
KESIMPTA injektionsvätska, lösning i förfylld injektionspenna 20 mg
Kvalitativ och kvantitativ sammansättning
Kesimpta 20 mg injektionsvätska, lösning i förfylld spruta
Varje förfylld spruta innehåller 20 mg ofatumumab i 0,4 ml lösning (50 mg/ml).
Kesimpta 20 mg injektionsvätska, lösning i förfylld injektionspenna
Varje förfylld injektionspenna innehåller 20 mg ofatumumab i 0,4 ml lösning (50 mg/ml).
Ofatumumab är en helt human monoklonal antikropp, som produceras i en murin cellinje (NS0) genom rekombinant DNA‑teknik.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Injektionsvätska, lösning (injektionsvätska)
Injektionsvätska, lösning (injektionsvätska) i förfylld injektionspenna (Sensoready‑penna)
Kliniska uppgifter
Terapeutiska indikationer
Kesimpta är indicerat för behandling av vuxna patienter med skovvis multipel skleros (RMS) med aktiv sjukdom som definieras av kliniska eller bilddiagnostiska fynd (se avsnitt Farmakodynamiska egenskaper).
Villkor
Neurologisten sairauksien hoitoon perehtyneen lääkärin on aloitettava hoito.
Dosering och administreringssätt
Behandling ska initieras av en läkare med erfarenhet av behandling av neurologiska sjukdomar.
Dosering
Rekommenderad dos är 20 mg ofatumumab administrerad som subkutan injektion med:
- initial dosering vecka 0, 1 och 2 följd av
- månadsvis dosering med start vecka 4.
Missade doser
Om patienten missar en injektion ska dosen administreras så snart som möjligt utan att vänta på nästa schemalagda dos. Efterföljande doser ska administreras med rekommenderat intervall.
Särskilda populationer
Vuxna över 55 år
Inga studier har utförts på MS‑patienter över 55 år. Baserat på de begränsade data som finns tillgängliga, anses inte dosjustering vara nödvändigt för patienter över 55 år (se avsnitt Farmakokinetiska egenskaper).
Nedsatt njurfunktion
Patienter med nedsatt njurfunktion förväntas inte behöva dosjustering (se avsnitt Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Patienter med nedsatt leverfunktion förväntas inte behöva dosjustering (se avsnitt Farmakokinetiska egenskaper).
Pediatrisk population
Säkerhet och effekt för Kesimpta för barn i åldern 0 till 18 år har ännu inte fastställts. Inga data finns tillgängliga.
Administreringssätt
Detta läkemedel är avsett för subkutan injektion, som administreras av patienten själv.
Vanliga injektionsställen för subkutana injektioner är buken, låret och utsidan av överarmen.
Den första injektionen ska utföras under överinseende av hälso- och sjukvårdspersonal (se avsnitt Varningar och försiktighet).
Uttömmande anvisningar om administrering återfinns i bipacksedeln.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Patienter med kraftigt nedsatt immunförsvar (se avsnitt Varningar och försiktighet).
Patienter med allvarlig aktiv infektion, tills denna gått tillbaka (se avsnitt Varningar och försiktighet).
Känd aktiv malignitet.
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Systemiska injektionsrelaterade reaktioner
Patienterna ska informeras om att systemiska injektionsrelaterade reaktioner (SIRR) kan uppkomma, vanligen inom 24 timmar och i huvudsak efter den första injektionen (se avsnitt Biverkningar). Symtom som oftast observerats i kliniska studier med RMS inkluderar feber, huvudvärk, myalgi, frossa, trötthet (fatigue), illamående och kräkningar och var övervägande (99,8 %) milda till måttliga i svårighetsgrad. Inga livshotande SIRR rapporterades i kliniska RMS-studier (se avsnitt Biverkningar).
Ytterligare SIRR rapporterade efter godkännande för försäljning inkluderar hudutslag, urtikaria, dyspné och angioödem (t.ex. svullnad i tungan, svalget eller larynx) och sällsynta fall som rapporterades som anafylaxi. Även om det fanns några fall som var allvarliga och resulterade i att behandlingen med ofatumumab avbröts, fanns det också allvarliga fall där patienter kunde fortsätta behandlingen med ofatumumab utan ytterligare incidenter.
Vissa SIRR-symtom kan vara kliniskt omöjliga att skilja från akuta överkänslighetsreaktioner av typ 1 (IgE-medierade). En överkänslighetsreaktion kan uppträda under vilken som helst injektion, även om den vanligtvis inte skulle uppstå vid den första injektionen. För efterföljande injektioner bör allvarligare symtom än tidigare upplevt, eller nya svåra symtom, föranleda övervägande av en potentiell överkänslighetsreaktion. Patienter med känd IgE-medierad överkänslighet mot ofatumumab får inte behandlas med ofatumumab (se avsnitt Kontraindikationer).
Endast begränsad nytta av premedicinering med steroider sågs i kliniska RMS‑studier. Om injektionsrelaterade reaktioner uppkommer kan de hanteras med symtomatisk behandling. Därför är premedicinering inte nödvändig.
Den första injektionen ska utföras under överinseende av hälso- och sjukvårdspersonal som genomgått lämplig utbildning (se avsnitt Dosering och administreringssätt).
Lokala reaktioner vid injektionsstället
Lokala reaktioner vid injektionsstället som observerats i kliniska studier innefattade erytem, svullnad, klåda och smärta (se avsnitt Biverkningar).
Infektioner
Det rekommenderas att patientens immunstatus utvärderas innan behandling initieras.
Baserat på dess verkningssätt samt tillgänglig klinisk erfarenhet, kan ofatumumab potentiellt öka risken för infektioner (se avsnitt Biverkningar).
Hos patienter med aktiv infektion ska administreringen skjutas upp, tills infektionen har gått tillbaka.
Ofatumumab får inte ges till patienter med kraftigt nedsatt immunförsvar (t.ex. patienter med betydande neutropeni eller lymfocytopeni).
