
OLUMIANT tabletti, kalvopäällysteinen 2 mg, 4 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Lääkäri
Potilas
Vaikuttavat aineet ja niiden määrät
Olumiant 1 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 1 mg barisitinibia.
Olumiant 2 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 2 mg barisitinibia.
Olumiant 4 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 4 mg barisitinibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti).
Kliiniset tiedot
Käyttöaiheet
Nivelreuma
Barisitinibi on tarkoitettu keskivaikean tai vaikean aktiivisen nivelreuman hoitoon aikuispotilaille, joilla vähintään yksi tautiprosessiin vaikuttava reumalääke (disease-modifying anti-rheumatic drug, DMARD) on tuottanut riittämättömän vasteen tai ollut huonosti siedetty. Barisitinibi-valmistetta voidaan käyttää ainoana lääkkeenä tai yhdessä metotreksaatin kanssa (saatavilla olevat tiedot eri yhdistelmistä, ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakodynamiikka).
Atooppinen ihottuma
Barisitinibi on tarkoitettu keskivaikean tai vaikean atooppisen ihottuman hoitoon aikuisille ja vähintään 2-vuotiaille pediatrisille potilaille, joille harkitaan systeemistä hoitoa.
Pälvikalju
Barisitinibi on tarkoitettu vaikean pälvikaljun hoitoon aikuispotilaille ja vähintään 12-vuotiaille nuorille potilaille (ks. kohta Farmakodynamiikka).
Lastenreuma (juveniili idiopaattinen artriitti)
Barisitinibi on tarkoitettu aktiivisen lastenreuman (juveniili idiopaattinen artriitti, JIA) hoitoon vähintään 2-vuotiaille potilaille, joilla vähintään yksi aiempi tavanomainen synteettinen tai biologinen tautiprosessiin vaikuttava reumalääke on tuottanut riittämättömän vasteen tai ollut huonosti siedetty:
- Juveniili idiopaattinen polyartriitti (polyartriitti reumatekijäpositiivinen [RF+] tai reumatekijänegatiivinen [RF-], laajeneva oligoartriitti),
- Entesiitteihin liittyvä artriitti, ja
- Lasten nivelpsoriaasi (juveniili nivelpsoriaasi).
Barisitinibi-valmistetta voidaan käyttää ainoana lääkkeenä tai yhdessä metotreksaatin kanssa.
Ehto
Valmisteen käyttöaiheissa mainittujen sairauksien hoitoon perehtyneen lääkärin on aloitettava hoito.
Annostus ja antotapa
Hoidon saa aloittaa tämän lääkevalmisteen käyttöaiheina olevien sairauksien diagnosointiin ja hoitoon perehtynyt lääkäri.
Annostus
Nivelreuma
Suositeltu barisitinibiannos on 4 mg kerran vuorokaudessa. 2 mg annosta kerran vuorokaudessa suositellaan potilaille, joilla on suurentunut tromboembolisten laskimotapahtumien, merkittävien sydän- ja verisuonitapahtumien ja syövän riski, ≥ 65-vuotiaille potilaille ja potilaille, joilla on anamneesissa krooninen tai toistuva infektio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). 4 mg annosta kerran vuorokaudessa voidaan harkita potilaille, joiden tautiaktiivisuutta ei ole saatu hallintaan 2 mg annoksella kerran vuorokaudessa. 2 mg annosta kerran vuorokaudessa on harkittava myös potilaille, joilla tautiaktiivisuus on saatu pitkäkestoisesti hallintaan 4 mg annoksella kerran vuorokaudessa ja joiden annosta halutaan pienentää (ks. kohta Farmakodynamiikka).
Atooppinen ihottuma
Aikuiset
Suositeltu barisitinibiannos on 4 mg kerran vuorokaudessa. 2 mg annosta kerran vuorokaudessa suositellaan potilaille, joilla on suurentunut tromboembolisten laskimotapahtumien, merkittävien sydän- ja verisuonitapahtumien ja syövän riski, ≥ 65-vuotiaille potilaille ja potilaille, joilla on anamneesissa krooninen tai toistuva infektio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). 4 mg annosta kerran vuorokaudessa voidaan harkita potilaille, joiden tautiaktiivisuutta ei ole saatu hallintaan 2 mg annoksella kerran vuorokaudessa. 2 mg annosta kerran vuorokaudessa on harkittava potilaille, joilla tautiaktiivisuus on saatu pitkäkestoisesti hallintaan 4 mg annoksella kerran vuorokaudessa ja joiden annosta halutaan pienentää (ks. kohta Farmakodynamiikka).
Barisitinibia voidaan käyttää yhdessä paikallisesti käytettävien kortikosteroidien kanssa tai ilman niitä. Barisitinibin teho saattaa voimistua, jos samanaikaisesti käytetään paikallisia kortikosteroidivalmisteita (ks. kohta Farmakodynamiikka). Paikallisesti käytettäviä kalsineuriinin estäjiä voidaan käyttää, mutta niiden käyttö on rajattava ainoastaan herkille ihoalueille kuten kasvoille, kaulalle, taivealueille ja sukuelinten alueelle.
Hoidon lopettamista on harkittava potilailla, joilla ei havaita hoitovastetta 8 viikon hoidon jälkeen.
Lapset ja nuoret (vähintään 2-vuotiaat)
Vähintään 30 kg painaville potilaille suositeltu barisitinibiannos on 4 mg kerran vuorokaudessa. Vähintään 10 kg mutta alle 30 kg painaville potilaille suositeltu annos on 2 mg kerran vuorokaudessa. Annoksen puolittamista on harkittava potilaille, joilla tautiaktiivisuus on saatu pitkäkestoisesti hallintaan suositusannoksella ja joiden annosta halutaan pienentää.
Barisitinibia voidaan käyttää yhdessä paikallisesti käytettävien kortikosteroidien kanssa tai ilman niitä. Paikallisesti käytettäviä kalsineuriinin estäjiä voidaan käyttää, mutta niiden käyttö on rajattava ainoastaan herkille ihoalueille kuten kasvoille, kaulalle, taivealueille ja sukuelinten alueelle.
Hoidon lopettamista on harkittava potilailla, joilla ei havaita hoitovastetta 8 viikon hoidon jälkeen.
Pälvikalju
Aikuiset
Suositeltu barisitinibiannos on 4 mg kerran vuorokaudessa. 2 mg annosta kerran vuorokaudessa suositellaan potilaille, joilla on suurentunut tromboembolisten laskimotapahtumien, merkittävien sydän- ja verisuonitapahtumien ja syövän riski, ≥ 65-vuotiaille potilaille ja potilaille, joilla on anamneesissa krooninen tai toistuva infektio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). 4 mg annosta kerran vuorokaudessa voidaan harkita potilaille, joiden tautiaktiivisuutta ei ole saatu hallintaan 2 mg annoksella kerran vuorokaudessa. 2 mg annosta kerran vuorokaudessa on harkittava myös potilaille, joilla tautiaktiivisuus on saatu pitkäkestoisesti hallintaan 4 mg annoksella kerran vuorokaudessa ja joiden annosta halutaan pienentää (ks. kohta Farmakodynamiikka).
Vakaan vasteen saavuttamisen jälkeen suositellaan hoidon jatkamista vähintään usean kuukauden ajan relapsin välttämiseksi. Hoidon hyöty-riskisuhde on arvioitava uudelleen säännöllisin väliajoin yksilöllisen tarpeen mukaan.
Hoidon lopettamista on harkittava potilailla, joilla ei havaita hoitovastetta 36 viikon hoidon jälkeen.
Nuoret (vähintään 12‑vuotiaat)
Suositeltu barisitinibiannos on 4 mg kerran vuorokaudessa potilaille, joiden paino on vähintään 30 kg. Katso kohdasta Annostus ja antotapaPediatriset potilaat tietoa alle 30 kg painaville potilaille. 2 mg annosta kerran vuorokaudessa on harkittava myös potilaille, joilla tautiaktiivisuus on saatu pitkäkestoisesti hallintaan 4 mg annoksella kerran vuorokaudessa ja joiden annosta halutaan pienentää.
Vakaan vasteen saavuttamisen jälkeen suositellaan hoidon jatkamista vähintään usean kuukauden ajan relapsin välttämiseksi. Hoidon hyöty-riskisuhde on arvioitava uudelleen säännöllisin väliajoin yksilöllisen tarpeen mukaan.
Hoidon lopettamista on harkittava potilailla, joilla ei havaita hoitovastetta 36 viikon hoidon jälkeen.
Lastenreuma (vähintään 2- ja alle 18-vuotiaat potilaat)
Suositeltu barisitinibiannos on 4 mg kerran vuorokaudessa vähintään 30 kg painaville potilaille. Potilaille, jotka painavat vähintään 10 kg ja alle 30 kg, suositeltu barisitinibiannos on 2 mg kerran vuorokaudessa.
Hoidon lopettamista on harkittava potilailla, joilla ei havaita hoitovastetta 12 viikon hoidon jälkeen.
Hoidon aloitus
Hoitoa ei saa aloittaa, jos potilaan absoluuttinen lymfosyyttimäärä on alle 0,5 x 109 solua/l, absoluuttinen neutrofiilimäärä on alle 1 x 109 solua/l tai hemoglobiinipitoisuus on alle 80 g/l. Hoito voidaan aloittaa, kun arvot kohenevat näitä raja-arvoja suuremmiksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annoksen pienentäminen
Jos potilas käyttää vahvoja orgaanisten anionien kuljettajaproteiini 3:n (OAT3:n) estäjiä, esim. probenesidia, tai potilaan kreatiniinipuhdistuma on 30–60 ml/min, pediatrisille potilaille suositeltu barisitinibiannos on pienennettävä puoleen ja aikuispotilaille suositeltu annos on 2 mg (ks. kohta Yhteisvaikutukset).
Erityisryhmät
Munuaisten vajaatoiminta
Suositeltu annos on 2 mg kerran vuorokaudessa, jos aikuispotilaan kreatiniinipuhdistuma on 30–60 ml/min. Pediatrisille potilaille, joiden kreatiniinipuhdistuma on 30–60 ml/min, suositeltu barisitinibiannos on pienennettävä puoleen. Barisitinibia ei suositella potilaille, joiden kreatiniinipuhdistuma on < 30 ml/min (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea maksan vajaatoiminta. Barisitinibia ei suositella potilaille, joilla on vaikea maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Iäkkäät
Vähintään 75-vuotiaiden potilaiden hoidosta on hyvin niukasti kliinistä kokemusta.
Pediatriset potilaat
Barisitinibin turvallisuutta ja tehoa alle 2 vuoden ikäisillä lapsilla, joilla on atooppinen ihottuma tai lastenreuma, ei ole vielä varmistettu. Tietoja ei ole saatavilla. Katso kohdasta Annostus ja antotapa yllä annostustiedot vähintään 2-vuotiaille lapsille.
Barisitinibin turvallisuutta ja tehoa alle 12-vuoden ikäisten lasten tai alle 30 kg painavien lasten pälvikaljun hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla. Katso yllä olevasta kohdasta Annostus ja antotapa. lisätietoa annostuksesta vähintään 12-vuotiaille ja vähintään 30 kg painaville nuorille.
Pediatrisille potilaille, jotka eivät pysty nielemään tabletteja, tabletit voidaan dispergoida veteen ennen antoa (ks. alla ”Vaihtoehtoinen annostelu lapsille”). Vaihtoehtoisesti voidaan käyttää oraalisuspensiota, jos sellainen on saatavilla.
Antotapa
Suun kautta.
Barisitinibi otetaan kerran vuorokaudessa ruoan kanssa tai ilman ruokaa. Se voidaan ottaa mihin kellonaikaan tahansa.
Vaihtoehtoinen annostelu lapsille
Pediatrisille potilaille, jotka eivät pysty nielemään kokonaista tablettia, voidaan harkita tabletin dispergoimista veteen. Vain vettä voidaan käyttää tabletin dispergoimiseen. Vain annokseen tarvittava määrä tabletteja dispergoidaan.
Jos jostain syystä kaikkea näin valmistettua suspensiota ei saada annosteltua, uutta tablettia ei pidä dispergoida ja annostella, vaan on odotettava seuraavaan aikataulun mukaiseen annoksenottoaikaan.
Ohjeet lääkevalmisteen dispergoimiseksi ennen annostelua, katso kohta Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Raskaus (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Barisitinibi-valmistetta tulee käyttää seuraaville potilasryhmille vain, jos muita soveltuvia hoitovaihtoehtoja ei ole käytettävissä: - 65-vuotiaat ja sitä vanhemmat; - potilaat, joilla on ollut ateroskleroottinen sydän- ja verisuonisairaus tai muita kardiovaskulaarisia riskitekijöitä (kuten nykyisin tupakoivat tai aiemmin pitkään tupakoineet); - potilaat, joilla on syöpään liittyviä riskitekijöitä (esim. he ovat sairastaneet tai sairastavat syöpää) |
JAK-estäjien käyttö 65-vuotiailla ja sitä vanhemmilla potilailla
Koska tofasitinibillä (toinen JAK:n estäjä) tehdyssä laajassa satunnaistetussa tutkimuksessa havaittiin suurentunut merkittävien sydän- ja verisuonitapahtumien, pahanlaatuisten kasvainten, vakavien infektioiden ja kokonaiskuolleisuuden riski yli 65-vuotiailla potilailla, barisitinibia tulee käyttää näille potilaille vain, jos sopivia hoitovaihtoehtoja ei ole käytettävissä.
Infektiot
Vakavia ja joskus kuolemaan johtaneita infektioita, mukaan lukien opportunistisia infektioita on raportoitu potilailla, jotka ovat saaneet muita JAK-estäjiä.
Barisitinibihoidon yhteydessä infektioiden kuten ylähengitystieinfektioiden esiintymistiheys suurenee verrattuna lumehoitoon (ks. kohta Haittavaikutukset). Kliinisissä nivelreumatutkimuksissa yhdistelmähoito metotreksaatin kanssa suurensi infektioiden esiintymistiheyttä verrattuna pelkkään barisitinibihoitoon.
Hoidon riskit ja hyödyt on punnittava tarkoin ennen barisitinibihoidon aloittamista, jos potilaalla on aktiivisia, kroonisia tai toistuvia infektioita (ks. kohta Annostus ja antotapa). Jos infektio syntyy, potilaan vointia on seurattava tarkoin ja hoito on tauotettava, mikäli potilas ei reagoi tavanomaiseen hoitoon. Hoitoa ei saa aloittaa uudelleen ennen infektion paranemista.
Koska infektioiden ilmaantuvuus on suurempi yleensä iäkkäillä ja diabeetikoilla, varovaisuutta on noudatettava hoidettaessa iäkkäitä ja diabetesta sairastavia potilaita. Yli 65-vuotiailla potilailla barisitinibia tulee käyttää vain, jos sopivia hoitovaihtoehtoja ei ole käytettävissä.
Tuberkuloosi
Potilaat on seulottava tuberkuloosin varalta ennen hoidon aloittamista. Barisitinibia ei saa antaa potilaille, joilla on aktiivinen tuberkuloosi. Tuberkuloosilääkitystä on harkittava ennen hoidon aloittamista, jos potilaalla on aiemmin hoitamaton, latentti tuberkuloosi.
Veriarvojen poikkeavuudet
Kliinisissä tutkimuksissa ilmoitettiin absoluuttisen neutrofiilimäärän laskua tasolle < 1 x 109/l, absoluuttisen lymfosyyttimäärän laskua tasolle < 0,5 x 109/l ja hemoglobiinipitoisuuden laskua tasolle < 80 g/l.
Hoitoa ei saa aloittaa tai hoito on tauotettava, jos rutiiniseurannassa todetaan, että potilaan absoluuttinen neutrofiilimäärä on < 1 x 109/l, absoluuttinen lymfosyyttimäärä on < 0,5 x 109/l tai hemoglobiinipitoisuus on < 80 g/l (ks. kohta Annostus ja antotapa).
Lymfosytoosin riski on suurentunut iäkkäillä nivelreumapotilailla. Harvinaisissa tapauksissa on ilmoitettu lymfoproliferatiivisia häiriöitä.
Virusten reaktivaatio
Kliinisissä tutkimuksissa ilmoitettiin virusten reaktivaatiota, mm. herpesvirusten reaktivaatiota (esim. vyöruusu, herpes simplex) (ks. kohta Haittavaikutukset). Kliinisissä nivelreumatutkimuksissa vyöruusua ilmoitettiin yleisemmin vähintään 65-vuotiailla potilailla, jotka olivat saaneet aiemmin sekä biologisia että synteettisiä tavanomaisia tautiprosessiin vaikuttavia reumalääkkeitä. Jos potilaalle kehittyy vyöruusu, barisitinibihoito on tauotettava, kunnes vyöruusu paranee.
