
ERLOTINIB KRKA tabletti, kalvopäällysteinen 150 mg
Vaikuttavat aineet ja niiden määrät
Erlotinib Krka 25 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 25 mg erlotinibia (hydrokloridina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 15,72 mg laktoosia.
Erlotinib Krka 100 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 100 mg erlotinibia (hydrokloridina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 62,89 mg laktoosia.
Erlotinib Krka 150 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 150 mg erlotinibia (hydrokloridina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 94,34 mg laktoosia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen
Kliiniset tiedot
Käyttöaiheet
Ei-pienisoluinen keuhkosyöpä (NSCLC)
Erlotinib Krka on tarkoitettu ensilinjan hoidoksi paikallisesti levinneen tai metastasoituneen ei-pienisoluisen keuhkosyövän (NSCLC) hoitoon potilaille, joilla on aktivoivia EGFR-mutaatioita.
Erlotinib Krka on myös tarkoitettu ylläpitohoitoon heti ensimmäisessä linjassa annetun solunsalpaajahoidon jälkeen paikallisesti levinnyttä tai metastasoitunutta ei-pienisoluista keuhkosyöpää sairastaville potilaille, joilla on aktivoivia EGFR-mutaatioita ja joiden tauti on stabiili ensimmäisessä linjassa annetun solunsalpaajahoidon jälkeen.
Erlotinib Krka on tarkoitettu myös paikallisesti levinneen tai metastasoituneen NSCLC:n hoitoon, kun vähintään yksi aikaisempi solunsalpaajahoito on osoittautunut tehottomaksi. Erlotinib Krka on tarkoitettu potilaille, joiden kasvaimessa ei ole aktivoivia EGFR-mutaatioita, silloin kun muiden hoitovaihtoehtojen ei katsota sopivan potilaalle.
Määrättäessä Erlotinib Krka -valmistetta on huomioitava tekijät, jotka vaikuttavat elinajan pidentymiseen.
Hoidosta ei ole osoitettu saatavan elinaikahyötyä eikä muitakaan kliinisesti merkittäviä vaikutuksia potilaille, joiden kasvaimet ovat epidermaalinen kasvutekijäreseptori (EGFR) -IHC-negatiivisia (ks. kohta Farmakodynamiikka).
Haimasyöpä
Erlotinib Krka on tarkoitettu yhdessä gemsitabiinin kanssa metastasoituneen haimasyövän hoitoon.
Määrättäessä Erlotinib Krka -valmistetta on huomioitava tekijät, jotka vaikuttavat elinajan pidentymiseen (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Paikallisesti edennyttä tautia sairastavilla potilailla elinaikahyötyä ei ole osoitettu.
Ehto
Valmistetta tulee käyttää vain syövän hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Erlotinib Krka -hoito tulee toteuttaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Ei-pienisoluinen keuhkosyöpä
EGFR-mutaatiotestaus on tehtävä hyväksyttyjen käyttöaiheiden mukaisesti (ks. kohta Käyttöaiheet).
Erlotinib Krka -valmisteen suositeltu vuorokausiannos on 150 mg, joka otetaan vähintään tunti ennen ateriaa tai kaksi tuntia aterian jälkeen.
Potilaat, joilla on haimasyöpä
Erlotinib Krkan suositeltu vuorokausiannos on 100 mg, joka otetaan vähintään tunti ennen ateriaa tai kaksi tuntia aterian jälkeen yhdistelmähoitona gemsitabiinin kanssa (ks. gemsitabiinin valmisteyhteenvedosta käyttö haimasyövän hoidossa). Jos potilaalla ei esiinny ihottumaa ensimmäisten 4–8 hoitoviikon aikana, Erlotinib Krka -hoidon jatkaminen on arvioitava uudelleen (ks. kohta Farmakodynamiikka).
Jos annoksen muuttaminen on tarpeen, annosta on pienennettävä 50 mg kerrallaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Erlotinib Krka -valmistetta on saatavana vahvuuksina 25 mg, 100 mg ja 150 mg.
Annoksen muuttaminen saattaa olla tarpeen, jos samanaikaisesti annetaan CYP3A4-substraatteja tai muita CYP3A4-entsyymin toimintaa muuttavia aineita (ks. kohta Yhteisvaikutukset).
Maksan vajaatoiminta
Erlotinibi eliminoituu metaboloitumalla maksassa ja erittymällä sappeen. Erlotinibialtistus oli samanlainen kohtalaista maksan vajaatoimintaa sairastavilla potilailla (Child–Pugh pistemäärä 7–9) verrattuna potilaisiin, joiden maksa toimi normaalisti. Varovaisuutta on kuitenkin syytä noudattaa, jos Erlotinib Krka -valmistetta annetaan maksan vajaatoimintaa sairastaville potilaille. Annoksen pienentämistä tai Erlotinib Krka -hoidon keskeyttämistä on harkittava, jos vakavia haittavaikutuksia esiintyy. Erlotinibin turvallisuutta ja tehoa ei ole tutkittu vaikeaa maksan toimintahäiriötä (ASAT/SGOT ja ALAT/SGPT > 5 x ULN) sairastavilla potilailla. Erlotinib Krka -valmisteen käyttöä ei suositella vaikeaa maksan toimintahäiriötä sairastaville potilaille (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Erlotinibin tehoa ja turvallisuutta ei ole tutkittu munuaisten vajaatoimintaa sairastavilla potilailla (seerumin kreatiniini > 1,5 x normaalialueen yläraja). Farmakokineettisten tietojen perusteella annoksen muuttaminen ei näytä olevan tarpeen, jos potilaalla on lievä tai kohtalainen munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka). Erlotinib Krkaa ei suositella potilaille, joilla on vaikea munuaisten vajaatoiminta.
Pediatriset potilaat
Erlotinibin turvallisuutta ja tehoa hyväksyttyihin käyttöaiheisiin ei ole varmistettu alle 18-vuotiaiden potilaiden hoidossa. Erlotinib Krka -valmistetta ei suositella pediatrisille potilaille.
Tupakoijat
Tupakoinnin on osoitettu vähentävän erlotinibialtistusta 50–60 %:lla. Korkeimmat siedetyt erlotinibiannokset ovat olleet 300 mg tupakoivilla NSCLC-potilailla. Potilailla, jotka jatkavat tupakointia, 300 mg:n annoksen ei todettu parantavan toisen linjan hoidon tehoa solunsalpaajahoidon epäonnistuttua verrattuna suositeltuun 150 mg:n annokseen. 300 mg:n ja 150 mg:n annosten turvallisuutta koskevat tiedot olivat verrannolliset. Suurempia erlotinibiannoksia saaneilla potilailla todettiin kuitenkin ihottuman, interstitiaalisen keuhkosairauden ja ripulin ilmaantuvuuden numeerista lisääntymistä. Tupakoitsijoita on neuvottava lopettamaan tupakointi (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset, Farmakodynamiikka ja Farmakokinetiikka).
Vasta-aiheet
Yliherkkyys erlotinibille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
EGFR-mutaatiostatuksen arviointi
Kun Erlotinib Krka -valmistetta harkitaan paikallisesti edenneen tai metastasoituneen ei-pienisoluisen keuhkosyövän ensilinjan hoitoon tai ylläpitohoitoon, on tärkeää, että potilaan EGFR-mutaatiostatus määritetään.
EGFR-mutaatiostatus on määritettävä paikallisen hoitokäytännön mukaan joko kudosnäytteestä saatavasta kasvaimen DNA:sta tai verinäytteestä (plasmasta) saatavasta verenkierrossa olevasta vapaasta DNA:sta (circulating free DNA, cfDNA) EGFR-mutaatiostatuksen määrittämiseen sopivaksi osoitetulla validoidulla, robustilla, luotettavalla ja herkällä testillä, jolla on ennalta määritelty kynnysarvo.
Jos käytetään plasmaan perustuvaa cfDNA-testiä ja aktivoivien mutaatioiden testitulos on negatiivinen, on tehtävä mahdollisuuksien mukaan kudostesti, koska plasmaan perustuvat testit voivat antaa väärän negatiivisen tuloksen.
Tupakoijat
Tupakoitsijoita tulisi kehottaa lopettamaan tupakointi, sillä tupakoitsijoilla plasman erlotinibipitoisuudet alenevat tupakoimattomiin verrattuna. Pitoisuuden pienentyminen on todennäköisesti kliinisesti merkityksellistä (ks. kohdat Annostus ja antotapa, Yhteisvaikutukset, Farmakodynamiikka ja Farmakokinetiikka).
Interstitiaalinen keuhkosairaus
Potilailla, jotka ovat saaneet erlotinibia ei-pienisoluisen keuhkosyövän, haimasyövän tai muiden pitkälle edenneiden kiinteiden kasvainten hoitoon, on raportoitu melko harvoin interstitiaalisen keuhkosairauden (ILD) kaltaisia tapahtumia ja myös niihin liittyneitä kuolemantapauksia. Ei-pienisoluisen keuhkosyövän hoitoa koskevassa keskeisessä BR.21-tutkimuksessa ILD:n ilmaantuvuus oli erlotinibi-ryhmässä sama (0,8 %) kuin plaseboryhmässä. Ei-pienisoluista keuhkosyöpää koskeneiden satunnaistettujen kontrolloitujen kliinisten tutkimusten meta-analyysissä (vertailuryhmien puuttumisen takia ei otettu mukaan faasin I ja yhden hoitoryhmän faasin II tutkimuksia), ILD:n kaltaisten tapahtumien ilmaantuvuus erlotinibi-hoitoa saaneilla potilailla oli 0,9 % verrattuna 0,4 %:iin vertailuryhmien potilailla. Haimasyöpätutkimuksessa ILD:n kaltaisten tapahtumien ilmaantuvuus oli erlotinibin ja gemsitabiinin yhdistelmähoitoa saaneessa ryhmässä 2,5 % ja plaseboa ja gemsitabiinia saaneessa ryhmässä 0,4 %. Raportoituja diagnooseja epäillyissä ILD:n kaltaisissa tapahtumissa olivat pneumoniitti, sädepneumoniitti, yliherkkyydestä johtuva pneumoniitti, interstitiaalinen pneumonia, interstitiaalinen keuhkosairaus, obliteroiva bronkioliitti, keuhkofibroosi, akuutti hengitysvaikeusoireyhtymä (ARDS), alveoliitti ja keuhkoinfiltraatio. Oireiden ilmaantumisajankohta vaihteli muutamasta vuorokaudesta useisiin kuukausiin erlotinibi-hoidon alkamisen jälkeen. Sekoittavat tai myötävaikuttavat tekijät olivat yleisiä, ja niitä olivat esimerkiksi samanaikainen tai aikaisempi solunsalpaajahoito, aikaisempi sädehoito, taustalla oleva parenkymaalinen keuhkosairaus, metastasoitunut keuhkosairaus ja keuhkoinfektiot. ILD:n ilmaantuvuus oli suurempi Japanissa tehdyissä tutkimuksissa mukana olleilla potilailla (noin 5 %; kuolleisuusaste 1,5 %).
Jos potilaalle ilmaantuu äkillisesti uusia ja/tai eteneviä selittämättömiä keuhko-oireita, kuten hengenahdistusta, yskää ja kuumetta, erlotinibi-hoito on keskeytettävä diagnostisen arvioinnin ajaksi.
