
POLIVY kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 30 mg, 140 mg
Vaikuttavat aineet ja niiden määrät
Polivy 30 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo kuiva-ainetta välikonsentraatiksi infuusionestettä varten, liuos sisältää 30 mg polatutsumabi-vedotiinia.
Käyttökuntoon saattamisen jälkeen yksi ml sisältää 20 mg polatutsumabi-vedotiinia.
Polivy 140 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo kuiva-ainetta välikonsentraatiksi infuusionestettä varten, liuos sisältää 140 mg polatutsumabi-vedotiinia.
Käyttökuntoon saattamisen jälkeen yksi ml sisältää 20 mg polatutsumabi-vedotiinia.
Polatutsumabi-vedotiini on vasta-aineen ja lääkkeen konjugaatti, joka koostuu CD79b:hen kohdistuvasta monoklonaalisesta vasta-aineesta (rekombinantti humanisoitu immunoglobuliini G1 [IgG1], joka tuotetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa), ja siihen kovalenttisesti konjugoidusta mikrotubulustoimintaan vaikuttavasta monometyyliauristatiini E:stä (MMAE).
Apuaine, jonka vaikutus tunnetaan
Yksi 30 mg:n injektiopullo Polivy-valmistetta sisältää 1,8 mg polysorbaattia 20.
Yksi 140 mg:n injektiopullo Polivy-valmistetta sisältää 8,4 mg polysorbaattia 20.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos (kuiva-aine välikonsentraattia varten).
Kliiniset tiedot
Käyttöaiheet
Polivy yhdistelmänä rituksimabin, syklofosfamidin, doksorubisiinin ja prednisonin (R-CHP) kanssa on tarkoitettu aiemmin hoitamatonta diffuusia suurisoluista B-solulymfoomaa sairastavien aikuispotilaiden hoitoon.
Polivy yhdistelmänä bendamustiinin ja rituksimabin (BR) kanssa on tarkoitettu uusiutunutta/refraktorista diffuusia suurisoluista B-solulymfoomaa sairastavien aikuispotilaiden hoitoon, kun hematopoieettinen kantasolusiirto ei sovellu potilaalle.
Ehto
Valmistetta saa määrätä vain syöpäpotilaiden hoitoon perehtynyt lääkäri ja sitä saa antaa vain syöpäpotilaiden hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa.
Annostus ja antotapa
Polivy-hoito on annettava vain syöpäpotilaiden diagnosointiin ja hoitoon perehtyneiden terveydenhoidon ammattilaisten valvonnassa.
Annostus
Diffuusi suurisoluinen B-solulymfooma
Aiemmin hoitamattomat potilaat
Suositeltu Polivy-annos on 1,8 mg/kg infuusiona laskimoon 21 vuorokauden välein 6 hoitosyklin ajan yhdistelmänä rituksimabin, syklofosfamidin, doksorubisiinin ja prednisonin (R-CHP-hoidon) kanssa. Polivy, rituksimabi, syklofosfamidi ja doksorubisiini voidaan antaa hoitosyklin 1. päivänä missä tahansa järjestyksessä prednisonin annon jälkeen. Prednisonia annetaan kunkin hoitosyklin 1.–5. päivänä. Hoitosyklit 7 ja 8 koostuvat rituksimabimonoterapiasta.
Ks. aiemmin hoitamatonta diffuusia suurisoluista B-solulymfoomaa sairastaville potilaille Polivy-valmisteen kanssa yhdistelmänä annettavien solunsalpaajien valmisteyhteenvedot.
Uusiutunutta tai refraktorista tautia sairastavat potilaat
Suositeltu Polivy-annos on 1,8 mg/kg infuusiona laskimoon 21 vuorokauden välein 6 hoitosyklin ajan yhdistelmänä bendamustiinin ja rituksimabin kanssa. Polivy, bendamustiini ja rituksimabi voidaan antaa missä tahansa järjestyksessä kunkin hoitosyklin 1. päivänä. Polivy-valmisteen kanssa annettaessa bendamustiinin suositeltu annos on 90 mg/m2/vrk kunkin hoitosyklin 1. päivänä ja 2. päivänä, ja rituksimabin suositeltu annos on 375 mg/m2 kunkin hoitosyklin 1. päivänä. Polivy-annoksia 1,8 mg/kg saavien potilaiden yli 240 mg:n kokonaisannoksesta on vähän kliinistä kokemusta, joten suositellaan, ettei annosta 240 mg/hoitosykli ylitetä.
Aiemmin hoitamatonta ja uusiutunutta tai refraktorista tautia sairastavat potilaat
Jos esilääkitystä ei ole vielä annettu, potilaalle annetaan ennen Polivy-hoitoa esilääkityksenä antihistamiinia ja kuumelääkitystä.
Annosten viivästyminen tai antamatta jääminen
Jos suunniteltu Polivy-annos jää antamatta, se pitää antaa mahdollisimman pian, ja hoitoaikataulua muutetaan siten, että antoväli säilyy 21 päivänä.
Annosmuutokset
Jos potilaalle kehittyy infuusioon liittyvä reaktio, Polivy-infuusion antonopeutta pitää hidastaa tai anto keskeyttää. Jos potilaalle ilmaantuu hengenvaarallinen reaktio, Polivy-infuusion anto pitää lopettaa heti pysyvästi.
Polivy-hoidon mahdolliset annosmuutokset aiemmin hoitamatonta diffuusia suurisoluista B‑solulymfoomaa ja uusiutunutta tai refraktorista tautia sairastaville potilaille ovat erilaiset.
Annosmuutokset perifeerisen neuropatian hoidossa (kohta Varoitukset ja käyttöön liittyvät varotoimet), ks. taulukko 1 jäljempänä.
Taulukko 1. Polivy-annosmuutokset perifeerisen neuropatian yhteydessä
| Käyttöaihe | Perifeerisen neuropatian vaikeusaste minkä tahansa hoitosyklin 1. päivänä | Annosmuutos |
| Aiemmin hoitamaton diffuusi suurisoluinen B‑solulymfooma | Aste 2a | Sensorinen neuropatia:
Motorinen neuropatia:
Noudata samanaikaisen sensorisen ja motorisen neuropatian yhteydessä rajoittavinta edellä mainittua annosmuutossuositusta. |
| Aste 3a | Sensorinen neuropatia:
Motorinen neuropatia:
Noudata samanaikaisen sensorisen ja motorisen neuropatian yhteydessä rajoittavinta edellä mainittua annosmuutossuositusta. | |
| Aste 4 | Lopeta Polivy-hoito. | |
| Uusiutunut/refraktorinen diffuusi suurisoluinen B‑solulymfooma | Aste 2–3 | Keskeytä Polivy-hoito, kunnes oireet lievenevät asteeseen ≤ 1. Jos oireet lievenevät asteeseen ≤ 1 viimeistään 14. päivänä, jatka Polivy-hoitoa pysyvästi pienennetyllä annoksella 1,4 mg/kg. Jos annos on aiemmin pienennetty tasolle 1,4 mg/kg, lopeta Polivy-hoito. Jos oireet eivät lievene asteeseen ≤ 1 viimeistään 14. päivänä, lopeta Polivy-hoito |
| Aste 4 | Lopeta Polivy-hoito |
a R-CHP-hoitoa voidaan jatkaa.
Annosmuutokset luuydinlaman hoidossa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), ks. taulukko 2 jäljempänä.
Taulukko 2. Polivy-valmisteen, solunsalpaajahoidon ja rituksimabin annosmuutokset luuydinlaman hoidossa
| Käyttöaihe | Luuydinlaman vaikeusaste minkä tahansa hoitosyklin 1. päivänä | Annosmuutos |
| Aiemmin hoitamaton diffuusi suurisoluinen B‑solulymfooma | Asteen 3–4 neutropenia | Keskeytä kaikki hoidot, kunnes absoluuttinen neutrofiilimäärä korjautuu arvoon > 1 000/mikrol. Jos absoluuttinen neutrofiilimäärä korjautuu arvoon > 1 000/mikrol viimeistään 7. päivänä, jatka kaikkia hoitoja annosta pienentämättä. Jos absoluuttinen neutrofiilimäärä korjautuu arvoon > 1 000/mikrol myöhemmin kuin 7. päivänä
|
| Asteen 3–4 trombosytopenia | Keskeytä kaikki hoidot, kunnes trombosyyttimäärä korjautuu arvoon > 75 000/mikrol. Jos trombosyyttimäärä korjautuu arvoon > 75 000/mikrol viimeistään 7. päivänä, jatka kaikkia hoitoja annosta pienentämättä. Jos trombosyyttimäärä korjautuu arvoon > 75 000/mikrol myöhemmin kuin 7. päivänä
| |
| Uusiutunut/refraktorinen diffuusi suurisoluinen B‑solulymfooma | Asteen 3–4 neutropenia1 | Keskeytä kaikki hoidot, kunnes absoluuttinen neutrofiilimäärä korjautuu arvoon > 1 000/mikrol. Jos absoluuttinen neutrofiilimäärä korjautuu arvoon > 1 000/mikrol viimeistään 7. päivänä, jatka kaikkia hoitoja annosta pienentämättä. Jos absoluuttinen neutrofiilimäärä korjautuu arvoon > 1 000/mikrol myöhemmin kuin 7. päivänä
|
| Asteen 3–4 trombosytopenia1 | Keskeytä kaikki hoidot, kunnes trombosyyttimäärä korjautuu arvoon > 75 000/mikrol. Jos trombosyyttimäärä korjautuu arvoon > 75 000/mikrol viimeistään 7. päivänä, jatka kaikkia hoitoja annosta pienentämättä. Jos trombosyyttimäärä korjautuu arvoon > 75 000/mikrol myöhemmin kuin 7. päivänä
|
1Jos pääasiallinen syy on lymfooma, bendamustiiniannosta ei tarvitse pienentää.
Annosmuutokset infuusioon liittyvien reaktioiden hoidossa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), ks. taulukko 3 jäljempänä.
Taulukko 3. Polivy-valmisteen annosmuutokset infuusioon liittyvien reaktioiden yhteydessä
| Käyttöaihe | Infuusioon liittyvän reaktion vaikeusaste minkä tahansa hoitosyklin 1. päivänä | Annosmuutos |
| Aiemmin hoitamaton ja uusiutunut/refraktorinen diffuusi suurisoluinen B‑solulymfooma | Asteen 1–3 infuusioon liittyvä reaktio | Keskeytä Polivy-infuusio, ja anna tukihoitoa. Asteen 3 hengityksen vinkumisen, bronkospasmin tai yleistyneen urtikarian ensimmäisellä ilmaantumiskerralla lopeta Polivy-hoito pysyvästi. Asteen 2 hengityksen vinkumisen tai urtikarian uusiutuessa tai minkä tahansa asteen 3 oireen uusiutuessa lopeta Polivy-hoito pysyvästi. Muissa tapauksissa infuusiota voidaan jatkaa, kun oireet häviävät täysin. Antonopeus on tällöin puolet (50 %) hitaampi kuin ennen hoidon keskeyttämistä. Jos infuusioon liittyviä oireita ei ilmaannu, infuusionopeutta voidaan lisätä 50 mg/h 30 minuutin välein. Seuraavassa hoitosyklissä Polivy-infuusio annetaan 90 minuutin kestoisena. Jos infuusioon liittyvää reaktiota ei ilmene, seuraavat infuusiot voidaan antaa 30 minuutin kestoisena. Anna kaikissa hoitosykleissä esilääkitys. |
| Asteen 4 infuusioon liittyvä reaktio | Lopeta Polivy-infuusio heti. Anna tukihoitoa. Lopeta Polivy-hoito pysyvästi. |
Erityiset potilasryhmät
Iäkkäät potilaat
Iältään ≥ 65-vuotiaiden potilaiden Polivy-annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Jos potilaan kreatiniinipuhdistuma (CrCl) on ≥ 30 ml/min, Polivy-annosta ei tarvitse muuttaa. Suositusannosta ei ole määritetty potilaille, joiden CrCl on < 30 ml/min, koska tietoja ei ole riittävästi.
