
MYRELEZ injektionsvätska, lösning i förfylld spruta 60 mg, 90 mg, 120 mg
not_interestedSaatavuushäiriö
Ei saatavilla
MYRELEZ injektioneste, liuos, esitäytetty ruisku
- 60 mg0,5 ml15.03.2025 - 30.09.2026
- 90 mg0,5 ml01.02.2025 - 30.09.2026
- 120 mg0,5 ml30.09.2024 - 30.09.2026
Saatavilla
Saman valmisteen muut pakkaukset ja/tai vaihtokelpoiset valmisteet / MYRELEZ injektioneste, liuos, esitäytetty ruisku 60 mg
SOMATULINE AUTOGEL injektioneste, liuos, esitäytetty ruisku
- 60 mg60 mg
Saman valmisteen muut pakkaukset ja/tai vaihtokelpoiset valmisteet / MYRELEZ injektioneste, liuos, esitäytetty ruisku 90 mg
SOMATULINE AUTOGEL injektioneste, liuos, esitäytetty ruisku
- 90 mg90 mg
Saman valmisteen muut pakkaukset ja/tai vaihtokelpoiset valmisteet / MYRELEZ injektioneste, liuos, esitäytetty ruisku 120 mg
SOMATULINE AUTOGEL injektioneste, liuos, esitäytetty ruisku
- 120 mg120 mg
Kvalitativ och kvantitativ sammansättning
Lanreotid (INN), 60 mg, 90 mg eller 120 mg (som acetat).
Varje förfylld spruta innehåller en övermättad lösning av lanreotidacetat motsvarande 0,246 mg lanreotidbas/mg lösning, vilket säkerställer en injektionsdos på 60 mg, 90 mg respektive 120 mg lanreotid.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Injektionsvätska, lösning i förfylld spruta.
Kliniska uppgifter
Terapeutiska indikationer
Myrelez är avsett för:
-
behandling av patienter med akromegali då cirkulerande nivåer av tillväxthormon (GH) och/eller insulinliknande tillväxtfaktor-1 (IGF‑1) förblir onormala efter kirurgiskt ingrepp och/eller strålbehandling eller hos patienter för vilka kirurgi och/eller strålbehandling inte är något alternativ
-
behandling av gastroenteropankreatiska neuroendokrina tumörer (GEP-NET), av grad 1 och vissa tumörer av grad 2 (Ki-67-värde upp till 10 %), med ursprungslokalisering i midgut, pankreas eller av okänt ursprung, undantaget primärtumör i hindgut, hos vuxna patienter med inoperabel, lokalt avancerad eller metastaserande sjukdom (se avsnitt Farmakodynamiska egenskaper)
- symtomlindring i samband med neuroendokrina tumörer (särskilt karcinoidtumörer).
Dosering och administreringssätt
Dosering
Akromegali
Rekommenderad startdos är 60 mg till 120 mg administrerat var 28:e dag.
Dosen kan ändras efter patientens reaktion (vilken bedöms enligt symptombild och/eller biokemisk verkan) eller patientens eventuella erfarenheter av somatostatinanaloger.
Som exempel ska den initiala dosen Myrelez till patienter som tidigare behandlats med lanreotid 30 mg givet var 14:e dag vara 60 mg var 28:e dag och för patienter tidigare behandlade med lanreotid 30 mg givet var 10:e dag ska den initiala dosen Myrelez vara 90 mg var 28:e dag.
Därefter ska dosen anpassas individuellt efter svaret från patienten (bedömt som en minskning av symtom och/eller minskade GH- och/eller IGF‑1-nivåer).
För patienter vars kliniska symtom och biokemiska parametrar inte adekvat kontrolleras kan dosen Myrelez höjas till upp till 120 mg med 28 dagars mellanrum.
Om fullständig kontroll erhålls (baserat på GH-nivåer under 1 ng/ml, normaliserade IGF‑1-nivåer och/eller symtombortfall) kan dosen minskas.
