstitiaalisen keuhkosairauden kaltaisia haittavaikutuksia (esim. pneumoniitti), mukaan lukien kuolemaan johtaneita tapahtumia, on raportoitu amivantamabihoitoa saaneilla potilailla (ks. kohta Haittavaikutukset). Potilasta on seurattava interstitiaaliseen keuhkosairauteen / pneumoniittiin viittaavien oireiden varalta (esim. hengenahdistus, yskä, kuume). Jos oireita kehittyy, Rybrevant-hoito on keskeytettävä näiden oireiden tutkimisen ajaksi. Epäilty interstitiaalinen keuhkosairaus tai interstitiaalisen keuhkosairauden kaltainen haittavaikutus on arvioitava ja asianmukainen hoito aloitettava tarpeen mukaan. Rybrevant-hoito on lopetettava pysyvästi potilailla, joilla on vahvistettu interstitiaalinen keuhkosairaus tai interstitiaalisen keuhkosairauden kaltainen haittavaikutus (ks. kohta Annostus ja antotapa).
Laskimotromboemboliset tapahtumat (VTE) käytettäessä samanaikaisesti latsertinibia
Laskimotromboembolisia tapahtumia (myös kuolemaan johtaneita), joita olivat mm. syvä laskimotukos ja keuhkoembolia, raportoitiin potilailla, jotka saivat Rybrevant-valmistetta yhdistelmänä latsertinibin kanssa (ks. kohta Haittavaikutukset). Kliinisten hoitosuositusten mukaisesti potilaille pitää antaa estolääkityksenä joko suun kautta otettavaa suoraa antikoagulanttia tai pienimolekyylistä hepariinia. K‑vitamiiniantagonistien käyttöä ei suositella.
Potilaita pitää seurata laskimotromboembolisten tapahtumien oireiden ja löydösten varalta. Potilaille, joilla ilmenee laskimotromboembolisia tapahtumia, on annettava hyytymisenestohoitoa kliinisen tarpeen mukaan. Jos laskimotromboembolisiin tapahtumiin liittyy kliinistä epävakautta, hoito pitää keskeyttää, kunnes potilaan tila on kliinisesti vakaa. Sen jälkeen kummankin lääkevalmisteen käyttöä voidaan jatkaa aiemmalla annoksella.
Jos laskimotromboembolisia tapahtumia ilmenee uudelleen asianmukaisesta hyytymisenestohoidosta huolimatta, Rybrevant-hoito pitää lopettaa. Latsertinibihoitoa voidaan jatkaa aiemmalla annoksella (ks. kohta Annostus ja antotapa).
Ihon ja kynsien reaktiot
Ihottumaa (mukaan lukien aknea muistuttava ihottuma), kutinaa, ihon kuivumista ja ihon haavaumia on ilmennyt amivantamabihoitoa saaneilla potilailla (ks. kohta Haittavaikutukset). Potilasta on ohjeistettava välttämään altistumista auringolle Rybrevant-hoidon aikana ja 2 kuukauden ajan hoidon jälkeen. Suojavaatetus ja laajakirjoisen UVA/UVB-aurinkovoiteen käyttö on suositeltavaa. Ihottuman varalta suositellaan ennalta ehkäiseviä toimia. Ne käsittävät hoidon alussa estohoidon jollakin suun kautta otettavalla antibiootilla (esim. 100 mg doksisykliiniä tai minosykliiniä kaksi kertaa päivässä) päivästä 1 alkaen 12 ensimmäisen hoitoviikon ajan sekä päänahan paikallishoidon antibioottivoiteella (esim. 1 % klindamysiini) suun kautta otettavan antibioottihoidon päättymisen jälkeen seuraavien 9 kuukauden ajan. Kasvoihin ja koko kehoon (päänahkaa lukuun ottamatta) suositellaan ei‑komedogeenista kosteusvoidetta (mieluiten keramidipohjainen tai muu ihoa pitkäkestoisesti kosteuttava koostumus, joka ei sisällä kuivattavia ainesosia), ja käsien ja jalkaterien pesemiseen suositellaan klooriheksidiiniliuosta päivästä 1 alkaen. Näitä toimia suositellaan jatkamaan koko hoidon ajan.
Hoidon alussa on suositeltavaa olla valmiina lääkemääräykset paikallisesti käytettäviä ja/tai suun kautta otettavia antibiootteja sekä paikallisesti käytettäviä kortikosteroideja varten, jotta estohoidosta huolimatta mahdollisesti kehittyvä ihottuma voidaan hoitaa viiveettä. Jos ihoreaktioita kehittyy, hoitona on annettava tukihoitoa, paikallisesti käytettäviä kortikosteroideja ja paikallisesti käytettäviä ja/tai suun kautta otettavia antibiootteja. Vaikeusasteen 3 tasoisten tai heikosti siedettyjen vaikeusasteen 2 tasoisten tapahtumien osalta hoitona on annettava myös systeemisiä antibiootteja ja suun kautta otettavia steroideja. Jos potilaalla ilmenee vaikea-asteinen ihottuma, jossa on epätyypillinen ilmiasu tai jakautuminen tai joka ei kohennu 2 viikon sisällä, hänet on lähetettävä viipymättä ihotautilääkärin arvioon. Rybrevant-valmisteen annosta on pienennettävä tai hoito on keskeytettävä tai lopetettava pysyvästi vaikeusasteen perusteella (ks. kohta Annostus ja antotapa).
Toksista epidermaalista nekrolyysia on raportoitu. Tällä lääkevalmisteella toteutettava hoito on lopetettava, jos toksisen epidermaalisen nekrolyysin esiintyminen vahvistetaan.
Silmien häiriöt
Silmien häiriöitä, mukaan lukien keratiitti, on ilmennyt amivantamabihoitoa saaneilla potilailla (ks. kohta Haittavaikutukset). Potilas, jolla ilmenee pahenevia silmäoireita, on lähetettävä viipymättä silmälääkärin arvioon. Hänen on keskeytettävä piilolinssien käyttö siihen saakka, kunnes oireet on arvioitu. Katso kohdasta Annostus ja antotapa annosmuutokset asteen 3 tai 4 tasoisten silmien häiriöiden kohdalla.
Natriumpitoisuus
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”. Tämä lääkevalmiste voidaan laimentaa 9 mg:n/ml (0,9 %) natriumkloridiliuoksella infuusiota varten. Tämä täytyy huomioida potilailla, joilla on ruokavalion natriumrajoitus (ks. kohta Käyttö- ja käsittelyohjeet).
Polysorbaattipitoisuus
Tämä lääkevalmiste sisältää 0,6 mg polysorbaatti 80:tä per ml, mikä vastaa 4,2 mg:aa per 7 ml:n injektiopullo. Polysorbaatit saattavat aiheuttaa yliherkkyysreaktioita.
Yhteisvaikutukset
Lääkkeiden yhteisvaikutustutkimuksia ei ole tehty. IgG1:n monoklonaalisena vasta-aineena muuttumattoman amivantamabin erittyminen munuaisten kautta ja maksaentsyymivälitteinen metabolia eivät todennäköisesti ole merkittäviä eliminaatioreittejä. Näin ollen lääkettä metaboloivien entsyymien variaatioiden ei odoteta vaikuttavan amivantamabin eliminaatioon. Amivantamabilla on korkea affiniteetti EGFR:n ja MET:n spesifisiin epitooppeihin, eikä sen siten odoteta muuttavan lääkkeitä metaboloivien entsyymien toimintaa.
Rokotukset
Rokotusten tehosta ja turvallisuudesta amivantamabia saavilla potilailla ei ole kliinistä tietoa. Vältä eläviä tai eläviä heikennettyjä taudinaiheuttajia sisältävien rokotteiden käyttöä potilaan saadessa amivantamabia.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi /ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä amivantamabihoidon aikana ja 3 kuukautta hoidon päättymisen jälkeen.
Raskaus
Ei ole olemassa tietoja ihmisistä, jotta voitaisiin arvioida amivantamabin raskauden aikaiseen käyttöön liittyviä riskejä. Eläimillä ei ole tehty lisääntymistä koskevia tutkimuksia tietojen keräämiseksi lääkkeeseen liittyvistä riskeistä. EGFR:n ja MET:n estäjämolekyylien anto tiineenä oleville eläimille sai aikaan alkion/sikiön heikentyneen kehityksen, alkioiden kuolleisuuden ja tiineyden keskeytymisten lisääntymistä. Näin ollen vaikutusmekanisminsa ja eläinmallien löydösten perusteella amivantamabi voi olla vahingollista sikiölle, kun sitä annetaan raskaana olevalle naiselle. Amivantamabia ei pidä antaa raskauden aikana, ellei naisen hoidosta saaman hyödyn katsota olevan merkittävämpi kuin mahdolliset sikiöön kohdistuvat riskit. Jos potilas tulee raskaaksi käyttäessään tätä lääkevalmistetta, potilaalle on ilmoitettava mahdollisista sikiölle koituvista riskeistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Imetys
Ei tiedetä, erittyykö amivantamabi ihmisillä äidinmaitoon. Ihmisen IgG-vasta-aineiden tiedetään erittyvän äidinmaitoon muutaman ensimmäisen päivän ajan synnytyksestä, ja pitoisuudet laskevat pieniksi nopeasti. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois tällä lyhyellä jaksolla heti synnytyksen jälkeen, vaikka IgG-vasta-aineet todennäköisesti hajoavat imeytymättä imetetyn lapsen ruoansulatuskanavassa. On päätettävä, lopetetaanko imetys vai pidättäydytäänkö amivantamabihoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Amivantamabin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoja. Vaikutuksia miesten ja naisten hedelmällisyyteen ei ole arvioitu eläinkokeissa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Rybrevant-valmisteella saattaa olla kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Ks. kohta Haittavaikutukset (esim. huimaus, väsymys/uupumus, näön heikentyminen). Jos potilaalle tulee hoitoon liittyviä oireita, mukaan lukien näkökykyyn liittyvät haittavaikutukset, jotka vaikuttavat hänen keskittymis- ja reaktiokykyynsä, on suositeltavaa, ettei potilas aja autoa tai käytä koneita ennen kuin tämä vaikutus loppuu.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Amivantamabimonoterapiaa koskevassa tietoaineistossa (N = 380) yleisimmät haittavaikutukset, mukaan lukien kaikki vaikeusasteet, olivat ihottuma (76 %), infuusioon liittyvät reaktiot (67 %), kynsitoksisuus (47 %), hypoalbuminemia (31 %), edeema (26 %), väsymys/uupumus (26 %), stomatiitti (24 %), pahoinvointi (23 %) ja ummetus (23 %). Vakavia haittavaikutuksia olivat interstitiaalinen keuhkosairaus (1,3 %), infuusioon liittyvä reaktio (1,1 %) ja ihottuma (1,1 %). Kolme prosenttia potilaista lopetti Rybrevant-hoidon haittavaikutusten takia. Yleisimpiä hoidon keskeyttämiseen johtaneita haittavaikutuksia olivat infuusioon liittyvä reaktio (1,1 %), interstitiaalinen keuhkosairaus (0,5 %) ja kynsitoksisuus (0,5 %).
Haittavaikutustaulukko
Taulukossa 7 esitetään yhteenveto haittavaikutuksista, joita on ilmennyt amivantamabia monoterapiana saaneilla potilailla.