Ennen barisitinibihoidon aloittamista potilaat on seulottava virushepatiittien varalta kliinisten suositusten mukaisella tavalla. Kliinisistä tutkimuksista suljettiin pois potilaat, joiden kohdalla oli näyttöä aktiivisesta hepatiitti B- tai hepatiitti C ‑infektiosta. Potilaat, joilla hepatiitti C ‑viruksen vasta-ainetesti oli positiivinen mutta hepatiitti C ‑viruksen RNA-testi negatiivinen, saivat osallistua tutkimuksiin. Tutkimuksiin saivat osallistua myös potilaat, joilla hepatiitti B ‑viruksen pinta-antigeenivasta-ainetesti ja hepatiitti B ‑viruksen ydinantigeenivasta-ainetesti olivat positiiviset mutta hepatiitti B ‑viruksen pinta-antigeenitesti oli negatiivinen. Tällaisia potilaita on seurattava hepatiitti B ‑viruksen (HBV) DNA:n ilmentymisen varalta. Jos HBV:n DNA:ta todetaan, on konsultoitava maksatauteihin erikoistunutta lääkäriä ja selvitettävä, onko hoidon keskeyttäminen aiheellista.
Rokotukset
Barisitinibihoitoa saavien potilaiden reagoinnista elävillä rokotteilla toteutettuihin rokotuksiin ei ole tietoa. Elävien, heikennettyjen rokotteiden anto barisitinibihoidon aikana tai juuri ennen hoitoa ei ole suositeltavaa. Kaikkien potilaiden, erityisesti pediatristen potilaiden, rokotukset suositellaan päivittämään ajan tasalle voimassa olevien rokotussuositusten mukaisesti ennen barisitinibihoidon aloittamista.
Lipidit
Barisitinibihoitoa saaneilla pediatrisilla potilailla ja aikuispotilailla ilmoitettiin annosriippuvaista veren lipidiarvojen suurenemista (ks. kohta Haittavaikutukset). Statiinihoito pienensi aikuisilla suurentuneet LDL (low density lipoprotein) -kolesteroliarvot hoitoa edeltävälle tasolle. Sekä pediatristen potilaiden että aikuispotilaiden lipidiarvot on arvioitava noin 12 viikon kuluttua barisitinibihoidon aloittamisesta. Tämän jälkeen potilaiden hoidossa on noudatettava hyperlipidemian kansainvälisiä kliinisiä hoitosuosituksia.
Maksan transaminaasiarvojen suureneminen
Barisitinibihoitoa saaneilla potilailla ilmoitettiin annosriippuvaista alaniinitransaminaasiarvojen (ALAT) ja aspartaattitransaminaasiarvojen (ASAT) suurenemista (ks. kohta Haittavaikutukset).
Kliinisissä tutkimuksissa ilmoitettiin ALAT- ja ASAT-arvojen suurenemista ≥ 5 ja ≥ 10 kertaa viitevälin ylärajan (ULN) suuruisiksi. Kliinisissä nivelreumatutkimuksissa yhdistelmähoito metotreksaatin kanssa johti maksan transaminaasiarvojen suurenemiseen useammin kuin pelkkä barisitinibihoito (ks. kohta Haittavaikutukset).
Jos rutiiniseurannassa todetaan ALAT- tai ASAT-arvojen suurenemista ja epäillään lääkkeen aiheuttamaa maksavauriota, hoito tauotetaan, kunnes tämä diagnoosi on suljettu pois.
Maligniteetit
Immunomoduloivat lääkevalmisteet saattavat suurentaa maligniteettien kuten lymfoomien riskiä. Lymfoomaa ja muita pahanlaatuisia kasvaimia on raportoitu potilailla, jotka ovat saaneet JAK-estäjiä, mukaan lukien barisitinibia.
Laajassa satunnaistetussa aktiivikontrolloidussa tutkimuksessa tofasitinibilla (toinen JAK-estäjä) 50-vuotiailla ja sitä vanhemmilla nivelreumapotilailla, joilla oli vähintään yksi sydän- ja verisuonitautien lisäriskitekijä, kasvoi pahanlaatuisten kasvainten, erityisesti keuhkosyövän, lymfooman ja ei-melanoottisten ihosyöpien esiintyvyys tofasitinibilla verrattuna TNF-estäjiin.
Yli 65-vuotiailla potilailla, nykyisin tupakoivilla tai aiemmin pitkään tupakoineilla potilailla tai potilailla, joilla on muita pahanlaatuisten kasvainten riskitekijöitä (esim. he ovat sairastaneet tai sairastavat syöpää), barisitinibia tulee käyttää vain, jos sopivia hoitovaihtoehtoja ei ole käytettävissä.
Kaikille potilaille suositellaan ihon säännöllistä tutkimista, erityisesti niille, joilla on ihosyövän riskitekijöitä.
Tromboemboliset laskimotapahtumat
Retrospektiivisessa havainnointitutkimuksessa havaittiin enemmän tromboembolisia laskimotapahtumia barisitinibilla hoidetuilla nivelreumapotilailla verrattuna TNF-estäjillä hoidettuihin potilaisiin (ks. kohta Haittavaikutukset).
Laajassa satunnaistetussa, aktiivikontrolloidussa tutkimuksessa tofasitinibilla (toinen JAK-estäjä) 50-vuotiailla ja sitä vanhemmilla nivelreumapotilailla, joilla oli vähintään yksi sydän- ja verisuonitautien lisäriskitekijä, havaittiin annosriippuvaisesti enemmän tromboembolisia laskimotapahtumia ja keuhkoemboliatapahtumia tofasitinibilla verrattuna TNF-estäjiin.
Potilailla, joilla on sydän- ja verisuonitautien tai pahanlaatuisten kasvainten riskitekijöitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet “Merkittävät sydän- ja verisuonitapahtumat” ja “Maligniteetit”), barisitinibia tulee käyttää vain, jos sopivia hoitovaihtoehtoja ei ole saatavilla.
Potilailla, joilla on tromboembolisten laskimotapahtumien riskitekijöitä, muita kuin sydän- ja verisuonitautien tai maligniteettien riskitekijöitä, barisitinibia tulee käyttää varoen. Tromboembolisten laskimotapahtumien riskitekijöihin, muita kuin sydän- ja verisuonisairaudet tai pahanlaatuiset kasvaimet, kuuluvat aiemmin sairastettu laskimotromboembolia, potilaan leikkaus tai immobilisaatio, yhdistelmäehkäisytablettien käyttö tai hormonikorvaushoito ja perinnöllinen hyytymishäiriö.
Potilaat tulee arvioida uudelleen säännöllisin väliajoin barisitinibihoidon aikana tromboembolisten laskimotapahtumien riskitekijöiden muutosten arvioimiseksi.
Potilaiden, joilla on tromboembolisten laskimotapahtumien merkkejä ja oireita, tila tulee arvioida viipymättä ja lopettaa barisitinibihoito, jos epäillään laskimotromboembolia, annoksesta tai käyttöaiheesta riippumatta.
Merkittävät sydän- ja verisuonitapahtumat (Major adverse cardiovascular events, MACE)
Retrospektiivisessä havainnointitutkimuksessa nivelreumapotilailla havaittiin korkeampi vakavien sydän- ja verisuonitapahtumien esiintyvyys barisitinibillä verrattuna TNF-estäjillä hoidettuihin potilaisiin.
Laajassa satunnaistetussa aktiivikontrolloidussa tutkimuksessa tofasitinibillä (toinen JAK-estäjä) 50-vuotiailla ja sitä vanhemmilla nivelreumapotilailla, joilla oli vähintään yksi kardiovaskulaarinen lisäriskitekijä, havaittiin enemmän vakavia kardiovaskulaarisia haittatapahtumia, joiksi määritellään kardiovaskulaarinen kuolema, ei-kuolemaan johtava sydäninfarkti ja ei-kuolemaan johtava aivohalvaus, tofasitinibillä (toinen JAK-estäjä) verrattuna TNF-estäjiin.
Tämän vuoksi yli 65-vuotiaille potilaille, nykyisin tupakoiville tai aiemmin pitkään tupakoineille potilaille ja potilaille, joilla on ollut ateroskleroottinen sydän- ja verisuonisairaus tai muita kardiovaskulaarisia riskitekijöitä, barisitinibia tulee käyttää vain, jos sopivia hoitovaihtoehtoja ei ole käytettävissä.
Laboratorioarvojen seuranta
| Taulukko 1. Laboratorioarvot ja seurantaohjeet | ||
| Laboratorioarvo | Toiminta | Seurantaohje |
| Lipidiarvot | Potilaita hoidetaan hyperlipidemian kansainvälisten kliinisten hoitosuositusten mukaisesti. | 12 viikon kuluttua hoidon aloituksesta ja tämän jälkeen hyperlipidemian kansainvälisten kliinisten hoitosuositusten mukaisesti |
| Absoluuttinen neutrofiilimäärä | Hoito on tauotettava, jos absoluuttinen neutrofiilimäärä on < 1 x 109/l. Hoito voidaan aloittaa uudelleen, kun absoluuttinen neutrofiilimäärä korjautuu tätä raja-arvoa suuremmaksi. | Ennen hoidon aloittamista ja tämän jälkeen rutiiniseurannan mukaisesti |
| Absoluuttinen lymfosyyttimäärä | Hoito on tauotettava, jos absoluuttinen lymfosyyttimäärä on < 0,5 x 109/l. Hoito voidaan aloittaa uudelleen, kun absoluuttinen lymfosyyttimäärä korjautuu tätä raja-arvoa suuremmaksi. | |
| Hemoglobiini (Hb) | Hoito on tauotettava, jos hemoglobiinipitoisuus on < 80 g/l. Hoito voidaan aloittaa uudelleen, kun hemoglobiinipitoisuus korjautuu tätä raja-arvoa suuremmaksi. | |
| Maksan transaminaasiarvot | Hoito on tauotettava, jos epäillään lääkkeen aiheuttamaa maksavauriota. | |
Immunosuppressoivat lääkevalmisteet
Tautiprosessiin vaikuttavien biologisten reumalääkkeiden, biologisten immunomodulaattorien ja muiden Janus-kinaasin (JAK) estäjien samanaikaista käyttöä ei suositella, sillä additiivisen immunosuppressoivan vaikutuksen mahdollisuutta ei voida sulkea pois.
Nivelreuman ja lastenreuman hoidossa barisitinibin käytöstä muiden vahvojen immunosuppressoivien lääkevalmisteiden kuin metotreksaatin (esim. atsatiopriinin, takrolimuusin, siklosporiinin) kanssa on niukasti tietoa. Varovaisuus on tarpeen tällaisia yhdistelmiä käytettäessä (ks. kohta Yhteisvaikutukset).
Atooppisen ihottuman ja pälvikaljun hoidossa käyttöä yhdessä siklosporiinin tai muiden voimakkaiden immunosuppressanttien kanssa ei ole tutkittu, eikä samanaikaista käyttöä suositella (ks. kohta Yhteisvaikutukset).
Yliherkkyys
Myyntiluvan myöntämisen jälkeen on raportoitu barisitinibin antoon liittyviä yliherkkyystapauksia. Jos mitään vakavia allergisia tai anafylaktisia reaktioita esiintyy, tulee hoito lopettaa välittömästi.
Divertikuliitti
Kliinisistä tutkimuksista ja markkinoille tulon jälkeisistä lähteistä on ilmoitettu divertikuliittitapauksia ja maha-suolikanavan perforaatiota (ks. kohta Haittavaikutukset). Barisitinibia on käytettävä varoen potilailla, joilla on divertikuliitti, ja etenkin potilailla, joille annetaan samaan aikaan pitkäaikaista hoitoa sellaisilla lääkevalmisteilla, joiden yhteydessä divertikuliitin riski on suurentunut. Tällaisia lääkkeitä ovat ei-steroidaaliset tulehduskipulääkkeet, kortikosteroidit ja opioidit. Potilaat, joille ilmaantuu uusia vatsan alueen ongelmiin viittaavia merkkejä ja oireita, on tutkittava nopeasti, jotta divertikuliitti tai maha-suolikanavan perforaatio voitaisiin tunnistaa varhain.
Hypoglykemia diabetekseen hoitoa saavilla potilailla
Ilmoituksia hypoglykemiasta on tehty JAK-estäjien, kuten barisitinibin, käytön aloittamisen jälkeen diabeteslääkitystä saavilla potilailla. Jos hypoglykemiaa ilmenee, diabeteslääkityksen annostusta voi olla tarpeen muuttaa.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Farmakodynaamiset yhteisvaikutukset
Immunosuppressoivat lääkevalmisteet
Yhdistelmähoitoa tautiprosessiin vaikuttavien biologisten reumalääkkeiden, biologisten immunomodulaattoreiden tai muiden JAK:n estäjien kanssa ei ole tutkittu. Kliinisissä tutkimuksissa barisitinibin käyttö nivelreuman ja lastenreuman hoidossa yhdessä vahvojen immunosuppressoivien lääkevalmisteiden kuten atsatiopriinin, takrolimuusin tai siklosporiinin kanssa oli rajallista, ja additiivisen immunosuppressoivan vaikutuksen riskiä ei voida sulkea pois. Atooppisen ihottuman ja pälvikaljun hoidossa valmisteen käyttöä yhdessä siklosporiinin tai muiden voimakkaiden immunosuppressanttien kanssa ei ole arvioitu, eikä samanaikaista käyttöä suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Muiden lääkevalmisteiden mahdollinen vaikutus barisitinibin farmakokinetiikkaan
Kuljettajaproteiinit
Barisitinibi on in vitro orgaanisten anionien kuljettajaproteiini 3:n (OATP3:n), P-glykoproteiinin (P-gp), rintasyöpäresistenssiproteiinin (BCRP) ja monilääke- ja toksisten aineiden ekstruusioproteiini (MATE) 2-K:n substraatti. Kliinisen farmakologian tutkimuksessa probenesidin (OAT3:n estäjä, jolla on vahva estopotentiaali) anto suurensi barisitinibin AUC(0-∞)-arvon noin 2-kertaiseksi, mutta ei vaikuttanut tmax- eikä Cmax-arvoon. Näin ollen potilaille, jotka käyttävät vahvan estopotentiaalin omaavia OAT3:n estäjiä kuten probenesidia, suositeltu barisitinibiannos on pienennettävä puoleen (ks. kohta Annostus ja antotapa). Estopotentiaaliltaan heikompien OAT3:n estäjien käyttöä ei ole arvioitu kliinisen farmakologian tutkimuksissa. Aihiolääke leflunomidi muuntuu nopeasti teriflunomidiksi, joka on heikko OAT3:n estäjä ja saattaa siten johtaa barisitinibialtistuksen suurenemiseen. Spesifisiä yhteisvaikutustutkimuksia ei ole tehty, joten varovaisuus on tarpeen, kun leflunomidia tai teriflunomidia annetaan samanaikaisesti barisitinibin kanssa. Ibuprofeenin ja diklofenaakin (OAT3:n estäjiä) samanaikainen käyttö voi johtaa barisitinibialtistuksen suurenemiseen, mutta niiden OAT3-toiminnan estopotentiaali on probenesidia heikompi eikä kliinisesti merkittäviä yhteisvaikutuksia siis ole odotettavissa. Barisitinibin käyttö yhdessä siklosporiinin (P-gp:n/BCRP:n estäjä) tai metotreksaatin (useiden kuljettajaproteiinien kuten OATP1B1-, OAT1-, OAT3-, BCRP-, MRP2-, MRP3- ja MRP4-kuljettajaproteiinien substraatti) kanssa ei vaikuttanut kliinisesti merkittävästi barisitinibialtistukseen.
Sytokromi P450 ‑entsyymit
Barisitinibi on in vitro sytokromi P450 ‑entsyymin (CYP) 3A4 substraatti, mutta alle 10 % annoksesta metaboloituu hapettumalla. Kliinisen farmakologian tutkimuksissa barisitinibin anto yhdessä ketokonatsolin (vahva CYP3A:n estäjä) kanssa ei vaikuttanut kliinisesti merkittävästi barisitinibin farmakokinetiikkaan. Barisitinibin anto yhdessä flukonatsolin (keskivahva CYP3A/CYP2C19/CYP2C9-toiminnan estäjä) tai rifampisiinin (vahva CYP3A:n indusori) kanssa ei muuttanut barisitinibialtistusta kliinisesti merkittävästi.
