
EVRENZO tabletti, kalvopäällysteinen 20 mg, 50 mg, 70 mg, 100 mg, 150 mg
Vaikuttavat aineet ja niiden määrät
Evrenzo 20 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 20 mg roksadustaattia.
Apuaineet, joiden vaikutus tunnetaan
Yksi 20 mg:n kalvopäällysteinen tabletti sisältää 40,5 mg laktoosia, 0,9 mg alluranpunaista AC alumiinilakkaa ja 0,21 mg soijalesitiiniä.
Evrenzo 50 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 50 mg roksadustaattia.
Apuaineet, joiden vaikutus tunnetaan
Yksi 50 mg:n kalvopäällysteinen tabletti sisältää 101,2 mg laktoosia, 1,7 mg alluranpunaista AC alumiinilakkaa ja 0,39 mg soijalesitiiniä.
Evrenzo 70 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 70 mg roksadustaattia.
Apuaineet, joiden vaikutus tunnetaan
Yksi 70 mg:n kalvopäällysteinen tabletti sisältää 141,6 mg laktoosia, 2,1 mg alluranpunaista AC alumiinilakkaa ja 0,47 mg soijalesitiiniä.
Evrenzo 100 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 100 mg roksadustaattia.
Apuaineet, joiden vaikutus tunnetaan
Yksi 100 mg:n kalvopäällysteinen tabletti sisältää 202,4 mg laktoosia, 2,8 mg alluranpunaista AC alumiinilakkaa ja 0,63 mg soijalesitiiniä.
Evrenzo 150 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 150 mg roksadustaattia.
Apuaineet, joiden vaikutus tunnetaan
Yksi 150 mg:n kalvopäällysteinen tabletti sisältää 303,5 mg laktoosia, 3,7 mg alluranpunaista AC alumiinilakkaa ja 0,84 mg soijalesitiiniä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Evrenzo on tarkoitettu aikuispotilaille krooniseen munuaissairauteen (CKD) liittyvän oireisen anemian hoitoon.
Ehto
Hoidon saa aloittaa vain anemian hoitoon perehtynyt lääkäri.
Annostus ja antotapa
Roksadustaattihoidon saa aloittaa vain anemian hoitoon perehtynyt lääkäri. Kaikki muut anemian aiheuttajat on arvioitava ennen Evrenzo-hoidon aloittamista sekä päätettäessä annoksen suurentamisesta.
Anemian oireet ja jälkiseuraukset voivat vaihdella iän, sukupuolen ja yleisen tautitaakan mukaan. Lääkärin on arvioitava yksittäisen potilaan taudinkulku ja kliininen tila. Anemian oireiden lisäksi tietyt kriteerit, kuten hemoglobiiniarvon (Hb) laskunopeus, aiempi vaste rautahoidolle ja punasolusiirron tarpeellisuuden riski voivat olla oleellisia arvioitaessa yksittäisen potilaan taudinkulkua ja kliinistä tilaa.
Annostus
Asianmukainen roksadustaattiannos otetaan suun kautta kolme kertaa viikossa. Annoksia ei saa ottaa peräkkäisinä päivinä.
Annos määritetään yksilöllisesti ja hoidolla pyritään saavuttamaan Hb-tavoitearvo 10–12 g/dl ja pitämään se tällä tasolla, kuten alla on kuvattu.
Roksadustaattihoitoa ei pidä jatkaa 24 viikkoa pidempään, jos kliinisesti merkittävää Hb-arvon nousua ei saavuteta. Vaihtoehtoiset syyt riittämättömälle vasteelle on selvitettävä ja hoidettava ennen Evrenzo-hoidon aloittamista uudelleen.
Aloitusannos hoidon aloittamisen yhteydessä
Rautavarastojen riittävyys on varmistettava ennen hoidon aloittamista.
Potilaat, joita ei parhaillaan hoideta erytropoieesiastimuloivalla aineella (ESA)
Kun anemian hoito aloitetaan potilaalle, joka ei ole aiemmin saanut ESA-hoitoa, roksadustaatin suositeltu aloitusannos on 70 mg kolme kertaa viikossa, kun potilaan paino on alle 100 kg ja 100 mg kolme kertaa viikossa, kun potilaan paino on vähintään 100 kg.
ESA-hoidosta siirtyvät potilaat
Potilaat, jotka saavat parhaillaan ESA-hoitoa, voidaan siirtää roksadustaattihoitoon, mutta jos ESA-hoitoa saavan dialyysipotilaan tila on muutoin vakaa, siirtämistä roksadustaattihoitoon tulee harkita vain pätevästä kliinisestä syystä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Sellaisten ESA-hoitoa saavien potilaiden siirtämistä roksadustaattihoitoon, joiden tila on muutoin vakaa ja jotka eivät saa dialyysihoitoa, ei ole tutkittu. Näiden potilaiden roksadustaattihoidosta päätettäessä päätöksen on perustuttava potilaan yksilölliseen hyöty-riskisuhteeseen.
Roksadustaatin suositeltu aloitusannos perustuu hoidon vaihtamista edeltävien 4 viikon aikana käytettyyn keskimääräiseen ESA-annokseen (ks. taulukko 1). Ensimmäisellä roksadustaattiannoksella korvataan hoitoaikataulun mukainen seuraava ESA-annos.
Taulukko 1. Kolme kertaa viikossa otettavan roksadustaatin aloitusannokset ESA-hoidosta siirtyville potilaille
| Darbepoetiini alfa -annos laskimoon tai ihon alle (mikrogrammaa/viikko) | Epoetiiniannos laskimoon tai ihon alle (IU/viikko) | Metoksipolyetyleeniglykoliepoetiini beeta -annos laskimoon tai ihon alle (mikrogrammaa/kuukausi) | Roksadustaattiannos (milligrammaa kolme kertaa viikossa) |
| < 25 | < 5 000 | < 80 | 70 |
| 25 – < 40 | 5 000 – ≤ 8 000 | 80 – ≤ 120 | 100 |
| 40 – ≤ 80 | > 8 000 – ≤ 16 000 | > 120 – ≤ 200 | 150 |
| > 80 | > 16 000 | > 200 | 200 |
| ESA: erytropoieesia stimuloiva aine | |||
Annoksen muuttaminen ja Hb-arvon seuranta
Yksilöllisesti määritetty ylläpitoannos on 20–400 mg kolme kertaa viikossa (ks. kohta Suurin suositeltu annos). Hb-arvoja tulee seurata kahden viikon välein, kunnes Hb-tavoitearvo 10–12 g/dl on saavutettu ja vakiintunut ja tämän jälkeen 4 viikon välein tai kliinisen tarpeen mukaan.
Roksadustaattiannosta voidaan sovittaa suurentamalla tai pienentämällä aloitusannosta asteittain 4 viikkoa hoidon aloittamisen jälkeen ja sitten 4 viikon välein, paitsi jos Hb-arvo nousee enemmän kuin 2 g/dl, jolloin annosta on välittömästi pienennettävä yhdellä annostasolla. Kun roksadustaattiannosta sovitetaan, on otettava huomioon potilaan senhetkinen Hb-arvo ja Hb-arvossa edellisten 4 viikon aikana tapahtuneet muutokset, ja noudatettava taulukossa 2 kuvatun annoksen säätöalgoritmin mukaisia annoksen säätövaiheita.
Annoksen asteittaisessa suurentamisessa tai pienentämisessä tulee noudattaa saatavilla olevien annosten järjestystä: 20 mg‑40 mg‑50 mg‑70 mg‑100 mg‑150 mg‑200 mg‑250 mg‑300 mg‑400 mg (vain dialyysihoitoa saaville CKD-potilaille).
Taulukko 2. Annoksen säätämistä koskevat säännöt
| Hb-arvon muutos edellisten 4 viikon aikana1 | Nykyinen Hb-arvo (g/dl): | |||
| < 10,5 | 10,5–11,9 | 12,0–12,9 | ≥ 13,0 | |
Muutos arvossa yli +1,0 g/dl | Ei muutosta | Pienennä annosta yhdellä annostasolla | Pienennä annosta yhdellä annostasolla | Keskeytä hoito, seuraa Hb-arvoa, ja kun Hb-arvo on < 12,0 g/dl, jatka hoitoa kaksi annostasoa pienemmällä annoksella |
Muutos arvossa välillä ‑1,0 – +1,0 g/dl | Suurenna annosta yhdellä annostasolla | Ei muutosta | Pienennä annosta yhdellä annostasolla | |
Muutos arvossa alle ‑1,0 g/dl | Suurenna annosta yhdellä annostasolla | Suurenna annosta yhdellä annostasolla | Ei muutosta | |
| Roksadustaattiannosta ei saa muuttaa useammin kuin 4 viikon välein, paitsi jos Hb-arvo nousee enemmän kuin 2 g/dl minkä tahansa 4 viikon jakson aikana, jolloin annosta on välittömästi pienennettävä yhdellä annostasolla. 1Hemoglobiiniarvon (Hb) muutos edellisten 4 viikon aikana = (nykyinen Hb-arvo) – (4 viikkoa aikaisemmin mitattu edellinen Hb-arvo). | ||||
Jos annosta täytyy pienentää entisestään, vaikka potilas saa jo pienintä annosta (20 mg kolme kertaa viikossa), ei annoksen pienentämistä 20 mg:sta saa tehdä tablettia murtamalla vaan pidentämällä annosväliä kahteen kertaan viikossa. Jos annosta täytyy vieläkin pienentää, annosväli voidaan pidentää yhteen kertaan viikossa.
Ylläpitoannos
Kun Hb-arvo on vakiintunut tavoitearvoon 10–12 g/dl, sen säännöllistä seurantaa on jatkettava ja annoksen muuttamissääntöjä noudatettava (ks. taulukko 2).
Potilaat, joille aloitetaan dialyysihoito roksadustaattihoidon aikana
Annosta ei tarvitse spesifisesti säätää CKD-potilaalle, jolle aloitetaan dialyysihoito roksadustaattihoidon aikana. Tavanomaisia annoksen muuttamissääntöjä (ks. taulukko 2) tulee noudattaa.
Roksadustaatin käyttö samanaikaisesti indusoijien tai inhibiittorien kanssa
Kun hoito aloitetaan tai lopetetaan potilaan saadessa samanaikaisesti voimakkaita CYP2C8:n estäjiä (esim. gemfibrotsiilia) tai indusoijia (esim. rifampisiinia) tai UGT1A9:n estäjiä (esim. probenesidia): Hb-arvon rutiiniseurantaa on jatkettava ja annoksen muuttamissääntöjä noudatettava (ks. taulukko 2 ja kohdat Yhteisvaikutukset ja Farmakokinetiikka).
Suurin suositeltu annos
Jos potilas ei saa dialyysihoitoa
Roksadustaattiannos ei saa olla yli 3 mg/kg tai 300 mg kolme kertaa viikossa, kumpi tahansaon pienempi.
Jos potilas saa dialyysihoitoa
Roksadustaattiannos ei saa olla yli 3 mg/kg tai 400 mg kolme kertaa viikossa, kumpi tahansa on pienempi.
Annoksen unohtuminen
Jos annos unohtuu ja seuraavan aikataulun mukaisen annoksen ottamiseen on yli 1 vuorokausi, unohtunut annos on otettava mahdollisimman pian. Jos seuraavan aikataulun mukaisen annoksen ottamiseen on enintään 1 vuorokausi, unohtunut annos on jätettävä väliin ja seuraava annos on otettava seuraavana aikataulun mukaisena päivänä. Molemmissa tapauksissa tämän jälkeen jatketaan normaalin antoaikataulun mukaisesti.
Iäkkäät
Aloitusannosta ei tarvitse muuttaa iäkkäille potilaille (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Aloitusannostasoa ei tarvitse sovittaa potilaille, joilla on lievä maksan vajaatoiminta (Child‑Pugh-luokka A) (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Varovaisuutta suositellaan määrättäessä roksadustaattia potilaille, joilla on keskivaikea maksan vajaatoiminta. Keskivaikeaa maksan vajaatoimintaa (Child‑Pugh-luokka B) sairastavan potilaan hoitoa aloitettaessa aloitusannos on pienennettävä puoleen tai annostasolle, joka on lähimpänä puolta aloitusannoksesta. Evrenzo-valmistetta ei suositella vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastaville potilaille, koska turvallisuutta ja tehoa tässä potilasryhmässä ei ole arvioitu (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Roksadustaatin turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Kalvopäällysteiset Evrenzo-tabletit tulee ottaa suun kautta ruoan kanssa tai ilman ruokaa. Tabletit tulee niellä kokonaisina, eikä niitä saa pureskella, rikkoa tai murskata, koska kliinisiä tietoja tästä ei ole, sekä tabletin valoherkän ytimen suojaamiseksi valohajoamiselta.
