
BEOVU injektioneste, liuos, esitäytetty ruisku 120 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Yleinen
Vaikuttavat aineet ja niiden määrät
Yksi millilitra injektionestettä (liuos) sisältää 120 mg brolusitsumabia*.
* Brolusitsumabi on humanisoitu, monoklonaalinen yksiketjuinen Fv-vasta-ainefragmentti (scFv-vasta-ainefragmentti), joka on valmistettu Escherichia coli ‑soluissa rekombinaatio-DNA-tekniikalla.
Beovu 120 mg/ml injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 19,8 mg brolusitsumabia 0,165 millilitrassa liuosta. Tämä määrä riittää yhteen 6 mg brolusitsumabia sisältävään 0,05 ml:n kerta-annokseen liuosta.
Beovu 120 mg/ml injektioneste, liuos
Yksi injektiopullo sisältää 27,6 mg brolusitsumabia 0,23 millilitrassa liuosta. Tämä määrä riittää yhteen 6 mg brolusitsumabia sisältävään 0,05 ml:n kerta-annokseen liuosta.
Apuaine, jonka vaikutus tunnetaan
Yksi esitäytetty ruisku sisältää 0,03 mg polysorbaatti 80:tä 0,165 millilitrassa liuosta. Tämä vastaa 0,01 mg polysorbaatti 80:tä per annos (0,05 ml).
Yksi injektiopullo sisältää 0,05 mg polysorbaatti 80:tä 0,23 millilitrassa liuosta. Tämä vastaa 0,01 mg polysorbaatti 80:tä per annos (0,05 ml).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste).
Kliiniset tiedot
Käyttöaiheet
Beovu on tarkoitettu aikuisille
- neovaskulaarisen (kostean) silmänpohjan ikärappeuman (AMD) hoitoon (ks. kohta Farmakodynamiikka)
- diabeettisesta makulaturvotuksesta (DME) johtuvan näön heikentymisen hoitoon (ks. kohta Farmakodynamiikka).
Ehto
Valmistetta saa antaa lääkäri, jolla on kokemusta lasiaiseen annettavista injektioista.
Annostus ja antotapa
Beovu-valmisteen antaa pätevä silmälääkäri, jolla on kokemusta injektioiden antamisesta silmän lasiaiseen.
Annostus
Kostea silmänpohjan ikärappeuma
Hoidon aloitus – latausvaihe
Suositeltu annos on 6 mg brolusitsumabia (0,05 ml liuosta) injektiona silmän lasiaiseen 4 viikon välein (kerran kuukaudessa) ensimmäisten 3 annoksen ajan. Tautiaktiivisuuden arviointia suositellaan 16 viikon (4 kuukauden) kuluttua hoidon aloittamisesta.
Vaihtoehtoisesti hoito voidaan aloittaa antamalla 6 mg brolusitsumabia (0,05 ml liuosta) 6 viikon välein ensimmäisten 2 annoksen ajan. Tautiaktiivisuuden arviointia suositellaan 12 viikon (3 kuukauden) kuluttua hoidon aloittamisesta. Kolmas annos voidaan antaa näöntarkkuuden ja/tai anatomisten parametrien perusteella viikolla 12 arvioidun tautiaktiivisuuden pohjalta.
Ylläpitohoito
Viimeisen latausannoksen jälkeen lääkäri voi muuttaa hoitovälejä yksilöllisesti näöntarkkuuden ja/tai anatomisten parametrien perusteella arvioimansa tautiaktiivisuuden pohjalta. Potilaille, joilla ei ole tautiaktiivisuutta, suositellaan hoitoa 12 viikon (3 kuukauden) välein. Potilaille, joilla on tautiaktiivisuutta, suositellaan hoitoa 8 viikon (2 kuukauden) välein. Jos potilaita hoidetaan treat-and-extend-hoito-ohjelman (ohjelma, jossa hoitoväliä voidaan pidentää asteittain silmän hoitovasteen perusteella) mukaisesti eikä merkkejä tautiaktiivisuudesta ole, hoitovälejä voidaan pidentää asteittain, kunnes tautiaktiivisuuden merkit uusiutuvat. Hoitoväliä tulee pidentää tai lyhentää enintään 4 viikkoa (1 kuukausi) kerrallaan (ks. kohta Farmakodynamiikka). Yli 20 viikon (5 kuukauden) hoitoväleistä on rajallisesti tietoa. Kahden Beovu-annoksen välinen hoitoväli ei saa olla lyhyempi kuin 8 viikkoa (2 kuukautta) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jos näkökykyä koskevat tulokset ja anatomiset tulokset viittaavat siihen, että potilas ei hyödy hoidon jatkamisesta, Beovu-hoito tulee lopettaa.
Diabeettinen makulaturvotus
Suositeltu annos on 6 mg brolusitsumabia (0,05 ml liuosta) injektiona silmän lasiaiseen 6 viikon välein ensimmäisten 5 annoksen ajan.
Tämän jälkeen lääkäri voi muuttaa hoitovälejä yksilöllisesti näöntarkkuuden ja/tai anatomisten parametrien perusteella arvioimansa tautiaktiivisuuden pohjalta. Potilaille, joilla ei ole tautiaktiivisuutta, suositellaan hoitoa 12 viikon (3 kuukauden) välein. Potilaille, joilla on tautiaktiivisuutta, suositellaan hoitoa 8 viikon (2 kuukauden) välein. Kun hoitoa on jatkettu 12 kuukauden ajan, potilaille, joilla ei ole tautiaktiivisuutta, voidaan harkita enintään 16 viikon (4 kuukauden) hoitovälejä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Jos näkökykyä koskevat tulokset ja anatomiset tulokset viittaavat siihen, että potilas ei hyödy hoidon jatkamisesta, Beovu-hoito tulee lopettaa.
Erityisryhmät
Iäkkäät potilaat
Annosta ei tarvitse muuttaa 65 vuotta täyttäneiden potilaiden kohdalla (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa munuaisten vajaatoimintapotilailla (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Brolusitsumabia ei ole tutkittu maksan vajaatoimintapotilailla. Annosta ei tarvitse muuttaa maksan vajaatoimintapotilailla (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Brolusitsumabin turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Beovu on tarkoitettu käytettäväksi vain silmän lasiaiseen.
Injektioneste on tarkastettava silmämääräisesti ennen antoa (ks. kohta Käyttö- ja käsittelyohjeet).
Intravitreaalinen injektiotoimenpide on toteutettava aseptisissa olosuhteissa. Tähän sisältyy kirurginen käsien desinfiointi ja steriilien käsineiden, steriilin liinan ja steriilin luomilevittimen (tai vastaavan) käyttö. Saatavilla on oltava steriilit parasenteesivälineet varmuuden vuoksi. Potilaan aiemmat yliherkkyysreaktiot on selvitettävä tarkoin ennen intravitreaalista toimenpidettä (ks. kohta Vasta-aiheet). Ennen injektiota silmänympärysiho, silmäluomi ja silmän pinta desinfioidaan laajakirjoisella paikallisella mikrobisidilla ja annetaan riittävä puudutus.
Injektioneula pistetään 3,5–4,0 mm limbuksesta posteriorisesti lasiaistilaan, vältetään horisontaalista meridiaania ja tähdätään silmämunan keskikohtaan. Annettava 0,05 ml:n tilavuus injisoidaan hitaasti. Kovakalvon pistoskohtaa vaihdetaan seuraavien injektioiden yhteydessä.
Potilaita on seurattava silmänpaineen kohoamisen varalta välittömästi intravitreaalisen injektion jälkeen. Sopiva seuranta voi sisältää näköhermon pään perfuusion tarkistuksen tai tonometrian. Steriilejä parasenteesivälineitä on tarvittaessa oltava saatavilla.
Intravitreaalisen injektion jälkeen potilaita on neuvottava viipymättä ilmoittamaan kaikista endoftalmiittiin viittaavista oireista (esim. silmäkipu, silmän punoitus, valoherkkyys, näön sumeneminen).
Esitäytetty ruisku
Esitäytetty ruisku on tarkoitettu vain yhtä käyttökertaa varten. Yhtä esitäytettyä ruiskua saa käyttää vain yhden silmän hoitoon.
Esitäytetyssä ruiskussa oleva tilavuus (0,165 ml) on suurempi kuin suositeltu annos (0,05 ml), joten osa esitäytetyssä ruiskussa olevasta tilavuudesta on hävitettävä ennen antoa.