Progressiv multifokal leukoencefalopati
John Cunningham‑virusinfektioner (JCV‑infektioner) som orsakar progressiv multifokal leukoencefalopati (PML) har observerats hos patienter som fått behandling med antikroppar mot CD20, andra MS‑behandlingar samt ofatumumab (i väsentligt högre doser vid onkologiska indikationer). Därför ska läkare vara uppmärksamma på eventuell PML i anamnesen och på kliniska symtom eller MRT‑fynd som kan tyda på PML. Vid misstanke om PML ska behandlingen med ofatumumab avbrytas, tills PML har uteslutits.
Reaktivering av hepatit B‑virus
Hepatit B‑reaktivering, som i vissa fall resulterat i fulminant hepatit, leversvikt och död, har förekommit hos patienter som behandlats med antikroppar mot CD20.
Patienter med aktiv HBV‑infektion bör inte behandlas med ofatumumab. Screening för HBV bör utföras hos alla patienter innan behandling inleds. Som ett minimum bör screeningen innefatta testning av hepatit B‑ytantigen (HBsAg) och hepatit B‑kärnantikropp (HBcAb). Dessa kan kompletteras med andra lämpliga markörer enligt lokala riktlinjer. Patienter med positiv hepatit B‑serologi (antingen HBsAg eller HBcAb) bör remitteras till en specialist på leversjukdomar innan behandling inleds. Dessa patienter bör också följas upp och behandlas enligt lokala medicinska riktlinjer för att förhindra hepatit B‑reaktivering.
Behandling av patienter med kraftigt nedsatt immunförsvar
Patienter med kraftigt nedsatt immunförsvar får inte behandlas förrän immunförsvaret normaliserats (se avsnitt Kontraindikationer).
Användning av andra immunsuppressiva läkemedel samtidigt med ofatumumab rekommenderas inte, förutom kortikosteroider för symtomatisk behandling av skov.
Vaccinationer
Alla vaccinationer bör administreras enligt riktlinjerna för vaccinationer. Levande eller levande försvagade vacciner bör administreras minst 4 veckor före initiering av behandling med ofatumumab, och inaktiverade vacciner bör, om möjligt, administreras minst 2 veckor före initiering av behandling med ofatumumab.
Ofatumumab kan påverka effekten av inaktiverade vacciner.
Säkerheten vid immunisering med levande eller levande försvagade vacciner efter behandling med ofatumumab har inte studerats. Vaccination med levande eller levande försvagade vacciner rekommenderas inte under behandling eller efter avslutad behandling, förrän B‑cellerna har återhämtat sig (se avsnitt Interaktioner). Mediantiden till B-cellåterhämtning till den nedre normalgränsen (LLN, definierad som 40 celler/mikroliter) eller till värdet vid studiestart är 24,6 veckor efter avslutad behandling, baserat på data från fas III-studier (se avsnitt Farmakodynamiska egenskaper).
Vaccination av spädbarn vars mödrar fått behandling med ofatumumab under graviditeten
Levande eller levande försvagade vacciner ska inte ges till spädbarn vars mödrar fått behandling med ofatumumab under graviditeten, förrän det har bekräftats att antalet B‑celler ligger inom normalvärdet. Depletion av B‑celler hos dessa spädbarn kan öka riskerna relaterade till levande eller levande försvagade vacciner.
Inaktiverade vacciner kan administreras vid behov innan det har bekräftats att antalet B‑celler ligger inom normalvärdet. Bedömning av immunsvaret på vaccinerna, inklusive konsultation av en kvalificerad specialist, bör dock övervägas för att säkerställa huruvida ett skyddande immunsvar har byggts upp (se avsnitt Fertilitet, graviditet och amning).
Hjälpämnen med känd effekt
Natrium
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per dos, dvs. är näst intill ”natriumfritt”.
Polysorbater
Detta läkemedel innehåller 0,08 mg polysorbat 80 per dos. Polysorbater kan orsaka allergiska reaktioner.
Interaktioner
Inga interaktionsstudier har utförts, eftersom inga interaktioner som involverar cytokrom P450‑enzymer, andra metaboliserande enzymer eller transportörer förväntas.
Vaccinationer
Säkerheten och förmågan att framkalla en primär eller anamnestisk (recall) respons på immunisering med levande, levande försvagade eller inaktiverade vacciner under behandling med ofatumumab har inte undersökts. Responsen på vaccinationer kan vara försvagad när antalet B‑celler är reducerat. Det rekommenderas att patienterna slutför vaccinationerna innan behandling med ofatumumab inleds (se avsnitt Varningar och försiktighet).
Övriga immunsuppressiva eller immunmodulerande läkemedel
Risken för additiva effekter relaterade till immunsystemet bör beaktas när immunsuppressiva läkemedel administreras samtidigt med ofatumumab.
Vid initiering av behandling med ofatumumab efter behandling med andra immunsuppressiva läkemedel med långvarig immuneffekt, liksom vid initiering av behandling med andra immunsuppressiva läkemedel med långvarig immuneffekt efter behandling med ofatumumab, bör dessa läkemedels verkningstid och verkningssätt tas i beaktande på grund av potentiella additiva immunsuppressiva effekter (se avsnitt Farmakodynamiska egenskaper).
Fertilitet, graviditet och amning
Fertila kvinnor
Fertila kvinnor ska använda en effektiv preventivmetod (metoder som resulterar i en graviditetsfrekvens lägre än 1 %) under behandling med Kesimpta och i 2 månader efter den sista dosen Kesimpta.
Graviditet
Det finns begränsade data från användning av ofatumumab hos gravida kvinnor. Ofatumumab kan passera placentan och orsaka depletion av B‑celler hos foster, baserat på fynd från djurstudier (se avsnitt Prekliniska säkerhetsuppgifter). Ingen teratogenicitet observerades efter intravenös administrering av ofatumumab till dräktiga apor under organogenesen.