Erlotinibilla ja gemsitabiinilla samanaikaisesti hoidettavia potilaita on tarkkailtava huolellisesti mahdollisen ILD:n kaltaisen toksisuuden kehittymisen vuoksi. Jos ILD diagnosoidaan, erlotinibi-hoito on lopetettava ja aloitettava asianmukainen hoito tarpeen mukaan (ks. kohta Haittavaikutukset).
Ripuli, elimistön kuivuminen, elektrolyyttihäiriö ja munuaisten vajaatoiminta
Ripulia on todettu noin 50 %:lla erlotinibi-hoitoa saaneista potilaista, mikä sisältää hyvin harvinaiset kuolemaan johtaneet tapaukset. Kohtalaista tai vaikeaa ripulia tulisi hoitaa esimerkiksi loperamidilla. Annoksen pienentäminen saattaa olla tarpeen joissakin tapauksissa. Kliinisissä tutkimuksissa annoksia pienennettiin 50 mg kerrallaan.
Annoksen pienentämistä 25 mg kerrallaan ei ole tutkittu. Jos potilaalla esiintyy vaikeaa tai jatkuvaa ripulia, pahoinvointia, ruokahaluttomuutta tai oksentelua, johon liittyy kuivumista, erlotinibi-hoito on keskeytettävä ja aloitettava asianmukainen hoito nestevajauksen korjaamiseksi (ks. kohta Haittavaikutukset). Hypokalemiaa ja munuaisten vajaatoimintaa (myös kuolemaan johtaneita tapauksia) on raportoitu harvoin. Jotkut tapauksista olivat seurausta ripulista, oksentelusta ja/tai anoreksiasta johtuvasta vakavasta elimistön kuivumisesta, kun taas osassa tapauksista sekoittavana tekijänä oli samanaikainen solunsalpaajahoito. Mikäli ripuli on vakavaa, jatkuvaa tai johtaa elimistön kuivumiseen, erlotinibi-hoito tulisi keskeyttää ja ryhtyä tehokkaisiin toimenpiteisiin potilaan nesteyttämiseksi laskimonsisäisesti. Erityisesti tulisi kiinnittää huomiota potilaisiin, joilla on näitä haittavaikutuksia pahentavia riskitekijöitä (etenkin samanaikaiset kemoterapiat ja muut lääkitykset, oireet tai sairaudet, tai muut ennalta altistavat tekijät mukaan lukien korkea ikä). Lisäksi munuaisten toimintaa ja seerumin elektrolyyttejä (mukaan lukien kalium) tulisi tarkkailla potilailla, joilla on elimistön kuivumisriski.
Maksatoksisuus
Erlotinibi-hoidon aikana on raportoitu vakavia lääkeaineen aiheuttamia maksavaurioita, mukaan lukien hepatiittia, akuuttia hepatiittia ja maksan vajaatoimintaa (myös kuolemaan johtaneita tapauksia). Riskitekijöitä voivat olla olemassa oleva maksasairaus tai samanaikaiset maksatoksiset lääkitykset. Maksan toiminnan testausta suositellaan säännöllisin väliajoin erlotinibihoidon aikana. Maksan toiminnan seurannan pitää olla tiheämpää potilailla, joilla on olemassa oleva maksan vajaatoiminta tai sappitietukos. Jos potilas raportoi maksavaurioon mahdollisesti viittaavia oireita, kliiniset tutkimukset ja maksan toimintaa mittaavat testit on tehtävä viipymättä. Erlotinibin anto on keskeytettävä, jos maksan toiminnassa tapahtuu vakavia muutoksia (ks. kohta Haittavaikutukset). Erlotinibin käyttöä ei suositella potilaille, joilla on vaikea maksan toimintahäiriö.
Ruoansulatuskanavan perforaatio
Erlotinibilla hoidetuilla potilailla on suurentunut riski saada ruoansulatuskanavan perforaatio, joka havaitaan harvoin (mukaan lukien muutamat kuolemaan johtaneet tapaukset). Riski on suurentunut potilailla, jotka saavat samanaikaisesti verisuonten kasvua estäviä lääkkeitä, kortikosteroideja, tulehduskipulääkkeitä ja/tai taksaanipohjaisia solunsalpaajia tai joilla on ollut divertikkeleitä, maha- tai pohjukaissuolihaava. Erlotinibi-hoito on lopetettava pysyvästi, jos potilaalle muodostuu ruoansulatuskanavan perforaatio (ks. kohta Haittavaikutukset).
Rakkulaiset ja hilseilyä tai ihon kuoriutumista aiheuttavat iho-oireet
Rakkulaisia ja hilseileviä tai ihon kuoriutumista aiheuttavia iho-oireita on raportoitu. Hyvin harvoissa tapauksissa on ollut viitteitä Stevens–Johnsonin oireyhtymästä tai toksisesta epidermaalisesta nekrolyysistä, jotka olivat joissakin tapauksissa kuolemaan johtavia (ks. kohta Haittavaikutukset). Erlotinibi-hoito on keskeytettävä tai lopetettava, jos potilaalle muodostuu vaikeita rakkulaisia, hilseileviä tai ihon kuoriutumista aiheuttavia iho-oireita. Potilaat, joilla on rakkulaisia ja hilseileviä tai ihon kuoriutumista aiheuttavia ihosairauksia, on testattava ihoinfektion varalta, ja heitä on hoidettava paikallisten hoitosuositusten mukaisesti.
Silmäoireet
Jos potilaalla on sarveiskalvontulehduksen akuutteja tai pahenemiseen viittaavia löydöksiä tai oireita, esimerkiksi silmätulehdusta, kyynelvuotoa, valoherkkyyttä, näön hämärtymistä, silmäkipua ja/tai silmän punoitusta, hänet on viipymättä ohjattava silmälääkärin arvioon. Jos diagnoosina on haavainen sarveiskalvontulehdus, erlotinibi-hoito on keskeytettävä tai lopetettava. Jos diagnoosina on sarveiskalvontulehdus, hoidon jatkamisen hyötyjä ja riskejä on arvioitava huolella. Erlotinibia on käytettävä varoen potilailla, joilla on aiemmin esiintynyt sarveiskalvontulehdusta, haavaista sarveiskalvontulehdusta tai vaikeaa kuivasilmäisyyttä. Piilolasien käyttö on myös riskitekijä sarveiskalvontulehdukselle ja haavautumille. Hyvin harvoissa tapauksissa erlotinibilla hoidetuilla potilailla on raportoitu sarveiskalvon perforaatiota tai haavautumia (ks. kohta Haittavaikutukset).
Yhteisvaikutukset muiden lääkevalmisteiden kanssa
Voimakkaat CYP3A4-induktorit voivat heikentää erlotinibin tehoa, kun taas voimakkaat CYP3A4-estäjät voivat lisätä toksisuutta. Tämän tyyppisten aineiden samanaikaista käyttöä on vältettävä (ks. kohta Yhteisvaikutukset).
Muut yhteisvaikutukset
Erlotinibille on ominaista liukoisuuden väheneminen, jos pH on yli 5. Ruoansulatuskanavan yläosan pH:ta muuttavat lääkkeet, kuten protonipumpun estäjät, H2-salpaajat ja antasidit, saattavat muuttaa erlotinibin liukoisuutta ja siten sen biologista hyötyosuutta. Annettaessa erlotinibia samanaikaisesti tämän tyyppisten lääkkeiden kanssa annoksen nosto ei todennäköisesti kompensoi altistuksen pienenemistä. Erlotinibin ja protonipumpun estäjien samanaikaista käyttöä on vältettävä. Erlotinibin käyttöä yhdessä H2-salpaajien ja antasidien kanssa ei ole tutkittu. Biologisen hyötyosuuden aleneminen on kuitenkin todennäköistä ja siksi tällaisten yhdistelmien käyttöä tulisi välttää (ks. kohta Yhteisvaikutukset). Jos antasidien käyttö on välttämätöntä erlotinibi-hoidon aikana, kyseinen lääke tulisi ottaa vähintään 4 tuntia ennen päivittäistä erlotinibi-annosta tai 2 tuntia sen jälkeen.
Apuaineet
Erlotinib Krka sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Erlotinibi ja muut CYP-substraatit
Erlotinibi on voimakas CYP1A1:n estäjä, kohtalainen CYP3A4:n ja CYP2C8:n estäjä sekä voimakas UGT1A1-entsyymin katalysoiman glukuronidaation estäjä in vitro.
Voimakkaan CYP1A1:n eston fysiologista merkitystä ei tunneta, sillä CYP1A1-entsyymin esiintyminen ihmiskudoksissa on hyvin harvinaista.
Annettaessa erlotinibia samanaikaisesti siprofloksasiinin kanssa, joka on kohtalainen CYP1A2:n inhibiittori, erlotinibialtistus (AUC) lisääntyi merkitsevästi (39 %), kun taas maksimipitoisuuden (Cmax) muutos ei ollut tilastollisesti merkitsevä. Myös altistus aktiiviselle metaboliitille lisääntyi (AUC noin 60 % ja Cmax 48 %). Näiden lisäysten kliinistä merkitystä ei tunneta. Varovaisuutta on noudatettava, kun erlotinibia annetaan yhdessä siprofloksasiinin tai potentin CYP1A2:n inhibiittorin (esim. fluvoksamiini) kanssa. Jos potilaalla esiintyy erlotinibiin liittyviä haittavaikutuksia, erlotinibiannosta voidaan alentaa.
Erlotinibin anto ennen tai samanaikaisesti tyypillisten CYP3A4:n substraattien (midatsolaamin ja erytromysiinin) kanssa ei muuttanut näiden puhdistumaa, mutta näytti vähentävän suun kautta otetun midatsolaamin biologista hyötyosuutta jopa 24 %. Eräässä toisessa kliinisessä tutkimuksessa erlotinibi ei vaikuttanut samanaikaisesti annetun CYP3A4/2C8-substraatin, paklitakselin, farmakokinetiikkaan. Merkittävät vaikutukset muiden CYP3A4:n substraattien puhdistumaan ovat siksi epätodennäköisiä.
Glukuronidaation esto voi aiheuttaa yhteisvaikutuksia sellaisten lääkevalmisteiden kanssa, jotka ovat UGT1A1:n substraatteja ja poistuvat yksinomaan tämän reitin kautta. Potilailla, joilla UGT1A1:n pitoisuus on alhainen tai joilla on perinnöllinen glukuronidaatiohäiriö (esim. Gilbertin tauti), saattaa esiintyä kohonneita seerumin bilirubiiniarvoja. Näitä potilaita hoidettaessa on noudatettava varovaisuutta.
Erlotinibi metaboloituu maksassa maksan sytokromientsyymien, pääasiassa CYP3A4:n ja vähäisemmässä määrin CYP1A2:n, välityksellä. Maksan ulkopuolella tapahtuva metabolia, CYP3A4-entsyymin välityksellä suolistossa, CYP1A1:n välityksellä keuhkoissa ja CYP1B1:n välityksellä kasvainkudoksessa, vaikuttaa mahdollisesti myös erlotinibin metaboliseen puhdistumaan. Yhteisvaikutuksia voi esiintyä sellaisten lääkeaineiden kanssa, jotka metaboloituvat näiden entsyymien välityksellä tai ovat niiden estäjiä tai induktoreita.
