
RETSEVMO tabletti, kalvopäällysteinen 40 mg, 80 mg
Vaikuttavat aineet ja niiden määrät
Retsevmo 40 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 40 mg selperkatinibia.
Retsevmo 80 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 80 mg selperkatinibia.
Retsevmo 120 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 120 mg selperkatinibia.
Retsevmo 160 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 160 mg selperkatinibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti).
Kliiniset tiedot
Käyttöaiheet
Retsevmo on tarkoitettu monoterapiaksi aikuispotilaille, joilla on
-
pitkälle edennyt RET‑fuusiopositiivinen ei‑pienisoluinen keuhkosyöpä, jota ei ole hoidettu aikaisemmin RET-estäjällä. Biomarkkereihin perustuva potilasvalinta, katso kohta Annostus ja antotapa.
Retsevmo on tarkoitettu monoterapiaksi aikuisille ja 2-vuotiaille ja sitä vanhemmille pediatrisille potilaille, joilla on
-
pitkälle edennyt RET‑fuusiopositiivinen kilpirauhassyöpä, joka on radiojodihoidolle refraktorinen (jos radiojodihoito on ollut tarkoituksenmukaista). Biomarkkereihin perustuva potilasvalinta, katso kohta Annostus ja antotapa.
-
pitkälle edennyt RET‑mutaatiopositiivinen medullaarinen kilpirauhassyöpä. Biomarkkereihin perustuva potilasvalinta, katso kohta Annostus ja antotapa.
-
pitkälle edennyt RET-fuusiopositiivinen kiinteä kasvain, silloin kun hoitovaihtoehtojen, joiden kohteena ei ole RET, kliininen hyöty on rajallinen tai niitä ei ole jäljellä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka). Biomarkkereihin perustuva potilasvalinta, katso kohta Annostus ja antotapa.
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Retsevmo-hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Potilasvalinta
Ennen Retsevmo-hoidon aloittamista RET‑geenimutaation (medullaarinen kilpirauhassyöpä) tai -fuusion (kaikki muut kasvaintyypit) esiintyminen on arvioitava CE-merkityllä IVD-diagnostiikkatestillä, jolla on vastaava käyttötarkoitus. Mikäli CE-merkittyä IVD-testiä ei ole saatavilla, arviointi tulee tehdä tähän käyttötarkoitukseen validoidulla vaihtoehtoisella testillä.
Annostus
Suositeltu Retsevmo‑annos vähintään 12-vuotiaille potilaille perustuu painoon:
-
alle 50 kg: 120 mg kahdesti vuorokaudessa.
-
vähintään 50 kg: 160 mg kahdesti vuorokaudessa
Vähintään 2-vuotiaiden ja alle 12-vuotiaiden pediatristen potilaiden suositeltu Retsevmo-annos perustuu seuraaviin kehon pinta-alan (BSA) luokkiin (Taulukko 1).
Taulukko 1 Suositeltu annos vähintään 2-vuotiaille ja alle 12-vuotiaille pediatrisille potilaille
Kehon pinta-ala | Suositeltu annos |
0,45–0,65 m2 | 40 mg kolmesti päivässä |
0,66–1,08 m2 | 80 mg kahdesti päivässä |
1,09–1,52 m2 | 120 mg kahdesti päivässä |
≥1,53 m2 | 160 mg kahdesti päivässä |
Retsevmoa ei suositella pediatrisille potilaille, joiden kehon pinta-ala on alle 0,45 m2.
Jos potilas oksentaa tai annos jää väliin, potilasta neuvotaan ottamaan seuraava annos tavanomaiseen aikaan; uutta annosta ei pidä ottaa.
Hoitoa jatketaan, kunnes tauti etenee tai ilmenee toksisuutta, joka ei ole hyväksyttävää.
Käytössä olevaa selperkatinibiannosta on pienennettävä 50 %, jos samanaikaisesti käytetään vahvaa CYP3A:n estäjää. Jos CYP3A:n estäjän käyttö lopetetaan, selperkatinibiannos on suurennettava (kyseisen estäjän 3–5 puoliintumisajan kuluttua) annokseen, jota käytettiin ennen estäjän käytön aloittamista.
Annosmuutokset
Joidenkin haittavaikutusten hoito voi edellyttää hoidon tauottamista ja/tai annoksen pienentämistä. Retsevmo‑annosmuutoksista esitetään yhteenveto taulukossa 2 ja taulukossa 3.
Taulukko 2 Suositellut Retsevmo‑annoksen muutokset haittavaikutusten yhteydessä painon ja BSA:n mukaan
Annosmuutos | Aikuiset ja nuoret ≥ 50 kg | Aikuiset ja nuoret | Pediatriset 2 - < 12 | Pediatriset 2 - < 12 | Pediatriset 2 - < 12 | Pediatriset 2 - < 12 |
Aloitusannos | 160 mg suun kautta kahdesti päivässä | 120 mg suun kautta kahdesti päivässä | 160 mg suun kautta kahdesti päivässä | 120 mg suun kautta kahdesti päivässä | 80 mg suun kautta kahdesti päivässä | 40 mg suun kautta kolmesti päivässä |
Ensimmäinen annoksen pienennys | 120 mg suun kautta kahdesti päivässä | 80 mg suun kautta kahdesti päivässä | 120 mg suun kautta kahdesti päivässä | 80 mg suun kautta kahdesti päivässä | 40 mg suun kautta kahdesti päivässä | 40 mg suun kautta kahdesti päivässä |
Toinen annoksen pienennys | 80 mg suun kautta kahdesti päivässä | 40 mg suun kautta kahdesti päivässä | 80 mg suun kautta kahdesti päivässä | 40 mg suun kautta kahdesti päivässä | 40 mg suun kautta kerran päivässä | 40 mg suun kautta kerran päivässä |
Kolmas annoksen pienennys* | 40 mg suun kautta kahdesti päivässä | Ei soveltuva | 40 mg suun kautta kahdesti päivässä | 40 mg suun kautta kerran päivässä | Lopetettava pysyvästi | Lopetettava pysyvästi |
*Selperkatinibihoito on lopetettava pysyvästi potilailla, jotka eivät siedä kolmea annoksen pienentämistä.
Pediatristen potilaiden annosmuutokset haittavaikutusten yhteydessä määritetään kulloinkin käytössä olevan annoksen perusteella noudattaen samaa vaiheittaista annostason muuttamista koskevaa lähestymistapaa kuin aikuisilla jäljempänä kuvattujen haittavaikutusten osalta (taulukko 3), ellei toisin mainita.
Taulukko 3 Suositellut annosmuutokset haittavaikutusten yhteydessä
Haittavaikutus |
| Annosmuutokset |
Alaniiniaminotransferaasi (ALAT)‑ tai aspartaattiaminotransferaasi (ASAT) ‑arvon suureneminen | Aste 3 tai 4 |
|
Yliherkkyys | Kaikki asteet |
|
QT‑ajan piteneminen | Aste 3 |
|
Aste 4 |
| |
Hypertensio | Aste 3 |
|
Aste 4 |
| |
Verenvuototapahtumat | Aste 3 |
|
Aste 4 |
| |
Interstitiaalinen keuhkosairaus (ILD)/keuhkokuume | Aste 2 |
|
Aste 3 tai 4 |
| |
Muut haittavaikutukset | Aste 3 tai 4 |
|
Erityisryhmät
Iäkkäät
Annosta ei tarvitse muuttaa iän perusteella (ks. kohta Farmakokinetiikka).
Hoidon aikana ilmenneissä haittatapahtumissa tai selperkatinibin tehossa ei yleisesti havaittu eroja ≥ 65‑vuotiaiden ja nuorempien potilaiden välillä. Tietoa on rajallisesti ≥ 75‑vuotiaista potilaista.
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen, jos potilaalla on lievä, keskivaikea tai vaikea munuaisten vajaatoiminta. Selperkatinibin antamisesta loppuvaiheen munuaisten vajaatoimintaa sairastaville tai dialyysihoidossa oleville potilaille ei ole tietoja (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Maksan vajaatoimintapotilaiden huolellinen seuranta on tärkeää. Annosta ei tarvitse muuttaa, jos potilaalla on lievä (Child–Pugh‑luokitus A) tai keskivaikea (Child–Pugh‑luokitus B) maksan vajaatoiminta. Aikuisilla, joilla on vaikea (Child–Pugh‑luokitus C) maksan vajaatoiminta, selperkatinibin annostus on 80 mg kahdesti vuorokaudessa (kohta Farmakokinetiikka). Selperkatinibin käyttöä ei ole tutkittu alle 18-vuotiailla potilailla, joilla on maksan vajaatoiminta. Hoito alle 18-vuotiailla potilailla, joilla on vaikea maksan vajaatoiminta, on aloitettava varoen. Annostuksen muuttamisen tulee perustua kliiniseen harkintaan ja yksilölliseen potilaan siedettävyyteen. Alle 12-vuotiailla pediatrisilla potilailla, joilla on vaikea maksan vajaatoiminta, suositeltu aloitusannos esitetään taulukossa 4.
Taulukko 4 Suositeltu aloitusannos alle 12-vuotiaille pediatrisille potilaille, joilla on vaikea maksan vajaatoiminta
Kehon pinta-ala | Suositeltu aloitusannos |
0,45–0,65 m2 | 40 mg kerran päivässä |
0,66–1,08 m2 | 40 mg kahdesti päivässä |
1,09–1,52 m2 | 40 mg kahdesti päivässä |
≥ 1,53 m2 | 80 mg kahdesti päivässä |
Pediatriset potilaat
Retsevmo‑valmistetta ei saa käyttää alle 2‑vuotiailla lapsilla, koska tietoja ei ole saatavilla.
Retsevmo on tarkoitettu vähintään 2‑vuotiaille ja sitä vanhemmille potilaille RET‑mutaatiopositiivisen medullaarisen kilpirauhassyövän,RET‑fuusiopositiivisen kilpirauhassyövän, ja RET-fuusiopositiivisten kiinteiden kasvainten hoitoon (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka). Vähintään 12-vuotiailla potilailla annostus perustuu painoon (ks. kohta Annostus ja antotapa). Vähintään 2-vuotiaiden ja alle 12-vuotiaiden potilaiden annostus perustuu kehon pinta-alaan. Perustuen prekliinisen tutkimuksen tuloksiin (ks. kohta Prekliiniset tiedot turvallisuudesta), nuorten potilaiden kasvuruston avoimia kasvulevyjä tulee seurata. Annoksen keskeyttämistä tai hoidon lopettamista tulee harkita kasvulevyn poikkeavuuksien vakavuuden ja yksilöllisen riski-hyötyarvion perusteella.
Antotapa
Retsevmo otetaan suun kautta.
Tabletit niellään kokonaisina tasaisen vaikutuksen varmistamiseksi (potilas ei saa murskata, pureskella eikä jakaa tablettia ennen nielemistä. Tabletit voidaan ottaa ruoan kanssa tai ilman. Jos suurempia tabletteja on vaikea niellä kokonaisina, potilaat voivat harkita useiden pienempien tablettien ottamista tarvittavan annoksen saavuttamiseksi.
Annokset on otettava suunnilleen samaan aikaan joka päivä.
Retsevmo on otettava ruokailun yhteydessä, jos protonipumpun estäjää käytetään samanaikaisesti (ks. kohta Yhteisvaikutukset).
Retsevmo on otettava 2 tuntia ennen tai 10 tuntia myöhemmin kuin H2‑reseptorisalpaaja (ks. kohta Yhteisvaikutukset).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Teho eri kasvaintyypeissä
Selperkatinibin hyöty on osoitettu yksihaaraisissa tutkimuksissa, joihin osallistui pieni joukko potilaita, joilla oli RET-geenifuusiota ilmentävä kasvain. Selperkatinibilla osoitettu hyöty perustui objektiiviseen hoitovasteeseen ja vasteen kestoon rajallisella määrällä kasvaintyyppejä. Teho voi vaihdella määrällisesti kasvaintyypin ja samanaikaisten genomimuutosten mukaan (ks. kohta Farmakodynamiikka). Tästä johtuen selperkatinibia tulee käyttää vain, jos ei ole olemassa tai jäljellä hoitovaihtoehtoja, joista on osoitettu olevan kliinistä hyötyä (toisin sanoen, tyydyttäviä hoitovaihtoehtoja ei ole).
Interstitiaalinen keuhkosairaus (ILD)/keuhkokuume
Selperkatinibilla hoidetuilla potilailla on ilmoitettu vakavia, henkeä uhkaavia tai kuolemaan johtaneita ILD/keuhkokuume -tapauksia (ks. kohta Haittavaikutukset). Potilaita tulee tarkkailla ILD:hen/keuhkokuumeeseen viittaavien keuhko-oireiden varalta. Selperkatinibihoito keskeytetään ja jokainen potilas, jolla on akuutteja tai pahenevia hengitystieoireita, jotka voivat viitata ILD:hen (esim. hengenahdistus, yskä ja kuume), tutkitaan ILD:n osalta ja hoidetaan tarkoituksenmukaisella tavalla. ILD:n/keuhkokuumeen vaikeusasteen perusteella selperkatinibiannos on keskeytettävä, pienennettävä tai lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Alaniiniaminotransferaasin (ALAT) tai aspartaattiaminotransferaasin (ASAT) suureneminen
Selperkatinibia saaneilla potilailla on ilmoitettu asteen ≥ 3 ALAT‑arvon suurenemista ja asteen ≥ 3 ASAT‑arvon suurenemista (ks. kohta Haittavaikutukset). ALAT‑ ja ASAT‑arvoa on seurattava ennen selperkatinibihoidon alkua, 2 viikon välein ensimmäisten 3 hoitokuukauden ajan, kerran kuukaudessa seuraavien 3 hoitokuukauden ajan ja muuten kliinisen tarpeen mukaan. ALAT‑ tai ASAT‑arvon suurenemisen asteesta riippuen selperkatinibiannosta saattaa olla tarpeen muuttaa (ks. kohta Annostus ja antotapa).
Hypertensio
Selperkatinibihoitoa saaneilla potilailla on ilmoitettu hypertensiota (ks. kohta Haittavaikutukset). Potilaan verenpaineen on oltava hallinnassa ennen selperkatinibihoidon aloitusta. Verenpainetta on seurattava selperkatinibihoidon aikana ja hoidettava tarvittaessa tavanomaisella verenpainelääkityksellä. Hypertension asteesta riippuen selperkatinibiannosta saattaa olla tarpeen muuttaa (ks. kohta Annostus ja antotapa). Selperkatinibihoito on lopetettava pysyvästi, jos lääketieteellisesti merkittävää hypertensiota ei saada hallintaan verenpainelääkityksellä.
QT‑ajan piteneminen
Selperkatinibihoitoa saaneilla potilailla on ilmoitettu QT‑ajan pitenemistä (ks. kohta Farmakodynamiikka). Selperkatinibia on käytettävä varoen, jos potilaalla on esim. synnynnäinen tai hankinnainen pitkän QT‑ajan oireyhtymä tai muu kliininen tila, joka altistaa rytmihäiriöille.
Potilaan QTcF‑ajan on oltava ≤ 470 ms (≤ 440 ms alle 12-vuotiailla potilailla) ja seerumin elektrolyyttiarvojen viitealueella ennen selperkatinibihoidon aloittamista. Kaikkien potilaiden EKG:tä ja seerumin elektrolyyttiarvoja on seurattava viikon kuluttua selperkatinibihoidon aloittamisesta, vähintään kerran kuukaudessa ensimmäisten 6 kuukauden ajan ja muuten kliinisen tarpeen mukaan. Seurantatiheyttä on muokattava riskitekijöiden (mm. ripulin, oksentelun ja/tai pahoinvoinnin) perusteella. Hypokalemia, hypomagnesemia ja hypokalsemia on korjattava ennen selperkatinibihoidon aloittamista ja hoidon aikana. QT-aikaa on seurattava EKG:llä useammin potilailla, joita hoidetaan samanaikaisesti lääkevalmisteilla, joiden tiedetään pidentävän QT-aikaa. Selperkatinibihoito voidaan joutua tauottamaan tai annosta voidaan joutua muuttamaan (ks. kohta Annostus ja antotapa).
Kilpirauhasen vajaatoiminta
Selperkatinibihoitoa saaneilla potilailla on ilmoitettu kilpirauhasen vajaatoimintaa (ks. kohta Haittavaikutukset). Kaikille potilaille suositellaan kilpirauhasen toiminnan lähtötason laboratoriomittausta. Potilaat, joilla on kilpirauhasen vajaatoiminta, tulee hoitaa tavanomaisen kliinisen hoitokäytännön mukaisesti ennen selperkatinibihoidon aloittamista. Kaikkia potilaita on tarkkailtava huolellisesti kilpirauhasen toimintahäiriön oireiden ja löydösten varalta selperkatinibihoidon aikana. Kilpirauhasen toimintaa on seurattava säännöllisesti selperkatinibihoidon aikana. Potilaita, joille kehittyy kilpirauhasen toimintahäiriö, hoidetaan tavanomaisen kliinisen hoitokäytännön mukaisesti. Näiden potilaiden vaste levotyroksiini (T4) -korvaushoidolle voi olla riittämätön, koska selperkatinibi voi estää levotyroksiinin muuttumista trijodityroniiniksi (T3) ja liotyroniinilisä voi olla tarpeen (ks. kohta Yhteisvaikutukset).
