
IBRANCE tabletti, kalvopäällysteinen 75 mg, 100 mg, 125 mg
Vaikuttavat aineet ja niiden määrät
IBRANCE 75 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 75 mg palbosiklibia.
IBRANCE 100 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 100 mg palbosiklibia.
IBRANCE 125 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 125 mg palbosiklibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
IBRANCE on tarkoitettu hormonireseptoripositiivisen ja HER2‑negatiivisen paikallisesti edenneen tai metastasoituneen rintasyövän hoitoon
- yhdessä aromataasinestäjän kanssa
- tai yhdessä fulvestrantin kanssa aiemmin hormonaalista hoitoa saaneille naisille (ks. kohta Farmakodynamiikka).
Pre‑ tai perimenopausaalisilla naisilla hormonaalinen hoito on yhdistettävä LHRH (luteinisoivaa hormonia vapauttava hormoni) -agonistin kanssa.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
IBRANCE-hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus
Suositeltu annostus on 125 mg palbosiklibia kerran vuorokaudessa 21 perättäisenä päivänä, minkä jälkeen palbosiklibihoidossa on 7 päivän tauko (hoito-ohjelma 3/1). Yksi hoitosykli on siten 28 vuorokautta. IBRANCE-hoitoa tulee jatkaa niin kauan kuin potilas hyötyy hoidosta kliinisesti tai kunnes ilmenee toksisuutta, joka ei ole hyväksyttävää.
Palbosiklibin kanssa yhdessä annettava aromataasinestäjä tulee antaa valmisteyhteenvedossa esitetyn suosituksen mukaisesti. Pre- tai perimenopausaalisten potilaiden hoidossa palbosiklibin ja aromataasinestäjän yhdistelmään tulee aina liittää LHRH-agonisti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Palbosiklibin kanssa yhdessä annettavan fulvestrantin suositusannos on 500 mg lihakseen ensimmäisen hoitosyklin päivinä 1 ja 15, toisen hoitosyklin päivänä 1 ja tämän jälkeen kerran kuukaudessa kunkin syklin päivänä 1. Tutustu fulvestranttivalmisteen valmisteyhteenvetoon. Pre‑ tai perimenopausaaliselle potilaalle tulee antaa LHRH-agonistia paikallisen hoitokäytännön mukaan jo ennen palbosiklibin ja fulvestrantin yhdistelmähoidon aloittamista, ja LHRH-agonistihoidon tulee jatkua yhdistelmähoidon keston ajan.
Potilasta on kehotettava ottamaan annos suurin piirtein samaan kellonaikaan joka päivä. Jos potilas oksentaa tai unohtaa annoksen, kyseisenä päivänä ei tule ottaa ylimääräistä tai unohtunutta annosta. Seuraava lääkemääräyksen mukainen annos tulee ottaa tavanomaiseen aikaan.
Annosmuutokset
IBRANCE-valmisteen annosmuutosten suositellaan perustuvan potilaan yksilölliseen turvallisuuteen ja sietokykyyn.
Joidenkin haittavaikutusten hallinta saattaa edellyttää annostelun tilapäistä keskeyttämistä, seuraavan hoitosyklin aloituksen viivästyttämistä ja/tai annoksen pienentämistä tai hoidon pysyvää lopettamista taulukoiden 1, 2 ja 3 annoksen pienentämistä koskevien ohjeiden mukaisesti (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Taulukko 1. Suositellut IBRANCE-annosmuutokset haittavaikutusten vuoksi
Annostaso | Annos |
Suositusannos | 125 mg/vrk |
Ensimmäinen alennettu annostaso | 100 mg/vrk |
Toinen alennettu annostaso | 75 mg/vrk* |
*Jos annosta olisi tarpeen pienentää edelleen alle 75 mg:aan/vrk, hoito tulee lopettaa. | |
Täydellinen verenkuva tulee määrittää ennen IBRANCE-hoidon aloittamista, jokaisen hoitosyklin alussa ja kahden ensimmäisen hoitosyklin aikana myös päivänä 15 sekä kliinisen tarpeen mukaan.
Jos kuuden ensimmäisen hoitosyklin aikana ilmenee vaikeusasteen ≤ 2 neutropeniaa, täydellinen verenkuva tulee määrittää 3 kuukauden välein ennen kunkin seuraavan hoitosyklin aloittamista sekä kliinisen tarpeen mukaan.
On suositeltavaa, että neutrofiilien absoluuttinen määrä (B-Neut) on ≥ 1,0 x 109/l ja verihiutalemäärä ≥ 50 x 109/l ennen IBRANCE-valmisteen antoa.
Taulukko 2. IBRANCE-annosmuutokset ja hoidon hallinta – hematologinen toksisuus
CTCAE-vaikeusaste | Annosmuutokset |
Vaikeusaste 1 tai 2 | Annosta ei tarvitse muuttaa. |
Vaikeusaste 3a | Hoitosyklin päivä 1: Keskeytä IBRANCE-hoito, kunnes haittavaikutus lievittyy vaikeusasteelle ≤ 2, ja määritä täydellinen verenkuva uudestaan 1 viikon kuluessa. Kun haittavaikutus lievittyy vaikeusasteelle ≤ 2, aloita seuraava hoitosykli samalla annoksella. Kahden ensimmäisen hoitosyklin päivä 15: Jos vaikeusasteen 3 haittavaikutus todetaan päivänä 15, jatka IBRANCE-hoitoa senhetkisellä annoksella hoitosyklin loppuun saakka ja määritä täydellinen verenkuva uudestaan päivänä 22. Jos päivänä 22 todetaan vaikeusasteen 4 haittavaikutus, ks. jäljempänä oleva annosmuutosohje koskien vaikeusasteen 4 toksisuutta. Harkitse annoksen pienentämistä, jos toipuminen vaikeusasteen 3 neutropeniasta pitkittyy (> 1 viikko) tai jos vaikeusasteen 3 neutropenia uusiutuu seuraavien hoitosyklien päivänä 1. |
Vaikeusasteen 3 B‑Neutb (≥ 0,5 − < 1,0 x 109/l) ja ≥ 38,5 °C kuume ja/tai infektio | Milloin tahansa: Keskeytä IBRANCE-hoito, kunnes haittavaikutus lievittyy vaikeusasteelle ≤ 2. Aloita hoito uudestaan yhtä tasoa pienemmällä annoksella. |
Vaikeusaste 4a | Milloin tahansa: Keskeytä IBRANCE-hoito, kunnes haittavaikutus lievittyy vaikeusasteelle ≤ 2. Aloita hoito uudestaan yhtä tasoa pienemmällä annoksella. |
Toksisuuden vaikeusasteet CTCAE (Common Terminology Criteria for Adverse Events) v4.0 ‑luokituksen mukaan. B‑Neut = neutrofiilien absoluuttinen määrä, LLN (lower limit of normal) = viitevälin alaraja a Taulukko koskee kaikkia muita hematologisia haittavaikutuksia lukuun ottamatta lymfopeniaa (ellei tähän liity kliinisiä tapahtumia esim. opportunisti-infektioita). b B‑Neut: vaikeusaste 1: ≥ 1,5 x 109/l – < LLN; vaikeusaste 2: ≥ 1,0 – < 1,5 x 109/l; vaikeusaste 3: ≥ 0,5 – < 1,0 x 109/l; vaikeusaste 4: < 0,5 x 109/l. | |
Taulukko 3. IBRANCE-annosmuutokset ja hoidon hallinta – muu kuin hematologinen toksisuus
CTCAE-vaikeusaste | Annosmuutokset |
Vaikeusaste 1 tai 2 | Annosta ei tarvitse muuttaa. |
Vaikeusasteen ≥ 3 muu kuin hematologinen toksisuus (jos toksisuus jatkuu huolimatta siihen annetusta hoidosta) | Keskeytä hoito, kunnes oireet lievittyvät
Aloita hoito uudestaan yhtä tasoa pienemmällä annoksella. |
Toksisuuden vaikeusasteet CTCAE (Common Terminology Criteria for Adverse Events) v4.0 ‑luokituksen mukaan. | |
IBRANCE-hoito tulee lopettaa pysyvästi, jos potilaalla todetaan vaikea interstitiaalinen keuhkosairaus (ILD)/pneumoniitti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Erityisryhmät
Iäkkäät potilaat
IBRANCE-annoksen muuttaminen ei ole tarpeen ≥ 65-vuotiaiden potilaiden hoidossa (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
IBRANCE-annoksen muuttaminen ei ole tarpeen, jos potilaalla on lievä tai keskivaikea maksan vajaatoiminta (Child-Pugh A ja B). Jos potilaalla on vaikea maksan vajaatoiminta (Child-Pugh C), IBRANCE-hoidon suositusannos on 75 mg kerran vuorokaudessa hoito-ohjelmalla 3/1 (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Munuaisten vajaatoiminta
IBRANCE-annoksen muuttaminen ei ole tarpeen, jos potilaalla on lievä, keskivaikea tai vaikea munuaisten vajaatoiminta (laskennallinen glomerulussuodosnopeus, eGFR ≥ 15 ml/min). Saatavilla olevat tiedot hemodialyysia tarvitsevien potilaiden hoidosta ovat riittämättömiä annossuositusten antamiseksi (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Ei ole asianmukaista käyttää IBRANCE-valmistetta pediatrisille potilaille rintasyövän hoitoon. IBRANCE-valmisteen tehoa lapsille ja < 18 vuoden ikäisille nuorille ei ole osoitettu. Saatavissa olevat tiedot on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka.
Antotapa
IBRANCE-tabletit otetaan suun kautta. Tabletit voidaan ottaa ruoan kanssa tai tyhjään mahaan (ks. kohta Farmakokinetiikka). Palbosiklibia ei tule ottaa yhdessä greippihedelmän tai greippimehun kanssa (ks. kohta Yhteisvaikutukset).
IBRANCE-tabletit on nieltävä kokonaisena (niitä ei saa pureskella, murskata eikä jakaa ennen nielemistä). Tablettia ei saa ottaa, jos se on rikkoutunut, murtunut tai se ei muutoin ole ehjä.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Mäkikuismaa sisältävien valmisteiden käyttö (ks. kohta Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Pre-/perimenopausaaliset potilaat
Aromataasinestäjien vaikutusmekanismista johtuen munasarjat tulee poistaa tai niiden toiminta tulee estää LHRH-agonistilla, kun IBRANCE-valmistetta annetaan yhdessä aromataasinestäjän kanssa pre-/perimenopausaalisille potilaille. Palbosiklibin ja fulvestrantin yhdistelmähoitoa on tutkittu pre-/perimenopausaalisten potilaiden hoidossa vain LHRH-agonistiin yhdistettynä.
Henkeä uhkaava viskeraalinen metastasointi
Palbosiklibin tehoa ja turvallisuutta ei ole tutkittu potilailla, joilla on henkeä uhkaava viskeraalinen metastasointi (ks. kohta Farmakodynamiikka).
Hematologiset häiriöt
Jos potilaalle kehittyy vaikeusasteen 3 tai 4 neutropenia, annostelun tilapäistä keskeyttämistä, annoksen pienentämistä tai seuraavan hoitosyklin aloituksen viivästyttämistä suositellaan. Potilasta tulee seurata asianmukaisesti (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Interstitiaalinen keuhkosairaus/pneumoniitti
Potilailla, joita hoidetaan IBRANCE-valmisteella hormonaaliseen hoitoon yhdistettynä, voi ilmetä vaikea, henkeä uhkaava tai kuolemaan johtava interstitiaalinen keuhkosairaus (ILD)/pneumoniitti.
