
ENHERTU kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 100 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Terveydenhuollon ammattilainen
Opas terveydenhuollon ammattilaisille lääkitysvirheiden välttämiseksi
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo kuiva-ainetta välikonsentraatiksi infuusionestettä varten sisältää 100 mg trastutsumabi-derukstekaania. Käyttökuntoon saattamisen jälkeen yksi 5 ml:n injektiopullo sisältää 20 mg/ml trastutsumabi-derukstekaania (ks. kohta Käyttö- ja käsittelyohjeet).
Trastutsumabi-derukstekaani on nisäkkäiden (kiinanhamsterin munasarja) soluissa tuotettava vasta-aine-lääkekonjugaatti, joka sisältää humanisoitua monoklonaalista HER2 IgG1 -vasta-ainetta (mAb), jolla on sama aminohapposekvenssi kuin trastutsumabilla. Se on kovalenttisesti sidottu DXd:hen (eksatekaanijohdannainen ja topoisomeraasi I:n estäjä) tetrapeptidipohjaisella pilkottavalla linkkerillä. Yhteen vasta-ainemolekyyliin on kiinnittynyt noin 8 derukstekaanimolekyyliä.
Apuaine, jonka vaikutus tunnetaan
Yksi 100 mg:n injektiopullo sisältää 1,5 mg polysorbaattia 80 (E433).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos.
Kliiniset tiedot
Käyttöaiheet
Rintasyöpä
HER2-positiivinen rintasyöpä
Enhertu on tarkoitettu monoterapiana sellaisten aikuispotilaiden hoitoon, joilla on leikkaushoitoon soveltumaton tai metastasoitunut HER2-positiivinen rintasyöpä, ja jotka ovat aiemmin saaneet vähintään yhtä anti-HER2-pohjaista hoitoa.
Heikosti HER2-positiivinen ja erittäin heikosti HER2-positiivinen rintasyöpä
Enhertu on tarkoitettu monoterapiana sellaisten aikuispotilaiden hoitoon, joilla on leikkaushoitoon soveltumaton tai metastasoitunut
- hormonireseptoripositiivinen (HR-positiivinen), heikosti HER2-positiivinen tai erittäin heikosti HER2-positiivinen rintasyöpä, ja jotka ovat saaneet vähintään yhtä endokriinista hoitoa metastasoituneeseen tautiin ja joille endokriinisen hoidon ei katsota soveltuvan seuraavan linjan hoidoksi (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
- heikosti HER2-positiivinen rintasyöpä, ja jotka ovat aiemmin saaneet solunsalpaajahoitoa metastasoituneeseen tautiin tai joilla tauti on uusiutunut adjuvanttisolunsalpaajahoidon aikana tai 6 kuukauden sisällä sen päättymisestä (ks. kohta Annostus ja antotapa).
Ei-pienisoluinen keuhkosyöpä
Enhertu on tarkoitettu monoterapiana sellaisten aikuispotilaiden hoitoon, joilla on pitkälle edennyt ei-pienisoluinen keuhkosyöpä ja joiden kasvaimissa on aktivoiva HER2 (ERBB2) -mutaatio, ja jotka tarvitsevat systeemistä hoitoa saatuaan platinapohjaista kemoterapiaa joko yhdessä immunoterapian kanssa tai ilman sitä.
Mahasyöpä
Enhertu on tarkoitettu monoterapiana sellaisten aikuispotilaiden hoitoon, joilla on pitkälle edennyt HER2-positiivinen mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooma, ja jotka ovat saaneet aiempaa trastutsumabipohjaista hoitoa.
Ehto
Valmistetta saa määrätä vain syöpäpotilaiden hoitoon perehtynyt lääkäri ja sitä saa antaa vain syöpäpotilaiden hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa.
Annostus ja antotapa
Enhertu-valmisteen määrää lääkäri, ja se on annettava sellaisen terveydenhuollon ammattilaisen valvonnassa, jolla on kokemusta syöpälääkkeiden antamisesta. Lääkitysvirheiden välttämiseksi on tärkeää varmistaa kunkin injektiopullon etiketistä, että valmisteltava ja annettava lääkevalmiste on Enhertu (trastutsumabi-derukstekaani) eikä trastutsumabi tai trastutsumabiemtansiini.
Enhertu-valmistetta ei saa korvata trastutsumabilla tai trastutsumabiemtansiinilla.
Potilaiden valinta
HER2-positiivinen rintasyöpä
Trastutsumabi-derukstekaania rintasyöpään saavilla potilailla on oltava dokumentoidusti HER2-positiivinen kasvain, joka määritellään immunohistokemiallisessa (IHC) määrityksessä 3+ pisteeksi (ICH 3+) tai suhdeluvuksi ≥ 2,0 in situ -hybridisaatiolla (ISH) tai fluoresenssin in situ -hybridisaatiolla (FISH), kun määritys tehdään CE-merkityllä in vitro -diagnostiikkaan (IVD) tarkoitetulla lääkinnällisellä laitteella. Jos CE-merkittyä IVD-laitetta ei ole saatavilla, HER2-status on määritettävä jollakin vaihtoehtoisella validoidulla testillä.
Heikosti HER2-positiivinen tai erittäin heikosti HER2-positiivinen rintasyöpä
Trastutsumabi-derukstekaania saavilla potilailla on oltava dokumentoidusti heikosti HER2-positiivinen kasvain, jonka määritelmänä on IHC 1+ tai IHC 2+ / ISH-negatiivisuus, tai erittäin heikosti HER2-positiivinen kasvain, jonka määritelmänä on IHC 0 ja solukalvon värjäytyminen (IHC > 0 < 1+), kun määritys tehdään CE-merkityllä IVD-laitteella. Jos CE-merkittyä IVD-laitetta ei ole saatavilla, HER2-status on määritettävä jollakin vaihtoehtoisella validoidulla testillä (ks. kohta Farmakodynamiikka).
Ei-pienisoluinen keuhkosyöpä
Trastutsumabi-derukstekaania pitkälle edenneeseen ei-pienisoluiseen keuhkosyöpään saavilla potilailla on oltava aktivoiva HER2 (ERBB2) -mutaatio, joka on määritetty CE-merkityllä in vitro -diagnostiikkaan (IVD) tarkoitetulla lääkinnällisellä laitteella. Jos CE-merkittyä IVD-laitetta ei ole saatavilla, HER2-mutaation status on määritettävä jollakin vaihtoehtoisella validoidulla testillä.
Mahasyöpä
Trastutsumabi-derukstekaania mahalaukun tai ruokatorvi-mahalaukkurajan syöpään saavilla potilailla on oltava dokumentoidusti HER2-positiivinen kasvain, joka määritellään immunohistokemiallisessa (IHC) määrityksessä 3+ pisteeksi (ICH 3+) tai suhdeluvuksi ≥ 2 in situ -hybridisaatiolla (ISH) tai fluoresenssin in situ -hybridisaatiolla (FISH), kun määritys tehdään CE-merkityllä in vitro -diagnostiikkaan (IVD) tarkoitetulla lääkinnällisellä laitteella. Jos CE-merkittyä IVD-laitetta ei ole saatavilla, HER2-status on määritettävä jollakin vaihtoehtoisella validoidulla testillä.
Annostus
Rintasyöpä
Suositeltu Enhertu-annos on 5,4 mg/kg laskimoinfuusiona kolmen viikon välein (21 vuorokauden sykli), kunnes sairaus etenee tai ilmaantuu ei-hyväksyttävissä olevaa toksisuutta.
Ei-pienisoluinen keuhkosyöpä
Suositeltu Enhertu-annos on 5,4 mg/kg laskimoinfuusiona kolmen viikon välein (21 vuorokauden sykli), kunnes sairaus etenee tai ilmaantuu ei-hyväksyttävissä olevaa toksisuutta.
Mahasyöpä
Suositeltu Enhertu-annos on 6,4 mg/kg laskimoinfuusiona kolmen viikon välein (21 vuorokauden sykli), kunnes sairaus etenee tai ilmaantuu ei-hyväksyttävissä olevaa toksisuutta.
Aloitusannos on annettava 90 minuuttia kestävänä infuusiona laskimoon. Jos potilas sieti aiemman infuusion hyvin, seuraavat Enhertu-annokset voidaan antaa 30 minuuttia kestävinä infuusioina.
Enhertu-valmisteen infuusionopeutta on hidastettava tai infuusio on keskeytettävä, jos potilaalla ilmenee infuusioon liittyviä oireita (ks. kohta Haittavaikutukset). Enhertu-hoito on lopetettava pysyvästi, mikäli potilaalla ilmenee vaikeita infuusioreaktioita.
Esilääkitys
Enhertu on emeettinen (ks. kohta Haittavaikutukset) eli se aiheuttaa pahoinvointia ja/tai oksentelua, mukaan lukien viivästynyttä pahoinvointia ja/tai oksentelua. Ennen jokaista Enhertu-annosta potilasta on esilääkittävä kahden tai kolmen lääkevalmisteen yhdistelmällä (esim. deksametasonilla ja joko 5-HT3-reseptorin salpaajalla ja/tai NK1-reseptorin salpaajalla, sekä muilla lääkevalmisteilla, mikäli aiheellista) solunsalpaajahoidon aiheuttaman pahoinvoinnin ja oksentelun ehkäisemiseksi.
Annosmuutokset
Haittavaikutusten hoito voi edellyttää väliaikaista infuusion keskeytystä, annoksen pienentämistä tai Enhertu-hoidon lopettamista taulukoissa 1 ja 2 annettujen ohjeiden mukaisesti.
Enhertu-annosta ei pidä suurentaa uudelleen annoksen pienentämisen jälkeen.
Taulukko 1: Annoksen pienentämisen aikataulu
| Annoksen pienentämisen aikataulu | Rintasyöpä ja ei-pienisoluinen keuhkosyöpä | Mahasyöpä |
| Suositeltu aloitusannos | 5,4 mg/kg | 6,4 mg/kg |
| Ensimmäinen pienennetty annos | 4,4 mg/kg | 5,4 mg/kg |
| Toinen pienennetty annos | 3,2 mg/kg | 4,4 mg/kg |
| Jos annosta on tarpeen yhä pienentää | Lopeta hoito | Lopeta hoito |
Taulukko 2: Annoksen muutokset haittavaikutustilanteissa
| Haittavaikutus | Vaikeusaste | Hoidon muutos | |
| Interstitiaalinen keuhkosairaus (ILD) / pneumoniitti | Oireeton ILD/pneumoniitti (aste 1) | Keskeytä Enhertu-hoito, kunnes haittavaikutus paranee asteeseen 0, ja sen jälkeen
| |
| Oireellinen ILD/pneumoniitti (aste 2 tai korkeampi) |
| ||
| Neutropenia | Aste 3 (alle 1,0–0,5 × 109/l) |
| |
| Aste 4 (alle 0,5 × 109/l) |
| ||
| Kuumeinen neutropenia | Absoluuttinen neutrofiilimäärä alle 1,0 × 109/l ja ruumiinlämpö yli 38,3 °C tai ruumiinlämpö vähintään 38 °C pidempään kuin yhden tunnin ajan. |
| |
| Vasemman kammion ejektiofraktio (LVEF) pienentynyt | LVEF on yli 45 % ja absoluuttinen lasku lähtötilanteesta on 10–20 % |
| |
| LVEF 40–45 % | Ja absoluuttinen lasku lähtötilanteesta on alle 10 % |
| |
| Ja absoluuttinen lasku lähtötilanteesta on 10–20 % |
| ||
| LVEF on alle 40 % tai absoluuttinen lasku lähtötilanteesta on yli 20 % |
| ||
| Oireellinen sydämen vajaatoiminta (CHF) |
| ||
Toksisuusasteet ovat National Cancer Institute -laitoksen haittavaikutusten yleisten termikriteerien version 5.0 mukaisia (NCI-CTCAE v.5.0).
Annoksen viivästyminen tai antamatta jääminen
Jos suunniteltu annos viivästyy tai jää antamatta, se on annettava mahdollisimman pian odottamatta seuraavaan suunniteltuun sykliin. Antoaikataulua on muutettava, jotta annosväli pysyy 3 viikossa. Infuusio on annettava annoksella ja nopeudella, jota potilas sieti viimeisimmässä infuusiossa.
Erityisryhmät
Iäkkäät
Enhertu-annosta ei tarvitse muuttaa 65-vuotiaille tai vanhemmille potilaille. Vähintään 75 vuoden ikäisistä potilaista on saatavilla niukasti tietoja.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa potilaille, joilla on lievä (kreatiniinipuhdistuma [CLcr] ≥ 60 ja < 90 ml/min) tai keskivaikea (CLcr ≥ 30 ja < 60 ml/min) munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka). Vaikeaa munuaisten vajaatoimintaa tai loppuvaiheen munuaissairautta sairastavien potilaiden mahdollista annosmuutostarvetta ei voida arvioida, koska vaikea munuaissairaus oli poissulkukriteeri kliinisissä tutkimuksissa. Potilailla, joilla on keskivaikea munuaisten vajaatoiminta, on havaittu korkeampi ilmaantuvuus asteen 1 ja 2 ILD/pneumoniitille, joka on johtanut lisääntyneeseen lääkityksen lopettamiseen. Lähtötilanteessa keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla, jotka saivat Enhertu-valmistetta 6,4 mg/kg, havaittiin enemmän vakavia haittavaikutuksia kuin potilailla, joiden munuaistoiminta oli normaali. Keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavia potilaita on tarkkailtava huolellisesti haittavaikutusten, mukaan lukien ILD:n/pneumoniitin, varalta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa potilaille, joiden kokonaisbilirubiini on ≤ 1,5 kertaa normaalin yläraja (ULN) aspartaattitransaminaasiarvosta (ASAT) riippumatta. Potilaiden, joiden kokonaisbilirubiini on > 1,5 kertaa ULN (ASAT-arvosta riippumatta), mahdollista annosmuutostarvetta ei voida arvioida, koska tietoja on vain vähän. Siksi näitä potilaita on tarkkailtava huolellisesti (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Enhertu-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Enhertu on tarkoitettu annettavaksi laskimoon. Terveydenhuollon ammattilaisen on saatettava se käyttökuntoon, laimennettava se ja annettava se infuusiona laskimoon. Enhertu-valmistetta ei saa antaa injektiona tai boluksena laskimoon.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Lääkitysvirheiden välttämiseksi on tärkeää varmistaa kunkin injektiopullon etiketistä, että valmisteltava ja annettava lääkevalmiste on Enhertu (trastutsumabi-derukstekaani) eikä trastutsumabi tai trastutsumabiemtansiini.
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Interstitiaalinen keuhkosairaus / pneumoniitti
Interstitiaalisen keuhkosairauden (ILD) ja/tai pneumoniitin tapauksia on raportoitu Enhertu-hoidon yhteydessä (ks. kohta Haittavaikutukset). Kuolemaan johtaneita tapauksia on havaittu. Potilaita on neuvottava ilmoittamaan välittömästi yskästä, hengenahdistuksesta, kuumeesta ja/tai kaikista uusista tai pahenevista hengitystieoireista. Potilaita on seurattava ILD:n/pneumoniitin merkkien ja oireiden varalta. ILD:n/pneumoniitin merkit on tutkittava viipymättä. Potilaat, joilla epäillään ILD:tä/pneumoniittia, on arvioitava kuvantamistutkimuksella, mieluiten tietokonetomografiakuvauksella (TT). Keuhkolääkärin konsultaatiota on harkittava. Oireettomassa (asteen 1) ILD:ssä/pneumoniitissa on harkittava kortikosteroidihoitoa (esim. ≥ 0,5 mg/kg/vrk prednisolonia tai vastaavaa). Enhertu-hoito on keskeytettävä, kunnes haittavaikutus paranee luokkaan 0, ja sitä voidaan jatkaa taulukon 2 ohjeiden mukaan (ks. kohta Annostus ja antotapa). Oireelliseen ILD:hen/pneumoniittiin (aste 2 tai korkeampi) on viipymättä aloitettava kortikosteroidihoito (esim. ≥ 1 mg/kg/vrk prednisolonia tai vastaavaa) ja sitä on jatkettava vähintään 14 vuorokautta, minkä jälkeen annostusta on pienennettävä asteittain vähintään 4 viikon kuluessa. Enhertu-hoito on lopetettava pysyvästi, jos potilaalla diagnosoidaan oireellinen (asteen 2 tai korkeampi) ILD/pneumoniitti (ks. kohta Annostus ja antotapa). Potilailla, joilla on aiemmin ollut ILD/pneumoniitti, tai potilailla, joilla on keskivaikea tai vaikea munuaisten vajaatoiminta, on suurempi riski uuden ILD:n/pneumoniitin kehittymiseen ja heitä on tarkkailtava huolellisesti (ks. kohta Annostus ja antotapa).
Neutropenia
Neutropeniatapauksia, myös kuolemaan johtanutta kuumeista neutropeniaa, raportoitiin kliinisissä Enhertu-tutkimuksissa. Täydellinen verenkuva on tarkistettava ennen Enhertu-hoidon aloittamista, ennen jokaista annosta ja kliinisen tarpeen mukaan. Neutropenian vaikeusasteen mukaan Enhertu-hoito voidaan joutua keskeyttämään tai annosta on pienennettävä (ks. kohta Annostus ja antotapa).
Vasemman kammion toimintahäiriö
Vasemman kammion ejektiofraktion (LVEF) pienentymistä on havaittu anti-HER2-hoitojen yhteydessä. LVEF tulee arvioida tavanomaisella sydämen toiminnan tutkimuksella (sydämen kaikukuvaus tai MUGA-tutkimus [tasapainotila-angiografia]) ennen Enhertu-hoidon aloittamista ja säännöllisin väliajoin hoidon aikana kliinisen tarpeen mukaan. LVEF:n pienentymistä on hoidettava keskeyttämällä Enhertu-hoito. Enhertu-hoito on lopetettava pysyvästi, jos LVEF on alle 40 % tai absoluuttinen lasku lähtötilanteesta on vahvistetusti yli 20 %. Enhertu-hoito on lopetettava pysyvästi, jos potilaalla on oireellinen kongestiivinen sydämen vajaatoiminta (CHF) (ks. taulukko 2 kohdassa Annostus ja antotapa).
