
LEQVIO injektioneste, liuos, esitäytetty ruisku 284 mg
Vaikuttavat aineet ja niiden määrät
Yksi esitäytetty ruisku sisältää inklisiraaninatriumia määrän, joka vastaa 284 mg inklisiraania 1,5 ml:ssa liuosta.
Yksi ml sisältää inklisiraaninatriumia määrän, joka vastaa 189 mg:aa inklisiraania.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste).
Kliiniset tiedot
Käyttöaiheet
Leqvio on tarkoitettu käytettäväksi ruokavaliohoidon lisänä aikuisille, joilla on primaarinen hyperkolesterolemia (heterotsygoottinen familiaalinen tai ei‑familiaalinen) tai sekamuotoinen dyslipidemia:
- yhdessä statiinin tai statiinin ja muiden lipidipitoisuuksia laskevien hoitojen kanssa, jos potilaan LDL‑kolesteroliarvotavoitetta ei saavuteta suurimmalla siedetyllä statiiniannoksella, tai
- ainoana lääkkeenä tai yhdessä muiden lipidipitoisuuksia laskevien hoitojen kanssa, jos potilas ei siedä statiineja tai statiinit ovat vasta‑aiheisia.
Annostus ja antotapa
Annostus
Suositeltu annos on 284 mg inklisiraania yhtenä injektiona ihon alle hoidon alussa, 3 kuukauden kuluttua ja tämän jälkeen 6 kuukauden välein.
Väliin jääneet annokset
Jos antoaikataulun mukainen annos viivästyy alle 3 kuukautta, annetaan väliin jäänyt inklisiraani-annos ja tämän jälkeen jatketaan potilaan alkuperäisen inklisiraani-hoidon aikataulun mukaisesti.
Jos antoaikataulun mukainen annos viivästyy yli 3 kuukautta, antoaikataulu aloitetaan alusta. Potilaalle annetaan inklisiraani-aloitusinjektio, seuraava injektio 3 kuukauden kuluttua ja tämän jälkeen injektiot 6 kuukauden välein.
Siirtyminen monoklonaalisten proproteiini-konvertaasi-subtilisiini-keksiini tyypin 9 (PCSK9)-vasta-aineiden käytöstä Leqvio-hoitoon
Inklisiraani-hoito voidaan aloittaa heti viimeisen monoklonaalisen PCSK9-vasta-aineannoksen jälkeen. Jotta LDL (pienitiheyksinen rasvaproteiini) ‑kolesterolipitoisuuden alentamiseen tähtäävän hoidon vaikutus säilyisi, on suositeltavaa antaa inklisiraani 2 viikon kuluessa viimeisestä monoklonaalisesta PCSK9-vasta-aineannoksesta.
Erityisryhmät
Iäkkäät potilaat
Annosta ei tarvitse muuttaa iäkkäillä potilailla (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä (Child–Pugh‑luokka A) tai keskivaikea (Child–Pugh‑luokka B) maksan vajaatoiminta. Vaikeaa (Child–Pugh‑luokka C) maksan vajaatoimintaa sairastavien potilaiden hoidosta ei ole tietoja (ks. kohta Farmakokinetiikka). Inklisiraanin käytössä on noudatettava varovaisuutta, jos potilaalla on vaikea maksan vajaatoiminta.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä, keskivaikea tai vaikea munuaisten vajaatoiminta tai loppuvaiheen munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka). Inklisiraanin käytöstä potilaille, joilla on vaikea munuaisten vajaatoiminta, on vain vähän kokemusta. Inklisiraanin käytössä on noudatettava varovaisuutta näillä potilailla. Varotoimet hemodialyysipotilailla, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Pediatriset potilaat
Inklisiraanin turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Ihon alle.
Inklisiraani on tarkoitettu annettavaksi injektiona ihon alle vatsan alueelle tai vaihtoehtoisesti olkavarteen tai reiteen. Injektiota ei pidä antaa ihoalueelle, jolla on aktiivinen ihosairaus tai ‑vaurio, kuten auringonpolttama, ihottuma, inflammaatio tai infektio.
Jokainen 284 mg:n annos annetaan yksittäisellä esitäytetyllä ruiskulla. Kaikki esitäytetyt ruiskut ovat kertakäyttöisiä.
Inklisiraani on tarkoitettu terveydenhuollon ammattilaisen annettavaksi.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Hemodialyysi
Hemodialyysin vaikutusta inklisiraanin farmakokinetiikkaan ei ole tutkittu. Inklisiraani eliminoituu munuaisteitse, joten hemodialyysia ei saa toteuttaa ennen kuin inklisiraanin antamisesta on kulunut vähintään 72 tuntia.
Natriumin määrä
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Inklisiraani ei ole lääkkeiden yleisten kuljettajaproteiinien substraatti ja vaikka in vitro -tutkimuksia ei ole tehty, se ei todennäköisesti ole sytokromi P450 -entsyymin substraatti. Inklisiraani ei ole sytokromi P450 ‑entsyymien eikä lääkkeiden yleisten kuljettajaproteiinien estäjä eikä indusori. Inklisiraanilla ei siis todennäköisesti ole kliinisesti merkittäviä yhteisvaikutuksia muiden lääkevalmisteiden kanssa. Perustuen rajallisiin saatavilla oleviin tietoihin kliinisesti merkittävät yhteisvaikutukset atorvastatiinin, rosuvastatiinin tai muiden statiinien kanssa eivät ole todennäköisiä.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja inklisiraanin käytöstä raskaana oleville naisille. Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi inklisiraanin käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö inklisiraani ja/tai sen metaboliitit ihmisillä äidinmaitoon. Olemassa olevat farmakokineettiset/toksikologiset tiedot koe-eläimistä ovat osoittaneet inklisiraanin erittyvän rintamaitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois.