CYP3A4:n voimakkaat estäjät vähentävät erlotinibin metaboliaa ja suurentavat erlotinibin pitoisuutta plasmassa. Kliinisessä tutkimuksessa, jossa erlotinibia annettiin yhtaikaa voimakkaan CYP3A4:n estäjän, ketokonatsolin (200 mg suun kautta kaksi kertaa vuorokaudessa 5 vuorokauden ajan) kanssa, erlotinibialtistus suureni (AUC 86 % ja Cmax 69 %). Siksi on syytä noudattaa varovaisuutta, jos erlotinibi yhdistetään voimakkaaseen CYP3A4:n estäjään, kuten sienilääkkeinä käytettäviin atsolijohdoksiin (ketokonatsoliin, itrakonatsoliin, vorikonatsoliin), proteaasinestäjiin, erytromysiiniin tai klaritromysiiniin. Erlotinibiannosta on pienennettävä tarvittaessa, varsinkin havaittaessa toksisia vaikutuksia.
CYP3A4:n voimakkaat induktorit tehostavat erlotinibin metaboliaa ja pienentävät merkittävästi erlotinibin pitoisuutta plasmassa. Kliinisessä tutkimuksessa, jossa erlotinibia annettiin yhtä aikaa voimakkaan CYP3A4:n induktorin, rifampisiinin (600 mg suun kautta kerran vuorokaudessa 7 vuorokauden ajan) kanssa, erlotinibin AUC:n mediaani pieneni 69 %. Rifampisiinin anto samanaikaisesti erlotinibin 450 mg:n kerta-annoksen kanssa johti erlotinibialtistuksen (AUC) keskiarvoon, joka oli 57,5 % erlotinibin 150 mg:n kerta-annoksen keskiarvosta, kun erlotinibi annettiin ilman rifampisiinihoitoa. Erlotinibin käyttöä samanaikaisesti CYP3A4:n induktorien kanssa tulee siksi välttää. Jos potilaita on tarpeen hoitaa samanaikaisesti Erlotinibilla ja potentilla CYP3A4:n induktorilla, kuten rifampisiinilla, annoksen lisäämistä 300 mg:aan tulisi harkita seuraten samalla tarkasti hoidon vaikutusta potilaiden turvallisuuteen (mukaan lukien munuaisten ja maksan toiminta sekä seerumin elektrolyytit). Jos potilaat sietävät hoitoa hyvin yli 2 viikon ajan, annoksen lisäämistä 450 mg:aan voidaan harkita tarkan turvallisuusseurannan jatkuessa. Myös muut induktorit, esimerkiksi fenytoiini, karbamatsepiini, barbituraatit tai mäkikuisma (hypericum perforatum) saattavat pienentää altistusta. Varovaisuutta on noudatettava, jos näitä lääkeaineita annetaan yhtaikaa erlotinibin kanssa. Mikäli mahdollista, olisi harkittava muita vaihtoehtoisia hoitoja, joilla ei ole voimakasta CYP3A4-entsyymiä indusoivaa vaikutusta.
Erlotinibi ja kumariiniantikoagulantit
Erlotinibilla hoidettavilla potilailla on raportoitu yhteisvaikutuksia kumariiniantikoagulanttien kanssa varfariini mukaan luettuna. Tästä on seurannut INR-arvojen (International Normalized Ratio) nousua ja lisääntyneitä verenvuototapahtumia, jotka ovat muutamissa tapauksissa johtaneet kuolemaan. Kumariiniantikoagulantteja saavia potilaita on tarkkailtava säännöllisesti tromboplastiiniajan tai INR-arvon muutosten havaitsemiseksi.
Erlotinibi ja statiinit
Erlotinibin ja statiinin yhtäaikainen käyttö saattaa lisätä riskiä statiinin aiheuttamalle myopatialle mukaan lukien rabdomyolyysi, jota on havaittu harvoin.
Erlotinibi ja tupakoijat
Farmakokineettisessä yhteisvaikutustutkimuksessa, jossa tupakoivia potilaita verrattiin tupakoimattomiin, havaittiin tupakoivilla potilailla merkittävä 2,8-kertainen alenema AUCinf-arvossa 24 tuntia erlotinibin annon jälkeen. Vastaava alenema tupakoivien potilaiden Cmax-arvossa oli 1,5‑kertainen sekä plasmapitoisuus pieneni 9-kertaisesti. Tämän vuoksi tupakointia jatkavia potilaita tulisi kannustaa lopettamaan tupakointi mahdollisimman aikaisessa vaiheessa ennen erlotinibi-hoidon aloittamista, koska muutoin plasman erlotinibipitoisuudet saattavat pienentyä. CURRENTS- tutkimuksesta saatujen tietojen perusteella suuremmasta 300 mg:n erlotinibiannoksesta ei havaittu aktiivisesti tupakoivilla potilailla olevan hyötyä suositeltuun 150 mg:n annokseen verrattuna. 300 mg:n ja 150 mg:n annosten turvallisuutta koskevat tiedot olivat verrannolliset. Suurempia erlotinibiannoksia saaneilla potilailla todettiin kuitenkin ihottuman, interstitiaalisen keuhkosairauden ja ripulin ilmaantuvuuden numeerista lisääntymistä (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet, Farmakodynamiikka ja Farmakokinetiikka).
Erlotinibi ja P-glykoproteiinin estäjät
Erlotinibi on lääkeaineita kuljettavan P-glykoproteiinin substraatti. P-glykoproteiinin estäjien, kuten siklosporiinin ja verapamiilin, käyttö samanaikaisesti erlotinibin kanssa saattaa johtaa erlotinibin muuttuneeseen jakautumiseen ja/tai eliminaatioon. Tämän yhteisvaikutuksen merkitystä esimerkiksi keskushermoston toksisuuden kannalta ei ole selvitetty. Tällaisissa tilanteissa on noudatettava varovaisuutta.
Erlotinibi ja pH:ta muuttavat lääkevalmisteet
Erlotinibille on ominaista liukoisuuden väheneminen, jos pH on yli 5. Ylemmän ruoansulatuskanavan pH:ta muuttavat lääkkeet saattavat vaikuttaa erlotinibin liukoisuuteen ja siten sen biologiseen hyötyosuuteen. Erlotinibin anto samanaikaisesti omepratsolin kanssa, joka on protonipumpun estäjä, vähensi erlotinibialtistusta (AUC) 46 % ja maksimipitoisuutta (Cmax) 61 %. Tmax-arvo tai puoliintumisaika eivät muuttuneet. Erlotinibin ja ranitidiini 300 mg:n (H2-reseptorin antagonisti) samanaikainen annostelu alensi erlotinibialtistusta (AUC) 33 %:lla ja maksimipitoisuutta (Cmax) 54 %:lla. Annoksen nostaminen ei todennäköisesti kompensoi altistuksen pienenemistä, jos erlotinibia annostellaan samanaikaisesti tämän tyyppisten lääkkeiden kanssa. Kun erlotinibi annosteltiin 2 tuntia ennen ranitidiini 150 mg:n annosta tai 10 tuntia sen jälkeen (kahdesti päivässä), erlotinibialtistus (AUC) aleni vain 15 %:lla ja maksimipitoisuus (Cmax) vain 17 %:lla. Antasidien vaikutusta erlotinibin imeytymiseen ei ole tutkittu, mutta imeytyminen saattaa huonontua ja siten johtaa plasmapitoisuuksien alenemiseen. Yhteenvetona voidaan todeta, että erlotinibin ja protonipumpun estäjien samanaikaista käyttöä on vältettävä. Jos antasidien käyttö on välttämätöntä erlotinibi-hoidon aikana, kyseinen lääke on otettava vähintään 4 tuntia ennen päivittäistä erlotinibi-annosta tai 2 tuntia sen jälkeen. Jos ranitidiinin käyttöä harkitaan, se on annosteltava porrastetusti, esimerkiksi erlotinibi otetaan joko vähintään 2 tuntia ennen ranitidiiniannosta tai 10 tuntia sen jälkeen.
Erlotinibi ja gemsitabiini
Ib-faasin tutkimuksessa gemsitabiini ei vaikuttanut merkittävästi erlotinibin farmakokinetiikkaan eikä erlotinibilla ollut merkittäviä vaikutuksia gemsitabiinin farmakokinetiikkaan.
Erlotinibi ja karboplatiini/paklitakseli
Erlotinibi nostaa platinapitoisuuksia. Kliinisessä tutkimuksessa erlotinibin käyttö samanaikaisesti karboplatiinin ja paklitakselin kanssa johti platinan AUC0-48-kokonaisarvon kasvuun 10,6 %:lla. Vaikka nousu on tilastollisesti merkitsevä, eron suuruusluokkaa ei pidetä kliinisesti merkittävänä. Kliinisessä käytössä saattaa olla muita samanaikaisesti vaikuttavia tekijöitä (kuten munuaisten vajaatoiminta), jotka johtavat lisääntyneeseen karboplatiinialtistukseen. Karboplatiinilla tai paklitakselilla ei ollut merkittäviä vaikutuksia erlotinibin farmakokinetiikkaan.
Erlotinibi ja kapesitabiini
Kapesitabiini saattaa nostaa erlotinibipitoisuuksia. Kun erlotinibia annettiin yhdessä kapesitabiinin kanssa, erlotinibin AUC kasvoi tilastollisesti merkitsevästi ja Cmax-arvo lähes merkitsevästi verrattuna toiseen tutkimukseen, jossa annettiin ainoastaan erlotinibia. Erlotinibilla ei ollut merkittäviä vaikutuksia kapesitabiinin farmakokinetiikkaan.
Erlotinibi ja proteasomin estäjät
Proteasomin estäjien, kuten bortetsomibin, voidaan vaikutusmekanisminsa takia odottaa vaikuttavan EGFR-estäjien, erlotinibi mukaan lukien, tehoon. Tätä vaikutusta tukevat vähäiset kliiniset tiedot sekä prekliiniset tutkimukset, joissa proteasomin on todettu aiheuttavan EGFR:n hajoamista.
Raskaus ja imetys
Raskaus
Erlotinibin käytöstä raskaana oleville naisille ei ole riittävästi tietoa. Eläinkokeissa ei ole havaittu merkkejä teratogeenisuudesta tai poikkeavasta synnytyksestä. Raskauteen liittyviä haittavaikutuksia ei kuitenkaan voida poissulkea, koska eläinkokeissa on havaittu lisääntynyttä alkion/sikiön kuolleisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista ihmiseen kohdistuvaa vaaraa ei tunneta.
Naiset, jotka voivat tulla raskaaksi
Naisia, jotka voivat tulla raskaaksi, on kehotettava välttämään raskaaksi tulemista erlotinibi-hoidon aikana. Asianmukaisia ehkäisymenetelmiä on käytettävä hoidon aikana ja vähintään 2 viikon ajan hoidon päättymisen jälkeen. Hoitoa voidaan jatkaa raskauden aikana vain, jos mahdollinen äidin saama hyöty on suurempi kuin sikiölle aiheutuva haitta.
