
BRUKINSA kapseli, kova 80 mg
Vaikuttavat aineet ja niiden määrät
Yksi kova kapseli sisältää 80 mg tsanubrutinibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, kova.
Kliiniset tiedot
Käyttöaiheet
BRUKINSA monoterapiana käytettynä on tarkoitettu sellaisten aikuispotilaiden hoitoon, joilla on Waldenströmin makroglobulinemia (WM) ja jotka ovat saaneet vähintään yhtä aiempaa hoitoa, tai ensilinjan hoitona potilaille, joille kemoimmunoterapia ei sovellu käytettäväksi.
BRUKINSA monoterapiana käytettynä on tarkoitettu sellaisten aikuispotilaiden hoitoon, joilla on marginaalivyöhykkeen lymfooma (MZL) ja jotka ovat saaneet vähintään yhtä aiempaa CD20-vasta-aineisiin perustuvaa hoitoa.
BRUKINSA monoterapiana käytettynä on tarkoitettu sellaisten aikuispotilaiden hoitoon, joilla on krooninen lymfosyyttinen leukemia (KLL).
BRUKINSA ja obinututsumabi yhdistelmänä on tarkoitettu sellaisten aikuispotilaiden hoitoon, joilla on refraktorinen tai relapsoitunut follikulaarinen lymfooma (FL) ja jotka ovat saaneet vähintään kahta aiempaa systeemistä hoitoa.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Tällä lääkevalmisteella toteutettava hoito on aloitettava ja sitä on valvottava lääkärin toimesta, joka on perehtynyt syöpälääkevalmisteiden käyttöön.
Annostus
Tsanubrutinibin suositeltu vuorokausiannos on 320 mg. Vuorokausiannos voidaan ottaa joko kerran vuorokaudessa (neljä 80 mg:n kapselia) tai kahteen 160 mg:n annokseen (kaksi 80 mg:n kapselia) jaettuna. Hoitoa on jatkettava, kunnes tauti etenee tai ilmaantuu ei-hyväksyttävissä olevaa toksisuutta.
BRUKINSA:n ja obinututsumabin yhdistelmä
Tsanubrutinibi on annettava ennen obinututsumabi-infuusiota. Suositeltu annos on 1 000 mg obinututsumabia laskimonsisäisesti syklin 1 päivinä 1, 8 ja 15 sekä sykleissä 2–6 jokaisen 28 päivän syklin päivänä 1. Lääkärin harkinnan mukaan obinututsumabia voidaan annostella 100 mg syklin 1 päivänä 1 ja 900 mg syklin 1 päivänä 2 sen sijaan, että annosteltaisiin 1 000 mg syklin 1 päivänä 1. Ylläpitohoito obinututsumabilla (yksi infuusio joka toinen kuukausi enintään kahden vuoden ajan) voidaan määrätä. Katso obinututsumabin valmisteyhteenvedosta lisätietoja annostelusta, mukaan lukien esilääkityksestä ennen jokaista infuusiota.
Annoksen muuttaminen haittavaikutusten myötä
Taulukossa 1 on esitetty suositellut tsanubrutinibin annoksen muutokset, jotka koskevat asteen 3 tai suurempia haittavaikutuksia.
Taulukko 1: Suositellut annoksen muutokset haittavaikutusten myötä
Haittavaikutus | Haittavaikutuksen esiintyminen | Annoksen muuttaminen (aloitusannos: 320 mg kerran vuorokaudessa tai 160 mg kahdesti vuorokaudessa) |
Vähintään asteen 3 ei-hematologiset toksisuudet Asteen 3 kuumeinen neutropenia Asteen 3 trombosytopenia, johon liittyy merkittävää verenvuotoa Asteen 4 neutropenia (joka kestää yli 10 peräkkäistä päivää) Asteen 4 trombosytopenia (joka kestää yli 10 peräkkäistä päivää) | Ensimmäinen | BRUKINSA-hoito keskeytetään Kun toksisuus on lievittynyt ≤ asteen 1 tasolle tai lähtötasolle: Jatketaan annoksella 320 mg kerran vuorokaudessa tai 160 mg kahdesti vuorokaudessa |
Toinen | BRUKINSA-hoito keskeytetään Kun toksisuus on lievittynyt ≤ asteen 1 tasolle tai lähtötasolle: Jatketaan annoksella 160 mg kerran vuorokaudessa tai 80 mg kahdesti vuorokaudessa | |
Kolmas | BRUKINSA-hoito keskeytetään Kun toksisuus on lievittynyt ≤ asteen 1 tasolle tai lähtötasolle: Jatketaan annoksella 80 mg kerran vuorokaudessa | |
Neljäs | BRUKINSA-hoito lopetetaan |
Oireetonta lymfosytoosia ei pidä katsoa haittavaikutukseksi. Näiden potilaiden on jatkettava BRUKINSA-hoitoa.
Katso obinututsumabin valmisteyhteenvedosta obinutustumabin annoksen muutokset, jotka koskevat haittavaikutuksia.
Annoksen muutokset muun samanaikaisen hoidon takia
Annoksen muutokset käytettäessä valmistetta CYP3A:n estäjien tai induktorien kanssa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakokinetiikka):
Taulukko 2: Suositellut annoksen muutokset, kun valmistetta annetaan samanaikaisesti muiden lääkevalmisteiden kanssa
CYP3A | Samanaikaisesti annettava lääkevalmiste | Suositeltu annos |
Estäjä | Voimakas CYP3A:n estäjä (esim. posakonatsoli, vorikonatsoli, ketokonatsoli, itrakonatsoli, klaritromysiini, indinaviiri, lopinaviiri, ritonaviiri, telapreviiri) | 80 mg kerran vuorokaudessa |
Kohtalainen CYP3A:n estäjä (esim. erytromysiini, siprofloksasiini, diltiatseemi, dronedaroni, flukonatsoli, verapamiili, aprepitantti, imatinibi, greippimehu, Sevillan appelsiinit eli pomeranssi) | 80 mg kahdesti vuorokaudessa | |
Induktori | Voimakas CYP3A:n induktori (esim. karbamatsepiini, fenytoiini, rifampisiini, mäkikuisma) Kohtalainen CYP3A:n induktori (esim. bosentaani, efavirentsi, etraviriini, modafiniili, nafsilliini) | Samanaikaista käyttöä on vältettävä; on harkittava vaihtoehtoisia lääkeaineita, joiden aiheuttama CYP3A:n induktio on heikompaa |
Väliin jäänyt annos:
Kaksinkertaista annosta ei pidä ottaa väliin jääneen annoksen korvaamiseksi. Jos annosta ei oteta aikataulun mukaisena aikana, seuraava annos on otettava normaalin aikataulun mukaisesti.
Erityiset potilasryhmät
Iäkkäät potilaat
Erilliset annoksen muutokset eivät ole tarpeen iäkkäille (≥ 65-vuotiaille) potilaille.
Munuaisten vajaatoiminta
Annoksen muuttamista ei suositella potilaille, joilla on lievä, kohtalainen tai vaikea munuaisten vajaatoiminta (Cockcroft-Gault-yhtälöllä arvioitu kreatiniinipuhdistuma ≥ 15 ml/min). Vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma < 30 ml/min) sairastavista potilaista ja loppuvaiheen munuaistautia (ESRD, n = 12) sairastavista potilaista on olemassa vain vähän tietoja. Potilaita, joilla on munuaisten vajaatoiminta (kreatiniinipuhdistuma < 30 ml/min) ja dialyysihoitoa saavia potilaita on seurattava haittavaikutusten varalta (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa potilaille, joilla on lievä (Child-Pugh-luokka A) tai kohtalainen (Child-Pugh-luokka B) maksan vajaatoiminta. Potilaat, joilla oli lievä tai kohtalainen maksan vajaatoiminta, saivat BRUKINSA-hoitoa kliinisissä tutkimuksissa. Suositeltu BRUKINSA-valmisteen annos vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastaville potilaille on 80 mg suun kautta kahdesti vuorokaudessa. BRUKINSA-valmisteen turvallisuutta ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla. Näitä potilaita on seurattava tarkoin haittavaikutusten varalta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
BRUKINSA-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
BRUKINSA-kapselit otetaan suun kautta. Kovat kapselit voidaan ottaa ruuan kanssa tai ilman ruokaa. Potilaita on neuvottava nielemään kapselit kokonaisena veden kanssa. Kapseleita ei saa avata, rikkoa eikä pureskella.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Verenvuoto
Vakavia ja kuolemaan johtaneita verenvuototapahtumia on esiintynyt BRUKINSA-hoitoa saaneilla potilailla. Potilailla on raportoitu asteen 3 tai sitä suurempia verenvuototapahtumia, kuten kallonsisäistä ja ruoansulatuskanavan verenvuotoa, hematuriaa ja veririntaa (ks. kohta Haittavaikutukset). Kaikenasteisia verenvuototapahtumia, mukaan lukien purppura ja petekiat, esiintyi potilailla, joilla oli hematologisia maligniteetteja. Verenvuototapahtumien mekanismia ei tunneta hyvin (ks. kohta Farmakokinetiikka).
BRUKINSA saattaa lisätä verenvuotoriskiä potilailla, jotka saavat verihiutaleiden muodostusta estävää hoitoa tai antikoagulanttihoitoa. Annoksen muuttaminen voi olla tarpeen asteen 3 tai sitä korkeampien haittavaikutusten takia suositellulla tavalla (ks. kohta Annostus ja antotapa). Varfariinia tai muita K-vitamiinin agonisteja ei pidä antaa samanaikaisesti BRUKINSA-hoidon kanssa. Potilaita on seurattava verenvuodon merkkien varalta ja täydellisen verenkuvan osalta on harkittava antikoagulanttihoidon tai verihiutale-estäjien riskejä ja hyötyjä silloin, kun niitä annetaan yhdessä BRUKINSA-hoidon kanssa. Tsanubrutinibin keskeyttämisen hyöty-riskisuhdetta on arvioitava 3–7 päivän ajan ennen leikkausta ja leikkauksen jälkeen leikkauksen tyypistä ja verenvuotoriskistä riippuen.
Infektiot
Kuolemaan johtaneita ja kuolemaan johtamattomia infektioita (mukaan lukien bakteeri-, virus- tai sieni-infektiot tai sepsis) ja opportunistisia infektioita (esim. herpesvirus-, kryptokokki-, aspergillus- ja pneumocystis jiroveci -infektiot) on esiintynyt potilailla, joita on hoidettu BRUKINSA-valmisteella. Potilailla esiintyi asteen 3 tai sitä korkeampia infektioita (ks. kohta Haittavaikutukset). Yleisin asteen 3 tai korkeampi infektio oli keuhkokuume. Hepatiitti B -viruksen (HBV) uudelleenaktivoitumisesta johtuvia infektioita on myös esiintynyt. Ennen BRUKINSA-hoidon aloittamista potilaan hepatiitti B-virusstatus on tutkittava. Maksasairauksiin erikoistuneen lääkärin konsultaatio on suositeltavaa ennen hoidon aloittamista potilaille, joille on positiivinen hepatiitti B-viruksen testitulos tai joilla on positiivinen B-hepatiitin serologia. Potilasta on seurattava ja hoidettava standardihoitokäytäntöjen mukaisesti B-hepatiitin uudelleenaktivoitumisen estämiseksi. Estolääkitystä on harkittava standardihoidon mukaisesti potilailla, joilla on suurentunut infektioriski. Potilaita on tarkkailtava infektion merkkien ja oireiden varalta ja hoidettava asianmukaisesti.
Sytopeniat
Asteen 3 tai 4 sytopenioita, mukaan lukien neutropenia, trombosytopenia ja laboratoriomittausten tulosten perusteella todettu anemia, raportoitiin BRUKINSA-hoitoa saaneilla potilailla (ks. kohta Haittavaikutukset). Täydellistä verenkuvaa on seurattava kuukausittain hoidon aikana (ks. kohta Annostus ja antotapa).
Toiset primaariset maligniteetit
BRUKINSA-valmisteella hoidetuilla potilailla, joilla on hematologisia maligniteetteja, on esiintynyt toisia primaarisia maligniteetteja, myös muita kuin ihon syöpiä. Yleisin toinen primaarinen maligniteetti oli ihosyöpä (tyvisolusyöpä ja ihon levyepiteelisyöpä). Potilaita on neuvottava käyttämään auringolta suojaavia toimenpiteitä.
Eteisvärinä ja lepatus
Eteisvärinää ja eteislepatusta on esiintynyt potilailla, joilla on hematologisia maligniteetteja ja joita on hoidettu BRUKINSA-valmisteella, erityisesti potilailla, joilla on sydämeen liittyviä riskitekijöitä, hypertensiota ja akuutteja infektioita ja jotka ovat iäkkäitä (≥ 65-vuotiaita). Eteisvärinän ja eteislepatuksen merkkejä ja oireita on seurattava ja hoidettava tarvittavin osin (ks. kohta Farmakodynamiikka).