On päätettävä lopetetaanko rintaruokinta vai lopetetaanko Leqvio‑hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Inklisiraanin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoja. Eläinkokeissa ei ole todettu vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Leqvio-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Ainoita inklisiraaniin liittyneitä haittavaikutuksia olivat injektiokohdan haittavaikutukset (8,2 %).
Haittavaikutustaulukko
Haittavaikutukset esitetään elinjärjestelmäluokittain (taulukko 1). Esiintymistiheysluokat määritellään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1 Inklisiraanihoitoa saaneilla potilailla ilmoitetut haittavaikutukset
Elinjärjestelmäluokka | Haittavaikutus | Yleisyysluokka |
Immuunijärjestelmä | Yliherkkyys, mukaan lukien anafylaktinen reaktio, angioödeema, ihottuma, urtikaria, kutina | Tuntematon |
Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan haittavaikutukset1 | Yleinen |
1 Ks. kohta ”Valikoitujen haittavaikutusten kuvaus”. | ||
Valikoitujen haittavaikutusten kuvaus
Injektiokohdan haittavaikutukset
Vaiheen III tutkimuksissa injektiokohdan haittavaikutuksia esiintyi 8,2 %:lla inklisiraania saaneista potilaista ja 1,8 %:lla lumelääkettä saaneista. Niiden potilaiden osuus, jotka keskeyttivät hoidon injektiokohdan haittavaikutusten vuoksi, oli 0,2 % inklisiraaniryhmässä ja 0,0 % lumeryhmässä. Kaikki näistä haittavaikutuksista olivat lieviä tai keskivaikeita ja ohimeneviä ja korjautuivat aiheuttamatta jälkitiloja. Inklisiraania saaneilla potilailla yleisimpiä injektiokohdan haittavaikutuksia olivat injektiokohdan reaktio (3,1 %), injektiokohdan kipu (2,2 %), injektiokohdan punoitus (1,6 %) ja injektiokohdan ihottuma (0,7 %).
Erityisryhmät
Iäkkäät
Vaiheen III tutkimuksissa inklisiraania sai 1 833 potilasta, joista 981 (54 %) oli täyttänyt 65 vuotta ja 239 (13 %) oli täyttänyt 75 vuotta. Turvallisuudessa ei kokonaisuudessaan havaittu eroja iäkkäiden potilaiden ja nuorempien potilaiden välillä.
Immunogeenisyys
Vaiheen III tutkimuksissa 1 830 potilaan näytteet tutkittiin lääkkeeseen kohdistuvien vasta‑aineiden varalta. Tällaisia vasta-aineita todettiin vahvistetusti 1,8 %:lla (33 potilaalla 1 830:stä) ennen inklisiraanin antoa ja 4,9 %:lla (90 potilaalla 1 830:stä) 18 kuukautta kestäneen inklisiraani-hoidon aikana. Inklisiraanin kliinisessä tehossa, turvallisuudessa ja farmakodynamiikassa ei havaittu kliinisesti merkittäviä eroja potilailla, joilla todettiin vasta‑aineita inklisiraanille.
Laboratorioarvot
Vaiheen III tutkimuksissa havaittiin veren transaminaasiarvojen suurenemista (1-3 kertaa normaaliarvojen ylärajan) yleisemmin inklisiraania saavilla potilailla (ALAT: 19,7% ja ASAT: 17,2%) kuin lumelääkettä saavilla potilailla (ALAT: 13,6% ja ASAT: 11,1%). Nämä arvojen nousut eivät ylittäneet kliinisesti merkitsevää kynnysarvoa (yli 3 kertaa normaaliarvojen ylärajaa), eivät aiheuttaneet oireita eikä niillä ei ollut yhteyttä haittavaikutuksiin tai muihin maksan vajaatoiminnan merkkeihin.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä, lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta.
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisesti merkittäviä haittatapahtumia ei havaittu, kun terveille vapaaehtoisille annettiin inklisiraania annoksina, jotka olivat enimmillään kolminkertaisia verrattuna terapeuttiseen annokseen. Inklisiraanin yliannostukseen ei ole spesifistä hoitoa. Yliannostustapauksessa potilasta hoidetaan oireenmukaisesti, ja tukitoimiin on ryhdyttävä tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: lipidejä muuntavat lääkeaineet, muut lipidejä muuntavat lääkeaineet, ATC-koodi: C10AX16
Vaikutusmekanismi
Inklisiraani on kolesterolipitoisuutta pienentävä, kaksijuosteinen siRNA-molekyyli, jonka koodaavaan juosteeseen konjugoitu triantennaarinen N-asetyyligalaktosamiini (GalNAc) edistää inklisiraanin soluunottoa maksasoluihin. Maksasoluissa inklisiraani hyödyntää RNA-interferenssimekanismia ja säätelee PCSK9:n (proprotein convertase subtilisin kexin type 9) mRNA:n katalyyttistä hajoamista. Tämä lisää LDL‑reseptorin kierrätystä ja ilmentymistä maksasolujen pinnalla, jolloin LDL‑kolesterolin soluunotto lisääntyy ja LDL‑kolesterolin määrä verenkierrossa laskee.