Imetys
Ei tiedetä, erittyykö erlotinibi ihmisen rintamaitoon. Erlotinibin vaikutusta maidontuotantoon tai sen erittymistä rintamaitoon ei ole tutkittu. Koska hoidosta imetettävälle lapselle mahdollisesti aiheutuvaa haittaa ei tunneta, äitejä on kehotettava lopettamaan imettäminen Erlotinib Krka -hoidon ajaksi ja vähintään 2 viikon ajaksi viimeisen annoksen jälkeen.
Hedelmällisyys
Eläinkokeissa ei ole havaittu hedelmällisyyden heikentymistä. Hedelmällisyyteen liittyviä haittavaikutuksia ei voida kuitenkaan poissulkea, koska eläinkokeissa on havaittu vaikutuksia lisääntymistoimintoihin (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista ihmiseen kohdistuvaa vaaraa ei tunneta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia hoidon vaikutuksesta ajokykyyn tai koneidenkäyttökykyyn ei ole tehty, mutta erlotinibi ei heikennä psyykkistä suorituskykyä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Erlotinibin turvallisuustiedot perustuvat yli 1 500 potilaaseen, jotka saivat monoterapiana vähintään yhden 150 mg:n annoksen erlotinibia, ja yli 300 potilaaseen, jotka saivat erlotinibia 100 mg:n tai 150 mg:n annoksina yhdessä gemsitabiinin kanssa.
Ei-pienisoluinen keuhkosyöpä (erlotinibi-monoterapiahoito)
Ensilinjan hoito potilailla, joilla on EGFR-mutaatioita
Avoimeen, satunnaistettuun faasin III tutkimukseen ML20650 osallistui 154 potilasta. Erlotinibin turvallisuutta ensilinjan hoidossa arvioitiin 75 NSCLC-potilaalla, joilla oli aktivoivia EGFR-mutaatioita kasvaimessa.
Tutkimuksessa ML20650 yleisimmin esiintyneet haittavaikutukset erlotinibilla hoidetuilla potilailla olivat ihottuma ja ripuli. Useimmat haittavaikutukset olivat astetta 1 ja 2 eivätkä vaatineet hoitoa. Tarkat tiedot ihottuman ja ripulin vaikeusasteista ja ilmaantuvuuksista kaikissa kliinisissä tutkimuksissa ovat jäljempänä kohdassa ”Valikoitujen haittavaikutusta kuvaus”.
Ylläpitohoito
Kahdessa muussa kaksoissokkoutetussa, satunnaistetussa ja plasebokontrolloidussa faasin III tutkimuksessa BO18192 (SATURN) ja BO25460 (IUNO) erlotinibia annettiin ylläpitohoitona ensilinjan solunsalpaajahoidon jälkeen. Nämä tutkimukset tehtiin yhteensä 1 532 potilaalla, joilla oli edennyt, uusiutunut tai metastaattinen ei-pienisoluinen keuhkosyöpä. Potilaat olivat saaneet ensilinjan standardihoitona platinapohjaista solunsalpaajahoitoa.
Tutkimuksissa BO18192 ja BO25460 yleisimmin esiintyneet haittavaikutukset erlotinibilla hoidetuilla potilailla olivat ihottuma ja ripuli.
Toisen linjan ja myöhempien linjojen hoito
Yleisimmin raportoituja haittavaikutuksia olivat ihottuma ja ripuli satunnaistetussa kaksoissokkotutkimuksessa (BR.21; erlotinibin annostelu toisessa linjassa). Useimpien oireiden vaikeusasteluokka oli 1/2, eivätkä ne vaatineet erityisiä toimenpiteitä.
Ihottuma ilmaantui keskimäärin 8 vuorokauden (mediaani) ja ripuli 12 vuorokauden (mediaani) kuluttua.
Haimasyöpä (erlotinibi-yhdistelmähoito gemsitabiinin kanssa)
Keskeisessä PA.3-haimasyöpätutkimuksessa, jossa potilaat saivat erlotinibia 100 mg:n annoksina yhdessä gemsitabiinin kanssa, yleisimmät haittavaikutukset olivat uupumus, ihottuma ja ripuli. Ihottuma ilmaantui keskimäärin 10 vuorokauden (mediaani) ja ripuli 15 vuorokauden (mediaani) kuluttua.
Haittavaikutustaulukko
Yhteenveto kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeen pelkästään erlotinibin tai erlotinibin ja solunsalpaajahoidon yhdistelmän käytössä havaittujen haittavaikutusten ilmaantuvuudesta esitetään taulukossa 1. Haittavaikutukset luetellaan MedDRA -elinjärjestelmäluokituksen mukaisesti. Kunkin haittavaikutuksen esiintymistiheys esitetään seuraavaan esitystavan mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1000, < 1/100), harvinainen (≥ 1/10 000, < 1/1000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Yhteenveto kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeen havaituista haittavaikutuksista yleisyysluokittain
| Infektiot | ||
| Hyvin yleinen | infektio* | |
| Aineenvaihdunta ja ravitsemus | ||
| Hyvin yleinen | ruokahaluttomuus, painon lasku | |
| Psyykkiset häiriöt | ||
| Hyvin yleinen | masennus | |
| Hermosto | ||
| Hyvin yleinen | neuropatia, päänsärky | |
| Silmät | ||
| Hyvin yleinen | silmän kuivuudesta johtuva sarveis- ja sidekalvon tulehdus | |
| Yleinen | sarveiskalvotulehdus, sidekalvotulehdus | |
| Melko harvinainen* | silmäripsien muutokset* | |
| Hyvin harvinainen | sarveiskalvon perforaatiot, sarveiskalvon haavauma, uveiitti | |
| Hengityselimet, rintakehä ja välikarsina | ||
| Hyvin yleinen | hengenahdistus, yskä | |
| Yleinen | nenäverenvuoto | |
| Melko harvinainen | interstitiaalinen keuhkosairaus* | |
| Ruoansulatuselimistö | ||
| Hyvin yleinen | ripuli*, pahoinvointi, oksentelu, suutulehdus, vatsakipu, dyspepsia, ilmavaivat | |
| Yleinen | ruoansulatuskanavan verenvuoto* | |
| Melko harvinainen | ruoansulatuskanavan perforaatio* | |
| Harvinainen | suoliston ilmakuplatauti | |
| Maksa ja sappi | ||
| Hyvin yleinen | poikkeavuudet maksan toimintakokeissa* | |
| Harvinainen | maksan vajaatoiminta*, hepatiitti | |
| Tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin) | akuutti hepatiitti | |
| Iho ja ihonalainen kudos | ||
| Hyvin yleinen | ihottuma*, kutina | |
| Yleinen | alopesia, ihon kuivuminen, kynnenvierustulehdus, karvatuppitulehdus, akne tai aknetyyppinen ihottuma, ihon haavaumat | |
| Melko harvinainen | hirsutismi, kulmakarvojen muutokset, hauraat ja irtoilevat kynnet, lievät ihoreaktiot, kuten hyperpigmentaatio | |
| Harvinainen | kämmenten ja jalkapohjien erytrodysestesia | |
| Hyvin harvinainen | Stevens–Johnsonin oireyhtymä / toksinen epidermaalinen nekrolyysi* | |
| Munuaiset ja virtsatiet | ||
| Yleinen | munuaisten vajaatoiminta | |
| Hyvin yleinen | munuaistulehdus, proteinuria | |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Hyvin yleinen | uupumus, kuume, vilunpuistatukset | |
* Ks. lisätietoja jäljempänä kohdassa ”Valikoitujen haittavaikutusten kuvaus”.
Valikoitujen haittavaikutusten kuvaus
Ihottuma
Ihottuma käsittää aknetyyppisen ihottuman. Ihottuma ilmenee yleensä lievänä tai keskivaikeana punoittavana ja märkänäppyläisenä ihottumana, joka voi ilmaantua auringolle altistuneille alueille tai pahentua näillä alueilla. Auringolle altistuvien potilaiden voi olla suositeltavaa käyttää suojaavaa vaatetusta ja/tai aurinkosuojavoidetta (esim. mineraaleja sisältävää aurinkosuojavoidetta).
Ripuli
Ripulista voi aiheutua elimistön kuivumista, hypokalemia ja munuaisten vajaatoiminta sekä kuolemantapauksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Taulukko 2. Yhteenveto kussakin kliinisessä tutkimuksessa havaituista ihottuman ja ripulin ilmaantuvuudesta ja vaikeusasteesta
| Tutkimus | Käyttöaihe | Ihottuma (%) | Ripuli (%) | ||||||||
| Vaikeusaste | Toimenpide | Vaikeusaste | Toimenpide | ||||||||
| Mikä tahansa | 3 | 4 | Lopetus1 | Muutos2 | Mikä tahansa | 3 | 4 | Lopetus1 | Muutos2 | ||
| ML20650 | Ei-pienisoluinen keuhkosyöpä | 80 | 9 | 0 | 1 | 11 | 57 | 4 | 0 | 1 | 7 |
| BO18192 | Ei-pienisoluinen keuhkosyöpä | 49,2 | 6.0 | 0 | 1 | 8,3 | 20,3 | 1,8 | 0 | <1 | 3 |
| BO25460 | Ei-pienisoluinen keuhkosyöpä | 39,4 | 5.0 | 0 | 0 | 5,6 | 24,2 | 2,5 | 0 | 0 | 2,8 |
| BR.21 | Ei-pienisoluinen keuhkosyöpä | 75 | 9 | 1 | 6 | 54 | 6 | 1 | 1 | ||
| PA.3 | Haimasyöpä | - | 5 | 1 | 2 | - | 5 | 1 | 2 | ||
1 Hoidon lopetus
2 Annosmuutos
Infektio
Nämä voivat olla vaikea-asteisia infektioita, joihin liittyy tai ei liity neutropeniaa, mukaan lukien keuhkokuume, sepsis ja selluliitti.
Silmäripsien muutokset
Muutoksia ovat mm. silmäripsien sisäänkasvaminen, liikakasvu ja paksuuntuminen.
Interstitiaalinen keuhkosairaus
Interstitiaaliseen keuhkosairauteen liittyy kuolemaan johtaneita tapauksia erlotinibia ei-pienisoluisen keuhkosyövän tai muiden pitkälle edenneiden kiinteiden kasvainten hoitoon saaneilla potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Japanilaisilla potilailla on havaittu tavanomaista suurempi ilmaantuvuus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ruoansulatuskanavan verenvuoto
Ruoansulatuskanavan verenvuotoihin liittyy myös kuolemaan johtaneita tapauksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kliinisissä tutkimuksissa joihinkin tapauksiin on liittynyt samanaikainen varfariinin käyttö ja joihinkin
samanaikainen tulehduskipulääkkeiden käyttö (ks. kohta Yhteisvaikutukset). Ruoansulatuskanavan perforaatioihin on liittynyt myös kuolemaan johtaneita tapauksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Poikkeavuudet maksan toimintakokeissa
Poikkeavuuksia ovat olleet suurentunut alaniiniaminotransferaasi- [ALAT], aspartaattiaminotransferaasi- [ASAT] ja bilirubiinipitoisuus. Tapaukset olivat vaikeusasteeltaan pääasiassa lieviä tai keskivaikeita, luonteeltaan ohimeneviä tai niihin liittyi maksametastaaseja.