Tuumorilyysioireyhtymä
Tuumorilyysioireyhtymää on raportoitu melko harvoin tsanubrutinibi-monoterapian yhteydessä, erityisesti potilailla, joita on hoidettu kroonisen lymfosyyttisen leukemian (KLL) takia (ks. kohta Haittavaikutukset). Oleelliset riskit (esim. korkea kasvainkuorma tai veren virtsahappopitoisuus) on arvioitava ja on ryhdyttävä asianmukaisiin varotoimenpiteisiin. Potilaita on seurattava tarkoin ja hoidettava asianmukaisesti.
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisymenetelmää BRUKINSA-hoitoa käyttäessään (ks. kohta Raskaus ja imetys).
BRUKINSA sisältää natriumia
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos, eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Tsanubrutinibi metaboloituu pääasiassa sytokromi P450-entsyymin 3A (CYP3A) välityksellä.
Lääkkeet, jotka voivat suurentaa tsanubrutinibin pitoisuutta plasmassa
BRUKINSA-hoidon ja voimakkaasti tai kohtalaisesti CYP3A:ta estävien lääkevalmisteiden samanaikainen käyttö voi lisätä tsanubrutinibialtistusta.
Voimakkaat CYP3A:n estäjät
Useiden itrakonatsolin (voimakas CYP3A:n estäjä) annosten samanaikainen anto terveille vapaaehtoisille nosti Cmax-pitoisuuden 2,6-kertaiseksi ja AUC-arvon 3,8-kertaiseksi. Vahvojen CYP3A:n estäjien, vorikonatsolin ja klaritromysiinin, useiden annosten samanaikainen anto potilaille, joilla oli B-solumaligniteetteja, johti tsanubrutinibialtistuksen suurenemiseen, vorikonatsolin kanssa annosnormalisoituna AUC0‑24h-arvon suurenemisen 3,30-kertaiseksi ja Cmax-arvon suurenemisen 3,29-kertaiseksi sekä klaritromysiinin kanssa annosnormalisoituna AUC0‑24h-arvon suurenemisen 1,92-kertaiseksi ja Cmax-arvon suurenemisen 2,01-kertaiseksi.
Jos voimakasta CYP3A:n estäjää on käytettävä (esim. posakonatsoli, vorikonatsoli, ketokonatsoli, itrakonatsoli, klaritromysiini, indinaviiri, lopinaviiri, ritonaviiri, telapreviiri), BRUKINSA-hoidon annosta on pienennettävä 80 mg:aan (yksi kapseli) estäjän käytön ajaksi. Potilaita on seurattava tarkoin toksisuuden varalta ja annoksen muuttamista koskevia ohjeita on noudatettava tarpeen mukaisesti (ks. kohta Annostus ja antotapa).
Kohtalaiset CYP3A:n estäjät
Kohtalaisten CYP3A:n estäjien, flukonatsolin ja diltiatseemin, toistuvien annosten samanaikainen anto potilaille, joilla oli B-solumaligniteetteja, johti tsanubrutinibialtistuksen suurenemiseen, flukonatsolin kanssa annosnormalisoituna AUC0‑24h-arvon suurenemisen 1,88-kertaiseksi ja Cmax-arvon suurenemisen 1,81-kertaiseksi sekä diltiatseemin kanssa annosnormalisoituna AUC0‑24h-arvon suurenemisen 1,62-kertaiseksi ja Cmax-arvon suurenemisen 1,62-kertaiseksi.
Jos kohtalaista CYP3A:n estäjää on käytettävä (esim. erytromysiini, siprafloksasiini, diltiatseemi, dronedaroni, flukonatsoli, verapamiili, aprepitantti, imatinibi, greippimehu, Sevillan appelsiinit eli pomeranssi), BRUKINSA-hoidon annosta on pienennettävä 160 mg:aan (kaksi kapselia) estäjän käytön ajaksi. Potilaita on seurattava tarkoin toksisuuden varalta ja annoksen muuttamista koskevia ohjeita on noudatettava tarpeen mukaisesti (ks. kohta Annostus ja antotapa).
Heikot CYP3A:n estäjät
Paasto-olosuhteissa tehdyt simulaatiot viittaavat siihen, että heikot CYP3A:n estäjät (esim. siklosporiini, fluvoksamiini ja simetidiini) voivat lisätä tsanubrutinibin AUC-arvoa 1,5-kertaiseksi. Annoksen muuttaminen ei ole tarpeen yhdistettäessä hoitoon heikkoja estäjiä. Potilaita on seurattava tarkoin toksisuuden varalta ja annoksen muuttamista koskevia ohjeita on noudatettava tarpeen mukaisesti.
Lääkkeet, jotka voivat pienentää tsanubrutinibin pitoisuutta plasmassa
Tsanubrutinibin ja voimakkaiden tai kohtalaisten CYP3A:n induktorien samanaikainen käyttö voi pienentää tsanubrutinibin pitoisuutta plasmassa.
CYP3A:n induktorit
Useiden rifampisiinin (voimakas CYP3A:n induktori) annosten samanaikainen anto vähensi tsanubrutinibin Cmax-pitoisuutta 92 % ja AUC-arvoa 93 % terveillä tutkittavilla. Samanaikaista antoa voimakkaiden CYP3A:n induktorien (esim. karbamatsepiini, fenytoiini, rifampisiini, mäkikuisma) ja kohtalaisten CYP3A:n induktorien (esim. bosentaani, efavirentsi, etraviriini, modafiniili, nafsilliini) kanssa on vältettävä (ks. kohta Annostus ja antotapa). Samanaikainen useiden rifabutiinin (kohtalainen CYP3A:n induktori) annosten anto vähensi tsanubrutinibin Cmax-pitoisuutta 48 % ja AUC-arvoa 44 % terveillä tutkittavilla. Tsanubrutinibin ja voimakkaiden CYP3A:n induktorien (esim. karbamatsepiini, fenytoiini, rifampisiini, mäkikuisma) tai kohtalaisten CYP3A:n induktorien (esim. bosentaani, efavirentsi, etraviriini, modafiniili, nafsilliini) samanaikaista käyttöä on vältettävä (ks. kohta Annostus ja antotapa). Heikkoja CYP3A:n induktoreja (esim. pioglitatsoni) voidaan käyttää varoen BRUKINSA-hoidon aikana.
Mahahappoa vähentävät lääkkeet
Tsanubrutinibin farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja, kun sitä annettiin samanaikaisesti mahahappoa vähentävien lääkkeiden (protonipumpun estäjät, H2- reseptorin antagonistit) kanssa.
Lääkkeet, joiden pitoisuuksiin plasmassa tsanubrutinibi voi vaikuttaa
Tsanubrutinibi on heikko CYP3A:n ja CYP2C19:n induktori. Samanaikainen tsanubrutinibin käyttö voi pienentää näiden substraattilääkevalmisteiden pitoisuuksia plasmassa.
CYP3A:n substraatit
Useiden tsanubrutinibiannosten samanaikainen anto pienensi midatsolaamin (CYP3A:n substraatti) Cmax-pitoisuutta 30 % ja AUC-arvoa 47 %. Kapean terapeuttisen indeksin omaavia lääkevalmisteita, joita CYP3A metaboloi (esim. alfentaniili, siklosporiini, dihydroergotamiini, ergotamiini, fentanyyli, pimotsidi, kinidiini, sirolimuusi ja takrolimuusi), on käytettävä varoen, koska tsanubrutinibi voi pienentää näiden lääkevalmisteiden plasma-altistusta.
CYP2C19:n substraatit
Useiden tsanubrutinibiannosten samanaikainen anto pienensi omepratsolin (CYP2C19:n substraatti) Cmax-pitoisuutta 20 % ja AUC-arvoa 36 %. Kapean terapeuttisen indeksin omaavia lääkevalmisteita, joita CYP2C19 metaboloi (esim. S-mefenytoiini), on käytettävä varoen, koska tsanubrutinibi voi pienentää näiden lääkkeiden plasma-altistusta.
Samanaikainen anto kuljetussubstraattien/-estäjien kanssa
Useiden tsanubrutinibiannosten samanaikainen anto suurensi digoksiinin (P-gp:n substraatti) Cmax-pitoisuutta 34 % ja AUC-arvoa 11 %. Rosuvastatiinin (BCRP:n substraatti) farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja, kun sitä annettiin samanaikaisesti tsanubrutinibin kanssa.
Suun kautta otettavien P-gp:n substraattien samanaikainen anto kapean terapeuttisen indeksin omaavien lääkkeiden (esim. digoksiini) on toteutettava varoen, koska tsanubrutinibi voi lisätä niiden pitoisuuksia.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi/raskaudenehkäisy naisilla
Eläinkokeiden perusteella BRUKINSA voi aiheuttaa haittaa sikiölle, kun sitä annetaan raskaana oleville naisille (ks. kohta Prekliiniset tiedot turvallisuudesta). Naisten on vältettävä raskaaksi tulemista BRUKINSA-hoidon aikana ja vähintään 1 kuukauden ajan hoidon päättymisen jälkeen. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä BRUKINSA-hoidon aikana ja vähintään 1 kuukauden ajan hoidon päättymisen jälkeen. Toistaiseksi ei tiedetä, heikentääkö tsanubrutinibi hormonaalisten ehkäisyvalmisteiden tehoa. Näin ollen hormonaalista ehkäisyä käyttävien naisten on käytettävä lisäksi jotakin estemenetelmää. Raskaustestiä suositellaan ennen hoidon aloittamista naisille, jotka voivat tulla raskaaksi.
Raskaus
BRUKINSA-valmistetta ei pidä käyttää raskauden aikana. Ei ole olemassa tietoja tsanubrutinibin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Imetys
Ei tiedetä, erittyvätkö tsanubrutinibi tai sen metaboliitit ihmisen rintamaitoon. Ei-kliinisiä tutkimuksia ei ole tehty. Imeväiseen kohdistuvia riskejä ei voida poissulkea. Rintaruokinta on lopetettava BRUKINSA-hoidon ajaksi.
Hedelmällisyys
Rotilla ei havaittu vaikutusta urosten tai naaraiden hedelmällisyyteen, mutta annoksella 300 mg/kg/vrk havaittiin morfologisia poikkeavuuksia spermassa ja implantaation jälkeisten keskenmenojen lisääntymistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
BRUKINSA-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Joillakin BRUKINSA-hoitoa saaneilla potilailla on raportoitu uupumusta, heitehuimausta ja asteniaa. Tämä on otettava huomioon potilaan ajokykyä ja koneiden käyttökykyä arvioitaessa.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Tsanubrutinibi-monoterapia
Yleisimmin esiintyneitä tsanubrutinibi-monoterapian haittavaikutuksia (≥ 20 %) olivat ylähengitystieinfektio§ (36 %), mustelmat§ (32 %), hemorragia/hematooma§ (30 %), neutropenia§ (30 %), tuki- ja liikuntaelimistön kipu§ (27 %), ihottuma§ (25 %), keuhkokuume§ (24 %), ripuli§ (21 %) ja yskä§ (21 %) (taulukko 3).
Yleisimmät asteen 3 tai sitä vakavammat tsanubrutinibi-monoterapian haittavaikutukset (> 3 %) olivat neutropenia§ (21 %), keuhkokuume§ (14 %), korkea verenpaine§ (8 %), trombosytopenia§ (6 %), anemia (6 %) ja hemorragia/hematooma§ (4 %).
1 550 potilaasta, joita hoidettiin tsanubrutinibilla, 4,8 % keskeytti hoidon haittavaikutuksista johtuen. Yleisimmin esiintynyt hoidon keskeyttämiseen johtanut haittavaikutus oli keuhkokuume (2,6 %). Annoksen pienentämiseen johtavia haittavaikutuksia esiintyi 5,0 %:lla potilaista. Yleisin tsanubrutinibiannoksen pienentämiseen johtanut haittavaikutus oli neutropenia§ (0,9 %), ripuli (0,7 %) ja keuhkokuume§ (0,6 %).
Tsanubrutinibi-obinututsumabi-yhdistelmähoito
Yleisimmin esiintyneitä tsanubrutinibi-obinututsumabi-yhdistelmähoidon haittavaikutuksia (≥ 20%) olivat trombosytopenia§ (37 %), neutropenia§ (31 %) ja väsymys§ (27 %) (taulukko 4).
Yleisimmät asteen 3 tai sitä vakavammat tsanubrutinibi-obinututsumabi-yhdistelmähoidon haittavaikutukset (> 3%) olivat neutropenia§ (25 %), trombosytopenia§ (16 %), keuhkokuume§ (15 %) ja anemia (5 %).
143 potilaasta, jotka saivat tsanubrutinibi-obinututsumabi-yhdistelmähoitoa, 4,9 % keskeytti hoidon haittavaikutuksista johtuen. Yleisimmin esiintynyt hoidon keskeyttämiseen johtanut haittavaikutus oli keuhkokuume (4,2 %). Annoksen pienentämiseen johtavia haittavaikutuksia esiintyi 7,7 %:lla potilaista. Yleisin tsanubrutinibiannoksen pienentämiseen johtanut haittavaikutus oli trombosytopenia§ (4,9 %) ja neutropenia (1,4 %).
