
XYDALBA kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 500 mg
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää dalbavansiinihydrokloridia määrän, joka vastaa 500 mg:aa dalbavansiinia.
Käyttökuntoon saattamisen jälkeen yksi millilitra liuosta sisältää 20 mg dalbavansiinia.
Laimennetun infuusionesteen lopullisen dalbavansiinipitoisuuden on oltava 1‑5 mg/ml (ks. kohta Käyttö- ja käsittelyohjeet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos (kuiva-aine välikonsentraattia varten).
Kliiniset tiedot
Käyttöaiheet
Xydalba on tarkoitettu ihon ja ihorakenteiden akuuttien bakteeri-infektioiden (ABSSSI) hoitoon aikuisille ja pediatrisille potilaille heti syntymästä lähtien (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Viralliset ohjeet koskien bakteerilääkkeiden tarkoituksenmukaista käyttöä on otettava huomioon.
Annostus ja antotapa
Annostus
Aikuiset
Dalbavansiinin suositeltu annos on 1 500 mg, joka annetaan joko 1 500 mg:n kertainfuusiona tai kahtena annoksena: 1 000 mg ja viikon kuluttua tästä 500 mg (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Pediatriset potilaat
Dalbavansiinin suositeltu annos on kerta-annos potilaan iän ja painon mukaan.
Dalbavansiiniannos pediatrisille potilaille
| Ikäryhmä | Annos (hoito kerta-annoksella) |
| Syntymästä 6:een ikävuoteen | 22,5 mg/kg (maksimi 1 500 mg) |
| 6 – < 18 vuotta | 18 mg/kg (maksimi 1 500 mg) |
Erityisryhmät
Iäkkäät
Annoksen muuttaminen ei ole tarpeen (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa aikuisille ja pediatrisille potilaille, joilla on lievä tai kohtalainen munuaisten vajaatoiminta (kreatiniinipuhdistuma ≥ 30–79 ml/min). Annosta ei tarvitse muuttaa aikuisille potilaille, jotka käyvät säännöllisessä hoitosuunnitelman mukaisessa hemodialyysissa (3 kertaa/viikko), ja dalbavansiinia voidaan antaa hemodialyysin ajoituksesta riippumatta.
Kroonista munuaisten vajaatoimintaa sairastaville aikuisille potilaille, joiden kreatiniinipuhdistuma on < 30 ml/min ja jotka eivät käy säännöllisessä hoitosuunnitelman mukaisessa hemodialyysissa, dalbavansiinin suositeltu annos on pienennettävä joko 1 000 mg:aan, joka annetaan kertainfuusiona, tai 750 mg:aan, jonka jälkeen viikon kuluttua annetaan 375 mg (ks. kohta Farmakokinetiikka).
Ei ole riittävästi tietoja, joiden perusteella voitaisiin suositella annoksen muuttamista 3 kuukauden – alle 18 vuoden ikäisille potilaille, joiden kreatiniinipuhdistuma on alle 30 ml/min/1,73 m2, ja alle 3 kuukauden ikäisille potilaille, joilla on munuaisten vajaatoiminta (ts. seerumin kreatiniiniarvo ≥ 2 kertaa normaalin yläraja tai virtsantuotto < 0,5 ml/kg/h) tai jotka tarvitsevat dialyysiä. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdassa Farmakokinetiikka, ei voida antaa suosituksia annostuksesta.
Maksan vajaatoiminta
Dalbavansiiniannoksen muuttamista ei suositella potilaille, joilla on lievä maksan vajaatoiminta (Child–Pugh A). Varovaisuutta on noudatettava määrättäessä dalbavansiinia potilaille, joilla on kohtalainen tai vaikea maksan vajaatoiminta (Child–Pugh B ja C), koska tietoja sopivan annostuksen määrittämiseksi ei ole saatavilla (ks. kohta Farmakokinetiikka).
Antotapa
Laskimoon.
Xydalba on saatettava käyttökuntoon ja edelleen laimennettava ennen sen antamista infuusiona laskimoon 30 minuutin aikana. Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Yliherkkyysreaktiot
Dalbavansiinia on annettava varoen potilaille, joiden tiedetään olevan yliherkkiä muille glykopeptideille, sillä ristiyliherkkyyttä saattaa esiintyä. Jos potilaalle ilmaantuu allerginen reaktio dalbavansiinille, antaminen on lopetettava ja allergisen reaktion asianmukainen hoito aloitettava.
Clostridioides (aiemmin Clostridium) difficile -bakteeriin liittyvä ripuli
Bakteerilääkkeisiin liittyvää koliittia ja pseudomembranoottista koliittia on raportoitu lähes kaikkia antibiootteja käytettäessä, ja vaikeusasteeltaan ne voivat vaihdella lievästä hengenvaaralliseen. Siksi koliitin mahdollisuus on syytä ottaa huomioon potilailla, joilla esiintyy ripulia dalbavansiinihoidon aikana tai sen jälkeen (ks. kohta Haittavaikutukset). Tällaisessa tapauksessa on harkittava dalbavansiinihoidon lopettamista ja tukitoimenpiteitä sekä spesifisen Clostridioides (aiemmin Clostridium) difficile -hoidon aloittamista. Näitä potilaita ei saa koskaan hoitaa peristaltiikkaa lamaavilla lääkevalmisteilla.
Infuusioon liittyvät reaktiot
Xydalba on annettava infuusiona laskimoon, ja infuusion kokonaiskeston on oltava 30 minuuttia, jotta infuusioon liittyvien reaktioiden riski jää mahdollisimman pieneksi. Glykopeptidejä sisältävien bakteerilääkkeiden nopea infuusio laskimoon voi aiheuttaa reaktioita, kuten ylävartalon punoitusta, nokkosihottumaa, kutinaa ja/tai ihottumaa. Nämä reaktiot voivat loppua, kun infuusio lopetetaan tai sitä hidastetaan.