Patienter som är väl kontrollerade med en somatostatinanalog kan behandlas med Myrelez 120 mg var 42:a till 56:e dag (6 till 8 veckor).
Långtidsuppföljning av symtom samt nivåer av tillväxthormon och IGF‑1 ska vara standardpraxis för alla patienter.
Behandling av gastroenteropankreatiska neuroendokrina tumörer, av grad 1 och vissa tumörer av grad 2 (Ki-67-värde upp till 10 %), med ursprungslokalisering i midgut, pankreas eller av okänt ursprung, undantaget primärtumör i hindgut, hos vuxna patienter med inoperabel, lokalt avancerad eller metastaserande sjukdom.
Rekommenderad dos är en injektion med Myrelez 120 mg administrerat var 28:e dag. Behandlingen med Myrelez bör fortsätta så länge som det är nödvändigt för tumörkontroll.
Symtomlindring i samband med neuroendokrina tumörer
Rekommenderad startdos är 60 mg till 120 mg administrerat var 28:e dag.
Dosen ska justeras enligt graden av symtomlindring som erhålls.
Nedsatt njur-/leverfunktion
På grund av lanreotids breda terapeutiska intervall, behövs ingen dosjustering hos patienter med nedsatt njur- eller leverfunktion (se avsnitt Farmakokinetiska egenskaper).
Äldre patienter
På grund av lanreotids breda terapeutiska intervall, behövs ingen dosjustering för äldre patienter (se avsnitt Farmakokinetiska egenskaper).
Pediatrisk population
Användning av Myrelez rekommenderas inte till barn och ungdomar på grund av avsaknad av data om säkerhet och effekt.
Administreringssätt
Myrelez ges som en djup subkutan injektion i den övre yttre kvadranten i sätesregionen eller i den övre delen av lårets utsida.
För patienter som får en fast dos av Myrelez kan läkemedlet ges antingen av patienten själv eller av en person som har fått övning i hur injektionen ges. Vid egeninjektion ska injektionen ges i övre delen av lårets utsida.
Det är upp till hälso- och sjukvårdspersonal att bestämma om patienten själv eller en person som har utbildats i hur injektionen ges ska administrera läkemedlet.
Oavsett injektionsställe ska huden inte lyftas upp och nålen ska sättas in snabbt med hela sin längd, vinkelrät mot huden.
Växla injektionsstället mellan höger och vänstra sida.
Kontraindikationer
Överkänslighet mot den aktiva substansen, somatostatin eller närbesläktade peptider eller mot något hjälpmedel som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Lanreotid kan reducera gallblåsans motilitet och leda till gallstensbildning, varför patienter periodvis kan behöva övervakas. Det har förekommit rapporter efter godkännandet om gallsten som medfört komplikationer, inklusive kolecystit, kolangit och pankreatit, som krävt kolecystektomi hos patienter som tar lanreotid. Vid misstänkt komplikation av kolelitiasis, avbryt behandling med lanreotid och sätt in lämplig behandling.
Farmakologiska studier på djur och människa visar att lanreotid, liksom somatostatin och dess analoger, hämmar insulin- och glukagonsekretionen. Därmed kan patienter som behandlas med lanreotid få hypoglykemi eller hyperglykemi. Blodglukosnivån ska kontrolleras när lanreotidbehandlingen påbörjas samt vid dosändring, och eventuell diabetesbehandling bör justeras därefter.
Viss minskning av sköldkörtelfunktionen har observerats vid lanreotidbehandling av akromegalipatienter, även om klinisk hypotyreos är sällsynt (< 1 %). Sköldkörtelfunktiontester ska göras när det är kliniskt motiverat.
Hos patienter utan underliggande hjärtproblem kan lanreotid leda till minskad hjärtfrekvens utan att nödvändigtvis passera tröskelvärdet för bradykardi. Hos patienter med preexisterande hjärtsjukdom kan sinusbradykardi uppkomma. Försiktighet ska iakttas när behandling med lanreotid påbörjas hos patienter med bradykardi (se avsnitt Interaktioner).