Kliinisissä tutkimuksissa (PALOMA‑1, PALOMA‑2, PALOMA‑3) 1,4 %:lla IBRANCE-hoitoa saaneista potilaista raportoitiin jonkin vaikeusasteen ILD/pneumoniitti. Vaikeusasteen 3 ILD/pneumoniitti todettiin 0,1 %:lla potilaista. Yhtään vaikeusasteen 4 tai kuolemaan johtanutta tapausta ei raportoitu. Valmisteen markkinoille tulon jälkeen on raportoitu ILD-/pneumoniittitapauksia, mukaan lukien kuolemaan johtaneita tapauksia (ks. kohta Haittavaikutukset).
Potilasta tulee tarkkailla ILD:hen/pneumoniittiin viittaavien keuhko-oireiden (esim. hypoksia, yskä, hengenahdistus) varalta. Jos potilaalla ilmenee uusia tai pahenevia hengitysoireita ja hänelle epäillään kehittyneen ILD/pneumoniitti, IBRANCE-hoito tulee keskeyttää välittömästi ja potilaan tila tulee arvioida. IBRANCE-hoito tulee lopettaa pysyvästi, jos potilaalla todetaan vaikea ILD tai pneumoniitti (ks. kohta Annostus ja antotapa).
Infektiot
IBRANCE-valmisteella on myelosuppressiivisia ominaisuuksia, joten se saattaa altistaa potilaan infektioille.
Infektioita on raportoitu satunnaistetuissa tutkimuksissa enemmän IBRANCE-hoitoa saaneilla potilailla kuin vertailuhoitoa saaneilla potilailla. Vaikeusasteen 3 infektioita todettiin 5,6 %:lla ja vaikeusasteen 4 infektioita 0,9 %:lla potilaista, jotka saivat IBRANCE-valmistetta osana eri yhdistelmähoitoja (ks. kohta Haittavaikutukset).
Potilaita tulee seurata infektion merkkien ja oireiden varalta ja hoitaa asianmukaisesti (ks. kohta Annostus ja antotapa).
Lääkärin on kehotettava potilasta ilmoittamaan heti, jos kuumetta ilmenee.
Laskimotromboembolia
Laskimotromboemboliatapauksia raportoitiin potilailla, jotka saivat IBRANCE-hoitoa (ks. kohta Haittavaikutukset). Potilaita tulee seurata syvän laskimotromboosin ja keuhkoembolian merkkien ja oireiden varalta ja hoitaa asianmukaisesti.
Maksan vajaatoiminta
IBRANCE-hoitoa tulee antaa varoen potilaille, joilla on keskivaikea tai vaikea maksan vajaatoiminta. Toksisuuden merkkejä tulee seurata tarkoin (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Munuaisten vajaatoiminta
IBRANCE-hoitoa tulee antaa varoen potilaille, joilla on keskivaikea tai vaikea munuaisten vajaatoiminta. Toksisuuden merkkejä tulee seurata tarkoin (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Samanaikainen hoito CYP3A4:n estäjillä tai induktoreilla
Voimakkaat CYP3A4:n estäjät voivat lisätä haittavaikutuksia (ks. kohta Yhteisvaikutukset). Voimakkaiden CYP3A:n estäjien samanaikaista käyttöä palbosiklibin kanssa tulee välttää. Samanaikaista käyttöä tulee harkita vasta sen jälkeen, kun mahdolliset hyödyt ja riskit on huolellisesti arvioitu. Jos samanaikaista käyttöä voimakkaan CYP3A:n estäjän kanssa ei voida välttää, IBRANCE-annos tulee pienentää 75 mg:aan kerran vuorokaudessa. Kun voimakkaan CYP3A:n estäjän käyttö lopetetaan, IBRANCE-annos tulee suurentaa (estäjän 3–5 puoliintumisajan jälkeen) takaisin määrään, jota potilas sai ennen kuin CYP3A:n estäjän käyttö aloitettiin (ks. kohta Yhteisvaikutukset).
CYP3A:n induktorien samanaikainen anto voi pienentää palbosiklibialtistusta ja siten lisätä riskiä tehon puutteesta. Siksi palbosiklibin käyttöä samanaikaisesti voimakkaiden CYP3A4:n induktorien kanssa tulee välttää. Käytettäessä samanaikaisesti palbosiklibia ja kohtalaisia CYP3A:n induktoreja annosten muuttaminen ei ole tarpeen (ks. kohta Yhteisvaikutukset).
Naiset, jotka voivat tulla raskaaksi, ja heidän partnerinsa
Naisten, jotka voivat tulla raskaaksi, ja heidän miespuolisten kumppaniensa on käytettävä luotettavaa raskauden ehkäisymenetelmää IBRANCE-hoidon aikana (ks. kohta Raskaus ja imetys).
Yhteisvaikutukset
Palbosiklibi metaboloituu ensisijaisesti CYP3A:n ja sulfotransferaasi 2A1 (SULT2A1) ‑entsyymin välityksellä. Palbosiklibi on CYP3A:n heikko ajasta riippuvainen estäjä in vivo.
Muiden lääkeaineiden vaikutukset palbosiklibin farmakokinetiikkaan
CYP3A:n estäjien vaikutus
Itrakonatsolin anto toistuvina 200 mg:n annoksina yhdessä palbosiklibin 125 mg:n kerta-annoksen kanssa suurensi palbosiklibin kokonaisaltistusta (AUCinf) noin 87 % ja huippupitoisuutta (Cmax) noin 34 % verrattuna pelkän palbosiklibin antoon 125 mg:n kerta-annoksena.
Muun muassa seuraavien voimakkaiden CYP3A:n estäjien samanaikaista käyttöä on vältettävä: klaritromysiini, indinaviiri, itrakonatsoli, ketokonatsoli, lopinaviiri/ritonaviiri, nefatsodoni, nelfinaviiri, posakonatsoli, sakinaviiri, telapreviiri, telitromysiini, vorikonatsoli sekä greippihedelmä ja greippimehu (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Annosten muuttaminen ei ole tarpeen, kun palbosiklibihoitoa annetaan samanaikaisesti heikkojen ja kohtalaisten CYP3A:n estäjien kanssa.
CYP3A:n induktorien vaikutus
Rifampisiinin anto toistuvina 600 mg:n annoksina yhdessä palbosiklibin 125 mg:n kerta-annoksen kanssa pienensi palbosiklibin AUCinf‑arvoa 85 % ja Cmax‑arvoa 70 % verrattuna pelkän palbosiklibin antoon 125 mg:n kerta-annoksena.
Muun muassa seuraavien voimakkaiden CYP3A:n induktorien samanaikaista käyttöä on vältettävä: karbamatsepiini, entsalutamidi, fenytoiini, rifampisiini ja mäkikuisma (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Kohtalaisen CYP3A:n induktorin modafiniilin anto toistuvina 400 mg:n päivittäisinä annoksina yhdessä IBRANCE 125 mg:n kerta-annoksen kanssa pienensi palbosiklibin AUCinf‑arvoa 32 % ja Cmax‑arvoa 11 % verrattuna pelkän IBRANCE-valmisteen antoon 125 mg:n kerta-annoksena. Annosten muuttaminen ei ole tarpeen, kun palbosiklibihoitoa annetaan samanaikaisesti kohtalaisten CYP3A:n induktorien kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Mahahapon eritystä vähentävien aineiden vaikutus
Toistuvina annoksina annettu protonipumpun estäjä rabepratsoli ei vaikuttanut paasto-olosuhteissa palbosiklibin imeytymisnopeuteen eikä imeytyvään määrään, kun sitä annettiin yhdessä IBRANCE 125 mg tabletin kerta-annoksen kanssa verrattuna yksinään annettuun IBRANCE 125 mg tabletin kerta-annokseen.
Koska H2‑reseptorin salpaajat ja paikallisesti vaikuttavat antasidit laskevat mahanesteen pH:ta vähemmän kuin protonipumpun estäjät, H2‑reseptorin salpaajilla ja antasideilla ei odoteta olevan kliinisesti merkittävää vaikutusta palbosiklibialtistukseen.
Palbosiklibin vaikutukset muiden lääkeaineiden farmakokinetiikkaan
Palbosiklibi on CYP3A:n heikko ajasta riippuvainen estäjä päivittäisillä 125 mg:n annoksilla, kun vakaa tila (steady state) on saavutettu. Palbosiklibin anto toistuvina annoksina yhdessä midatsolaamin kanssa suurensi midatsolaamin AUCinf‑arvoa 61 % ja Cmax‑arvoa 37 % verrattuna midatsolaamin antoon yksinään.
Herkkien CYP3A:n substraattien, joilla on kapea terapeuttinen indeksi (esim. alfentaniili, siklosporiini, dihydroergotamiini, ergotamiini, everolimuusi, fentanyyli, pimotsidi, kinidiini, sirolimuusi ja takrolimuusi), annosta voidaan joutua pienentämään annettaessa niitä samanaikaisesti IBRANCE-valmisteen kanssa, koska IBRANCE saattaa suurentaa niiden altistusta.
Yhteisvaikutus palbosiklibin ja letrotsolin välillä
Tiedot rintasyöpäpotilaiden kliinisen tutkimuksen yhteisvaikutuksia arvioivasta osiosta osoittivat, että palbosiklibin ja letrotsolin välillä ei ollut yhteisvaikutuksia annettaessa näitä sisältäviä lääkevalmisteita samanaikaisesti.
Tamoksifeenin vaikutus palbosiklibialtistukseen
Tiedot yhteisvaikutustutkimuksesta, johon otettiin terveitä miespuolisia tutkittavia, osoittivat palbosiklibialtistusten olevan toisiinsa verrattavissa, kun palbosiklibia annettiin kerta-annoksena joko yksinään tai toistuvien tamoksifeeniannosten kanssa.
Yhteisvaikutus palbosiklibin ja fulvestrantin välillä
Tiedot rintasyöpäpotilaiden kliinisestä tutkimuksesta osoittivat, että palbosiklibin ja fulvestrantin välillä ei ollut kliinisesti merkittäviä yhteisvaikutuksia annettaessa näitä sisältäviä lääkevalmisteita samanaikaisesti.
Yhteisvaikutus palbosiklibin ja suun kautta otettavien ehkäisyvalmisteiden välillä
Palbosiklibin ja suun kautta otettavien ehkäisyvalmisteiden välillä ei ole tehty yhteisvaikutustutkimuksia (ks. kohta Raskaus ja imetys).
In vitro ‑tutkimukset kuljettajaproteiinien kanssa
In vitro -tutkimuksista saadut tiedot viittaavat siihen, että palbosiklibi estää suoliston P‑glykoproteiinin (P‑gp) ja rintasyövän resistenssiproteiinin (breast cancer resistance protein, BCRP) välittämää kuljetusta. Siksi palbosiklibin anto yhdessä sellaisten lääkeaineiden kanssa, jotka ovat joko P‑gp:n substraatteja (esim. digoksiini, dabigatraani, kolkisiini) tai BCRP:n substraatteja (esim. pravastatiini, rosuvastatiini, fluvastatiini, sulfasalatsiini), saattaa lisätä näiden terapeuttista vaikutusta ja haittavaikutuksia.
In vitro ‑tietojen perusteella palbosiklibi saattaa estää orgaanisen kationin kuljettaja 1:n (organic cationic transporter 1, OCT1) soluunottoa ja siten lisätä altistusta lääkevalmisteille, jotka ovat tämän kuljettajaproteiinin substraatteja (esim. metformiini).
