
FULVESTRANT STADA injektioneste, liuos, esitäytetty ruisku 250 mg
Vaikuttavat aineet ja niiden määrät
Yksi esitäytetty ruisku sisältää 250 mg fulvestranttia 5 millilitrassa liuosta.
Yksi millilitra liuosta sisältää 50 mg fulvestranttia.
Apuaineet, joiden vaikutus tunnetaan
Yksi millilitra liuosta sisältää 100 mg etanolia (96 %), 100 mg bentsyylialkoholia ja 150 mg bentsyylibentsoaattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos, esitäytetty ruisku.
Kliiniset tiedot
Käyttöaiheet
Fulvestrant Stada on tarkoitettu:
-
monoterapiana estrogeenireseptoripositiivisen paikallisesti edenneen tai metastasoituneen rintasyövän hoitoon postmenopausaalisille naisille:
- jotka eivät ole aiemmin saaneet hormonaalista hoitoa tai
- joiden sairaus on uusiutunut joko liitännäishoitona annetun antiestrogeenihoidon aikana tai sen jälkeen, tai joiden sairaus on edennyt antiestrogeenihoidon aikana.
- yhdistelmänä palbosiklibin kanssa hormonireseptoripositiivisen, ihmisen epidermaalisen kasvutekijän reseptorin 2 (HER2) suhteen negatiivisen paikallisesti edenneen tai metastasoituneen rintasyövän hoitoon aiemmin endokriinistä hoitoa saaneille naisille (ks. kohta Farmakodynamiikka).
Pre- tai perimenopausaalisilla naisilla palbosiklibia sisältävään yhdistelmähoitoon on yhdistettävä luteinisoivan hormonin vapauttajahormonin (LHRH) agonisti.
Annostus ja antotapa
Annostus
Aikuiset naiset (mukaan lukien ikääntyneet)
Suositusannos on 500 mg yhden kuukauden välein. Lisäksi annetaan 500 mg:n annos kahden viikon kuluttua aloitusannoksesta.
Kun fulvestranttia käytetään yhdistelmänä palbosiklibin kanssa, on tutustuttava myös palbosiklibin valmisteyhteenvetoon.
Ennen hoidon aloittamista fulvestrantin ja palbosiklibin yhdistelmällä ja koko yhdistelmähoidon ajan on premenopausaalisia ja perimenopausaalisia naisia hoidettava LHRH-agonisteilla paikallisen kliinisen käytännön mukaisesti.
Erityisryhmät
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa potilaille, joilla on lievä tai keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma ≥ 30 ml/min). Turvallisuutta ja tehoa ei ole tutkittu potilailla, joilla on vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma < 30 ml/min). Siksi varovaisuutta tulee noudattaa näillä potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa potilaille, joilla on lievä tai keskivaikea maksan vajaatoiminta. Fulvestrant Stada -valmistetta tulisi kuitenkin käyttää varoen kyseisille potilaille, koska fulvestrantille altistuminen saattaa lisääntyä. Tietoa ei ole valmisteen käytöstä potilaille, joilla on vaikea maksan vajaatoiminta (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Fulvestrant Stada -valmisteen turvallisuutta ja tehoa vastasyntyneistä alle 18-vuoden ikäisten lasten hoidossa ei ole varmistettu. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta.
Antotapa
Fulvestrant Stada annetaan kahtena peräkkäisenä hitaana lihaksensisäisenä 5 ml:n injektiona (1‑2 minuuttia/injektio) kumpaankin pakaralihakseen (gluteaalinen alue).
Jos Fulvestrant Stada injisoidaan dorsogluteaaliselle alueelle, on varovaisuutta noudatettava iskiashermon läheisyyden vuoksi.
Tarkemmat anto-ohjeet, ks. kohta Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Raskaus ja imetys (ks. kohta Raskaus ja imetys).
Vaikea maksan vajaatoiminta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Varoitukset ja käyttöön liittyvät varotoimet
Fulvestrantin käytössä tulee noudattaa varovaisuutta potilailla, joilla on lievä tai keskivaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa, Vasta-aiheet ja Farmakokinetiikka).
Fulvestrantin käytössä tulee noudattaa varovaisuutta potilailla, joilla on vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma alle 30 ml/min).
Lihaksensisäisestä antoreitistä johtuen fulvestranttia tulee käyttää varoen potilaille, joilla on verenvuototaipumus tai trombosytopenia, sekä potilaille, jotka käyttävät antikoagulaatiolääkkeitä.
Tromboembolisia tapahtumia esiintyy yleisesti edennyttä rintasyöpää sairastavilla naisilla, ja niitä on havaittu fulvestrantilla tehdyissä kliinisissä tutkimuksissa (ks. kohta Haittavaikutukset). Tämä tulee ottaa huomioon määrättäessä fulvestranttia riskipotilaille.
Fulvestrantti-injektion yhteydessä on raportoitu injektiokohtaan liittyviä reaktioita, kuten iskiasta, hermosärkyä, neuropaattista kipua ja perifeeristä neuropatiaa. Injisoitaessa fulvestranttia dorsogluteaaliselle alueelle on varovaisuutta noudatettava iskiashermon läheisyyden vuoksi (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Fulvestrantin pitkäaikaisaikaisvaikutuksista luustoon ei ole tietoa. Fulvestrantin vaikutusmekanismin vuoksi osteoporoosiriski on mahdollinen.
Fulvestrantin tehoa ja turvallisuutta (monoterapiana tai yhdistelmänä palbosiklibin kanssa) ei ole tutkittu potilailla, joilla on kriittinen viskeraalinen tauti.
Kun fulvestranttia käytetään yhdessä palbosiklibin kanssa, on tutustuttava myös palbosiklibin valmisteyhteenvetoon.
Interferenssi estradiolin vasta-ainemäärityksissä
Fulvestrantin ja estradiolin rakenteellisen samankaltaisuuden vuoksi fulvestrantti saattaa häiritä vasta‑aineisiin perustuvia estradiolimäärityksiä, mikä saattaa johtaa virheellisesti kohonneisiin estradiolipitoisuuksiin.
Pediatriset potilaat
Fulvestrant Stada -valmistetta ei suositella käytettäväksi lasten eikä nuorten hoitoon, sillä valmisteen turvallisuutta ja tehoa näillä potilailla ei ole varmistettu (ks. kohta Farmakodynamiikka).
Apuaineet
Etanoli 96 %
Tämä lääkevalmiste sisältää 12,4 tilavuusprosenttia etanolia (alkoholia) eli 1 000 mg:aa per 500 mg:aa fulvestranttia, mikä vastaa 25 ml:aa olutta tai 10 ml:aa viiniä per annos.
Alkoholimäärällä ei todennäköisesti ole vaikutusta aikuisiin.
Tämän lääkevalmisteen sisältämä alkoholi saattaa muuttaa muiden lääkkeiden vaikutusta.
Haitallinen alkoholismissa.
Bentsyylialkoholi
Tämä lääkevalmiste sisältää 1 000 mg:aa bentsyylialkoholia per 500 mg:aa fulvestranttia. Bentsyylialkoholi saattaa aiheuttaa allergisia reaktioita.
Suuria tilavuuksia tulee käyttää varoen ja vain, jos käyttö on välttämätöntä. Tämä koskee erityisesti potilaita, joilla on heikentynyt maksan tai munuaisten toiminta ja siten kumuloitumisen ja toksisuuden riski (metabolinen asidoosi).
Bentsyylibentsoaatti
Tämä lääkevalmiste sisältää 1 500 mg:aa bentsyylibentsoaattia per 500 mg:aa fulvestranttia.
Yhteisvaikutukset
Fulvestrantin ja midatsolaamin (CYP3A4:n substraatti) kliininen yhteisvaikutustutkimus osoitti, että fulvestrantti ei estä CYP3A4-entsyymiä. Fulvestrantin kliiniset yhteisvaikutustutkimukset rifampisiinin (CYP3A4-indusori) ja ketokonatsolin (CYP3A4-estäjä) kanssa eivät osoittaneet kliinisesti merkittävää muutosta fulvestrantin puhdistumassa. Annoksen muuttaminen ei siksi ole tarpeen potilaille, jotka saavat samanaikaisesti fulvestranttia ja CYP3A4-estäjää tai -indusoria.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä fulvestranttihoidon aikana ja kahden vuoden ajan viimeisestä annoksesta.
Raskaus
Fulvestrantin käyttö on vasta-aiheista raskauden aikana (ks. kohta Vasta-aiheet). Fulvestrantin on osoitettu läpäisevän istukan lihakseen annetun kerta-annoksen jälkeen rotalla ja kanilla. Eläinkokeissa on havaittu lisääntymistoksisuutta, kuten sikiön epämuodostumien ja sikiökuolemien esiintyvyyden lisääntymistä (ks. kohta Prekliiniset tiedot turvallisuudesta). Jos potilas tulee raskaaksi Fulvestrant Stada -hoidon aikana, potilaalle on kerrottava mahdollisesta sikiölle aiheutuvasta vaarasta ja keskenmenon riskistä.
Imetys
Rintaruokinta on lopetettava fluvestranttihoidon ajaksi. Fulvestrantti erittyy imettävien rottien maitoon. Ei tiedetä, erittyykö fulvestrantti ihmisen rintamaitoon. Ottaen huomioon imetettävälle lapselle fulvestrantin käytöstä mahdollisesti aiheutuvat vakavat haittavaikutukset valmisteen käyttö on vasta-aiheinen rintaruokinnan aikana (ks. kohta Vasta-aiheet).