Maksan vajaatoiminta
Polivy-valmisteen antamista keskivaikeaa tai vaikeaa maksan vajaatoimintaa (bilirubiinipitoisuus suurempi kuin 1,5 × viitearvojen yläraja [ULN]) sairastaville potilaille pitää välttää.
Polivy-valmistetta lievää maksan vajaatoimintaa (bilirubiinipitoisuus suurempi kuin ULN tai enintään 1,5 × ULN tai aspartaattiaminotransferaasipitoisuus [ASAT] suurempi kuin ULN) sairastaville potilaille annettaessa aloitusannosta ei tarvitse muuttaa.
Lievää maksan vajaatoimintaa [määritelty ASAT- tai ALAT-arvoksi > 1,0–2,5 × ULN tai kokonaisbilirubiinipitoisuus > 1,0–1,5 × ULN] sairastavassa tutkitussa potilasjoukossa altistus konjugoimattomalle monometyyliauristatiini E:lle lisääntyi enintään 40 %, mitä ei katsottu kliinisesti merkittäväksi.
Pediatriset potilaat
Turvallisuutta ja tehoa lapsille ja alle 18 vuoden ikäisille nuorille ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Polivy annetaan laskimoon.
Polivy-aloitusannos annetaan 90 minuutin kestoisena infuusiona laskimoon. Potilaita tarkkaillaan infuusioon liittyvien reaktioiden tai yliherkkyysreaktioiden havaitsemiseksi infuusion aikana ja vähintään 90 minuutin ajan aloitusannoksen päättymisen jälkeen.
Jos potilas sieti edellisen Polivy-infuusion hyvin, seuraava annos voidaan antaa 30 minuutin kestoisena infuusiona, ja potilasta tarkkaillaan infuusion aikana ja vähintään 30 minuutin ajan infuusion päättymisen jälkeen.
Polivy on saatettava käyttökuntoon ja laimennettava edelleen terveydenhoidon ammattilaisen valvonnassa aseptiikkaa noudattaen. Valmiste annetaan infuusiona laskimoon sille tarkoitetun infuusioletkun (varustettu steriilillä, pyrogeenittömällä, niukasti proteiineja sitovalla letkun sisällä olevalla tai siihen kiinnitetyllä suodattimella, jonka huokoskoko on 0,2 tai 0,22 mikrometriä) ja katetrin kautta. Polivy-valmistetta ei saa antaa laskimoon paineella eikä boluksena.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Valmisteen käsittelyssä tai annossa huomioitavat varotoimet
Polivy sisältää sytotoksisen komponentin, joka on kovalenttisesti sitoutunut monoklonaaliseen vasta-aineeseen. Noudata asianmukaisia käsittely- ja hävittämistapoja (ks. kohta Käyttö- ja käsittelyohjeet).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiiviset vaikea-asteiset infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Luuydinlama
Polivy-hoitoa saaneilla potilailla on raportoitu vakavaa ja vaikea-asteista neutropeniaa ja kuumeista neutropeniaa jo ensimmäisestä hoitosyklistä lähtien. Kliinisessä kehitysohjelmassa vaadittiin profylaktisen granulosyyttiryhmiä stimuloivan kasvutekijän (G-CSF) antamista, joten sen antamista pitää harkita. Polivy-hoidon aikana voi ilmetä myös asteen 3 tai 4 trombosytopeniaa tai anemiaa. Täydellistä verenkuvaa seurataan ennen jokaista Polivy-annosta. Potilaille, joilla on asteen 3 tai asteen 4 neutropeniaa ja/tai trombosytopeniaa, harkitaan tiheämpää laboratorioseurantaa ja/tai Polivy-hoidon antoajankohdan siirtämistä myöhemmäksi tai hoidon lopettamista (ks. kohta Annostus ja antotapa).
Perifeerinen neuropatia
Polivy-hoitoa saaneilla potilailla on raportoitu perifeeristä neuropatiaa jo ensimmäisestä hoitosyklistä lähtien, ja sen riski suurenee seuraavien annosten myötä. Jos potilaalla on perifeeristä neuropatiaa jo ennestään, se saattaa pahentua. Polivy-hoidon yhteydessä raportoitu perifeerinen neuropatia on pääasiassa sensorista perifeeristä neuropatiaa. Motorista ja sensomotorista perifeeristä neuropatiaa on kuitenkin myös raportoitu. Potilaita seurataan perifeerisen neuropatian oireiden havaitsemiseksi. Oireita ovat mm. hypestesia, hyperestesia, parestesiat, dysestesia, neuropaattinen kipu, polttava tunne, lihasheikkous ja kävelyn häiriöt. Jos potilaalle ilmaantuu perifeeristä neuropatiaa tai jo olemassa oleva perifeerinen neuropatia pahenee, voi olla tarpeen siirtää Polivy-annoksen antoajankohtaa myöhemmäksi, pienentää annosta tai lopettaa hoito (ks. kohta Annostus ja antotapa).
Infektiot
Polivy-hoitoa saaneilla potilailla on raportoitu vakavia, hengenvaarallisia tai kuolemaan johtaneita infektioita, mukaan lukien opportunistisia infektioita, kuten keuhkokuumetta (mukaan lukien pneumocystis jirovecii ‑keuhkokuume ja muiden sienten aiheuttama keuhkokuume), bakteremiaa, sepsistä, herpesinfektioita ja sytomegalovirusinfektioita (ks. kohta Haittavaikutukset). Piilevien infektioiden reaktivaatiota on raportoitu. Potilaita on seurattava tarkoin hoidon aikana bakteeri-, sieni- tai virusinfektioiden havaitsemiseksi, ja jos oireita tai löydöksiä ilmenee, potilaan on hakeuduttava lääkäriin. Infektioiden estohoitoa koko Polivy-hoidon ajan pitää harkita. Jos potilaalla on aktiivinen vaikea-asteinen infektio, Polivy-hoitoa ei anneta. Jos potilaalle kehittyy vakavia infektioita, Polivy-hoito ja muu samanaikainen solunsalpaajahoito lopetetaan.
HI-virus
Polivy-hoitoa ei ole tutkittu HIV-potilailla. CYP3A:n estäjien samanaikainen käyttö, ks. kohta Yhteisvaikutukset.
Rokotukset
Hoidon aikana ei pidä antaa eläviä tai eläviä heikennettyjä taudinaiheuttajia sisältäviä rokotteita. Eläviä rokotteita äskettäin saaneilla potilailla ei ole tehty tutkimuksia.
Progressiivinen multifokaalinen leukoenkefalopatia (PML)
Polivy-hoidon yhteydessä on raportoitu progressiivista multifokaalista leukoenkefalopatiaa (ks. kohta Haittavaikutukset). Potilaita on seurattava tarkoin progressiiviseen multifokaaliseen leukoenkefalopatiaan viittaavien neurologisten, kognitiivisten tai käyttäytymisen muutosten ilmaantumisen tai pahenemisen havaitsemiseksi. Jos progressiivista multifokaalista leukoenkefalopatiaa epäillään, Polivy-hoito ja samanaikainen solunsalpaajahoito keskeytetään. Jos diagnoosi varmistuu, nämä hoidot lopetetaan pysyvästi.
Tuumorilyysioireyhtymä
Potilailla, joilla on suuri kasvaintaakka ja nopeasti kasvava kasvain, saattaa olla tavanomaista suurempi tuumorilyysioireyhtymän riski. Ennen Polivy-hoidon aloittamista on ryhdyttävä sopiviin paikallisten hoitosuositusten mukaisiin toimenpiteisiin / aloitettava estohoito. Potilaita on seurattava Polivy-hoidon aikana tarkoin tuumorilyysioireyhtymän havaitsemiseksi.
Infuusioon liittyvät reaktiot
Polivy-hoidosta voi aiheutua infuusioon liittyviä reaktioita, myös vaikea-asteisia reaktioita. Infuusioon liittyvät reaktiot voivat ilmetä viivästyneesti vielä 24 tuntia Polivy-hoidon saamisen jälkeen. Potilaalle annetaan antihistamiinia ja kuumetta alentavaa lääkettä ennen Polivy-valmisteen annostelua, ja potilaan vointia tarkkaillaan koko infuusion ajan. Jos infuusioon liittyvä reaktio ilmaantuu, keskeytetään infuusion anto ja aloitetaan asianmukainen hoito (ks. kohta Annostus ja antotapa).
Alkio- ja sikiötoksisuus
Raskaana olevalle naiselle annettu Polivy-valmiste voi vaikutusmekanisminsa ja prekliinisten tutkimusten perusteella vahingoittaa sikiötä (ks. kohta Prekliiniset tiedot turvallisuudesta). Raskaana olevalle naiselle pitää kertoa sikiölle aiheutuvasta riskistä.
Naispotilaita, jotka voivat tulla raskaaksi, pitää neuvoa käyttämään tehokasta ehkäisyä Polivy-hoidon aikana ja vähintään 9 kuukauden ajan viimeisen annoksen jälkeen (ks. kohta Raskaus ja imetys). Miespotilaita, joiden naiskumppani voi tulla raskaaksi, pitää neuvoa käyttämään tehokasta ehkäisyä Polivy-hoidon aikana ja vähintään 6 kuukauden ajan viimeisen annoksen jälkeen (ks. kohta Raskaus ja imetys).
Hedelmällisyys
Prekliinisissä tutkimuksissa polatutsumabi-vedotiinista on aiheutunut kivestoksisuutta, joten se voi heikentää miesten lisääntymistoimintoja ja hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta). Polivy-hoitoa saavia miehiä kehotetaan siksi hakeutumaan siemennesteen talteenottoon ja säilytykseen ennen hoitoa (ks. kohta Raskaus ja imetys).
Iäkkäät potilaat
Tutkimuksessa GO39942 Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneista 435:stä aiemmin hoitamatonta diffuusia suurisoluista B‑solulymfoomaa sairastavasta potilaasta 227 (52,2 %) oli ≥ 65‑vuotiaita. Iältään ≥ 65-vuotiailla potilailla vakavien haittavaikutusten ilmaantuvuus oli 39,2 % ja iältään < 65-vuotiailla potilailla se oli 28,4 %. Vakavien haittavaikutusten ilmaantuvuus oli R-CHOP-hoitoa (rituksimabia, syklofosfamidia, doksorubisiinia, vinkristiiniä ja prednisonia) saaneen hoitohaaran iäkkäillä potilailla samankaltainen.
Tutkimuksessa GO29365 Polivy-hoitoa yhdistelmänä bendamustiinin ja rituksimabin (BR-hoidon) kanssa saaneista 151:stä aiemmin hoidettua diffuusia suurisoluista B-solulymfoomaa sairastavasta potilaasta 103 potilasta (68 %) oli ≥ 65-vuotiaita. Iältään ≥ 65-vuotiailla potilailla vakavien haittavaikutusten ilmaantuvuus oli samankaltainen (55 %) kuin < 65-vuotiailla potilailla (56 %). Kliinisissä Polivy-tutkimuksissa ei ollut mukana riittävästi ≥ 65-vuotiaita potilaita sen selvittämiseksi, onko heidän vasteensa erilainen nuorempiin potilaisiin verrattuna.
