
XELJANZ oraaliliuos 1 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Lääkäri
Opas annostukseen, annosteluun, hoidon seurantaan ja haittavaikutusten hallintaan.
Potilas
Vaikuttavat aineet ja niiden määrät
Yksi millilitra oraaliliuosta sisältää tofasitinibisitraattia määrän, joka vastaa 1 mg tofasitinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi millilitra oraaliliuosta sisältää 2,39 mg propyleeniglykolia.
Yksi millilitra oraaliliuosta sisältää 0,9 mg natriumbentsoaattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet
Lääkemuoto
Oraaliliuos
Kliiniset tiedot
Käyttöaiheet
Tofasitinibi on tarkoitettu aktiivisen juveniilin idiopaattisen polyartriitin (JIA) (reumatekijäpositiivinen [RF+] tai reumatekijänegatiivinen [RF-]) ja laajeneva oligoartriitti), ja lasten psoriaasiartriitin hoitoon vähintään 2-vuotiaille potilaille, jotka eivät ole saaneet riittävää vastetta aiemmalle tautiprosessia hidastavalle reumalääkehoidolle (DMARD).
Tofasitinibi voidaan antaa yhdessä metotreksaatin (MTX) kanssa tai monoterapiana potilaille, jotka eivät siedä metotreksaattia tai joille metotreksaattihoito ei ole tarkoituksenmukaista.
Ehto
Hoito on aloitettava ja toteutettava käyttöaiheissa mainittujen sairauksien diagnosointiin ja hoitoon perehtyneen erikoislääkärin seurannassa.
Annostus ja antotapa
Hoito tulee aloittaa ja sitä tulee seurata tofasitinibin käyttöaiheina olevien sairauksien diagnosointiin ja hoitoon perehtyneen erikoislääkärin valvonnassa.
Annostus
Tofasitinibia voidaan käyttää monoterapiana tai yhdessä MTX:n kanssa.
Kaksivuotiaiden ja sitä vanhempien potilaiden painonmukainen suositusannos:
Taulukko 1: Tofasitinibiannos kaksivuotiaille ja vanhemmille potilaille, joilla on idiopaattinen juveniili polyartriitti ja lasten psoriaasiartriitti
Paino (kg) | Annos |
10 ‑ < 20 | 3,2 mg (3,2 ml oraaliliuosta) kaksi kertaa vuorokaudessa |
20 ‑ < 40 | 4 mg (4 ml oraaliliuosta) kaksi kertaa vuorokaudessa |
≥ 40 | 5 mg (5 ml oraaliliuosta tai 5 mg kalvopäällysteinen tabletti) kaksi kertaa vuorokaudessa |
Vähintään 40 kg painavat potilaat, joita hoidetaan tofasitinibioraaliliuoksella (5 ml) kaksi kertaa vuorokaudessa, voivat siirtyä saamaan kalvopäällysteisiä tofasitinibitabletteja (5 mg) kaksi kertaa vuorokaudessa. Alle 40 kg painavat potilaat eivät voi siirtyä tofasitinibia sisältävästä oraaliliuoshoidosta tablettihoitoon.
Annoksen muuttaminen
Annoksen muuttaminen ei ole tarpeen, jos valmistetta käytetään yhdessä MTX:n kanssa.
Hoidon keskeyttäminen ja lopettaminen
Saatavilla olevat tiedot viittaavat siihen, että kliinistä paranemista on havaittavissa 18 viikon kuluessa tofasitinibihoidon aloittamisesta. Hoidon jatkamista on harkittava tarkasti, jos potilaalla ei havaita kliinistä paranemista tämän ajan kuluessa.
Jos potilaalle kehittyy vakava infektio, tofasitinibihoito on keskeytettävä siihen saakka, kunnes infektio on saatu hallintaan.
Hoito voi olla tarpeen keskeyttää annosriippuvaisten laboratorioarvojen poikkeamien, kuten lymfopenian, neutropenian ja anemian, hoitamiseksi. Alla olevissa taulukoissa 2, 3 ja 4 on esitetty suositukset hoidon keskeyttämisestä tai lopettamisesta laboratorioarvojen poikkeamien vaikeusasteen perusteella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hoidon aloittamista ei suositella pediatrisille potilaille, joiden absoluuttinen lymfosyyttimäärä (B-Lymf) on alle 0,75 x 109/l.
Taulukko 2. Matala absoluuttinen lymfosyyttimäärä
Matala absoluuttinen lymfosyyttimäärä (B-Lymf) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | |
Laboratorioarvo | Suositus |
B-Lymf ≥ 0,75 | Annos pidetään ennallaan. |
B-Lymf 0,50–0,75 | Jos lymfosyyttimäärä pysyy pitkään tällä välillä (kaksi peräkkäistä arvoa tälle välille rutiinimäärityksessä), annosta on pienennettävä tai hoito on keskeytettävä, kunnes B-Lymf on yli 0,75. Tofasitinibi 5 mg -annoksia kaksi kertaa vuorokaudessa käyttävien potilaiden hoito on keskeytettävä. Kun B-Lymf on yli 0,75, jatketaan kliinisesti tarkoituksenmukaista hoitoa. |
B-Lymf < 0,5 | Jos laboratorioarvo varmistuu 7 päivän kuluessa tehdyssä uusintamäärityksessä, hoito on lopetettava. |
Hoidon aloittamista ei suositella pediatrisille potilaille, joiden absoluuttinen neutrofiilien määrä (B-Neut) on alle 1,2 x 109/l.
Taulukko 3. Matala absoluuttinen neutrofiilien määrä
Matala absoluuttinen neutrofiilien määrä (B-Neut) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | |
Laboratorioarvo | Suositus |
B-Neut > 1,0 | Annos pidetään ennallaan. |
B-Neut 0,5–1,0 | Jos neutrofiilimäärä pysyy pitkään tällä välillä (kaksi peräkkäistä arvoa tälle välille rutiinimäärityksessä), annosta on pienennettävä tai hoito on keskeytettävä, kunnes B-Neut on yli 1,0. Tofasitinibi 5 mg -annoksia kaksi kertaa vuorokaudessa käyttävien potilaiden hoito on keskeytettävä. Kun B-Neut on yli 1,0, jatketaan kliinisesti tarkoituksenmukaista hoitoa. |
B-Neut < 0,5 | Jos laboratorioarvo varmistuu 7 päivän kuluessa tehdyssä uusintamäärityksessä, hoito on lopetettava. |
Hoidon aloittamista ei suositella pediatrisille potilaille, joiden hemoglobiiniarvo on alle 100 g/l.
Taulukko 4. Matala hemoglobiiniarvo
Matala hemoblogiiniarvo (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | |
Laboratorioarvo | Suositus |
Laskenut ≤ 20 g/l lähtötasosta ja pitoisuus on ≥ 90 g/l | Annos pidetään ennallaan. |
Laskenut > 20 g/l lähtötasosta tai pitoisuus on < 80 g/l (varmistettu uusintamäärityksellä) | Hoito on keskeytettävä, kunnes hemoglobiiniarvo on korjautunut normaaliksi. |
Yhteisvaikutukset
Jos potilas käyttää voimakkaita sytokromin P450 (CYP) 3A4 estäjiä (esim. ketokonatsolia) tai jos potilas käyttää samanaikaisesti yhtä tai useampaa lääkevalmistetta, joista aiheutuu sekä CYP3A4:n toiminnan kohtalainen estyminen että CYP2C19:n toiminnan voimakas estyminen (esim. flukonatsoli) (ks. kohta Yhteisvaikutukset), tofasitinibin kokonaisvuorokausiannos on pienennettävä seuraavasti: yksi 5 mg kalvopäällysteinen tabletti kerran vuorokaudessa tai painoon perustuva vastaava määrä kerran vuorokaudessa potilailla, jotka saavat 5 mg kalvopäällysteisiä tabletteja, tai painoon perustuvan vastaavan määrän, kahdesti vuorokaudessa.
Erityisryhmät
Iäkkäät
Tofasitinibioraaliliuoksen turvallisuutta ja tehokkuutta ei ole varmistettu iäkkäillä potilailla.
Maksan vajaatoiminta
Taulukko 5. Maksan vajaatoimintaa sairastavien potilaiden annoksen muuttaminen
Maksan vajaatoiminnan aste | Luokitus | Eri oraaliliuoksen annoksen muuttaminen potilaille, joilla on maksan vajaatoiminta |
Lievä | Child–Pugh A | Annosta ei tarvitse muuttaa. |
Keskivaikea | Child–Pugh B | Annos on pienennettävä 5 mg:aan tai painoon perustuvaan vastaavaan määrään kerran vuorokaudessa, kun annos potilaille, joiden maksan toiminta on normaali, on 5 mg tai painoon perustuva vastaava määrä kaksi kertaa vuorokaudessa (ks. kohta Farmakokinetiikka). |
Vaikea | Child–Pugh C | Tofasitinibia ei pidä käyttää potilaille, joilla on vaikea maksan vajaatoiminta (ks. kohta Vasta-aiheet). |
Munuaisten vajaatoiminta
Taulukko 6. Munuaisten vajaatoimintaa sairastavien potilaiden annoksen muuttaminen
Munuaisten vajaatoiminnan aste | Kreatiniinipuhdistuma | Eri oraaliliuoksen annoksen muuttaminen potilaille, joilla on munuaisten vajaatoiminta |
Lievä | 50–80 ml/min | Annosta ei tarvitse muuttaa. |
Keskivaikea | 30–49 ml/min | Annosta ei tarvitse muuttaa. |
Vaikea (mukaan lukien hemodialyysia saavat potilaat) | < 30 ml/min | Annos on pienennettävä 5 mg:aan tai painoon perustuvaan vastaavaan määrään kerran vuorokaudessa, kun annos potilaille, joiden munuaisten toiminta on normaali, on 5 mg tai painoon perustuva vastaava määrä kaksi kertaa vuorokaudessa. Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden hoitoa on jatkettava pienennetyllä annoksella myös hemodialyysin jälkeen (ks. kohta Farmakokinetiikka). |
Pediatriset potilaat (alle 2-vuotiaat lapset)
Tofasitinibin turvallisuutta ja tehoa alle kahden vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Suun kautta.
Tofasitinibioraaliliuos annetaan käyttämällä mukana tulevaa pulloon kiinnitettyä sovitinta ja suun kautta annosteluun käytettävää ruiskua.
Tofasitinibi annetaan suun kautta ruokailun yhteydessä tai tyhjään mahaan.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Aktiivinen tuberkuloosi, vakavat infektiot, kuten sepsis tai opportunisti-infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Vaikea maksan vajaatoiminta (ks. kohta Annostus ja antotapa).
- Raskaus ja imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Tofasitinibia tulee käyttää seuraaville potilasryhmille vain, jos soveltuvia hoitovaihtoehtoja ei ole käytettävissä:
|
Samanaikainen käyttö muiden lääkehoitojen kanssa
Tofasitinibia ei ole tutkittu yhdessä biologisten lääkkeiden, kuten TNF:n estäjien, interleukiini-1R:n (IL-1R) estäjien, IL-6R:n estäjien, CD20-antigeenin monoklonaalisten vasta-aineiden, IL‑17:n estäjien, IL‑12/IL‑23:n estäjien, anti-integriinien, selektiivisten kostimulaation modulaattorien ja voimakkaiden immunosuppressiivisten lääkeaineiden, kuten atsatiopriinin, 6-merkaptopuriinin, siklosporiinin ja takrolimuusin, kanssa. Näiden samanaikaista käyttöä pitää välttää johtuen voimistuneen immunosuppression ja lisääntyneen infektioriskin mahdollisuudesta.
Kliinisissä nivelreumatutkimuksissa esiintyi haittavaikutuksia enemmän käytettäessä tofasitinibia yhdistelmänä metotreksaatin kanssa kuin käytettäessä tofasitinibia monoterapiana.
Tofasitinibin samanaikaista käyttöä yhdessä fosfodiesteraasi 4:n estäjien kanssa ei ole tutkittu tofasitinibilla tehdyissä kliinisissä tutkimuksissa.
Laskimotromboembolia
Tofasitinibia käyttävillä potilailla on havaittu vakavia laskimotromboembolioita, mukaan lukien keuhkoembolioita (KE), joista osa johti potilaan kuolemaan, kallonsisäisiä sinustrombooseja ja syviä laskimotukoksia (SLT) (ks. taulukko 8 kohdassa Haittavaikutukset). Myyntiluvan myöntämisen jälkeisessä satunnaistetussa turvallisuustutkimuksessa oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä, tofasitinibilla havaittiin annosriippuvaisesti suurentunut laskimotromboembolian riski verrattuna TNF:n estäjiin (ks. kohdat Haittavaikutukset ja Farmakodynamiikka).
Tutkimuksessa tehtiin eksploratiivinen post hoc -analyysi potilaista, joilla oli tunnettuja laskimotromboembolian riskitekijöitä. Laskimotromboembolioita havaittiin yleisimmin tofasitinibihoitoa saaneilla potilailla, joilla 12 kuukauden hoidon jälkeen D-dimeeripitoisuus oli ≥ 2× ULN, kuin niillä, joiden D-dimeeripitoisuus oli < 2× ULN. TNF:n estäjähoitoa saaneilla potilailla tämä havainto ei ollut selkeä. Vähäinen laskimotromboemboliatapahtumien määrä ja rajoitettu D-dimeeritestien saatavuus (tutkittiin ainoastaan tutkimuksen alussa, 12 kuukauden kohdalla ja tutkimuksen lopussa) rajoittavat näiden tulosten tulkintaa. Potilailla, joilla ei ollut laskimotromboemboliaa tutkimuksen aikana, keskimääräiset D-dimeeritasot olivat merkittävästi alhaisempia 12 hoitokuukauden kohdalla verrattuna lähtötasoon kaikissa hoitohaaroissa. D-dimeeritasoja ≥ 2× ULN havaittiin kuitenkin 12 kuukauden kohdalla noin 30 %:lla potilaista, joilla ei ollut laskimotromboembolisia tapahtumia. Tämä osoittaa, ettei D-dimeeritesti ollut tässä tutkimuksessa kovin spesifinen.
Potilaille, joilla on merkittävien sydän- tai verisuonitapahtumien tai syövän riskitekijöitä (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet ”Merkittävät sydän- ja verisuonitapahtumat, mukaan lukien sydäninfarkti” ja ”Syövät ja lymfoproliferatiiviset sairaudet”), tulisi käyttää tofasitinibia vain, jos soveltuvia hoitovaihtoehtoja ei ole käytettävissä.
Tofasitinibin käytössä on noudatettava varovaisuutta, jos potilaalla on muita kuin merkittäviin sydän- ja verisuonitapahtumiin ja syöpään liittyviä laskimotromboembolian riskitekijöitä. Muita kuin merkittäviin sydän- ja verisuonitapahtumiin ja syöpään liittyviä laskimotromboembolian riskitekijöitä ovat aiempi laskimotromboembolia, potilaalle tehtävä suuri leikkaus, immobilisaatio, yhdistelmäehkäisytablettien tai hormonikorvausvalmisteiden käyttö, periytyvä hyytymishäiriö. Potilaat on tutkittava säännöllisesti uudelleen tofasitinibihoidon aikana laskimotromboembolian riskin muutosten arvioimiseksi.