Tiedot edustavat amivantamabialtistusta 380 potilaalla, joilla oli paikallisesti edennyt tai etäpesäkkeinen ei-pienisoluinen keuhkosyöpä, platinapohjaisen solunsalpaajahoidon epäonnistumisen jälkeen. Potilaat saivat 1 050 mg (alle 80 kg:n painoiset potilaat) tai 1 400 mg (vähintään 80 kg:n painoiset potilaat) amivantamabia. Amivantamabialtistuksen mediaani oli 4,1 kuukautta (vaihteluväli: 0,0–39,7 kuukautta).
Kliinisten tutkimusten aikana havaitut haittavaikutukset on lueteltu alla esiintymistiheysluokan mukaisesti. Esiintymistiheydet on luokiteltu seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on esitetty kussakin esiintymistiheysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 7 Amivantamabia monoterapiana saaneilla potilailla ilmenneet haittavaikutukset | |||
| Elinjärjestelmä Haittavaikutus | Esiintymistiheysluokka | Mikä tahansa vaikeusaste (%) | Vaikeusasteet 3–4 (%) |
| Aineenvaihdunta ja ravitsemus | |||
| Hypoalbuminemia* (ks. kohta Farmakodynamiikka) | Hyvin yleinen | 31 | 2† |
| Ruokahalun heikkeneminen | 16 | 0,5† | |
| Hypokalsemia | 10 | 0,3† | |
| Hypokalemia | Yleinen | 9 | 2 |
| Hypomagnesemia | 8 | 0 | |
| Hermosto | |||
| Huimaus* | Hyvin yleinen | 13 | 0,3† |
| Silmät | |||
| Näön heikentyminen* | Yleinen | 3 | 0 |
| Silmäripsien kasvu* | 1 | 0 | |
| Muut silmien häiriöt* | 6 | 0 | |
| Keratiitti | Melko harvinainen | 0,5 | 0 |
| Uveiitti | 0,3 | 0 | |
| Hengityselimet, rintakehä ja välikarsina | |||
| Interstitiaalinen keuhkosairaus* | Yleinen | 3 | 0,5† |
| Ruoansulatuselimistö | |||
| Ripuli | Hyvin yleinen | 11 | 2† |
| Stomatiitti* | 24 | 0,5† | |
| Pahoinvointi | 23 | 0,5† | |
| Ummetus | 23 | 0 | |
| Oksentelu | 12 | 0,5† | |
| Vatsakipu* | Yleinen | 9 | 0,8† |
| Peräpukamat | 3,7 | 0 | |
| Maksa ja sappi | |||
| Alaniiniaminotransferaasiarvon nousu | Hyvin yleinen | 15 | 2 |
| Aspartaattiaminotransferaasiarvon nousu | 13 | 1 | |
| Veren alkalisen fosfataasin nousu | 12 | 0,5† | |
| Iho ja ihonalainen kudos | |||
| Ihottuma* | Hyvin yleinen | 76 | 3† |
| Kynsitoksisuus* | 47 | 2† | |
| Ihon kuivuminen* | 19 | 0 | |
| Kutina | 18 | 0 | |
| Ihon haavauma | Melko harvinainen | 0,8 | 0 |
| Toksinen epidermaalinen nekrolyysi | 0,3 | 0,3† | |
| Luusto, lihakset ja sidekudos | |||
| Lihaskipu | Hyvin yleinen | 11 | 0,3† |
| Yleisoireet ja antopaikassa todettavat haitat | |||
| Edeema* | Hyvin yleinen | 26 | 0,8† |
| Väsymys/uupumus* | 26 | 0,8† | |
| Kuume | 11 | 0 | |
| Vammat, myrkytykset ja hoitokomplikaatiot | |||
| Infuusioon liittyvä reaktio | Hyvin yleinen | 67 | 2 |
| * Yhdistetyt termit † Vain vaikeusasteen 3 tapahtumia | |||
Turvallisuusprofiilin yhteenveto
Amivantamabin, karboplatiinin ja pemetreksedin yhdistelmähoitoa koskevassa tietoaineistossa (N = 301) yleisimmin esiintyneet minkä tahansa vaikeusasteen haittavaikutukset olivat ihottuma (83 %), neutropenia (57 %), kynsitoksisuus (53 %), infuusioon liittyvät reaktiot (51 %), väsymys/uupumus (43 %), stomatiitti (39 %), pahoinvointi (43 %), trombosytopenia (40 %), ummetus (40 %), edeema (40 %), ruokahalun heikkeneminen (33 %), hypoalbuminemia (32 %), alaniiniaminotransferaasiarvon nousu (26 %), aspartaattiaminotransferaasiarvon nousu (23 %), oksentelu (22 %) ja hypokalemia (20 %). Vakavia haittavaikutuksia olivat ihottuma (2,7 %), laskimotromboembolia (2,3 %), trombosytopenia (2,3 %) ja interstitiaalinen keuhkosairaus (2,0 %). Kahdeksan prosenttia potilaista keskeytti Rybrevant-hoidon haittavaikutusten vuoksi. Yleisimmin esiintyneitä hoidon keskeyttämiseen johtaneita haittavaikutuksia olivat infuusioon liittyvät reaktiot (2,7 %), ihottuma (2,3 %), interstitiaalinen keuhkosairaus (2,3 %) ja kynsitoksisuus (1,0 %).
Taulukossa 8 esitetään yhteenveto haittavaikutuksista, joita ilmeni amivantamabin ja solunsalpaajahoidon yhdistelmää saaneilla potilailla.
Tiedot edustavat altistusta amivantamabin, karboplatiinin ja pemetreksedin yhdistelmälle 301 potilaalla, joilla oli paikallisesti edennyt tai etäpesäkkeinen ei-pienisoluinen keuhkosyöpä. Potilaat saivat amivantamabia 1 400 mg (< 80 kg painavat potilaat) tai 1 750 mg (≥ 80 kg painavat potilaat) viikoittain 4 viikon ajan. Viikosta 7 alkaen potilaat saivat amivantamabia 1 750 mg (< 80 kg painavat potilaat) tai 2 100 mg (≥ 80 kg painavat potilaat) 3 viikon välein. Mediaani altistusaika amivantamabin, karboplatiinin ja pemetreksedin yhdistelmälle oli 7,7 kuukautta (vaihteluväli 0,0−28,1 kuukautta).
Kliinisten tutkimusten aikana havaitut haittavaikutukset on lueteltu alla esiintymistiheysluokan mukaisesti. Esiintymistiheydet on luokiteltu seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on esitetty kussakin esiintymistiheysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 8: Amivantamabin, karboplatiinin ja pemetreksedin yhdistelmää saaneilla potilailla ilmenneet haittavaikutukset | |||
Elinjärjestelmä Haittavaikutus | Esiintymistiheysluokka | Mikä tahansa vaikeusaste (%) | Vaikeusasteet 3-4 (%) |
| Veri ja imukudos | |||
| Neutropenia | Hyvin yleinen | 57 | 39 |
| Trombosytopenia | 40 | 12 | |
| Aineenvaihdunta ja ravitsemus | |||
| Ruokahalun heikkeneminen | Hyvin yleinen | 33 | 1,3 |
| Hypoalbuminemia* | 32 | 3,7 | |
| Hypokalemia | 20 | 6,6 | |
| Hypomagnesemia | 13 | 1,3 | |
| Hypokalsemia | 12 | 1,0 | |
| Hermosto | |||
| Huimaus* | Yleinen | 10 | 0,3 |
| Verisuonisto | |||
| Laskimotromboembolia* | Hyvin yleinen | 14 | 3,0 |
| Silmät | |||
| Muut silmien häiriöt* | Yleinen | 7,3 | 0 |
| Näön heikentyminen* | 3,0 | 0 | |
| Silmäripsien kasvaminen | Melko harvinainen | 0,3 | 0 |
| Keratiitti | 0,3 | 0 | |
| Uveiitti | 0,3 | 0 | |
| Hengityselimet, rintakehä ja välikarsina | |||
| Interstitiaalinen keuhkosairaus* | Yleinen | 2,3 | 1,7 |
| Ruoansulatuselimistö | |||
| Pahoinvointi | Hyvin yleinen | 43 | 1,0 |
| Ummetus | 40 | 0,3 | |
| Stomatiitti* | 39 | 3,0 | |
| Oksentelu | 22 | 2,0 | |
| Ripuli | 19 | 2,3 | |
| Vatsakipu* | Yleinen | 11 | 0,3 |
| Peräpukamat | 9,3 | 0,7 | |
| Maksa ja sappi | |||
| Alaniiniaminotransferaasiarvon nousu | Hyvin yleinen | 26 | 4,3 |
| Aspartaattiaminotransferaasiarvon nousu | 23 | 0,7 | |
| Veren alkalisen fosfataasin nousu | Yleinen | 10 | 0,3 |
| Iho ja ihonalainen kudos | |||
| Ihottuma* | Hyvin yleinen | 83 | 14 |
| Kynsitoksisuus* | 53 | 4,3 | |
| Ihon kuivuminen* | 16 | 0 | |
| Kutina | 10 | 0 | |
| Ihon haavauma | Yleinen | 3,7 | 0,7 |
| Luusto, lihakset ja sidekudos | |||
| Lihaskipu | Yleinen | 5,0 | 0,7 |
| Yleisoireet ja antopaikassa todettavat haitat | |||
| Väsymys/uupumus* | Hyvin yleinen | 43 | 4,7 |
| Edeema* | 40 | 1,3 | |
| Kuume | 14 | 0 | |
| Vammat, myrkytykset ja hoitokomplikaatiot | |||
| Infuusioon liittyvä reaktio | Hyvin yleinen | 51 | 3,0 |
| * Yhdistetyt termit | |||
Turvallisuusprofiilin yhteenveto
Amivantamabin ja latsertinibin yhdistelmää koskevassa tietoaineistossa (N = 421) yleisimmät minkä tahansa vaikeusasteen haittavaikutukset olivat ihottuma (89 %), kynsitoksisuus (71 %), infuusioon liittyvät reaktiot (63 %), hypoalbuminemia (48 %), maksatoksisuus (47 %), edeema (47 %), stomatiitti (43 %), laskimotromboembolia (37 %), parestesiat (latsertinibi) (34 %), väsymys/uupumus (32 %), ripuli (29 %), ummetus (29 %), ihon kuivuminen (26 %), kutina (24 %), ruokahalun heikkeneminen (24 %), hypokalsemia (21 %), pahoinvointi (21 %) ja muut silmien häiriöt (21 %). Yleisimpiä vakavia haittavaikutuksia olivat laskimotromboembolia (11 %), keuhkokuume (4,0 %), ihottuma (3,1 %), interstitiaalinen keuhkosairaus / pneumoniitti (2,9 %), maksatoksisuus (2,4 %), COVID‑19-tauti (2,4 %) sekä infuusioon liittyvät reaktiot ja pleuraeffuusio (2,1 %). Kaksikymmentäkolme prosenttia potilaista lopetti Rybrevant-hoidon haittavaikutusten vuoksi. Yleisimmät Rybrevant-hoidon lopettamiseen johtaneet haittavaikutukset olivat ihottuma (5,5 %), infuusioon liittyvät reaktiot (4,5 %), kynsitoksisuus (3,6 %), interstitiaalinen keuhkosairaus (2,9 %) ja laskimotromboembolia (2,9 %).