Övergående depletion av perifera B‑celler samt lymfocytopeni har rapporterats hos spädbarn, vars mödrar exponerats för andra antikroppar mot CD20 under graviditeten. Det är inte känt hur länge depletionen av B‑celler varar hos spädbarn som exponerats för ofatumumab in utero, och inte heller vilken inverkan depletionen har på säkerheten och effekten av vacciner (se avsnitt Varningar och försiktighet och Farmakodynamiska egenskaper).
Behandling med ofatumumab ska undvikas under graviditet, såvida inte den potentiella nyttan för modern uppväger den potentiella risken för fostret.
För att bidra till att fastställa effekterna av ofatumumab hos gravida kvinnor uppmanas hälso- och sjukvårdspersonal att rapportera alla graviditeter och komplikationer som förekommer under behandlingen eller inom 2 månader efter den sista dosen ofatumumab till det lokala ombudet för innehavaren av godkännande för försäljning. Därigenom kan patienterna följas upp genom PRIM‑programmet (PRegnancy outcomes Intensive Monitoring). Därtill ska alla biverkningar som gäller graviditet rapporteras till (se detaljer nedan).
webbplats: www.fimea.fi
Säkerhets‐ och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Amning
Det finns begränsade data om användning av ofatumumab hos kvinnor som ammar. Det finns inga data om ofatumumabs effekt på mjölkproduktion. Hos människa sker utsöndring av IgG‑antikroppar i bröstmjölk under de första dagarna efter förlossningen, och utsöndringen minskar sedan till låga koncentrationer kort därefter. Publicerade data tyder på att antikroppar i bröstmjölk inte når blodcirkulationen hos nyfödda och spädbarn i betydande mängder.
I en observationsstudie har det rapporterats att koncentrationen av ofatumumab i bröstmjölk i allmänhet var låg, med Cavg och Cmax på mindre än 0,02 µg/ml i bröstmjölk hos behandlade ammande kvinnor.
I samma observationsstudie hade fem spädbarn med befintliga B-celler normala nivåer. Åtta spädbarn fick levande vacciner under/efter exponering via amning utan komplikationer. Spädbarn (upp till 24 månaders ålder) uppvisade inga avvikelser avseende infektioner, antibiotikaanvändning, sjukhusinläggningar eller förseningar i utveckling.
En risk för det ammade barnet kan därmed inte uteslutas under de första dagarna efter födseln. Därefter kan ofatumumab användas under amning om det är kliniskt motiverat. Om patienten behandlats med ofatumumab fram till de sista månaderna av graviditeten, kan amning dock inledas omedelbart efter förlossningen.
Fertilitet
Det finns inga data om ofatumumabs effekt på fertiliteten hos människa.
Icke‑kliniska data tydde inte på några potentiella risker för människa, baserat på fertilitetsparametrar för hanar och honor som utvärderats hos apor.
Effekter på förmågan att framföra fordon och använda maskiner
Kesimpta har ingen eller försumbar effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Summering av säkerhetsprofilen
De viktigaste och vanligaste rapporterade biverkningarna är övre luftvägsinfektioner (39,4 %), systemiska injektionsrelaterade reaktioner (20,6 %), lokala reaktioner vid injektionsstället (10,9 %) och urinvägsinfektioner (11,9 %) (se avsnitt Varningar och försiktighet och underavsnittet ”Beskrivning av utvalda biverkningar” nedan för ytterligare information).
Tabell över biverkningar
Biverkningar som har rapporterats i samband med användning av ofatumumab i pivotala kliniska RMS‑studier och efter godkännande för försäljning listas i tabell 1 (i enlighet med MedDRAs organklassificeringssystem). Inom varje organsystemklass rangordnas biverkningarna efter frekvens med den oftast förekommande biverkningen först. Inom varje frekvensområde presenteras biverkningarna efter fallande allvarlighetsgrad. Frekvenskategorierna definieras enligt följande konvention: mycket vanliga (≥1/10); vanliga (≥1/100, <1/10); mindre vanliga (≥1/1 000, <1/100); sällsynta (≥1/10 000, <1/1 000); mycket sällsynta (<1/10 000); ingen känd frekvens (kan inte beräknas från tillgängliga data).
Tabell 1 Tabell över biverkningar
Infektioner och infestationer | |
Mycket vanliga | Övre luftvägsinfektioner1 Urinvägsinfektioner2 |
Vanliga | Oral herpes |
Immunsystemet | |
Ingen känd frekvens | Överkänslighetsreaktioner3 |
Magtarmkanalen | |
Vanliga | Illamående, kräkningar4 |
Lever och gallvägar | |
Vanliga | Förhöjda leverenzymer5 |
Allmänna symtom och/eller symtom vid administreringsstället | |
Mycket vanliga | Reaktioner vid injektionsstället (lokala) |
Undersökningaroch provtagningar | |
Vanliga | Minskat immunoglobulin M i blodet |
Skador och förgiftningar och behandlingskomplikationer | |
Mycket vanliga | Injektionsrelaterade reaktioner (systemiska) |
1 Gruppering av rekommenderade termer beaktades vid bestämning av biverkningsfrekvensen och innefattar följande: nasofaryngit, övre luftvägsinfektion, influensa, sinuit, faryngit, rinit, viral övre luftvägsinfektion, tonsillit, akut sinuit, faryngotonsillit, laryngit, streptokockorsakad faryngit, viral rinit, bakteriell sinuit, bakteriell tonsillit, viral faryngit, viral tonsillit, kronisk sinuit, herpes i näsan, trakeit. 2 Gruppering av rekommenderade termer beaktades vid bestämning av biverkningsfrekvensen och innefattar följande: urinvägsinfektion, cystit, escherichiaorsakad urinvägsinfektion, asymtomatisk bakteriuri, bakteriuri. 3 Rapporterades efter godkännande för försäljning (se avsnitt Varningar och försiktighet). 4 Illamående och kräkningar har rapporterats i samband med systemiska injektionsrelaterade reaktioner (se nedan och avsnitt Varningar och försiktighet). 5 Gruppering av rekommenderade termer beaktades för bestämning av biverkningsfrekvens och innefattar följande: förhöjt alaninaminotransferas, förhöjt aspartataminotransferas, förhöjda leverenzymer, förhöjda transaminaser, förhöjt leverfunktionstest, onormal leverfunktion, onormalt leverfunktionstest | |
Beskrivning av utvalda biverkningar
Infektioner
I de kliniska RMS‑studierna i fas III var den totala frekvensen infektioner och allvarliga infektioner hos patienter som behandlades med ofatumumab liknande frekvensen hos de som behandlades med teriflunomid (51,6 % mot 52,7 % respektive 2,5 % mot 1,8 %). Två patienter (0,2 %) avbröt och 11 patienter (1,2 %) gjorde tillfälligt uppehåll i studiebehandlingen på grund av en allvarlig infektion.