Farmakodynaamiset vaikutukset
Kun inklisiraania annettiin 284 mg:n kerta-annos ihon alle, LDL‑kolesterolipitoisuus pieneni 14 vuorokauden kuluessa valmisteen annosta. LDL-kolesterolipitoisuus pieneni keskimäärin 49–51 % 30–60 vuorokauden kuluessa valmisteen annosta. Päivän 180 kohdalla LDL‑kolesterolipitoisuus oli edelleen noin 53 % pienempi kuin lähtötilanteessa.
Kliininen teho ja turvallisuus
Kliinisissä tutkimuksissa ja eräissä julkaisuissa 284 mg:n inklisiraani-annoksen katsottiin olevan vastaava ja verrattavissa oleva 300 mg:n annokseen inklisiraania natriumsuolana.
Inklisiraanin tehoa arvioitiin kolmessa vaiheen III tutkimuksessa potilailla, joilla oli ateroskleroottinen sydän- ja verisuonitauti (sepelvaltimotauti, aivoverisuonisairaus tai ääreisvaltimotauti), erityisen suuri riski sairastua sydän-ja verisuonitautiin (tyypin 2 diabetes, familiaalinen hyperkolesterolemia tai vähintään 20 %:n riski sairastaa kardiovaskulaaritapahtuma 10 vuoden kuluessa Framingham Risk Score ‑pisteytyksellä tai vastaavalla arvioituna) ja/tai familiaalinen hyperkolesterolemia (FH). Potilaat käyttivät suurinta siedettyä statiiniannosta, sekä mahdollisesti muuta lipidejä muuntavaa hoitoa, joilla ei kuitenkaan oltu saavutettu hoitotavoitetta LDL-pitoisuuksien pienenemisessä. Noin 17 % potilaista ei sietänyt statiineja. Potilaille annetiin 284 mg inklisiraania tai lumelääkettä injektiona ihon alle päivänä 1, päivänä 90, päivänä 270 ja päivänä 450. Potilaita seurattiin päivään 540 asti.
Inklisiraanin vaikutusta kardiovaskulaarisairastavuuteen ja ‑kuolleisuuteen ei ole vielä selvitetty.
Vaiheen III tutkimusten yhdistelmäanalyysin perusteella ihon alle annettu inklisiraani pienensi LDL‑kolesterolipitoisuuksia 50–55 % jo päivänä 90 (kuva 1), ja vaikutus säilyi pitkäaikaishoidon aikana. Suurin LDL‑kolesterolipitoisuuden saavutettiin päivän 150 kohdalla toisen antokerran jälkeen. LDL-kolesterolipitoisuuden pienenemisellä hieman, mutta tilastollisesti merkitsevästi enemmän (enintään 65 %) oli yhteys alempiin lähtötilanteen LDL-kolesterolipitoisuuksiin (noin < 2 mmol/l [< 77 mg/dl]), korkeampiin lähtötilanteen PCSK9-pitoisuuksiin ja suurempiin statiiniannoksiin.
Kuva 1 LDL‑kolesterolipitoisuuden keskimääräinen muutos (%) lähtötilanteesta potilailla, joilla oli primaarinen hyperkolesterolemia tai sekamuotoinen dyslipidemia, inklisiraani-hoidon vs. lumehoidon yhteydessä (yhdistettyjen tietojen analyysi)
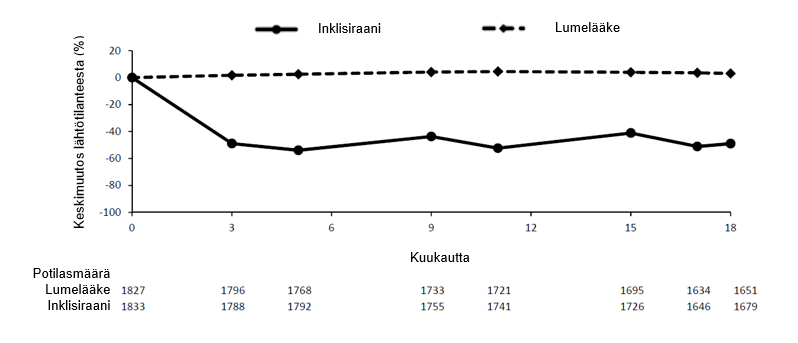
Ateroskleroottinen sydän- ja verisuonitauti ja erityisen suuri sydän-ja verisuonitautiriski
Kaksi tutkimusta toteutettiin potilailla, joilla oli ateroskleroottinen sydän- ja verisuonitauti tai erityisen suuri riski sairastua sydän-ja verisuonitautiin (ORION‑10 ja ORION‑11). Potilaat käyttivät suurinta siedettyä statiiniannosta, sekä mahdollisesti muuta lipidejä muuntavaa hoitoa (esim. etsetimibi), joilla ei kuitenkaan oltu saavutettu hoitotavoitetta LDL-pitoisuuksien pienenemisessä. LDL-kolesterolipitoisuuden pienenemisen oletetaan vähentävän merkittäviä sydän-ja verisuonitapahtumia, joten molemmissa tutkimuksissa rinnakkaisina ensisijaisina päätetapahtumina olivat LDL-kolesterolipitoisuuden prosentuaalinen muutos lähtötilanteesta päivään 510 mennessä verrattuna lumelääkkeeseen sekä ajan suhteen korjattu LDL-kolesterolipitoisuuden prosentuaalinen muutos lähtötilanteesta päivinä 90–540. Näin pyrittiin arvioimaan LDL-kolesterolipitoisuuteen kohdistuvaa kokonaisvaikutusta ajan mittaan.