Verihiutaleiden määrän alenemista† (laboratorioarvojen perusteella) havaittiin 65 %:lla (kaikki asteet) ja 12 %:lla (aste 3 tai 4) potilaista, joille annettiin tsanubrutinibi-obinututsumabi-yhdistelmähoitoa, verrattuna 43 %:iin (kaikki asteet) ja 11 %:iin (aste 3 tai 4) potilaista, joille annettiin vain obinututsumabia. Kaikilla asteilla ja asteella 3 tai 4 verihiutaleiden määrän alenemista raportoitiin 39 %:lla ja 7,8 %:lla potilaista, joille annettiin tsanubrutinibi-monoterapiaa.
Taulukoitu luettelo haittavaikutuksista
Turvallisuusprofiili perustuu yhdistettyihin tietoihin 1 550:stä kliinisissä tutkimuksissa BRUKINSA-hoitoa saaneesta potilaasta, joilla oli B-solumaligniteetteja, mukaan lukien potilaat, joilla oli krooninen lymfosyyttinen leukemia (N = 938), Waldenströmin makroglobulinemia (N = 249), manttelisolulymfooma (N = 140), marginaalivyöhykkeen lymfooma (N = 93), follikulaarinen lymfooma (N = 59) ja muun tyyppisiä B-solumaligniteetteja (N = 71). Potilaiden altistusajan mediaani oli 34,41 kuukautta.
Tsanubrutinibi-obinututsumabi-yhdistelmähoidon turvallisuusprofiili perustuu ROSEWOOD-tutkimuksen tietoihin 143 potilaalta, joilla on follikulaarinen lymfooma ja joita hoidettiin BRUKINSA:n ja obinututsumabin yhdistelmällä. Potilaiden altistusajan mediaani oli 12,35 kuukautta.
BRUKINSA-hoitoa monoterapiana tai yhdistelmähoitona obinututsumabin kanssa B-solumaligniteettien hoitoon saaneilla potilailla esiintyneet haittavaikutukset on lueteltu taulukossa 3 ja taulukossa 4 elinjärjestelmäluokan ja esiintymistiheyden perusteella ryhmiteltyinä. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3: Kliinisissä tutkimuksissa raportoidut tsanubrutinibimonoterapian haittavaikutukset potilailla, joilla oli B-solumaligniteetteja (n = 1 550)
MedDRA elinjärjestelmäluokka | MedDRA termit | Kaikki asteet*(%) | Aste 3 tai vakavampi (%) |
Infektiot | Ylähengitystieinfektio§ | Hyvin yleinen (36) | 2 |
Keuhkokuume§ # | Hyvin yleinen (24) | 14 | |
Keuhkokuume | Hyvin yleinen (15) | 8 | |
Alahengitystieinfektio | Yleinen (5) | < 1 | |
Virtsatieinfektio | Hyvin yleinen (14) | 2 | |
Bronkiitti | Yleinen (4) | < 1 | |
Hepatiitti B:n uudelleenaktivoituminen | Melko harvinainen (< 1) | < 1 | |
Veri ja imukudos | Neutropenia§ | Hyvin yleinen (30) | 21 |
Febriili neutropenia | Yleinen (2) | 2 | |
Trombosytopenia§ | Hyvin yleinen (18) | 6 | |
Anemia§ | Hyvin yleinen (16) | 6 | |
Aineenvaihdunta ja ravitsemus | Tuumorilyysioireyhtymä§# | Melko harvinainen (< 1) | < 1 |
Hermosto | Huimaus§ | Hyvin yleinen (12) | < 1 |
Sydän | Eteisvärinä ja -lepatus | Yleinen (5) | 2 |
Verisuonisto | Mustelmat§ | Hyvin yleinen (32) | < 1 |
Kontuusio | Hyvin yleinen (20) | 0 | |
Petekiat | Yleinen (7) | < 1 | |
Purppura | Yleinen (5) | < 1 | |
Ekkymoosi | Yleinen (3) | < 1 | |
Hemorragia/Hematooma§ # | Hyvin yleinen (30) | 3 | |
Hematuria | Hyvin yleinen (11) | < 1 | |
Nenäverenvuoto | Yleinen (8) | < 1 | |
Suoliston verenvuoto | Melko harvinainen (< 1) | < 1 | |
Korkea verenpaine§ | Hyvin yleinen (17) | 8 | |
Hengityselimet, rintakehä ja välikarsina | Yskä§ | Hyvin yleinen (21) | < 1 |
Ruoansulatuskanava | Ripuli | Hyvin yleinen (21) | 2 |
Ummetus | Hyvin yleinen (14) | < 1 | |
Iho ja ihonalainen kudos | Ihottuma§ | Hyvin yleinen (25) | < 1 |
Kutina | Yleinen (8) | < 1 | |
Yleistynyt kesivä ihottuma | Tuntematon | Tuntematon | |
Luusto, lihakset ja sidekudos | Tuki- ja liikuntaelimistön kipu§ | Hyvin yleinen (27) | 2 |
Nivelkipu | Hyvin yleinen (15) | < 1 | |
Selkäkipu | Hyvin yleinen (12) | < 1 | |
Yleisoireet ja antopaikassa todettavat haitat | Väsymys§ | Hyvin yleinen (18) | 1 |
Väsymys | Hyvin yleinen (14) | 1 | |
Heikkous | Yleinen (4) | < 1 | |
Perifeerinen edeema | Yleinen (9) | < 1 | |
Tutkimukset† | Neutrofiilien määrän lasku†± | Hyvin yleinen (52) | 22 |
Trombosyyttien lasku†± | Hyvin yleinen (39) | 8 | |
Hemoglobiinin lasku†± | Hyvin yleinen (26) | 4 |
* Asteet arvioitiin National Cancer Instituten haittavaikutusten Common Terminology Criteria for Adverse Events (NCI-CTCAE)-terminologiakriteerien version 4.03 perusteella.
† Laboratoriomittausten perusteella
± Prosenttiosuudet perustuvat niiden potilaiden määrään, joilta on saatavilla arvio lähtötasolla sekä vähintään yksi arvio lähtötason jälkeen.
§ Sisältää useita haittavaikutuksia koskevat termit
# Sisältää kuolemaan johtaneet tapahtumat.
Taulukko 4: Kliinisessä tutkimuksessa BGB‑3111‑212 raportoidut tsanubrutinibi-obinututsumabi-yhdistelmähoidon haittavaikutukset potilailla, joilla oli follikulaarinen lymfooma (n = 143)
MedDRA elinjärjestelmäluokka | MedDRA-termit | Kaikki asteet*(%) | Aste 3 tai vakavampi (%) |
Infektiot | Ylähengitystieinfektio§ | Hyvin yleinen (14) | < 1 |
Keuhkokuume§# | Hyvin yleinen (20) | 15 | |
Keuhkokuume | Hyvin yleinen (13) | 11 | |
Alahengitystieinfektio | Yleinen (4) | < 1 | |
Virtsatieinfektio§ | Yleinen (10) | 2 | |
Bronkiitti | Yleinen (2) | 0 | |
Veri ja imukudos | Trombosytopenia§ | Hyvin yleinen (37) | 16 |
Neutropenia§ | Hyvin yleinen (31) | 25 | |
Anemia§ | Hyvin yleinen (12) | 5 | |
Hermosto | Huimaus§ | Yleinen (4) | 0 |
Sydän | Eteisvärinä ja -lepatus§ | Yleinen (3) | 1 |
Verisuonisto | Hemorragia/Hematooma§# | Hyvin yleinen (16) | < 1 |
Nenäverenvuoto | Yleinen (5) | 0 | |
Hematuria | Yleinen (< 1) | 0 | |
Mustelmat§ | Hyvin yleinen (15) | 0 | |
Kontuusio | Hyvin yleinen (8) | 0 | |
Petekiat | Yleinen (6) | 0 | |
Purppura | Yleinen (2) | 0 | |
Ekkymoosi | Yleinen (1) | 0 | |
Korkea verenpaine§ | Yleinen (4) | < 1 | |
Hengityselimet, rintakehä ja välikarsina | Yskä§ | Hyvin yleinen (13) | 0 |
Ruoansulatuselimistö | Ripuli | Hyvin yleinen (19) | 3 |
Ummetus | Hyvin yleinen (13) | 0 | |
Iho ja ihonalainen kudos | Ihottuma§ | Hyvin yleinen (10) | 0 |
Kutina | Yleinen (7) | 0 | |
Yleistynyt kesivä ihottuma | Tuntematon | Tuntematon | |
Luusto, lihakset ja sidekudos | Tuki- ja liikuntaelimistön kipu§ | Hyvin yleinen (18) | 2 |
Selkäkipu | Hyvin yleinen (11) | < 1 | |
Nivelkipu | Yleinen (4) | 0 | |
Yleisoireet ja antopaikassa todettavat haitat | Väsymys§ | Hyvin yleinen (27) | 1 |
Väsymys | Hyvin yleinen (15) | 0 | |
Heikkous | Yleinen (12) | < 1 | |
Perifeerinen edeema | Yleinen (2) | 0 | |
Tutkimukset†± | Trombosyyttien lasku†± | Hyvin yleinen (65) | 12 |
Neutrofiilien määrän lasku†± | Hyvin yleinen (48) | 18 | |
Hemoglobiinin lasku†± | Hyvin yleinen (31) | < 1 |
* Haittavaikutusten asteet määritettiin National Cancer Instituten Common Terminology Criteria for Adverse Events (NCI-CTCAE) -terminologiakriteerien version 5.0 perusteella.
† Laboratoriomittausten perusteella
§ Sisältää useita haittavaikutuksia koskevat termit
# Sisältää kuolemaan johtaneet tapahtumat.
± Prosenttiosuudet perustuvat niiden potilaiden määrään, joilta on saatavilla arvio lähtötasolla sekä vähintään yksi arvio lähtötason jälkeen.
Muut erityiset potilasryhmät
Iäkkäät potilaat
BRUKINSA-monoterapiaa saaneista 1 550:stä potilaasta 61,3 % oli vähintään 65-vuotiaita. Asteen 3 tai sitä vakavampien haittavaikutusten esiintymistiheys oli hieman suurempi iäkkäillä potilailla, jotka saivat tsanubrutinibihoitoa (69,6 %:lla ≥ 65-vuotiaista potilaista ja 62,7 %:lla alle 65-vuotiaista potilaista). ≥ 65-vuotiaiden potilaiden ja nuorempien potilaiden välillä ei havaittu kliinisesti merkittäviä eroja turvallisuudessa.
BRUKINSA:n ja obinututsumabin yhdistelmähoitoa saaneista 143 potilaasta 42,0 % oli vähintään 65-vuotiaita. Asteen 3 tai sitä vakavampien haittavaikutusten esiintymistiheys oli hieman suurempi iäkkäillä potilailla, jotka saivat tsanubrutinibi-obinututsumabi-yhdistelmähoitoa (70,0 %:lla ≥ 65-vuotiaista potilaista ja 62,7 %:lla alle 65-vuotiaista potilaista). ≥ 65-vuotiaiden potilaiden ja nuorempien potilaiden välillä ei havaittu kliinisesti merkittäviä eroja turvallisuudessa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
BRUKINSA-valmisteelle ei ole olemassa spesifistä vastalääkettä. Jos potilas saa yliannoksen, hänen vointiaan on seurattava tarkoin ja hänelle on annettava asianmukaista tukihoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset aineet, Brutonin tyrosiinikinaasin estäjät, ATC-koodi: L01EL03.
Vaikutusmekanismi
Tsanubrutinibi on Brutonin tyrosiinikinaasin (BTK) estäjä. Tsanubrutinibi muodostaa kovalenttisen sidoksen kysteiinijäännöksen kanssa BTK:n aktiivisessa kohdassa, mikä johtaa BTK:n aktiivisuuden estämiseen. BTK on B-soluantigeenireseptori (BCR)- ja sytokiinireseptorireittien signalointimolekyyli. B-soluissa BTK-signalointi johtaa B-solujen proliferaatioon, liikenteeseen, kemotaksikseen ja adheesioon tarvittavien reittien aktivoitumiseen.
Farmakodynaamiset vaikutukset
BTK-sitoutumisaste perifeerisen veren mononukleaarisoluissa (PBMC) ja imusolmukebiopsioissa
Vakaan tilan BTK-sitoutumisasteen mediaani perifeerisen veren mononukleaarisoluissa säilyi 100 %:n tasolla 24 tunnin aikana 320 mg:n kokonaisvuorokausiannoksella potilailla, joilla oli B-solumaligniteetteja. Vakaan tilan BTK-sitoutumisasteen mediaani imusolmukkeissa oli 94–100 % suositellun annoksen jälkeen.
Vaikutus QT/QTc-aikaan ja sydämen elektrofysiologiaan
Suositelluilla annoksilla (320 mg kerran vuorokaudessa tai 160 mg kahdesti vuorokaudessa) ei havaittu kliinisesti merkittäviä vaikutuksia QTc-aikaan. Kerta-annoksella, joka oli 1,5 kertaa suositeltu enimmäisannos (480 mg), tsanubrutinibi ei pidentänyt QT-aikaa kliinisesti merkittävässä määrin (≥ 10 ms).