Yhteisvaikutus palbosiklibin ja statiinien välillä
Palbosiklibin samanaikainen käyttö sellaisten statiinien kanssa, jotka ovat CYP3A4:n ja/tai BCRP:n substraatteja, voi lisätä rabdomyolyysin riskiä plasman statiinipitoisuuden kohoamisen vuoksi. Palbosiklibin ja simvastatiinin tai atorvastatiinin samanaikaisen käytön jälkeen on raportoitu rabdomyolyysiä, mukaan lukien kuolemaan johtaneita tapauksia.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehillä ja naisilla
Tätä lääkevalmistetta saavien naisten, jotka voivat tulla raskaaksi, ja heidän miespuolisten kumppaniensa on käytettävä asianmukaisia raskauden ehkäisymenetelmiä (esim. kahta erilaista estemenetelmää) hoidon aikana ja vähintään 3 viikon (naiset) tai 14 viikon (miehet) ajan hoidon päättymisen jälkeen (ks. kohta Yhteisvaikutukset).
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja palbosiklibin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). IBRANCE-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Palbosiklibin vaikutusta maidontuotantoon, sen erittymistä rintamaitoon tai vaikutuksia imeväiseen ei ole tutkittu kliinisesti tai eläinkokeissa. Ei tiedetä, erittyykö palbosiklibi ihmisen rintamaitoon. Palbosiklibihoidon aikana ei saa imettää.
Hedelmällisyys
Prekliinisissä lisääntymistutkimuksissa ei ilmennyt vaikutuksia naarasrottien kiimakiertoon eikä uros‑ ja naarasrottien paritteluun ja hedelmällisyyteen. Ihmisen hedelmällisyyteen liittyviä kliinisiä tietoja ei ole kuitenkaan saatu. Prekliinisissä turvallisuustutkimuksissa todettujen urosten lisääntymiselimiin liittyvien löydösten (kivesten siementiehyiden rappeutuminen, lisäkivesten hypospermia [siemennesteen liian vähäinen määrä], siittiöiden liikkuvuuden ja siittiötiheyden väheneminen sekä eturauhasen erityksen väheneminen) perusteella palbosiklibihoito saattaa vaarantaa miehen hedelmällisyyden (ks. kohta Prekliiniset tiedot turvallisuudesta). Siten miesten kannattaa harkita siemennesteen talteenottoa ennen IBRANCE-hoidon aloittamista.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
IBRANCE-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. IBRANCE voi kuitenkin aiheuttaa väsymystä, joten autoa ajettaessa ja koneita käytettäessä on noudatettava varovaisuutta.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
IBRANCE-hoidon yleinen turvallisuusprofiili perustuu yhdistettyihin tietoihin satunnaistetuista kliinisistä tutkimuksista, joissa oli 872 potilasta. Potilailla oli hormonireseptoripositiivinen ja HER2‑negatiivinen edennyt tai metastasoitunut rintasyöpä, ja he saivat palbosiklibin ja hormonaalisen hoidon yhdistelmähoitoa (527 potilasta sai letrotsolia ja 345 potilasta sai fulvestranttia).
Satunnaistetuissa kliinisissä tutkimuksissa palbosiklibia saaneilla potilailla ilmoitetut yleisimmät (≥ 20 %) minkä tahansa vaikeusasteen haittavaikutukset olivat neutropenia, infektiot, leukopenia, väsymys, pahoinvointi, suutulehdus, anemia, ripuli, hiustenlähtö ja trombosytopenia. Yleisimmät (≥ 2 %) palbosiklibiin liittyneet vaikeusasteen ≥ 3 haittavaikutukset olivat neutropenia, leukopenia, infektiot, anemia, kohonnut ASAT (aspartaattiaminotransferaasi) ‑arvo, väsymys ja kohonnut ALAT (alaniiniaminotransferaasi) ‑arvo.
Annosta pienennettiin tai muutettiin jonkin haittavaikutuksen vuoksi 38,4 %:lla potilaista, jotka saivat IBRANCE-hoitoa satunnaistetuissa kliinisissä tutkimuksissa osana jotain yhdistelmähoitoa.
Hoito lopetettiin pysyvästi jonkin haittavaikutuksen vuoksi 5,2 %:lla potilaista, jotka saivat IBRANCE-hoitoa satunnaistetuissa kliinisissä tutkimuksissa osana jotain yhdistelmähoitoa.
Haittavaikutustaulukko
Taulukossa 4 luetellut haittavaikutukset ovat peräisin 3 satunnaistetun tutkimuksen yhdistetyistä tiedoista. Yhdistettyjen tietojen perusteella palbosiklibihoidon mediaanikesto lopullisen kokonaiselinaika-analyysin ajankohtana oli 14,8 kuukautta.
Taulukossa 5 luetellut laboratorioarvojen poikkeamat ovat peräisin 3 satunnaistetun tutkimuksen yhdistetyistä tiedoista.
Haittavaikutukset on lueteltu elinjärjestelmien ja esiintymistiheyksien mukaan. Esiintymistiheysluokat on määritelty seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10) ja melko harvinaiset (≥ 1/1 000, < 1/100). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 4. Haittavaikutukset perustuen 3 satunnaistetun tutkimuksen yhdistettyihin tietoihin (n = 872) ja valmisteen markkinoille tulon jälkeiseen käyttöön | |||
Elinjärjestelmäluokka Esiintymistiheys Suositeltu termia | Kaikki vaikeusasteet n (%) | Vaikeusaste 3 n (%) | Vaikeusaste 4 n (%) |
Infektiot Hyvin yleiset | |||
| Infektiotb | 516 (59,2) | 49 (5,6) | 8 (0,9) |
Veri ja imukudos Hyvin yleiset | |||
| Neutropeniac | 716 (82,1) | 500 (57,3) | 97 (11,1) |
| Leukopeniad | 424 (48,6) | 254 (29,1) | 7 (0,8) |
| Anemiae | 258 (29,6) | 45 (5,2) | 2 (0,2) |
| Trombosytopeniaf | 194 (22,2) | 16 (1,8) | 4 (0,5) |
| Yleiset | |||
| Kuumeinen neutropenia | 12 (1,4) | 10 (1,1) | 2 (0,2) |
| Aineenvaihdunta ja ravitsemus Hyvin yleiset | |||
| Ruokahalun heikkeneminen | 152 (17,4) | 8 (0,9) | 0 (0,0) |
| Hermosto | |||
| Yleiset | |||
| Makuhäiriö | 79 (9,1) | 0 (0,0) | 0 (0,0) |
| Silmät | |||
| Yleiset | |||
| Näön hämärtyminen | 48 (5,5) | 1 (0,1) | 0 (0,0) |
| Kyynelerityksen lisääntyminen | 59 (6,8) | 0 (0,0) | 0 (0,0) |
| Silmien kuivuminen | 36 (4,1) | 0 (0,0) | 0 (0,0) |
| Verisuonisto | |||
| Yleiset | |||
| Laskimotromboemboliaj | 28 (3,2) | 11 (1,3) | 7 (0,8) |
Hengityselimet, rintakehä ja välikarsina Yleiset | |||
| Nenäverenvuoto | 77 (8,8) | 0 (0,0) | 0 (0,0) |
| ILD/pneumoniittii | 12 (1,4) | 1 (0,1) | 0 (0,0) |
Ruoansulatuselimistö Hyvin yleiset | |||
| Suutulehdusg | 264 (30,3) | 8 (0,9) | 0 (0,0) |
| Pahoinvointi | 314 (36,0) | 5 (0,6) | 0 (0,0) |
| Ripuli | 238 (27,3) | 9 (1,0) | 0 (0,0) |
| Oksentelu | 165 (18,9) | 6 (0,7) | 0 (0,0) |
Iho ja ihonalainen kudos Hyvin yleiset | |||
| Ihottumah | 158 (18,1) | 7 (0,8) | 0 (0,0) |
| Hiustenlähtö | 234 (26,8) | N/A | N/A |
| Ihon kuivuminen | 93 (10,7) | 0 (0,0) | 0 (0,0) |
| Yleiset | |||
| Käsi-jalkaoireyhtymä | 16 (1,8) | 0 (0,0) | 0 (0,0) |
| Melko harvinaiset | |||
| Ihon lupus erythematosus (Cutaneous Lupus Erythematosus, CLE) | 1 (0,1) | 0 (0,0) | 0 (0,0) |
| Erythema multiforme | 1 (0,1) | 0 (0,0) | 0 (0,0) |
Yleisoireet ja antopaikassa todettavat haitat Hyvin yleiset | |||
| Väsymys | 362 (41,5) | 23 (2,6) | 2 (0,2) |
| Voimattomuus | 118 (13,5) | 14 (1,6) | 1 (0,1) |
| Kuume | 115 (13,2) | 1 (0,1) | 0 (0,0) |
Tutkimukset Hyvin yleiset | |||
| Kohonnut ALAT-arvo | 92 (10,6) | 18 (2,1) | 1 (0,1) |
| Kohonnut ASAT-arvo | 99 (11,4) | 25 (2,9) | 0 (0,0) |
| Yleiset | |||
| Veren kohonnut kreatiniinipitoisuus | 57 (6,5) | 3 (0,3) | 2 (0,2) |
ALAT = alaniiniaminotransferaasi, ASAT = aspartaattiaminotransferaasi, ILD = interstitiaalinen keuhkosairaus, n = potilaiden lukumäärä, N/A (not applicable) = ei sovellu
a Suositellut termit MedDRA 17.1 ‑version mukaan.
b Infektiot sisältää kaikki suositellut termit, jotka kuuluvat elinjärjestelmäluokkaan ”Infektiot”.
c Neutropenia sisältää seuraavat suositellut termit: neutropenia, neutrofiilimäärän väheneminen.
d Leukopenia sisältää seuraavat suositellut termit: leukopenia, valkosolumäärän väheneminen.
e Anemia sisältää seuraavat suositellut termit: anemia, hemoglobiinin lasku, hematokriitin lasku.
f Trombosytopenia sisältää seuraavat suositellut termit: trombosytopenia, verihiutalemäärän väheneminen.
g Suutulehdus sisältää seuraavat suositellut termit: aftainen suutulehdus, huulitulehdus, kielitulehdus, kielikipu, suun haavautuminen, limakalvotulehdus, suukipu, suunielun oireet, suunielun kipu, suutulehdus.
h Ihottuma sisältää seuraavat suositellut termit: ihottuma, makulopapulaarinen ihottuma, kutiava ihottuma, erytematoottinen ihottuma, papulaarinen ihottuma, ihotulehdus, aknea muistuttava ihottuma, äkillinen toksinen ihottuma.
i ILD/pneumoniitti sisältää kaikki ilmoitetut suositellut termit, jotka kuuluvat interstitiaalista keuhkosairautta (tarkasti) kuvaaviin standardoituihin MedDRA-termeihin.
j Laskimotromboembolia sisältää seuraavat suositellut termit: keuhkoembolia, embolia, syvä laskimotromboosi, perifeerinen embolia, tromboosi.
Taulukko 5. Laboratorioarvojen poikkeamat perustuen 3 satunnaistetun tutkimuksen
yhdistettyihin tietoihin (n = 872)
| IBRANCE + letrotsoli tai IBRANCE + fulvestrantti | Vertailuhoito (pelkkä letrotsoli tai fulvestrantti) | |||||
| Laboratorioarvojen poikkeamat | Kaikki vaikeus-asteet % | Vaikeus-aste 3 % | Vaikeus-aste 4 % | Kaikki vaikeus-asteet % | Vaikeus-aste 3 % | Vaikeus-aste 4 % |
| Valkosolumäärän väheneminen | 97,4 | 41,8 | 1,0 | 26,2 | 0,2 | 0,2 |
| Neutrofiilimäärän väheneminen | 95,6 | 57,5 | 11,7 | 17,0 | 0,9 | 0,6 |
| Veren kohonnut kreatiniinipitoisuus | 95,5 | 1,6 | 0,3 | 86,8 | 0,0 | 0,0 |
| Anemia | 80,1 | 5,6 | N/A | 42,1 | 2,3 | N/A |
| Verihiutalemäärän väheneminen | 65,2 | 1,8 | 0,5 | 13,2 | 0,2 | 0,0 |
| Kohonnut ASAT-arvo | 55,5 | 3,9 | 0,0 | 43,3 | 2,1 | 0,0 |
| Kohonnut ALAT-arvo | 46,1 | 2,5 | 0,1 | 33,2 | 0,4 | 0,0 |
ASAT = aspartaattiaminotransferaasi, ALAT = alaniiniaminotransferaasi, n = potilaiden lukumäärä, N/A = ei sovellu
Huom.: Laboratoriotulokset on luokiteltu NCI CTCAE (National Cancer Institute, Common Terminology Criteria for Adverse Events) ‑vaikeusasteluokituksen v4.0 mukaan.