Toksisuus alkiolle ja sikiölle
Enhertu voi aiheuttaa sikiölle haittaa, jos sitä annetaan raskaana olevalle naiselle. Myyntiluvan saamisen jälkeisissä ilmoituksissa HER2-reseptorin antagonisti trastutsumabin käyttö raskauden aikana aiheutti lapsiveden niukkuutta, joka ilmeni kuolemaan johtaneena keuhkojen hypoplasiana, luuston epämuodostumina ja vastasyntyneiden kuolemina. Eläimistä tehtyjen löydösten ja vaikutusmekanismin perusteella Enhertu-valmisteen topoisomeraasi I -estäjäkomponentti DXd voi myös aiheuttaa haittaa alkiolle ja sikiölle, kun sitä annetaan raskaana olevalle naiselle (ks. kohta Raskaus ja imetys).
Naisille, jotka voivat tulla raskaaksi, on tehtävä raskaustesti ennen Enhertu-hoidon aloittamista. Potilaalle on kerrottava mahdollisista sikiöön kohdistuvista riskeistä. Naisia, jotka voivat tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisyä hoidon aikana ja vähintään 7 kuukauden ajan viimeisen Enhertu-annoksen jälkeen. Miespotilaita, joiden kumppani voi tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisyä Enhertu-hoidon aikana ja vähintään 4 kuukauden ajan viimeisen Enhertu-annoksen jälkeen (ks. kohta Raskaus ja imetys).
Potilaat, joilla on keskivaikea tai vaikea maksan vajaatoiminta
Tietoja on niukasti potilaista, joilla on keskivaikea maksan vajaatoiminta, ja vaikeaa maksan vajaatoimintaa sairastavista potilaista ei ole lainkaan tietoja. Koska topoisomeraasi I -estäjän, DXd:n, metabolia ja sappieritys ovat sen ensisijaisia eliminaatioreittejä, Enhertu-valmistetta on annettava varoen potilaille, joilla on keskivaikea tai vaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Yhteisvaikutukset
Samanaikainen ritonaviirin (OATP1B:n, CYP3A:n ja P-gp:n estäjä) tai itrakonatsolin (voimakas CYP3A:n ja P-gp:n estäjä) käyttö aiheutti noin 10–20 %:n altistuksen lisääntymisen trastutsumabi-derukstekaanille ja vapautuvalle topoisomeraasi I -estäjälle, DXd:lle, mitä ei pidetä kliinisesti merkittävänä. Annosta ei tarvitse muuttaa annettaessa trastutsumabi-derukstekaania samanaikaisesti CYP3A:n, OATP1B:n tai P-gp-kuljettajaproteiinien estäjien kanssa (ks. kohta Farmakokinetiikka).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / miesten ja naisten ehkäisy
Naisille, jotka voivat tulla raskaaksi, on tehtävä raskaustesti ennen Enhertu-hoidon aloittamista.
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä Enhertu-hoidon aikana ja vähintään 7 kuukauden ajan viimeisen annoksen jälkeen.
Miespotilaiden, joiden kumppani voi tulla raskaaksi, on käytettävä tehokasta ehkäisyä Enhertu-hoidon aikana ja vähintään 4 kuukauden ajan viimeisen annoksen jälkeen.
Raskaus
Ei ole olemassa tietoja Enhertu-valmisteen käytöstä raskaana oleville naisille. HER2-reseptorin antagonisti trastutsumabi voi kuitenkin aiheuttaa sikiölle haittaa, jos sitä annetaan raskaana olevalle naiselle. Myyntiluvan saamisen jälkeisissä ilmoituksissa trastutsumabin käyttö raskauden aikana aiheutti lapsiveden niukkuutta, joka ilmeni joissakin tapauksissa kuolemaan johtaneena keuhkojen hypoplasiana, luuston epämuodostumina ja vastasyntyneiden kuolemina. Eläimistä tehtyjen löydösten ja vaikutusmekanismin perusteella Enhertu-valmisteen topoisomeraasi I -estäjäkomponentin, DXd:n, voi odottaa aiheuttavan haittaa alkiolle ja sikiölle, kun sitä annetaan raskaana olevalle naiselle (ks. kohta Prekliiniset tiedot turvallisuudesta).
Enhertu-valmisteen antamista raskaana oleville naisille ei suositella, ja potilaille on kerrottava mahdollisista sikiöön kohdistuvista riskeistä ennen raskaaksi tuloa. Jos nainen tulee raskaaksi, hänen on otettava yhteyttä lääkäriin välittömästi. Jos nainen tulee raskaaksi Enhertu-hoidon aikana tai 7 kuukauden sisällä viimeisestä Enhertu-annoksesta, tiivistä seurantaa suositellaan.
Imetys
Ei tiedetä, erittyykö trastutsumabi-derukstekaani ihmisen rintamaitoon. Ihmisen IgG erittyy ihmisen rintamaitoon, eikä imeväiseen kohdistuvaa imeytymisen ja vakavien haittavaikutusten mahdollisuutta tunneta. Siksi naiset eivät saa imettää Enhertu-hoidon aikana tai 7 kuukauteen viimeisen annoksen jälkeen. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja/tai Enhertu-hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Trastutsumabi-derukstekaanilla ei ole tehty erityisiä hedelmällisyystutkimuksia. Eläintoksisuustutkimusten tulosten perusteella Enhertu voi heikentää miesten suvunjatkamiskykyä ja hedelmällisyyttä. Ei tiedetä, kulkeutuvatko trastutsumabi-derukstekaani tai sen metaboliitit siemennesteeseen. Ennen hoidon aloittamista miespotilaita tulee kehottaa hakeutumaan sukusolujen varastointia koskevaan neuvontaan. Miespotilaat eivät saa pakastaa tai luovuttaa spermaa hoitojakson aikana tai vähintään 4 kuukauteen viimeisen Enhertu-annoksen jälkeen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Enhertu-valmisteella voi olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaita on kehotettava varovaisuuteen ajaessaan tai käyttäessään koneita, mikäli heillä ilmenee väsymystä, päänsärkyä tai huimausta Enhertu-hoidon aikana (ks. kohta Haittavaikutukset).
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Enhertu 5,4 mg/kg
Yhdistetyssä turvallisuuspopulaatiossa on arvioitu potilaat, jotka saivat vähintään yhden Enhertu-annoksen 5,4 mg/kg (n = 2335) eri kasvaintyypeissä tehdyissä kliinisissä tutkimuksissa. Hoidon mediaanikesto tässä turvallisuuspopulaatiossa oli 9,0 kuukautta (vaihteluväli: 0,7–45,1 kuukautta).
Yleisimpiä haittavaikutuksia olivat pahoinvointi (71,1 %), uupumus (55,3 %), oksentelu (37,3 %), hiustenlähtö (36,1 %), anemia (35,9 %) neutropenia (35,1 %), ummetus (31,7 %), ruokahalun heikentyminen (30,6 %), ripuli (30,1 %), transaminaasiarvojen nousu (26,6 %), tuki- ja liikuntaelinkipu (23,6 %), trombosytopenia (23,1 %) ja leukopenia (21,5 %).
Yleisimpiä National Cancer Institute -laitoksen haittavaikutusten yleisten termikriteerien (NCI-CTCAE v.5.0) mukaisia asteen 3 tai 4 haittavaikutuksia olivat neutropenia (18,0 %), anemia (10,5 %), uupumus (7,8 %), leukopenia (6,0 %), trombosytopenia (5,4 %), pahoinvointi (4,9 %), lymfopenia (3,9 %), hypokalemia (3,8 %), transaminaasiarvojen nousu (3,5 %), ripuli (2,5 %), oksentelu (2,4 %), ruokahalun heikentyminen (1,8 %), pneumonia (1,3 %) ja ejektiofraktion pienentyminen (1,0 %). Asteen 5 haittavaikutuksia ilmeni 1,4 %:lla potilaista, mukaan lukien ILD/pneumoniitti (1,1 %).
Lääkkeenanto keskeytettiin haittavaikutusten vuoksi 32,6 %:lla Enhertu-hoitoa saaneista potilaista. Yleisimpiä hoidon keskeytykseen liittyviä haittavaikutuksia olivat neutropenia (12,4 %), uupumus (4,7 %), anemia (4,6 %), leukopenia (3,2 %), ylähengitysteiden infektio (3,0 %), ILD/pneumoniitti (2,6 %), trombosytopenia (2,4 %) ja pneumonia (2,0 %). Annosta pienennettiin 20,3 %:lla Enhertu-hoitoa saaneista potilaista. Yleisimpiä annoksen pienennykseen liittyviä haittavaikutuksia olivat uupumus (5,1 %), pahoinvointi (4,8 %) neutropenia (3,5 %) ja trombosytopenia (2,3 %). Hoito lopetettiin haittavaikutusten vuoksi 11,7 %:lla Enhertu-hoitoa saaneista potilaista. Yleisin pysyvään hoidon lopetukseen liittyvä haittavaikutus oli ILD/pneumoniitti (8,4 %).
Enhertu 6,4 mg/kg
Yhdistetyssä turvallisuuspopulaatiossa on arvioitu potilaat, jotka saivat vähintään yhden Enhertu-annoksen 6,4 mg/kg (n = 1 133) eri kasvaintyypeissä tehdyissä kliinisissä tutkimuksissa. Hoidon mediaanikesto tässä turvallisuuspopulaatiossa oli 5,1 kuukautta (vaihteluväli: 0,4–41,0 kuukautta).
Yleisimpiä haittavaikutuksia olivat pahoinvointi (64,3 %), uupumus (57,3 %), anemia (47,9 %), ruokahalun heikentyminen (46,8 %), neutropenia (45,9 %), oksentelu (34,7 %), ripuli (33,0 %), trombosytopenia (32,9 %), leukopenia (31,2 %), hiustenlähtö (29,0 %), ummetus (28,2 %) ja transaminaasiarvojen nousu (26,4 %).
Yleisimpiä National Cancer Institute -laitoksen haittavaikutusten yleisten termikriteerien mukaisia asteen 3 tai 4 haittavaikutuksia olivat neutropenia (28,4 %), anemia (22,8 %), leukopenia (12,3 %), trombosytopenia (10,8 %), uupumus (8,6 %), hypokalemia (5,8 %), pansytopenia (5,6 %), pahoinvointi (5,6 %), lymfopenia (5,5 %), ruokahalun heikentyminen (5,3 %), transaminaasiarvojen nousu (3,6 %), pneumonia (3,0 %), kuumeinen neutropenia (2,6 %), oksentelu (2,6 %), ripuli (1,9 %), painon lasku (1,7 %), vatsakipu (1,5 %), veren alkalisen fosfataasin arvon nousu (1,2 %), veren bilirubiiniarvon nousu (1,2 %), interstitiaalinen keuhkosairaus (ILD, 1,1 %) ja ejektiofraktion pienentyminen (1,1 %). Asteen 5 haittavaikutuksia ilmeni 2,2 %:lla potilaista, mukaan lukien ILD (1,6 %).
Lääkkeenanto keskeytettiin haittavaikutusten vuoksi 40,7 %:lla Enhertu-hoitoa saaneista potilaista. Yleisimpiä hoidon keskeytykseen liittyviä haittavaikutuksia olivat neutropenia (14,7 %), anemia (8,5 %), uupumus (6,0 %), ILD (4,7 %), leukopenia (3,9 %), pneumonia (3,3 %), trombosytopenia (3,2 %), ruokahalun heikentyminen (2,7 %) ja ylähengitysteiden infektio (2,6 %). Annosta pienennettiin 29,1 %:lla Enhertu-hoitoa saaneista potilaista. Yleisimpiä annoksen pienennykseen liittyviä haittavaikutuksia olivat uupumus (8,4 %), neutropenia (6,4 %), pahoinvointi (5,6 %), ruokahalun heikentyminen (4,1 %) ja trombosytopenia (3,8 %). Hoito lopetettiin haittavaikutusten vuoksi 13,8 %:lla Enhertu-hoitoa saaneista potilaista. Yleisin pysyvään hoidon lopetukseen liittyvä haittavaikutus oli ILD (10,1 %).
Enhertu-hoitoa 6,4 mg/kg:n annoksella saaneista mahasyöpää sairastavista potilaista (n = 546) 19,2 %:lle tehtiin verensiirto 28 vuorokauden kuluessa anemian tai trombosytopenian alkamisesta. Verensiirrot tehtiin pääasiassa anemian vuoksi.
Haittavaikutustaulukko
Haittavaikutukset, joita ilmeni vähintään yhden Enhertu-annoksen kliinisissä tutkimuksissa saaneilla potilailla, on esitetty taulukossa 3. Haittavaikutukset on lueteltu MedDRA-elinjärjestelmäluokan (SOC) ja yleisyysluokan mukaisesti. Yleisyysluokat ovat hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3: Trastutsumabi-derukstekaaniannoksilla 5,4 mg/kg ja 6,4 mg/kg hoidetuilla potilailla esiintyneet haittavaikutukset eri kasvaintyypeissä
Elinjärjestelmäluokka Yleisyysluokka
| 5,4 mg/kg Haittavaikutus | 6,4 mg/kg Haittavaikutus |
|---|---|---|
| Infektiot | ||
| Hyvin yleinen | ylähengitysteiden infektioa | ylähengitysteiden infektioa |
| Yleinen | pneumonia | pneumonia |
| Veri ja imukudos | ||
| Hyvin yleinen | anemiab, neutropeniac, trombosytopeniad, leukopeniae | anemiab, neutropeniac, trombosytopeniad, leukopeniae, lymfopeniaf |
| Yleinen | lymfopeniaf, kuumeinen neutropenia, pansytopeniag | kuumeinen neutropenia, pansytopeniag |
| Aineenvaihdunta ja ravitsemus | ||
| Hyvin yleinen | hypokalemiah, ruokahalun heikentyminen | hypokalemiah, ruokahalun heikentyminen |
| Yleinen | kuivuminen | kuivuminen |
| Hermosto | ||
| Hyvin yleinen | päänsärkyi | |
| Yleinen | huimaus, dysgeusia | huimaus, päänsärkyi, dysgeusia |
| Silmät | ||
| Yleinen | silmien kuivuminen, näön sumeneminenj | silmien kuivuminen, näön sumeneminenj |
| Hengityselimet, rintakehä ja välikarsina | ||
| Hyvin yleinen | interstitiaalinen keuhkosairausk, yskä | interstitiaalinen keuhkosairausk, yskä |
| Yleinen | hengenahdistus, nenäverenvuoto | hengenahdistus, nenäverenvuoto |
| Ruoansulatuselimistö | ||
| Hyvin yleinen | pahoinvointi, oksentelu, ummetus, ripuli, vatsakipul, stomatiittim, dyspepsia | pahoinvointi, oksentelu, ripuli, ummetus, vatsakipul, stomatiittim |
| Yleinen | vatsan turvotus, gastriitti, ilmavaivat | dyspepsia, vatsan turvotus, gastriitti, ilmavaivat |
| Maksa ja sappi | ||
| Hyvin yleinen | transaminaasiarvojen nousun | transaminaasiarvojen nousun |
| Iho ja ihonalainen kudos | ||
| Hyvin yleinen | hiustenlähtö | hiustenlähtö |
| Yleinen | ihottumao, kutina, ihon hyperpigmentaatiop | ihottumao, kutina, ihon hyperpigmentaatiop |
| Luusto, lihakset ja sidekudos | ||
| Hyvin yleinen | tuki- ja liikuntaelinkipuq | tuki- ja liikuntaelinkipuq |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Hyvin yleinen | uupumusr, kuume | uupumusr, kuume, perifeerinen turvotus |
| Yleinen | perifeerinen turvotus | |
| Tutkimukset | ||
| Hyvin yleinen | ejektiofraktion pienentyminens, painon lasku | ejektiofraktion pienentyminens, painon lasku |
| Yleinen | veren alkalisen fosfataasin nousu, veren bilirubiiniarvon nousut, veren kreatiniiniarvon nousu | veren alkalisen fosfataasin nousu, veren bilirubiiniarvon nousut, veren kreatiniiniarvon nousu |
| Vammat, myrkytykset ja hoitokomplikaatiot | ||
| Yleinen | infuusioon liittyvät reaktiotu | |
| Melko harvinainen | infuusioon liittyvät reaktiotu | |
a Sisältää influenssan, influenssan kaltaisen sairauden, nasofaryngiitin, faryngiitin, sinuiitin, nuhan, kurkunpäätulehduksen ja ylähengitysteiden infektion.
b Kaikille kasvaintyypeille annoksella 5,4 mg/kg sisältää anemian, hemoglobiiniarvon laskun, punasolumäärän laskun ja hematokriittiarvon laskun. Kaikille kasvaintyypeille annoksella 6,4 mg/kg sisältää anemian, veren hemoglobiiniarvon laskun, hematokriittiarvon laskun ja veren punasolumäärän laskun.
c Sisältää neutropenian ja neutrofiilimäärän laskun.
d Sisältää trombosytopenian ja verihiutalemäärän laskun.
e Sisältää leukopenian ja valkosolumäärän laskun.
f Sisältää lymfopenian ja lymfosyyttimäärän laskun.
g Haittavaikutus määriteltiin pansytopeniaksi, jos tutkittava täytti kaikki kolme kriteeriä, jotka olivat hemoglobiiniarvo < 100 g/l ja CTCAE-luokituksen mukaan vähintään vaikeusasteen 2 haittavaikutus, neutrofiiliarvo < 1,5 x 109/l ja CTCAE-luokituksen mukaan vähintään vaikeusasteen 1 haittavaikutus sekä verihiutalearvo < 100 x 109/l ja CTCAE-luokituksen mukainen haittavaikutuksen vaikeusasteen määritys oli tehty, kun perusteena olivat tiedot samalta laboratorionäytteenottopäivältä, ja/tai jos oli käytetty suositeltua termiä pansytopenia.
h Sisältää hypokalemian ja veren kaliumarvon laskun.
i Kaikille kasvaintyypeille annoksella 5,4 mg/kg sisältää päänsäryn, sinuspäänsäryn ja migreenin. Kaikille kasvaintyypeille annoksella 6,4 mg/kg sisältää päänsäryn ja migreenin.
j Sisältää näön sumenemisen ja näön heikentymisen.