Kliininen teho ja turvallisuus
Potilaat, joilla on Waldenströmin makroglobulinemia (WM)
BRUKINSA-valmisteen turvallisuutta ja tehoa Waldenströmin makroglobulinemiassa arvioitiin satunnaistetussa, avoimessa monikeskustutkimuksessa, jossa verrattiin tsanubrutinibia ja ibrutinibia (ASPEN-tutkimus, BGB‑3111‑302) potilailla, jotka eivät olleet aiemmin saanut BTK:n estäjähoitoa. Tutkimukseen mukaan soveltuvat potilaat olivat vähintään 18-vuotiaita ja heillä oli kliininen ja varma relapsoineen/refraktorisen Waldenströmin makroglobulinemian diagnoosi, tai he eivät olleet saaneet aiemmin hoitoa siinä kohtaa, kun heidän hoitava lääkärinsä katsoi, ettei tavanomaisen kemoimmunoterapian käyttö sovellu potilaalle. Potilaiden oli täytettävä vähintään yksi Seventh International Workshop on Waldenström’s Macroglobulinemia (IWWM) -konsensuspaneelin hoitokriteeri, ja heillä oli mitattavissa oleva sairaus, jonka määritelmänä oli seerumin IgM-pitoisuus > 0,5 g/dl. Potilaat, joilla oli MYD88-mutaatio (MYD88MUT) määritettiin kohorttiin 1 (N = 201), ja heidät satunnaistettiin suhteessa 1:1 saamaan joko tsanubrutinibia 160 mg kahdesti vuorokaudessa (haara A) tai ibrutinibia 420 mg kerran vuorokaudessa (haara B) taudin etenemiseen tai kohtuuttomaan toksisuuteen asti. Tutkittavat, joilla todettiin olevan MYD88-villityyppi (MYD88WT) geenisekvenssoinnin perusteella (sen arvioitiin esiintyvän noin 10 %:lla tutkimukseen osallistuneista tutkittavista potilaista), otettiin mukaan kohorttiin 2 (N = 28), ja he saivat tsanubrutinibia 160 mg kahdesti vuorokaudessa kolmannessa, ei- satunnaistetussa tutkimushaarassa (haara C).
Kohortissa 1 (MYD88MUT) mediaani-ikä oli 70 vuotta (vaihteluväli 38–90 vuotta). 71 % ibrutinibilla hoidetuista potilaista ja 60 % tsanubrutinibilla hoidetuista potilaista oli yli 65-vuotiaita. 33 % tsanubrutinibihaarassa olleista potilaista ja 22 % ibrutinibihaarassa olleista potilaista oli yli 75-vuotiaita. 67 % oli miehiä ja 91 % valkoihoisia. Tutkimuksen alussa 44 %:lla ibrutinibihaarassa olleista potilaista ja 46 %:lla tsanubrutinibihaarassa olleista potilailla oli korkeita International Prognostic Scoring System (IPSS)-pistemääriä. 164 potilaalla oli relapsoinut tai refraktorinen tauti; aiempien hoitojen mediaanimäärä oli 1 (vaihteluväli 1–8).
Ensisijainen tulosmittari oli täydellisen vasteen (CR) tai erittäin hyvän osittaisen vasteen (VGPR) määrä, jonka riippumaton arviointitoimikunta (IRC) arvioi mukautetuilla kuudennessa IWWM -konsensuspaneelissa päivitetyillä vastekriteereillä. Kohortin 1 toissijaisia päätetapahtumia ovat merkittävä vaste (MRR), vasteen kesto, tutkijan määrittämä täydellinen vaste tai erittäin hyvä osittainen vaste ja etenemisvapaa elossaolo (PFS).
Erittäin hyvän osittaisen vasteen tai täydellisen vasteen määrän ensisijaisen päätetapahtuman osalta paremmuuden testaaminen edellytti relapsoituneen/refraktorisen taudin analyysijoukon testaamista ennen hoitoaikeen mukaisen (ITT) analyysijoukon testaamista. Seurannan mediaaniaika oli 19,4 kuukautta. Relapsoituneen/refraktorisen taudin potilaista 19,8 % saavutti erittäin hyvän osittaisen vasteen tai täydellisen vasteen ibrutinibihaarassa ja tsanubrutinibihaarassa 28,9 %. Ensisijaisen tehoa mittaavan päätetapahtuman tulos ei ollut merkitsevä relapsoituneen/refraktorisen taudin analyysijoukossa (kaksipuolinen p-arvo = 0,1160). Taulukossa 5 on esitetty yhteenveto riippumattoman arviointitoimikunnan arvioimista vasteista relapsoituneen/refraktorisen taudin analyysijoukon ja hoitoaikeen mukaisen analyysijoukon osalta. Vasteita havaittiin kaikissa tsanubrutinibia käyttäneissä alaryhmissä, myös MYD88WT -potilailla (kohortti 2), joilla erittäin hyvän osittaisen vasteen tai täydellisen vasteen määrä oli 26,9 % ja merkittävän vasteen määrä 50 %.
Taulukko 5: Ensisijainen analyysi riippumattoman arviointitoimikunnan määrittämästä taudin vasteesta (ASPEN-tutkimus)
Vasteen luokka | Relapsoituneen/refraktorisen taudin analyysijoukko | Hoitoaikeen mukainen analyysijoukko | ||
Ibrutinibi N = 81 | Tsanubrutinibi N = 83 | Ibrutinibi N = 99 | Tsanubrutinibi N = 102 | |
Seuranta-ajan mediaani, kuukautta (vaihteluväli) | 18,79 (0,5; 30,0) | 18,73 (0,4; 28,7) | 19,38 (0,5; 31,1) | 19,47 (0,4; 31,2) |
Täydellinen vaste | 0 (0,0) | 0 (0,0) | 0 (0,0) | 0 (0,0) |
Erittäin hyvä osittainen vaste | 16 (19,8) | 24 (28,9) | 19 (19,2) | 29 (28,4) |
Osittainen vaste | 49 (60,5) | 41 (49,4) | 58 (58,6) | 50 (49,0) |
Erittäin hyvän osittaisen vasteen tai täydellisen vasteen määrä, n (%) | 16 (19,8) | 24 (28,9) | 19 (19,2) | 29 (28,4) |
95 %:n luottamusväli a | (11,7; 30,1) | (19,5; 39,9) | (12,0; 28,3) | (19,9; 38,2) |
Riskiero (%) b | 10,7 | 10,2 | ||
95 %:n luottamusväli a | (-2,5; 23,9) | (-1,5; 22,0) | ||
p-arvo c | 0,1160 |
| ||
Merkittävä vaste (osittainen vaste tai parempi), n (%) | 65 (80,2) | 65 (78,3) | 77 (77,8) | 79 (77,5) |
95 %:n luottamusväli a | (69,9; 88,3) | (67,9; 86,6) | (68,3;85,5) | (68,1; 85,1) |
Riskiero (%) b | -3,5 | -0,5 | ||
95 %:n luottamusväli | (-16,0; 9,0) | (-12,2; 11,1) | ||
Merkittävän vasteen kesto |
|
|
|
|
Tapahtumavapaa osuus 18 kuukauden kohdalla d, % (95 %:n luottamusväli) | 85,6 (73,1; 92,6) | 87,0; (72,5, 94,1) | 87,9 (77,0; 93,8) | 85,2 (71,7, 92,6) |
Prosenttiosuudet perustuvat N:ään.
a 2-puolinen Clopper-Pearsonin 95 %:n luottamusväli.
b Mantel-Haenszelin yleinen riskiero ja 95 %:n luottamusväli laskettiin käyttäen normaaliapproksimaatiota ja Saton keskivirhettä, joka on ositettu ositetekijöiden osalta IRT:n (ositteet CXCR4 WT ja UNK on yhdistetty) ja ikäryhmän (≤ 65 ja > 65) mukaisesti. Ibrutinibi on vertailuryhmä.
c Perustuu CMH-testiin, joka on ositettu ositetekijöiden osalta IRT:n (ositteet CXCR4 WT ja UNK on yhdistetty) ja ikäryhmän (≤ 65 ja > 65) mukaisesti.
d Tapahtumavapaat osuudet on arvioitu Kaplan-Meierin menetelmällä siten, että 95 %:n luottamusvälit on arvioitu Greenwoodin kaavan avulla.
Päivitettyjen tietojen katkaisuajankohtaan mennessä kertyneiden tietojen pohjalta tutkijan arvioinnin mukaisesti määritetty etenemisvapaan elossaolon tapahtumavapaa osuus oli 30 kuukauden kohdalla 77,6 % ibrutinibia saaneilla ja 84,9 % tsanubrutinibia saaneilla. Arvioitu yleinen riskitiheyksien suhde oli 0,734 (95 %:n luottamusväli: 0,380; 1,415).
Potilaat, joilla on marginaalivyöhykkeen lymfooma (MZL)
Tsanubrutinibin tehoa arvioitiin vaiheen 2 avoimessa yhden haaran monikeskustutkimuksessa, jossa oli 68 MZL-potilasta, jotka olivat saaneet vähintään yhtä aiempaa CD20-vasta-aineisiin perustuvaa hoitoa (MAGNOLIA-tutkimus, BGB‑3111‑214). 26 potilaalla (38,2 %) potilaalla oli ekstranodaalinen MZL, 26:lla (38,2 %) nodaalinen MZL, 12:lla (17,6 %) pernaan liittyvä MZL ja 4 potilaalla (6 %) alatyyppi oli tuntematon. Tsanubrutinibia annettiin 160 mg kahdesti vuorokaudessa, kunnes tauti eteni tai ilmaantui ei-hyväksyttävissä olevaa toksisuutta. Potilaiden mediaani-ikä oli 70 vuotta (vaihteluväli 37–95) ja heistä 53 % oli miehiä. Alkuperäisestä diagnoosista kulunut mediaaniaika oli 61,5 kuukautta (vaihteluväli 2,0–353,6). Aiempien hoitojen mediaanimäärä oli 2 (vaihteluväli 1–6). 27,9 %:lla potilaista oli 3 tai useampi systeeminen hoitolinja; 98,5 % (n=67) potilaista oli saanut aikaisemmin rituksimabipohjaista solunsalpaajahoitoa ja 85,3 % (n=58) oli saanut aikaisemmin hoitoa alkyloivilla aineilla; 5,9 % potilaista (n=4) oli saanut aikaisemmin kantasolusiirron. Kuudellakymmennelläkolmella (92,6 %) potilaalla lähtötilanteen ECOG-status oli 0 tai 1. Kahdellakymmenelläkahdella (32,4 %) potilaalla oli refraktorinen tauti tutkimuksen alussa.
Hoitovaste oli vuoden 2014 Lugano-kriteerien mukainen, ja ensisijainen päätetapahtuma oli kokonaisvasteosuus, jonka riippumaton arviointitoimikunta (IRC) arvioi (taulukko 6).
Taulukko 6: Riippumattoman arviointitoimikunnan määrittämät tehokkuustulokset potilailla, joilla on MZL (MAGNOLIA-tutkimus)
| Tutkimus BGB‑3111‑214 (N = 66)a |
ORR (95 % CI) | 68 % (55,6; 79,1) |
CR | 26 % |
PR | 42 % |
Mediaani-DoR kuukausina (95 % CI) | NE (25,0, NE) |
DOR tapahtumavapaa osuus b 24 kuukauden kohdalla, % (95% CI) | 72,9 (54,4, 84,9) |
Tutkimuksen mediaaniseuranta-aika kuukausina (Min., Maks.) | 28,04 (1,64, 32,89) |
a Tehoa ei voitu arvioida kahdella potilaalla tutkimuksessa BGB‑3111‑214 johtuen MZL:n muuntumisesta diffuusiksi suurisoluisesti B-solulymfoomaksi, mikä varmistui näytteiden keskitetyssä arvioinnissa.
b Tapahtumavapaat osuudet arvioitiin Kaplan-Meierin menetelmällä, 95 % luottamusvälit arvioitiin käyttämällä Greenwoodin kaavaa.
ORR: kokonaisvasteosuus, CR: täydellinen vaste, PR: osittainen vaste, DoR: vasteen kesto, CI: luottamusväli, NE: ei arvioitavissa
Tutkimuksessa BGB‑3111‑214 mediaaniaika vasteen saavuttamiseen oli 2,79 kuukautta (vaihteluväli 1,7–11,1 kuukautta). Kun tutkimuksen seuranta-ajan mediaani oli 28,04 kuukautta (vaihteluväli 1,64–32,89 kuukautta), vasteen keston (DOR) mediaania IRC:n arvioimana ei ollut saavutettu (95 % CI 25,0 kuukautta – NE) ja yhteensä 72,9 %:n (95 % CI 54,4–84,9) vasteen saaneista arvioitiin olevan tapahtumavapaa 24 kuukauden kohdalla ensimmäisen vasteen jälkeen.
Kokonaisvasteosuudet olivat samanlaiset eri MZL-alatyyppien kohdalla (ekstranodaalinen, nodaalinen, pernaan liittyvä).