Hedelmällisyys
Fulvestrantin vaikutuksia ihmisen hedelmällisyyteen ei ole tutkittu.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Fulvestrantin käytöllä ei ole lainkaan tai juuri lainkaan vaikutusta ajokykyyn tai koneidenkäyttökykyyn. Fulvestranttihoidon aikana on kuitenkin raportoitu hyvin yleisesti voimattomuutta. Potilaiden, joilla esiintyy voimattomuutta, tulisi noudattaa varovaisuutta autolla ajon tai koneiden käytön aikana.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Monoterapia
Tämä kohta sisältää tiedot kaikista kliinisissä tutkimuksissa tai markkinoille tulon jälkeisessä seurannassa havaituista sekä omaehtoisesti ilmoitetuista haittavaikutuksista. Fulvestranttimonoterapiaa koskevassa yhdistetyssä tietoaineistossa yleisimmin raportoituja haittavaikutuksia olivat pistokohdan reaktiot, voimattomuus, pahoinvointi ja maksaentsyymien nousu (ALAT, ASAT, AFOS).
Taulukossa 1 seuraavat lääkkeen haittavaikutusten yleisyysluokat on laskettu fulvestrantti 500 mg ‑hoitoryhmän perusteella tutkimusten, joissa verrattiin fulvestrantin 500 mg:n annosta fulvestrantin 250 mg:n annokseen [CONFIRM- (tutkimus D6997C00002), FINDER 1- (tutkimus D6997C00004), FINDER 2- (tutkimus D6997C00006) ja NEWEST-tutkimus (tutkimus D6997C00003)], tai pelkästään FALCON-tutkimuksen (tutkimus D699BC00001), jossa verrattiin fulvestrantin 500 mg:n annosta anastrotsolin 1 mg:n annokseen, yhdistetyistä turvallisuusanalyyseistä.
Jos esiintyvyydet yhdistetyissä turvallisuusanalyyseissä ja FALCON-tutkimuksessa olivat erilaiset, esiintyvyys on ilmoitettu suuremman esiintyvyyden mukaisesti. Taulukossa 1 olevat esiintyvyydet perustuvat kaikkiin raportoituihin tapauksiin riippumatta tutkijan syy-yhteysarvioista. Fulvestrantti 500 mg -hoidon mediaanikesto koko yhdistetyssä tietoaineistossa (edellä mainitut tutkimukset ja FALCON mukaan lukien) oli 6,5 kuukautta.
Taulukoitu luettelo haittavaikutuksista
Haittavaikutukset on lueteltu alla yleisyys- ja elinjärjestelmäluokituksen mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100 ja < 1/10), melko harvinainen (≥ 1/1 000 ja < 1/100). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1 Fulvestranttimonoterapiaa saaneilla potilailla ilmoitetut lääkkeen aiheuttamat haittavaikutukset
| Haittavaikutukset elinjärjestelmä- ja yleisyysluokituksen mukaan | ||
| Infektiot | Yleinen | Virtsatieinfektiot |
| Veri ja imukudos | Yleinen | Vähentynyt verihiutaleiden määräe |
| Immuunijärjestelmä | Hyvin yleinen | Yliherkkyysreaktiote |
| Melko harvinainen | Anafylaktiset reaktiot | |
| Aineenvaihdunta ja ravitsemus | Yleinen | Anoreksiaa |
| Hermosto | Yleinen | Päänsärky |
| Verisuonisto | Hyvin yleinen | Kuumat aallote |
| Yleinen | Laskimotromboemboliata | |
| Ruoansulatuselimistö | Hyvin yleinen | Pahoinvointi |
| Yleinen | Oksentelu, ripuli | |
| Maksa ja sappi | Hyvin yleinen | Maksaentsyymiarvojen kohoaminen (ALAT, ASAT, AFOS)a |
| Yleinen | Bilirubiiniarvon kohoaminena | |
| Melko harvinainen | Maksan vajaatoimintac,f, hepatiittif, gamma-GT-arvon kohoaminenf | |
| Iho ja ihonalainen kudos | Hyvin yleinen | Ihottumae |
| Luusto, lihakset ja sidekudos | Hyvin yleinen | Nivel- ja muskuloskeletaalinen kipud |
| Yleinen | Selkäkipua | |
| Sukupuolielimet ja rinnat | Yleinen | Emätinverenvuotoe |
| Melko harvinainen | Emätinkandidiaasif, valkovuotof | |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Voimattomuusa, pistokohdan reaktiotb |
| Yleinen | Perifeerinen neuropatiae, iskiase | |
| Melko harvinainen | Pistokohdan verenvuotof, pistokohdan hematoomaf, hermosärkyc, f | |
a Sisältää haittavaikutukset, joihin fulvestrantin myötävaikutusta ei voida tarkalleen arvioida taustalla olevan sairauden takia.
b Termi pistokohdan reaktiot ei sisällä termejä pistokohdan verenvuoto, pistokohdan hematooma, iskias, hermosärky eikä perifeerinen neuropatia.
c Haittavaikutusta ei havaittu laajoissa kliinisissä tutkimuksissa (CONFIRM, FINDER 1, FINDER 2, NEWEST). Yleisyys on laskettu käyttämällä 95 % luottamusvälin ylärajalle määritettyä arvoa. Arvon on laskettu olevan 3/560 (560 on laajoihin kliinisiin tutkimuksiin osallistuneiden potilaiden määrä), joka vastaa yleisyysluokitusta ’melko harvinainen’.
d Sisältää nivelsäryn ja harvinaisemman muskuloskeletaalisen kivun, lihaskivun ja kivun raajoissa.
e Yleisyysluokitukset yhdistetyssä turvallisuusaineistossa ja FALCON-tutkimuksessa ovat erilaiset.
f Lääkkeeseen liittyvää haittavaikutusta ei havaittu FALCON-tutkimuksessa.
Valikoitujen haittavaikutusten kuvaus
Jäljempänä esitetyt kuvaukset perustuvat turvallisuusanalyysisarjaan, joka sisälsi 228 potilasta, jotka olivat saaneet ainakin yhden (1) annoksen fulvestranttia, ja 232 potilasta, jotka olivat saaneet ainakin yhden (1) annoksen anastrotsolia vaiheen 3 FALCON-tutkimuksessa.
Nivel- ja muskuloskeletaalinen kipu
FALCON-tutkimuksessa nivel- ja muskuloskeletaalista kipua haittavaikutuksena ilmoittaneiden potilaiden määrä oli fulvestranttihaarassa 65 (31,2 %) ja anastrotsolihaarassa 48 (24,1 %). Fulvestranttihaaran 65 potilaasta 40 % (26/65) ilmoitti nivel- ja muskuloskeletaalista kipua ensimmäisen hoitokuukauden aikana ja 66,2 % (43/65) ensimmäisten kolmen hoitokuukauden aikana. Yksikään potilas ei ilmoittanut tapahtumia, joiden CTCAE-kriteerien mukainen vaikeusaste oli ≥ 3 tai jotka edellyttivät annoksen pienentämistä, annostuksen keskeyttämistä tai hoidon lopettamista näiden haittavaikutusten vuoksi.
Yhdistelmähoito palbosiklibin kanssa
Yhdistelmänä palbosiklibin kanssa käytetyn fulvestrantin kokonaisturvallisuusprofiili perustuu satunnaistetussa PALOMA-3-tutkimuksessa saatuihin tietoihin 517 potilaasta, joilla oli hormonireseptoripositiivinen ja HER2-negatiivinen edennyt tai metastasoitunut rintasyöpä (ks. kohta Farmakodynamiikka). Fulvestranttia yhdistelmänä palbosiklibin kanssa saaneilla potilailla ilmoitettuja yleisimpiä (≥ 20 %) minkä tahansa vaikeusasteen haittavaikutuksia olivat neutropenia, leukopenia, infektiot, väsymys, pahoinvointi, anemia, suutulehdus, ripuli, trombosytopenia ja oksentelu. Yleisimpiä (≥ 2 %) haittavaikutuksia, joiden vaikeusaste oli ≥ 3, olivat neutropenia, leukopenia, infektiot, anemia, ASAT‑arvon kohoaminen, trombosytopenia ja väsymys.
Taulukossa 2 on esitetty PALOMA-3-tutkimuksen yhteydessä ilmoitetut haittavaikutukset.
Fulvestranttialtistuksen mediaanikesto oli fulvestrantin ja palbosiklibin yhdistelmän hoitohaarassa 11,2 kuukautta ja fulvestrantin ja lumelääkkeen yhdistelmän hoitohaarassa 4,8 kuukautta. Fulvestrantin ja palbosiklibin yhdistelmän hoitohaarassa palbosiklibialtistuksen mediaanikesto oli 10,8 kuukautta.