Maksatoksisuus
Polivy-hoitoa saaneilla potilailla on esiintynyt maksasoluvaurioon sopivaa vakavaa maksatoksisuutta, mukaan lukien transaminaasi- ja/tai bilirubiinipitoisuuksien kohoamista (ks. kohta Haittavaikutukset). Potilaalla ennestään oleva maksasairaus, lähtötilanteessa koholla olevat maksaentsyymipitoisuudet ja muiden lääkevalmisteiden samanaikainen käyttö voivat lisätä tätä riskiä. Maksaentsyymi- ja bilirubiinipitoisuuksia pitää seurata (ks. kohta Annostus ja antotapa).
Infuusiokohdan ekstravasaatiovamma
Polatutsumabi-vedotiinin antamisen jälkeen on tuntien – viikkojen kuluessa ekstravasaation tapahtumisesta ilmennyt iho- ja pehmytkudosvammoja (ks. kohta Haittavaikutukset). Varmista ennen Polivy-valmisteen antamisen aloittamista hyvä laskimoyhteys ja seuraa annon aikana infuusiokohtaa mahdollisen ekstravasaation varalta. Jos ekstravasaatio tapahtuu, lopeta infuusio, seuraa kudosvauriota ja hoida paikallisten kliinisten ohjeistusten mukaisesti.
Loppuosa annoksesta voidaan kliinisen päätöksen perusteella antaa toiseen raajaan.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Tämä lääkevalmiste sisältää polysorbaattia 20. Yksi injektiopullo Polivy 30 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos sisältää 1,8 mg polysorbaattia 20. Yksi injektiopullo Polivy 140 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos sisältää 8,4 mg polysorbaattia 20, joka vastaa 1,2 mg/ml. Polysorbaatti 20 saattaa aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Varsinaisia polatutsumabi-vedotiinia koskevia kliinisiä yhteisvaikutustutkimuksia ihmisillä ei ole tehty.
Yhteisvaikutukset samanaikaisesti käytettävien CYP3A4:n estäjien, substraattien tai induktorien ja P‑gp:n estäjien kanssa
Polatutsumabi-vedotiinista vapautuvasta monometyyliauristatiini E:stä (MMAE) tehdyt fysiologiaan perustuvat farmakokineettiset mallisimulaatiot (PBPK-mallinnukset) osoittavat, että voimakkaat CYP3A4:n ja P-gp:n estäjät (esim. ketokonatsoli) saattavat suurentaa konjugoimattoman monometyyliauristatiini E:n aika-pitoisuuskäyrän alle jäävää pinta-alaa (AUC-arvoa) 48 %. Varovaisuuteen kehotetaan käytettäessä samanaikaisesti CYP3A4:n estäjiä. Voimakkaita CYP3A4:n estäjiä (esim. bosepreviiri, klaritromysiini, kobisistaatti, indinaviiri, itrakonatsoli, nefatsodoni, nelfinaviiri, posakonatsoli, ritonaviiri, sakinaviiri, telapreviiri, telitromysiini, vorikonatsoli) samanaikaisesti käyttäviä potilaita pitää seurata tarkemmin toksisuuden oireiden havaitsemiseksi.
Konjugoimattoman monometyyliauristatiini E:n ei odoteta muuttavan sellaisten samanaikaisesti käytettävien lääkkeiden AUC-arvoa, jotka ovat CYP3A4:n substraatteja (esim. midatsolaami).
Voimakkaat CYP3A4:n induktorit (esim. rifampisiini, karbamatsepiini, fenobarbitaali, fenytoiini, mäkikuisma [Hypericum perforatum]) voivat pienentää altistusta konjugoimattomalle monometyyliauristatiini E:lle.
Yhteisvaikutukset käytettäessä rituksimabia, bendamustiinia, syklofosfamidia ja doksorubisiinia yhdistelmänä polatutsumabi-vedotiinin kanssa
Polatutsumabi-vedotiinin samanaikainen käyttö ei vaikuta rituksimabin, bendamustiinin, syklofosfamidin ja doksorubisiinin farmakokinetiikkaan. Rituksimabin samanaikaiseen käyttöön liittyy populaatiofarmakokineettisen analyysin perusteella vasta-aineeseen konjugoidun monometyyliauristatiini E:n (acMMAE) 24 % suurentunut plasman AUC-arvo ja konjugoimattoman monometyyliauristatiini E:n (MMAE) 37 % pienentynyt plasman AUC-arvo. Vasta-aineeseen konjugoidun monometyyliauristatiini E:n ja konjugoitumattoman monometyyliauristatiini E:n AUC-arvot plasmassa Polivy-valmisteen ja R-CHP-hoidon yhdistelmää käytettäessä ovat muiden Polivy-tutkimusten mukaiset. Annosta ei tarvitse muuttaa.
Bendamustiini ei vaikuta vasta-aineeseen konjugoidun monometyyliauristatiini E:n (acMMAE) eikä konjugoimattoman monometyyliauristatiini E:n (MMAE) AUC-arvoon plasmassa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi/Ehkäisy miehille ja naisille
Naiset
Naisia, jotka voivat tulla raskaaksi, pitää neuvoa käyttämään tehokasta ehkäisyä polatutsumabi-vedotiinihoidon aikana ja vähintään 9 kuukauden ajan viimeisen annoksen jälkeen.
Miehet
Miespotilaita, joiden naiskumppani voi tulla raskaaksi, pitää neuvoa käyttämään tehokasta ehkäisyä polatutsumabi-vedotiinihoidon aikana ja vähintään 6 kuukauden ajan viimeisen annoksen jälkeen.
Raskaus
Polivy-hoitoa saavista raskaana olevista naisista ei ole tietoja. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Raskaana olevalle naiselle annettu polatutsumabi-vedotiini voi vaikutusmekanisminsa ja prekliinisten tutkimusten perusteella vahingoittaa sikiötä. Naiset, jotka voivat tulla raskaaksi, tutkitaan ennen hoitoa raskauden mahdollisuuden poissulkemiseksi. Polivy-hoitoa ei suositella raskauden aikana eikä naisille, jotka voivat tulla raskaaksi eivätkä käytä ehkäisyä, paitsi jos mahdolliset hyödyt äidille ovat sikiölle aiheutuvia mahdollisia riskejä suuremmat.
Imetys
Ei tiedetä, erittyvätkö polatutsumabi-vedotiini tai sen metaboliitit ihmisen rintamaitoon. Imetettävään lapseen kohdistuvia riskejä ei voida poissulkea. Naisten on lopetettava imettäminen Polivy-hoidon ajaksi ja vähintään 3 kuukaudeksi viimeisen annoksen jälkeen.
Hedelmällisyys
Prekliinisissä tutkimuksissa polatutsumabi-vedotiinista on aiheutunut kivestoksisuutta, joten se voi heikentää miesten lisääntymistoimintoja ja hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Tällä lääkkeellä hoitoa saavia miehiä kehotetaan siksi hakeutumaan siemennesteen talteenottoon ja säilytykseen ennen hoitoa. Polivy-hoitoa saavia miehiä kehotetaan olemaan siittämättä lasta hoidon aikana ja 6 kuukauden ajan viimeisen annoksen jälkeen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Polivy-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Polivy-hoidon aikana voi esiintyä infuusioon liittyviä reaktioita, perifeeristä neuropatiaa, uupumusta ja huimausta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Polivy-valmisteen turvallisuutta on arvioitu tutkimuksessa GO39942 (POLARIX) 435 potilaalla. Kohdassa Haittavaikutukset kuvatut haittavaikutukset tunnistettiin:
- kliinisessä pivotaalitutkimuksessa GO39942 (POLARIX) hoidon ja seurannan aikana aiemmin hoitamatonta diffuusia suurisoluista B-solulymfoomaa sairastavilla potilailla, jotka saivat Polivy-valmisteen ja R-CHP-hoidon (n = 435) yhdistelmää tai R‑CHOP-hoitoa (n = 438). Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa ryhmässä 91,7 % sai kuusi Polivy-hoitosykliä kun taas R-CHOP-hoitoa saaneessa ryhmässä 88,5 % potilaista sai kuusi hoitosykliä vinkristiiniä.
Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneilla aiemmin hoitamatonta diffuusia suurisoluista B-solulymfoomaa sairastavilla potilailla:
- yleisimmin (≥ 30 %) raportoituja haittavaikutuksia olivat perifeerinen neuropatia (52,9 %), pahoinvointi (41,6 %), neutropenia (38,4 %) ja ripuli (30,8 %)
- vakavia haittavaikutuksia raportoitiin 24,1 %:lla potilaista, jotka saivat Polivy-valmisteen ja R-CHP-hoidon yhdistelmää
- yleisimpiä vakavia haittavaikutuksia, joita raportoitiin ≥ 5 %:lla potilaista, olivat kuumeinen neutropenia (10,6 %) ja keuhkokuume (5,3 %)
- hoidon lopettamiseen johtanut haittavaikutus, jota esiintyi > 1 %:lla Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneista potilaista, oli keuhkokuume (1,1 %).
Polivy-valmisteen turvallisuutta on tutkimuksessa GO29365 arvioitu 151 potilaalla. Kohdassa Haittavaikutukset kuvatut haittavaikutukset tunnistettiin:
- kliinisessä pivotaalitutkimuksessa GO29365 aiemmin hoidettujen diffuusia suurisoluista B-solulymfoomaa sairastavien potilaiden (n = 151) hoidon ja seurannan aikana. Tässä on mukana esivaiheen potilaat (n = 6), satunnaistetut potilaat (n = 39) ja jatkovaiheen kohortin potilaat (n = 106), jotka saivat Polivy-valmisteen ja BR-hoidon yhdistelmää. Näitä potilaita verrattiin satunnaistettuihin potilaisiin (n = 39), jotka saivat pelkästään BR-hoitoa. Hoitohaarojen potilaat saivat 5 hoitosykliä (mediaani), kun taas vertailuhaaran satunnaistetut potilaat saivat 3 hoitosykliä (mediaani).
Polivy-valmisteen ja BR-hoidon yhdistelmällä aiemmin hoidettua diffuusia suurisoluista B‑solulymfoomaa sairastavilla potilailla:
- yleisimmin (≥ 30 %) raportoituja haittavaikutuksia (kaikki vaikeusasteet) aiemmin Polivyn ja BR-hoidon yhdistelmää saaneilla diffuusia suurisoluista B-solulymfoomaa sairastavilla potilailla, olivat neutropenia (45,7 %), ripuli (35,8 %), pahoinvointi (33,1 %), trombosytopenia (32,5 %), anemia (31,8 %) ja perifeerinen neuropatia (30,5 %)
- vakavia haittavaikutuksia raportoitiin 41,7 %:lla potilaista, jotka saivat Polivy-hoidon ja BR-hoidon yhdistelmää
- yleisimpiä vakavia haittavaikutuksia, joita raportoitiin ≥ 5 %:lla potilaista, olivat kuumeinen neutropenia (10,6 %), sepsis (9,9 %), keuhkokuume (8,6 %) ja kuume (7,9 %)
- hoidon lopettamiseen johtanut haittavaikutus, jota esiintyi > 5 %:lla Polivy-valmisteen ja BR-hoidon yhdistelmää saaneista potilaista, oli trombosytopenia (7,9 %).