Potilaat, joilla on krooninen lymfosyyttinen leukemia (KLL)
BRUKINSA-valmisteen tehoa KLL-potilailla arvioitiin kahdessa satunnaistetussa ja kontrolloidussa tutkimuksessa.
SEQUOIA-tutkimus (BGB‑3111‑304): Kansainvälinen vaiheen 3 avoin, satunnaistettu tutkimus tsanubrutinibista verrattuna bendamustiinin ja rituksimabin yhdistelmään verrattuna potilailla, joilla on aiemmin hoitamaton KLL
SEQUOIA-tutkimus (BGB‑3111‑304) on avoin, satunnaistettu, aktiivikontrolloitu vaiheen 3 monikeskustutkimus tsanubrutinibimonoterapiasta verrattuna bendamustiinin ja rituksimabin yhdistelmään 479 potilaalla, joilla on aiemmin hoitamaton KLL ilman 17p-deleetiota (del(17p)) (haarat A ja B; kohortti 1). Tutkimuksen haara C (kohortti 2) on yhden hoitohaaran monikeskustutkimus tsanubrutinibimonoterapiasta 110 potilaalla, joilla on aiemmin hoitamaton KLL ja keskitetysti vahvistettu del(17p).
Molempiin kohortteihin otettiin vähintään 65-vuotiaita potilaita sekä 18–65-vuotiaita potilaita, joille fludarabiinilla, syklofosfamidilla ja rituksimabilla (FCR) toteutettava kemoimmunoterapia ei ollut soveltunut.
Demografiset ja lähtötilanteen ominaisuudet olivat yleisesti ottaen tasapainossa kohortin 1 haaran A (tsanubrutinibi) ja haaran B (bendamustiini ja rituksimabi) välillä. Molemmissa haaroissa mediaani-ikä oli 70,0 vuotta. Vähintään 75-vuotiaita oli hieman suurempi osuus haarassa A (26,1 %) kuin haarassa B (22,3 %) ja 65–75-vuotiaita oli hieman suurempi osuus haarassa A (55,2 %) kuin haarassa B (58,4 %). Kohortissa 1 92,7 %:lla potilaista lähtötilanteen ECOG-toimintakykyluokka oli 0 tai 1 (93,7 %:lla haarassa A ja 91,6 %:lla haarassa B). Kohortissa 2 (haaran C tsanubrutinibi) 87,3 %:lla potilaista lähtötilanteen ECOG-toimintakykyluokka oli 0 tai 1.
Demografiset ja lähtötilanteen ominaisuudet olivat myös yleisesti ottaen samanlaisia kohortin 1 haarassa A (tsanubrutinibi) ja kohortin 2 haarassa C (tsanubrutini).
Kohortissa 1 satunnaistaminen ositettiin iän (alle 65 vuotta tai vähintään 65 vuotta), Binet’n vaiheen (C, A tai B), immunoglobuliinin vaihtelevan osan raskasketjun (IGHV) mutaatiostatuksen (mutatoitunut tai ei-mutatoitunut) ja maantieteellisen alueen (Pohjois-Amerikka, Eurooppa tai Aasia-Tyynimeri) suhteen. Yhteensä 479 potilasta satunnaistettiin (hoitoaikeen mukainen [ITT] analyysijoukko), 241 saamaan tsanubrutinibia jatkuvana monoterapiana ja 238 saamaan kuuden hoitojakson verran bendamustiinia ja rituksimabia (BR-hoitoa).
Kohortissa 1 tsanubrutinibihaarassa A olevat potilaat saivat 160 mg kahdesti vuorokaudessa, kunnes tauti eteneni tai ilmaantui ei-hyväksyttävissä olevaa toksisuutta. Haarassa B olevat potilaat saivat bendamustiinia annoksella 90 mg/m2/vrk kunkin hoitojakson ensimmäisten kahden vuorokauden ajan ja rituksimabia annoksella 375 mg/m2 hoitojakson 1 ajan sekä annoksella 500 mg/m2 hoitojaksojen 2–6 ajan. Kukin hoitojakso oli noin 28 vuorokauden pituinen. Kohortissa 2 (haara C) potilaat saivat tsanubrutinibia 160 mg kahdesti vuorokaudessa kunnes tauti eteni tai ilmaantui ei-hyväksyttävissä olevaa toksisuutta.
Kohortissa 1 ensisijaisena päätetapahtumana oli etenemisvapaa elinaika (PFS), jonka arvioi riippumaton keskitetty arviointitoimikunta (IRC). Toissijaisiin päätetapahtumiin sisältyi mm. IRC-toimikunnan arvioinnin perusteella määritetty kokonaisvasteen osuus.
Kohortissa 1 etenemisvapaan elinajan seurannan mediaanikesto oli 25,0 kuukautta (vaihteluväli 0,0–41,4). Etenemisvapaan elinajan suhteellinen osuus (PFS osuus) 24 kuukauden kohdalla oli 85,5 % (95 %:n luottamusväli: 80,1; 89,6) tsanubrutinibia saaneilla ja 69,5 % (95 %:n luottamusväli: 62,4; 75,5) BR-hoitoa saaneilla. Kohortissa 2 etenemisvapaan elinajan seurannan mediaanikesto oli 27,9 kuukautta (vaihteluväli: 1,0–38,8) ja etenemisvapaan elinajan suhteellinen osuus 24 kuukauden kohdalla oli 88,9 % (95 %:n luottamusväli: 81,3; 93,6). IRC-toimikunnan arvioima kokonaisvasteosuus (ORR) kohortissa 2 oli 90,0 % (95 %:n luottamusväli: 82,8; 94,9). IRC-toimikunnan arvioon perustuva mediaaniaika vähintään osittaiseen vasteeseen oli 2,89 kuukautta (vaihteluväli 1,8; 14,2) kohortin 1 tsanubrutinibihaarassa ja 2,86 kuukautta (vaihteluväli: 1,9; 13,9) kohortin 2 tsanubrutinibihaarassa.
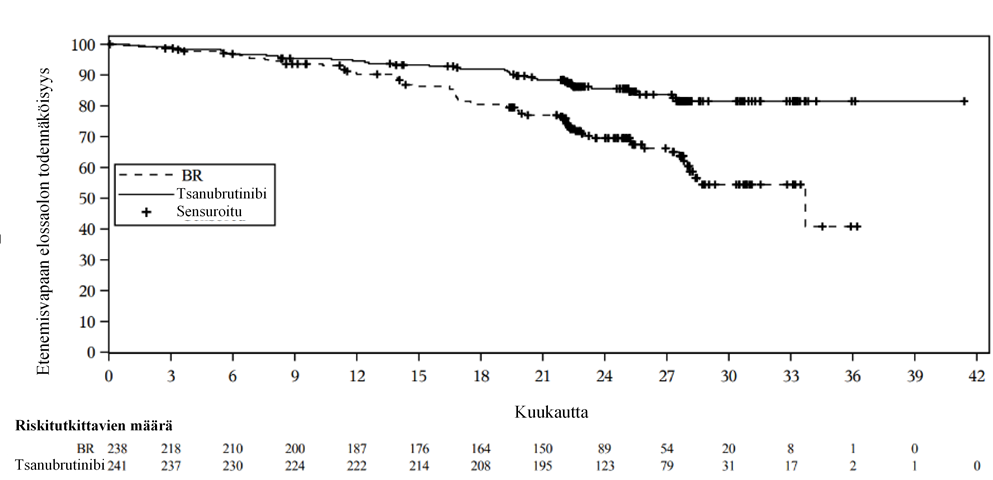
Kohortin 1 tehoa koskevat tulokset on esitetty taulukossa 7. Kohortin 1 molempien haarojen etenemisvapaan elinajan Kaplan-Meier-kuvaajat on esitetty kuvassa 1.
Taulukko 7: Tehoa koskevat tulokset SEQUOIA-tutkimuksesta
| Kohortti 1* Potilaat, joilla ei ollut Del(17p) -deleetiota | |
Päätetapahtuma | Tsanubrutinibi (N = 241) | Bendamustiini + rituksimabi (N = 238) |
Etenemisvapaa elinaika† |
|
|
Tapahtumien määrä, n (%) | 36 (14,9) | 71 (29,8) |
Taudin eteneminen, n (%) | 27 (11,2) | 59 (24,8) |
Kuolema, n (%) | 9 (3,7) | 12 (5,0) |
Mediaani (95 %:n luottamusväli), kuukautta a | NE (NE, NE) | 33,7 (28,1; NE) |
Riskitiheyksien suhde (95 %:n luottamusväli) b | 0,42 (0,28; 0,63) | |
P-arvo c | < 0,0001 | |
Kokonaisvasteosuus † % (95 %:n luottamusväli) | 94,6 % (91,0; 97,1) | 85,3 % (80,1; 89,5) |
Vasteen saaneiden kokonaisosuus: CR+CRi+nPR+PR+PR-L, CR: täydellinen vaste, CRi: täydellinen vaste ja vaillinainen hematopoieettinen toipuminen, nPR: nodulaarinen osittainen vaste, PR: osittainen vaste, PR-L: osittainen vaste lymfosytoomaan, CI: luottamusväli, NE: ei arvioitavissa, etenemisvapaan elinajan seurannan mediaanikesto oli 25,0 kuukautta (95 %:n luottamusväli: 24,6; 25,2).
* ITT-analyysijoukko
† Riippumattoman keskitetyn arviointitoimikunnan arvioimana.
a Perustuu Kaplan-Meier-estimointiin.
b Perustuu ositettuun Coxin regressiomalliin, jossa bendamustiini + rituksimabi oli vertailuryhmänä.
c Perustuu ositettuun log-rank-testiin.
Päivitetyssä ad hoc -analyysissä, jossa etenemisvapaan elinajan (PFS) seuranta-ajan mediaanikesto oli 33,5 kuukautta, tutkijan arvioima PFS pysyi samana kuin ensisijaisessa analyysissä, jossa HR oli 0,33 (95 %:n luottamusväli: 0,22–0,48, kuvaileva P <0,0001) tsanubrutinibihaarassa bendamustiini + rituksimabi -haaraan nähden. PFS:n mediaania ei saavutettu tsanubrutinibihaarassa, ja bendamustiini + rituksimabi -haarassa se oli 39,2 kuukautta. 36 kuukauden kuluttua satunnaistamisesta 83,6 % tsanubrutinibihoitoa saaneista potilaista ja 55,1 % bendamustiini + rituksimabi -hoitoa saaneista potilaista arvioitiin olevan etenemisvapaita ja elossa. Seuranta-ajan mediaanikeston ollessa 35,8 kuukautta kokonaiselossaolon (OS) mediaania ei saavutettu kummassakaan haarassa. 36 kuukauden kokonaiselossaolon arvio oli tsanubrutinibihaarassa 90,9 % (95 %:n luottamusväli: 86,3–94,0) ja bendamustiini + rituksimabi -haarassa 89,5 % (95 %:n luottamusväli: 84,2–93,1).
Kuva 1: Kaplan-Meier-kuvaaja IRC-toimikunnan arvioimasta etenemisvapaasta elossaolosta SEQUOIA-tutkimuksen kohortissa 1 (ITT-joukko)
ALPINE-tutkimus (BGB‑3111‑305): Vaiheen 3 satunnaistettu tutkimus tsanubrutinibista verrattuna ibrutinibiin potilailla, joilla on uusiutunut/hoitoon reagoimaton (R/R) KLL
ALPINE-tutkimus (BGB‑3111‑305) on avoin, satunnaistettu, vaiheen 3 aktiivikontrolloitu monikeskustutkimus. Siihen otettiin mukaan 652 potilasta, joilla oli uusiutunut tai hoitoon reagoimaton KLL vähintään yhden aiemman systeemisen hoidon jälkeen. Potilaat satunnaistettiin saamaan joko tsanubrutinibia 160 mg suun kautta kahdesti vuorokaudessa tai ibrutinibia 420 mg suun kautta kerran vuorokaudessa. Hoitoa jatkettiin kunnes tauti eteni tai ilmaantui ei-hyväksyttävissä olevaa toksisuutta.
Satunnaistaminen ositettiin iän (alle 65-vuotiaat ja vähintään 65-vuotiaat), maantieteellisen alueen (Kiina tai muu kuin Kiina), hoitoon reagoimattomuuden statuksen (kyllä tai ei) ja del(17 p)/TP53-mutaatiostatuksen (esiintyy tai ei esiinny) suhteen.
Lähtötilanteen demografiset ja taudin ominaisuudet olivat yleisesti ottaen tasapainossa hoitohaarojen välillä ITT-analyysijoukossa ja ensimmäisten 415 satunnaisten potilaan osalta.
ITT-analyysijoukossa mediaani-ikä oli 67,0 vuotta tsanubrutinibihaarassa ja 68,0 vuotta ibrutinibihaarassa. Suurimmalla osalla molempien haarojen potilaista lähtötilanteen ECOG-toimintakykyluokka oli 0 tai 1 (97,9 %:lla tsanubrutibihaarassa; 96,0 %:lla ibrutinibihaarassa). Ensimmäisillä 415 potilaalla havaittiin samanlaiset demografiset ja lähtötilanteen ominaisuudet. Aiempien systeemisen hoidon hoitolinjojen mediaanimäärä oli 1,0 sekä tsanubrutinibihaarassa (vaihteluväli 1–6) että ibrutinibihaarassa (vaihteluväli 1–8), sekä ITT-analyysijoukossa että ensimmäisillä 415 satunnaistetulla potilaalla.