Tabletit pitää ottaa vähintään 1 tunnin kuluttua fosfaatin sitojien (lantaania lukuun ottamatta) tai muiden moniarvoisia kationeja, kuten kalsiumia, rautaa, magnesiumia tai alumiinia, sisältävien lääkevalmisteiden ottamisesta (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
Vasta-aiheet
- yliherkkyys vaikuttavalle aineelle, maapähkinälle, soijalle tai kohdassa Apuaineet mainituille apuaineille
- raskauden viimeinen kolmannes (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Raskaus ja imetys)
- imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Sydän- ja verisuonitapahtumien ja kuoleman riski
Yleisesti roksadustaattihoitoon liittyvän sydän- ja verisuonitapahtumien ja kuoleman riskin on arvioitu olevan samaa luokkaa ESA-hoitoon liittyvän sydän- ja verisuonitapahtumien ja kuoleman riskin kanssa näiden hoitojen suorasta vertailusta saatujen tietojen perusteella (ks. kohta Farmakodynamiikka). Tätä riskiä ei voitu arvioida riittävän luotettavasti lumelääkkeeseen nähden potilailla, joilla oli CKD:hen liittyvä anemia ja jotka eivät saaneet dialyysihoitoa, joten näiden potilaiden roksadustaattihoidosta päätettäessä on käytettävä samanlaista harkintaa kuin ennen ESA-hoidon määräämistä . Lisäksi on tunnistettu useita tälle riskille mahdollisesti altistavia tekijöitä, kuten hoitovasteen puute ja ESA-hoitoa saavien, tilaltaan vakaiden dialyysipotilaiden siirtäminen roksadustaattihoitoon (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka). Jos hoitovastetta ei saavuteta, ei roksadustaattihoitoa pidä jatkaa 24 viikkoa pidempään (ks. kohta Annostus ja antotapa). Jos ESA-hoitoa saavan dialyysipotilaan tila on muutoin vakaa, siirtämistä roksadustaattihoitoon tulee harkita vain pätevästä kliinisestä syystä (ks. kohta Annostus ja antotapa). Tätä riskiä ei voitu arvioida ESA-hoitoa saavilla potilailla, joiden tila on vakaa, joilla on CKD:hen liittyvä anemia ja jotka eivät saa dialyysihoitoa, sillä näitä potilaita ei ole tutkittu. Näiden potilaiden roksadustaattihoidosta päätettäessä päätöksen on perustuttava potilaan yksilölliseen hyöty-riskisuhteeseen.
Tromboottiset verisuonitapahtumat
Tromboottisten verisuonitapahtumien, mukaan lukien syvän laskimotukoksen (SLT), keuhkoembolian (KE) ja aivoinfarktin raportoitua riskiä ja roksadustaattihoidon hyötyjä on punnittava huolellisesti etenkin niiden potilaiden kohdalla, joilla on tromboottisten verisuonitapahtumien riskitekijöitä (esim. lihavuus tai aiemmin esiintynyt tromboottinen verisuonitapahtuma). Suurin osa syvistä laskimotukos-, keuhkoembolia- ja aivoinfarktitapahtumista oli vakavia. Kuolemaan johtaneita aivoinfarkteja on raportoitu.
Hb-arvojen nopeaa kohoamista on havaittu joidenkin aivoverenkiertohäiriöiden yhteydessä.
Kliinisissä tutkimuksissa dialyysihoitoa saavilla CKD-potilailla veritien tromboosien (VAT) raportoitiin olevan hyvin yleisiä (ks. kohta Haittavaikutukset).
Näillä potilailla, jotka saivat roksadustaattihoitoa, veritien tromboosit olivat yleisimpiä ensimmäisten 12 viikon aikana hoidon aloittamisen jälkeen ja etenkin jos Hb-arvo oli yli 12 g/dl ja jos se oli noussut yli 2 g/dl neljän viikon ajanjaksolla. Hb-arvojen tiivistä seurantaa ja annoksen muuttamista annoksen muuttamissääntöjen (ks. taulukko 2) mukaisesti suositellaan tällaisten arvojen välttämiseksi.
Potilaat, joilla ilmenee tromboottiseen verisuonitapahtumaan viittaavia merkkejä ja oireita, on viipymättä arvioitava ja hoidettava asianmukaisesti. Hoidon keskeyttämis- tai lopettamispäätöksen on perustuttava yksilöllisen hyöty-riskisuhteen arviointiin.
Kouristuskohtaukset
Kliinisissä tutkimuksissa roksadustaattia saavilla potilailla raportoitiin kouristuskohtausten olevan yleisiä (ks. kohta Haittavaikutukset). Roksadustaattia on käytettävä varoen, jos potilaalla on aiemmin esiintynyt kouristuskohtauksia, epilepsiaa tai sairauksia, joihin liittyy alttius kouristuskohtauksille, kuten keskushermoston infektioita (CNS). Hoidon keskeyttämis- tai lopettamispäätöksen on perustuttava potilaan yksilölliseen hyöty-riskisuhteeseen.
Vakavat infektiot
Yleisimmin raportoituja vakavia infektioita olivat keuhkokuume ja virtsatieinfektiot. Potilaat, joilla ilmenee infektioon viittaavia merkkejä ja oireita, on arvioitava viipymättä ja hoidettava asianmukaisesti.
Sepsis
Sepsis oli yksi yleisimmin raportoiduista vakavista infektioista, ja siihen liittyi myös kuolemantapauksia. Potilaat, joilla ilmenee sepsikseen (esim. infektio, joka etenee elimistössä ja johon liittyy matala verenpaine ja elinten toimintahäiriön mahdollisuus) viittaavia merkkejä ja oireita, on arvioitava viipymättä ja hoidettava asianmukaisesti.
Kilpirauhasen sekundaarinen vajaatoiminta
Roksadustaatin käytön yhteydessä on raportoitu sekundaarista hypotyreoosia (ks. kohta Haittavaikutukset). Nämä reaktiot olivat palautuvia, kun roksadustaatin käyttö lopetettiin. Kilpirauhasen toiminnan seurantaa suositellaan kliinisen tarpeen mukaan.
Riittämätön hoitovaste
Jos potilaan vaste roksadustaattihoidolle on riittämätön, syyt on selvitettävä. Ravinnepuutokset on korjattava. Samanaikaiset infektiot, piilevä verenhukka, hemolyysi, vaikea alumiinimyrkytys, taustalla olevat hematologiset sairaudet tai luuydinfibroosi saattavat myös heikentää erytropoieettista vastetta. Retikulosyyttilaskennan sisällyttämistä arviointiin on syytä harkita. Jos vasteen puuttumisen tyypilliset syyt on suljettu pois ja potilaalla on retikulosytopenia, on harkittava luuydintutkimusta. Jos riittämättömälle hoitovasteelle ei löydy hoidettavissa olevaa syytä, Evrenzo-hoitoa ei pidä jatkaa 24 viikkoa pidempään.
Maksan vajaatoiminta
Varovaisuutta on noudatettava annettaessa roksadustaattia potilaille, joilla on keskivaikea maksan vajaatoiminta (Child-Pugh-luokka B). Evrenzo-valmistetta ei suositella potilaille, joilla on vaikea maksan vajaatoiminta (Child-Pugh-luokka C) (ks. kohta Farmakokinetiikka.).
Raskaus ja ehkäisy
Roksadustaattihoitoa ei pidä aloittaa naisille, jotka suunnittelevat raskaaksi tulemista tai ovat raskaana, eikä tilanteessa, jossa krooniseen munuaissairauteen liittyvä anemia diagnosoidaan raskauden aikana. Tällaisissa tapauksissa tulee mahdollisuuksien mukaan aloittaa vaihtoehtoinen hoito. Jos nainen tulee raskaaksi roksadustaattihoidon aikana, hoito on keskeytettävä ja aloitettava mahdollisuuksien mukaan vaihtoehtoinen hoito. Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä hoidon aikana sekä ainakin viikon ajan viimeisen Evrenzo-annoksen jälkeen (ks. kohdat Vasta-aiheet ja Raskaus ja imetys).
Väärinkäyttö
Väärinkäyttö voi johtaa punasolujen veren tilavuusosuuden liialliseen suurenemiseen. Tähän voi liittyä henkeä uhkaavia sydän- ja verisuonijärjestelmän komplikaatioita.
Apuaineet
Laktoosi
Evrenzo sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Alluranpunainen AC alumiinilakka
Evrenzo sisältää alluranpunaista AC alumiinilakkaa, joka saattaa aiheuttaa allergisia reaktioita.
Soijalesitiini
Evrenzo sisältää soijalesitiiniä ja on vasta-aiheinen potilaille, jotka ovat allergisia maapähkinälle tai soijalle (ks. kohta Vasta-aiheet).
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutus roksadustaattiin
Fosfaatin sitojat ja muut moniarvoisia kationeja sisältävät valmisteet
Kun roksadustaattia annettiin terveille tutkittaville samanaikaisesti fosfaatin sitojien sevelameerikarbonaatin tai kalsiumasetaatin kanssa, roksadustaatin AUC-arvo pieneni 67 % ja vastaavasti 46 %, ja Cmax-arvo pieneni 66 % ja vastaavasti 52 %. Roksadustaatti saattaa muodostaa kelaatteja sellaisten moniarvoisten kationien kanssa, joita on fosfaatin sitojissa tai muissa valmisteissa, jotka sisältävät kalsiumia, rautaa, magnesiumia tai alumiinia. Fosfaatin sitojien antaminen eri aikaan (vähintään 1 tunnin erolla) ei vaikuttanut CKD-potilaiden roksadustaattialtistukseen kliinisesti merkitsevässä määrin. Roksadustaatti pitää ottaa vähintään 1 tunnin kuluttua fosfaatin sitojien tai muiden moniarvoisia kationeja sisältävien lääkevalmisteiden tai ravintolisien ottamisesta (ks. kohta Annostus ja antotapa). Tämä rajoitus ei koske lantaanikarbonaattia, sillä roksadustaatin ja lantaanikarbonaatin samanaikainen anto ei muuttanut plasman roksadustaattialtistusta kliinisesti merkittävästi.
CYP2C8- tai UGT1A9-aktiivisuuden muuntajat
Roksadustaatti on CYP2C8:n ja UGT1A9:n substraatti. Kun roksadustaattia annettiin terveille tutkittaville samanaikaisesti gemfibrotsiilin (CYP2C8:n ja OATP1B1:n estäjä) tai probenesidin (UGT:n ja OAT1/OAT3:n estäjä) kanssa, roksadustaatin AUC-arvo suureni 2,3‑kertaiseksi ja Cmax-arvo 1,4‑kertaiseksi. Hb-arvoja on seurattava aloitettaessa tai lopetettaessa samanaikaista hoitoa gemfibrotsiililla, probenesidilla, muilla voimakkailla CYP2C8:n estäjillä tai indusoijilla tai muilla voimakkailla UGT1A9:n estäjillä. Roksadustaattiannosta on muutettava Hb-arvon seurantaan perustuvien annoksen muuttamissääntöjen mukaisesti (ks. taulukko 2).
Roksadustaatin vaikutukset muihin lääkevalmisteisiin
OATP1B1:n tai BCRP:n substraatit
Roksadustaatti on BCRP:n ja OATP1B1:n estäjä. Näillä kuljettajaproteiineilla on tärkeä rooli suolistossa ja maksassa tapahtuvassa statiinien imeytymisessä ja effluksissa. Kun terveille tutkittaville annettiin 200 mg roksadustaattia samanaikaisesti simvastatiinin kanssa, simvastatiinin AUC-arvo suureni 1,8‑kertaiseksi ja Cmax-arvo 1,9‑kertaiseksi, ja simvastatiinihapon (simvastatiinin aktiivisen metaboliitin) AUC-arvo suureni 1,9‑kertaiseksi ja Cmax-arvo 2,8‑kertaiseksi. Simvastatiinin ja simvastatiinihapon pitoisuudet suurenivat myös, kun simvastatiinia annettiin 2 tuntia ennen roksadustaatin antoa tai 4 tai 10 tuntia roksadustaatin jälkeen. Kun 200 mg:n roksadustaattiannoksen kanssa annettiin samanaikaisesti rosuvastatiinia, rosuvastatiinin AUC-arvo suureni 2,9‑kertaiseksi ja Cmax-arvo 4,5‑kertaiseksi. Kun 200 mg:n roksadustaattiannoksen kanssa annettiin samanaikaisesti atorvastatiinia, atorvastatiinin AUC-arvo suureni 2,0‑kertaiseksi ja Cmax-arvo 1,3‑kertaiseksi.