Kliinisissä tutkimuksissa todettujen haittavaikutusten yhteenvetotaulukko
Taulukossa 4 esitetään 586:lla Polivy-hoitoa saaneella potilaalla todetut haittavaikutukset. Haittavaikutukset esitetään seuraavassa taulukossa MedDRA-elinjärjestelmäluokittain ja esiintymistiheysluokittain. Kunkin haittavaikutuksen esiintymistiheysluokka perustuu seuraavaan esitystapaan: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 4. Polivy-hoitoa kliinisissä tutkimuksissa saaneilla potilailla todetut haittavaikutukset
| Infektiot | |
| Hyvin yleinen | keuhkokuumea, ylähengitysteiden infektio |
| Yleinen | sepsisa, herpesvirusinfektioa, sytomegalovirusinfektio, virtsatieinfektioc |
| Veri ja imukudos | |
| Hyvin yleinen | kuumeinen neutropenia, neutropenia, trombosytopenia, anemia, leukopenia |
| Yleinen | lymfopenia, pansytopenia |
| Aineenvaihdunta ja ravitsemus | |
| Hyvin yleinen | hypokalemia, heikentynyt ruokahalu |
| Yleinen | hypokalsemia, hypoalbuminemia |
| Hermosto | |
| Hyvin yleinen | perifeerinen neuropatia |
| Yleinen | huimaus |
| Silmät | |
| Melko harvinainen | näön sumeneminenb |
| Hengityselimet, rintakehä ja välikarsina | |
| Hyvin yleinen | yskä |
| Yleinen | pneumoniitti, hengenahdistusc |
| Ruoansulatuselimistö | |
| Hyvin yleinen | ripuli, pahoinvointi, ummetus, oksentelu, mukosiittic, vatsakipu |
| Iho ja ihonalainen kudos | |
| Hyvin yleinen | alopesiac |
| Yleinen | kutina, ihoinfektiotc, ihottumac, kuiva ihoc |
| Luusto, lihakset ja sidekudos | |
| Yleinen | nivelkipu, lihaskipuc |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Hyvin yleinen | kuume, uupumus, astenia |
| Yleinen | perifeerinen turvotusc, vilunväreet |
| Melko harvinainen | infuusiokohdan ekstravasaatio |
| Tutkimukset | |
| Hyvin yleinen | painon lasku |
| Yleinen | suurentunut lipaasipitoisuusb, hypofosfatemia |
| Maksa ja sappi | |
| Yleinen | suurentunut transaminaasipitoisuus |
| Vammat, myrkytykset ja hoitokomplikaatiot | |
| Hyvin yleinen | infuusioon liittyvä reaktio |
a Haittavaikutukseen liittyi kuolemantapauksia
b Haittavaikutuksia havaittiin vain uusiutunutta tai refraktorista diffuusia suurisoluista B-solulymfoomaa sairastavilla.
c Haittavaikutuksia havaittiin vain aiemmin hoitamatonta diffuusia suurisoluista B-solulymfoomaa sairastavilla.
Lueteltuja haittavaikutuksia havaittiin sekä aiemmin hoitamatonta diffuusia suurisoluista B-solulymfoomaa että uusiutunutta tai refraktorista diffuusia suurisoluista B-solulymfoomaa sairastavilla, paitsi jos alaviitteissä mainitaan toisin.
Harvinaiset ja hyvin harvinaiset haittavaikutukset: ei ole
Valikoitujen haittavaikutusten kuvaus
Luuydinlama
Lumelääkekontrolloidussa tutkimuksessa GO39942 (POLARIX) 0,5 % Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneen haaran potilaista lopetti tutkimushoidon neutropenian vuoksi. R-CHOP-haarassa yksikään potilas ei lopettanut tutkimushoitoa neutropenian vuoksi. Trombosytopeniatapahtumat johtivat tutkimushoidon lopettamiseen 0,2 %:lla potilaista Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa, mutta ei yhdelläkään potilaalla R‑CHOP-haarassa. Yksikään potilas ei lopettanut hoitoa anemian vuoksi Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa eikä R-CHOP-hoitoa saaneessa haarassa.
Avoimessa tutkimuksessa GO29365 Polivy-valmisteen ja BR-hoidon yhdistelmää saaneista potilaista 4 % lopetti Polivy-hoidon neutropenian vuoksi verrattuna 2,6 %:iin BR-hoitoa saaneista potilaista, jotka lopettivat hoidon neutropenian vuoksi. Trombosytopenia johti hoidon lopettamiseen Polivy-valmisteen ja BR-hoidon yhdistelmää saaneissa haaroissa 7,9 %:lla potilaista ja BR-hoitoa saaneessa haarassa 5,1 %:lla potilaista. Yksikään potilas ei lopettanut hoitoa anemian vuoksi Polivy-valmisteen ja BR-hoidon yhdistelmää saaneessa haarassa eikä BR-hoitoa saaneessa haarassa. Polivy-valmisteen ja BR-hoidon yhdistelmää saaneessa haarassa vähintään vaikeusasteen 3 neutropeniaa raportoitiin 40,4 %:lla, trombosytopeniaa 25,8 %:lla ja anemiaa 12,6 %:lla potilaista.
Perifeerinen neuropatia
Lumekontrolloidussa tutkimuksessa GO39942 (POLARIX) Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa raportoitiin asteen 1 perifeeristä neuropatiaa 39,1 %:lla potilaista, asteen 2 perifeeristä neuropatiaa 12,2 %:lla potilaista ja asteen 3 perifeeristä neuropatiaa 1,6 %:lla potilaista. R-CHOP-hoitohaarassa raportoitiin asteen 1 perifeeristä neuropatiaa 37,2 %:lla potilaista, asteen 2 perifeeristä neuropatiaa 15,5 %:lla potilaista ja asteen 3 perifeeristä neuropatiaa 1,1 %:lla potilaista. Asteiden 4–5 perifeerisiä neuropatiatapahtumia ei raportoitu Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa eikä R-CHOP-hoitoa saaneessa haarassa. Perifeerisen neuropatian vuoksi tutkimushoidon lopetti 0,7 % potilaista Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa verrattuna 2,3 %:iin R-CHOP-hoitohaarassa. Annosta oli pienennetty perifeerisen neuropatian vuoksi 4,6 %:lla Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneista potilaista, kun taas vastaava luku R-CHOP-hoitoa saaneessa haarassa oli 8,2 %. Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa ensimmäisen perifeerisen neuropatiatapahtuman alkamiseen kuluneen ajan mediaani oli 2,27 kuukautta verrattuna 1,87 kuukauteen R-CHOP-hoitohaarassa. Perifeerinen neuropatia hävisi kliiniseen katkaisupäivämäärään mennessä 57,8 %:lla potilaista verrattuna 66,9 %:iin R-CHOP-hoitoa saaneessa haarassa. Perifeerisen neuropatian häviämiseen kuluneen ajan mediaani oli 4,04 kuukautta Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa verrattuna 4,6 kuukauteen R-CHOP-hoitoa saaneessa haarassa.
Avoimessa tutkimuksessa GO29365 Polivy-valmisteen ja BR-hoidon yhdistelmää saaneessa haarassa asteen 1 perifeeristä neuropatiaa raportoitiin 15,9 %:lla potilaista ja asteen 2 perifeeristä neuropatiaa raportoitiin 12,6 %:lla potilaista. BR-hoitohaarassa asteen 1 perifeerisiä neuropatiatapahtumia raportoitiin 2,6 %:lla potilaista ja asteen 2 perifeerisiä neuropatiatapahtumia raportoitiin 5,1 %:lla potilaista. Asteen 3 perifeeristä neuropatiaa raportoitiin yksi tapahtuma Polivy-valmisteen ja BR-hoidon yhdistelmää saaneissa haaroissa, mutta perifeeristä neuropatiaa ei raportoitu yhdelläkään potilaalla BR-hoitohaarassa. Asteen 4–5 perifeeristä neuropatiaa ei raportoitu Polivy-valmisteen ja BR-hoidon yhdistelmää saaneissa haaroissa eikä BR-hoitoa saaneessa haarassa. Perifeerisen neuropatian seurauksena 2,6 % potilaista lopetti Polivy-hoidon ja 2,0 %:lla potilaista Polivy-annosta pienennettiin. BR-hoitoa saaneessa haarassa yksikään potilas ei lopettanut hoitoa eikä yhdenkään annosta pienennetty perifeerisen neuropatian vuoksi. Polivy-valmisteen ja BR-hoidon yhdistelmää saaneissa haaroissa ensimmäinen perifeerisen neuropatian tapahtuman ilmenemiseen kuluneen ajan mediaani oli 1,6 kuukautta, ja 39,1 % potilaista, joilla perifeerisen neuropatian tapahtumia ilmaantui, raportoi tapahtuman hävinneen.
Infektiot
Lumekontrolloidussa tutkimuksessa GO39942 (POLARIX) infektioita, mukaan lukien keuhkokuumetta ja muun tyyppisiä infektioita, raportoitiin 49,7 %:lla potilaista Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa ja 42,7 %:lla potilaista R-CHOP-hoitohaarassa. Asteen 3-4 infektioita ilmaantui 14,0 %:lle potilaista Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa ja 11,2 %:lle potilaista R-CHOP-hoitohaarassa. Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa vakavia infektioita raportoitiin 14,0 %:lla potilaista ja kuolemaan johtaneita infektioita raportoitiin 1,1 %:lla potilaista. R-CHOP-hoitohaarassa vakavia infektioita raportoitiin 10,3 %:lla potilaista ja kuolemaan johtaneita infektioita raportoitiin 1,4 %:lla potilaista. Hoidon keskeytti infektion vuoksi 7 potilasta (1,6 %) Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa verrattuna 10 potilaaseen (2,3 %) R-CHOP-hoitohaarassa.
Avoimessa tutkimuksessa GO29365 infektioita, mukaan lukien keuhkokuumetta ja muun tyyppisiä infektioita, raportoitiin 48,3 %:lla potilaista Polivy-valmisteen ja BR-hoidon yhdistelmää saaneissa haaroissa ja 51,3 %:lla potilaista BR-hoitohaarassa. Polivy-valmisteen ja BR-hoidon yhdistelmää saaneissa haaroissa raportoitiin vakavia infektioita 27,2 %:lla potilaista ja kuolemaan johtaneita infektioita 6,6 %:lla potilaista. BR-hoitohaarassa vakavia infektioita raportoitiin 30,8 %:lla potilaista ja kuolemaan johtaneita infektioita raportoitiin 10,3 %:lla potilaista. Neljä potilasta (2,6 %) Polivy-valmisteen ja BR-hoidon yhdistelmää saaneissa haaroissa lopetti hoidon infektion vuoksi verrattuna kahteen potilaaseen (5,1 %) BR-hoitohaarassa.
Progressiivinen multifokaalinen leukoenkefalopatia (PML)
Lumekontrolloidussa tutkimuksessa GO39942 (POLARIX) ei raportoitu progressiivista multifokaalista leukoenkefalopatiaa.
Avoimessa tutkimuksessa GO29365 yhdellä Polivy-valmisteen sekä bendamustiinin ja obinututsumabin yhdistelmää saaneella potilaalla todettiin progressiivinen multifokaalinen leukoenkefalopatia, joka johti potilaan kuolemaan. Potilas oli aiemmin saanut kolmea hoitolinjaa, joihin kuului anti-CD20-vasta-aineita.
Maksatoksisuus
Lumekontrolloidussa tutkimuksessa GO39942 (POLARIX) maksatoksisuutta raportoitiin 10,6 %:lla potilaista Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa ja 7,3 %:lla potilaista R-CHOP-hoitohaarassa. Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa valtaosa oli asteen 1−2 tapahtumia (8,7 %), asteen 3 tapahtumia raportoitiin 1,8 %:lla potilaista. Asteen 4 tai 5 tapahtumia ei todettu. Vakavia maksatoksisuustapahtumia raportoitiin 1 potilaalla (0,2 %), ja ne olivat korjautuvia.