Vahvat CYP3A4:n indusorit
Vahvojen CYP3A4:n indusorien samanaikaista käyttöä on vältettävä, sillä yhdistelmän käyttöön liittyy selperkatinibin tehon heikkenemisen riski (ks. kohta Yhteisvaikutukset).
Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy naisilla ja miehillä
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä hoidon aikana ja vähintään yhden viikon ajan viimeisen selperkatinibiannoksen jälkeen. Miesten, joiden kumppani voi tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään yhden viikon ajan viimeisen selperkatinibiannoksen jälkeen (ks. kohta Raskaus ja imetys).
Hedelmällisyys
Ei‑kliinisten turvallisuuslöydösten perusteella Retsevmo saattaa heikentää miehen ja naisen hedelmällisyyttä (ks. kohdat Raskaus ja imetys ja Prekliiniset tiedot turvallisuudesta). Sekä miesten että naisten on hakeuduttava hedelmällisyyden säilyttämistä koskevaan neuvontaan ennen hoitoa.
Yliherkkyys
Selperkatinibihoitoa saaneilla potilailla on ilmoitettu yliherkkyyttä. Suurin osa tapahtumista havaittiin ei‑pienisoluista keuhkosyöpää sairastavilla potilailla, joita oli aiemmin hoidettu PD‑1- tai PD‑L1‑immunoterapialla (ks. kohta Haittavaikutukset). Yliherkkyyden oireisiin ja löydöksiin kuului kuume, ihottuma ja nivelkipu tai lihaskipu sekä samanaikainen verihiutalemäärän pieneneminen tai aminotransferaasiarvojen suureneminen.
Jos yliherkkyyttä ilmenee, selperkatinibihoito on tauotettava ja aloitettava steroidihoito. Yliherkkyysreaktion asteesta riippuen selperkatinibiannosta saattaa olla tarpeen muuttaa (ks. kohta Annostus ja antotapa). Steroidihoitoa jatketaan, kunnes potilaan tavoiteannos on saavutettu, minkä jälkeen steroidiannosta pienennetään vähitellen. Selperkatinibihoito on lopetettava pysyvästi, jos yliherkkyys uusiutuu.
Verenvuodot
Selperkatinibia saaneilla potilailla on ilmoitettu vakavia, myös kuolemaan johtaneita, verenvuototapahtumia (ks. kohta Haittavaikutukset).
Selperkatinibihoito on lopetettava pysyvästi, jos potilaalle ilmaantuu henkeä uhkaava tai vaikea uusiutuva verenvuoto (ks. kohta Annostus ja antotapa).
Tuumorilyysioireyhtymä (TLS)
Selperkatinibilla hoidetuilla potilailla on havaittu TLS-tapauksia. TLS:n riskitekijöitä ovat korkea kasvainkuormitus, olemassa oleva krooninen munuaisten vajaatoiminta, vähävirtsaisuus, nestehukka, hypotensio ja hapan virtsa. Näitä potilaita tulee seurata tarkasti ja hoitaa kliinisen aiheen mukaan. Asianmukaista ennaltaehkäisyä, mukaan lukien nesteytys, tulee harkita.
Reisiluun pään (lonkan) epifysiolyysi pediatrisilla potilailla
Reisiluun pään epifysiolyysiä on ilmoitettu selperkatinibia saaneilla pediatrisilla potilailla (< 18-vuoden ikäiset) (ks. kohta Haittavaikutukset). Potilaita on seurattava reisiluun pään epifysiolyysiin viittaavien oireiden varalta, ja heitä on hoidettava lääketieteellisesti ja kirurgisesti asianmukaisella tavalla.
Vakavat ihoreaktiot (SCARs)
Stevens-Johnsonin oireyhtymää (SJS), joka voi olla hengenvaarallinen tai johtaa kuolemaan, on raportoitu selperkatinibihoidon yhteydessä (ks. kohta Haittavaikutukset). Potilaita tulee neuvoa tunnistamaan vakavien ihoreaktioiden oireet ja hakeutumaan välittömästi lääkärin hoitoon, jos he havaitsevat viitteellisiä oireita tai löydöksiä. Jos ilmenee oireita ja löydöksiä, jotka viittaavat näihin reaktioihin, selperkatinibihoito on lopetettava välittömästi ja harkittava vaihtoehtoista hoitoa (tarvittaessa). Jos potilaalle on kehittynyt vakava ihoreaktio, kuten SJS, selperkatinibin käytön aikana, selperkatinibihoitoa ei saa missään vaiheessa aloittaa uudelleen kyseiselle potilaalle.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti, eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset selperkatinibin farmakokinetiikkaan
Selperkatinibi metaboloituu CYP3A4‑välitteisesti. CYP3A4‑entsyymitoimintaan vaikuttavat lääkevalmisteet saattavat siis vaikuttaa selperkatinibin farmakokinetiikkaan.
Selperkatinibi on P‑glykoproteiinin (P‑gp) ja rintasyöpäresistenssiproteiinin (BCRP) substraatti in vitro. Nämä kuljettajaproteiinit eivät kuitenkaan näytä rajoittavan suun kautta otetun selperkatinibin imeytymistä, sillä suun kautta otetun selperkatinibin biologinen hyötyosuus on 73 %, ja P‑gp:n estäjä rifampisiinin samanaikainen anto suurensi altistusta selperkatinibille minimaalisesti (selperkatinibin AUC0‑24‑arvo suureni noin 6,5 % ja Cmax‑arvo noin 19 %).
Aineet, jotka saattavat suurentaa plasman selperkatinibipitoisuuksia
Kun selperkatinibia annettiin 160 mg kerta‑annos yhdessä itrakonatsolin (vahva CYP3A:n estäjä) kanssa, selperkatinibin Cmax‑arvo suureni 30 % ja AUC‑arvo 130 % verrattuna tilanteeseen, jossa selperkatinibi annettiin yksinään. Jos voimakasta CYP3A:n ja/tai P‑gp:n estäjää (mm. ketokonatsolia, itrakonatsolia, vorikonatsolia, ritonaviiria, sakinaviiria, telitromysiiniä, posakonatsolia tai nefatsodonia) on käytettävä samanaikaisesti, selperkatinibiannosta on pienennettävä (ks. kohta Annostus ja antotapa).
Aineet, jotka saattavat pienentää plasman selperkatinibipitoisuuksia
Kun rifampisiinia (vahva CYP3A:n indusori) annettiin samanaikaisesti, selperkatinibin AUC‑arvo pieneni noin 87 % ja Cmax‑arvo noin 70 % verrattuna tilanteeseen, jossa selperkatinibi annettiin yksinään. Näin ollen vahvojen CYP3A4:n indusorien (mm. karbamatsepiinin, fenobarbitaalin, fenytoiinin, rifabutiinin, rifampisiinin ja mäkikuisman [Hypericum perforatum]) samanaikaista käyttöä on vältettävä.
Selperkatinibin vaikutukset muiden lääkevalmisteiden farmakokinetiikkaan (pitoisuuden suureneminen plasmassa)
Herkät CYP2C8:n substraatit
Selperkatinibi suurensi repaglinidin (CYP2C8:n substraatti) Cmax‑arvoa noin 91 % ja AUC‑arvoa noin 188 %. Näin ollen samanaikaista käyttöä herkkien CYP2C8:n substraattien (esim. amodiakiini, serivastatiini, entsalutamidi, paklitakseli, repaglinidi, torasemidi, sorafenibi, rosiglitatsoni, buprenorfiini, seleksipagi, dasabuviiri ja montelukasti) kanssa on vältettävä.
Herkät CYP3A4:n substraatit
Selperkatinibi suurensi midatsolaamin (CYP3A4:n substraatti) Cmax‑arvoa noin 39 % ja AUC‑arvoa noin 54 %. Näin ollen samanaikaista käyttöä herkkien CYP3A4:n substraattien (esim. alfentaniili, avanafiili, buspironi, konivaptaani, darifenasiini, darunaviiri, ebastiini, lomitapidi, lovastatiini, midatsolaami, naloksegoli, nisoldipiini, sakinaviiri, simvastatiini, tipranaviiri, triatsolaami, vardenafiili) kanssa on vältettävä.
Samanaikainen anto mahan pH‑arvoon vaikuttavien lääkevalmisteiden kanssa
Selperkatinibin liukoisuus on pH‑riippuvaista; liukoisuus vähenee pH‑arvon suurenemisen myötä. Selperkatinibin farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja, kun samanaikaisesti annettiin useita vuorokausiannoksia ranitidiinia (H2‑reseptorisalpaaja) 2 tuntia selperkatinibiannoksen jälkeen.
Samanaikainen anto protonipumpun estäjien kanssa
Kun useita vuorokausiannoksia omepratsolia (protonipumpun estäjä) annettiin samanaikaisesti, selperkatinibin AUC0‑INF‑arvo ja Cmax‑arvo pienenivät, kun selperkatinibi annettiin tyhjään mahaan. Kun omepratsolia annettiin useita vuorokausiannoksia samanaikaisesti, selperkatinibin AUC0‑INF‑arvo ja Cmax‑arvo eivät muuttuneet merkitsevästi, kun Retsevmo annettiin ruokailun yhteydessä.
Samanaikainen anto kuljettajaproteiinien substraattien kanssa
Selperkatinibi estää munuaisten MATE1‑kuljettajaproteiinia (multidrug and toxin extrusion protein 1). Selperkatinibilla voi olla in vivo ‑yhteisvaikutuksia MATE1:n kliinisesti merkittävien substraattien kuten kreatiniinin kanssa (ks. kohta Farmakokinetiikka).
Selperkatinibi on P‑gp:n ja BCRP:n estäjä in vitro. In vivo, selperkatinibi suurensi dabigatraanin, P-gp:n substraatin Cmax-arvoa 43 % ja AUC-arvoa 38 %, siksi herkkien P‑gp:n substraattien (esim. feksofenadiini, dabigatraanieteksilaatti, kolkisiini, saksagliptiini) käytössä on noudatettava varovaisuutta, ja erityisesti niillä, joilla on kapea terapeuttinen alue (esim. digoksiini) (ks. kohta Farmakokinetiikka). Selperkatinibi suurensi rosuvastatiinin, BCRP-substraatti, Cmax-arvoa 71 % ja AUC‑arvoa 80 % in vivo. Varovaisuutta on noudatettava käytettäessä samanaikaisesti BCRP‑substraattia (esim. rosuvastatiini, pratsosiini) (ks. kohta Farmakokinetiikka).
Lääkevalmisteet, jotka voivat olla vähemmän tehokkaita, kun niitä annetaan selperkatinibin kanssa
Selperkatinibi voi estää dejodinaasi D2 -entsyymiä ja siten vähentää levotyroksiinin (T4) muuttumista trijodityroniiniksi (T3). Potilaiden vaste levotyroksiini-korvaushoidolle voi siksi olla riittämätön ja liotyroniinilisä voi olla tarpeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy naisilla ja miehillä
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä hoidon aikana ja vähintään yhden viikon ajan viimeisen selperkatinibiannoksen jälkeen. Miesten, joiden kumppani voi tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään yhden viikon ajan viimeisen selperkatinibiannoksen jälkeen.
Raskaus
Selperkatinibin käytöstä raskaana oleville naisille ei ole saatavilla tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Retsevmo‑valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä ehkäisyä. Valmistetta tulee käyttää raskauden aikana vain, jos hoidon mahdollinen hyöty oikeuttaa sikiöön mahdollisesti kohdistuvan riskin.
Imetys
Ei tiedetä, erittyykö selperkatinibi ihmisillä äidinmaitoon. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. Imetys on lopetettava Retsevmo‑hoidon ajaksi ja vähintään yhdeksi viikoksi viimeisen annoksen jälkeen.
Hedelmällisyys
Selperkatinibin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Eläimillä tehtyjen tutkimusten löydösten perusteella Retsevmo‑hoito saattaa heikentää miesten ja naisten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta). Sekä miesten että naisten on hakeuduttava hedelmällisyyden säilyttämistä koskevaan neuvontaan ennen hoitoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Retsevmo‑valmisteella voi olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaita on kehotettava olemaan varovaisia ajaessaan tai käyttäessään koneita, jos heillä esiintyy uupumusta tai huimausta Retsevmo‑hoidon aikana (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yhteenvedossa esitetään selperkatinibihoitoa saaneiden potilaiden haittavaikutusten yhdistetty esiintyvyys suurenevilla annoksilla toteutetussa, avoimessa, vaiheen 1/2 monikeskustutkimuksessa (LIBRETTO‑001) ja kahdessa avoimessa, satunnaistetussa vaiheen 3 vertailevassa monikeskustutkimuksessa (LIBRETTO‑431 ja LIBRETTO‑531). Yleisimmät (≥ 1,0 %) vakavat haittavaikutukset ovat keuhkokuume (5,3 %), verenvuoto (2,4 %), vatsakipu (2,1 %), veren natriumpitoisuuden pieneneminen (2,0 %), ripuli (1,5 %), yliherkkyys (1,4 %), oksentelu (1,3 %), veren kreatiniiniarvon suureneminen (1,3 %), kuume (1,3 %), virtsateiden infektiot (1,3 %), ALAT-arvon suureneminen (1,0 %) ja ASAT-arvon suureneminen (1,0 %).
Retsevmo‑hoito oli lopetettava pysyvästi hoidon aikana ilmenneiden haittatapahtumien takia 8,8 %:lla potilaista (mikä tahansa haittatapahtuma riippumatta sen yhteydestä Retsevmo‑hoitoon). Yleisimpiä hoidon pysyvään lopettamiseen johtaneita haittavaikutuksia (vähintään 3 potilaalla) olivat ALAT‑arvon suureneminen (0,7 %), uupumus (0,5 %), ASAT‑arvon suureneminen (0,4 %), veren bilirubiiniarvon suureneminen (0,3 %), keuhkokuume (0,3 %), trombosytopenia (0,3 %), verenvuoto (0,3 %) ja yliherkkyys (0,3 %).
Haittavaikutustaulukko
LIBRETTO‑001-, LIBRETTO‑431- ja LIBRETTO‑531-tutkimuksissa selperkatinibihoitoa saaneilla potilailla ilmoitettujen haittavaikutusten yhdistetyt esiintyvyydet ja vaikeusasteet on esitetty taulukossa 5.
Haittavaikutukset on luokiteltu MedDRA-elinjärjestelmäluokan ja yleisyyden mukaan.
Yleisyysluokat määritellään seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Selperkatinibihoidon keston mediaani oli 30,09 kk (LIBRETTO‑001), 16,7 kk (LIBRETTO‑431) ja 14,9 kk (LIBRETTO‑531).
Taulukko 5 Haittavaikutukset selperkatinibia saaneilla potilailla (N = 1188)
Elinjärjestelmä (MedDRA) | Haittavaikutus | Esiintymistiheys, kaikki asteet | Esiintymistiheys, aste ≥ 3 |
|---|---|---|---|
Infektiot | Virtsatieinfektioa | Hyvin yleinen | Yleinen |
Keuhkokuumeb | Hyvin yleinen | Yleinen | |
Immuunijärjestelmäc | Yliherkkyysd | Yleinen | Yleinen |
Umpieritys | Kilpirauhasen vajaatoiminta | Hyvin yleinen | - |
Aineenvaihdunta ja ravitsemus | Ruokahalun heikkeneminen | Hyvin yleinen | Melko harvinainen |
Hermosto | Päänsärkye | Hyvin yleinen | Yleinen |
Huimausf | Hyvin yleinen | Melko harvinainen | |
Sydän | QT‑ajan piteneminen EKG‑tutkimuksessag | Hyvin yleinen | Yleinen |
Verisuonisto | Hypertensioh | Hyvin yleinen | Hyvin yleinen |
Verenvuotoi | Hyvin yleinen | Yleinen | |
Hengityselimet, rintakehä ja välikarsina | Interstitiaalinen keuhko-sairaus/keuhkokuumej | Yleinen | Melko harvinainen |
Kylothorax | Yleinen | Melko harvinainen | |
Ruoansulatuselimistö | Ripulik | Hyvin yleinen | Yleinen |
Suun kuivuusl | Hyvin yleinen | Melko harvinainen | |
Vatsakipum | Hyvin yleinen | Yleinen | |
Ummetus | Hyvin yleinen | Melko harvinainen | |
Pahoinvointi | Hyvin yleinen | Yleinen | |
Oksentelun | Hyvin yleinen | Yleinen | |
Suutulehduso | Hyvin yleinen | Melko harvinainen | |
Kyloperitoneump | Yleinen | Melko harvinainen | |
Iho ja ihonalainen kudos | Ihottumaq | Hyvin yleinen | Yleinen |
Stevens-Johnsonin oireyhtymär | Tuntematon | Tuntematon | |
Luusto, lihakset ja sidekudos | Reisiluun pään (lonkan) epifysiolyysis | Yleinen | Yleinen |
Sukupuolielimet ja rinnat | Erektiohäiriöt | Hyvin yleinen | Melko harvinainen |
Yleisoireet ja antopaikassa todettavat haitat | Turvotus/edeemau | Hyvin yleinen | Yleinen |
Uupumusv | Hyvin yleinen | Yleinen | |
Kuume | Hyvin yleinen | Melko harvinainen | |
Tutkimuksetw | ASAT‑arvon suureneminen | Hyvin yleinen | Hyvin yleinen |
ALAT‑arvon suureneminen | Hyvin yleinen | Hyvin yleinen | |
Kalsiumarvon pieneneminen | Hyvin yleinen | Yleinen | |
Lymfosyyttiarvon pieneneminen | Hyvin yleinen | Hyvin yleinen | |
Valkosoluarvon pieneneminen | Hyvin yleinen | Yleinen | |
Albumiiniarvon pieneneminen | Hyvin yleinen | Yleinen | |
Kreatiniiniarvon suureneminen | Hyvin yleinen | Yleinen | |
Natriumarvon pieneneminen | Hyvin yleinen | Hyvin yleinen | |
Alkalisen fosfataasiarvon suureneminen | Hyvin yleinen | Yleinen | |
Verihiutalearvon pieneneminen | Hyvin yleinen | Yleinen | |
Kokonaisbilirubiiniarvon suureneminen | Hyvin yleinen | Yleinen | |
Neutrofiiliarvon pieneneminen | Hyvin yleinen | Yleinen | |
Hemoglobiiniarvon pieneneminen | Hyvin yleinen | Yleinen | |
Magnesiumarvon pieneneminen | Hyvin yleinen | Yleinen | |
Kaliumarvon pieneneminen | Hyvin yleinen | Yleinen |
a Virtsatieinfektio sisältää virtsatieinfektion, virtsarakkotulehduksen, urosepsiksen, E. colin aiheuttaman virtsatieinfektion, E. colin aiheuttaman pyelonefriitin, munuaistulehduksen, nitriitin esiintymisen virtsassa, pyelonefriitin, virtsaputkitulehduksen, bakteerin aiheuttaman virtsatietulehduksen ja sienen aiheuttaman urogenitaalielinten infektion.