Taulukossa 9 esitetään yhteenveto amivantamabin ja latsertinibin yhdistelmähoitoa saaneilla potilailla esiintyneistä haittavaikutuksista.
Tiedot kuvastavat 421:n paikallisesti edennyttä tai etäpesäkkeistä ei-pienisoluista keuhkosyöpää sairastavan potilaan altistusta amivantamabin ja latsertinibin yhdistelmälle. Potilaat saivat amivantamabia 1 050 mg (< 80 kg:n painoiset potilaat) tai 1 400 mg (≥ 80 kg:n painoiset potilaat) kerran viikossa 4 viikon ajan ja sen jälkeen 2 viikon välein. Amivantamabin ja latsertinibin yhdistelmää saaneessa ryhmässä altistuksen mediaani oli 18,5 kuukautta (vaihteluväli: 0,2–31,4 kuukautta).
Kliinisissä tutkimuksissa havaitut haittavaikutukset on lueteltu jäljempänä esiintyvyysluokittain seuraavan esitystavan mukaan: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on esitetty kussakin esiintyvyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 9: Amivantamabin ja latsertinibin yhdistelmää saaneilla potilailla ilmenneet amivantamabin haittavaikutukset | |||
Elinjärjestelmä Haittavaikutus | Esiintymistiheysluokka | Mikä tahansa vaikeusaste (%) | Vaikeusasteet 3–4 (%) |
| Aineenvaihdunta ja ravitsemus | |||
| Hypoalbuminemia* | Hyvin yleinen | 48 | 5 |
| Ruokahalun heikkeneminen | 24 | 1,0 | |
| Hypokalsemia | 21 | 2,1 | |
| Hypokalemia | 14 | 3,1 | |
| Hypomagnesemia | Yleinen | 5,0 | 0 |
| Hermosto | |||
| Parestesiat*‡ | Hyvin yleinen | 34 | 1,7 |
| Huimaus* | 13 | 0 | |
| Verisuonisto | |||
| Laskimotromboembolia* | Hyvin yleinen | 37 | 11 |
| Silmät | |||
| Muut silmien häiriöt* | Hyvin yleinen | 21 | 0,5 |
| Näön heikentyminen* | Yleinen | 4,5 | 0 |
| Keratiitti | 2,6 | 0,5 | |
| Silmäripsien kasvaminen* | 1,9 | 0 | |
| Hengityselimet, rintakehä ja välikarsina | |||
| Interstitiaalinen keuhkosairaus / pneumoniitti* | Yleinen | 3,1 | 1,2 |
| Ruoansulatuselimistö | |||
| Stomatiitti* | Hyvin yleinen | 43 | 2,4 |
| Ripuli | 29 | 2,1 | |
| Ummetus | 29 | 0 | |
| Pahoinvointi | 21 | 1,2 | |
| Oksentelu | 12 | 0,5 | |
| Vatsakipu* | 11 | 0 | |
| Peräpukamat | Yleinen | 10 | 0,2 |
| Maksa ja sappi | |||
| Maksatoksisuus† | Hyvin yleinen | 47 | 9 |
| Iho ja ihonalainen kudos | |||
| Ihottuma* | Hyvin yleinen | 89 | 27 |
| Kynsitoksisuus* | 71 | 11 | |
| Ihon kuivuminen* | 26 | 1,0 | |
| Kutina | 24 | 0,5 | |
| Käsi-jalkaoireyhtymä | Yleinen | 6 | 0,2 |
| Ihon haavauma | 5 | 0,7 | |
| Nokkosihottuma | 1,2 | 0 | |
| Luusto, lihakset ja sidekudos | |||
| Lihaskrampit | Hyvin yleinen | 17 | 0,5 |
| Lihaskipu | 13 | 0,7 | |
| Yleisoireet ja antopaikassa todettavat haitat | |||
| Edeema* | Hyvin yleinen | 47 | 2,9 |
| Väsymys/uupumus* | 32 | 3,8 | |
| Kuume | 12 | 0 | |
| Vammat, myrkytykset ja hoitokomplikaatiot | |||
| Infuusioon liittyvä reaktio | Hyvin yleinen | 63 | 6 |
* yhdistetyt termit ‡ arvioitu vain latsertinibin haittavaikutuksena. † Yleisimmät haittavaikutukset olivat suurentunut ALAT-arvo (36 %), suurentunut ASAT-arvo (29 %) ja suurentunut veren alkalisen fosfataasin pitoisuus (12 %). | |||
Valikoitujen haittavaikutusten kuvaus
Infuusioon liittyvät reaktiot
Amivantamabimonoterapiaa saaneilla potilailla infuusioon liittyviä reaktioita ilmeni 67 %:lla potilaista. Infuusioon liittyvistä reaktioista 98 % oli vaikeusasteen 1–2 tasoisia. Infuusioon liittyvistä reaktioista 99 % ilmeni ensimmäisen infuusion kohdalla; mediaaniaika niiden alkamiseen oli 60 minuuttia, ja suurin osa ilmeni 2 tunnin kuluessa infuusion alkamisesta. Yleisimmin esiintyviä merkkejä ja oireita olivat vilunväristykset, hengenahdistus, pahoinvointi, punoitus, epämukava tunne rintakehässä ja oksentelu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Amivantamabia yhdistelmänä karboplatiinin ja pemetreksedin kanssa saaneilla potilailla infuusioon liittyviä reaktioita ilmeni 50 %:lla potilaista. Infuusioon liittyvistä reaktioista yli 94 % oli vaikeusasteen 1–2 tasoisia. Valtaosa infuusioon liittyvistä reaktioista ilmeni ensimmäisen infuusion kohdalla; mediaaniaika niiden alkamiseen oli 60 minuuttia (vaihteluväli 0–7 tuntia), ja suurin osa ilmeni 2 tunnin kuluessa infuusion alkamisesta.
Infuusioon liittyviä reaktioita voi toisinaan ilmetä, kun amivantamabihoito aloitetaan uudelleen hoidon oltua pitkään, yli 6 viikkoa, keskeytettynä.
Infuusioon liittyviä reaktioita ilmaantui 63 %:lle potilaista, jotka saivat hoitoa amivantamabin ja latsertinibin yhdistelmällä. Yhdeksänkymmentäneljä prosenttia infuusioon liittyvistä reaktioista oli vaikeusasteen 1–2 tasoisia. Valtaosa infuusioon liittyvistä reaktioista ilmaantui ensimmäisen infuusion yhteydessä, ja niiden ilmaantumiseen kuluneen ajan mediaani oli 1 tunti. Suurin osa ilmaantui 2 tunnin kuluessa infuusion alkamisesta. Yleisimpiä oireita ja löydöksiä olivat vilunväristykset, hengenahdistus, pahoinvointi, punoitus, epämukava tunne rintakehässä ja oksentelu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Infuusioon liittyvä reaktio voi toisinaan ilmetä aloitettaessa amivantamabin käyttö uudelleen yli 6 viikon hoitotauon jälkeen.
Vaiheen 2 avoimessa monikeskustutkimuksessa ei-pienisoluista keuhkosyöpää sairastaville potilaille annettiin 8 mg deksametasonia suun kautta kaksi kertaa päivässä kumpanakin ensimmäistä Rybrevant-infuusiota edeltävänä kahtena päivänä sekä 8 mg suun kautta ensimmäisen infuusion antopäivänä 60 minuuttia ennen infuusiota (yhteensä 5 annosta) laskimoon annettavan deksametasonin lisäksi. Kun hoitoon lisättiin suun kautta annettava deksametasoni, infuusioon liittyvien reaktioiden ilmaantuvuudeksi ensimmäisen infuusion antopäivänä raportoitiin 22,5 % eikä vaikeusasteen ≥ 3 infuusioon liittyviä reaktioita raportoitu (ks. kohta Annostus ja antotapa).
Interstitiaalinen keuhkosairaus
Interstitiaalista keuhkosairautta tai interstitiaalisen keuhkosairauden kaltaisia reaktioita on raportoitu amivantamabin ja muiden EGFR:n estäjien käytön yhteydessä. Interstitiaalista keuhkosairautta tai pneumoniittia raportoitiin 2,6 %:lla amivantamabimonoterapiaa saaneista potilaista, 2,3 %:lla amivantamabia yhdistelmänä karboplatiinin ja pemetreksedin kanssa saaneista potilaista ja 3,1 %:lla amivantamabin ja latsertinibin yhdistelmähoitoa saaneista potilaista, mukaan lukien 1 (0,2 %) kuolemaan johtanut tapaus. Kliinisestä tutkimuksesta poissuljettiin potilaat, joilla oli sairaushistoriassa interstitiaalinen keuhkosairaus, lääkkeen aikaansaama interstitiaalinen keuhkosairaus, säteilyn aiheuttama pneumoniitti, joka on edellyttänyt steroidihoitoa, tai merkkejä kliinisesti aktiivisesta interstitiaalisesta keuhkosairaudesta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Laskimotromboemboliset (VTE) tapahtumat käytettäessä samanaikaisesti latsertinibia
Kun Rybrevant-valmistetta käytettiin yhdistelmänä latsertinibin kanssa, laskimotromboembolisia tapahtumia, mukaan lukien syviä laskimotukoksia (SLT) ja keuhkoembolioita, raportoitiin 37 %:lla Rybrevant-valmisteen ja latsertinibin yhdistelmää saaneista 421 potilaasta. Useimmat tapauksista olivat asteen 1 tai 2 tapahtumia. Asteen 3–4 tapahtumia ilmeni 11 %:lla Rybrevant-valmisteen ja latsertinibin yhdistelmähoitoa saaneista potilaista, ja 0,5 % Rybrevant-valmisteen ja latsertinibin yhdistelmähoitoa saaneista potilaista kuoli. Tietoja estolääkityksenä käytettävistä antikoagulanteista ja laskimotromboembolisten tapahtumien hoidosta, ks. kohdat Annostus ja antotapa ja 4.4.
Rybrevant-valmisteen ja latsertinibin yhdistelmähoitoa saaneilla potilailla laskimotromboembolisen tapahtuman ensimmäiseen ilmenemiseen kuluneen ajan mediaani oli 84 vuorokautta. Laskimotromboemboliset tapahtumat johtivat Rybrevant-hoidon lopettamiseen 2,9 %:lla potilaista.
Ihon ja kynsien reaktiot
Ihottumaa (mukaan lukien aknea muistuttava ihottuma), kutinaa ja ihon kuivumista ilmeni 76 %:lla potilaista, joita hoidettiin amivantamabilla yksinään. Useimmat tapaukset olivat vaikeusasteen 1 tai 2 tasoisia. Vaikeusasteen 3 tasoisia ihottumatapahtumia ilmeni 3 %:lla potilaista. Amivantamabihoidon keskeyttämiseen johtanutta ihottumaa ilmeni 0,3 %:lla potilaista. Ihottuma kehittyi yleensä ensimmäisten 4 hoitoviikon aikana, ja sen alkamiseen kuluvan ajan mediaani oli 14 vuorokautta. Kynsitoksisuutta on ilmennyt amivantamabihoitoa saaneilla potilailla. Tapahtumista useimmat olivat vaikeusasteen 1 tai 2 tasoisia. Vaikeusasteen 3 tasoista kynsitoksisuutta ilmeni 1,8 %:lla potilaista.