Toisessa tutkimuksessa raportoitiin kahdessa tapauksessa vakavaa maksatoksisuutta (hepatosellulaarinen vaurio ja maksan rasvoittuminen), joka oli korjautuvaa.
Maha-suolikanavan toksisuus
Lumekontrolloidussa tutkimuksessa GO39942 (POLARIX) maha-suolikanavan toksisuustapahtumia raportoitiin 76,1 %:lla potilaista Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa verrattuna 71,9 %:iin potilaista R-CHOP-hoitohaarassa. Valtaosa tapahtumista oli asteen 1–2 tapahtumia, ja asteen ≥ 3 tapahtumia raportoitiin 9,7 %:lla potilaista Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saaneessa haarassa, ja 8,2 %:lla potilaista R-CHOP-hoitohaarassa. Yleisimmät maha-suolikanavan toksisuuteen liittyvät tapahtumat olivat pahoinvointi ja ripuli.
Avoimessa tutkimuksessa GO29365 maha-suolikanavan toksisuuteen liittyviä tapahtumia raportoitiin 72,8 %:lla potilaista Polivy-valmisteen ja BR-hoidon yhdistelmää saaneissa haaroissa ja 66,7 %:lla potilaista BR-hoitohaarassa. Valtaosa tapahtumista oli asteen 1–2 tapahtumia, ja asteen 3–4 tapahtumia raportoitiin 16,5 %:lla potilaista Polivy-valmisteen ja BR-hoidon yhdistelmää saaneissa haaroissa ja 12,9 %:lla potilaista BR-hoitohaarassa. Yleisimmät maha-suolikanavan toksisuuteen liittyvät tapahtumat olivat ripuli ja pahoinvointi.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Ihmisellä tehdyistä kliinisistä tutkimuksista ei ole kokemusta yliannoksesta. Suurin tähän mennessä tutkittu annos on 2,4 mg/kg, joka annettiin infuusiona laskimoon. Siihen liittyi yleisemmin ja vaikeampiasteisia perifeerisen neuropatian tapahtumia. Jos potilas saa yliannoksen, infuusion antaminen keskeytetään heti ja potilasta seurataan tarkoin.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet; muut antineoplastiset lääkeaineet; monoklonaaliset vasta-aineet ATC-koodi: L01FX14
Vaikutusmekanismi
Polatutsumabi-vedotiini on CD79b‑kohdennettu vasta-ainelääkekonjugaatti, joka valikoivasti vapauttaa voimakasta mitoosia estävää ainetta (monometyyliauristatiini E eli MMAE) B-soluihin ja siten tappaa pahanlaatuiset B-solut. Polatutsumabi-vedotiinimolekyyli koostuu humanisoidusta monoklonaalisesta immunoglobuliini G1 ‑vasta-aineesta, ja siihen katkaistavan linkkerin välityksellä kovalenttisesti sitoutuneesta monometyyliauristatiini E:stä. Monoklonaalinen vasta-aine sitoutuu suurella affiniteetilla ja erittäin selektiivisesti CD79b-antigeeniin, joka on B‑solureseptorin pintakomponentti. CD79b-antigeenin ilmentyminen rajoittuu B‑solulinjassa normaaleihin soluihin (plasmasoluja lukuun ottamatta) ja pahanlaatuisiin B-soluihin. Diffuuseista suurisoluisista B‑solulymfoomista > 95 % ilmentää sitä. CD79b-antigeeniin sitouduttuaan polatutsumabi-vedotiini siirtyy nopeasti solun sisään, ja lysosomaaliset proteaasit katkaisevat linkkerin, jolloin monometyyliauristatiini E vapautuu solun sisään. Monometyyliauristatiini E sitoutuu mikrotubuluksiin ja tappaa jakautuvat solut estämällä solunjakautumisen ja indusoimalla apoptoosin.
Farmakodynaamiset vaikutukset
Sydämen elektrofysiologia
Polatutsumabi-vedotiinia suositusannoksina B-solusyöpien hoitoon aiemmin saaneilla potilailla tehdyistä kahdesta avoimesta tutkimuksesta saatujen EKG-tietojen perusteella polatutsumabi-vedotiini ei pidentänyt keskimääräistä QTc-aikaa kliinisesti merkittävästi.
Kliininen teho ja turvallisuus
Aiemmin hoitamaton diffuusi suurisoluinen B-solulymfooma
Polivy-valmisteen tehoa arvioitiin kansainvälisessä, satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (POLARIX, GO39942) 879 potilaalla, joilla oli aiemmin hoitamaton diffuusi suurisoluinen B-solulymfooma.
Mukaan soveltuneet potilaat olivat iältään 18–80-vuotiaita. Heidän IPI-pisteensä olivat 2–5 ja ECOG-toimintakykynsä oli 0–2. Histologioita olivat diffuusi suurisoluinen B-solulymfooma (tarkemmin määrittelemätön), aktivoituneen B-solun kaltainen (ABC), itukeskussoluperäinen (GCB), high-grade B‑solulymfooma (määrittämätön, double hit, triple hit) ja muut suurisoluisen B‑solulymfooman alatyypit (EBV-positiivinen, T‑soluvaltainen/histiosyyttivaltainen). Potilailla ei ollut tiedossa olevaa keskushermoston lymfoomaa tai asteen > 1 perifeeristä neuropatiaa.
Potilaat satunnaistettiin suhteessa 1:1 saamaan Polivy-valmisteen ja R-CHP-hoidon yhdistelmää tai R‑CHOP-hoitoa kuuden 21 päivän pituisen hoitosyklin ajan ja sen jälkeen kummassakin hoitohaarassa lisäksi kaksi hoitosykliä pelkästään rituksimabia. Potilaat ositettiin seuraavasti: IPI-pisteet (2 vs 3–5), sairauteen liittyy tai ei liity suuri tautimassa (leesio ≥ 7,5 cm) ja maantieteellinen alue.
Polivy-valmistetta annettiin laskimoon annoksina 1,8 mg/kg hoitosyklien 1–6 päivänä 1. R-CHP-hoitoa tai R-CHOP-hoitoa annettiin hoitosyklien 1–6 päivästä 1 alkaen, minkä jälkeen hoitosyklien 7−8 päivänä 1 annettiin pelkästään rituksimabia. Näissä hoitohaaroissa hoito annettiin seuraavasti:
- Polivy-valmisteen ja R-CHP-hoidon yhdistelmää saanut haara: Polivy 1,8 mg/kg, rituksimabi 375 mg/m², syklofosfamidi 750 mg/m², doksorubisiini 50 mg/m² ja prednisoni 100 mg/vrk suun kautta kunkin hoitosyklin päivinä 1–5
- R-CHOP-hoitohaara: rituksimabi 375 mg/m², syklofosfamidi 750 mg/m², doksorubisiini 50 mg/m², vinkristiini 1,4 mg/m² ja prednisoni 100 mg/vrk suun kautta kunkin hoitosyklin päivinä 1–5.
Nämä kaksi hoitoryhmää olivat lähtötilanteen demografisten ja sairautta koskevien ominaisuuksien suhteen yleisesti ottaen tasapainossa. Iän mediaani oli 65 vuotta (vaihteluväli 19–80 vuotta), 53,6 % potilaista oli valkoihoisia ja 53,8 % oli miehiä. 43,8 %:lla sairauteen liittyi suuri tautimassa, 38,0 %:lla oli IPI-pisteet 2, 62,0 %:lla oli IPI-pisteet 3–5 ja 88,7 %:lla oli levinneisyysasteen 3 tai 4 tauti. Suurimmalla osalla potilaista (84,2 %) oli diffuusi suurisoluinen B-solulymfooma (mukaan lukien tarkemmin määrittelemätön, aktivoituneen B-solun kaltainen ja itukeskussoluperäinen).
Potilaista 211:llä lähtösolutulosta ei raportoitu. Lähtösolun osalta arvioitavissa olleen potilasjoukon (n = 668) geeniekspressioprofiloinnin mukaan 33,1 %:lla potilaista oli aktivoituneen B‑solun kaltainen diffuusi suurisoluinen B-solulymfooma ja 52,7 %:lla potilaista oli itukeskussoluperäinen diffuusi suurisoluinen B-solulymfooma.
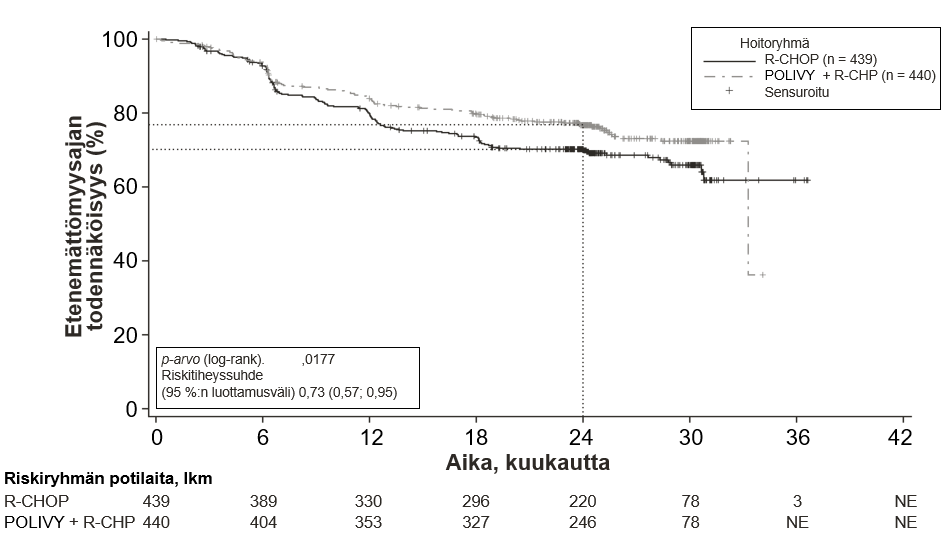
Tutkimuksen ensisijainen päätetapahtuma oli tutkijan arvioima etenemättömyysaika. Seuranta-ajan keston mediaani oli 28,2 kuukautta. Yhteenveto tehon tuloksista esitetään taulukossa 5 ja kuvassa 1.