Interaktioner
De farmakologiska gastrointestinala effekterna av lanreotid kan medföra minskad intestinal absorption av andra, samtidigt administrerade läkemedel, däribland ciklosporin. Samtidig administrering av ciklosporin och lanreotid kan minska den relativa biotillgängligheten av ciklosporin och en justering av dosen ciklosporin kan bli nödvändig för att bibehålla de terapeutiska nivåerna.
Interaktion med starkt proteinbundna läkemedel är inte sannolik då lanreotid endast binder till serumproteiner i måttlig utsträckning.
Begränsade publicerade data tyder på att samtidig administrering av somatostatinanaloger och bromokriptin kan öka tillgängligheten av bromokriptin.
Samtidig administration av hjärtfrekvenssänkande läkemedel (t.ex. betablockerare) kan förstärka den hjärtfrekvensminskning som normalt ses med lanreotid. Dosjustering av sådan samtidig medicinering kan bli nödvändig.
Begränsade publicerade data tyder på att somatostatinanaloger kan minska metabolisk clearance av substanser som metaboliseras av cytokrom‑P450-enzymer, vilket kan bero på tillväxthormonhämningen. Eftersom det inte kan uteslutas att lanreotid kan ha denna effekt ska andra läkemedel som metaboliseras av CYP3A4 och som har ett lågt terapeutiskt index (t.ex. kinidin, terfenadin) användas med försiktighet.
Fertilitet, graviditet och amning
Graviditet
Det finns en begränsad mängd data (färre än 300 graviditeter) från användning av lanreotid hos gravida kvinnor.
Djurstudier har påvisat reproduktionstoxicitet men inga tecken på teratogena effekter (se avsnitt Prekliniska säkerhetsuppgifter). Den potentiella risken för människor är okänd.
Som en försiktighetsåtgärd bör användning av Myrelez undvikas under graviditet.
Amning
Det är inte känt om detta läkemedel utsöndras i bröstmjölk.
En risk för det nyfödda barnet/spädbarnet kan inte uteslutas. Myrelez ska inte användas under amning.
Fertilitet
Nedsatt fertilitet orsakad av hämning av GH-utsöndring observerades hos honråttor vid doser avsevärt högre än terapeutiska doser hos människa.
Effekter på förmågan att framföra fordon och använda maskiner
Myrelez har mindre eller måttlig effekt på förmågan att framföra fordon och använda maskiner. Inga studier om effekter på förmågan att framföra fordon och använda maskiner har utförts.
Yrsel har dock rapporterats för Myrelez (se avsnitt Biverkningar). Om en patient drabbas ska han/hon inte köra bil eller använda maskiner.
Biverkningar
Biverkningar som rapporterats av patienter med akromegali och GEP‑NET och som behandlats med lanreotid i kliniska prövningar finns listade under motsvarande organsystem enligt följande klassificering:
Mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10), mindre vanliga (≥ 1/1 000, < 1/100), ingen känd frekvens (kan inte beräknas från tillgängliga data).
De vanligaste förväntade biverkningarna vid behandling med lanreotid är gastrointestinala besvär (vanligast rapporterade är diarré och buksmärtor, oftast lindriga eller måttliga och övergående), kolelitiasis (ofta asymtomatisk) och reaktion vid injektionsstället (smärta, knutor och förhårdnader).