Övre luftvägsinfektioner
I dessa studier fick 39,4 % av patienterna som behandlades med ofatumumab övre luftvägsinfektioner, jämfört med 37,8 % av patienterna som behandlades med teriflunomid. Infektionerna var i huvudsak lindriga till medelsvåra och utgjordes oftast av nasofaryngit, övre luftvägsinfektion och influensa.
Systemiska injektionsrelaterade reaktioner
I de kliniska RMS‑studierna i fas III rapporterades SIRR hos 20,6 % av patienterna som fick behandling med ofatumumab.
Incidensen av SIRR var högst vid den första injektionen (14,4 %) och minskade signifikant vid de efterföljande injektionerna (4,4 % vid andra, <3 % från och med den tredje injektionen). SIRR var i huvudsak (99,8 %) lindriga till medelsvåra. Två (0,2 %) MS‑patienter som fick behandling med ofatumumab rapporterade allvarliga men inte livshotande injektionsrelaterade reaktioner. De vanligaste rapporterade symtomen (≥2 %) innefattade feber, huvudvärk, myalgi, frossa och trötthet. Ytterligare rapporterade symtom var illamående (1,7 %) och kräkningar (0,6 %).
Lokala reaktioner vid injektionsstället
I de kliniska RMS‑studierna i fas III rapporterades lokala reaktioner vid injektionsstället hos 10,9 % av patienterna som fick behandling med ofatumumab.
Lokala reaktioner vid injektionsstället var mycket vanliga. De var alla lindriga till medelsvåra och av typen icke‑allvarliga. De vanligaste rapporterade symtomen (≥2 %) innefattade erytem, smärta, klåda och svullnad.
Laboratorieavvikelser
Immunglobuliner
Under de kliniska RMS‑studierna i fas III observerades en minskning i medelvärdet för immunglobulin M (IgM) (30,9 % minskning efter 48 veckor och 38,8 % minskning efter 96 veckor) och ingen koppling till risken för infektioner, inklusive allvarliga infektioner, kunde påvisas.
Hos 14,3 % av patienterna resulterade behandling med ofatumumab i en minskning av IgM ner till ett värde under 0,34 g/l.
Ofatumumab associerades med en övergående minskning i medelnivåerna av immunglobulin G (IgG) på 4,3 % efter 48 veckors behandling, men en förhöjning på 2,2 % efter 96 veckor.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till (se detaljer nedan).
webbplats: www.fimea.fi
Säkerhets‐ och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Doser upp till 700 mg har administrerats i kliniska studier med MS‑patienter, utan dosbegränsande toxicitet. I händelse av överdosering rekommenderas att patienten övervakas för tecken eller symtom på biverkningar och att lämplig symtomatisk behandling sätts in enligt behov.
Ofatumumab har tidigare använts för indikationer relaterade till kronisk lymfatisk leukemi (KLL), i doser upp till 2 000 mg administrerade som intravenös infusion. Ofatumumab administrerat som subkutan injektion har inte utvärderats för dessa indikationer; det är inte godkänt för och får inte användas som behandling vid onkologiska indikationer.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Immunsuppressiva medel, monoklonala antikroppar, ATC‑kod: L04AG12
Verkningsmekanism
Ofatumumab är en helt human monoklonal immunglobulin G1‑antikropp (IgG1‑antikropp) mot CD20. CD20‑molekylen är ett transmembrant fosfoprotein som uttrycks på B‑lymfocyter från pre‑B‑ till moget B‑lymfocytstadium. CD20‑molekylen uttrycks också på en liten andel aktiverade T‑celler. En subkutan administreringsväg för ofatumumab och efterföljande frisättning/absorption från vävnaden möjliggör en gradvis interaktion med B-celler.
Bindningen av ofatumumab till CD20 inducerar lysering av CD20‑positiva B‑celler huvudsakligen genom komplementberoende cytotoxicitet (CDC) och, i lägre grad, genom antikroppsberoende cellmedierad cytotoxicitet (ADCC). Ofatumumab har också visats inducera cellysering i celler med såväl starkt som svagt uttryck av CD20. T‑celler som uttrycker CD20 blir också reducerade av ofatumumab.
Farmakodynamisk effekt
Depletion av B‑celler
I de kliniska RMS‑studierna där man använde ofatumumab 20 mg var 4:e vecka, efter en inledande dosregim på 20 mg dag 1, 7 och 14, resulterade administreringen i en snabb och långvarig minskning av antalet B‑celler till under LLN (definierad som 40 celler/mikroliter) så tidigt som två veckor efter att behandlingen sattes in. Innan underhållsfasen inleddes vid vecka 4 nåddes totala B‑cellsnivåer på <10 celler/mikroliter hos 94 % av patienterna, vilket ökade till 98 % av patienterna vid vecka 12, och de kvarstod så länge som 120 veckor (dvs. medan studiebehandlingen pågick).