Munuaisten vajaatoiminta
Tietoa dalbavansiinin tehosta ja turvallisuudesta potilailla, joiden kreatiniinipuhdistuma on < 30 ml/min, on niukasti. Simulaatioiden perusteella annoksen muuttaminen on tarpeen kroonista munuaisten vajaatoimintaa sairastavilla aikuisilla potilailla, joiden kreatiniinipuhdistuma on < 30 ml/min ja jotka eivät saa säännöllistä hemodialyysihoitoa (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka). Ei ole riittävästi tietoja, joiden perusteella voitaisiin suositella annoksen muuttamista 3 kuukauden – 18 vuoden ikäisille potilaille, joiden kreatiniinipuhdistuma on alle 30 ml/min/1,73 m2, ja alle 3 kuukauden ikäisille potilaille, joilla on munuaisten vajaatoiminta (ts. seerumin kreatiniiniarvo ≥ 2 kertaa normaalin yläraja tai virtsantuotto < 0,5 ml/kg/h) tai jotka tarvitsevat dialyysiä.
Seka-infektiot
Seka-infektioissa, joissa potilaalla epäillään olevan gramnegatiivisia bakteereja, hänelle on annettava myös sopivaa gramnegatiivisiin bakteereihin kohdistuvaa bakteerilääkettä tai -lääkkeitä (ks. kohta Farmakodynamiikka).
Ei-herkät organismit
Antibioottien käyttö saattaa edistää ei-herkkien mikrobien liikakasvua. Jos superinfektio kehittyy hoidon aikana, asianmukaisiin toimenpiteisiin on ryhdyttävä.
Kliinisten tietojen rajallisuus
Saatavilla on vain vähän tietoa yli kahtena annoksena (viikon välein) annetun dalbavansiinin turvallisuudesta ja tehosta. Tärkeimmissä ihon ja ihorakenteiden akuuttien bakteeri-infektioiden tutkimuksissa hoidettuja infektiotyyppejä olivat vain seuraavat: selluliitti/ruusu, absessit ja haava-infektiot. Dalbavansiinin käyttämisestä vaikeasti immuunipuutteisten potilaiden hoidossa ei ole kokemusta.
Apuaineet
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Reseptorien seulontatutkimuksesta in vitro saadut tulokset eivät viittaa todennäköiseen yhteisvaikutukseen muiden terapeuttisten kohteiden kanssa eivätkä kliinisesti relevanttien farmakodynaamisten yhteisvaikutusten mahdollisuuteen (ks. kohta Farmakodynamiikka).
Dalbavansiinilla ei ole tehty kliinisiä lääkkeiden yhteisvaikutustutkimuksia.
Muiden lääkevalmisteiden mahdollinen vaikutus dalbavansiinin farmakokinetiikkaan
Dalbavansiini ei metaboloidu CYP-entsyymien kautta in vitro, joten samanaikaisesti annetut CYP:n indusorit tai estäjät eivät todennäköisesti vaikuta dalbavansiinin farmakokinetiikkaan.
Ei tiedetä, onko dalbavansiini maksan sisään- ja uloskuljetusproteiinien substraatti. Samanaikainen antaminen näiden kuljettajaproteiinien estäjien kanssa saattaa lisätä altistusta dalbavansiinille. Esimerkkejä tällaisista kuljettajaproteiinien estäjistä ovat tehostetut proteaasin estäjät, verapamiili, kinidiini, itrakonatsoli, klaritromysiini ja siklosporiini.
Dalbavansiinin mahdollinen vaikutus muiden lääkevalmisteiden farmakokinetiikkaan
Dalbavansiinin yhteisvaikutuspotentiaali CYP-entsyymien kautta metaboloituvien lääkevalmisteiden kanssa on odotettavasti pieni, sillä se ei ole CYP-entsyymien estäjä eikä indusori in vitro. Tietoja dalbavansiinista CYP2C8:n estäjänä ei ole.
Ei tiedetä, onko dalbavansiini kuljettajaproteiinien estäjä. Lisääntynyttä altistusta sellaisten kuljettajaproteiinien substraateille, jotka ovat herkkiä kuljettajaproteiinien aktiivisuuden estolle, kuten statiineille ja digoksiinille, ei voida sulkea pois, jos niitä annetaan yhdessä dalbavansiinin kanssa.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja dalbavansiinin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Xydalba-valmisteen käyttöä ei suositella raskauden aikana, ellei mahdollinen odotettavissa oleva hyöty selvästi oikeuta sikiölle mahdollisesti aiheutuvan haitan.
Imetys
Ei tiedetä, erittyykö dalbavansiini ihmisen rintamaitoon. Dalbavansiini erittyy kuitenkin imettävien rottien maitoon ja saattaa erittyä ihmisen rintamaitoon. Dalbavansiini ei imeydy suun kautta hyvin; vaikutusta rintamaitoa saavan imeväisen maha-suolikanavan flooraan tai suun flooraan ei kuitenkaan voida sulkea pois. On päätettävä, lopetetaanko rintaruokinta vai jatketaanko sitä tai lopetetaanko Xydalba-hoito vai jatketaanko sitä, ottaen huomioon rintaruokinnan hyödyt lapselle ja hoidon hyödyt äidille.
Hedelmällisyys
Eläinkokeissa on havaittu hedelmällisyyden heikentymistä (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmisille ei tunneta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Xydalba-valmisteella voi olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn, koska pienellä määrällä potilaita on raportoitu heitehuimausta (ks. kohta Haittavaikutukset).
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
2 473 potilasta sai dalbavansiinia faasin 2/3 kliinisissä tutkimuksissa joko 1 500 mg kertainfuusiona tai 1 000 mg ja viikon kuluttua 500 mg. Yleisimmät haittavaikutukset, joita esiintyi ≥ 1 prosentilla dalbavansiinilla hoidetuista potilaista, olivat pahoinvointi (2,4 %), ripuli (1,9 %) ja päänsärky (1,3 %). Nämä haittavaikutukset olivat vaikeusasteeltaan yleensä lieviä tai kohtalaisia.
Taulukoitu luettelo haittavaikutuksista (taulukko 1)
Dalbavansiinin faasin 2/3 kliinisissä tutkimuksissa on havaittu seuraavia haittavaikutuksia. Nämä haittavaikutukset on luokiteltu elinjärjestelmän ja yleisyyden mukaan. Yleisyysluokat on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000).