Biverkningsprofilen är likartad för alla indikationer.
| Organklass | Mycket vanliga (≥ 1/10) | Vanliga (≥ 1/100, < 1/10) | Mindre vanliga (≥ 1/1 000, < 1/100) | Erfarenhet av säkerhet efter marknadsföring (ingen känd frekvens) | |
| Infektioner och infestationer | Abscess vid injektionsstället | ||||
| Metabolism och nutrition | Hypoglykemi, minskad aptit**, hyperglykemi, diabetes mellitus | ||||
| Psykiska störningar | Sömnlöshet* | ||||
| Centrala och perifera nervsystemet | Yrsel, huvudvärk*, letargi** | ||||
| Hjärtat | Sinusbradykardi | ||||
| Blodkärl | Värmevallningar* | ||||
| Magtarmkanalen | Diarré, lös avföring*, buksmärta | Illamående, kräkningar, förstoppning, gasbildning, bukdistension, obehagskänsla i buken*, dyspepsi, steatorré** | Missfärgad avföring* | Pankreatit | |
| Lever och gallvägar | Kolelitiasis | Gallgångsdilatation* | Kolecystit, kolangit | ||
| Muskuloskeletala systemet och bindväv | Muskuloskeletal smärta**, myalgi** | ||||
| Hud och subkutan vävnad | Alopeci, hypotrikos* | ||||
| Allmänna symtom och/eller symtom vid administreringsstället | Asteni, trötthet, reaktion vid injektionsstället (smärta, svullnad, förhårdnad, knöl, klåda) | ||||
| Undersökningar | Förhöjt ALAT*, onormalt ASAT*, onormalt ALAT*, förhöjt bilirubin i blodet*, förhöjt blodglukos*, förhöjt glykosylerat hemoglobin*, viktminskning, minskad mängd pankreasenzymer** | Förhöjt ASAT*, förhöjt alkaliskt fostafas i blodet*, onormalt bilirubin i blodet*, minskad mängd natrium i blodet* | |||
| Immunsystemet | Allergiska reaktioner (omfattande angioödem, anafylaxi, överkänslighet) | ||||
* baserat på poolade studier genomförda på patienter med akromegali
** baserat på poolade studier genomförda på patienter med GEP‑NET
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Vid överdosering ska symtomatisk behandling sättas in.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Hypofys- och hypotalamushormoner samt analoger; somatostatin och analoger, ATC-kod: H01C B03
Verkningsmekanism
Lanreotid är en oktapeptid härledd från endogent somatostatin. Liksom somatostatin hämmar lanreotid ett flertal endokrina, neuroendokrina, exokrina och parakrina funktioner. Lanreotid har hög affinitet till de humana somatostatinreceptorerna (SSTR) 2 och 5 och minskad bindningsaffinitet för humana SSTR 1, 3 och 4. Aktiviteten vid humana SSTR 2 och 5 anses vara den primära mekanismen för tillväxthormonhämning. Lanreotid är mer aktiv än naturligt somatostatin och uppvisar en längre verkningstid.
Lanreotid hämmar liksom somatostatin exokrin sekretion inklusive den basala utsöndringen av motilin, matsmältningshämmande peptid och pankreaspolypeptid, men har ingen signifikant effekt på fastande sekretin eller gastrinutsöndring. Utöver detta minskar lanreotid även plasmanivåerna av kromogranin A och 5-HIAA (5-hydroxiindolättiksyra) i urin hos GEP-NET-patienter med förhöjda nivåer av dessa tumörmarkörer. Lanreotid hämmar i stor utsträckning måltidsinducerad ökning av övre tarmkäxets arteriella blodflöde och det portala venösa blodflödet. Lanreotid reducerar signifikant prostaglandin E1-stimulerad tarmsekretion av vatten, natrium, kalium och klorid. Lanreotid reducerar prolaktinnivåer hos långtidsbehandlade patienter med akromegali.
I en öppen studie administrerades lanreotid 120 mg var 28:e dag i 48 veckor till 90 tidigare obehandlade akromegalipatienter som diagnostiserats med hypofysemakroadenom.