Myös muiden statiinien kanssa on odotettavissa yhteisvaikutuksia. Jos statiineja annetaan samanaikaisesti roksadustaatin kanssa, potilasta on seurattava statiineihin liittyvien haittavaikutusten ja statiiniannoksen pienentämistarpeen varalta. Katso lisätietoja statiinin valmisteyhteenvedosta, kun valitset potilaalle sopivaa statiiniannosta.
Roksadustaatti saattaa suurentaa muiden sellaisten valmisteiden altistusta plasmassa, jotka ovat BCRP:n tai OATP1B1:n substraatteja. Potilasta on seurattava samanaikaisesti käytettyjen lääkevalmisteiden aiheuttamien haittavaikutusten varalta, ja annosta on säädettävä tarvittaessa.
Roksadustaatti ja erytropoieesia stimuloivat aineet (ESA)
Roksadustaatin ja ESA-hoitojen samanaikaista antoa ei suositella, koska tämän yhdistelmän käyttöä ei ole tutkittu.
Raskaus ja imetys
Raskaus, naiset, jotka voivat tulla raskaaksi, ja ehkäisy
Ei ole olemassa tietoja roksadustaatin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Roksadustaatti on vasta-aiheista 3. raskauskolmanneksen aikana (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Roksadustaatin käyttöä ei suositella 1. ja 2. raskauskolmanneksen aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jos nainen tulee raskaaksi Evrenzo-hoidon aikana, hoito on keskeytettävä ja vaihdettava mahdollisuuksien mukaan vaihtoehtoiseen hoitoon (ks. kohta Vasta-aiheet).
Imetys
Ei tiedetä, erittyykö/erittyvätkö roksadustaatti/metaboliitit ihmisen rintamaitoon. Olemassa olevat tiedot koe-eläimistä ovat osoittaneet roksadustaatin erittyvän rintamaitoon (yksityiskohdat, ks. kohta Prekliiniset tiedot turvallisuudesta). Evrenzo on vasta-aiheista rintaruokinnan aikana (ks. kohdat Vasta-aiheet ja Prekliiniset tiedot turvallisuudesta).
Hedelmällisyys
Eläimillä tehdyissä tutkimuksissa roksadustaatilla ei ollut vaikutusta urosten tai naaraiden hedelmällisyyteen. Urosrottien lisääntymiselimissä havaittiin kuitenkin muutoksia. Roksadustaatin mahdollisia vaikutuksia miesten hedelmällisyyteen ei tällä hetkellä tunneta. Emolle toksisilla annoksilla todettiin alkiokuolemien lisääntymistä (ks. kohta Prekliiniset tiedot turvallisuudesta). Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä hoidon aikana ja ainakin 1 viikko viimeisen Evrenzo-annoksen jälkeen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Roksadustaatilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Evrenzo-hoidon aikana on raportoitu kouristuskohtauksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Siksi potilaiden on noudatettava varovaisuutta ajaessaan autoa tai käyttäessään koneita.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Evrenzo-valmisteen turvallisuutta arvioitiin 3 542 dialyysistä riippumattomalla (NDD) ja 3 353 dialyysistä riippuvaisella (DD) aneemisella CKD-potilaalla, jotka olivat saaneet vähintään yhden annoksen roksadustaattia.
Yleisimpiä (≥ 10 %) roksadustaattiin liittyviä haittavaikutuksia ovat hypertensio (13,9 %), veritien tromboosi (12,8 %), ripuli (11,8 %), perifeerinen turvotus (11,7 %), hyperkalemia (10,9 %) ja pahoinvointi (10,2 %).
Yleisimpiä (≥ 1 %) roksadustaattiin liittyviä vakavia haittavaikutuksia olivat sepsis (3,4 %), hyperkalemia (2,5 %), hypertensio (1,4 %) ja syvä laskimotukos (1,2 %).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa ja/tai markkinoille tulon jälkeen havaitut haittavaikutukset on esitetty tässä kohdassa yleisyysluokittain.
Yleisyysluokat määritellään seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 3. Haittavaikutukset
| MedDRA-elinjärjestelmäluokka (SOC) | Yleisyysluokka | Haittavaikutus |
| Infektiot | Yleinen | Sepsis |
| Veri ja imukudos | Yleinen | Trombosytopenia |
| Umpieritys | Tuntematon | Sekundaarinen hypotyreoosi |
| Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Hyperkalemia |
| Psyykkiset häiriöt | Yleinen | Unettomuus |
| Hermosto | Yleinen | Kouristuskohtaukset, päänsärky |
| Melko harvinainen | Aivoinfarkti | |
| Verisuonisto | Hyvin yleinen | Hypertensio, veritien tromboosi (VAT)1 |
| Yleinen | Syvä laskimotukos (SLT) | |
| Hengityselimet, rintakehä ja välikarsina | Melko harvinainen | Keuhkoembolia |
| Ruoansulatuselimistö | Hyvin yleinen | Pahoinvointi, ripuli |
| Yleinen | Ummetus, oksentelu | |
| Maksa ja sappi | Melko harvinainen | Hyperbilirubinemia |
| Iho ja ihonalainen kudos | Tuntematon | Yleistynyt eksfoliatiivinen dermatiitti (DEG) |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Perifeerinen turvotus |
| Tutkimukset | Tuntematon | Veren tyreotropiinin (TSH) määrän vähentyminen, veren kuparipitoisuuden nousu |
| 1Tätä haittavaikutusta esiintyi CKD-potilailla, jotka saivat dialyysihoitoa roksadustaattihoidon aikana. | ||
Valikoitujen haittavaikutusten kuvaus
Tromboottiset verisuonitapahtumat
SLT-tapahtumat olivat melko harvinaisia CKD-potilailla, jotka eivät saaneet dialyysihoitoa. Niitä esiintyi 1,0 %:lla (0,6 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) roksadustaattiryhmän potilaista ja 0,2 %:lla (0,2 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) lumeryhmän potilaista. Dialyysihoidossa olevilla CKD-potilailla SLT-tapahtumia esiintyi 1,3 %:lla (0,8 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) roksadustaattiryhmän potilaista ja 0,3 %:lla (0,1 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) ESA-ryhmän potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
CKD-potilailla, jotka eivät saaneet dialyysihoitoa, keuhkoembolioita todettiin 0,4 %:lla (0,2 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) roksadustaattiryhmän potilaista verrattuna 0,2 %:iin (0,1 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) lumeryhmän potilaista. Dialyysihoidossa olevilla CKD-potilailla keuhkoembolioita todettiin 0,6 %:lla (0,3 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) roksadustaattiryhmän potilaista verrattuna 0,5 %:iin (0,3 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) ESA-ryhmän potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Dialyysihoidossa olevilla CKD-potilailla veritien trombooseja todettiin 12,8 %:lla (7,6 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) roksadustaattiryhmän potilaista verrattuna 10,2 %:iin (5,4 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) ESA-ryhmän potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Dialyysihoidossa olevilla CKD-potilailla iskeemisten keskushermoston verisuonitapahtumien kokonaisilmaantuvuus oli suurempi roksadustaattiryhmässä (3,9 %) kuin lumeryhmässä (2,4 %) ja seurannassa mukautettu ilmaantuvuus oli suurempi roksadustaattiryhmässä (2,3) kuin lumeryhmässä (1,8). Aivoinfarkteja esiintyi 0,2 % enemmän roksadustaattiryhmässä kuin lumeryhmässä (0,6 % vs. 0,4 %).
Dialyysihoidossa olevilla CKD-potilailla iskeemisten keskushermoston verisuonitapahtumien kokonaisilmaantuvuus oli samaa luokkaa roksadustaattihoitoa saaneessa ryhmässä (4,8 %) ja vaikuttavaa vertailuhoitoa saaneessa ryhmässä (4,2 %). Tapahtumien ilmaantuvuus 100 potilasaltistusvuotta kohden oli roksadustaattihoitoa saaneessa ryhmässä 2,8 ja vaikuttavaa vertailuhoitoa saaneessa ryhmässä 2,2. Iskeemisiä aivohalvauksia esiintyi 0,2 % enemmän roksadustaattiryhmässä kuin vaikuttavaa vertailuhoitoa saaneessa ryhmässä (0,8 % vs. 0,6 %).
Kouristuskohtaukset
CKD-potilailla, jotka eivät saaneet dialyysihoitoa, kouristuskohtauksia esiintyi 1,1 %:lla (0,6 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) roksadustaattiryhmän potilaista ja 0,2 %:lla (0,2 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) lumeryhmän potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Dialyysihoidossa olevilla CKD-potilailla kouristuskohtauksia esiintyi 2,0 %:lla (1,2 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) roksadustaattiryhmän potilaista ja 1,6 %:lla (0,8 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) ESA-ryhmän potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sepsis
CKD-potilailla, jotka eivät saaneet dialyysihoitoa, sepsis todettiin 2,1 %:lla (1,3 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) roksadustaattiryhmän potilaista verrattuna 0,4 %:iin (0,3 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) lumeryhmän potilaista. Dialyysihoidossa olevilla potilailla sepsis todettiin 3,4 %:lla (2,0 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) roksadustaattiryhmän potilaista verrattuna 3,4 %:iin (1,8 potilasta, joilla esiintyi tapahtumia 100 potilasvuoden altistusta kohden) ESA-ryhmän potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ihoreaktiot
Markkinoille tulon jälkeisen seurannan aikana on raportoitu yleistynyttä eksfoliatiivista dermatiittia, joka kuuluu vakaviin ihon haittavaikutuksiin (SCAR), ja sillä on osoitettu olevan yhteys roksadustaattihoitoon (esiintymistiheys tuntematon).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Terveillä tutkittavilla roksadustaatin 5 mg/kg (enintään 510 mg) supraterapeuttisten kerta-annosten antoon liittyi sykkeen ohimenevää nousua, lievän tai keskivaikean tuki- ja liikuntaelimistön kivun esiintyvyyden nousua, päänsärkyä, sinustakykardiaa ja harvemmin matalaa verenpainetta, mutta mikään näistä löydöksistä ei ollut vakava. Roksadustaatin yliannostus voi suurentaa Hb-arvon tavoitetasoa (10–12 g/dl) korkeammaksi. Tämä tulee korjata keskeyttämällä roksadustaattihoito tai pienentämällä roksadustaatin annosta (ks. kohta Annostus ja antotapa) sekä seuraamalla potilasta huolellisesti ja hoitamalla kliinisen tarpeen mukaan. Roksadustaatti ja sen metaboliitit eivät poistu hemodialyysissä merkittävässä määrin (ks. kohta Farmakokinetiikka).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: anemialääkkeet, muut anemialääkkeet, ATC-koodi: B03XA05.
Vaikutusmekanismi
Roksadustaatti on hypoksiassa indusoituvan tekijän prolyylihydroksylaasin estäjä (HIF‑PHI). HIF‑PH-entsyymien aktiivisuus kontrolloi solunsisäisiä HIF-pitoisuuksia. HIF on transkriptiotekijä, joka säätelee erytropoieesiin osallistuvien geenien ilmentymistä. HIF-reitin aktivaatiolla on tärkeä rooli hypoksian aiheuttamassa adaptiivisessa vasteessa, joka lisää punasolujen tuotantoa. HIF-PH:n palautuvan eston kautta roksadustaatti stimuloi koordinoitua erytropoieettista vastetta, joka käsittää plasman endogeenisen erytropoietiinin (EPO) pitoisuuksien nousun, raudan kuljetusproteiinien säätelyn ja hepsidiinipitoisuuden (raudan säätelyproteiini, jonka pitoisuus kasvaa CKD:hen liittyvässä tulehduksessa) pienenemisen. Tämä parantaa raudan biologista hyötyosuutta, lisää hemoglobiinin tuotantoa ja suurentaa punasolujen massaa.
Farmakodynaamiset vaikutukset
Vaikutukset QTc-aikaan ja syketiheyteen
Perusteellisessa QT-tutkimuksessa (TQT) ei todettu QTc-välin pidentymistä, kun terveille tutkittaville annettiin terapeuttinen 2,75 mg/kg kerta-annos ja supraterapeuttinen 5 mg/kg (enintään 510 mg) kerta-annos roksadustaattia. Samassa perusteellisessa QT-tutkimuksessa todettiin lumelääkkeen suhteen korjattua syketiheyden nousua, joka oli enintään 9–10 lyöntiä minuutissa 8–12 tuntia 2,75 mg/kg annoksen jälkeen ja 15–18 lyöntiä minuutissa 6–12 tuntia 5 mg/kg annoksen jälkeen.
Kliininen teho ja turvallisuus
Kehitysohjelma CKD-potilaiden anemian hoidossa
Roksadustaatin tehoa ja turvallisuutta arvioitiin vähintään 52 viikon ajan maailmanlaajuisessa vaiheen 3 tutkimusohjelmassa, joka kattoi 8 satunnaistettua ja monikeskustutkimusta dialyysistä riippumattomilla (NDD) ja dialyysistä riippuvaisilla (DD) aneemisilla CKD-potilailla (ks. taulukko 4).