Valikoitujen haittavaikutusten kuvaus
Minkä tahansa vaikeusasteen neutropeniaa raportoitiin 716 potilaalla (82,1 %), jotka saivat IBRANCE-hoitoa osana jotain yhdistelmähoitoa. Vaikeusasteen 3 neutropeniaa raportoitiin 500 potilaalla (57,3 %) ja vaikeusasteen 4 neutropeniaa 97 potilaalla (11,1 %) (ks. taulukko 4).
Kolmessa satunnaistetussa kliinisessä tutkimuksessa mediaaniaika ensimmäisen minkä tahansa vaikeusasteen neutropenian ilmaantumiseen oli 15 päivää (12–700 päivää). Vaikeusasteen ≥ 3 neutropenian mediaanikesto oli 7 päivää.
Kuumeista neutropeniaa on raportoitu 0,9 %:lla potilaista, jotka saivat IBRANCE-valmisteen ja fulvestrantin yhdistelmähoitoa, ja 1,7 %:lla potilaista, jotka saivat palbosiklibin ja letrotsolin yhdistelmähoitoa.
IBRANCE-hoitoa kliinisessä ohjelmassa saaneista noin 2 %:lla on raportoitu kuumeista neutropeniaa.
Pediatriset potilaat
Palbosiklibin ja solunsalpaajahoidon yhdistelmää on arvioitu A5481092-tutkimuksessa 79 pediatrisella potilaalla, joilla oli kiinteitä kasvaimia, mukaan lukien uusiutunut tai hoitoon reagoimaton Ewingin sarkooma (EWS) (ks. kohta Farmakodynamiikka). Palbosiklibin turvallisuusprofiili oli tässä pediatrisessa potilasryhmässä yhdenmukainen aikuisen potilasjoukon tunnetun turvallisuusprofiilin kanssa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Palbosiklibin yliannostuksessa saattaa ilmetä sekä maha-suolikanavan (esim. pahoinvointi, oksentelu) että hematologista (esim. neutropenia) toksisuutta. Potilaalle tulee antaa yleistä elintoimintoja tukevaa hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut syöpälääkkeet, proteiinikinaasin estäjät, ATC-koodi: L01EF01
Vaikutusmekanismi
Palbosiklibi on hyvin selektiivinen sykliinistä riippuvaisten kinaasien (cyclin-dependent kinases, CDK) 4 ja 6 reversiibeli estäjä. Sykliini D1 ja CDK4/6 ovat useiden soluproliferaatioon johtavien viestintäreittien alavirrassa.
Farmakodynaamiset vaikutukset
Palbosiklibi vähentää soluproliferaatiota estämällä CDK4/6:n toimintaa ja siten solusyklin etenemistä G1‑faasista S‑faasiin. Kun palbosiklibia testattiin molekyylitasolla tyypitetyssä rintasyövän solulinjasarjassa, sillä todettiin merkittävää aktiivisuutta luminaalisissa rintasyövissä, etenkin estrogeenireseptori (ER) ‑positiivisissa rintasyövissä. Tutkituissa solulinjoissa retinoblastoomaproteiinin (RB) puute liittyi palbosiklibin heikentyneeseen aktiivisuuteen. Tuoreilla kasvainnäytteillä tehdyssä jatkotutkimuksessa ei kuitenkaan todettu yhteyttä RB1:n ilmentymisen ja kasvainvasteen välillä. Yhteyttä ei myöskään todettu, kun vastetta palbosiklibille tutkittiin in vivo potilasperäisissä siirremalleissa. Saatavilla olevat kliiniset tiedot on raportoitu kliinistä tehoa ja turvallisuutta koskevassa osiossa (ks. kohta Farmakodynamiikka).
Sydämen elektrofysiologia
Palbosiklibin vaikutusta sykkeen mukaan korjattuun QT‑aikaan (QTc) arvioitiin 77 edennyttä rintasyöpää sairastavan potilaan farmakokineettisista tiedoista käyttäen ajan suhteen kaltaistettua EKG‑muutosta lähtötilanteesta. Palbosiklibi ei pidentänyt QTc‑aikaa kliinisesti merkittävässä määrin suositusannoksella 125 mg kerran vuorokaudessa (hoito-ohjelma 3/1).
Kliininen teho ja turvallisuus
Satunnaistettu vaiheen 3 PALOMA‑2‑tutkimus: IBRANCE yhdistettynä letrotsoliin
Palbosiklibin ja letrotsolin yhdistelmähoidon tehoa verrattiin lumelääkkeen ja letrotsolin yhdistelmään kansainvälisessä, satunnaistetussa monikeskustutkimuksessa, joka oli kaksoissokkoutettu ja lumekontrolloitu. Tutkimus toteutettiin rinnakkaisryhmien asetelmalla (potilaiden siirtymistä tutkimushaarasta toiseen ei sallittu). Tutkimukseen osallistuneilla naisilla oli leikkaus- tai sädehoitoon soveltumaton paikallisesti edennyt ER‑positiivinen ja HER2‑negatiivinen rintasyöpä, jota ei voitu hoitaa kuratiivisesti, tai metastasoitunut ER‑positiivinen ja HER2‑negatiivinen rintasyöpä. Potilaat eivät olleet saaneet aikaisempaa systeemistä hoitoa edenneeseen rintasyöpään.
Yhteensä 666 postmenopausaalista potilasta satunnaistettiin suhteessa 2:1 saamaan joko palbosiklibin ja letrotsolin yhdistelmähoitoa tai lumelääkkeen ja letrotsolin yhdistelmää. Tutkimuspotilaat ositettiin seuraavien tekijöiden mukaan: kasvaimen sijainti (viskeraalinen vs muu kuin viskeraalinen), taudista vapaa aika (disease free interval, DFI) (neo)adjuvanttihoidon päättymisestä sairauden uusiutumiseen (de novo metastasoitunut vs DFI ≤ 12 kuukautta vs DFI > 12 kuukautta) ja syövän aiemmat (neo)adjuvanttihoidot (aiempi hormonaalinen hoito vs ei aiempaa hormonaalista hoitoa). Potilaat, joilla oli oireita aiheuttavia viskeraalisia metastaaseja ja riski henkeä uhkaaviin komplikaatioihin lyhyellä aikavälillä (mukaan lukien potilaat, joilla keuhko- tai sydänpussin tai vatsakalvon nestekertymä oli hyvin runsasta eikä hallittavissa, sekä potilaat, joilla oli keuhkojen lymfangiitti tai tuumorimassaa yli 50 % maksan tilavuudesta), eivät täyttäneet tutkimuksen sisäänottokriteerejä.
Määrättyä tutkimushoitoa jatkettiin, kunnes ensimmäisen kerran ilmeni jokin seuraavista: tauti eteni objektiivisesti arvioiden, oireet pahenivat, toksisuus ei ollut hyväksyttävissä, potilas kuoli tai perui osallistumissuostumuksensa. Potilaiden siirtymistä tutkimushaarasta toiseen (cross-over) ei sallittu.
Potilaiden lähtötilanteen demografiset ominaisuudet ja ennusteeseen liittyvät tekijät olivat keskenään tasapainossa palbosiklibin ja letrotsolin yhdistelmää tai lumelääkkeen ja letrotsolin yhdistelmää saaneiden tutkimusryhmien välillä. Tutkimuspotilaiden mediaani ikä oli 62 vuotta (vaihteluväli 28–89 vuotta). Ennen edenneen rintasyövän diagnoosia 48,3 % potilaista oli saanut solunsalpaajahoitoa ja 56,3 % hormonaalista hoitoa (neo)adjuvanttihoitona, kun taas 37,2 % potilaista ei ollut saanut aiempaa systeemistä hoitoa (neo)adjuvanttiasetelmassa. Valtaosalla potilaista (97,4 %) syöpä oli lähtötilanteessa metastasoitunut; 49,2 %:lla potilaista oli viskeraalisia metastaaseja ja 23,6 %:lla potilaista pelkästään luustometastaaseja.
Tutkimuksen ensisijainen päätetapahtuma oli tutkijoiden RECIST (Response Evaluation Criteria in Solid Tumours) ‑kriteereiden mukaisesti (v. 1.1) arvioima taudin etenemisestä vapaa elinaika (progression-free survival, PFS). Tehon toissijaisia päätetapahtumia olivat objektiivinen vaste (objective response, OR), kliinisen hyödyn saaneiden osuus (clinical benefit rate, CBR), turvallisuus ja muutokset elämänlaadussa.
Kun tiedonkeruu katkaistiin 26.2.2016, tutkimuksessa saavutettiin taudin etenemisestä vapaan elinajan piteneminen, mikä oli tutkimuksen ensisijainen tavoite. Riskitiheyksien suhde (hazard ratio, HR) oli 0,576 (95 %:n luottamusväli 0,46–0,72) palbosiklibin ja letrotsolin yhdistelmähoidon eduksi; ositetun log-rank-testin yksisuuntainen p‑arvo oli < 0,000001. Ensisijaisen ja toissijaisten päätetapahtumien päivitetty analyysi tehtiin, kun seurantaa oli jatkettu vielä 15 kuukautta (tiedonkeruu katkaistiin 31.5.2017). Taudin etenemisestä vapaata elinaikaa koskevia tapahtumia todettiin yhteensä 405: 245 tapahtumaa palbosiklibin ja letrotsolin yhdistelmää saaneilla (55,2 %:lla potilaista) ja 160 tapahtumaa vertailuhoitoa saaneilla potilailla (72,1 %:lla potilaista).
Taulukossa 6 on esitetty PALOMA-2-tutkimuksen tehoa koskevat tulokset tiedonkeruun kahdesta eri ajankohdasta (ensimmäinen ja päivitetty analyysi) sekä tutkijoiden arvioimana että riippumattomasti arvioituna.