Harkitse D-dimeeritasojen testaamista noin 12 hoitokuukauden jälkeen nivelreumapotilailla, joilla on tunnettuja laskimotromboembolian riskitekijöitä. Jos D-dimeeritestitulos on ≥ 2× ULN, varmistu ennen päätöstä hoidon jatkamisesta, että kliiniset hyödyt ylittävät tofasitinibihoidon jatkamisen riskit.
Potilas, jolla on laskimotromboembolian merkkejä ja oireita, on viipymättä tutkittava, ja tofasitinibihoito on lopetettava sen annostuksesta tai käyttöaiheesta riippumatta.
Verkkokalvon laskimotukos
Tofasitinibihoitoa saaneilla potilailla on raportoitu verkkokalvon laskimotukoksia (ks. kohta Haittavaikutukset). Potilaita pitää neuvoa hakeutumaan viipymättä lääkäriin, jos heille ilmaantuu verkkokalvon laskimotukokseen viittaavia oireita.
Vakavat infektiot
Tofasitinibihoitoa saavilla potilailla on raportoitu vakavia ja toisinaan kuolemaan johtaneita bakteerien, mykobakteerien, invasiivisten sienten, virusten tai muiden opportunististen patogeenien aiheuttamia infektioita (ks. kohta Haittavaikutukset). Aasian maantieteellisellä alueella on suurempi opportunisti-infektioiden riski (ks. kohta Haittavaikutukset). Kortikosteroideja saavat nivelreumapotilaat saattavat olla alttiita infektioille.
Tofasitinibihoitoa ei pidä aloittaa, jos potilaalla on aktiivinen infektio, paikalliset infektiot mukaan lukien.
Hoidon riskit ja hyödyt pitää arvioida ennen tofasitinibihoidon aloittamista,
-
jos potilaalla on toistuvia infektioita
-
jos potilaalla on aiemmin ollut jokin vakava infektio tai opportunisti-infektio
-
jos potilas on asunut tai matkustanut alueilla, joilla esiintyy endeemisiä mykooseja
-
jos potilaalla on perussairauksia, joiden vuoksi hän voi olla alttiimpi infektioille.
Potilasta pitää seurata tarkoin tofasitinibihoidon aikana ja sen jälkeen infektioiden merkkien ja oireiden havaitsemiseksi. Hoito on keskeytettävä, jos potilaalle kehittyy vakava infektio, opportunisti-infektio tai sepsis. Jos potilaalle kehittyy tofasitinibihoidon aikana uusi infektio, hänelle on tehtävä viipymättä kattavat immuunipuutteisiin potilaisiin sovellettavat diagnostiset kokeet, hoito asianmukaisilla mikrobilääkkeillä on aloitettava ja potilaan tilaa on seurattava tarkoin.
Infektioiden ilmaantuvuus on iäkkäillä ja diabetespotilailla yleensä tavanomaista suurempi, joten iäkkäiden ja diabetespotilaiden hoidossa on noudatettava varovaisuutta (ks. kohta Haittavaikutukset). Tofasitinibihoitoa voidaan käyttää 65-vuotiaille ja sitä vanhemmille potilaille vain, jos muita soveltuvia hoitovaihtoehtoja ei ole käytettävissä (ks. kohta Farmakodynamiikka).
Infektioriski saattaa suurentua lymfopenian vaikeusasteen pahentuessa, joten potilaan yksilöllisen infektioriskin arvioinnissa pitää ottaa huomioon lymfosyyttimäärä. Lymfopeniaan liittyvät hoidon lopettamista ja lymfopenian seurantaa koskevat kriteerit esitetään kohdassa Annostus ja antotapa.
Tuberkuloosi
Hoidon riskit ja hyödyt pitää arvioida ennen tofasitinibihoidon aloittamista,
-
jos potilas on altistunut tuberkuloosille
-
jos potilas on asunut tai matkustanut alueilla, joilla esiintyy endeemistä tuberkuloosia.
Potilas on tutkittava ja testattava piilevän ja aktiivisen infektion toteamiseksi ennen tofasitinibihoidon aloittamista sekä soveltuvien ohjeiden mukaisesti hoidon aikana.
Piilevää tuberkuloosia sairastavat potilaat (positiivinen testitulos) on hoidettava tavanomaisella mykobakteerilääkityksellä ennen tofasitinibihoidon aloittamista.
Tuberkuloosilääkitystä on myös harkittava ennen tofasitinibihoidon aloittamista, jos potilaan testitulos on negatiivinen, mutta potilaalla on aiemmin ollut piilevä tai aktiivinen tuberkuloosi eikä sen riittävästä hoidosta voida varmistua, tai jos potilaan testitulos on negatiivinen, mutta hänellä on tuberkuloosi-infektion riskitekijöitä. Tuberkuloosin hoitoon perehtyneen terveydenhuollon ammattilaisen konsultointia suositellaan päätöksenteon tueksi, kun selvitetään tuberkuloosihoidon tarkoituksenmukaisuutta yksittäiselle potilaalle. Potilasta on seurattava tarkoin tuberkuloosin merkkien ja oireiden kehittymisen havaitsemiseksi. Tämä koskee myös potilaita, joilla piilevän tuberkuloosin testitulos ennen hoidon aloittamista oli negatiivinen.
Virusten uudelleenaktivoituminen
Tofasitinibia saaneilla potilailla on havaittu virusten uudelleenaktivoitumista ja herpes-virusten (esim. Herpes zoster) uudelleenaktivoitumista (ks. kohta Haittavaikutukset).
Vyöruusun (Herpes zoster) riski vaikuttaa olevan muita suurempi tofasitinibihoitoa saaneilla
-
japanilaisilla tai korealaisilla potilailla
-
potilailla, joiden B-Lymf on alle 1,0 x 109/l (ks. kohta Annostus ja antotapa)
-
potilailla, jotka ovat pitkään sairastaneet nivelreumaa ja jotka ovat aiemmin saaneet kahta tai useampaa tautiprosessia hidastavaa biologista reumalääkettä (bDMARD).
Tofasitinibin vaikutusta kroonisen virushepatiitin uudelleenaktivoitumiseen ei tiedetä. Kliinisiin tutkimuksiin ei otettu mukaan potilaita, jotka osoittautuivat seulonnassa hepatiitti B- tai C-viruksen osalta positiivisiksi. Virushepatiitin seulonta pitää tehdä kliinisten ohjeistojen mukaisesti ennen tofasitinibihoidon aloittamista.
Ainakin yksi vahvistettu etenevä multifokaalinen leukoenkefalopatia (PML) -tapaus on raportoitu nivelreumapotilailla, jotka ovat saaneet tofasitinibia valmisteen markkinoille tulon jälkeen. PML voi johtaa kuolemaan ja se on otettava huomioon tehtäessä erotusdiagnoosia immunosuppressiopotilailla, joilla on uusia tai pahenevia neurologisia oireita.
Merkittävät sydän- ja verisuonitapahtumat (mukaan lukien sydäninfarkti)
Tofasitinibia käyttävillä potilailla on todettu merkittäviä sydän- ja verisuonitapahtumia (MACE).
Myyntiluvan myöntämisen jälkeen tehdyssä valmisteen turvallisuutta koskeneessa satunnaistetussa tutkimuksessa tutkittiin vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Tutkimuksessa sydäninfarktien ilmaantuvuus lisääntyi tofasitinibilla hoidetuilla potilailla TNF:n estäjillä hoidettuihin potilaisiin verrattuna (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). 65-vuotiaille ja sitä vanhemmille potilaille, pitkään tupakoineille tai aiemmin pitkään tupakoineille sekä potilaille, joilla on aiemmin ollut ateroskleroottinen valtimotauti tai joilla on muita sydän- ja verisuonitapahtumien riskitekijöitä, tofasitinibia tulisi käyttää vain, jos soveltuvia hoitovaihtoehtoja ei ole käytettävissä (ks. kohta Farmakodynamiikka).
Syövät ja lymfoproliferatiiviset sairaudet
Tofasitinibi voi vaikuttaa elimistön syöpää torjuviin puolustusmekanismeihin.
Myyntiluvan myöntämisen jälkeen tehdyssä valmisteen turvallisuutta koskeneessa satunnaistetussa tutkimuksessa tutkittiin vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Tutkimuksessa tofasitinibilla hoidetuilla potilailla syövän (erityisesti ei-melanoottisen ihosyövän, keuhkosyövän ja lymfoomien) ilmaantuvuus lisääntyi TNF:n estäjillä hoidettuihin potilaisiin verrattuna (ks. kohdat Haittavaikutukset ja Farmakodynamiikka).
Tofasitinibilla hoidetuilla potilailla on todettu ei-melanoottista ihosyöpää, keuhkosyöpää ja lymfoomia myös muissa kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeen.
Kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeen tofasitinibilla hoidetuilla potilailla on havaittu myös muita syöpiä, muun muassa rintasyöpää, melanoomaa, eturauhassyöpää ja haimasyöpää.
65-vuotiaille ja sitä vanhemmille potilaille, pitkään tupakoineille tai aiemmin pitkään tupakoineille sekä potilaille, joilla on muita syöpään liittyviä riskitekijöitä (aktiivinen tai aiemmin sairastettu syöpä, muu kuin onnistuneesti hoidettu ei-melanoottinen ihosyöpä), tofasitinibia tulisi käyttää vain, jos soveltuvia hoitovaihtoehtoja ei ole käytettävissä (ks. kohta Farmakodynamiikka).
Säännöllistä ihon tutkimista suositellaan kaikille potilaille, erityisesti niille, joilla on tavanomaista suurempi ihosyöpäriski (ks. taulukko 7 kohdassa Haittavaikutukset).
Interstitiaalinen keuhkosairaus
Varovaisuutta suositellaan myös kroonista keuhkosairautta sairastavien tai aiemmin sairastaneiden potilaiden hoidossa, koska he saattavat olla alttiimpia infektioille. Tofasitinibihoitoa saaneilla potilailla on raportoitu kliinisissä nivelreumatutkimuksissa ja valmisteen markkinoille tulon jälkeen interstitiaalista keuhkosairautta (joka on toisinaan johtanut potilaan kuolemaan), mutta Janus-kinaasi (JAK) inhibition merkitystä näissä tapahtumissa ei tiedetä. Aasialaisilla nivelreumapotilailla tiedetään olevan tavanomaista suurempi interstitiaalisen keuhkosairauden riski, joten tämän potilasryhmän hoidossa on noudatettava varovaisuutta.
Maha-suolikanavan perforaatio
Kliinisissä tutkimuksissa on raportoitu maha-suolikanavan perforaatioita, mutta Janus-kinaasin (JAK) eston merkitystä näiden tapausten ilmaantumiseen ei kuitenkaan tiedetä. Tofasitinibia on käytettävä varoen, jos potilaalla voi olla suurentunut maha-suolikanavan perforaatioiden riski (esim. divertikuliittia aiemmin sairastaneet potilaat, kortikosteroideja ja/tai ei-steroidaalisia tulehduskipulääkkeitä samanaikaisesti käyttävät potilaat). Jos potilaalle ilmaantuu uusia vatsaoireita ja -löydöksiä, potilas on tutkittava viipymättä, jotta maha-suolikanavan perforaatio voidaan todeta varhaisvaiheessa.
Luunmurtumat
Tofasitinibihoitoa saaneilla potilailla on havaittu luunmurtumia.
Tofasitinibin käytössä potilaille, joilla on tunnettuja luunmurtumien riskitekijöitä, kuten iäkkäille potilaille, naispotilaille sekä kortikosteroideja käyttäville potilaille, on noudatettava varovaisuutta käyttöaiheesta ja annostuksesta riippumatta.
Maksaentsyymit
Joillakin potilailla havaittiin tofasitinibihoidon yhteydessä tavanomaista yleisemmin maksaentsyymipitoisuuksien kohoamista (ks. kohta Haittavaikutukset maksaentsyymikokeet). Tofasitinibihoidon aloittamista on harkittava tarkkaan, jos potilaan alaniiniaminotransferaasi- (ALAT) tai aspartaattiaminotransferaasipitoisuus (ASAT) on kohonnut, sekä erityisesti silloin, kun tofasitinibihoito on aloitettu yhdessä potentiaalisten maksatoksisten lääkevalmisteiden, kuten MTX:n, kanssa. Hoidon aloittamisen jälkeen suositellaan seuraamaan maksa-arvoja säännöllisesti sekä tutkimaan kohonneiden maksaentsyymipitoisuuksien mahdollinen syy, jotta mahdollinen lääkkeestä aiheutuva maksavaurio tunnistetaan. Jos lääkkeen epäillään aiheuttaneen maksavaurion, tofasitinibin anto on keskeytettävä, kunnes tämä diagnoosi on suljettu pois.
Yliherkkyys
Myyntiin tulon jälkeen on ilmoitettu yliherkkyystapauksista, jotka liittyvät tofasitinibin antoon. Allergisia reaktioita kuten angioedeema ja urtikaria; vakavia reaktioita on esiintynyt. Jos potilaalle kehittyy vakava allerginen tai anafylaktinen reaktio, tofasitinibihoito on heti keskeytettävä.
Laboratoriokokeiden tulokset
Lymfosyytit
Tofasitinibihoitoon liittyi suurentunut lymfopenian ilmaantuvuus verrattuna lumelääkkeeseen. Lymfosyyttimäärään alle 0,75 x 109/l liittyi suurentunut vakavien infektioiden ilmaantuvuus. Tofasitinibihoidon aloittamista tai jatkamista ei suositella, jos potilaan lymfosyyttimäärä on varmistetusti alle 0,75 x 109/l. Lymfosyyttimäärä on määritettävä lähtötilanteessa, ja sen jälkeen sitä on seurattava kolmen kuukauden välein. Lymfosyyttimäärän perusteella suositellut muutokset hoitoon, ks. kohta Annostus ja antotapa.
Neutrofiilit
Tofasitinibihoitoon liittyi suurentunut neutropenian (neutrofiilejä alle 2,0 x 109/l) ilmaantuvuus verrattuna lumelääkkeeseen. Tofasitinibihoidon aloittamista ei suositella aikuisille, jos potilaan B-Neut on alle 1,0 x 109/l, ja pediatrisille potilaille, jos B-Neut on alle 1,2 x 109/l. B-Neut on määritettävä lähtötilanteessa ja uudelleen 4−8 viikon kuluttua hoidon aloittamisesta sekä tämän jälkeen 3 kuukauden välein. Neutrofiilien absoluuttisen määrän perusteella suositellut muutokset hoitoon, ks. kohta Annostus ja antotapa.
Hemoglobiini
Tofasitinibihoidon on todettu laskevan hemoglobiiniarvoja. Tofasitinibihoidon aloittamista ei suositella aikuisille, jos potilaan hemoglobiiniarvon on alle 90 g/l, ja pediatrisille potilaille, jos hemoglobiiniarvo on alle 100 g/l. Hemoglobiiniarvo on määritettävä lähtötilanteessa ja uudelleen 4–8 viikon kuluttua hoidon aloittamisesta sekä tämän jälkeen 3 kuukauden välein. Hemoglobiiniarvon perusteella suositellut muutokset hoitoon, ks. kohta Annostus ja antotapa.