Ihottumaa (mukaan lukien aknea muistuttava ihottuma) ilmeni 83 %:lla potilaista, joita hoidettiin amivantamabilla yhdessä karboplatiinin ja pemetreksedin kanssa. Useimmat tapaukset olivat vaikeusasteen 1 tai 2 tasoisia. Vaikeusasteen 3 tasoisia ihottumatapahtumia ilmeni 14 %:lla potilaista. Amivantamabihoidon keskeyttämiseen johtanutta ihottumaa ilmeni 2,3 %:lla potilaista. Ihottuma kehittyi yleensä ensimmäisten 4 hoitoviikon aikana, ja sen alkamiseen kuluvan ajan mediaani oli 14 vuorokautta. Kynsitoksisuutta on ilmennyt amivantamabihoitoa yhdessä karboplatiinin ja pemetreksedin kanssa saaneilla potilailla. Tapahtumista useimmat olivat vaikeusasteen 1 tai 2 tasoisia. Vaikeusasteen 3 tasoista kynsitoksisuutta ilmeni 4,3 %:lla potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ihottumaa (mukaan lukien aknea muistuttava ihottuma) ilmeni 89 %:lla amivantamabin ja latsertinibin yhdistelmähoitoa saaneista potilaista. Useimmat tapauksista olivat asteen 1 tai 2 tapahtumia. Asteen 3 ihottumaa ilmeni 27 %:lla potilaista. Ihottumaa, joka johti amivantamabihoidon lopettamiseen, ilmeni 5,5 %:lla potilaista. Yleensä ihottuma kehittyi neljän ensimmäisen hoitoviikon aikana, ja ihottuman ilmenemiseen kuluneen ajan mediaani oli 14 vuorokautta. Amivantamabin ja latsertinibin yhdistelmähoitoa saaneilla potilailla ilmeni kynsitoksisuutta. Useimmat tapauksista olivat asteen 1 tai 2 tapahtumia. Asteen 3 kynsitoksisuutta ilmeni 11 %:lla potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilailla, jotka saivat hoitona Rybrevant-valmisteen ja latsertinibin yhdistelmää, tehtiin vaiheen 2 tutkimus estohoidon käytön arvioimiseksi. Estohoito käsitti suun kautta otettavan antibiootin, päänahkaan paikallisesti käytettävän antibiootin, kosteusvoiteen kasvoihin ja koko kehoon (päänahkaa lukuun ottamatta) ja antiseptisen aineen käsiin ja jalkateriin (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Asteen ≥ 2 dermatologisten haittavaikutusten ilmaantuvuuden osoitettiin vähentyneen ensimmäisten 12 hoitoviikon aikana verrattuna tavanomaiseen kliinisessä hoitokäytännössä käytettävään dermatologiseen hoitoon (38,6 % vs. 76,5 %, p < 0,0001). Lisäksi päänahkaan liittyvät asteen ≥ 2 haittavaikutukset vähenivät ensimmäisten 12 hoitoviikon aikana (8,6 % vs. 29,4 %), ja myös annosta pienennettiin (7,1 % vs. 19,1 %), hoito keskeytettiin (15,7 % vs. 33,8 %) ja hoito lopetettiin (1,4 % vs. 4,4 %) dermatologisten haittavaikutusten vuoksi harvemmin.
Silmien häiriöt
Silmien häiriöitä, mukaan lukien keratiitti (0,5 %), ilmeni 9 %:lla potilaista, joita hoidettiin amivantamabilla yksinään. Muita raportoituja haittavaikutuksia olivat silmäripsien kasvaminen, näön heikentyminen ja muut silmien häiriöt. Kaikki tapahtumat olivat vaikeusasteen 1–2 tasoisia.
Silmien häiriöitä, mukaan lukien keratiitti (0,3 %), ilmeni 11 %:lla potilaista, joita hoidettiin amivantamabilla yhdessä karboplatiinin ja pemetreksedin kanssa. Muita raportoituja haittavaikutuksia olivat silmäripsien kasvaminen, näön heikentyminen, uveiitti ja muut silmien häiriöt. Kaikki tapahtumat olivat vaikeusasteen 1–2 tasoisia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Silmien häiriöitä, mukaan lukien keratiitti (2,6 %), ilmeni amivantamabin ja latsertinibin yhdistelmähoitoa saaneilla potilailla. Muita raportoituja haittavaikutuksia olivat silmäripsien kasvaminen, näön heikkeneminen ja muut silmien häiriöt. Useimmat tapauksista olivat asteen 1–2 tapahtumia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Erityisryhmät
Iäkkäät
Amivantamabin käytöstä 75-vuotiailla ja sitä vanhemmilla potilailla on vähän kliinisiä tietoja (ks. kohta Farmakodynamiikka). Turvallisuudessa ei yleisesti havaittu eroja ≥ 65-vuotiaiden ja < 65-vuotiaiden potilaiden välillä.
Immunogeenisuus
Kuten kaikkien terapeuttisten proteiinien, myös tämän valmisteen käytön yhteydessä on olemassa immunogeenisuuden mahdollisuus. Kliinisissä tutkimuksissa, joissa oli mukana potilaita, joilla oli paikallisesti edennyt tai etäpesäkkeinen ei-pienisoluinen keuhkosyöpä, ja joissa heitä hoidettiin amivantamabilla, neljä (0,2 %) 1 862:sta Rybrevant-hoitoa saaneesta tutkittavasta, joilla amivantamabivasta-aineet olivat arvioitavissa, hoidosta aiheutuva amivantamabivasta-aineita koskeva tulos oli positiivinen. Ei ole olemassa näyttöä siitä, että amivantamabivasta-aineet aiheuttaisivat farmakokineettisen profiilin, tehoprofiilin tai turvallisuusprofiilin muutoksia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Siedettyä enimmäisannosta ei määritelty kliinisessä tutkimuksessa, jossa potilaat saivat enintään 2 100 mg laskimoon. Amivantamabin yliannostukseen ei ole olemassa tunnettua spesifistä vastalääkettä. Yliannostuksen sattuessa Rybrevant-hoito on lopetettava, potilasta on seurattava haittatapahtumien merkkien tai oireiden varalta ja asianmukaiset tukitoimenpiteet on otettava käyttöön välittömästi siihen saakka, kunnes kliininen toksisuus on lievittynyt tai hävinnyt.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: monoklonaaliset vasta-aineet ja vasta‑ainekonjugoidut lääkkeet, ATC-koodi: L01FX18.
Vaikutusmekanismi
Amivantamabi on vähän fukoosia sisältävä, täysin humaani IgG1-pohjainen EGFR-MET-bispesifinen vasta-aine. Sillä on immuunisoluihin kohdentuvaa aktiivisuutta, joka kohdentuu kasvaimiin, joissa on aktivoivia EGFR:n mutaatioita, kuten eksonin 19 deleetioita, eksonin 21 L858R-substituutioita ja eksonin 20 insertiomutaatioita. Amivantamabi sitoutuu EGFR:n ja MET:n ekstrasellulaarisiin domeeneihin.
Amivantamabi häiritsee EGFR:n ja MET:n signalointitoimintoja estämällä ligandin sitoutumisen ja tehostamalla EGFR:n ja MET:n hajoamista estäen näin kasvaimen kasvun ja etenemisen. EGFR:n ja MET:n esiintyminen kasvainsolujen pinnalla mahdollistaa myös hoidon kohdentamisen näihin soluihin immuuniefektorisoluvälitteisesti, jolloin luonnolliset tappajasolut ja makrofagit voivat tuhota kasvainsoluja vasta-aineriippuvaisen solutoksisuuden (ADCC) ja trogosytoosin kautta.
Farmakodynaamiset vaikutukset
Albumiini
Amivantamabi pienensi seerumin albumiinipitoisuutta tyypillisesti ensimmäisten 8 viikon aikana (ks. kohta Haittavaikutukset); kyseessä on MET:n eston farmakodynaaminen vaikutus. Tämän jälkeen albumiinipitoisuus vakautui amivantamabihoidon loppuajaksi.
Kliininen teho ja turvallisuus
Aiemmin hoitamaton ei-pienisoluinen keuhkosyöpä, jossa on EGFR:n eksonin 19 deleetioita tai eksonin 21 L858R-substituutiomutaatioita (MARIPOSA)
NSC3003 (MARIPOSA) on satunnaistettu, avoin, aktiivikontrolloitu, vaiheen 3 monikeskustutkimus, jossa arvioidaan Rybrevant-valmisteen ja latsertinibin yhdistelmähoidon tehoa ja turvallisuutta verrattuna osimertinibimonoterapiaan, kun niitä käytetään ensilinjan hoitona potilaille, joilla on EGFR-mutaatiopositiivinen, paikallisesti edennyt tai etäpesäkkeinen ei‑pienisoluinen keuhkosyöpä, joka ei ole hoidettavissa kuratiivisella hoidolla. Edellytyksenä oli, että potilasnäytteistä oli paikallisessa testauksessa tunnistettu jompikumpi kahdesta yleisestä EGFR-mutaatiosta (eksonin 19 deleetio tai eksonin 21 L858R-substituutiomutaatio). Kaikkien potilaiden kasvainkudosnäytteet (94 %) ja/tai plasmanäytteet (6 %) testattiin paikallisesti EGFR-geenin mutaatiostatuksen (eksonin 19 deleetio ja/tai eksonin 21 L858R-substituutiomutaatio) määrittämiseksi käyttämällä polymeraasiketjureaktiota (PCR, polymerase chain reaction) 65 %:lla ja uuden sukupolven sekvensointia (NGS, next generation sequencing) 35 %:lla potilaista.
Yhteensä 1 074 potilasta satunnaistettiin (2:2:1) saamaan Rybrevant-valmisteen ja latsertinibin yhdistelmähoitoa, osimertinibimonoterapiaa tai latsertinibimonoterapiaa, kunnes sairaus etenee tai ilmenee toksisuutta, joka ei ole hyväksyttävissä. Rybrevant-valmistetta annettiin laskimoon 1 050 mg (alle 80 kg painaville potilaille) tai 1 400 mg (vähintään 80 kg painaville potilaille) kerran viikossa 4 viikon ajan ja sen jälkeen joka toinen viikko viikosta 5 alkaen. Latsertinibia annettiin 240 mg suun kautta kerran vuorokaudessa. Osimertinibia annettiin 80 mg suun kautta kerran vuorokaudessa. Satunnaistaminen ositettiin EGFR-mutaatiotyypin (eksonin 19 deleetio tai eksonin 21 L858R), etnisen taustan (aasialainen tai muu kuin aasialainen) ja aiempien aivoetäpesäkkeiden (kyllä tai ei) mukaan.