Esitäytetyn ruiskun koko tilavuuden injisointi voi johtaa yliannokseen. Ilman poistamiseksi ylimääräisen lääkevalmisteen mukana, paina mäntää hitaasti, kunnes männän kuperan kumisen kärjen reuna on 0,05 ml:n annosmerkin kohdalla (mikä vastaa 50 μl, eli 6 mg brolusitsumabia).
Injektiopullo
Injektiopullo on tarkoitettu vain yhtä käyttökertaa varten. Yhtä injektiopulloa saa käyttää vain yhden silmän hoitoon.
Injektiopullossa oleva tilavuus (0,23 ml) on suurempi kuin suositeltu annos (0,05 ml), joten osa injektiopullossa olevasta tilavuudesta on hävitettävä ennen antoa.
Injektiopullon koko tilavuuden injisointi voi johtaa yliannokseen. Poista ilma ruiskusta varovasti ylimääräisen lääkevalmisteen mukana, ja säädä annos 0,05 ml:n annosmerkin kohdalle (mikä vastaa 50 μl, eli 6 mg brolusitsumabia).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiivinen okulaarinen tai periokulaarinen infektio tai sen epäily.
Aktiivinen silmänsisäinen inflammaatio.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Endoftalmiitti, silmänsisäinen inflammaatio, traumaattinen kaihi, verkkokalvon irtauma, verkkokalvon repeämä, verkkokalvon vaskuliitti ja/tai verkkokalvon verisuonitukos
Silmän lasiaiseen annettavien injektioiden, myös Beovu-injektioiden, yhteydessä on esiintynyt endoftalmiittia, silmänsisäistä inflammaatiota, traumaattista kaihia, verkkokalvon irtaumaa ja verkkokalvon repeämää (ks. kohta Haittavaikutukset). Beovu-valmistetta annettaessa on aina käytettävä asianmukaista aseptista injektiotekniikkaa.
Potilaita on neuvottava ilmoittamaan viipymättä edellä mainittuihin tapahtumiin viittaavista oireista.
Silmänsisäinen inflammaatio, mukaan lukien verkkokalvon vaskuliitti ja/tai verkkokalvon verisuonitukos
Beovu-valmisteen käytön yhteydessä on ilmoitettu silmänsisäistä inflammaatiota, mukaan lukien verkkokalvon vaskuliittia ja/tai verkkokalvon verisuonitukoksia (ks. kohdat Vasta-aiheet ja Haittavaikutukset). Potilailla, joille kehittyi hoidon aikana vasta‑aineita, todettiin enemmän silmänsisäisiä inflammaatiotapauksia. Selvitysten jälkeen havaittiin, että verkkokalvon vaskuliitti ja/tai verkkokalvon verisuonitukos ovat immuunivälitteisiä tapahtumia. Silmänsisäinen inflammaatio, mukaan lukien verkkokalvon vaskuliitti ja/tai verkkokalvon verisuonitukos, voi ilmaantua ensimmäisen intravitreaalisen injektion jälkeen ja milloin tahansa hoidon aikana. Useimmiten näitä tapahtumia on havaittu hoidon alussa.
Kliinisten tutkimusten perusteella näitä tapahtumia ilmaantui useammin Beovu-hoitoa saaneille naispotilaille kuin miespotilaille (esim. 5,3 %:lle naispotilaista ja 3,2 %:lle miespotilaista HAWK- ja HARRIER -tutkimuksissa), ja japanilaisille potilaille.
Beovu-hoito on lopetettava, jos potilaalle ilmaantuu näitä tapahtumia, ja hoitotoimiin on ryhdyttävä viipymättä. Niiden Beovu-hoitoa saaneiden potilaiden vointia on seurattava tarkasti, joilla on aiemmin ollut silmänsisäinen inflammaatio ja/tai verkkokalvon verisuonitukos (ensimmäistä brolusitsumabi-injektiota edeltävien 12 kuukauden aikana). Näillä potilailla on suurentunut riski verkkokalvon vaskuliitin ja/tai verkkokalvon verisuonitukoksen ilmaantumiselle.
Ylläpitohoidossa kahden Beovu-annoksen väli ei saa olla lyhyempi kuin 8 viikkoa ottaen huomioon, että silmänsisäisen inflammaation (mukaan lukien verkkokalvon vaskuliitin) ja verkkokalvon verisuonitukoksen ilmaantuvuus oli suurempi neovaskulaarista silmänpohjan ikärappeumaa sairastavilla potilailla, jotka saivat Beovu-ylläpitohoitoa kliinisessä tutkimuksessa 4 viikon välein, kuin Beovu-ylläpitohoitoa 8 tai 12 viikon välein vaiheen III kliinisissä avaintutkimuksissa (HAWK ja HARRIER) saaneilla potilailla.
Silmänpaineen nousu
Ohimenevää silmänpaineen nousua on havaittu 30 minuutin kuluessa silmän lasiaiseen annettavien endoteelikasvutekijän (VEGF) estäjien annosta, mukaan lukien brolusitsumabi (ks. kohta Haittavaikutukset). Erityinen varovaisuus on tarpeen, jos potilaalla on huonossa hoitotasapainossa oleva glaukooma (Beovu-valmistetta ei saa injisoida, jos silmänpaine on ≥ 30 mmHg). Sekä silmänpainetta että näköhermon pään perfuusiota on tarkkailtava ja hoidettava asianmukaisesti.
Molempien silmien hoito
Molempiin silmiin samanaikaisesti annettavan brolusitsumabihoidon turvallisuutta ja tehoa ei ole tutkittu.
Immunogeenisuus
Kyseessä on proteiinilääke, joten brolusitsumabihoitoon voi liittyä immunogeenisuutta (ks. kohta Haittavaikutukset). Potilaita on ohjeistettava kertomaan lääkärille, jos heille ilmaantuu oireita kuten silmäkipua tai lisääntynyttä epämukavuuden tunnetta silmässä, silmien punaisuuden pahenemista, näön sumenemista tai huononemista, näkökentässä näkyvien pienten hiukkasten lisääntymistä tai lisääntynyttä valoherkkyyttä (ks. kohta Haittavaikutukset).
Muiden VEGF:n estäjien samanaikainen käyttö
Beovu-valmisteen ja muiden VEGF:n estäjälääkevalmisteiden samanaikaisesta käytöstä samaan silmään ei ole tietoa. Brolusitsumabia ei saa antaa rinnakkain muiden VEGF:n estäjälääkevalmisteiden kanssa (ei systeemisten eikä silmään annettavien) (ks. kohta Yhteisvaikutukset).
Hoidon tauottaminen
Silmän lasiaiseen annettavia VEGF:n estäjähoitoja käytettäessä, annos on jätettävä väliin eikä hoitoa saa jatkaa ennen seuraavaa sovittua hoitokertaa, jos todetaan jokin seuraavista:
- parhaan lasikorjatun näöntarkkuuden (best corrected visual acuity, BCVA) heikkeneminen ≥ 30 kirjainta verrattuna edelliseen näöntarkkuuden arviointiin;
- verkkokalvoreikä;
- verkkokalvonalainen verenvuoto, joka ulottuu verkkokalvon keskikuoppaan (fovea) tai jossa verenvuodon laajuus on ≥ 50 % leesion kokonaisalasta;
- suunniteltu tai tehty silmäleikkaus edeltävien tai seuraavien 28 päivän aikana.
Verkkokalvon pigmenttiepiteelin repeämä
Riskitekijöitä, jotka suurentavat verkkokalvon pigmenttiepiteelin repeämän kehittymisriskiä kostean silmänpohjan ikärappeuman VEGF:n estäjähoidon jälkeen, ovat mm. kookas ja/tai korkea verkkokalvon pigmenttiepiteelin irtauma. Brolusitsumabihoitoa aloitettaessa on noudatettava varovaisuutta, jos potilaalla on näitä verkkokalvon pigmenttiepiteelin repeämän riskitekijöitä.
Regmatogeeninen verkkokalvon irtauma tai makulareikä
Hoito on lopetettava, jos potilaalla on regmatogeeninen verkkokalvon irtauma tai asteen 3 tai 4 makulareikä.