Taulukko 2 PALOMA-3-tutkimuksessa todetut haittavaikutukset (N = 517)
Elinjärjestelmä Yleisyys Suositeltu termia | Fulvestrantti + Palbosiklibi (N = 345) | Fulvestrantti + lumelääke (N = 172) | ||
Kaikki vaikeusasteet n (%) | Vaikeusaste ≥ 3 n (%) | Kaikki vaikeusasteet n (%) | Vaikeusaste ≥ 3 n (%) | |
| Infektiot | ||||
| Hyvin yleinen | ||||
| Infektiotb | 188 (54,5) | 19 (5,5) | 60 (34,9) | 6 (3,5) |
| Veri ja imukudos | ||||
| Hyvin yleinen | ||||
| Neutropeniac | 290 (84,1) | 240 (69,6) | 6 (3,5) | 0 |
| Leukopeniad | 207 (60,0) | 132 (38,3) | 9 (5,2) | 1 (0,6) |
| Anemiae | 109 (31,6) | 15 (4,3) | 24 (14,0) | 4 (2,3) |
| Trombosytopeniaf | 88 (25,5) | 10 (2,9) | 0 | 0 |
| Melko harvinainen | ||||
| Kuumeinen neutropenia | 3 (0,9) | 3 (0,9) | 0 | 0 |
| Aineenvaihdunta ja ravitsemus | ||||
| Hyvin yleinen | ||||
| Vähentynyt ruokahalu | 60 (17,4) | 4 (1,2) | 18 (10,5) | 1 (0,6) |
| Hermosto | ||||
| Yleinen | ||||
| Makuhäiriöt | 27 (7,8) | 0 | 6 (3,5) | 0 |
| Silmät | ||||
| Yleinen | ||||
| Lisääntynyt kyynelnesteen eritys | 25 (7,2) | 0 | 2 (1,2) | 0 |
| Näön hämärtyminen | 24 (7,0) | 0 | 3 (1,7) | 0 |
| Kuivat silmät | 15 (4,3) | 0 | 3 (1,7) | 0 |
| Hengityselimet, rintakehä ja välikarsina | ||||
| Yleinen | ||||
| Nenäverenvuoto | 25 (7,2) | 0 | 4 (2,3) | 0 |
| Ruoansulatuselimistö | ||||
| Hyvin yleinen | ||||
| Pahoinvointi | 124 (35,9) | 2 (0,6) | 53 (30,8) | 1 (0,6) |
| Suutulehdusg | 104 (30,1) | 3 (0,9) | 24 (14,0) | 0 |
| Ripuli | 94 (27,2) | 0 | 35 (20,3) | 2 (1,2) |
| Oksentelu | 75 (21,7) | 2 (0,6) | 28 (16,3) | 1 (0,6) |
| Iho ja ihonalainen kudos | ||||
| Hyvin yleinen | ||||
| Alopesia | 67 (19,4) | Ei oleellinen | 11 (6,4) | Ei oleellinen |
| Ihottumah | 63 (18,3) | 3 (0,9) | 10 (5,8) | 0 |
| Yleinen | ||||
| Kuiva iho | 28 (8,1) | 0 | 3 (1,7) | 0 |
| Yleisoireet ja antopaikassa todettavat haitat | ||||
| Hyvin yleinen | ||||
| Väsymys | 152 (44,1) | 9 (2,6) | 54 (31,4) | 2 (1,2) |
| Kuume | 47 (13,6) | 1 (0,3) | 10 (5,8) | 0 |
| Yleinen | ||||
| Voimattomuus | 27 (7,8) | 1 (0,3) | 13 (7,6) | 2 (1,2) |
| Tutkimukset | ||||
| Hyvin yleinen | ||||
| Kohonnut ASAT-arvo | 40 (11,6) | 11 (3,2) | 13 (7,6) | 4 (2,3) |
| Yleinen | ||||
| Kohonnut ALAT-arvo | 30 (8,7) | 7 (2,0) | 10 (5,8) | 1 (0,6) |
ALAT = alaniiniaminotransferaasi; ASAT = aspartaattiaminotransferaasi; N/n = potilaiden määrä.
a Suositellut termit on lueteltu MedDRA-version 17.1 mukaisesti.
b Infektiot sisältävät kaikki suositellut termit, jotka kuuluvat elinjärjestelmäluokkaan Infektiot.
c Neutropenia sisältää seuraavat suositellut termit: neutropenia, vähentynyt neutrofiilien määrä.
d Leukopenia sisältää seuraavat suositellut termit: leukopenia, vähentynyt valkosolujen määrä.
e Anemia sisältää seuraavat suositellut termit: anemia, hemoglobiiniarvon pieneneminen, hematokriitin pieneneminen.
f Trombosytopenia sisältää seuraavat suositellut termit: trombosytopenia, vähentynyt verihiutaleiden määrä.
g Suutulehdus sisältää seuraavat suositellut termit: aftainen suutulehdus, huulitulehdus, kielitulehdus, glossodynia, suun haavauma, limakalvotulehdus, suukipu, suunieluvaiva, suunielukipu, suutulehdus.
h Ihottuma sisältää seuraavat suositellut termit: ihottuma, makulopapulaarinen ihottuma, kutiava ihottuma, punoittava ihottuma, näppyläinen ihottuma, dermatiitti, aknetyyppinen dermatiitti, toksinen ihottuma.
Valikoitujen haittavaikutusten kuvaus
Neutropenia
PALOMA-3-tutkimuksessa, jossa annettiin fulvestranttia yhdistelmänä palbosiklibin kanssa, ilmoitettiin minkä tahansa vaikeusasteen neutropeniaa 290 potilaalla (84,1 %). Vaikeusasteen 3 neutropeniaa ilmoitettiin 200 potilaalla (58,0 %) ja vaikeusasteen 4 neutropeniaa 40 potilaalla (11,6 %). Fulvestrantin ja lumelääkkeen yhdistelmän hoitohaarassa (n = 172) minkä tahansa vaikeusasteen neutropeniaa ilmoitettiin 6 potilaalla (3,5 %). Fulvestrantin ja lumelääkkeen yhdistelmän hoitohaarassa ei ollut ilmoitettu yhtään vaikeusasteen 3 ja 4 neutropeniatapausta.
Potilailla, jotka saivat fulvestranttia yhdistelmänä palbosiklibin kanssa, mediaaniaika minkä tahansa vaikeusasteen neutropenian ensimmäiseen ilmaantumiseen oli 15 vuorokautta (vaihteluväli: 13–512 vuorokautta) ja vaikeusasteen ≥ 3 neutropenian mediaanikesto oli 16 vuorokautta. Kuumeista neutropeniaa ilmoitettiin kolmella (0,9 %) potilaista, jotka saivat fulvestranttia yhdistelmänä palbosiklibin kanssa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‐haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Fulvestranttivalmisteen yliannostuksesta ihmisillä on yksittäisiä raportteja. Yliannostustapauksissa suositellaan potilaan oireenmukaista supportiivista hoitoa. Eläinkokeissa ei ole havaittu muita kuin suoraan tai epäsuorasti antiestrogeenivaikutukseen liittyviä vaikutuksia silloin, kun fulvestranttia annettiin suurina annoksina (ks. kohta Prekliiniset tiedot turvallisuudesta).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Endokrinologiset lääkeaineet, antiestrogeenit, ATC-koodi: L02BA03
Vaikutusmekanismi ja farmakodynaamiset vaikutukset
Fulvestrantti on kilpaileva estrogeenireseptorin (ER) antagonisti, joka sitoutuu estrogeenireseptoreihin yhtä voimakkaasti kuin estradioli. Fulvestrantti salpaa estrogeenien troofiset vaikutukset täysin ilman partiaalista (estrogeenin kaltaista) agonistivaikutusta. Vaikutusmekanismiin liittyen estrogeenireseptoriproteiinin määrä vähenee (down-regulation). Primaarista rintasyöpää sairastavia postmenopausaalisia naisia koskevat kliiniset tutkimukset ovat osoittaneet, että fulvestrantti vähentää estrogeenireseptoriproteiinia (down-regulation) estrogeenireseptoripositiivisissa kasvaimissa voimakkaammin kuin plasebo. Myös progesteronireseptorien ekspressio väheni merkitsevästi, kuten oli oletettavissa, koska fulvestrantilla ei ole omaa estrogeeniagonistivaikutusta. On myös osoitettu, että rintasyöpäkasvaimien postmenopausaalisessa neoadjuvanttihoidossa fulvestrantti 500 mg vähentää ER:n ja proliferaation merkkiaineen Ki67:n ilmentymistä enemmän kuin fulvestrantti 250 mg.
Kliininen teho ja turvallisuus edenneessä rintasyövässä
Monoterapia
Vaiheen 3 kliininen tutkimus tehtiin 736:lla edennyttä rintasyöpää sairastavalla postmenopausaalisella naisella, joilla syöpä oli uusiutunut adjuvanttihoitona annetun hormonaalisen hoidon aikana tai sen jälkeen tai joilla syöpä oli edennyt hormonaalisen hoidon jälkeen. Tässä tutkimuksessa oli mukana 423 potilasta, joilla syöpä oli uusiutunut tai edennyt antiestrogeenihoidon aikana (AE-alaryhmä), ja 313 potilasta, joilla syöpä oli uusiutunut tai edennyt aromataasinestäjähoidon aikana (AI-alaryhmä). Tässä tutkimuksessa verrattiin fulvestrantti 500 mg:n (n = 362) tehoa ja turvallisuutta fulvestrantti 250 mg:n (n = 374) tehoon ja turvallisuuteen. Ensisijainen päätetapahtuma oli etenemisvapaa elinaika (Progression-free survival, PFS). Tärkeimpiä toissijaisia tehokkuuden päätetapahtumia olivat objektiivinen hoitovasteprosentti (ORR), kliininen hyötyprosentti (CBR) ja kokonaiselinaika (OS). Taulukossa 3 on yhteenveto CONFIRM-tutkimuksen tehokkuustuloksista.