Aiemmin BTK:n estäjällä hoidetut potilaat suljettiin pois tutkimuksesta 305, ja tsanubrutinibin käytöstä aiemman BCL 2:n estäjähoidon jälkeen on saatavilla vain vähän tietoja.
652 potilaan kokonaismäärästä 327 määritettiin saamaan tsanubrutinibimonoterapiaa ja 325 ibrutinibimonoterapiaa. Tehon arviointi perustuu ennalta määritettyyn ITT-joukon 415 ensimmäisen potilaan tiedoilla tehtyyn välianalyysiin. Näistä potilasta 207 oli satunnaistettu saamaan tsanubrutinibimonoterapiaa ja 208 ibrutinibimonoterapiaa. Tehoa koskevat tulokset on esitetty taulukossa 8.
Ensisijaisena päätetapahtumana oli kokonaisvasteosuus (ORR, määritelmänä vähintään osittainen vaste).
Ennalta määriteltynä ORR-välianalyysin ajankohtana, joka tehtiin ensimmäisten 415 satunnaistetun potilaan tiedoilla, tsabrutinibi osoittautui yhdenveroiseksi ibrutinibin kanssa (yksipuolinen p < 0,0001) ja paremmaksi kuin ibrutinibi (kaksipuolinen p = 0,0006) tutkimussuunnitelmassa ennalta määritellyssä ensisijaisessa päätetapahtumassa eli tutkijan arvioimassa kokonaisvasteosuudessa. Myös IRC-toimikunnan määrittämän vasteen perusteella todettiin tsanubrutinibin yhdenveroisuus ibrutinibin kanssa (yksipuolinen p < 0,0001). ORR-loppuanalyysissä tutkijan arvioima ORR oli edelleen suurempi tsabrutinibihaarassa (79,5 %) kuin ibrutinibihaarassa (71,1 %, deskriptiivinen p = 0,0133). Myös IRC-toimikunnan määrittämä ORR oli merkittävästi suurempi tsabrutinibihaarassa kuin ibrutinibihaarassa, ja tsabrutinibin (80,4 %) todettiin olevan parempi kuin ibrutinibi (72,9 %, kaksipuolinen p = 0,0264).
Taulukko 8: Tehoa koskevat tulokset ALPINE-tutkimuksessa (ennalta määritetty ensimmäisten 415 satunnaistetun potilaan välianalyysi) tutkijan (tutkimussuunnitelmassa määritetty ensisijainen päätetapahtuma) ja IRC-toimikunnan arvioimina
| Tutkijan arvioima (tutkimussuunnitelmassa määritetty ensisijainen päätetapahtuma) | IRC-toimikunnan arvioima | ||
Päätetapahtuma | Tsanubrutinibi (N=207) | Ibrutinibi (N=208) | Tsanubrutinibi (N=207) | Ibrutinibi (N=208) |
Kokonaisvasteosuus§ (ORR) n (%) | 162 (78,3) | 130 (62,5) | 158 (76,3) | 134 (64,4) |
(95 %:n luottamusväli) | (72,0; 83,7) | (55,5; 69,1) | (69,6; 81,9) | (57,5; 70,9) |
Vasteiden suhteellinen määrä a (95 %:n luottamusväli) | 1,25 (1,10; 1,41) | 1,17 (1,04; 1,33) | ||
Yhdenveroisuus b | yksipuolinen p-arvo < 0,0001 | yksipuolinen p-arvo < 0,0001 | ||
Paremmuus c | kaksipuolinen p-arvo 0,0006 | kaksipuolinen p-arvo 0,0121 | ||
Vasteen kesto d: 12 kuukauden tapahtumavapaa osuus % (95 %:n luottamusväli) | 89,8 (78,1; 95,4) | 77,9 (64,7; 86,7) | 90,3 (82,3; 94,8) | 78,0 (66,1; 86,2) |
Kokonaisvasteosuus: (ORR): CR+CRi+nPR+PR, CR: täydellinen vaste, CRi: täydellinen vaste ja vaillinainen hemapoieettinen toipuminen, nPR: nodulaarinen osittainen vaste, PR: osittainen vaste
Tutkijan arvioima vasteen mediaanikesto ei ollut vielä mitattavissa tsanubrutinibihaarassa välianalyysin kohdalla, tutkimuksen seuranta-ajan mediaanikesto oli 15,31 kuukautta (vaihteluväli: 0,1; 23,1) tsanubrutinibihaarassa ja 15,43 kuukautta (vaihteluväli: 0,1; 26,0) ibrutinibihaarassa.
§ Hypoteesin testaus ORR:n yhdenvertaisuudelle ensimmäisillä 415 satunnaistetulla potilaalla välianalyysin ajankohtana ainoastaan yksipuolisella merkitsevyystasolla 0,005.
a Vasteiden suhteellinen määrä: arvioitu suhde, joka muodostuu kokonaisvasteesta tsanubrutinibihaarassa jaettuna ibrutinibihaaran kokonaisvasteella.
b Ositettu testi, jossa vertailtiin nollahypoteesin suhteelliseen vasteiden määrään 0,8558.
c Ositettu Cochran-Mantel-Haenszelin testi.
d Kaplan-Meierin estimaatti.
Vasteen saamiseen kuluva mediaaniaika tutkijan arvioimana ORR-välianalyysin ajankohtana, ensimmäisillä 415 satunnaistetulla potilaalla oli 5,59 kuukautta (vaihteluväli: 2,7; 14,1) tsanubrutinibihaarassa ja 5,65 kuukautta (vaihteluväli: 2,8; 16,7) ibrutinibihaarassa. IRC-toimikunnan arvioimat tulokset olivat samansuuntaisia (5,55 kuukautta tsanubrutinibihaarassa ja 5,63 kuukautta ibrutinibihaarassa). ORR-loppuanalyysissa, joka tehtiin kaikkien 652 satunnaistetun potilaan tiedoilla, vasteen saamiseen kuluva mediaaniaika oli ennallaan (5,59 kuukautta tsanubrutinibihaarassa vs. 5,65 kuukautta ibrutinibihaarassa tutkijan arvioimana ja 5,52 kuukautta tsanubrutinibihaarassa vs. 5,62 kuukautta ibrutinibihaarassa IRC-toimikunnan arvioimana.)
Potilailla, joilla oli del(17p)-mutaatio ensimmäisten 415 satunnaistetun potilaan joukossa, tutkijan arvioiman ORR oli 83,3 % (95 %:n luottamusväli 62,5; 95,3; 20/24 potilaalla) tsanubrutinibihaarassa ja 53,8 % (95 %:n luottamusväli 33,3; 73,4; 14/26 potilaalla) ibrutinibihaarassa. IRC-toimikunnan arvion perusteella ORR oli 79,2 % (95 %:n luottamusväli 57,8; 92,9; 19/24 potilaalla) tsanubrutinibihaarassa ja 61,5 % (95 %:n luottamusväli 40,6; 79,8; 16/26 potilaalla) ibrutinibihaarassa. ORR-loppuanalyysissä, joka tehtiin kaikkien 652 satunnaistetun potilaan tiedoilla, tutkijan arvioima ORR oli 86,7 % (95 %:n luottamusväli 73,2; 94,9; 39/45 potilaalla, joilla oli del(17p)-mutaatio) tsanubrutinibihaarassa ja 64,0 % (95 %:n luottamusväli 49,2; 77,1; 32/50 potilaalla, joilla oli del(17p)-mutaatio) ibrutinibihaarassa.
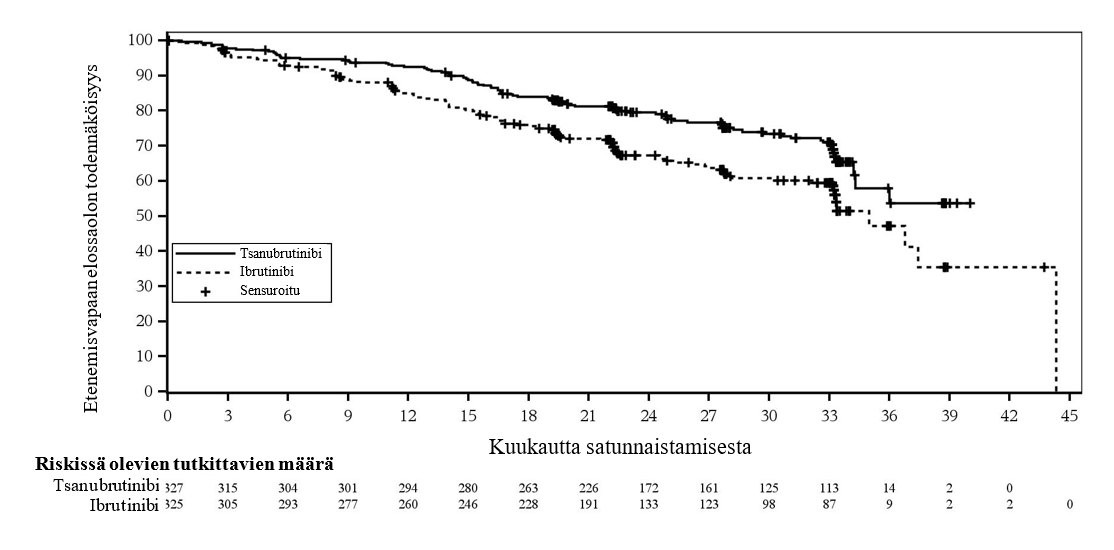
Yhteensä 652 potilasta osallistui tutkimukseen ennalta määriteltynä PFS-loppuanalyysin ajankohtana (katkaisuajankohta 8.8.2022). PFS-seuranta-ajan mediaani oli tutkijan arvioimana 28,1 kuukautta ja IRC-toimikunnan arvioimana 30,7 kuukautta. Tsanubrutinibi osoittautui PFS:n suhteen paremmaksi kuin ibrutinibi tutkijan ja IRC-toimikunnan arvioimana. PFS-analyysin tehoa koskevat tulokset on esitetty taulukossa 8 ja PFS-analyysin Kaplan Meyer ‑kuvaaja IRC-toimikunnan arvioimana on esitetty kuvassa 2.
Taulukko 9: Tehoa koskevat tulokset ALPINE-tutkimuksessa (ennalta määritetty ensimmäisten 652 satunnaistetun potilaan PFS-loppuanalyysi) tutkijan ja IRC-toimikunnan arvioimana (katkaisuajankohta 8.8.2022)
| Tutkijan arvioima | Riippumattomasti arvioitu | ||
Päätetapahtuma | Tsanubrutinibi (N = 327) | Ibrutinibi (N = 325) | Tsanubrutinibi (N = 327) | Ibrutinibi (N = 325) |
Etenemisvapaa elinaika |
|
| ||
Tapahtumien määrä, n (%) | 87 (26,6) | 118 (36,3) | 88 (26,9) | 120 (36,9) |
Riskitiheyksien suhdea (95 %:n luottamusväli) | 0,65 (0,49, 0,86) | 0.65 (0.49, 0.86) | ||
Kaksipuolinen p-arvob | 0,0024 | 0.0024 | ||
a Perustuu ositettuun Coxin regressiomalliin, jossa ibrutinibi oli vertailuryhmänä.
b Perustuu ositettuun log-rank-testiin.
Kuva 2: Kaplan-Meier-kuvaaja riippumattomasti keskitetysti arvioidusta etenemisvapaasta elinajasta (ITT) (katkaisuajankohta 8.8.2022)
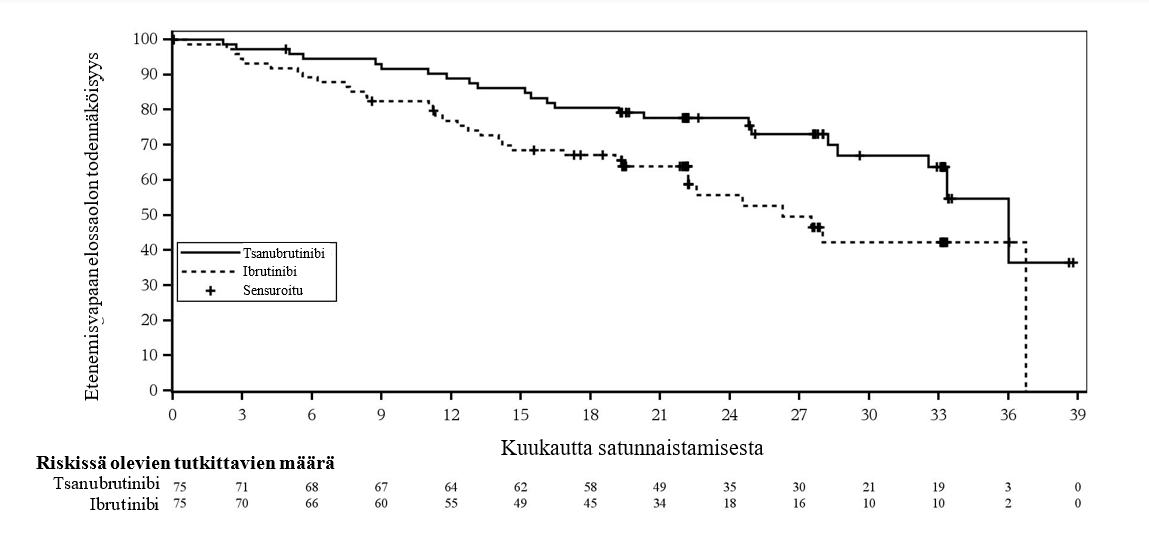
Potilailla joilla oli del(17p)-/TP53-mutaatio, etenemisvapaan elinajan riskitiheyksien suhde tutkijan arvioimana oli 0,53 (95 %:n luottamusväli, 0,31, 0,88). Riippumattoman arvioinnin perusteella riskitiheyksien suhde oli 0,52 (95 %:n luottamusväli, 0,30, 0,88) (kuva 3).