Intravitreaalisen käytön systeemiset vaikutukset
Systeemisiä haittatapahtumia, mukaan lukien muualla kuin silmässä ilmaantuvia verenvuotoja ja valtimoiden tromboemboliatapahtumia on ilmoitettu lasiaiseen injektoitavan VEGF:n estäjähoidon jälkeen. On olemassa teoreettinen riski, että nämä voivat liittyä VEGF:n estoon. On vain rajallisesti tietoa sellaisten silmänpohjan ikärappeumaa tai diabeettista makulaturvotusta sairastavien potilaiden hoidosta, joilla on ollut edeltävien 3 kuukauden aikana aivohalvaus (stroke), ohimeneviä aivoverenkierron häiriöitä tai sydäninfarkti. Varovaisuutta on noudatettava hoidettaessa tällaisia potilaita.
Populaatiot, joista on vain rajallisesti tietoa
Beovu-hoidosta on vain rajallisesti kokemusta diabetesta sairastavilla potilailla, joilla hemoglobiini A1c (HbA1c) on suurempi kuin 10 %:a tai joilla on proliferatiivinen diabeettinen retinopatia. Kokemusta Beovu-hoidosta ei ole myöskään diabetesta sairastavilla potilailla, joilla on hallitsematon verenpainetauti. Hoitavan lääkärin tulee ottaa tietojen puuttuminen huomioon hoidettaessa tällaisia potilaita.
Natriumpitoisuus
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Polysorbaatti 80:n pitoisuus
Tämä lääkevalmiste sisältää 0,01 mg polysorbaatti 80:tä per annos (0,05 ml). Polysorbaatit saattavat aiheuttaa allergisia reaktioita. Potilaita on neuvottava kertomaan lääkärille, jos heillä on allergioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä brolusitsumabihoidon aikana ja brolusitsumabihoidon päätyttyä vielä vähintään yhden kuukauden ajan viimeisen annoksen jälkeen.
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja brolusitsumabin käytöstä raskaana oleville naisille. Tiineillä jaavanmakakeilla toteutetussa tutkimuksessa ei havaittu viitteitä lisääntymistoksisuutta koskevista haitoista. Ei ole tehty riittäviä eläinkokeita lisääntymistoksisuuden selvittämiseksi (ks. kohta Prekliiniset tiedot turvallisuudesta). Vaikka silmään annon jälkeinen systeeminen altistus on valmisteen vaikutusmekanismin vuoksi hyvin alhainen, alkion- ja sikiönkehitykseen kohdistuvat riskit ovat mahdollisia. Siksi brolusitsumabia ei saa käyttää raskauden aikana, ellei hoidon mahdollinen hyöty ylitä sikiöön mahdollisesti kohdistuvia riskejä.
Imetys
Ei tiedetä, erittyykö brolusitsumabi ihmisen rintamaitoon. Lisääntymistoksisuutta koskeneessa tutkimuksessa brolusitsumabia ei havaittu jaavanmakakien rintamaidossa eikä imeväisten seerumissa (ks. kohta Prekliiniset tiedot turvallisuudesta). Rintaruokittuun vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Brolusitsumabihoitoa ei suositella rintaruokinnan aikana, eikä rintaruokintaa pidä aloittaa ennen kuin brolusitsumabihoidon päättymisestä ja viimeisestä annoksesta on kulunut vähintään yksi kuukausi. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko brolusitsumabihoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Lisääntymistä tai hedelmällisyyttä koskevia tutkimuksia ei ole tehty. VEGF-toiminnan eston on osoitettu vaikuttavan munarakkuloiden kehitykseen, keltarauhasen toimintaan ja hedelmällisyyteen. VEGF:n estäjien vaikutusmekanismin vuoksi naisen hedelmällisyyteen kohdistuvat riskit ovat mahdollisia.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Beovu-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Vaikutus johtuu silmän lasiaiseen annettavan injektion ja toimenpiteeseen liittyvän silmätutkimuksen jälkeen mahdollisesti esiintyvistä ohimenevistä näköhäiriöistä. Potilaiden on oltava ajamatta ja käyttämättä koneita, kunnes näkö on korjautunut riittävästi.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Kostea silmänpohjan ikärappeuma
Kostean silmänpohjan ikärappeuman osalta turvallisuuspopulaatio koostui kaikkiaan 1 088 potilaasta, jotka saivat brolusitsumabia kahdessa vaiheen III tutkimuksessa. Näistä potilaista 730 sai hoitoa suositellulla 6 mg:n annoksella.
Yleisimmin ilmoitettuja haittavaikutuksia olivat heikentynyt näöntarkkuus (7,3 %), kaihi (7,0 %), sidekalvon verenvuoto (6,3 %) ja lasiaiskellujat (5,1 %).
Vakavimpia haittavaikutuksia olivat sokeus (0,8 %), endoftalmiitti (0,7 %), verkkokalvovaltimon tukos (0,8 %) ja verkkokalvon irtauma (0,7 %).
Diabeettinen makulaturvotus
Diabeettisen makulaturvotuksen osalta turvallisuuspopulaatio koostui kaikkiaan 558 potilaasta, jotka saivat brolusitsumabia kahdessa vaiheen III tutkimuksessa. Näistä potilaista 368 sai hoitoa suositellulla 6 mg:n annoksella.
Yleisimmin ilmoitetut haittavaikutukset olivat kaihi (9,0 %), sidekalvon verenvuoto (6,5 %) ja silmänpaineen nousu (5,4 %).
Vakavimpia haittavaikutuksia olivat kaihi (9,0 %), verkkokalvon verisuonitukos (1,1 %), verkkokalvovaltimon tukos (0,8 %) ja endoftalmiitti (0,5 %).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa esiintyneet Beovu-valmisteen annon jälkeiset haittavaikutukset on esitetty yhteenvetona taulukossa 1.
Haittavaikutukset (taulukko 1) esitetään MedDRA-elinjärjestelmäluokittain. Kunkin elinjärjestelmäluokan haittavaikutukset esitetään yleisyysjärjestyksessä yleisimmistä alkaen. Kunkin haittavaikutuksen kohdalla mainittava yleisyysluokka perustuu seuraavaan käytäntöön: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Kunkin yleisyysluokan haittavaikutukset esitetään vakavuusjärjestyksessä vakavimmista alkaen.
Taulukko 1 Haittavaikutusten yleisyydet kliinisissä tutkimuksissa
| Elinjärjestelmäluokka | Yleisyysluokka* |
| Immuunijärjestelmä | |
| Yliherkkyys (mukaan lukien urtikaria, ihottuma, kutina, punoitus) | Yleinen |
| Silmät | |
| Heikentynyt näöntarkkuus | Yleinen |
| Verkkokalvoverenvuoto | Yleinen |
| Uveiitti | Yleinen |
| Iridosykliitti | Yleinen |
| Iriitti | Yleinen |
| Verkkokalvon verisuonitukos | Yleinen |
| Lasiaisen verenvuoto | Yleinen |
| Lasiaisen irtauma | Yleinen |
| Verkkokalvon repeämä | Yleinen |
| Kaihi | Yleinen |
| Sidekalvon verenvuoto | Yleinen |
| Lasiaiskellujat | Yleinen |
| Silmäkipu | Yleinen |
| Silmänpaineen nousu | Yleinen |
| Sidekalvotulehdus | Yleinen |
| Verkkokalvon pigmenttiepiteelin repeämä | Yleinen |
| Näön sumentuminen | Yleinen |
| Sarveiskalvon abraasio | Yleinen |
| Keratitis punctata | Yleinen |
| Sokeus | Melko harvinainen |
| Endoftalmiitti | Melko harvinainen |
| Verkkokalvon irtauma | Melko harvinainen |
| Sidekalvon verekkyys | Melko harvinainen |
| Lisääntynyt kyynelnesteen eritys | Melko harvinainen |
| Poikkeava tuntemus silmässä | Melko harvinainen |
| Verkkokalvon pigmenttiepiteelin irtauma | Melko harvinainen |
| Lasiaistulehdus | Melko harvinainen |
| Etukammion tulehdus | Melko harvinainen |
| Etukammion valotie | Melko harvinainen |
| Sarveiskalvon turvotus | Melko harvinainen |
| Verkkokalvon vaskuliitti | Melko harvinainen |
| Silmän kovakalvon tulehdus (skleriitti)** | Melko harvinainen |
*Yleisyysluokka on joko kosteaa silmänpohjan ikärappeumaa koskeneiden vaiheen III tutkimusten yhdistettyjen tietojen tai diabeettista makulaturvotusta koskeneiden vaiheen III avaintutkimusten yhdistettyjen tietojen mukainen sen mukaan, kumpi ilmaantuvuuksista oli suurempi. **mukaan lukien silmän kovakalvon pintaosan tulehdus (episkleriitti) | |
Tiettyjen haittavaikutusten kuvaus
Immunogeenisuus
Potilaalle saattaa kehittyä immuunivaste Beovu-hoidon yhteydessä.