Lipidien seuranta
Tofasitinibihoitoon liittyi lipidien, kuten kokonaiskolesteroli-, LDL-kolesteroli- ja HDL-kolesterolipitoisuuden, suurenemista. Suurimmat poikkeamat havaittiin yleensä 6 viikon kuluessa hoidon aloittamisesta. Lipidit on määritettävä 8 viikon kuluttua tofasitinibihoidon aloittamisesta. Potilaan hyperlipidemia on hoidettava kliinisten suositusten mukaisesti. Tofasitinibiin liittyvä kohonnut kokonais- ja LDL-kolesterolipitoisuus voidaan saada pienennettyä hoitoa edeltävälle tasolle statiinihoidolla.
Hypoglykemia diabetekseen hoitoa saavilla potilailla
Diabeteslääkkeitä käyttävillä potilailla on raportoitu tofasitinibin käytön aloittamisen jälkeen hypoglykemiaa. Jos hypoglykemia ilmenee, diabeteslääkkeen annosta voi olla tarpeen muuttaa.
Rokotukset
Kaikkien potilaiden, erityisesti idiopaattista juveniilia polyartriittia ja lasten psoriaasiartriittia sairastavien potilaiden, rokotukset suositellaan päivittämään ajan tasalle voimassa olevien rokotussuositusten mukaisesti ennen tofasitinibihoidon aloittamista. Elävien rokotteiden antamista tofasitinibihoidon aikana ei suositella. Päätettäessä elävien rokotteiden antamisesta ennen tofasitinibihoidon aloittamista on otettava huomioon kyseisen potilaan olemassa oleva immunosuppressio.
Profylaktisen zoster-rokotteen antoa on harkittava rokotussuositusten mukaisesti. Erityistä huomiota on kiinnitettävä potilaisiin, jotka ovat pitkään sairastaneet nivelreumaa, ja jotka ovat aiemmin saaneet kahta tai useampaa tautiprosessia hidastavaa biologista reumalääkettä (bDMARD). Jos annetaan elävää zoster-rokotetta, sitä voidaan antaa vain potilaille, joiden tiedetään sairastaneen vesirokon tai joiden tiedetään olevan vesirokkovirukselle (varicella zoster virus, VZV) seropositiivisia. Jos vesirokon sairastaminen ei ole varmuudella todennettavissa, suositellaan VZV-vasta-aineiden testaamista.
Rokotus elävillä rokotteilla on annettava vähintään 2 viikkoa, mutta mieluiten 4 viikkoa, ennen kuin hoito tofasitinibilla aloitetaan tai voimassaolevien immuniteettia muuntavia lääkevalmisteita koskevien rokotusohjeiden mukaisesti. Tietoja elävien rokotteiden välityksellä saaduista sekundaarisista infektioista tofasitinibia saavilla potilailla ei ole saatavilla.
Lääkevalmisteen sisältämät apuaineet
Propyleeniglykoli
Tämä lääkevalmiste sisältää 2,39 mg propyleeniglykolia per ml.
Esimerkkejä altistumisesta propyleeniglykolille päivittäisten annosten perusteella (ks. kohta Annostus ja antotapa) ovat seuraavat:
-
3,2 mg annos kaksi kertaa vuorokaudessa XELJANZ 1 mg/ml oraaliliuosta annettuna lapselle, joka painaa 10 kg – < 20 kg, saisi aikaan propyleeniglykolialtistuksen 1,53 mg/kg/vrk.
-
4 mg annos kaksi kertaa vuorokaudessa XELJANZ 1 mg/ml oraaliliuosta annettuna lapselle, joka painaa 20 kg – < 40 kg, saisi aikaan propyleeniglykolialtistuksen 0,96 mg/kg/vrk.
-
5 mg annos kaksi kertaa vuorokaudessa XELJANZ 1 mg/ml oraaliliuosta annettuna lapselle, joka painaa ≥ 40 kg, saisi aikaan propyleeniglykolialtistuksen 0,60 mg/kg/vrk.
Natriumbentsoaatti
Tämä valmiste sisältää 0,9 mg natriumbentsoaattia per ml.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per ml, eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Muiden lääkevalmisteiden mahdollinen vaikutus tofasitinibin farmakokinetiikkaan
Tofasitinibi metaboloituu CYP3A4-entsyymin välityksellä, joten yhteisvaikutukset CYP3A4-entsyymiä estävien tai indusoivien lääkevalmisteiden kanssa ovat todennäköisiä. Tofasitinibialtistus suurenee, kun sitä käytetään yhdessä voimakkaiden CYP3A4:n estäjien (esim. ketokonatsolin) kanssa tai kun yhden tai useamman lääkevalmisteen samanaikainen käyttö aiheuttaa CYP3A4:n kohtalaisen estymisen ja CYP2C19:n voimakkaan estymisen (esim. flukonatsoli) (ks. kohta Annostus ja antotapa).
Tofasitinibialtistus vähenee, jos samaan aikaan annetaan voimakkaita CYP:n induktoreja (esim. rifampisiinia). Pelkät CYP2C19:n estäjät tai P-glykoproteiinin estäjät eivät todennäköisesti muuta tofasitinibin farmakokinetiikkaa merkittävästi.
Ketokonatsolin (voimakas CYP3A4:n estäjä), flukonatsolin (kohtalainen CYP3A4:n ja voimakas CYP2C19:n estäjä), takrolimuusin (heikko CYP3A4:n estäjä) ja siklosporiinin (kohtalainen CYP3A4:n estäjä) samanaikainen käyttö suurensi tofasitinibin AUC-arvoa. Rifampisiini (voimakas CYP:n induktori) puolestaan pienensi tofasitinibin AUC-arvoa. Tofasitinibin samanaikainen käyttö voimakkaiden CYP:n induktorien (esim. rifampisiinin) kanssa saattaa johtaa kliinisen vasteen häviämiseen tai heikkenemiseen (ks. kuva 1). Voimakkaiden CYP3A4:n induktorien ja tofasitinibin samanaikaista käyttöä ei suositella. Ketokonatsolin ja flukonatsolin samanaikainen käyttö suurensi tofasitinibin Cmax-arvoa, kun taas takrolimuusi, siklosporiini ja rifampisiini pienensivät tofasitinibin Cmax-arvoa. Metotreksaattiannosten 15–25 mg samanaikainen käyttö kerran viikossa ei vaikuttanut nivelreumapotilailla tofasitinibin farmakokinetiikkaan (ks. kuva 1).
Kuva 1. Muiden lääkevalmisteiden vaikutus tofasitinibin farmakokinetiikkaan
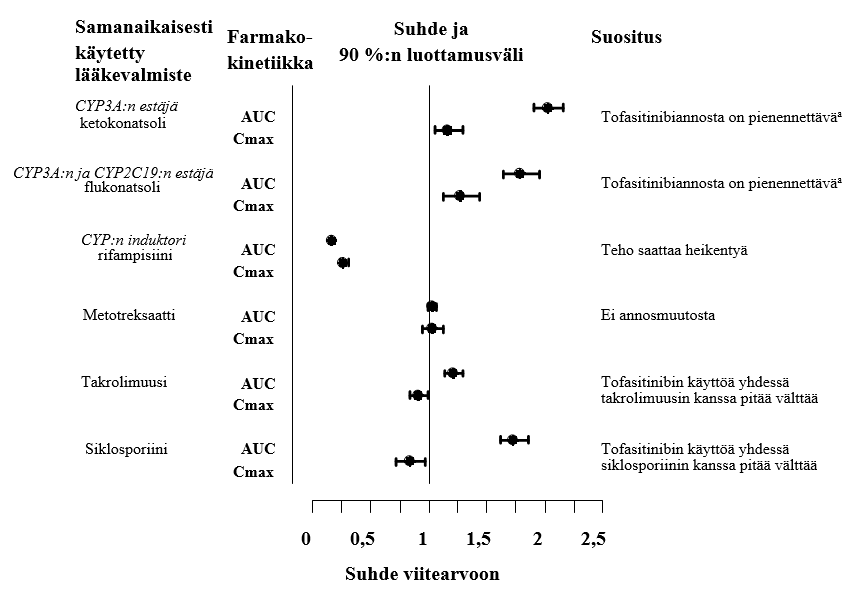
Huom.: Vertailuryhmä sai pelkästään tofasitinibia.
a Tofasitinibiannos on pienennettävä annokseen 5 mg kalvopäällysteinen tabletti vuorokaudessa tai painoon perustuva vastaava määrä oraaliliuosta potilailla, jotka saivat 5 mg tai painoon perustuvan vastaavan määrän kaksi kertaa vuorokaudessa (ks. kohta Annostus ja antotapa).
Tofasitinibin mahdollinen vaikutus muiden lääkevalmisteiden farmakokinetiikkaan
Tofasitinibin samanaikainen käyttö ei vaikuttanut terveillä vapaaehtoisilla naisilla ehkäisytablettien, levonorgestreelin ja etinyyliestradiolin, farmakokinetiikkaan.
Tofasitinibin samanaikainen käyttö metotreksaattiannosten 15–25 mg kerran viikossa kanssa pienensi nivelreumapotilailla metotreksaatin AUC-arvoa 10 % ja huippupitoisuutta (Cmax) 13 %. Metotreksaattialtistus ei pienene siinä määrin, että se edellyttäisi muutoksia metotreksaatin yksilölliseen annostukseen.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Raskaus
Tofasitinibin käytöstä raskaana oleville naisille ei ole olemassa riittäviä ja hyvin kontrolloituja tutkimuksia. Tofasitinibin on osoitettu olevan teratogeeninen rotilla ja kaniineilla, ja sen on osoitettu vaikuttavan synnytykseen sekä peri-/postnataaliseen kehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Varotoimenpiteenä tofasitinibin käyttö raskauden aikana on vasta-aiheista (ks. kohta Vasta-aiheet).
Naiset, jotka voivat tulla raskaaksi/raskauden ehkäisy naisilla
Naisia, jotka voivat tulla raskaaksi, pitää neuvoa käyttämään tehokasta ehkäisymenetelmää tofasitinibihoidon aikana ja vähintään neljä viikkoa viimeisen annoksen jälkeen.
Imetys
Julkaistujen tietojen perusteella tofasitinibi erittyy ihmisen rintamaitoon. Tofasitinibin vaikutuksia imetettävään vauvaan ei julkaistusta kirjallisuudesta ja valmisteen markkinoille tulon jälkeen saatujen tietojen perusteella tunneta, ja tiedot rajoittuvat pieneen lukumäärään tapauksia, joissa haittavaikutuksiin ei liittynyt syy-yhteyttä. Riskiä imetettävälle lapselle ei voida sulkea pois. Varotoimenpiteenä tofasitinibin käyttö imetyksen aikana on vasta-aiheista (ks. kohta Vasta-aiheet).
Hedelmällisyys
Muodollisia tutkimuksia mahdollisista vaikutuksista ihmisen hedelmällisyyteen ei ole tehty. Tofasitinibi heikensi naarasrottien, mutta ei urosrottien, hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tofasitinibilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Nivelreuma
Yleisimpiä vakavia haittavaikutuksia olivat vakavat infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Pitkäaikaisturvallisuutta koskeneessa koko altistetussa potilasjoukossa tofasitinibihoidon yhteydessä yleisimmin raportoituja vakavia infektioita ovat olleet keuhkokuume (1,7 %), vyöruusu (Herpes zoster) (0,6 %), virtsatieinfektiot (0,4 %), selluliitti (0,4 %), divertikuliitti (0,3 %) ja umpilisäketulehdus (0,2 %). Tofasitinibihoidon yhteydessä raportoituja opportunisti-infektioita ovat olleet tuberkuloosi ja muut mykobakteeri-infektiot, Cryptococcus-infektiot, histoplasmoosi, ruokatorven kandidiaasi, usean dermatomin alueella esiintyvä vyöruusu (Herpes zoster), sytomegalovirusinfektio, BK-virusinfektio ja listerioosi. Joidenkin potilaiden tautimuoto on ollut pikemminkin disseminoitunut kuin paikallinen. Muita vakavia infektioita, joita ei ole raportoitu kliinisissä tutkimuksissa, saattaa myös esiintyä (esim. koksidioidomykoosia).
Kaksoissokkoutettujen lume- tai metotreksaattikontrolloitujen kliinisten tutkimusten kolmen ensimmäisen kuukauden aikana yleisimmin raportoituja haittavaikutuksia olivat päänsärky (3,9 %), ylähengitysteiden infektiot (3,8 %), ylähengitysteiden virusinfektio (3,3 %), ripuli (2,9 %), pahoinvointi (2,7 %) ja hypertensio (2,2 %).
Kaksoissokkoutettujen lume- tai metotreksaattikontrolloitujen tutkimusten kolmen ensimmäisen kuukauden aikana hoidon haittavaikutusten vuoksi keskeyttäneiden potilaiden osuus oli 3,8 % tofasitinibihoitoa saaneista potilaista. Yleisimpiä hoidon keskeyttämiseen johtaneita infektioita kontrolloitujen kliinisten tutkimusten kolmen ensimmäisen kuukauden aikana olivat vyöruusu (Herpes zoster) (0,19 %) ja keuhkokuume (0,15 %).
Haittavaikutustaulukko
Seuraavassa taulukossa luetellut haittavaikutukset ovat nivelreuma- ja nivelpsoriaasipotilaille ja haavaista paksusuolitulehdusta sairastaville potilaille tehdyistä kliinisistä tutkimuksista, ja ne on esitetty elinjärjestelmäluokittain (System Organ Class = SOC) ja esiintyvyyden mukaan, ja ne on määritelty seuraavan esitystavan mukaisesti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000) tai tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutusten vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 7. Haittavaikutukset
Elinjärjestelmäluokka | Yleiset | Melko harvinaiset | Harvinaiset | Hyvin harvinaiset | Tuntematon |
Infektiot | Keuhkokuume Influenssa Vyöruusu (Herpes zoster) Virtsatieinfektio Sinuiitti Keuhkoputkitulehdus Nasofaryngiitti Nielutulehdus | Tuberkuloosi Divertikuliitti Pyelonefriitti Selluliitti Yskänrokko (Herpes simplex) Virusperäinen gastroenteriitti Virusinfektio | Sepsis Urosepsis Disseminoitunut tuberkuloosi Bakteremia Pneumocystis jirovecii‑keuhkokuume Pneumokokkikeuhkokuume Bakteerikeuhkokuume Sytomegalovirusinfektio Bakteeriperäinen artriitti | Keskushermoston tuberkuloosi Kryptokokki-meningiitti Nekrotisoiva faskiitti Enkefaliitti Stafylokokkibakteremia Mycobacterium avium ‑kompleksi-infektio Atyyppinen mykobakteeri-infektio |
|
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) |
| Keuhkosyöpä Ei-melanoottiset ihosyövät | Lymfooma |
|
|
Veri ja imukudos | Lymfopenia Anemia | Leukopenia Neutropenia |
|
|
|
Immuunijärjestelmä |
|
|
|
| Yliherkkyys* Angioedeema* Urtikaria* |
Aineenvaihdunta ja ravitsemus |
| Dyslipidemia Hyperlipidemia Elimistön kuivuminen |
|
|
|
Psyykkiset häiriöt |
| Unettomuus |
|
|
|
Hermosto | Päänsärky | Parestesiat |
|
|
|
Sydän |
| Sydäninfarkti |
|
|
|
Verisuonisto | Hypertensio | Laskimotromboembolia** |
|
|
|
Hengityselimet, rintakehä ja välikarsina | Yskä | Hengenahdistus Nenän sivuonteloiden tukkoisuus |
|
|
|
Ruoansulatuselimistö | Vatsakipu Oksentelu Ripuli Pahoinvointi Gastriitti Dyspepsia |
|
|
|
|
Maksa ja sappi |
| Rasvamaksa Suurentunut maksaentsyymi-pitoisuus Suurentunut transaminaasi-pitoisuus Suurentunut gammaglutamyyli-transferaasipitoisuus | Poikkeavat tulokset maksan toimintakokeissa |
|
|
Iho ja ihonalainen kudos | Ihottuma Akne | Eryteema Kutina |
|
|
|
Luusto, lihakset ja sidekudos | Nivelkipu | Nivelten turpoaminen Jännetulehdus | Tuki- ja liikuntaelimistön kipu |
|
|
Yleisoireet ja antopaikassa todettavat haitat | Raajojen turvotus | Kuume Uupumus |
|
|
|
Tutkimukset | Suurentunut veren kreatiinikinaasipitoisuus | Suurentunut veren kreatiniinipitoisuus Suurentunut veren kolesterolipitoisuus Suurentunut LDL-pitoisuus Painon nousu |
|
|
|
Vammat ja myrkytykset |
| Nivelsiteen nyrjähdys Lihasvenähdys |
|
|
|
* Spontaanista haittavaikutusraportoinnista saatu tieto
** Laskimotromboembolia sisältää keuhkoembolian, syvän laskimotukoksen, verkkokalvon laskimotukoksen ja kallonsisäisen sinustromboosin.