b Keuhkokuumeisiin sisältyvät keuhkokuume, keuhkoinfektio, aspiraatiokeuhkokuume, empyeema, keuhkojen konsolidaatio, keuhkopussin infektio, bakteerikeuhkokuume, stafylokokki-keuhkokuume, epätyypillinen keuhkokuume, keuhkopaise, pneumocystis jirovecii -keuhkokuume, pneumokokkikeuhkokuume, respiratory syncytial virus (RSV) -keuhkokuume, infektioon liittyvä pleuraeffuusio ja viruskeuhkokuume.
c Yliherkkyysreaktioille oli tunnusomaista makulopapulaarinen ihottuma, jota edelsi usein kuume ja siihen liittyvä nivelkipu/lihaskipu potilaan ensimmäisen hoitojakson aikana (tyypillisesti päivinä 7–21).
d Yliherkkyyteen sisältyy lääkeyliherkkyys ja yliherkkyys.
e Päänsärky sisältää päänsäryn, sinus- ja jännityspäänsäryn.
f Huimaus sisältää heitehuimauksen, huimauksen, presynkopeen ja asentohuimauksen.
g QT-ajan piteneminen EKG-tutkimuksessa sisältää QT-ajan pitenemisen ja epänormaalin QT-ajan EKG-tutkimuksessa.
h Hypertensioon sisältyy hypertensio ja kohonnut verenpaine.
i Verenvuotoon sisältyy nenäverenvuoto, veriyskä, ruhjevamma, hematuria, peräsuolen verenvuoto, emättimen verenvuoto, aivoverenvuoto, traumaattinen hematooma, verivirtsa, sidekalvon verenvuoto, mustelmat, ienverenvuoto, hematokeesit, hiussuonipurkaumat, verirakkulat, spontaani hematooma, vatsan hematooma, peräaukon verenvuoto, angina bullosa -verenvuoto, disseminoitunut intravaskulaarinen koagulaatio, silmän verenvuoto, mahaverenvuoto, maha-suolikanavan verenvuoto, kallonsisäinen verenvuoto, ihonalainen verenvuoto, peräpukamaverenvuoto, maksan hematooma, vatsansisäinen verenvuoto, verenvuoto suussa tai ruokatorvessa, lantion hematooma, periorbitaalinen hematooma tai verenvuoto, verenvuoto nielusta, keuhkojen ruhje, purppura, retroperitoneaalinen hematooma, ihon verenvuoto, lukinkalvonalainen verenvuoto, suolen divertikkelin verenvuoto, silmän hematooma, verioksennukset, verenvuoto, aivohaveri, maksan verenvuoto, kurkunpään verenvuoto, alemman maha-suolikanavan verenvuoto, meleena, menorragia, positiivinen piilevä veren näytetulos, toimenpiteen jälkeinen verenvuoto, postmenopausaalinen verenvuoto, verkkokalvon verenvuoto, kovakalvon verenvuoto, subduraalinen verenvuoto, traumaattinen hemothorax, tuumorin verenvuoto, ylemmän maha-suolikanavan verenvuoto, kohtuverenvuoto, suonen pistokohdan hematooma, verinivel ja hematooma.
j Interstitiaalinen keuhkosairaus/keuhkokuume sisältää interstitiaalisen keuhkosairauden, keuhkotulehduksen, säteilykeuhkotulehduksen, restriktiivisen keuhkosairauden, akuutin hengitysvaikeusoireyhtymän, alveoliitin, bronkioliitin, langerhansin solujen histiosytoosin, keuhkojen säteilyvaurion, kystisen keuhkosairauden, keuhkoinfiltraatit ja keuhkojen varjostuman.
k Ripuli sisältää ripulin, peräaukon inkontinenssin, kiireellisen ulostamisen, tiheät suolen liikkeet ja maha-suolikanavan hypermotiliteetin.
l Suun kuivumiseen sisältyy suun kuivuminen ja limakalvojen kuivuus.
m Vatsakipu sisältää vatsakivun, ylävatsakivun, tuntemuksia vatsassa, alavatsakivun ja maha-suolikanavan kivun.
n Oksentelu sisältää oksentelun, yökkäilyn ja regurgitaation.
o Suutulehdus sisältää suutulehduksen, suun haavaumat, suun limakalvon tulehdusreaktion ja suun limakalvon rakkulat.
p Kyloperitoneum sisältää kyloperitoneumin ja ascites chylouksen (MedDRA LLTs).
q Ihottuma sisältää ihottuman, makulopapulaarisen ihottuman, dermatiitin, ihon hilseilyn, makulaarisen ihottuman, punoittavan ihottuman, urtikarian, allergisen dermatiitin, hilseilevän ihottuman, papulaarisen ihottuman, morbilliformisen ihottuman, kutisevan ihottuman, vesikulaarisen ihottuman, perhosihottuman, follikulaarisen ihottuman, yleistyneen ihottuman, märkärakkulaisen ihottuman ja ihoreaktion.
r Markkinoille tulon jälkeisistä ilmoituksista.
s Reisiluun pään epifysiolyysiä on havaittu yleisesti (6,4 %) selperkatinibilla hoidetuilla (n = 47) pediatrisilla potilailla (< 18-vuoden ikäiset).
t Erektiohäiriöitä on havaittu hyvin yleisesti (12,4 %) selperkatinibilla hoidetuilla miespotilailla kliinisissä tutkimuksissa (n = 986).
u Edeemaan sisältyy perifeerinen edeema, kasvojen edeema, periorbitaalinen edeema, turvonneet kasvot, paikallinen edeema, perifeerinen turvotus, yleistynyt edeema, silmäluomen edeema, silmän turvotus, lymfedeema, sukupuolielinten edeema, kivespussin turvotus, angioedeema, silmäedeema, edeema, kivespussin edeema, ihon edeema, turvotus, silmäkuopan edeema, kivesten turvotus, vulvovaginaalinen turvotus, silmäkuopan turvotus, peniksen turvotus, periorbitaalinen turvotus ja silmäluomen turvotus.
v Uupumus sisältää uupumuksen/väsymyksen, voimattomuuden ja huonovointisuuden.
w Perustuu laboratorioarviointeihin. Prosenttiosuus lasketaan niiden potilaiden lukumäärän perusteella, joilla on lähtötilanteen arviointi ja vähintään yksi lähtötilanteen jälkeinen arvio nimittäjänä.
Valikoitujen haittavaikutusten kuvaus selperkatinibia saavilla potilailla
Aminotransferaasiarvojen suureneminen (ASAT‑ tai ALAT‑arvon suureneminen)
Laboratorioarvioinnin perusteella ALAT‑arvon suurenemista ilmoitettiin 59,4 %:lla potilaista ja ASAT‑arvon suurenemista 61 %:lla potilaista. Asteen 3 tai 4 ALAT‑arvon suurenemista ilmoitettiin 14,1 %:lla potilaista ja asteen 3 tai 4 ASAT‑arvon suurenemista 9,5 %:lla potilaista.
LIBRETTO‑001-tutkimuksessa ensimmäiseen suurenemiseen kuluneen ajan mediaani oli ASAT‑arvon kohdalla 4,7 viikkoa (vaihteluväli: 0,7–227,9) ja ALAT‑arvon kohdalla 4,4 viikkoa (vaihteluväli: 0,9–186,1). LIBRETTO‑431-tutkimuksessa ensimmäiseen suurenemiseen kuluneen ajan mediaani oli ASAT‑arvon kohdalla 5,1 viikkoa (vaihteluväli: 0,7–88,1) ja ALAT‑arvon kohdalla 5,1 viikkoa (vaihteluväli: 0,7–110,9). LIBRETTO‑531-tutkimuksessa ensimmäiseen suurenemiseen kuluneen ajan mediaani oli ASAT‑arvon kohdalla 6,1 viikkoa (vaihteluväli: 0,1–85,1) ja ALAT‑arvon kohdalla 6,1 viikkoa (vaihteluväli: 0,1–85,1).
On suositeltavaa muuttaa annosta, jos potilaalla esiintyy asteen 3 tai 4 ALAT‑ tai ASAT‑arvon suurenemista (ks. kohta Annostus ja antotapa).
QT‑ajan piteneminen
Niillä 837 potilaalla, joille tehtiin LIBRETTO‑001-tutkimuksessa EKG-tutkimus, löydösten arvioinnissa todettiin, että 8,1 %:lla potilaista QTcF‑enimmäisaika oli lähtötilanteen jälkeen > 500 ms ja 21,6 %:lla QTcF-ajan suurin pitenemä lähtötilanteesta oli > 60 ms. Niistä 156 potilaasta, joille tehtiin LIBRETTO‑431-tutkimuksessa EKG-tutkimus, 5,1 %:lla lähtötilanteen jälkeinen QTcF‑enimmäisaika oli > 500 ms ja 16,7 %:lla QTcF-ajan suurin pitenemä lähtötilanteesta oli > 60 ms. Niistä 191 potilaasta, joille tehtiin LIBRETTO‑531-tutkimuksessa EKG-tutkimus, 3,7 %:lla lähtötilanteen jälkeinen QTcF‑enimmäisaika oli > 500 ms ja 17,8 %:lla QTcF-ajan suurin pidentymä lähtötilanteesta oli > 60 ms.
LIBRETTO‑001-, LIBRETTO‑431- ja LIBRETTO‑531-tutkimuksissa ei ilmoitettu kääntyvien kärkien takykardiaa, asteen ≥ 3 rytmihäiriöitä tai kliinisesti merkittäviä hoidon aikana ilmenneitä rytmihäiriöitä, kammiotakykardiaa, kammiovärinää tai kammiolepatusta. Äkkikuolemia ja kuolemaan johtaneita sydänpysähdyksiä ilmoitettiin potilailla, joilla oli merkittävä sydänanamneesi. Kaikkien tutkimusten potilaista yhteensä kaksi (0,2 %) lopetti selperkatinibihoidon QT‑ajan pidentymisen vuoksi. Retsevmo‑hoito voidaan joutua tauottamaan tai annosta voidaan joutua muuttamaan (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Hypertensio
Niillä 837 potilaalla, joilta mitattiin LIBRETTO‑001-tutkimuksessa verenpaine, systolinen paine suureni lähtötilanteesta enintään 32 mmHg (mediaani; vaihteluväli: −15, +100). Diastolista verenpainetta koskevat tulokset olivat samaa luokkaa, mutta suurenemat olivat pienempiä.
LIBRETTO‑001-tutkimuksessa vain 10,3 %:lla potilaista lähtötilanteen aste pysyi ennallaan hoidon aikana ja 40,7 %:lla se suureni 1 asteella, 38,5 %:lla 2 asteella ja 9,8 %:lla 3 asteella. Hoidosta johtuvaa hypertensiota ilmoitettiin 44,8 %:lla potilaista, joilla oli anamneesissa hypertensiota (28,2 %:lla aste 3, 4), ja 41,7 %:lla potilaista, joilla ei ollut anamneesissa hypertensiota (14,1 %:lla aste 3, 4).
Niistä 154:stä selperkatinibihoitoa saaneesta potilaasta, joilta mitattiin LIBRETTO‑431-tutkimuksessa verenpaine, 23,4 %:lla lähtötilanteen aste pysyi ennallaan hoidon aikana ja 49,4 %:lla se suureni 1 asteella, 22,7 %:lla 2 asteella ja 3,3 %:lla 3 asteella.
Niistä 192:sta selperkatinibihoitoa saaneesta potilaasta, joilta mitattiin LIBRETTO‑531-tutkimuksessa verenpaine, 20,8 %:lla lähtötilanteen aste pysyi ennallaan hoidon aikana ja 43,8 %:lla se suureni 1 asteella, 27,6 %:lla 2 asteella ja 6,8 %:lla 3 asteella.
LIBRETTO‑001-tutkimuksessa yhteensä 19,8 %:lla potilaista, LIBRETTO‑431-tutkimuksessa 20,3 %:lla potilaista ja LIBRETTO‑531-tutkimuksessa 19,2 %:lla potilaista esiintyi hoidon aikana ilmennyttä asteen 3 hypertensiota (määritelmä: systolinen maksimiverenpaine yli 160 mmHg). Hoidon aikana ilmennyttä asteen 4 hypertensiota raportoitiin LIBRETTO‑001-tutkimuksessa 0,1 %:lla potilaista; LIBRETTO‑431- ja LIBRETTO‑531-tutkimuksissa sitä ei raportoitu lainkaan.
LIBRETTO‑001-tutkimuksessa kaksi potilasta (0,2 %) lopetti hoidon pysyvästi hypertension takia, LIBRETTO‑431- ja LIBRETTO‑531-tutkimuksissa ei yksikään. Annoksen muuttaminen on suositeltavaa, jos potilaalle kehittyy hypertensio (ks. kohta Annostus ja antotapa). Selperkatinibihoito on lopetettava pysyvästi, jos lääketieteellisesti merkitsevää hypertensiota ei saada hallintaan verenpainelääkityksellä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yliherkkyys
Yliherkkyyden oireita ja löydöksiä olivat kuume, ihottuma ja nivelkipu tai lihaskipu sekä samanaikainen verihiutalearvon pieneneminen tai aminotransferaasiarvon suureneminen.
LIBRETTO‑001-tutkimuksessa 24,0 % (201/837) selperkatinibilla hoidetuista potilaista oli saanut aiemmin PD‑1- tai PD‑L1-immunoterapiaa. Yliherkkyyttä esiintyi yhteensä 5,7 %:lla (48/837) selperkatinibia saaneista potilaista. Asteen 3 yliherkkyyttä esiintyi 1,9 %:lla (16/837) potilaista. Näistä 48 potilaasta, joilla esiintyi yliherkkyyttä LIBRETTO‑001-tutkimuksessa, 54,2 %:lla (26/48) oli ei‑pienisoluinen keuhkosyöpä ja he olivat saaneet aiempaa PD‑1- tai PD‑L1-immunoterapiaa. LIBRETTO‑001-tutkimuksessa asteen 3 yliherkkyyttä esiintyi 3,5 %:lla (7/201) potilaista, jotka olivat saaneet aiempaa PD‑1- tai PD‑L1-immunoterapiaa. LIBRETTO‑001-tutkimuksessa yliherkkyyden ilmaantumiseen kuluneen ajan mediaani oli 1,9 viikkoa (vaihteluväli: 0,7–203,9 viikkoa): 1,7 viikkoa potilailla, jotka olivat saaneet aiempaa PD‑1- tai PD‑L1-immunoterapiaa ja 4,4 viikkoa potilailla, jotka eivät olleet saaneet aiempaa anti-PD-1/PD-L1-immunoterapiaa.
LIBRETTO‑431-tutkimukseen otettiin potilaita, joilla oli pitkälle edennyt tai etäpesäkkeinen ei‑pienisoluinen keuhkosyöpä. Yliherkkyyttä esiintyi yhteensä 1,9 %:lla (3/158) selperkatinibia saaneista potilaista. Asteen 3 yliherkkyyttä esiintyi 0,6 %:lla (1/158) potilaista. LIBRETTO‑001- ja LIBRETTO‑431-tutkimusten yhdistetyn analyysin perusteella niistä selperkatinibia saaneista potilaista, joilla oli ei-pienisoluinen keuhkosyöpä ja jotka olivat saaneet aiempaa anti‑PD‑1/PD‑L1-immunoterapiaa (N = 205), yliherkkyyttä esiintyi 16,6 %:lla ja asteen ≥ 3 yliherkkyyttä 5,9 %:lla.