Taulukko 1.
| Elinjärjestelmä | Yleinen | Melko harvinainen | Harvinainen |
| Infektiot | vulvovaginaalinen mykoottinen infektio, virtsatieinfektio, sieni-infektio, Clostridioides (aiemmin Clostridium) difficile -koliitti, suun kandidiaasi | ||
| Veri ja imukudos | anemia, trombosytoosi, eosinofilia, leukosytopenia, neutropenia | ||
| Immuunijärjestelmä | anafylaktoidinen reaktio | ||
| Aineenvaihdunta ja ravitsemus | ruokahalun väheneminen | ||
| Psyykkiset häiriöt | unettomuus | ||
| Hermosto | päänsärky | dysgeusia, heitehuimaus | |
| Verisuonisto | punoitus (flushing), flebiitti | ||
| Hengityselimet, rintakehä ja välikarsina | yskä | bronkospasmi | |
| Ruoansulatuselimistö | pahoinvointi, ripuli | ummetus, vatsakipu, dyspepsia, vatsavaivat, oksentelu | |
| Iho ja ihonalainen kudos | kutina, nokkosihottuma, ihottuma | ||
| Sukupuolielimet ja rinnat | ulkosynnyttimien ja emättimen kutina | ||
| Yleisoireet ja antopaikassa todettavat haitat | infuusioon liittyvät reaktiot | ||
| Tutkimukset | suurentunut veren laktaattidehydrogenaasiarvo, suurentunut ALAT-arvo, suurentunut ASAT-arvo, suurentunut veren virtsahappopitoisuus, poikkeavuudet maksan toimintakokeissa, suurentuneet transaminaasiarvot, suurentunut veren AFOS-arvo, suurentunut verihiutaleiden määrä, ruumiinlämmön kohoaminen, suurentuneet maksan entsyymiarvot, suurentunut gammaglutamyylitransferaasiarvo |
Valikoitujen haittavaikutusten kuvaus
Farmakologiseen ryhmään liittyvät haittavaikutukset
Ototoksisuus on liitetty glykopeptidien (vankomysiinin ja teikoplaniinin) käyttöön; samanaikaisesti ototoksisella lääkevalmisteella, kuten aminoglykosidilla, hoidetuilla potilailla riski voi olla suurentunut.
Pediatriset potilaat
Dalbavansiinin turvallisuutta arvioitiin yhdessä faasin 3 kliinisessä tutkimuksessa 169 pediatrisella potilaalla, joiden ikä vaihteli vastasyntyneestä < 18 vuoden ikäisiin, ja joilla oli ihon ja ihorakenteiden akuutteja bakteeri-infektioita (tai epäilty tai vahvistettu sepsis, jos potilaan ikä oli alle 3 kuukautta). Potilaita hoidettiin dalbavansiinilla (91 potilasta sai kerta-annoksen dalbavansiinia ja 78 potilasta, jotka kaikki olivat vähintään 3 kuukauden ikäisiä, sai kaksi annosta dalbavansiinia). Näistä 169 pediatrisesta potilaasta 58 oli nuoria (vähintään 12-vuotiaita), 49 oli 6 – < 12 vuoden ikäisiä lapsia, 35 oli 2 – < 6 vuoden ikäisiä lapsia, 17 oli 3 kuukauden – < 2 vuoden ikäisiä lapsia ja 10 oli alle 3 kuukauden ikäisiä. Lisäksi faasin 1 avoimessa farmakokineettisessä tutkimuksessa dalbavansiinin kerta-annosta arvioitiin kahdeksalla alle 3 kuukauden ikäisellä potilaalla. Näissä kahdessa kliinisessä tutkimuksessa oli 18 alle 3 kuukauden ikäistä lasta, mukaan lukien 3 keskosta ja 5 täysiaikaista vastasyntynyttä. Dalbavansiinin turvallisuutta koskevat löydökset näillä pediatrisilla potilailla olivat yleisesti ottaen samankaltaisia kuin aikuisilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Dalbavansiinin yliannostuksen hoidosta ei ole saatavissa erityistä tietoa, koska kliinisissä tutkimuksissa ei ole havaittu annosta rajoittavaa toksisuutta. Faasin 1 tutkimuksissa terveille vapaaehtoisille on annettu enintään 1 500 mg:n kerta-annoksia ja 4 500 mg:n kumulatiivisia annoksia korkeintaan 8 viikon jakson aikana. Merkkejä toksisuudesta ei havaittu, eivätkä laboratoriokokeet antaneet aihetta kliinisiin toimenpiteisiin. Faasin 3 tutkimuksissa potilaille on annettu enintään 1 500 mg:n kerta-annoksia.
Dalbavansiinin yliannostuksen hoito koostuu seurannasta ja yleisistä tukitoimenpiteistä. Vaikka saatavilla ei ole tietoja erityisesti hemodialyysin käytöstä yliannostuksen hoidossa, on huomioitava, että eräässä maksan vajaatoimintaa sairastavilla potilailla tehdyssä faasin 1 tutkimuksessa alle 6 % suositellusta dalbavansiiniannoksesta oli poistunut 3 tunnin hemodialyysin jälkeen.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: systeemiset bakteerilääkkeet, peptidoglykaanibakteerilääkkeet, ATC-koodi: J01XA04.
Vaikutusmekanismi
Dalbavansiini on bakterisidinen lipoglykopeptidi.
Se vaikuttaa sille herkkiin grampositiivisiin bakteereihin estämällä soluseinämän synteesiä: se sitoutuu kantapeptidin terminaaliseen D‑alanyyli-D-alaniiniin soluseinämän naskentissa peptidoglykaanissa estäen disakkaridin alayksiköitä muodostamasta ristisidoksia (transpeptidaatio ja transglykosylaatio), mikä johtaa bakteerisolujen kuolemaan.
Resistenssimekanismi
Kaikki gramnegatiiviset bakteerit ovat luontaisesti resistenttejä dalbavansiinille.