Mahan pH-arvoon vaikuttavat aineet
Mahan pH-arvon suurentaminen omepratsolilla ei vaikuttanut kliinisesti merkittävästi barisitinibialtistukseen.
Barisitinibin mahdollinen vaikutus muiden lääkevalmisteiden farmakokinetiikkaan
Kuljettajaproteiinit
Barisitinibi ei estänyt in vitro OAT1-, OAT2-, OAT3-, orgaanisten kationien kuljettajaproteiini (OCT) 2-, OATP1B1-, OATP1B3-, BCRP-, MATE1- ja MATE2-K-toimintaa kliinisesti merkittävissä konsentraatiossa. Barisitinibi saattaa olla kliinisesti merkittävä OCT1:n estäjä, toistaiseksi ei kuitenkaan tunneta selektiivisiä OCT1:n substraatteja, joihin kohdistuvia kliinisesti merkittäviä yhteisvaikutuksia voitaisiin ennakoida. Kliinisen farmakologian tutkimuksissa ei todettu kliinisesti merkittäviä altistuksen muutoksia, kun barisitinibia annettiin yhdessä digoksiinin (P-gp:n substraatti) tai metotreksaatin (useiden kuljettajaproteiinien substraatti) kanssa.
Sytokromi P450 ‑entsyymit
Kliinisen farmakologian tutkimuksissa barisitinibin anto yhdessä simvastatiinin, etinyyliestradiolin tai levonorgestreelin (CYP3A:n substraatteja) kanssa ei vaikuttanut kliinisesti merkittävästi näiden lääkevalmisteiden farmakokinetiikkaan.
Raskaus ja imetys
Raskaus
JAK/STAT-reitin on todettu osallistuvan solujen adheesioon ja solujen polariteettiin, mikä voi vaikuttaa alkion varhaiskehitykseen. Ei ole olemassa riittäviä tietoja barisitinibin käytöstä raskaana oleville naisille. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Barisitinibi oli teratogeeninen rotalla ja kaniinilla. Eläintutkimukset viittaavat siihen, että suurempia annoksia käytettäessä barisitinibilla voi olla haitallinen vaikutus luuston kehitykseen in utero.
Barisitinibi on vasta-aiheinen raskauden aikana (ks. kohta Vasta-aiheet). Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 1 viikon ajan hoidon jälkeen. Jos potilas tulee raskaaksi barisitinibihoidon aikana, vanhemmille on kerrottava sikiöön mahdollisesti kohdistuvasta riskistä.
Imetys
Ei tiedetä, erittyvätkö barisitinibi tai sen metaboliitit ihmisellä äidinmaitoon. Eläintutkimuksista saadut saatavilla olevat farmakodynamiikan/toksikologian tiedot ovat osoittaneet, että barisitinibi erittyy maitoon (ks. kohta Prekliiniset tiedot turvallisuudesta).
Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois, eikä barisitinibia saa käyttää imetyksen aikana. On päätettävä, lopetetaanko imetys vai lopetetaanko barisitinibihoito ottaen huomioon imetyksestä aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Eläintutkimusten tulokset viittaavat siihen, että barisitinibihoito saattaa heikentää naaraiden hedelmällisyyttä hoidon aikana; hoito ei kuitenkaan vaikuttanut urosten siittiötuotantoon (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Barisitinibilla ei ole haitallista vaikutusta ajokykyyn ja koneiden käyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitettuja barisitinibin haittavaikutuksia ovat LDL-kolesteroliarvojen suurentuminen (26,0 %), ylähengitystieinfektiot (16,9 %), päänsärky (5,2 %), herpes simplex (3,2 %) ja virtsatieinfektiot (2,9 %). Nivelreumapotilailla esiintyi melko harvoin vakavaa keuhkokuumetta (0,2 %) ja vakavaa vyöruusua (0,2 %).
Haittavaikutustaulukko
Arvioitu esiintymistiheys: Hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000). Ellei toisin mainita, taulukossa 2 mainittavat esiintymistiheydet perustuvat kliinisissä tutkimuksissa aikuisilla ja/tai markkinoille tulon jälkeen ilmoitettuihin nivelreuman, atooppisen ihottuman ja pälvikaljun hoidon yhdistettyihin tietoihin; jos esiintymistiheydet eroavat käyttöaiheissa merkittävästi, asia mainitaan alaviitteessä taulukon alla. Haittavaikutukset esitetään kussakin esiintymistiheysluokassa vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 2. Haittavaikutukset | |||
| Elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen |
| Infektiot | Ylähengitystieinfektiot | Keuhkokuumed Vyöruusub Virtsatieinfektiot Gastroenteriitti Herpes simplex Karvatuppitulehdusg | |
| Veri ja imukudos | Trombosytoosi, > 600 x 109/la, d | Neutropenia, < 1 x 109/la | |
| Immuunijärjestelmä | Kasvojen turvotus Urtikaria | ||
| Aineenvaihdunta ja ravitsemus | Hyperkolesterolemiaa | Hypertriglyseridemiaa | |
| Hermosto | Päänsärky | ||
| Verisuonisto | Syvä laskimotukosb | ||
| Hengityselimet, rintakehä ja välikarsina | Keuhkoveritulppaf | ||
| Ruoansulatuselimistö | Vatsakipud Pahoinvointid | Divertikuliitti | |
| Maksa ja sappi | ALAT-arvon suureneminen, ≥ 3 x ULNa,d | ASAT-arvon suureneminen, ≥ 3 x ULNa,e | |
| Iho ja ihonalainen kudos | Ihottuma Aknec | ||
| Tutkimukset | Kreatiinikinaasiarvon suureneminen, > 5 x ULNa, c | Painonnousu | |
a Mukana laboratorioseurannassa todetut muutokset (ks. teksti jäljempänä).
b Vyöruusun ja syvän laskimotukoksen esiintymistiheydet perustuvat kliinisiin nivelreumatutkimuksiin.
c Kliinisissä nivelreumatutkimuksissa aknea ja tasolle > 5 x viitevälin yläraja suurentuneita kreatiinikinaasipitoisuuksia esiintyi melko harvoin.
d Atooppista ihottumaa koskeneissa kliinisissä tutkimuksissa pahoinvointia ja tasolle ≥ 3 x viitevälin yläraja suurentuneita ALAT-arvoja esiintyi melko harvoin. Pälvikaljua koskeneissa kliinisissä tutkimuksissa vatsakipua esiintyi melko harvoin. Atooppista ihottumaa ja pälvikaljua koskeneissa kliinisissä tutkimuksissa keuhkokuumetta ja trombosytoosia (> 600 x 109 solua/l) esiintyi melko harvoin.
e Pälvikaljua koskeneissa kliinisissä tutkimuksissa tasolle ≥ 3 x viitevälin yläraja suurentuneita ASAT‑arvoja esiintyi usein.
f Keuhkoveritulpan esiintymistiheys perustuu nivelreumaa ja atooppista ihottumaa koskeneisiin kliinisiin tutkimuksiin.
g Karvatuppitulehdusta havaittiin pälvikaljuutta koskeneissa kliinisissä tutkimuksissa. Tulehdus esiintyi yleensä päänahan alueella, jolla tapahtui hiusten takaisinkasvua.
Tiettyjen haittavaikutusten kuvaus
Ruoansulatuselimistö
Kliinisissä nivelreumatutkimuksissa aiemmin hoitamattomilla potilailla pahoinvoinnin esiintyminen 52 viikon aikana oli yleisempää metotreksaatti- ja barisitinibiyhdistelmähoitoryhmässä (9,3 %) kuin pelkkää metotreksaattia saaneilla (6,2 %) tai pelkkää barisitinibihoitoa saaneilla (4,4 %). Nivelreuman, atooppisen ihottuman ja pälvikaljun kliinisistä tutkimuksista saatujen yhdistettyjen tietojen perusteella pahoinvointia esiintyi eniten ensimmäisten 2 hoitoviikon aikana.
Vatsakipu oli yleensä lievää ja ohimenevää, eikä siihen liittynyt ruoansulatuskanavan infektioperäisiä eikä tulehduksellisia häiriöitä, eivätkä tapaukset johtaneet hoidon keskeyttämiseen.
Infektiot
Nivelreuman, atooppisen ihottuman ja pälvikaljun kliinisistä tutkimuksista saatujen yhdistettyjen tietojen perusteella useimmat infektiot olivat lieviä tai keskivaikeita. Molempia annoksia arvioineissa tutkimuksissa infektioita ilmoitettiin 31,0 %:lla 4 mg:n annoksia saaneista, 25,7 %:lla 2 mg:n annoksia saaneista ja 26,7 %:lla lumeryhmäläisistä. Kliinisissä nivelreumatutkimuksissa yhdistelmähoito metotreksaatin kanssa lisäsi infektioiden esiintymistiheyttä verrattuna pelkkään barisitinibihoitoon. Vyöruusun esiintymistiheys oli yleinen nivelreumassa, hyvin harvinainen atooppisessa ihottumassa ja melko harvinainen pälvikaljussa. Atooppisen ihottuman kliinisissä tutkimuksissa antibioottihoitoa edellyttäneitä ihoinfektioita esiintyi vähemmän barisitinibiryhmässä kuin lumelääkeryhmässä.
Vakavien infektioiden esiintymistiheys oli samanlainen barisitinibilla ja lumelääkkeellä. Vakavien infektioiden esiintymistiheys pysyi vakaana pitkäaikaisen altistuksen aikana. Vakavien infektioiden kokonaisilmaantuvuus kliinisessä tutkimusohjelmassa oli nivelreumassa 3,2 vakavaa infektiota 100:aa potilasvuotta kohti, atooppisessa ihottumassa 2,1 vakavaa infektiota 100:aa potilasvuotta kohti ja pälvikaljussa 0,8 vakavaa infektiota 100:aa potilasvuotta kohti. Nivelreumapotilailla esiintyi melko harvoin vakavaa keuhkokuumetta ja vakavaa vyöruusua.
Maksan transaminaasiarvojen suureneminen
Annosriippuvaista ALAT- ja ASAT-arvojen suurenemista ilmoitettiin yli 16 viikkoa kestäneissä tutkimuksissa. ALAT-/ASAT-arvojen keskimääräinen suureneminen pysyi stabiilina ajan mittaan. Useimmissa tapauksissa maksan transaminaasiarvojen suureneminen ≥ 3 x viitevälin ylärajan oli oireetonta ja ohimenevää.
Nivelreumapotilailla barisitinibin käyttäminen potentiaalisesti maksatoksisten lääkevalmisteiden, kuten metotreksaatin, kanssa johti useammin näiden arvojen suurenemiseen.
Lipidiarvojen suureneminen
Nivelreuman, atooppisen ihottuman ja pälvikaljun kliinisistä tutkimuksista saatujen yhdistettyjen tietojen perusteella barisitinibihoidon yhteydessä esiintyi annosriippuvaista lipidiarvojen kuten kokonaiskolesteroli-, LDL-kolesteroli- ja HDL (high density lipoprotein) -kolesteroliarvojen suurenemista. LDL/HDL-suhde ei muuttunut. Arvojen suurenemista todettiin 12 viikon kohdalla, ja arvot pysyivät tämän jälkeen stabiilisti lähtötasoa suurempina, myös pitkäkestoisessa nivelreuman jatkotutkimuksessa. Kokonaiskolesterolin ja LDL-kolesterolin keskiarvot suurenivat viikolle 52 asti atooppista ihottumaa tai pälvikaljua sairastavilla potilailla. Kliinisissä nivelreumatutkimuksissa barisitinibihoito yhdistettiin annosriippuvaiseen triglyseridiarvojen suurenemiseen. Triglyseridiarvot eivät suurentuneet atooppisen ihottuman ja pälvikaljun kliinisissä tutkimuksissa.
Statiinihoito pienensi suurentuneet LDL-kolesteroliarvot hoitoa edeltävälle tasolle.
Kreatiinikinaasi
Barisitinibihoito yhdistettiin annosriippuvaiseen kreatiinikinaasiarvojen suurenemiseen. Kreatiinikinaasin keskiarvo oli suurentunut viikon 4 kohdalla, ja arvot pysyivät tämän jälkeen lähtötasoa suurempina. Kaikissa käyttöaiheissa kreatiinikinaasiarvojen suureneminen tasolle > 5 x normaali viitevälin yläraja, oli useimmiten ohimenevää eikä edellyttänyt hoidon lopettamista.
Kliinisissä tutkimuksissa ei todettu vahvistettuja rabdomyolyysitapauksia.
Neutropenia
Neutrofiilimäärän keskiarvon pienenemistä todettiin 4 viikon kohdalla ja arvot pysyivät lähtötasoa matalampina ajan mittaan. Neutropenian ja vakavien infektioiden esiintymisen välillä ei ollut selvää yhteyttä. Kliinisissä tutkimuksissa hoito kuitenkin keskeytettiin, jos absoluuttinen neutrofiiliarvo oli < 1 x 109/l.
Trombosytoosi
Annosriippuvaista trombosyyttimäärän keskiarvojen suurenemista havaittiin ja se pysyi vakaana, lähtötasoa suurempana ajan mittaan.
Pediatriset potilaat
Lastenreuma
Lastenreuman kliinisessä tutkimusohjelmassa barisitinibille (mikä tahansa annos) altistui yhteensä 220 potilasta (ikä vähintään 2 ja alle 18 vuotta), mikä vastaa 326 potilasaltistusvuotta.
Kliinisen lastenreumatutkimuksen lumekontrolloidussa, kaksoissokkoutetussa, satunnaistetussa aktiivisen lääkkeen lopettamisen jälkeisessä vaiheessa barisitinibia saaneilla pediatrisilla potilailla (n = 82) päänsärky oli hyvin yleistä (11 %), neutropenia (< 1 000 solua/mm3) yleistä (2,4 %, yksi potilas) ja keuhkoembolia yleistä (1,2 %, yksi potilas).
Atooppinen ihottuma pediatrisilla potilailla
Lasten ja nuorten hoidon turvallisuusarviointi perustuu vaiheen III BREEZE-AD-PEDS tutkimuksen turvallisuustietoihin. Tutkimuksessa 466 potilasta (2–18-vuotiaita) sai jotakin barisitinibiannosta. Pediatrisilla potilailla todettu turvallisuusprofiili oli yleisesti ottaen verrattavissa aikuisten turvallisuusprofiiliin. Neutropenia (< 1 x 109 solua/l) oli yleisempää (1,7 %) kuin aikuisilla.
Pälvikalju nuorilla potilailla
Vaiheen III BRAVE-AA-PEDS tutkimuksessa barisitinibille (mikä tahansa annos) altistui yhteensä 245 potilasta (ikä vähintään 12 vuotta ja alle 18 vuotta). Näistä potilaista 85 hoidettiin 4 mg:n annoksella lumekontrolloidun hoitojakson aikana. Nuorilla potilailla todettu turvallisuusprofiili oli yleisesti ottaen verrattavissa aikuisten turvallisuusprofiiliin. Akne (10,6 %) ja neutropenia (3,6 %) (< 1 x 109 solua/l) olivat yleisempiä kuin aikuisilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa on annettu aikuispotilaille enintään 40 mg kerta-annoksia ja toistuvina annoksina enintään 20 mg annoksia päivittäin 10 päivän ajan ilman annosta rajoittavaa toksisuutta. Spesifistä toksisuutta ei todettu. Terveillä vapaaehtoisilla saadut tiedot 40 mg kerta-annoksen farmakokinetiikasta viittaavat siihen, että oletettavasti yli 90 % annetusta annoksesta eliminoituu 24 tunnissa. Yliannostustapauksessa on suositeltavaa seurata potilaan vointia haittavaikutusten oireiden ja löydösten varalta. Jos haittavaikutuksia kehittyy, potilasta hoidetaan asianmukaisesti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, Januskinaasin (JAK) estäjät, ATC-koodi: L04AF02
Vaikutusmekanismi
Barisitinibi on selektiivinen, reversiibeli Janus-kinaasien 1 ja 2 (JAK1 ja JAK2) estäjä. Eristetyillä entsyymeillä tehdyissä kokeissa barisitinibi esti JAK1:n, JAK2:n, tyrosiinikinaasi 2:n ja JAK3:n toimintaa. IC50-pitoisuudet olivat 5,9 nM (JAK1), 5,7 nM (JAK2), 53 nM (tyrosiinikinaasi 2) ja > 400 nM (JAK3).