LIBRETTO‑531-tutkimukseen otettiin potilaita, joilla oli pitkälle edennyt tai etäpesäkkeinen medullaarinen kilpirauhassyöpä. Yliherkkyyttä esiintyi yhdellä (0,5 %; 1/193) selperkatinibia saaneista potilaista. Tällä yhdellä potilaalla yliherkkyys oli astetta 3.
Retsevmo‑hoito voidaan joutua tauottamaan tai annosta voidaan joutua muuttamaan (ks. kohta Annostus ja antotapa).
Verenvuodot
LIBRETTO‑001-, LIBRETTO‑431- ja LIBRETTO‑531-tutkimuksissa asteen ≥ 3 verenvuototapahtumia esiintyi 2,5 %:lla selperkatinibia saaneista potilaista. LIBRETTO‑001-tutkimuksessa neljällä potilaalla (0,5 %) verenvuototapahtuma johti kuolemaan. Näistä kaksi oli aivoverenvuotoja, ja yksi trakeostoomakohdan verenvuoto ja yksi veriyskä. LIBRETTO‑431- ja LIBRETTO‑531-tutkimuksissa ei ilmoitettu kuolemaan johtaneita verenvuototapahtumia selperkatinibia saaneilla potilailla. Verenvuototapahtuman ilmaantumiseen kuluneen ajan mediaani oli LIBRETTO‑001-tutkimuksessa 34,1 viikkoa (vaihteluväli: 0,1–234,6 viikkoa), LIBRETTO‑431-tutkimuksessa 16,8 viikkoa (vaihteluväli: 1,1–94,1 viikkoa) ja LIBRETTO‑531-tutkimuksessa 10,7 viikkoa (vaihteluväli: 1,0–124,1 viikkoa).
Selperkatinibihoito on lopetettava pysyvästi, jos potilaalle ilmaantuu henkeä uhkaava tai vaikea uusiutuva verenvuoto (ks. kohta Annostus ja antotapa).
Lisätietoa erityisryhmistä
Pediatriset potilaat
LIBRETTO‑001‑tutkimuksessa oli kolme < 18‑vuotiasta potilasta (vaihteluväli: 15–17 v), joilla oli RET-mutaatiopositiivinen medullaarinen kilpirauhassyöpä. LIBRETTO‑121‑tutkimuksessa oli yksitoista < 18‑vuotiasta potilasta (vaihteluväli: 2–17 v), joilla oli RET‑mutaatiopositiivinen medullaarinen kilpirauhassyöpä, kolmetoista < 18-vuotiasta potilasta (vaihteluväli: 6–17 v), joilla oli RET-fuusiopositiivinen kilpirauhassyöpä, sekä kuusi < 18-vuotiasta potilasta (vaihteluväli: 5-15 vuotta), joilla oli RET-muuntunut kiinteä kasvain, joista kolmella oli RET-fuusiopositiivinen kiinteä kasvain.
LIBRETTO‑531-tutkimuksessa oli yksi 12‑vuotias potilas, jolla oli RET‑mutaatiopositiivinen medullaarinen kilpirauhassyöpä. Reisiluun pään epifysiolyysitapauksia on ilmoitettu selperkatinibihoitoa saaneilla < 18-vuotiailla potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Alle 18 vuoden ikäisten lasten hoidossa ei ole havaittu muita erityisiä turvallisuutta koskevia löydöksiä.
LIBRETTO-121 haittavaikutustaulukko
Selperkatinibilla hoitoa saaneilla ≤ 21‑vuotiailla potilailla LIBRETTO‑121‑tutkimuksessa ilmoitettujen haittavaikutusten esiintyvyys ja vaikeusaste on esitetty taulukossa 6.
Haittavaikutukset on luokiteltu MedDRA-elinjärjestelmäluokan ja yleisyyden mukaan. Yleisyysluokat on määritelty seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100 – < 1/10); melko harvinainen (≥ 1/1 000 – < 1/100); harvinainen (≥ 1/10 000 – < 1/1 000); hyvin harvinainen (< 1/10 000); ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Selperkatinibihoidon keston mediaani oli 30,26 kuukautta.
Taulukko 6 Haittavaikutukset selperkatinibia saaneilla potilailla LIBRETTO-121-tutkimuksessa (N= 36)
Elinjärjestelmä (MedDRA) | Haittavaikutus | Esiintymistiheys, kaikki asteet# | Esiintymistiheys, aste ≥ 3 |
Infektiot | Virtsatieinfektioa | Hyvin yleinen | Yleinen |
Keuhkokuumeb | Yleinen | Yleinen | |
Immuunijärjestelmäc | Yliherkkyysd | Hyvin yleinen | Yleinen |
Umpieritys | Kilpirauhasen vajaatoimintae | Hyvin yleinen | - |
Aineenvaihdunta ja ravitsemus | Ruokahalun heikkeneminen | Yleinen | - |
Hermosto | Päänsärky | Hyvin yleinen | - |
Huimaus | Hyvin yleinen | - | |
Sydän | QT‑ajan piteneminen EKG‑tutkimuksessaf | Hyvin yleinen | Yleinen |
Verisuonisto | Verenvuotog | Hyvin yleinen | - |
Hypertensio | Yleinen | Yleinen | |
Ruoansulatuselimistö | Ripulih | Hyvin yleinen | Yleinen |
Pahoinvointi | Hyvin yleinen | Yleinen | |
Vatsakipui | Hyvin yleinen | - | |
Oksentelu | Hyvin yleinen | Yleinen | |
Ummetus | Hyvin yleinen | Yleinen | |
Suutulehdus | Hyvin yleinen | - | |
Suun kuivuus | Yleinen | - | |
Kyloperitoneum | Yleinen | - | |
Iho ja ihonalainen kudos | Ihottumaj | Hyvin yleinen | - |
Luusto, lihakset ja sidekudos | Luustolihassärkyk | Hyvin yleinen | - |
Reisiluun pään (lonkan) epifysiolyysi | Yleinen | Yleinen | |
Sukupuolielimet ja rinnat | Erektiohäiriöl | Hyvin yleinen | Yleinen |
Yleisoireet ja antopaikassa todettavat haitat | Kuume | Hyvin yleinen | - |
Uupumusm | Hyvin yleinen | - | |
Turvotus/edeeman | Hyvin yleinen | - | |
Painon nousu | Hyvin yleinen | Hyvin yleinen | |
Tutkimukseto | Kalsiumarvon pieneneminen | Hyvin yleinen | Hyvin yleinen |
ALAT‑arvon suureneminen | Hyvin yleinen | Yleinen | |
Albumiiniarvon pieneneminen | Hyvin yleinen | - | |
ASAT‑arvon suureneminen | Hyvin yleinen | Yleinen | |
Alkalisen fosfataasiarvon suureneminen | Hyvin yleinen | - | |
Valkosoluarvon pieneneminen | Hyvin yleinen | Yleinen | |
Neutrofiiliarvon pieneneminen | Hyvin yleinen | Yleinen | |
Hemoglobiiniarvon pieneneminen | Hyvin yleinen | Hyvin yleinen | |
Kokonaisbilirubiiniarvon suureneminen | Hyvin yleinen | Yleinen | |
Magnesiumarvon pieneneminen | Hyvin yleinen | Yleinen | |
Verihiutalearvon pieneneminen | Hyvin yleinen | Yleinen | |
Lymfosyyttiarvon pieneneminen | Hyvin yleinen | Hyvin yleinen | |
Kaliumarvon pieneneminen | Hyvin yleinen | Yleinen | |
Kreatiniiniarvon suureneminen | Hyvin yleinen | Yleinen | |
Natriumarvon pieneneminen | Hyvin yleinen | - |
# LIBRETTO-121-tutkimuksen 36 potilaasta kolmekymmentäyksi oli alle 18-vuotiaita ja viisi oli 18–21-vuotiaita.
a Virtsatieinfektio sisältää virtsatieinfektion ja virtsarakkotulehduksen
b Keuhkokuumeisiin sisältyvät keuhkokuume ja respiratory syncytial virus (RSV) -keuhkokuume
c Yliherkkyysreaktioille oli tunnusomaista makulopapulaarinen ihottuma, jota edelsi usein kuume ja siihen liittyvä nivelkipu/lihaskipu potilaan ensimmäisen hoitojakson aikana (tyypillisesti päivinä 7–21).
d Yliherkkyyteen sisältyy lääkeyliherkkyys ja yliherkkyys.
e Kilpirauhasen vajaatoiminta sisältää kilpirauhasen vajaatoiminnan, veren tyreotropiinihormonin pitoisuuden suurenemisen ja veren tyreoglobuliinipitoisuuden suurenemisen.
f QT-ajan piteneminen EKG-tutkimuksessa sisältää QT-ajan pitenemisen ja pidentyneen QRS-heilahduksen EKG-tutkimuksessa.
g Verenvuoto sisältää nenäverenvuodon, hematurian, aktivoidun osittaisen tromboplastiiniajan pitenemisen, peräaukon verenvuodon, verivirtsaisuuden, veriyskän, menorragian ja suun verenvuodon.
h Ripuli sisältää ripulin ja peräaukon inkontinenssin.
i Vatsakipu sisältää vatsakivun, ylävatsakivun ja vatsavaivat.
j Ihottuma sisältää makulopapulaarisen ihottuman, ihottuman, punoittavan ihottuman ja urtikarian.
k Luustolihassärky sisältää nivelkivun, raajojen kivun, selkäkivun, ei-sydänperäisen rintakivun, luukivun, rinnan luustolihaskivun, luustolihaskivun ja niskakivun.
l Erektiohäiriö selperkatinibilla hoidetuilla miespotilailla. Nimittäjä on mukautettu miehille ominaisen tapahtuman vuoksi (n = 19).
m Uupumus sisältää väsymyksen, astenian ja huonovointisuuden.
n Edeemaan sisältyy kasvojen edeema, raajojen edeema, periorbitaalinen edeema, yleistynyt edeema, paikallinen edeema ja turvotus.
o Perustuu laboratoriotutkimuksiin. Prosenttiosuus on laskettu käyttäen nimittäjänä niiden potilaiden lukumäärää, joille oli tehty lähtötilanteen arviointi ja vähintään yksi lähtötilanteen jälkeinen arviointi.
Iäkkäät
LIBRETTO‑001-tutkimuksessa selperkatinibia saaneista potilaista 24,7 % oli ≥ 65–74‑vuotiaita, 8,6 % oli 75–84‑vuotiaita ja 1,0 % oli ≥ 85‑vuotiaita. LIBRETTO‑431-tutkimuksessa selperkatinibia saaneista potilaista 26,6 % oli ≥ 65–74‑vuotiaita, 9,5 % oli 75–84‑vuotiaita ja 1,3 % oli ≥ 85‑vuotiaita. LIBRETTO‑531-tutkimuksessa selperkatinibia saaneista potilaista 20,2 % oli ≥ 65–74‑vuotiaita ja 5,2 % oli 75–84‑vuotiaita, eikä kukaan ollut ≥ 85‑vuotias. LIBRETTO‑001-tutkimuksessa ilmoitettujen vakavien haittatapahtumien yleisyys oli suurempi ≥ 65–74‑vuotiailla (58,0 %), 75–84‑vuotiailla (62,5 %) ja ≥ 85‑vuotiailla potilailla (100,0 %) kuin < 65‑vuotiailla (46,7 %). LIBRETTO‑431-tutkimuksessa ilmoitettujen vakavien haittatapahtumien yleisyys oli suurempi ≥ 65–74‑vuotiailla (38,1 %), 75–84‑vuotiailla (46,7 %) ja ≥ 85‑vuotiailla potilailla (50,0 %) kuin < 65‑vuotiailla (31,3 %). LIBRETTO‑531-tutkimuksessa ilmoitettujen vakavien haittatapahtumien yleisyys oli suurempi 75–84‑vuotiailla potilailla (50 %) kuin < 65‑vuotiailla (20,8 %) ja 65–74‑vuotiailla potilailla (17,9 %).
LIBRETTO‑001-tutkimuksessa selperkatinibihoidon lopettamiseen johtaneiden haittatapahtumien yleisyys oli suurempi ≥ 65–74‑vuotiailla (10,1 %), 75–84‑vuotiailla (19,4 %) ja ≥ 85‑vuotiailla potilailla (37,5 %) kuin < 65‑vuotiailla (7,6 %). LIBRETTO‑431-tutkimuksessa selperkatinibihoidon lopettamiseen johtaneiden haittatapahtumien yleisyys oli suurempi ≥ 65–74‑vuotiailla (14,3 %) ja 75–84‑vuotiailla (20,0 %) kuin < 65‑vuotiailla (7,1 %). Tutkimuksessa yksikään ≥ 85‑vuotiaista potilaista ei keskeyttänyt selperkatinibihoitoa haittatapahtuman takia. LIBRETTO‑531-tutkimuksessa selperkatinibihoidon lopettamiseen johtaneiden haittatapahtumien yleisyys oli suurempi 75–84‑vuotiailla (10 %) ja ≥ 65–74‑vuotiailla potilailla (7,7 %) kuin < 65‑vuotiailla (3,5 %).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‑haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostuksen oireita ei ole todennettu. Jos yliannostusta epäillään, potilaalle on annettava elintoimintoja tukevaa hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: syöpälääkkeet ja immuunivasteen muuntajat, antineoplastiset lääkeaineet, proteiinikinaasin estäjät, ATC‑koodi: L01EX22
Vaikutusmekanismi
Selperkatinibi on RET (rearranged during transfection) ‑reseptorityrosiinikinaasin estäjä. Selperkatinibi esti villin tyypin RET‑kinaasia ja useita mutatoituneita RET‑isoformeja sekä VEGFR1‑ ja VEGFR3‑kinaaseja; IC50‑arvojen vaihteluväli oli 0,92–67,8 nM. Muissa entsyymikokeissa selperkatinibi esti myös FGFR1‑, FGFR2‑ ja FGFR3 ‑kinaaseja suurempina pitoisuuksina, jotka olivat kuitenkin kliinisesti saavutettavissa. Sitoutumistutkimuksessa havaittiin merkitsevää antagonistista sitoutumista (> 50 %) serotoniinin (5‑HT) kuljettajaproteiiniin (70,2 % antagonismi) ja α2C‑adrenoreseptoriin (51,7 % antagonismi), kun selperkatinibin pitoisuus oli 1 µM. Pitoisuus 1 µM on noin 7 kertaa suurempi kuin sitoutumaton enimmäispitoisuus plasmassa tehokkaan selperkatinibiannoksen käytön yhteydessä.
Tietyt RET‑geenin pistemutaatiot tai kromosomien uudelleenjärjestymät, joihin liittyy RET‑geenin in‑frame ‑fuusio eri partnerien kanssa, voivat johtaa konstitutiivisesti aktivoituneisiin kimeerisiin RET‑fuusioproteiineihin. Fuusioproteiinit voivat toimia onkogeenisinä ajureina edistämällä kasvainsolulinjojen soluproliferaatiota. In vitro- ja in vivo ‑kasvainmalleissa selperkatinibilla osoitettiin olevan antituumori-vaikutusta soluissa, joissa RET‑proteiini on konstitutiivisesti aktiivinen geenifuusioiden ja ‑mutaatioiden seurauksena (mm. CCDC6‑RET, KIF5B‑RET, RET V804M ja RET M918T). Lisäksi selperkatinibilla osoitettiin olevan vaikutusta kasvaimeen hiirillä, joille oli implantoitu intrakraniaalisesti potilaalta peräisin oleva RET‑fuusiopositiivinen kasvain.
Farmakodynaamiset vaikutukset
Sydämen elektrofysiologia
Perusteellisessa QT‑tutkimuksessa, johon osallistui 32 tervettä tutkittavaa ja jossa käytettiin positiivista vertailuvalmistetta, ei todettu suurta QTcF‑ajan muutosta (> 20 ms), kun selperkatinibipitoisuudet olivat samaa luokkaa kuin hoitoannostuksen käytössä. Altistus‑vasteanalyysi osoitti, että hoitotasoa suuremmat pitoisuudet voivat johtaa QTc‑ajan pitenemiseen > 20 ms:lla. Selperkatinibia saaneilla potilailla ilmoitettiin QT‑ajan pitenemistä. Tästä syystä hoidon tauottaminen tai annoksen muuttaminen saattaa olla tarpeen (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Kliininen teho ja turvallisuus
Retsevmo‑valmisteen tehoa arvioitiin vaiheen 1/2 avoimessa yksihaaraisessa kliinisessä monikeskustutkimuksessa (LIBRETTO‑001) aikuispotilailla, joilla oli pitkälle edennyt RET‑fuusiopositiivinen ei‑pienisoluinen keuhkosyöpä, RET‑fuusiopositiivinen kilpirauhassyöpä tai muu RET-fuusiopositiivinen kiinteä kasvain, sekä aikuisilla ja nuorilla potilailla, joilla oli RET‑mutaatiopositiivinen medullaarinen kilpirauhassyöpä. Retsevmo-valmisteen teho RET‑fuusiopositiivisessa ei-pienisoluisessa keuhkosyövässä vahvistettiin vaiheen 3 LIBRETTO‑431-tutkimuksessa (ks. kohtaAiemmin hoitamaton RET-fuusiopositiivinen ei-pienisoluinen keuhkosyöpä). Retsevmo-valmisteen teho RET‑mutaatiopositiivisessa medullaarisessa kilpirauhassyövässä vahvistettiin vaiheen 3 LIBRETTO‑531-tutkimuksessa (ks. kohta RET‑mutaatiopositiivinen medullaarinen kilpirauhassyöpä, jota ei ole hoidettu vandetanibilla ja kabotsantinibilla).