En tumörvolymminskning på ≥ 20 % konstaterades hos 63 % av patienterna (95 % KI: 52 %–73 %). Vid vecka 48 var den genomsnittliga procentuella minskningen av tumörvolymen 26,8 %. GH-nivåerna var lägre än 2,5 ug/l hos 77,8 % av patienterna och IGF-1-nivåerna normaliserades hos 50 % av patienterna. Normaliserade IGF-1-nivåer i kombination med GH-nivåer under 2,5 ug/l observerades hos 43,5 % av patienterna. De flesta av patienterna rapporterade en lindring av akromegalisymtom såsom trötthet, kraftiga svettningar, artralgi och svullnad i mjukvävnad. Både tidig och fortsatt minskning av tumörvolymen samt lägre koncentrationer av tillväxthormon och IGF‑1 sågs från vecka 12. Patienter som förväntades kräva kirurgi eller strålbehandling av hypofysen under studieperioden exkluderades ur studien.
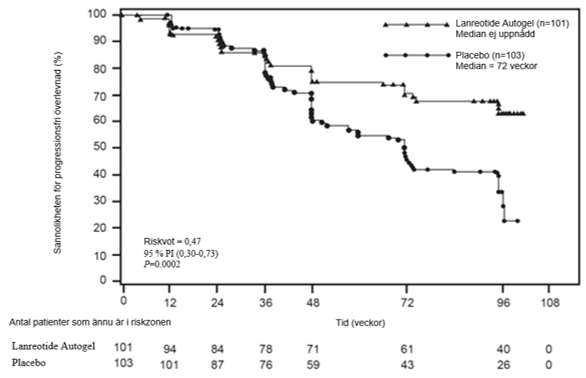
I en randomiserad, dubbelblind, placebokontrollerad, fas III multicenterstudie som pågick under en bestämd tid på 96 veckor, undersöktes den antiproliferativa effekten av lanreotid hos patienter med gastroenteropankreatiska neuroendokrina tumörer.
Patienter randomiserades i förhållandet 1:1 till att få antingen lanreotid 120 mg var 28:e dag (n = 101) eller placebo (n = 103). Randomiseringen stratifierades för tidigare behandling vid påbörjandet av studien och för progression vid baslinjen, mätt med RECIST 1.0 (Response Evaluation Criteria in Solid Tumors), under en 3 till 6 månaders screeningfas.
Patienterna hade metastaserande och/eller lokalt avancerad inoperabel sjukdom med histologiskt bekräftad väl eller måttligt differentierad tumör främst lokaliserad i pankreas (44,6 % patienter), midgut (35,8 %), hindgut (6,9 %) eller med annan/okänd primär lokalisering (12,7 %).
69 % av patienterna med GEP‑NET hade tumörgrad 1 (G1), definierad antingen av proliferationsindex Ki-67 ≤ 2 % (50,5 % av den totala patientpopulationen) eller ett mitotiskt index < 2 mitoser/10 HPF (18,5 % av den totala patientpopulationen) och 30 % av patienterna med GEP-NET hade tumörer i det lägre intervallet av grad 2 (G2) (definierad som ett Ki-67-värde > 2 % - ≤ 10 %). För 1 % av patienterna fanns ingen information om grad. Studien exkluderade patienter med GEP-NET G2 med högre cellulär proliferationsvärde (Ki-67 > 10 % - ≤ 20 %) och GEP neuroendokrina karcinom G3 (Ki-67-index > 20 %).
Totalt hade 52,5 % av patienterna en tumörbörda i levern ≤ 10 %, 14,5 % hade en tumörbörda i levern > 10 och ≤ 25 % och 33 % hade en tumörbörda i levern > 25 %.
Det primära effektmåttet var progressionsfri överlevnad (PFS), mätt som tid till antingen sjukdomsprogression enligt RECIST 1.0 eller död inom 96 veckor efter första administrering. PFS-analysen gjordes med en oberoende central granskning av röntgenbilder för bedömning av progression.