Kostea silmänpohjan ikärappeuma
Kun Beovu‑valmistetta oli annettu 88 viikon ajan, hoidon aikana kehittyneitä brolusitsumabia torjuvia vasta‑aineita todettiin 23–25 %:lla potilaista.
Diabeettinen makulaturvotus
Kun Beovu‑valmistetta oli annettu 96 viikon ajan, hoidon aikana kehittyneitä brolusitsumabia torjuvia vasta‑aineita todettiin 16–23 %:lla potilaista.
Kosteaa silmänpohjan ikärappeumaa tai diabeettista makulaturvotusta sairastavilla potilailla, joille kehittyi hoidon aikana vasta‑aineita, todettiin haittavaikutuksina enemmän silmänsisäisiä inflammaatiotapauksia. Selvitysten jälkeen havaittiin, että verkkokalvon vaskuliitti ja/tai verkkokalvon verisuonitukos, tyypillisesti silmänsisäisen inflammaation yhteydessä, ovat Beovu-altistukseen liittyviä immuunivälitteisiä haittatapahtumia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Brolusitsumabia torjuvat vasta-aineet eivät vaikuttaneet kliiniseen tehoon.
Lääkeaineryhmään liittyvät haittavaikutukset
Lasiaiseen annetun VEGF:n estäjähoidon jälkeen on olemassa teoreettinen valtimoiden tromboemboliatapahtumien, mukaan lukien aivohalvauksen (stroke) ja sydäninfarktin, riski. Brolusitsumabilla tehdyissä kliinisissä tutkimuksissa tromboembolisten tapahtumien esiintymistiheys silmänpohjan ikärappeumaa (AMD) tai diabeettista makulaturvotusta (DME) sairastavilla potilailla oli alhainen. Esiintymistiheydessä ei ollut merkittäviä huomattavia eroja brolusitsumabi- ja vertailuvalmistehoitoryhmien välillä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: http://www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 Fimea
Yliannostus
Yliannostus suositustilavuutta suuremmalla injektiotilavuudella voi suurentaa silmänpainetta. Yliannostapauksessa onkin seurattava silmänpainetta ja aloitettava asianmukainen hoito, mikäli tämä on hoitavan lääkärin mielestä aiheellista.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: silmätautien lääkkeet, uudissuonittumisen estoon käytettävät lääkkeet, ATC-koodi: S01LA06
Vaikutusmekanismi
Brolusitsumabi on humanisoitu monoklonaalinen yksiketjuinen Fv-vasta-ainefragmentti (scFv-vasta-ainefragmentti), jonka molekyylipaino on ~26 kDa.
Endoteelikasvutekijä A ‑reitin (VEGF‑A-reitin) kautta tapahtuvan signaloinnin lisääntymisellä on yhteys patologiseen angiogeneesiin silmässä ja retinaturvotukseen. Brolusitsumabi sitoutuu suurella affiniteetilla VEGF‑A-isoformeihin (esim. VEGF110, VEGF121 ja VEGF165) ja estää siten VEGF‑A:ta sitoutumasta VEGFR‑1- ja VEGFR‑2-reseptoreihinsa. VEGF‑A:n sitoutumista estämällä brolusitsumabi vähentää endoteelisolujen proliferaatiota, mikä vähentää patologista uudissuonimuodostusta ja vähentää verisuonten läpäisevyyttä.
Farmakodynaamiset vaikutukset
Kostea silmänpohjan ikärappeuma
HAWK- ja HARRIER-tutkimuksissa veren ja nesteen tihkumiseen liittyviä suonikalvon uudissuonittumista (CNV) karakterisoivia anatomisia parametreja hyödynnettiin hoitopäätösten pohjana olevissa tautiaktiivisuuden arvioinneissa. Keskeisen makulan paksuuden (CST) pienenemistä ja intraretinaalisen/subretinaalisen nesteen (IRF/SRF) tai verkkokalvon pigmenttiepiteelin alapuolisen (sub‑RPE) nesteen vähenemistä todettiin Beovu-hoitoa saaneilla potilailla jo 4 viikon kuluttua hoidon aloituksesta ja viikoille 48 ja 96 asti.
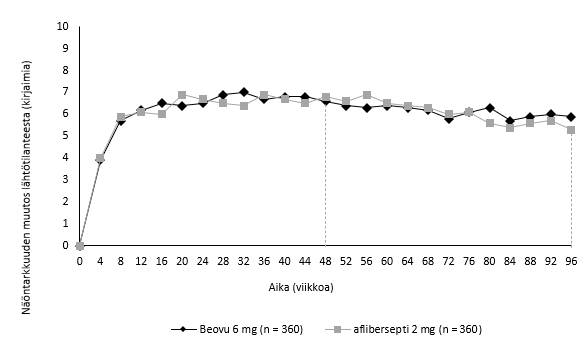
Viikolla 16 ero keskeisen makulan paksuuden pienenemisessä oli molemmissa tutkimuksissa tilastollisesti merkitsevä verrattaessa Beovu-hoitoa saaneita afliberseptihoitoa saaneisiin (HAWK: ‑161 vs. ‑134 mikronia; HARRIER: ‑174 vs. ‑134 mikronia). Keskeisen makulan paksuuden pieneneminen lähtötasoon verrattuna oli tilastollisesti merkitsevää myös viikolla 48 (HAWK: ‑173 vs. ‑144 mikronia; HARRIER: ‑194 vs. ‑144 mikronia), ja säilyi molempien tutkimusten päättymiseen viikolle 96 asti (HAWK: ‑175 vs. ‑149 mikronia; HARRIER: ‑198 vs. ‑155 mikronia).
Viikolla 16 ero niiden potilaiden prosenttiosuuksissa, joilla esiintyi intraretinaalista/subretinaalista nestettä, oli tilastollisesti merkitsevä Beovu-ryhmän ja afliberseptiryhmän välillä molemmissa tutkimuksissa (HAWK: 34 % vs. 52 %; HARRIER: 29 % vs. 45 %). Tämä ero oli tilastollisesti merkitsevä myös viikolla 48 (HAWK: 31 % vs. 45 %; HARRIER: 26 % vs. 44 %), ja säilyi molempien tutkimusten päättymiseen viikolle 96 asti (HAWK: 24 % vs. 37 %; HARRIER: 24 % vs. 39 %).
Viikolla 16 ero niiden potilaiden prosenttiosuuksissa, joilla esiintyi verkkokalvon pigmenttiepiteelin alapuolista nestettä, oli tilastollisesti merkitsevä Beovu-ryhmän ja afliberseptiryhmän välillä molemmissa tutkimuksissa (HAWK: 19 % vs. 27 %; HARRIER: 16 % vs. 24 %). Tämä ero oli tilastollisesti merkitsevä myös viikolla 48 (HAWK: 14 % vs. 22 %; HARRIER: 13 % vs. 22 %), ja säilyi molempien tutkimusten päättymiseen viikolle 96 asti (HAWK: 11 % vs. 15 %; HARRIER: 17 % vs. 22 %).
Näissä tutkimuksissa Beovu-hoitoa saaneilla potilailla todettiin suonikalvon neovaskularisaatioleesioiden koon pienenemistä jo viikolla 12 sekä viikoilla 48 ja 96 hoidon aloittamisen jälkeen.
Diabeettinen makulaturvotus
KESTREL- ja KITE-tutkimuksissa olennaisia anatomisia parametreja hyödynnettiin hoitopäätösten pohjana olevissa tautiaktiivisuuden arvioinneissa. Keskeisen makulan paksuuden pienenemistä ja intraretinaalisen/subretinaalisen nesteen vähenemistä todettiin Beovu-hoitoa saaneilla potilailla jo 4 viikon kuluttua hoidon aloituksesta ja viikolle 52 asti. Nämä muutokset säilyivät viikolle 100 asti.