Taulukko 3 Yhteenveto CONFIRM-tutkimuksen tuloksista ensisijaisen päätetapahtuman (PFS) ja tärkeimpien toissijaisten tehokkuuden päätetapahtumien suhteen
| Muuttuja | Arvion tyyppi; hoidon vertailu | Fulvestrantti 500 mg (N = 362) | Fulvestrantti 250 mg (N = 374) | Vertailu ryhmien välillä (fulvestrantti 500 mg/fulvestrantti 250 mg) | ||||
| Riskisuhde | 95 % CI | p-arvo | ||||||
| PFS | K-M-mediaani (kk); riskisuhde | |||||||
| Kaikki potilaat | 6,5 | 5,5 | 0,80 | 0,68; 0,94 | 0,006 | |||
| -AE-alaryhmä (n = 423) | 8,6 | 5,8 | 0,76 | 0,62; 0,94 | 0,013 | |||
| -AI-alaryhmä (n = 313)a | 5,4 | 4,1 | 0,85 | 0,67; 1,08 | 0,195 | |||
| OSb | K-M-mediaani (kk); riskisuhde | |||||||
| Kaikki potilaat | 26,4 | 22,3 | 0,81 | 0,69; 0,96 | 0,016c | |||
| -AE-alaryhmä (n = 423) | 30,6 | 23,9 | 0,79 | 0,63; 0,99 | 0,038c | |||
| -AI-alaryhmä (n = 313)a | 24,1 | 20,8 | 0,86 | 0,67; 1,11 | 0,241c | |||
| Muuttuja | Arvion tyyppi; hoidon vertailu | Fulvestrantti 500 mg (N = 362) | Fulvestrantti 250 mg (N = 374) | Vertailu ryhmien välillä (fulvestrantti 500 mg/fulvestrantti 250 mg) | ||||
| absoluuttinen ero (%) | 95 % CI | |||||||
| ORRd | % potilaista, joilla OR; absoluuttinen ero (%) | |||||||
| Kaikki potilaat | 13,8 | 14,6 | -0,8 | -5,8; 6,3 | ||||
| -AE-alaryhmä (n = 296) | 18,1 | 19,1 | -1,0 | -8,2; 9,3 | ||||
| -AI-alaryhmä (n = 205)a | 7,3 | 8,3 | -1,0 | -5,5; 9,8 | ||||
| CBRe | % potilaista, joilla CB; absoluuttinen ero (%) | |||||||
| Kaikki potilaat | 45,6 | 39,6 | 6,0 | -1,1; 13,3 | ||||
| -AE-alaryhmä (n = 423) | 52,4 | 45,1 | 7,3 | -2,2; 16,6 | ||||
| -AI-alaryhmä (n = 313)a | 36,2 | 32,3 | 3,9 | -6,1; 15,2 | ||||
a Fulvestrantti on tarkoitettu käytettäväksi potilaille, joiden sairaus on uusiutunut tai edennyt antiestrogeenihoidon aikana. AI-alaryhmän tulokset eivät ole vakuuttavia.
b OS (kokonaiselinaika) on esitetty lopullisille elinaika-analyyseille, kun 75 % potilaista oli kuollut.
c p-arvo ilman monivertailukorjausta ensimmäisen kokonaiselinaika-analyysin (50 % potilaista kuollut) ja päivitetyn kokonaiselinaika-analyysin (75 % potilaista kuollut) välillä
d ORR (objektiivinen hoitovasteprosentti) arvioitiin potilailla, joiden vaste voitiin arvioida lähtötilanteessa (eli ne, joiden sairaus oli mitattavissa lähtötilanteessa: 240 potilasta fulvestrantti 500 mg:n ryhmässä ja 261 potilasta fulvestrantti 250 mg:n ryhmässä).
e Potilaat, joilla parhaana objektiivisena vasteena täydellinen vaste, osittainen vaste tai stabiili tauti ≥ 24 viikon ajan.
PFS: Progression-free survival eli etenemisvapaa elinaika; ORR: Objective response rate eli objektiivinen hoitovasteprosentti; OR: Objective response eli objektiivinen hoitovaste; CBR: Clinical benefit rate eli kliininen hyötyprosentti; CB: Clinical benefit eli kliininen hyöty; OS: Overall survival eli kokonaiselinaika; K-M: Kaplan-Meier; CI: Confidence interval eli luottamusväli; AI: aromataasinestäjä AE: antiestrogeeni.
Postmenopausaalisilla naisilla, joilla oli ER-positiivinen ja/tai PgR-positiivinen paikallisesti edennyt tai metastasoitunut rintasyöpä ja jotka eivät olleet aiemmin saaneet mitään hormonihoitoa, tehtiin vaiheen 3 satunnaistettu, kaksoissokkoutettu, kaksoislume-, monikeskustutkimus, jossa verrattiin fulvestranttivalmisteen 500 mg:n annosta anastrotsolin 1 mg:n annokseen. Yhteensä 462 potilasta satunnaistettiin suhteessa 1:1 saamaan joko fulvestranttia 500 mg tai anastrotsolia 1 mg.
Satunnaistaminen ositettiin sairauden levinneisyyden (paikallisesti edennyt tai metastasoitunut), edenneeseen tautiin aiemmin annetun solunsalpaajahoidon ja mitattavissa olevan sairauden mukaan.
Tutkimuksen ensisijainen tehoa koskeva päätemuuttuja oli tutkijalääkärin arvioima etenemisvapaa elinaika (PFS), joka arvioitiin RECIST 1.1 -kriteerien (Response Evaluation Criteria in Solid Tumours) mukaisesti. Keskeisiä toissijaisia tehoa koskevia päätemuuttujia olivat kokonaiselinaika (OS) ja objektiivinen hoitovasteprosentti (ORR).
Tähän tutkimukseen osallistuneiden potilaiden mediaani-ikä oli 63 vuotta (vaihteluväli 36−90). Useimmilla (87,0 %) potilailla oli lähtötilanteessa metastasoitunut tauti. 55,0 %:lla potilaista oli lähtötilanteessa sisäelinmetastaasi. Yhteensä 17,1 % potilaista oli saanut aiemmin solunsalpaajahoitoa edenneeseen tautiin ja 84,2 %:lla potilaista oli mitattavissa oleva sairaus.
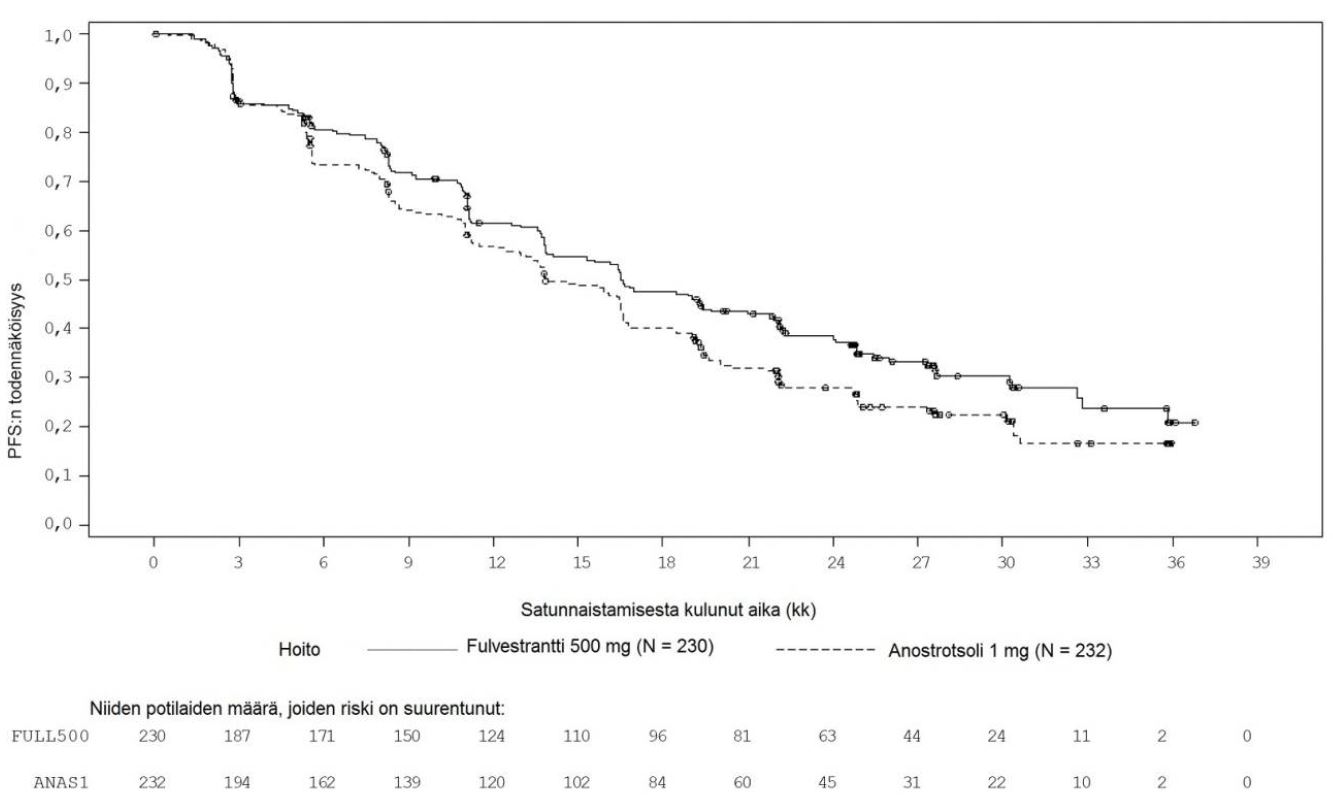
Suurimmassa osassa etukäteen määritellyistä potilaiden alaryhmistä havaittiin yhdenmukaiset tulokset. Alaryhmässä, jossa potilaalla ei ollut sisäelinmetastaasia (n = 208), riskisuhde fulvestranttihaarassa verrattuna anastrotsolihaaraan oli 0,592 (95 %:n luottamusväli: 0,419; 0,837). Alaryhmässä, jossa potilailla oli sisäelinmetastaasi (n = 254), riskisuhde fulvestranttihaarassa verrattuna anastrotsolihaaraan oli 0,993 (95 %:n luottamusväli 0,740; 1,331). FALCON-tutkimuksen tehoa koskevat tulokset on esitetty taulukossa 4 ja kuvassa 1.