LIBRETTO‑001-tutkimus sisälsi kaksi osaa: vaihe 1 (annoksen suurentaminen) ja vaihe 2 (tutkimuksen laajennus). Vaiheessa 1 ensisijaisena tavoitteena oli määrittää suositeltu selperkatinibiannos vaihetta 2 varten. Vaiheessa 2 ensisijaisena tavoitteena oli arvioida selperkatinibin vaikutusta kasvaimeen määrittämällä objektiivinen hoitovaste (ORR) riippumattoman arviointitoimikunnan arvioimana. Tutkimukseen otettiin potilaita, joilla oli RECIST‑kriteerien version 1.1 mukainen mitattavissa oleva tai ei-mitattavissa oleva tauti ja näyttö kasvaimen RET‑geenimuutoksesta. Potilas soveltui tutkimukseen keskushermostoetäpesäkkeistä huolimatta, jos tila oli stabiili. Sen sijaan tutkimuksesta suljettiin pois potilaat, joilla oli oireinen keskushermoston primaarikasvain, etäpesäkkeitä, leptomeningeaalinen karsinomatoosi tai selkäydinkompressio. Tutkimuksesta suljettiin pois myös potilaat, joilla oli jokin muu tiedossa oleva primaarinen ajurimuutos kuin RET, kliinisesti merkittävä aktiivinen sydän‑ ja verisuonitauti, anamneesissa sydäninfarkti tai QTcF‑aika > 470 ms.
Tutkimuksen vaiheessa 2 potilaat saivat Retsevmo‑valmistetta 160 mg suun kautta kahdesti vuorokaudessa, kunnes ilmeni toksisuutta, jota ei voitu hyväksyä, tai taudin etenemistä. RET‑geenimuutos määritettiin prospektiivisesti paikallisissa laboratorioissa NGS‑menetelmällä (uuden sukupolven sekvensointi), PCR‑menetelmällä (polymeraasiketjureaktio) tai FISH‑menetelmällä (fluoresenssi in situ ‑hybridisaatio). Ensisijainen tehon tulosmuuttuja oli objektiivinen hoitovaste (ORR), jonka arvioi sokkoutettu riippumaton arviointitoimikunta RECIST v1.1 ‑kriteerien perusteella. Toissijaisia tehon tuloksia olivat vasteen kesto (DOR), etenemisvapaa elossaoloaika (PFS) ja kokonaiselossaoloaika (OS).
Aiemmin hoitamaton RET-fuusiopositiivinen ei-pienisoluinen keuhkosyöpä
LIBRETTO-431
Retsevmo-valmisteen teho RET-fuusiopositiivisen ei-pienisoluisen keuhkosyövän hoidossa vahvistettiin LIBRETTO‑431-monikeskustutkimuksessa, joka oli vaiheen 3 satunnaistettu, avoin vertailututkimus. Siinä selperkatinibihoitoa verrattiin platinapohjaisen valmisteen ja pemetreksedin yhdistelmään, jota annettiin joko pembrolitsumabin kanssa tai ilman pembrolitsumabia. Tutkimuksen potilailla oli pitkälle edennyt tai etäpesäkkeinen RET-fuusiopositiivinen ei-pienisoluinen keuhkosyöpä. Tutkimukseen otettiin aikuispotilaita, joilla oli histologisesti varmistettu paikallisesti levinnyt tai etäpesäkkeinen ei-pienisoluinen keuhkosyöpä, joka ei soveltunut leikkaushoitoon ja johon ei ollut etäpesäkkeisessä vaiheessa aiemmin annettu systeemistä hoitoa. Tutkimukseen otettiin myös potilaita, jotka olivat saaneet adjuvantti- tai neoadjuvanttihoitoa, jos systeemisen hoidon viimeisestä annoksesta oli satunnaistamisvaiheessa kulunut vähintään 6 kuukautta. Potilaat saivat 160 mg selperkatinibia kahdesti vuorokaudessa (aloitusannos) tai platinapohjaisen valmisteen ja pemetreksedin yhdistelmää joko pembrolitsumabin kanssa tai ilman pembrolitsumabia. Potilaat stratifioitiin maantieteellisen alueen (Itä-Aasia vs. muut alueet) ja tutkijan arvioiman lähtötilanteen aivometastaasistatuksen (ei todettu tai ei tiedossa vs. todettu) mukaan sekä sen mukaan, oliko tutkija (ennen satunnaistamista) aikonut hoitaa potilasta pembrolitsumabilla vai ilman sitä. Ensisijainen tehon tulosmuuttuja oli etenemisvapaa elossaoloaika (PFS), jonka arvioi sokkoutettu riippumaton arviointitoimikunta RECIST 1.1 ‑kriteerien perusteella. Toissijaisia tehon tulosmuuttujia olivat kokonaiselossaoloaika (OS), objektiivinen hoitovaste (ORR) / vasteen kesto (DOR) / taudin hallintavaste (DCR) sokkoutetun riippumattoman arviointitoimikunnan arvioimana, kallonsisäinen objektiivinen hoitovaste (ORR) / vasteen kesto (DOR) sokkoutetun riippumattoman arviointitoimikunnan arvioimana sekä keuhko-oireiden pahenemiseen kulunut aika NSCLC‑oirearviointikyselyn (SAQ) mukaan.
LIBRETTO‑431-tutkimuksen lähtöryhmien mukaisen populaation (ITT‑populaatio) tutkimukseen otetuista ja satunnaistetuista 261 potilaasta 212 stratifioitiin ITT‑pembrolitsumabipopulaatioon sen perusteella, että tutkija oli (ennen satunnaistamista) aikonut hoitaa potilasta pembrolitsumabilla. ITT‑pembrolitsumabipopulaatiossa 129 potilasta sai selperkatinibia ja 83 potilasta sai platinapohjaisen valmisteen ja pemetreksedin yhdistelmää ja pembrolitsumabia. ITT‑pembrolitsumabipopulaatiossa potilaiden iän mediaani oli 61,5 vuotta (vaihteluväli: 31–84 vuotta). 53,3 % potilaista oli naisia. 41,3 % potilaista oli valkoihoisia, 56,3 % oli aasialaisia ja 1 % oli mustaihoisia. 67,9 % potilaista ei ollut koskaan tupakoinut. ITT‑pembrolitsumabipopulaatiossa 93 %:lla potilaista oli etäpesäkkeinen tauti ja 20,3 %:lla oli lähtötilanteessa etäpesäkkeitä keskushermostossa. ECOG-toimintakykyluokaksi ilmoitettiin 0–1 (96,7 %:lla) tai 2 (3,3 %:lla). Fuusiopartnereista yleisin oli KIF5B (44,8 %) ja seuraavaksi yleisin CCDC6 (9,9 %). Tutkimuksessa saavutettiin sen ensisijainen päätetapahtuma, etenemisvapaan elossaoloajan (PFS) piteneminen, sekä ITT‑pembrolitsumabipopulaatiossa että ITT‑populaatiossa. Ensisijaiset tehotulokset ITT-pembrolitsumabipopulaatiossa aiempaa hoitoa saamattomilla potilailla, joilla oli RET-fuusiopositiivinen ei-pienisoluinen keuhkosyöpä, esitetään taulukossa 7 ja kuvassa 1.
Taulukko 7 LIBRETTO-431: Tehotietojen yhteenveto (sokkoutetun riippumattoman arviointitoimikunnan arviointi, ITT-pembrolitsumabipopulaatio)
| Selperkatinibi | Vertailuhaara (platinapohjainen valmiste + pemetreksedi ja pembrolitsumabi) |
|---|---|---|
Etenemisvapaa elossaoloaika | N = 129 | N = 83 |
Mediaani [kk] (95 % lv) | 24,84 (16,89–Ei arv.) | 11,17 (8,77–16,76) |
Riskitiheyssuhde (95 % lv) | 0,465 (0,309–0,699) | |
Stratifioidun logrank-testin p-arvo | 0,0002 | |
24 kuukauden PFS-osuus (%) (95 % lv) | 54,2 (43,6–63,6) | 31,6 (20,1–43,7) |
Objektiivinen vaste (täydellinen vaste + osittainen vaste) |
|
|
% (95 % lv ) | 83,7 (76,2–89,6) | 65,1 (53,8–75,2) |
Täydellinen vaste n (%) | 9 (7,0) | 5 (6,0) |
Osittainen vaste n (%) | 99 (76,7) | 49 (59,0) |
Vasteen kesto* |
|
|
Mediaani [kk] (95 % lv) | 24,18 (17,94–Ei arv.) | 11,47 (9,66–23,26) |
Potilaiden osuus (%), joilla vasteen kesto |
|
|
24 kuukautta (95 % lv) | 59,6 (47,5–69,8) | 22,8 (6,3–45,5) |
lv = luottamusväli; Ei arv. = ei arvioitavissa
*Seurannan keston mediaani oli selperkatinibihaarassa 17,97 kuukautta (25.; 75. persentiili: 12,32; 21,03) ja vertailuhaarassa 14,55 kuukautta (25.; 75. prosenttipiste: 9,69; 20,73).
Tiedon katkaisupäivä: 1.5.2023
Kuva 1. LIBRETTO-431: Etenemisvapaan elossaolon Kaplan–Meier-kuvaaja (sokkoutetun riippumattoman arviointitoimikunnan arviointi, ITT-pembrolitsumabipopulaatio)
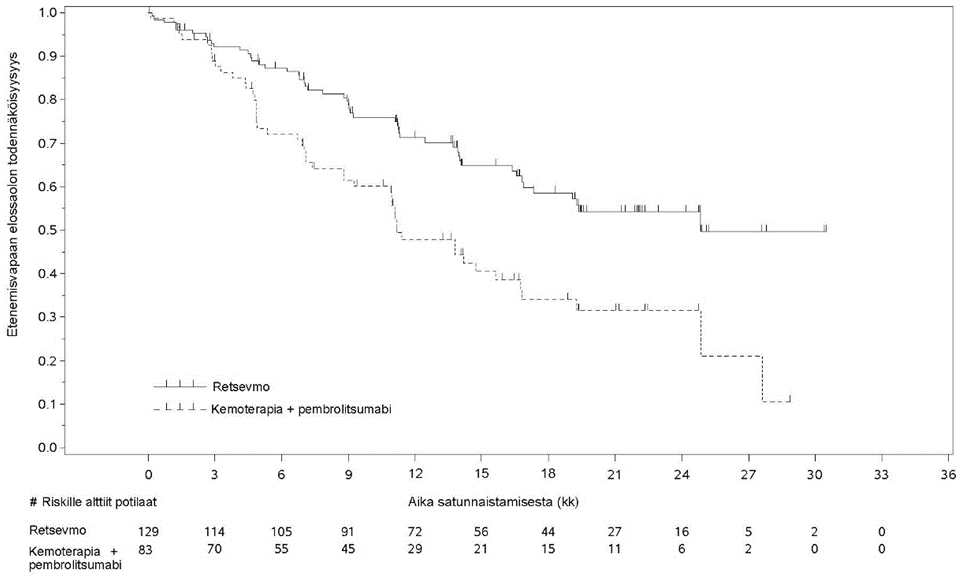
Tiedon katkaisupäivä: 1.5.2023
Tietoa kokonaiselossaolosta (OS) ei ollut riittävästi etenemisvapaan elossaolon (PFS) primaarianalyysin ajankohtana. Päivitetyn kuvailevan kokonaiselossaolon välianalyysin ajankohtana (43 % ennalta määritetyistä lopulliseen kokonaiselossaolon analyysiin tarvittavista tapahtumista, tiedon katkaisupäivä 1. toukokuuta 2024) ITT-populaatiossa todettiin kahdessa haarassa 75 tapahtumaa ja riskitiheyssuhde (HR) oli 1,259 ([95 %:n luottamusväli: 0,777-2,040]; p = 0,3496). 30 kuukauden kohdalla arvioitu kokonaiselossaolo oli 71 % (95 %:n luottamusväli: 63-78) selperkatinibiryhmässä ja 76 % (95 %:n luottamusväli: 66-84) kontrolliryhmässä. Kokonaiselossaoloon on voinut vaikuttaa eroavaisuudet etenemisen jälkeisissä hoidoissa. Niistä 68 kontrollihaaran potilaasta, joilla sairaus eteni, 50 potilasta (74 %) sai selperkatinibia sairauden edetessä. Niistä 71 selperkatinibihaaran potilaasta, joiden sairaus eteni, 16 (23 %) sai solunsalpaajahoitoa ja/tai immuunivasteen estäjähoitoa ja 44 (62 %) jatkoi selperkatinibin käyttöä.
ITT-pembrolitsumabipopulaatiossa selperkatinibi viivästytti merkitsevästi potilaan raportoimien ei-pienisoluisen keuhkosyövän oireiden pahenemista NSCLC-SAQ-kyselylomakkeen kokonaispistemäärän perusteella mitattuna (suureneminen ≥ 2 pistettä) vertailuhaaraan verrattuna (HR: 0,34 [95 % lv: 0,20–0,55]; mediaaniaikaa ei saavutettu selperkatinibihaarassa, vertailuhaarassa se oli 1,9 kuukautta [95 % lv: 0,7; 6,6]). Lisäksi selperkatinibihoito viivästytti merkitsevästi vahvistettua fyysisen toimintakyvyn heikkenemistä ja edisti yleisen elämänlaadun säilymistä ajan mittaan.
LIBRETTO-001
LIBRETTO-001-tutkimukseen otettiin mukaan 362 RET-fuusiopositiivista ei-pienisoluista keuhkosyöpää sairastavaa potilasta. Näistä 69 ei ollut aiemmin saanut hoitoa. Mediaani-ikä oli 63 vuotta (vaihteluväli: 23–92 vuotta). 62,3 % potilaista oli naisia. 69,6 % potilaista oli valkoisia, 18,8 % oli aasialaisia, 5,8 % oli mustia ja 69,6 % ei ollut koskaan tupakoinut. Useimmilla potilailla (98,6 %) oli metastaattinen sairaus tutkimukseen otettaessa ja 23,2 %:lla oli lähtötilanteessa keskushermoston etäpesäkkeitä tutkijan arvioiden mukaan. ECOG-toimintakykyluokaksi ilmoitettiin 0–1 (94,2 %) tai 2 (5,8 %). Yleisin fuusiopartneri oli KIF5B (69,6 %), jota seurasi CCDC6 (14,5 %) ja sitten NCOA4 (1,4 %). Aiemmin hoitamattoman RET-fuusiopositiivisen ei-pienisoluisen keuhkosyövän hoidon tehotuloksista on yhteenveto taulukossa 8.
Taulukko 8 LIBRETTO-001: Objektiivinen vaste ja vasteen kesto
| Tehoarviointiin soveltuvat potilaat Riippumattoman arviointitoimikunnan arvio |
|---|---|
N | 69 |
Objektiivinen vaste (täydellinen vaste + osittainen vaste) |
|
% (95 % lv) | 82,6 (71,6–90,7) |
Täydellinen vaste n (%) | 5 (7,2) |
Osittainen vaste n (%) | 52 (75,4) |
Vasteen kesto (kk)* |
|
Mediaani, 95 % lv | 20,23 (15,4–29,5) |
Potilaiden osuus (%), joilla vasteen kesto |
|
≥ 6 kuukautta (95 % lv) | 87,5 (75,5–93,8) |
≥ 12 kuukautta (95 % lv) | 66,7 (52,4–77,6) |
lv = luottamusväli
*Seurannan keston mediaani oli 37,09 kuukautta (25., 75. prosenttipiste: 24,0, 45,1)
Tiedon katkaisupäivä: 13.1.2023
Aiemmin hoidettu RET-fuusiopositiivinen ei-pienisoluinen keuhkosyöpä
LIBRETTO‑001-tutkimuksessa yhteensä 247 potilasta oli saanut aiemmin platinapohjaista kemoterapiaa. Mediaani-ikä oli 61 vuotta (vaihteluväli: 23–81 vuotta). 56,7 % potilaista oli naisia. 43,7 % potilaista oli valkoisia, 47,8 % aasialaisia, 4,9 % mustia ja 66,8 % ei ollut koskaan tupakoinut. Useimmilla potilailla (98,8 %) oli metastaattinen sairaus tutkimukseen otettaessa ja 31,2 %:lla oli lähtötilanteessa keskushermoston etäpesäkkeitä tutkijan arvion mukaan. ECOG-toimintakykyluokaksi ilmoitettiin 0–1 (97,1 %) tai 2 (2,8 %). Yleisin fuusiopartneri oli KIF5B (61,9 %), jota seurasi CCDC6 (21,5 %) ja sitten NCOA4 (2,0 %). Aikaisempien systeemisten hoitojen mediaanimäärä oli 2 (vaihteluväli: 1–15) ja 43,3 % (n = 107/247) sai 3 tai useampaa aikaisempaa systeemistä hoitoa. Aikaisemmat hoidot sisälsivät anti-PD1/PD L1 ‑hoidon (58,3 %), multikinaasi-inhibiittorin (MKI) (31,6 %) ja taksaanit (34,8 %). 41,3 %:lla oli muuta systeemistä hoitoa. Taulukossa 9 on yhteenveto tehotuloksista aiemmin hoidetuilla RET-fuusiopositiivisilla ei-pienisoluista keuhkosyöpää sairastavilla potilailla.