Återhämtning av B‑celler
Data från kliniska RMS‑studier i fas III tyder på att mediantiden till återhämtning av B‑cellsnivåerna till LLN eller utgångsläget är 24,6 veckor efter att behandlingen avslutats. Farmakokinetisk B‑cellsmodellering och ‑simulation av återhämtning av B‑celler bekräftar dessa data och förutspår att mediantiden till återhämtning av B‑cellsnivåerna till LLN är 23 veckor efter att behandlingen avslutats.
Immunogenicitet
I RMS‑studier i fas III var den totala incidensen av behandlingsinducerade anti‑läkemedelsantikroppar (ADA) 0,2 % (2 av 914) hos patienter som behandlades med ofatumumab, och inga patienter med ADA som förstärker eller neutraliserar behandlingen identifierades. Effekten av positiva ADA-titrar på farmakokinetik, säkerhetsprofil eller B‑cellskinetik kan inte bedömas med tanke på den låga förekomsten av ADA i samband med ofatumumab.
Klinisk effekt och säkerhet
Effekten och säkerheten av ofatumumab utvärderades i två randomiserade, dubbelblinda, aktivt kontrollerade pivotala studier i fas III med identisk design (studie 1 [ASCLEPIOS I] och studie 2 [ASCLEPIOS II]) med patienter i åldrarna 18 till 55 år med skovvis multipel skleros (RMS), vars funktionsnedsättningsstatus vid screeningen gav EDSS‑poäng (Expanded Disability Status Scale) från 0 till 5,5 och som hade haft minst ett dokumenterat skov inom det senaste året eller två skov under de två senaste åren eller gadoliniumförstärkande (Gd‑positiva) lesioner vid MRT‑undersökningar inom det senaste året. Både nydiagnostiserade patienter och patienter som bytte från sin nuvarande behandling inkluderades.
I de två studierna randomiserades 927 respektive 955 patienter med RMS i förhållande 1:1 till att få antingen subkutana injektioner med ofatumumab 20 mg var 4:e vecka med början vid vecka 4, efter en initial dosregim på tre doser om 20 mg per vecka under de första 14 dagarna (dag 1, 7 och 14), eller teriflunomid 14 mg kapslar oralt en gång per dag. Patienterna fick även matchande placebo som motsvarade den andra behandlingsarmen för att säkerställa blindning (dubbelplacebo‑design).
Behandlingstiden för enskilda patienter varierade utgående från när kriterierna för avslutning av studien uppfylldes. För båda studierna var medianbehandlingstiden 85 veckor, och 33,0 % av patienterna i ofatumumabgruppen respektive 23,2 % av patienterna i teriflunomidgruppen fick behandling i mer än 96 veckor.
Demografi och utgångskarakteristika var välbalanserade mellan behandlingsarmarna och de båda studierna (se tabell 2). Medelåldern var 38 år, den genomsnittliga sjukdomstiden var 8,2 år från att det första symtomet uppträdde, och den genomsnittliga EDSS‑poängen var 2,9; 40 % av patienterna hade inte tidigare fått sjukdomsmodifierande behandling och 40 % hade Gd‑förstärkande T1‑lesioner vid MRT‑undersökningen vid studiestart.
Det primära effektmåttet för båda studierna var den årliga frekvensen av bekräftade skov (ARR) baserat på EDSS‑poäng. Sekundära huvudeffektmått innefattade tiden till förvärrad funktionsnedsättning på EDSS‑skalan (bekräftad vid 3 och 6 månader), definierad som en ökning av EDSS‑poängen om ≥1,5, ≥1 eller ≥0,5 hos patienter med EDSS‑poäng vid studiestart på 0, 1 till 5 respektive ≥5,5. Ytterligare sekundära huvudeffektmått innefattade antalet Gd‑förstärkande T1‑lesioner per MRT‑undersökning, den årliga frekvensen av nya eller förstorade T2‑lesioner och koncentrationen av Neurofilament light (NfL) i serum. Sekundära huvudeffektmått relaterade till funktionsnedsättning utvärderades i en metaanalys av kombinerade data från ASCLEPIOS studie 1 och studie 2 enligt studieprotokollen.
Tabell 2 Demografi och utgångskarakteristika
| Karakteristika | Studie 1 (ASCLEPIOS I) | Studie 2 (ASCLEPIOS II) | ||
| Ofatumumab (N = 465) | Teriflunomid (N = 462) | Ofatumumab (N = 481) | Teriflunomid (N = 474) | |
| Ålder (medel ± standardavvikelse; år) | 39 ± 9 | 38 ± 9 | 38 ± 9 | 38 ± 9 |
| Kön (kvinna; %) | 68,4 | 68,6 | 66,3 | 67,3 |
| Sjukdomstid från MS‑diagnos (medel/median; år) | 5,77/3,94 | 5,64/3,49 | 5,59/3,15 | 5,48/3,10 |
| Andel patienter som tidigare fått sjukdomsmodifierande behandling (%) | 58,9 | 60,6 | 59,5 | 61,8 |
| Antal skov under de senaste 12 månaderna | 1,2 | 1,3 | 1,3 | 1,3 |
| EDSS‑poäng (medel/median) | 2,97/3,00 | 2,94/3,00 | 2,90/3,00 | 2,86/2,50 |
| Medeltal av totala volymen av T2‑lesioner (cm3) | 13,2 | 13,1 | 14,3 | 12,0 |
| Patienter med Gd+ T1‑lesioner (%) | 37,4 | 36,6 | 43,9 | 38,6 |
| Antal Gd+ T1‑lesioner (medel) | 1,7 | 1,2 | 1,6 | 1,5 |
Effektresultaten för båda studierna sammanfattas i tabell 3, figur 1 och figur 2.
I båda fas III‑studierna uppvisade ofatumumab i jämförelse med teriflunomid en signifikant minskning av den årliga skovfrekvensen på 50,5 % respektive 58,4 %.