Kuva 3: Kaplan-Meier-kuvaaja riippumattomasti keskitetysti arvioidusta etenemisvapaasta elinajasta potilailla, joilla on Del(17p)/TP53-mutaatio (ITT) (katkaisuajankohta 8.8.2022)
Seurannan arvioitu mediaani oli 32,8 kuukautta, joten kokonaiselinajan mediaania ei saavutettu kummassakaan haarassa, sillä 17%:lla potilaista ilmeni tapahtuma.
Potilaat, joilla on follikulaarinen lymfooma
Tsanubrutinibi-obinututsumabi-yhdistelmähoidon tehoa obinututsumabihoitoon verrattuna arvioitiin ROSEWOOD-tutkimuksessa (BGB‑3111‑212), joka on vaiheen 2 satunnaistettu, avoin monikeskustutkimus. Tutkimukseen kirjattiin kaikkiaan 217 potilasta, joilla oli relapsoitunut (määritelmänä taudin eteneminen viimeisimmän hoitojakson päättymisen jälkeen) tai refraktorinen (määritelmänä täydellisen tai osittaisen vasteen puuttuminen viimeisimmällä hoitojaksolla) asteen 1–3a follikulaarinen lymfooma, jotka olivat saaneet vähintään kahta aiempaa systeemistä hoitoa, mukaan lukien CD20-vasta-aineisiin perustuvaa ja alkyloiviin aineisiin perustuvaa yhdistelmähoitoa. Potilaat satunnaistettiin suhteessa 2:1 saamaan joko tsanubrutinibia 160 mg suun kautta kahdesti vuorokaudessa taudin etenemiseen tai kohtuuttomaan toksisuuteen asti yhdistettynä obinututsumabiin, jota annettiin 1 000 mg laskimonsisäisesti, (haara A) tai vain obinututsumabia (haara B). Obinututsumabia annettiin ensimmäisen syklin päivinä 1, 8 ja 15, sitten syklien 2–6 päivänä 1. Jokainen sykli kesti 28 päivää. Potilaat saivat valinnaista obinututsumabi-ylläpitohoitoa yhden infuusion joka toisessa syklissä, korkeintaan 20 annosta.
Obinututsumabi-haaraan satunnaistetut potilaat saivat siirtyä toiseen haaraan ja saada tsanubrutinibin ja obinututsumabin yhdistelmää, mikäli ilmeni taudin etenemistä tai vasteen puuttumista (määrityksenä vakaa tauti parhaana vasteena) 12 syklin jälkeen.
Satunnaistaminen ositettiin aiempien hoitolinjojen määrän (2–3 ja ˃ 3), rituksimabi-refraktorisen tilan (kyllä ja ei) ja maantieteellisen alueen (Kiina ja muut maat/alueet) mukaan.
Lähtötilanteen demografiset ja taudin ominaisuudet olivat yleisesti ottaen tasapainossa tsanubrutinibi-yhdistelmähaaran ja obinututsumabi-monoterapiahaaran välillä 217 satunnaistetun potilaan osalta. Potilaiden mediaani-ikä oli 64 vuotta (vaihteluväli 31–88), 49,8 oli miehiä ja 64,1 % oli valkoihoisia. Suurimmalla osalla (97,2 %) potilaista lähtötilanteen ECOG-toimintakykyluokka oli 0 tai 1.
Seulontahetkellä suurin osa potilaista oli Ann Arbor -tasolla III tai IV (179 potilasta [82,5 %]). 88 potilaalla (40,6 %) oli laaja sairaus (määritelmänä > 1 lähtötason kohdeleesion läpimitta > 5 cm). 123 potilasta (56,7 %) täytti GELF-kriteerit.
Aiempien syöpähoitojen mediaanimäärä oli kolme linjaa (vaihteluväli 2–11 linjaa). Kaikki 217 potilasta saivat > 2 aiempaa hoitolinjaa, joihin kuului rituksimabihoitoa (monoterapiana tai yhdistettynä sädehoitoon), ja 217 potilaasta 59 (27,2 %) sai > 3 aiempaa hoitolinjaa. 217 potilaasta 114 (52,5 %) oli refraktorisia rituksimabille (määritelmänä vasteen puuttuminen tai taudin eteneminen aiemman rituksimabia sisältävän hoidon aikana [monoterapia tai yhdistettynä sädehoitoon] tai taudin eteneminen kuuden kuukauden kuluessa viimeisestä rituksimabiannoksesta, induktio- tai ylläpitohoitotilanteessa). 12 (5,5 %) potilasta sai aiempaa obinututsumabia.
Kaikkiaan 217 potilaasta 145 satunnaistettiin tsanubrutinibi-yhdistelmähoitohaaraan ja 72 satunnaistettiin obinututsumabi-monoterapiahaaraan. Seuranta-ajan mediaani käy ilmi taulukosta 10. Tsanubrutinibi-altistuksen keston mediaani oli 12,4 kuukautta tietojen katkaisuajankohtana 31.12.2024.
Obinututsumabi-monoterapiahaaraan satunnaistetusta 72 potilaasta 36 siirtyi yhdistelmähoitoon.
Ensisijainen päätetapahtuma oli kokonaisvasteosuus (määritelmänä osittainen vaste tai täydellinen vaste), joka on määritetty riippumattoman keskitetyn arvion mukaisesti NHL:n Lugano-luokittelua käyttäen. Tärkeimpiä toissijaisia päätetapahtumia olivat muun muassa vasteen kesto (DOR), etenemisvapaa elinaika (PFS) ja kokonaiselinaika (OS).
Tehoa koskevat tulokset on esitetty taulukossa 10 ja kuvassa 4.
Taulukko 10: Tehoa koskevat tulokset ICR-toimikunnan arvioimana (ROSEWOOD-tutkimus)
| Tsanubrutinibi + obinututsumabi | Obinututsumabi | Tsanubrutinibi + obinututsumabi | Obinututsumabi |
|---|---|---|---|---|
Tietojen katkaisuajankohta | 31.12.2024 | 25.6.2022 | ||
Seuranta-ajan mediaani | 36,83 | 31,52 | 20,21 | 20,40 |
Kokonaisvasteosuus, n (%) (95 %:n luottamusvälia) |
102 (70,3) (62,2; 77,6) |
32 (44,4) (32,7; 56,6) |
100 (69,0) (60,8; 76,4) |
33 (45,8) (34,0; 58.0) |
P-arvob | 0,0003 | 0,0012 | ||
CR | 61 (42,1) | 14 (19,4) | 57 (39,3) | 14 (19,4) |
PR | 41 (28,3) | 18 (25,0) | 43 (297) | 19 (26,4) |
Vasteen kesto (kuukausina) |
|
|
|
|
Mediaani (95 %:n luottamusväli)c | 32,9 (19,6; 43,1) | 14,0 (9,2; 26,5) | NE (25,3; NE) | 14 (9,2; 25,1) |
Etenemisvapaa elinaika (kuukausina) |
|
|
|
|
Mediaani (95 %:n luottamusväli)c | 22,1 (16,1; 34,0) | 10,3 (6,5; 13,8) | 28,0 (16,1; NE) | 10,4 (6,5; 13,8) |
Kokonaisvasteosuus: CR + PR, CR: täydellinen vaste, PR: osittainen vaste
a Perustuu Clopper-Pearson-estimointiin.
b Cochran-Mantel-Haenszelin testi ositettuna rituksimabi-refraktorisen tilan, aiempien hoitolinjojen määrän ja maantieteellisen alueen mukaan IRT:n mukaisesti.
c Mediaanit arvioitu Kaplan-Meier-menetelmällä; 95 %:n luottamusväli arvioitu Brookmeyerin ja Crowleyn menetelmällä.
d DOR-asteet arvioitu Kaplan-Meier-menetelmällä; 95 %:n luottamusväli käyttäen Greenwoodin mallia. DOR:ää ei kontrolloitu tyypin I virheen osalta, ja luottamusvälit ovat luonteeltaan nimellisiä.
Kuva 4:Kaplan-Meier-kuvaaja riippumattomasti keskitetysti arvioidusta etenemisvapaasta elinajasta (ITT)
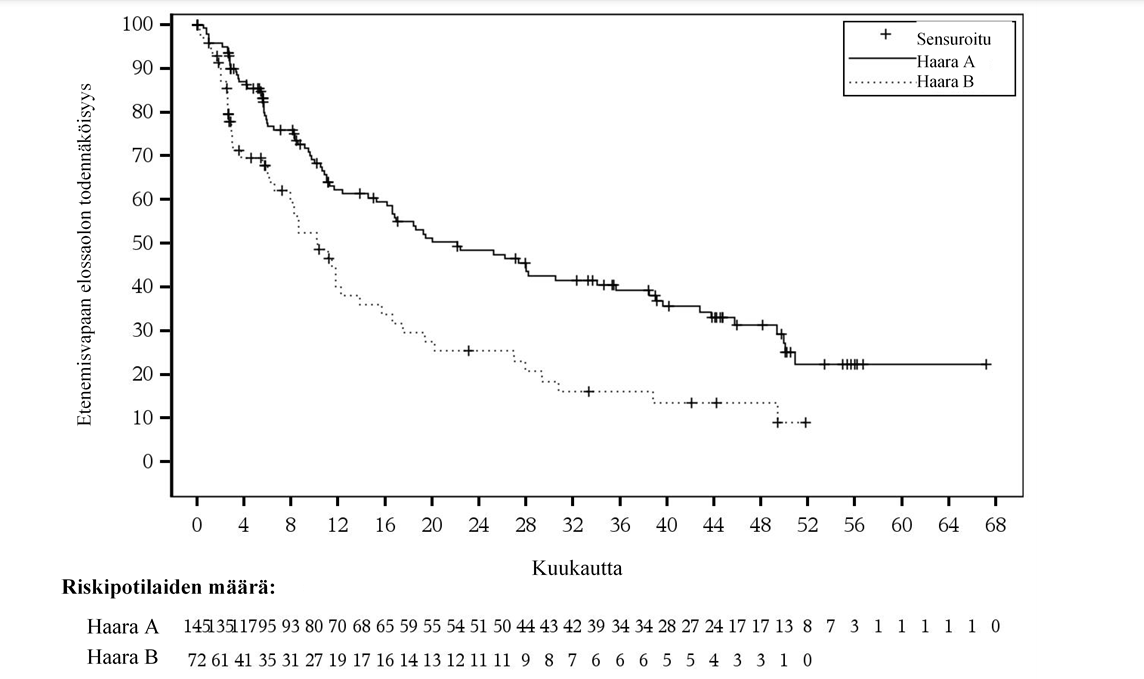
Haara A, Tsanubrutinibi + obinututsumabi; haara B, obinututsumabi
Kokonaiselinaika
31.12.2024 mennessä 51 potilasta (35,2 %) yhdistelmähoitohaarassa ja 33 potilasta (45,8 %) obinututsumabi-monoterapiahaarassa kuoli. 18 kuukauden kohdalla kokonaiselinaika oli 84,1 % (95 %:n luottamusväli: 76,6; 89,3) yhdistelmähoitohaarassa ja 71,5 % (95 %:n luottamusväli: 59,0; 80,8) obinututsumabi-monoterapiahaarassa. Kokonaiselinaika-analyysi saattaa olla sulautunut 36 potilaan (50,0 %) osalta, jotka siirtyivät obinututsumabi-monoterapiahaarasta yhdistelmähoitohaaraan.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset BRUKINSA-valmisteen käytöstä kaikissa pediatrisissa potilasryhmissä lymfoplasmasyyttisen lymfooman hoidossa ja kypsien B-solukasvainten hoidossa (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Tsanubrutinibin maksimipitoisuus plasmassa (Cmax) ja plasman lääkepitoisuuden aikakäyrän alle jäävä pinta-ala (AUC) lisääntyvät suhteessa annokseen annosalueella 40–320 mg (0,13–1 kertaa suositeltu kokonaisvuorokausiannos). Toistuvan yhden viikon aikaisen annon jälkeen havaittiin vähäistä tsanubrutinibin systeemistä kertymistä.