Vaiheen 3–5 kroonista munuaissairautta sairastavilla NDD-potilailla tehdyistä tutkimuksista kolme oli kaksoissokkoutettua ja lumekontrolloitua tutkimusta (ALPS, 1517‑CL‑0608; ANDES, FGCL‑4592‑060; OLYMPUS, D5740C00001) ja yksi tutkimuksista oli avoin, ESA-kontrolloitu tutkimus (DOLOMITES, 1517‑CL‑0610), jossa vertailuvalmisteena käytettiin darbepoetiini alfaa. Kaikissa NDD-tutkimuksissa tehoa ja turvallisuutta arvioitiin potilailla jotka eivät olleet aiemmin saaneet ESA-hoitoja, korjaamalla Hb-arvo ja pitämällä se sitten tavoitealueella 10–12 g/dl (Hb-arvon korjausasetelma).
Neljässä avoimessa, ESA-kontrolloidussa DD-tutkimuksessa (vertailuvalmiste: epoetiini alfa ja/tai darbepoetiini alfa), joissa potilaat saivat hemodialyysi- tai peritoneaalidialyysihoitoa, tehoa ja turvallisuutta arvioitiin erilaisissa asetelmissa:
- Hb-arvon korjausasetelmassa (HIMALAYAS, FGCL‑4592‑063).
- ESA-hoidon vaihtamisasetelmassa, jossa potilaat siirrettiin ESA-hoidosta, jotta Hb-arvo pysyisi tavoitealueella (PYRENEES, 1517‑CL‑0613; SIERRAS, FGCL‑4592‑064).
- tai asetelmassa, jossa yhdistyivät Hb-arvon korjaus ja ESA-hoidosta siirtyminen (ROCKIES, D5740C00002).
NDD-tutkimuksiin osallistuneilla potilailla oli asteen 3–5 CKD, eivätkä he saaneet dialyysihoitoa. Keskimääräinen Hb-arvo oli ≤ 10,0 g/dl kaikilla muilla paitsi DOLOMITES-tutkimukseen (1517‑CL‑0610) osallistuneilla potilailla, sillä tässä tutkimuksessa keskimääräinen Hb-arvo sai olla ≤ 10,5 g/dl. Ferritiiniarvojen oli oltava tasolla ≥ 30 ng/ml (ALPS, 1517‑CL‑0608; ANDES, FGCL‑4592‑060), ≥ 50 ng/ml (OLYMPUS, D5740C00001) tai ≥ 100 ng/ml (DOLOMITES, 1517‑CL‑0610). Lukuun ottamatta tutkimusta (OLYMPUS, D5740C00001), jossa ESA-hoitoja ei sallittu satunnaistamista edeltävän 6 viikon jakson aikana, eivätkä potilaat olleet saaneet käyttää mitään ESA-hoitoja 12 viikon sisällä satunnaistamisesta.
DD-tutkimuksiin osallistuneiden potilaiden oli oltava dialyysihoidossa: PYRENEES-tutkimuksessa (1517‑CL‑0613) vaatimuksena oli stabiili DD, joka määriteltiin yli 4 kuukautta kestäneeksi dialyysiksi, ja HIMALAYAS-tutkimuksessa (FGCL‑4592‑063) äskettäin aloitettu (ID) DD, joka määriteltiin ≥ 2 viikkoa mutta ≤ 4 kuukautta kestäneeksi dialyysiksi. SIERRAS-tutkimukseen (FGCL‑4592‑064) ja ROCKIES-tutkimukseen (D5740C00002) osallistui sekä stabiilia (noin 80–90 %) että äskettäin aloitettua (noin 10–20 %) dialyysihoitoa saavia, dialyysistä riippuvaisia potilaita. Ferritiiniarvon oli oltava kaikilla potilailla ≥ 100 ng/ml. Kaikki potilaat olivat tarvinneet ESA-hoitoa laskimoon tai ihon alle vähintään 8 viikon ajan ennen satunnaistamista, paitsi HIMALAYAS-tutkimuksessa (FGCL‑4592‑063), johon ei otettu potilaita, jotka olivat saaneet mitä tahansa ESA-hoitoa satunnaistamista edeltävien 12 viikon aikana.
Roksadustaattihoitoa annettiin kohdassa Annostus ja antotapa kuvattujen annostusohjeiden periaatteiden mukaisesti.
Potilaiden demografiset tiedot ja kaikki lähtötilanteen ominaisuudet olivat vertailukelpoisia eri tutkimusten roksadustaatti- javerrokkiryhmien välillä. Mediaani-ikä satunnaistamisvaiheessa oli 55–69 vuotta, ja potilaista 16,6–31,1 % kuului 65–74-vuotiaiden ikäryhmään ja 6,8–35 % vähintään 75-vuotiaiden ikäryhmään. Naispotilaiden prosenttiosuus oli 40,5–60,7 %. Potilaat olivat rodultaan useimmiten valkoihoisia, tummaihoisia tai afroamerikkalaisia ja aasialaisia. Yleisimpiä kroonisen munuaissairauden etiologioita olivat diabeettinen nefropatia ja hypertensiivinen nefropatia. Hb-arvojen mediaani oli 8,60–10,78 g/dl. Lähtötilanteen rauta-arvot olivat kunnossa noin 50–60 %:lla NDD-potilaista ja 80–90 %:lla DD-potilaista.
Seitsemästä vaiheen 3 tutkimuksesta saadut tiedot yhdistettiin kahteen erilliseen ryhmään (kolme NDD- ja neljä DD-tutkimusta) (ks. taulukko 4).
NDD-pooliin sisältyi kolme lumekontrolloitua NDD-tutkimusta (2 386 potilasta, jotka saivat roksadustaattia, ja 1 884 potilasta, jotka saivat lumelääkettä). NDD-poolin analyyseihin ei sisällytetty vaiheen 3 ESA-kontrolloidusta, NDD-potilailla tehdystä DOLOMITES-tutkimuksesta (1517‑CL‑0610; 323 potilasta, jotka saivat roksadustaattia, ja 293 potilasta, jotka saivat darbepoetiini alfaa) saatuja tietoja, koska kyseinen tutkimus oli ainoa NDD-potilailla tehty avoin, aktiivikontrolloitu tutkimus.
DD-pooliin sisältyi neljä ESA-kontrolloitua DD-tutkimusta (2 354 potilasta, jotka saivat roksadustaattia, ja 2 360 potilasta, jotka saivat ESA-hoitoa [epoetiini alfaa ja/tai darbepoetiini alfaa]). DD-pooli jaettiin vielä kahden erilaisen hoitoasetelman mukaisesti kahteen osajoukkoon:
- DD-potilaat, jotka olivat saaneet dialyysihoitoa yli 2 viikon ja alle 4 kuukauden ajan, määriteltiin dialyysin äskettäin aloittaneiksi (ID), dialyysistä riippuvaisiksi potilaiksi (ID DD -pooli) Hb-arvon korjausasetelmassa.
- DD-potilaat, jotka olivat dialyysihoidossa vielä tämän neljän kuukauden kynnyksen jälkeen, määriteltiin stabiilissa dialyysihoidossa oleviksi, dialyysistä riippuvaisiksi potilaiksi (stabiili DD -pooli) ESA-hoidon vaihtamisasetelmassa.
Taulukko 4. Yleiskatsaus roksadustaatin vaiheen 3 kehitysohjelmaan kroonista munuaissairautta sairastavilla aneemisilla potilailla
| NDD-potilailla tehdyt tutkimukset | |||||
| Lumekontrolloidut tutkimukset (NDD-pooli) | ESA‑kontrolli (darbepoetiini alfa) | ||||
| Asetelma | Hb-arvon korjaus | ||||
| Tutkimus | ALPS (1517‑CL‑0608) | ANDES (FGCL‑4592‑060) | OLYMPUS (D5740C00001) | DOLOMITES (1517‑CL‑0610) | |
Satunnaistettu (roksadustaatti/vertailuvalmiste) | 594 (391/203) | 916 (611/305) | 2760 (1384/1376) | 616 (323/293) | |
| DD-potilailla tehdyt tutkimukset | |||||
| ESA‑kontrolloidut tutkimukset (DD-pooli) (epoetiini alfa tai darbepoetiini alfa) | |||||
| Asetelma | ESA-hoidon vaihtaminen | Hb-arvon korjaus | ESA-hoidon vaihtaminen ja Hb-arvon korjaus | ||
| Tutkimus | PYRENEES (1517‑CL‑0613) | SIERRAS (FGCL‑4592‑064) | HIMALAYAS (FGCL‑4592‑063) | ROCKIES (D5740C00002) | |
Satunnaistettu (roksadustaatti/vertailuvalmiste) | 834 (414/420) | 740 (370/370) | 1039 (522/517) | 2101 (1048/1053) | |
| DD: dialyysistä riippuvainen; ESA: erytropoieesia stimuloiva aine; Hb: hemoglobiini; NDD: dialyysistä riippumaton. | |||||
NDD CKD -potilaat
Tulokset tehosta
Hb-arvot hoidon aikana
Kliinisissä tutkimuksissa roksadustaatti oli tehokas Hb-tavoitearvon (10–12 g/dl) saavuttamisessa ja ylläpidossa aneemisilla CKD-potilailla, jotka eivät saaneet dialyysihoitoa (ks. kuva 1).
Kuva 1. Keskimääräinen (SE) Hb-arvo (g/dl) ajan mittaan viikkoon 52 asti (FAS); NDD-pooli (Hb-arvon korjaus)
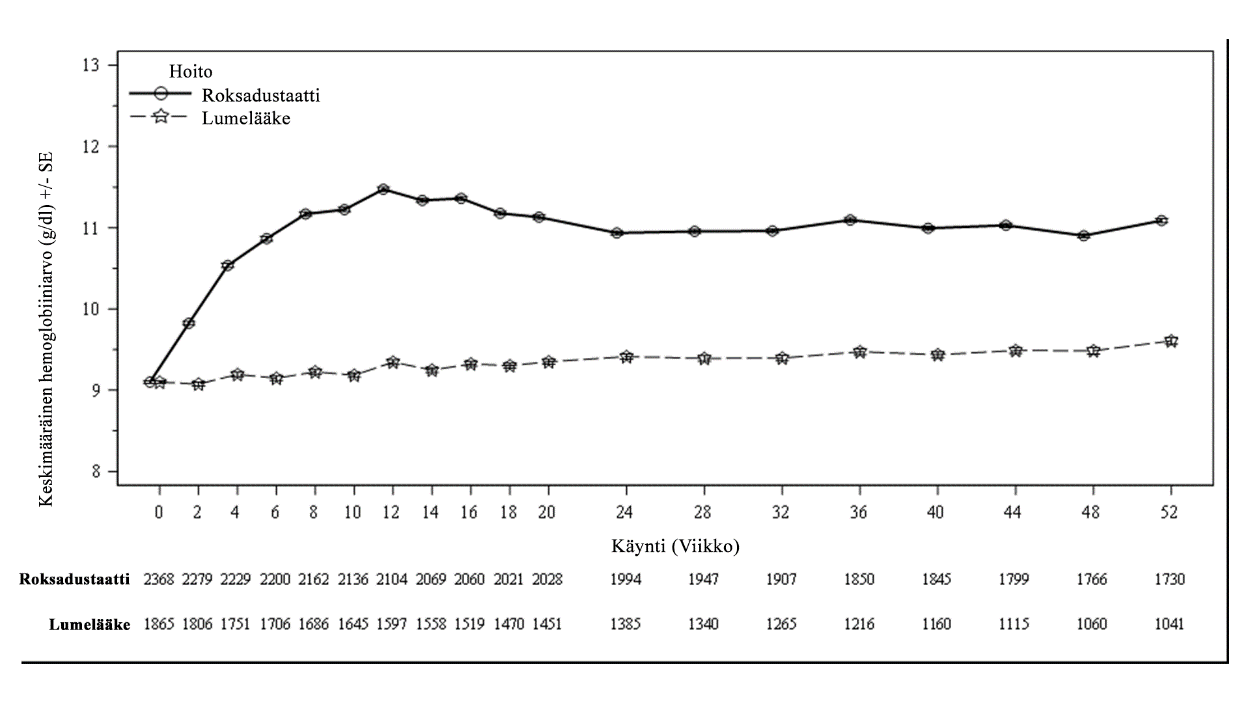
FAS: koko analysoitava joukko; Hb: hemoglobiini; NDD: dialyysistä riippumaton; SE: keskivirhe.