Maksan vajaatoiminta
Tähän liittyy kuolemaan johtaneita tapauksia. Riskitekijöitä voivat olla mm. potilaalla jo ennestään
oleva maksasairaus tai samanaikaiset maksatoksiset lääkitykset (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Stevens–Johnsonin oireyhtymä / toksinen epidermaalinen nekrolyysi
Näihin liittyy kuolemaan johtaneita tapauksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet
Terveet tutkimushenkilöt ovat sietäneet erlotinibia oraalisina kerta-annoksina, joissa on ollut enintään 1 000 mg ja syöpäpotilaat annoksina, joissa on ollut enintään 1 600 mg erlotinibia. Terveille tutkimushenkilöille annetut toistuvat 200 mg:n annokset kahdesti vuorokaudessa olivat huonosti siedettyjä jo muutaman päivän hoidon jälkeen. Näistä tutkimuksista saatujen tulosten perusteella suositellun annoksen ylittäminen saattaa aiheuttaa vaikeita haittavaikutuksia, kuten ripulia, ihottumaa ja mahdollisesti maksan aminotransferaasiarvojen nousua.
Hoito
Jos yliannostusta epäillään, erlotinibi-lääkitys on keskeytettävä ja aloitettava oireenmukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: solunsalpaajat, proteiinikinaasin estäjät, ATC-koodi: L01EB02
Vaikutusmekanismi
Erlotinibi on ihmisen epidermaalisen kasvutekijäreseptorin tyypin 1 (EGFR, joka tunnetaan myös nimellä HER1) tyrosiinikinaasin estäjä. Erlotinibi estää voimakkaasti EGFR:n solunsisäistä fosforylaatiota. EGFR ilmentyy terveiden solujen ja syöpäsolujen pinnalla. Ei-kliinisissä koemalleissa EGFR-fosfotyrosiinin esto johtaa solun kasvun pysähtymiseen ja/tai solukuolemaan.
EGFR-mutaatiot saattavat aiheuttaa antiapoptoottisen ja proliferatiivisen signaalinvälitysreitin jatkuvan aktivaation. EGFR-mutaatiopositiivisissa kasvaimissa erlotinibin voimakas vaikutus perustuu erlotinibin voimakkaaseen kiinnittymiseen ATP:n sitoutumispaikkaan EGFR:n mutatoituneella kinaasialueella. Signaalinvälitysreitin estäminen estää solujen lisääntymisen ja käynnistää solukuolemat sisäisen apoptoottisen mekanismin kautta. Kasvaimen regressio havaitaan hiirimallissa, joka ilmentää näitä aktivoivia EGFR-mutaatioita.
Kliininen teho
Ensilinjan hoito NSCLC-potilaille, joilla on aktivoivia EGFR-mutaatioita (erlotinibi-monoterapiahoito)
Erlotinibin teho osoitettiin avoimessa, satunnaistetussa faasin III tutkimuksessa (ML20650, EURTAC), jossa ensilinjan NSCLC-potilailla oli aktivoivia EGFR-mutaatioita kasvaimissa. Tutkimukseen osallistui kaukaasialaisia potilaita, joilla oli metastasoitunut tai paikallisesti levinnyt NSCLC (aste IIIB ja IV). Potilaat, joiden kasvaimessa oli mutaatioita EGFR:n tyrosiinikinaasialueella (eksonissa 19 deleetio tai eksonissa 21 mutaatio), eivät olleet aiemmin saaneet solunsalpaajahoitoa tai muuta systeemistä kasvainkemoterapiaa levinneeseen tautiin. Potilaat satunnaistettiin suhteessa 1:1 kahteen ryhmään, jotka saivat joko erlotinibi 150 mg -valmistetta tai enintään neljä hoitosykliä platinapohjaista yhdistelmähoitoa.
Ensisijainen päätetapahtuma oli tutkijoiden määrittelemä PFS. Tehoa kuvaavat tulokset esitetään taulukossa 3.
Kuva 1: Kaplan–Meier-arvio tutkijoiden määrittelemästä taudin etenemisvapaa elinajasta (PFS) tutkimuksessa ML20650 (EURTAC) (cut-off: huhtikuussa 2012)
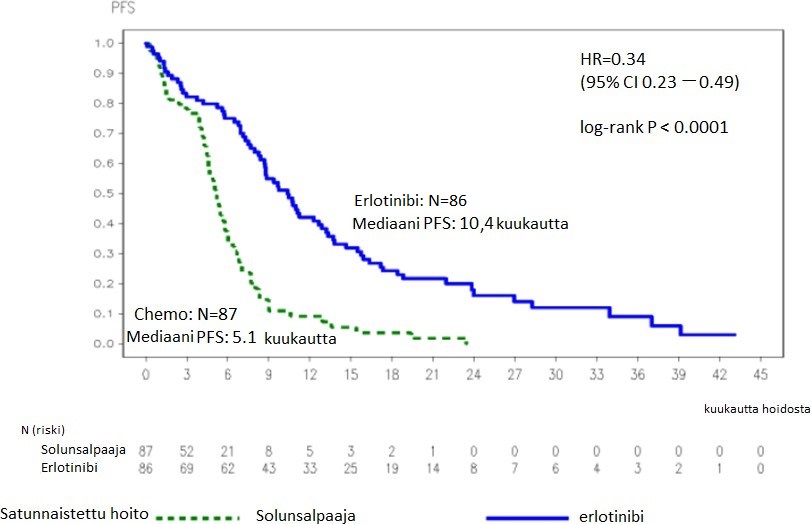
Taulukko 3: Tehoa kuvaavat tulokset tutkimuksessa ML20650 (EURTAC) erlotinibi verrattuna solunsalpaajahoitoon
Erlotinibi | Solun- salpaajahoito | Riskisuhde (95 % CI) | p-arvo | ||
Suunniteltu välianalyysi (35 % OS maturity) (n =153) Cut-off date: Aug 2010 | n = 77 | n = 76 | |||
Ensisijainen päätetapahtuma: | 9,4 | 5,2 | 0,42 | p < 0,0001 | |
taudin etenemisvapaa elinaika | |||||
(PFS, mediaani, kuukausina)* | |||||
tutkijoiden määrittelemä ** | |||||
[0,27-0,64] | |||||
Riippumaton arviointilautakunta ** | 10,4 | 5,4 | 0,47 | p =0,003 | |
[0,27-0,78] | |||||
Paras vastetaso (CR/PR) | 54,5 % | 10,5 % | p < 0,0001 | ||
Kokonaiselinaika (OS) (kuukausina) | 22,9 | 18,8 | 0,80 [0,47-1,37] | p = 0,4170 | |
Eksplora-tiivinen analyysi (40 % OS maturity) (n = 173) Cut-off date: Jan 2011 | n = 86 | n = 87 | |||
PFS (mediaani, kuukausina), tutkijoiden määrittelemä | 9,7 | 5,2 | 0,37 [0,27-0,54] | p < 0,0001 | |
Paras vastetaso (CR/PR) | 58,1 % | 14,9 % | p < 0,0001 | ||
OS (kuukausina) | 19,3 | 19,5 | 1,04 [0,65-1,68] | p = 0,8702 | |
Päivitetty analyysi (62 % OS maturity) (n = 173) Cut-off date: April 2012 | n = 86 | n = 87 | |||
PFS (mediaani, kuukausina) | 10,4 | 5,1 | 0,34 [0,23-0,49] | p < 0,0001 | |
OS*** (kuukausina) | 22,9 | 20,8 | 0,93 [0,64-1,36] | p = 0,7149 |
CR = complete response = täydellinen vaste; PR = partial response = osittainen vaste
* Taudin etenemisen tai kuoleman riskissä havaittiin 58 %:n väheneminen.
** Tutkijoiden ja riippumattoman arviointilautakunnan arvioiden yhtäpitävyys oli 70 %.
*** Suurta vaihtovuorisuutta havaittiin 82 %:lla niillä solunsalpaajahaaran potilaista, jotka olivat saaneet jatkohoitoa EGFR:n tyrosiinikinaasin estäjällä. Näistä potilasta kaikki paitsi kaksi olivat saaneet erlotinibia jatkohoitona.
Ylläpitohoito NSCLC-potilaille ensilinjan solunsalpaajan jälkeen (erlotinibi-monoterapiahoito)
Erlotinibin ylläpitohoidon tehoa ja turvallisuutta NSCLC-potilailla on tutkittu satunnaistetussa, kaksoissokkoutetussa ja plasebokontrolloidussa tutkimuksessa (BO18192, SATURN) ensilinjan solunsalpaajan jälkeen. Tutkimukseen osallistui 889 paikallisesti levinnyttä tai metastasoitunutta ei- pienisoluista keuhkosyöpää sairastavaa potilasta, joilla tauti ei ollut edennyt platinapohjaisen yhdistelmähoidon 4 syklin jälkeen. Potilaat satunnaistettiin kahteen ryhmään (1:1), jotka saivat suun kautta kerran päivässä joko erlotinibi 150 mg -valmistetta tai plasebovalmistetta taudin etenemiseen asti. Ensisijaiseen päätetapahtumaan kuului taudin etenemisvapaa elinaika (PFS) kaikilla tutkimuspotilailla. Tutkimushaarojen välillä väestötilastolliset tekijät ja taudin piirteet olivat lähtötilanteessa hyvin tasapainossa. Tutkimuksesta poissuljettiin potilaat, joilla oli ECOG PS > 1 ja merkittävä maksan tai munuaisten vajaatoiminta.
Tässä tutkimuksessa kokonaispotilasjoukon todettiin hyötyneen hoidosta ensisijaisena päätetapahtumana olleen etenemisvapaan elinajan suhteen (HR = 0,71, p < 0,0001) sekä toissijaisena päätetapahtumana olleen kokonaiselinajan suhteen (HR = 0,81, p = 0,0088). Suurin hyöty havaittiin kuitenkin ennalta määritellyssä eksploratiivisessa analyysissä potilaista, joilla oli aktivoivia EGFR- mutaatioita (n = 49), missä todettiin merkittävä hyöty etenemisvapaan elinajan osalta (HR = 0,10, 95 % luottamusväli 0,04–0,25; p < 0,0001) ja kokonaiselinajan riskisuhteen osalta, joka oli 0,83 (95 %:n luottamusväli 0,34–2,02). EGFR-mutaatiopositiivisessa potilasjoukossa 67 % plaseboa saaneista potilaista sai toisen tai myöhemmän linjan hoitona EGFR-tyrosiinikinaasin estäjiä.
Tutkimukseen BO25460 (IUNO) osallistui 643 potilasta, joilla oli pitkälle edennyt NSCLC ja joiden kasvaimissa ei ollut aktivoivia EGFR-mutaatioita (eksonissa 19 deleetio tai eksonissa 21 pistemutaatio [L858R]) ja joiden tauti ei ollut edennyt neljän hoitosyklin ajan annetun platinapohjaisen solunsalpaajahoidon jälkeen.
Tutkimuksen tavoite oli verrata kokonaiselinaikaa ensilinjan ylläpitohoitona annetun erlotinibihoidon ja taudin etenemisen aikana annetun erlotinibihoidon välillä. Tutkimus ei saavuttanut ensisijaista päätetapahtumaa. Kokonaiselinaika käytettäessä erlotinibi-valmistetta ensilinjan ylläpitohoitona ei ollut parempi verrattuna erlotinibi-valmisteen käyttöön toisen linjan hoitona, kun potilaiden kasvaimessa ei ollut aktivoivaa EGFR-mutaatiota (HR = 1,02, 95 %:n luottamusväli 0,85–1,22, p = 0,82). Toissijaisessa päätetapahtumassa eli etenemisvapaassa elinajassa ei todettu eroa erlotinibi-valmisteella ja plasebolla toteutetun ylläpitohoidon yhteydessä (HR = 0,94, 95 %:n luottamusväli 0,80–1,11; p = 0,48).