Kliininen teho ja turvallisuus
Kostea silmänpohjan ikärappeuma
Beovu-valmisteen tehoa ja turvallisuutta arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa vaiheen III monikeskustutkimuksessa (HAWK‑ ja HARRIER‑tutkimukset) potilailla, joilla oli neovaskulaarinen (kostea) silmänpohjan ikärappeuma. Näissä tutkimuksissa yhteensä 1 817 potilasta sai hoitoa kahden vuoden ajan (1 088 potilasta sai Beovu-hoitoa ja 729 sai vertailuvalmiste afliberseptia). Potilaiden ikä vaihteli 50 vuodesta 97 vuoteen; ikäkeskiarvo oli 76 vuotta.
Molemmissa tutkimuksissa kolme ensimmäistä annosta annettiin kerran kuukaudessa (viikoilla 0, 4 ja 8), minkä jälkeen brolusitsumabiryhmän potilaat saivat hoitoa 12 viikon välein ja annosväliä voitiin muuttaa 8 viikkoon tautiaktiivisuuden perusteella. Lääkäri arvioi tautiaktiivisuuden ensimmäisen 12‑viikkoisen annosvälin aikana (viikoilla 16 ja 20) ja tämän jälkeen jokaisella sovitulla 12 viikon välein toteutetulla hoitokäynnillä. Jos potilaalla todettiin tautiaktiivisuutta (esim. huonontunut näöntarkkuus, suurentunut keskeisen makulan paksuus ja/tai intraretinaalista/subretinaalista nestettä tai nestettä verkkokalvon pigmenttiepiteelin alla) jollakin näistä käynneistä, hänen kohdallaan siirryttiin 8 viikon annosväliin. Vertailuvalmiste afliberseptia annettiin 8 viikon välein kuukauden välein annettujen kolmen ensimmäisen annoksen jälkeen.
Tulokset
Tutkimusten ensisijainen tehon päätetapahtuma oli parhaan lasikorjatun näöntarkkuuden (best corrected visual acuity, BCVA) muutos lähtötilanteesta viikolle 48, kun mittarina oli early treatment diabetic retinopathy study ‑tutkimuksen (ETDRS) kirjainpistemäärä; ensisijaisena tavoitteena oli osoittaa, että Beovu‑hoito oli vähintään samanarvoinen (non-inferior) kuin aflibersepti. Molemmissa tutkimuksissa Beovu‑hoito (antoväli 12 viikkoa tai 8 viikkoa) oli teholtaan vähintään samanarvoinen kuin aflibersepti (2 mg; antoväli 8 viikkoa). Ensimmäisen vuoden aikana todettu näöntarkkuuden paraneminen säilyi toisen vuoden aikana.
Molempien tutkimusten tarkemmat tulokset esitetään taulukossa 2 ja kuvassa 1 jäljempänä.
Taulukko 2 Näöntarkkuustulokset vaiheen III tutkimusten viikoilla 48 ja 96 – HAWK‑ ja HARRIER-tutkimukset
| HAWK | HARRIER | ||||||
| Tehotulosmuuttuja | Viikko | Beovu (n = 360) | Aflibersepti 2 mg (n = 360) | Ero (95 % lv), brolusitsumabi – aflibersepti | Beovu (n = 370) | Aflibersepti 2 mg (n = 369) | Ero (95 % lv), brolusitsumabi – aflibersepti |
| Parhaan lasikorjatun näöntarkkuuden (mittari: ETDRS-kirjainpistemäärä) keskimuutos lähtötilanteesta | 48 | 6,6 (SE = 0,71) | 6,8 (SE = 0,71) | ‑0,2 (‑2,1; 1,8) P < 0,0001 a) | 6,9 (SE = 0,61) | 7,6 (SE = 0,61) | -0,7 (-2,4; 1,0) P < 0,0001 a) |
| 36 – 48 b) | 6,7 (SE = 0,68) | 6,7 (SE = 0,68) | 0,0 (‑1,9; 1,9) P < 0,0001 a) | 6,5 (SE = 0,58) | 7,7 (SE = 0,58) | ‑1,2 (‑2,8; 0,4) P = 0,0003 a) | |
| 96 | 5,9 (SE = 0,78) | 5,3 (SE = 0,78) | 0,5 (‑1,6; 2,7) | 6,1 (SE = 0,73) | 6,6 (SE = 0,73) | ‑0,4 (‑2,5; 1,6) | |
| Potilaat, joiden näkö parani vähintään 15 kirjaimen verran (%) | 48 | 33,6 | 25,4 | 8,2 (2,2; 15,0) | 29,3 | 29,9 | ‑0,6 (‑7,1; 5,8) |
| 96 | 34,2 | 27,0 | 7,2 (1,4; 13,8) | 29,1 | 31,5 | ‑2,4 (‑8,8; 4,1) | |
| Potilaat, joiden näöntarkkuus heikkeni (paras lasikorjattu näöntarkkuus huononi ≥ 15 kirjaimen verran) (%) | 48 | 6,4 | 5,5 | 0,9 (‑2,7; 4,3) | 3,8 | 4,8 | ‑1,0 (‑3,9; 2,2) |
| 96 | 8,1 | 7,4 | 0,7 (‑3,6; 4,6) | 7,1 | 7,5 | ‑0,4 (‑3,8; 3,3) | |
Parhaan lasikorjatun näöntarkkuuden (best corrected visual acuity, BCVA) kohdalla puuttuvat tiedot imputoitiin LOCF-menetelmällä eli käyttämällä analyysissä tuoreinta havaintoa. ETDRS: early treatment diabetic retinopathy study ‑tutkimus. SE: keskivirhe (standard error). a) P-arvo koskee vähintään samanarvoisuuden (non-inferiority) hypoteesia, jossa vähintään samanarvoisuuden raja-arvo on 4,0 kirjainta. b) Tärkeä toissijainen päätetapahtuma, jossa otetaan huomioon Beovu-hoidon ja afliberseptihoidon antoajankohtien erot. | |||||||
Kuva 1 Näöntarkkuuden keskimuutos lähtötilanteesta viikolle 96 HAWK- ja HARRIER-tutkimuksissa
HAWK
HARRIER
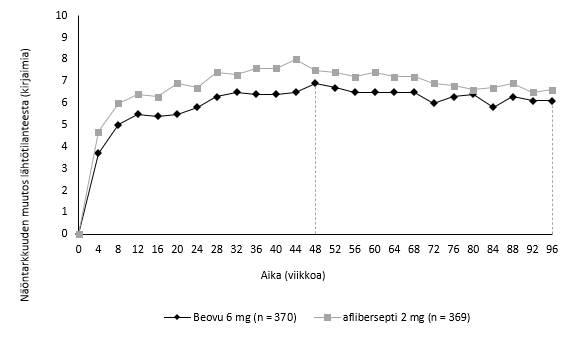
HAWK-tutkimuksessa näöntarkkuuden paranemisen saavuttaneen Beovu-ryhmän potilaista 56 % sai hoitoa 12 viikon annosvälein viikolla 48. HARRIER-tutkimuksessa vastaava osuus oli 51 %. Viikolla 96 vastaavat osuudet olivat 45 % (HAWK) ja 39 % (HARRIER). Potilaista, joiden katsottiin soveltuvan saamaan hoitoa 12 viikon välein ensimmäisen 12 viikon hoitojakson aikana, 85 % ja 82 % jatkoi 12 viikon annosvälein viikolle 48 asti. Potilaista, jotka saivat hoitoa 12 viikon välein viikolla 48, 82 % ja 75 % jatkoi 12 viikon annosvälein viikolle 96 asti.
Molemmissa tutkimuksissa hoitovaikutukset arviointikelpoisissa alaryhmissä (ryhmittelyperusteena esim. ikä, sukupuoli, etninen tausta, lähtötilanteen näöntarkkuus, verkkokalvon paksuus lähtötilanteessa, vauriotyyppi, vaurioiden koko, nestestatus) vastasivat yleisesti koko populaatioissa todettuja tuloksia.
Tautiaktiivisuuden arviointi perustui näöntarkkuuden ja/tai anatomisten parametrien muutoksiin; anatomisia parametrejä olivat mm. keskeisen makulan paksuus ja/tai intraretinaalinen/subretinaalinen neste tai neste verkkokalvon pigmenttiepiteelin alla. Tautiaktiivisuutta arvioitiin koko tutkimusten ajan. Tautiaktiivisuuden anatomiset parametrit olivat Beovu-ryhmässä viikolla 48 ja viikolla 96 pienemmät kuin afliberseptiryhmässä (ks. kohta ”Farmakodynaamiset vaikutukset”).