Taulukko 4 Ensisijaisen tehoa koskevan päätemuuttujan (PFS) ja keskeisten toissijaisten tehoa koskevien päätemuuttujien (tutkijalääkärin arvio, hoitoaiepopulaatio) tulosten yhteenveto – FALCON-tutkimus
Fulvestrantti 500 mg (N = 230) | Anastrotsoli 1 mg (N = 232) | |
| Etenemisvapaa elinaika | ||
| PFS-tapahtumien määrä (%) | 143 (62,2 %) | 166 (71,6 %) |
| PFS, riskisuhde (95 %:n luottamusväli) ja p-arvo | HR 0,797 (0,637 - 0,999) p = 0,0486 | |
| PFS, mediaani [kk (95 %:n luottamusväli)] | 16,6 (13,8; 21,0) | 13,8 (12,0; 16,6) |
| OS-tapahtumien määrä* | 67 (29,1 %) | 75 (32,3 %) |
| OS, riskisuhde (95 %:n luottamusväli) ja p-arvo | HR 0,875 (0,629 – 1,217) p = 0,4277 | |
| ORR** | 89 (46,1 %) | 88 (44,9 %) |
| ORR, kerroinsuhde (95 %:n luottamusväli) ja p-arvo | OR 1,074 (0,716 – 1,614) p = 0,7290 | |
| Mediaani-DoR (kk) | 20,0 | 13,2 |
| CBR | 180 (78,3 %) | 172 (74,1 %) |
| CBR, kerroinsuhde (95 %:n luottamusväli) ja p-arvo | OR 1,253 (0,815 – 1,932) p = 0,3045 | |
*(31 % potilaista kuollut) – ei lopullinen OS-analyysi
**potilailla, joilla oli mitattavissa oleva sairaus
Kuva 1 Etenemisvapaan elinajan (tutkijalääkärin arvio, hoitoaiepopulaatio) Kaplan-Meier-kuvaaja – FALCON-tutkimus
Vaiheen 3 kliinisiä tutkimuksia on tehty kaksi, ja niihin osallistui yhteensä 851 edennyttä rintasyöpää sairastavaa postmenopausaalista naista, joilla syöpä oli uusiutunut adjuvanttihoitona annetun hormonaalisen hoidon aikana tai jälkeen tai joilla syöpä oli edennyt hormonaalisen hoidon jälkeen. Tutkimukseen osallistuneista 77 prosentilla oli estrogeenireseptoripositiivinen rintasyöpä. Näissä tutkimuksissa verrattiin fulvestratin kerran kuukaudessa annettavan 250 mg annoksen turvallisuutta ja tehoa anastrotsolin (aromataasin estäjän) 1 mg:n vuorokausiannokseen. Fulvestrantin 250 mg:n kuukausiannos osoittautui vähintään yhtä tehokkaaksi kuin anastrotsoli arvioitaessa etenemisvapaata elinaikaa, objektiivista vastetta ja aikaa potilaan kuolemaan. Näissä päätetapahtumissa ei ollut tilastollisesti merkitsevää eroa kahden hoitoryhmän välillä. Ensisijainen päätetapahtuma oli etenemisvapaa elinaika. Yhdistetty analyysi kummastakin tutkimuksesta osoitti, että 83 %:lla fulvestranttivalmistetta saaneista potilaista ja 85 %:lla anastratsolia saaneista potilaista syöpä eteni. Yhdistetty analyysi kummastakin tutkimuksesta osoitti, että fulvesrantti 250 mg:n ja anastratsolin välinen etenemisvapaan elinajan riskisuhde oli 0,95 (95 %:n luottamusväli 0,82–1,10). Objektiivinen hoitovasteprosentti fulvestrantti 250 mg:lla oli 19,2 % ja anastratsolilla 16,5 %. Mediaaniaika potilaan kuolemaan oli fulvestranttivalmisteella hoidetuilla potilailla 27,4 kuukautta ja anastratsolilla hoidetuilla 27,6 kuukautta. Kuolemaan kuluneen ajan riskisuhde fulvestrantti 250 mg:n ja anastratsolin välillä oli 1,01 (95 %:n luottamusväli 0,86–1,19).
Yhdistelmähoito palbosiklibin kanssa
Vaiheen 3 kansainvälisessä, satunnaistetussa, kaksoissokkoutetussa, rinnakkaisryhmillä toteutetussa monikeskustutkimuksessa verrattiin fulvestranttivalmisteen 500 mg:n annosta yhdistelmänä palbosiklibin 125 mg:n annoksen kanssa fulvestranttivalmisteen 500 mg:n annokseen yhdistelmänä lumelääkkeen kanssa. Tutkimukseen osallistuneilla naisilla oli hormonireseptoripositiivinen ja HER2‑negatiivinen paikallisesti edennyt rintasyöpä, jota ei voitu hoitaa resektiolla tai parantavassa tarkoituksessa annettavalla sädehoidolla, tai menopausaaliseen statukseen katsomatta heillä oli metastasoitunut rintasyöpä ja heidän tautinsa oli edennyt aiemman, (neo-)adjuvanttihoitona tai metastaattiseen tautiin annetun endokriinisen hoidon jälkeen.
Yhteensä 521 pre- tai peri- ja postmenopausaalista naista, joiden tauti oli edennyt endokriinisen liitännäishoidon aikana tai 12 kuukauden kuluessa sen päättymisestä tai aiemman edenneeseen tautiin annetun endokriinisen hoidon aikana tai yhden kuukauden kuluessa siitä, satunnaistettiin suhteessa 2:1 saamaan fulvestranttivalmistetta yhdessä palbosiklibin kanssa tai fulvestranttivalmistetta yhdessä lumelääkkeen kanssa ja ositettiin dokumentoidun aiemmalle hormonihoidolle osoitetun herkkyyden, tutkimukseen osallistumisen vaiheessa todetun menopausaalisen statuksen (pre- tai perimenopausaalinen vs. postmenopausaalinen) ja sisäelinmetastaasien olemassaolon mukaan. Pre- tai perimenopausaalisille naisille annettiin gosereliinia, joka on LHRH-agonisti. Potilaat, joilla oli edennyt tai metastasoitunut, oireinen sisäelimiin levinnyt tauti ja joilla oli lyhyellä aikavälillä hengenvaarallisten komplikaatioiden riski (mukaan lukien potilaat, joilla oli massiivisia hallitsemattomia effuusioita [pleuraalisia, perikardiaalisia tai peritoneaalisia], pulmonaalinen lymfangiitti ja joilla oli metastaaseja yli 50 %:ssa maksasta), eivät soveltuneet osallistumaan tutkimukseen.
Potilaat saivat heille määrättyä hoitoa siihen asti, kunnes objektiivisesti todettiin taudin eteneminen, oireet pahenivat, ilmeni toksisuutta, jota ei voitu hyväksyä, potilas kuoli tai peruutti suostumuksensa, sen mukaan, mikä näistä tapahtui ensin. Siirtyminen tutkimusryhmästä toiseen ei ollut sallittua.
Fulvestranttivalmistetta yhdessä palbosiklibin kanssa saaneiden hoitohaaran ja fulvestranttivalmistetta yhdessä lumelääkettä saaneiden hoitohaaran potilaat vastasivat toisiaan lähtötilanteen demografisten tietojen ja prognostisten ominaisuuksien osalta. Tähän tutkimukseen osallistuneiden potilaiden mediaani-ikä oli 57 vuotta (vaihteluväli 29, 88). Suurin osa kunkin hoitohaaran potilaista oli valkoihoisia, heillä oli todettu dokumentoidusti herkkyys aiemmalle hormonihoidolle ja he olivat postmenopausaalisia. Noin 20 % potilaista oli pre- tai perimenopausaalisia. Kaikki potilaat olivat saaneet aiemmin systeemistä hoitoa ja useimmat potilaat kussakin hoitohaarassa olivat saaneet aiemmin solunsalpaajahoitoa ensisijaisen diagnoosinsa vuoksi. Yli puolella (62 %:lla) potilaista ECOG-suorituskykypistemäärä oli 0, potilaista 60 %:lla oli sisäelinmetastaaseja ja 60 % oli saanut useampaa kuin yhtä hormonihoitoa ensisijaisen diagnoosinsa vuoksi.
Tutkimuksen ensisijainen päätemuuttuja oli tutkijalääkärin arvioima etenemisvapaa elinaika, joka arvioitiin RECIST 1.1 -kriteerien mukaisesti. Tukena käytetyt PFS-analyysit perustuivat riippumattomaan keskitetysti toteutettuun radiologiseen arvioon. Toissijaisia päätemuuttujia olivat OR, CBR, kokonaiselinaika (OS), turvallisuus ja kipuun liittyvän päätemuuttujan osalta aika tilan huononemiseen (time-to-deterioration, TTD).
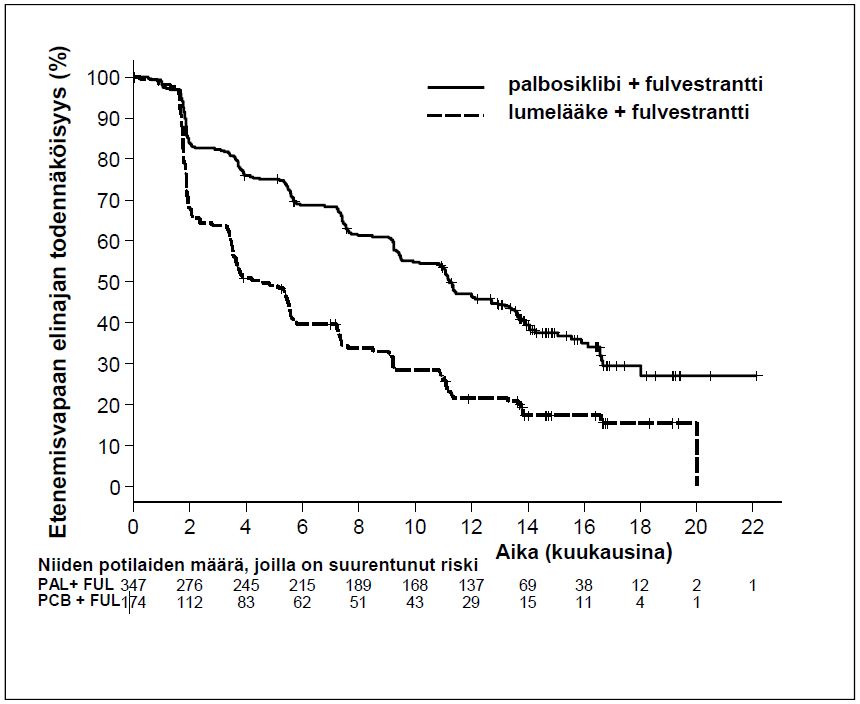
Välianalyysissä, joka tehtiin 82 %:sta suunniteltuja PFS-tapahtumia, todettiin, että tutkimuksessa saavutettiin sen ensisijainen päätemuuttuja, tutkijalääkärin arvioima etenemisvapaan elinajan piteneminen; tulokset ylittivät etukäteen määritellyn tehoa koskevan Haybittle-Peto-raja-arvon (α = 0,00135), mikä osoitti etenemisvapaan elinajan tilastollisesti merkitsevän pitenemisen ja kliinisesti merkittävän hoitovaikutuksen. Taulukossa 5 on esitetty tehoa koskevien tietojen tuoreempi päivitys.