Lähtötilanteen demografiset tiedot ja sairauden ominaisuudet olivat samankaltaiset eri hoitohaaroissa. Iän mediaani oli 63 vuotta (vaihteluväli: 25–88 vuotta), 45 % potilaista oli ≥ 65-vuotiaita, 62 % oli naisia ja 59 % oli aasialaisia ja 38 % valkoihoisia. Lähtötilanteen ECOG (Eastern Cooperative Oncology Group) ‑toimintakykyluokka oli 0 (34 %) tai 1 (66 %), 69 % potilaista ei ollut koskaan tupakoinut, 41 %:lla oli aiemmin ollut aivoetäpesäkkeitä ja 90 %:lla todettiin alkuperäisen diagnosoinnin yhteydessä levinneisyysasteen IV syöpä. EGFR-mutaatioista 60 % oli eksonin 19 deleetioita ja 40 % eksonin 21 L858R-substituutiomutaatioita.
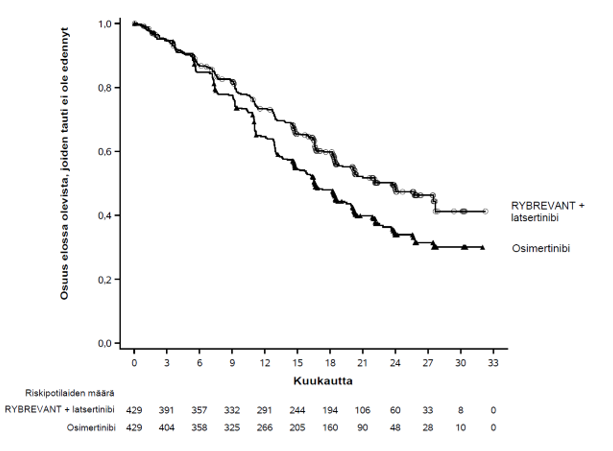
Rybrevant-valmisteen ja latsertinibin yhdistelmän käytössä todettiin sokkoutetun riippumattoman keskitetyn arvioinnin (BICR) perusteella tilastollisesti merkitsevä etenemättömyysajan piteneminen.
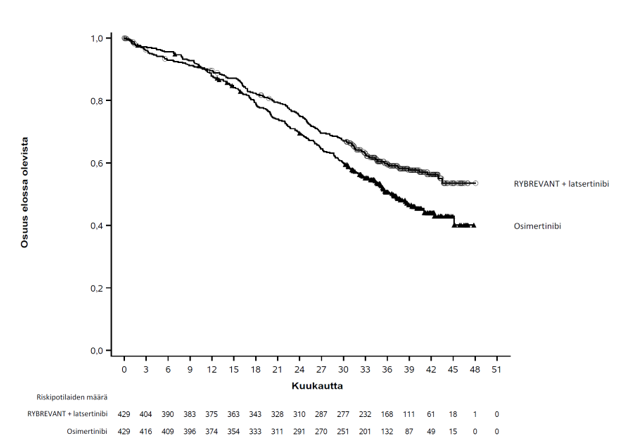
Kokonaiselinajan loppuanalyysissa todettiin kokonaiselinajan tilastollisesti merkitsevä piteneminen Rybrevant-valmisteen ja latsertinibin yhdistelmän käytössä osimertinibiin verrattuna (ks. taulukko 10 ja kuva 2).
| Taulukko 10. Tehoa koskevat tulokset MARIPOSA-tutkimuksesta | ||
Rybrevant + latsertinibi (N = 429) | Osimertinibi (N = 429) | |
| Etenemättömyysaika (PFS)a | ||
| Tapahtumien lukumäärä | 192 (45 %) | 252 (59 %) |
| Mediaani, kuukautta (95 %:n luottamusväli) | 23,7 (19,1–27,7) | 16,6 (14,8–18,5) |
| Riskitiheyksien suhde (95 %:n luottamusväli); p‑arvo | 0,70 (0,58–0,85); p = 0,0002 | |
| Kokonaiselinaika (OS) | ||
| Tapahtumien lukumäärä | 173 (40 %) | 217 (51 %) |
| Mediaani, kuukautta (95 %:n luottamusväli) | NE (42,9–NE) | 36,7 (33,4–41,0) |
| Riskitiheyksien suhde (95 %:n luottamusväli); p‑arvo | 0,75 (0,61–0,92); p = 0,0048 | |
| Objektiivinen hoitovaste (ORR)a, b | ||
| ORR, % (95 %:n luottamusväli) | 80 % (76–84 %) | 77 % (72–81 %) |
| Vasteen kesto (DOR)a, b | ||
| Mediaani (95 %:n luottamusväli), kuukautta | 25,8 (20,3–33,9) | 18,1 (14,8–20,1) |
PFS = Progression-free survival; OS = Overall survival; ORR = Objective response rate; DOR = Duration of response; NE = ei arvioitavissa (not estimable). Etenemättömyysaikaa koskevien tietojen tiedonkeruun päättymispäivä oli 11.8.2023 ja seurannan mediaani oli 22,0 kuukautta. Vasteen kestoa ja objektiivista hoitovastetta koskevien tietojen tiedonkeruun päättymispäivä oli 13.5.2024 ja seurannan mediaani oli 31,3 kuukautta. Kokonaiselinaikaa koskevien tietojen tiedonkeruun päättymispäivä oli 4.12.2024 ja seurannan mediaani oli 37,8 kuukautta. a Sokkoutettu riippumaton keskitetty arvio (BICR) RECIST v1.1 ‑kriteerien mukaan. b Perustuu varmistettuihin vasteen saaneisiin. | ||
Kuva 1. Kaplan–Meierin käyrä, joka kuvaa sokkoutetun riippumattoman keskitetyn arvion (BICR) mukaista etenemättömyysaikaa aiemmin hoitamattomilla ei‑pienisoluista keuhkosyöpää sairastavilla potilailla
Kuva 2. Kaplan–Meierin käyrä, joka kuvaa aiemmin hoitamattomien ei‑pienisoluista keuhkosyöpää sairastavien potilaiden kokonaiselinaikaa
MARIPOSA-tutkimuksessa ennalta määritettyjä päätetapahtumia olivat kallonsisäinen objektiivinen hoitovaste ja vasteen kesto riippumattoman keskitetyn arvion mukaan. Alaryhmässä, jossa potilailla oli lähtötilanteessa kallonsisäisiä leesioita, Rybrevant-valmisteen ja latsertinibin yhdistelmähoidolla saavutettiin vastaava kallonsisäinen objektiivinen hoitovaste kuin verrokkivalmisteella. Tutkimussuunnitelman mukaisesti kaikille potilaille MARIPOSA-tutkimuksessa tehtiin useita aivojen magneettikuvauksia kallonsisäisen vasteen ja vasteen keston arvioimiseksi. Tulosten yhteenveto esitetään taulukossa 11.
| Taulukko 11. Kallonsisäinen objektiivinen hoitovaste ja vasteen kesto sokkoutetun riippumattoman keskitetyn arvion (BICR) mukaan tutkittavilla, joilla oli lähtötilanteessa kallonsisäisiä leesioita – MARIPOSA | ||
Rybrevant + latsertinibi (N = 180) | Osimertinibi (N = 186) | |
| Kallonsisäisen kasvaimen vasteen arviointi | ||
| Kallonsisäinen objektiivinen hoitovaste (täydellinen vaste + osittainen vaste), % (95 %:n luottamusväli) | 78 % (71–84 %) | 77 % (71–83 %) |
| Täydellinen vaste | 64 % | 59 % |
| Kallonsisäisen vasteen kesto | ||
| Vasteen saaneiden lkm | 140 | 144 |
| Mediaani, kk (95 %:n luottamusväli) | 35,0 (20,4–NE) | 25,1 (22,1–31,2) |
NE = ei arvioitavissa. Kallonsisäistä objektiivista hoitovastetta ja vasteen kestoa koskevien tietojen tiedonkeruun päättymispäivä oli 4.12.2024 ja seurannan mediaani oli 37,8 kuukautta. | ||
Aiemmin hoidettu ei-pienisoluinen keuhkosyöpä, jossa on EGFR:n eksonin 19 deleetioita tai eksonin 21 L858R-substituutiomutaatioita (MARIPOSA‑2)
MARIPOSA‑2 on vaiheen 3 satunnaistettu (2:2:1) avoin monikeskustutkimus, jossa on mukana paikallisesti edennyttä tai etäpesäkkeistä ei-pienisoluista keuhkosyöpää sairastavia potilaita, joiden keuhkosyövässä on EGFR:n eksonin 19 deleetioita tai eksonin 21 L858R-substituutiomutaatioita (mutaatiotestaus voitiin tehdä paikallisesti edenneen tai etäpesäkkeisen taudin diagnoosiajankohtana tai sen jälkeen; kun EGFR-mutaatiostatus oli aiemmin todettu, testausta ei tarvinnut uusia tutkimukseen mukaan tullessa) ja joilla aiempi jotakin kolmannen sukupolven EGFR:n tyrosiinikinaasin estäjää sisältänyt hoito oli epäonnistunut. Tutkimukseen satunnaistettiin yhteensä 657 potilasta, joista 263 sai karboplatiinia ja pemetreksediä (CP) ja 131 sai Rybrevant-valmistetta yhdessä karboplatiinin ja pemetreksedin kanssa (Rybrevant‑CP). Lisäksi 263 potilasta satunnaistettiin saamaan erillisessä tutkimushaarassa Rybrevant-valmistetta yhdessä latsertinibin, karboplatiinin ja pemetreksedin kanssa. Rybrevant-valmistetta annettiin 1 400 mg (potilaille, joiden paino oli < 80 kg) tai 1 750 mg (potilaille, joiden paino oli ≥ 80 kg) laskimoon kerran viikossa 4 viikon ajan ja sen jälkeen 1 750 mg:n annoksina (potilaille, joiden paino oli < 80 kg) tai 2 100 mg:n annoksina (potilaille, joiden paino oli ≥ 80 kg) 3 viikon välein viikosta 7 alkaen, kunnes sairaus eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Karboplatiinia annettiin laskimoon 3 viikon välein enimmillään 12 viikkoa pitoisuus-aikakäyrän alle jäävän pinta-alan ollessa 5 mg/ml/min (AUC 5). Pemetreksediä annettiin 500 mg/m2 laskimoon 3 viikon välein, kunnes sairaus eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä.
Potilaat ositettiin osimertinibihoitolinjan (ensilinja tai toinen linja), aiempien aivoissa todettujen etäpesäkkeiden (kyllä tai ei) ja aasialaisen etnisen taustan (kyllä tai ei) perusteella.
Rybrevant‑CP-haaraan tai CP-haaraan satunnaistettujen 394 potilaan iän mediaani oli 62 vuotta (vaihteluväli: 31–85 vuotta), ja 38 % potilaista oli ≥ 65-vuotiaita; 60 % oli naisia, 48 % oli aasialaisia ja 46 % oli valkoihoisia. Lähtötilanteen ECOG (Eastern Cooperative Oncology Group) ‑toimintakykyluokka oli 0 (40 %) tai 1 (60 %); 66 % ei ollut koskaan tupakoinut; 45 %:lla oli anamneesissa etäpesäkkeitä aivoissa ja 92 %:lla oli alkuvaiheen diagnoosin ajankohtana levinneisyysasteen IV syöpä.