Taulukko 5 Tutkimukseen GO39942 (POLARIX) perustuva yhteenveto tehosta aiemmin hoitamatonta diffuusia suurisoluista B-solulymfoomaa sairastavilla potilailla
Polivy + R-CHP N = 440 | R-CHOP N = 439 | |
| Ensisijainen päätetapahtuma | ||
| Etenemättömyysaika1,* | ||
| Potilaita, joilla oli tapahtumia, lkm (%) | 107 (24,3 %) | 134 (30,5 %) |
| Riskitiheyssuhde (HR) (95 %:n luottamusväli) | 0,73 (0,57; 0,95) | |
| p-arvo3,** | 0,0177 | |
| 2 vuoden etenemättömyysajan estimaatti (%) | 76,7 | 70,2 |
| (95 %:n luottamusväli) | (72,65; 80,76) | (65,80; 74,61) |
| Keskeiset toissijaiset päätetapahtumat | ||
| Elossaolo ilman tapahtumia (EFSeff)1 | ||
| Potilaita, joilla oli tapahtuma, lkm (%) | 112 (25,5 %) | 138 (31,4 %) |
| Riskitiheyssuhde (HR) (95 %:n luottamusväli) | 0,75 (0,58; 0;96) | |
| p-arvo3,** | 0,0244 | |
| Objektiivinen vasteluku hoidon päättyessä2 | ||
| Vasteen saaneita (%) (täydellinen vaste, osittainen vaste) | 376 (85,5 %) | 368 (83,8 %) |
| Vasteluvun ero (%) (95 %:n luottamusväli) | 1,63 (-3,32; 6,57) | |
| Täydellinen vaste (%)2,* | ||
| Vasteen saaneita (%) | 343 (78,0 %) | 325 (74,0 %) |
| Vasteluvun ero (%) (95 %:n luottamusväli) | 3,92 (-1,89; 9,70) | |
| Osittainen vaste (%) | 33 (7,5 %) | 43 (9,8 %) |
| 95 %:n Clopper-Pearsonin luottamusväli | [5,22; 10,37] | [7,18; 12,97] |
EFSeff: Elossaoloa ilman tapahtumia koskeva teho: käytetään kuvastamaan tehosta johtuvaa elossaoloa ilman tapahtumia ja määritellään ajaksi satunnaistamispäivämäärästä minkä tahansa seuraavista varhaisimpaan ilmaantumiseen: sairauden eteneminen/uusiutuminen, mistä tahansa syystä aiheutunut kuolema, tutkijan määrittelemä tehon ensisijainen syy, muu kuin sairauden eteneminen/uusiutuminen, joka johti tutkimussuunnitelmassa mainitsemattoman lymfoomahoidon aloittamiseen, jos hoidon päättymisen jälkeen otettiin kudosnäyte ja siinä oli nähtävissä jäännöstauti riippumatta siitä, aloitettiinko tutkimussuunnitelmassa mainitsematon lymfoomahoito vai ei
1) tutkijan arvio
2) sokkoutetun riippumattoman keskitetyn arvioijatahon arvio
3) Log-rank-testi, ositettu
*Lugano 2014 ‑vastekriteerien mukaan
**Ositettu IPI-pisteiden (2 vs 3–5), sairauteen liittyvän suuren tautimassan (kyllä/ei), maantieteellisen alueen mukaan
Välianalyysissa elossaolon keskeinen toissijainen päätetapahtuma oli keskeneräinen eikä tilastollista eroa ollut (ositettu riskitiheyssuhde 0,94 [95 %:n luottamusväli 0,65; 1,37]; p = 0,7524).
Kuva 1. Kaplan–Meierin käyrä tutkijan arvioimasta etenemättömyysajasta tutkimuksessa GO39942 (POLARIX)
Uusiutunut tai refraktorinen diffuusi suurisoluinen B-solulymfooma
Polivy-valmisteen tehoa tutkittiin kansainvälisessä, avoimessa monikeskustutkimuksessa (GO29365), jossa oli mukana 80 potilaan satunnaistettu kohortti, johon kuuluneet potilaat olivat aiemmin saaneet hoitoa diffuusiin suurisoluiseen B-solulymfoomaan. Potilaat satunnaistettiin suhteessa 1:1 saamaan kuuden 21 päivää kestäneen hoitosyklin ajan Polivy-valmisteen ja BR-hoidon yhdistelmää tai pelkästään BR-hoitoa. Potilaat ositettiin sen mukaan, oliko aiempaan hoitoon saadun vasteen kesto ≤ 12 kuukautta vai > 12 kuukautta.
Mukaan soveltuneille potilaille ei voitu tehdä autologista hematopoieettista kantasolusiirtoa ja heillä oli ollut relapsi tai hoitoon reagoimaton tauti vähintään yhden systeemisen solunsalpaajahoidon jälkeen. Tutkimukseen ei otettu mukaan potilaita, jotka olivat aiemmin saaneet allogeenisen hematopoieettisen kantasolusiirron tai joilla oli keskushermoston lymfooma, transformoitunut indolentti lymfooma, asteen 3b follikulaarinen lymfooma, merkittävä sydän- ja verisuonisairaus tai keuhkosairaus, aktiivisia infektioita, ASAT tai ALAT > 2,5 × ULN tai kokonaisbilirubiinipitoisuus ≥ 1,5 × ULN, kreatiniinipitoisuus > 1,5 × ULN (tai CrCl < 40 ml/min), paitsi jos nämä johtuivat perussairautena sairastetusta lymfoomasta.
Polivy annettiin laskimoon annoksella 1,8 mg/kg hoitosyklin 1 päivänä 2 ja hoitosyklien 2–6 päivänä 1. Bendamustiini annettiin laskimoon annoksina 90 mg/m2 hoitosyklin 1 päivinä 2 ja 3 sekä hoitosyklien 2–6 päivinä 1 ja 2. Rituksimabia annettiin annoksella 375 mg/m2 hoitosyklien 1–6 päivänä 1.
Niistä 80 potilaasta, jotka satunnaistettiin saamaan Polivy-valmisteen ja BR-hoidon yhdistelmää (n = 40) tai pelkästään BR-hoitoa (n = 40), valtaosa oli valkoihoisia (71 %) ja miehiä (66 %). Iän mediaani oli 69 vuotta (vaihteluväli: 30−86 vuotta). 80 potilaasta 64 potilaan (80 %) ECOG-toimintakykystatus oli 0−1, ja 80 potilaasta 14 potilaan (18 %) ECOG-toimintakykystatus oli 2. Valtaosalla potilaista (98 %) oli tarkemmin määrittelemätön diffuusi suurisoluinen B-solulymfooma. Kaiken kaikkiaan 48 %:lla potilaista oli aktivoituneen B-solun kaltainen (ABC) diffuusi suurisoluinen B-solulymfooma ja 40 %:lla oli itukeskussoluperäinen (GCB) diffuusi suurisoluinen B-solulymfooma. Pääasiallisia syitä siihen, että potilaat eivät soveltuneet hematopoieettiseen kantasolusiirtoon, olivat ikä (40 %), riittämätön vaste salvage-hoitoon (26 %) ja aiemmin epäonnistunut siirto (20 %). Aiempien hoitojen lukumäärän mediaani oli 2 (vaihteluväli: 1–7), ja 29 % (n = 23) oli saanut yhtä aiempaa hoitoa, 25 % (n = 20) oli saanut kahta aiempaa hoitoa ja 46 % (n = 37) oli saanut kolmea tai useampaa aiempaa hoitoa. Satunnaistetussa vaiheen II tutkimuksessa yhtä potilasta lukuun ottamatta kukaan polatutsumabi-vedotiinin ja BR-hoidon yhdistelmää saaneen hoitohaaran potilaista ei ollut aiemmin saanut bendamustiinihoitoa. 80 %:lla potilaista oli hoitoon reagoimaton tauti. Potilailla, jotka saivat polatutsumabi-vedotiinin ja BR-hoidon yhdistelmää, ja joiden CD3+ -lymfosyyttimäärä määritettiin, CD3+ -lymfosyyttien absoluuttinen määrä oli > 200 solua/mikrolitra 95 %:lla potilaista (n=134) kun lymfosyytit määritettiin ennen hoitoa, 79 %:lla potilaista (n=72) kun lymfosyytit määritettiin hoidon päättymisen jälkeen ja 83 %:lla potilaista (n=18) kun lymfosyytit määritettiin 6 kuukautta hoidon päättymisen jälkeen.
Tutkimuksen ensisijainen päätetapahtuma oli täydellisen vasteen saaneiden potilaiden lukumäärä hoidon päättyessä (6–8 viikkoa hoitosyklin 6 päivän 1 tai viimeisen tutkimushoidon jälkeen); täydellisen vasteen saaneiden potilaiden lukumäärä perustui riippumattoman arviointitoimikunnan PET-TT-tutkimuksen avulla tekemään arvioon.
Taulukko 6. Tutkimukseen GO29365 perustuva yhteenveto hoidon tehosta diffuusiin suurisoluiseen B-solulymfoomaan aiemmin hoitoa saaneilla potilailla
Polivy + bendamustiini + rituksimabi N = 40 | Bendamustiini + rituksimabi N = 40 | |
| Havainnointiajan mediaani 22 kuukautta | ||
| Ensisijainen päätetapahtuma | ||
| Täydellisen vasteen saaneiden lukumäärä* (riippumattoman arviointitoimikunnan arvio) hoidon päättyessä** | ||
| Vasteen saaneita (%) | 16 (40,0) | 7 (17,5) |
| Vasteluvun ero (%) (95 %:n luottamusväli) | 22,5 (2,6; 40,2) | |
| p-arvo (Cochran-Mantel-Haenszelin khiin neliö ‑testi***) | 0,0261 | |
| Keskeiset toissijaiset ja eksploratiiviset päätetapahtumat | ||
| Vasteen kesto (tutkijan arvio) | ||
Analyysiin mukaan otettujen potilaiden lukumäärä Niiden potilaiden lukumäärä (%), joilla esiintyi tapahtuma | 28 17 (60,7) | 13 11 (84,6) |
Vasteen kestoajan mediaani (95 %:n luottamusväli), kuukautta Riskitiheyssuhde (HR) (95 %:n luottamusväli) | 10,3 (5,6; NE) | 4,1 (2,6; 12,7) |
| 0,44 (0,20; 0,95) | ||
| p-arvo (log-rank-testi, ositettu***) | 0,0321 | |
| Kokonaisvasteluku* (tutkijan arvio) hoidon päättyessä** | ||
| Vasteen saaneita (%) (täydellinen vaste, osittainen vaste) | 19 (47,5) | 7 (17,5) |
| Vasteluvun ero (%) (95 %:n luottamusväli) | 30,0 (9,5; 47,4) | |
| p-arvo (Cochran-Mantel-Haenszelin khiin neliö ‑testi***) | 0,0036 | |
| Täydellinen vaste (%) | 17 (42,5) | 6 (15,0) |
| Vasteluvun ero (%) (95 %:n luottamusväli) | 27,5 (7,7; 44,7) | |
| p-arvo (Cochran-Mantel-Haenszelin khiin neliö ‑testi***) | 0,0061 | |
Osittainen vaste (%) 95 %:n Clopper-Pearsonin luottamusväli | 2 (5,0) [0,6; 16,9] | 1 (2,5) [0,06; 13,2] |
| Paras kokonaisvasteluku* (tutkijan arvio) | ||
| Vasteen saaneita (%) (täydellinen vaste, osittainen vaste) | 28 (70,0) | 13 (32,5) |
| Vasteluvun ero (%) (95 %:n luottamusväli) | 37,5 (15,6; 54,7) | |
| Täydellinen vaste (%) | 23 (57,5) | 8 (20,0) |
| 95 %:n Clopper-Pearsonin luottamusväli | [40,9; 73,0] | [9,1; 35,7] |
Osittainen vaste (%) 95 %:n Clopper-Pearsonin luottamusväli | 5 (12,5) [4,2; 26,8] | 5 (12,5) [4,2; 26,8] |
NE = ei arvioitavissa (not evaluable)
*Muokattujen Lugano 2014 ‑kriteerien mukaan: täydellinen vaste edellytti PET-TT-kuvauksella tehdyn varmistuksen luuytimestä. PET-TT-kuvaukseen perustuva osittainen vaste edellytti sekä PET-TT-kriteerien että TT-kriteerien täyttymistä.