Taulukko 9 LIBRETTO‑001: Objektiivinen vaste ja vasteen kesto
| Tehoarviointiin soveltuvat potilaat Riippumattoman arviointitoimikunnan arvio |
|---|---|
N | 247 |
Objektiivinen vaste (täydellinen vaste + osittainen vaste) |
|
% (95 % lv) | 61,5 (55,2–67,6) |
Täydellinen vaste, n (%) | 20 (8,1) |
Osittainen vaste, n (%) | 132 (53,4) |
Vasteen kesto (kk)* |
|
Mediaani (95 % lv) | 31,6 (20,4–42,3) |
Potilaiden osuus (%), joilla vasteen kesto |
|
≥ 6 kuukautta (95 % lv) | 87,0 (80,4–91,5) |
≥ 12 kuukautta (95 % lv) | 73,0 (65,0–79,5) |
lv = luottamusväli
*Seurannan keston mediaani oli 39,52 kuukautta (25., 75. prosenttipiste: 24,6, 45,0)
Tiedon katkaisupäivä: 13.1.2023
Keskushermostovaste RET‑fuusiopositiivisessa ei‑pienisoluisessa keuhkosyövässä
LIBRETTO‑431-tutkimuksessa sokkoutetun riippumattoman arviointitoimikunnan arvioima keskushermoston objektiivinen hoitovaste (ORR) oli 82,4 % (14/17; 95 % lv: 56,6–96,2) niillä 17 potilaalla, joilla oli lähtötilanteessa mitattavissa olevia etäpesäkkeitä aivoissa ja jotka saivat selperkatinibihoitoa, kun taas ITT‑pembrolitsumabipopulaation vertailuhaaran 12 potilaalla se oli 58,3 % (7/12; 95 % lv: 27,7–84,4). Täydellinen vaste (CR) todettiin selperkatinibihaarassa 6 potilaalla 17:stä (35,3 %) ja vertailuhaarassa 2 potilaalla 12:sta (16,7 %). Vasteen keston (DOR) seurannan mediaanin ollessa selperkatinibihaarassa 9,92 kuukautta (95 % lv: 7,66–18,10) ja vertailuhaarassa 12,68 kuukautta (95 % lv: 2,79–ei arv.) vasteen keston mediaania ei saavutettu selperkatinibihaarassa (95 % lv: 7,62–ei arv.) ja vertailuhaarassa se oli 13,4 kuukautta (95 % lv: 3,45–ei arv.). 192 potilaalla, joista oli saatavilla lähtötilanteen kallonsisäiset kuvantamistulokset, syyn mukaan spesifioitu keskushermoston taudin etenemiseen kuluneen ajan riskitiheyssuhde oli sokkoutetun riippumattoman arviointitoimikunnan arvioimana 0,28 (95 % lv: 0,12–0,68) (150 potilaalla, joilla ei ollut lähtötilanteessa kallonsisäisiä etäpesäkkeitä, riskitiheyssuhde oli 0,17 [95 % lv: 0,04–0,69], ja 42 potilaalla, joilla oli lähtötilanteessa kallonsisäisiä etäpesäkkeitä, riskitiheyssuhde oli 0,61 [95 % lv: 0,19–1,92]). Selperkatinibihaarassa 8 potilaalla (6,7 %) ja vertailuhaarassa 13 potilaalla (18,1 %) todettiin ensimmäinen keskushermostoetenemistapahtuma.
LIBRETTO‑001-tutkimuksessa riippumattoman arviointitoimikunnan arvioima keskushermoston objektiivinen vasteprosentti oli 84,6 % (22/26; 95 % lv: 65,1–95,6) 26 potilaalla, joilla oli mitattavissa oleva sairaus. Täydellinen vaste havaittiin 7 (26,9 %) potilaalla ja osittainen vaste 15 (57,5 %) potilaalla. Keskushermostovasteen keston (CNS DOR) mediaani oli 9,36 kuukautta (95 % lv: 7,4–15,3).
Aiemmin systeemisesti hoitamaton RET-fuusiopositiivinen kilpirauhassyöpä
LIBRETTO‑001‑tutkimukseen otetuista potilaista, joilla oli RET‑fuusiopositiivinen kilpirauhassyöpä ja jotka eivät olleet saaneet aiemmin muuta systeemistä hoitoa kuin radiojodia, 24 potilaalla oli mahdollisuus seurantaan vähintään 6 kuukauden ajan, ja heidän katsottiin soveltuvan tehoarviointiin. Mediaani-ikä oli 60,5 vuotta (vaihteluväli: 20–84 vuotta). Potilaista 58,3 % oli miehiä. Potilaista 75 % oli valkoihoisia. ECOG-toimintakykyluokaksi ilmoitettiin 0–1 (95,8 %) tai 2 (4,2 %). Potilaista 100 %:lla oli etäpesäkkeinen tauti. 22 potilasta 24:stä potilaasta (91,7 %) oli saanut radiojodia ennen tutkimukseen ottamista ja heidät katsottiin radiojodille refraktoriseksi. Eri histologiatyyppejä 24 potilaalla olivat: papillaarinen (n = 23) ja huonosti erilaistunut (n = 1). Yleisin fuusiopartneri oli CCDC6 (62,5 %) ja toiseksi yleisin NCOA4 (29,2 %). Taulukossa 10 on yhteenveto tehotuloksista RET-fuusiopositiivista kilpirauhassyöpää sairastavilla potilailla, jotka eivät olleet saaneet aiemmin systeemistä hoitoa.
Taulukko 10 LIBRETTO‑001: Objektiivinen vaste ja vasteen kesto
| Tehoarviointiin soveltuvat potilaat Riippumattoman arviointitoimikunnan arvio |
|---|---|
N | 24 |
Objektiivinen vaste (täydellinen vaste + osittainen vaste) |
|
% (95 % lv) | 95,8 (78,9–99,9) |
Täydellinen vaste, n (%) | 5 (20,8) |
Osittainen vaste, n (%) | 18 (75,0) |
Vasteen kesto (kk)* |
|
Mediaani (95 % lv) | Ei arv. (42,8–Ei arv.) |
Potilaiden osuus (%), joilla vasteen kesto |
|
≥ 12 kuukautta (95 % lv) | 100,0 (100,0–100,0) |
≥ 24 kuukautta (95 % lv) | 94,4 (66,6–99,2) |
≥ 36 kuukautta (95 % lv) | 88,9 (62,4–97,1) |
lv = luottamusväli; Ei arv. = ei arvioitavissa
*Seurannan keston mediaani oli 17,81 kuukautta (25., 75. prosenttipiste: 32,3; 62,5)
Tiedon katkaisupäivä: 14.2.2025
Aiemmin hoidettu RET‑fuusiopositiivinen kilpirauhassyöpä
LIBRETTO‑001‑tutkimukseen otetuista potilaista, joilla oli RET‑fuusiopositiivinen kilpirauhassyöpä ja jotka olivat saaneet aiemmin jotain muuta systeemistä hoitoa kuin radiojodia, 41 potilaalla oli mahdollisuus seurantaan vähintään 6 kuukauden ajan, ja heidän katsottiin soveltuvan tehoarviointiin. Iän mediaani oli 58 vuotta (vaihteluväli: 25–88 vuotta). Potilaista 43,9 % oli miehiä. Potilaista 58,5 % oli valkoihoisia, 29,3 % aasialaisia ja 7,3 % mustaihoisia. ECOG‑toimintakykyluokaksi ilmoitettiin 0–1 (92,7 %) tai 2 (7,3 %). Potilaista 100 %:lla oli etäpesäkkeinen tauti. Potilaiden aiempien systeemisten hoitojen mediaanimäärä oli 3 (vaihteluväli: 1–7). Yleisimpiä aiempia hoitoja olivat radiojodi (73,2 %) ja multikinaasi-inhibiittori (85,4 %). Potilaista 9,8 % oli saanut muuta systeemistä hoitoa. Eri histologiatyyppejä 41 potilaalla olivat: papillaarinen (n = 31), huonosti erilaistunut (n = 5), anaplastinen (n= 4) ja Hürthlen solu (n = 1). Yleisin fuusiopartneri oli CCDC6 (61,0 %) ja toiseksi yleisin NCOA4 (19,5 %).
Aiemmin hoidetun RET‑fuusiopositiivisen kilpirauhassyövän hoidon tehotuloksista on yhteenveto taulukossa 11.
Taulukko 11 LIBRETTO-001: Objektiivinen vaste ja vasteen kesto
| Tehoarviointiin soveltuvat potilaat Riippumattoman arviointitoimikunnan arvio |
|---|---|
N | 41 |
Objektiivinen vaste (täydellinen vaste + osittainen vaste) |
|
% (95 % lv) | 85,4 (70,8–94,4) |
Täydellinen vaste, n (%) | 5 (12,2) |
Osittainen vaste, n (%) | 30 (73,2) |
Vasteen kesto (kk)* |
|
Mediaani (95 % lv) | 26,7 (12,1–Ei arv.) |
Potilaiden osuus (%), joilla vasteen kesto |
|
≥ 12 kk (95 % lv) | 71,7 (52,4–84,2) |
≥ 24 kk (95 % lv) | 50,7 (30,4–67,8) |
lv = luottamusväli; Ei arv. = ei arvioitavissa
*Seurannan keston mediaani oli 33,87 kk (25., 75. prosenttipiste: 12,9, 44,8)
Tiedon katkaisupäivä: 13.1.2023
RET-mutaatiopositiivinen medullaarinen kilpirauhassyöpä, jota ei ole hoidettu vandetanibilla ja kabotsantinibilla
LIBRETTO-531
Retsevmo-valmisteen teho RET-mutaatiopositiivisen medullaarisen kilpirauhassyövän hoidossa vahvistettiin LIBRETTO‑531-monikeskustutkimuksessa, joka oli vaiheen 3 satunnaistettu, avoin vertailututkimus. Siinä selperkatinibihoitoa verrattiin lääkärin valinnan mukaan joko kabotsantinibihoitoon tai vandetanibihoitoon potilailla, joilla oli progressiivinen, pitkälle edennyt RET-mutaatiopositiivinen medullaarinen kilpirauhassyöpä, johon ei ollut aiemmin annettu kinaasinestäjähoitoa. Tutkimukseen otettiin aikuisia ja nuoria potilaita, joilla oli histologisesti varmistettu, leikkaushoitoon soveltumaton, paikallisesti levinnyt tai etäpesäkkeinen medullaarinen kilpirauhassyöpä ja jotka eivät olleet aiemmin saaneet kinaasinestäjähoitoa. Potilaat saivat 160 mg selperkatinibia kahdesti vuorokaudessa (aloitusannos) tai lääkärin valinnan mukaan joko kabotsantinibia (140 mg kerran vuorokaudessa) tai vandetanibia (300 mg kerran vuorokaudessa). Potilaat stratifioitiin RET-mutaation (M918T vs. muu) mukaan ja vertailuhaaraan satunnaistetut potilaat myös aiotun hoidon (kabotsantinibi vs. vandetanibi) mukaan. Ensisijainen tehon tulosmuuttuja oli etenemisvapaa elossaoloaika (PFS), jonka arvioi sokkoutettu riippumaton arviointitoimikunta RECIST 1.1 ‑kriteerien perusteella. Tärkeimmät toissijaiset tehon tulosmuuttujat olivat elossaoloaika ilman hoidon epäonnistumista (TFFS) ja komparatiivinen siedettävyys, ja muita toissijaisia tehon tulosmuuttujia olivat kokonaiselossaoloaika (OS) ja objektiivinen hoitovaste (ORR) / vasteen kesto (DOR) sokkoutetun riippumattoman arviointitoimikunnan arvioimana.
LIBRETTO‑531-tutkimuksen lähtöryhmien mukaisen populaation (ITT‑populaatio) tutkimukseen otetuista ja satunnaistetuista 291 potilaasta 193 satunnaistettiin selperkatinibihaaraan ja 98 vertailuhaaraan. Vertailuhaaraan satunnaistetuista 98 potilaasta 73 kuului kabotsantinibiositteeseen ja 25 vandetanibiositteeseen. ITT-populaatiossa potilaiden iän mediaani oli 55 vuotta (vaihteluväli: 12–84 vuotta). 37,1 % potilaista oli naisia. 69,4 % potilaista oli valkoihoisia, 27,7 % oli aasialaisia ja 2,9 % oli mustaihoisia. Useimmilla potilailla (77 %) oli etäpesäkkeinen tauti tutkimukseenottovaiheessa. ECOG-toimintakykyluokaksi ilmoitettiin 0–1 (98,3 %:lla) tai 2 (1 %:lla). Yleisin mutaatiotyyppi oli M918T (62,5 %). Tutkimuksessa saavutettiin ensisijainen päätetapahtuma, joka oli etenemisvapaan elossaoloajan pidentyminen ITT-populaatiossa. Yhteenveto tehotuloksista ITT‑populaatiossa esitetään taulukossa 12 ja kuvassa 2.
Taulukko 12 LIBRETTO-531: Tehotietojen yhteenveto (sokkoutetun riippumattoman arviointitoimikunnan arvio, ITT-populaatio)
Selperkatinibi | Vertailuhaara (kabotsantinibi tai vandetanibi) | |
|---|---|---|
Etenemisvapaa elossaoloaika (PFS) | N = 193 | N = 98 |
Mediaani [kk] (95 % lv) | Ei arv. (Ei arv.– Ei arv.) | 16,76 (12,22–25,10) |
Riskitiheyssuhde (95 % lv) | 0,280 (0,165–0,475) | |
Stratifioidun logrank-testin p-arvo | < 0,0001 | |
30 kuukauden PFS-osuus (%) (95 % lv) | 76,4 (66,5–83,8) | 24,8 (6,9–48,3) |
Elossaolo ilman hoidon epäonnistumista (TFFS)* | N = 193 | N = 98 |
Mediaani [kk] (95 % lv) | Ei arv. (Ei arv.– Ei arv.) | 13,93 (11,27–25,10) |
Riskitiheyssuhde (95 % lv) | 0,254 (0,153–0,423) | |
Stratifioidun logrank-testin p-arvo | < 0,0001 | |
30 kuukauden TFFS-osuus (%) (95 % lv) | 75,8 (65,9–83,2) | 25,3 (7,2–48,8) |
Objektiivinen vaste (täydellinen vaste + osittainen vaste) | ||
% (95 % lv) | 69,4 (62,4–75,8) | 38,8 (29,1–49,2) |
Täydellinen vaste n (%) | 23 (11,9) | 4 (4,1) |
Osittainen vaste n (%) | 111 (57,5) | 34 (34,7) |
Vasteen kesto# | ||
Mediaani [kk] (95 % lv) | Ei arv. (Ei arv.– Ei arv.) | 16,56 (10,41– Ei arv.) |
Potilaiden osuus (%), joilla vasteen kesto | ||
≥ 24 kuukautta (95 % lv) | 79,1 (66,9–87,2) | Ei arv. (Ei arv. – Ei arv.) |
lv = luottamusväli; Ei arv. = ei arvioitavissa
*Elossaolo ilman hoidon epäonnistumista (TFFS) määriteltiin ajaksi satunnaistamisesta ensimmäiseen seuraavista: dokumentoitu radiologinen taudin eteneminen RECIST 1.1 ‑kriteerien mukaan, liiallinen hoidon keskeyttämiseen johtanut toksisuus tutkijan arvioimana, kuolema mistä tahansa syystä.
#Seurannan keston mediaani oli selperkatinibihaarassa 11,14 kuukautta (25.; 75. prosenttipiste: 5,62; 16,62) ja vertailuhaarassa 12,81 kuukautta (25. ja 75. prosenttipiste: 6,34; 15,51).
Tiedon katkaisupäivä: 22.5.2023
Kuva 2. LIBRETTO-531: Etenemisvapaan elossaolon Kaplan-Meier-kuvaaja (sokkoutetun riippumattoman arviointitoimikunnan arviointi, ITT-populaatio)
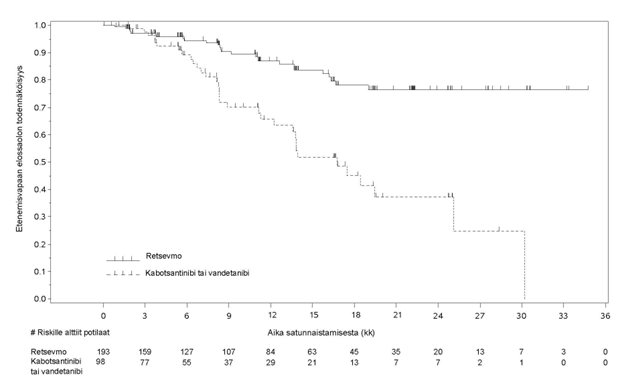
Tiedon katkaisupäivä: 22.5.2023
Etenemisvapaan elossaolon (PFS) primaarianalyysin ajankohtana kahdessa tutkimushaarassa oli todettu yhteensä 18 kokonaiselossaoloajan (OS) tapahtumaa. ITT‑populaatiossa kokonaiselossaoloajan riskitiheyssuhde (HR) oli 0,374 (95 % lv: 0,147–0,949). Sensuroitujen osuus oli selperkatinibihaarassa 95,9 % ja vertailuhaarassa 89,8 %.