Hb-arvon tärkeimmät tehon päätetapahtumat NDD CKD -potilailla
NDD-potilailla, jotka tarvitsivat anemiahoitoa Hb-arvon korjaamiseen, ensimmäisten 24 viikon aikana Hb-vasteen saaneiden potilaiden osuus oli suurempi roksadustaattia saaneessa ryhmässä (80,2 %) kuin lumeryhmässä (8,7 %). Hb-arvo suureni tilastollisesti merkitsevästi lähtöarvosta viikoille 28 ja 36 roksadustaattiryhmän potilailla (1,91 g/dl) verrattuna lumeryhmään (0,14 g/dl), ja 95 %:n luottamusvälin alaraja oli yli 1. NDD-tutkimuksissa Hb-arvo suureni vähintään 1 g/dl mediaaniajassa 4,1 viikkoa (ks. taulukko 5).
Avoimessa, ESA-kontrolloidussa, dialyysistä riippumattomilla potilailla tehdyssä DOLOMITES (1517-CL-0610) -tutkimuksessa ensimmäisten 24 viikon aikana Hb-vasteen saavuttaneiden potilaiden osuus oli samanarvoinen roksadustaattia saaneessa ryhmässä (89,5 %) kuin darbepoetiini alfaa saaneessa ryhmässä (78 %) (ks. taulukko 5).
Taulukko 5. Hb-arvon tärkeimmät tehon päätetapahtumat (NDD)
| Populaatio | NDD CKD -potilaat | |||
| Asetelma | Hb-arvon korjaus | Hb-arvon korjaus | ||
| Päätetapahtuma/ parametri | NDD-pooli (FAS) | DOLOMITES (PPS) 1517‑CL‑0610 | ||
Roksadustaatti n = 2 368 | Lumelääke n = 1 865 | Roksadustaatti n = 286 | Darbepoetiini alfa n = 273 | |
| Hb-vasteen1 saavuttaneiden potilaiden osuus | ||||
Hoitovasteen saavuttaneet, n (%) [95 %:n CI] | 1 899 (80,2) [78,5; 81,8] | 163 (8,7) [7,5; 10,1] | 256 (89,5) [85,4; 92,8] | 213 (78,0) [72,6; 82,8] |
| Osuuksien ero [95 %:n CI] | 71,5 [69,40; 73,51] | 11,51 [5,66; 17,36] | ||
| Ristitulosuhde [95 %:n CI] | 40,49 [33,01; 49,67] | 2,48 [1,53; 4,04] | ||
| P-arvo | < 0,0001 | ND | ||
| Hb-arvon muutos lähtötilanteesta (g/dl)2 | ||||
| Keskiarvo (SD) lähtötilanteessa | 9,10 (0,74) | 9,10 (0,73) | 9,55 (0,76) | 9,54 (0,69) |
| CFB-keskiarvo (SD) | 1,85 (1,07) | 0,17 (1,08) | 1,85 (1,08) | 1,84 (0,97) |
| LS-keskiarvo | 1,91 | 0,14 | 1,85 | 1,84 |
| LS-keskiarvojen ero [95 %:n CI] | 1,77 [1,69; 1,84] | 0,02 [‑0,13; 0,16] | ||
| P-arvo | < 0,0001 | 0,844 | ||
| CFB: muutos lähtötilanteesta; CI: luottamusväli; CKD: krooninen munuaissairaus; FAS: koko analysoitava joukko; Hb: hemoglobiini; LS: pienin neliösumma; ND: ei tehty; NDD: dialyysistä riippumaton; PPS: tutkimussuunnitelman mukainen joukko; SD: keskihajonta. 1Hb-vaste ensimmäisten 24 viikon aikana 2Hb-arvon muutos lähtötilanteesta viikoilla 28–36 | ||||
DD CKD -potilaat
Hb-arvot hoidon aikana
Kliinisissä tutkimuksissa roksadustaatti oli tehokas Hb-tavoitearvon (10–12 g/dl) saavuttamisessa ja ylläpidossa dialyysihoitoa saavilla CKD-potilailla aiemmasta ESA-hoidosta riippumatta (ks. kuvat 2 ja 3).
Kuva 2. Keskimääräinen (SE) Hb-arvo viikkoon 52 asti (FAS); ID DD -osajoukko (Hb-arvon korjaus)
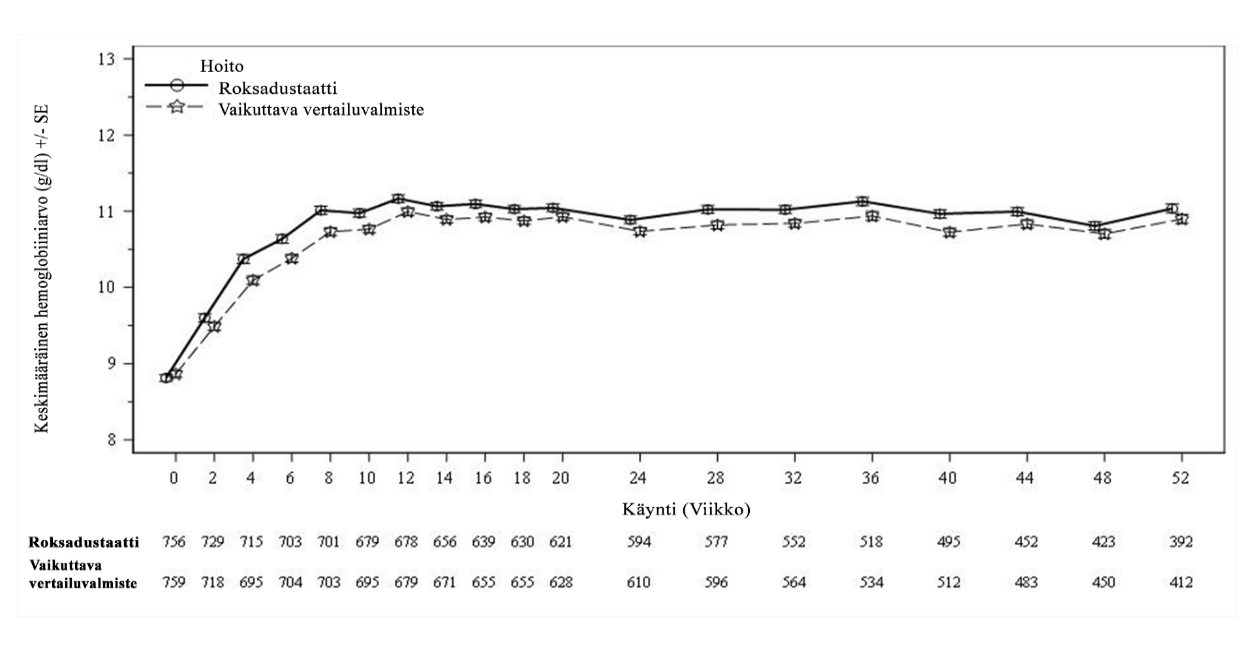
DD: dialyysistä riippuvainen; FAS: koko analysoitava joukko; Hb: hemoglobiini; ID: dialyysin äskettäin aloittanut; SE: keskivirhe.
Kuva 3. Keskimääräinen (SE) Hb-arvo (g/dl) ajan mittaan viikkoon 52 asti (FAS); stabiili DD-osajoukko (ESA-hoidon vaihtaminen)
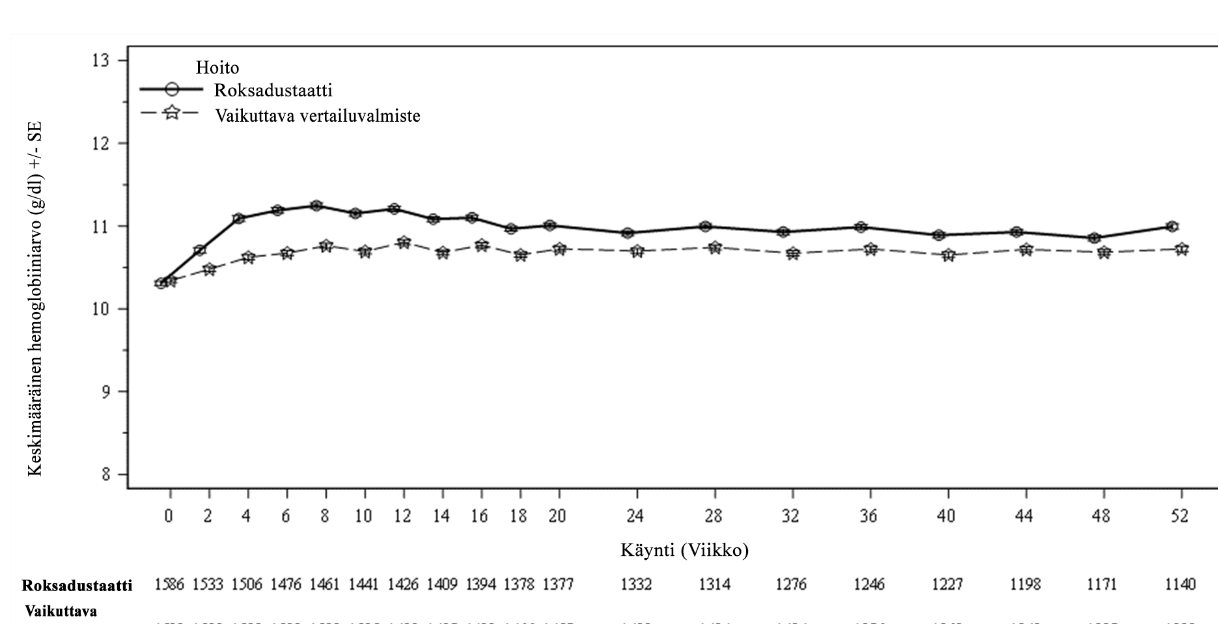
DD: dialyysistä riippuvainen; ESA: erytropoieesia stimuloiva aine; FAS: koko analysoitava joukko; Hb: hemoglobiini; SE: keskivirhe.
Hb-arvon tärkeimmät tehon päätetapahtumat DD CKD -potilailla
DD-potilailla, jotka tarvitsivat anemiahoitoa Hb-arvon korjaamiseen ja ESA-hoidosta siirtyneillä potilailla, Hb-arvo suureni roksadustaattiryhmässä lähtötilanteesta viikoille 28–36. Tämä nousu oli verrattavissa ESA-ryhmässä todettuun Hb-arvon nousuun ja se oli ennalta määrättyä -0,75 g/dl samanarvoisuusmarginaalia suurempi. Ensimmäisten 24 viikon aikana Hb-vasteen saavuttaneiden potilaiden osuus oli samaa luokkaa roksadustaatti- ja ESA-ryhmissä (ks. taulukko 6).
Taulukko 6. Hb-arvon tärkeimmät tehon päätetapahtumat (DD)
| Populaatio | DD-potilaat | |||
| Asetelma | Hb-arvon korjaus | ESA-hoidon vaihtaminen | ||
Päätetapahtuma/ parametri | ID DD -pooli (FAS/PPS) | Stabiili DD -pooli (PPS) | ||
Roksadustaatti n = 756 | ESA n = 759 | Roksadustaatti n = 1 379 | ESA n = 1 417 | |
| Hb-arvon muutos lähtötilanteesta (g/dl) | ||||
| Keskiarvo (SD) lähtötilanteessa | 8,77 (1,20) | 8,82 (1,20) | 10,32 (0,99) | 10,37 (0,99) |
| CFB-keskiarvo (SD) | 2,37 (1,57) | 2,12 (1,46) | 0,65 (1,15) | 0,36 (1,23) |
| LS-keskiarvo | 2,17 | 1,89 | 0,58 | 0,28 |
| LS-keskiarvojen ero [95 %:n CI] | 0,28 [0,110; 0,451] | 0,30 [0,228; 0,373] | ||
| P-arvo | 0,0013 | < 0,0001 | ||
| Hb-vasteen saavuttaneiden potilaiden osuus 1, 2 | ||||
Hoitovasteen saavuttaneet, n (%) [95 %:n CI] | 453 (59,9) [56,3; 63,4] | 452 (59,6) [56,0; 63,1] | 978 (70,9) [68,4; 73,3] | 959 (67,7) [65,2; 70,1] |
| Osuuksien ero [95 %:n CI] | 0,3 [‑4,5; 5,1] | 2,7 [‑0,7; 6,0] | ||
| kertoimiensuhde [95 %:n CI] | ND | ND | ||
| P-arvo | ND | ND | ||
| CFB: muutos lähtötilanteesta; CI: luottamusväli; CKD: krooninen munuaissairaus; DD: dialyysistä riippuvainen; ESA: erytropoieesia stimuloiva aine; FAS: koko analysoitava joukko; Hb: hemoglobiini; ID: dialyysin äskettäin aloittanut; LS: pienin neliösumma; ND: ei tehty; PPS: tutkimussuunnitelman mukainen joukko; SD: keskihajonta. 1Hb-arvo tavoitealueella 10,0–12,0 g/dl viikoilla 28–36 ilman varahoitoa 6 viikon sisällä ennen tätä 8 viikon pituista arviointijaksoa ja sen aikana. 2ID DD -poolin tiedot analysoitiin vain viikoilta 28–52. | ||||
Varahoito, punasolusiirto ja laskimoon annettava rauta
Roksadustaattihoidon vaikutukset varahoidon, punasolusiirron ja laskimoon annettavan raudan tarpeeseen on esitetty taulukossa 7 (NDD) ja taulukossa 8 (DD). Kliinisissä tutkimuksissa roksadustaatti pienensi hepsidiinin (raudan metabolian säätelijä) määrää, pienensi ferritiinin määrää ja suurensi seerumin rautapitoisuutta transferriinisaturaation ollessa vakaa. Kaikkia näitä parametreja arvioitiin rauta-arvojen indikaattoreina ajan mittaan.