ORION‑10 oli kaksoissokkoutettu, satunnaistettu, lumekontrolloitu, 18 kuukauden pituinen monikeskustutkimus, johon osallistuneilla 1 561 potilaalla oli ateroskleroottinen sydän- ja verisuonitauti.
Ikäkeskiarvo lähtötilanteessa oli 66 v (vaihteluväli 35–90 v), 60 % tutkittavista oli 65 vuotta täyttäneitä, 31 % oli naisia, 86 % oli valkoihoisia, 13 % oli mustia, 1 % oli aasialaisia ja 14 % oli latinalaisamerikkalaisia. Lähtötilanteen LDL-kolesteroliarvon keskiarvo oli 2,7 mmol/l (105 mg/dl). Tutkittavista 69 % sai suuren intensiteetin statiinihoitoa, 19 % sai keskisuuren intensiteetin statiinihoitoa, 1 % sai pienen intensiteetin statiinihoitoa ja 11 % ei käyttänyt statiineja. Yleisimmin käytetyt statiinit olivat atorvastatiini ja rosuvastatiini.
Inklisiraani-hoito johti tilastollisesti merkitsevään -52 prosenttiyksikön keskimääräiseen muutokseen LDL-kolesterolitasossa päivänä 510 verrattuna lumelääkeryhmään (95 % lv ‑56 %, ‑49 %; p < 0,0001) (taulukko 2).
Ajan suhteen korjattu keskimääräinen LDL-kolesterolin muutos päivien 90-540 välillä oli tilastollisesti merkitsevä -54 prosenttiyksikköä verrattuna lumelääkeryhmään (95 % lv ‑56 %, ‑51 %; p < 0,0001). Muita tuloksia, ks. taulukko 2.
Taulukko 2 Lipidiparametrien prosentuaalinen keskimääräinen muutos lähtötilanteesta ja ero lumelääkkeeseen nähden päivän 510 kohdalla ORION‑10-tutkimuksessa
| Hoitoryhmä | LDL-kolesteroli | Kokonaiskolesteroli | Ei‑HDL-kolesteroli | Apo‑B | Lp(a)* |
| Lähtötilanteen keskiarvo mg/dl** | 105 | 181 | 134 | 94 | 122 |
| Päivä 510 (prosentuaalinen keskimuutos lähtötilanteesta) | |||||
| Lumelääke (n = 780) | 1 | 0 | 0 | ‑2 | 4 |
| Inklisiraani (n = 781) | ‑51 | ‑34 | ‑47 | ‑45 | ‑22 |
Ero lumelääkkeeseen nähden (pienimmän neliösumman keskiarvo) (95 % lv) | ‑52 (‑56; ‑49) | ‑33 (‑35; ‑31) | ‑47 (‑50; ‑44) | ‑43 (‑46; ‑41) | ‑26 (‑29; ‑22) |
*Päivänä 540; Lp(a)-arvojen mediaanimuutos (%) **Lähtötilanteen keskiarvo Lp(a)-arvoille nmol/l | |||||
Päivän 510 kohdalla LDL-kolesterolin tavoitearvon < 1,8 mmol/l (< 70 mg/dl) oli saavuttanut 84 % inklisiraania saaneista, ateroskleroottista sydän- ja verisuonitautia sairastavista potilaista ja 18 % lumelääkettä saaneista potilaista.
LDL‑kolesterolipitoisuuden prosentuaalinen pieneneminen lähtötilanteen ja päivän 510 välillä sekä ajan suhteen korjattu LDL‑kolesterolipitoisuuden prosentuaalinen pieneneminen lähtötilanteesta päivinä 90–540 olivat johdonmukaisia ja tilastollisesti merkitseviä (p < 0,0001) kaikissa alaryhmissä riippumatta lähtötilanteen demografisista tiedoista, lähtötilanteen tautitiedoista (mm. sukupuoli, ikä, painoindeksi, etninen tausta ja statiinien käyttö lähtötilanteessa), samanaikaisista sairauksista ja maantieteellisestä alueesta.
ORION‑11 oli kansainvälinen, kaksoissokkoutettu, satunnaistettu, lumekontrolloitu, 18 kuukauden pituinen monikeskustutkimus 1 617 potilaalla, joilla oli ateroskleroottinen sydän- ja verisuonitauti tai erityisen suuri riski sairastua sydän- ja verisuonitautiin. Yli 75 % potilaista sai entuudestaan suuren intensiteetin statiinihoitoa, 87 %:lla potilaista oli ateroskleroottinen sydän- ja verisuonitauti ja 13 % potilaista oli erityisen suuressa riskissä sairastua sydän-ja verisuonitautiin.
Ikäkeskiarvo lähtötilanteessa oli 65 v (vaihteluväli 20–88 v), 55 % tutkittavista oli 65 vuotta täyttäneitä, 28 % oli naisia, 98 % oli valkoihoisia, 1 % oli mustia, 1 % oli aasialaisia ja 1 % oli latinalaisamerikkalaisia. Lähtötilanteen LDL-kolesteroliarvon keskiarvo oli 2,7 mmol/l (105 mg/dl). Tutkittavista 78 % sai suuren intensiteetin statiinihoitoa, 16 % sai keskisuuren intensiteetin statiinihoitoa, 0,4 % sai pienen intensiteetin statiinihoitoa ja 5 % ei käyttänyt statiineja. Yleisimmin käytetyt statiinit olivat atorvastatiini ja rosuvastatiini.