k Kaikille kasvaintyypeille annoksella 5,4 g/kg interstitiaalinen keuhkosairaus sisältää seuraavat tapahtumat, jotka arvioitiin ILD:ksi: akuutti hengitysvajaus (n = 2), alveoliitti (n = 2), bronkiektasia (n = 1), taudin eteneminen (n = 1), yliherkkyyspneumoniitti (n = 1), idiopaattinen interstitiaalinen pneumonia (n = 1), interstitiaalinen keuhkosairaus (n = 109), alahengitysteiden infektio (n = 1), keuhkosairaus (n = 1), keuhkoinfiltraatio (n = 1), keuhkovarjostuma (n = 4), lymfangiitti (n = 1), organisoituva pneumonia (n = 9), pneumonia (n = 9), bakteeriperäinen pneumonia (n = 2), sieniperäinen pneumonia (n = 1), pneumoniitti (n = 136), keuhkofibroosi (n = 2), keuhkotiivistymä (n = 1), keuhkotoksisuus (n = 3), sädepneumoniitti (n = 4) ja hengitysvajaus (n = 5). Kaikille kasvaintyypeille annoksella 6,4 g/kg interstitiaalinen keuhkosairaus sisältää tapahtumat, jotka arvioitiin ILD:ksi: alveoliitti (n = 1), interstitiaalinen keuhkosairaus (n = 68), keuhkovarjostuma (n = 2), organisoituva pneumonia (n = 4), pneumonia (n = 1), pneumoniitti (n = 98), keuhkotoksisuus (n = 1), sädepneumoniitti (n = 1) ja hengitysvajaus (n = 5).
l Sisältää epämukavan tunteen vatsassa, ruoansulatuskanavan kivun, vatsakivun, alavatsakivun ja ylävatsakivun.
m Kaikille kasvaintyypeille annoksella 5,4 mg/kg sisältää stomatiitin, suun limakalvon haavauman, suun haavaumat, suun limakalvon eroosion ja suun äkilliset limakalvo-oireet. Kaikille kasvaintyypeille annoksella 6,4 mg/kg sisältää stomatiitin, suun limakalvon haavauman ja suun haavaumat.
n Sisältää transaminaasiarvojen nousun, alaniiniaminotransferaasiarvon nousun, aspartaattiaminotransferaasiarvon nousun, gamma-glutamyylitransferaasiarvon nousun, poikkeavan maksan toiminnan, poikkeavat maksa-arvot, maksa-arvojen nousun ja hypertransaminasemian.
o Kaikille kasvaintyypeille annoksella 5,4 g/kg sisältää ihottuman, märkärakkulaisen ihottuman, makulopapulaarisen ihottuman, papulaarisen ihottuman, makulaarisen ihottuman ja kutisevan ihottuman. Kaikille kasvaintyypeille annoksella 6,4 g/kg sisältää ihottuman, märkärakkulaisen ihottuman, makulopapulaarisen ihottuman, papulaarisen ihottuman ja kutisevan ihottuman.
p Kaikille kasvaintyypeille annoksella 5,4 mg/kg sisältää ihon hyperpigmentaation, ihon värjäytymisen ja pigmentaatiohäiriön. Kaikille kasvaintyypeille annoksella 6,4 mg/kg sisältää hyperpigmentaation ja pigmentaatiohäiriön.
q Sisältää selkäkivun, lihaskivun, raajakivun, tuki- ja liikuntaelinkivun, lihaskouristukset, luukivun, niskakivun, tuki- ja liikuntaelinperäisen rintakivun ja epämukavan tunteen raajassa.
r Sisältää voimattomuuden, uupumuksen, huonovointisuuden ja letargian.
s Kaikille kasvaintyypeille annoksella 5,4 mg/kg ejektiofraktion pienentyminen sisältää LVEF:n pienentymisen laboratorioparametrit (n = 312) ja/tai seuraavat suositellut termit: ejektiofraktion pienentyminen (n = 99), sydämen vajaatoiminta (n = 5), akuutti sydämen vajaatoiminta (n = 1), krooninen sydämen vajaatoiminta (n = 1), kongestiivinen sydämen vajaatoiminta (n = 1) ja vasemman kammion toimintahäiriö (n = 3). Kaikille kasvaintyypeille annoksella 6,4 mg/kg ejektiofraktion pienentyminen sisältää LVEF:n pienentymisen laboratorioparametrit (n = 125) ja/tai seuraavat suositellut termit: ejektiofraktion pienentyminen (n = 20), vasemman kammion toimintahäiriö (n = 1), sydämen vajaatoiminta (n = 2), akuutti sydämen vajaatoiminta (n = 1) ja kongestiivinen sydämen vajaatoiminta (n = 1).
t Kaikille kasvaintyypeille annoksella 5,4 mg/kg sisältää veren bilirubiiniarvon nousun, hyperbilirubinemian, konjugoituneen bilirubiinin arvon nousun ja veren konjugoimattoman bilirubiinin arvon nousun. Kaikille kasvaintyypeille annoksella 6,4 mg/kg sisältää veren bilirubiiniarvon nousun, hyperbilirubinemian ja veren konjugoituneen bilirubiinin arvon nousun.
u Kaikille kasvaintyypeille annoksella 5,4 mg/kg infuusioon liittyvät reaktiot sisältävät infuusioon liittyvän reaktion (n = 23) ja yliherkkyyden (n = 2). Kaikille kasvaintyypeille annoksella 6,4 mg/kg infuusioon liittyviä reaktioita ovat infuusioon liittyvä reaktio (n = 6) ja yliherkkyys (n = 1). Kaikkien infuusioon liittyvien reaktioiden aste oli 1 tai 2.
Kuvaus valikoiduista haittavaikutuksista
Interstitiaalinen keuhkosairaus / pneumoniitti
Tutkija raportoi ILD:tä, pneumoniittia, organisoituvaa pneumoniaa ja akuuttia interstitiaalista pneumoniittia eri kasvaintyypeillä tehdyissä kliinisissä tutkimuksissa 13,3 %:lla potilaista (n = 2335), joita hoidettiin Enhertu-annoksella 5,4 mg/kg. ILD/pneumoniitti vahvistettiin arvioinnissa 12,2 %:lla potilaista, ja se johti lääkityksen lopettamiseen 8,4 %:lla potilaista ja lääkkeen annon keskeyttämiseen 2,6 %:lla potilaista. Useimmat ILD-/pneumoniittitapaukset olivat asteen 1 (2,9 %) ja asteen 2 (7,5 %) tapahtumia. Asteen 3 tapahtumia ilmeni 0,7 %:lla ja asteen 4 tapahtumia ilmeni yhdessä tapauksessa. Asteen 5 (kuolemaan johtavia) tapahtumia ilmeni 1,1 %:lla potilaista. Mediaaniaika ensimmäiseen esiintymään oli 5,5 kuukautta (vaihteluväli: –0,3–31,5), mukaan lukien kaksi potilasta, joilla arvioitiin olleen ILD jo ennestään. 280 vuorokauden mediaaniseurannan kohdalla toipumista ei raportoitu 30,8 %:lla potilaista, joilla arvioitiin olleen ILD/pneumoniitti (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Tutkija raportoi ILD:tä, pneumoniittia, organisoituvaa pneumoniaa ja akuuttia interstitiaalista pneumoniittia eri kasvaintyypeillä tehdyissä kliinisissä tutkimuksissa 16,9 %:lla potilaista (n = 1 133), joita hoidettiin Enhertu-annoksella 6,4 mg/kg. ILD/pneumoniitti vahvistettiin arvioinnissa 15,4 %:lla potilaista, ja se johti lääkityksen lopettamiseen 10,1 %:lla potilaista ja lääkkeen annon keskeyttämiseen 4,7 %:lla potilaista. Useimmat ILD-/pneumoniittitapaukset olivat asteen 1 (4,1 %) ja asteen 2 (8,6 %) tapahtumia. Asteen 3 tapahtumia ilmeni 1,1 %:lla, ja asteen 4 tapahtumia ilmeni yksi tapaus. Asteen 5 (kuolemaan johtavia) tapahtumia ilmeni 1,6 %:lla potilaista. Mediaaniaika ensimmäiseen esiintymään oli 4,1 kuukautta (vaihteluväli: -0,5–21,0), mukaan lukien kaksi potilasta, joilla arvioitiin olleen ILD jo ennestään. 251 vuorokauden (mediaani) seurannan kohdalla toipumista ei raportoitu 37,4 %:lla potilaista, joilla arvioitiin olleen ILD/pneumoniitti (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Neutropenia
Neutropeniaa raportoitiin 35,1 %:lla potilaista (n = 2335), joita hoidettiin Enhertu-annoksella 5,4 mg/kg eri kasvaintyypeillä tehdyissä kliinisissä tutkimuksissa. 18,0 % tapauksista oli asteen 3 tai 4 tapahtumia. Mediaaniaika ensimmäiseen ilmenemiseen oli 42 vuorokautta (vaihteluväli: 1 vuorokausi – 31,9 kuukautta), ja ensimmäisen tapahtuman mediaanikesto oli 21 vuorokautta (vaihteluväli: 1 vuorokausi – 17,1 kuukautta). Kuumeista neutropeniaa raportoitiin 1,0 %:lla potilaista, ja < 0,1 % tapauksista oli asteen 5 tapahtumia (ks. kohta Annostus ja antotapa).
Neutropeniaa raportoitiin 45,9 %:lla potilaista (n = 1 133), joita hoidettiin Enhertu-annoksella 6,4 mg/kg eri kasvaintyypeillä tehdyissä kliinisissä tutkimuksissa. 28,4 % tapauksista oli asteen 3 tai 4 tapahtumia. Mediaaniaika ensimmäiseen ilmenemiseen oli 16 vuorokautta (vaihteluväli: 1 vuorokausi – 24,8 kuukautta), ja ensimmäisen tapahtuman mediaanikesto oli 9 vuorokautta (vaihteluväli: 1 vuorokausi – 17,2 kuukautta). Kuumeista neutropeniaa raportoitiin 2,6 %:lla potilaista, ja tapauksista 0,1 % oli asteen 5 tapahtumia (ks. kohta Annostus ja antotapa).
Vasemman kammion toimintahäiriö
LVEF:n pienentymistä raportoitiin eri kasvaintyypeillä tehdyissä kliinisissä tutkimuksissa Enhertu-annoksella 5,4 mg/kg 108 potilaalla (4,6 %; n = 2335), joista 14:llä (0,6 %) oli asteen 1 tapahtuma, 80:llä (3,4 %) asteen 2 tapahtuma, 13:lla (0,6 %) asteen 3 tapahtuma ja yhdellä (< 0,1 %) asteen 4 tapahtuma. Laboratorioparametreihin (sydämen kaikukuvaus tai MUGA-tutkimus) perustuva LVEF:n pienentymisen esiintyvyys oli 296/2075 (14,3 %) asteen 2 osalta ja 15/2075 (0,7 %) asteen 3 osalta. Enhertu-hoitoa ei ole tutkittu potilailla, joiden LVEF oli alle 50 % ennen hoidon aloittamista (ks. kohta Annostus ja antotapa).
Vasemman kammion toimintahäiriö johti hoidon keskeytykseen 27 potilaalla 2335:stä (1,2 %). Mediaaniaika vaikea-asteisimmin alentuneeseen LVEF:ään oli 4,8 kuukautta, ja mediaaniaika vaikea-asteisimmin alentuneesta LVEF:stä toipumiseen (≥ 90 % lähtötasosta) oli 6,3 kuukautta.
Enhertu-annoksella 6,4 mg/kg LVEF:n pienentymistä raportoitiin eri kasvaintyypeillä tehdyissä kliinisissä tutkimuksissa 23 potilaalla (2,0 %; n = 1 133), joista 1:llä (0,1 %) oli asteen 1 tapahtuma, 16:lla (1,4 %) asteen 2 tapahtuma, ja 6:lla (0,5 %) asteen 3 tapahtuma. Laboratorioparametreihin (kaikukardiografia tai MUGA-skannaus) perustuva LVEF:n pienentymisen esiintyvyys oli 114/953 (12,0 %) asteen 2 osalta ja 11/953 (1,2 %) asteen 3 osalta.
Vasemman kammion toimintahäiriö johti hoidon keskeytykseen 6 potilaalla 1 133:sta (0,5 %). Mediaaniaika vaikea-asteisimmin alentuneeseen LVEF:ään oli 5,5 kuukautta, ja mediaaniaika vaikea-asteisimmin alentuneesta LVEF:stä toipumiseen (≥ 90 % lähtötasosta) oli 2,8 kuukautta.
Infuusioon liittyvät reaktiot
Potilaista, jotka saivat Enhertu-hoitoa annoksella 5,4 mg/kg eri kasvaintyypeillä tehdyissä kliinisissä tutkimuksissa (n = 2335), infuusioon liittyviä reaktioita raportoitiin 25 potilaalla (1,1 %). Suurin osa reaktioista oli vaikeusasteeltaan 1 tai 2. Viisi infuusioon liittyvää reaktiota (0,2 %) johti lääkkeen annon keskeytykseen, ja 1 tapahtuma (< 0,1 %) johti hoidon lopettamiseen.
Potilaista, jotka saivat Enhertu-hoitoa annoksella 6,4 mg/kg eri kasvaintyypeillä tehdyissä kliinisissä tutkimuksissa (n = 1 133), infuusioon liittyviä reaktioita raportoitiin 7 potilaalla (0,6 %). Kaikki reaktiot olivat vaikeusasteeltaan 1 tai 2. Asteen 3 reaktioita ei raportoitu. Yksi infuusioon liittyvä reaktio (0,1 %) johti lääkkeen annon keskeytykseen. Hoidon lopettamiseen johtaneita tapahtumia ei esiintynyt.
Immunogeenisuus
Kuten kaikkien terapeuttisten proteiinien kohdalla, immunogeenisuuden mahdollisuus on olemassa. Kliinisissä tutkimuksissa arvioituja annoksia 5,4 mg/kg ja 6,4 mg/kg käytettäessä 2,2 %:lle (70/3124) arviointikelpoisista potilaista kehittyi vasta-aineita trastutsumabi-derukstekaanille Enhertu-hoidon jälkeen. Hoidon aikana ilmaantuneiden neutraloivien trastutsumabi-derukstekaanin vasta-aineiden ilmaantuvuus oli 0,1 % (3/3124). Vasta-aineiden kehittymisellä ei ollut ilmeistä vaikutusta Enhertu-valmisteen farmakokinetiikkaan, turvallisuuteen ja/tai tehoon.
Pediatriset potilaat
Turvallisuutta tälle potilasryhmälle ei ole määritetty.
Iäkkäät
Eri kasvaintyypeillä tehdyissä kliinisissä tutkimuksissa (Enhertu-annos 5,4 mg/kg) potilaista (n = 2335) 28,9 % oli vähintään 65-vuotiaita ja 6,3 % vähintään 75-vuotiaita. Vähintään 65-vuotiailla potilailla esiintyi enemmän asteen 3–4 haittavaikutuksia (48,4 %) kuin alle 65-vuotiailla potilailla (43,2 %) ja siten enemmän haittavaikutuksista johtuvaa hoidon lopettamista. Kuolemaan johtaneiden haittavaikutusten ilmaantuvuus oli vähintään 65-vuotiailla potilailla 2,4 % ja alle 65-vuotiailla potilailla 1 %.
Enhertu-hoitoa annoksella 6,4 mg/kg saaneista 1 133:sta eri kasvaintyypeillä tehtyjen kliinisten tutkimusten potilaasta 39,6 % oli vähintään 65-vuotiaita ja 7,9 % vähintään 75-vuotiaita. Asteen 3–4 haittavaikutusten ilmaantuvuus vähintään 65-vuotiailla potilailla oli 60,8 % ja nuoremmilla potilailla 61,1 %. Vähintään 75-vuotiailla potilailla esiintyi enemmän asteen 3–4 haittavaikutuksia (64,4 %) kuin alle 75-vuotiailla potilailla (60,7 %). Vähintään 75-vuotiailla potilailla esiintyi enemmän vakavia haittavaikutuksia (34,4 %) ja kuolemaan johtaneita tapahtumia (4,4 %) kuin alle 75-vuotiailla potilailla (21,2 % ja 1,6 %). Tietoja, joiden avulla voidaan arvioida turvallisuutta vähintään 75-vuotiaille potilaille, on niukasti.
Etniset erot
Kliinisissä tutkimuksissa ei havaittu oleellisia eroja altistuksessa ja tehossa eri etnisten potilasryhmien välillä. Enhertu-hoitoa 6,4 mg/kg:n annoksella saaneilla aasialaisilla potilailla esiintyi ei-aasialaisiin potilaisiin verrattuna enemmän (≥ 10 %:n ero) neutropeniaa (58,3 % vs. 29,4 %), anemiaa (55,2 % vs. 38,3 %), leukopeniaa (46,7 % vs. 10,5 %) ja trombosytopeniaa (43,1 % vs. 19,3 %). Aasialaisista potilaista 3,4 %:lla esiintyi verenvuototapahtuma 14 vuorokauden kuluessa trombosytopenian alkamisesta verrattuna 0,8 %:iin ei-aasialaisista potilaista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Suurinta siedettyä trastutsumabi-derukstekaaniannosta ei ole määritetty. Kliinisissä tutkimuksissa suurin kokeiltu kerta-annos oli 8,0 mg/kg. Yliannostustapauksissa potilaita on seurattava tarkasti haittavaikutusten merkkien tai oireiden varalta, ja heille on annettava oireenmukaista hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, ihmisen epidermaalisen kasvutekijän reseptori 2:n (HER2) estäjät, ATC-koodi: L01FD04
Vaikutusmekanismi
Enhertu, trastutsumabi-derukstekaani, on HER2-reseptoriin kohdistuva vasta-aine-lääkekonjugaatti. Vasta-aine on humanisoitu anti-HER2 IgG1, joka on liitetty topoisomeraasi I:n estäjä derukstekaaniin (DXd) tetrapeptidipohjaisella pilkottavalla linkkerillä. Vasta-aine-lääkekonjugaatti on stabiili plasmassa. Vasta-aineosan tehtävänä on sitoutua tiettyjen kasvainsolujen pinnalla ilmentyvään HER2-reseptoriin. Sitoutumisen jälkeen trastutsumabi-derukstekaanikompleksi kulkeutuu solun sisälle, jossa se käy läpi linkkerin pilkkomisen syöpäsoluissa yli-ilmentyvien lysosomaalisten entsyymien toimesta. Vapautunut DXd on solukalvot läpäisevä ja aiheuttaa DNA-vaurioita ja apoptoottisen solukuoleman. DXd, eksatekaanijohdannainen, on noin 10 kertaa voimakkaampi kuin SN-38, irinotekaanin aktiivinen metaboliitti.