Janus-kinaasit (JAK) ovat entsyymejä, jotka välittävät solun pintareseptoreista solunsisäisiä signaaleja useille sytokiineille ja kasvutekijöille, jotka osallistuvat hematopoieesiin, tulehdukseen ja immuunitoimintaan. Solunsisäisessä signalointireitissä JAK-entsyymit fosforyloivat ja aktivoivat STAT-transkriptiotekijöitä (signal transducer and activator of transcription), jotka käynnistävät geenien ilmentymisen solussa. Barisitinibi moduloi näitä signalointireittejä estämällä osittain JAK1- ja JAK2-entsyymitoimintaa, mikä vähentää STAT-transkriptiotekijöiden fosforylaatiota ja aktivoitumista.
Farmakodynaamiset vaikutukset
IL-6:n indusoiman STAT3:n fosforylaation esto
Barisitinibin anto johti annosriippuvaiseen IL-6:n indusoiman STAT3:n fosforylaation estoon terveiden henkilöiden kokoveressä. Suurin estovaikutus todettiin 2 tunnin kohdalla lääkkeen annosta, ja tilanne palautui lähelle lähtötasoa 24 tuntiin mennessä.
Immunoglobuliinit
IgG-, IgM-, ja IgA-immunoglobuliinien pitoisuuksien keskiarvot seerumissa pienenivät 12 viikon kuluessa barisitinibihoidon aloittamisesta ja pysyivät vakaalla, lähtöarvoa pienemmällä tasolla vähintään 104 viikkoon asti. Useimmilla potilailla immunoglobuliinipitoisuudet pysyivät muutoksesta huolimatta normaalilla viitealueella.
Lymfosyytit
Absoluuttisen lymfosyyttimäärän keskiarvo suureni 1 viikon kuluessa barisitinibihoidon aloittamisesta, palasi lähtötasolle viikkoon 24 mennessä ja pysyi sitten vakaana vähintään 104 viikkoon asti. Useimmilla potilailla lymfosyyttimäärät pysyivät muutoksesta huolimatta normaalilla viitealueella.
C-reaktiivinen proteiini
Nivelreumapotilailla todettiin C-reaktiivisen proteiinin (CRP) pitoisuuksien pienenemistä seerumissa jo 1 viikon kuluttua hoidon aloittamisesta. Muutos säilyi koko lääkkeen käytön ajan.
Kreatiniini
Kliinisissä tutkimuksissa barisitinibi johti seerumin kreatiniinipitoisuuksien suurenemiseen keskimäärin 3,8 µmol/l kahden hoitoviikon jälkeen. Tämän jälkeen kreatiniinipitoisuudet pysyivät vakaina. Ilmiö voi johtua siitä, että barisitinibi estää kreatiniinin erittymistä munuaistubuluksissa. Seerumin kreatiniinipitoisuuteen perustuvat glomerulusten suodatusnopeuden (GFR) arviot saattavat siis pienentyä hiukan, vaikka varsinaista munuaistoiminnan heikkenemistä ei tapahdu eikä munuaishaittavaikutuksia esiinny. Pälvikaljua sairastavilla potilailla seerumin kreatiniinin keskiarvon suureneminen jatkui viikolle 52 asti. Atooppista ihottumaa ja pälvikaljua sairastavilla potilailla barisitinibihoitoon liittyi kystatiini C ‑pitoisuuden (jota käytetään myös glomerulusten suodatusnopeuden arviointiin) pienenemistä viikolla 4. Arvot eivät pienentyneet sen jälkeen enempää.
Ihon in vitro ‑mallit
Ihmisen ihon in vitro ‑mallissa, jota käsiteltiin proinflammatorisilla sytokiineilla (IL-4, IL-13, IL-31), barisitinibi vähensi epidermiksen keratinosyyttien pSTAT3:n ilmentämistä ja lisäsi filaggriinin ilmentämistä. Filaggriiniproteiini osallistuu ihon läpäisyesteen toimintaan ja atooppisen ihottuman patogeneesiin.
Rokotteita koskeva tutkimus
Barisitinibin vaikutusta inaktivoituja osia sisältävien rokotteiden humoraaliseen vasteeseen arvioitiin tutkimuksessa, jossa stabiilia 2 mg:n tai 4 mg:n barisitinibihoitoa saaneet 106 nivelreumapotilasta saivat inaktivoitua pneumokokki- tai tetanus-rokotetta. Suurin osa näistä potilaista (n = 94) sai samaan aikaan metotreksaattihoitoa. Yhteensä ryhmän pneumokokki-rokotetta saaneista potilaista 68 % (95 % lv: 58,4 %, 76,2 %) saavutti riittävän IgG-immuunivasteen. Tetanus-rokotetta saaneista potilaista 43,1 % (95 % lv: 34 %, 52,8 %) saavutti riittävän IgG-immuunivasteen.
Kliininen teho
Nivelreuma
Kerran vuorokaudessa otettavan barisitinibin tehoa ja turvallisuutta arvioitiin neljässä vaiheen III satunnaistetussa, kaksoissokkoutetussa monikeskustutkimuksessa aikuisilla potilailla, joilla oli vuoden 2010 ACR/EULAR-kriteereillä diagnosoitu keskivaikea tai vaikea, aktiivinen nivelreuma (taulukko 3). Heillä tuli olla lähtötilanteessa vähintään 6 aristavaa ja 6 turvonnutta niveltä. Kaikki potilaat, jotka suorittivat nämä tutkimukset loppuun, saivat siirtyä pitkäkestoiseen jatkotutkimukseen, jossa hoitoa jatkettiin enintään 7 vuotta.
| Taulukko 3. Kliinisen tutkimuksen yhteenveto | |||
| Tutkimuksen nimi (Kesto) | Populaatio (Määrä) | Hoitoryhmät | Keskeiset tulosmuuttujat (tiivistelmä) |
RA-BEGIN (52 viikkoa) | Ei aiempaa metotreksaattihoitoa1 (584)
|
|
|
RA-BEAM (52 viikkoa) | Metotreksaatilla riittämätön vaste2 (1305)
|
Kaikilla potilailla taustahoitona metotreksaatti |
|
RA-BUILD (24 viikkoa) | Tavanomaisella reumalääkkeellä riittämätön vaste3 (684)
|
Taustahoitona tavanomainen reumalääke,5 jos tavanomainen reumalääke käytössä vakaana annoksena tutkimukseenottohetkellä |
|
RA-BEACON (24 viikkoa) | TNF:n estäjillä riittämätön vaste4 (527)
|
Taustahoitona tavanomainen reumalääke5 |
|
Lyhenteet: x 1 = kerran vuorokaudessa; ACR = American College of Rheumatology; SDAI = Simplified Disease Activitity Index; HAQ-DI = Health Assessment Questionnaire‑Disability Index; mTSS = modified Total Sharp Score
1 Potilaat olivat saaneet alle 3 annosta metotreksaattia eivätkä olleet saaneet mitään muita tavanomaisia tai biologisia tautiprosessiin vaikuttavia reumalääkkeitä.
2 Potilaalla riittämätön vaste metotreksaattiin (+/- muihin tavanomaisiin tautiprosessiin vaikuttaviin reumalääkkeisiin); ei aiempaa biologista lääkitystä.
3 ≥ 1 tavanomainen tautiprosessiin vaikuttava reumalääke tuottanut potilaalle riittämättömän vasteen tai ollut huonosti siedetty; ei aiempaa biologista lääkitystä.
4 ≥ 1 biologinen tautiprosessiin vaikuttava reumalääke (mukaan lukien vähintään 1 TNF:n estäjä) tuottanut potilaalle riittämättömän vasteen tai ollut huonosti siedetty.
5 Yleisimpiä samanaikaisesti käytettyjä tavanomaisia tautiprosessiin vaikuttavia reumalääkkeitä olivat metotreksaatti, hydroksiklorokiini, leflunomidi ja sulfasalatsiini.
Kliininen vaste
Kaikissa tutkimuksissa barisitinibi 4 mg x 1 ‑hoitoa saaneet saavuttivat tilastollisesti merkitsevästi suuremman ACR20-, ACR50- ja ACR70-vasteen viikolla 12 kuin lume-, metotreksaatti- tai adalimumabihoitoa saaneet (ks. taulukko 4). Teho alkoi kaikilla mittareilla mitattuna nopeasti ja jo viikolla 1 saavutettiin merkitsevästi parempi hoitovaste. Potilailla todettiin jatkuva, pitkäkestoinen hoitovaste; ACR20/50/70-vasteet säilyivät vähintään 2 vuoden ajan, myös pitkäkestoisessa jatkotutkimuksessa.
Barisitinibi 4 mg ‑hoito paransi merkitsevästi ACR-vasteen kaikkien yksittäisten osa-alueiden tuloksia sekä ainoana lääkkeenä että yhdessä tavanomaisten reumalääkkeiden kanssa käytettynä (mm. aristavien ja turvonneiden nivelten määrät, potilaan ja lääkärin yleisarviot, HAQ-DI, kivun arviointi ja CRP) verrattuna lumehoitoon, metotreksaattiin tai adalimumabiin.
Merkittäviä teho- ja turvallisuuseroja ei havaittu, kun potilaat ryhmiteltiin alaryhmiin barisitinibin ohella käytetyn reumalääketyypin perusteella.
Remissio ja vähäinen tautiaktiivisuus
Tilastollisesti merkitsevästi suurempi osuus barisitinibi 4 mg ‑hoitoa saaneista kuin lume- tai metotreksaattihoitoa saaneista saavutti remission (SDAI ≤ 3,3 ja CDAI ≤ 2,8) tai vähäisen tautiaktiivisuuden tai remission (DAS28-lasko tai DAS28-hsCRP ≤ 3,2 ja DAS28-lasko tai DAS28-hsCRP < 2,6) viikoilla 12 ja 24 (taulukko 4).
Lumehoitoa paremmat remissioprosentit todettiin jo viikolla 4. Remissioprosentit ja vähäisen tautiaktiivisuuden prosenttiosuudet säilyivät vähintään 2 vuoden ajan. Tiedot pitkäkestoisesta jatkotutkimuksesta enintään 6 vuoden seurannan ajalta osoittivat vähäisen tautiaktiivisuuden/remission jatkuvan.
| Taulukko 4: Vaste, remissio ja fyysinen toimintakyky | ||||||||||||
| Tutkimus | RA-BEGIN Ei aiempaa metotreksaattihoitoa | RA-BEAM Metotreksaatilla riittämätön vaste | RA-BUILD Tavanomaisella reumalääkkeellä riittämätön vaste / huono siedettävyys | RA-BEACON TNF:n estäjällä riittämätön vaste / huono siedettävyys | ||||||||
Hoito ryhmä | MTX | BARI 4 mg | BARI 4 mg + MTX | Lume | BARI 4 mg | ADA 40 mg 2 viikon välein | Lume | BARI 2 mg | BARI 4 mg | Lume | BARI 2 mg | BARI 4 mg |
| N | 210 | 159 | 215 | 488 | 487 | 330 | 228 | 229 | 227 | 176 | 174 | 177 |
| ACR20: | ||||||||||||
| Viikko 12 | 59 % | 79 %*** | 77 %*** | 40 % | 70 %***† | 61 %*** | 39 % | 66 %*** | 62 %*** | 27 % | 49 %*** | 55 %*** |
| Viikko 24 | 62 % | 77 %** | 78 %*** | 37 % | 74 %***† | 66 %*** | 42 % | 61 %*** | 65 %*** | 27 % | 45 %*** | 46 %*** |
| Viikko 52 | 56 % | 73 %*** | 73 %*** | 71 %†† | 62 % | |||||||
| ACR50: | ||||||||||||
| Viikko 12 | 33 % | 55 %*** | 60 %*** | 17 % | 45 %***†† | 35 %*** | 13 % | 33 %*** | 34 %*** | 8 % | 20 %** | 28 %*** |
| Viikko 24 | 43 % | 60 %** | 63 %*** | 19 % | 51 %*** | 45 %*** | 21 % | 41 %*** | 44 %*** | 13 % | 23 %* | 29 %*** |
| Viikko 52 | 38 % | 57 %*** | 62 %*** | 56 %† | 47 % | |||||||
| ACR70: | ||||||||||||
| Viikko 12 | 16 % | 31 %*** | 34 %*** | 5 % | 19 %***† | 13 %*** | 3 % | 18 %*** | 18 %*** | 2 % | 13 %*** | 11 %** |
| Viikko 24 | 21 % | 42 %*** | 40 %*** | 8 % | 30 %***† | 22 %*** | 8 % | 25 %*** | 24 %*** | 3 % | 13 %*** | 17 %*** |
| Viikko 52 | 25 % | 42 %*** | 46 %*** | 37 % | 31 % | |||||||
| DAS28-hsCRP ≤ 3,2: | ||||||||||||
| Viikko 12 | 30 % | 47 %*** | 56 %*** | 14 % | 44 %***†† | 35 %*** | 17 % | 36 %*** | 39 %*** | 9 % | 24 %*** | 32 %*** |
| Viikko 24 | 38 % | 57 %*** | 60 %*** | 19 % | 52 %*** | 48 %*** | 24 % | 46 %*** | 52 %*** | 11 % | 20 %* | 33 %*** |
| Viikko 52 | 38 % | 57 %*** | 63 %*** | 56 %† | 48 % | |||||||
| SDAI ≤ 3,3: | ||||||||||||
| Viikko 12 | 6 % | 14 %* | 20 %*** | 2 % | 8 %*** | 7 %*** | 1 % | 9 %*** | 9 %*** | 2 % | 2 % | 5 % |
| Viikko 24 | 10 % | 22 %** | 23 %*** | 3 % | 16 %*** | 14 %*** | 4 % | 17 %*** | 15 %*** | 2 % | 5 % | 9 %** |
| Viikko 52 | 13 % | 25 %** | 30 %*** | 23 % | 18 % | |||||||
| CDAI ≤ 2,8: | ||||||||||||
| Viikko 12 | 7 % | 14 %* | 19 %*** | 2 % | 8 %*** | 7 %** | 2 % | 10 %*** | 9 %*** | 2 % | 3 % | 6 % |
| Viikko 24 | 11 % | 21 %** | 22 %** | 4 % | 16 %*** | 12 %*** | 4 % | 15 %*** | 15 %*** | 3 % | 5 % | 9 %* |
| Viikko 52 | 16 % | 25 %* | 28 %** | 22 % | 18 % | |||||||
| HAQ-DI-pisteiden pienin kliinisesti merkittävä muutos (HAQ-DI-pisteet pienentyneet ≥ 0,30): | ||||||||||||
| Viikko 12 | 60 % | 81 %*** | 77 %*** | 46 % | 68 %*** | 64 %*** | 44 % | 60 %*** | 56 %** | 35 % | 48 %* | 54 %*** |
| Viikko 24 | 66 % | 77 %* | 74 % | 37 % | 67 %***† | 60 %*** | 37 % | 58 %*** | 55 %*** | 24 % | 41 %*** | 44 %*** |
| Viikko 52 | 53 % | 65 %* | 67 %** | 61 % | 55 % | |||||||
Huom. Vasteen saavuttaneiden osuudet kunakin ajankohtana perustuvat kyseiseen hoitoon alun perin satunnaistettujen potilaiden määrään (N). Jos potilas keskeytti tutkimuksen tai sai varahoitoa, tämän jälkeen katsottiin, että hän ei ollut saavuttanut vastetta.
Lyhenteet: ADA = adalimumabi; MTX = metotreksaatti; BARI = barisitinibi
* p ≤ 0,05; ** p ≤ 0,01; *** p ≤ 0,001 verrattuna lumeeseen (RA-BEGIN-tutkimuksessa verrattuna metotreksaattiin).
† p ≤ 0,05; †† p ≤ 0,01; ††† p ≤ 0,001 verrattuna adalimumabiin.
Radiologinen vaste
Barisitinibihoidon vaikutusta rakenteellisten nivelvaurioiden etenemiseen arvioitiin radiologisesti RA‑BEGIN-, RA‑BEAM- ja RA‑BUILD-tutkimuksissa. Arvioinnissa käytettiin muokattuja Sharpin kokonaispisteitä (mTSS, modified Total Sharp Score) ja tämän mittarin eri osa-alueita, eroosiopisteitä ja nivelraon kaventumispisteitä.