**6–8 viikkoa hoitosyklin 6 päivän 1 tai viimeisen tutkimushoidon jälkeen
*** Ositettu vasteen kestolla ennen hoitoa (≤ 12 kuukautta vs > 12 kuukautta)
Kokonaiselossaolo (OS) oli eksploratiivinen päätetapahtuma, joka ei ollut tyypin I virheen suhteen kontrolloitu. Polivy-valmisteen sekä bendamustiinin ja rituksimabin yhdistelmää saaneen hoitohaaran kokonaiselossaolon mediaani oli 12,4 kuukautta (95 %:n luottamusväli: 9,0; ei arvioitavissa) verrattuna 4,7 kuukauteen (95 %:n luottamusväli: 3,7; 8,3) vertailuhaarassa. Kokonaiselossaolon riskitiheyssuhteen (HR) vakioimaton estimaatti oli 0,42. Lähtötilanteen kovariaattien vaikutusta selvitettäessä kokonaiselossaolon riskitiheyssuhteeksi vakioitiin 0,59. Kovariaatteja olivat primaari refraktorinen status, aiempien hoitolinjojen lukumäärä, IPI-luokka ja aiempi kantasolusiirto.
Tutkijan arvioima etenemättömyysaika (PFS) oli eksploratiivinen päätetapahtuma, joka ei ollut tyypin I virheen suhteen kontrolloitu. Polivy-valmisteen sekä bendamustiinin ja rituksimabin yhdistelmää saaneen hoitohaaran taudin etenemättömyysajan mediaani oli 7,6 kuukautta (95 %:n luottamusväli: 6,0; 17,0) verrattuna 2,0 kuukauteen (95 %:n luottamusväli: 1,5; 3,7) vertailuhaarassa. Etenemättömyysajan riskitiheyssuhteen vakioimaton estimaatti oli 0,34.
Immunogeenisuus
Immuunivaste on samoin kuin muiden proteiinilääkevalmisteiden käytössä mahdollinen myös polatutsumabi-vedotiinihoitoa saaneilla potilailla. GO39442-tutkimuksessa (POLARIX) 1,4 %:lla (6/427) potilaista ja GO29365-tutkimuksessa 5,2 %:lla (12/233) potilaista testitulos polatutsumabi-vedotiinin vasta-aineille oli positiivinen, mutta yhdelläkään näistä potilaista neutraloivien vasta-aineiden testitulos ei ollut positiivinen. Polatutsumabi-vedotiinin vasta-aineille positiivisen testituloksen saaneiden potilaiden lukumäärä oli pieni, joten immunogeenisuuden mahdollisesta vaikutuksesta tehoon tai turvallisuuteen ei voida tehdä päätelmiä.
Immunogeenisuusmäärityksen tulokset ovat hyvin riippuvaisia monista tekijöistä, kuten määrityksen herkkyydestä ja spesifisyydestä, määritysmenetelmästä, näytteen käsittelystä, näytteenottoajankohdasta, samanaikaisista lääkityksistä ja perussairaudesta. Tämän vuoksi polatutsumabi-vedotiinin vasta-aineiden ilmaantuvuuden vertaaminen muiden valmisteiden vasta-aineiden ilmaantuvuuteen voi olla harhaanjohtavaa.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Polivy-valmisteen käytöstä kypsien B-solukasvainten hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Vasta-aineeseen konjugoidun monometyyliauristatiini E:n (acMMAE) altistus plasmassa lisääntyi annosriippuvaisesti, kun polatutsumabi-vedotiinin annos oli 0,1–2,4 mg/kg. Ensimmäisen 1,8 mg/kg polatutsumabi-vedotiiniannoksen jälkeen vasta-aineeseen konjugoidun monometyyliauristatiini E:n keskimääräinen huippupitoisuus (Cmax) oli 803 (± 233) ng/ml ja pitoisuus-aikakäyrän alle jäävä pinta-ala nollasta äärettömään (AUCinf) oli 1860 (± 966) vrk•ng/ml. Populaatiofarmakokineettinen analyysi osoitti, että vasta-aineeseen konjugoidun monometyyliauristatiini E:n AUC-arvo suureni hoitosyklissä 3 noin 30 % hoitosyklin 1 AUC-arvoon verrattuna ja saavutti yli 90 % hoitosyklin 6 AUC-arvosta. Vasta-aineeseen konjugoidun monometyyliauristatiini E:n terminaalinen puoliintumisaika hoitosyklissä 6 oli noin 12 vuorokautta (95 %:n luottamusväli 8,1–19,5 vuorokautta). Vasta-aineeseen konjugoidun monometyyliauristatiini E:n ennustettu pitoisuus hoitosyklin 6 lopussa on populaatiofarmakokineettisen analyysin perusteella noin 80 % teoreettisesta vakaan tilan arvosta.
Altistus konjugoimattomalle monometyyliauristatiini E:lle, joka on polatutsumabi-vedotiinin sytotoksinen komponentti, suureni annosriippuvaisesti, kun polatutsumabi-vedotiinin annos oli 0,1−2,4 mg/kg. Monometyyliauristatiini E:n pitoisuus plasmassa noudatti muodostumisnopeuden rajoittamaa kinetiikkaa. Ensimmäisen 1,8 mg/kg polatutsumabi-vedotiiniannoksen jälkeen Cmax oli 6,82 (± 4,73) ng/ml, aika huippupitoisuuteen plasmassa oli noin 2,5 vuorokautta ja terminaalinen puoliintumisaika oli noin 4 vuorokautta. Plasmassa altistus konjugoimattomalle monometyyliauristatiini E:lle on < 3 % altistuksesta vasta-aineeseen konjugoidulle monometyyliauristatiini E:lle. Populaatiofarmakokineettisen analyysin perusteella altistus (AUC-arvo) konjugoimattomalle monometyyliauristatiini E:lle vähenee kolmen viikon välein toistuvassa annossa.
Populaatiofarmakokineettisiin simulaatioihin perustuva post-hoc-analyysi ennusti yli 100 kg painoisen potilaan altistuksen konjugoitumattomalle monometyyliauristatiini E:lle suurenevan enintään 55 %.
Imeytyminen
Polivy annetaan infuusiona laskimoon. Muita antoreittejä ei ole tutkittu.
Jakautuminen
Vasta-aineeseen konjugoidun monometyyliauristatiini E:n keskusjakautumistilavuuden populaatioestimaatti oli 3,15 l, mikä on lähes plasman tilavuus. Monometyyliauristatiini E sitoutuu kohtalaisesti (71–77 %) in vitro ihmisen plasman proteiineihin. Monometyyliauristatiini E ei jakaudu merkittävästi ihmisen veren punasoluihin in vitro: veren ja plasman välinen suhde on 0,79:0,98.
In vitro ‑tiedot osoittavat, että monometyyliauristatiini E on P-gp:n substraatti, joka ei kuitenkaan kliinisesti merkittävinä pitoisuuksina estä P-gp:tä.
Biotransformaatio
Polatutsumabi-vedotiini oletettavasti kataboloituu potilaissa, minkä seurauksena muodostuu pieniä peptidejä, aminohappoja, konjugoimatonta monometyyliauristatiini E:tä ja konjugoimattomaan monometyyliauristatiini E:hen liittyviä kataboliitteja. Monometyyliauristatiini E:n metaboliittien pitoisuutta ihmisen plasmassa ei ole mitattu.
In vitro ‑tutkimukset osoittavat, että monometyyliauristatiini E on CYP3A4/5:n substraatti, mutta ei indusoi pääasiallisia CYP-entsyymejä. Monometyyliauristatiini E on heikko CYP3A4/5:n aikariippuvainen estäjä, mutta kliinisesti merkittävinä pitoisuuksina se ei estä CYP3A4/5:tä kilpailevasti.
Monometyyliauristatiini E ei estä CYP1A2-, CYP2B6-, CYP2C8-, CYP2C9-, CYP2C19- tai CYP2D6-entsyymejä.
Eliminaatio
Konjugaatti (acMMAE) eliminoituu populaatiofarmakokineettisen analyysin perusteella pääasiassa epäspesifisen lineaarisen puhdistumareitin kautta nopeudella 0,9 l/vrk. In vivo ‑tutkimukset polatutsumabi-vedotiinia (radioaktiivisesti merkitty MMAE) saaneilla rotilla osoittavat, että valtaosa radioaktiivisuudesta erittyy ulosteisiin ja pieni osa radioaktiivisuudesta erittyy virtsaan.
Pediatriset potilaat
Polatutsumabi-vedotiinin farmakokinetiikkaa ei ole tutkittu pediatrisilla potilailla (< 18-vuotiailla).
Iäkkäät potilaat
Ikä ei populaatiofarmakokineettisten analyysien perusteella vaikuttanut vasta-aineeseen konjugoidun monometyyliauristatiini E:n ja konjugoimattoman monometyyliauristatiini E:n farmakokinetiikkaan 19–89-vuotiailla potilailla. Vasta-aineeseen konjugoidun monometyyliauristatiini E:n ja konjugoimattoman monometyyliauristatiini E:n farmakokinetiikassa ei populaatiofarmakokineettisten analyysien perusteella havaittu merkittäviä eroja < 65-vuotiaiden (n = 394) ja ≥ 65-vuotiaiden (n = 495) potilaiden välillä.
Munuaisten vajaatoiminta
Lievää (CrCl 60–89 ml/min, n = 361) tai keskivaikeaa (CrCl 30–59 ml/min, n = 163) munuaisten vajaatoimintaa sairastavilla potilailla altistus vasta-aineeseen konjugoidulle monometyyliauristatiini E:lle ja konjugoimattomalle monometyyliauristatiini E:lle on populaatiofarmakokineettisten analyysien perusteella samankaltainen kuin potilailla, joiden munuaisten toiminta on normaali (CrCl ≥ 90 ml/min, n = 356). Vaikean munuaisten vajaatoiminnan (CrCl 15–29 ml/min, n = 4) vaikutuksesta farmakokinetiikkaan ei ole riittävästi tietoa. Loppuvaiheen munuaissairautta ja/tai dialyysihoitoa saavista potilaista ei ole tietoja saatavissa.
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa sairastavilla potilailla (ASAT- tai ALAT-arvo > 1,0–2,5 × viitearvojen yläraja [ULN] tai kokonaisbilirubiinipitoisuus > 1,0–1,5 × ULN, n = 133) altistus vasta-aineeseen konjugoidulle monometyyliauristatiini E:lle on populaatiofarmakokineettisten analyysien perusteella samankaltainen, kun taas konjugoimattoman monometyyliauristatiini E:n AUC-arvo on enintään 40 % suurempi kuin potilailla, joiden maksan toiminta on normaali (n = 737).
Keskivaikean maksan vajaatoiminnan (kokonaisbilirubiinipitoisuus > 1,5–3 × ULN, n = 11) vaikutuksesta farmakokinetiikkaan ei ole riittävästi tietoa. Vaikeaa maksan vajaatoimintaa sairastavista tai maksansiirron saaneista potilaista on vähän tietoja saatavissa.
Prekliiniset tiedot turvallisuudesta
Systeeminen toksisuus
Sekä rotilla että cynomolgus-apinoilla monometyyliauristatiini E:n ja polatutsumabi-vedotiinin antamiseen liittyviä vallitsevia systeemisiä toksisuuksia olivat mm. korjautuva luuydintoksisuus ja siihen liittyvät vaikutukset perifeerisen veren soluihin.
Genotoksisuus
Polatutsumabi-vedotiinilla ei ole tehty erityisiä mutageenisuustutkimuksia. Monometyyliauristatiini E ei ollut mutageeninen bakteerien käänteismutaatiomäärityksessä (Amesin testi) eikä hiiren lymfoomamutaatiotestissä (L5178Y mouse lymphoma forward mutation assay).
Monometyyliauristatiini E oli todennäköisesti aneugeenisen mekanismin kautta genotoksinen rotan luuytimen mikrotumakokeessa. Monometyyliauristatiini E on mikrotubuluksia hajottava aine, joten tämä mekanismi on yhdenmukainen sen farmakologisen vaikutuksen kanssa.