Valikoitujen haittavaikutusten kuvaus
Laskimotromboembolia
Nivelreuma
Laajassa (N = 4 362), satunnaistetussa myyntiluvan myöntämisen jälkeisessä turvallisuustutkimuksessa oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Siinä havaittiin, että TNF:n estäjiin verrattuna tofasitinibihoitoa saaneilla potilailla laskimotromboembolian ilmaantuvuus oli suurempi ja annosriippuvainen (ks. kohta Farmakodynamiikka). Valtaosa näistä tapahtumista oli vakavia, ja osa johti potilaan kuolemaan. Keuhkoembolioitten ilmaantumistiheys (95 %:n luottamusväli) oli 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 0,17 (0,08–0,33), 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 0,50 (0,32–0,74) ja TNF:n estäjiä saaneilla 0,06 (0,01–0,17) potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden. Riskitiheyksien suhde (hazard ratio, HR) keuhkoembolian suhteen oli 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 2,93 (0,79–10,83) ja 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 8,26 (2,49–27,43) (ks. kohta Farmakodynamiikka) verrattuna TNF:n estäjiin. Tofasitinibihoitoa saaneista potilaista, joilla todettiin keuhkoembolia, suurimmalla osalla (97 %) oli laskimotromboembolian riskitekijöitä.
Kaikki infektiot
Nivelreuma
Kontrolloiduissa vaiheen 3 kliinisissä tutkimuksissa 0–3 kuukauden aikana infektioiden esiintyvyys oli tofasitinibimonoterapiaa 5 mg kaksi kertaa vuorokaudessa (yhteensä 616 potilasta) saaneilla potilailla 16,2 % (100 potilasta) ja 10 mg kaksi kertaa vuorokaudessa (yhteensä 642 potilasta) saaneilla potilailla 17,9 % (115 potilasta) verrattuna 18,9 %:iin (23 potilasta) lumeryhmässä (yhteensä 122 potilasta). Kontrolloiduissa vaiheen 3 kliinisissä tutkimuksissa, joissa peruslääkityksenä oli tautiprosessia hidastava reumalääkehoito (DMARD), 0–3 kuukauden aikana infektioiden esiintyvyys oli tofasitinibia 5 mg kaksi kertaa vuorokaudessa ja DMARD-hoitoa saaneilla potilailla (yhteensä 973 potilasta) 21,3 % (207 potilasta), ja tofasitinibia 10 mg kaksi kertaa vuorokaudessa ja DMARD-hoitoa saaneilla potilailla (yhteensä 969 potilasta) 21,8 % (211 potilasta) verrattuna 18,4 %:iin (103 potilasta) lumevalmisteen ja DMARD-hoidon yhdistelmää saaneessa ryhmässä (yhteensä 559 potilasta).
Yleisimmin raportoituja infektioita olivat ylähengitysteiden infektiot (3,7 %) ja nasofaryngiitti (3,2 %).
Pitkäaikaisen turvallisuuden selvittämisessä mukana olleilla kaikilla altistetuilla potilailla (yhteensä 4 867 potilasta) infektioiden kokonaisilmaantumistiheys tofasitinibihoidon yhteydessä oli 46,1 potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden (5 mg kaksi kertaa vuorokaudessa saaneilla 43,8 potilasta, joilla oli tapahtumia, ja 10 mg kaksi kertaa vuorokaudessa saaneilla vastaavasti 47,2). Monoterapiaa 5 mg kaksi kertaa vuorokaudessa saaneilla potilailla ilmaantumistiheys oli 48,9 potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden ja 10 mg kaksi kertaa vuorokaudessa saaneilla vastaavasti 41,9 (yhteensä 1 750 potilasta). Yhdistelmähoitona DMARD-lääkkeitä saaneet potilaat: 5 mg kaksi kertaa vuorokaudessa saaneiden potilaiden ilmaantumistiheys oli 41,0 potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden ja 10 mg kaksi kertaa vuorokaudessa saaneilla vastaavasti 50,3 (yhteensä 3 117 potilasta).
Vakavat infektiot
Nivelreuma
6 kuukautta ja 24 kuukautta kestäneissä kontrolloiduissa kliinisissä tutkimuksissa tofasitinibimonoterapiaa 5 mg kaksi kertaa vuorokaudessa saaneessa ryhmässä vakavien infektioiden määrä oli 1,7 potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden. Tofasitinibimonoterapiaa 10 mg kaksi kertaa vuorokaudessa saaneessa ryhmässä määrä oli 1,6 potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden, vastaavasti lumeryhmässä 0 ja metotreksaattiryhmässä 1,9.
6, 12 tai 24 kuukautta kestäneissä tutkimuksissa tofasitinibia 5 mg kaksi kertaa vuorokaudessa yhdistelmänä DMARD-hoidon kanssa saaneilla potilailla vakavien infektioiden määrä oli 3,6 potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden, tofasitinibia 10 mg kaksi kertaa vuorokaudessa yhdistelmänä DMARD-hoidon kanssa saaneilla potilailla vastaavasti 3,4 ja lumelääkettä yhdistelmänä DMARD-hoidon kanssa saaneilla potilailla 1,7.
Pitkäaikaisen turvallisuuden selvittämisessä mukana olleilla kaikilla altistetuilla potilailla vakavien infektioiden kokonaisilmaantumistiheys oli tofasitinibia 5 mg kaksi kertaa vuorokaudessa saaneilla 2,4 potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden ja tofasitinibia 10 mg kaksi kertaa vuorokaudessa saaneilla vastaavasti 3,0. Yleisimpiä vakavia infektioita olivat mm. keuhkokuume, vyöruusu (Herpes zoster), virtsatieinfektio, selluliitti, gastroenteriitti ja divertikuliitti. Opportunisti-infektioita on raportoitu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Laajassa (N = 4 362), satunnaistetussa myyntiluvan myöntämisen jälkeisessä turvallisuustutkimuksessa oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Siinä havaittiin, että TNF:n estäjiin verrattuna tofasitinibihoitoa saaneilla potilailla vakavien infektioiden ilmaantuvuus oli suurempi ja annosriippuvainen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vakavien infektioiden ilmaantumistiheys (95 %:n luottamusväli) oli 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 2,86 (2,41–3,37), 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 3,64 (3,11–4,23) ja TNF:n estäjiä saaneilla 2,44 (2,02–2,92) potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden. Riskitiheyksien suhde (hazard ratio, HR) vakavien infektioiden osalta oli 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 1,17 (0,92–1,50) ja 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 1,48 (1,17–1,87) verrattuna TNF:n estäjiin.
Virusten uudelleenaktivoituminen
Tofasitinibilla hoidetuilla potilailla, jotka ovat japanilaisia tai korealaisia, tai joilla on ollut nivelreuma pitkään ja jotka olivat aiemmin saaneet kahta tai useampaa tautiprosessia hidastavaa biologista reumalääkettä (bDMARD), tai potilailla, joiden absoluuttinen lymfosyyttien määrä (B-Lymf) on alle 1,0 x 109/l, tai joiden tofasitinibiannos on 10 mg kaksi kertaa vuorokaudessa, saattaa olla suurentunut vyöruusun (Herpes zoster) riski (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Laajassa (N = 4 362) satunnaistetussa myyntiluvan myöntämisen jälkeisessä turvallisuutta koskeneessa tutkimuksessa oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Siinä havaittiin vyöruusuun (Herpes zoster) liittyvien tapahtumien lisääntyminen tofasitinibihoitoa saaneilla potilailla TNF:n estäjiä saaneisiin verrattuna. Vyöruusun (Herpes zoster) ilmaantumistiheys (95 %:n luottamusväli) oli 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 3,75 (3,22, 4,34) potilasta, 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 3,94 (3,38, 4,57) potilasta ja TNF:n estäjiä saaneilla 1,18 (0,90, 1,52) potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden.
Laboratoriokokeet
Lymfosyytit
Kontrolloiduissa kliinisissä nivelreumatutkimuksissa varmistettua B-Lymf:n laskua alle 0,5 x 109/l todettiin 0,3 %:lla potilaista ja B-Lymf 0,50–0,75 x 109/l todettiin 1,9 %:lla potilaista yhdistetyssä ryhmässä (annoksia 5 mg kaksi kertaa vuorokaudessa ja 10 mg kaksi kertaa vuorokaudessa saaneet potilaat).
Pitkäaikaisen turvallisuuden selvittämisessä mukana olleilla nivelreumapotilailla varmistettua B-Lymf:n laskua alle 0,5 x 109/l todettiin yhteensä 1,3 %:lla potilaista ja B-Lymf 0,50–0,75 x 109/l todettiin 8,4 %:lla potilaista yhdistetyssä ryhmässä (annoksia 5 mg kaksi kertaa vuorokaudessa ja 10 mg kaksi kertaa vuorokaudessa saaneet potilaat).
Varmistettuun B-Lymf:n laskuun alle 0,75 x 109/l liittyi suurentunut vakavien infektioiden ilmaantuvuus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Neutrofiilit
Kontrolloiduissa kliinisissä nivelreumatutkimuksissa varmistettua B-Neut-arvon laskua alle 1,0 x 109/l esiintyi 0,08 %:lla potilaista yhdistetyssä ryhmässä (annoksia 5 mg kaksi kertaa vuorokaudessa ja 10 mg kaksi kertaa vuorokaudessa saaneet potilaat). Varmistettua B-Neut-arvon laskua alle 0,5 x 109/l ei havaittu missään hoitoryhmässä. Neutropenian ja vakavien infektioiden ilmaantuvuuden välillä ei esiintynyt selkeää yhteyttä.
Pitkäaikaisen turvallisuuden selvittämisessä mukana olleilla nivelreumapotilailla varmistetun B-Neut-arvon laskun ilmaantumistapa ja ilmaantuvuus olivat yhdenmukaiset kontrolloiduissa kliinisissä tutkimuksissa tehtyjen havaintojen kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksaentsyymikokeet
Varmistettua maksaentsyymien nousua yli 3-kertaiseksi viitevälin ylärajaan nähden havaittiin nivelreumapotilailla melko harvoin. Jos näiden potilaiden maksaentsyymit olivat koholla, hoitoon tehtävät muutokset (esim. samanaikaisesti käytetyn tautiprosessia hidastavan reumalääkkeen (DMARD) annoksen pienentäminen, tofasitinibihoidon keskeyttäminen tai tofasitinibiannoksen pienentäminen) pienensivät maksaentsyymipitoisuutta tai normalisoivat pitoisuuden.
Nivelreumaa koskeneen vaiheen 3 monoterapiatutkimuksen kontrolloidussa osiossa (0–3 kuukautta) (tutkimus I, ks. kohta Farmakodynamiikka) ALAT-arvojen nousua yli 3‑kertaisiksi viitevälin ylärajaan nähden havaittiin 1,65 %:lla lumelääkettä saaneista, 0,41 %:lla 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista ja 0 %:lla 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista potilaista. Tässä tutkimuksessa ASAT-arvojen nousua yli 3‑kertaisiksi viitevälin ylärajaan nähden havaittiin 1,65 %:lla lumelääkettä saaneista, 0,41 %:lla 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista ja 0 %:lla 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista potilaista.
Nivelreumaa koskeneessa vaiheen 3 monoterapiatutkimuksessa (0–24 kuukautta) (tutkimus VI, ks. kohta Farmakodynamiikka) ALAT-arvojen havaittiin suurentuneen yli 3‑kertaisiksi viitearvojen ylärajaan nähden 7,1 %:lla metotreksaattia saaneista, 3,0 %:lla 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista ja 3,0 %:lla 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista potilaista. Tässä tutkimuksessa ASAT-arvojen nousua yli 3-kertaisiksi viitearvojen ylärajaan nähden havaittiin 3,3 %:lla metotreksaattia saaneista, 1,6 %:lla 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista ja 1,5 %:lla 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista potilaista.
Nivelreumaa koskeneiden vaiheen 3 tutkimusten, joissa peruslääkityksenä oli tautiprosessia hidastava reumalääkehoito (DMARD), kontrolloidussa osiossa (0–3 kuukautta) (tutkimukset II–V, ks. kohta Farmakodynamiikka) ALAT-arvojen nousua yli 3‑kertaisiksi viitevälin ylärajaan nähden havaittiin 0,9 %:lla lumelääkettä saaneista, 1,24 %:lla 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista ja 1,14 %:lla 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista potilaista. Näissä tutkimuksissa ASAT-arvojen nousua yli 3‑kertaisiksi viitevälin ylärajaan nähden havaittiin 0,72 %:lla lumelääkettä saaneista, 0,5 %:lla 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista ja 0,31 %:lla 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista potilaista.
Nivelreumaa koskeneissa pitkäkestoisissa monoterapiajatkotutkimuksissa ALAT-arvojen havaittiin suurentuneen yli 3‑kertaisiksi viitearvojen ylärajaan nähden 1,1 %:lla 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista ja vastaavasti 1,4 %:lla 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista potilaista. ASAT-arvojen nousua yli 3-kertaisiksi viitearvojen ylärajaan nähden havaittiin alle 1 %:lla molemmissa sekä 5 mg että 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneissa potilasryhmissä.
Nivelreumaa koskeneissa pitkäkestoisissa jatkotutkimuksissa, joissa peruslääkityksenä oli tautiprosessia hidastava reumalääkehoito (DMARD), ALAT-arvojen havaittiin suurentuneen yli 3‑kertaisiksi viitearvojen ylärajaan nähden 1,8 %:lla 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista ja vastaavasti 1,6 %:lla 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista potilaista. ASAT-arvojen nousua yli 3-kertaisiksi viitearvojen ylärajaan nähden havaittiin alle 1 %:lla molemmissa sekä 5 mg että 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneissa potilasryhmissä.