Staphylococcus- ja Enterococcus-lajien dalbavansiiniresistenssiin vaikuttaa välillisesti VanA-genotyyppi, joka saa aikaan kohdepeptidin modifikaation naskentissa soluseinämässä. In vitro ‑tutkimusten perusteella muut vankomysiinin resistenssigeenien luokat eivät vaikuta dalbavansiinin aktiivisuuteen.
Dalbavansiinin MIC-arvot ovat suurempia vankomysiiniherkkyydeltään alentuneilla stafylokokeilla (VISA) kuin vankomysiinille täysin herkillä kannoilla. Jos ne isolaatit, joilla on suuremmat dalbavansiinin MIC-arvot, edustavat stabiileja fenotyyppejä ja korreloivat muiden glykopeptidien resistenssin kanssa, todennäköinen mekanismi olisi glykopeptidikohteiden määrän kasvu naskentissa peptidoglykaanissa.
Dalbavansiinin ja muiden antibioottiryhmien välillä ei havaittu ristiresistenssiä in vitro -tutkimuksissa. Metisilliiniresistenssillä ei ole vaikutusta dalbavansiinin aktiivisuuteen.
Yhteisvaikutukset muiden bakteerilääkkeiden kanssa
In vitro -tutkimuksissa ei ole havaittu antagonismia dalbavansiinin ja muiden yleisesti käytettävien antibioottien (eli kefepiimin, keftatsidiimin, keftriaksonin, imipeneemin, meropeneemin, amikasiinin, atstreonaamin, siprofloksasiinin, piperasilliinin/tatsobaktaamin ja trimetopriimin/sulfametoksatsolin) välillä, kun niitä on testattu 12:ta gramnegatiivista patogeenilajia vastaan (ks. kohta Yhteisvaikutukset).
Herkkyystestauksen raja-arvot
European Committee on Antimicrobial Susceptibility Testing (EUCAST) on asettanut dalbavansiinia koskevat raja-arvot herkkyystesteille pienimmälle bakteerin kasvua estävälle pitoisuudelle (MIC), ja ne on lueteltu täällä: https://www.ema.europa.eu/documents/other/minimum-inhibitory-concentration-mic-breakpoints_en.xlsx
Farmakokinetiikan ja farmakodynamiikan suhde
Bakterisidinen aktiivisuus stafylokokkeja vastaan in vitro on aikariippuvainen dalbavansiinin seerumipitoisuuksilla, jotka vastaavat ihmisiltä mitattua seerumipitoisuutta käytettäessä suositeltua annosta. Dalbavansiinin farmakokinetiikan ja farmakodynamiikan in vivo -suhdetta S. aureus -lajin osalta tutkittiin käyttäen neutropeenista eläininfektiomallia. Se osoitti, että dalbavansiinin antibakteerinen aktiivisuus korreloi ilmeisesti parhaiten vapaan lääkeaineen plasmapitoisuus-aikakäyrän alle jäävän alueen ja pienimmän bakteerien kasvua estävän pitoisuuden suhteen (fAUC/MIC) kanssa.
Kliininen teho spesifisiä patogeeneja vastaan
Teho ihon ja ihorakenteiden akuutteja bakteeri-infektioita (ABSSSI) aiheuttavia dalbavansiinille herkkiä patogeeneja vastaan on osoitettu kliinisissä tutkimuksissa in vitro:
- Staphylococcus aureus
- Streptococcus pyogenes
- Streptococcus agalactiae
- Streptococcus dysgalactiae
- Streptococcus anginosus -ryhmä (mukaan lukien S. anginosus, S. intermedius ja S. constellatus).
Antibakteerinen aktiivisuus muita relevantteja patogeeneja vastaan
Kliinistä tehoa seuraavia patogeeneja vastaan ei ole osoitettu, vaikka in vitro -tutkimukset viittaavat siihen, että ne ovat herkkiä dalbavansiinille hankinnaisten resistenssimekanismien puuttuessa:
- ryhmän G streptokokit
- Clostridium perfringens
- Peptostreptococcus-lajit.
Pediatriset potilaat
Xydalba-valmistetta on arvioitu yhdessä faasin 3 avoimessa, satunnaistetussa, vertailuvalmisteella kontrolloidussa kliinisessä tutkimuksessa pediatrisilla potilailla, joiden ikä vaihteli vastasyntyneestä < 18 vuoden ikäisiin ja joilla oli ihon ja ihorakenteiden akuutteja bakteeri-infektioita tai joilla oli epäilty tai vahvistettu sepsis, jos potilas oli alle 3 kuukauden ikäinen. Tutkimukseen osallistui 169 potilasta, joita hoidettiin dalbavansiinilla (91 potilasta sai kerta-annoksen dalbavansiinia ja 78 potilasta, jotka kaikki olivat vähintään 3 kuukauden ikäisiä, sai kaksi annosta dalbavansiinia), ja 30 potilasta, joita hoidettiin vertailuvalmisteella. Seitsemällä potilaalla 10:stä alle 3 kuukauden ikäisestä potilaasta oli epäilty tai vahvistettu sepsis tutkimukseen rekisteröitäessä. Ensisijaisena tavoitteena oli arvioida dalbavansiinin turvallisuutta ja siedettävyyttä, ja toissijaisia tavoitteita olivat tehon ja farmakokinetiikan arvioinnit. Tehon päätetapahtuma oli kuvaileva. Kliininen paranemisprosentti tutkimuksen lopussa (mITT) oli kerta-annoksen dalbavansiinia saaneessa ryhmässä 95,2 % (79/83), kaksi dalbavansiiniannosta saaneessa ryhmässä 97,3 % (72/74) ja vertailuvalmistetta saaneessa ryhmässä 100 % (30/30).