Barisitinibi 4 mg ‑hoito esti tilastollisesti merkitsevästi rakenteellisten nivelvaurioiden etenemistä (taulukko 5). Eroosiopisteiden ja nivelraon kaventumispisteiden analyysitulokset vastasivat kokonaispistemäärien tuloksia. Niiden potilaiden osuus, joilla ei todettu lainkaan radiologista taudin etenemistä (mTSS-pisteiden muutos ≤ 0), oli barisitinibi 4 mg ‑ryhmässä merkitsevästi suurempi kuin lumeryhmässä viikoilla 24 ja 52.
| Taulukko 5. Radiologiset muutokset | |||||||||
| Tutkimus | RA-BEGIN Ei aiempaa metotreksaattihoitoa | RA-BEAM Metotreksaatilla riittämätön vaste | RA-BUILD Tavanomaisella reumalääkkeellä riittämätön vaste / huono siedettävyys | ||||||
| Hoitoryhmä | MTX | BARI 4 mg | BARI 4 mg + MTX | Lumea | BARI 4 mg | ADA 40 mg 2 viikon välein | Lume | BARI 2 mg | BARI 4 mg |
| mTSS-pisteet, keskimuutos lähtötilanteesta: | |||||||||
| Viikko 24 | 0,61 | 0,39 | 0,29* | 0,90 | 0,41*** | 0,33*** | 0,70 | 0,33* | 0,15** |
| Viikko 52 | 1,02 | 0,80 | 0,40** | 1,80 | 0,71*** | 0,60*** | |||
| Potilaat, joilla ei radiologista taudin etenemistäb: | |||||||||
| Viikko 24 | 68 % | 76 % | 81 %** | 70 % | 81 %*** | 83 %*** | 74 % | 72 % | 80 % |
| Viikko 52 | 66 % | 69 % | 80 %** | 70 % | 79 %** | 81 %** | |||
Lyhenteet: ADA = adalimumabi; MTX = metotreksaatti; BARI = barisitinibi
a Lumehoidon tiedot viikolta 52 perustuvat lineaariseen ekstrapolointiin.
b ”Ei taudin etenemistä” määriteltiin tilanteeksi, jossa mTSS-pisteiden muutos oli ≤ 0.
* p ≤ 0,05; ** p ≤ 0,01; *** p ≤ 0,001 verrattuna lumeeseen (RA-BEGIN-tutkimuksessa verrattuna metotreksaattiin).
Fyysisen toimintakyvyn vaste ja terveyteen liittyvät hoitotulokset
Barisitinibi 4 mg -hoidon käyttö joko ainoana hoitona tai yhdessä tavanomaisten tautiprosessiin vaikuttavien reumalääkkeiden kanssa johti merkitsevään fyysisen toimintakyvyn paranemiseen (mittausperusteena HAQ-DI) ja kivun lievittymiseen (mittausperusteena VAS-asteikko 0–100) verrattuna kaikkiin vertailuhoitoihin (lume-, metotreksaatti- ja adalimumabihoitoon). Potilaiden tilanteen kohenemista todettiin jo viikolla 1, ja RA-BEGIN- ja RA-BEAM-tutkimuksissa se säilyi 52 viikkoon asti.
RA-BEAM- ja RA-BUILD-tutkimuksissa barisitinibi 4 mg -hoito vähensi merkitsevästi aamuisen niveljäykkyyden keston keskiarvoa ja vaikeusastetta verrattuna lume- tai adalimumabihoitoon, kun asiaa arvioitiin päivittäin täytettävillä sähköisillä potilaspäiväkirjoilla.
Barisitinibihoitoa saaneet potilaat ilmoittivat kaikissa tutkimuksissa potilaan raportoiman elämänlaadun paranemista, kun mittarina käytettiin SF 36-mittarin (Short Form (36) Health Survey) fyysisen osion pisteitä, ja uupumuksen lievittymistä, kun mittarina käytettiin FACIT-F -pisteitä (Functional Assessment of Chronic Illness Therapy-Fatigue).
Barisitinibi 4 mg vs. 2 mg
4 mg ja 2 mg annosten tehoerot olivat selvimmät potilailla, joilla biologinen tautiprosessiin vaikuttava reumalääke oli tuottanut riittämättömän vasteen tai ollut huonosti siedetty (RA‑BEACON). Tässä potilasryhmässä todettiin, että barisitinibi 4 mg kohensi tilastollisesti merkitsevästi ACR-vasteen eri osa-alueita eli turvonneiden nivelten määrää, aristavien nivelten määrää ja laskoa verrattuna lumehoitoon viikolla 24, kun taas barisitinibi 2 mg ei eronnut lumehoidosta. Lisäksi sekä RA‑BEACON- että RA‑BUILD-tutkimuksessa todettiin, että teho alkoi nopeammin ja efektikoko oli yleisesti ottaen suurempi 4 mg annosryhmässä kuin 2 mg annosryhmässä.
Pitkäkestoisessa jatkotutkimuksessa RA‑BEAM-, RA‑BUILD- ja RA‑BEACON-tutkimusten potilaat, joilla saavutettiin pitkäkestoinen vähäinen tautiaktiivisuus tai remissio (CDAI ≤ 10) vähintään 15 kuukauden barisitinibi 4 mg x 1 ‑hoidon jälkeen, satunnaistettiin uudelleen kaksoissokkoutetusti suhteessa 1:1 joko jatkamaan hoitoa annoksella 4 mg x 1 tai pienentämään annosta tasolle 2 mg x 1. CDAI-pisteiden perusteella arvioitu vähäinen tautiaktiivisuus tai remissio säilyi valtaosalla potilaista:
- Viikolla 12: 451/498 (91 %) 4 mg annosta jatkaneista vrt. 405/498 (81 %) pienempään 2 mg annokseen siirtyneistä (p ≤ 0,001)
- Viikolla 24: 434/498 (87 %) 4 mg annosta jatkaneista vrt. 372/498 (75 %) pienempään 2 mg annokseen siirtyneistä (p ≤ 0,001)
- Viikolla 48: 400/498 (80 %) 4 mg annosta jatkaneista vrt. 343/498 (69 %) pienempään 2 mg annokseen siirtyneistä (p ≤ 0,001)
- Viikolla 96: 347/494 (70 %) 4 mg annosta jatkaneista vrt. 297/496 (60 %) pienempään 2 mg annokseen siirtyneistä (p ≤ 0,001)
Jos vähäinen tautiaktiivisuus tai remissio menetettiin annoksen pienentämisen jälkeen, tauti saatiin valtaosalla potilaista uudelleen hallintaan, kun 4 mg annos otettiin jälleen käyttöön.
Atooppinen ihottuma aikuisilla
Barisitinibi-valmisteen tehoa ja turvallisuutta monoterapiana tai yhdessä paikallisten kortikosteroidien kanssa arvioitiin kolmessa vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa 16 viikon pituisessa tutkimuksessa (BREEZE‑AD1, ‑AD2 ja ‑AD7). Tutkimuksiin osallistui 1 568 potilasta, joilla oli keskivaikea tai vaikea atooppinen ihottuma, jonka määritelmänä olivat IGA-pistemäärä (Investigator’s Global Assessment) ≥ 3, EASI-pistemäärä (Eczema Area and Severity Index) ≥ 16 ja BSA-arvo (body surface area; affisioituneen alueen osuus kehon pinta-alasta) ≥ 10 %. Tutkimukseen soveltuvat potilaat olivat yli 18-vuotiaita, ja paikalliset lääkevalmisteet olivat tuottaneet heillä riittämättömän vasteen tai olleet huonosti siedettyjä. Potilaat saivat käyttää varahoitoa (johon kuului paikallisesti tai systeemisesti käytettäviä valmisteita). Tällöin katsottiin, että kyseinen potilas ei ollut saavuttanut vastetta. BREEZE‑AD7‑tutkimuksen lähtötilanteessa kaikki potilaat saivat samanaikaista paikallista kortikosteroidihoitoa, ja potilaat saivat käyttää paikallisia kalsineuriinin estäjiä.Kaikki potilaat, jotka olivat näissä tutkimuksissa mukana loppuun asti, saivat siirtyä pitkäkestoiseen jatkotutkimukseen (BREEZE-AD3), jossa hoitoa jatkettiin enintään 4 vuotta.
Vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa BREEZE‑AD4‑tutkimuksessa arvioitiin barisitinibin tehoa yhdessä paikallisten kortikosteroidivalmisteiden kanssa 52 viikon ajan 463 potilaalla, joilla oli keskivaikea tai vaikea atooppinen ihottuma ja joilla peroraalinen siklosporiinihoito oli epäonnistunut, huonosti siedetty tai vasta-aiheinen.
Lähtötilanteen tiedot
Lumekontrolloiduissa vaihen III tutkimuksissa (BREEZE-AD1, -AD2, -AD7 ja -AD4) kaikkien hoitoryhmien yhdistetyissä tiedoissa potilaista 37 % oli naisia, 64 % oli valkoihoisia, 31 % oli aasialaisia ja 0,6 % oli tummaihoisia. Potilaiden ikäkeskiarvo oli 35,6 vuotta. Näissä tutkimuksissa 42−51 %:lla potilaista lähtötilanteen IGA-pistemäärä oli 4 (vaikea atooppinen ihottuma), ja 54−79 % potilaista oli saanut aiempaa systeemistä hoitoa atooppiseen ihottumaan. Lähtötilanteessa EASI-pistekeskiarvo oli 29,6−33,5; kutinan NRS-pistemäärän (Itch Numerical Rating Scale) viikoittainen keskiarvo oli 6,5−7,1; DLQI-keskiarvo (Dermatology Life Quality Index) oli välillä 13,6−14,9; ja HADS-yhteispisteiden (Hospital Anxiety and Depression Scale) keskiarvo oli 10,9−12,1.
Kliininen hoitovaste
Tutkimukset, joissa 16 viikon monoterapia (BREEZE-AD1, -AD2) ja yhdistelmähoito paikallisen kortikosteroidin kanssa (BREEZE-AD7)
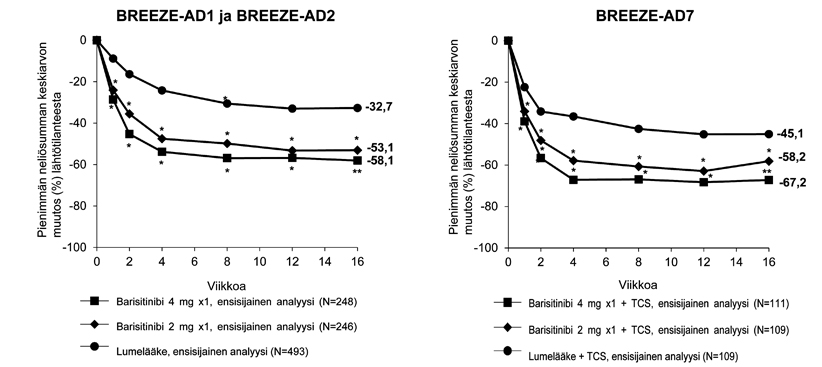
Merkitsevästi suurempi osuus barisitinibi 4 mg ‑ryhmään satunnaistetuista potilaista saavutti IGA-pistemäärän 0 tai 1 (ensisijainen päätetapahtuma), EASI-75-vasteen tai kutinan lievittyminen ≥ 4 pisteellä (kutinan NRS-arvo) verrattuna lumelääkkeeseen viikolla 16 (taulukko 6). Kuvassa 1 esitetään EASI-pistemäärän keskimääräinen prosentuaalinen muutos lähtötilanteesta viikolle 16.
Kutinan NRS-arvo koheni ≥ 4‑pistettä merkitsevästi suuremmalla osuudella barisitinibi 4 mg ‑ryhmään satunnaistetuista kuin lumelääkettä saaneista tutkittavista ensimmäisen hoitoviikon aikana tutkimuksissa BREEZE-AD1 ja -AD2, ja jo viikolla 2 tutkimuksessa BREEZE-AD7; p < 0,002.
Hoidon vaikutukset eri alaryhmissä (määrittelyperusteet: paino, ikä, sukupuoli, etninen tausta, taudin vaikeusaste ja aiemmat hoidot, mm. immunosuppressanttihoito) vastasivat koko tutkimuspopulaatiossa havaittuja tuloksia.
| Taulukko 6. Barisitinibin teho viikolla 16 (koko analyysipopulaatioa) | |||||||||
| Monoterapia | Yhdistelmähoito paikallisen kortikosteroidin (TCS) kanssa | ||||||||
| Tutkimus | BREEZE-AD1 | BREEZE-AD2 | BREEZE-AD7 | ||||||
| Hoitoryhmä | Lumelääke | BARI 2 mg | BARI 4 mg | Lumelääke | BARI 2 mg | BARI 4 mg | Lumelääke +TCS | BARI + TCS | BARI + TCS |
| N | 249 | 123 | 125 | 244 | 123 | 123 | 109 | 109 | 111 |
IGA 0 tai 1, vasteen saavuttaneet (%)b,c | 4,8 | 11,4** | 16,8** | 4,5 | 10,6** | 13,8** | 14,7 | 23,9 | 30,6** |
EASI-75, vasteen saavuttaneet (%)c | 8,8 | 18,7** | 24,8** | 6,1 | 17,9** | 21,1** | 22,9 | 43,1* | 47,7** |
| Kutinan NRS (≥ 4 pisteen parannus), vasteen saavuttaneet (%)c, d | 7,2 | 12,0 | 21,5** | 4,7 | 15,1** | 18,7** | 20,2 | 38,1* | 44,0** |
BARI = barisitinibi
* tilastollisesti merkitsevä verrattuna lumelääkkeeseen, ei korjattu kertautuvuuden suhteen;
** tilastollisesti merkitsevä verrattuna lumelääkkeeseen, korjattu kertautuvuuden suhteen
a Koko analyysipopulaatio käsitti kaikki satunnaistetut potilaat.
b Vasteen saavuttaminen määriteltiin tilanteeksi, jossa potilaan IGA-pistemäärä oli 0 tai 1 (”puhdas” tai ”lähes puhdas”) ja IGA‑pistemäärä pieneni ≥ 2 pisteen verran asteikolla 0–4.
c Vastetta saavuttamattomien imputointi: Jos potilas sai varahoitoa tai hänestä puuttui tietoja, katsottiin, että hän ei saavuttanut vastetta.
d Esitetyt tulokset koskevat arviointikelpoisten potilaiden osajoukkoa (potilaat, joilla kutinan NRS-pistemäärä ≥ 4 lähtötilanteessa).
Kuva 1. EASI-pistemäärän prosentuaalinen keskimuutos lähtötilanteesta (koko analyysipopulaatio)a
TCS = Yhdistelmähoito paikallisen kortikosteroidin kanssa
* tilastollisesti merkitsevä verrattuna lumelääkkeeseen, ei korjattu kertautuvuuden suhteen
** tilastollisesti merkitsevä verrattuna lumelääkkeeseen, korjattu kertautuvuuden suhteen
a Koko analyysipopulaatio (FAS) käsitti kaikki satunnaistetut potilaat. Varahoidon käytön jälkeen tai lääkevalmisteen käytön pysyvän lopettamisen jälkeen kerätyt tiedot katsottiin puuttuviksi tiedoiksi. Pienimmän neliösumman keskiarvot perustuvat toistomittausten sekamallin analyyseihin (Mixed Model with Repeated Measures, MMRM).
Vasteen säilyminen
Vasteen säilymisen arvioimiseksi 1 398 tutkittavalle, jotka saivat barisitinibia 16 viikon ajan BREEZE‑AD1-tutkimuksessa (N = 566), BREEZE‑AD2-tutkimuksessa (N = 540) tai BREEZE‑AD7-tutkimuksessa (N = 292), tarjottiin mahdollisuus osallistua pitkäkestoiseen jatkotutkimukseen (BREEZE‑AD3). Tietoja on saatavilla enintään 4 vuoden (216 viikon) kumulatiivisesta hoidosta. Vasteen havaittiin jatkuvan niillä potilailla, jotka saivat vähintään jonkun vasteen (IGA-pistemäärä 0, 1 tai 2) barisitinibin aloituksen jälkeen.