Matalatiheyksinen lipoproteiini (LDL) -kolesteroli
Roksadustaattihoidon vaikutukset LDL-kolesteroliin on esitetty taulukoissa 7 ja 8. Keskimääräiset LDL- ja korkeatiheyksinen lipoproteiini (HDL) -kolesteroliarvot laskivat roksadustaattia saaneilla potilailla verrattuna lumelääkettä tai ESA-hoitoa saaneisiin potilaisiin. Vaikutus LDL-kolesteroliarvoon oli voimakkaampi johtaen LDL/HDL-suhteen pienenemiseen ja oli todettavissa statiinien käytöstä riippumatta.
Taulukko 7. Muut tehon päätetapahtumat: varahoidon käyttö, raudan antaminen laskimoon kuukausittain ja LDL-kolesteroliarvon muutos lähtötilanteesta (NDD)
| Populaatio | NDD CKD -potilaat | ||||
| Interventio | Korjaus | Korjaus | |||
| Päätetapahtuma/parametri | NDD-pooli (FAS) | DOLOMITES (1517‑CL‑0610) | |||
Roksadustaatti n = 2 368 | Lumelääke n = 1 865 | Roksadustaatti n = 322 | Darbepoetiini alfa n = 292 | ||
| Varahoitoa saaneiden potilaiden määrä, n (%)1 | 211 (8,9) | 580 (31,1) | ND | ||
| RBC | 118 (5,0) | 240 (12,9) | |||
| i.v.-rauta | 50 (2,1) | 90 (4,8) | |||
| ESA | 48 (2,0) | 257 (13,8) | |||
| IR | 10,4 | 41,0 | |||
| Riskitiheyksien suhde | 0,19 | ND | |||
| 95 %:n CI | 0,16; 0,23 | ||||
| P-arvo | < 0,0001 | ||||
| i.v.-rautaa saaneiden potilaiden määrä, n (%)2 | ND | 20 (6,2) | 37 (12,7) | ||
| IR | 9,9 | 21,2 | |||
| Riskitiheyksien suhde | 0,45 | ||||
| 95 %:n CI | 0,26; 0,78 | ||||
| P-arvo | 0,004 | ||||
| LDL-kolesteroliarvon muutos lähtötilanteesta (mmol/l) viikoille 12–283 | |||||
| ANCOVA-analyysi | |||||
| LS-keskiarvo | ‑0,446 | 0,066 | ‑0,356 | 0,047 | |
| 95 %:n CI | ‑0,484; ‑0,409 | 0,017; 0,116 | ‑0,432; ‑0,280 | ‑0,033; 0,127 | |
| LS-keskiarvojen ero | ‑0,513 | ‑0,403 | |||
| 95 %:n CI | ‑0,573; ‑0,453 | ‑0,510; ‑0,296 | |||
| P-arvo | < 0,0001 | < 0,001 | |||
NDD-poolille annetut p-arvot ovat nimellisiä p-arvoja. 3 OLYMPUS-tutkimuksessa (D5740C00001) LDL-kolesteroliarvon muutos lähtötilanteesta arvioitiin vain viikolle 24 asti. | |||||
Taulukko 8. Muut tehon päätetapahtumat: varahoidon käyttö, raudan antaminen laskimoon kuukausittain ja LDL-kolesteroliarvon muutos lähtötilanteesta (DD)
| Populaatio | DD CKD -potilaat | |||
| Interventio | Korjaus | Hoidon vaihtaminen | ||
Päätetapahtuma/ parametri | ID DD -pooli (FAS) | Stabiili DD -pooli (FAS) | ||
Roksadustaatti n = 756 | ESA n = 759 | Roksadustaatti n = 1 586 | ESA n = 1 589 | |
| i.v.-raudan keskimääräinen kuukausittainen annos (mg) viikkojen 28–52 aikana1 | ||||
| n | 606 | 621 | 1414 | 1486 |
| Keskiarvo (SD) | 53,57 (143,097) | 70,22 (173,33) | 42,45 (229,80) | 61,99 (148,02) |
| LDL-kolesteroliarvon muutos lähtötilanteesta (mmol/l) viikoille 12–28 | ||||
| ANCOVA-analyysi | ||||
| LS-keskiarvo | ‑0,610 | ‑0,157 | ‑0,408 | ‑0,035 |
| 95 %:n CI | ‑0,700; ‑0,520 | ‑0,245; ‑0,069 | ‑0,449; ‑0,368 | ‑0,074; 0,003 |
LS-keskiarvojen ero (R-vertailuvalmiste) | ‑0,453 | ‑0,373 | ||
| 95 %:n CI | ‑0,575; ‑0,331 | ‑0,418; ‑0,328 | ||
| P-arvo | < 0,0001 | < 0,0001 | ||
| ID DD ja stabiilille DD -poolille annetut p-arvot ovat nimellisiä p-arvoja. ANCOVA: kovarianssianalyysi; CI: luottamusväli; CKD: krooninen munuaistauti; DD: dialyysistä riippuvainen; ESA: erytropoieesia stimuloiva aine; FAS: koko analysoitava joukko; ID: dialyysin äskettäin aloittanut; i.v.: laskimonsisäinen; LDL: matalatiheyksinen lipoproteiini; LS: pienin neliösumma; R: roksadustaatti. 1PYRENEES-tutkimuksen (1517‑CL‑0613) ajanjakso oli viikolle 36 asti ja ROCKIES- tutkimuksen (D5740C0002) ajanjakso oli viikosta 36 tutkimuksen päättymiseen asti. | ||||
Dialyysitutkimuksessa SIERRAS (FGCL-4592-064) merkitsevästi pienempi osuus roksadustaattiryhmän potilaista sai hoidon aikana punasolusiirron verrattuna epoetiini alfaa saaneeseen ryhmään (12,5 % vs. 21,1 %). ROCKIES-tutkimuksessa (D5740C00002) numeerinen vähenemä ei ollut tilastollisesti merkitsevä (9,8 % vs. 13,2 %).
Potilaiden raportoimat tulokset (ei dialyysiä)
DOLOMITES-tutkimuksessa (1517‑CL‑0610) roksadustaatin samanarvoisuus darbepoetiiniin nähden vahvistettiin SF‑36 PF- ja SF‑36 VT -arviointien osalta.
Potilaiden raportoimat tulokset (dialyysi)
PYRENEES-tutkimuksessa (1517‑CL‑0613) roksadustaatin samanarvoisuus ESA-hoitoihin nähden vahvistettiin SF‑36 PF- ja SF‑36 VT ‑muutoksina lähtötilanteesta viikoille 12–28.
Kliininen turvallisuus
Yhdistettyjen, varmistettujen sydän- ja verisuonitapahtumien meta-analyysi
Vaiheen 3 tutkimusohjelmassa arvioitujen, merkittävien kardiovaskulaaristen haittatapahtumien (MACE; mistä tahansa syystä johtuvan kuolleisuuden [ACM], sydäninfarkti, aivohalvaus), sekä MACE+-tapahtumien (mistä tahansa syystä johtuvan kuolleisuus, sydäninfarktin, aivohalvauksen ja joko epästabiilista rasitusrintakivusta tai kongestiivisesta sydämen vajaatoiminnasta johtuva sairaalahoito) meta-analyysi suoritettiin 8 984 potilaalla.
MACE-, MACE+- ja ACM-tulokset esitetään kolmen tietojoukon osalta yhdistettyä riskitiheyksien suhdetta (HR) ja sen 95 %:n luottamusväliä (CI) käyttäen. Tietojoukot ovat:
- NDD-potilaiden yhdistetty, lumekontrolloitu Hb-korjauksen tietojoukko (koostuu tutkimuksiin OLYMPUS [D5740C00001], ANDES [FGCL-4592-060] ja ALPS [1517-CL-0608] osallistuneista potilaista; ks. taulukko 4)
- NDD- ja ID-DD-potilaiden yhdistetty, ESA-kontrolloitu Hb-korjauksen tietojoukko (koostuu tutkimuksiin DOLOMITES [1517-CL-0610] ja HIMALAYAS [FGCL-4592-063] osallistuneista potilaista sekä tutkimuksiin SIERRAS [FGCL-4592-064] ja ROCKIES [D5740C00002] osallistuneista ID-DD-potilaista; ks. taulukko 4)
- ESA-hoitoon siirtyneiden, voinniltaan vakaiden DD-potilaiden yhdistetty, ESA-kontrolloitu tietojoukko (koostuu tutkimukseen PYRENEES [1517-CL-0613] osallistuneista potilaista sekä tutkimuksiin ROCKIES [D5740C00002] ja SIERRAS [FGCL-4592-064] osallistuneista DD-potilaista, joiden vointi oli vakaa; ks. taulukko 4).
MACE, MACE+ ja ACM dialyysistä riippumattomien CKD-potilaiden lumekontrolloidussa Hb-korjauksen joukossa
NDD-potilailla tehtyihin hoidon aikaisiin MACE-, MACE+- ja ACM-analyyseihin sisältyivät kaikki tiedot tutkimushoidon aloittamisesta siihen asti, kun hoidon jälkeisen seurannan päättymisestä oli kulunut 28 vuorokautta. Hoidon aikaisissa analyyseissa käytettiin Coxin mallia, joka oli painotettu käänteisesti sensurointitodennäköisyyden mukaan (IPCW-menetelmä) ja jolla pyrittiin korjaamaan roksadustaattia ja lumelääkettä saaneiden ryhmien seuranta-ajan erot, mukaan lukien riskin suurenemiseen ja hoidon ennenaikaiseen keskeyttämiseen myötävaikuttavat tunnistetut tekijät sekä etenkin arvioitua glomerulusten suodatusnopeutta (eGFR) ja Hb-arvoa määrittävät tekijät lähtötilanteessa ja ajan kuluessa. Ei ole varmuutta siitä, liittyykö tämän mallin käyttöön vielä muita sekoittavia tekijöitä. Hoidon aikaisissa analyyseissa riskitiheyksien suhteet olivat 1,26, 1,17 ja 1,16 (ks. taulukko 9). ITT-analyyseihin sisältyivät kaikki tiedot tutkimushoidon aloittamisesta hoidon lopettamista seuraavan turvallisuusseurannan päättymiseen asti. ITT-analyysi on otettu mukaan sen havainnollistamiseksi, että riski jakautuu epätasaisesti ja suosii lumelääkettä hoidon aikaisessa analyysissa. ITT-analyysit kuitenkin yleensä osoittavat tutkimuslääkehoidon vaikutuksen heikentymistä, eikä virheellisen tuloksen mahdollisuutta voida täysin poissulkea näiden ITT-analyysien kohdalla etenkään siksi, koska ESA-varahoito otettiin käyttöön tutkimushoidon lopettamisen jälkeen. Riskitiheyksien suhteet olivat 1,10, 1,07 ja 1,08 ja 95 %:n luottamusvälien ylärajat olivat vastaavasti 1,27, 1,21 ja 1,26.