Farmakokinetiikka
Dalbavansiinin farmakokinetiikkaa on luonnehdittu terveillä tutkittavilla, potilailla ja erityisryhmillä. Systeemiset altistukset dalbavansiinille ovat annosriippuvaisia 140–1 120 mg:n kerta-annosten jälkeen, mikä osoittaa dalbavansiinin lineaarisen farmakokinetiikan. Dalbavansiinin kumuloitumista ei havaittu terveillä aikuisilla sen jälkeen, kun heille oli annettu toistuvasti laskimonsisäisiä infuusioita kerran viikossa enintään 8 viikon ajan (1 000 mg päivänä 1, minkä jälkeen annettiin enintään 7 viikottaista 500 mg:n annosta).
Keskimääräinen terminaalisen eliminaation puoliintumisaika (t1/2) oli 372 tuntia (vaihteluväli 333–405 tuntia). Dalbavansiinin farmakokinetiikkaa voidaan parhaiten kuvata kolmitilamallilla (α- ja β-jakautumisvaiheet, jota seuraa terminaalinen eliminaatiovaihe). Jakautumisen puoliintumisaika (t1/2β), joka muodostaa suurimman osan kliinisesti relevantista pitoisuus-aikaprofiilista, vaihteli 5 vuorokaudesta 7 vuorokauteen ja on yhdenmukainen kerran viikossa suoritetun annostelun kanssa.
Alla olevassa taulukossa 2 esitetään dalbavansiinin arvioidut farmakokineettiset parametrit kahden annoksen hoidon ja kerta-annoshoidon jälkeen.
Taulukko 2.
| Dalbavansiinin farmakokineettisten parametrien keskiarvo (keskihajonta) aikuisilla farmakokineettisella populaatioanalyysilla määritettyinä1 | ||
| Parametri | Kahden annoksen hoito2 | Kerta-annoshoito3 |
| Cmax (mg/l) | Päivä 1: 281 (52) | Päivä 1: 411 (86) |
| Päivä 8: 141 (26) | ||
| AUC0–vrk14 (mg•h/l) | 18 100 (4 600) | 20 300 (5 300) |
| CL (l/h) | 0,048 (0,0086) | 0,049 (0,0096) |
1 Lähde: DAL-MS-01. 2 1 000 mg päivänä 1 + 500 mg päivänä 8; tutkimuksen DUR001-303 tutkittavat, joiden farmakokineettinen näyte oli arvioitavissa. 3 1 500 mg; tutkimuksen DUR001-303 tutkittavat, joiden farmakokineettinen näyte oli arvioitavissa. | ||
Kuvassa 1 esitetään dalbavansiinipitoisuudet plasmassa ajan funktiona kahden annoksen hoidon ja kerta-annoshoidon jälkeen.
Kuva 1. Dalbavansiinipitoisuudet plasmassa ajan funktiona tyypillisellä ihon ja ihorakenteiden akuuttia bakteeri-infektiota sairastavalla aikuisella potilaalla (simulaatio, jossa käytetään populaatiofarmakokineettistä mallia) sekä kerta-annoshoidossa että kahden annoksen hoidossa
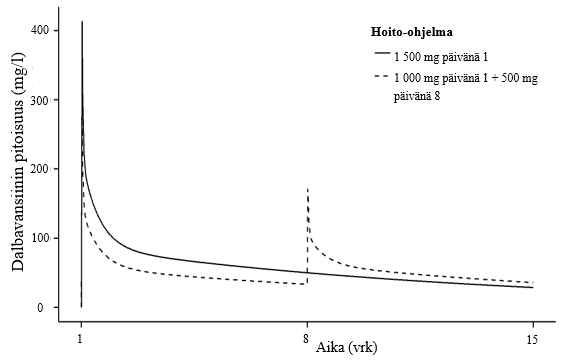
Jakautuminen
Vakaan tilan puhdistuma ja jakautumistilavuus terveiden tutkittavien ja infektiopotilaiden välillä ovat vertailukelpoisia. Vakaan tilan jakautumistilavuus oli samanlainen kuin solunulkoisen nesteen tilavuus. Dalbavansiini sitoutuu palautuvasti ihmisen plasman proteiineihin, pääasiassa albumiiniin. Dalbavansiinin sitoutumisaste plasman proteiineihin on 93 %, eikä se muutu lääkkeen pitoisuuden, munuaisten vajaatoiminnan tai maksan vajaatoiminnan mukaan. Kun terveille vapaaehtoisille annettiin laskimoon 1 000 mg:n kerta-annos, ihon rakkulanesteen AUC-arvo kohosi (sitoutunut ja vapaa dalbavansiini) noin 60 prosenttiin verrattuna plasman AUC-arvoon annoksen jälkeisenä päivänä 7.
Biotransformaatio
Metaboliitteja ei ole havaittu merkittävässä määrin ihmisen plasmassa. Hydroksidalbavansiini- ja mannosyyliaglykoni-nimisiä metaboliitteja on havaittu virtsassa (< 25 % annetusta annoksesta). Näiden metaboliittien tuottamisesta huolehtivia aineenvaihduntareittejä ei ole tunnistettu, mutta koska aineenvaihdunnan osuus dalbavansiinin kokonaiseliminaatiosta on suhteellisen pieni, dalbavansiinin metabolian estämisen tai induktion aiheuttavia lääkkeiden yhteisvaikutuksia ei ole odotettavissa. Hydroksidalbavansiinin ja mannosyyliaglykonin antibakteerinen aktiivisuus on huomattavasti pienempi kuin dalbavansiinin vastaava aktiivisuus.
Eliminaatio
Sen jälkeen, kun terveille tutkittaville oli annettu 1 000 mg:n kerta-annos dalbavansiinia, tästä annoksesta keskimäärin 19–33 % erittyi virtsaan dalbavansiinina ja 8–12 % sen metaboliittina, hydroksidalbavansiinina. Noin 20 % annetusta annoksesta erittyi ulosteisiin.