Annoksen pienentäminen
Pitkäaikaisessa BREEZE-AD3-jatkotutkimuksessa potilaat, joilla oli puhdas, lähes puhdas iho tai lievä tauti (IGA 0, 1 tai 2) ja jotka saivat barisitinibia 4 mg kerran vuorokaudessa, satunnaistettiin uudelleen viikolla 52 jatkamaan annoksella 4 mg kerran vuorokaudessa tai pienentämään annokseen 2 mg kerran vuorokaudessa. Potilaista, jotka pienensivät annoksen 2 mg:aan, 37 %:lla oli IGA 0-, 1- tai 2 -vaste ja 52 %:lla EASI75-vaste viikolla 200. Tämän ryhmän potilaista 47 %:lla kutinan NRS-arvo oli parantunut ≥ 4 pisteellä viikolla 52 ja 40 %:lla viikolla 68. Relapsoivien (IGA ≥ 3) potilaiden osuus oli pienempi niiden potilaiden alaryhmässä, joilla oli puhdas tai lähes puhdas iho (IGA 0 tai 1) annoksen pienentämisen alussa. Potilaista, jotka relapsoivat (IGA ≥ 3) annoksen pienentämisen jälkeen, suurin osa sai sairauden hallintaan barisitinibi 4 mg:n uudelleenaloituksen jälkeen.
Elämänlaatu/potilaiden raportoimat hoitotulokset atooppisen ihottuman hoidossa
Molemmissa monoterapiatutkimuksissa (BREEZE‑AD1 ja BREEZE‑AD2) ja tutkimuksessa, jossa käytettiin samanaikaisesti paikallista kortikosteroidihoitoa (BREEZE‑AD7), barisitinibi 4 mg ‑hoito paransi merkitsevästi potilaiden raportoimia hoitovasteita; se muun muassa pienensi kutinan NRS-pistemäärää, paransi nukkumista (mittarina ADSS), lievitti ihon kipua (ihon kivun NRS-pistemäärä), paransi elämänlaatua (DLQI) sekä ahdistuneisuus- ja masennusoireita (HADS), joita ei korjattu kertautuvuuden suhteen, viikolla 16 verrattuna lumelääkkeeseen (ks. taulukko 7).
| Taulukko 7. Elämänlaatu/potilaiden raportoimat hoitotulokset barisitinibimonoterapian yhteydessä sekä barisitinibin ja paikallisen kortikosteroidin yhdistelmän käytön yhteydessä viikolla 16 (koko analyysipopulaatio)a | |||||||||
| Monoterapia | Yhdistettynä paikalliseen kortikosteroidihoitoon (TCS) | ||||||||
| Tutkimus | BREEZE-AD1 | BREEZE-AD2 | BREEZE-AD7e | ||||||
| Hoitoryhmä | Lumelääke | BARI 2 mg | BARI 4 mg | Lumelääke | BARI 2 mg | BARI 4 mg | Lumelääke + TCS | BARI + TCS | BARI + TCS |
| N | 249 | 123 | 125 | 244 | 123 | 123 | 109 | 109 | 111 |
| ADSS kohta kaksi (≥ 2 pisteen parannus), vasteen saavuttaneet (%)c,d | 12,8 | 11,4 | 32,7* | 8,0 | 19,6 | 24,4* | 30,6 | 61,5* | 66,7* |
| Ihon kivun NRS-pistemäärän muutos, keskiarvo (keskivirhe)b | -0,84 (0,24) | -1,58 (0,29) | -1,93** (0,26) | -0,86 (0,26) | -2,61** (0,30) | -2,49** (0,28) | -2,06 (0,23) | -3,22* (0,22) | -3,73* (0,23) |
| DLQI-arvon muutos, keskiarvo (keskivirhe)b | -2,46 (0,57) | -4,30* (0,68) | -6,76* (0,60) | -3,35 (0,62) | -7,44* (0,71) | -7,56* (0,66) | -5,58 (0,61) | -7,50* (0,58) | -8,89* (0,58) |
| HADS-arvon muutos, keskiarvo (keskivirhe)b | -1,22 (0,48) | -3,22*
| -3,56* (0,52) | -1,25 (0,57) | -2,82 (0,66) | -3,71* (0,62) | -3,18 (0,56) | -4,75* (0,54) | -5,12* (0,54) |
BARI = Barisitinibi
* tilastollisesti merkitsevä verrattuna lumelääkkeeseen, ei korjattu kertautuvuuden suhteen
** tilastollisesti merkitsevä verrattuna lumelääkkeeseen, korjattu kertautuvuuden suhteen
a Koko analyysipopulaatio (FAS) käsitti kaikki satunnaistetut potilaat.
b Esitetyt tulokset kuvaavat pienimmän neliösumman keskiarvon muutosta lähtötilanteesta (keskivirhe). Varahoidon käytön jälkeen tai lääkevalmisteen käytön pysyvän lopettamisen jälkeen kerätyt tiedot katsottiin puuttuviksi tiedoiksi. Pienimmän neliösumman keskiarvot perustuvat toistomittausten sekamallin analyyseihin (Mixed Model with Repeated Measures, MMRM).
c ADSS, kohta kaksi: kutinan aiheuttamien yöllisten heräämiskerrat.
d Vastetta saavuttamattomien imputointi: Jos potilas sai varahoitoa tai hänestä puuttui tietoja, katsottiin, että hän ei saavuttanut vastetta. Tulokset esitetään niiltä potilailta, jotka soveltuivat arviointiin (potilaat, joilla ADSS kohta kaksi lähtötilanteessa ≥ 2 pistettä).
e Kaikki potilaat saivat samanaikaisesti paikallista kortikosteroidihoitoa ja potilaiden oli sallittua käyttää paikallisia kalsineuriinin estäjiä.
Kliininen hoitovaste potilailla, joita on aiemmin hoidettu siklosporiinilla tai joilla hoito on vasta-aiheinen (BREEZE-AD4-tutkimus)
Tutkimukseen otettiin yhteensä 463 potilasta, joilla peroraalisella siklosporiinihoidolla ei saatu riittävää hoitovastetta (n = 173), hoito oli huonosti siedetty (n = 75) tai vasta-aiheinen (n = 126). Ensisijainen päätetapahtuma oli EASI-75-vasteen saavuttaneiden potilaiden osuus viikolla 16. Taulukossa 8 esitetään yhteenveto ensisijaisen päätetapahtuman tiedoista ja joidenkin tärkeimpien toissijaisten päätetapahtumien tiedoista viikolla 16.
| Taulukko 8: Barisitinibin teho käytettynä yhdessä paikallisen kortikosteroidihoidon kanssaa BREEZE-AD4-tutkimuksen viikolla 16 (koko analyysipopulaatio)b | |||
| Tutkimus | BREEZE-AD4 | ||
| Hoitoryhmä | Lumelääkea | BARI 2 mga | BARI 4 mga |
| N | 93 | 185 | 92 |
EASI-75, vasteen saavuttaneet (%)c | 17,2
| 27,6
| 31,5**
|
IGA 0 tai 1, vasteen saavuttaneet (%)c,e | 9,7 | 15,1 | 21,7* |
| Kutinan NRS-pistemäärä, (≥ 4 pisteen parannus), vasteen saavuttaneet (%)c,f | 8,2 | 22,9* | 38,2** |
| DLQI-keskiarvon muutos (keskivirhe)d | -4,95 (0,752) | -6,57 (0,494) | -7,95* (0,705) |
BARI = Barisitinibi
* tilastollisesti merkitsevä verrattuna lumelääkkeeseen, ei korjattu kertautuvuuden suhteen
** tilastollisesti merkitsevä verrattuna lumelääkkeeseen, korjattu kertautuvuuden suhteen
a Kaikki potilaat käyttivät samanaikaisesti paikallista kortikosteroidihoitoa, ja kalsineuriinin estäjien paikallinen käyttö sallittiin.
b Koko analyysipopulaatio (FAS) käsitti kaikki satunnaistetut potilaat.
c Vastetta saavuttamattomien imputointi: Jos potilas sai varahoitoa tai hänestä puuttui tietoja, katsottiin, että hän ei saavuttanut vastetta.
d Varahoidon käytön tai lääkevalmisteen käytön pysyvän lopettamisen jälkeen kerätyt tiedot katsottiin puuttuviksi tiedoiksi. Pienimmän neliösumman keskiarvot perustuvat toistomittausten
sekamallin analyyseihin (Mixed Model with Repeated Measures, MMRM).
e Vasteen saavuttaminen määriteltiin tilanteeksi, jossa potilaan IGA-pistemäärä oli 0 tai 1 (”puhdas” tai ”lähes puhdas”) ja IGA‑pistemäärä pieneni ≥ 2 pisteen verran asteikolla 0–4.
f Tulokset esitetään niiltä potilailta, jotka soveltuivat arviointiin (potilaat, joiden lähtötason kutinan NRS-pistemäärä ≥ 4).
Pälvikalju aikuisilla
Kerran vuorokaudessa otettavan barisitinibin tehoa ja turvallisuutta arvioitiin yhdessä vaiheen II/III adaptiivisessa tutkimuksessa (BRAVE‑AA1) ja yhdessä vaiheen III tutkimuksessa (BRAVE‑AA2). BRAVE‑AA1‑tutkimuksen vaihetta III edustava osa ja vaiheen III BRAVE‑AA2‑tutkimus olivat satunnaistettuja, kaksoissokkoutettuja, lumekontrolloituja tutkimuksia. Ne kestivät 36 viikkoa, ja niihin kuului enintään 200 viikon jatkovaihe. Molemmissa vaiheen III tutkimuksissa potilaat satunnaistettiin suhteessa 2:2:3 saamaan lumelääkettä, barisitinibia 2 mg tai barisitinibia 4 mg. Tutkimuksiin soveltuvat potilaat olivat 18–60‑vuotiaita (miespotilaat) tai 18–70‑vuotiaita (naispotilaat), ja heillä oli parhaillaan vaikea pälvikaljuepisodi (pälvikalju kattoi ≥ 50 % päänahasta), joka oli kestänyt yli 6 kk. Tutkimukseen eivät soveltuneet potilaat, joiden meneillään oleva episodi oli kestänyt yli 8 vuotta, ellei pälvikaljuläiskien alueella ollut havaittu hiusten takaisinkasvua edellisten 8 vuoden aikana. Ainoat sallitut samanaikaiset pälvikaljuhoidot olivat finasteridi (tai muut 5‑alfareduktaasin estäjät), suun kautta otettava tai paikallisesti käytettävä minoksidiili sekä bimatoprostisilmätipat ripsien kasvun edistämiseen, jos annos oli tutkimukseenottohetkellä vakaa.
Molemmissa tutkimuksissa arvioitiin ensisijaisena päätetapahtumana niiden tutkittavien osuutta, joiden Severity of Alopecia Tool (SALT) ‑pistemäärä oli viikon 36 kohdalla ≤ 20 (päänahasta vähintään 80 % hiusten peittämää). Lisäksi molemmissa tutkimuksissa tarkasteltiin kliinikon nelipisteisellä asteikolla arvioimaa kulmakarvojen ja silmäripsien lähtöä (ClinRO Measure for Eyebrow Hair Loss™, ClinRO Measure for Eyelash Hair Loss™).
Lähtötilanteen tiedot
BRAVE‑AA1‑tutkimuksen vaihetta III edustavaan osaan ja vaiheen III BRAVE‑AA2‑tutkimukseen osallistui 1 200 aikuispotilasta. Kaikkien hoitoryhmien yhdistetyissä tiedoissa potilaiden ikäkeskiarvo oli 37,5 vuotta ja potilaista 61 % oli naisia. Pälvikaljun keskikesto ilmaantumisesta oli 12,2 vuotta ja meneillään olevan hiustenlähtöepisodin keskikesto oli 3,9 vuotta. Tutkimusten yhdistetty SALT‑pisteiden mediaani oli 96 (vastaa hiustenlähtöä 96 %:ssa päänahasta), ja noin 44 %:lla potilaista ilmoitettiin kaikkien ihokarvojen puuttuminen (alopecia universalis). Tutkimusten yhdistetyissä tiedoissa 69 %:lla potilaista kulmakarvojen lähtö oli lähtötilanteessa merkittävää tai täydellistä ja 58 %:lla potilaista silmäripsien lähtö oli merkittävää tai täydellistä, minkä määritelmänä oli kulmakarvoja ja silmäripsiä koskeva kliinikon ilmoittama ClinRO Measure -pistemäärä 2 tai 3. Noin 90 % potilaista oli saanut vähintään yhtä pälvikaljuhoitoa jossain vaiheessa ennen tutkimukseenottoa ja 50 % potilaista oli käyttänyt vähintään yhtä systeemistä immunosuppressanttia. Potilaista vain 4,3 %:n ilmoitettiin käyttäneen tutkimusten aikana sallittua samanaikaista pälvikaljunhoitoa.
Kliininen hoitovaste
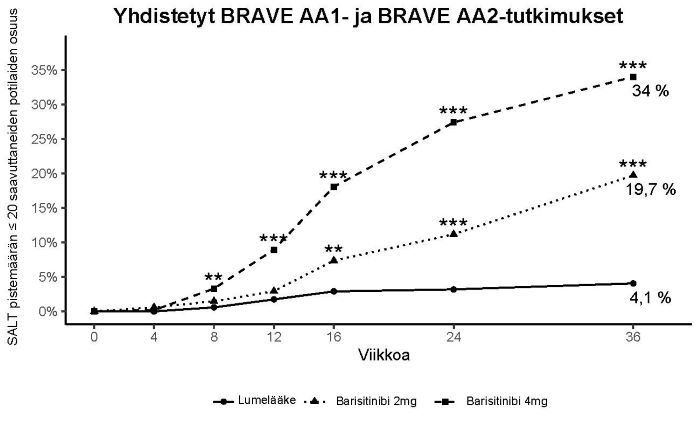
Molemmissa tutkimuksissa merkitsevästi suurempi osuus barisitinibi 4 mg kerran vuorokaudessa ‑ryhmään satunnaistetuista potilaista saavutti SALT‑pistemäärän ≤ 20 verrattuna lumelääkettä saaneisiin potilaisiin viikolla 36. BRAVE‑AA1‑tutkimuksessa SALT‑pistemäärä ≤ 20 saavutettiin aikaisimmillaan viikolla 8 ja BRAVE‑AA2‑tutkimuksessa aikaisimmillaan viikolla 12. Teho oli johdonmukainen useimpien toissijaisten päätetapahtumien kohdalla (taulukko 9). Kuvassa 2 on esitetty SALT‑pistemäärän ≤ 20 saavuttaneiden potilaiden osuus viikolle 36 asti.
Hoidon vaikutukset eri alaryhmissä (sukupuoli, ikä, paino, glomerulusten laskennallinen suodatusnopeus, etninen tausta, maantieteellinen sijainti, taudin vaikeusaste ja meneillään olevan pälvikaljuepisodin kesto) vastasivat koko tutkimuspopulaatiossa viikolla 36 havaittuja tuloksia.
| Taulukko 9. Barisitinibin teho yhdistetyissä tutkimuksissa viikolle 36 asti (viikon 36 yhdistetty tehopopulaatioa) | |||
|---|---|---|---|
| BRAVE‑AA1‑tutkimuksen (vaiheen II/III tutkimuksen vaihetta III edustavan osan) ja BRAVE‑AA2‑tutkimuksen (vaiheen III tutkimuksen) yhdistetyt tiedot* | |||
Lumelääke N = 345 | Barisitinibi 2 mg N = 340 | Barisitinibi 4 mg N = 515 | |
| SALT‑pistemäärä ≤ 20 viikolla 36 | 4,1 % | 19,7 %** | 34,0 %** |
| SALT‑pistemäärä ≤ 20 viikolla 24 | 3,2 % | 11,2 % | 27,4 %** |
| Kulmakarvojen lähtö, kliinikon ilmoittama tulos (ClinRO Measure for Eyebrow Hair Loss) 0 tai 1 viikolla 36 ja ≥ 2 pisteen parannus lähtötilanteestab | 3,8 % | 15,8 % | 33,0 %** |
| Silmäripsien lähtö, kliinikon ilmoittama tulos (ClinRO Measure for Eyelash Hair Loss) 0 tai 1 viikolla 36 ja ≥ 2 pisteen parannus lähtötilanteestab | 4,3 % | 12,0 % | 33,9 %** |
| Pälvikaljua koskevaksi muokatun Skindex16‑mittarin tunneosa‑alueen pistemäärän muutos, keskiarvo (keskivirhe)c | −11,33 (1,768) | −19,89 (1,788) | −23,81 (1,488) |
| Pälvikaljua koskevaksi muokatun Skindex16‑mittarin toimintakykyosa‑alueen pistemäärän muutos, keskiarvo (keskivirhe)c | −9,26 (1,605) | −13,68 (1,623) | −16,93 (1,349) |
a Viikon 36 yhdistetty tehopopulaatio: kaikki BRAVE‑AA1‑tutkimuksen vaihetta III edustavaan osaan ja BRAVE‑AA2‑tutkimukseen otetut potilaat.
* Yhdistetyn analyysin tulokset vastaavat yksittäisten tutkimusten tuloksia.