Tutkimuksen BO25460 (IUNO) tietojen perusteella erlotinibi-valmistetta ei suositella käytettäväksi ensilinjan ylläpitohoitona potilaille, joilla ei ole aktivoivaa EGFR-mutaatiota.
NSCLC-hoito, kun vähintään yksi aikaisempi solunsalpaajahoito on osoittautunut tehottomaksi (erlotinibi-monoterapiahoito)
Erlotinibin teho ja turvallisuus toisessa ja kolmannessa hoitolinjassa osoitettiin satunnaistetussa plasebokontrolloidussa kaksoissokkotutkimuksessa (BR.21), johon osallistuneilla 731 potilaalla oli paikallisesti levinnyt tai metastasoitunut ei-pienisoluinen keuhkosyöpä. Potilaat olivat saaneet aikaisemmin vähintään yhden solunsalpaajahoidon, joka oli osoittautunut tehottomaksi. Potilaat satunnaistettiin kahteen ryhmään (2:1), jotka saivat joko erlotinibia 150 mg tai plasebovalmistetta suun kautta kerran päivässä. Tutkimuksen tulosmuuttujat olivat kokonaiselinaika, taudin etenemisvapaa elinaika (PFS), hoitovaste, vasteen kesto, aika ennen keuhkosyöpään liittyvien oireiden (yskä, hengenahdistus ja kipu) pahenemista ja turvallisuus. Ensisijainen tulosmuuttuja oli elinaika.
Demografiset ominaisuudet olivat hyvin tasapainossa hoitoryhmien välillä. Noin kaksi kolmasosaa potilaista oli miehiä, ja ECOG-luokituksen mukainen toimintakyky oli lähtötilanteessa noin kolmanneksella potilaista 2 ja 9 %:lla potilaista 3. Erlotinibi-ryhmän potilaista 93 % ja plaseboryhmän potilaista 92 % oli saanut aikaisemmin platinapohjaista solunsalpaajahoitoa, ja erlotinibi-ryhmän potilaista 36 % ja plaseboryhmästä 37 % oli saanut taksaanihoitoa.
Korjattu riskisuhde kuolemalle (hazard ratio, HR) erlotinibi-ryhmässä suhteessa plaseboryhmään oli 0,73 (95 %:n luottamusväli 0,60–0,87) (p = 0,001). Erlotinibi-ryhmän potilaista 31,2 % ja plaseboryhmän potilaista 21,5 % oli elossa 12 kuukauden kuluttua. Kokonaiselinajan mediaani oli erlotinibi-ryhmässä 6,7 kuukautta (95 %:n luottamusväli 5,5–7,8 kuukautta) ja plaseboryhmässä 4,7 kuukautta (95 %:n luottamusväli 4,1–6,3 kuukautta).
Hoidon vaikutusta kokonaiselinaikaan tutkittiin erilaisissa potilaiden alaryhmissä. Erlotinibin vaikutus kokonaiselinaikaan oli samanlainen seuraavissa potilasryhmissä: lähtötason suorituskykyluokka (ECOG) 2-3 (riskisuhde 0,77; 95 %:n luottamusväli 0,6–1,0) tai 0–1 (riskisuhde 0,73; 95 %:n luottamusväli 0,6–0,9), mies (riskisuhde 0,76; 95 %:n luottamusväli 0,6-0,9) tai nainen (riskisuhde 0,80; 95 %:n luottamusväli 0,6–1,1), alle 65-vuotiaat potilaat (riskisuhde 0,75; 95 %:n luottamusväli 0,6–0,9) tai tätä vanhemmat potilaat (riskisuhde 0,79; 95 %:n luottamusväli 0,6–1,0), yksi aikaisempi hoitojakso (riskisuhde 0,76; 95 %:n luottamusväli 0,6–1,0) tai useampi kuin yksi hoitojakso (riskisuhde 0,75; 95 %:n luottamusväli 0,6–1,0), valkoihoiset (riskisuhde 0,79; 95 %:n luottamusväli 0,6–1,0) tai aasialaiset potilaat (riskisuhde 0,61; 95 %:n luottamusväli 0,4–1,0), adenokarsinoomapotilaat (riskisuhde 0,71; 95 %:n luottamusväli 0,6–0,9) tai levyepiteelikarsinoomapotilaat (riskisuhde 0,67; 95 %:n luottamusväli 0,5–0,9). Sen sijaan vaikutusta kokonaiselinaikaan ei havaittu potilailla, joilla oli histologialtaan muu kasvain kuin adenokarsinooma tai levyepiteelikarsinooma (riskisuhde 1,04; 95 %:n luottamusväli 0,7-1,5). Erlotinibin vaikutus kokonaiselinaikaan oli samanlainen potilailla, joilla oli diagnoosia tehtäessä alle asteen IV sairaus (riskisuhde 0,65; 95 %:n luottamusväli 0,5-0,8) kuin potilailla, joilla oli asteen IV sairaus (riskisuhde 0,92; 95%:n luottamusväli 0,7-1,2). Potilaat, jotka eivät olleet koskaan tupakoineet, hyötyivät erlotinibista enemmän (riskisuhde 0,42; 95 %:n luottamusväli 0,28–0,64) verrattuna nykyisiin tupakoitsijoihin tai aikaisemmin tupakoineisiin potilaisiin (riskisuhde 0,87; 95 %:n luottamusväli 0,71–1,05).
Niillä 45 %:lla potilaista, joiden kasvainten EGFR-status tunnettiin, riskisuhde oli EGFR-positiivisten potilaiden osalta 0,68 (95 %:n luottamusväli 0,49–0,94) ja EGFR-negatiivisten osalta 0,93 (95 %:n luottamusväli 0,63–1,36). EGFR-status määritettiin immunohistokemiallisesti käyttäen EGFR pharmDx -systeemiä. EGFR-negatiiviseksi määriteltiin tapaukset, joissa alle 10 % kasvainsoluista värjäytyi. Muilla 55 %:lla potilaista, joilla EGFR-statusta ei tunnettu, riskisuhde oli 0,77 (95 %:n luottamusväli 0,61–0,98).
Taudin etenemisvapaan elinajan mediaani oli erlotinibi-ryhmässä 9,7 viikkoa (95 %:n luottamusväli 8,4–12,4 viikkoa) ja plaseboryhmässä 8,0 viikkoa (95 %:n luottamusväli 7,9–8,1 viikkoa).
RECIST-kriteereihin perustuva objektiivinen hoitovaste oli erlotinibi-ryhmässä 8,9 % (95 %:n luottamusväli 6,4–12,0). Ensimmäiset 330 potilasta arvioitiin keskitetysti (hoitovaste 6,2 %), ja tutkijat arvioivat 401 potilasta (hoitovaste 11,2 %).
Vasteen keston mediaani oli 34,3 viikkoa, vaihteluväli oli 9,7–57,6+ viikkoa. Täydellinen vaste, osittainen vaste tai stabiili tauti todettiin erlotinibi-ryhmässä 44,0 %:lla ja plaseboryhmässä 27,5 %:lla potilaista (p = 0,004).
Erlotinibin aikaansaama elinaikahyöty tuli esiin myös potilailla, joilla ei saavutettu objektiivista tuumorivastetta (RECIST-kriteerien perusteella). Tästä oli osoituksena se, että riskisuhde kuolemalle oli 0,82 (95 %:n luottamusväli 0,68–0,99) potilailla, joiden paras hoitovaste oli stabiili tai etenevä tauti.
Erlotinibilla oli suotuisa vaikutus oireisiin, sillä se viivästytti merkitsevästi yskän, hengenahdistuksen ja kivun pahenemista plasebovalmisteeseen verrattuna.
Kaksoissokkoutetussa, satunnaistetussa faasin III tutkimuksessa (MO22162, CURRENTS) verrattiin kahta erlotinibi-annosta (300 mg versus 150 mg) paikallisesti levinnyttä tai metastasoitunutta ei- pienisoluista keuhkosyöpää (NSCLC) sairastavilla tupakoivilla (keskimäärin 38 askivuotta) potilailla toisen linjan hoidossa, kun solunsalpaajahoito oli epäonnistunut. 300 mg:n erlotinibi-annoksesta ei osoitettu etenemisvapaan elinajan suhteen suurempaa hyötyä suositeltuun annokseen nähden (7,00 viikkoa [300 mg:n annos] vs 6,86 viikkoa [suositeltu annos]).
Kaikki toissijaiset tehon päätetapahtumat olivat yhdenmukaisia ensisijaisen päätetapahtuman kanssa, eikä kokonaiselinajassa havaittu eroa 300 mg ja 150 mg erlotinibia vuorokaudessa saaneiden potilaiden välillä (riskisuhde [HR] 1,03, 95 %:n luottamusväli 0,80–1,32). 300 mg:n ja 150 mg:n annosten turvallisuutta koskevat tiedot olivat verrannolliset. Suurempia erlotinibiannoksia saaneilla potilailla todettiin kuitenkin ihottuman, interstitiaalisen keuhkosairauden ja ripulin ilmaantuvuuden numeerista lisääntymistä. CURRENTS-tutkimuksesta saatujen tietojen perusteella suuremmasta 300 mg:n erlotinibiannoksesta ei havaittu aktiivisesti tupakoivilla potilailla olevan hyötyä suositeltuun 150 mg:n annokseen verrattuna.
Potilaita ei valikoitu tähän tutkimukseen EGFR-mutaatiostatuksen perusteella. Ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakokinetiikka.
Haimasyöpä (erlotinibi yhdistelmähoitona gemsitabiinin kanssa PA.3-tutkimuksessa)
Erlotinibin ja gemsitabiinin yhdistelmän tehoa ja turvallisuutta arvioitiin paikallisesti levinneen, leikattavaksi soveltumattoman tai metastasoituneen haimasyövän ensivaiheen hoitona satunnaistetussa, plasebokontrolloidussa kaksoissokkotutkimuksessa. Potilaat satunnaistettiin saamaan joko erlotinibia tai plaseboa kerran vuorokaudessa jatkuvan hoito-ohjelman mukaan sekä gemsitabiinia laskimoon [1 000 mg/m2, 1. hoitojakso – 8 viikkoa: 1., 8., 15., 22., 29., 36. ja 43. päivänä; 2. hoitojakso ja sitä seuraavat jaksot – 4 viikkoa: 1., 8. ja 15. päivänä (hyväksytty annos ja hoito-ohjelma haimasyövässä, ks. gemsitabiinin valmisteyhteenveto)]. Erlotinibi tai plasebo otettiin suun kautta kerran vuorokaudessa taudin etenemiseen tai sietämättömien toksisten vaikutusten ilmaantumiseen asti. Tutkimuksen ensisijainen tulosmuuttuja oli elinaika.