In vitro -tutkimukset osoittavat, että trastutsumabi-derukstekaanin vasta-aineosa, jolla on sama aminohapposekvenssi kuin trastutsumabilla, sitoutuu myös FcγRIIIa:han ja komplementti C1q:hun. Vasta-aine välittää vasta-aineesta riippuvaista soluvälitteistä sytotoksisuutta (ADCC) ihmisen rintasyöpäsoluissa, jotka yli-ilmentävät HER2:ta. Lisäksi vasta-aine estää signaalinvälitystä fosfatidyyli-inositoli-3-kinaasi (PI3-K) -reitin kautta ihmisen rintasyöpäsoluissa, jotka yli-ilmentävät HER2:ta.
Kliininen teho
HER2-positiivinen rintasyöpä
DESTINY-Breast03 (NCT03529110)
Enhertu-valmisteen tehoa ja turvallisuutta tutkittiin DESTINY-Breast03-tutkimuksessa, joka oli avoin, aktiivikontrolloitu, satunnaistettu, kaksihaarainen vaiheen 3 monikeskustutkimus. Tutkimukseen otettiin potilaita, joilla oli HER2-positiivinen, leikkaushoitoon soveltumaton tai metastasoitunut rintasyöpä, ja jotka olivat saaneet aikaisemmin trastutsumabi- ja taksaanihoitoa metastasoituneeseen tautiin tai joilla tauti uusiutui adjuvanttihoidon aikana tai 6 kuukauden sisällä sen päättymisestä.
Tutkimukseen osallistuvien potilaiden arkistoiduista rintakasvainnäytteistä oli osoitettava HER2-positiivisuus, jonka määritelmänä oli HER2 IHC 3+ tai ISH-positiivisuus. Tutkimukseen ei otettu potilaita, joilla oli ollut aiemmin steroidihoitoa edellyttävä ILD/pneumoniitti tai joilla oli ILD/pneumoniitti seulontavaiheessa, joilla oli hoitamattomia ja oireellisia aivometastaaseja, joilla oli aikaisemmin ollut kliinisesti merkittävä sydänsairaus, tai joiden metastasoitunutta tautia oli hoidettu aiemmin anti-HER2-vasta-aine-lääkekonjugaatilla. Potilaat satunnaistettiin 1:1 saamaan joko Enhertu-valmistetta 5,4 mg/kg (N = 261) tai trastutsumabiemtansiinia 3,6 mg/kg (N = 263) infuusiona laskimoon kolmen viikon välein. Satunnaistaminen ositettiin hormonireseptoristatuksen, aikaisemman pertutsumabihoidon ja anamneesissa olevan sisäelimiin levinneen taudin mukaisesti. Hoitoa jatkettiin, kunnes tauti eteni, tutkittava kuoli, tutkittava peruutti suostumuksensa tai toksisuutta ei voitu hyväksyä.
Ensisijainen tehon tulosmittari oli etenemisvapaa elinaika (PFS), jonka arvioi sokkoutettu riippumaton keskitetty arvioijataho (BICR) kiinteiden kasvainten hoitovasteen arviointikriteerien (RECIST v1.1) mukaisesti. Kokonaiselossaoloaika (OS) oli tärkeä toissijainen tehon tulosmittari. Muita toissijaisia päätetapahtumia olivat tutkijan arviointiin perustuva PFS, vahvistettu objektiivinen vaste (ORR) ja vasteen kesto (DOR).
Potilaiden demografiset ominaisuudet ja lähtötilanteen sairausominaisuudet olivat tasapainossa hoitohaarojen välillä. Lähtötilanteen väestötiedot ja sairausominaisuudet 524 satunnaistetulla potilaalla olivat: mediaani-ikä 54 vuotta (vaihteluväli: 20–83); vähintään 65-vuotiaita (20,2 %); naisia (99,6 %); aasialaisia (59,9 %), valkoihoisia (27,3 %), mustia tai afrikkalaisamerikkalaisia (3,6 %); Eastern Cooperative Oncology Group (ECOG) -toimintakyky 0 (62,8 %) tai 1 (36,8 %); hormonireseptoristatus (positiivinen: 51,9 %); sisäelimiin levinnyt tauti (73,3 %); olemassa olevat aivometastaasit (lähtötilanteessa 15,6 %); 48,3 % potilaista oli saanut yhden linjan aikaisempaa systeemistä hoitoa metastasoituneeseen tautiin ja 9,5 % potilaista ei ollut aiemmin saanut hoitoa metastasoituneeseen tautiin. 61,1 %:a potilaista oli aikaisemmin hoidettu pertutsumabilla.
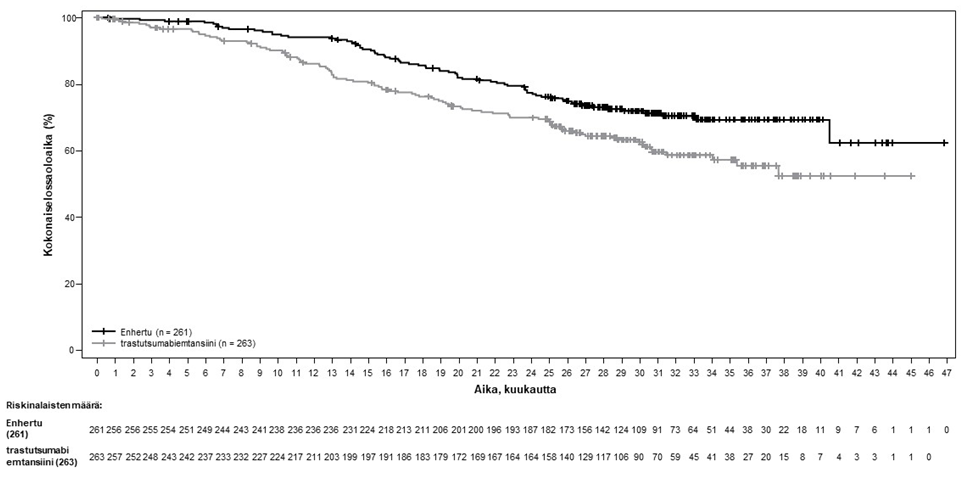
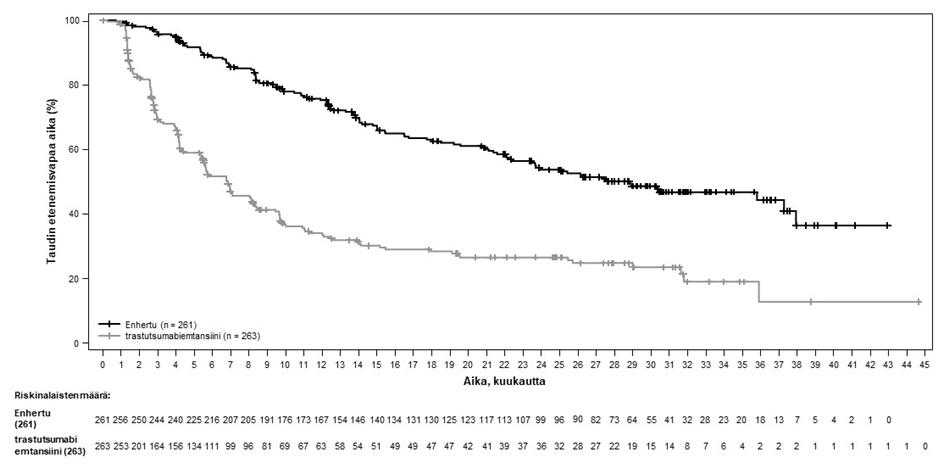
Etenemisvapaata elinaikaa (PFS) koskeva ennalta määritetty välianalyysi perustui 245 tapahtumaan (73 % lopulliseen analyysiin suunniteltujen tapahtumien kokonaismäärästä). Tutkimus osoitti tilastollisesti merkitsevän paranemisen BICR:n arvioiman PFS:n suhteen Enhertu-hoitoon satunnaistetuilla potilailla verrattuna trastutsumabiemtansiinilla hoidettuihin potilaisiin. BICR:n mukaiset PFS-tiedot ensisijaisesta analyysista (tiedonkeruun katkaisu 21. toukokuuta 2021) ja päivitetyt OS-, ORR- ja DOR-tulokset tiedonkeruun katkaisusta 25. heinäkuuta 2022 on esitetty taulukossa 4.
Taulukko 4: DESTINY-Breast03-tutkimuksen tehon tulokset
| Tehon parametri | Enhertu N = 261 | trastutsumabiemtansiini N = 263 |
| Etenemisvapaa elinaika (PFS) BICR:n mukaana | ||
| Tapahtumien lukumäärä (%) | 87 (33,3) | 158 (60,1) |
| Mediaani, kuukautta (95 %:n CI) | ES (18,5, EA) | 6,8 (5,6, 8,2) |
| Hasardisuhde (95 %:n CI) | 0,28 (0,22, 0,37) | |
| p-arvo | p < 0,000001† | |
| Kokonaiselossaoloaika (OS)b | ||
| Tapahtumien lukumäärä (%) | 72 (27,6) | 97 (36,9) |
| Mediaani, kuukautta (95 %:n CI) | ES (40,5, EA) | ES (34,0, EA) |
| Hasardisuhde (95 %:n CI) | 0,64 (0,47, 0,87) | |
| p-arvoc | p = 0,0037 | |
| PFS BICR:n mukaan (päivitetty)b | ||
| Tapahtumien lukumäärä (%) | 117 (44,8) | 171 (65,0) |
| Mediaani, kuukautta (95 %:n CI) | 28,8 (22,4, 37,9) | 6,8 (5,6, 8,2) |
| Hasardisuhde (95 %:n CI) | 0,33 (0,26, 0,43) | |
| Vahvistettu objektiivinen vaste (ORR) BICR:n mukaanb | ||
| n (%) | 205 (78,5) | 92 (35,0) |
| 95 %:n CI | (73,1, 83,4) | (29,2, 41,1) |
| Täydellinen vaste n (%) | 55 (21,1) | 25 (9,5) |
| Osittainen vaste n (%) | 150 (57,5) | 67 (25,5) |
| Vasteen kesto BICR:n mukaanb | ||
| Mediaani, kuukautta (95 %:n CI) | 36,6 (22,4, EA) | 23,8 (12,6, 34,7) |
CI = luottamusväli; EA = ei arvioitavissa; ES = ei saavutettu
† Esitettiin 6 desimaalilla
a Tiedonkeruun katkaisu 21. toukokuuta 2021
b Tiedonkeruun katkaisu 25. heinäkuuta 2022 ennalta suunniteltua kokonaiselossaoloajan väliaika-analyysia varten
c P-arvo perustuu stratifioituun log-rank-testiin; ylitti 0,013:n tehokkuusrajan.
Kuva 1: Kaplan-Meier-kuvaaja kokonaiselossaoloajasta (tiedonkeruun katkaisu 25. heinäkuuta 2022)
Kuva 2: Kaplan-Meier-kuvaaja etenemisvapaasta elinajasta BICR-arvioinnin mukaan (tiedonkeruun katkaisu 25. heinäkuuta 2022)
Samankaltaisia PFS-tuloksia havaittiin ennalta määritetyissä alajoukoissa, mukaan lukien aikaisempi pertutsumabihoito, hormonireseptoristatus ja sisäelimiin levinnyt tauti.
DESTINY-Breast02 (NCT03523585)
Enhertu-valmisteen tehoa ja turvallisuutta arvioitiin DESTINY-Breast02-tutkimuksessa, joka oli vaiheen 3 satunnaistettu, avoin, aktiivikontrolloitu monikeskustutkimus. Tutkimukseen otettiin mukaan potilaita, joilla oli leikkaushoitoon soveltumaton tai metastasoitunut HER2-positiivinen rintasyöpä, ja jotka olivat resistenttejä tai reagoivat huonosti aiempaan T-DM1-hoitoon. Tutkimukseen osallistuvien potilaiden arkistoiduista rintakasvainnäytteistä oli osoitettava HER2-positiivisuus, jonka määritelmänä oli HER2 IHC 3+ tai ISH-positiivisuus. Tutkimukseen ei otettu potilaita, joilla oli ollut aiemmin steroidihoitoa edellyttävä ILD/pneumoniitti tai joilla oli ILD/pneumoniitti seulontavaiheessa. Tutkimukseen ei myöskään otettu mukaan potilaita, joilla oli hoitamattomia tai oireellisia aivometastaaseja, tai joilla oli aikaisemmin ollut kliinisesti merkittävä sydänsairaus.
Potilaat satunnaistettiin 2:1 saamaan joko Enhertu-valmistetta 5,4 mg/kg (n = 406) infuusiona laskimoon kolmen viikon välein tai lääkärin valitsemaa hoitoa (n = 202, trastutsumabi yhdessä kapesitabiinin kanssa tai lapatinibi yhdessä kapesitabiinin kanssa). Satunnaistaminen ositettiin hormonireseptoristatuksen, aikaisemman pertutsumabihoidon ja anamneesissa olevan sisäelimiin levinneen taudin mukaisesti. Hoitoa jatkettiin, kunnes tauti eteni, tutkittava kuoli, tutkittava peruutti suostumuksensa tai toksisuutta ei voitu hyväksyä.
Ensisijainen tehon tulosmittari oli etenemisvapaa elinaika (PFS), jonka arvioi sokkoutettu riippumaton keskitetty arvioijataho (BICR) RECIST v1.1 -kriteerien mukaisesti. Kokonaiselossaoloaika (OS) oli tärkeä toissijainen tehon tulosmittari. Muita toissijaisia päätetapahtumia olivat tutkijan arviointiin perustuva PFS, vahvistettu objektiivinen vaste (ORR) ja vasteen kesto (DOR).
Potilaiden väestötiedot ja taudin lähtötilanteen ominaisuudet olivat samankaltaisia hoitohaarojen välillä. 608 satunnaistetun potilaan mediaani-ikä oli 54 vuotta (vaihteluväli: 22–88 vuotta); naisia (99,2 %); valkoihoisia (63,2 %) aasialaisia (29,3 %), mustaihoisia tai afroamerikkalaisia (2,8 %); Eastern Cooperative Oncology Group (ECOG) -toimintakyky 0 (57,4 %) tai 1 (42,4 %); hormonireseptoristatus (positiivinen: 58,6 %); sisäelimiin levinnyt tauti (78,3 %); lähtötilanteessa olemassa olevat aivometastaasit (18,1 %), ja 4,9 % potilaista oli saanut yhden linjan aikaisempaa systeemistä hoitoa metastasoituneeseen tautiin.
Yhteenveto tehon tuloksista on esitetty taulukossa 5 ja kuvissa 3 ja 4.
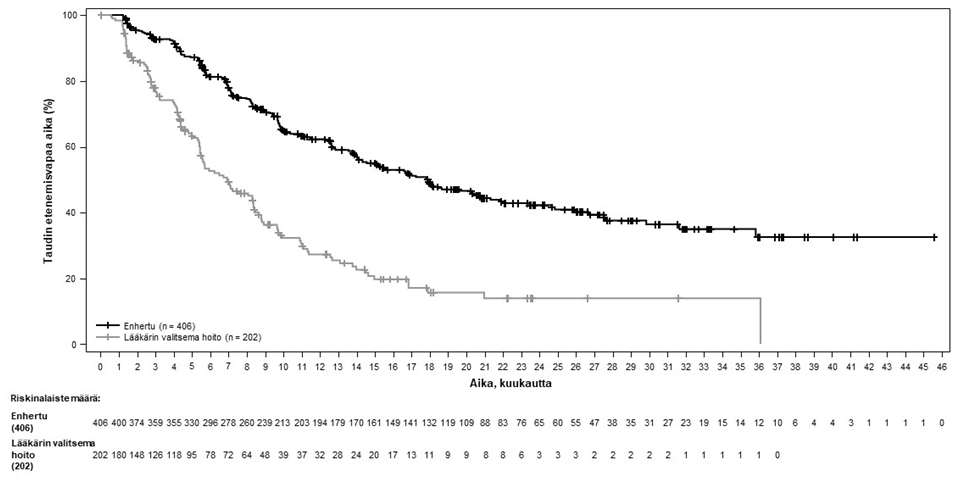
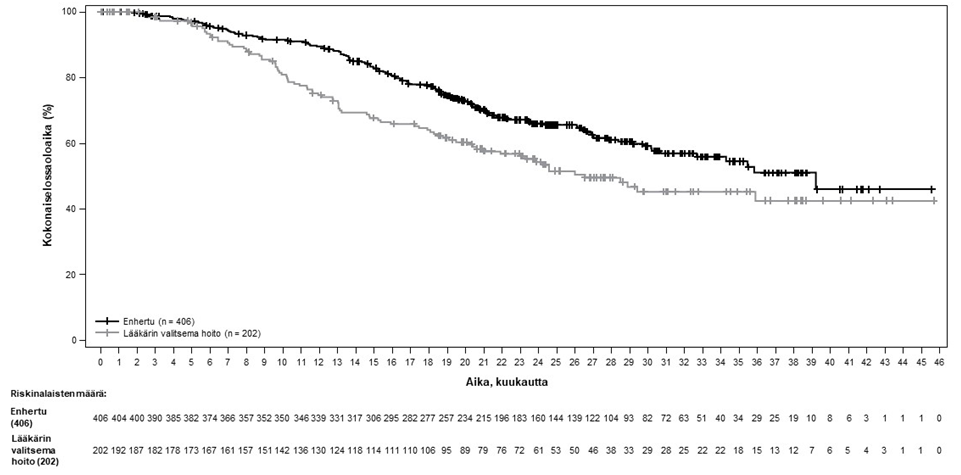
Taulukko 5: DESTINY-Breast02-tutkimuksen tehon tulokset
| Tehon parametri | Enhertu N = 406 | Lääkärin valitsema hoito N = 202 |
|---|---|---|
| PFS BICR:n mukaan | ||
| Tapahtumien lukumäärä (%) | 200 (49,3) | 125 (61,9) |
| Mediaani, kuukautta (95 %:n CI) | 17,8 (14,3, 20,8) | 6,9 (5,5, 8,4) |
| Hasardisuhde (95 %:n CI) | 0,36 (0,28, 0,45) | |
| p-arvo | p < 0,000001† | |
| Kokonaiselossaoloaika (OS) | ||
| Tapahtumien lukumäärä (%) | 143 (35,2) | 86 (42,6) |
| Mediaani, kuukautta (95 %:n CI) | 39,2 (32,7, EA) | 26,5 (21,0, EA) |
| Hasardisuhde (95 %:n CI) | 0,66 (0,50, 0,86) | |
| p-arvoa | p = 0,0021 | |
| PFS tutkijan arvion mukaan | ||
| Tapahtumien lukumäärä (%) | 206 (50,7) | 152 (75,2) |
| Mediaani, kuukautta (95 %:n CI) | 16,7 (14,3, 19,6) | 5,5 (4,4, 7,0) |
| Hasardisuhde (95 %:n CI) | 0,28 (0,23, 0,35) | |
| Vahvistettu objektiivinen vaste (ORR) BICR:n mukaan | ||
| n (%) | 283 (69,7) | 59 (29,2) |
| 95 %:n CI | (65,0, 74,1) | (23,0, 36,0) |
| Täydellinen vaste n (%) | 57 (14,0) | 10 (5,0) |
| Osittainen vaste n (%) | 226 (55,7) | 49 (24,3) |
| Vasteen kesto BICR:n mukaan | ||
| Mediaani, kuukautta (95 %:n CI) | 19,6 (15,9, EA) | 8,3 (5,8, 9,5) |
CI = luottamusväli; EA = ei arvioitavissa
† Esitettiin 6 desimaalilla
a P-arvo perustuu stratifioituun log-rank-testiin: ylitti 0,004:n tehokkuusrajan.