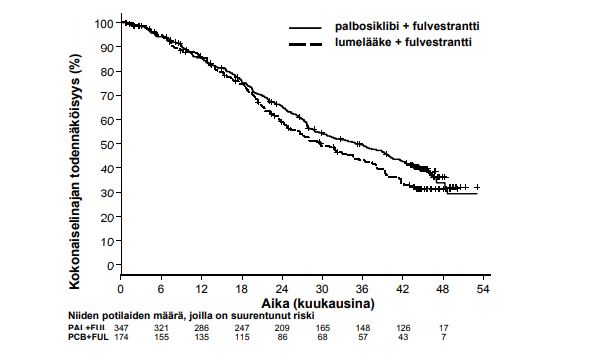
Kun seuranta-ajan mediaani oli 45 kuukautta, tehtiin lopullinen kokonaiselinaika-analyysi 310 tapahtuman (60 % satunnaistetuista potilaista) perusteella. Palbosiklibin ja fulvestrantin yhdistelmän hoitohaaran ja lumelääkkeen ja fulvestrantin yhdistelmän hoitohaaran välillä havaittiin 6,9 kuukauden ero kokonaiselinajan mediaanissa. Tämä tulos ei ollut tilastollisesti merkitsevä etukäteen määritetyllä merkitsevyystasolla, joka oli 0,0235 (yksitahoinen). Lumelääkkeen ja fulvestrantin yhdistelmän hoitohaarassa 15,5 % satunnaistetuista potilaista sai palbosiklibia ja muita CDK:n estäjiä seuraavana hoitona taudin etenemisen jälkeen.
PALOMA3-tutkimuksen tutkijalääkärin arvioimista etenemisvapaata elinaikaa ja lopullista kokonaiselinaikaa koskevista tiedoista saadut tulokset on esitetty taulukossa 5. Niitä kuvaavat Kaplan-Meier-kuvaajat on esitetty kuvissa 2 ja 3.
Taulukko 5 Tehoa koskevat tulokset – PALOMA-3-tutkimus (tutkijalääkärin arvio, hoitoaiepopulaatio)
Päivitetty analyysi (tietojenkeruu päättynyt 23.10.2015) | ||
Fulvestrantti ja palbosiklibi (N = 347) | Fulvestrantti ja lumelääke (N = 174) | |
| Etenemisvapaa elinaika | ||
| Mediaani [kk (95 %:n luottamusväli)] | 11,2 (9,5; 12,9) | 4,6 (3,5; 5,6) |
| Riskisuhde (95 %:n luottamusväli) ja p-arvo | 0,497 (0,398; 0,620), p < 0,000001 | |
| Toissijaiset päätemuuttujat* | ||
| OR [% (95 %:n luottamusväli)] | 26,2 (21,7; 31,2) | 13,8 (9,0; 19,8) |
| OR (mitattavissa oleva sairaus) [% (95 %:n luottamusväli)] | 33,7 (28,1; 39,7) | 17,4 (11,5; 24,8) |
| CBR [% (95 %:n luottamusväli)] | 68,0 (62,8; 72,9) | 39,7 (32,3; 47,3) |
Lopullinen kokonaiselinaika (OS) (viimeinen tiedonkeruupäivä 13.4.2018) | ||
| Tapahtumien määrä (%) | 201 (57,9) | 109 (62,6) |
| Mediaani [kk (95 %:n luottamusväli)] | 34,9 (28,8; 40,0) | 28,0 (23,6; 34,6) |
| Riskisuhde (95 %:n luottamusväli) ja p-arvo† | 0,814 (0,644; 1,029) p = 0,0429†* | |
CBR = kliininen hyötyprosentti; N = potilaiden määrä; OR = objektiivinen hoitovaste;
Toissijaisia päätemuuttujia koskevat tulokset perustuvat RECIST 1.1 -kriteerien mukaisesti vahvistettuihin ja vahvistamattomiin vasteisiin
* Ei tilastollisesti merkitsevä.
† Yksitahoinen p-arvo, joka on saatu satunnaistamisen mukaan ositetulla log-rank-testillä, jossa stratifiointitekijät olivat sisäelinmetastaasien olemassaolo ja herkkyys aiemmalle endokriiniselle hoidolle.
Kuva 2. Etenemisvapaan elinajan (tutkijalääkärin arvio, hoitoaiepopulaatio) Kaplan-Meier-kuvaaja – PALOMA-3-tutkimus (viimeinen tiedonkeruupäivä 23.10.2015)
FUL = fulvestrantti; PAL = palbosiklibi; PCB = lumelääke
Taudin etenemisen tai kuoleman riskin vähenemä todettiin fulvestranttivalmistetta yhdessä palbosiklibin kanssa saaneiden haarassa kaikissa yksittäisissä potilaiden alaryhmissä, jotka oli määritelty stratifiointitekijöiden ja lähtötilanteen ominaisuuksien mukaan. Vähenemä oli ilmeinen pre- tai perimenopausaalisilla naisilla (riskisuhde 0,46 [95 %:n luottamusväli: 0,28; 0,75]) ja postmenopausaalisilla naisilla (riskisuhde 0,52 [95 %:n luottamusväli: 0,40; 0,66]) sekä potilailla, joilla tauti oli metastasoitunut sisäelimiin (riskisuhde 0,50 [95 %:n luottamusväli: 0,38; 0,65], ja potilailla, joilla tauti oli metastasoitunut muualle kuin sisäelimiin (riskisuhde 0,48 [95 %:n luottamusväli: 0,33; 0,71]). Hyöty todettiin myös metastasoituneen taudin aiemmista hoitolinjoista riippumatta, olipa hoitolinjoja ollut nolla (riskisuhde 0,59 [95 %:n luottamusväli: 0,37; 0,93]), yksi (riskisuhde 0,46 [95 %:n luottamusväli: 0,32; 0,64]), kaksi (riskisuhde 0,48 [95 %:n luottamusväli: 0,30; 0,76]) tai vähintään kolme (riskisuhde 0,59 [95 %:n luottamusväli: 0,28; 1,22]).
Kuva 3. Kokonaiselinajan Kaplan-Meier-kuvaaja (hoitoaiepopulaatio) – PALOMA3-tutkimus (viimeinen tiedonkeruupäivä 13.4.2018)
FUL = fulvestrantti; PAL = palbosiklibi; PCB = lumelääke
Taulukossa 6 on esitetty muut arvioidut tehoa koskevat mittarit (OR ja TTR) alaryhmistä, joissa potilailla oli tai ei ollut viskeraalista tautia.
Taulukko 6 PALOMA-3-tutkimuksen tehoa koskevat tulokset viskeraalisen ja ei-viskeraalisen taudin yhteydessä (hoitoaiepopulaatio)
| Viskeraalinen tauti | Ei-viskeraalinen tauti | |||
Fulvestrantti ja palbosiklibi (N = 206) | Fulvestrantti ja lumelääke (N = 105) | Fulvestrantti ja palbosiklibi (N = 141) | Fulvestrantti ja lumelääke (N = 69) | |
| OR [% (95 %:n luottamusväli)] | 35,0 (28,5; 41,9) | 13,3 (7,5; 21,4) | 13,5 (8,3; 20,2) | 14,5 (7,2; 25,0) |
TTR*, mediaani [kk (vaihteluväli)] | 3,8 (3,5; 16,7) | 5,4 (3,5; 16,7) | 3,7 (1,9; 13,7) | 3,6 (3,4; 3,7) |
*Vahvistettuihin ja vahvistamattomiin vasteisiin perustuvat vastetulokset.
N = potilaiden määrä; OR = objektiivinen hoitovaste; TTR = aika ensimmäiseen kasvainvasteeseen (time to first tumor response).
Potilaiden ilmoittamat oireet arvioitiin käyttämällä EORTC-järjestön (European Organization for Research and Treatment of Cancer) elämänlaatukyselyä (QLQ)-C30 ja sen rintasyöpämoduulia (EORTC QLQ-BR23). Fulvestranttivalmistetta yhdessä palbosiklibin kanssa saaneiden haarassa yhteensä 335 potilasta ja fulvestranttivalmistetta yhdessä lumelääkkeen kanssa saaneiden haarassa 166 potilasta vastasi kyselyyn lähtötilanteessa ja ainakin yhdellä lähtötilanteen jälkeisellä tutkimuskäynnillä.
Aika tilan huononemiseen oli määritelty etukäteen ajaksi, joka kului lähtötilanteesta siihen, että kipuoireiden pistemäärä suureni lähtötilanteesta ensimmäisen kerran vähintään 10 pisteellä. Palbosiklibin lisääminen fulvestranttihoitoon johti oireiden suhteen saavutettuun hyötyyn, sillä kipuoireiden suhteen todettu aika tilan huononemiseen piteni merkitsevästi verrattuna lumelääkkeen kanssa annettuun fulvestranttihoitoon (mediaani 8,0 kuukautta verrattuna 2,8 kuukauteen; riskisuhde 0,64 [95 %:n luottamusväli: 0,49; 0,85]; p < 0,001).