Erityisryhmät
Munuaisten vajaatoiminta
Dalbavansiinin farmakokinetiikkaa arvioitiin 28 aikuisella tutkittavalla, joilla oli eriasteista munuaisten vajaatoimintaa, ja 15 vastaavalla verrokilla, joiden munuaiset toimivat normaalisti. Sen jälkeen kun 500 mg:n tai 1 000 mg:n kerta-annos dalbavansiinia oli annettu, keskimääräinen puhdistuma plasmassa (CLT) väheni 11 %, 35 % ja 47 % lievää (CLCR 50–79 ml/min), kohtalaista (CLCR 30–49 ml/min) ja vaikeaa (CLCR < 30 ml/min) munuaisten vajaatoimintaa sairastavilla potilailla, tässä järjestyksessä, verrattuna terveisiin tutkittaviin, joiden munuaiset toimivat normaalisti. Keskimääräinen AUC-arvo oli noin 2 kertaa suurempi niillä tutkittavilla, joiden kreatiniinipuhdistuma oli < 30 ml/min. Näissä farmakokineettisissä dalbavansiinitutkimuksissa havaittiin vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla keskimääräinen plasman CLT-arvon lasku ja siihen liittyvä AUC0-∞-arvon nousu, jonka kliinistä merkitystä ei ole varmistettu. Tutkittavilla, joilla on loppuvaiheen munuaissairaus ja jotka saivat säännöllisesti suunnitelman mukaista munuaisdialyysihoitoa (3 kertaa/viikko), dalbavansiinin farmakokinetiikka oli samanlainen kuin lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla tutkittavilla, ja alle 6 % annetusta annoksesta on poistunut 3 tunnin kuluttua hemodialyysista. Annostusohjeet munuaisten vajaatoimintaa sairastaville aikuisille potilaille, ks. kohta Annostus ja antotapa.
Ei ole riittävästi tietoja, joiden perusteella voitaisiin suositella annoksen muuttamista 3 kuukauden – alle 18 vuoden ikäisille potilaille, joiden kreatiniinipuhdistuma on alle 30 ml/min/1,73 m2. Alle 3 kuukauden ikäisiä potilaita, joilla on munuaisten vajaatoiminta (ts. seerumin kreatiniiniarvo ≥ 2 kertaa normaalin yläraja tai virtsantuotto < 0,5 ml/kg/h) tai jotka tarvitsivat dialyysiä, ei otettu mukaan kliinisiin tutkimuksiin. Niillä 18 pediatrisella potilaalla, jotka olivat alle 3 kuukauden ikäisiä ja jotka otettiin mukaan kliinisiin tutkimuksiin, normalisoidun kreatiniinipuhdistuman vaihteluväli (Schwartzin vieritestausyhtälön perusteella) oli 34–118 ml/min/1,73 m2. Farmakokineettisiä havainnointitietoja ei ole saatavilla vaikeaa munuaisten vajaatoimintaa sairastavista pediatrisista potilaista. Populaatiofarmakokineettisen mallinnuksen perusteella dalbavansiinin ennustettu keskimääräinen AUC-arvo vaikeaa munuaisten vajaatoimintaa (CLCR ≤ 30 ml/min/1,73 m2) sairastavilla pediatrisilla tutkittavilla oli noin 13–30 % suurempi kuin samaa annosta saaneilla pediatrisilla potilailla, joiden munuaiset toimivat normaalisti.
Maksan vajaatoiminta
Dalbavansiinin farmakokinetiikkaa arvioitiin 17 tutkittavalla, joilla oli lievä, kohtalainen tai vaikea maksan vajaatoiminta, verraten sitä 9 vastaavaan terveeseen tutkittavaan, joiden maksa toimi normaalisti. Keskimääräinen AUC ei muuttunut tutkittavilla, joilla oli lievä maksan vajaatoiminta, verrattuna tutkittaviin, joiden maksa toimi normaalisti, mutta keskimääräinen AUC-arvo laski 28 % kohtalaista ja 31 % vaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla. Kohtalaista ja vaikeaa maksan vajaatoimintaa sairastavien tutkittavien alentuneen altistuksen syytä tai kliinistä merkitystä ei tiedetä. Annostusohjeet maksan vajaatoimintaa sairastaville potilaille, ks. kohta Annostus ja antotapa.
Sukupuoli
Dalbavansiinin farmakokinetiikassa ei ole havaittu kliinisesti merkittäviä sukupuoleen liittyviä eroja terveillä tutkittavilla eikä infektiopotilailla. Annoksen muuttamista sukupuolen perusteella ei suositella.
Iäkkäät
Dalbavansiinin farmakokinetiikka ei muuttunut merkitsevästi iän myötä; sen vuoksi annoksen muuttaminen iän perusteella ei ole tarpeen (ks. kohta Annostus ja antotapa). Dalbavansiinista iäkkäiden hoidossa on vain vähän kokemusta: 276 potilasta iältään ≥ 75 vuotta osallistui faasin 2/3 kliinisiin tutkimuksiin; näistä 173 sai dalbavansiinia. Kliinisissä tutkimuksissa oli mukana enintään 93-vuotiaita potilaita.
Pediatriset potilaat
Dalbavansiinin farmakokinetiikkaa on arvioitu 219:lla yksittäisellä pediatrisella potilaalla (ikä 4 vuorokautta – 17 vuotta, mukaan lukien vastasyntyneet keskoset [sikiöikä 32 – < 37 viikkoa; n = 3] ja täysiaikaisia vastasyntyneitä [sikiöikä 37–40 viikkoa; n = 5]). Mallin perusteella ennustettu dalbavansiinin AUC0–120h-arvo plasmassa vastasyntyneillä keskosilla syntymän hetkellä (sikiöikä 32 – < 37 viikkoa) oli noin 62 % aikuisten potilaiden arvosta, kun taas vanhemmissa pediatrisissa ryhmissä AUC0–120h-arvo oli 84–96 % aikuispotilaiden arvosta. Kaikissa pediatrisissa ikäryhmissä niiden potilaiden prosenttiosuus, jotka saavuttivat farmakokinetiikan/farmakodynamiikan tavoitteet lääkkeen in vivo -aktiivisuuden osalta, oli yli 90 %, kun MIC-arvot olivat enintään 0,5 mg/l.