Ero niiden potilaiden prosenttiosuuksissa, joilla oli tautiaktiivisuutta viikolla 16, oli tilastollisesti merkitsevä Beovu-ryhmän ja afliberseptiryhmän välillä (24 % vs. 35 % HAWK-tutkimuksessa, p = 0,0013; 23 % vs. 32 % HARRIER-tutkimuksessa, p = 0,0021).
Molemmissa tutkimuksissa Beovu-hoito paransi kliinisesti merkittävästi potilaiden raportoimia hoitotuloksia (ennalta määritelty toissijainen tehon päätetapahtuma) verrattuna lähtötilanteeseen, kun raportointimenetelmänä käytettiin National Eye Institute Visual Function Questionnaire ‑kyselyä (NEI VFQ‑25‑kysely). Näiden muutosten suuruusluokka oli samankaltainen kuin julkaistuissa tutkimuksissa havaittu suuruusluokka, joka vastasi parhaan lasikorjatun näöntarkkuuden paranemista 15 kirjaimen verran. Potilaiden raportoimien hoitotulosten paraneminen säilyi toisen vuoden aikana.
Beovu-ryhmän ja afliberseptiryhmän välillä ei todettu kliinisesti merkittäviä eroja lähtötilanteen ja viikon 48 välillä NEI VFQ‑25 ‑kokonaispisteissä eikä tämän mittarin eri osa-asteikoilla (yleinen näkökyky, silmän kipu, lähinäköä vaativat toiminnot, kaukonäköä vaativat toiminnot, sosiaalinen toimintakyky, psyykkinen hyvinvointi, vaikeudet roolitoiminnoista suoriutumisessa, riippuvuus hoitajasta, ajokyky, värinäkö ja perifeerinen näkö).
HAWK‑ ja HARRIER‑tutkimuksissa Beovu annettiin 4 viikon välein (kuukausittain) ensimmäisten 3 annoksen ajan (latausvaihe) ja sen jälkeen 12:n tai 8 viikon välein (ylläpitohoito). Näiden tutkimusten Beovu‑ryhmien tulokset toistettiin populaatiofarmakokineettistä/‑farmakodynaamista mallinnusta hyödyntäneessä simulaatiotutkimuksessa, jossa Beovu‑valmistetta annettiin 6 viikon välein ensimmäisten 2:n tai 3 annoksen ajan (latausvaihe) ja sen jälkeen 12:n tai 8 viikon välein (ylläpitohoito).
Ylläpitovaiheen treat-and-extend-annosteluohjelmaa tutkittiin TALON-tutkimuksessa, joka oli 64-viikkoinen, kaksihaarainen, satunnaistettu, kaksoissokkoutettu, vaiheen IIIb monikeskustutkimus, jossa arvioitiin Beovun tehoa ja turvallisuutta verrattuna aflibersepti 2 mg:aan neovaskulaarista silmänpohjan ikärappeumaa sairastavilla potilailla.
737 potilasta satunnaistettiin suhteessa 1:1 jompaankumpaan hoitohaaraan, saamaan joko 6 mg brolusitsumabia tai 2 mg afliberseptiä. Molempiin hoitoryhmiin kuuluville potilaille annettiin yksi injektio 4 viikon välein ensimmäisten 3 injektion ajan ja sen jälkeen yksi injektio 8 viikon kuluttua. Tämän jälkeen hoitovälejä oli joko 8 viikon välein, 12 viikon välein tai 16 viikon välein aina viikkoon 60 tai 62 asti.
Keskimääräinen muutos BCVA:ssa lähtötilanteesta viikolla 64 oli +4,7 ETDRS-kirjainta Beovulla vs. +4,9 ETDRS-kirjainta 2 mg:n afliberseptilla.
Hoitovälien tulokset viikolla 64 on esitetty taulukossa 3.
Taulukko 3 Viimeinen hoitoväli, jolla ei tautiaktiivisuutta: potilasosuus viikolla 64
| Hoitohaara | ||
| Hoitoväli (viikkoa) | Brolusitsumabi 6 mg n=366 | Aflibersepti 2 mg n=368 |
| 4 | 23,2 % | 41,8 % |
| 8 | 26,0 % | 22,0 % |
| 12 | 22,4 % | 23,9 % |
| 16 | 28,4 % | 12,2 % |
255 potilasta, jotka suorittivat TALON-tutkimuksen, otettiin mukaan 56 viikkoa kestäneeseen TALON-tutkimuksen avoimeen, yhden hoitohaaran jatkotutkimukseen, ja heitä hoidettiin brolusitsumabi treat-and-extend-annosteluohjelmalla ilman latausvaihetta ja enintään 20 viikon hoitovälillä.
Viikolla 56 yli 50 % 237 henkilöstä, jotka olivat saaneet vähintään 2 injektiota, oli hoitovälillä 16 viikkoa (24,9 %) tai 20 viikkoa (28,7 %), eikä heillä ollut tautiaktiivisuutta ja näöntarkkuus säilyi koko tutkimuksen ajan.
Diabeettinen makulaturvotus
Beovu-valmisteen tehoa ja turvallisuutta arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa vaiheen III monikeskustutkimuksessa (KESTREL‑ ja KITE‑tutkimukset) potilailla, joilla oli diabeettisesta makulaturvotuksesta johtuva näön heikkeneminen. Näissä tutkimuksissa yhteensä 926 potilasta sai hoitoa kahden vuoden ajan (558 potilasta sai brolusitsumabia ja 368 aflibersepti 2 mg ‑hoitoa). Potilaiden ikä vaihteli 23 vuodesta 87 vuoteen; ikäkeskiarvo oli 63 vuotta.
Molemmissa tutkimuksissa viiden ensimmäisen annoksen jälkeen (annokset viikoilla 0, 6, 12, 18 ja 24) brolusitsumabiryhmän potilaat saivat hoitoa 12 viikon välein ja annosväliä voitiin muuttaa 8 viikkoon tautiaktiivisuuden perusteella. Lääkäri arvioi tautiaktiivisuuden ensimmäisen 12‑viikkoisen annosvälin aikana (viikoilla 32 ja 36) ja tämän jälkeen jokaisella sovitulla hoitokäynnillä. Jos potilaalla todettiin tautiaktiivisuutta (esim. huonontunut näöntarkkuus, suurentunut keskeisen makulan paksuus) jollakin näistä käynneistä, hänen kohdallaan siirryttiin 8 viikon annosväliin. Jos potilaalla ei todettu KITE-tutkimuksen toisen vuoden aikana tautiaktiivisuutta, voitiin siirtyä 16 viikon annosväliin. Vertailuvalmiste afliberseptia annettiin 8 viikon välein kuukauden välein annettujen viiden ensimmäisen annoksen jälkeen.
Tulokset
Tutkimusten ensisijainen tehon päätetapahtuma oli parhaan lasikorjatun näöntarkkuuden (best corrected visual acuity, BCVA) muutos lähtötilanteesta viikolle 52, kun mittarina oli early treatment diabetic retinopathy study ‑tutkimuksen (ETDRS) kirjainpistemäärä; ensisijaisena tavoitteena oli osoittaa, että Beovu‑hoito oli vähintään samanarvoinen (non-inferior) kuin aflibersepti 2 mg. Molemmissa tutkimuksissa Beovu‑hoito (antoväli 12 viikkoa tai 8 viikkoa) oli teholtaan vähintään samanarvoinen kuin aflibersepti 2 mg (antoväli 8 viikkoa).
KESTREL- ja KITE-tutkimusten tulokset myös osoittivat, että Beovu-hoito oli vähintään samanarvoinen kuin aflibersepti 2 mg tärkeimmän toissijaisen päätetapahtuman osalta (BCVA:n keskimuutos lähtötilanteesta viikolta 40 viikolle 52 saakka).
Ensimmäisen vuoden aikana todettu näöntarkkuuden paraneminen säilyi toisen vuoden aikana.
Molempien tutkimusten tarkemmat tulokset esitetään taulukossa 4 ja kuvassa 2 jäljempänä.