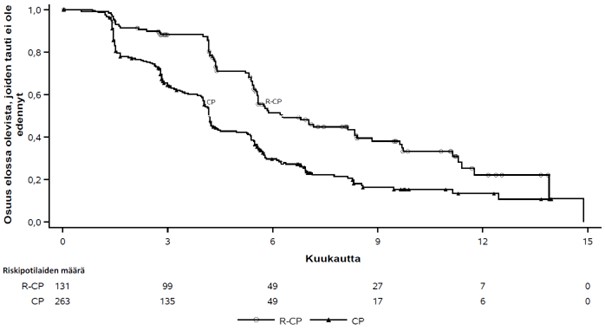
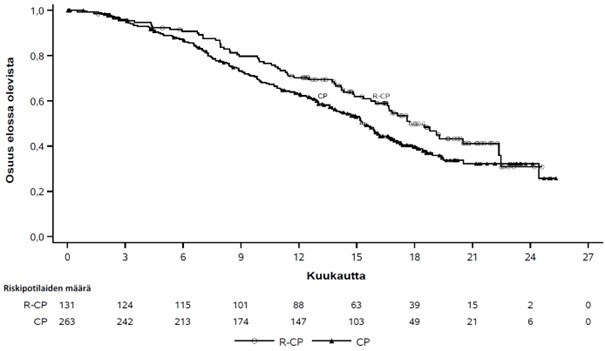
Rybrevant-valmisteen, karboplatiinin ja pemetreksedin yhdistelmän osoitettiin pidentäneen etenemättömyysaikaa (PFS) tilastollisesti merkitsevästi karboplatiiniin ja pemetreksediin verrattuna; riskitiheyksien suhde (HR) oli 0,48 (95 %:n luottamusväli: 0,36–0,64; p < 0,0001). Kokonaiselinajan toisen välianalyysin ajankohtana, jolloin seurannan mediaani oli Rybrevant‑CP-ryhmässä noin 18,6 kuukautta ja CP-ryhmässä noin 17,8 kuukautta, kokonaiselinajan riskitiheyksien suhde (HR) oli 0,73 (95 %:n luottamusväli: 0,54–0,99; p = 0,0386), joka ei ollut tilastollisesti merkitsevä (testattu ennalta asetetulla merkitsevyystasolla 0,0142).
Yhteenveto tehoa koskevista tuloksista esitetään taulukossa 12.
| Taulukko 12 Tehoa koskevat tulokset MARIPOSA-2-tutkimuksessa | ||
Rybrevant+ karboplatiini+ pemetreksedi (N = 131) | Karboplatiini+ pemetreksedi (N = 263) | |
| Etenemättömyysaika (PFS)a | ||
| Tapahtumien lukumäärä (%) | 74 (57) | 171 (65) |
| Mediaani, kuukautta (95 %:n luottamusväli) | 6,3 (5,6–8,4) | 4,2 (4,0–4,4) |
| Riskitiheyksien suhde (HR) (95 %:n luottamusväli); p‑arvo | 0,48 (0,36–0,64); p < 0,0001 | |
| Kokonaiselinaika (OS) | ||
| Tapahtumien lukumäärä (%) | 65 (50) | 143 (54) |
| Mediaani, kuukautta (95 %:n luottamusväli) | 17,7 (16,0–22,4) | 15,3 (13,7–16,8) |
| Riskitiheyksien suhde (HR) (95 %:n luottamusväli); p‑arvob | 0,73 (0,54–0,99); p = 0,0386
| |
| Objektiivinen vasteosuusa | ||
| Kokonaisvasteosuus (ORR), % (95 %:n luottamusväli) | 64 % (55–72 %) | 36 % (30–42 %) |
| Ristitulosuhde (OR) (95 %:n luottamusväli); p-arvo | 3,10 (2,00–4,80); p < 0,0001 | |
| Vasteen kesto a | ||
| Mediaani (95 %:n luottamusväli), kuukautta | 6,90 (5,52–NE) | 5,55 (4,17–9,56) |
| Potilaita, joiden vasteen kesto ≥ 6 kuukautta | 31,9 % | 20,0 % |
NE = ei arvioitavissa (not estimable) Etenemättömyysaikaa, vasteen kestoa ja kokonaisvasteosuutta koskevat tulokset on saatu katkaisuajankohdan 10. heinäkuuta 2023 tiedoista, kun näiden päätetapahtumien hypoteesin testaus ja loppuanalyysi tehtiin. Kokonaiselinaikaa koskevat tulokset on saatu katkaisuajankohdan 26. huhtikuuta 2024 tiedoista tehdystä toisesta kokonaiselinajan välianalyysista. a Sokkoutettu riippumaton keskitetty arviointi b p-arvoa verrataan kaksitahoiseen merkitsevyystasoon 0,0142. Siten kokonaiselinajan tulokset eivät ole toisen välianalyysin osalta merkitseviä. | ||
Kuva 3 Aiemmin hoidettua ei-pienisoluista keuhkosyöpää sairastavien potilaiden sokkoutettuun riippumattomaan keskitettyyn arviointiin perustuvan etenemättömyysajan Kaplan-Meier-käyrä
Etenemättömyysaikaa koskeva hyöty vertailtaessa Rybrevant‑CP-hoitoa CP-hoitoon oli yhdenmukainen kaikissa ennalta määritellyissä analysoiduissa alaryhmissä, joita olivat mm. etninen tausta, ikä, sukupuoli, aiempi tupakointitausta ja keskushermoston etäpesäkestatus tutkimukseen mukaan tullessa.
Kuva 4 Aiemmin hoidettua ei-pienisoluista keuhkosyöpää sairastavien potilaiden kokonaiselinajan Kaplan-Meier-käyrä
Kallonsisäisiin etäpesäkkeisiin liittyvää tehoa koskevat tiedot
Potilaat, joilla oli oireettomia tai aiemmin hoidettuja ja stabiileja kallonsisäisiä etäpesäkkeitä, soveltuivat satunnaistettaviksi MARIPOSA‑2-tutkimukseen.
Rybrevant‑CP-hoitoon liittyi kallonsisäisen kokonaisvasteosuuden numeerinen lisäys (Rybrevant‑CP-hoidossa 23,3 % vs. CP-hoidossa 16,7 %, ristitulosuhde (OR) 1,52; 95 %:n luottamusväli (0,51–4,50) ja kallonsisäisen vasteen keston piteneminen (Rybrevant‑CP-hoidossa 13,3 kuukautta [95 %:n luottamusväli: 1,4 – ei arvioitavissa] verrattuna 2,2 kuukauteen CP-haarassa [95 %:n luottamusväli: 1,4 – ei arvioitavissa]). Rybrevant-CP-haaran seuranta-ajan mediaani oli noin 18,6 kuukautta.
Aiemmin hoitamaton ei-pienisoluinen keuhkosyöpä, jossa on eksonin 20 insertiomutaatioita (PAPILLON)
PAPILLON on vaiheen 3 satunnaistettu, avoin monikeskustutkimus, jossa Rybrevant-valmisteen, karboplatiinin ja pemetreksedin yhdistelmästä koostuvaa hoitoa verrataan pelkkään solunsalpaajahoitoon (karboplatiini ja pemetreksedi) aiemmin hoitamattomilla paikallisesti edennyttä tai etäpesäkkeistä ei-pienisoluista keuhkosyöpää sairastavilla potilailla, joilla on aktivoivia EGFR:n eksonin 20 insertiomutaatioita. Kaikkien 308 potilaan kasvainkudos- (92,2 %) ja/tai plasmanäytteet (7,8 %) testattiin paikallisesti EGFR:n eksonin 20 insertiomutaatiostatuksen selvittämiseksi. Testauksessa käytettiin 55,5 %:lla potilaista uuden sukupolven sekvensointia (NGS) ja 44,5 %:lla potilasta PCR-määritystä (polymeraasiketjureaktiomääritys). Testaus tehtiin myös keskitetysti käyttämällä AmoyDx® LC10 ‑kudostestiä, Thermo Fisher Oncomine Dx Target Test -testiä ja Guardant 360® CDx ‑plasmatestiä.
Potilaat, joilla oli seulonnassa etäpesäkkeitä aivoissa, joita oli hoidettu definitiivisesti soveltuivat osallistumaan tutkimukseen, kun heidän tilansa oli kliinisesti stabiili, he olivat oireettomia eivätkä olleet saaneet kortikosteroidihoitoa vähintään kahteen viikkoon ennen satunnaistamista.
Rybrevant-valmistetta annettiin 1 400 mg (potilaille, joiden paino oli < 80 kg) tai 1 750 mg (potilaille, joiden paino oli ≥ 80 kg) laskimoon kerran viikossa 4 viikon ajan ja sen jälkeen 1 750 mg:n annoksina (potilaille, joiden paino oli < 80 kg) tai 2 100 mg:n annoksina (potilaille, joiden paino oli ≥ 80 kg) 3 viikon välein viikosta 7 alkaen, kunnes sairaus eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Karboplatiinia annettiin laskimoon 3 viikon välein enimmillään 12 viikkoa pitoisuus-aikakäyrän alle jäävän pinta-alan ollessa 5 mg/ml/min (AUC 5). Pemetreksediä annettiin 500 mg/m2 laskimoon 3 viikon välein, kunnes sairaus eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Satunnaistaminen ositettiin ECOG-toimintakykyluokan (0 tai 1) ja aiempien etäpesäkkeiden aivoissa (kyllä tai ei) mukaan. Karboplatiini- ja pemetreksedihaaraan satunnaistettujen potilaiden, joiden sairauden oli varmistettu edenneen, sallittiin siirtyä Rybrevant-monoterapiaan. Yhteensä 308 tutkittavaa satunnaistettiin (1:1) saamaan Rybrevant-valmistetta yhdistelmänä karboplatiinin ja pemetreksedin kanssa (N = 153) tai karboplatiinia ja pemetreksediä (N = 155). Iän mediaani oli 62 vuotta (vaihteluväli: 27–92 vuotta), ja 39 % tutkittavista oli ≥ 65-vuotiaita; 58 % oli naisia, 61 % oli aasialaisia ja 36 % oli valkoihoisia. Lähtötilanteen ECOG (Eastern Cooperative Oncology Group) ‑toimintakykyluokka oli 0 (35 %) tai 1 (64 %); 58 % ei ollut koskaan tupakoinut; 23 %:lla oli anamneesissa etäpesäkkeitä aivoissa ja 84 %:lla oli alkuvaiheen diagnoosin ajankohtana levinneisyysasteen IV syöpä.
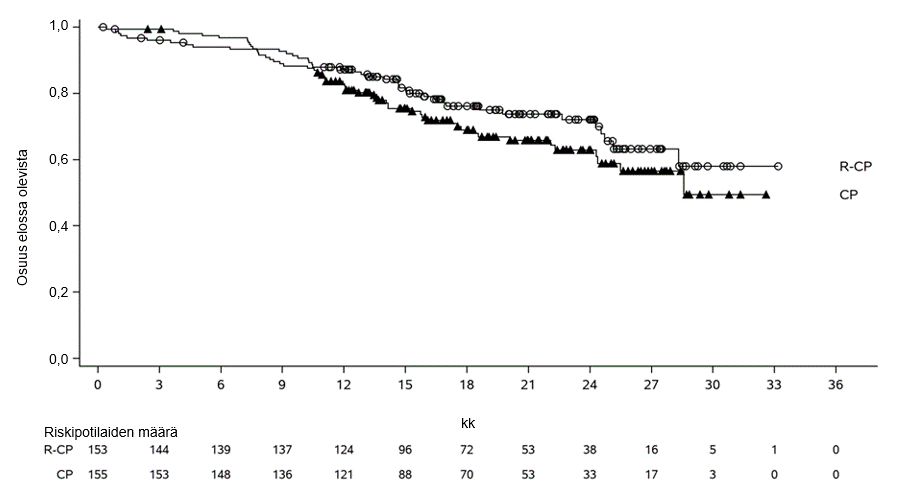
PAPILLON-tutkimuksen ensisijainen päätetapahtuma oli etenemättömyysaika (PFS) sokkoutetulla riippumattomalla keskitetyllä arvioinnilla (BICR) arvioituna. Seuranta-ajan mediaani oli 14,9 (vaihteluväli: 0,3−27,0) kuukautta.