Taulukko 6. PALOMA-2-tutkimuksen tehoa koskevat tulokset ensimmäisen ja päivitetyn analyysin perusteella (koko tutkimuspopulaatio [intent-to-treat, ITT]) | |||||||
Ensimmäinen analyysi (tiedonkeruu katkaistu 26.2.2016) | Päivitetty analyysi (tiedonkeruu katkaistu 31.5.2017) | ||||||
IBRANCE + letrotsoli (n = 444) | lumelääke + letrotsoli (n = 222) | IBRANCE + letrotsoli (n = 444) | lumelääke + letrotsoli (n = 222) | ||||
Taudin etenemisestä vapaa elinaika (PFS) tutkijoiden arvioimana | |||||||
Tapahtumien lukumäärä (%) | 194 (43,7) | 137 (61,7) | 245 (55,2) | 160 (72,1) | |||
Mediaani PFS [kk (95 %:n luottamusväli)] | 24,8 (22,1–NE) | 14,5 (12,9–17,1) | 27,6 (22,4–30,3) | 14,5 (12,3–17,1) | |||
Riskitiheyksien suhde (95 %:n luottamusväli) ja p-arvo | 0,576 (0,463–0,718), p < 0,000001 | 0,563 (0,461–0,687), p < 0,000001 | |||||
Taudin etenemisestä vapaa elinaika (PFS) riippumattomasti arvioituna | |||||||
Tapahtumien lukumäärä (%) | 152 (34,2) | 96 (43,2) | 193 (43,5) | 118 (53,2) | |||
Mediaani PFS [kk (95 %:n luottamusväli)] | 30,5 (27,4–NE) | 19,3 (16,4–30,6) | 35,7 (27,7–38,9) | 19,5 (16,6–26,6) | |||
Riskitiheyksien suhde (95 %:n luottamusväli) ja yksisuuntainen p-arvo | 0,653 (0,505–0,844), p = 0,000532 | 0,611 (0,485–0,769), p = 0,000012 | |||||
Objektiivisen vasteen saaneiden osuus, ORR* [% (95 %:n luottamusväli)] | 46,4 (41,7–51,2) | 38,3 (31,9–45,0) | 47,5 (42,8–52,3) | 38,7 (32,3–45,5) | |||
Objektiivisen vasteen saaneiden osuus, ORR*(mitattavissa oleva tauti) [% (95 %:n luottamusväli)] | 60,7 (55,2–65,9) | 49,1 (41,4–56,9) | 62,4 (57,0–67,6) | 49,7 (42,0–57,4) | |||
Kliinisen hyödyn saaneiden osuus, CBR* [% (95 %:n luottamusväli)] | 85,8 (82,2–88,9) | 71,2 (64,7–77,0) | 85,6 (82,0–88,7) | 71,2 (64,7–77,0) | |||
n = potilaiden lukumäärä, NE (not estimable) = ei arvioitavissa *Toissijaisten päätetapahtumien tulokset perustuvat RECIST 1.1 ‑kriteerien mukaan vahvistettuihin ja vahvistamattomiin vasteisiin. | |||||||
Kuvassa 1 on esitetty taudin etenemisestä vapaata elinaikaa (PFS) kuvaavat Kaplan–Meierin käyrät päivitetystä analyysistä (tiedonkeruu katkaistu 31.5.2017).
Kuva 1. Taudin etenemisestä vapaata elinaikaa kuvaavat Kaplan–Meierin käyrät PALOMA-2-tutkimuksessa, 31.5.2017 (tutkijoiden arvio, koko tutkimuspopulaatio [ITT])
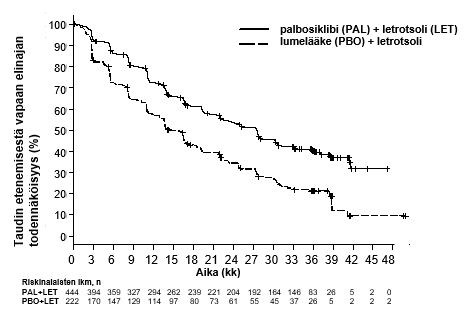
Taudin etenemisestä vapaata elinaikaa koskevan hoitovaikutuksen johdonmukaisuutta koko potilasjoukossa tutkittiin ennalta määritellyin alaryhmäanalyysein, jotka perustuivat ennusteeseen liittyviin tekijöihin ja lähtötilanteen ominaisuuksiin. Sekä ensimmäisessä että päivitetyssä analyysissä todettiin, että palbosiklibin ja letrotsolin yhdistelmähoito pienensi taudin etenemisen tai kuoleman riskiä kaikissa ositustekijöiden ja lähtötilanteen ominaisuuksien mukaan määritellyissä potilasalaryhmissä.
Myös päivitetyssä analyysissä (tiedonkeruu katkaistu 31.5.2017) todettiin pienempi riski seuraavissa alaryhmissä: (1) potilaat, joilla oli viskeraalisia metastaaseja (HR = 0,62; 95 %:n luottamusväli 0,47–0,81; taudin etenemisestä vapaan elinajan mediaani [mPFS] 19,3 kk vs 12,3 kk), ja potilaat, joilla ei ollut viskeraalisia metastaaseja (HR = 0,50; 95 %:n luottamusväli 0,37–0,67; mPFS 35,9 kk vs 17,0 kk) sekä (2) potilaat, joilla oli vain luustometastaaseja (HR = 0,41; 95 %:n luottamusväli 0,26–0,63; mPFS 36,2 kk vs 11,2 kk) ja potilaat, joilla metastasointi ei ollut rajoittunut luustoon (HR = 0,62; 95 %:n luottamusväli 0,50–0,78; mPFS 24,2 kk vs 14,5 kk). Palbosiklibin ja letrotsolin yhdistelmähoidon havaittiin myös pienentävän taudin etenemisen tai kuoleman riskiä 512 potilaan alaryhmässä, jossa kasvaimet oli todettu immunohistokemiallisessa (IHC) määrityksessä RB (retinoblastoomaproteiini) -positiivisiksi (HR = 0,543; 95 %:n luottamusväli 0,433–0,681; mPFS 27,4 kk vs 13,7 kk). Kun palbosiklibin ja letrotsolin yhdistelmähoitoa verrattiin lumelääkkeen ja letrotsolin yhdistelmään 51 potilaan alaryhmässä, jossa kasvaimet oli todettu IHC-määrityksessä RB-negatiivisiksi, tilastollisesti merkitsevää eroa ei todettu (HR = 0,868; 95 %:n luottamusväli 0,424–1,777; mPFS 23,2 kk vs 18,5 kk).
Taulukossa 7 on esitetty päivitetyn analyysin (tiedonkeruu katkaistu 31.5.2017) tehoa koskevia tuloksia (ORR ja vasteeseen kulunut aika, TTR [time to response]) kahdessa potilasalaryhmässä (viskeraalisia metastaaseja vs ei viskeraalisia metastaaseja).
Taulukko 7. PALOMA-2-tutkimuksen tehoa koskevia tuloksia potilailla, joilla tautiin liittyi vs ei liittynyt viskeraalisia metastaaseja (ITT-populaatio, tiedonkeruu katkaistu 31.5.2017)
Viskeraalisia metastaaseja | Ei viskeraalisia metastaaseja | |||
IBRANCE + letrotsoli (n = 214) | lumelääke + letrotsoli (n = 110) | IBRANCE + letrotsoli (n = 230) | lumelääke + letrotsoli (n = 112) | |
Objektiivisen vasteen saaneiden osuus, ORR [% (95 %:n luottamusväli)] | 59,8 (52,9–66,4) | 46,4 (36,8–56,1) | 36,1 (29,9–42,7) | 31,3 (22,8–40,7) |
Vasteeseen kulunut aika, TTR mediaani [kk (vaihteluväli)] | 5,4 (2,0–30,4) | 5,3 (2,6–27,9) | 3,0 (2,1–27,8) | 5,5 (2,6–22,2) |
n = potilaiden lukumäärä Objektiivisten vasteiden osuus perustuu RECIST 1.1 ‑kriteerien mukaan vahvistettuihin ja vahvistamattomiin vasteisiin. | ||||
Päivitetyn analyysin ajankohtana mediaaniaika satunnaistamisesta toisen tutkimushoidon jälkeisen hoidon aloittamiseen oli 38,8 kuukautta palbosiklibin ja letrotsolin yhdistelmähoitoa saaneilla ja 28,8 kuukautta lumelääkettä ja letrotsolia saaneilla (HR = 0,73; 95 %:n luottamusväli 0,58–0,91).
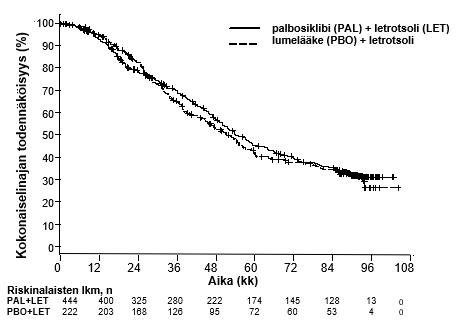
PALOMA-2-tutkimusta koskevan kokonaiselinajan lopullisen analyysin tulokset on esitetty taulukossa 8. Kun seuranta-ajan mediaani oli 90 kuukautta, kokonaiselinajan lopulliset tulokset eivät olleet tilastollisesti merkitseviä. Kokonaiselinaikaa kuvaavat Kaplan–Meierin käyrät on esitetty kuvassa 2.
Taulukko 8. PALOMA-2 (ITT-populaatio) – kokonaiselinajan lopulliset tulokset
Lopullinen kokonaiselinaika (OS) (tiedonkeruu katkaistu 15. marraskuuta 2021) | ||
IBRANCE + letrotsoli (n = 444) | lumelääke + letrotsoli (n = 222) | |
Tapahtumien lukumäärä (%) | 273 (61,5) | 132 (59,5) |
Seurantaan jäävien tutkittavien lukumäärä (%) | 112 (25,2) | 43 (19,4) |
Mediaani OS (kk [95 %:n luottamusväli]) | 53,9 (49,8–60,8) | 51,2 (43,7–58,9) |
Riskitiheyksien suhde (95 %:n luottamusväli) ja p‑arvo† | 0,956 (0,777–1,177), p = 0,6755†* | |
* Ei tilastollisesti merkitsevä.
† Log-rank-testin kahdensuuntainen p-arvo, joka on ositettu satunnaistamisajankohtaan perustuvaan tietoon taudin sijainnista (viskeraalinen vs ei-viskeraalinen).
Kuva 2. Kokonaiselinaikaa kuvaavat Kaplan–Meierin käyrät PALOMA‑2-tutkimuksessa (koko tutkimuspopulaatio, ITT)
Satunnaistettu vaiheen 3 PALOMA‑3-tutkimus: IBRANCE yhdistettynä fulvestranttiin
Palbosiklibin ja fulvestrantin yhdistelmähoidon tehoa verrattiin lumelääkkeen ja fulvestrantin yhdistelmään kansainvälisessä, satunnaistetussa ja kaksoissokkoutetussa monikeskustutkimuksessa. Tutkimus toteutettiin rinnakkaisryhmien asetelmalla (potilaiden siirtymistä tutkimushaarasta toiseen ei sallittu). Tutkimukseen osallistuneilla naisilla oli leikkaus- tai sädehoitoon soveltumaton paikallisesti edennyt hormonireseptoripositiivinen ja HER2‑negatiivinen rintasyöpä, jota ei voitu hoitaa kuratiivisesti, tai metastasoitunut hormonireseptoripositiivinen ja HER2‑negatiivinen rintasyöpä. Potilas voitiin ottaa tutkimukseen menopaussistatuksesta riippumatta. Rintasyövän tuli olla edennyt aiemman, joko (neo)adjuvanttiasetelmassa tai metastasoituneeseen tautiin annetun, hormonaalisen hoidon aikana tai jälkeen.
Tutkimukseen osallistui yhteensä 521 pre-, peri‑ tai postmenopausaalista naista, joiden rintasyöpä oli edennyt joko hormonaalisen adjuvanttihoidon aikana tai 12 kuukauden kuluessa sen päättymisestä tai edenneeseen rintasyöpään aiemmin annetun hormonaalisen hoidon aikana tai 1 kuukauden kuluessa sen päättymisestä. Potilaat satunnaistettiin suhteessa 2:1 saamaan joko palbosiklibin ja fulvestrantin yhdistelmähoitoa tai lumelääkkeen ja fulvestrantin yhdistelmää. Tutkimuspotilaat ositettiin seuraavien tekijöiden mukaan: dokumentoitu herkkyys aiemmalle hormonaaliselle hoidolle, menopaussistatus (pre-/peri- vs postmenopausaalinen) tutkimukseen ottohetkellä ja viskeraalisten metastaasien esiintyminen. Pre- ja perimenopausaalisille naisille annettiin LHRH-agonisti gosereliinia. Potilaat, joilla oli oireita aiheuttavia viskeraalisia metastaaseja ja riski henkeä uhkaaviin komplikaatioihin lyhyellä aikavälillä (mukaan lukien potilaat, joilla keuhko- tai sydänpussin tai vatsakalvon nestekertymä oli hyvin runsasta eikä hallittavissa, sekä potilaat, joilla oli keuhkojen lymfangiitti tai tuumorimassaa yli 50 % maksan tilavuudesta), eivät täyttäneet tutkimuksen sisäänottokriteerejä.