Taulukko 9. Kardiovaskulaarinen turvallisuus ja kuolleisuus lumekontrolloidussa Hb-korjauksen NDD-poolissa
| MACE | MACE+ | ACM | ||||
Roksadustaatti n = 2 386 | Lumelääke n = 1 884 | Roksadustaatti n = 2 386 | Lumelääke n = 1 884 | Roksadustaatti n = 2 386 | Lumelääke n = 1 884 | |
| Hoidon aikana | ||||||
| Potilaat, joilla esiintyi tapahtumia (%) | 344 (14,4) | 166 (8,8) | 448 (18,8) | 242 (12,8) | 260 (10,9) | 122 (6,5) |
| FAIR | 8,7 | 6,8 | 11,6 | 10,1 | 6,4 | 5,0 |
| HR (95 %:n CI) | 1,26 (1,02; 1,55) | 1,17 (0,99; 1,40) | 1,16 (0,90; 1,50) | |||
| ITT | ||||||
| Potilaat, joilla esiintyi tapahtumia (%) | 480 (20,1) | 350 (18,6) | 578 (24,2) | 432 (22,9) | 400 (16,8) | 301 (16) |
| FAIR | 10,6 | 10,3 | 13,2 | 13,2 | 8,3 | 8,1 |
| HR (95 %:n CI) | 1,10 (0,96; 1,27) | 1,07 (0,94; 1,21) | 1,08 (0,93; 1,26) | |||
| ACM: mistä tahansa syystä johtuva kuolleisuus; ACM on MACE:n/MACE+:n komponentti. CI: luottamusväli; FAIR (follow-up adjusted incidence rate): seurannassa mukautettu ilmaantuvuus (sellaisten potilaiden määrä, joilla esiintyi tapahtuma/100 potilasvuotta); HR: riskitiheyksien suhde; ITT: hoitoaikeen mukainen; MACE: merkittävä kardiovaskulaarinen haittatapahtuma (kuolema, ei kuolemaan johtava sydäninfarkti ja/tai aivohalvaus); MACE+: merkittävä kardiovaskulaarinen haittatapahtuma, mukaan lukien epästabiilista rasitusrintakivusta ja/tai kongestiivisesta sydämen vajaatoiminnasta johtuva sairaalahoito. | ||||||
MACE, MACE+ ja ACM dialyysistä riippumattomien ja äskettäin aloitetusta dialyysistä riippuvaisten CKD-potilaiden ESA-kontrolloidussa Hb-korjauksen joukossa
Hb-korjauksen asetelmassa NDD- ja ID-DD-potilaiden lähtötilanteen ominaisuudet ja hoidon keskeytysprosentit olivat samankaltaiset roksadustaattia saaneiden ja ESA-hoitoa saaneiden potilaiden yhdistetyissä ryhmissä. Hoidon aikana todettujen MACE-, MACE+- ja ACM-tapahtumien analyysien perusteella riskitiheyksien suhteet olivat 0,79, 0,78 ja 0,78 ja riskitiheyksien suhteiden 95 %:n luottamusvälien ylärajat olivat vastaavasti 1,02, 0,98 ja 1,05 (ks. taulukko 10). Hoidon aikana tehdyistä analyyseista ei saatu näyttöä siitä, että roksadustaattiin liittyisi suurempi kardiovaskulaarinen turvallisuusriski tai kuolleisuusriski kuin ESA-hoitoihin Hb-arvon korjausta tarvitsevilla CKD-potilailla.
Taulukko 10. Kardiovaskulaarinen turvallisuus ja kuolleisuus ESA-kontrolloidussa Hb-korjauksen poolissa
| MACE | MACE+ | ACM | ||||
| Roksadustaatti n = 1 083 | ESA n = 1 059 | Roksadustaatti n = 1 083 | ESA n = 1 059 | Roksadustaatti n = 1 083 | ESA n = 1 059 | |
| Hoidon aikana | ||||||
| Potilaat, joilla esiintyi tapahtumia (%) | 105 (9,7) | 136 (12,8) | 134 (12,4) | 171 (16,1) | 74 (6,8) | 99 (9,3) |
| IR | 6,5 | 8,2 | 8,3 | 10,3 | 4,6 | 6,0 |
| HR (95 %:n CI) | 0,79 (0,61; 1,02) | 0,78 (0,62; 0,98) | 0,78 (0,57; 1,05) | |||
| ACM: mistä tahansa syystä johtuva kuolleisuus: ACM on MACE:n/MACE+:n komponentti. CI: luottamusväli; ESA: erytropoieesia stimuloiva aine; HR: riskitiheyksien suhde; IR: ilmaantuvuus (sellaisten potilaiden määrä, joilla esiintyi tapahtuma/100 potilasvuotta); MACE: merkittävä kardiovaskulaarinen haittatapahtuma (kuolema, ei kuolemaan johtava sydäninfarkti ja/tai aivohalvaus); MACE+: merkittävä kardiovaskulaarinen haittatapahtuma, mukaan lukien epästabiilista rasitusrintakivusta ja/tai kongestiivisesta sydämen vajaatoiminnasta johtuva sairaalahoito. | ||||||
MACE, MACE+ ja ACM ESA-kontrolloidussa, ESA-hoidosta siirtyneessä, voinniltaan vakaassa, dialyysistä riippuvaisessa CKD-potilaiden joukossa
ESA-hoidosta siirtyneillä, voinniltaan vakailla DD-potilailla hoidon aikana todettujen MACE-, MACE+- ja ACM-tapahtumien analyysien perusteella riskitiheyksien suhteet olivat 1,18, 1,03 ja 1,23 ja riskitiheyksien suhteiden 95 %:n luottamusvälien ylärajat olivat vastaavasti 1,38, 1,19 ja 1,49 (ks. taulukko 11). Taulukon 11 tulosten tulkinnassa on syytä noudattaa varovaisuutta, sillä roksadustaattihoitoon määrätyt potilaat siirrettiin ESA-hoidosta tutkimuksen alussa, ja mihin tahansa uuteen hoitoon siirtymisen riski verrattuna vanhan hoidon jatkamiseen Hb-arvon ollessa vakaa saattaa vaikuttaa havaittuihin tuloksiin, jolloin hoitojen tehoestimaatteja ei voida luotettavasti vertailla.
Taulukko 11. Kardiovaskulaarinen turvallisuus ja kuolleisuus ESA-kontrolloidussa, ESA-hoidosta siirtyneessä, voinniltaan vakaassa DD-poolissa
| MACE | MACE+ | ACM | ||||
| Roksadustaatti n = 1 594 | ESA n = 1 594 | Roksadustaatti n = 1 594 | ESA n = 1 594 | Roksadustaatti n = 1 594 | ESA n = 1 594 | |
| Hoidon aikana | ||||||
Potilaat, joilla esiintyi tapahtumia (%) | 297 (18,6) | 301 (18,9) | 357 (22,4) | 403 (25,3) | 212 (13,3) | 207 (13,0) |
| IR | 10,4 | 9,2 | 12,5 | 12,3 | 7,4 | 6,3 |
| HR (95 %:n CI) | 1,18 (1,00; 1,38) | 1,03 (0,90; 1,19) | 1,23 (1,02; 1,49) | |||
| ACM: mistä tahansa syystä johtuva kuolleisuus: ACM on MACE:n/MACE+:n komponentti. CI: luottamusväli; ESA: erytropoieesia stimuloiva aine; HR: riskitiheyksien suhde; IR: ilmaantuvuus (sellaisten potilaiden määrä, joilla esiintyi tapahtuma/100 potilasvuotta); MACE: merkittävä kardiovaskulaarinen haittatapahtuma (kuolema, ei kuolemaan johtava sydäninfarkti ja/tai aivohalvaus); MACE+: merkittävä kardiovaskulaarinen haittatapahtuma, mukaan lukien epästabiilista rasitusrintakivusta ja/tai kongestiivisesta sydämen vajaatoiminnasta johtuva sairaalahoito. | ||||||
Farmakokinetiikka
Plasman roksadustaattialtistus (plasman lääkeainepitoisuus-aikakuvaajan alle jäänyt pinta-ala [AUC] ja lääkeaineen huippupitoisuus plasmassa [Cmax]) on suhteessa annokseen suositellulla terapeuttisella annosalueella. Kun lääkettä otetaan kolme kertaa viikossa, plasman roksadustaattipitoisuudet saavuttavat vakaan tilan viikossa (3 annosta) minimaalisella kerääntymisellä. Roksadustaatin farmakokinetiikka ei muutu ajan kuluessa.
Imeytyminen
Huippupitoisuus plasmassa (Cmax) saavutetaan yleensä 2 tuntia paastotilassa otetun annoksen jälkeen.
Roksadustaatin ottaminen ruoan kanssa pienensi Cmax-arvoa 25 %, mutta ei vaikuttanut AUC-arvoon paastotilaan verrattuna. Roksadustaatti voidaan siis ottaa joko ruoan kanssa tai ilman ruokaa (ks. kohta Annostus ja antotapa).
Jakautuminen
Roksadustaatti sitoutuu voimakkaasti (noin 99-prosenttisesti) ihmisen plasman proteiineihin, pääasiassa albumiiniin. Roksadustaatin veri-plasmasuhde on 0,6. Näennäinen jakautumistilavuus vakaassa tilassa on 24 l.
Biotransformaatio
In vitro -tietojen perusteella roksadustaatti on CYP2C8- ja UGT1A9-entsyymien sekä BCRP:n, OATP1B1:n, OAT1:n ja OAT3:n substraatti. Roksadustaatti ei ole OATP1B3:n eikä P‑gp:n substraatti.Roksadustaatti metaboloituu pääasiassa hydroksiroksadustaatiksi ja roksadustaatti‑O‑glukuronidiksi. Muuttumattomassa muodossa oleva roksadustaatti oli merkittävin ihmisen plasmassa kiertävä komponentti, eikä minkään ihmisen plasmassa havaitun metaboliitin osuus kokonaisaltistuksesta lääkkeeseen liittyville yhdisteille ollut yli 10 %. Ihmiselle spesifisiä metaboliitteja ei havaittu.
Eliminaatio
Roksadustaatin keskimääräinen efektiivinen puoliintumisaika (t1/2) on CKD-potilailla noin 15 tuntia. Roksadustaatin näennäinen kokonaispuhdistuma (CL/F) on 1,1 l/h CKD-potilailla, jotka eivät saa dialyysihoitoa, ja 1,4 l/h CKD-potilailla, jotka saavat dialyysihoitoa. Roksadustaatti ja sen metaboliitit eivät poistu hemodialyysissä merkittävässä määrin.
Kun radiomerkittyä roksadustaattia annettiin suun kautta terveille tutkittaville, 96 % radioaktiivisuudesta voitiin mitata (50 % ulosteesta, 46 % virtsasta). Annoksesta 28 % erittyi ulosteeseen muuttumattomassa muodossa olevana roksadustaattina. Alle 2 % annoksesta erittyi virtsaan muuttumattomassa muodossa olevana roksadustaattina.
Erityisryhmät
Iän, sukupuolen, painon ja rodun vaikutukset
Roksadustaatin farmakokinetiikassa ei todettu ikään (≥ 18), sukupuoleen, rotuun, painoon, munuaistoimintaan (eGFR) tai dialyysihoitoon liittyviä kliinisesti merkittäviä eroja aikuispotilailla, joilla oli CKD:stä johtuva anemia.
Hemodialyysi
Dialyysistä riippuvaisilla CKD-potilailla ei havaittu merkittäviä eroja farmakokineettisten parametrien arvoissa, kun roksadustaatti annettiin 2 tuntia ennen hemodialyysiä tai 1 tunti sen jälkeen. Dialyysi on merkityksetön reitti roksadustaatin kokonaispuhdistuman kannalta.
Maksan vajaatoiminta
Roksadustaatin 100 mg:n kerta-annoksen jälkeen roksadustaatin keskimääräinen AUC-arvo oli 23 % suurempi ja keskimääräinen Cmax-arvo 16 % pienempi tutkittavilla, joilla oli keskivaikea maksan vajaatoiminta (Child‑Pugh-luokka B) ja normaali munuaistoiminta verrattuna tutkittaviin, joiden maksa ja munuaiset toimivat normaalisti. Tutkittavilla, joilla oli keskivaikea maksan vajaatoiminta (Child‑Pugh-luokka B) ja normaali munuaistoiminta, havaittiin sitoutumattoman roksadustaatin AUCinf-arvon suurenemista (+70 %) terveisiin tutkittaviin verrattuna.
Roksadustaatin farmakokinetiikkaa ei ole tutkittu vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavilla henkilöillä.
Lääkkeiden yhteisvaikutukset
In vitro -tietojen perusteella roksadustaatti on CYP2C8:n, BCRP:n, OATP1B1:n ja OAT3:n estäjä (ks. kohta Yhteisvaikutukset). Roksadustaatin samanaikainen anto ei vaikuttanut rosiglitatsonin (kohtalaisen herkkä CYP2C8:n substraatti) farmakokinetiikkaan. Roksadustaatti saattaa olla UGT1A1:n estäjä suolistossa mutta ei maksassa, eikä sen ole havaittu estävän muita metaboloivia CYP-entsyymejä tai kuljettajaproteiineja eikä indusoivan CYP-entsyymejä kliinisesti merkittävillä pitoisuuksilla.