Den prespecificerade metaanalysen av kombinerade data visade att ofatumumab i jämförelse med teriflunomid signifikant minskade risken för 3 månaders bekräftad progression av funktionsnedsättning med 34,3 % och risken för 6 månaders bekräftad progression av funktionsnedsättning med 32,4 % (se figur 1).
Ofatumumab jämfört med teriflunomid minskade signifikant antalet Gd‑förstärkande T1‑lesioner med 95,9 % och frekvensen av nya eller förstorade T2‑lesioner med 83,5 % (värdena representerar medeltal av minskningarna för de kombinerade studierna).
Ofatumumab jämfört med teriflunomid minskade signifikant NfL-koncentrationerna från den första bedömningen efter 3 månader (se tabell 3 och figur 2).
En liknande effekt av ofatumumab på de viktigaste effektresultaten jämfört med teriflunomid observerades i de två fas III-studierna i explorativa subgrupper definierade efter kön, ålder, kroppsvikt, tidigare steroidfri MS-behandling samt funktionsnedsättning vid studiestart och sjukdomsaktivitet.
Tabell 3 Översikt av de viktigaste resultaten från fas III‑studier av RMS
| Effektmått | Studie 1 (ASCLEPIOS I) | Studie 2 (ASCLEPIOS II) | ||
Ofatumumab 20 mg (n = 465) | Teriflunomid 14 mg (n = 462) | Ofatumumab 20 mg (n = 481) | Teriflunomid 14 mg (n = 474) | |
| Effektmått baserade på separata studier | ||||
| Årlig skovfrekvens (ARR) (primärt effektmått)1 | 0,11 | 0,22 | 0,10 | 0,25 |
| Frekvensreduktion | 50,5 % (p <0,001) | 58,4 % (p <0,001) | ||
| Medeltal av antalet Gd‑förstärkande T1‑lesioner per MRT‑undersökning | 0,0115 | 0,4555 | 0,0317 | 0,5172 |
| Relativ reduktion | 97,5 % (p <0,001) | 93,9 % (p <0,001) | ||
| Antalet nya eller förstorade T2‑lesioner per år | 0,72 | 4,00 | 0,64 | 4,16 |
| Relativ reduktion | 81,9 % (p <0,001) | 84,6 % (p <0,001) | ||
| NfL efter 3 månader (pg/ml) | 8,80 | 9,41 | 8,92 | 10,02 |
| Relativ reduktion | 7 % (p = 0,011) | 11 % (p <0,001) | ||
| Effektmått baserade på prespecificerade metaanalyser | ||||
Andel patienter med en 3 månaders bekräftad progression av funktionsnedsättning2 Riskreduktion | 10,9 % ofatumumab jämfört med 15,0 % teriflunomid 34,3 % (p = 0,003) | |||
Andel patienter med en 6 månaders bekräftad progression av funktionsnedsättning2 Riskreduktion | 8,1 % ofatumumab jämfört med 12,0 % teriflunomid 32,4 % (p = 0,012) | |||
1 Bekräftade skov (förenade med en kliniskt relevant förändring i EDSS). 2 Kaplan–Meier‑skattningar efter 24 månader. 3 och 6 månaders bekräftad progression av funktionsnedsättning bedömdes utifrån prospektivt planerad analys av kombinerade data från de två fas III‑studierna och definierad som en kliniskt betydelsefull ökning i EDSS som kvarstått i minst 3 respektive 6 månader. En kliniskt betydelsefull ökning i EDSS definieras som en ökning på minst 1,5 poäng om EDSS‑poängen vid studiestart var 0, en ökning på minst 1,0 poäng om EDSS‑poängen vid studiestart var 1,0–5,0 poäng och en ökning på minst 0,5 poäng om EDSS‑poängen vid studiestart var 5,5 poäng eller högre. | ||||
Figur 1 Tid till första 3‑månaders bekräftad progression av funktionsnedsättning per behandling (ASCLEPIOS studie 1 och studie 2 kombinerade, hela analyspopulationen)
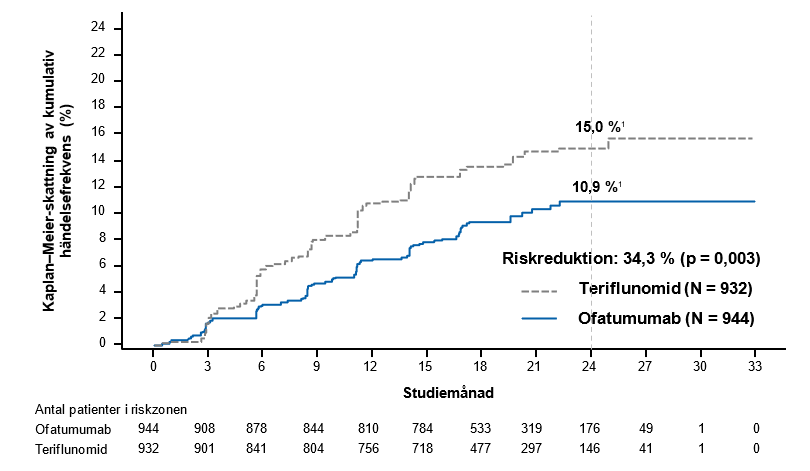
1 Värdena som visas på kurvorna representerar Kaplan–Meier‑skattningar av risken för händelsen vid 24 månader (markerad med den vertikala streckade linjen).