Määrättyä tutkimushoitoa jatkettiin, kunnes ensimmäisen kerran ilmeni jokin seuraavista: tauti eteni objektiivisesti arvioiden, oireet pahenivat, toksisuus ei ollut enää hyväksyttävissä, potilas kuoli tai perui osallistumissuostumuksensa. Potilaiden siirtymistä hoitoryhmästä toiseen (cross-over) ei sallittu.
Potilaiden lähtötilanteen demografiset ominaisuudet ja ennusteeseen liittyvät tekijät olivat keskenään tasapainossa palbosiklibin ja fulvestrantin yhdistelmähoitoa sekä lumelääkkeen ja fulvestrantin yhdistelmää saaneiden tutkimusryhmien välillä. Tutkimuspotilaiden mediaani ikä oli 57 vuotta (vaihteluväli 29–88 vuotta). Kummassakin tutkimushaarassa suurin osa potilaista oli valkoihoisia, dokumentoidusti herkkiä aiemmalle hormonaaliselle hoidolle ja postmenopausaalisia. Noin 20 % potilaista oli pre‑/perimenopausaalisia. Kaikki potilaat olivat saaneet aiempaa systeemistä hoitoa, ja useimmat potilaat kummassakin tutkimushaarassa olivat saaneet aiemmin solunsalpaajahoitoa ensimmäiseen diagnoosiin. Yli puolella (62 %) potilaista ECOG (Eastern Cooperative Oncology Group) -suorituskykyluokka oli 0, 60 %:lla oli viskeraalisia metastaaseja, ja 60 % oli saanut useampaa kuin yhtä aiempaa hormonaalista hoitoa ensimmäiseen diagnoosiin.
Tutkimuksen ensisijainen päätetapahtuma oli tutkijoiden RECIST v1.1 -kriteereiden mukaisesti arvioima taudin etenemisestä vapaa elinaika (PFS). Taudin etenemisestä vapaan elinajan arviointia tukevat lisäanalyysit perustuivat keskitetysti tehtyyn riippumattomaan radiologiseen arvioon. Toissijaisia päätetapahtumia olivat objektiivinen vaste (OR), kliinisen hyödyn saaneiden osuus (CBR), kokonaiselinaika (OS), turvallisuus ja aika oireiden pahenemiseen (time to deteroration, TTD) kivun osalta.
Tutkimuksen ensisijainen päätetapahtuma, taudin etenemisestä vapaan elinajan piteneminen tutkijoiden arvioimana, saavutettiin välianalyysissa, joka tehtiin kun 82 % tutkimussuunnitelman mukaisista PFS-tapahtumista oli todettu. Tulokset ylittivät ennalta määritellyn tehoa mittaavan Haybittle-Peto-menetelmän rajan (α = 0,00135), mikä osoittaa tilastollisesti merkitsevää taudin etenemisestä vapaan elinajan pitenemistä ja kliinisesti merkittävää hoitotehoa.
Taulukossa 9 on esitetty tehoa koskevien tietojen viimeisin päivitys.
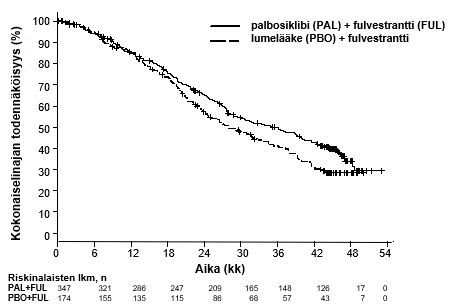
Kun seuranta-ajan mediaani oli 45 kuukautta, tehtiin lopullinen kokonaiselinaika-analyysi, joka perustui 310 tapahtumaan (60 % tutkimukseen satunnaistetuista potilaista). Kokonaiselinajan mediaaneissa todettiin 6,9 kuukauden ero palbosiklibin ja fulvestrantin yhdistelmähoitoa saaneiden ja lumelääkkeen ja fulvestrantin yhdistelmää saaneiden välillä. Tulos ei ollut tilastollisesti merkitsevä ennalta määritellyllä merkitsevyystasolla 0,0235 (yksisuuntainen). Lumelääkkeen ja fulvestrantin yhdistelmää saaneista satunnaistetuista potilaista 15,5 % sai taudin etenemisen jälkeen palbosiklibiä ja muita CDK-estäjiä.
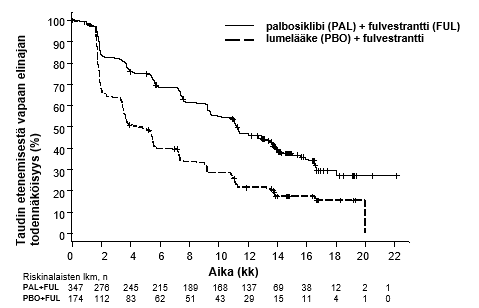
Taulukossa 9 on esitetty PALOMA-3-tutkimuksen taudin etenemisestä vapaata elinaikaa (PFS, tutkijoiden arvio) ja lopullista kokonaiselinaikaa (OS) koskevat tiedot. Vastaavat Kaplan–Meierin käyrät esitetään kuvissa 3 (PFS) ja 4 (OS).
Taulukko 9. PALOMA-3-tutkimuksen tehoa koskevat tulokset (tutkijoiden arvio, koko tutkimuspopulaatio [intent-to-treat, ITT])
Päivitetty analyysi (tietojen kerääminen katkaistu 23.10.2015) | |||
IBRANCE + fulvestrantti (n = 347) | lumelääke + fulvestrantti (n = 174) | ||
Taudin etenemisestä vapaa elinaika, PFS | |||
Tapahtumien lukumäärä (%) | 200 (57,6) | 133 (76,4) | |
Mediaani [kk (95 %:n luottamusväli)] | 11,2 (9,5−12,9) | 4,6 (3,5−5,6) | |
Riskitiheyksien suhde (95 %:n luottamusväli) ja p‑arvo | 0,497 (0,398–0,620), p < 0,000001 | ||
Toissijaiset tehon päätetapahtumat | |||
Objektiivisen vasteen saaneiden osuus, ORR [% (95 %:n luottamusväli)] | 26,2 (21,7−31,2) | 13,8 (9,0–19,8) | |
Objektiivisen vasteen saaneiden osuus, ORR (mitattavissa oleva tauti), [% (95 %:n luottamusväli)] | 33,7 (28,1–39,7) | 17,4 (11,5–24,8) | |
Kliinisen hyödyn saaneiden osuus, CBR [% (95 %:n luottamusväli)] | 68,0 (62,8–72,9) | 39,7 (32,3–47,3) | |
Kokonaiselinaika, OS (lopullinen analyysi, tiedonkeruu katkaistu 13.4.2018) | |||
Tapahtumien lukumäärä (%) | 201 (57,9) | 109 (62,6) | |
Mediaani [kk (95 %:n luottamusväli)] | 34,9 (28,8–40,0) | 28,0 (23,6–34,6) | |
Riskitiheyksien suhde (95 %:n luottamusväli) ja p-arvo† | 0,814 (0,644–1,029) p = 0,0429†* | ||
n = potilaiden lukumäärä | |||
Toissijaisen päätetapahtuman tulokset perustuvat RECIST 1.1 ‑kriteerien mukaan vahvistettuihin ja vahvistamattomiin vasteisiin.
* Ei tilastollisesti merkitsevä.
† Log-rank-testin yhdensuuntainen p-arvo, joka on ositettu satunnaistamisajankohtaan perustuvaan tietoon viskeraalimetastaasien esiintymisestä ja herkkyydestä aiemmalle hormonaaliselle hoidolle.
Kuva 3. Taudin etenemisestä vapaata elinaikaa kuvaavat Kaplan–Meierin käyrät PALOMA-3-tutkimuksessa (tutkijoiden radiologinen arvio, koko tutkimuspopulaatio [ITT]) (tiedonkeruu katkaistu 23.10.2015)
Palbosiklibin ja fulvestrantin yhdistelmähoidon havaittiin pienentävän taudin etenemisen tai kuoleman riskiä kaikissa ositustekijöiden ja lähtötilanteen ominaisuuksien mukaan määritellyissä potilasalaryhmissä. Tämä todettiin selvästi pre‑/perimenopausaalisilla (HR = 0,46; 95 %:n luottamusväli 0,28–0,75) ja postmenopausaalisilla (HR = 0,52; 95 %:n luottamusväli 0,40–0,66) potilailla sekä potilailla, joilla oli viskeraalisia metastaaseja (HR = 0,50; 95 %:n luottamusväli 0,38–0,65) ja joilla ei ollut viskeraalisia metastaaseja (HR = 0,48; 95 %:n luottamusväli 0,33–0,71). Hyöty havaittiin myös riippumatta siitä, kuinka monta aiempaa hoitoa potilas oli saanut metastasoituneeseen tautiin: 0 (HR = 0,59; 95 %:n luottamusväli 0,37–0,93), 1 (HR = 0,46; 95 %:n luottamusväli 0,32–0,64), 2 (HR = 0,48; 95 %:n luottamusväli 0,30–0,76) tai ≥ 3 (HR = 0,59; 95 %:n luottamusväli 0,28–1,22).
Kuva 4. Kokonaiselinaikaa kuvaavat Kaplan–Meierin käyrät PALOMA-3-tutkimuksessa (koko tutkimuspopulaatio, ITT) (tiedonkeruu katkaistu 13.4.2018)
Taulukossa 10 on esitetty tehoa koskevia tuloksia (ORR ja TTR) kahdesta potilasalaryhmästä (viskeraalisia metastaaseja vs ei viskeraalisia metastaaseja).
Taulukko 10. PALOMA-3-tutkimuksen tehoa koskevia tuloksia, kun tautiin liittyi vs ei liittynyt viskeraalisia metastaaseja (ITT-populaatio) | ||||
Viskeraalisia metastaaseja | Ei viskeraalisia metastaaseja | |||
IBRANCE + fulvestrantti (n = 206) | lumelääke + fulvestrantti (n = 105) | IBRANCE + fulvestrantti (n = 141) | lumelääke + fulvestrantti (n = 69) | |
Objektiivisen vasteen saaneiden osuus, ORR [% (95 %:n luottamusväli)] | 35,0 (28,5−41,9) | 13,3 (7,5−21,4) | 13,5 (8,3−20,2) | 14,5 (7,2−25,0) |
Vasteeseen kulunut aika, TTR mediaani [kk (vaihteluväli)] | 3,8 (3,5−16,7) | 5,4 (3,5−16,7) | 3,7 (1,9−13,7) | 3,6 (3,4−3,7) |
n = potilaiden lukumäärä Objektiivisten vasteiden osuus perustuu RECIST 1.1 ‑kriteerien mukaan vahvistettuihin ja vahvistamattomiin vasteisiin. | ||||
Potilaiden raportoimia oireita arvioitiin elämän laatua selvittävällä EORTC (European Organisation for Research and Treatment of Cancer) QLQ‑C30-kyselylomakkeella ja sen rintasyöpää koskevalla osiolla EORTC QLQ‑BR23. Palbosiklibin ja fulvestrantin yhdistelmähoitoa saaneista 335 potilasta ja lumelääkkeen ja fulvestrantin yhdistelmää saaneista 166 potilasta vastasi kyselylomakkeeseen lähtötilanteessa ja ainakin yhdellä vastaanottokäynnillä sen jälkeen.