Vaikutukset postmenopausaaliseen endometriumiin
Prekliiniset tiedot eivät viittaa siihen, että fulvestrantilla olisi stimuloiva vaikutus postmenopausaaliseen endometriumiin (ks. kohta Prekliiniset tiedot turvallisuudesta). Kaksi viikkoa kestänyt tutkimus, johon osallistui terveitä postmenopausaalisia vapaaehtoisia naisia, joita hoidettiin etinyyliestradiolin 20 μg:n vuorokausiannoksilla, osoitti, että estrogeenihoitoa edeltävästi annettu fulvestrantti 250 mg vähensi postmenopausaalisen endometriumin stimulaatiota merkitsevästi enemmän kuin plasebo (arvioitiin endometriumin paksuuden ultraäänitutkimuksella).
Korkeintaan 16 viikkoa kestänyt neoadjuvanttihoito joko fulvestrantti 500 mg:lla tai fulvestrantti 250 mg:lla hoidetuilla rintasyöpäpotilailla ei aiheuttanut kliinisesti merkittäviä muutoksia endometriumin paksuuteen, mikä osoittaa agonistisen vaikutuksen puuttumista. Näyttöä endometriumiin kohdistuvista haittavaikutuksista ei havaittu tutkituissa rintasyöpäpotilaissa. Tietoa endometriumin morfologiasta ei ole saatavilla.
Kahdessa lyhytkestoisessa tutkimuksessa (1 ja 12 viikkoa) premenopausaalisilla naisilla, joilla oli hyvänlaatuinen gynekologinen sairaus, ei havaittu merkittäviä eroja endometriumin paksuudessa (mitattiin ultraäänellä) fulvestrantti- ja plaseboryhmien välillä.
Vaikutukset luustoon
Fulvestrantin pitkäaikaisvaikutuksista luustoon ei ole tietoa. Korkeintaan 16 viikkoa kestävä neoadjuvanttihoito joko fulvestrantti 500 mg:lla tai fulvestrantti 250 mg:lla hoidetuilla rintasyöpäpotilailla ei aiheuttanut kliinisesti merkittäviä muutoksia seerumin luun aineenvaihdunnan merkkiaineisiin.
Pedriatriset potilaat
Fulvestrant Stada -valmistetta ei ole tarkoitettu käytettäväksi lapsille. Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset fulvestrantin käytöstä rintasyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Avoimessa vaiheen 2 tutkimuksessa arvioitiin fulvestrantin turvallisuutta, tehoa ja farmakokinetiikkaa kolmellakymmenellä 1–8-vuotiaalla tytöllä, joilla oli McCune–Albrightin oireyhtymään liittyvä progressiivinen ennenaikainen murrosikä. Lapsipotilaat saivat fulvestranttia lihakseen 4 mg/kg/kk. Tässä 12 kk:n pituisessa tutkimuksessa arvioitiin useita McCune–Albrightin oireyhtymään liittyviä päätetapahtumia ja todettiin emätinvuotojen esiintymistiheyden pienentymistä ja luustoiän etenemisen hidastumista. Fulvestrantin vakaan tilan minimipitoisuudet lapsilla olivat tässä tutkimuksessa samankaltaiset kuin aikuisilla (ks. kohta Farmakokinetiikka). Tässä suppeassa tutkimuksessa ei havaittu uusia turvallisuuteen liittyviä ongelmia. 5 vuoden tietoja ei kuitenkaan ole vielä saatavilla.
Farmakokinetiikka
Imeytyminen
Pitkävaikutteisen lihakseen annetun fulvestrantti-injektion jälkeen fulvestrantti imeytyy hitaasti, ja huippupitoisuudet plasmassa (Cmax) saavutetaan noin 5 päivän kuluttua annosta. Annettaessa fulvestranttia 500 mg:n annoksella saavutetaan ensimmäisen kuukauden aikana altistustasot, jotka ovat vakaan tilan tasolla tai sen lähellä (keskimääräinen [variaatiokerroin]: AUC 475 [33,4 %] ng·vrk/ml; Cmax 25,1 [35,3 %] ng/ml; Cmin 16,3 [25,9 %] ng/ml). Vakaassa tilassa fulvestranttipitoisuudet vaihtelevat verrattain vähän, ja ero huippupitoisuuden ja pienimmän pitoisuuden välillä on korkeintaan noin 3-kertainen. Lihakseen annetun injektion jälkeen altistus on suunnilleen annosriippuvaista annosvälillä 50–500 mg.
Jakautuminen
Fulvestrantti jakautuu laajalle ja nopeasti. Laaja teoreettinen jakautumistilavuus vakaassa tilassa (Vdss) on noin 3–5 l/kg. Tästä voidaan päätellä, että pääosa lääkkeestä jakautuu ekstravaskulaaritilaan. Fulvestrantin sitoutumisaste plasman proteiineihin on korkea (99 %). VLDL (very low density lipoprotein)-, LDL (low density lipoprotein)- ja HDL (high density lipoprotein)- lipoproteiinien fraktiot ovat tärkeimpiä sitoutumiskohtia. Kilpailevaa proteiineihin sitoutumista koskevia yhteisvaikutustutkimuksia ei tehty. Sukupuolihormoneja sitovan globuliinin (SHBG) merkitystä ei ole selvitetty.
Biotransformaatio
Fulvestrantin metaboliaa ei ole täysin selvitetty, mutta se sisältää yhdistelmiä useista mahdollisista endogeenisten steroidien biotransformaatioreiteistä. Antiestrogeenimallien avulla arvioituna tunnistetut metaboliitit (mukaan lukien 17-ketoni-, sulfoni-, 3-sulfaatti-, 3- ja 17‑glukuronidimetaboliitit) ovat aktiivisuudeltaan heikompia tai fulvestrantin kaltaisia. Tutkimukset, joissa on käytetty ihmisen maksapreparaatteja ja rekombinantteja ihmisen entsyymejä, osoittavat, että CYP3A4 on ainut fulvestrantin oksidaatioon osallistuva P-450-isoentsyymi, mutta metaboloituminen näyttäisi tapahtuvan in vivo useammin muuta reittiä kuin P450-entsyymien kautta. In vitro ‑tutkimustulosten mukaan fulvestrantti ei estä CYP450 isoentsyymejä.
Eliminaatio
Fulvestrantti eliminoituu pääasiassa metaboloituneessa muodossa. Erittyminen tapahtuu pääasiassa ulosteiden mukana. Virtsan mukana poistuva määrä on alle 1 %. Fulvestrantin puhdistuma on nopeaa, 11±1,7 ml/min/kg, mikä viittaa erittymiseen paljolti maksan kautta. Terminaalinen puoliintumisaika (t1/2) lihakseen annetun injektion jälkeen määräytyy imeytymisnopeuden mukaan, ja sen arvioidaan olevan 50 vuorokautta.
Erityisryhmät
Vaiheen 3 tutkimustulosten populaatioanalyysissä ei havaittu eroja fulvestrantin farmakokineettisessä profiilissa iän (vaihteluväli 33–89 vuotta), painon (40–127 kg) tai rodun suhteen.
Munuaisten vajaatoiminta
Lievä tai keskivaikea munuaisten vajaatoiminta ei vaikuttanut fulvestrantin farmakokinetiikkaan kliinisesti merkitsevästi.
Maksan vajaatoiminta
Fulvestrantin farmakokinetiikkaa on arvioitu kliinisessä kerta-annostutkimuksessa naisilla, joilla oli lievä tai keskivaikea maksan vajaatoiminta (Child-Pugh-luokka A tai B). Tutkimuksessa käytettiin lyhytkestoisen lihaksensisäisen injektiovalmisteen suurta annosta. Maksan vajaatoimintapotilailla todettiin 2,5-kertaiset AUC-arvot terveisiin naisiin verrattuna. On odotettavissa, että fulvestranttia saavat potilaat sietävät hyvin tämän suuruisen lisäaltistumisen. Vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavia naisia ei tutkittu.
Pediatriset potilaat
Fulvestrantin farmakokinetiikkaa arvioitiin kliinisessä tutkimuksessa kolmellakymmenellä tytöllä, joilla oli McCune–Albrightin oireyhtymään liittyvä progressiivinen ennenaikainen murrosikä (ks. kohta Farmakodynamiikka). Pediatriset potilaat olivat iältään 1–8-vuotiaita ja saivat fulvestranttia lihakseen 4 mg/kg/kk. Vakaan tilan minimipitoisuuden (Cmin, ss) geometrinen keskiarvo oli 4,2 ng/ml (keskihajonta 0,9 ng/ml) ja AUCss:n geometrinen keskiarvo oli 3 680 ng·h/ml (keskihajonta 1 020 ng·h/ml). Näiden rajallisten tietojen perusteella fulvestrantin vakaan tilan minimipitoisuudet lapsilla näyttävät olevan samankaltaisia kuin aikuisilla.
Prekliiniset tiedot turvallisuudesta
Fulvestrantin akuutti toksisuus on vähäistä.
Fulvestrantti oli moniannostutkimuksissa hyvin siedetty kaikilla eläinlajeilla. Vehikkelin katsotaan aiheuttaneen paikallisia reaktioita mukaan lukien myosiittia ja granuloomia pistokohdassa, mutta verrattuna kontrolliin (suolaliuos), myosiitin vaikeusasteen havaittiin lisääntyvän kaneilla käytettäessä fulvestranttia. Toksisuustutkimuksissa, joissa valmistetta annettiin useana annoksena lihakseen rotille ja koirille, fulvestrantin antiestrogeenivaikutus oli syynä lähes kaikkiin havaittuihin vaikutuksiin, erityisesti naaraiden lisääntymisjärjestelmään kohdistuviin vaikutuksiin, mutta myös muihin hormoniriippuvaisiin elimiin kohdistuviin vaikutuksiin kummallakin sukupuolella. Useisiin eri kudoksiin liittyvää valtimotulehdusta havaittiin joillakin koirilla jatkuvan (12 kuukauden) annostelun jälkeen.