Taulukko 3.
| Populaatiofarmakokineettiseen analyysiin perustuvat dalbavansiinin simuloidut keskimääräiset (keskihajonta) farmakokineettiset parametrit pediatrisilla potilailla ja aikuisilla1 | ||||||||
| Parametri | Vastasyntynyt keskonen | Täysiaikainen vastasyntynyt | Nuori imeväinen | Imeväinen | Pikkulapsi | Lapsi | Nuori | Aikuinen |
| Ikä | Sikiöikä 26 – < 37 viikkoa | Syntymä – 1 kuukausi | 1 kuukausi – < 3 kuukautta | 3 kuukautta – < 2 vuotta | 2 vuotta – < 6 vuotta | 6 vuotta – < 12 vuotta | 12 vuotta – < 18 vuotta | ≥ 18 vuotta |
| Annos | 22,5 mg/kg | 22,5 mg/kg | 22,5 mg/kg | 22,5 mg/kg | 22,5 mg/kg | 18 mg/kg | 18 mg/kg | 1 500 mg |
| Cmax (mg/l) | 228 (88) | 305 (130) | 305 (130) | 306 (130) | 303 (130) | 258 (110) | 250 (110) | 417 (110) |
| AUC0-120h (mg•h/l) | 6 480 (2 000) | 8 930 (2 900) | 9 040 (3 000) | 9 470 (3 100) | 10 100 (3 300) | 8 850 (2 900) | 9 030 (3 100) | 10 500 (3 100) |
| 1 Lähde: DAL-MS-03 | ||||||||
Prekliiniset tiedot turvallisuudesta
Dalbavansiinin toksisuutta on arvioitu sen jälkeen, kun valmistetta on annettu päivittäin laskimonsisäisesti noin 3 kuukauden ajan rotille ja koirille. Annoksesta riippuvaan toksisuuteen sisältyivät seerumin laboratorioarvot ja munuaisten ja maksan vaurioita osoittavat histologiset merkit, alentuneet punasoluarvot ja injektiokohdan ärsytys. Vain koirilla havaittiin annosriippuvaisesti infuusiosta johtuvia reaktioita, joista tyypillisiä olivat ihon turvotus ja/tai punoitus (ei liity injektiokohtaan), limakalvojen kalpeus, syljeneritys, oksentelu, sedaatio sekä lievä verenpaineen aleneminen ja sydämen lyöntitiheyden lisääntyminen. Nämä infuusioreaktiot olivat ohimeneviä (korjaantuivat 1 tunnin kuluessa annostelusta) ja johtuivat histamiinin vapautumisesta. Dalbavansiinin toksisuusprofiili nuorilla rotilla oli yhdenmukainen aiemmin täysikasvuisilla rotilla samansuuruisilla annoksilla (mg/kg/vrk) havaitun toksisuusprofiilin kanssa.
Rotilla ja kaneilla tehdyissä lisääntymistoksisuustutkimuksissa ei ilmennyt viitteitä teratogeenisista vaikutuksista. Kun altistukset rotilla olivat noin 3 kertaa kliinistä altistusta suurempia, havaittiin hedelmällisyyden alenemista ja alkiokuolemien ilmaantuvuuden lisääntymistä, sikiön painon ja luuston kovettumisen vähenemistä sekä vastasyntyneiden kuolleisuuden lisääntymistä. Kaneilla ilmeni emon toksisuuteen liittyviä keskenmenoja altistuksen ollessa ihmisellä käytettyä terapeuttista annosaluetta pienempi.
Pitkäaikaisia karsinogeenisuustutkimuksia ei ole tehty. Dalbavansiini ei ollut mutageeninen eikä klastogeeninen useissa in vitro- ja invivo -geenitoksisuustesteissä.
Farmaseuttiset tiedot
Apuaineet
Mannitoli (E421)
Laktoosimonohydraatti
Suolahappo (pH:n säätämiseen)
Natriumhydroksidi (pH:n säätämiseen)
Yhteensopimattomuudet
Natriumkloridiliuokset voivat saada aikaan saostumista, eikä niitä saa käyttää käyttökuntoon saattamiseen tai laimentamiseen (ks. kohta Käyttö- ja käsittelyohjeet).
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden tai infuusioliuosten kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Kuiva-aine: 4 vuotta
Xydalba-valmisteen on osoitettu säilyvän kemiallisesti ja fysikaalisesti stabiilina sekä käyttökuntoon saatettuna liuoksena että laimennettuna liuoksena 48 tuntia enintään 25 °C:n lämpötilassa. Käyttöön liittyvä kokonaisaika käyttökuntoon saattamisesta antoon ei saa ylittää 48 tuntia.
Mikrobiologiselta kannalta valmiste on käytettävä välittömästi. Jos valmistetta ei käytetä välittömästi, käytönaikainen säilytysaika ja säilytysolosuhteet ovat käyttäjän vastuulla, mutta ne ovat kuitenkin yleensä enintään 24 tuntia 2–8 °C:n lämpötilassa, ellei käyttökuntoon saattaminen tai laimentaminen ole tapahtunut kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa. Ei saa jäätyä.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
XYDALBA kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
500 mg (L:kyllä) 1 kpl (976,01 €)
PF-selosteen tieto
Kertakäyttöinen 48 ml:n injektiopullo (tyypin I lasia), jossa on elastomeerista valmistettu tulppa ja vihreä napsautuskorkki.
Jokainen pakkaus sisältää yhden injektiopullon.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen tai vaaleankeltainen jauhe.
Käyttö- ja käsittelyohjeet
Xydalba saatetaan käyttökuntoon lisäämällä siihen steriiliä injektionesteisiin käytettävää vettä, minkä jälkeen valmiste laimennetaan 50 mg/ml:n (5-prosenttiseen) glukoosi-infuusionesteeseen.
Xydalba-injektiopullot ovat kertakäyttöisiä.
Ohjeet käyttökuntoon saattamiseen ja laimentamiseen
Xydalba-valmisteen käyttökuntoon saattamisessa ja laimentamisessa on käytettävä aseptista tekniikkaa.
1. Jokaisen injektiopullon sisältö on saatettava käyttökuntoon lisäämällä siihen hitaasti 25 ml injektionesteisiin käytettävää vettä.