Yhteenveto tehoa koskevista tuloksista esitetään taulukossa 13.
| Taulukko 13 Tehoa koskevat tulokset PAPILLON-tutkimuksessa | ||
Rybrevant+ karboplatiini+ pemetreksedi (N = 153) | Karboplatiini+ pemetreksedi (N = 155) | |
| Etenemättömyysaika (PFS) a | ||
| Tapahtumien lukumäärä | 84 (55 %) | 132 (85 %) |
| Mediaani, kuukautta (95 %:n luottamusväli) | 11,4 (9,8–13,7) | 6,7 (5,6–7,3) |
| Riskitiheyksien suhde (HR) (95 %:n luottamusväli); p‑arvo | 0,395 (0,29–0,52); p < 0,0001 | |
| Objektiivinen vasteosuusa, b | ||
| Kokonaisvasteosuus (ORR), % (95 %:n luottamusväli) | 73 % (65–80 %) | 47 % (39–56 %) |
| Ristitulosuhde (OR) (95 %:n luottamusväli); p‑arvo | 3,0 (1,8–4,8); p < 0,0001 | |
| Täydellinen vaste | 3,9 % | 0,7 % |
| Osittainen vaste | 69 % | 47 % |
| Kokonaiselinaika (OS)c | ||
| Tapahtumien lukumäärä | 40 | 52 |
| Kokonaiselinajan mediaani, kuukautta (95 %:n luottamusväli) | NE (28,3–NE) | 28,6 (24,4–NE) |
| Riskitiheyksien suhde (HR) (95 %:n luottamusväli); p‑arvo | 0,756 (0,50–1,14); p = 0,1825 | |
| NE = ei arvioitavissa (not estimable) a Sokkoutettu riippumaton keskitetty arviointi RECIST v1.1 ‑kriteerien mukaan b Perustuu Kaplan‑Meierin estimaattiin. c Perustuu päivitettyyn kokonaiselinaikaan, kun seuranta-ajan mediaani on 20,9 kuukautta. Kokonaiselinajan analyysiä ei säädetty hoidosta toiseen siirtymisestä (crossover) aiheutuvan mahdollisen sekoittavan vaikutuksen mukaan (78 [50,3 %] potilasta karboplatiinin ja pemetreksedin yhdistelmää saaneessa hoitohaarassa, jotka saivat sen jälkeen Rybrevant-monoterapiahoitoa). | ||
Kuva 5 Aiemmin hoitamatonta ei-pienisoluista keuhkosyöpää sairastavien potilaiden sokkoutettuun riippumattomaan keskitettyyn arviointiin perustuvan etenemättömyysajan Kaplan-Meier-käyrä
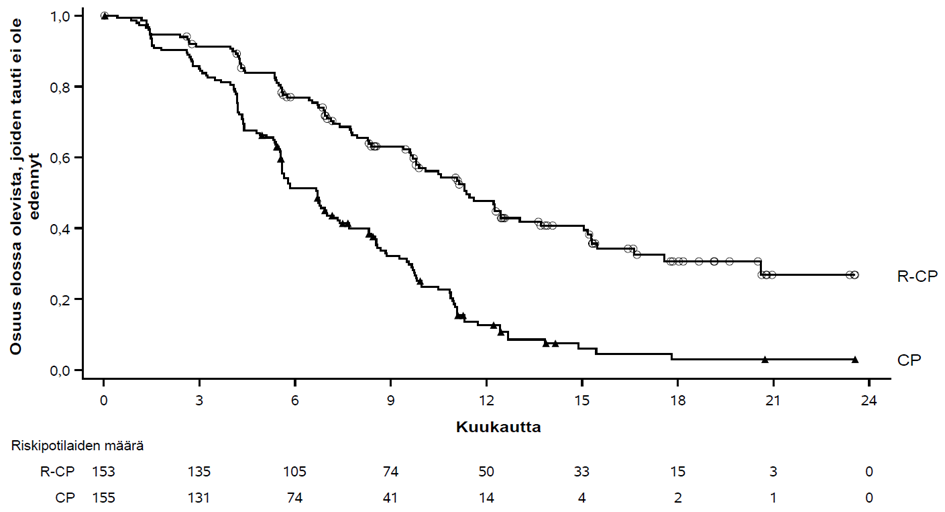
Rybrevant-valmisteen, karboplatiinin ja pemetreksedin yhdistelmän hyöty etenemättömyysajan suhteen verrattuna karboplatiiniin ja pemetreksediin oli yhdenmukainen kaikissa ennalta määritellyissä alaryhmissä: etäpesäkkeitä aivoissa potilaiden tullessa mukaan tutkimukseen (kyllä tai ei), ikä (< 65 tai ≥ 65), sukupuoli (mies tai nainen), etninen tausta (aasialainen tai muu kuin aasialainen), paino (< 80 kg tai ≥ 80 kg), ECOG-toimintakykyluokka (0 tai 1) ja aiempi tupakointitausta (kyllä tai ei).
Kuva 6 Aiemmin hoitamatonta ei-pienisoluista keuhkosyöpää sairastavien potilaiden kokonaiselinajan Kaplan-Meier-käyrä
Aiemmin hoidettu ei-pienisoluinen keuhkosyöpä, jossa on eksonin 20 insertiomutaatioita (CHRYSALIS)
CHRYSALIS on avoin, useassa kohortissa tehty monikeskustutkimus, jossa arvioitiin Rybrevant-valmisteen turvallisuutta ja tehoa paikallisesti edennyttä tai etäpesäkkeistä ei-pienisoluista keuhkosyöpää sairastavien potilaiden hoidossa. Tehoa on arvioitu 114 potilaalla, joilla oli paikallisesti edennyt tai etäpesäkkeinen ei-pienisoluinen keuhkosyöpä ja EGFR:n eksonin 20 insertiomutaatioita ja joiden tauti oli edennyt platinapohjaisen solunsalpaajahoidon aikana tai sen jälkeen. Tutkittavien seurannan mediaanikesto oli 12,5 kuukautta. EGFR:n eksonin 20 insertiomutaatioiden määrittäminen kasvainkudoksesta (93 %) ja/tai plasmanäytteistä (10 %) toteutettiin paikallisesti uuden sukupolven sekvensoinnilla (NGS) 46 %:lla potilaista ja/tai polymeraasiketjureaktiomäärityksellä (PCR) 41 %:lla potilaista; 4 %:lla tapaa ei ilmoitettu. Potilaat, joilla oli hoitamattomia aivoetäpesäkkeitä tai joilla oli aiemmin ollut pitkäaikaishoitoa steroideilla tai muilla immunosuppressiivisilla lääkeaineilla vaatinut interstitiaalinen keuhkosairaus kahden edellisen vuoden aikana, eivät soveltuneet mukaan tutkimukseen. Rybrevant-valmistetta annettiin laskimoon 1 050 mg alle 80 kg:n painoisille potilaille ja 1 400 mg vähintään 80 kg:n painoisille potilaille kerran viikossa 4 viikon ajan ja sen jälkeen 2 viikon välein viikosta 5 alkaen kliinisen hyödyn häviämiseen saakka tai kunnes ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Ensisijaisena tehon päätetapahtumana oli tutkijan arvioima kokonaisvasteosuus (ORR), jonka määritelmänä oli täydellinen vaste (CR) tai osittainen vaste (PR) RECIST v1.1 -kriteerien perusteella. Tämän lisäksi ensisijainen päätetapahtuma arvioitiin sokkoutetulla riippumattomalla keskitetyllä arvioinnilla (BICR). Toissijaisiin tehon päätetapahtumiin sisältyi vasteen kesto (DOR).
Mediaani-ikä oli 62 vuotta (vaihteluväli: 36–84). 41 % potilaista oli ≥ 65-vuotiaita. 61 % oli naisia, 52 % aasialaisia ja 37 % valkoihoisia. Aiempien hoitojen mediaanilukumäärä oli 2 (vaihteluväli: 1–7 hoitoa). Lähtötilanteessa 29 %:lla tutkittavista oli Eastern Cooperative Oncology Group (ECOG) ‑toimintakykyluokka 0 ja 70 %:lla oli ECOG-toimintakykyluokka 1. 57 % ei ollut tupakoinut koskaan, 100 %:lla oli levinneisyysasteen IV syöpä ja 25 % oli saanut aiempaa hoitoa aivoetäpesäkkeisiin. Eksonissa 20 olevia insertioita havaittiin kahdeksassa eri aminohappotähteessä. Yleisimpiä tähteitä olivat A767 (22 %), S768 (16 %), D770 (12 %) ja N771 (11 %).
Tehoa koskevista tuloksista on esitetty yhteenveto taulukossa 14.
| Taulukko 14 Tehoa koskevat tulokset CHRYSALIS-tutkimuksessa |
Tutkijan arviointi (N=114) | |
| Kokonaisvasteosuusa, b(95 %:n luottamusväli) | 37 % (28-46 %) |
| Täydellinen vaste | 0 % |
| Osittainen vaste | 37 % |
| Vasteen kesto | |
| Mediaanic (95 %:n luottamusväli), kuukautta | 12,5 (6,5-16,1) |
| Potilaat, joiden vasteen kesto oli ≥ 6 kuukautta | 64 % |
a Vahvistettu vaste
b Tutkijan arvioima kokonaisvasteosuus ja vasteen kesto vastasivat sokkoutettua riippumatonta keskitettyä arviointia; kokonaisvasteosuus oli sokkoutetussa riippumattomassa keskitetyssä arvioinnissa 43 % (34-53 %); 3 %:lla oli täydellinen vaste ja 40 %:lla osittainen vaste. Vasteen keston mediaani oli sokkoutetussa riippumattomassa keskitetyssä arvioinnissa 10,8 kuukautta (95 %:n luottamusväli: 6,9-15,0) ja potilaita, joilla vasteen kesto oli ≥ 6 kuukautta, oli sokkoutetussa riippumattomassa keskitetyssä arvioinnissa 55 %.
c Kaplan-Meierin estimaatin perusteella.
Kasvaimia vastaan kohdistuvaa aktiivisuutta havaittiin tutkituissa mutaatioiden alatyypeissä.
Iäkkäät
Tehossa ei yleisesti havaittu eroja ≥ 65-vuotiaiden ja < 65-vuotiaiden potilaiden välillä.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Rybrevant-valmisteen käytöstä ei-pienisoluisen keuhkosyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Rybrevant-monoterapiaa koskevien tietojen perusteella amivantamabin pitoisuus-aikakäyrän alle jäävä pinta-ala (AUC1 viikko) lisääntyy suhteellisesti annosalueella 350–1750 mg.