Potilaiden demografiset ja sairauteen liittyvät ominaisuudet olivat lähtötilanteessa samanlaiset molemmissa hoitoryhmissä, erlotinibia 100 mg:n annoksina ja gemsitabiinia tai plaseboa ja gemsitabiinia saaneessa ryhmässä, lukuun ottamatta hieman suurempaa naisten osuutta erlotinibi/gemsitabiini-haarassa verrattuna plasebo/gemsitabiini-haaraan:
Lähtötilanne | Erlotinibi | Plasebo |
Naiset | 51 % | 44 % |
ECOG-toimintakykyluokka lähtötilanteessa (PS) = 0 | 31 % | 32 % |
ECOG-toimintakykyluokka lähtötilanteessa (PS) = 1 | 51 % | 51 % |
ECOG-toimintakykyluokka lähtötilanteessa (PS) = 2 | 17 % | 17 % |
Metastasoitunut tauti lähtötilanteessa | 77 % | 76 % |
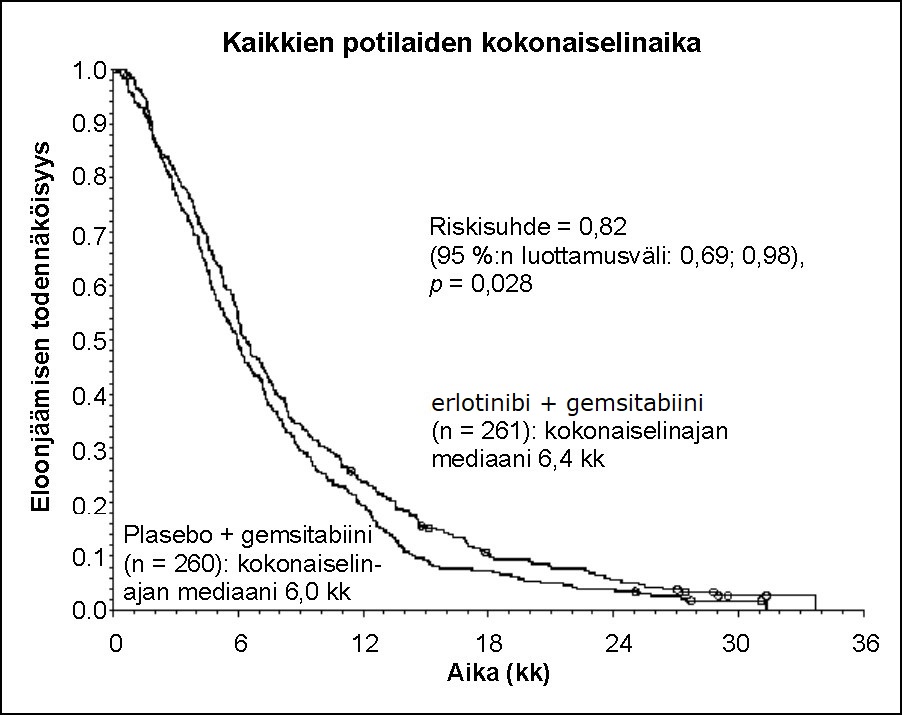
Elinajan arviointi tehtiin hoitoaikeen (”intent-to-treat”) mukaisessa potilasjoukossa, ja se perustui elinajan seurantatietoihin. Tulokset on esitetty seuraavassa taulukossa (metastasoitunutta ja paikallisesti edennyttä tautia koskevat tulokset on saatu eksploratiivisesta alaryhmäanalyysistä).
Tulosmuuttuja | Erlotinibi (kk) | Plasebo (kk) | Ero (Δ) (kk) | Luottamus -väli erolle (Δ) | Riski- suhde | Luottamusväli riskisuhteelle | p-arvo |
Kaikki potilaat | |||||||
Kokonaiselinajan mediaani | 6,4 | 6,0 | 0,41 | -0,54–1,64 | 0,82 | 0,69–0,98 | 0,028 |
Kokonaiselinajan keskiarvo | 8,8 | 7,6 | 1,16 | -0,05–2,34 | |||
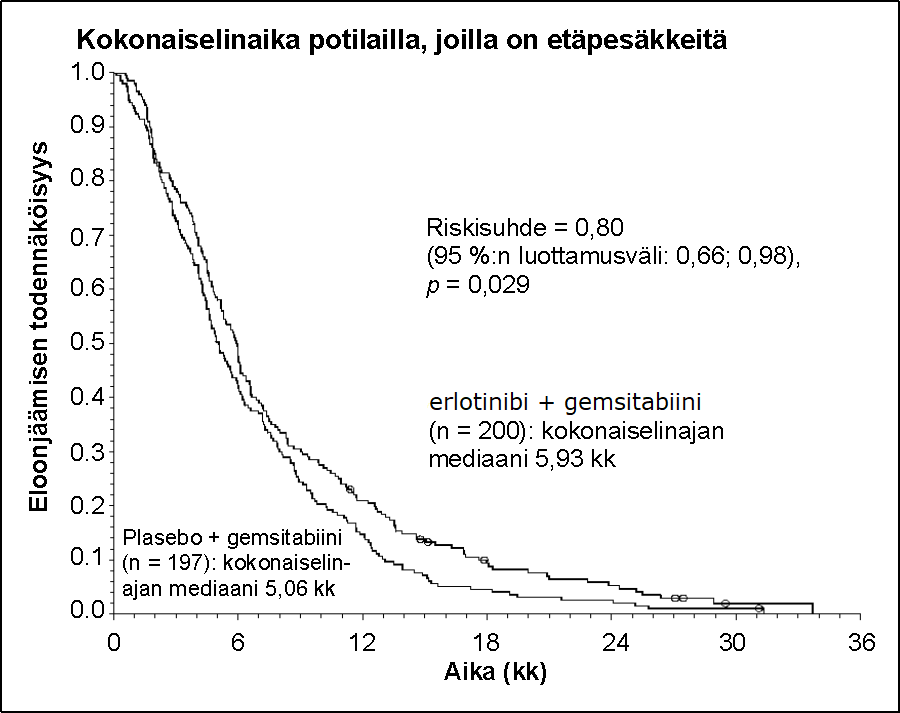
Potilaat, joilla metastasoitunut tauti | |||||||
Kokonaiselinajan mediaani | 5,9 | 5,1 | 0,87 | -0,26–1,56 | 0,80 | 0,66–0,98 | 0,029 |
Kokonaiselinajan keskiarvo | 8,1 | 6,7 | 1,43 | 0,17–2,66 | |||
Potilaat, joilla paikallisesti edennyt tauti | |||||||
Kokonaiselinajan mediaani | 8,5 | 8,2 | 0,36 | -2,43–2,96 | 0,93 | 0,65–1,35 | 0,713 |
Kokonaiselinajan keskiarvo | 10,7 | 10,5 | 0,19 | -2,43–2,69 | |||
Post-hoc-analyysin mukaan potilaat, joiden kliininen tila on lähtötilanteessa suotuisa (alhainen kivun intensiteetti, hyvä elämänlaatu ja hyvä suorituskyky), saattavat hyötyä erlotinibi-hoidosta muita potilaita enemmän. Hyöty tulee parhaiten esille, jos potilaan kivun intensiteettiä mittaava pistemäärä lähtötilanteessa on alhainen.
Post-hoc-analyysissä erlotinibia saaneilla potilailla, joille kehittyi ihottuma, kokonaiselinajan mediaani oli pidempi verrattuna potilaisiin, joille ei kehittynyt ihottumaa (kokonaiselinajan mediaani 7,2 kk vs 5 kk; riskisuhde 0,61).
Erlotinibia saavista potilaista 90 %:lle kehittyi ihottuma ensimmäisten 44 päivän aikana. Aika (mediaani) ihottuman ilmaantumiselle oli 10 päivää.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset erlotinibin käytöstä kaikkien pediatristen potilasryhmien hoidossa ei-pienisoluisessa keuhkosyövässä ja haimasyövässä (ks. kohta Annostus ja antotapa Pediatriset potilaat).
Farmakokinetiikka
Imeytyminen
Suun kautta annetun erlotinibin huippupitoisuus plasmassa saavutetaan noin 4 tunnin kuluttua annostelusta. Terveillä vapaaehtoisilla tehdyssä tutkimuksessa absoluuttisen hyötyosuuden arvioitiin olevan 59 %. Ruoka saattaa suurentaa lääkeainealtistusta oraalisen annoksen jälkeen.
Jakautuminen
Erlotinibin näennäisen jakautumistilavuuden keskiarvo on 232 l. Aine jakautuu myös ihmisen kasvainkudokseen. Erlotinibin pitoisuus kasvainkudoksessa oli keskimäärin 1 185 ng/g tutkimuksessa, jossa neljälle potilaalle (kolmella ei-pienisoluinen keuhkosyöpä ja yhdellä kurkunpään syöpä) annettiin erlotinibia 150 mg/vrk suun kautta ja kasvainnäytteet otettiin 9. hoitopäivänä kirurgisesti poistetuista kasvaimista. Tämä vastasi keskimäärin 63 %:a (vaihteluväli 5–161 %) vakaan tilan aikana mitatusta huippupitoisuudesta plasmassa. Tärkeimpien aktiivisten metaboliittien pitoisuus kasvainkudoksessa oli keskimäärin 160 ng/g, mikä vastasi keskimäärin 113 %:a (vaihteluväli 88–130 %) todetusta vakaan tilan huippupitoisuudesta plasmassa. Plasman proteiineihin sitoutumisaste on noin 95 %. Erlotinibi sitoutuu seerumin albumiiniin ja happamaan alfa-1-glykoproteiiniin (AAG).
Biotransformaatio
Erlotinibi metaboloituu ihmisen maksassa maksan sytokromientsyymien, pääasiassa CYP3A4:n ja vähäisemmässä määrin CYP1A2:n, välityksellä. Maksan ulkopuolella tapahtuva metabolia, CYP3A4-entsyymin välityksellä suolistossa, CYP1A1:n välityksellä keuhkoissa ja CYP1B1:n välityksellä kasvainkudoksessa, vaikuttaa mahdollisesti myös erlotinibin metaboliseen puhdistumaan.
Kolme päämetaboliareittiä on tunnistettu: 1) toisen tai molempien sivuketjujen O-demetylaatio ja sitä seuraava hapettuminen karboksyylihapoiksi, 2) asetyleeniosan hapettuminen ja sitä seuraava hydrolyysi aryylikarboksyylihapoksi ja 3) fenyyliasetyleeniosan aromaattinen hydroksylaatio. Ei- kliinisissä in vitro -analyyseissä ja in vivo -kasvainmalleissa jommankumman sivuketjun O-demetylaation kautta muodostuneilla erlotinibin päämetaboliiteilla, OSI-420:llä ja OSI-413:lla, on erlotinibiin verrattava teho. Niiden pitoisuus plasmassa on < 10 % erlotinibin pitoisuudesta, ja niillä on samanlainen farmakokinetiikka kuin erlotinibilla.
Eliminaatio
Erlotinibi erittyy pääasiassa metaboliitteina ulosteeseen (> 90 %), ja munuaisten kautta eliminoituu vain pieni osa (noin 9 %) oraalisesta annoksesta. Alle 2 % suun kautta annetusta annoksesta erittyy lähtöaineena. Populaatiofarmakokineettisessä analyysissä erlotinibia ainoana lääkkeenä saaneilla 591 potilaalla näennäisen puhdistuman keskiarvo oli 4,47 l/h ja puoliintumisajan mediaani 36,2 tuntia. Siten vakaan tason pitoisuus plasmassa saavutetaan todennäköisesti noin 7–8 vuorokaudessa.