Inklisiraani-hoito johti tilastollisesti merkitsevään -50 prosenttiyksikön keskimääräiseen muutokseen LDL-kolesterolitasossa päivänä 510 verrattuna lumelääkeryhmään (95 % lv ‑53 %, ‑47 %; p < 0,0001) (taulukko 3).
Ajan suhteen korjattu keskimääräinen LDL-kolesterolin muutos päivien 90-540 välillä oli tilastollisesti merkitsevä -49 prosenttiyksikköä verrattuna lumelääkeryhmään (95 % lv ‑52 %, ‑47 %; p < 0,0001). Muita tuloksia, ks. taulukko 3.
Taulukko 3 Lipidiparametrien prosentuaalinen keskimääräinen muutos lähtötilanteesta ja ero lumelääkkeeseen nähden päivän 510 kohdalla ORION‑11-tutkimuksessa
| Hoitoryhmä | LDL-kolesteroli | Kokonaiskolesteroli | Ei‑HDL-kolesteroli | Apo‑B | Lp(a)* |
| Lähtötilanteen keskiarvo mg/dl** | 105 | 185 | 136 | 96 | 107 |
| Päivä 510 (prosentuaalinen keskimuutos lähtötilanteesta) | |||||
| Lumelääke (n = 807) | 4 | 2 | 2 | 1 | 0 |
| Inklisiraani (n = 810) | ‑46 | ‑28 | ‑41 | ‑38 | ‑19 |
Ero lumelääkkeeseen nähden (pienimmän neliösumman keskiarvo) (95 % lv) | ‑50 (‑53; ‑47) | ‑30 (‑32; ‑28) | ‑43 (‑46; ‑41) | ‑39 (‑41; ‑37) | ‑19 (‑21; ‑16) |
*Päivänä 540; Lp(a)-arvojen mediaanimuutos (%) **Lähtötilanteen keskiarvo Lp(a)-arvoille nmol/l | |||||
Päivän 510 kohdalla LDL-kolesterolin tavoitearvon < 1,8 mmol/l (< 70 mg/dl) oli saavuttanut 82 % inklisiraani-hoitoa saaneista, ateroskleroottista sydän- ja verisuonitautia sairastavista potilaista ja 16 % lumelääkettä saaneista potilaista. Erityisen suuren riskin potilaista LDL-kolesterolin tavoitearvon < 2,6 mmol/l (< 100 mg/dl) saavutti 78 % inklisiraani-hoitoa saaneista potilaista ja 31 % lumelääkettä saaneista potilaista.
LDL‑kolesterolipitoisuuden prosentuaalinen pieneneminen lähtötilanteen ja päivän 510 välillä sekä ajan suhteen korjattu LDL‑kolesterolipitoisuuden prosentuaalinen pieneneminen lähtötilanteesta päivinä 90–540 olivat johdonmukaisia ja tilastollisesti merkitseviä (p < 0,05) kaikissa alaryhmissä riippumatta lähtötilanteen demografisista tiedoista, lähtötilanteen tautitiedoista (mm. sukupuoli, ikä, painoindeksi, etninen tausta ja statiinien käyttö lähtötilanteessa), samanaikaisista sairauksista ja maantieteellisestä alueesta.
Heterotsygoottinen familiaalinen hyperkolesterolemia
ORION‑9 oli kansainvälinen, kaksoissokkoutettu, satunnaistettu, lumekontrolloitu, 18 kuukauden pituinen monikeskustutkimus, johon osallistuneilla 482 potilaalla oli heterotsygoottinen familiaalinen hyperkolesterolemia (HeFH). Kaikki potilaat käyttivät suurimpia siedettyjä statiiniannoksia, sekä mahdollisesti muuta lipidejä muuntavaa hoitoa (esim. etsetimibi), joilla ei kuitenkaan oltu saavutettu hoitotavoitetta LDL-pitoisuuksien pienenemisessä. Heterotsygoottisen familiaalisen hyperkolesterolemian diagnoosi perustui joko genotyypitykseen tai kliinisiin kriteereihin (”varma familiaalinen hyperkolesterolemia” joko Simon Broome- tai WHO/Dutch Lipid Network ‑kriteereillä arvioituna).
Rinnakkaisia ensisijaisia päätetapahtumia olivat LDL-kolesterolipitoisuuden prosentuaalinen muutos lähtötilanteen ja päivän 510 välisenä aikana verrattuna lumelääkkeeseen ja ajan suhteen korjattu LDL-kolesterolipitoisuuden prosentuaalinen muutos lähtötilanteesta päivinä 90–540. Näin pyrittiin arvioimaan kokonaisvaikutusta LDL-kolesterolipitoisuuteen ajan mittaan. Tärkeimmät toissijaiset päätetapahtumat olivat LDL-kolesterolipitoisuuden absoluuttinen muutos lähtötilanteen ja päivän 510 välisenä aikana, ajan suhteen korjattu LDL-kolesterolipitoisuuden absoluuttinen muutos lähtötilanteesta päivinä 90–540 ja PCSK9-, kokonaiskolesteroli-, Apo B- ja ei-HDL-kolesterolipitoisuuksien prosentuaalinen muutos lähtötilanteen ja päivän 510 välisenä aikana. Muita toissijaisia päätetapahtumia olivat yksilöllinen vaste inklisiraani-hoidolle ja niiden potilaiden osuus, jotka saavuttivat ateroskleroottisen sydän- ja verisuonitaudin riskiään vastaavat yleiset lipiditavoitearvot.