Laajassa (N = 4 362) satunnaistetussa myyntiluvan myöntämisen jälkeisessä turvallisuutta koskeneessa tutkimuksessa oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Siinä ALAT-arvojen kohoamista vähintään 3‑kertaiseksi viitearvojen ylärajaan nähden havaittiin 6,01 %:lla 5 mg tofasitibinia kaksi kertaa vuorokaudessa saaneista, 6,54 %:lla 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista ja 3,77 %:lla TNF:n estäjiä saaneista potilaista. ASAT-arvojen kohoamista vähintään 3‑kertaisiksi viitearvojen ylärajaan nähden havaittiin 3,21 %:lla 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista, 4,57 %:lla 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneista ja 2,38 %:lla TNF:n estäjiä saaneista potilaista.
Lipidit
Lipidiarvojen (kokonais-, LDL- ja HDL-kolesteroli, triglyseridit) kohoamista tutkittiin nivelreumapotilailla tehdyissä kontrolloiduissa kaksoissokkoutetuissa kliinisissä tutkimuksissa ensimmäisen kerran 1 kuukauden kuluttua tofasitinibihoidon aloittamisesta. Arvojen suurenemista havaittiin kyseisenä ajankohtana, ja ne pysyivät sen jälkeen vakaina.
Kontrolloiduissa kliinisissä nivelreumatutkimuksissa lipidien muutoksia esiintyi lähtötilanteesta tutkimuksen päättymiseen saakka (6–24 kuukautta) seuraavasti:
-
LDL-kolesterolipitoisuuden keskiarvo oli suurentunut 12 kuukauden hoidon jälkeen 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneiden ryhmässä 15 % ja 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneiden ryhmässä 20 % ja 24 kuukauden hoidon jälkeen pitoisuus oli suurentunut 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneiden ryhmässä 16 % ja 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneiden ryhmässä 19 %.
-
HDL-kolesterolipitoisuuden keskiarvo oli suurentunut 12 kuukauden hoidon jälkeen 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneiden ryhmässä 17 % ja 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneiden ryhmässä 18 % ja 24 kuukauden hoidon jälkeen pitoisuus oli suurentunut 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneiden ryhmässä 19 % ja 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneiden ryhmässä 20 %.
Lipidipitoisuudet palasivat tofasitinibihoidon lopettamisen jälkeen hoitoa edeltäneelle tasolle.
Sekä keskimääräisen LDL-kolesterolipitoisuuden suhde HDL-kolesterolipitoisuuteen (LDL/HDL) että apolipoproteiini B:n suhde ApoA1:een (ApoB/ApoA1) pysyivät tofasitinibihoitoa saaneilla potilailla pääasiassa ennallaan.
Kontrolloidussa kliinisessä nivelreumatutkimuksessa kohonnut LDL-kolesterolipitoisuus ja ApoB vastasivat statiinihoitoon ja pienenivät hoitoa edeltäneelle tasolle.
Pitkäaikaisen turvallisuuden selvittämisessä mukana olleilla nivelreumapotilailla kohonneet lipidiparametrit olivat yhdenmukaisia kontrolloiduissa kliinisissä tutkimuksissa tehtyjen havaintojen kanssa.
Laajassa (N = 4 362) satunnaistetussa myyntiluvan myöntämisen jälkeisessä turvallisuutta koskeneessa tutkimuksessa oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Seuraavassa esitetään yhteenveto lähtötilanteen lipidiparametrien muutoksista lähtötilanteesta 24 kuukauteen saakka:
-
LDL-kolesterolipitoisuuden keskiarvo oli suurentunut 12 kuukauden hoidon jälkeen 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla potilailla 13,80 %, 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla potilailla 17,04 % ja TNF:n estäjiä saaneilla potilailla 5,50 %. 24 kuukauden hoidon jälkeen pitoisuus oli suurentunut 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla potilailla 12,71 %, 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla potilailla 18,14 % ja TNF:n estäjiä saaneilla potilailla 3,64 %,
-
HDL-kolesterolipitoisuuden keskiarvo oli suurentunut 12 kuukauden hoidon jälkeen 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla potilailla 11,71 %, 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla potilailla 13,63 % ja TNF:n estäjiä saaneilla potilailla 2,82 %. 24 kuukauden hoidon jälkeen pitoisuus oli suurentunut 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla potilailla 11,58 %, 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla potilailla 13,54 % ja TNF:n estäjiä saaneilla potilailla 1,42 %.
Sydäninfarkti
Nivelreuma
Laajassa (N = 4 362) satunnaistetussa myyntiluvan myöntämisen jälkeisessä turvallisuutta koskeneessa tutkimuksessa oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Sydäninfarktin (kuolemaan johtamaton) ilmaantumistiheys (95 %:n luottamusväli) oli 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 0,37 (0,22; 0,57), 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 0,33 (0,19; 0,53) ja TNF:n estäjiä saaneilla 0,16 (0,07; 0,31) potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden. Tutkimuksessa raportoitiin muutamista kuolemaan johtaneista sydäninfarkteista, ja niiden ilmaantumistiheys oli tofasitinibilla hoidetuilla potilailla samankaltainen kuin TNF:n estäjillä hoidetuilla potilailla (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka). Tutkimuksessa edellytettiin vähintään 1 500 potilaan seurantaa kolmen vuoden ajan.
Syövät ei-melanoottista ihosyöpää lukuun ottamatta
Nivelreuma
Laajassa (N = 4 362) satunnaistetussa myyntiluvan myöntämisen jälkeisessä turvallisuutta koskeneessa tutkimuksessa oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Keuhkosyövän ilmaantumistiheys (95 %:n luottamusväli) oli 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 0,23 (0,12; 0,40), 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 0,32 (0,18; 0,51) ja TNF:n estäjiä saaneilla 0,13 (0,05; 0,26) potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka). Tutkimuksessa edellytettiin vähintään 1 500 potilaan seurantaa kolmen vuoden ajan.
Lymfoomien ilmaantumistiheys (95 %:n luottamusväli) oli 5 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 0,07 (0,02; 0,18), 10 mg tofasitinibia kaksi kertaa vuorokaudessa saaneilla 0,11 (0,04; 0,24) ja TNF:n estäjiä saaneilla 0,02 (0,00; 0,10) potilasta, joilla oli tapahtumia, 100 potilasvuotta kohden (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Pediatriset potilaat
Idiopaattinen juveniili polyartriitti ja lasten psoriaasiartriitti
Lastenreumaa sairastavilla potilailla vaiheen 3 pivotaalitutkimuksessa (tutkimus JIA‑I) ja pitkäkestoisessa jatkotutkimuksessa, jossa oli mukana idiopaattista juveniilia polyartriittia, lasten psoriaasiartriittia, entesiittiin liittyvää artriittia ja systeemistä juveniilia idiopaattista artriittia sairastavia potilaita, todetut haittavaikutukset olivat yleisesti yhdenmukaisia tyypiltään ja esiintyvyydeltään aikuisilla nivelreumapotilailla havaittujen haittavaikutusten kanssa, lukuun ottamatta joitakin infektioita (ylähengitystieinfektio ja virusinfektio) ja maha-suolikanavan häiriöitä tai yleisiä häiriöitä (vatsakipu ja kuume), jotka olivat yleisempiä lastenreumapotilailla. Metotreksaatti oli yleisin samanaikaisesti käytetty csDMARD (156 potilasta 157:stä csDMARD-valmisteella hoidetusta potilaasta käytti päivänä 1 metotreksaattia). Samaan aikaan muiden csDMARD-valmisteiden kanssa käytetyn tofasitinibin turvallisuusprofiilista ei ole riittävästi tietoja.
Infektiot
Tutkimuksen JIA-I kaksoissokkoutetussa osassa infektio oli yleisimmin raportoitu haittavaikutus (44,3 %). Infektiot olivat yleensä vaikeusasteeltaan lieviä tai kohtalaisia.
Tutkimuksessa JIA‑I vakavia infektioita raportoitiin 3:lla tofasitinibia saaneella potilaalla (1,3 %); tapahtumien ilmaantumistiheys oli 2,43 potilasta 100 potilasvuotta kohden. Herpes zoster raportoitiin 2 potilaalla (0,9 %); tapahtumien ilmaantumistiheys oli 1,63 potilasta 100 potilasvuotta kohden. Kumpikin tapahtuma rajoittui yhteen dermatomiin, eivätkä ne olleet vakavia.
Pitkäkestoisessa jatkotutkimuksessa vakavia infektioita ilmeni 14:lla tofasitinibia saaneella potilaalla (4,6 %); tapahtumien ilmaantumistiheys oli 1,50 potilasta 100 potilasvuotta kohden. Yleisimpiä vakavia infektioita olivat herpes zoster ja pneumonia. Herpes zoster ‑tapahtumia raportoitiin 6 potilaalla (2,0 %); tapahtumien ilmaantumistiheys oli 0,64 potilasta 100 potilasvuotta kohden. Näistä 2 potilaalla (0,7 %) ilmeni vakavia, useisiin dermatomeihin levinneitä herpes zoster‑tapahtumia; tapahtumien ilmaantumistiheys oli 0,21 tapahtumaa 100 potilasvuotta kohden. Lisäksi 3 herpes zoster ‑tapahtumaa (2 vakavaa ja 1 ei‑vakava) rajoittui yhteen dermatomiin. Yksi ei‑vakava herpes zoster ‑tapahtuma oli levinnyt kahteen vierekkäiseen dermatomiin ja ilmeni potilaalla, jolla oli anamneesissa ei‑vakava vesirokko, joka oli raportoitu tutkimuksessa 5,8 vuotta aikaisemmin.
Maksatapahtumat
Lastenreumaa koskevaan pivotaalitutkimukseen osallistuneiden potilaiden ASAT- ja ALAT-arvojen piti olla alle 1,5 kertaa normaalin ylärajan (ULN), jotta heidät voitiin ottaa mukaan tutkimukseen. Tutkimuksessa JIA‑I ALAT-arvon raportoitiin kohonneen > 5 × ULN kahdella peräkkäisellä käynnillä yhdellä tofasitinibia saaneella potilaalla (0,4 %), joka sai peruslääkityksenä metotreksaattihoitoa. Tapahtuman todettiin liittyvän tofasitinibiin, ja se korjautui metotreksaatti- ja tofasitinibihoidon keskeyttämisen jälkeen. Pitkäkestoisessa jatkotutkimuksessa ASAT- tai ALAT-arvon raportoitiin kohonneen > 5 × ULN kahdella peräkkäisellä käynnillä kahdella potilaalla (0,66 %); kummankaan tapahtuman ei todettu liittyneen tofasitinibiin. Yksikään tapahtuma kummassakaan tutkimuksessa ei täyttänyt Hyn lain kriteereitä.
Laboratoriokokeet
Muutokset laboratoriokokeiden arvoissa lastenreumapotilailla kliinisessä kehitysohjelmassa olivat yhdenmukaisia aikuisilla nivelreumapotilailla nähtyjen arvojen kanssa. Lastenreuman pivotaalitutkimuksessa olevilta potilailta edellytettiin verihiutalemäärää ≥ 100 000 solua/mm3 jotta heidät voitiin ottaa mukaan tutkimukseen. Näin ollen tietoja ei ole saatavilla lastenreumapotilaista, joiden verihiutalemäärä on <100 000 solua/mm3 ennen tofasitinibihoidon aloittamista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
Yliannostus
Yliannostustapauksessa suositellaan potilaan seuraamista haittavaikutusten merkkien ja oireiden havaitsemiseksi. Tofasitinibin yliannoksen hoitoon ei ole spesifistä vasta-ainetta. Hoidon on oltava oireenmukaista ja elintoimintoja tukevaa.
Terveistä vapaaehtoisista koehenkilöistä aina 100 mg:n kerta-annoksiin saakka saadut farmakokineettiset tiedot viittaavat siihen, että yli 95 % otetusta annoksesta oletetaan eliminoituvan 24 tunnin kuluessa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, Januskinaasin (JAK) estäjät, ATC-koodi: L04AF01
Vaikutusmekanismi
Tofasitinibi on voimakas, selektiivinen JAK-perheen estäjä. Tofasitinibi estää entsyymimäärityksissä kinaaseja JAK1, JAK2, JAK3 ja vähäisemmässä määrin kinaasia TyK2. Tofasitinibi on kuitenkin erittäin selektiivinen muiden ihmisen genomin kinaasien suhteen. Ihmisen soluissa tofasitinibi estää lähinnä kinaaseihin JAK3 ja/tai JAK1 liittyvää heterodimeeristen sytokiinireseptorien signaalinvälitystä, joka on funktionaalisesti selektiivisempää kuin sytokiinireseptorien JAK2-kinaasiparien kautta tapahtuva signaalinvälitys. Tofasitinibin aiheuttama JAK1:n ja JAK3:n estyminen heikentää interleukiinien (IL-2, -4, -6, -7, -9, -15, -21) sekä tyypin I ja tyypin II interferonien signaalinvälitystä, mikä muuttaa immuunivastetta ja tulehdusvastetta.
Farmakodynaamiset vaikutukset
Tofasitinibihoitoon liittyi nivelreumapotilaiden pisimmillään 6 kuukautta kestäneen hoidon aikana verenkierrossa olevien luonnollisten tappajasolujen (NK) CD16/56+‑annosriippuvaista vähenemistä, ja niiden vähenemisen arvioitiin olleen suurinta noin 8−10 viikon kuluttua hoidon aloittamisen jälkeen. Nämä muutokset hävisivät tavallisesti 2–6 viikon kuluttua hoidon lopettamisen jälkeen. Tofasitinibihoitoon liittyi annosriippuvaista B-solumäärän lisääntymistä. T-lymfosyyttien ja T‑lymfosyyttien alaryhmien (CD3+, CD4+ ja CD8+) määrän muutokset verenkierrossa olivat vähäisiä ja epäjohdonmukaisia.
Pitkäaikaishoidossa (tofasitinibihoidon keston mediaani noin 5 vuotta) CD4+-määrän todettiin vähentyneen lähtötilanteesta 28 % (mediaani) ja CD8+-määrän 27 % (mediaani). Lyhytkestoisessa käytössä havaitusta vähenemisestä poiketen luonnollisten tappajasolujen CD16/56+ määrän todettiin lisääntyneen lähtötilanteesta 73 % (mediaani). CD19-positiivisten B-solujen määrän ei todettu enää lisääntyneen pitkäkestoisen tofasitinibihoidon jälkeen. Kaikkien näiden lymfosyyttialaryhmien muutokset palautuivat hoidon tilapäisen keskeyttämisen jälkeen lähemmäs lähtötilannetta. Lymfosyyttialaryhmien määrien ja vakavien tai opportunisti-infektioiden tai vyöruusun (Herpes zoster) välisestä yhteydestä ei ollut näyttöä (lymfosyyttien absoluuttisen määrän seuranta, ks. kohta Annostus ja antotapa).
IgG-, IgM- ja IgA-kokonaispitoisuuden muutokset seerumissa olivat nivelreumapotilaiden 6 kuukautta kestäneen tofasitinibihoidon aikana vähäisiä, annoksesta riippumattomia ja samankaltaisia kuin lumelääkkeen yhteydessä, mikä viittaa siihen, ettei systeemistä humoraalista suppressiota esiinny.