** Tilastollisesti merkitsevä, korjattu kertautuvuuden suhteen kunkin yksittäisen tutkimuksen graafisen testauksen suunnitelman mukaisesti.
b Potilaita, joiden kulmakarvojen lähtöä koskeva kliinikon ilmoittama tulos (ClinRO Measure for Eyebrow Hair Loss) oli lähtötilanteessa ≥ 2: 236 (lumelääke), 240 (barisitinibi 2 mg), 349 (barisitinibi 4 mg). Potilaita, joiden silmäripsien lähtöä koskeva kliinikon ilmoittama tulos (ClinRO Measure for Eyelash Hair loss) oli lähtötilanteessa ≥ 2: 186 (lumelääke), 200 (barisitinibi 2 mg), 307 (barisitinibi 4 mg). Molempien kliinikon ilmoittamien tulosten kohdalla käytetään nelipisteistä vasteasteikkoa (0–3), jolla pistemäärä 0 tarkoittaa ”ei karvojenlähtöä” ja 3 ”ei havaittavissa olevia kulmakarvoja/silmäripsiä”.
c Pälvikaljua koskevaksi muokatun Skindex16‑mittarin viikon 36 analyysin otoskoot ovat n = 256 (lumelääke), 249 (barisitinibi 2 mg), 392 (barisitinibi 4 mg).
Kuva 2: SALT‑pistemäärän ≤ 20 saavuttaneiden potilaiden osuus viikolle 36 asti
** p‑arvo, barisitinibi vs. lumelääke ≤ 0,01; *** p‑arvo, barisitinibi vs. lumelääke ≤ 0,001.
Teho viikkoon 52 asti
SALT‑pistemäärän ≤ 20 saavuttaneiden barisitinibihoitoa saaneiden potilaiden osuuden suureneminen jatkui viikon 36 jälkeen, ja osuus barisitinibi 4 mg ‑hoitoa saaneista potilaista oli viikon 52 kohdalla 39,0 %. Viikon 52 tulokset alaryhmistä, joiden määrittelyperusteena oli taudin vaikeusaste ja episodin kesto lähtötilanteessa, vastasivat viikon 36 tuloksia ja koko tutkimuspopulaatiossa havaittuja tuloksia.
Annoksen laskua arvioinut osatutkimus
BRAVE‑AA2‑tutkimuksessa potilaat, jotka olivat saaneet barisitinibia 4 mg kerran vuorokaudessa satunnaistamisesta lähtien ja jotka saavuttivat SALT‑pistemäärän ≤ 20 viikolla 52, satunnaistettiin uudelleen kaksoissokkoutetusti jatkamaan 4 mg kerran vuorokaudessa ‑hoitoa tai saamaan pienennettyä annosta 2 mg kerran vuorokaudessa. Tulosten mukaan vaste säilyi viikolle 76 asti 96 %:lla potilaista, jotka jatkoivat barisitinibi 4 mg ‑hoitoa, ja 74 %:lla potilaista, jotka satunnaistettiin uudelleen barisitinibi 2 mg ‑hoitoon.
Lastenreuma
Lastenreuman barisitinibihoidon kliininen kehitysohjelma koostui yhdestä päättyneestä vaiheen III avaintutkimuksesta (JUVE-BASIS) ja yhdestä meneillään olevasta pitkäaikaisesta avoimesta turvallisuutta koskevasta jatkotutkimuksesta (JUVE-X).
JUVE-BASIS oli enimmillään 44‑viikkoinen lumekontrolloitu tutkimus, johon sisältyi satunnaistettu, kaksoissokkoutettu vaihe, jossa osa potilaista siirtyi barisitinibista lumelääkkeeseen (double‑blind withdrawal, DBW). Tutkimuksessa arvioitiin kerran vuorokaudessa annostellun barisitinibin tehoa ja turvallisuutta vähintään 2- ja alle 18‑vuotiailla lastenreumaa sairastavilla potilailla, joilla vähintään yksi tavanomainen synteettinen tai biologinen tautiprosessiin vaikuttava reumalääke oli tuottanut riittämättömän vasteen tai ollut huonosti siedetty. Potilailla oli joko juveniili idiopaattinen polyartriitti (reumatekijäpositiivinen tai -negatiivinen), leviävää oligoartriittityyppiä edustava lastenreuma, entesiitteihin liittyvä lastenreuma tai lasten nivelpsoriaasi ILAR:in (International League of Associations for Rheumatology) kriteerien mukaisesti luokiteltuna. JUVE-BASIS‑tutkimukseen osallistuneet potilaat soveltuivat JUVE-X‑tutkimukseen.
JUVE-BASIS‑tutkimuksessa potilaat saivat avointa barisitinibihoitoa kerran vuorokaudessa noin 12 viikon ajan lähtötilanteesta. Potilaat, jotka olivat vähintään 2- ja alle 9-vuotiaita, saivat 2 mg päivittäin ja potilaat, jotka olivat vähintään 9- ja alle 18-vuotiaita, saivat 4 mg päivittäin, jotta saavutettiin vastaava altistuminen kuin aikuisilla 4 mg:n annoksella. Viikolla 12 hoitovaste arvioitiin potilaskohtaisesti (PedACR30‑kriteerien perusteella). Vähintään PedACR30‑vasteen saavuttaneet potilaat satunnaistettiin (suhteessa 1:1) saamaan lumelääkettä tai jatkamaan saman barisitinibiannoksen käyttöä 32 viikkoa kestäneessä kaksoissokkoutetussa, lumekontrolloidussa vaiheessa. Potilaille, jotka eivät saavuttaneet PedACR30‑vastetta, annettiin mahdollisuus osallistua JUVE-X‑tutkimukseen.
JUVE-BASIS‑tutkimuksen ensisijainen tehon päätetapahtuma oli taudin pahenemisvaiheen alkamiseen kulunut aika siinä vaiheessa, jolloin osa potilaista oli siirretty barisitinibista lumelääkkeeseen.
Lähtötilanteen tiedot
JUVE‑BASIS-tutkimukseen otettiin yhteensä 220 potilasta. Näistä potilaista 163 (74,4 %) soveltui satunnaistettaviksi DBW‑vaiheen barisitinibihoitoon (n = 82) tai lumehoitoon (n = 81). 144 potilaalla oli juveniili idiopaattinen polyartriitti, 16 potilaalla leviävää oligoartriittityyppiä edustava lastenreuma, 50 potilaalla entesiitteihin liittyvä idiopaattinen lastenreuma ja 10 potilaalla lasten nivelpsoriaasi.
JUVE-BASIS‑tutkimuksessa ikäkeskiarvo oli 13 vuotta (keskihajonta 3,0) ja 69,1 % tutkittavista oli tyttöjä. Potilaiden määrä eri ikäryhmissä oli seuraava: vähintään 2- ja alle 6-vuotiaat: n = 6; vähintään 6- ja alle 9-vuotiaat: n = 9; vähintään 9- ja alle 12-vuotiaat: n = 30, ja vähintään 12- ja alle 18-vuotiaat: n = 175.
Lastenreumadiagnoosista kuluneen ajan keskiarvo (kaikki tutkimuksen potilaat) oli 4 vuotta. Samanaikaisten hoitojen käyttö DBW‑vaiheessa oli hoitoryhmissä samaa luokkaa (yleisimpiin samanaikaisiin tavanomaisiin, synteettisiin tautiprosessiin vaikuttaviin reumalääkkeisiin kuuluivat metotreksaatti, sulfasalatsiini ja leflunomidi). Yhteensä 127 potilasta (57,7 %) käytti lähtötilanteessa metotreksaattia.
Kliininen vaste
JUVE-BASIS‑tutkimuksessa taudin pahenemisvaiheen ilmaantumiseen kulunut aika oli barisitinibia saaneiden potilaiden ryhmässä merkitsevästi pidempi kuin lumelääkettä saaneilla (kuva 3). Lisäksi barisitinibia saaneista potilaista useampi saavutti PedACR‑arvon 30/50/70/90/100 lumelääkkeeseen verrattuna vaiheessa, jolloin osa potilaista oli siirretty barisitinibista lumelääkkeeseen.
Kuva 3. Taudin pahenemisvaiheen alkamiseen kulunut aika siinä vaiheessa, jolloin osa potilaista oli siirretty barisitinibista lumelääkkeeseen (DBW-vaihe)
Ei ol. = ei oleellinen; HR = riskitiheyssuhde; lv = luottamusväli
*a HR stratifioitiin lastenreumaluokkien mukaan (polyartriitti ja leviävää oligoartriittityyppiä edustava lastenreuma vs. entesiitteihin liittyvä lastenreuma ja lasten nivelpsoriaasi).
*b P-arvo perustuu logrank-testiin, joka stratifioitiin lastenreumaluokkien mukaan (polyartriitti ja leviävää oligoartriittityyppiä edustava lastenreuma vs. entesiitteihin liittyvä lastenreuma ja lasten nivelpsoriaasi).
Pahenemisvaiheen ilmaantumiseen kulunut aika ja PedACR-tulokset olivat yleisesti yhdenmukaiset kaikissa lastenreuman alatyypeissä ja taustaominaisuuksien mukaan arvioituna (ikä, maantieteellinen alue, paino, aiempi biologisten lääkkeiden käyttö, samanaikainen metotreksaatti- tai kortikosteroidihoito) sekä koko tutkimuspopulaatioon verrattuna.
Atooppinen ihottuma pediatrisilla potilailla
Barisitinibin tehoa ja turvallisuutta yhdessä paikalliskortikosteroidien kanssa arvioitiin yhdessä satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa, 16 viikon pituisessa vaiheen III tutkimuksessa (BREEZE-AD-PEDS). Tutkimukseen osallistui 483 potilasta, joilla oli keskivaikea tai vaikea atooppinen ihottuma, jonka määritelmänä olivat IGA-pistemäärä ≥ 3, EASI-pistemäärä ≥ 16 ja BSA-arvo ≥ 10 %. Tutkimukseen soveltuvat potilaat olivat 2– alle 18-vuotiaita, paikalliset lääkevalmisteet olivat tuottaneet heillä riittämättömän vasteen tai olleet huonosti siedettyjä ja heille harkittiin systeemistä hoitoa. Kaikille potilaille määrättiin samanaikaisesti mietoa tai keskivahvaa paikalliskortikosteroidia, ja potilaille sallittiin paikallishoito kalsineuriinin estäjällä tutkimuksen aikana. Potilaat satunnaistettiin suhteessa 1:1:1:1 saamaan lumehoitoa tai tutkittavaa pieni-, keskisuuri- tai suuriannoksista barisitinibihoitoa (jolloin saavutettiin vastaava altistus kuin aikuisilla [1 mg:n, 2 mg:n ja 4 mg:n annoksilla] atooppisen ihottuman hoidossa). Tutkimukseen sisältyy meneillään oleva pitkäkestoinen 4 vuoden jatkovaihe.
Lähtötilanteen tiedot
Kaikkien hoitoryhmien yhdistetyissä tiedoissa potilaista 76 % oli valkoihoisia, 15 % oli aasialaisia, 3 % oli tummaihoisia ja 50 % oli tyttöjä. Ikäkeskiarvo oli 12 vuotta. Potilaista 72 % oli vähintään 10-vuotiaita ja 28 % alle 10-vuotiaita. Populaatiosta 14 % oli enintään 6-vuotiaita (6 vuotta [N = 28], 5 vuotta [N = 11], 4 vuotta [N = 16], 3 vuotta [N = 8], 2 vuotta [N = 5]). Tässä tutkimuksessa 38 %:lla potilaista lähtötilanteen IGA-pistemäärä oli 4 (vaikea atooppinen ihottuma), ja 42 % potilaista oli saanut aiempaa systeemistä hoitoa atooppiseen ihottumaan. Lähtötilanteessa EASI-pistemäärän vaihteluväli oli 12,2−70,8 ja kutinan NRS-pistemäärän viikoittainen keskiarvo vähintään 10-vuotiailla oli 5,5 (keskihajonta = 2,6).
Kliininen vaste
Tilastollisesti merkitsevästi suurempi osuus 4 mg:n barisitinibiannosta vastaavan altistuksen ryhmään satunnaistetuista potilaista saavutti IGA-pistemäärän 0 tai 1 (ensisijainen päätetapahtuma), EASI75-vasteen tai kutinan lievittymisen ≥ 4 pisteellä (kutinan NRS-arvo) verrattuna lumelääkkeeseen viikolla 16 (taulukko 10). Kuvassa 4 esitetään IGA-pistemäärän 0 tai 1 saavuttamiseen kulunut aika.
Hoidon vaikutukset eri alaryhmissä (määrittelyperusteet: paino, ikä, sukupuoli, etninen tausta, taudin vaikeusaste ja aiemmat hoidot, ml. immunosuppressanttihoito) vastasivat koko tutkimuspopulaatiossa havaittuja tuloksia.
| Taulukko 10. Barisitinibin teho pediatrisilla potilailla viikolla 16a | ||
Tutkimus | BREEZE-AD-PEDS | |
Hoitoryhmä | Lumelääke | BARI 4 mg ‑annosta vastaava altistus |
N | 122 | 120 |
IGA 0 tai 1, vasteen saavuttaneet (%)b,c | 16,4 | 41,7** |
EASI75, vasteen saavuttaneet (%)c | 32,0 | 52,5** |
Kutinan NRS (≥ 4 pisteen parannus), vasteen saavuttaneet (%)c,d | 16,4 | 35,5** |
BARI = barisitinibi
** Tilastollisesti merkitsevä verrattuna lumelääkkeeseen, korjattu kertautuvuuden suhteen.
a Lähtöryhmien mukainen (intent to treat, ITT) populaatio (kaikki satunnaistetut potilaat).
b Vasteen saavuttaminen määriteltiin tilanteeksi, jossa potilaan IGA-pistemäärä oli 0 tai 1 (”puhdas” tai ”lähes puhdas”) ja IGA-pistemäärä pieneni ≥ 2 pisteen verran asteikolla 0–4.
c Vastetta saavuttamattomien imputointi: Jos potilas sai varahoitoa tai hänestä puuttui tietoja, katsottiin, että hän ei saavuttanut vastetta.
d Esitetyt tulokset koskevat arviointikelpoisten potilaiden osajoukkoa (≥ 10-vuotiaat potilaat, joilla kutinan NRS-pistemäärä lähtötilanteessa ≥ 4; BARI 4 mg ‑annosta vastaava altistus: N = 62; lumelääke: N = 55).
Kuva 4. IGA-pistemäärän 0 tai 1 sekä ≥ 2 pisteen paraneminen saavuttamiseen kulunut aika pediatrisilla potilailla viikolle 16 asti
NRI = vastetta saavuttamattomien imputointi; * p < 0,05; ** p < 0,01; *** p < 0,001 vs. lumelääke (nimellinen p-arvo; logistinen regressioanalyysi); † Tilastollisesti merkitsevä ja korjattu kertautuvuuden suhteen
Kutinan NRS-arvo parani ≥ 4 pistettä merkitsevästi suuremmalla osuudella 4 mg:n barisitinibiannosta vastaavan altistuksen ryhmään satunnaistetuista verrattuna lumelääkettä saaneisiin jo viikolla 4 (korjattu kertautuvuuden suhteen).
Samanaikaisen paikalliskortikosteroidihoidon tarve väheni. Tämän osoitti käytettyjen paikalliskortikosteroidien grammamäärän pienenemän mediaani 4 mg:n barisitinibiannosta vastaavan altistuksen ryhmässä verrattuna lumeryhmään 16 viikon aikana ja paikalliskortikosteroidittomien päivien suurempi mediaanimäärä 4 mg:n barisitinibiannosta vastaavan altistuksen ryhmässä (25 päivää) verrattuna lumeryhmään (11 päivää) 16 viikon aikana.
Pälvikalju nuorilla potilailla
Kerran vuorokaudessa annostellun barisitinibin tehoa ja turvallisuutta arvioitiin yhdessä vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa 36 viikon tutkimuksessa (BRAVE‑AA‑PEDS). Potilaat satunnaistettiin saamaan lumelääkettä, barisitinibia 2 mg tai 4 mg suhteessa 1:1:1. Tutkimukseen soveltuvat potilaat olivat vähintään 12-vuotiaita ja alle 18‑vuotiaita nuoria, joiden paino oli vähintään 30 kg ja joilla oli parhaillaan yli 6 kuukautta kestänyt vaikea pälvikaljuepisodi (pälvikalju kattoi ≥ 50 % päänahasta).