Ikäkeskiarvo lähtötilanteessa oli 55 v (vaihteluväli 21–80 v), 22 % tutkittavista oli 65 vuotta täyttäneitä, 53 % oli naisia, 94 % oli valkoihoisia, 3 % oli mustia, 3 % oli aasialaisia ja 3 % oli latinalaisamerikkalaisia. Lähtötilanteen LDL-kolesteroliarvon keskiarvo oli 4,0 mmol/l (153 mg/dl). Tutkittavista 74 % sai suuren intensiteetin statiinihoitoa, 15 % sai keskisuuren intensiteetin statiinihoitoa ja 10 % ei käyttänyt statiineja. Potilaista 52 % sai etsetimibihoitoa. Yleisimmin käytetyt statiinit olivat atorvastatiini ja rosuvastatiini.
Inklisiraani-hoito johti tilastollisesti merkitsevään -48 prosenttiyksikön keskimääräiseen muutokseen LDL-kolesterolitasossa päivänä 510 verrattuna (95 % lv ‑54 %, ‑42 %; p < 0,0001) (taulukko 4).
Ajan suhteen korjattu keskimääräinen LDL-kolesterolin muutos päivien 90-540 välillä oli tilastollisesti merkitsevä -44 prosenttiyksikköä verrattuna lumelääkeryhmään (95 % lv ‑48 %, ‑40 %; p < 0,0001). Muita tuloksia, ks. taulukko 4.
Taulukko 4 Lipidiparametrien prosentuaalinen keskimääräinen muutos lähtötilanteesta ja ero lumelääkkeeseen nähden päivän 510 kohdalla ORION‑9-tutkimuksessa
| Hoitoryhmä | LDL-kolesteroli | Kokonais-kolesteroli | Ei‑HDL-kolesteroli | Apo‑B | Lp(a)* |
| Lähtötilanteen keskiarvo mg/dl** | 153 | 231 | 180 | 124 | 121 |
| Päivä 510 (prosentuaalinen keskimuutos lähtötilanteesta) | |||||
| Lumelääke (n = 240) | 8 | 7 | 7 | 3 | 4 |
| Inklisiraani (n = 242) | ‑40 | ‑25 | ‑35 | ‑33 | ‑13 |
Ero lumelääkkeeseen nähden (pienimmän neliösumman keskiarvo) (95 % lv) | ‑48 (‑54; ‑42) | ‑32 (‑36; ‑28) | ‑42 (‑47; ‑37) | ‑36 (‑40; ‑32) | ‑17 (‑22; ‑12) |
*Päivänä 540; Lp(a)-arvojen mediaanimuutos (%) **Lähtötilanteen keskiarvo Lp(a)-arvoille nmol/l | |||||
Päivän 510 kohdalla LDL-kolesterolin tavoitearvon < 1,8 mmol/l (< 70 mg/dl) saavutti 52,5 % inklisiraani-hoitoa saaneista ja 1,4 % lumelääkettä saaneista potilaista, joilla oli ateroskleroottinen sydän- ja verisuonitauti. Potilaista, joilla oli erityisen suuri riski sairastua ateroskleroottiseen sydän- ja verisuonitautiin, LDL-kolesterolitavoitearvon < 2,6 mmol/l (< 100 mg/dl) saavutti 66,9 % inklisiraani-hoitoa saaneista potilaista ja 8,9 % lumelääkettä saaneista.
LDL-kolesterolipitoisuuden prosentuaalinen muutos lähtötilanteen ja päivän 510 välisenä aikana sekä ajan suhteen korjattu LDL-kolesterolipitoisuuden prosentuaalinen muutos lähtötilanteesta päivinä 90–540 olivat johdonmukaiset ja tilastollisesti merkitsevät (p < 0,05). Muutos havaittiin kaikissa alaryhmissä riippumatta lähtötilanteen demografisista tiedoista, lähtötilanteen tautitiedoista (mm. sukupuoli, ikä, painoindeksi, etninen tausta ja statiinien käyttö lähtötilanteessa), samanaikaisista sairauksista ja maantieteellisestä alueesta.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset inklisiraanin käytöstä kohonneiden kolesterolipitoisuuksien hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Kun inklisiraania annettiin kerta-annos ihon alle, systeeminen altistus inklisiraanille suureni suunnilleen annoksen mukaisesti 24 mg:n ja 756 mg:n välillä. Suositeltua hoito-ohjelmaa (284 mg) käytettäessä huippupitoisuudet plasmassa saavutettiin noin 4 tunnin kuluessa annoksen antamisesta. Cmax-keskiarvo oli 509 ng/ml. Pitoisuudet pienenivät havaitsemisrajan alittavalle tasolle 48 tunnin kuluessa annosta. Plasman pitoisuus-aikakäyrän alle jäävän pinta-alan keskiarvo antohetkestä äärettömään ekstrapoloituna oli 7 980 ng*h/ml. Kun inklisiraania annettiin toistuvasti ihon alle, farmakokineettiset löydökset olivat samankaltaiset kuin kerta-annoksen jälkeen.
Jakautuminen
Inklisiraani sitoutuu 87‑prosenttisesti proteiineihin in vitro plasmassa kliinisesti todettavilla pitoisuuksilla. Kun terveille aikuisille annettiin 284 mg:n kerta-annos inklisiraania ihon alle, näennäinen jakautumistilavuus oli noin 500 litraa. Non-kliiniseen aineistoon perustuen inklisiraanin on todettu kulkeutuvan suurelta osin ja selektiivisesti maksaan, joka on kolesterolipitoisuuksien pienenemisen kohde-elin.