Nivelreumapotilaiden tofasitinibihoidon jälkeen C‑reaktiivisen proteiinin (CRP) pitoisuuden seerumissa havaittiin pienentyneen nopeasti, ja pitoisuus pysyi pienentyneenä koko hoidon ajan. Tofasitinibihoidon yhteydessä havaitut CRP-muutokset eivät korjautuneet täysin 2 viikon kuluessa hoidon lopettamisesta, mikä viittaa siihen, että farmakodynaaminen aktiivisuus kestää puoliintumisaikaa pidempään.
Rokotteita koskevat tutkimukset
Kontrolloidussa kliinisessä tutkimuksessa, jossa nivelreumapotilaille aloitettiin tofasitinibihoito 10 mg:n annoksina kaksi kertaa vuorokaudessa tai lumehoito, influenssarokotteeseen vasteen saaneiden lukumäärä oli sekä tofasitinibiryhmässä (57 %) että lumehoitoa saaneessa ryhmässä (62 %) samankaltainen. Pneumokokki polysakkaridirokotteeseen vasteen saaneiden osuudet olivat seuraavat: 32 % sekä tofasitinibi- että metotreksaattihoitoa saaneista potilaista, 62 % tofasitinibimonoterapiaa saaneista potilaista, 62 % metotreksaattimonoterapiaa saaneista ja 77 % lumehoitoa saaneista. Tämän kliinistä merkitystä ei tiedetä, mutta samankaltaisia tuloksia saatiin pitkäkestoista tofasitinibihoitoa annoksina 10 mg kaksi kertaa vuorokaudessa saaneilla potilailla tehdyssä erillisessä influenssa- ja pneumokokki polysakkaridirokotetutkimuksessa.
Peruslääkityksenä metotreksaattia saaneilla nivelreumapotilailla tehtiin kontrolloitu tutkimus, jossa heille annettiin heikennettyä elävää herpesvirusta sisältävää rokotetta 2–3 viikkoa ennen kuin heille aloitettiin 12 viikon tofasitinibihoito annoksina 5 mg kaksi kertaa vuorokaudessa tai lumehoito. Sekä tofasitinibi- että lumehoitoa saaneilla potilailla havaittiin 6 viikon hoidon jälkeen näyttöä humoraalisista ja soluvälitteisistä vasteista VZV:lle. Vasteet olivat samankaltaisia kuin 50-vuotiailla ja vanhemmilla terveillä vapaaehtoisilla. Erään potilaan, joka ei ollut sairastanut vesirokkoa ja jolla ei ollut hoidon alkaessa vesirokon vasta-aineita, havaittiin 16 päivää rokotuksen jälkeen vesirokkoviruskannan aiheuttama levinnyt infektio. Tofasitinibihoito lopetettiin, ja potilas toipui saatuaan hoitoa tavanomaisilla viruslääkevalmisteiden annoksilla. Kyseinen potilas sai myöhemmin voimakkaan, mutta viivästyneen, humoraalisen ja soluvasteen rokotteeseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kliininen teho ja turvallisuus
Kliininen vaste
Tofasitinibin käyttöä lastenreuman hoitoon selvittävä vaiheen 3 ohjelma käsitti yhden loppuun saatetun vaiheen 3 tutkimuksen (tutkimus JIA-I [A3921104]) ja yhden käynnissä olevan pitkäaikaisen jatkotutkimuksen (A3921145). Näihin tutkimuksiin otettiin mukaan seuraavat lastenreumaa sairastavien potilaiden alaryhmät: potilaat, joilla oli joko RF+ tai RF- polyartriitti, laajeneva oligoartriitti, systeeminen JIA, johon liittyi aktiivinen artriitti eikä senhetkisiä systeemisiä oireita (viitataan nimellä pJIA-tietoaineisto) ja kaksi erillistä alaryhmää potilaita, joilla oli lasten nivelpsoriaasi ja entesiittiin liittyvä artriitti (ERA). Kuitenkin vain ne potilaat, joilla oli joko RF+ tai RF- polyartriitti tai laajeneva oligoartriitti otettiin mukaan pJIA:n tehopopulaatioon. Epäselviä tuloksia on saatu alaryhmässä, jossa potilailla oli systeeminen JIA, johon liittyi aktiivinen artriitti eikä senhetkisiä systeemisiä oireita. Potilaat, joilla oli lasten nivelpsoriaasi, sisällytettiin mukaan erillisenä tehoalaryhmänä. ERA-potilaita ei otettu mukaan tehoanalyysiin.
Kaikki soveltuvat potilaat tutkimuksessa JIA-I saivat avoimen vaiheen hoitona 5 mg kalvopäällysteisiä tofasitinibitabletteja kaksi kertaa vuorokaudessa tai painoon perustuvan vastaavan määrän tofasitinibioraaliliuosta kaksi kertaa vuorokaudessa 18 viikon ajan (ns. sisäänajovaihe [run-in phase]); potilaat, jotka saivat vähintään JIA ACR30 -vasteen avoimen hoitojakson lopussa, satunnaistettiin (1:1) saamaan joko vaikuttavaa ainetta sisältäviä 5 mg:n kalvopäällysteisiä tofasitinibitabletteja tai tofasitinibioraaliliuosta tai lumelääkettä 26 viikon pituisen kaksoissokkoutetun, lumekontrolloidun vaiheen aikana. Potilaat, jotka eivät saaneet JIA ACR30 -vastetta avoimen hoitojakson sisäänajovaiheen aikana tai joilla oli yksittäinen taudin pahenemisjakso milloin tahansa, poistettiin tutkimuksesta. Kaikkiaan 225 potilasta osallistui avoimen hoitojakson sisäänajovaiheeseen. Näistä 173 (76,9 %) potilasta soveltui satunnaistettavaksi kaksoissokkoutettuun vaiheeseen saamaan joko vaikuttavaa ainetta sisältäviä 5 mg kalvopäällysteisiä tofasitinibitabletteja tai painoon perustuvan vastaavan määrän tofasitinibioraaliliuosta (n=88) kaksi kertaa vuorokaudessa tai lumelääkettä (n=85). Kaksoissokkoutetun vaiheen aikana 58 (65,9 %) potilasta tofasitinibiryhmässä ja 58 (68,2 %) potilasta lumelääkeryhmässä sai metotreksaattia. Tämä oli sallittua tutkimussuunnitelmassa, mutta sitä ei edellytetty.
Tutkimuksen kaksoissokkoutettuun vaiheeseen satunnaistettiin 133 pJIA-potilasta [RF+ tai RF- polyartriitti ja laajeneva oligoartriitti] ja 15 lasten nivelpsoriaasipotilasta ja heidät sisällytettiin jäljempänä esitettyihin tehoanalyyseihin.
Merkit ja oireet
Tutkimuksessa JIA-I merkitsevästi pienemmällä osuudella pJIA-potilaista, joita hoidettiin 5 mg kalvopäällysteisillä tofasitinibitableteilla kaksi kertaa vuorokaudessa tai painoon perustuvalla vastaavalla määrällä tofasitinibioraaliliuosta kaksi kertaa vuorokaudessa, oireet pahenivat äkillisesti (flare) viikolla 44, verrattuna lumelääkkeellä hoidettuihin potilaisiin. Merkitsevästi suurempi osuus pJIA-potilaista, joita hoidettiin 5 mg kalvopäällysteisillä tofasitinibitableteilla kaksi kertaa vuorokaudessa tai tofasitinibioraaliliuoksella, sai JIA ACR30-, 50-, ja 70-vasteet viikolla 44 verrattuna lumelääkkeellä hoidettuihin potilaisiin (taulukko 8).
Tulokset olivat 5 mg tofasitinibia kaksi kertaa vuorokaudessa käytettäessä taudin äkillisen pahenemisen ja JIA ACR30/50/70 -vasteiden suhteen paremmat kuin lumelääkettä käytettäessä, lastenreuman seuraavien alatyyppien osalta: RF+ polyartriitti, RF- polyartriitti, laajeneva oligoartriitti, ja lasten nivelpsoriaasi JIA -alatyypit. Tulokset olivat yhdenmukaisia koko tutkimuspopulaation tulosten kanssa.
Tulokset olivat 5 mg tofasitinibia kaksi kertaa vuorokaudessa käytettäessä taudin äkillisen pahenemisen ja JIA ACR30/50/70 -vasteiden suhteen paremmat kuin lumelääkettä käytettäessä pJIA-potilailla, jotka saivat tofasitinibia 5 mg kaksi kertaa vuorokaudessa samanaikaisesti metotreksaatin kanssa päivänä 1 [n=101 (76 %)] ja kun tofasitinibia käytettiin monoterapiana [n=32 (24 %)]. Lisäksi tulokset olivat 5 mg tofasitinibia kaksi kertaa vuorokaudessa käytettäessä taudin äkillisen pahenemisen ja JIA ACR30/50/70 -vasteiden suhteen paremmat kuin lumelääkettä käytettäessä pJIA-potilailla, joita oli aikaisemmin hoidettu bDMARD-valmisteella [n=39 (29 %)] ja pJIA-potilailla, joita ei ollut aikaisemmin hoidettu bDMARD-valmisteella [n=94 (71 %)].
Tutkimuksessa JIA-I avoimen hoitojakson sisäänajovaiheen viikolla 2 JIA ACR30 -vaste pJIA-potilailla oli 45,03 %.
Taulukko 8: Primaariset ja sekundaariset tehon päätetapahtumat pJIA-potilailla viikolla 44* tutkimuksessa JIA-I (kaikki p-arvot <0,05)
Primaarinen päätetapahtuma (Tyypin I virhe kontrolloitu) | Hoitoryhmä | Esiintyvyysluku | Ero (%) lumelääkkeeseen (95 % luottamusväli) |
Taudin äkillinen paheneminen | Tofasitinibi 5 mg kaksi kertaa vuorokaudessa (N=67) | 28 % | -24,7 (-40,8, -8,5) |
Lumelääke (N=66) | 53 % | ||
Toissijaiset päätetapahtumat (Tyypin I virhe kontrolloitu) | Hoitoryhmä | Vasteluku | Ero (%) lumelääkkeeseen (95 % luottamusväli |
JIA ACR30 | Tofasitinibi 5mg kaksi kertaa vuorokaudessa (N=67)) | 72 % | 24,7 (8,50, 40,8) |
Lumelääke (N=66) | 47 % | ||
JIA ACR50 | Tofasitinibi 5mg kaksi kertaa vuorokaudessa (N=67) | 67 % | 20,2 (3,72, 36,7) |
Lumelääke (N=66) | 47 % | ||
JIA ACR70 | Tofasitinibi 5mg kaksi kertaa vuorokaudessa (N=67) | 55 % | 17,4 (0,65, 34,0) |
Lumelääke (N=66) | 38 % | ||
Toissijainen päätetapahtuma (Tyypin I virhe kontrolloitu) | Hoitoryhmä | LS keskiarvo (SEM) | Ero (%) lumelääkkeeseen (95 % luottamusväli |
Muutos kaksoissokkoutetusta lähtötilanteesta CHAQ-toimintakyvyttömyysindeksissä | Tofasitinibi 5mg kaksi kertaa vuorokaudessa (N=67; n=46) | -0,11 (0,04) | -0,11 (-0,22, -0,01) |
Lumelääke (N=66; n=31) | 0,00 (0,04) |
ACR = American College of Rheumatology; CHAQ = childhood health assessment questionnaire -kyselylomake; LS = pienimmät neliösummat; n = niiden potilaiden lukumäärä, joilla havaintoja käynnillä; N = potilaiden kokonaismäärä; JIA = lastenreuma; SEM = keskiarvon keskivirhe
* 26 viikon kaksoissokkoutettu vaihe on viikon 18 satunnaistamispäivästä viikkoon 44 .
Tyypin I virheen suhteen kontrolloidut päätetapahtumat testataan tässä järjestyksessä: taudin paheneminen, JIA ACR50, JIA ACR30, JIA ACR70, CHAQ-toimintakyvyttömyysindeksi
Tutkimuksen JIA-I kaksoissokkoutetussa vaiheessa kussakin JIA ACR -vasteen osa-alueessa todettiin enemmän paranemista avoimen hoitojakson lähtötilanteesta (päivä 1) viikolla 24 ja viikolla 44 pJIA-potilailla, joita hoidettiin tofasitinibioraaliliuoksella 5 mg kaksi kertaa vuorokaudessa tai painon mukaisella vastaavalla määrällä kaksi kertaa vuorokaudessa, verrattuna lumelääkettä saaneisiin.
Fyysinen toimintakyky ja terveyteen liittyvä elämänlaatu
Fyysisen toimintakyvyn muutoksia tutkimuksessa JIA-I mitattiin CHAQ-toimintakyvyttömyysindeksillä. Keskimääräinen muutos kaksoissokkoutetusta lähtötilanteesta CHAQ-toimintakyvyttömyysindeksissä pJIA-potilaista oli merkitsevästi pienempi ryhmässä, joka sai 5 mg kalvopäällysteisiä tofasitinibitabletteja kaksi kertaa vuorokaudessa tai painoon perustuvan vastaavan määrän tofasitinibioraaliliuosta kaksi kertaa vuorokaudessa verrattuna lumelääkkeeseen viikolla 44 (taulukko 8). Keskimääräinen muutos kaksoissokkoutetusta lähtötilanteesta CHAQ-toimintakyvyttömyysindeksissä oli parempi käytettäessä tofasitinibia 5 mg kaksi kertaa vuorokaudessa verrattuna lumelääkkeeseen, kaikissa seuraavissa ryhmissä: RF+ polyartriitti, RF- polyartriitti, laajeneva oligoartriitti, ja lasten nivelpsoriaasi JIA -alatyypit. Muutokset olivat yhdenmukaisia koko tutkimuspopulaatiossa.
Pitkäkestoinen jatkotutkimus
Pitkäkestoisessa jatkotutkimuksessa 152 pJIA-potilasta tutkimuksesta JIA‑I sai avoimena hoitona tofasitinibia 5 mg kaksi kertaa vuorokaudessa enintään 7,5 vuoden ajan. Potilaista ne, jotka keskeyttivät osallistumisensa tutkimuksen JIA‑I avoimen vaiheen aikana (avoin tofasitinibi) ja ne, jotka osallistuivat kaksoissokkoutettuun vaiheeseen ja saivat tofasitinibia 5 mg kaksi kertaa vuorokaudessa (kaksoissokkoutettu tofasitinibi–tofasitinibi) tai lumelääkettä (kaksoissokkoutettu tofasitinibi–lumelääke), voitiin ottaa mukaan pitkäkestoiseen jatkotutkimukseen, jos he täyttivät sisäänottokriteerit. Useimmat tutkimuksen JIA‑I kaksoissokkoutettuun vaiheeseen osallistuneista potilaista osallistuivat pitkäkestoiseen jatkotutkimukseen (64/67 kaksoissokkoutettu tofasitinibi–tofasitinibi ‑ryhmästä ja 64/66 kaksoissokkoutettu tofasitinibi–lumelääke ‑ryhmästä). Tofasitinibille altistumisen mediaani oli 46,3 kuukautta (vaihteluväli: 1,0–88,4) kaksoissokkoutettu tofasitinibi–tofasitinibi ‑ryhmässä ja 43,8 kuukautta (vaihteluväli: 1,5–89,0) kaksoissokkoutettu tofasitinibi–lumelääke ‑ryhmässä. Taulukossa 9 esitetään kaksoissokkoutettu tofasitinibi–tofasitinibi- ja kaksoissokkoutettu tofasitinibi–lumelääke ‑ryhmien JIA ACR ‑vasteet kuukauden 45 kohdalla havaintojen ja muokatun mNRI-imputoinnin perusteella (potilaat, jotka keskeyttivät osallistumisensa riittämättömän kliinisen vasteen tai haittatapahtuman vuoksi, katsottiin hoitoon vastaamattomiksi).