Populaatiofarmakokineettisen mallin simulaatioiden perusteella AUC1 viikko ‑arvo oli noin 2,8 kertaa suurempi 2 viikon välein tapahtuvan annostelun viidennen annoksen jälkeen ja 2,6 kertaa suurempi 3 viikon välein tapahtuvan annostelun neljännen annoksen jälkeen. Amivantamabin vakaan tilan pitoisuudet saavutettiin sekä 3 viikon että 2 viikon välein tapahtuvassa annostelussa viikkoon 13 mennessä, ja systeeminen kertymä oli 1,9‑kertainen.
Jakautuminen
Populaatiofarmakokineettisen analyysin yksittäisten farmakokineettisten parametrien estimaattien perusteella amivantamabin kokonaisjakautumistilavuuden geometrinen keskiarvo (variaatiokerroin, %) on 5,12 l (27,8 %) Rybrevant-valmisteen suositellun annoksen annon jälkeen.
Eliminaatio
Populaatiofarmakokineettisen analyysin yksittäisten farmakokineettisten parametrien estimaattien perusteella amivantamabin lineaarisen puhdistuman geometrinen keskiarvo (variaatiokerroin, %) on 0,266 l/vrk (30,4 %) ja lineaariseen puhdistumaan liittyvän terminaalisen puoliintumisajan geometrinen keskiarvo on 13,7 vrk (31,9 %).
Erityiset potilasryhmät
Iäkkäät potilaat
Iän perusteella (21–88 vuotta) ei havaittu kliinisesti merkittäviä eroja amivantamabin farmakokinetiikassa.
Munuaisten vajaatoiminta
Amivantamabin farmakokinetiikassa ei havaittu kliinisesti merkittävää vaikutusta potilailla, joilla oli lievä (60 ≤ kreatiniinipuhdistuma [CrCl] < 90 ml/min), kohtalainen (29 ≤ CrCl < 60 ml/min) tai vaikea (15 ≤ CrCl < 29 ml/min) munuaisten vajaatoiminta. Vaikeaa munuaisten vajaatoimintaa sairastavista potilaista on vähän tietoja (n = 1), mutta siitä ei ole näyttöä, että näiden potilaiden annosta olisi tarpeen muuttaa. Loppuvaiheen munuaissairauden (CrCl < 15 ml/min) vaikutusta amivantamabin farmakokinetiikkaan ei tunneta.
Maksan vajaatoiminta
Maksan toiminnan muutoksilla ei todennäköisesti ole mitään vaikutusta amivantamabin eliminaatioon, koska IgG1-pohjaiset molekyylit, kuten amivantamabi, eivät metaboloidu maksareittien kautta.
Amivantamabin farmakokinetiikassa ei havaittu kliinisesti merkittävää vaikutusta lievän maksan vajaatoiminnan ([kokonaisbilirubiini ≤ ULN ja ASAT > ULN] tai [ULN < kokonaisbilirubiini ≤ 1,5 x ULN]) tai kohtalaisen maksan vajaatoiminnan (1,5 × ULN < kokonaisbilirubiini ≤ 3 × ULN ja ASAT mikä tahansa) seurauksena. Kohtalaista maksan vajaatoimintaa sairastavista potilaista on vähän tietoja (n = 1), mutta siitä ei ole näyttöä, että näiden potilaiden annosta olisi tarpeen muuttaa. Vaikean maksan vajaatoiminnan (kokonaisbilirubiini > 3 kertaa ULN) vaikutusta amivantamabin farmakokinetiikkaan ei tunneta.
Pediatriset potilaat
Rybrevant-valmisteen farmakokinetiikkaa pediatrisilla potilailla ei ole tutkittu.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Karsinogeenisuus ja mutageenisuus
Eläinkokeita ei ole tehty amivantamabin karsinogeenisuuden selvittämiseksi. Rutiininomaiset genotoksisuus- ja karsinogeenisuustutkimukset eivät yleensä sovellu käytettäväksi biologisten lääkkeiden kohdalla, koska suuret proteiinit eivät voi diffundoitua soluihin eivätkä voi olla vuorovaikutuksessa DNA:n tai kromosomaalisen materiaalin kanssa.
Lisääntymistoksisuus
Eläinkokeita ei ole tehty lisääntymiseen ja sikiön kehitykseen kohdistuvien vaikutusten arvioimiseksi. Vaikutusmekanisminsa perusteella amivantamabi voi kuitenkin aiheuttaa sikiölle vaurioita tai kehityksellisiä poikkeavuuksia. Kuten kirjallisuudessa on raportoitu, alkion/sikiön tai emon EGFR-signaloinnin vähentyminen, puuttuminen tai häiriintyminen voi estää implantaation, aiheuttaa alkion/sikiön keskenmenon tiineyden eri vaiheissa (istukan kehitykseen kohdistuvien vaikutusten kautta), aiheuttaa kehityksellisiä poikkeavuuksia eri elimissä tai aiheuttaa eloonjääneen sikiön ennenaikaisen kuoleman. Vastaavasti MET:n tai sen ligandin hepatosyyttikasvutekijän poistogeenisuus oli letaali alkiolle istukan vaikeiden kehityspuutosten seurauksena. Sikiöillä esiintyi myös vikoja useiden elinten lihasten kehittymisessä. Ihmisen IgG1:n tiedetään pääsevän istukan läpi. Näin ollen amivantamabi voi mahdollisesti siirtyä äidiltä kehittyvään sikiöön.
Farmaseuttiset tiedot
Apuaineet
Etyleenidiamiinitetraetikkahapon (EDTA) dinatriumsuoladihydraatti
L-histidiini
L-histidiinihydrokloridimonohydraatti
L-metioniini
Polysorbaatti 80 (E433)
Sakkaroosi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta
Laimentamisen jälkeen
Kemiallinen ja fysikaalinen käytön aikainen säilyvyys on osoitettu 10 tunnin ajalta 15–25 °C:n lämpötilassa huoneenvalossa. Mikrobiologian kannalta valmiste on käytettävä välittömästi, ellei laimennusmenetelmä poista mikrobikontaminaation riskiä. Jos valmistetta ei käytetä välittömästi, käyttökuntoon saattamisen jälkeiset säilytysajat ja olosuhteet ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2–8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
RYBREVANT infuusiokonsentraatti, liuosta varten
350 mg (L:ei) 1 kpl (7 ml) (1611,11 €)
PF-selosteen tieto
7 ml konsentraattia tyypin 1 lasisessa injektiopullossa, jossa on elastomeerinen suljin, alumiinisinetti ja repäisykorkki, ja joka sisältää 350 mg amivantamabia. Pakkauskoko: 1 injektiopullo.
Valmisteen kuvaus:
Liuos on väritön tai vaaleankeltainen, ja sen pH-arvo on 5,7 ja osmolaliteetti noin 310 mOsm/kg.
Käyttö- ja käsittelyohjeet
Valmista liuos laskimoinfuusiota varten aseptisella tekniikalla seuraavasti:
Valmistus
- Määritä tarvittava annos ja tarvittava Rybrevant-injektiopullojen määrä potilaan lähtötilanteen painon perusteella (ks. kohta Annostus ja antotapa). Yksi injektiopullo sisältää 350 mg amivantamabia.
- 2 viikon välein tapahtuvassa annossa < 80 kg:n painoisille potilaille annetaan 1 050 mg ja ≥ 80 kg:n painoisille potilaille annetaan 1 400 mg kerran viikossa. Yhteensä annoksia annetaan neljä. Viikosta 5 alkaen annoksia annetaan 2 viikon välein.
- 3 viikon välein tapahtuvassa annossa < 80 kg:n painoisille potilaille annetaan 1 400 mg kerran viikossa. Yhteensä annoksia annetaan neljä. Viikosta 7 alkaen annetaan 1 750 mg 3 viikon välein. ≥ 80 kg:n painoisille potilaille annetaan 1 750 mg kerran viikossa. Yhteensä annoksia annetaan neljä. Viikosta 7 alkaen annetaan 2 100 mg 3 viikon välein.
- Tarkista, että Rybrevant-liuos on väritöntä tai vaaleankeltaista. Älä käytä liuosta, jos siinä on värimuutoksia tai näkyviä hiukkasia.
- Poista 250 ml:n infuusiopussista (joko 5 % glukoosiliuosta tai 9 mg/ml [0,9 %] natriumkloridiliuosta injektiota varten) tilavuus, joka vastaa tarvittavaa lisättävän Rybrevant-liuoksen tilavuutta, ja hävitä se (hävitä 7 ml laimenninta infuusiopussista kutakin injektiopulloa kohden). Infuusiopussien on oltava valmistettu polyvinyylikloridista (PVC), polypropeenista (PP), polyeteenistä (PE) tai polyolefiiniseoksesta (PP+PE).
- Vedä 7 ml Rybrevant-valmistetta kustakin injektiopullosta ja lisää se sen jälkeen infuusiopussiin. Yksi injektiopullo sisältää 0,5 ml:n ylitäytön riittävän ulosvedettävän tilavuuden varmistamiseksi. Lopullisen infuusiopussissa olevan tilavuuden on oltava 250 ml. Hävitä mahdollisesti injektiopulloon jäänyt käyttämätön osa.
- Käännä pussi varovasti ylösalaisin liuoksen sekoittamiseksi. Älä ravista.
- Tutki liuos silmämääräisesti hiukkasten ja värimuutosten varalta ennen antoa. Älä käytä liuosta, jos siinä näkyy värimuutoksia tai näkyviä hiukkasia.
Anto
- Anna laimennettu liuos laskimoinfuusiona infuusiovälineillä, jossa on virtauksensäädin ja in-line-tyyppinen, steriili, ei-pyrogeeninen, vähän proteiinia sitova polyeetterisulfoni(PES)suodatin (huokoskoko 0,22 tai 0,2 mikrometriä). Antovälineiden on oltava valmistettu polyuretaanista (PU), polybutadieenistä (PBD), PVC:stä, PP:stä tai PE:stä.
- Suodattimella varustetut antovälineet on esitäytettävä joko 5-prosenttisella glukoosiliuoksella tai 0,9-prosenttisella natriumkloridiliuoksella ennen kunkin Rybrevant-infuusion aloittamista.
- Älä infusoi Rybrevant-valmistetta samanaikaisesti samalla laskimoletkulla muiden lääkeaineiden kanssa.
- Laimennettu liuos on annettava 10 tunnin sisällä (mukaan lukien infuusioaika) huoneenlämmössä (15–25 °C) ja huoneenvalossa.
- Ensimmäisen annoksen kohdalla ilmenevien infuusioon liittyvien reaktioiden yleisyyden takia amivantamabi on infusoitava perifeerisen laskimon kautta viikolla 1 ja viikolla 2. Seuraavilla viikoilla infuusioon liittyvien reaktioiden riski on pienempi, joten infuusio voidaan antaa keskuslaskimokatetrin kautta. Ks. infuusionopeudet kohdasta Annostus ja antotapa.
Hävittäminen
Tämä lääkevalmiste on tarkoitettu vain kertakäyttöön. Käyttämätön lääkevalmiste, jota ei ole annettu 10 tunnin sisällä, on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
RYBREVANT infuusiokonsentraatti, liuosta varten
350 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FX18
Valmisteyhteenvedon muuttamispäivämäärä
20.02.2026
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com