Tsanubrutinibin vakaan tilan päivittäisen AUC-arvon geometrinen keskiarvo (%CV) on 2,099 (42 %) ng·h/ml 160 mg:n kahdesti vuorokaudessa annon jälkeen ja 1,917 (59 %) 320 mg:n kerran vuorokaudessa annon jälkeen. Tsanubrutinibin vakaan tilan Cmax-pitoisuuden geometrinen keskiarvo (%CV) on 299 (56 %) ng/ml 160 mg:n kahdesti vuorokaudessa annon jälkeen ja 533 (55 %) 320 mg:n kerran vuorokaudessa annon jälkeen.
Imeytyminen
Tsanubrutinibin tmax-ajan mediaani on 2 tuntia. Tsanubrutinibin AUC-arvossa tai Cmax-pitoisuudessa ei havaittu kliinisesti merkittäviä eroja rasvapitoisen aterian (noin 1 000 kilokaloria, 50 % kokonaiskaloripitoisuudesta peräisin rasvasta) annon jälkeen terveillä tutkittavilla.
Jakautuminen
Tsanubrutinibin ilmeisen vakaan tilan jakautumistilavuuden geometrinen keskiarvo (%CV) terminaalivaiheen aikana (Vz/F) oli 522 l (71 %). Tsanubrutinibin sitoutuminen plasman proteiineihin on noin 94-prosenttista ja veren ja plasman välinen suhde oli 0,7–0,8.
Metabolia
Tsanubrutinibi metaboloituu pääasiassa sytokromi P450(CYP)3A:n välityksellä.
Eliminaatio
Tsanubrutinibin keskimääräinen puoliintumisaika (t½) on noin 2–4 tuntia 160 mg:n tai 320 mg:n kerta-annoksen suun kautta annon jälkeen. Tsanubrutinibin ilmeisen oraalisen puhdistuman geometrinen keskiarvo (%CV) terminaalivaiheen aikana oli 128 (61 %) L/h. Terveille tutkittaville annetun 320 mg:n radioleimatun tsanubrutinibin kerta-annoksen jälkeen noin 87 % annoksesta erittyi ulosteeseen (38 % muuttumattomana) ja 8 % virtsaan (alle 1 % muuttumattomana).
Erityiset potilasryhmät
Iäkkäät potilaat
Iällä (19–90 vuotta; keskimääräinen ikä 65±12,5 vuotta) ei ollut kliinisesti merkittävää vaikutusta tsanubrutinibin farmakokinetiikkaan populaatiofarmakokineettisen analyysin (N = 1 291) perusteella.
Sukupuoli
Sukupuolella (872 miestä ja 419 naista) ei ollut kliinisesti merkittävää vaikutusta tsanubrutinibin farmakokinetiikkaan populaatiofarmakokineettisen analyysin perusteella.
Rotu
Rodulla (964 valkoihoista, 237 aasialaista, 30 tummaihoista ja 25 muuksi luokiteltua) ei ollut kliinisesti merkittävää vaikutusta tsanubrutinibin farmakokinetiikkaan populaatiofarmakokineettisen analyysin perusteella.
Kehonpaino
Kehonpainolla (36–149 kg, keskimääräinen paino 76,5±16,9 kg) ei ollut kliinisesti merkittävää vaikutusta tsanubrutinibin farmakokinetiikkaan populaatiofarmakokineettisen analyysin (N = 1 291) perusteella.
Munuaisten vajaatoiminta
Tsanubrutinibi eliminoituu vain vähän munuaisten kautta. Populaatiofarmakokineettisen analyysin perusteella lievällä, kohtalaisella tai vaikealla munuaisten vajaatoiminnalla (kreatiniinipuhdistuma [CrCl] ≥ 15 ml/min Cockcroft-Gault-yhtälöllä arvioituna) ei ollut vaikutusta tsanubrutinibialtistukseen. Analyysi perustui 362 potilaaseen, joilla munuaisten toiminta oli normaalia, 523 lievää munuaisten vajaatoimintaa sairastavaan potilaaseen, 303 kohtalaista munuaisten vajaatoimintaa sairastavaan potilaaseen, 11 vaikeaa munuaisten vajaatoimintaan sairastavaan potilaaseen ja yhteen loppuvaiheen munuaistautia (ESRD) sairastavaan potilaaseen. ESRD:n (kreatiniinipuhdistuma < 15 ml/min) ja dialyysin vaikutuksia tsanubrutibin farmakinetiikkaan ei tunneta.
Maksan vajaatoiminta
Tsanubrutinibin kokonais-AUC-arvo suureni 11 % lievää maksan vajaatoimintaa (Child-Pugh-luokka A) sairastavilla tutkittavilla, 21 % kohtalaista maksan vajaatoimintaa (Child-Pugh-luokka B) sairastavilla tutkittavilla ja 60 % vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavilla tutkittavilla verrattuna tutkittaviin, joiden maksan toiminta oli normaalia. Tsanubrutinibin sitomaton AUC-arvo suureni 23 % lievää maksan vajaatoimintaa (Child-Pugh-luokka A) sairastavilla tutkittavilla, 43 % kohtalaista maksan vajaatoimintaa (Child-Pugh-luokka B) sairastavilla tutkittavilla ja 194 % vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavilla tutkittavilla verrattuna tutkittaviin, joiden maksan toiminta oli normaalia. Child-Pugh-luokan pistemäärän, lähtötason seerumin albumiinin, lähtötason seerumin bilirubiinin ja lähtötason protrombiiniajan ja sitomattoman tsanubrutinibin AUC-arvon välillä havaittiin merkittävä korrelaatio.
In vitro -tutkimukset
CYP-entsyymit
Tsanubrutinibi on heikko CYP2B6:n ja CYP2C8:n induktori. Tsanubrutinibi ei ole CYP1A2:n induktori.
Samanaikainen anto kuljetussubstraattien/estäjien kanssa
Tsanubrutinibi on todennäköisesti P-gp:n substraatti. Tsanubrutinibi ei ole OAT1:n, OAT3:n, OCT2:n, OATP1B1:n eikä OATP1B3:n substraatti tai estäjä.
Farmakodynaamiset yhteisvaikutukset
In vitro -tutkimus osoitti, että tsanubrutinibin ja rituksimabin välisen farmakodynaamisen yhteisvaikutuksen mahdollisuus on pieni ja tsanubrutinibi ei todennäköisesti häiritse CD20-vasta-aineiden aikaansaamaan ADCC-vaikutusta.
In vitro-, ex vivo- ja eläinkokeet osoittivat, että tsanubrutinibilla ei ollut vaikutuksia tai oli vain vähäisiä vaikutuksia verihiutaleiden aktivoitumiseen, glykoproteiiniekspressioon ja veritulppien muodostumiseen.
Prekliiniset tiedot turvallisuudesta
Yleinen toksisuus
Tsanubrutinibin yleiset toksikologiset profiilit määriteltiin suun kautta Sprague-Dawley-rotilla enintään 6 kuukauden pituisen hoidon osalta ja beaglekoirillaenintään 9 kuukauden pituisen hoidon osalta.
Rotilla toistetun annoksen tutkimuksissa, joissa hoito kesti enintään 6 kuukautta, testituotteeseen liittyvä kuolleisuus havaittiin annoksella 1000 mg/kg/vrk (81x kliininen AUC) histopathologisin löydöksin ruoansulatuskanavassa. Muut löydökset liittyivät pääasiassa haimaan (atrofia, fibroplasia, hemorragia ja/tai tulehduksellinen soluinfiltraatio) annoksilla ≥ 30 mg/kg/vrk (3x kliininen AUC), nenää/suuta/silmiä ympäröivään ihoon (tulehduksellinen soluinfiltraatio, eroosio/haavauma) annoksesta 300 mg/kg/vrk (16x kliininen AUC)ja keuhkoihin (makrofageja keuhkorakkuloissa) annoksella 300 mg/kg/vrk . Kaikki nämä löydökset katosivat täysin tai osittain 6 viikon toipumisen jälkeen, paitsi haimalöydökset, joita ei pidetty kliinisesti relevantteina.
Koirilla toistetun annoksen tutkimuksissa, joissa hoito kesti enintään 9 kuukautta, testituotteeseen liittyvät löydökset havaittiin lähinnä ruoansulatuskanavassa (pehmeät/vetiset/limaiset ulosteet) ja ihossa (ihottuma, punainen värimuutos ja paksuuntuminen/hilseily) mesentrisissä, mandibulaarisissa ja suoliston imusolmukkeissa ja pernassa (lymfoidi depleetio tai erytrofagosytoosi) annoksilla alkaen 10 mg/kg/vrk (kolminkertainen kliiniseen AUC-arvoon nähden) annokseen 100 mg/kg/vrk saakka(18x kliininen AUC). Kaikki nämä löydökset katosivat täysin tai osittain 6 viikon toipumisen jälkeen
Karsinogeenisuus/genotoksisuus
Karsinogeenisuustutkimuksia ei ole tehty tsanubrutinibilla.
Tsanubrutinibi ei ollut mutageeninen bakteerien mutageenisuusanalyysissä eikä klastogeeninen kromosomien aberraatioanalyysissä nisäkkäiden (CHO) soluissa eikä klastogeeninen in vivo luuytimen mikrotuma-analyysissä rotilla.
Kehitys- ja lisääntymistoksisuus
Yhdistetty urosten ja naarasten hedelmällisyyttä ja varhaisvaiheen alkion kehitystä selvittävä tutkimus tehdiin rotilla suun kautta annettavan tsanubrutinibin annoksilla 30, 100 ja 300 mg/kg/vrk. Urosten tai naarasten hedelmällisyyteen kohdistuvia vaikutuksia ei havaittu, mutta suurimmalla testatulla annoksella havaittiin morfologisia poikkeavuuksia siemennesteessä ja implantaation jälkeisten keskenmenojen lisääntymistä. Annos 100 mg/kg/vrk on noin 13-kertaa suurempi kuin ihmisen terapeuttinen altistus.
Alkion ja sikiön kehitystä koskevia toksisuustutkimuksia on tehty sekä rotilla että kaniineilla. Tsanubrutinibia annettiin suun kautta tiineille rotille organogeneesin aikana annoksilla 30, 75 ja 150 mg/kg/vrk. Sydämen epämuodostumia (2- tai 3-kammioisia sydämiä, joiden esiintymistiheys oli 0,3–1,5 %) havaittiin kaikilla annostasoilla ilman emoon kohdistuvaa toksisuutta. Annos 30 mg/kg/vrk on noin viisi kertaa suurempi kuin ihmisen terapeuttinen altistus.
Tsanubrutinibin anto tiineille kaniineille organogeneesin aikana annoksilla 30, 70 ja 150 mg/kg/vrk aiheutti implantaation jälkeisen keskenmenon suurimmalla annoksella. Annos 70 mg/kg on noin 25 kertaa suurempi kuin ihmisen terapeuttinen altistus, ja siihen liittyi emotoksisuutta.
Ennen syntymää ja syntymän jälkeen tehdyssä kehitystoksisuustutkimuksessa tsanubrutinibia annettiin rotille suun kautta annoksilla 30, 75 ja 150 mg/kg/vrk implantaatiosta alkaen vieroitukseen saakka. Keskisuuren ja suuren annoksen saaneiden ryhmien jälkeläisillä ilmeni ruumiinpainon vähentymistä vierottamista edeltävänä aikana, ja kaikissa annosryhmissä ilmeni haitallisia silmälöydöksiä (esim. kaihi, ulkoneva silmä). 30 mg/kg/vrk:n annos on noin viisi kertaa suurempi kuin ihmisen terapeuttinen altistus.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Mikrokiteinen selluloosa
Kroskarmelloosinatrium
Natriumlauryylisulfaatti (E487)
Vedetön, kolloidinen piidioksidi
Magnesiumstearaatti
Kapselin kuori
Liivate
Titaanidioksidi (E171)
Painomuste
Sellakkalasite (E904)
Musta rautaoksidi (E172)
Propyleeniglykoli (E1520)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BRUKINSA kapseli, kova
80 mg (L:ei) 120 kpl (5373,23 €)
PF-selosteen tieto
HDPE-purkki, jossa on lapsiturvallinen polypropeenisuljin. Yksi pahvipakkaus sisältää yhden purkin, jossa on 120 kovaa kapselia.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen läpinäkymätön kova kapseli, jonka pituus on 22 mm ja jossa on mustalla musteella merkintä ”ZANU 80”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BRUKINSA kapseli, kova
80 mg 120 kpl
- Ylempi erityiskorvaus (100 %). Tsanubrutinibi: Aikuisten kroonisen lymfaattisen leukemian, marginaalivyöhykkeen lymfooman ja Waldenströmin makroglobulinemian hoito erityisin edellytyksin (1542).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Tsanubrutinibi: Aikuisten kroonisen lymfaattisen leukemian ja Waldenströmin makroglobulinemian hoito erityisin edellytyksin (3086).
ATC-koodi
L01EL03
Valmisteyhteenvedon muuttamispäivämäärä
15.07.2026
Yhteystiedot
Gävlegatan 16
113 30 Stockholm
Sweden
beonemedicines.se
nordics@beonemed.com