Farmakokinetiikka erityisryhmissä
Populaatiofarmakokineettisen analyysin perusteella ei havaittu merkittävää yhteyttä ennustetun näennäisen puhdistuman ja potilaan iän, painon, sukupuolen tai etnisen taustan välillä. Potilaaseen liittyviä tekijöitä, jotka korreloivat erlotinibin farmakokinetiikkaan, olivat seerumin kokonaisbilirubiini, AAG ja tämänhetkinen tupakointi. Seerumin kohonneisiin kokonaisbilirubiini- ja AAG-pitoisuuksiin liittyi hidastunut erlotinibin puhdistuma. Näiden erojen kliininen merkitys on epäselvä. Tupakoitsijoilla erlotinibin puhdistuma oli sen sijaan nopeutunut. Tämä havainto vahvistettiin tupakoimattomilla ja tupakoivilla terveillä vapaaehtoisilla koehenkilöillä tehdyssä farmakokineettisessä tutkimuksessa, jossa annettiin 150 mg:n kerta-annos erlotinibia suun kautta. Cmax-arvon geometrinen keskiarvo oli 1056 ng/ml tupakoimattomilla ja 689 ng/ml tupakoitsijoilla; tupakoitsijoiden ja tupakoimattomien keskiarvojen suhde oli 65,2 % (95 %:n luottamusväli: 44,3–95,9; p = 0,031). AUC0-inf-arvon geometrinen keskiarvo oli 18726 ng•h/ml tupakoimattomilla ja 6718 ng•h/ml tupakoitsijoilla; keskiarvojen suhde oli 35,9 % (95 %:n luottamusväli: 23,7–54,3; p < 0,0001). C24h-arvon geometrinen keskiarvo oli 288 ng/ml tupakoimattomilla ja 34,8 ng/ml tupakoitsijoilla; keskiarvojen suhde oli 12,1 % (95 %:n luottamusväli: 4,82–30,2; p = 0,0001).
Faasin III NSCLC pivotaalitutkimuksessa tupakoitsijoilla erlotinibin alhaisin pitoisuus plasmassa vakaassa tilassa oli 0,65 µg/ml (n = 16), joka oli noin 2 kertaa alhaisempi kuin tupakoinnin lopettaneilla tai potilailla, jotka eivät olleet koskaan tupakoineet (1,28 µg/ml, n = 108). Tämä vaikutus oli seurausta erlotinibin näennäisen plasmapuhdistuman 24 %:n noususta. Faasin I annosvastetutkimuksessa tupakoivien NSCLC-potilaiden farmakokineettiset analyysit tehtiin vakaassa tilassa. Analyysit osoittivat, että erlotinibialtistuma kasvoi suhteessa annokseen, kun erlotinibin annosta nostettiin 150 mg:sta korkeimpaan siedettyyn 300 mg:n annokseen. Vakaan tilan pitoisuus plasmassa oli tupakoitsijoilla 1,22 µg/ml (n = 17) 300 mg:n annoksella. Ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakodynamiikka.
Farmakokineettisten tutkimustulosten perusteella tupakoitsijoita tulisi kehottaa lopettamaan tupakointi erlotinibi-hoidon ajaksi, sillä muutoin plasman lääkeainepitoisuudet saattavat pienentyä.
Populaatiofarmakokineettisen analyysin perusteella opioidi näytti suurentavan altistusta noin 11 %.
Toisessa populaatiofarmakokineettisessä analyysissä erlotinibia koskevat tiedot oli saatu 204 haimasyöpäpotilaalta, jotka saivat yhdistelmähoitona erlotinibia ja gemsitabiinia. Tämä analyysi osoitti, että erlotinibin puhdistumaan vaikuttavat kovariaatit olivat haimasyöpätutkimuksen potilailla hyvin samankaltaiset kuin aikaisemmassa farmakokineettisessä analyysissä potilailla, jotka saivat erlotinibia ainoana lääkeaineena. Uusia kovariaattivaikutuksia ei havaittu. Gemsitabiinin samanaikainen käyttö ei vaikuttanut erlotinibin puhdistumaan plasmasta.
Munuaisten vajaatoiminta
Erlotinibi ja sen metaboliitit eivät erity merkittävässä määrin munuaisten kautta, sillä vain alle 9 % kerta-annoksesta erittyy virtsaan. Populaatiofarmakokineettisessä analyysissä erlotinibipuhdistuman ja kreatiniinipuhdistuman välillä ei havaittu kliinisesti merkittävää yhteyttä. Tutkimustuloksia ei ole potilaista, joilla kreatiniinipuhdistuma oli alle 15 ml/min.
Maksan vajaatoiminta
Erlotinibi poistuu pääasiassa maksan kautta. Kiinteitä kasvaimia ja kohtalaista maksan vajaatoimintaa sairastavilla potilailla (Child–Pugh pistemäärä 7–9), erlotinibin AUC0-t-arvon geometrinen keskiarvo oli 27 000 ng•h/ml ja Cmax-arvon 805 ng/ml. Potilailla, joiden maksa toimi normaalisti mukaan lukien potilaat, joilla oli primaarinen maksasyöpä tai maksametastaaseja, vastaavat arvot olivat 29 300 ng•h/ml ja 1 090 ng/ml. Vaikka Cmax-arvo oli tilastollisesti merkitsevästi pienempi kohtalaista maksan vajaatoimintaa sairastavilla potilailla, eroa ei pidetä kliinisesti merkittävänä. Vaikean maksan vajaatoiminnan vaikutusta erlotinibin farmakokinetiikkaan ei ole tutkittu. Populaatiofarmakokineettisessä analyysissä seerumin kohonneisiin kokonaisbilirubiinipitoisuuksiin liittyi hitaampi erlotinibin puhdistuma.
Iäkkäät potilaat
Ei ole tutkittu erikseen iäkkäillä potilailla.
Pediatriset potilaat
Ei ole tutkittu erikseen pediatrisilla potilailla.
Prekliiniset tiedot turvallisuudesta
Pitkäaikaisen annostelun vaikutukset, joita havaittiin vähintään yhdellä eläinlajilla tai yhdessä tutkimuksessa, kohdistuivat sarveiskalvoon (atrofia, haavaumat), ihoon (karvatuppien degeneraatio ja tulehdus, punoitus ja karvanlähtö), munasarjoihin (atrofia), maksaan (maksanekroosi), munuaisiin (munuaisten papillanekroosi ja munuaistiehyiden laajentuma) ja ruoansulatuselimistöön (hidastunut mahalaukun tyhjeneminen ja ripuli). Punasoluarvot olivat alentuneet ja valkosoluarvot, pääasiassa neutrofiiliarvo, olivat koholla. Lisäksi havaittiin hoitoon liittyviä ALAT-, ASAT- ja bilirubiiniarvojen nousuja. Nämä löydökset tehtiin altistuksen ollessa selvästi kliinisesti merkittävän altistustason alapuolella.
Vaikutusmekanisminsa perusteella erlotinibi saattaa olla teratogeeninen. Lisääntymistoimintoihin kohdistuvaa toksisuutta selvittelevissä tutkimuksissa rotille ja kaniineille annettiin lähes maksimaalisia siedettyjä annoksia ja/tai emolle toksisia annoksia. Tutkimuksissa havaittiin lisääntymistoimintoihin kohdistuvia (alkiotoksisuutta rotilla ja alkion resorptiota sekä sikiötoksisuutta kaniineilla) ja kehitykseen kohdistuvia (jälkeläisten kasvun hidastumista ja elinajan lyhenemistä rotilla) toksisia vaikutuksia, mutta teratogeenisuutta tai fertiliteetin heikkenemistä ei havaittu. Nämä löydökset tehtiin kliinisesti merkittävällä altistustasolla.
Erlotinibilla saatiin negatiivinen tulos tavanomaisissa genotoksisuustutkimuksissa. Tulokset olivat negatiivisia kaksi vuotta kestäneissä rottien ja hiirten karsinogeenisuustutkimuksissa, vaikka altistukset ylittivät ihmisen terapeuttisen altistuksen (2 ja 10 kertaa korkeampi perustuen Cmax- ja/tai AUC-arvoihin).
Rotilla havaittiin lievä fototoksinen ihoreaktio UV-säteilyaltistuksen jälkeen.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Laktoosimonohydraatti
Mikrokiteinen selluloosa (E460)
Hydroksipropyyliselluloosa (E463)
Natriumlauryylisulfaatti
Natriumtärkkelysglykolaatti (tyyppi A)
Kalsiumsilikaatti (E522)
Magnesiumstearaatti (E470b)
Kalvopäällyste
Hypromelloosi (E464)
Propyleeniglykoli (E1520)
Titaanidioksidi (E171)
Talkki (E553b)
Punainen rautaoksidi (E172) – vain 100 mg
Keltainen rautaoksidi (E172) – vain 25 mg ja 100 mg
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Säilytä läpipainopakkauksessa. Herkkä kosteudelle.
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ERLOTINIB KRKA tabletti, kalvopäällysteinen
150 mg (L:kyllä) 30 fol (128,55 €)
PF-selosteen tieto
Läpipainopakkaus (OPA/Alu/PVC//paperi/Alu): 30 ja 60 kalvopäällysteistä tablettia, rasiassa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Erlotinib Krka 25 mg kalvopäällysteiset tabletit
Haalean keltaiset, pyöreät, hieman kaksoiskuperat, kalvopäällysteiset tabletit, joissa on viistotut reunat, ja joiden toiselle puolelle on kaiverrettu merkintä 25. Tabletin mitat: halkaisija noin 6 mm.
Erlotinib Krka 100 mg kalvopäällysteiset tabletit
Haalean vaaleanpunaiset, oranssiin vivahtavat, pyöreät, hieman kaksoiskuperat, kalvopäällysteiset tabletit, joissa on viistotut reunat, ja joiden toiselle puolelle on kaiverrettu merkintä 100. Tabletin mitat: halkaisija noin 11 mm.
Erlotinib Krka 150 mg kalvopäällysteiset tabletit
Valkoiset tai melkein valkoiset, pyöreät, kaksoiskuperat, kalvopäällysteiset tabletit, joissa on viistotut reunat, ja joiden toiselle puolelle on kaiverrettu merkintä 150. Tabletin mitat: halkaisija noin 12 mm.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ERLOTINIB KRKA tabletti, kalvopäällysteinen
150 mg 30 fol
- Ylempi erityiskorvaus (100 %). Erlotinibi: Paikallisesti levinneen tai etäpesäkkeitä lähettäneen ei-pienisoluisen keuhkosyövän hoito erityisin edellytyksin, etäpesäkkeitä lähettäneen haimasyövän hoito erityisin edellytyksin (191).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Erlotinibi: Eräiden syöpäsairauksien hoito erityisin edellytyksin (325).
ATC-koodi
L01EB02
Valmisteyhteenvedon muuttamispäivämäärä
11.06.2024
Yhteystiedot
Tekniikantie 14
02150 Espoo
Suomi
020-7545330
www.krka.biz
info.fi@krka.biz