Aika oireiden pahenemiseen (TTD) oli ennalta määritelty ajankohdaksi, jolloin kipuoireen muutosta kuvaavat pisteet ensimmäisen kerran nousisivat lähtötilanteesta ≥ 10 pisteellä. Palbosiklibin lisääminen fulvestranttihoitoon johti hyötyihin oireiden hallinnassa pidentämällä merkitsevästi aikaa kipuoireen pahenemiseen verrattuna lumelääkkeen ja fulvestrantin yhdistelmään (mediaani 8,0 kk vs 2,8 kk; HR = 0,64; 95 %:n luottamusväli 0,49–0,85; p < 0,001).
Pediatriset potilaat
A5481092-tutkimuksen avoimessa satunnaistetussa vaiheen 2 osiossa palbosiklibista sekä irinotekaanista ja temotsolomidista koostuvan yhdistelmän tehoa verrattiin pelkkään irinotekaaniin ja temotsolomidiin sellaisten uusiutunutta tai hoitoon reagoimatonta Ewingin sarkoomaa sairastavien pediatristen (2 – < 18 vuoden ikäisten) ja nuorten aikuisten (18–20 vuoden ikäisten) potilaiden hoidossa, joille ei ollut tavanomaista hoitoa saatavissa.
Ennalta määritelty välianalyysi tehtiin 33:n tapahtumatonta elossaoloa (event free survival, EFS) koskevan tapahtuman perusteella (61,1 prosenttia 54 tutkittavasta). Palbosiklibin, irinotekaanin ja temotsolomidin yhdistelmän havaittu riskitiheyksien suhde (HR) verrattuna pelkkään irinotekaanin ja temotsolomidin yhdistelmään oli 2,03 (95 %:n luottamusväli: 0,902–4,572; ositettu yksisuuntainen p‑arvo = 0,9621).
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset IBRANCE‑valmisteen käytöstä rintasyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Palbosiklibin farmakokinetiikkaa karakterisoitiin terveillä vapaaehtoisilla ja potilailla, joilla oli kiinteitä kasvaimia, mukaan lukien edennyt rintasyöpä.
Imeytyminen
Palbosiklibin huippupitoisuus (Cmax) havaitaan yleensä 4–12 tunnin (huippupitoisuuden saavuttamiseen kuluva aika [Tmax]) kuluttua IBRANCE-tablettien annostelusta suun kautta. Suun kautta otetun 125 mg:n palbosiklibiannoksen absoluuttinen biologinen hyötyosuus on keskimäärin 46 %. Systeeminen altistus (pitoisuus/aika-kuvaajan pinta-ala, AUC) ja Cmax suurenevat yleisesti suhteessa annokseen annosvälillä 25–225 mg. Vakaa tila (steady-state) saavutettiin 8 päivän kuluessa palbosiklibin toistuvalla kerran vuorokaudessa annostelulla. Toistuvasti kerran vuorokaudessa annosteltuna palbosiklibi kertyy elimistöön kertymissuhteen mediaanin ollessa 2,4 (vaihteluväli 1,5–4,2).
Ruoan vaikutus
Palbosiklibin AUCinf suureni 22 % ja Cmax suureni 26 %, kun IBRANCE-tabletit otettiin runsasrasvaisen, runsaskalorisen ruoan kanssa (noin 800–1 000 kaloria, joista 150 kaloria proteiineista, 250 kaloria hiilihydraateista ja 500–600 kaloria rasvasta), ja vastaavasti 9 % ja 10 %, kun IBRANCE-tabletit otettiin kohtalaisesti rasvaa ja tavanomaisen määrän kaloreja sisältävän ruoan kanssa (noin 500–700 kaloria, joista 75–105 kaloria proteiineista, 250–350 kaloria hiilihydraateista ja 175–245 kaloria rasvasta) verrattuna IBRANCE-tablettien ottamiseen yön yli paastoamisen jälkeen. Tulosten perusteella palbosiklibitabletit voidaan ottaa ruokailun yhteydessä tai tyhjään mahaan.
Jakautuminen
Palbosiklibi sitoutui ihmisen plasman proteiineihin in vitro ~85‑prosenttisesti pitoisuudesta riippumatta. Sitoutumattoman palbosiklibin osuus (fu; keskiarvo) ihmisen plasmassa in vivo oli sitä suurempi, mitä heikompi potilaan maksan toimintakyky oli. Vastaavaa selvää muutostrendiä sitoutumattoman palbosiklibin osuudessa (fu; keskiarvo) ei todettu verrattaessa eri vaikeusasteen munuaisten vajaatoiminta sairastavia potilaita. Palbosiklibin sisäänotto ihmisen maksasoluihin in vitro tapahtui pääasiassa passiivisen diffuusion kautta. Palbosiklibi ei ole OATP1BI:n eikä OATP1B3:n substraatti.
Biotransformaatio
In vitro‑ ja in vivo -tutkimukset viittaavat siihen, että palbosiklibi metaboloituu ihmisillä suuressa määrin maksassa. Kun ihmisille annettiin 125 mg:n kerta-annos [14C]‑palbosiklibia suun kautta, palbosiklibin tärkeimmät ensisijaiset metaboliareitit olivat oksidaatio ja sulfonaatio sekä vähäisempinä myötävaikuttavina metaboliareitteinä asylaatio ja glukuronidaatio. Lääkkeen pääkomponentti plasmassa oli palbosiklibi.
Suurin osa lääkeaineesta erittyi metaboliitteina. Lääkkeen pääkomponentti ulosteessa oli palbosiklibin sulfamiinihappokonjugaatti, joka vastasi 25,8 %:a annetusta annoksesta. Ihmisen maksasoluilla, maksan sytosolisella ja S9:n fraktioilla sekä rekombinantti-sulfotransferaasientsyymeillä (SULT) tehdyt in vitro ‑tutkimukset viittaavat siihen, että palbosiklibi metaboloituu pääasiassa CYP3A:n ja SULT2A1:n välityksellä.
Eliminaatio
Edennyttä rintasyöpää sairastavilla potilailla suun kautta otetun palbosiklibin näennäisen puhdistuman (CL/F) geometrinen keskiarvo oli 63 l/h ja eliminaation puoliintumisaika plasmassa keskimäärin 28,8 tuntia. Kun kuudelle terveelle miespuoliselle tutkittavalle annettiin suun kautta kerta-annos [14C]‑palbosiklibia, 92 % (mediaani) radioaktiivisesti leimatusta annoksesta oli poistunut elimistöstä 15 päivässä; suurin osa (74 %) annoksesta erittyi ulosteisiin ja 17 % virtsaan. [14C]‑palbosiklibista erittyi 2 % muuttumattomana ulosteisiin ja 7 % virtsaan.
Palbosiklibi ei estä CYP1A2-, 2A6-, 2B6-, 2C8-, 2C9-, 2C19- ja 2D6-entsyymejä in vitro. Se ei myöskään kliinisesti merkityksellisinä pitoisuuksina indusoi CYP1A2‑, 2B6‑, 2C8‑ ja 3A4‑entsyymejä.
In vitro -tutkimukset viittaavat siihen, ettei palbosiklibi kliinisesti merkityksellisinä pitoisuuksina juurikaan estä orgaanisia anionin kuljettajia (organic anion transporters) OAT1:tä ja OAT3:a, orgaanista kationin kuljettajaa (organic cation transporter) OCT2:ta, orgaanisia anionia kuljettavia polypeptidejä OATP1B1:tä ja OATP1B3:a eikä sappisuolapumppua (bile salt export pump, BSEP).
Erityisryhmät
Ikä, sukupuoli ja paino
Sukupuoli ei vaikuttanut palbosiklibialtistukseen 183 syöpäpotilaan (50 miestä ja 133 naista, joiden ikä oli 22–89 vuotta ja paino 38–123 kg) populaatiofarmakokineettisessä analyysissa. Myöskään ikä tai paino ei vaikuttanut altistukseen kliinisesti merkittävässä määrin.
Pediatriset potilaat
Palbosiklibialtistus lapsilla, nuorilla ja nuorilla aikuisilla, joilla oli uusiutuneita tai hoitoon reagoimattomia kiinteitä kasvaimia, oli kaikissa ikäryhmissä (≤ 6 vuoden ikäiset, > 6 – < 12 vuoden ikäiset, ≥ 12 – < 18 vuoden ikäiset ja ≥ 18 vuoden ikäiset) samankaltainen koko annosalueella 55–95 mg/m2 (kehon pinta-alan perusteella normalisoitu palbosiklibiannostus), kun annos annettiin suun kautta kerran päivässä päivinä 1–14, mitä seurasi 7 päivän hoitotauko. Vakaan tilan palbosiklibialtistus pediatrisessa potilasjoukossa annoksen 75 mg/m2 kerran päivässä yhteydessä oli samankaltainen kuin aikuisilla tutkittavilla oli havaittu hyväksytyn annoksen 125 mg kerran päivässä yhteydessä (annos annettiin päivinä 1–21, mitä seurasi 7 päivän hoitotauko).
Maksan vajaatoiminta
Palbosiklibin farmakokineettiseen tutkimukseen otettiin henkilöitä, joilla oli normaali maksan toimintakyky tai eri vaikeusasteen maksan vajaatoiminta. Tulokset osoittavat, että sitoutumattoman palbosiklibin altistus (AUCinf) pieneni 17 % lievää maksan vajaatoimintaa (Child-Pugh A) sairastavilla ja suureni 34 % keskivaikeaa (Child‑Pugh B) ja 77 % vaikeaa (Child-Pugh C) maksan vajaatoimintaa sairastavilla verrattuna henkilöihin, joiden maksan toiminta oli normaali. Sitoutumattoman palbosiklibin huippupitoisuus (Cmax) suureni 7 % lievää, 38 % keskivaikeaa ja 72 % vaikeaa maksan vajaatoimintaa sairastavilla verrattuna henkilöihin, joiden maksan toiminta oli normaali. Lievä maksan vajaatoiminta ei myöskään vaikuttanut palbosiklibin farmakokinetiikkaan populaatiofarmakokineettisessä analyysissa, jossa mukana olleista edennyttä syöpää sairastavasta 183 potilaasta 40 potilaalla oli lievä maksan vajaatoiminta perustuen NCI (National Cancer Institute) -luokitukseen (bilirubiini enintään yhtä suuri kuin viitevälin yläraja-arvo [Upper Limit of Normal, ULN] ja aspartaattiaminotransferaasi [ASAT] > ULN tai bilirubiini > 1–1,5 kertaa ULN ja mikä tahansa ASAT-arvo).