Suun kautta otettu lääkehiili tai omepratsoli eivät vaikuta roksadustaatin farmakokinetiikkaan kliinisesti merkitsevästi. Klopidogreeli ei vaikuta roksadustaatin altistukseen CKD-potilailla.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen toksisuutta selvittävät tutkimukset
26 viikon pituisessa jaksottaisessa toistuvan altistuksen tutkimuksessa, jossa käytettiin Sprague-Dawley- tai Fisher-rottia, roksadustaatti aiheutti histopatologisia löydöksiä kuten aorttaläpän ja atrioventrikulaarisen (AV) läpän valvulopatioita annoksina, joilla saavutettiin noin 4–6 kertaiset AUC-kokonaisarvot ihmisille suositeltuun enimmäisannokseen (MRHD) verrattuna. Näitä löydöksiä havaittiin eloon jääneissä eläimissä lopetusvaiheessa ja myös eläimissä, jotka oli lopetettu aikaisemmin huonokuntoisuuden takia. Löydökset eivät myöskään olleet täysin korjaantuvia, sillä niitä todettiin eläimillä myös 30 vuorokauden toipumisjakson lopussa.
Liiallista farmakologista vaikutusta, joka on johtanut liialliseen erytropoieesiin, on havaittu toistuvan altistuksen toksisuutta selvittäneissä tutkimuksissa terveillä eläimillä.
Rotilla havaittiin hematologisia muutoksia, kuten verenkierrossa olevien verihiutaleiden vähenemistä ja aktivoidun partiaalisen tromboplastiiniajan ja protrombiiniajan pitenemistä, kun altistus oli noin kaksinkertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna. Trombeja todettiin luuytimessä (rotilla systeeminen altistus oli noin 7-kertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna), munuaisissa (rotilla systeeminen altistus oli noin 5–6-kertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna), keuhkoissa (rotilla systeeminen altistus oli noin 8-kertainen ja makakeilla noin kaksinkertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna) ja sydämessä (rotilla systeeminen altistus oli noin 4–6-kertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna).
Turvallisuus aivoille
26 viikon pituisessa jaksottaisesti toistuvan altistuksen tutkimuksessa, jossa käytettiin Sprague-Dawley-rottia, yhdellä eläimellä todettiin histologisessa tutkimuksessa aivojen nekroosia ja glioosia altistuksella, joka oli noin 6-kertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna. Kun Fisher-rottia hoidettiin yhtä kauan, yhteensä neljällä eläimellä todettiin aivojen/hippokampuksen nekroosia altistuksella, joka oli noin 3–5-kertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna.
Kun makakeille annettiin jaksottaisesti roksadustaattia 22 tai 52 viikon ajan, niillä ei todettu samankaltaisia löydöksiä systeemisillä altistuksilla, jotka olivat noin kaksinkertaisia MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna.
Karsinogeenisuus ja mutageenisuus
Roksadustaatti ei ollut mutageeninen Amesin mutageenisuustestissä in vitro, ihmisen ääreisveren lymfosyyteillä tehdyssä kromosomipoikkeavuustestissä in vitro eikä hiiren mikrotumatestissä in vivo annoksella, joka oli 40 kertaa suurempi kuin ihmisen vastaavaan annokseen perustuva MRHD-annos.
Hiirillä ja rotilla tehdyissä karsinogeenisuustutkimuksissa eläimet saivat roksadustaattia kliinisen annostusohjelman mukaisesti kolme kertaa viikossa. Roksadustaatin puhdistuma jyrsijöillä on nopea, joten systeemiset altistukset eivät olleet annosteluvaiheen aikana jatkuvia. Näin ollen muuhun kuin hoidon kohteeseen kohdistuvien (off-target) karsinogeenisten vaikutusten mahdollisuus on saatettu aliarvioida.
Hiirillä tehdyssä, 2 vuotta kestäneessä karsinogeenisuustutkimuksessa havaittiin keuhkojen bronkoalveolaaristen karsinoomien ilmaantuvuuden merkitsevää nousua sekä pientä että suurta annosta saaneissa ryhmissä (systeemiset altistukset olivat noin 1‑kertaisia ja noin kolminkertaisia MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna). Suurta annosta saaneessa ryhmässä naarailla todettiin ihonalaiskudoksen fibrosarkoomien merkitsevää lisääntymistä (systeemiset altistukset olivat noin kolminkertaisia MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna).
Rotilla tehdyssä, 2 vuotta kestäneessä karsinogeenisuustutkimuksessa havaittiin maitorauhasadenoomien ilmaantuvuuden merkitsevää nousua keskikokoista annosta saaneessa ryhmässä (systeeminen altistus oli alle 1‑kertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna). Tämä löydös ei kuitenkaan riippunut annoksesta, ja tämän tyyppisten kasvainten ilmaantuvuus oli pienempi suurinta testattua annosta saaneessa ryhmässä (systeeminen altistus oli noin 2‑kertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna), joten sitä ei pidetty tutkittavaan lääkkeeseen liittyvänä.
Kliinisissä tutkimuksissa samankaltaisia löydöksiä ei havaittu hiirillä ja rotilla tehdyissä karsinogeenisuustutkimuksissa.
Lisääntymis- ja kehitystoksisuus
Roksadustaatti ei vaikuttanut hoidettujen uros- tai naarasrottien paritteluun tai hedelmällisyyteen altistuksella, joka oli noin 4‑kertainen MRHD-annoksella saavutettuun ihmisen altistukseen verrattuna. NOAEL-annoksella urosrotilla todettiin kuitenkin lisäkivesten ja rakkularauhasten (nestettä sisältävien) painon pienentymistä, mutta se ei vaikuttanut urosten hedelmällisyyteen. Kaikkien urosten lisääntymiselimiin liittyvien löydösten NOEL-annos oli 1,6‑kertainen MRHD-annokseen verrattuna. Naarasrotilla elinkelvottomien alkioiden määrä ja implantaation jälkeiset menetykset lisääntyivät tällä annostasolla verrokkeihin nähden.
Rotilla ja kaneilla tehtyjen lisääntymis- ja kehitystoksisuustutkimusten tulokset osoittivat sikiöiden tai poikasten keskimääräisen painon laskua, istukan keskimääräisen painon nousua, keskenmenoja ja poikaskuolemia.
Tiineillä Sprague-Dawley-rotilla, joille roksadustaattia annettiin päivittäin implantaatiosta kovan suulaen sulkeutumiseen asti (tiineyspäivät 7–17), todettiin sikiöiden painon laskua ja luustomuutosten lisääntymistä altistuksella, joka oli noin 6-kertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna. Roksadustaatti ei vaikuttanut implantaation jälkeiseen sikiöiden eloonjääntiin.
Tiineille Uuden-Seelannin kaneille annettiin roksadustaattia päivittäin tiineyspäivästä 7 tiineyspäivään 19, ja tiineyspäivänä 29 tehtiin keisarileikkaus. Alkioihin ja sikiöihin kohdistuvia löydöksiä ei todettu, kun roksadustaatin systeeminen altistus oli enintään noin 3‑kertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna. Yksi naaras sai kuitenkin keskenmenon altistuksella, joka oli noin 1‑kertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna, ja kaksi naarasta altistuksella, joka oli noin 3‑kertainen MRHD-annoksella saavutettuun AUC-kokonaisarvoon verrattuna. Keskenmenon saaneet naaraat olivat laihoja.
Sprague‑Dawley-rotilla tehdyssä peri-/postnataalisessa kehitystutkimuksessa tiineille naaraille annettiin roksadustaattia päivittäin tiineyspäivästä 7 imetyspäivään 20 asti. Imetysaikana niiden naaraiden poikasilla, joiden roksadustaattialtistus oli noin 2‑kertainen MRHD-annoksella saavutettuun Cmax-kokonaisarvoon verrattuna, todettiin korkeaa kuolleisuutta vieroitusta edeltävässä vaiheessa, ja poikaset lopetettiin vieroitusvaiheessa. Elossaolo 21 päivää syntymän jälkeen (imetysindeksi) oli merkitsevästi heikompi niiden naaraiden poikasilla, jotka olivat saaneet roksadustaattia annoksina, joilla saavutettu systeeminen altistus oli noin 3‑kertainen verrattuna ihmisen altistukseen MRHD-annoksella, kun vertailukohtana olivat verrokkipoikueisiin syntyneet poikaset.
Tutkimuksessa, jossa poikasia vaihdettiin emojen välillä, suurimmat vaikutukset rotanpoikasten elinkelpoisuuteen havaittiin niillä poikasilla, jotka olivat altistuneet roksadustaatille vain syntymän jälkeen. Syntymään asti roksadustaatille altistuneiden poikasten elinkelpoisuus oli heikompi kuin poikasten, jotka eivät olleet altistuneet roksadustaatille.
Tutkimuksessa, jossa roksadustaatille altistumattomien rottien poikaset vaihdettiin roksadustaattia saaneille sijaisemoille (ihmisen vastaava annos oli noin 2‑kertainen MRHD- annokseen verrattuna), roksadustaattia todettiin poikasten plasmassa, mikä osoittaa lääkkeen erittyvän maitoon. Näiden emojen maidossa oli roksadustaattia. Roksadustaattia sisältävälle maidolle altistuneiden poikasten eloonjäänti oli heikompaa (85,1 %) verrattuna hoitamattomien emojen poikasiin, jotka siirrettiin hoitamattomille sijaisemoille (98,5 % jäi eloon). Roksadustaatille imetysaikana altistuneiden, eloonjääneiden poikasten keskimääräinen paino oli myös pienempi kuin verrokkien (ei altistusta in utero – ei altistusta maidon kautta).
Kardiovaskulaarinen turvallisuus
Kardiovaskulaarista turvallisuutta selvittäneessä farmakologisessa tutkimuksessa apinoilla, jotka saivat 100 mg/kg kerta-annoksen roksadustaattia, todettiin syketiheyden nousua. Vaikutusta hERG:hen tai EKG:hen. Muut rotilla tehdyt turvallisuusfarmakologiset tutkimukset ovat osoittaneet roksadustaatin pienentävän perifeeristä kokonaisvastusta, jota seuraa sykkeen refleksinomainen kiihtyminen, altistuksella, joka on noin kuusinkertainen MRHD-annoksella saavutettuun altistukseen verrattuna.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Laktoosimonohydraatti
Mikrokiteinen selluloosa (E460 (i))
Kroskarmelloosinatrium (E468)
Povidoni (E1201)
Magnesiumstearaatti (E470b)
Kalvopäällyste
Poly(vinyylialkoholi) (E1203)
Talkki (E553b)
Makrogoli (E1521)
Alluranpunainen AC alumiinilakka (E129)
Titaanidioksidi (E171)
Lesitiini (soija) (E322)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
EVRENZO tabletti, kalvopäällysteinen
20 mg (L:kyllä) 12 x 1 fol (86,12 €)
50 mg (L:kyllä) 12 x 1 fol (203,49 €)
70 mg (L:kyllä) 12 x 1 fol (278,48 €)
100 mg (L:kyllä) 12 x 1 fol (390,95 €)
150 mg (L:kyllä) 12 x 1 fol (578,44 €)
PF-selosteen tieto
Yksittäispakatut PVC-/alumiiniläpipainopakkaukset kartonkikotelossa.
Pakkauskoot
12 x 1 kalvopäällysteistä tablettia
36 x 1 kalvopäällysteistä tablettia
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Evrenzo 20 mg tabletit
Punaisia, soikeita tabletteja (noin 8 mm × 4 mm), joissa on toisella puolella painatus "20".
Evrenzo 50 mg tabletit
Punaisia, soikeita tabletteja (noin 11 mm × 6 mm), joissa on toisella puolella painatus "50".
Evrenzo 70 mg tabletit
Punaisia, pyöreitä tabletteja (noin 9 mm), joissa on toisella puolella painatus "70".
Evrenzo 100 mg tabletit
Punaisia, soikeita tabletteja (noin 14 mm × 7 mm), joissa on toisella puolella painatus "100".
Evrenzo 150 mg tabletit
Punaisia, mantelinmuotoisia tabletteja (noin 14 mm × 9 mm), joissa on toisella puolella painatus "150".
Käyttö- ja käsittelyohjeet
Ei erityisvaatimuksia hävittämisen suhteen.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
EVRENZO tabletti, kalvopäällysteinen
20 mg 12 x 1 fol
50 mg 12 x 1 fol
70 mg 12 x 1 fol
100 mg 12 x 1 fol
150 mg 12 x 1 fol
- Ylempi erityiskorvaus (100 %). Munuaisten vajaatoimintaan liittyvä vaikea anemia (138).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Roksadustaatti: Aikuisten krooniseen munuaissairauteen liittyvän oireisen anemian hoito erityisin edellytyksin (3066).
ATC-koodi
B03XA05
Valmisteyhteenvedon muuttamispäivämäärä
30.03.2026
Yhteystiedot
Hatsinanpuisto 8
02600 Espoo
09 8560 6000
www.astellas.fi