Kuva 3: Kaplan-Meier-kuvaaja etenemisvapaasta elinajasta BICR-arvioinnin mukaan
Kuva 4: Kaplan-Meier-kuvaaja kokonaiselossoloajasta
DESTINY-Breast01 (NCT03248492)
Enhertu-valmisteen tehoa ja turvallisuutta tutkittiin DESTINY-Breast01-tutkimuksessa, joka oli avoin, yksihaarainen, vaiheen 2 monikeskustutkimus. Siihen otettiin potilaita, joilla oli HER2-positiivinen, leikkaushoitoon soveltumaton ja/tai metastasoitunut rintasyöpä ja jotka olivat saaneet vähintään kahta anti-HER2-pohjaista hoitoa, kuten trastutsumabiemtansiinia (100 %), trastutsumabia (100 %) ja pertutsumabia (65,8 %). Arkistoiduista rintakasvainnäytteistä oli todettava HER2-positiivisuus, jonka määritelmänä oli HER2 IHC 3+ tai ISH-positiivisuus. Tutkimukseen ei otettu potilaita, joilla oli aiemmin hoidettu ILD tai joilla oli ILD seulontavaiheessa, joilla oli hoitamattomia tai oireellisia aivometastaaseja tai joilla oli ollut kliinisesti merkittävä sydänsairaus. Tutkimukseen otetuilla potilailla oli ainakin yksi mitattavissa oleva kiinteiden kasvaimien vastearviointikriteerien (RECIST v1.1) mukainen leesio. Enhertu-valmistetta annettiin laskimoinfuusiona 5,4 mg/kg kolmen viikon välein sairauden etenemiseen, kuolemaan, suostumuksen peruuttamiseen tai ei hyväksyttävissä olevan toksisuuden ilmaantumiseen saakka. Ensisijainen tehon tulosmittari oli RECIST v1.1 -kriteerien mukaisesti vahvistettu objektiivinen vasteprosentti (ORR) hoitoaikeen mukaisessa (ITT) ryhmässä riippumattomasti ja keskitetysti arvioituna (ICR). Toissijainen tehon tulosmittari oli vasteen kesto (DOR).
DESTINY-Breast01-tutkimukseen otettujen 184 potilaan lähtötilanteen väestö- ja sairausominaisuudet olivat: mediaani-ikä 55 vuotta (vaihteluväli: 28–96); vähintään 65 vuotta (23,9 %); naiset (100 %); valkoihoiset (54,9 %), aasialaiset (38,0 %), mustat tai afrikkalaisamerikkalaiset (2,2 %); Eastern Cooperative Oncology Group (ECOG) -toimintakyky 0 (55,4 %) tai 1 (44,0 %); hormonireseptoristatus (positiivinen: 52,7 %); sisäelimiin levinnyt tauti (91,8 %); aiemmin hoidetut ja stabiilit aivometastaasit (13,0 %); aiempien hoitojen mediaanimäärä metastaasitapauksissa: 5 (vaihteluväli: 2–17); kohdeleesioiden halkaisijoiden summa (< 5 cm: 42,4 %, ≥ 5 cm: 50,0 %).
Aiempi analyysi (seurannan mediaanikesto 11,1 kuukautta [vaihteluväli: 0,7–19,9 kuukautta]) osoitti, että vahvistettu objektiivinen vasteprosentti oli 60,9 % (95 %:n CI: 53,4; 68,0). Potilaista 6,0 % saavutti täydellisen vasteen ja 54,9 % osittaisen vasteen; 36,4 %:lla oli vakaa tauti, 1,6 %:lla etenevä tauti ja 1,1 % ei ollut arvioitavissa. Tässä vaiheessa vasteen mediaanikesto oli 14,8 kuukautta (95 %:n CI: 13,8; 16,9), ja 81,3 %:lla hoitoon vastanneista potilaista vaste kesti ≥ 6 kuukautta (95 %:n CI: 71,9; 87,8). Tehon tulokset päivitetystä tiedonkeruun katkaisuhetkestä, jolloin seurannan mediaanikesto oli 20,5 kuukautta (vaihteluväli: 0,7–31,4 kuukautta) on esitetty taulukossa 6.
Taulukko 6: DESTINY-Breast01-tutkimuksen tehon tulokset (hoitoaikeen mukainen analyysijoukko)
DESTINY-Breast01 N = 184 | |
| Vahvistettu objektiivinen vasteprosentti (95 %:n CI)*† | 61,4 % (54,0; 68,5) |
| Täydellinen vaste (CR) | 6,5 % |
| Osittainen vaste (PR) | 54,9 % |
| Vasteen kesto‡ | |
| Mediaani, kuukautta (95 %:n CI) | 20,8 (15,0; ES) |
| % joissa vasteen kesto ≥ 6 kuukautta (95 %:n CI) § | 81,5 % (72,2; 88,0) |
ORR 95 %:n CI on laskettu Clopper-Pearsonin menetelmällä
CI = luottamusväli
95 %:n CI:t on laskettu Brookmeyer-Crowleyn menetelmällä
*Vahvistetut vasteet (sokkoutettu, riippumaton, keskitetty arviointi) määriteltiin CR- tai PR-vasteen kirjaukseksi vahvistettuna toistetulla kuvantamisella vähintään 4 viikkoa sen käynnin jälkeen, jolloin vaste ensimmäisen kerran havaittiin.
†184 potilaasta 35,9 %:lla oli vakaa tauti, 1,6 %:lla etenevä tauti ja 1,1 % ei ollut arvioitavissa.
‡Mukaan lukien 73 potilasta, joiden tiedot sensuroitiin
§Perustuu Kaplan-Meierin arviointiin
ES = ei saavutettu
Johdonmukainen antituumoriaktiivisuus havaittiin kaikissa ennalta määritetyissä alaryhmissä, jotka perustuivat aiempaan pertutsumabihoitoon ja hormonireseptoristatukseen.
Heikosti HER2-positiivinen ja erittäin heikosti HER2-positiivinen rintasyöpä
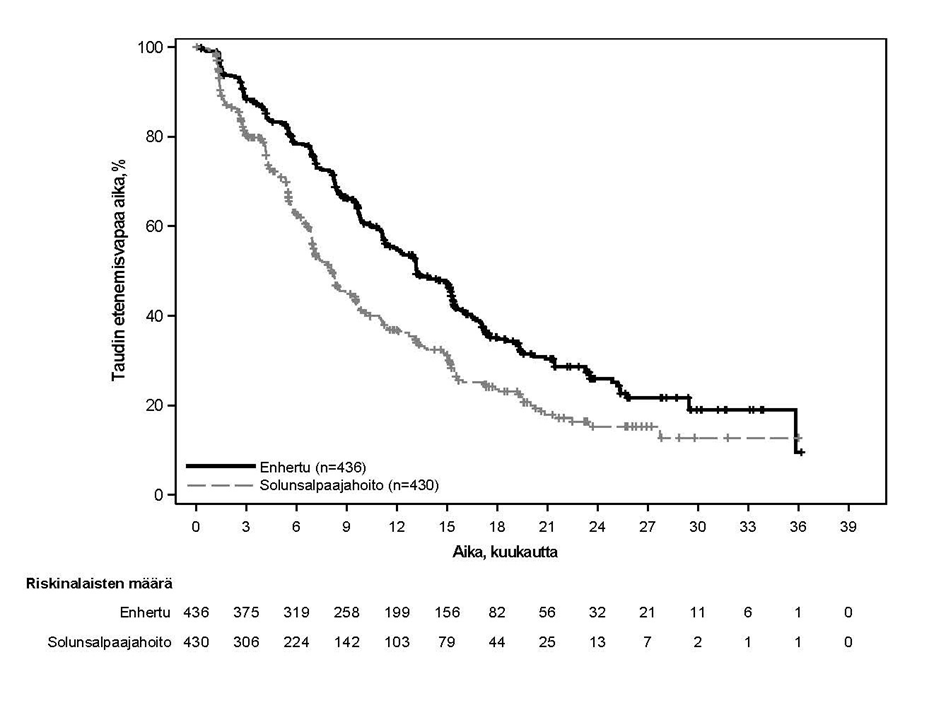
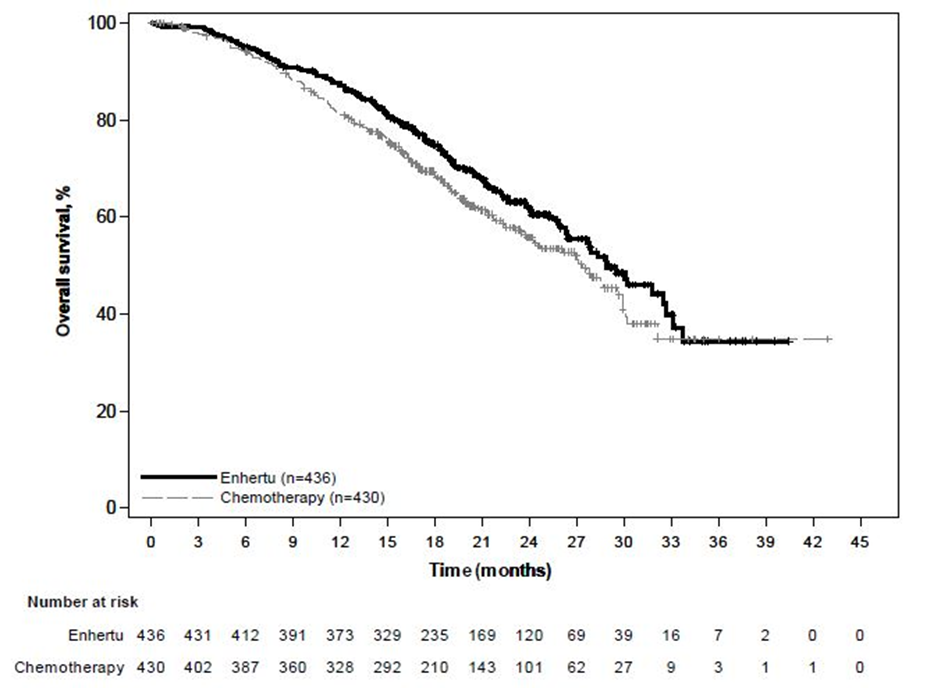
DESTINY-Breast06 (NCT04494425)
Enhertu-valmisteen tehoa ja turvallisuutta tutkittiin DESTINY-Breast06-tutkimuksessa, joka oli vaiheen 3 satunnaistettu, avoin monikeskustutkimus ja johon satunnaistettiin 866 aikuispotilasta, joilla oli pitkälle edennyt tai metastasoitunut HR-positiivinen rintasyöpä, jossa keskuslaboratorion suorittamalla PATHWAY/VENTANA anti-HER-2/neu (4B5) -testillä määritetty HER2:n ilmentymä oli heikko (IHC 1+ tai IHC 2+ / ISH-) tai erittäin heikko. Erittäin heikko HER2-positiivisuus (IHC 0 ja solukalvon värjäytyminen, tutkimuksessa IHC > 0 < 1+) määritellään heikoksi, epätäydelliseksi solukalvon HER2-värjäytymiseksi enintään 10 %:ssa syöpäsoluista. Potilaat soveltuivat mukaan, jos heidän tautinsa oli edennyt (a) vähintään kahden metastasoituneeseen tautiin annetun endokriinisen hoitolinjan jälkeen tai (b) yhden metastasoituneeseen tautiin annetun endokriinisen hoitolinjan jälkeen, ja taudin osoitettiin edenneen 24 kuukauden kuluessa endokriinisen adjuvanttihoidon aloittamisesta tai 6 kuukauden kuluessa metastasoituneeseen tautiin annetun ensilinjan endokriinisen hoidon ja CDK4/6-estäjän yhdistelmän aloittamisesta. Aiempaa neoadjuvantti- tai adjuvanttisolunsalpaajahoitoa saaneet potilaat soveltuivat tutkimukseen mukaan, jos tauditon jakso oli kestänyt yli 12 kuukautta. Tutkimukseen ei otettu potilaita, jotka olivat saaneet aiemmin solunsalpaajahoitoa pitkälle edenneeseen tai metastasoituneeseen tautiin tai joilla oli ollut aiemmin steroidihoitoa vaativa ILD/pneumoniitti tai joilla oli seulontavaiheessa ILD/pneumoniitti tai huonossa hoitotasapainossa oleva tai merkittävä sydän- ja verisuonisairaus, hoitamattomia ja oireellisia aivometastaaseja tai joiden ECOG-toimintakyky oli > 1.
Potilaat satunnaistettiin 1:1 saamaan joko Enhertu-valmistetta 5,4 mg/kg (N = 436) infuusiona laskimoon kolmen viikon välein tai lääkärin valitsemaa yhtä solunsalpaajaa (N = 430, kapesitabiini 60 %, nab-paklitakseli 24 % tai paklitakseli 16 %). Satunnaistaminen ositettiin seuraavasti: aiempi CDK4/6-estäjähoito (kyllä tai ei), aiempi taksaanihoito ei-metastasoituneeseen tautiin (kyllä tai ei) ja kasvainnäytteiden HER2 IHC -status (IHC 2+ / ISH-, IHC 1+, IHC > 0 < 1+). Enhertu-hoitoa jatkettiin, kunnes tauti eteni, tutkittava kuoli, tutkittava peruutti suostumuksensa tai toksisuutta ei voitu hyväksyä.
Ensisijainen tehon päätetapahtuma oli heikosti HER2-positiivista rintasyöpää sairastavien potilaiden etenemisvapaa elinaika (PFS), jonka arvioi sokkoutettu riippumaton keskitetty arvioijataho (BICR) RECIST v1.1 -kriteerien mukaisesti. Tärkeitä toissijaisia tehon päätetapahtumia olivat BICR:n RECIST v1.1 -kriteerien mukaisesti arvioima PFS kokonaispopulaatiossa (heikosti HER2-positiivinen ja erittäin heikosti HER2-positiivinen), kokonaiselossaoloaika (OS) heikosti HER2-positiivisilla potilailla ja OS kokonaispopulaatiossa. Muita toissijaisia päätetapahtumia olivat vahvistettu objektiivinen vaste (ORR) ja vasteen kesto (DOR).
Kokonaispopulaatiossa potilaiden demografiset ominaisuudet ja lähtötilanteen kasvaimen ominaisuudet olivat samankaltaisia hoitohaarojen välillä. Satunnaistettujen 866 potilaan mediaani-ikä oli 57 vuotta (vaihteluväli: 28–87), ja potilaista 31 % oli vähintään 65-vuotiaita, 99,9 % naisia, 53 % valkoihoisia, 35 % aasialaisia ja 1 % mustaihoisia tai afrikkalaisamerikkalaisia. Potilaiden lähtötilanteen ECOG-toimintakyky oli 0 (59 %) tai 1 (39 %), 18 %:lla potilaista HER2-tulos oli IHC > 0 < 1+, 55 %:lla potilaista IHC 1+ ja 27 %:lla potilaista IHC 2+ / ISH-; 67 %:lla potilaista oli maksametastaaseja, 32 %:lla keuhkometastaaseja, 8 %:lla aivometastaaseja ja 3 %:lla ainoastaan luustometastaaseja. Metastasoituneeseen tautiin hoitoa saaneiden potilaiden aiempien endokriinisten hoitolinjojen mediaani oli 2 (vaihteluväli: 1–5), ja 17 % potilaista oli saanut yhtä ja 68 % potilaista kahta aiempaa endokriinista hoitoa. Potilaista 89 % oli saanut aiemmin endokriinista hoitoa yhdistelmänä CDK4/6-estäjän kanssa metastasoituneeseen tautiin, 47 % oli saanut aiemmin antrasykliinejä ja 41 % oli saanut aiemmin taksaaneja ei-metastasoituneeseen tautiin.
Yhteenveto tehon tuloksista on esitetty taulukossa 7 sekä kuvissa 5 ja 6.