Komparatiivista siedettävyyttä arvioitiin 242 potilaalla (selperkatinibihaara, N = 161; vertailuhaara N = 81). Selperkatinibihaarassa todettiin tilastollisesti merkitsevästi pienempi sellaisen hoitoajan osuus, jona potilaat ilmoittivat haittavaikutusten suurta häiritsevyyttä (”high side effect bother”) (8 %), verrattuna vertailuhaaraan (24 %) (95 % lv: −23 %; −10 %, p < 0,0001), kun suureksi häiritsevyydeksi käsitettiin syöpähoidon toiminnallisen arvioinnin (Functional Assessment of Cancer Therapy) kohdan GP5 vastaukset 3 ”Quite a bit” (’Melko paljon’) ja 4 ”Very much” (’Erittäin paljon’).
Myöhemmässä kokonaiselossaoloajan analyysissa, jonka data lock ‑päivä oli 11.3.2024, kahdessa tutkimushaarassa todettiin 26 tapahtumaa, ja riskitiheyssuhde oli 0,275 (95 % lv: 0,124–0,608). Tässä analyysissa etenemisvapaan elossaolon riskitiheyssuhde oli 0,202 (95 % lv: 0,128–0,320) ja objektiivinen vaste oli selperkatinibihaarassa 82,4 % ja vertailuhaarassa 43,9 %.
LIBRETTO-001
LIBRETTO-001-tutkimukseen otetusta 324 potilaasta, joilla oli RET-mutaatiopositiivinen medullaarinen kilpirauhassyöpä, 143 potilasta ei ollut aiemmin saanut kabotsantinibi- ja vandetanibihoitoa. Näistä 116 ei ollut aiemmin saanut muuta systeemistä hoitoa ja 27 oli aiemmin saanut muuta systeemistä hoitoa. Potilaiden, jotka eivät olleet aiemmin saaneet kabotsantinibia ja vandetanibia, iän mediaani oli 57 vuotta (vaihteluväli: 15–87 vuotta). Potilaista 2 (1,4 %) oli alle 18-vuotiaita, 58,0 % oli miehiä. Potilaista 86,7 % oli valkoihoisia, 5,6 % aasialaisia ja 1,4 % mustaihoisia. Useimmilla potilailla (97,9 %) oli metastaattinen sairaus tutkimukseen ottamisen yhteydessä. ECOG-toimintakykyluokaksi ilmoitettiin 0-1 (95,9 %) tai 2 (4,2 %). Yleisin mutaatio oli M918T (60,1 %), jota seurasivat solunulkoiset kysteiinimutaatiot (23,8 %). Potilaiden, joilla oli RET-mutaatiopositiivinen medullaarinen kilpirauhassyöpä ja jotka eivät olleet saaneet kabotsantinibi- ja vandetanibihoitoa, hoidon tehotuloksista on yhteenveto taulukossa 13.
Taulukko 13 LIBRETTO-001: Objektiivinen vaste ja vasteen kesto
| Tehoarviointiin soveltuvat potilaat Riippumattoman arviointitoimikunnan arvio |
N | 143 |
Objektiivinen vaste (täydellinen vaste + osittainen vaste) |
|
% (95 % lv) | 82,5 (75,3–88,4) |
Täydellinen vaste, n (%) | 34 (23,8) |
Osittainen vaste, n (%) | 84 (58,7) |
Vasteen kesto (kk)* |
|
Mediaani (95 % lv) | Ei arv. (51,3–Ei arv.) |
Vasteen kesto (%) |
|
≥ 12 kuukautta (95 % lv) | 91,4 (84,6–95,3) |
≥ 24 kuukautta (95 % lv) | 84,1 (75,9–89,7) |
lv = luottamusväli; Ei arv. = ei arvioitavissa
*Seurannan keston mediaani oli 39,4 kuukautta (25., 75. prosenttipiste: 32,3, 45,4).
Tiedon katkaisupäivä 13.1.2023
Aiemmin hoidettu RET‑mutaatiopositiivinen medullaarinen kilpirauhassyöpä
LIBRETTO‑001‑tutkimukseen otetuista potilaista, joilla oli RET‑mutaatiopositiivinen medullaarinen kilpirauhassyöpä, 152 potilasta oli aiemmin hoidettu kabotsantinibillä ja/tai vandetanibilla, ja heidän katsottiin soveltuvan tehoarviointiin. Iän mediaani oli 58 vuotta (vaihteluväli 17–90: vuotta); 1 potilas (0,7 %) oli < 18‑vuotias. Potilaista 63,8 % oli miehiä. Potilaista 90,1 % oli valkoihoisia, 1,3 % aasialaisia ja 1,3 % mustaihoisia. ECOG‑toimintakykyluokaksi ilmoitettiin 0–1 (92,7 %) tai 2 (7,2 %). Potilaista 98,0 %:lla oli etäpesäkkeinen tauti. Yleisin mutaatio oli M918T (65,1 %) ja toiseksi yleisimpiä olivat ekstrasellulaariset kysteiinimutaatiot (15,8 %). Potilaista 100 % (n = 152) oli saanut aiempaa systeemistä hoitoa (aiempien systeemisten hoitojen mediaanimäärä 2) ja 27,6 % (n = 42) oli saanut vähintään 3:a aiempaa systeemistä hoitoa.
Aiemmin hoidetun RET‑mutaatiopositiivisen medullaarisen kilpirauhassyövän hoidon tehotuloksista on yhteenveto taulukossa 14.
Taulukko 14 LIBRETTO-001: Objektiivinen vaste ja vasteen kesto
| Tehoarviointiin soveltuvat potilaat Riippumattoman arviointitoimikunnan arvio |
N | 152 |
Objektiivinen vaste (täydellinen vaste + osittainen vaste) |
|
% (95 % lv) | 77,6 (70,2–84,0) |
Täydellinen vaste, n (%) | 19 (12,5) |
Osittainen vaste, n (%) | 99 (65,1) |
Vasteen kesto (kk)* |
|
Mediaani (95 % lv) | 45,3 (33,6–Ei arv.) |
Vasteen kesto (%) |
|
≥ 12 kuukautta (95 % lv) | 83,0 (74,6–88,8) |
≥ 24 kuukautta (95 % lv) | 66,4 (56,3–74,7) |
lv = luottamusväli; Ei arv. = ei arvioitavissa
*Seurannan keston mediaani oli 38,3 kk (25., 75. prosenttipiste: 23,0, 46,1)
Tiedon katkaisupäivä 13.1.2023
Muut RET-fuusiopositiiviset kiinteät kasvaimet
Tehoa arvioitiin 75 potilaalla, joilla oli muita RET-fuusiopositiivisia kasvaimia kuin ei-pienisoluinen keuhkosyöpä tai kilpirauhassyöpä. Potilaiden sairaus oli edennyt, tai he olivat saaneet aiempaa systeemistä hoitoa, tai heillä ei ollut jäljellä tyydyttäviä hoitovaihtoehtoja. Mediaani-ikä oli 59 vuotta (vaihteluväli: 21–92 vuotta). Potilaista 50,7 % oli naisia, 60,0 % valkoihoisia, 34,7 % aasialaisia ja 4,0 % tummaihoisia. ECOG-toimintakykyluokka oli 0–1 (90,6 %) tai 2 (9,3 %), ja 96,0 %:lla potilaista oli metastaattinen sairaus. 69 potilasta (92,0 %) oli saanut aiempaa systeemistä hoitoa. Aiempien systeemisten hoitojen mediaanimäärä oli 2 (vaihteluväli: 0-9) ja 36,0 % oli saanut 3:a tai useampaa aiempaa systeemistä hoitoa. Kukaan potilaista ei ollut saanut aiempaa hoitoa selektiivisellä RET-estäjällä. Yleisimmät syövät olivat paksusuolisyöpä (29,3 %), haimasyöpä (24,0 %), sylkirauhasten syöpä (6,7 %), sarkooma (6,7 %) ja sappitiehyeiden syöpä (6,7 %). Yleisimmät fuusiopartnerit olivat NCOA4 (38,7 %), CCDC6 (20,0 %) ja KIF5B (8,0 %). Taulukoissa 15 ja 16 on yhteenveto tehotuloksista RET-fuusiopositiivisille kiinteille kasvaimille (muu kuin ei-pienisoluinen keuhkosyöpä tai kilpirauhassyöpä).
Taulukko 15 LIBRETTO‑001: Objektiivinen vaste ja vasteen kesto
| Tehoarviointiin soveltuvat potilaat Riippumattoman arviointitoimikunnan arvio |
|---|---|
N | 75 |
Objektiivinen vaste (täydellinen vaste + osittainen vaste) | |
% (95 % lv) | 46,7 (35,1–58,6) |
Täydellinen vaste, n (%) | 4 (5,3) |
Osittainen vaste, n (%) | 31 (41,3) |
Vasteen kesto (kk)* | |
Mediaani (95 % lv) | 24,54 (11,2–49,1) |
Vasteen kesto (%) | |
≥ 6 kuukautta (95 % lv) | 82,0 (64,2–91,5) |
≥ 12 kuukautta (95 % lv) | 68,6 (49,3–81,8) |
≥ 24 kuukautta (95 % lv) | 52,5 (32,6–69,0) |
≥ 36 kuukautta (95 % lv) | 43,3 (24,0–61,1) |
lv = luottamusväli; Ei arv. = ei arvioitavissa
*Seurannan keston mediaani oli 32,23 kk (25., 75. prosenttipiste: 13,3; 50,8)
Tiedon katkaisupäivä 14.2.2025
Taulukko 16 LIBRETTO‑001: Objektiivinen vaste ja vasteen kesto kasvaintyypeittäin
Kasvaintyyppi | Potilaat (N = 75) | Objektiivinen vaste (Riippumattoman arviointitoimikunnan arvio) | Vasteen keston vaihteluväli (kk) | |
|---|---|---|---|---|
n (%) | 95 % lv | |||
Paksu- ja peräsuolisyöpä | 22 | 10 (45,5) | 24,4–67,8 | 4,63–36,14+ |
Haimasyöpä | 18 | 9 (50,0) | 26,0–74,0 | 2,50–52,14 |
Sylkirauhasten syöpä | 5 | 3 (60,0) | 14,7–94,7 | 5,72–37,19 |
Sappitiehyeiden syöpä | 5 | 2 (40,0) | 5,3–85,3 | 7,36–14,82 |
Sarkooma | 5 | 2 (40,0) | 5,3–85,3 | 3,71–56,51+ |
Ihokarsinooma | 3 | 1 (33,3) | 0,8–90,6 | 27,14 |
Tuntematon, primaarikasvain | 3 | 1 (33,3) | 0,8–90,6 | 9,23 |
Rintasyöpä | 2 | osittainen vaste, täydellinen vaste | 15,8–100,0 | 2,30+ –17,28 |
Ksantogranulooma | 2 | Ei arv., Ei arv.a | 0,0–84,2 | Ei soveltuva |
Karsinoidi | 1 | osittainen vaste | 2,5–100,0 | 49,08 |
Munasarjasyöpä | 1 | osittainen vaste | 2,5–100,0 | 28,55+ |
Keuhkon karsinosarkooma | 1 | Ei arv. | 0,0–97,5 | Ei soveltuva |
Peräsuolen neuroendokriininen kasvain | 1 | Ei arv. | 0,0–97,5 | Ei soveltuva |
Ohutsuolisyöpä | 1 | täydellinen vaste | 2,5–100,0 | 24,54 |
Neuroendokriininen kasvain | 1 | osittainen vaste | 2,5–100,0 | 23,13 |
Pienisoluinen keuhkosyöpä | 1 | stabiili sairaus | 0,0–97,5 | Ei soveltuva |
Maha-ruokatorviliitos | 1 | stabiili sairaus | 0,0–97,5 | Ei soveltuva |
Haiman neuroendokriini | 1 | osittainen vaste | 2,5–100,0 | 17,51+ |
Mahalaukku | 1 | stabiili sairaus | 0,0–97,5 | Ei soveltuva |
+ tarkoittaa jatkuvaa vastetta.
a Yhden ksantogranuloomapotilaan sairautta ei voitu arvioida riippumattoman arviointitoimikunnan toimesta, koska sairaus oli vain ihossa. Tutkijan arvioon perustuen kyseinen potilas sai täydellisen vasteen.
lv = luottamusväli, Ei arv. = ei arvioitavissa
Tiedon katkaisupäivä 14.2.2025
RET-fuusiopositiivisen syövän harvinaisuudesta johtuen potilaita tutkittiin useista kasvaintyypeistä, ja joitakin kasvaintyyppejä oli vain rajoitetulla potilasmäärällä. Tämä aiheuttaa epävarmuutta kasvaintyyppikohtaisen objektiivisen vasteen arviointiin. Koko populaation objektiivinen vaste ei välttämättä kuvasta odotettavissa olevaa vastetta tietyssä kasvaintyypissä.
Pediatriset potilaat
Marraskuun 8. päivään 2024 mennessä LIBRETTO‑121‑tutkimuksessa teho arvioitiin 36 potilaalla, joiden ikä oli 2–20 vuotta. LIBRETTO-121 on vaiheen 1/2 tutkimus pediatrisilla potilailla, joilla on pitkälle edennyt kiinteä kasvain tai primaarinen keskushermostokasvain aktivoivalla RET-muutoksella. 31 potilasta 36:sta oli alle 18-vuotiaita. 36:sta potilaasta 14 potilaalla oli RET-mutaatiopositiivinen medullarinen kilpirauhassyöpä, 1 potilaalla RET-fuusiopositiivinen medullaarinen kilpirauhassyöpä, 15 potilaalla RET-fuusiopositiivinen kilpirauhassyöpä, 3 potilaalla RET-fuusiopositiivinen muu kiinteä kasvain ja 3 potilaalla RET-mutaatiopositiivinen muu kiinteä kasvain.
RET-mutaatiopositiivista medullaarista kilpirauhassyöpää sairastavista 14 potilaasta 11 oli alle 18-vuotiaita, joista 5 oli alle 12-vuotiaita. Riippumattoman arviointitoimikunnan arvion mukaan kaikkien 14 potilaan objektiivinen vasteosuus oli 57,1 % (95 %:n luottamusväli: 28,9–82,3). Viidellä potilaalla todettiin varmistettu täydellinen vaste ja kolmella potilaalla todettiin varmistettu osittainen vaste.
Näiden 14 potilaan vasteen keston mediaania (kuukausina) ei voitu määrittää (95 %:n luottamusväli: 24,9 – ei voida määrittää). Potilaiden osuus (%), joilla vasteen kesto oli ≥ 24 kuukautta, oli 100 % (95 %:n luottamusväli: 100–100) ja ≥ 30 kuukauden kohdalla 83,3 % (95 %:n luottamusväli: 27,3–97,5).
RET‑fuusiopositiivista kilpirauhassyöpää sairastavista 15 potilaasta 13 oli alle 18‑vuotiaita, joista 3 oli alle 12‑vuotiaita. Näistä 15 potilaasta 7 oli saanut aiemmin radiojodihoitoa. Riippumattoman arviointitoimikunnan arvion mukaan kaikkien 15 potilaan objektiivinen vasteosuus oli 53,3 % (95 %:n luottamusväli: 26,6–78,7). Neljällä potilaalla todettiin varmistettu täytellinen vaste ja neljällä potilaalla varmistettu osittainen vaste. Näiden 15 potilaan vasteen keston mediaania (kuukausina) ei voitu määrittää (95 %:n luottamusväli: ei voida määrittää–ei voida määrittää). Niiden potilaiden osuus (%), joilla vasteen kesto oli ≥ 42 kuukautta, oli 100 % (95 %:n luottamusväli: 100–100).
Kaikki kolme potilasta, joilla oli RET‑fuusiopositiivisia muita kiinteitä kasvaimia, olivat alle 18‑vuotiaita, joista yksi oli alle 12‑vuotias. Näistä kolmesta potilaasta kahdella todettiin varmistettu osittainen vaste.
Kaikki kolme potilasta, joilla oli RET‑mutaatiopositiivisia muita kiinteitä kasvaimia, olivat alle 18‑vuotiaita, joista kaksi oli alle 12‑vuotiaita. Näistä kolmesta potilaasta yhdelläkään ei todettu hoitovastetta selperkatinibihoitoon.
Euroopanlääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset selperkatinibi-valmisteen käytöstä 6 kk ikäisillä ja sitä nuoremmilla potilailla, joilla on kiinteitä kasvaimia (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Selperkatinibin farmakokinetiikkaa arvioitiin potilailla, joilla oli paikallisesti edennyt tai etäpesäkkeinen kiinteä kasvain. Potilaille annettiin 160 mg selperkatinibia kahdesti vuorokaudessa, ellei toisin mainita. Selperkatinibin vakaan tilan AUC‑arvo ja Cmax‑arvo suurenivat lineaarisesta supra-additiiviseen tapaan annosalueella 20 mg kerran vuorokaudessa – 240 mg kahdesti vuorokaudessa.
Vakaa tila saavutettiin noin 7 vuorokaudessa, ja kumuloitumissuhteen mediaani annostuksella 160 mg kahdesti vuorokaudessa oli 3,4‑kertainen. Keskiarvo selperkatinibin vakaan tilan [variaatiokerroin (CV%)] Cmax‑arvolle oli 2 980 (53 %) ng/ml ja AUC0‑24h‑arvon keskiarvo oli 51 600 (58 %) ng*h/ml.