Taulukko 9: pJIA-potilaiden tehon päätetapahtumat pitkäkestoisen jatkotutkimuksen kuukauden 45 kohdalla (havaintoihin ja muokattuun mNRI-imputointiin perustuvat analyysit)
| Tofasitinibi–tofasitinibi (kaksoissokkoutettu) (N = 64) | Tofasitinibi–lumelääke (kaksoissokkoutettu) (N = 64) | ||
Havainnot | mNRI | Havainnot | mNRI | |
JIA ACR30, n / N (%) | 33 / 34 (97,1) | 33 / 43 (76,7) | 34 / 37 (91,9) | 34 / 49 (69,4) |
JIA ACR50, n / N (%) | 32 / 34 (94,1) | 32 / 43 (74,4) | 34 / 37 (91,9) | 34 / 49 (69,4) |
JIA ACR70, n / N (%) | 29 / 34 (85,3) | 29 / 43 (67,4) | 32 / 37 (86,5) | 32 / 49 (65,3) |
JIA ACR90, n / N (%) | 19 / 34 (55,9) | 19 / 43 (44,2) | 26 / 37 (70,3) | 26 / 49 (53,1) |
JIA ACR, inaktiivinen tauti, n / N (%) | 13 / 36 (36,1) | 13 / 45 (28,9) | 17 / 39 (43,6) | 17 / 51 (33,3) |
ACR = American College of Rheumatology; JIA = juveniili idiopaattinen artriitti; mNRI = muokattu imputointi, jossa katsotaan, että potilaat, joista tietoja puuttuu, eivät ole saaneet vastetta; n = niiden potilaiden lukumäärä, joilla havaintoja käynnillä; N = potilaiden kokonaismäärä. | ||||
Pitkäkestoiset kontrolloidut nivelreumaa koskevat turvallisuustiedot
Tutkimus ORAL Surveillance (A3921133) oli laaja (N = 4 362), satunnaistettu vaikuttavalla vertailuvalmisteella kontrolloitu myyntiluvan myöntämisen jälkeinen turvallisuuden seurantatutkimus, johon osallistui vähintään 50-vuotiaita nivelreumapotilaita. Potilailla oli vähintään yksi muu sydän- ja verisuonitapahtumien lisäriskitekijä (sydän- ja verisuonitapahtumien riskitekijöiksi määritellään tupakointi, diagnosoitu hypertensio, diabetes mellitus, ennenaikainen sepelvaltimotauti suvussa, aiempi sepelvaltimotauti, mukaan lukien aiempi revaskularisaatiotoimenpide, sepelvaltimon ohitusleikkaus, sydäninfarkti, sydämenpysähdys, epästabiili angina pectoris, akuutti sepelvaltimo-oireyhtymä ja nivelreumaan liittyvä nivelenulkoinen sairaus, esim. kyhmyt, Sjögrenin oireyhtymä, kroonisesta sairaudesta aiheutunut anemia, keuhkomanifestaatiot). Suurimmalla osalla (yli 90 %) tofasitinibihoitoa saaneista potilaista, jotka tupakoivat tai olivat aiemmin tupakoineet, tupakointi oli jatkunut yli 10 vuoden ajan. Tupakoinnin keston mediaani oli tupakoivilla 35,0 vuotta ja aiemmin tupakoineilla 39,0 vuotta. Potilailla oli oltava vakiintunut metotreksaattiannostus tutkimuksen alkaessa. Annoksen muuttaminen oli sallittua.
Potilaat satunnaistettiin suhteessa 1:1:1 avoimeen hoitoon, joka oli 10 mg tofasitinibia kaksi kertaa vuorokaudessa, 5 mg tofasitinibia kaksi kertaa vuorokaudessa tai TNF:n estäjähoito (TNF:n estäjä oli joko 50 mg etanerseptiä kerran viikossa tai 40 mg adalimumabia joka toinen viikko). Ensisijaiset päätetapahtumat olivat todettu syöpä (ei-melanoottista ihosyöpää lukuun ottamatta) ja todettu merkittävä sydän- ja verisuonitapahtuma (MACE); päätetapahtumat varmistettiin keskitetysti (adjudicated endpoints) ja niiden kumulatiivinen ilmaantuvuus ja tilastollinen arviointi olivat sokkoutettuja. Tutkimuksen voimalaskelma perustui tapahtumiin (event-powered study), ja sen ohella edellytettiin vähintään 1 500 potilaan seuraamista 3 vuoden ajan. Tutkimushoito 10 mg:lla tofasitinibia kaksi kertaa vuorokaudessa lopetettiin, ja potilaiden hoidoksi vaihdettiin 5 mg kaksi kertaa vuorokaudessa laskimotromboemboliatapahtumien annosriippuvaisen signaalin vuoksi. Tofasitinibia 10 mg kahdesti vuorokaudessa saaneen ryhmän osalta ennen annoksen vaihtamista ja sen jälkeen kerätyt tiedot analysoitiin alun perin satunnaistetussa hoitoryhmässä.
Tutkimus ei täyttänyt yhdistettyjen tofasitinibiannosten ja TNF:n estäjien väliselle ensisijaiselle vertailulle asetettua non-inferiority-kriteeriä, koska riskitiheyksien suhteelle asetettu 95 %:n luottamusvälin yläraja ylitti etukäteen määritetyn non-inferiority-kriteerin 1,8 todettujen MACE-tapahtumien ja todettujen syöpien osalta (ei-melanoottista ihosyöpää lukuun ottamatta).
Jäljempänä on esitetty tulokset todettujen MACE-tapahtumien, todettujen syöpien (ei-melanoottista ihosyöpää lukuun ottamatta) ja valikoitujen muiden tapahtumien osalta.
MACE (mukaan lukien sydäninfarkti) ja laskimotromboembolia
Muiden kuin kuolemaan johtaneiden sydäninfarktien määrän todettiin lisääntyneen tofasitinibilla hoidetuilla potilailla TNF:n estäjiä saaneisiin potilaisiin verrattuna. TNF:n estäjiin verrattuna tofasitinibihoitoa saaneilla potilailla laskimotromboemboliatapahtumien ilmaantuvuuden todettiin olevan suurempi ja annosriippuvainen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Taulukko 10: MACE:n, sydäninfarktin ja laskimotromboembolian ilmaantumistiheys ja riskitiheyksien suhde
| Tofasitinibi 5 mg kahdesti vuorokaudessa | Tofasitinibi 10 mg kahdesti vuorokaudessaa | Molemmat tofasitinibiannoksetb | TNF:n estäjä (TNFi) |
MACEc | ||||
IR (95 % CI) / 100 potilasvuotta | 0,91 (0,67, 1,21) | 1,05 (0,78, 1,38) | 0,98 (0,79, 1,19) | 0,73 (0,52, 1,01) |
HR (95 % CI) vs TNFi | 1,24 (0,81, 1,91) | 1,43 (0,94, 2,18) | 1,33 (0,91, 1,94) |
|
Kuolemaan johtanut sydäninfarktic | ||||
IR (95 % CI) / 100 potilasvuotta | 0,00 (0,00, 0,07) | 0,06 (0,01, 0,18) | 0,03 (0,01, 0,09) | 0,06 (0,01, 0,17) |
HR (95 % CI) vs TNFi | 0,00 (0,00, Inf) | 1,03 (0,21, 5,11) | 0,50 (0,10, 2,49) |
|
Muu kuin kuolemaan johtanut sydäninfarktic | ||||
IR (95 % CI) / 100 potilasvuotta | 0,37 (0,22, 0,57) | 0,33 (0,19, 0,53) | 0,35 (0,24, 0,48) | 0,16 (0,07, 0,31) |
HR (95% CI) vs TNFi | 2,32 (1,02, 5,30) | 2,08 (0,89, 4,86) | 2,20 (1,02, 4,75) |
|
Laskimotromboemboliad | ||||
IR (95 % CI) / 100 potilasvuotta | 0,33 (0,19; 0,53) | 0,70 (0,49; 0,99) | 0,51 (0,38; 0,67) | 0,20 (0,10; 0,37) |
HR (95% CI) vs TNFi | 1,66 (0,76; 3,63) | 3,52 (1,74; 7,12) | 2,56 (1,30; 5,05) |
|
Keuhkoemboliad | ||||
IR (95 % CI) / 100 potilasvuotta | 0,17 (0,08; 0,33) | 0,50 (0,32; 0,74) | 0,33 (0,23; 0,46) | 0,06 (0,01; 0,17) |
HR (95% CI) vs TNFi | 2,93 (0,79; 10,83) | 8,26 (2,49; 27,43) | 5,53 (1,70; 18,02) |
|
Syvä laskimotukosd | ||||
IR (95 % CI) / 100 potilasvuotta | 0,21 (0,11; 0,38) | 0,31 (0,17; 0,51) | 0,26 (0,17; 0,38) | 0,14 (0,06; 0,29) |
HR (95% CI) vs TNFi | 1,54 (0,60; 3,97) | 2,21 (0,90; 5,43) | 1,87 (0,81; 4,30) |
|
a Tofasitinibia 10 mg kahdesti vuorokaudessa saaneiden ryhmässä on tietoja potilaista, joiden tofasitinibiannos vaihdettiin 10 mg:sta kahdesti vuorokaudessa 5 mg:aan kahdesti vuorokaudessa tutkimuksessa tehdyn muutoksen seurauksena. | ||||
Tutkimuksessa määritettiin seuraavat sydäninfarktin (kuolemaan johtava ja muu kuin kuolemaan johtava) kehittymistä ennakoivat tekijät käyttämällä Coxin monimuuttujamallia takautuvasti (backward selection): ikä ≥ 65 vuotta, miespuolinen, nykyinen tai aiempi tupakointi, potilaan sairastama diabetes ja sepelvaltimotauti (johon sisältyy sydäninfarkti, sepelvaltimotauti, stabiili angina pectoris tai sepelvaltimotoimenpide) (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Syövät
Tofasitinibilla hoidetuilla potilailla havaittiin syöpien (ei-melanoottista ihosyöpää lukuun ottamatta), erityisesti keuhkosyövän ja lymfooman lisääntymistä TNF-estäjillä hoidettuihin potilaisiin verrattuna. Lisäksi tofasitinibilla hoidetuilla potilailla havaittiin ei‑melanoottisen ihosyövän lisääntymistä TNF-estäjillä hoidettuihin potilaisiin verrattuna.
Taulukko 11: Syöpien ilmaantumistiheys ja riskitiheyksien suhdea
| Tofasitinibi 5 mg kahdesti vuorokaudessa | Tofasitinibi 10 mg kahdesti vuorokaudessab | Molemmat tofasitinibiannoksetc | TNF:n estäjä (TNFi) |
Syövät NMSC:tä lukuun ottamatta | ||||
IR (95 % CI) / 100 potilasvuotta | 1,13 (0,87, 1,45) | 1,13 (0,86, 1,45) | 1,13 (0,94, 1,35) | 0,77 (0,55, 1,04) |
HR (95 % CI) vs TNFi | 1,47 (1,00, 2,18) | 1,48 (1,00, 2,19) | 1,48 (1,04, 2,09) |
|
Keuhkosyöpä | ||||
IR (95 % CI) / 100 potilasvuotta | 0,23 (0,12, 0,40) | 0,32 (0,18, 0,51) | 0,28 (0,19, 0,39) | 0,13 (0,05, 0,26) |
HR (95 % CI) vs TNFi | 1,84 (0,74, 4,62) | 2,50 (1,04, 6,02) | 2,17 (0,95, 4,93) |
|
Lymfooma | ||||
IR (95 % CI) / 100 potilasvuotta | 0,07 (0,02, 0,18) | 0,11 (0,04, 0,24) | 0,09 (0,04, 0,17) | 0,02 (0,00, 0,10) |
HR (95 % CI) vs TNFi | 3,99 (0,45, 35,70) | 6,24 (0,75, 51,86) | 5,09 (0,65, 39,78) |
|
NMSC | ||||
IR (95 % CI) / 100 potilasvuotta | 0,61 (0,41; 0,86) | 0,69 (0,47; 0,96) | 0,64 (0,50; 0,82) | 0,32 (0,18; 0,52) |
HR (95 % CI) vs TNFi | 1,90 (1,04; 3,47) | 2,16 (1,19; 3,92) | 2,02 (1,17; 3,50) |
|
a Tiedot syövistä (ei-melanoottista ihosyöpää lukuun ottamatta), keuhkosyövästä ja lymfoomasta perustuvat hoidon aikana tai hoidon lopettamisen jälkeen tutkimuksen loppuun mennessä esiintyneisiin tapahtumiin. Tiedot ei-melanoottisesta ihosyövästä perustuvat hoidon aikana tai 28 päivän kuluessa hoidon lopettamisesta esiintyneisiin tapahtumiin. | ||||
Tutkimuksessa määritettiin seuraavat syövän kehittymistä (ei-melanoottista ihosyöpää lukuun ottamatta) ennakoivat tekijät käyttämällä Coxin monimuuttujamallia takautuvasti (backward selection): ikä ≥ 65 vuotta, nykyinen tai aiempi tupakointi (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Kuolleisuus
Tofasitinibihoitoa saaneiden potilaiden kuolleisuuden havaittiin lisääntyneen TNF:n estäjiin verrattuna. Kuolleisuus johtui pääasiassa sydän- ja verisuonitapahtumista, infektioista ja syövistä.