Taulukko 7: DESTINY-Breast06-tutkimuksen tehon tulokset
| Tehon parametri | Heikosti HER2-positiivinen | Kokonaispopulaatio (heikosti HER2-positiivinen ja erittäin heikosti HER2-positiivinen) | ||
| Enhertu (N = 359) | Solunsalpaajahoito (N = 354) | Enhertu (N = 436) | Solunsalpaajahoito (N = 430) | |
| Etenemisvapaa elinaika BICR:n mukaan | ||||
| Tapahtumien lukumärä (%) | 225 (62,7) | 232 (65,5) | 269 (61,7) | 271 (63,0) |
| Mediaani, kuukautta (95 %:n CI) | 13,2 (11,4, 15,2) | 8,1 (7,0, 9,0) | 13,2 (12,0, 15,2) | 8,1 (7,0, 9,0) |
| Hasardisuhde (95 %:n CI) | 0,62 (0,52, 0,75) | 0,64 (0,54, 0,76) | ||
| p-arvo | < 0,0001 | < 0,0001 | ||
| Kokonaiselossaoloaika* | ||||
| Tapahtumien lukumäärä (%) | 136 (37,9) | 146 (41,2) | 161 (36,9) | 174 (40,5) |
| Mediaani, kuukautta (95 %:n CI) | 28,9 (25,7, 33,7) | 27,1 (23,5, 29,9) | 28,9 (26,4, 32,7) | 27,4 (23,9, 29,9) |
| Hasardisuhde (95 %:n CI) | 0,83 (0,66, 1,05) | 0,81 (0,66, 1,01) | ||
| Vahvistettu objektiivinen vaste BICR:n mukaan† | ||||
| n (%) | 203 (56,5) | 114 (32,2) | 250 (57,3) | 134 (31,2) |
| 95 %:n CI | 51,2, 61,7 | 27,4, 37,3 | 52,5, 62,0 | 26,8, 35,8 |
| Vasteen kesto BICR:n mukaan† | ||||
| Mediaani, kuukautta (95 %:n CI) | 14,1 (11,8, 15,9) | 8,6 (6,7, 11,3) | 14,3 (12,5, 15,9) | 8,6 (6,9, 11,5) |
Tiedonkeruun katkaisupäivä: 18. maaliskuuta 2024
CI = luottamusväli
*Ensimmäinen suunniteltu välianalyysi
†Tuloksia ei kontrolloitu tyypin 1 virheen suhteen, joten ne on tulkittava kuvaileviksi
Etenemisvapaan elinajan (PFS) suhteen havaittiin johdonmukaista hyötyä useissa ennalta määritetyissä alaryhmissä, mukaan lukien HER2:n ilmentyminen (IHC > 0 < 1+, IHC 1+, IHC 2+ / ISH-), aiempi CDK4/6-estäjähoito (kyllä tai ei), aiempi taksaanien käyttö ei-metastasoituneeseen tautiin (kyllä tai ei) ja aiempien metastasoituneeseen tautiin saatujen endokriinisten hoitolinjojen lukumäärä.
Erittäin heikosti HER2-positiivisten potilaiden alaryhmässä (n = 152) PFS:n mediaani oli 13,2 kuukautta (95 %:n luottamusväli: 9,8, 17,3) Enhertu-hoitoon satunnaistetuilla potilailla (n = 76) ja 8,3 kuukautta (95 %:n luottamusväli: 5,8, 15,2) solunsalpaajaan satunnaistetuilla potilailla; hasardisuhde 0,78 (95 %:n luottamusväli: 0,50, 1,21). OS:n mediaani oli 29,5 kuukautta (95 %:n luottamusväli: 27,9, ei arvioitavissa) Enhertu-hoitoon satunnaistetuilla potilailla ja 27,4 kuukautta (95 %:n luottamusväli: 19,4, ei arvioitavissa) solunsalpaajaan satunnaistetuilla potilailla; hasardisuhde 0,75 (95 %:n luottamusväli: 0,43, 1,29). Vahvistettu objektiivinen vasteprosentti oli 61,8 % (95 %:n luottamusväli: 50,0, 72,8) Enhertu-hoitoon satunnaistetuilla potilailla ja 26,3 % (95 %:n luottamusväli: 16,9, 37,7) solunsalpaajaan satunnaistetuilla potilailla. Vasteen mediaanikesto oli 14,3 kuukautta (95 %:n luottamusväli: 9,2, 20,7) Enhertu-hoitoon satunnaistetuilla potilailla ja 14,1 kuukautta (95 %:n luottamusväli: 5,9, ei arvioitavissa) solunsalpaajaan satunnaistetuilla potilailla.
Kuva 5: Kaplan-Meier-kuvaaja etenemisvapaasta elinajasta (kokonaispopulaatio)
Kuva 6: Kaplan-Meier-kuvaaja kokonaiselossaoloajasta (kokonaispopulaatio)
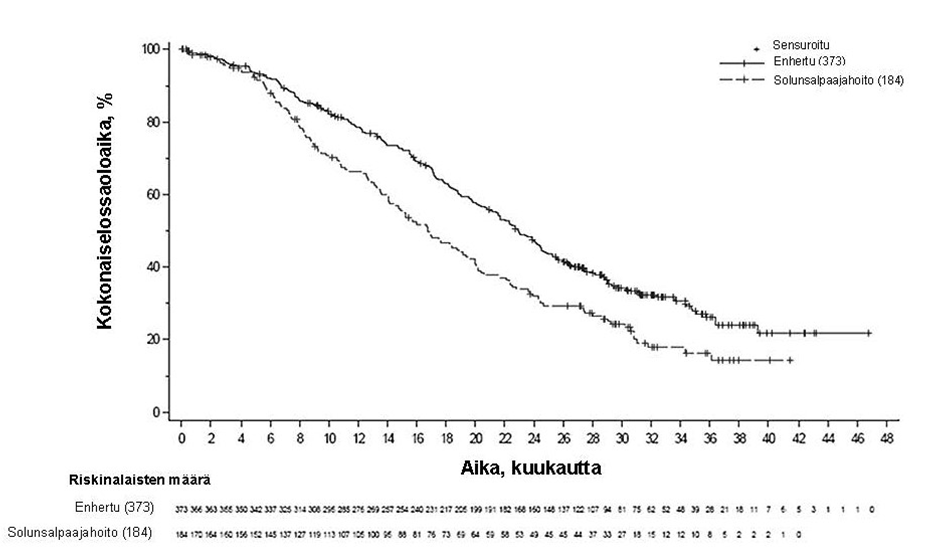
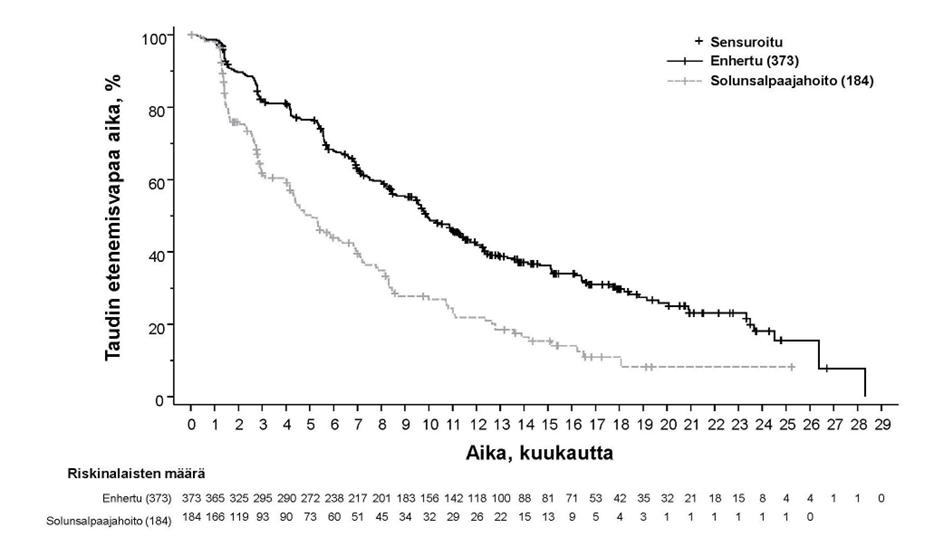
DESTINY-Breast04 (NCT03734029)
Enhertu-valmisteen tehoa ja turvallisuutta tutkittiin DESTINY-Breast04-tutkimuksessa, joka oli vaiheen 3, satunnaistettu, avoin, monikeskustutkimus ja johon otettiin 557 aikuispotilasta, joilla oli leikkaushoitoon soveltumaton tai metastasoitunut heikosti HER2-positiivinen-rintasyöpä. Tutkimuksessa oli 2 kohorttia, joista toinen käsitti 494 hormonireseptoripositiivista (HR+) potilasta ja toinen 63 hormonireseptorinegatiivista (HR-) potilasta. Heikon HER2-positiivisuuden määritelmänä oli IHC 1+ (joka määriteltiin heikoksi, osittaiseksi solukalvon värjäytymiseksi yli 10 %:ssa syöpäsoluista) tai IHC 2+ / ISH- keskuslaboratorion suorittamassa PATHWAY/VENTANA anti-HER-2/neu (4B5) -testissä. Mukaan otettavien potilaiden oli pitänyt saada solunsalpaajaa metastasoituneeseen tautiin tai heidän tautinsa oli pitänyt uusiutua adjuvanttisolunsalpaajahoidon aikana tai 6 kuukauden sisällä sen päättymisestä. Lisäksi osallistumiskriteerien mukaan HR+-potilaiden oli pitänyt saada vähintään yhtä hormonihoitoa ja heidän piti satunnaistamisen yhteydessä olla soveltumattomia lisähormonihoitoihin. Potilaat satunnaistettiin 2:1 saamaan joko Enhertu-valmistetta 5,4 mg/kg (N = 373) infuusiona laskimoon kolmen viikon välein tai lääkärin valitsemaa solunsalpaajaa (N = 184, eribuliini 51,1 %, kapesitabiini 20,1 %, gemsitabiini 10,3 %, nab-paklitakseli 10,3 %, paklitakseli 8,2 %). Satunnaistaminen ositettiin seuraavasti: kasvainnäytteiden HER2 IHC -status (IHC 1+ tai IHC 2+ / ISH-), metastasoituneeseen tautiin annettujen solunsalpaajahoitolinjojen lukumäärä (1 tai 2) sekä HR-status ja aiempi CDK4/6-estäjähoito (HR+ ja aiempi CDK4/6-estäjähoito, HR+ ilman aiempaa CDK4/6-estäjähoitoa tai HR-). Hoitoa jatkettiin, kunnes tauti eteni, tutkittava kuoli, tutkittava peruutti suostumuksensa tai toksisuutta ei voitu hyväksyä. Tutkimukseen ei otettu potilaita, joilla oli ollut aiemmin steroidihoitoa vaativa ILD/pneumoniitti tai joilla oli seulontavaiheessa ILD/pneumoniitti tai kliinisesti merkittävä sydänsairaus. Tutkimukseen ei myöskään otettu potilaita, joilla oli hoitamattomia tai oireellisia aivometastaaseja tai joiden ECOG-toimintakyky oli > 1.
Ensisijainen tehon päätetapahtuma oli HR-positiivista rintasyöpää sairastavien potilaiden etenemisvapaa elinaika (PFS), jonka arvioi sokkoutettu riippumaton keskitetty arvioijataho (BICR) RECIST v1.1 -kriteerien mukaisesti. Tärkeitä toissijaisia tehon päätetapahtumia olivat BICR:n RECIST v1.1 -kriteerien mukaisesti arvioima PFS kokonaispopulaatiossa (kaikki satunnaistetut HR-positiiviset ja HR-negatiiviset potilaat), kokonaiselossaoloaika (OS) HR-positiivisilla potilailla ja OS kokonaispopulaatiossa. Muita toissijaisia päätetapahtumia olivat vahvistettu objektiivinen vaste (ORR), vasteen kesto (DOR) ja potilaiden raportoimat tulokset (PRO).
Potilaiden demografiset ominaisuudet ja lähtötilanteen kasvaimen ominaisuudet olivat samankaltaisia hoitohaarojen välillä. Satunnaistettujen 557 potilaan mediaani-ikä oli 57 vuotta (vaihteluväli: 28–81), ja potilaista 23,5 % oli vähintään 65-vuotiaita, 99,6 % naisia ja 0,4 % miehiä, 47,9 % valkoihoisia, 40,0 % aasialaisia ja 1,8 % mustaihoisia tai afrikkalaisamerikkalaisia. Potilaiden lähtötilanteen ECOG-toimintakyky oli 0 (54,8 %) tai 1 (45,2 %). 57,6 %:lla potilaista HER2-tulos oli IHC 1+ ja 42,4 %:lla potilaista IHC 2+ / ISH-, 88,7 % potilaista oli HR-positiivisia ja 11,3 % HR-negatiivisia, ja 69,8 %:lla potilaista oli maksametastaaseja, 32,9 %:lla keuhkometastaaseja ja 5,7 %:lla aivometastaaseja. Potilaista 46,3 % oli aiemmin saanut antrasykliiniä (neo)adjuvanttihoitona ja 19,4 % oli aiemmin saanut antrasykliiniä paikallisesti edenneeseen ja/tai metastasoituneeseen tautiin. Metastasoituneeseen tautiin hoitoa saaneiden potilaiden aiempien systeemisten hoitolinjojen mediaani oli 3 (vaihteluväli: 1–9), ja 57,6 % potilaista oli saanut 1:tä ja 40,9 % potilaista 2:ta aiempaa solunsalpaajahoitoa. Tauti oli edennyt varhain (neoadjuvantti- tai adjuvanttihoidon aikana) 3,9 %:lla potilaista. HR-positiivisten potilaiden aiempien hormonihoitolinjojen mediaani oli 2 (vaihteluväli: 0–9), ja 70 % potilaista oli saanut aiemmin CDK4/6-estäjähoitoa.
Yhteenveto tehon tuloksista on esitetty taulukossa 8 sekä kuvissa 7 ja 8.
Taulukko 8: DESTINY-Breast04-tutkimuksen tehon tulokset
| Tehon parametri | HR-positiivisten potilaiden kohortti | Kokonaispopulaatio (HR-positiivisten ja HR-negatiivisten potilaiden kohortit) | ||
Enhertu (N = 331) | Solunsalpaajahoito (N = 163) | Enhertu (N = 373) | Solunsalpaajahoito (N = 184) | |
| Kokonaiselossaoloaika | ||||
| Tapahtumien lukumäärä (%) | 126 (38,1) | 73 (44,8) | 149 (39,9) | 90 (48,9) |
| Mediaani, kuukautta (95 %:n CI) | 23,9 (20,8, 24,8) | 17,5 (15,2, 22,4) | 23,4 (20,0, 24,8) | 16,8 (14,5, 20,0) |
| Hasardisuhde (95 %:n CI) | 0,64 (0,48, 0,86) | 0,64 (0,49, 0,84) | ||
| p-arvo | 0,0028 | 0,001 | ||
| Etenemisvapaa elinaika BICR:n mukaan | ||||
| Tapahtumien lukumäärä (%) | 211 (63,7) | 110 (67,5) | 243 (65,1) | 127 (69,0) |
| Mediaani, kuukautta (95 %:n CI) | 10,1 (9,5, 11,5) | 5,4 (4,4, 7,1) | 9,9 (9,0, 11,3) | 5,1 (4,2, 6,8) |
| Hasardisuhde (95 %:n CI) | 0,51 (0,40, 0,64) | 0,50 (0,40, 0,63) | ||
| p-arvo | < 0,0001 | < 0,0001 | ||
| Vahvistettu objektiivinen vaste BICR:n mukaan* | ||||
| n (%) | 175 (52,6) | 27 (16,3) | 195 (52,3) | 30 (16,3) |
| 95 %:n CI | 47,0, 58,0 | 11,0, 22,8 | 47,1, 57,4 | 11,3, 22,5 |
| Täydellinen vaste n (%) | 12 (3,6) | 1 (0,6) | 13 (3,5) | 2 (1,1) |
| Osittainen vaste n (%) | 164 (49,2) | 26 (15,7) | 183 (49,1) | 28 (15,2) |
| Vasteen kesto BICR:n mukaan* | ||||
| Mediaani, kuukautta (95 %:n CI) | 10,7 (8,5, 13,7) | 6,8 (6,5, 9,9) | 10,7 (8,5, 13,2) | 6,8 (6,0, 9,9) |
CI = luottamusväli
*Perustuu sähköisestä tapauskertomuslomakkeesta saatuihin HR-positiivisia potilaita koskeviin tietoihin (Enhertu-ryhmä N = 333 ja solunsalpaajahoitoryhmä N = 166).
Kokonaiselossaoloajan (OS) ja etenemisvapaan elinajan (PFS) suhteen havaittiin johdonmukaista hyötyä kaikissa ennalta määritetyissä alaryhmissä, mukaan lukien hormonireseptoristatus, aiempi CDK4/6-estäjähoito, aiempien solunsalpaajahoitojen lukumäärä ja HER2 IHC -status (IHC 1+ ja IHC 2+ / ISH-). HR-negatiivisten potilaiden alaryhmässä OS:n mediaani oli 18,2 kuukautta (95 %:n luottamusväli: 13,6, ei arvioitavissa) Enhertu-hoitoon satunnaistetuilla potilailla verrattuna 8,3 kuukauteen (95 %:n luottamusväli: 5,6, 20,6) solunsalpaajaan satunnaistetuilla potilailla; hasardisuhde 0,48 (95 %:n luottamusväli: 0,24, 0,95). PFS:n mediaani oli 8,5 kuukautta (95 %:n luottamusväli: 4,3, 11,7) Enhertu-hoitoon satunnaistetuilla potilailla ja 2,9 kuukautta (95 %:n luottamusväli: 1,4, 5,1) solunsalpaajaan satunnaistetuilla potilailla; hasardisuhde 0,46 (95 %:n luottamusväli: 0,24, 0,89).
Päivitetyssä kuvailevassa analyysissa, joka käsitti 32 kuukauden pituisen mediaaniseuranta-ajan, OS:n parannukset olivat yhdenmukaisia ensisijaisen analyysin kanssa. Hasardisuhde kokonaispopulaatiossa oli 0,69 (95 %:n luottamusväli: 0,55, 0,86), ja OS:n mediaani oli 22,9 kuukautta Enhertu-ryhmässä (95 %:n luottamusväli: 21,2, 24,5) vs. 16,8 kuukautta solunsalpaajahoitoryhmässä (95 %:n luottamusväli: 14,1, 19,5). Kaplan-Meierin käyrä päivitetylle OS-analyysille on esitetty kuvassa 7.