2. Ei saa ravistaa. Vaahtoamisen välttämiseksi pyörittele välillä injektiopulloa ja kääntele sitä välillä ylösalaisin varovasti, kunnes sen sisältö on täysin liuennut. Käyttökuntoon saattaminen voi kestää 5 minuuttia.
3. Käyttökuntoon saatettu konsentraatti injektiopullossa sisältää 20 mg/ml dalbavansiinia.
4. Käyttökuntoon saatetun konsentraatin on oltava kirkas, väritön tai keltainen liuos, jossa ei ole näkyviä hiukkasia.
5. Käyttökuntoon saatettu konsentraatti on edelleen laimennettava 50 mg/ml:n (5-prosenttisella) glukoosi-infuusioliuoksella.
6. Käyttökuntoon saatetun konsentraatin laimentamista varten tarvittava määrä 20 mg/ml:n konsentraattia siirretään injektiopullosta infuusionestepussiin tai -pulloon, jossa on 50 mg/ml:n (5-prosenttista) glukoosi-infuusioliuosta. Esimerkki: 25 ml konsentraattia sisältää 500 mg dalbavansiinia.
7. Laimentamisen jälkeen infuusioliuoksen lopullisen pitoisuuden on oltava 1–5 mg/ml dalbavansiinia.
8. Infuusioliuoksen on oltava kirkas, väritön tai keltainen liuos, jossa ei ole näkyviä hiukkasia.
9. Jos hiukkasia tai värjääntymistä havaitaan, liuos on hävitettävä.
Xydalba-valmistetta ei saa sekoittaa muiden lääkevalmisteiden tai infuusioliuosten kanssa. Natriumkloridia sisältävät liuokset voivat saada aikaan saostumista, ja niitä EI saa käyttää käyttökuntoon saattamiseen tai laimentamiseen. Käyttökuntoon saatetun Xydalba-konsentraatin yhteensopivuus on varmistettu ainoastaan 50 mg/ml:n (5-prosenttisen) glukoosi-infuusioliuoksen kanssa.
Jos samalla infuusioletkulla annetaan muita lääkevalmisteita Xydalba-valmisteen lisäksi, letku on huuhdeltava 5-prosenttisella glukoosi-infuusioliuoksella aina ennen Xydalba-infuusion antoa ja sen jälkeen.
Käyttö pediatrisille potilaille
Pediatrisilla potilailla Xydalba-annos vaihtelee lapsen iän ja painon perusteella 1 500 mg:n enimmäisannokseen asti. Siirrä lapsen painoon perustuva tarvittava annos käyttökuntoon saatettua dalbavansiiniliuosta yllä olevien ohjeiden mukaisesti injektiopullosta infuusionestepussiin tai -pulloon, jossa on 50 mg/ml:n (5‑prosenttista) glukoosi-infuusioliuosta. Laimennetun liuoksen lopullisen dalbavansiinipitoisuuden on oltava 1–5 mg/ml.
Alla olevassa taulukossa 4 kerrotaan, miten valmistetaan infuusioliuos, jonka lopullinen pitoisuus on 2 mg/ml tai 5 mg/ml (riittää useimpiin tilanteisiin) ja joka annetaan ruiskupumpulla siten, että saavutetaan 22,5 mg/kg:n annos syntymästä korkeintaan 12 kuukauden ikäisille ja 1–12 kg painaville pediatrisille potilaille. Liuos voidaan valmistaa muuhunkin pitoisuuteen, mutta lopullisen pitoisuuden on oltava välillä 1–5 mg/ml dalbavansiinia. Tarkista laskelmat taulukosta 4. Esitetyt arvot ovat likimääräisiä. Huomaa, että taulukko EI sisällä kaikkia mahdollisia laskennallisia annoksia kaikille ikäryhmille, mutta sitä voidaan käyttää likimääräisen tilavuuden arviointiin laskelmien tarkistamista varten.
Taulukko 4. Xydalba-valmisteen valmistelu (infuusion lopullinen pitoisuus 2 mg/ml tai 5 mg/ml, annetaan ruiskupumpulla) syntymästä korkeintaan 12 kuukauden ikäisille pediatrisille potilaille (annos 22,5 mg/kg)
| Potilaan paino (kg) | Annos (mg), jolla saavutetaan 22,5 mg/kg | Käyttökuntoon saatetun dalbavansiiniliuoksen tilavuus (20 mg/ml), joka vedetään injektiopullosta (ml) | Laimentimen (50 mg/ml (5 %) glukoosiliuos) tilavuus, joka lisätään sekoittamista varten (ml) | Dalbavansiini-infuusioliuoksen lopullinen pitoisuus | Ruiskupumpulla annettava kokonaistilavuus (ml) |
| 1 | 22,5 | 10 ml | 90 ml | 2 mg/ml | 11,3 |
| 2 | 45,0 | 22,5 | |||
| 3 | 67,5 | 33,8 | |||
| 4 | 90,0 | 45,0 | |||
| 5 | 112,5 | 56,3 | |||
| 6 | 135,0 | 67,5 | |||
| 7 | 157,5 | 78,8 | |||
| 8 | 180,0 | 90,0 | |||
| 9 | 202,5 | 20 ml | 60 ml | 5 mg/ml | 40,5 |
| 10 | 225,0 | 45,0 | |||
| 11 | 247,5 | 49,5 | |||
| 12 | 270,0 | 54,0 |
Hävittäminen
Hävitä käyttökuntoon saatetun liuoksen mahdollinen käyttämättä jäänyt osa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
XYDALBA kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
500 mg 1 kpl
- Ei korvausta.
ATC-koodi
J01XA04
Valmisteyhteenvedon muuttamispäivämäärä
21.10.2025
Yhteystiedot
P.O. Box 1452
SE-25114 Helsingborg
Sweden
+46 42 13 57 70
www.advanzpharma.com
medicalinformation@advanzpharma.com
General enquiries: InfoAbcur@advanzpharma.com
Telefon: +46 42 13 57 70
Medical enquiries, Adverse events, product complaints: medicalinformation@advanzpharma.com
Telefon: +358 800 416231