Koirilla tehdyissä tutkimuksissa, joissa lääke annettiin suun kautta ja laskimoon, havaittiin sydämeen ja verenkiertoelimiin kohdistuvia vaikutuksia (vähäinen ST-nousu EKG:ssä [suun kautta], sinuspysähdys yhdellä koiralla [laskimoon]). Näitä esiintyi, kun fulvestranttiannokset olivat suuremmat potilaiden saamiin annoksiin verrattuna (Cmax > 15-kertainen), ja niillä on todennäköisesti vain vähäinen merkitys käytettäessä kliinisiä annoksia ihmisillä.
Fulvestrantilla ei ollut genotoksista vaikutusta.
Kuten antiestrogeenivaikutusten perusteella voidaan olettaa, fulvestrantin havaittiin vaikuttavan lisääntymiseen ja alkion-/sikiönkehitykseen annoksilla, jotka vastaavat kliinisessä käytössä olevia annoksia. Naarasrotilla havaittiin tilapäisesti heikentynyttä naaraiden hedelmällisyyttä ja alkioiden selviytymistä, synnytyshäiriöitä ja sikiöepämuodostumien lisääntymistä (mm. tarsaalifleksuuraa). Fulvestranttia saaneiden kanien tiineys keskeytyi. Istukan paino ja implantaation jälkeinen sikiökuolleisuus lisääntyi. Kanin sikiömuutosten esiintyvyys kasvoi (lantiokaaren sijoittuminen taaksepäin ja 27 presakraalinikamaa).
Kahden vuoden onkogeenisuustutkimus rotilla (fulvestrantti-injektio lihakseen) osoitti hyvänlaatuisten munasarjan granuloosasolukasvaimien lisääntymistä naarasrotilla käytettäessä suuria 10 mg/rotta/15 vrk -annoksia, sekä kivesten leydiginsolukasvainten lisääntymistä urosrotilla. Hiirillä tehdyssä kahden vuoden onkogeenisuustutkimuksessa (suun kautta anto kerran vuorokaudessa) sekä hyvänlaatuiset että pahanlaatuiset munasarjojen sukupienakasvaimet lisääntyivät 150 ja 500 mg/kg vuorokausiannoksilla. Näiden löydösten vaikutuksettomalla annostasolla systeeminen altistus (AUC) ihmisillä odotettuun altistukseen nähden oli naarasrotilla suunnilleen 1,5-kertainen ja urosrotilla 0,8‑kertainen ja sekä uros- että naarashiirillä 0,8-kertainen. Näiden kasvainten syntyä selittävät lääkkeen farmakologiaan liittyvät endokriinisen takaisinkytkennän muutokset gonadotropiinipitoisuuksissa, joita antiestrogeenit aiheuttavat hedelmällisessä iässä oleville eläimille. Siksi näitä havaintoja ei pidetä merkityksellisinä, kun fulvestrantilla hoidetaan edennyttä rintasyöpää sairastavia postmenopausaalisia naisia.
Ympäristö ön kohdistuvien riskien arviointi
Ympäristöriskejä arvioivat tutkimukset ovat osoittaneet, että fulvestrantti saattaa aiheuttaa ympäristöhaittoja vesistöille (ks. kohta Käyttö- ja käsittelyohjeet).
Farmaseuttiset tiedot
Apuaineet
Etanoli (96 %)
Bentsyylialkoholi
Bentsyylibentsoaatti
Risiiniöljy, puhdistettu
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
4 vuotta
Säilytys
Säilytä ja kuljeta kylmässä (2 °C–8 °C).
Säilytä esitäytetty ruisku alkuperäispakkauksessa. Herkkä valolle.
Lämpötilapoikkeamia 2 °C–8 °C ulkopuolelle on rajoitettava. Valmisteen säilyttämistä yli 30 °C lämpötiloissa on vältettävä, eikä valmistetta saa säilyttää yli 28 päivää olosuhteissa, joissa keskimääräinen säilytyslämpötila on alle 25 °C (mutta yli 2 °C–8 °C). Valmiste on palautettava ohjeiden mukaisiin säilytysolosuhteisiin (säilytä ja kuljeta kylmässä (2 °C–8 °C)) välittömästi lämpötilapoikkeamien jälkeen. Lämpötilapoikkeamilla on kumulatiivinen vaikutus valmisteen laatuun, eikä 28 päivän säilytysajanjaksoa poikkeavissa olosuhteissa saa ylittää Fulvestrant Stada -valmisteen 4 vuoden kestoajan puitteissa (ks. kohta Kestoaika). Altistuminen alle 2 °C lämpötiloille ei vahingoita valmistetta, jos sitä ei säilytetä alle -20 °C:ssa.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
FULVESTRANT STADA injektioneste, liuos, esitäytetty ruisku
250 mg (L:ei) 2 x 5 ml (73,23 €)
PF-selosteen tieto
Esitäytetty ruisku koostuu seuraavista osista: tyypin I kirkkaasta lasista valmistettu sylinteri, jossa on muovinen ruiskun korkki, bromobutyyliä oleva männän kumitulppa ja polypropeenia olevat mäntä ja männän ulosvetoeste. Ruisku sisältää 5 ml injektionestettä.
Fulvestrant Stada -valmistetta on saatavilla neljässä eri pakkauksessa:
- Pakkaus, jossa on yksi esitäytetty ruisku, yksi hypoderminen, steriili turvaneula (BD SafetyGlide) ja pakkausseloste.
- Pakkaus, jossa on kaksi esitäytettyä ruiskua, kaksi hypodermista, steriiliä turvaneulaa (BD SafetyGlide) ja pakkausseloste.
- Pakkaus, jossa on neljä esitäytettyä ruiskua, neljä hypodermista, steriiliä turvaneulaa (BD SafetyGlide) ja pakkausseloste.
- Pakkaus, jossa on kuusi esitäytettyä ruiskua, kuusi hypodermista, steriiliä turvaneulaa (BD Safety Glide) ja pakkausseloste.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas, väritön tai keltainen, öljyinen ja paksu liuos, jossa ei ole näkyviä partikkeleita.
Käyttö- ja käsittelyohjeet
Valmisteen käsittely ja hävittäminen tulee tapahtua antineoplastisten valmisteiden tavoin noudattaen paikallisia ohjeita. Raskaana olevan terveydenhuollonammattilaisen ei tule käsitellä tai antaa Fulvestrant Stada 250 mg injektioneste, liuos, esitäytetty ruisku -valmistetta.
Käyttöohjeet
Anna injektio noudattamalla paikallisia ohjeita suuren tilavuuden lihaksensisäisille injektioille.
HUOM.: Iskiashermon läheisyyden vuoksi on varovaisuutta noudatettava, jos Fulvestrant Stada annetaan dorsogluteaaliselle alueelle (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitus: Turvaneulaa (BD SafetyGlide Shielding Hypodermic Needle) ei saa autoklavoida ennen käyttöä. Kädet tulee pitää neulan takana aina käytön ja hävityksen aikana.
Tee seuraavat toimenpiteet kummallekin ruiskulle:
- Ota lasiruisku pakkauksesta ja tarkista, ettei ruisku ole vioittunut.
- Poista turvaneulan (BD SafetyGlide) ulompi pakkausmateriaali.
- Ennen antoa on parenteraalisista liuoksista tarkistettava silmämääräisesti, ettei liuos sisällä partikkeleita tai ole värjäytynyt.
- Pidä ruisku pystyasennossa.
- Ota toisella kädellä kiinni suojakorkista, kierrä varovasti korkkia ja poista korkki. Älä koske ruiskun kärkeen, jotta se säilyy steriilinä (ks. kuva 1).
Kuva 1
- Aseta turvaneula Luer-Lock-liittimeen ja kierrä kunnes se on tiukasti kiinni (ks. kuva 2).
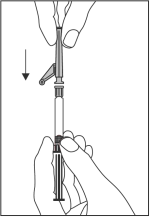
Kuva 2
- Tarkista, että neula on lukittunut Luer-Lock-liittimeen ennen kuin pidät neulaa muussa kuin pystyasennossa.
- Välttääksesi vahingoittamasta neulan kärkeä poista neulan suojus vetämällä se suoraan pois neulasta.
- Vie esitäytetty ruisku antopaikalle.
- Poista neulan hylsy.
- Poista ylimääräinen ilma ruiskusta.
-
Anna pakaralihakseen (gluteaaliselle alueelle) hitaasti (1-2 minuuttia/injektio). Käytön helpottamiseksi neulan viistokärki ja vipuvarsi on asetettu samalle puolelle (ks. kuva 3).

Kuva 3 -
Työnnä heti injektion jälkeen vipuvartta yhdellä sormen painalluksella aktivoidaksesi neulansuojausmekanismin (ks. kuva 4).
HUOM.: Aktivoi poispäin itsestäsi ja muista henkilöistä. Odota, kunnes kuulet naksahduksen, ja varmista silmämääräisesti, että neulan kärki on täysin peitossa.
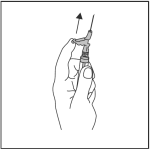
Kuva 4
Hävittäminen
Esitäytetyt ruiskut on tarkoitettu vain kertakäyttöön.
Tämä lääkevalmiste voi aiheuttaa vaaraa vesistöille. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti (ks. kohta Prekliiniset tiedot turvallisuudesta).
Korvattavuus
FULVESTRANT STADA injektioneste, liuos, esitäytetty ruisku
250 mg 2 x 5 ml
- Ylempi erityiskorvaus (100 %). Fulvestrantti: Rintasyövän hoito erityisin edellytyksin (192).
- Peruskorvaus (40 %).
ATC-koodi
L02BA03
Valmisteyhteenvedon muuttamispäivämäärä
07.08.2023
Yhteystiedot
PL 1310, Puolikkotie 8, 02230 Espoo (käyntiosoite)
00101 Helsinki
0207 416 888