Kuva 7: Kaplan-Meier-kuvaaja kokonaiselossaoloajasta (kokonaispopulaatio) (päivitetty analyysi))
Kuva 8: Kaplan-Meier-kuvaaja etenemisvapaasta elinajasta BICR-arvioinnin mukaan (kokonaispopulaatio)
Ei-pienisoluinen keuhkosyöpä
DESTINY-Lung02 (NCT04644237)
Enhertu-valmisteen tehoa ja turvallisuutta tutkittiin DESTINY-Lung02-tutkimuksessa, joka oli vaiheen 2 satunnaistettu tutkimus, jossa arvioitiin kahta annostasoa. Hoidon annostus oli sokkoutettu potilaille ja tutkijoille. Tutkimukseen otettiin aikuispotilaita, joilla oli metastasoitunut HER2-mutantti ei-pienisoluinen keuhkosyöpä, ja jotka olivat saaneet vähintään yhtä hoitoa, joka sisälsi platinapohjaista kemoterapiaa. Paikalliset laboratoriot määrittivät aktivoivan HER2 (ERBB2) -mutaation prospektiivisesti kasvainkudoksessa käyttämällä validoitua testiä, kuten seuraavan sukupolven sekvensointia, polymeraasiketjureaktiota tai massaspektrometriaa. Potilaat satunnaistettiin 2:1 saamaan Enhertu-valmistetta 5,4 mg/kg tai 6,4 mg/kg kolmen viikon välein. Satunnaistaminen ositettiin aiemman PD-1:n estäjillä ja/tai PD-1-ligandin estäjillä saadun hoidon mukaan (kyllä vs. ei). Hoitoa annettiin, kunnes tauti eteni, tutkittava kuoli, tutkittava perui suostumuksensa tai toksisuutta ei voitu hyväksyä. Tutkimukseen ei otettu potilaita, joilla oli ollut aiemmin steroidihoitoa vaativa ILD/pneumoniitti tai joilla oli seulontavaiheessa ILD/pneumoniitti tai kliinisesti merkittävä sydänsairaus. Tutkimukseen ei myöskään otettu potilaita, joilla oli hoitamattomia ja oireellisia aivometastaaseja tai joiden ECOG-toimintakyky oli > 1.
Ensisijainen tehon päätetapahtuma oli vahvistettu objektiivinen vasteprosentti (ORR), jonka arvioi sokkoutettu riippumaton keskitetty arvioijataho (BICR) RECIST v1.1 -kriteerien mukaisesti. Toissijainen tehon päätetapahtuma oli vasteen kesto (DOR).
Demografiset ja lähtötilanteen sairausominaisuudet 5,4 mg/kg:n ryhmään otettujen 102 potilaan osalta olivat: mediaani-ikä 59,4 vuotta (vaihteluväli 31–84), nainen (63,7 %), aasialainen (63,7 %), valkoihoinen (22,5 %) tai muu (13,7 %); ECOG-toimintakyky 0 (28,4 %) tai 1 (71,6 %); mutaatio ERBB2-kinaasidomeenissa (97,1 %) tai solun ulkoisessa domeenissa (2,9 %); HER2-mutaatio eksonissa 19 tai eksonissa 20 (96,1 %); vakaita aivometastaaseja (34,3 %); aikaisempi tupakointi (46,1 %) (kukaan tutkittavista ei tupakoinut nykyisin); aikaisemmin tehty keuhkoresektio (21,6 %). Metastaasien suhteen 32,4 % oli saanut useampaa kuin kahta aikaisempaa systeemistä hoitoa, 100 % sai platinapohjaista hoitoa, 73,5 % sai hoitoa PD-1:n/PD-1-ligandin estäjillä, ja 50,0 % oli aikaisemmin saanut platinapohjaisen hoidon ja PD-1:n/PD-1-ligandin estäjähoidon yhdistelmää.
Yhteenveto tehon tuloksista on esitetty taulukossa 9. Seurannan mediaanikesto oli 11,5 kuukautta (tiedonkeruun katkaisuhetki: 23. joulukuuta 2022).
Taulukko 9: DESTINY-Lung02-tutkimuksen tehon tulokset
| Tehon parametri | DESTINY-Lung02 5,4 mg/kg N = 102 |
| Vahvistettu objektiivinen vasteprosentti (ORR) BICR:n mukaan | |
| n (%) | 50 (49,0) |
| (95 %:n luottamusväli)* | (39,0, 59,1) |
| Täydellinen vaste (CR) n (%) | 1 (1,0) |
| Osittainen vaste (PR) n (%) | 49 (48,0) |
| Vasteen kesto | |
| Mediaani, kuukautta (95 %:n luottamusväli) † | 16,8 (6,4, EA) |
*95 %:n luottamusväli on laskettu Clopper-Pearsonin menetelmällä
EA = ei arvioitavissa
†95 %:n luottamusväli on laskettu Brookmeyer-Crowleyn menetelmällä
Mahasyöpä
DESTINY-Gastric04 (NCT04704934)
Enhertu-valmisteen tehoa ja turvallisuutta tutkittiin DESTINY-Gastric04-tutkimuksessa, joka oli vaiheen 3, satunnaistettu, avoin, aktiivikontrolloitu monikeskustutkimus. Tutkimukseen otettiin mukaan aikuispotilaita, joilla oli paikallisesti edennyt, leikkaushoitoon soveltumaton tai metastasoitunut HER2-positiivinen mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooma, joka oli edennyt trastutsumabipohjaisen hoidon aikana tai sen jälkeen. Potilaat satunnaistettiin 1:1 saamaan joko Enhertu-valmistetta (N = 246) tai ramusirumabin ja paklitakselin yhdistelmää (N = 248). Satunnaistaminen ositettiin HER2-statuksen (IHC 3+ tai IHC 2+ / ISH-positiivinen), maantieteellisen alueen (Aasia [lukuun ottamatta Manner-Kiinaa] vs. Länsi-Eurooppa vs. Manner-Kiina / muu maailma) ja ensilinjan hoidon aikana tapahtuneeseen taudin etenemiseen kuluneen ajan (< 6 kuukautta tai ≥ 6 kuukautta) mukaan. Kasvainnäytteistä piti paikallisesti tai keskitetysti vahvistaa HER2-positiivisuus, jonka määritelmä oli IHC 3+ tai IHC 2+ / ISH-positiivisuus. Tutkimukseen ei otettu mukaan potilaita, joilla oli ollut aiemmin steroidihoitoa edellyttänyt ILD/pneumoniitti tai joilla oli ILD/pneumoniitti seulontavaiheessa, joilla oli ollut aiemmin kliinisesti merkittävä sydänsairaus tai joilla oli aktiivisia aivometastaaseja. Hoitoa annettiin, kunnes tauti eteni, tutkittava kuoli tai ilmaantui toksisuutta, jota ei voitu hyväksyä. Ensisijainen tehoa koskevan hoitotuloksen mittari oli kokonaiselossaoloaika (OS). Toissijaisia hoitotuloksen mittareita olivat etenemisvapaa elinaika (PFS), vahvistettu objektiivinen vasteprosentti (ORR) ja vasteen kesto (DOR).
Potilaiden väestötiedot ja taudin lähtötilanteen ominaisuudet olivat samankaltaisia hoitohaarojen välillä. DESTINY-Gastric04-tutkimukseen mukaan otettujen 494 potilaan mediaani-ikä oli 63,7 vuotta (vaihteluväli 21,1–87,0), ja potilaista 79,4 % oli miehiä, 49,8 % oli valkoihoisia, 40,1 % oli aasialaisia ja 0,4 % oli mustaihoisia tai afroamerikkalaisia. Potilaiden ECOG-toimintakyky oli joko 0 (37,4 %) tai 1 (61,9 %). Potilaista 61,1 %:lla oli mahalaukun adenokarsinooma ja 38,9 %:lla ruokatorvi-mahalaukkurajan adenokarsinooma. HER2-status oli 84 %:lla potilaista IHC 3+ ja 16 %:lla IHC 2+ / ISH-positiivinen. Potilaista 70 %:lla oli metastaaseja kahdessa tai useammassa paikassa, 61,7 %:lla oli maksametastaaseja, 6,9 %:lla oli aivometastaaseja ja 15,6 % potilaista oli aiemmin saanut immunoterapiaa.
Yhteenveto tehon tuloksista on esitetty taulukossa 10 ja kuvassa 9.
Taulukko 10: DESTINY-Gastric04-tutkimuksen tehon tulokset
| Tehon parametri | Enhertu N = 246 | Ramusirumabi + paklitakseli N = 248 |
|---|---|---|
| Kokonaiselossaoloaika (OS) | ||
| Tapahtumien lukumäärä (%) | 124 (50,4) | 142 (57,3) |
| Mediaani, kuukautta (95 %:n CI) | 14,7 (12,1, 16,6) | 11,4 (9,9, 15,5) |
| Hasardisuhde (95 %:n CI)* | 0,70 (0,55, 0,90) | |
| p-arvo† | p = 0,0044 | |
| Tutkijan arvioima etenemisvapaa elinaika (PFS) | ||
| Tapahtumien lukumäärä (%) | 166 (67,5) | 156 (62,9) |
| Mediaani, kuukautta (95 %:n CI) | 6,7 (5,6, 7,1) | 5,6 (4,9, 5,8) |
| Hasardisuhde (95 %:n CI)* | 0,74 (0,59, 0,92) | |
| p-arvo† | p = 0,0074 | |
| Tutkijan arvioima vahvistettu objektiivinen vasteprosentti (ORR)†† | ||
| n (%) | 104 (44,3) | 69 (29,1) |
| 95 %:n CI | (37,8, 50,9) | (23,4, 35,3) |
| p-arvo§ | p = 0,0006 | |
| Täydellinen vaste n (%) | 7 (3,0) | 3 (1,3) |
| Osittainen vaste n (%) | 97 (41,3) | 66 (27,8) |
| Tutkijan arvioima vasteen kesto (DOR) | ||
| Mediaani, kuukautta (95 %:n CI) | 7,4 (5,7, 10,1) | 5,3 (4,1, 5,7) |
CI = luottamusväli
*Kaksisuuntainen p-arvo, joka perustuu ositettuun log-rank-testiin ja ositettuun Coxin malliin seuraavien IRT-ositustekijöiden mukaan korjattuna: HER2-status (IHC 3+ tai IHC 2+ / ISH+).
†Perustuu HER2-statuksen (IHC 3+ tai IHC 2+ / ISH+) mukaan ositettuun log-rank-testiin.
††ORR-kelpoisiksi laskettiin ne tutkittavat, jotka satunnaistettiin viimeistään 77 päivää (2 × 6 viikkoa – 1 viikko) ennen välianalyysin tiedonkeruun katkaisua. Vahvistettu ORR laskettiin käyttämällä kelpoisten tutkittavien lukumäärää nimittäjänä: Enhertu = 235, ramusirumabi + paklitakseli = 237.
§p-arvo ORR:n erolle perustuu Cochran-Mantel-Haenszelin testiin seuraavan ositustekijän mukaan korjattuna: HER2-status (IHC 3+ tai IHC 2+ / ISH+).
Kuva 9: Kaplan-Meier-kuvaaja kokonaiselossaoloajasta (koko analyysijoukko)
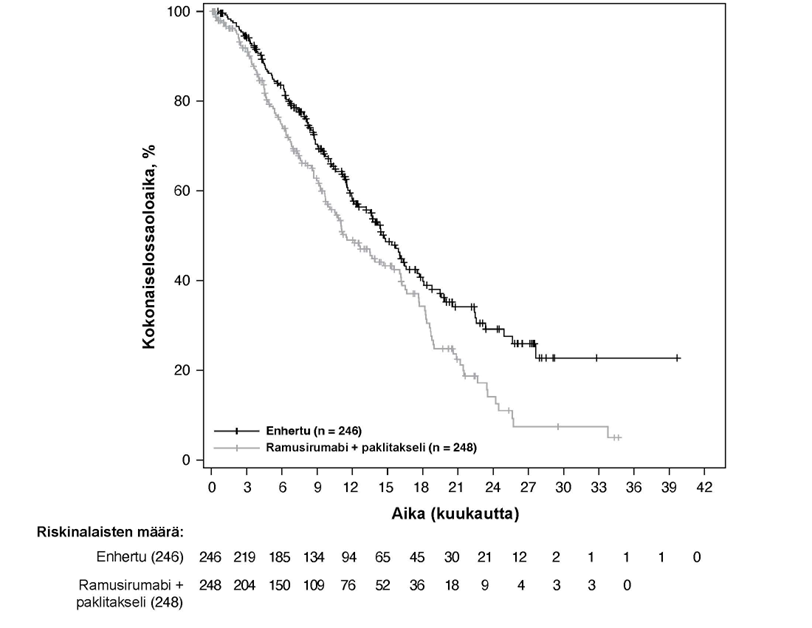
DESTINY-Gastric02 (NCT04014075)
Enhertu-valmisteen tehoa ja turvallisuutta tutkittiin DESTINY-Gastric02-tutkimuksessa, joka oli vaiheen 2, avoin, yksihaarainen monikeskustutkimus, joka toteutettiin Euroopassa ja Yhdysvalloissa. Tutkimukseen otettiin potilaita, joilla oli paikallisesti edennyt tai metastasoitunut HER2-positiivinen mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooma, ja joiden tauti oli edennyt aiemman trastutsumabipohjaisen hoidon aikana. Kasvainnäytteistä oli keskitetysti vahvistettu HER2-positiivisuus, jonka määritelmä oli IHC 3+ tai IHC 2+ / ISH-positiivisuus. Tutkimukseen ei otettu potilaita, joilla oli ollut aiemmin steroidihoitoa edellyttänyt ILD/pneumoniitti tai joilla oli ILD/pneumoniitti seulontavaiheessa, joilla oli ollut aiemmin kliinisesti merkittävä sydänsairaus tai joilla oli aktiivisia aivometastaaseja. Enhertu-valmistetta annettiin laskimoinfuusiona 6,4 mg/kg kolmen viikon välein, kunnes tauti eteni, tutkittava kuoli, tutkittava peruutti suostumuksensa tai toksisuutta ei voitu hyväksyä. Ensisijainen tehon tulosmittari oli vahvistettu objektiivinen vasteprosentti (ORR), jonka arvioi riippumaton keskitetty arvioijataho (ICR) RECIST v1.1 -kriteerien mukaisesti. Toissijaisia päätetapahtumia olivat vasteen kesto (DOR) ja kokonaiselossaoloaika (OS).
DESTINY-Gastric02-tutkimukseen otetun 79 potilaan demografiset ja lähtötilanteen sairausominaisuudet olivat: mediaani-ikä 61 vuotta (vaihteluväli 20–78), miehiä 72 %, valkoihoisia 87 %, aasialaisia 5,0 % ja mustaihoisia tai afroamerikkalaisia 1,0 %. Potilaiden ECOG-toimintakyky oli joko 0 (37 %) tai 1 (63 %). Potilaista 34 %:lla oli mahalaukun adenokarsinooma ja 66 %:lla ruokatorvi-mahalaukkurajan adenokarsinooma. HER2-status oli 86 %:lla potilaista IHC 3+ ja 13 %:lla IHC 2+ / ISH-positiivinen. Potilaista 63 %:lla oli maksametastaaseja.
Taulukossa 11 on yhteenveto tehon tuloksista (ORR ja DOR).
Taulukko 11: DESTINY-Gastric02-tutkimuksen tehon tulokset (koko analyysijoukko*)
| Tehon parametri | DESTINY-Gastric02 N = 79 |
| Tiedonkeruun katkaisuhetki 8. marraskuuta 2021 | |
Vahvistettu objektiivinen vasteprosentti† % (95 %:n CI)‡ | 41,8 (30,8, 53,4) |
| Täydellinen vaste (%) | 4 (5,1) |
| Osittainen vaste (%) | 29 (36,7) |
Vasteen kesto Mediaani§, kuukautta (95 %:n CI)¶ | 8,1 (5,9, EA) |
EA = Ei arvioitavissa
*Sisältää kaikki potilaat, jotka saivat vähintään yhden annoksen Enhertu-valmistetta
†Riippumattoman, keskitetyn arvioijatahon arvioima
‡Laskettu Clopper-Pearsonin menetelmällä
§Perustuu Kaplan-Meierin arviointiin
¶Laskettu Brookmeyerin ja Crowleyn menetelmällä
DESTINY-Gastric01 (NCT03329690)
Enhertu-valmisteen tehoa ja turvallisuutta tutkittiin DESTINY-Gastric01-tutkimuksessa, joka oli vaiheen 2, avoin, satunnaistettu monikeskustutkimus, joka toteutettiin Japanissa ja Etelä-Koreassa. Tähän arviointia tukevaan tutkimukseen otettiin aikuisia potilaita, joilla oli paikallisesti edennyt tai metastasoitunut HER2-positiivinen mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooma, ja joiden tauti oli edennyt vähintään kahden aiemman (mukaan lukien trastutsumabia, fluoropyrimidiinia ja platinaa sisältäneen) hoidon aikana. Potilaat satunnaistettiin 2:1 saamaan joko Enhertu-valmistetta (N = 126) tai lääkärin valinnan mukaan joko irinotekaania (N = 55) tai paklitakselia (N = 7). Kasvainnäytteiden HER2-positiivisuus oli vahvistettava keskitetysti, ja sen määritelmä oli IHC 3+ tai IHC 2+ / ISH-positiivisuus. Tutkimukseen ei otettu potilaita, joilla oli ollut aiemmin steroidihoitoa edellyttänyt ILD/pneumoniitti tai joilla oli ILD/pneumoniitti seulontavaiheessa, joilla oli ollut aiemmin kliinisesti merkittävä sydänsairaus tai joilla oli aktiivisia aivometastaaseja. Hoitoa jatkettiin, kunnes tauti eteni, tutkittava kuoli, tutkittava peruutti suostumuksensa tai toksisuutta ei voitu hyväksyä. Ensisijainen tehon tulosmittari oli vahvistamaton objektiivinen vasteprosentti (ORR), jonka arvioi riippumaton keskitetty arvioijataho (ICR) RECIST v1.1 -kriteerien mukaisesti. Toissijaisia tulosmittareita olivat kokonaiselossaoloaika (OS), etenemisvapaa elinaika (PFS), vasteen kesto (DOR) ja vahvistettu ORR.
Potilaiden demografiset ja lähtötilanteen sairausominaisuudet olivat samankaltaisia hoitohaarojen välillä. Potilaiden (N = 188) mediaani-ikä oli 66 vuotta (vaihteluväli 28–82), ja potilaista 76 % oli miehiä ja 100 % aasialaisia. Potilaiden ECOG-toimintakyky oli joko 0 (49 %) tai 1 (51 %). Potilaista 87 %:lla oli mahalaukun adenokarsinooma ja 13 %:lla ruokatorvi-mahalaukkurajan adenokarsinooma. HER2-status oli 76 %:lla potilaista IHC 3+ ja 23 %:lla IHC 2+ / ISH-positiivinen. Potilaista 54 %:lla oli maksametastaaseja ja 29 %:lla keuhkometastaaseja. Kohdeleesioiden halkaisijoiden summa oli 47 %:lla potilaista < 5 cm, 30 %:lla potilaista ≥ 5 – < 10 cm ja 17 %:lla potilaista ≥ 10 cm. Potilaista 55 % oli saanut kahta aiempaa ja 45 % vähintään kolmea aiempaa hoitoa paikallisesti edenneeseen tai metastasoituneeseen tautiin.