Biotransformaatio
Inklisiraania metaboloivat ensisijaisesti nukleaasit, jotka pilkkovat inklisiraanin eripituisiksi lyhyemmiksi, inaktiivisiksi nukleotideiksi. Inklisiraani ei ole lääkkeiden yleisten kuljettajaproteiinien substraatti ja vaikka in vitro-tutkimuksia ei ole tehty, se ei todennäköisesti ole sytokromi P450 -entsyymin substraatti.
Eliminaatio
Inklisiraanin lopullisen eliminaation puoliintumisaika on noin 9 tuntia, eikä toistuva anto johda kumuloitumiseen. Inklisiraanista 16 % eliminoituu munuaisten kautta.
Lineaarisuus/ei‑lineaarisuus
Vaiheen I kliinisessä tutkimuksessa todettiin, että inklisiraanialtistus suureni suunnilleen annoksen mukaisesti, kun inklisiraania annettiin ihon alle 24–756 mg:n annoksina. Kumuloitumista tai aikariippuvaisia muutoksia ei havaittu, kun inklisiraania annettiin toistuvina annoksina ihon alle.
Farmakokineettiset/farmakodynaamiset suhteet
Vaiheen I kliinisessä tutkimuksessa havaittiin, että inklisiraanin farmakokineettisten parametrien ja LDL-kolesteroliin kohdistuneiden farmakodynaamisten vaikutusten välillä ei ollut yhteyttä. Inklisiraani kulkeutuu selektiivisesti maksasoluihin, joissa se integroituu osaksi RNA:n indusoimaa vaimentamiskompleksia (RISC). Tämä johtaa pitkäkestoiseen vaikutukseen, joka on pidempi kuin plasmasta todetun 9 tunnin pituisen eliminaation puoliintumisajan perusteella voitaisiin olettaa. LDL-kolesterolipitoisuuksien pieneneminen oli voimakkainta 284 mg:n annoksen käytön yhteydessä, eivätkä suuremmat annokset tuottaneet voimakkaampia vaikutuksia.
Erityisryhmät
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa koskeneen spesifisen tutkimuksen tietoihin perustuvassa farmakokinetiikan analyysissä todettiin, että inklisiraanin Cmax-arvo suureni noin 2,3‑kertaiseksi ja AUC-arvo (käyrän alle jäävä pinta-ala) noin 1,6‑kertaiseksi potilailla, joilla oli lievä (kreatiniinipuhdistuma [CrCL] 60 ml/min – 89 ml/min) munuaisten vajaatoiminta, verrattuna potilaisiin, joiden munuaistoiminta on normaali. Keskivaikean (CrCL 30 ml/min - 59 ml/min) munuaisten vajaatoiminnan yhteydessä Cmax-arvo suureni noin 2,0‑kertaiseksi ja AUC-arvo noin 1,8‑kertaiseksi, ja vaikean (CrCL 15 ml/min - 29 ml/min) munuaisten vajaatoiminnan yhteydessä Cmax-arvo suureni noin 3,3‑kertaiseksi ja AUC-arvo noin 2,3‑kertaiseksi verrattuna potilaisiin, joiden munuaistoiminta on normaali. Vaikka altistukset plasmassa olivat tilapäisesti suurempia 48 tunnin ajan, LDL-kolesterolipitoisuuksien pieneneminen oli samaa luokkaa kaikissa munuaistoimintaryhmissä. Populaatiofarmakodynamiikan mallinnuksen perusteella annoksen muuttamista ei suositella, jos potilaalla on loppuvaiheen munuaisten vajaatoiminta. Farmakokinetiikkaa, farmakodynamiikkaa ja turvallisuutta koskevien tietojen perusteella annosta ei tarvitse muuttaa, jos potilaalla on lievä, keskivaikea tai vaikea munuaisten vajaatoiminta. Hemodialyysin vaikutuksia inklisiraanin farmakokinetiikkaan ei ole tutkittu. Inklisiraani eliminoituu munuaisteitse, joten hemodialyysia ei saa toteuttaa ennen kuin Leqvio-valmisteen annosta on kulunut vähintään 72 tuntia.
Maksan vajaatoiminta
Maksan vajaatoimintaa koskeneen spesifisen tutkimuksen tietoihin perustuvassa farmakokinetiikan analyysissä todettiin, että lievää (Child‑Pugh‑luokka A) maksan vajaatoimintaa sairastavilla inklisiraanin Cmax-arvo suureni noin 1,1‑kertaiseksi ja AUC-arvo noin 1,3‑kertaiseksi verrattuna potilaisiin, joiden maksatoiminta oli normaali. Keskivaikean (Child‑Pugh‑luokka B) maksan vajaatoiminnan yhteydessä Cmax-arvo suureni noin 2,1‑kertaiseksi ja AUC-arvo noin 2,0‑kertaiseksi verrattuna potilaisiin, joiden maksatoiminta oli normaali. Vaikka inklisiraanialtistukset plasmassa olivat tilapäisesti suurempia, inklisiraania saaneilla todettu LDL-kolesterolipitoisuuksien pieneneminen oli samaa luokkaa riippumatta siitä, oliko potilailla normaali maksatoiminta vai lievä maksan vajaatoiminta. Keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla lähtötilanteen PCSK9‑pitoisuudet olivat huomattavasti pienemmät ja LDL-kolesterolipitoisuuksien pieneneminen oli vähäisempää kuin potilailla, joilla maksa toimi normaalisti. Annosmuutokset eivät ole tarpeen, jos potilaalla on lievä tai keskivaikea maksan vajaatoiminta (Child–Pugh‑luokka A tai B). Inklisiraania ei ole tutkittu potilailla, joilla on vaikea maksan vajaatoiminta (Child–Pugh‑luokka C).