Taulukko 4 Näöntarkkuustulokset vaiheen III tutkimusten viikoilla 52 ja 100 – KESTREL‑ ja KITE-tutkimukset
| KESTREL | KITE | ||||||
| Tehotulos | Viikko | Beovu (n = 189) | Aflibersepti 2 mg (n = 187) | Ero (95 % lv) brolusitsumabi – aflibersepti | Beovu (n = 179) | Aflibersepti 2 mg (n = 181) | Ero (95 % lv) brolusitsumabi – aflibersepti |
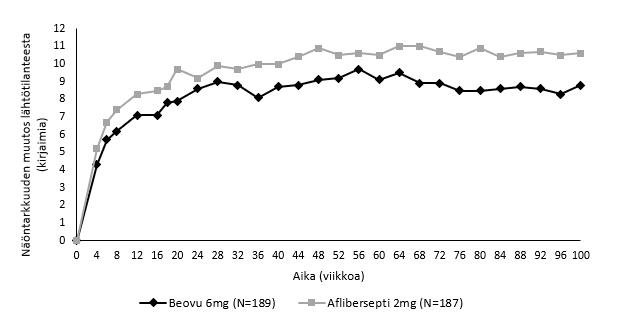
| BCVA:n muutos lähtötilanteesta (mittarina ETDRS-kirjainpistemäärä); pienimmän neliösumman menetelmällä laskettu keskiarvo (keskivirhe) | 52 | 9,2 (0,57) | 10,5 (0,57) | ‑1,3 (‑2,9; 0,3) P < 0,001a | 10,6 (0,66) | 9,4 (0,66) | 1,2 (‑0,6; 3,1) P < 0,001a |
| 40–52 | 9,0 (0,53) | 10,5 (0,53) | ‑1,5 (‑3.0; 0,0) P < 0,001a | 10,3 (0,62) | 9,4 (0,62) | 0,9 (‑0,9; 2,6) P < 0,001a | |
| 100 | 8,8 (0,75) | 10,6 (0,75) | ‑1,7 (‑3,8; 0,4) | 10,9 (0,85) | 8,4 (0,85) | 2,6 (0,2; 4,9) | |
| Vähintään 15 kirjaimen BCVA-paranema verrattuna lähtötilanteeseen tai BCVA ≥ 84 kirjainta (%) | 52 | 36,0 | 40,1 | ‑4,1 (‑13,3; 5,9) | 46,8 | 37,2 | 9,6 (‑0,4; 20,2) |
| 100 | 39,2 | 42,2 | ‑3,0 (‑12,5; 6,3) | 50,4 | 36,9 | 13,6 (3,3; 23,5) | |
BCVA: paras lasikorjattu näöntarkkuus; jos tutkittavaan silmään aloitettiin muu diabeettisen makulaturvotuksen hoito, tämän jälkeiset BCVA-arvot sensuroitiin ja korvattiin viimeisellä arvolla ennen kyseisen hoidon aloitusta. ETDRS: early treatment diabetic retinopathy study ‑tutkimus. LS: pienin neliösumma (least-square). SE: keskivirhe (standard error). a) P-arvo koskee vähintään samanarvoisuuden (non-inferiority) hypoteesia, jossa vähintään samanarvoisuuden raja-arvo on 4,0 kirjainta. | |||||||
Kuva 2 Näöntarkkuuden keskimuutos lähtötilanteesta viikolle 100 KESTREL- ja KITE-tutkimuksissa
KESTREL
KITE
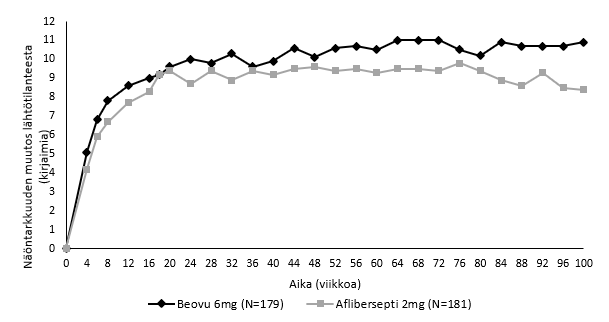
KESTREL-tutkimuksessa näöntarkkuuden paranemisen saavuttaneen Beovu-ryhmän potilaista 55 % sai hoitoa 12 viikon annosvälein viikolla 52; viikolla 100 vastaava luku oli 44 %. KITE-tutkimuksessa näöntarkkuuden paranemisen saavuttaneen Beovu-ryhmän potilaista 50 % sai hoitoa 12 viikon annosvälein viikolla 52 ja 37 % 12 viikon / 16 viikon annosvälein viikolla 100. Kummassakin tutkimuksessa niistä potilaista, joiden katsottiin soveltuvan saamaan hoitoa 12 viikon välein ensimmäisen 12 viikon hoitojakson aikana, noin 70 % jatkoi vähintään 12 viikon annosvälein viikolle 100 asti. KITE‑tutkimuksessa 25 % potilaista sai viikon 100 kohdalla Beovu‑hoitoa 16 viikon annosvälein.
Molemmissa tutkimuksissa hoitovaikutukset arviointikelpoisissa alaryhmissä (ryhmittelyperusteena esim. ikä, sukupuoli, lähtötilanteen HbA1c-arvo, lähtötilanteen näöntarkkuus, keskeisen makulan paksuus lähtötilanteessa, diabeettisen makulaturvotuksen vauriotyyppi, diabeettisen makulaturvotuksen kesto diagnoosista, verkkokalvon nestestatus) vastasivat yleisesti koko populaatioissa todettuja tuloksia.
KESTREL- ja KITE‑tutkimuksissa tautiaktiivisuutta arvioitiin koko tutkimusten ajan. Tautiaktiivisuuden arviointi perustui näöntarkkuuden ja/tai anatomisten parametrien muutoksiin; anatomisia parametrejä olivat mm. keskeisen makulan paksuus ja/tai intraretinaalisen/subretinaalisen nesteen esiintyminen. Keskeisen makulan paksuuden pienenemät lähtötilanteesta säilyivät viikolle 100. Viikon 100 kohdalla niiden potilaiden osuus, joilla esiintyi intraretinaalista/subretinaalista nestettä, oli pienempi Beovu‑hoitoa saaneilla (42 % KESTREL‑tutkimuksessa ja 41 % KITE‑tutkimuksessa) kuin aflibersepti 2 mg ‑hoitoa saaneilla (54 % KESTREL‑tutkimuksessa ja 57 % KITE‑tutkimuksessa).
KESTREL- ja KITE-tutkimuksissa arvioitiin diabeettisen retinopatian vaikeusastepisteet (diabetic rethinopathy severity score, DRSS). DRSS-pisteet olivat arvioitavissa lähtötilanteessa 98,1 %:lla potilaista sekä KESTREL- että KITE-tutkimuksessa. Poolatun analyysin perusteella Beovu oli vähintään samanarvoinen kuin aflibersepti 2 mg potilasosuudessa, jolla DRSS-pisteet parantuivat vähintään 2 pykälän verran lähtötilanteesta viikolla 52, kun vähintään samanarvoisuuden raja-arvo oli 10 %. Arvioidut osuudet olivat Beovun osalta 28,9 % ja aflibersepti 2 mg:n osalta 24,9 %, ja hoitojen ero oli tällöin 4,0 % (95 % lv: [-0,6; 8,6]). Viikon 100 kohdalla potilasosuus, jolla DRSS‑pisteet parantuivat ≥ 2 pykälän verran lähtötilanteesta viikolle 100, oli KESTREL‑tutkimuksessa Beovu‑hoitoa saaneista 32,8 % ja aflibersepti 2 mg ‑hoitoa saaneista 29,3 % ja KITE‑tutkimuksessa Beovu‑hoitoa saaneista 35,8 % ja aflibersepti 2 mg ‑hoitoa saaneista 31,1 %.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Beovu-valmisteen käytöstä neovaskulaarisen silmänpohjan ikärappeuman ja diabeettisen makulaturvotuksen hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Beovu annetaan suoraan lasiaiseen, ja se vaikuttaa paikallisesti silmässä.
Imeytyminen ja jakautuminen
Kun neovaskulaarista silmänpohjan ikärappeumaa sairastaville potilaille annettiin brolusitsumabia silmän lasiaiseen (6 mg/silmä), vapaan brolusitsumabin plasman Cmax-arvon geometrinen keskiarvo oli 49,0 ng/ml (vaihteluväli 8,97–548 ng/ml) ja se saavutettiin 1 vuorokaudessa.