Karsinogeenisuus
Polatutsumabi-vedotiinilla ja/tai monometyyliauristatiini E:llä ei ole tehty erityisiä karsinogeenisuustutkimuksia.
Hedelmällisyyden heikkeneminen
Polatutsumabi-vedotiinilla ei ole tehty erityisiä hedelmällisyyttä koskevia eläinkokeita. Rotilla tehdyn 4 viikkoa kestäneen toksisuuskokeen tulokset osoittavat, että polatutsumabi-vedotiini voi heikentää urosten lisääntymistoimintoja ja hedelmällisyyttä. Kivesten siementiehyen rappeuma ei korjautunut 6 viikkoa kestäneen hoidottoman jakson aikana. Löydökset korreloivat annoksia ≥ 2 mg/kg saaneiden urosten kivesten vähentyneen painon kanssa sekä pienten ja/tai pehmeiden kivesten silmin havaittavien löydösten kanssa niiden eläinten avauksessa, jotka eivät toipuneet eläinkokeen toimenpiteistä.
Lisääntymistoksisuus
Polatutsumabi-vedotiinilla ei ole tehty erityisiä teratogeenisuutta koskevia eläinkokeita. Tiineille rotille 0,2 mg/kg:n suuruisina annoksina annettu monometyyliauristatiini E ‑hoito aiheutti alkiokuolleisuutta ja sikiöille epämuodostumia (kuten kielen työntymistä ulos suusta, raajojen virhekiertymää, vatsahalkioita ja alaleuan synnynnäistä puuttumista). Monometyyliauristatiini E ‑annoksista 0,2 mg/kg rotille aiheutuva systeeminen altistus (AUC) on noin 50 % AUC-arvosta potilailla, jotka saavat suositellun Polivy-annoksen 1,8 mg/kg 21 vuorokauden välein.
Farmaseuttiset tiedot
Apuaineet
Meripihkahappo
Natriumhydroksidi (pH:n säätöön)
Sakkaroosi
Polysorbaatti 20 (E 432)
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa eikä laimentaa muilla lääkevalmisteilla, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
30 kuukautta.
Liuotettu kuiva-aine eli välikonsentraatti
Välikonsentraatti pitää mikrobiologiselta kannalta käyttää heti. Jos välikonsentraattia ei käytetä heti, käytönaikaiset säilytysajat ja ‑olosuhteet ovat käyttäjän vastuulla eivätkä saisi tavallisesti ylittää 24 tuntia jääkaapissa (2 °C – 8 °C), paitsi jos kuiva-aine on liuotettu kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa. Välikonsentraatin käytönaikaiseksi kemialliseksi ja fysikaaliseksi säilyvyydeksi on osoitettu enintään 72 tuntia jääkaapissa (2 °C – 8 °C) ja enintään 24 tuntia huoneenlämpötilassa (9 °C – 25 °C).
Laimennettu liuos
Laimennettu infuusioliuos pitää mikrobiologiselta kannalta käyttää heti. Jos liuosta ei käytetä heti, käytönaikaiset säilytysajat ja ‑olosuhteet ovat käyttäjän vastuulla eivätkä saisi tavallisesti ylittää 24 tuntia jääkaapissa (2 °C – 8 °C), paitsi jos liuos on laimennettu kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa. Käyttövalmiiksi laimennetun liuoksen kemialliset ja fysikaaliset säilyvyysajat esitetään taulukossa 7. Jos säilytysaika ylittää taulukossa 7 mainitut rajat, hävitä laimennettu Polivy-liuos.
Taulukko 7. Käyttövalmiiksi laimennetun infuusioliuoksen osoitetut kemialliset ja fysikaaliset säilyvyysajat
| Infuusioliuoksen valmistamiseen käytettävä liuotin | Laimennetun infuusionliuoksen säilytysolosuhteet1 |
| 9 mg/ml (0,9 %) natriumkloridiliuos | Enintään 72 tuntia jääkaapissa (2 °C – 8 °C) tai enintään 4 tuntia huoneenlämmössä (9 °C – 25 °C) |
| 4,5 mg/ml (0,45 %) natriumkloridiliuos | Enintään 72 tuntia jääkaapissa (2 °C – 8 °C) tai enintään 8 tuntia huoneenlämmössä (9 °C – 25 °C) |
| 5 % glukoosiliuos | Enintään 72 tuntia jääkaapissa (2 °C – 8 °C) tai enintään 8 tuntia huoneenlämmössä (9 °C – 25 °C) |
| 1 Valmisteen säilyvyyden varmistamiseksi mainittuja säilytysaikoja ei saa ylittää. | |
Säilytys
Säilytä jääkaapissa (2 °C – 8 ºC).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Välikonsentraatin ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
POLIVY kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
30 mg (L:ei) 1 kpl (2466,82 €)
140 mg (L:ei) 1 kpl (10849,60 €)
PF-selosteen tieto
Polivy 30 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos:
6 ml:n injektiopullo (väritön tyypin 1 lasi), jossa on tulppa (fluorihartsilaminaattia) ja alumiinisinetti sekä irti napsautettava muovikorkki. Injektiopullo sisältää 30 mg polatutsumabi-vedotiinia. Pakkauskoko: yksi injektiopullo.
Polivy 140 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos:
20 ml:n injektiopullo (väritön tyypin 1 lasi), jossa on tulppa (fluorihartsilaminaattia) ja alumiinisinetti sekä irti napsautettava muovikorkki. Injektiopullo sisältää 140 mg polatutsumabi-vedotiinia. Pakkauskoko: yksi injektiopullo.
Valmisteen kuvaus:
Valkoinen tai harmahtavan valkoinen kylmäkuivattu kuiva-ainekakku.
Käyttö- ja käsittelyohjeet
Yleiset varotoimet
Polivy sisältää sytotoksisen komponentin. Annetaan sytostaattien käyttöön perehtyneen lääkärin valvonnassa. Noudatetaan antineoplastisten ja sytotoksisten lääkkeiden asianmukaisia käsittely- ja hävittämisohjeita.
Käyttökuntoon saatettu valmiste ei sisällä säilytysainetta ja on tarkoitettu vain yhteen käyttökertaan. Tämän lääkevalmisteen kaikessa käsittelyssä noudatetaan asianmukaista aseptista tekniikkaa.
Polivy on ennen antoa saatettava käyttökuntoon käyttämällä steriiliä injektionesteisiin käytettävää vettä ja laimennettava edelleen infuusiopussiin, joka sisältää 9 mg/ml (0,9 %) natriumkloridi-injektionestettä, 4,5 mg/ml (0,45 %) natriumkloridi-injektionestettä tai 5-prosenttista glukoosiliuosta.
Välikonsentraatti ja infuusioliuos eivät saa jäätyä eikä niitä saa altistaa suoralle auringonvalolle.
Ohjeet kuiva-aineen liuottamiseen eli välikonsentraatin valmistamiseen
- Polivy 30 mg: Käytä steriiliä ruiskua, ja injisoi hitaasti 1,8 ml steriiliä injektionesteisiin käytettävää vettä 30 mg Polivy-valmistetta sisältävään injektiopulloon, jolloin saadaan kerta-annos 20 mg/ml polatutsumabi-vedotiinia sisältävää liuosta. Kohdista nestesuihku injektiopullon seinään eikä suoraan kylmäkuivattuun kuiva-ainekakkuun.
- Polivy 140 mg: Käytä steriiliä ruiskua, ja injisoi hitaasti 7,2 ml steriiliä injektionesteisiin käytettävää vettä 140 mg Polivy-valmistetta sisältävään injektiopulloon, jolloin saadaan kerta-annos 20 mg/ml polatutsumabi-vedotiinia sisältävää liuosta. Kohdista nestesuihku injektiopullon seinään eikä suoraan kylmäkuivattuun kuiva-ainekakkuun.
- Pyörittele injektiopulloa varovasti, kunnes kuiva-aine on liuennut täysin. Ei saa ravistaa.
- Tarkista, ettei välikonsentraatti ole värjäytynyttä eikä siinä ole hiukkasia. Välikonsentraatti on väritöntä tai hieman ruskeaa, kirkasta tai hieman opalisoivaa, eikä siinä saa olla näkyviä hiukkasia. Jos välikonsentraatti on värjäytynyttä tai sameaa tai jos siinä on näkyviä hiukkasia, älä käytä liuosta.
Laimennusohjeet
- Polivy on laimennettava lopulliseen pitoisuuteen 0,72–2,7 mg/ml infuusiopussiin, jonka tilavuus on vähintään 50 ml ja joka sisältää 9 mg/ml natriumkloridiliuosta, 4,5 mg/ml natriumkloridiliuosta tai 5-prosenttista glukoosiliuosta.
-
Laske tarvittava välikonsentraatin liuostilavuus (20 mg/ml) tarvittavan annoksen perusteella (ks. jäljempänä):

- Vedä injektiopullosta steriiliin ruiskuun tarvittava tilavuus Polivy-välikonsentraattia ja laimenna se infuusiopussiin. Hävitä injektiopulloon käyttämättä jäävä valmiste.
- Sekoita varovasti kääntelemällä infuusiopussia hitaasti. Ei saa ravistaa.
- Tarkista, ettei infuusiopussissa ole hiukkasia. Hävitä, jos havaitset hiukkasia.
Vältä käyttövalmiiksi sekoitetun infuusioliuoksen kuljettamista, koska ravistelu voi aiheuttaa kokkaroitumista. Jos käyttövalmiiksi sekoitettua infuusioliuosta kuljetetaan, poista infuusiopussista ilma ja rajoita kuljetus huoneenlämmössä (9 °C – 25 °C) 30 minuuttiin tai jääkaapissa (2 °C – 8 °C) 24 tuntiin. Jos ilma poistetaan, tarvitaan infuusiovälineet, joissa on ilmareiällä varustettu neula (spike), jotta varmistetaan tarkka annostelu infuusion aikana. Laimennetun valmisteen kokonaissäilytysaika ja kuljetusaika yhteensä eivät saa ylittää taulukossa 7 mainittua säilytysaikaa (ks. kohta Kestoaika).
Polivy on annettava sille tarkoitetun infuusioletkun (varustettu steriilillä, pyrogeenittömällä, niukasti proteiineja sitovalla letkun sisällä olevalla tai siihen kiinnitetyllä suodattimella, jonka huokoskoko on 0,2 tai 0,22 mikrometriä) sekä katetrin kautta.
Polivy on yhteensopiva infuusiopussien kanssa, joissa valmisteen kanssa kosketuksissa oleva materiaali on polyvinyylikloridia (PVC) tai polyolefiineja, kuten polyeteeniä (PE) tai polypropeenia. Yhteensopimattomuutta ei ole havaittu myöskään käytettäessä infuusiovälineitä tai ‑tarvikkeita, joissa valmisteen kanssa kosketuksissa oleva materiaali on PVC:tä, polyeteeniä, polyuretaania, polybutadieenia, akryylinitriilibutadieenistyreeniä, polykarbonaattia, polyeetteriuretaania, fluorattua eteeni-propeenia tai polytetrafluorieteeniä, tai käytettäessä suodatinkalvoja, jotka on valmistettu polyeetterisulfonista tai polysulfonista.
Hävittäminen
Polivy on tarkoitettu vain yhtä käyttökertaa varten.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
POLIVY kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
30 mg 1 kpl
140 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FX14
Valmisteyhteenvedon muuttamispäivämäärä
26.03.2026
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com