Tabell 1: Resultat för effekt i fas III-studien
Mediantid för progressionsfri överlevnad (veckor) | Riskkvot (95 % KI) | Minskning i risk för progression eller död | p-värde | |
Lanreotid (n = 101) | Placebo (n = 103) | |||
| > 96 veckor | 72,00 veckor (95 % KI: 48,57, 96,00) | 0,470 (0,304, 0,729) | 53 % | 0,0002 |
Figur 1: Kaplan‑Meier-kurvor över progressionsfri överlevnad
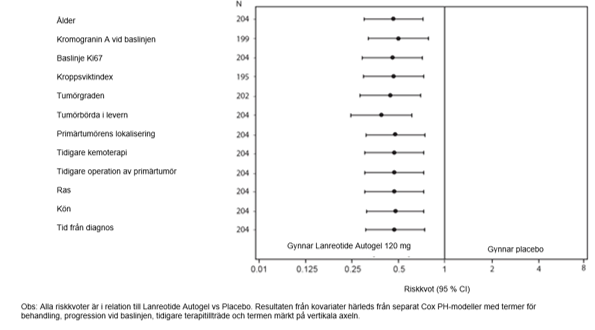
Den gynnsamma effekten av lanreotid med sänkt risk för progression eller död var konsekvent oberoende primärtumören lokalisering, tumörbörda i levern, tidigare kemoterapi, baslinjen för Ki-67, tumörgrad eller andra redan specificerade egenskaper som visas i figur 2.
I den totala studiepopulationen observerades kliniskt relevant fördel med lanreotidbehandling hos patienter med primärtumörer i pankreas och midgut samt tumörer av annan/okänd lokalisering. Det begränsade antalet patienter med tumörer i hindut (14/204) bidrog till svårigheter att tolka resultaten i denna subgrupp. Tillgängliga data tyder inte på någon fördel med lanreotid hos dessa patienter.
Figur 2: Resultat från kovariatanalys av PFS med Cox Proportional Hazards-modell
Crossover från placebo till lanreotidbehandling förekom i hos 45,6 % (47/103) av patienterna i den öppna förlängningsstudien.
Pediatrisk population
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för referensläkemedlet som innehåller lanreotid för alla grupper av den pediatriska populationen för akromegali och gigantism (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt). Europeiska läkemedelsmyndigheten har tagit med gastroenteropankreatiska neuroendokrina tumörer (med undantag för neuroblastom, neuroganglioblastom och feokromocytom) i listan över undantag för en kategori av läkemedel.
Farmakokinetiska egenskaper
Efter intravenös administrering till friska försökspersoner visade lanreotids farmakokinetiska parametrar begränsad extravaskulär distribution med en distributionsvolym vid steady-state på 16,1 liter. Totalt clearance var 23,7 l/timme, terminal halveringstid var 1,14 timmar och genomsnittlig residenstid var 0,68 timmar.
I studier som utvärderade utsöndring, utsöndrades mindre än 5 % av lanreotid i urinen och mindre än 0,5 % återfanns oförändrad i avföringen, vilket tyder på viss utsöndring via galla.
Efter djup subkutan administrering av lanreotid 60 mg, 90 mg och 120 mg till friska frivilliga ökade lanreotidkoncentrationen för att uppnå en genomsnittlig maximal serumkoncentration på 4,25, 8,39 respektive 6,79 ng/ml. Dessa värden av Cmax uppnås under den första dagen efter en administreringstid på 8, 12 respektive 7 timmar (medianvärden). Från den högsta nivån minskar koncentrationen av lanreotid i serum långsamt enligt en första ordningens kinetik med en terminal halveringstid på 23,3, 27,4 respektive 30,1 dagar. 4 veckor efter administreringen var genomsnittliga lanreotidnivåer i serum 0,9, 1,11 respektive 1,69 ng/ml. Absolut biotillgänglighet var 73,4, 69,0 respektive 78,4 %.