Muut erityisryhmät
Populaatiofarmakodynamiikan analyysissä arvioitiin 4 328 potilaan tietoja. Iän, painon, sukupuolen, etnisen taustan ja kreatiniinipuhdistuman ei todettu vaikuttavan merkitsevästi inklisiraanin farmakodynamiikkaan. Annoksen muuttamista ei suositella näiden demografisten tietojen perusteella.
Prekliiniset tiedot turvallisuudesta
Rotilla ja apinoilla toteutetuissa toistuvan altistuksen aiheuttamaa toksisuutta koskevissa tutkimuksissa NOAEL-annoksiksi (pitoisuus, jonka yhteydessä ei todettu mitään haitallista vaikutusta) määritettiin suurimmat ihon alle annetut annokset, joiden aiheuttamat altistukset ylittivät merkittävästi maksimaalisen altistuksen ihmisillä. Mikroskoopilla tehtyihin havaintoihin toksisuutta koskevista tutkimuksista sisältyi vakuolien muodostumista apinoiden maksasoluissa sekä rottien ja apinoiden munuaisissa. Näillä havainnoilla ei katsottu olevan yhteyttä kliinisiin laboratorioarvoihin eikä niiden katsottu olevan haitallisia.
Inklisiraani ei ollut karsinogeeninen Sprague‑Dawley-rotilla eikä TgRasH2‑hiirillä, joille annetut inklisiraaniannokset olivat riittävästi suurempia kuin kliiniset annokset.
Inklisiraanin ei todettu olevan mutageeninen eikä klastogeeninen testisarjassa, johon sisältyi bakteereilla tehty mutageenisuuskoe, ihmisen ääreisveren lymfosyyttien kromosomipoikkeavuustutkimus in vitro ja rotan luuytimen mikrotumakoe in vivo.
Rotilla ja kaniineilla toteutetuissa lisääntymistutkimuksissa ei havaittu näyttöä inklisiraanin aiheuttamista sikiöhaitoista suurimmillakaan annetuilla annoksilla, jotka tuottivat huomattavasti ihmisen enimmäisaltistusta suuremman altistuksen.
Inklisiraani ei vaikuttanut hedelmällisyyteen eikä lisääntymistuloksiin uros- ja naarasrotilla, jotka altistettiin inklisiraanille ennen tiineyttä ja tiineyden aikana. Näihin annoksiin liittyvä systeeminen altistus oli moninkertainen verrattuna ihmisen altistukseen kliinisiä annoksia käytettäessä.
Inklisiraania on havaittu imettävien rottien maidossa. Inklisiraanin systeemisestä imeytymisestä ei kuitenkaan ole havaittu näyttöä imettävien rottien vastasyntyneillä poikasilla.
Farmaseuttiset tiedot
Apuaineet
Injektionesteisiin käytettävä vesi
Natriumhydroksidi (pH:n säätelyyn) (E524)
Väkevä fosforihappo (pH:n säätelyyn) (E338)
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita. Ei saa jäätyä.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LEQVIO injektioneste, liuos, esitäytetty ruisku
284 mg (L:ei) 1 kpl (turvamekanismi, 1,5 ml (189 mg/ml)) (2554,25 €)
PF-selosteen tieto
Esitäytetty ruisku
1,5 ml liuosta esitäytetyssä ruiskussa (tyypin I lasia), jossa on mäntätulppa (bromobutyyli, FluroTec-päällysteinen kumi), neula ja jäykkä neulansuojus.
Pakkauksessa on yksi esitäytetty ruisku
Esitäytetty ruisku, jossa on neulan turvamekanismi
1,5 ml liuosta esitäytetyssä ruiskussa (tyypin I lasia), jossa on mäntätulppa (bromobutyyli, FluroTec-päällysteinen kumi), neula ja jäykkä neulansuojus sekä neulan turvamekanismi.
Pakkauksessa on yksi esitäytetty ruisku, jossa on neulan turvamekanismi.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Liuos on kirkasta, väritöntä tai vaaleankeltaista, eikä siinä ole käytännössä lainkaan hiukkasia.
Käyttö- ja käsittelyohjeet
Leqvio on tarkastettava silmämääräisesti ennen antoa. Liuoksen pitäisi olla kirkasta, väritöntä tai vaaleankeltaista, eikä siinä pitäisi olla käytännössä lainkaan hiukkasia. Jos liuoksessa on näkyviä hiukkasia, liuosta ei saa käyttää.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
LEQVIO injektioneste, liuos, esitäytetty ruisku
284 mg 1 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Alirokumabi, evolokumabi ja inklisiraani familiaalisen hyperkolesterolemian hoidossa (aikuiset): Familiaalisen hyperkolesterolemian hoito erityisin edellytyksin (388), Alirokumabi, evolokumabi ja inklisiraani: Hyperkolesterolemian ja sekamuotoisen dyslipidemian hoito erityisin edellytyksin (3015).
ATC-koodi
C10AX16
Valmisteyhteenvedon muuttamispäivämäärä
21.05.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com