Figur 2 NfL-koncentrationer i serum efter behandling (ASCLEPIOS studie 1 och studie 2 kombinerade, hela analyspopulationen)
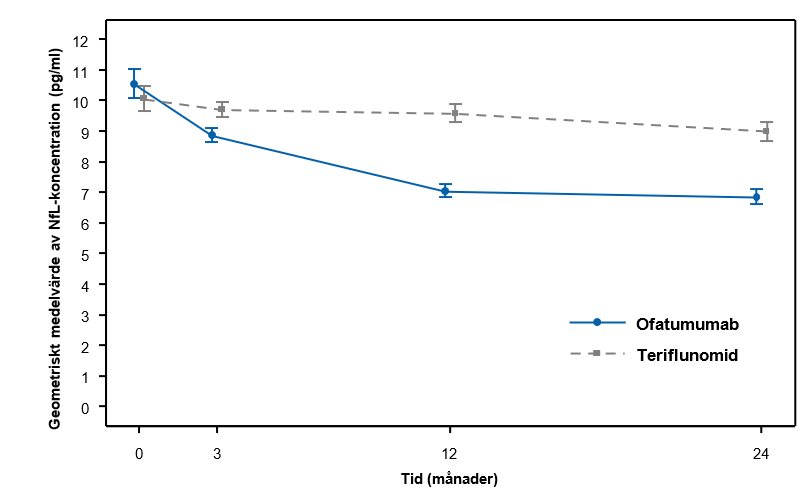
Linjediagrammen representerar de justerade geometriska medelvärdena med 95 % CI vid varje tidpunkt som kommer från modellen med upprepade mätningar. Geometriska medelvärden vid baslinjen härleds som exponentierat aritmetiskt medelvärde av naturlig logaritm av råvärden för NfL-koncentrationer i serum.
I fas III‑studierna var andelen patienter med biverkningar (83,6 % vs. 84,2 %) och biverkningar som ledde till avbrytande (5,7 % vs. 5,2 %) likartade i ofatumumab- och teriflunomidgrupperna.
Pediatrisk population
Europeiska läkemedelsmyndigheten har senarelagt kravet att skicka in studieresultat för Kesimpta för en eller flera grupper av den pediatriska populationen för behandling av multipel skleros (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Farmakokinetiska egenskaper
Absorption
Efter subkutan administrering har ofatumumab en förlängd frisättnings-/absorptionsprofil (Tmax på 4,3 dagar) och absorberas i huvudsak via lymfsystemet.
En subkutan dos om 20 mg per månad resulterar i en genomsnittlig AUCtau på 483 mikrogram*timme/ml och ett genomsnittligt Cmax på 1,43 mikrogram/ml vid steady state.
Distribution
Distributionsvolymen vid steady state uppskattades vara 5,42 liter efter upprepad subkutan administrering av ofatumumab med en dos om 20 mg.
Metabolism
Ofatumumab är ett protein vars förmodade metabolismväg är nedbrytning till små peptider och aminosyror via ubikvitära proteolytiska enzymer.
Eliminering
Ofatumumab elimineras på två sätt: via en målmedierad rutt som har samband med bindning till B‑celler och en måloberoende rutt medierad av ospecifik endocytos följd av intracellulär katabolism, liksom för andra IgG‑molekyler. Närvaro av B‑celler vid utgångsläget resulterar i en större andel av målmedierad clearance av ofatumumab vid behandlingsstarten. Dosering av ofatumumab leder till potent depletion av B‑celler, vilket resulterar i minskat totalclearance.
Halveringstiden vid steady state uppskattades vara cirka 11 dagar efter upprepad subkutan administrering av ofatumumab med en dos om 20 mg.
Linjäritet/icke‑linjäritet
Ofatumumab hade icke‑linjär farmakokinetik relaterad till dess minskande clearance över tid.
Särskilda populationer
Vuxna över 55 år
Inga farmakokinetiska studier har utförts med ofatumumab till patienter över 55 år, på grund av begränsad klinisk erfarenhet (se avsnitt Dosering och administreringssätt).
Pediatrisk population
Inga farmakokinetiska studier har utförts med ofatumumab hos pediatriska patienter yngre än 18 år.
Kön
Kön hade en ringa effekt (12 %) på central distributionsvolym för ofatumumab i en populationsanalys mellan olika studier, där högre värden för Cmax och AUC iakttogs hos kvinnliga patienter (48 % av patienterna i denna analys var män och 52 % var kvinnor). Dessa effekter anses inte vara kliniskt relevanta och ingen dosjustering rekommenderas.
Kroppsvikt
Baserat på resultaten av en tvärstudieanalys av populationen identifierades kroppsvikt som en kovariat för exponering (Cmax och AUC) för ofatumumab hos RMS-patienter. Kroppsvikt påverkade dock inte de säkerhets- och effektmått som utvärderats i de kliniska studierna. Därför är dosjustering inte nödvändig.
Nedsatt njurfunktion
Inga specifika studier av ofatumumab hos patienter med nedsatt njurfunktion har utförts.
Patienter med lindrigt nedsatt njurfunktion ingick i de kliniska studierna. Det finns ingen erfarenhet av patienter med måttligt eller kraftigt nedsatt njurfunktion. Eftersom ofatumumab inte utsöndras via urinen förväntas dock inte patienter med nedsatt njurfunktion behöva dosändringar.
Nedsatt leverfunktion
Inga studier av ofatumumab hos patienter med nedsatt leverfunktion har utförts.
Eftersom levermetabolismen av monoklonala antikroppar som ofatumumab är försumbar, förväntas nedsatt leverfunktion inte inverka på dess farmakokinetik. Därför förväntas inte patienter med nedsatt leverfunktion behöva dosändringar.
Prekliniska säkerhetsuppgifter
Gängse studier avseende allmäntoxicitet och säkerhetsfarmakologi visade inte några särskilda risker för människa.
Varken karcinogenicitets- eller mutagenicitetsstudier har utförts med ofatumumab. Som en antikropp förväntas inte ofatumumab interagera direkt med DNA.
Utvecklingsstudier på embryo/foster och pre‑/postnatala utvecklingsstudier (ePPND‑studier) på apor, visade att exponering för intravenöst ofatumumab under dräktighetstiden inte orsakade någon toxicitet hos den dräktiga honan, ingen teratogenicitet och inga biverkningar med avseende på utvecklingen hos embryo/foster eller den pre-/postnatala utvecklingen.