Efter djup subkutan administrering av lanreotid 60 mg, 90 mg och 120 mg till patienter med akromegali ökade lanreotidkoncentrationen för att uppnå en genomsnittlig maximal serumkoncentration på 1,6, 3,5 respektive 3,1 ng/ml. Dessa Cmax-värden uppnås under den första dagen efter en administreringstid på 6, 6 respektive 24 timmar. Från den högsta nivån minskar koncentrationen av lanreotid i serum långsamt enligt en första ordningens kinetik och 4 veckor efter administreringen var genomsnittliga lanreotidnivåer i serum 0,7, 1,0 respektive 1,4 ng/ml.
Steady state-nivåer av lanreotid i serum uppnåddes i genomsnitt efter 4 injektioner var 4:e vecka. Efter upprepad dosering var 4:e vecka var de genomsnittliga Cmax-värdena vid steady state 3,8, 5,7 och 7,7 ng/ml för 60, 90 respektive 120 mg och de genomsnittliga Cmin-värdena som erhålls är 1,8, 2,5 och 3,8 ng/ml. Fluktuationsindexet för topp/dalvärden var måttligt, varierande från 81 till 108 %.
Linjär farmakokinetisk frisättningsprofil sågs efter djup subkutan administrering av lanreotid 60, 90 och 120 mg hos patienter med akromegali.
I en populationsfarmakokinetisk analys med 290 GEP‑NET-patienter som fick lanreotid 120 mg, sågs initialt en snabb frisättning, genomsnittligt Cmax på 7,49 ± 7,58 ng/ml uppnåddes inom den första dagen efter en enda injektion. Steady state-koncentrationer uppnåddes efter 5 injektioner med lanreotid 120 mg var 28:e dag och varade fram till den sista uppmätningen (upp till 96 veckor efter den första injektionen). Vid steady state var genomsnittligt Cmax 13,9 ± 7,44 ng/ml och de genomsnittliga lägsta värdena 6,56 ± 1,99 ng/ml. Genomsnittliga terminal halveringstid var 49,8 ± 28,0 dagar.
Nedsatt njur-/leverfunktion
Personer med gravt nedsatt njurfunktion visar en cirka 2-faldig minskning i total serumclearance för lanreotid och därmed en ökning av halveringstiden och AUC. Hos patienter med måttligt till svårt nedsatt leverfunktion, observerades en minskning av clearance (30 %). Distributionsvolym och genomsnittlig residenstid ökade hos personer med alla grader av leverinsufficiens.
I den populationsfarmakokinetiska analysen av 165 GEP-NET-patienter med milt till måttligt nedsatt njurfunktion (106 respektive 59) som behandlades med Myrelez sågs ingen effekt på clearance av lanreotid. GEP-NET-patienter med svårt nedsatt njurfunktion har inte studerats.
Inga GEP-NET-patienter med nedsatt leverfunktion (enligt Child‑Pugh score) studerades.
Det är inte nödvändigt att ändra startdos för patienter med njur- eller leverfunktion, eftersom serumkoncentrationer av lanreotid i dessa populationer förväntas vara väl inom intervallet för serumkoncentrationer som tolereras väl hos friska personer.
Äldre patienter
Äldre personer visar en ökning av halveringstid och genomsnittlig residenstid jämfört med friska yngre personer. Det är inte nödvändigt att ändra startdos för äldre patienter eftersom serumkoncentrationer för lanreotid i denna population förväntas vara väl inom intervallet för serumkoncentrationer som tolereras väl hos friska personer.
I en populationsfarmakokinetisk analys av 122 GEP-NET-patienter i åldern 65 till 85 år, observerades ingen effekt av ålder på lanreotids clearance eller distributionsvolym.
Prekliniska säkerhetsuppgifter
I toxikologiska studier sågs effekter endast vid höga exponeringar/vid exponeringar avsevärt högre än klinisk exponering. Dessa effekter bedöms därför sakna klinisk relevans.
I studier som undersöker risk för cancerutveckling utförda på råttor och möss, har inga systematiska neoplastiska förändringar observerats vid doser som överstiger de som uppnås hos människa vid terapeutiska doser. Ökad förekomst av subkutana tumörer observerades vid injektionsstället, sannolikt beroende av den ökade dosfrekvensen hos djur (dagligen) jämfört med månatlig dosering hos människor och kan därför saknas klinisk relevans.