Tehon tulokset (tiedonkeruun katkaisuhetki: 3. kesäkuuta 2020) Enhertu-hoidolle (n = 126) vs. lääkärin valitsemalle kemoterapialle (n = 62) olivat vahvistetun objektiivisen vasteprosentin (ORR) osalta 39,7 % (95 %:n luottamusväli: 31,1, 48,8) vs. 11,3 % (95 %:n luottamusväli: 4,7, 21,9). Täydellisen vasteen saavuttaneiden osuudet olivat vastaavasti 7,9 % vs. 0 % ja osittaisen vasteen saavuttaneiden osuudet 31,7 % vs. 11,3 %. Muut tehon tulokset Enhertu-hoidolle vs. lääkärin valitsemalle kemoterapialle olivat: vasteen keston (DOR) mediaani: 12,5 kuukautta (95 %:n luottamusväli: 5,6, ei arvioitavissa) vs. 3,9 kuukautta (95 %:n luottamusväli: 3,0, 4,9). Etenemisvapaan elinajan (PFS) mediaani oli 5,6 kuukautta (95 %:n luottamusväli: 4,3, 6,9) vs. 3,5 kuukautta (95 %:n luottamusväli: 2,0, 4,3; hasardisuhde = 0,47 [95 %:n luottamusväli: 0,31, 0,71]). Kokonaiselossaoloajan analyysi, joka oli ennalta määritetty 133 kuolemaan, osoitti Enhertu-hoidon elossaolohyödyn verrattuna lääkärin valitsemaa hoitoa saavien ryhmään (hasardisuhde = 0,60). Kokonaiselossaoloajan mediaani oli Enhertu-ryhmässä 12,5 kuukautta (95 %:n luottamusväli: 10,3, 15,2) ja lääkärin valitsemaa hoitoa saavien ryhmässä 8,9 kuukautta (95 %:n luottamusväli: 6,4, 10,4).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Enhertu-valmisteen käytöstä rintasyövän, ei-pienisoluisen keuhkosyövän ja mahasyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Tämä lääkevalmiste on saanut ns. ehdollisen myyntiluvan. Se tarkoittaa, että lääkevalmisteesta odotetaan uutta tietoa.
Euroopan lääkevirasto arvioi vähintään kerran vuodessa tätä lääkevalmistetta koskevat uudet tiedot, ja tarvittaessa tämä valmisteyhteenveto päivitetään.
Farmakokinetiikka
Imeytyminen
Trastutsumabi-derukstekaani annetaan laskimoon. Muita antoreittejä ei ole tutkittu.
Jakautuminen
Populaatiofarmakokineettisen analyysin perusteella trastutsumabi-derukstekaanin ja topoisomeraasi I:n estäjä DXd:n sentraalisen jakautumistilavuuden (Vc) arvioitiin olevan 2,68 l ja 28,0 l, vastaavasti.
Ihmisen plasmassa DXd:n proteiiniin sitoutuminen in vitro oli keskimäärin noin 97 %.
DXd:n veri-plasmapitoisuussuhde in vitro oli noin 0,6.
Biotransformaatio
Lysosomaaliset entsyymit vapauttavat DXd:n katkaisemalla trastutsumabi-derukstekaanin solun sisällä.
Humanisoidun monoklonaalisen HER2 IgG1 -vasta-aineen odotetaan hajoavan pieniksi peptideiksi ja aminohapoiksi katabolisten reittien kautta samalla tavalla kuin endogeeninen IgG.
Ihmisen maksan mikrosomien in vitro -metaboliatutkimukset osoittavat, että DXd metaboloituu pääasiassa CYP3A4:n vaikutuksesta oksidatiivisten reittien kautta.
Eliminaatio
Kun trastutsumabi-derukstekaania annettiin laskimoon metastasoitunutta HER2-positiivista, heikosti HER2-positiivista rintasyöpää tai HER2-mutanttia ei-pienisoluista keuhkosyöpää sairastaville potilaille, trastutsumabi-derukstekaanin puhdistuman laskettiin populaatiofarmakokineettisessä analyysissa olevan 0,4 l/vrk ja DXd:n puhdistuman 18,4 l/h. Potilailla, joilla oli paikallisesti edennyt tai metastasoitunut mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooma, trastutsumabi-derukstekaanin puhdistuma oli noin 20 % suurempi kuin metastasoitunutta HER2-positiivista rintasyöpää sairastavilla potilailla. Trastutsumabi-derukstekaanin ja vapautuneen DXd:n näennäinen eliminaation puoliintumisaika (t1/2) oli syklissä 3 noin 7 vuorokautta. Kohtalaista trastutsumabi-derukstekaanin kertymistä havaittiin (noin 35 % syklissä 3 verrattuna sykliin 1).
Kun DXd:tä annettiin rotille laskimoon, tärkein erittymisreitti oli sapen kautta ulosteisiin. DXd oli runsain komponentti virtsassa, ulosteessa ja sappinesteessä. Kun apinoille annettiin yksittäinen laskimoinfuusio trastutsumabi-derukstekaania (6,4 mg/kg), muuttumaton vapautunut DXd oli runsain komponentti virtsassa ja ulosteessa. DXd:n erittymistä ei tutkittu ihmisillä.
In vitro-yhteisvaikutukset
Enhertu-valmisteen vaikutukset muiden lääkevalmisteiden farmakokinetiikkaan
In vitro -tutkimukset osoittavat, että DXd ei estä merkittäviä CYP450-entsyymejä, kuten CYP1A2, -2B6, -2C8, -2C9, -2C19, -2D6 ja -3A. In vitro -tutkimukset osoittavat, että DXd ei estä OAT1-, OAT3-, OCT1-, OCT2-, OATP1B1-, OATP1B3-, MATE1-, MATE2-K-, P-gp-, BCRP- tai BSEP-kuljettajaproteiineja.
Muiden lääkevalmisteiden vaikutukset Enhertu-valmisteen farmakokinetiikkaan
DXd oli in vitro P-gp-, OATP1B1-, OATP1B3-, MATE2-K-, MRP1- ja BCRP-substraatti.
Kliinisesti merkittäviä yhteisvaikutuksia ei odoteta esiintyvän sellaisten lääkevalmisteiden kanssa, jotka ovat MATE2-K, MRP1-, P-gp-, OATP1B- tai BCRP-kuljettajaproteiinien estäjiä (ks. kohta Yhteisvaikutukset).
Lineaarisuus/ei-lineaarisuus
Laskimoon annetun trastutsumabi-derukstekaanin ja vapautuneen DXd:n altistus suureni suhteessa annokseen annosalueella 3,2–8,0 mg/kg (noin 0,6–1,5 kertaa suositeltu annos) ja vaihtelu tutkittavien välillä oli vähäistä tai kohtalaista. Populaatiofarmakokineettisen analyysin perusteella tutkittavien välinen vaihtelu trastutsumabi-derukstekaanin ja DXd:n eliminaatiopuhdistumissa oli 24 % ja 28 %, vastaavasti, ja sentraalisissa jakautumistilavuuksissa 16 % ja 55 %, vastaavasti. Trastutsumabi-derukstekaanin ja DXd:n AUC-arvojen (seerumipitoisuuden vs. ajan käyrän alapuolinen pinta-ala) vaihtelu tutkittavien välillä oli noin 8 % ja 14 %, vastaavasti.
Erityisryhmät
Populaatiofarmakokineettisen analyysin perustella ikä (20–96 vuotta), rotu, etnisyys, sukupuoli ja paino eivät vaikuttaneet kliinisesti merkittävästi trastutsumabi-derukstekaanille tai vapautuneelle DXd:lle altistukseen.
Iäkkäät
Populaatiofarmakokineettinen analyysi osoitti, että ikä (vaihteluväli 20–96 vuotta) ei vaikuttanut trastutsumabi-derukstekaanin farmakokinetiikkaan.
Munuaisten vajaatoiminta
Erityistä munuaisten vajaatoimintatutkimusta ei ole tehty. Populaatiofarmakokineettisen analyysin perusteella, kun siihen sisällytettiin lievää (kreatiniinipuhdistuma [CLcr] ≥ 60 ja < 90 ml/min) tai keskivaikeaa (CLcr ≥ 30 ja < 60 ml/min) munuaisten vajaatoimintaa sairastavia potilaita (Cockcroft-Gaultin kaavalla arvioituna), lievä tai keskivaikea munuaisten vajaatoiminta ei vaikuttanut vapautuneen DXd:n farmakokinetiikkaan verrattuna normaaliin munuaisten toimintaan (CLcr ≥ 90 ml/min).
Maksan vajaatoiminta
Erityistä maksan vajaatoimintatutkimusta ei ole tehty. Populaatiofarmakokineettisen analyysin perusteella trastutsumabi-derukstekaanin farmakokinetiikan muutosten vaikutus potilaisiin, joiden kokonaisbilirubiini on ≤ 1,5 kertaa ULN (ASAT-arvosta riippumatta), ei ole kliinisesti merkittävä. Potilaista, joiden kokonaisbilirubiini on > 1,5–3 kertaa ULN (ASAT-arvosta riippumatta), on vain vähän tietoja johtopäätösten tekemiseen, ja potilaista, joiden kokonaisbilirubiini on > 3 kertaa ULN (ASAT-arvosta riippumatta), ei ole saatavilla lainkaan tietoja (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Ei ole tehty tutkimuksia, joissa tutkittaisiin trastutsumabi-derukstekaanin farmakokinetiikkaa lapsilla tai nuorilla.
Prekliiniset tiedot turvallisuudesta
Eläimillä toksisia vaikutuksia havaittiin trastutsumabi-derukstekaanin annon jälkeen lymfaattisissa ja hematopoieettisissa elimissä, suolistossa, munuaisissa, keuhkoissa, kiveksissä ja ihossa, kun topoisomeraasi I -estäjän (DXd) altistustaso oli pienempi kuin kliininen plasma-altistus. Näillä eläimillä vasta-aine-lääkekonjugaatin (ADC) altistustasot olivat kliinisen plasma-altistuksen tasolla tai sitä suurempia.
DXd oli klastogeeninen sekä rotan luuytimen mikrotumamäärityksessä in vivo että kiinanhamsterin keuhkokromosomin poikkeavuusmäärityksessä in vitro, eikä se ollut mutageeninen bakteerien käänteismutaatiomäärityksessä in vitro.
Trastutsumabi-derukstekaanilla ei ole tehty karsinogeenisuustutkimuksia.
Trastutsumabi-derukstekaanilla ei ole tehty erityisiä hedelmällisyystutkimuksia. Yleisten eläintoksisuustutkimusten tulosten perusteella trastutsumabi-derukstekaani voi heikentää miesten suvunjatkamiskykyä ja hedelmällisyyttä.
Trastutsumabi-derukstekaanilla ei ole tehty eläinten lisääntymis- tai kehitystoksisuustutkimuksia. Yleisten eläintoksisuustutkimusten tulosten perusteella trastutsumabi-derukstekaani ja DXd olivat toksisia nopeasti jakautuville soluille (lymfaattiset/hematopoieettiset elimet, suolisto tai kivekset) ja DXd oli genotoksinen, mikä viittaa alkiotoksisuuden ja teratogeenisuuden mahdollisuuteen.
Farmaseuttiset tiedot
Apuaineet
L-histidiini
L-histidiinihydrokloridimonohydraatti
Sakkaroosi
Polysorbaatti 80 (E433)
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
NaCl-infuusionestettä ei saa käyttää käyttökuntoon saattamiseen tai laimentamiseen, koska se voi saada aikaan hiukkasten muodostumista.
Kestoaika
Avaamaton injektiopullo
4 vuotta.
Käyttökuntoon saatettu liuos
Käytönaikaiseksi kemialliseksi ja fysikaaliseksi säilyvyydeksi on osoitettu enintään 48 tuntia 2–8 ºC:ssa.
Mikrobiologiselta kannalta valmiste on käytettävä välittömästi. Jos sitä ei käytetä välittömästi, käytönaikaiset säilytysajat ja käyttöä edeltävät olosuhteet ovat käyttäjän vastuulla, eivätkä ne tavallisesti saa olla yli 24 tuntia lämpötilassa 2–8 ºC, ellei käyttökuntoon saattaminen ole tapahtunut kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Laimennettu liuos
On suositeltavaa käyttää laimennettu liuos välittömästi. Jos sitä ei käytetä välittömästi, käyttökuntoon saatettua, laimennettua liuosta infuusiopusseissa, joissa on 5-prosenttista glukoosiliuosta, voidaan säilyttää huoneenlämmössä (≤ 30 ºC) enintään 4 tuntia (tähän aikaan sisältyy valmisteluun ja infuusioon käytetty aika) tai jääkaapissa lämpötilassa 2–8 ºC enintään 24 tuntia valolta suojattuna.
Säilytys
Säilytä jääkaapissa (2–8 °C).
Ei saa jäätyä.
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ENHERTU kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
100 mg (L:ei) 1 kpl (1889,57 €)
PF-selosteen tieto
Enhertu toimitetaan 10 ml:n tyypin 1 keltaisessa borosilikaattilasi-injektiopullossa, joka on suljettu fluorohartsilaminoidulla butyylikumitulpalla ja keltaisella poimutetulla polypropeeni-/alumiininapsautuskorkilla.
Yksi pahvikotelo sisältää 1 injektiopullon.
Valmisteen kuvaus:
Valkoinen tai kellertävänvalkoinen kylmäkuivattu jauhe.
Käyttö- ja käsittelyohjeet
Lääkitysvirheiden välttämiseksi on tärkeää varmistaa injektiopullojen etiketeistä, että valmisteltava ja annettava lääkevalmiste on Enhertu (trastutsumabi-derukstekaani) eikä trastutsumabi tai trastutsumabiemtansiini.
Kemoterapialääkevalmisteiden valmistelussa on käytettävä asianmukaisia toimenpiteitä. Käyttökuntoon saattamisessa ja laimennuksessa on käytettävä asianmukaista aseptista tekniikkaa.
Käyttökuntoon saattaminen
- Saatettava käyttökuntoon välittömästi ennen laimennusta.
- Täyteen annokseen saatetaan tarvita enemmän kuin yksi injektiopullo. Laske annos (mg), käyttökuntoon saatetun Enhertu-liuoksen tarvittava kokonaismäärä ja tarvittavien Enhertu-injektiopullojen määrä (ks. kohta Annostus ja antotapa).
- Saata jokainen 100 mg:n injektiopullo käyttökuntoon käyttämällä steriiliä ruiskua. Injektoi hitaasti 5 ml injektionesteisiin käytettävää vettä kuhunkin injektiopulloon, jotta lopulliseksi pitoisuudeksi tulee 20 mg/ml.
- Pyöritä injektiopulloa varovasti, kunnes aine on liuennut täysin. Älä ravista.
- Mikrobiologiselta kannalta valmiste on käytettävä välittömästi. Jos valmistetta ei käytetä välittömästi, käytönaikaiseksi kemialliseksi ja fysikaaliseksi säilyvyydeksi on osoitettu enintään 48 tuntia 2–8 ºC:ssa. Säilytä käyttökuntoon saatettuja Enhertu-injektiopulloja jääkaapissa lämpötilassa 2–8 ºC suojattuna valolta. Ei saa jäätyä.
- Käyttökuntoon saatettu valmiste ei sisällä säilöntäaineita, ja se on tarkoitettu vain kertakäyttöön.
Laimennus
- Vedä laskettu määrä injektiopullo(i)sta steriiliin ruiskuun. Tarkista käyttökuntoon saatettu liuos hiukkasten ja värimuutosten varalta. Liuoksen on oltava kirkasta ja väritöntä tai vaaleankeltaista. Älä käytä liuosta, jos siinä näkyy hiukkasia tai jos liuos on sameaa tai väärän väristä.
- Laimenna käyttökuntoon saatetun Enhertu-valmisteen laskettu tilavuus infuusiopussissa, jossa on 100 ml 5-prosenttista glukoosi-infuusionestettä, liuosta. Älä käytä fysiologista keittosuolaliuosta (ks. kohta Yhteensopimattomuudet). Polyvinyylikloridista tai polyolefiinista (etyleenin ja polypropeenin kopolymeeri) valmistettua infuusiopussia suositellaan.
- Käännä infuusiopussi varovasti ylösalaisin, jotta liuos sekoittuu perusteellisesti. Älä ravista.
- Suojaa infuusiopussi valolta peittämällä se.
- Jos valmistetta ei käytetä välittömästi, sitä saa säilyttää huoneenlämmössä (≤ 30 ºC) enintään 4 tunnin ajan (tähän aikaan sisältyy valmisteluun ja infuusioon käytetty aika) tai jääkaapissa lämpötilassa 2–8 ºC enintään 24 tunnin ajan suojattuna valolta. Ei saa jäätyä.
- Hävitä injektiopulloon jäänyt käyttämätön liuos.
Antaminen
- Jos valmisteltua infuusioliuosta säilytettiin jääkaapissa (2–8 ºC), on suositeltavaa antaa liuoksen lämmetä huoneenlämpöiseksi valolta suojattuna ennen potilaalle antamista.
- Enhertu on annettava vain laskimoinfuusiona 0,20 tai 0,22 mikronin letkunsisäisen polyeetterisulfoni (PES)- tai polysulfoni (PS) -suodattimen avulla.
- Aloitusannos on annettava 90 minuuttia kestävänä laskimoinfuusiona. Jos potilas sieti aiemman infuusion hyvin, seuraavat Enhertu-annokset voidaan antaa 30 minuuttia kestävinä infuusioina. Ei saa antaa laskimoinjektiona tai boluksena (ks. kohta Annostus ja antotapa).
- Suojaa infuusiopussi valolta peittämällä se.
- Enhertu-valmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa eikä muita lääkevalmisteita saa antaa saman laskimoyhteyden kautta.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ENHERTU kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
100 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FD04
Valmisteyhteenvedon muuttamispäivämäärä
21.11.2025
Yhteystiedot
Amagerfælledvej 106, 2.
2300 Copenhagen S
Denmark
+358 9 3540 7081
www.daiichi-sankyo.eu
info@daiichi-sankyo.dk