I dessa studier påvisades ofatumumab i blodet hos fostren och ungarna, vilket bekräftade överföring via placenta och att exponeringen för ofatumumab hos foster kvarstår postnatalt (lång halveringstid för den monoklonala antikroppen). Exponering för ofatumumab under dräktighetstiden ledde till förväntad depletion av CD20‑positiva B‑celler hos moderdjuren och deras foster och ungar, i kombination med minskad mjältvikt (utan histologiskt korrelat) hos fostren och ett minskat humoralt immunsvar på KLH (keyhole limpet haemocyanin) hos ungar vid höga doser. Alla dessa förändringar var reversibla under den 6 månader långa postnatala perioden. Hos ungar observerades tidig postnatal mortalitet vid en dos som var 160 gånger högre än den terapeutiska dosen (baserat på AUC) och den berodde sannolikt på potentiella infektioner till följd av immunmodulering. NOAEL (No Observed Adverse Effect Level) relaterad till den farmakologiska aktiviteten av ofatumumab hos ungar i ePPND‑studien, leder till en minst 22‑faldig AUC‑baserad säkerhetsmarginal när exponeringen på NOAEL‑nivån hos moderdjuren jämförs med human exponering vid den terapeutiska dosen 20 mg per månad.
I en studie specifikt inriktad på fertilitet hos apor, var fertilitetsmåtten hos hanar och honor opåverkade.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
L‑arginin
Natriumacetattrihydrat
Natriumklorid
Polysorbat 80 (E433)
Dinatriumedetatdihydrat
Saltsyra (för justering av pH)
Vatten för injektionsvätskor
Inkompatibiliteter
Då blandbarhetsstudier saknas får detta läkemedel inte blandas med andra läkemedel.
Hållbarhet
3 år
Särskilda förvaringsanvisningar
Kesimpta 20 mg injektionsvätska, lösning i förfylld spruta
Förvaras i kylskåp (2 °C–8 °C). Får ej frysas.
Vid behov kan Kesimpta förvaras i rumstemperatur (högst 30 °C) under en enda period på upp till 7 dagar. Om det inte används under denna period kan Kesimpta sedan återföras till kylskåpet i högst 7 dagar.
Förvara den förfyllda sprutan i ytterkartongen. Ljuskänsligt.
Kesimpta 20 mg injektionsvätska, lösning i förfylld injektionspenna
Förvaras i kylskåp (2 °C–8 °C). Får ej frysas.
Vid behov kan Kesimpta förvaras i rumstemperatur (högst 30 °C) under en enda period på upp till 7 dagar. Om det inte används under denna period kan Kesimpta sedan återföras till kylskåpet i högst 7 dagar.
Förvara den förfyllda injektionspennan i ytterkartongen. Ljuskänsligt.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
KESIMPTA injektioneste, liuos, esitäytetty kynä
20 mg (L:ei) 1 kpl (0,4 ml, 50 mg/ml) (1256,14 €)
PF-selosteen tieto
Kesimpta 20 mg injektionsvätska, lösning i förfylld spruta
Kesimpta tillhandahålls i en glasspruta för engångsbruk försedd med en nål av rostfritt stål, en kolvpropp och ett styvt nålskydd. Sprutan har en sprutkolv och ett stickskydd.
Kesimpta finns i enkelförpackningar innehållande 1 förfylld spruta och i flerpack som innehåller 3 (3 förpackningar med 1) förfyllda sprutor.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Kesimpta 20 mg injektionsvätska, lösning i förfylld injektionspenna
Kesimpta tillhandahålls i en glasspruta för engångsbruk försedd med en nål av rostfritt stål, en kolvpropp och ett styvt nålskydd. Sprutan är insatt i en autoinjektor.
Kesimpta finns i enkelförpackningar innehållande 1 förfylld injektionspenna och i flerpack som innehåller 3 (3 förpackningar med 1) förfyllda injektionspennor.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Läkemedlets utseende:
Lösningen är klar till lätt opaliserande och färglös till svagt brungul.
Särskilda anvisningar för destruktion och övrig hantering
Anvisningar om hantering av den förfyllda sprutan
Den förfyllda sprutan ska tas ut ur kylskåpet ca 15 till 30 minuter före injektionen så att den hinner uppnå rumstemperatur. Den förfyllda sprutan ska förvaras i originalkartongen tills den ska användas, och nålskyddet ska inte tas bort förrän strax innan injektionen ges. Före användning ska lösningen inspekteras visuellt genom sprutans fönster. Den förfyllda sprutan ska inte användas om vätskan innehåller synliga partiklar eller är grumlig.
Uttömmande anvisningar om administrering återfinns i bipacksedeln.
Anvisningar om hantering av den förfyllda injektionspennan
Den förfyllda injektionspennan ska tas ut ur kylskåpet ca 15 till 30 minuter före injektionen så att den hinner uppnå rumstemperatur. Den förfyllda injektionspennan ska förvaras i originalkartongen tills den ska användas, och skyddshatten ska inte tas bort förrän strax innan injektionen ges. Före användning ska lösningen inspekteras visuellt genom injektionspennans fönster. Den förfyllda injektionspennan ska inte användas om vätskan innehåller synliga partiklar eller är grumlig.
Uttömmande anvisningar om administrering återfinns i bipacksedeln.
Destruktion
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
KESIMPTA injektioneste, liuos, esitäytetty kynä
20 mg 1 kpl
- Ylempi erityiskorvaus (100 %). Dimetyylifumaraatti, diroksimeelifumaraatti, glatirameeriasetaatti, interferoni beeta, ofatumumabi ja ponesimodi: Aaltoilevan tai aaltoilevaan läheisesti rinnastettavan MS-taudin hoito erityisin edellytyksin (157).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Dimetyylifumaraatti, diroksimeelifumaraatti, glatirameeriasetaatti, interferoni beeta, ofatumumabi ja ponesimodi: Aaltoilevan ja aaltoilevaan läheisesti rinnastettavan MS-taudin hoito erityisin edellytyksin (303).
Atc-kod
L04AG12
Datum för översyn av produktresumén
19.06.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com