Lanreotid visade inte någon genotoxisk potential i in vitro och in vivo standardtester.
Lanreotid var inte teratogent hos råttor och kaniner. Embryo-fostertoxicitet observerades hos råttor (ökad förlust före implantation) och hos kaniner (ökad förlust efter implantation). Reproduktionsstudier på dräktiga råttor som gavs 30 mg/kg varannan vecka genom subkutan injektion (5 gånger den humana dosen baserat på jämförelse i kroppsyta) resulterade i minskad embryo‑/fosteröverlevnad. Studier på dräktiga kaniner som gavs subkutana injektioner på 0,45 mg/kg/dag (två gånger den humana terapeutiska exponeringen vid den maximala rekommenderade dosen på 120 mg, baserat på jämförelser av relativ kroppsyta) visade minskad fosteröverlevnad och ökade avvikelser i skelett/mjukvävnad hos foster.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Vatten för injektionsvätskor.
Koncentrerad ättiksyra (för pH-justering).
Inkompatibiliteter
Ej relevant.
Hållbarhet
3 år.
Efter att skyddspåsen av aluminium har öppnats ska läkemedlet användas omedelbart.
Särskilda förvaringsanvisningar
Förvaras i kylskåp (2 °C–8 °C) i originalförpackningen. Ljuskänsligt.
Läkemedlet kan åter läggas in i kylen (antalet temperaturvariationer får inte överstiga tre) för förvaring och senare användning, förutsatt att det förvarats i oöppnad skyddspåse vid högst 40 °C i sammanlagt högst 72 timmar.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
MYRELEZ injektioneste, liuos, esitäytetty ruisku
60 mg (L:ei) 0,5 ml (turvaneula) (427,50 €)
90 mg (L:ei) 0,5 ml (turvaneula) (565,39 €)
120 mg (L:ei) 0,5 ml (turvaneula) (681,33 €)
PF-selosteen tieto
Myrelez tillhandahålls i en förfylld spruta (av polypropen, försluten med en kolvpropp av termoplastisk elastomergummi och förseglad med ett lock av polypropen). Sprutan ligger i ett plasttråg förseglat i en aluminiumpåse tillsammans med en separat förpackad automatiskt stickskyddande nål för engångsbruk. Båda artiklarna är samförpackade i en kartong.
Kartong med en 0,5 ml spruta och en skicksskyddande nål (1,2 mm × 20 mm) i samma förpackning.
Flerpack med 3 kartonger, som var och en innehåller en 0,5 ml spruta och en stickskyddande nål (1,2 mm × 20 mm) i samma förpackning.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Läkemedlets utseende:
Vit till svagt gul halvfast beredning praktiskt taget fri från främmande partiklar.
Särskilda anvisningar för destruktion och övrig hantering
Injektionsvätskan i den förfyllda sprutan är klar att användas.
Ska användas omedelbart efter öppnande. Endast för engångsbruk. Använd inte om påsen är skadad eller tidigare öppnad.
Det är viktigt att läkemedlet injiceras exakt enligt anvisningarna i bipacksedeln.
Den använda injektionssprutan ska kastas i behållare avsedd för stickande/skärande avfall.
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
MYRELEZ injektioneste, liuos, esitäytetty ruisku
60 mg 0,5 ml
90 mg 0,5 ml
120 mg 0,5 ml
- Ylempi erityiskorvaus (100 %). Pahanlaatuiset kasvaimet, joita ei ole edellä erikseen mainittu (130).
- Peruskorvaus (40 %).
Atc-kod
H01CB03
Datum för översyn av produktresumén
06.03.2026
Yhteystiedot
Unit 17, Northwood House, Northwood Crescent, Northwood
D09 V504 Dublin 9
Ireland
+358 800 416231
www.advanzpharma.com
medicalinformation@advanzpharma.com