In vivo -tutkimukset osoittavat, että selperkatinibi on lievä P-gp:n estäjä.
In vitro ‑tutkimukset osoittavat, että kliinisesti merkittävät selperkatinibipitoisuudet eivät estä eivätkä indusoi CYP1A2‑, CYP2B6‑, CYP2C9‑, CYP2C19‑ eivätkä CYP2D6‑toimintaa.
In vitro ‑tutkimukset osoittavat, että kliinisesti merkittävät selperkatinibipitoisuudet estävät MATE1‑ ja BCRP‑toimintaa, mutta eivät OAT1‑, OAT3‑, OCT1‑, OCT2‑, OATP1B1‑, OATP1B3‑, BSEP‑ eivätkä MATE2‑K‑toimintaa. Selperkatinibi saattaa suurentaa seerumin kreatiniinipitoisuutta estämällä MATE1‑toimintaa ja vähentämällä täten kreatiniinin erittymistä munuaistubulusten kautta.
Selperkatinibin kova kapseli- ja kalvopäällysteinen tabletti -annosmuodot ovat biologisesti samanarvoisia.
Imeytyminen
Suun kautta annetun 160 mg annoksen jälkeen Retsevmo imeytyi nopeasti; tmax oli noin 2 tuntia. Suun kautta annetun selperkatinibin absoluuttisen biologisen hyötyosuuden geometrinen keskiarvo oli 73,2 % (vaihteluväli: 60,2–81,5 %).
Ruoan vaikutus
Tyhjään mahaan otetun selperkatinibin AUC‑ ja Cmax‑arvoon verrattuna selperkatinibin AUC‑arvo suureni 9 % ja Cmax‑arvo pieneni 14 %, kun terveille tutkittaville annettiin 160 mg kerta‑annos suun kautta runsasrasvaisen aterian yhteydessä. Muutoksia ei pidetty kliinisesti merkittävinä. Selperkatinibi voidaan siis ottaa ruoan kanssa tai ilman ruokaa.
Jakautuminen
Selperkatinibin jakautumistilavuuden (Vss/F) näennäinen keskiarvo (CV %) oli populaatiofarmakokinetiikan analyysin perusteella 203,1 l (69 %), kun selperkatinibia annettiin suun kautta aikuispotilaille. Selperkatinibi sitoutuu ihmisellä 96‑prosenttisesti plasman proteiineihin in vitro, eikä sitoutuminen ole pitoisuudesta riippuvaista. Veri–plasma‑pitoisuussuhde on 0,7.
Biotransformaatio
Selperkatinibi metaboloituu lähinnä CYP3A4‑välitteisesti. Kun terveille tutkittaville annettiin suun kautta 160 mg kerta‑annos radioaktiivisesti merkittyä [14C]‑selperkatinibia, plasmasta mitatuista radioaktiivisista komponenteista 86 % oli muuttumatonta selperkatinibia.
Eliminaatio
Selperkatinibin puhdistuman (CL/F) näennäinen keskiarvo (CV%) oli 5,5 l/h (45 %) ja puoliintumisaika 26,5 tuntia, kun selperkatinibia annettiin suun kautta aikuispotilaille. Kun terveille tutkittaville annettiin suun kautta 160 mg kerta‑annos radioaktiivisesti merkittyä [14C]‑selperkatinibia, 69 % annetusta radioaktiivisuudesta erittyi ulosteeseen (14 % muuttumatonta) ja 24 % virtsaan (11,5 % muuttumatonta).
Erityisryhmät
Ikä, sukupuoli ja paino
Iällä (vaihteluväli: 2–92 v) tai sukupuolella ei ollut kliinisesti merkittävää vaikutusta Retsevmo‑valmisteen farmakokinetiikkaan. Selperkatinibin näennäinen jakautumistilavuus (Vss/F) ja näennäinen puhdistuma (CL/F) suurenevat kehon painon kasvaessa (9,6–179 kg).
Jos potilas on vähintään 12-vuotias ja painaa < 50 kg, Retsevmo‑hoito on aloitettava annostuksella 120 mg kahdesti vuorokaudessa, ja jos potilas painaa ≥ 50 kg, Retsevmo‑hoito on aloitettava annostuksella 160 mg kahdesti vuorokaudessa.
Vähintään 2-vuotiaiden ja alle 12-vuotiaiden pediatristen potilaiden Retsevmo-hoito tulee aloittaa kehon pinta-alan perusteella määritetyllä annoksella (taulukko 1). Retsevmoa ei suositella pediatrisille potilaille, joiden kehon pinta-ala on alle 0,45 m2.
Maksan vajaatoiminta
Selperkatinibin AUC0‑∞‑arvo suureni 7 % tutkittavilla, joilla oli lievä maksan vajaatoiminta, ja 32 % tutkittavilla, joilla oli keskivaikea maksan vajaatoiminta (Child–Pugh‑luokitus). Näin ollen altistus selperkatinibille (AUC) lievää tai keskivaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla (Child–Pugh‑luokitus A tai B) on verrattavissa altistukseen terveillä tutkittavilla 160 mg annoksen käytön yhteydessä.
Selperkatinibin AUC0‑∞‑arvo suureni 77 % tutkittavilla, joilla oli vaikea maksan vajaatoiminta (Child–Pugh‑luokitus C). Selperkatinibin turvallisuudesta vaikeaa maksan vajaatoimintaa sairastavilla on niukasti kliinistä tietoa. Tästä syystä annoksen muuttamista suositellaan, jos potilaalla on vaikea maksan vajaatoiminta (kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Kun kliinisen farmakologian tutkimuksessa annettiin selperkatinibia 160 mg kerta‑annoksena, altistus (AUC) ei muuttunut lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavilla tutkittavilla. Loppuvaiheen munuaisten vajaatoimintaa (eGFR < 15 ml/min) sairastavia ja dialyysihoitoa saavia potilaita ei ole tutkittu.
Pediatriset potilaat
Selperkatinibialtistukset vähintään 2-vuotiailla pediatrisilla potilailla ovat verrattavissa aikuisten potilaiden altistuksiin suositelluilla annoksilla.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen tutkimuksia toteutettiin nuorilla ja kesken-/täysikasvuisilla rotilla ja kesken-/täysikasvuisilla minisioilla toksisuuden selvittämiseksi. Rotalla ja minisialla yleisiä toksisuuden kohde‑elimiä olivat hematopoieettinen järjestelmä, imukudokset, kieli, haima, ruoansulatuskanava, epifyysilevy ja urosten lisääntymiselinten kudokset. Yleensä kyseisten elinten toksisuudet olivat korjautuvia lukuun ottamatta kivesten toksisuutta nuorilla ja kesken-/täysikasvuisilla eläimillä ja muutoksia epifyysilevyissä nuorilla rotilla. Korjautuvaa toksisuutta havaittiin minisioilla vain munasarjoissa. Suuria annoksia käytettäessä ruoansulatuskanavan toksisuus aiheutti minisikojen sairastavuutta altistuksilla, jotka olivat yleisesti ottaen pienempiä kuin ihmisillä määritetyt altistukset suositusannosta käytettäessä. Yhdessä minisikatutkimuksessa naarailla havaittiin lievää, korjautuvaa QTc‑ajan pitenemisen lisääntymistä; muutos oli noin 12 % verrattuna verrokkeihin ja 7 % verrattuna annosta edeltäviin arvoihin. Vain rotilla havaittuja toksisuuden kohde‑elimiä olivat etuhammas, maksa, emätin, keuhkot ja Brunnerin rauhanen sekä usean kudoksen mineralisoituminen, johon liittyi hyperfosfatemia. Vain näissä elimissä rotilla esiintyneet toksisuudet olivat korjautuvia.
Toksisuus nuorilla
Selperkatinibi-altistus, joka oli noin 0,5–2-kertainen aikuisten ihmisten altistukseen verrattuna, aiheutti kuolleisuutta alle 21 päivän ikäisillä rotilla. Vertailukelpoinen altistus siedettiin 21 päivän ikäisillä ja sitä vanhemmilla rotilla.
Nuorilla ja kesken-/täysikasvuisilla rotilla ja kesken-/täysikasvuisilla minisioilla, joilla oli avoimet kasvulevyt, ilmeni mikroskooppisia hypertrofisia, hyperplastisia ja dysplastisia muutoksia rustoisissa kasvulevyissä (physis) selperkatinibin annon jälkeen. Nuorilla rotilla kasvulevyjen dysplasia oli peruuttamaton ja siihen liittyi reisiluun pituuden väheneminen ja luun mineraalitiheyden väheneminen. Luuston muutoksia havaittiin altistustasoilla, jotka vastasivat aikuispotilaiden altistusta suositeltua annostusta 160 mg kahdesti vuorokaudessa käytettäessä.
Nuorilla urosrotilla, joille annettiin selperkatinibia ja joiden annettiin saavuttaa lisääntymisikä annostelun lopettamisen jälkeen, ilmeni lisääntymiskyvyn heikkenemistä, kun ne paritettiin hoitamattomien naarasrottien kanssa. Kun altistustaso oli noin 3,4-kertainen verrattuna aikuisten tehokkaaseen altistukseen, havaittiin hedelmällisyyden ja parittelun indeksien laskua, lisääntynyttä ennen ja jälkeen alkion kiinnittymisen tapahtuvaa menetystä ja elävien alkioiden lukumäärän vähenemistä.
Genotoksisuus
Selperkatinibi ei ole genotoksinen hoitoannoksia käytettäessä. Rotan mikrotumatestissä in vivo selperkatinibi oli positiivinen, kun pitoisuudet olivat > 7‑kertaisia verrattuna Cmax‑arvoon käytettäessä ihmisen annosta 160 mg kahdesti vuorokaudessa. In vitro ‑mikrotumatestissä ihmisen ääreisveren lymfosyyteillä havaittiin moniselitteinen vaste, kun pitoisuus oli noin 485‑kertainen verrattuna Cmax‑arvoon käytettäessä ihmisen annosta.
Mutageenisuus
Selperkatinibi ei aiheuttanut mutaatioita bakteereilla tehdyssä mutageenisuuskokeessa.
Karsinogeenisuus
Selperkatinibin karsinogeenisuutta arvioitiin 2 vuotta kestäneessä tutkimuksessa rotilla. Joillakin naarasrotilla havaittiin emättimen kasvaimia plasma-altistustasoilla, jotka olivat samanlaisia kuin on havaittu aikuisilla potilailla, joita hoidettiin annoksella 160 mg kahdesti vuorokaudessa. Naarasrottien lisääntymiselimissä ei havaittu preneoplastisia muutoksia. Näiden löydösten kliininen merkitys on tuntematon. Selperkatinibi ei ollut karsinogeeninen urosrotilla tässä tutkimuksessa.
Selperkatinibi ei ollut karsinogeeninen uros- tai naarashiirillä kuuden kuukauden tutkimuksessa.
Alkiotoksisuus/teratogeenisuus
Eläinten lisääntymistutkimusten tietojen ja selperkatinibin vaikutusmekanismin perusteella selperkatinibin anto raskaana olevalle naiselle voi aiheuttaa haittaa sikiölle. Alkiokuolemia ja epämuodostumia todettiin, kun tiineille rotille annettiin selperkatinibia organogeneesin aikana ja kun emon altistukset olivat suunnilleen samaa luokkaa kuin ihmisten suositusannosten (160 mg kahdesti vuorokaudessa) yhteydessä.
Lisääntymistoksisuus
Rotilla ja minisioilla toteutettujen tutkimusten tulokset viittaavat siihen, että selperkatinibi voi heikentää miesten ja naisten hedelmällisyyttä.
Urosrotilla toteutetussa hedelmällisyystutkimuksessa havaittiin annosriippuvaista itusolukatoa ja spermatidiretentiota subkliinisillä AUC‑perusteisilla altistustasoilla (0,2‑kertaisia verrattuna kliiniseen altistukseen ihmisen suositusannoksen käytössä). Vaikutuksiin liittyi elinten painon lasku, siittiöiden liikkuvuuden huononeminen ja poikkeavien siittiöiden määrän suureneminen, kun AUC‑perusteiset altistustasot olivat noin kaksinkertaiset verrattuna kliiniseen altistukseen ihmisen suositusannoksen käytössä. Urosrottien hedelmällisyystutkimuksen mikroskooppilöydökset olivat yhdenmukaisia rotilla ja minisioilla toteutetuissa toistuvan altistuksen tutkimuksissa havaittujen vaikutusten kanssa. Annosriippuvaiseen, korjautumattomaan kivesten degeneraatioon liittyi luminaalisten siittiöiden väheneminen lisäkiveksissä subkliinisillä AUC‑perusteisilla altistustasoilla (0,1–0,4‑kertaisia verrattuna kliiniseen altistukseen ihmisen suositusannoksen käytössä).
Naarasrotilla toteutetussa hedelmällisyyttä ja alkion varhaiskehitystä koskeneessa tutkimuksessa havaittiin kiimakiertojen määrän vähenemistä sekä alkiokuolemia AUC‑perusteisilla altistustasoilla, jotka olivat suunnilleen samaa luokkaa kuin kliininen altistus ihmisen suositusannoksen käytössä. Rotilla toteutetuissa toistuvan altistuksen tutkimuksissa havaittiin korjautuvaa emättimen epiteelisolujen muuttumista limaa erittäviksi soluiksi ja yksittäisten solujen sarveistumista sekä kiimakiertojen muutoksia kliinisesti merkittävillä AUC‑perusteisilla altistustasoilla. Minisioilla havaittiin keltarauhasten vähenemistä ja/tai keltarauhaskystia subkliinisillä AUC‑perusteisilla kliinisillä altistustasoilla (0,07–0,3‑kertaisia verrattuna kliiniseen altistukseen ihmisen suositusannoksen käytössä).
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa
Mannitoli
Kroskarmelloosinatrium
Hydroksipropyyliselluloosa
Natriumstearyylifumaraatti
Kalvopäällyste
Retsevmo 40 mg kalvopäällysteinen tabletti
Polyvinyylialkoholi
Titaanidioksidi (E171)
Makrogoli
Talkki
Musta rautaoksidi (E172)
Retsevmo 80 mg kalvopäällysteinen tabletti
Polyvinyylialkoholi
Titaanidioksidi (E171)
Makrogoli
Talkki
Punainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Retsevmo 120 mg kalvopäällysteinen tabletti
Polyvinyylialkoholi
Titaanidioksidi (E171)
Makrogoli
Talkki
Musta rautaoksidi (E172)
Punainen rautaoksidi (E172)
Retsevmo 160 mg kalvopäällysteinen tabletti
Polyvinyylialkoholi
Titaanidioksidi (E171)
Makrogoli
Talkki
Punainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
RETSEVMO tabletti, kalvopäällysteinen
40 mg (L:ei) 56 fol (1360,58 €)
80 mg (L:ei) 56 fol (2618,63 €)
PF-selosteen tieto
30, 56 tai 60 kalvopäällysteistä tablettia alumiinifoliokannella sinetöidyissä kylmämuovattavassa alumiinifolioläpipainopakkauksessa (CFAF, cold formable aluminium foil).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Retsevmo 40 mg kalvopäällysteinen tabletti
Vaalean harmaa, pyöreä tabletti, jonka toisella puolella on merkintä ”5340” ja toisella puolella ”Ret 40”. Tabletin halkaisija on noin 6 mm.
Retsevmo 80 mg kalvopäällysteinen tabletti
Tummanpunaisen purppura, pyöreä tabletti, jonka toisella puolella on merkintä ”6082” ja toisella puolella ”Ret 80”. Tabletin halkaisija on noin 7,3 mm.
Retsevmo 120 mg kalvopäällysteinen tabletti
Vaalean purppura, pyöreä tabletti, jonka toisella puolella on merkintä ”6120” ja toisella puolella ”Ret 120”. Tabletin halkaisija on noin 8,75 mm.
Retsevmo 160 mg kalvopäällysteinen tabletti
Vaaleanpunainen, pyöreä tabletti, jonka toisella puolella on merkintä ”5562” ja toisella puolella ”Ret 160”. Tabletin halkaisija on noin 9,75 mm.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
RETSEVMO tabletti, kalvopäällysteinen
40 mg 56 fol
80 mg 56 fol
- Ylempi erityiskorvaus (100 %). Selperkatinibi: Keuhkosyövän ja kilpirauhassyövän hoito erityisin edellytyksin (1550).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Selperkatinibi: Aikuisten pitkälle edenneen RET-fuusiopositiivisen ei-pienisoluisen keuhkosyövän ja kilpirauhassyövän hoito sekä pitkälle edenneen RET-mutaatiopositiivisen medullaarisen kilpirauhassyövän hoito aikuisille ja vähintään 12-vuotiaille nuorille erityisin edellytyksin (3072).
ATC-koodi
L01EX22
Valmisteyhteenvedon muuttamispäivämäärä
04.06.2026
Yhteystiedot
 OY ELI LILLY FINLAND AB
OY ELI LILLY FINLAND AB Mannerheimintie 117
00280 Helsinki
09 854 5250
www.lilly.com/fi
medinfo_finland@lilly.com
Lääketietopalvelu puh. 0800-140 240