Taulukko 12: Kuolleisuuden ilmaantumistiheys ja riskitiheyksien suhdea
| Tofasitinibi 5 mg kahdesti vuorokaudessa | Tofasitinibi 10 mg kahdesti vuorokaudessa b | Molemmat tofasitinibi-annoksetc | TNF-estäjä (TNFi) |
Kuolleisuus (kaikki syyt) |
|
|
|
|
IR (95 % CI) / 100 potilasvuotta | 0,50 (0,33; 0,74) | 0,80 (0,57; 1,09) | 0,65 (0,50; 0,82) | 0,34 (0,20; 0,54) |
HR (95 % CI) vs. TNFi | 1,49 (0,81; 2,74) | 2,37 (1,34; 4,18) | 1,91 (1,12; 3,27) |
|
Kuolemaan johtaneet infektiot |
|
|
|
|
IR (95 % CI) / 100 potilasvuotta | 0,08 (0,02; 0,20) | 0,18 (0,08; 0,35) | 0,13 (0,07; 0,22) | 0,06 (0,01; 0,17) |
HR (95 % CI) vs. TNFi | 1,30 (0,29; 5,79) | 3,10 (0,84; 11,45) | 2,17 (0,62; 7,62) |
|
Kuolemaan johtaneet sydän- ja verisuonitapahtumat |
|
|
|
|
IR (95 % CI) / 100 potilasvuotta | 0,25 (0,13; 0,43) | 0,41 (0,25; 0,63) | 0,33 (0,23; 0,46) | 0,20 (0,10; 0,36) |
HR (95 % CI) vs. TNFi | 1,26 (0,55; 2,88) | 2,05 (0,96; 4,39) | 1,65 (0,81; 3,34) |
|
Kuolemaan johtaneet syövät |
|
|
|
|
IR (95 % CI) / 100 potilasvuotta | 0,10 (0,03; 0,23) | 0,00 (0,00; 0,08) | 0,05 (0,02; 0,12) | 0,02 (0,00; 0,11) |
HR (95 % CI) vs. TNFi | 4,88 (0.57; 41,74) | 0 (0,00; Inf) | 2,53 (0,30; 21,64) |
|
a Hoidon aikana tai 28 päivän kuluessa hoidon lopettamisesta ilmaantuneiden tapahtumien perusteella.
b Tofasitinibia 10 mg kahdesti vuorokaudessa saaneiden potilaiden ryhmässä on tietoja potilaista, joiden tofasitinibiannos vaihdettiin 10 mg:sta kahdesti vuorokaudessa 5 mg:aan kahdesti vuorokaudessa tutkimuksessa tehdyn muutoksen seurauksena.
c Tofasitinibiannos 5 mg kahdesti vuorokaudessa ja annos 10 mg kahdesti vuorokaudessa.
Lyhenteet: TNF = tuumorinekroositekijä, IR = ilmaantumistiheys (incidence rate), HR = riskitiheys (hazard ratio), CI = luottamusväli (confidence interval), Inf = ääretön (infinity).
Farmakokinetiikka
Tofasitinibin farmakokineettiselle profiilille on tyypillistä nopea imeytyminen (huippupitoisuus plasmassa saavutetaan 0,5–1 tunnissa), nopea eliminaatio (puoliintumisaika ~3 tuntia) ja suhteessa annokseen suureneva systeeminen altistus. Vakaan tilan pitoisuudet saavutetaan 24–48 tunnissa, ja kumuloituminen kaksi kertaa vuorokaudessa tapahtuvan annon yhteydessä on hyvin vähäistä.
Imeytyminen ja jakautuminen
Tofasitinibi imeytyy hyvin, ja sen oraalinen biologinen hyötyosuus on 74 %.Tofasitinibin anto runsasrasvaisen aterian yhteydessä ei muuttanut AUC-arvoa, mutta Cmax pieneni 32 %. Kliinisissä tutkimuksissa tofasitinibi annettiin aterioista riippumatta.
Laskimoon tapahtuneen annon jälkeen jakautumistilavuus on 87 l. Kiertävästä tofasitinibista plasman proteiineihin sitoutuneena on noin 40 %. Tofasitinibi sitoutuu pääasiassa albumiiniin eikä se vaikuta sitoutuvan α1-happamaan glykoproteiiniin. Tofasitinibi jakautuu samassa määrin veren punasoluihin ja plasmaan.
Biotransformaatio ja eliminaatio
Tofasitinibin puhdistumamekanismit ovat maksametabolia noin 70 % ja kanta-aineen erittyminen munuaisten kautta noin 30 %. Tofasitinibin metabolia on pääasiassa CYP3A4-välitteinen, ja CYP2C19 osallistuu siihen vähäisessä määrin. Ihmisellä radiomerkityllä lääkeaineella tehdyssä tutkimuksessa yli 65 % verenkierrossa olevasta kokonaisradioaktiivisuudesta vastasi muuttumatonta vaikuttavaa ainetta ja loput 35 % vastasi kahdeksaa metaboliittia, joista kukin vastasi alle 8 % kokonaisradioaktiivisuudesta. Kaikkia metaboliitteja on havaittu eri eläinlajeilla. Niiden vaikutuksen JAK1/3:n estoon oletetaan olevan alle kymmenkertainen tofasitinibiin verrattuna. Molekyylirakenteen samanaikaisesta muuntumisesta (stereo conversion) ihmisnäytteissä ei havaittu näyttöä. Tofasitinibin farmakologinen aktiivisuus liittyy kantamolekyyliin. In vitro, tofasitinibi on MDR1:n substraatti, mutta se ei toimi substraattina rintasyövän resistenssiproteiinille (BCRP), OATP1B1/1B3:lle tai OCT1/2:lle.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastavien tutkittavien AUC oli 37 % (lievä vajaatoiminta, kreatiniinipuhdistuma 50–80 ml/min), 43 % (kohtalainen vajaatoiminta, kreatiniinipuhdistuma 30–49 ml/min) ja 123 % (vaikea vajaatoiminta, kreatiniinipuhdistuma < 30 ml/min) suurempi verrattuna tutkittaviin, joiden munuaisten toiminta oli normaali (ks. kohta Annostus ja antotapa). Loppuvaiheen munuaistautia (ESRD) sairastavilla tutkittavilla dialyysihoidon merkitys tofasitinibin kokonaispuhdistuman kannalta oli suhteellisen vähäinen. Kun loppuvaiheen munuaistautia sairastaville tutkittaville annettiin 10 mg:n kerta-annos, muuna kuin dialyysipäivänä mitatun pitoisuuden keskimääräinen AUC-arvo oli noin 40 % (90 %:n luottamusvälit: 1,5–95 %) suurempi verrattuna tutkittaviin, joiden munuaisten toiminta oli normaali. Tofasitinibia ei tutkittu kliinisissä tutkimuksissa potilailla, joiden kreatiniinipuhdistuma (Cockcroft-Gaultin kaavalla arvioituna) oli lähtötilanteessa alle 40 ml/min (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa (Child–Pugh A) sairastavien tutkittavien AUC oli 3 %, ja kohtalaista maksan vajaatoimintaa (Child–Pugh B) sairastavien potilaiden AUC oli 65 % suurempi verrattuna tutkittaviin, joiden maksan toiminta oli normaali. Tofasitinibia ei tutkittu kliinisissä tutkimuksissa tutkittavilla, joilla oli vaikeaa maksan vajaatoimintaa (Child–Pugh C) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet) tai jotka olivat seulonnassa todettu HBV- tai HCV-positiivisiksi.
Yhteisvaikutukset
Tofasitinibi ei ole CYP-entsyymien (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ja CYP3A4) estäjä eikä induktori, eikä myöskään UGT:n estäjä (UGT1A1, UGT1A4, UGT1A6, UGT1A9 ja UGT2B7). Tofasitinibi ei ole MDR1:n, OATP1B1/1B3:n, OCT2:n, OAT1/3:n tai MRP:n estäjä kliinisesti merkittävinä pitoisuuksina.
Farmakokinetiikka pediatrisilla potilailla, joilla on lastenreuma
Populaatiofarmakokineettinen analyysi perustui tuloksiin ryhmistä, joissa potilaat saivat 5 mg:n kalvopäällysteisiä tofasitinibitabletteja kaksi kertaa vuorokaudessa tai painoon perustuvan vastaavan määrän tofasitinibioraaliliuosta kaksi kertaa vuorokaudessa. Analyysi osoitti, että tofasitinibin puhdistuma väheni ja jakautumistilavuus pieneni, kun JIA-potilaiden paino aleni. Saatavilla olevat tulokset osoittivat, että tofasitinibialtistuksessa (AUC) ei ollut kliinisesti merkittäviä eroja iän, rodun, sukupuolen, potilastyypin tai taudin lähtötilanteen vaikeusasteen perusteella. Tutkittavien välisen vaihtelun (% variaatiokerroin) (AUC:ssa) arvioitiin olevan noin 24 %.
Prekliiniset tiedot turvallisuudesta
Non-kliinisissä tutkimuksissa havaittiin vaikutuksia immuunisysteemiin ja hematopoieettiseen järjestelmään. Näiden katsottiin liittyvän tofasitinibin farmakologisiin ominaisuuksiin (JAK-kinaasin estoon). Kliinisesti merkityksellisillä annoksilla havaittiin immuunivasteen heikentymisestä aiheutuvia toissijaisia vaikutuksia, kuten bakteeri- ja virusinfektioita sekä lymfoomaa. Lymfooma havaittiin 3 aikuisella apinalla 8:sta, kun altistus oli kuusin- tai kolminkertainen tofasitinibin kliiniseen altistukseen verrattuna (sitoutumattoman aineen AUC ihmisellä annosten 5 mg tai 10 mg kaksi kertaa vuorokaudessa yhteydessä). Lymfoomia ei havaittu yhdelläkään nuorella apinalla 14:stä, kun tofasitinibialtistus oli viisin- tai 2,5-kertainen annoksista 5 mg tai 10 mg kaksi kertaa vuorokaudessa aiheutuvaan kliiniseen altistukseen nähden. Lymfoomien osalta apinoille haitaton annos (No Observable Adverse Effect Level, NOAEL) oli suunnilleen 1- tai 0,5-kertainen verrattuna annoksista 5 mg tai 10 mg kaksi kertaa vuorokaudessa aiheutuvaan kliiniseen altistukseen. Muita löydöksiä ihmisen altistusta suuremmilla annoksilla olivat vaikutukset maksaan ja maha-suolikanavaan.
Tofasitinibi ei ole mutageeninen eikä genotoksinen geenimutaatioita ja kromosomipoikkeavuuksia selvittäneiden in vitro- ja in vivo ‑koesarjojen tulosten perusteella.
Tofasitinibin karsinogeenisuutta tutkittiin kuusi kuukautta kestäneessä karsinogeenisuustutkimuksessa rasH2-siirtogeenisillä hiirillä ja kaksi vuotta kestäneessä karsinogeenisuustutkimuksessa rotilla. Tofasitinibi ei ollut karsinogeeninen hiirillä 38- tai 19-kertaisella altistuksella verrattuna annoksista 5 mg tai 10 mg kaksi kertaa vuorokaudessa aiheutuvaan kliiniseen altistukseen. Rotilla havaittiin hyvänlaatuisia kivesten interstitiaalisolujen (Leydigin solujen) kasvaimia: rotilla havaitut hyvänlaatuiset leydiginsolukasvaimet eivät liity ihmisen leydiginsolukasvainriskiin. Hibernoomia (ruskean rasvakudoksen syöpää) havaittiin naarasrotilla altistuksella, joka oli vähintään 83- tai 41-kertainen verrattuna annoksista 5 mg tai 10 mg kaksi kertaa vuorokaudessa aiheutuvaan kliiniseen altistukseen. Hyvänlaatuisia kateenkorvakasvaimia havaittiin naarasrotilla 187- tai 94-kertaisella altistuksella verrattuna annoksista 5 mg tai 10 mg kaksi kertaa vuorokaudessa aiheutuvaan kliiniseen altistukseen.
Tofasitinibin osoitettiin olevan teratogeeninen rotilla ja kaniineilla, ja sen on osoitettu vaikuttavan rotilla naaraiden hedelmällisyyteen (tiineyksien vähenemistä, keltarauhasten lukumäärän vähenemistä, implantaatiokohtien vähenemistä, elinkykyisten sikiöiden vähenemistä ja varhaisvaiheen resorptioiden lisääntymistä), synnytykseen sekä peri-/postnataaliseen kehitykseen. Tofasitinibi ei vaikuttanut uroksen hedelmällisyyteen, siittiöiden liikkuvuuteen eikä siittiöpitoisuuteen. Tofasitinibi erittyi imettävien rottien maitoon pitoisuuksina, jotka olivat noin kaksinkertaisia seerumissa 1–8 tuntia annoksen antamiseen jälkeen havaittuihin pitoisuuksiin nähden. Nuorilla rotilla ja apinoilla tehdyissä tutkimuksissa ei todettu tofasitinibiin liittyviä vaikutuksia urosten eikä naaraiden luiden kehitykseen, kun altistukset olivat samansuuruisia kuin ihmisille hyväksyttyjä annoksia käytettäessä.
Nuorilla eläimillä tehdyissä tutkimuksissa ei havaittu tofasitinibiin liittyviä löydöksiä jotka viittaisivat siihen, että pediatriset potilaat olisivat herkempiä kuin aikuiset. Nuorilla rotilla tehdyssä hedelmällisyystutkimuksessa ei saatu näyttöä kehitystoksisuudesta, vaikutuksista seksuaaliseen kypsymiseen, eikä näyttöä lisääntymistoksisuudesta (parittelu ja hedelmällisyys) sukukypsyyden saavuttamisen jälkeen. Yhden kuukauden pituisessa nuorilla rotilla tehdyssä tutkimuksessa ja 39 viikon pituisessa nuorilla apinoilla tehdyssä tutkimuksessa havaittiin tofasitinibiin liittyviä vaikutuksia immuunivasteen ja hematologisiin parametreihin, jotka olivat yhdenmukaisia JAK1/3:n ja JAK2:n estymisen kanssa. Nämä vaikutukset olivat palautuvia ja yhdenmukaisia niiden vaikutusten kanssa, joita havaittiin aikuisilla eläimillä vastaavilla altistuksilla.
Farmaseuttiset tiedot
Apuaineet
Viinirypälemakuaine [sisältää propyleeniglykolia (E1520), glyserolia (E422), ja luonnollisia makuaineita]
Kloorivetyhappo
Maitohappo (E270)
Puhdistettu vesi
Natriumbentsoaatti (E211)
Sukraloosi (E955)
Ksylitoli (E967)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
2 vuotta.
Kestoaika ensimmäisen avaamisen jälkeen
Hävitettävä 60 vuorokauden kuluttua pullon ensimmäisestä avaamisesta.
Säilytys
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Säilytä alkuperäispullossa ja -pakkauksessa. Herkkä valolle.
Säilytys ensimmäisen avaamisen jälkeen, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
XELJANZ oraaliliuos
1 mg/ml (L:ei) 240 ml (750,43 €)
PF-selosteen tieto
Valkoiset 250 ml:n HDPE-pullot, jotka sisältävät 240 ml oraaliliuosta. Pullo on suljettu lapsiturvallisella polypropeenikorkilla, jossa on PP-vuoraus, sinetöitynä alumiinikalvolla, joka on kiinnitetty paikalleen lämmittämällä. Suun kautta tapahtuvaa annostelua varten pakkauksessa on 5 ml:n ruisku, jossa on asteikko 3,2 ml, 4 ml ja 5 ml.
Pullon suljinjärjestelmä sisältää myös pientiheyspolyeteenistä (LDPE) valmistetun, pulloon painettavan sovittimen (PIBA, press-in bottle adapter).
Pakkauskoko: jokainen pakkaus sisältää yhden pullon, yhden pulloon painettavan sovittimen ja yhden ruiskun suun kautta antoa varten.
Valmisteen kuvaus:
Kirkas, väritön liuos.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
XELJANZ oraaliliuos
1 mg/ml 240 ml
- Alempi erityiskorvaus (65 %). Tofasitinibi: Tulehduksellisten nivel- ja suolistosairauksien hoito erityisin edellytyksin (291).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Tofasitinibi: Aikuisten nivelreuman tai nivelpsoriaasin ja keskivaikean tai vaikean haavaisen paksusuolitulehduksen hoito erityisin edellytyksin (3005).
ATC-koodi
L04AF01
Valmisteyhteenvedon muuttamispäivämäärä
11.06.2026
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com