Munuaisten vajaatoiminta
Palbosiklibin farmakokineettiseen tutkimukseen otettiin henkilöitä, joilla oli normaali munuaisten toimintakyky tai eri vaikeusasteen munuaisten vajaatoiminta. Tulokset osoittavat, että palbosiklibin kokonaisaltistus (AUCinf) suureni 39 % lievää munuaisten vajaatoimintaa (eGFR 60–89 ml/min) sairastavilla ja 42 % keskivaikeaa (eGFR 30–59 ml/min) ja 31 % vaikeaa (eGFR < 30 ml/min) munuaisten vajaatoimintaa sairastavilla verrattuna henkilöihin, joiden munuaisten toiminta oli normaali (eGFR ≥ 90 ml/min). Palbosiklibin huippupitoisuus (Cmax) suureni 17 % lievää, 12 % keskivaikeaa ja 15 % vaikeaa munuaisten vajaatoimintaa sairastavilla verrattuna henkilöihin, joiden munuaisten toiminta oli normaali. Lievä tai keskivaikea munuaisten vajaatoiminta ei myöskään vaikuttanut palbosiklibin farmakokinetiikkaan populaatiofarmakokineettisessä analyysissa, jossa mukana olleista edennyttä syöpää sairastavasta 183 potilaasta 73 potilaalla oli lievä ja 29 potilaalla keskivaikea munuaisten vajaatoiminta. Palbosiklibin farmakokinetiikkaa ei ole tutkittu hemodialyysia tarvitsevilla potilailla.
Etniset ryhmät
Farmakokineettisessä tutkimuksessa, johon otettiin terveitä vapaaehtoisia, suun kautta annostellun palbosiklibikerta-annoksen AUCinf-arvo oli japanilaisilla tutkittavilla 30 % suurempi ja Cmax-arvo 35 % suurempi kuin ei-aasialaisilla tutkittavilla. Tämä tutkimustulos ei kuitenkaan toistunut johdonmukaisesti myöhemmissä tutkimuksissa, joissa japanilaiset tai aasialaiset rintasyöpäpotilaat saivat useita palbosiklibiannoksia. Palbosiklibiannoksen muuttamista aasialaisille potilaille ei pidetä tarpeellisena perustuen analyysiin, jossa on huomioitu kumulatiivisia farmakokineettisia tietoja sekä turvallisuutta ja tehoa koskevia tietoja sekä aasialaisista että ei-aasialaisista potilaista.
Prekliiniset tiedot turvallisuudesta
Kerta-annoksen ja/tai toistuvan annostelun jälkeen ensisijaisia kohde-elinlöydöksiä olivat rottien ja koirien hemolymfopoieettiseen järjestelmään ja urosten lisääntymiselimiin sekä vain rottien luustoon ja aktiivisessa kasvuvaiheessa oleviin etuhampaisiin kohdistuneet vaikutukset. Näitä systeemisiä toksisuuksia havaittiin yleensä silloin, kun altistus oli AUC-arvon perusteella kliinisesti merkityksellinen. 12 viikon lääkkeettömän jakson jälkeen hemolymfopoieettiseen järjestelmään, urosten lisääntymiselimiin ja etuhampaisiin kohdistuneet vaikutukset olivat korjautuneet joko osittain tai kokonaan, kun taas vaikutukset luustoon eivät olleet korjautuneet. Lisäksi tunnistettiin verenkiertoelimistöön kohdistuneita vaikutuksia (korjatun QT‑ajan [QTc] piteneminen, syketiheyden harveneminen, sykevälin (RR) vaihtelun lisääntyminen ja systolisen verenpaineen nousu) telemetriaoperoiduilla koirilla, kun altistus oli Cmax‑arvon perusteella vähintään 4-kertainen ihmisen kliiniseen altistukseen verrattuna.
Karsinogeenisuus
Palbosiklibin karsinogeenisuutta on arvioitu 6 kuukautta kestäneessä tutkimuksessa siirtogeenisillä hiirillä ja 2 vuotta kestäneessä tutkimuksessa rotilla. Palbosiklibi ei aiheuttanut syöpää siirtogeenisille hiirille, kun sen annostus oli enintään 60 mg/kg/vrk (suurin annos, jolla vaikutusta ei havaittu [No Observed Effect Level, NOEL], oli AUC‑arvon perusteella noin 11‑kertainen ihmisen kliiniseen altistukseen verrattuna). Palbosiklibiin liittyviin kasvainlöydöksiin rotilla lukeutui keskushermoston mikrogliasolukasvainten ilmaantuvuuden lisääntyminen uroksilla, kun annostus oli 30 mg/kg/vrk. Naarasrotilla ei todettu kasvainlöydöksiä millään annostuksella tutkittaessa aina 200 mg:aan/kg/vrk asti. Suurin palbosiklibin annos, jolla karsinogeenisia vaikutuksia ei havaittu (NOEL), oli urosrotilla 10 mg/kg/vrk (AUC‑arvon perusteella noin 2‑kertainen ihmisen kliiniseen altistukseen verrattuna) ja naarasrotilla 200 mg/kg/vrk (AUC‑arvon perusteella noin 4‑kertainen ihmisen kliiniseen altistukseen verrattuna). Urosrotilla havaittujen kasvainlöydösten merkitystä ihmisille ei tunneta.
Genotoksisuus
Palbosiklibi ei ollut mutageeninen bakteerien käänteismutaatiotestissä (Amesin testi), eikä se aiheuttanut rakenteellisia kromosomipoikkeavuuksia ihmisen lymfosyyteillä in vitro tehdyssä kromosomipoikkeavuustestissä.
Kun annos oli ≥ 100 mg/kg/vrk, palbosiklibi indusoi aneugeenisen mekanismin välityksellä mikrotumia kiinanhamsterin munasarjan soluissa in vitro ja urosrottien luuytimessä. Eläinten altistus suurimmalla annoksella, jolla aneugeenisuutta ei havaittu (NOEL), oli AUC‑arvon perusteella noin 7‑kertainen ihmisen kliiniseen altistukseen verrattuna.
Hedelmällisyyden heikkeneminen
Palbosiklibi ei vaikuttanut naarasrottien paritteluun tai hedelmällisyyteen testiannoksilla
≤ 300 mg/kg/vrk (AUC‑arvon perusteella noin 3‑kertainen ihmisen kliiniseen altistukseen verrattuna). Naaraan lisääntymiskudoksiin kohdistuneita haittavaikutuksia ei havaittu toistuvan altistuksen toksisuustutkimuksissa, kun annos rotalle oli enintään 300 mg/kg/vrk (AUC‑arvon perusteella noin 5‑kertainen ihmisen kliiniseen altistukseen verrattuna) ja annos koiralle enintään 3 mg/kg/vrk (AUC‑arvon perusteella noin 3-kertainen ihmisen kliiniseen altistukseen verrattuna).
Rotilla ja koirilla todettujen prekliinisten löydösten perusteella palbosiklibi saattaa heikentää miehen lisääntymiskykyä ja hedelmällisyyttä. Palbosiklibiin liittyneitä löydöksiä kiveksissä, lisäkiveksissä, eturauhasessa ja siementiehyissä olivat kyseisten elinten painon lasku, näiden surkastuminen tai rappeutuminen, hypospermia, solujätteet tiehyissä, siittiöiden liikkuvuuden ja siittiötiheyden väheneminen sekä sekreetion väheneminen. Näitä löydöksiä havaittiin rotilla ja/tai koirilla, kun altistus oli AUC‑arvon perusteella rotalla ≥ 9‑kertainen ja koiralla subterapeuttinen ihmisen kliiniseen altistukseen verrattuna. Urosten lisääntymiselimiin kohdistuneiden vaikutusten havaittiin osittain korjaantuvan, rotalla 4 viikon ja koiralla 12 viikon lääkkeettömän jakson jälkeen. Vaikka urosten lisääntymiselimissä todettiin edellä mainittuja löydöksiä, vaikutuksia urosrottien paritteluun tai hedelmällisyyteen ei ilmennyt, kun AUC‑arvon perusteella arvioitu altistustaso oli 13-kertainen ihmisen kliiniseen altistukseen verrattuna.
Kehitystoksisuus
Palbosiklibi estää reversiibelisti sykliinistä riippuvaisia kinaaseja 4 ja 6 (CDK4/6), jotka osallistuvat solusyklin säätelyyn. Palbosiklibin käyttö raskauden aikana voi siten aiheuttaa sikiöhaittojen riskin. Palbosiklibi oli sikiötoksinen tiineissä eläimissä. Annoksella ≥ 100 mg/kg/vrk todettiin rottien luustomuutosten (kaulakylkiluu seitsemännen kaulanikaman kohdalla) ilmaantuvuuden lisääntymistä. Rottaemojen toksisella annoksella 300 mg/kg/vrk (AUC‑arvon perusteella 3‑kertainen ihmisen kliiniseen altistukseen verrattuna) havaittiin sikiöiden painon laskua ja kaniiniemojen toksisella annoksella 20 mg/kg/vrk (AUC‑arvon perusteella 4‑kertainen ihmisen kliiniseen altistukseen verrattuna) havaittiin luustomuutosten (myös eturaajojen pienissä varvasluissa) ilmaantuvuuden lisääntymistä. Varsinaista sikiöaltistusta ja siirtymistä istukan läpi ei ole tutkittu.
Farmaseuttiset tiedot
Apuaineet
Tablettiydin
Mikrokiteinen selluloosa
Kolloidinen piidioksidi
Krospovidoni
Magnesiumstearaatti
Meripihkahappo
Kalvopäällyste
Hypromelloosi (E464)
Titaanidioksidi (E171)
Triasetiini
Indigokarmiinialumiinilakka (E132)
Punainen rautaoksidi (E172) (vain 75 mg:n ja 125 mg:n tabletit)
Keltainen rautaoksidi (E172) (vain 100 mg:n tabletit)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Säilytä alkuperäisessä läpipainopakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
IBRANCE tabletti, kalvopäällysteinen
75 mg (L:ei) 21 fol (1871,77 €)
100 mg (L:ei) 21 fol (1871,77 €)
125 mg (L:ei) 21 fol (1871,77 €)
PF-selosteen tieto
PVC/OPA/Al/PVC/Al-läpipainolevy sisältäen 7 kalvopäällysteistä tablettia (jokaisessa kolossa 1 kalvopäällysteinen tabletti). Pakkaus sisältää 21 kalvopäällysteistä tablettia (pakkauksessa 3 läpipainolevyä) tai 63 kalvopäällysteistä tablettia (pakkauksessa 9 läpipainolevyä).
PVC/OPA/Al/PVC/Al-läpipainolevy sisältäen 7 kalvopäällysteistä tablettia (jokaisessa kolossa 1 kalvopäällysteinen tabletti) taskupakkauksessa. Pakkaus sisältää 21 kalvopäällysteistä tablettia (kotelossa 3 taskupakkausta).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
IBRANCE 75 mg kalvopäällysteiset tabletit
Pyöreä (halkaisija 10,3 mm), vaaleanvioletti kalvopäällysteinen tabletti, jonka toisella puolella on merkintä ”Pfizer” ja vastakkaisella puolella ”PBC 75”.
IBRANCE 100 mg kalvopäällysteiset tabletit
Soikea (15,0 x 8,0 mm), vihreä kalvopäällysteinen tabletti, jonka toisella puolella on merkintä ”Pfizer” ja vastakkaisella puolella ”PBC 100”.
IBRANCE 125 mg kalvopäällysteiset tabletit
Soikea (16,2 x 8,6 mm), vaaleanvioletti kalvopäällysteinen tabletti, jonka toisella puolella on merkintä ”Pfizer” ja vastakkaisella puolella ”PBC 125”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
IBRANCE tabletti, kalvopäällysteinen
75 mg 21 fol
100 mg 21 fol
125 mg 21 fol
- Ylempi erityiskorvaus (100 %). Palbosiklibi: Hormonireseptoripositiivisen ja HER2-negatiivisen paikallisesti edenneen tai metastasoituneen rintasyövän hoito erityisin edellytyksin (1501).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Palbosiklibi: Hormonireseptoripositiivisen ja HER2-negatiivisen paikallisesti edenneen tai etäpesäkkeitä lähettäneen rintasyövän hoito erityisin edellytyksin (394).
ATC-koodi
L01EF01
Valmisteyhteenvedon muuttamispäivämäärä
02.04.2025
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com