Biotransformaatio ja eliminaatio
Brolusitsumabi on monoklonaalinen vasta-ainefragmentti, eikä metaboliaa koskevia tutkimuksia ole tehty. Brolusitsumabi on yksiketjuinen vasta‑ainefragmentti, joten vapaan brolusitsumabin oletetaan eliminoituvan kohdemolekyylivälitteisen jakautumisen kautta sitoutumalla vapaisiin endogeenisiin VEGF‑molekyyleihin, munuaisteitse passiivisen eliminaation kautta ja proteolyysin kautta tapahtuvan metabolian kautta.
Silmän lasiaiseen annettujen injektioiden jälkeen brolusitsumabin eliminaation näennäinen systeeminen puoliintumisaika oli 4,3 ± 1,9 vrk. Useimmilla potilailla pitoisuudet olivat yleisesti kvantitointirajan (< 0,5 ng/ml) lähellä tai sen alapuolella noin 4 viikon kuluttua lääkkeen annosta. Brolusitsumabi ei kumuloitunut seerumiin, kun sitä annettiin silmän lasiaiseen 4 viikon välein.
Erityisryhmät
Iäkkäät potilaat
Silmän lasiaiseen annettujen injektioiden jälkeen systeemisessä farmakokinetiikassa ei todettu merkittäviä eroja tutkimuksessa, johon osallistui 22 iältään 65–74-vuotiasta potilasta, 18 iältään 75–84-vuotiasta potilasta ja 3 iältään ≥ 85-vuotiasta potilasta.
Munuaisten vajaatoiminta
Brolusitsumabin systeemistä farmakokinetiikkaa arvioitiin neovaskulaarista silmänpohjan ikärappeumaa sairastavilla potilailla, joiden munuaistoiminta oli normaali (≥ 90 ml/min [n = 21]) tai jotka sairastivat lievää (60 – < 90 ml/min [n = 22]) tai keskivaikeaa (30 – < 60 ml/min [n = 7]) munuaisten vajaatoimintaa. Vaikka systeemisen puhdistuman arvot potilailla, jotka sairastivat lievää tai keskivaikeaa munuaisten vajaatoimintaa olivat yleisesti pienempiä kuin potilailla, joiden munuaistoiminta oli normaali, ei lievällä tai keskivaikealla munuaisten vajaatoiminnalla havaittu merkittävää vaikutusta brolusitsumabin systeemisen kokonaisaltistukseen. Potilaita, joilla oli vaikea (< 30 ml/min) munuaisten vajaatoiminta, ei arvioitu.
Maksan vajaatoiminta
Brolusitsumabia ei ole tutkittu maksan vajaatoimintapotilailla. Lievä tai keskivaikea maksan vajaatoiminta ei oletettavasti vaikuta brolusitsumabin systeemiseen kokonaisaltistukseen, sillä metabolia tapahtuu proteolyysin kautta eikä riipu maksan toiminnasta.
Prekliiniset tiedot turvallisuudesta
Brolusitsumabin karsinogeenisuutta ja mutageenisuutta ei ole arvioitu tutkimuksissa.
Tiineille jaavanmakakeille brolusitsumabia annettin injektioina lasiaiseen neljän viikon välein annostasoilla, joilla saatiin aikaan maksimaalinen systeeminen altistus – kuusinkertainen verrattuna suositeltuun enimmäisannokseen ihmisellä (perusteena seerumin Cmax). Vaikutuksia alkion- tai sikiönkehitykseen, tiineyteen, synnytyksiin tai jälkeläisten elossaoloon, kasvuun tai postnataaliseen kehitykseen ei havaittu. Farmakologisen vaikutuksensa perusteella brolusitsumabia on kuitenkin pidettävä mahdollisesti teratogeenisena ja alkio-sikiötoksisena.
Farmaseuttiset tiedot
Apuaineet
Natriumsitraatti
Sakkaroosi
Polysorbaatti 80
Natriumhydroksidi (pH:n säätöä varten)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Esitäytetty ruisku: 2 vuotta
Injektiopullo: 2 vuotta
Säilytys
Esitäytetty ruisku
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä esitäytetty ruisku sinetöidyssä kuplapakkauksessa ja ulkopakkauksessa. Herkkä valolle. Avaamatonta kuplapakkausta voidaan säilyttää huoneenlämmössä (alle 25 °C) enintään 24 tunnin ajan ennen käyttöä.
Injektiopullo
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Avaamatonta injektiopulloa voidaan säilyttää huoneenlämmössä (alle 25 °C) enintään 24 tunnin ajan ennen käyttöä.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BEOVU injektioneste, liuos, esitäytetty ruisku
120 mg/ml (L:ei) 1 kpl (828,91 €)
PF-selosteen tieto
Esitäytetty ruisku
0,165 ml steriiliä liuosta esitäytetyssä ruiskussa (tyypin I lasia), jossa on bromobutyylikumista valmistettu männän kärki ja jonka ruiskun korkkiin kuuluu peukaloinnin paljastava jäykkä, valkoinen sinetti sekä harmaa bromobutyylikuminen kärjen suojakorkki, jossa on Luer lock ‑liitin. Esitäytetyssä ruiskussa on männän tappi ja violetti sormituki, ja se on pakattu sinetöityyn kuplapakkaukseen.
Pakkauskoko: 1 esitäytetty ruisku.
Injektiopullo
0,23 ml steriiliä liuosta lasisessa 2 ml injektiopullossa, jossa on pinnoitettu kumitulppa ja joka on sinetöity alumiinikorkilla, jossa on violetista muovista tehty nostolevy.
Pakkauskoko: 1 injektiopullo ja 1 tylppäkärkinen suodatinneula (18G x 1½″, 1,2 mm x 40 mm, 5 μm).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai hieman opaalinhohtoinen, väritön tai hieman ruskeankellertävä vesiliuos.
Käyttö- ja käsittelyohjeet
Esitäytetty ruisku
Esitäytetty ruisku sisältää suositeltua 6 mg:n annosta suuremman annoksen. Esitäytetystä ruiskusta vedettävissä olevaa tilavuutta (0,165 ml) ei saa käyttää kokonaan. Liuosylimäärä on poistettava ennen injektiota. Esitäytetyn ruiskun koko liuosmäärän injisointi voi johtaa yliannokseen. Poista ilmakuplat ja ylimääräinen lääkevalmiste painamalla mäntää hitaasti, kunnes männän kuperan kumisen kärjen reuna on ruiskussa olevan mustan annosviivan kohdalla (vastaa 0,05 ml eli 6 mg brolusitsumabia).
Liuos on tarkastettava silmämääräisesti jääkaapista ottamisen yhteydessä ja ennen antoa. Jos valmisteessa näkyy hiukkasia tai sameutta, esitäytettyä ruiskua ei saa käyttää ja on ryhdyttävä asianmukaisiin vaihtotoimiin.
Esitäytetty ruisku on steriili ja tarkoitettu vain yhtä käyttökertaa varten. Älä käytä tätä lääkettä, jos pakkaus tai esitäytetty ruisku on vahingoittunut tai viimeinen käyttöpäivämäärä on ohi. Pakkausselosteessa on tarkat käyttöohjeet.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Injektiopullo
Injektiopullo sisältää suositeltua 6 mg:n annosta suuremman annoksen. Injektiopullosta vedettävissä olevaa tilavuutta (0,23 ml) ei saa käyttää kokonaan. Liuosylimäärä on poistettava ennen injektiota. Injektiopullon koko tilavuuden injisointi voi johtaa yliannokseen. Injektion annos on asetettava 0,05 ml:n annosmerkin kohdalle, mikä vastaa 6 mg brolusitsumabia.
Liuos on tarkastettava silmämääräisesti jääkaapista ottamisen yhteydessä ja ennen antoa. Jos valmisteessa näkyy hiukkasia tai sameutta, injektiopulloa ei saa käyttää, ja on ryhdyttävä asianmukaisiin vaihtotoimiin.
Injektiopullon sisältö ja suodatinneula ovat steriilit ja ne on tarkoitettu vain yhtä käyttökertaa varten. Älä käytä tätä lääkettä, jos pakkaus, injektiopullo ja/tai suodatinneula on vahingoittunut tai viimeinen käyttöpäivämäärä on ohi. Pakkausselosteessa on tarkat käyttöohjeet.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BEOVU injektioneste, liuos, esitäytetty ruisku
120 mg/ml 1 kpl
- Ei korvausta.
ATC-koodi
S01LA06
Valmisteyhteenvedon muuttamispäivämäärä
05.05.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com