Tutkimukseen eivät soveltuneet potilaat, joiden meneillään oleva episodi oli kestänyt yli 8 vuotta, ellei pälvikaljuläiskien alueella ollut havaittu hiusten takaisinkasvua edellisten 8 vuoden aikana. Ainoat sallitut samanaikaiset pälvikaljuhoidot olivat suun kautta otettava tai paikallisesti käytettävä minoksidiili ja bimatoprostisilmätipat ripsien kasvun edistämiseen, jos annos oli tutkimukseenottohetkellä vakaa. Tutkimukseen sisältyy käynnissä oleva pitkäaikainen jatko‑osa, jonka kesto on enintään 2 vuotta.
Tutkimuksen ensisijainen päätetapahtuma oli niiden tutkittavien osuus, jotka saavuttivat viikon 36 kohdalla SALT (Severity of Alopecia Tool) ‑pistemäärän ≤ 20 (päänahasta vähintään 80 % hiusten peittämää). Lisäksi tutkimuksessa tarkasteltiin kliinikon nelipisteisellä asteikolla arvioimaa kulmakarvojen ja silmäripsien lähtöä (ClinRO Measure for Eyebrow Hair Loss™, ClinRO Measure for Eyelash Hair Loss™).
Lähtötilanteen tiedot
BRAVE‑AA‑PEDS‑tutkimuksen lumekontrolloituun nuorilla toteutettavaan osaan osallistui 257 nuorta potilasta. Kaikkien hoitoryhmien yhdistetyissä tiedoissa potilaiden ikäkeskiarvo oli 14,7 vuotta, ja potilaista 49 % oli tyttöjä. Pälvikaljun keskikesto ilmaantumisesta oli 6,4 vuotta ja meneillään olevan hiustenlähtöepisodin keskikesto oli 3,2 vuotta. Tutkimusten yhdistetty SALT-pisteiden mediaani oli 100 (vastaa hiustenlähtöä 100 %:ssa päänahasta), ja noin 54 %:lla potilaista ilmoitettiin kaikkien ihokarvojen puuttuminen (alopecia universalis). Potilaista 65 %:lla kulmakarvojen lähtö oli lähtötilanteessa merkittävää tai täydellistä, ja 57 %:lla potilaista silmäripsien lähtö oli merkittävää tai täydellistä, minkä määritelmänä oli kulmakarvoja ja silmäripsiä koskeva kliinikon ilmoittama ClinRO Measure -pistemäärä 2 tai 3. Kaikki potilaat olivat saaneet vähintään yhtä pälvikaljuhoitoa jossain vaiheessa ennen tutkimukseenottoa ja 52 % potilaista oli käyttänyt vähintään yhtä systeemistä hoitoa. Potilaista vain 3,1 %:n ilmoitettiin käyttäneen tutkimusten aikana sallittua samanaikaista pälvikaljunhoitoa.
Kliininen hoitovaste
Merkitsevästi suurempi osuus barisitinibi 4 mg kerran vuorokaudessa -ryhmään satunnaistetuista potilaista saavutti SALT-pistemäärän ≤ 20 verrattuna lumelääkettä saaneisiin potilaisiin viikolla 36, ja ero oli havaittavissa jo viikosta 12 alkaen. Barisitinibi 4 mg -annoksen teho oli johdonmukainen kaikkien keskeisten toissijaisten päätetapahtumien kohdalla (taulukko 11), mikä ilmeni numeerisesti valtaosassa toissijaisista päätetapahtumista, mukaan lukien kliinikon ja potilaan raportoimat tulokset. Kuvassa 5 on esitetty SALT-pistemäärän ≤ 20 saavuttaneiden potilaiden osuus viikolle 36 asti.
Hoidon vaikutukset eri alaryhmissä (sukupuoli, etninen tausta, maantieteellinen sijainti, taudin vaikeusaste ja meneillään olevan pälvikaljuepisodin kesto) vastasivat koko tutkimuspopulaatiossa viikolla 36 havaittuja tuloksia.
| Taulukko 11. Barisitinibin teho nuorilla potilailla viikolle 36 asti | |||
| BRAVE-AA-PEDS | |||
Lumelääke N = 88 | Barisitinibi 2 mg N = 84 | Barisitinibi 4 mg N = 85 | |
| SALT-pistemäärä ≤ 20 viikolla 36 | 4,5 % | 27,4 %** | 42,4 %** |
| SALT-pistemäärä ≤ 20 viikolla 24 | 3,4 % | 16,7 %** | 31,8 %** |
| SALT-pistemäärä ≤ 10 viikolla 36 | 2,3 % | 21,4 %** | 36,5 %** |
| Kulmakarvojen lähtö, kliinikon ilmoittama tulos (ClinRO Measure for Eyebrow Hair Loss) 0 tai 1 viikolla 36 ja ≥ 2 pisteen parannus lähtötilanteestaa | 0 % | 24,1 %* | 50,0 %* |
| Silmäripsien lähtö, kliinikon ilmoittama tulos (ClinRO Measure for Eyelash Hair Loss) 0 tai 1 viikolla 36 ja ≥ 2 pisteen parannus lähtötilanteestaa | 14,0 % | 25,5 % | 42,9 %* |
| Pälvikaljua koskevaksi muokatun Skindex-16-mittarin tunneosa-alueen pistemäärän muutos, LSM (keskivirhe)b | -3,98 (2,63) | -12,87 (2,55)* | -18,22 (2,60)* |
ClinRO = kliinikon raportoima tulosmuuttuja; LSM = pienimmän neliösumman keskiarvo
* Tilastollisesti merkitsevä verrattuna lumelääkkeeseen ilman monivertailukorjausta.
** Tilastollisesti merkitsevä verrattuna lumelääkkeeseen monivertailukorjauksella.
ᵃ Potilaat, joilla kulmakarvojen lähtöä kuvaava ClinRO‑pistemäärä oli ≥ 2 lähtötilanteessa: 60 (lumelääke), 54 (barisitinibi 2 mg), 54 (barisitinibi 4 mg). Potilaat, joilla ripsien lähtöä kuvaava ClinRO‑pistemäärä oli ≥ 2 lähtötilanteessa: 50 (lumelääke), 47 (barisitinibi 2 mg), 49 (barisitinibi 4 mg). Molemmissa ClinRO‑mittareissa käytetään nelipisteistä vasteasteikkoa (0–3), jolla pistemäärä 0 tarkoittaa ”ei karvojenlähtöä” ja 3 ”ei havaittavissa olevia kulmakarvoja/silmäripsiä”.
ᵇ Pälvikaljua koskevaksi muokatun Skindex-16-mittarin viikon 36 analyysin otoskoot ovat: n = 87 (lumelääke), 84 (barisitinibi 2 mg), 85 (barisitinibi 4 mg).
Kuva 5: SALT-pistemäärän ≤ 20 saavuttaneiden nuorten potilaiden osuus viikolle 36 asti
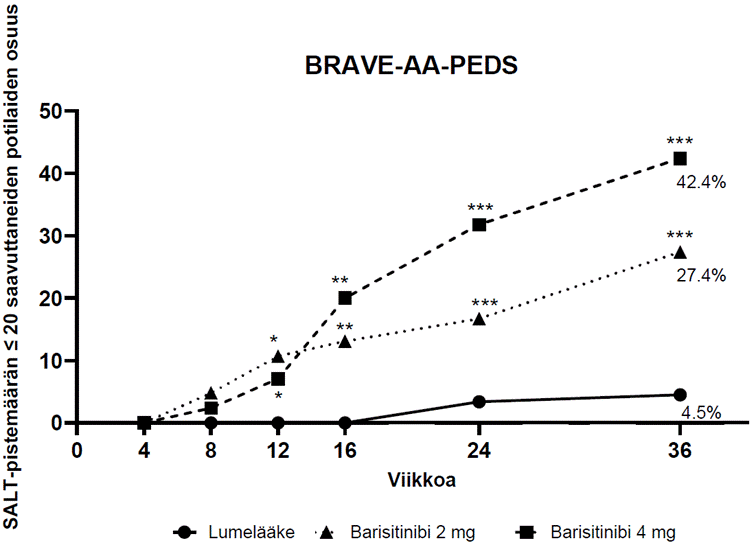
*p‑arvo, barisitinibi vs. lumelääke ≤ 0,05; **p‑arvo, barisitinibi vs. lumelääke ≤ 0,01, ***p‑arvo, barisitinibi vs. lumelääke ≤ 0,001.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset barisitinibin käytöstä kroonisen idiopaattisen artriitin ja pälvikaljun hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Barisitinibin tehoa 12 mg/vrk asti on arvioitu 71 potilaalla, joilla on CANDLE (krooninen atyyppinen neutrofiilinen dermatoosi, johon liittyy lipodystrofiaa ja lämmönnousua, n = 10), CANDLEen liittyvät sairaudet (CANDLE RC, n = 9), SAVI (interferonigeenin stimulaattoriin liittyvä vaskulopatia, Stimulator of interferon gene-Associated Vasculopathy, joka alkaa vauvaiässä, n = 8), nuorten dermatomyosiitti (JDM, n = 5) ja Aicardi Goutièresin oireyhtymä (AGS, n = 39). Kokonaispotilasaltistusvuosia oli 251. Metodologisten puutteiden vuoksi ei voitu tehdä varmoja johtopäätöksiä barisitinibin tehosta näillä potilailla. Vaikka turvallisuusmallit osoittivat samankaltaisuutta aikuisten käyttöaiheiden kanssa, haittatapahtumien esiintymistiheys oli yleensä korkeampi. AGS-populaatiossa havaittiin kolme kuolemaa. On epäselvää, liittyivätkö nämä kuolemat barisitinibihoitoon.
Barisitinibin tehoa ja turvallisuutta arvioitiin 29 potilaalla, joiden ikä oli vähintään 2- ja alle 18-vuotta, ja joilla oli aktiivinen lastenreumaan liittyvä uveiitti tai krooninen anteriorinen vasta-ainepositiivinen uveiitti. Potilaille, joilla oli riittämätön vaste metotreksaatille (n = 10) annettiin barisitinibia (n = 5) tai adalimumabia (n = 5). Kaikille potilaille, joilla oli riittämätön vaste biologiselle taudin etenemiseen vaikuttavalle reumalääkkeelle (n = 19), annettiin barisitinibia. Barisitinibiannos oli 2 mg kerran vuorokaudessa vähintään 2- ja alle 9-vuotiaille potilaille ja 4 mg kerran vuorokaudessa vähintään 9- ja alle 18-vuotiaille potilaille. Adalimumabiannos oli 20 mg (jos potilas painoi < 30 kg) tai 40 mg (jos potilas painoi ≥ 30 kg) kahden viikon välein.
Ensisijainen päätetapahtuma oli niiden potilaiden osuus, joilla tulehdustaso (etukammion solut) laski SUN-kriteerien (standardisation of uveitis nomenclature) mukaan 2 askelta tai nollaan viikkoon 24 mennessä silmässä, jossa tilanne oli lähtötilanteessa vaikein. Kahdeksan (33,3 %) potilasta sai vasteen barisitinibille (7 potilasta ryhmästä, jonka potilailla oli biologiselle taudin etenemiseen vaikuttavalle reumalääkkeelle riittämätön vaste ja 1 potilas ryhmästä, jonka potilailla oli metotreksaatille riittämätön vaste), mutta hoitovaste kahden kohortin välillä ei ollut tilastollisesti merkitsevä.
Farmakokinetiikka
Kun barisitinibi annettiin suun kautta, todettiin, että systeeminen altistus suureni suhteessa annokseen hoitoannosalueella. Barisitinibin farmakokinetiikka on ajan suhteen lineaarinen.
Imeytyminen
Suun kautta otettu barisitinibi imeytyy nopeasti, ja sen tmax-ajan mediaani on noin 1 tunti (vaihteluväli 0,5–3,0 h) ja absoluuttinen biologinen hyötyosuus noin 79 % (variaatiokerroin [CV] = 3,94 %). Farmakokineettiset tutkimukset ovat osoittaneet, että barisitinibitabletit ja oraalisuspensio ovat bioekvivalentteja AUC- ja Cmax-arvojen perusteella.
Ruoan nauttiminen tablettien kanssa pienensi altistusta jopa 14 %, pienensi Cmax-arvoa jopa 18 % ja pidensi tmax-aikaa 0,5 h. Vastaavasti oraalisuspension samanaikainen anto ruoan kanssa johti Cmax-arvon pienenemiseen 33 %:lla ja tmax-ajan viivästymiseen 2 tunnilla paastotilaan verrattuna. Lääkkeen anto aterian yhteydessä ei vaikuttanut kliinisesti merkittävästi altistukseen.
Jakautuminen
Laskimoinfuusiona annetun lääkkeen jakautumistilavuuden keskiarvo oli 76 l, mikä kuvaa barisitinibin jakautuvan kudoksiin. Noin 50 % barisitinibista sitoutuu plasman proteiineihin.
Biotransformaatio
Barisitinibin metabolia on CYP3A4-välitteistä, ja on todettu, että alle 10 % annoksesta biotransformoituu. Plasmassa ei todettu kvantifioitavissa olevia metaboliitteja. Kliinisen farmakologian tutkimuksessa barisitinibi erittyi lähinnä muuttumattomana vaikuttavana aineena virtsaan (69 %) ja ulosteeseen (15 %), ja vain 4 vähäistä hapettumismetaboliittia todettiin (3 virtsassa [noin 5 % annoksesta] ja 1 ulosteessa [noin 1 % annoksesta]). Barisitinibi on CYP3A4:n, OAT3:n, P-gp:n, BCRP:n ja MATE2-K:n substraatti ja saattaa olla kliinisesti merkittävä OCT1-kuljettajaproteiinien estäjä in vitro; (ks. kohta Yhteisvaikutukset). Barisitinibi ei estä kuljettajaproteiinien OAT1, OAT2, OAT3, OCT2, OATP1B1, OATP1B3, BCRP, MATE1 ja MATE2-K toimintaa kliinisesti merkittävissä konsentraatiossa.
Eliminaatio
Barisitinibin tärkein eliminaatioreitti on glomerulussuodatuksen ja aktiivisen sekreetion kautta OAT3-, P-gp-, BCRP- ja MATE2-K-välitteisesti tapahtuva munuaispuhdistuma. Kliinisen farmakologian tutkimuksessa noin 75 % annetusta annoksesta erittyi virtsaan ja noin 20 % annoksesta poistui ulosteen mukana.
Nivelreumapotilailla näennäisen puhdistuman (CL/F) keskiarvo oli 9,42 l/h (CV = 34,3 %) ja puoliintumisajan keskiarvo 12,5 h (CV = 27,4 %). Vakaassa tilassa Cmax on nivelreumapotilailla 1,4-kertaisesti suurempi ja AUC-arvo 2,0-kertaisesti suurempi kuin terveillä henkilöillä.
Atooppista ihottumaa sairastaneilla potilailla näennäisen puhdistuman (CL/F) keskiarvo oli 11,2 l/h (CV = 33,0 %) ja puoliintumisajan keskiarvo 12,9 h (CV = 36,0 %). Vakaassa tilassa Cmax ja AUC ovat atooppista ihottumaa sairastavilla potilailla 0,8-kertaiset verrattuna nivelreumapotilailla todettuihin arvoihin.
Pälvikaljupotilailla näennäisen puhdistuman (CL/F) keskiarvo oli 11,0 l/h (CV = 36,0 %) ja puoliintumisajan keskiarvo 15,8 h (CV = 35,0 %). Vakaassa tilassa Cmax ja AUC ovat pälvikaljupotilailla 0,9-kertaiset verrattuna nivelreumapotilailla todettuihin arvoihin.
Munuaisten vajaatoiminta
Munuaistoiminnan todettiin vaikuttavan merkitsevästi barisitinibialtistukseen. AUC-arvo oli lievää munuaisten vajaatoimintaa sairastavilla keskimäärin 1,41-kertainen (90 % luottamusväli [lv] 1,15‑1,74) ja keskivaikeaa munuaisten vajaatoimintaa sairastavilla keskimäärin 2,22-kertainen (90 % lv 1,81‑2,73) verrattuna henkilöihin, joiden munuaistoiminta oli normaali. Cmax-arvo oli lievää munuaisten vajaatoimintaa sairastavilla keskimäärin 1,16-kertainen (90 % lv 0,92–1,45) ja keskivaikeaa munuaisten vajaatoimintaa sairastavilla keskimäärin 1,46-kertainen (90 % lv 1,17–1,83) verrattuna henkilöihin, joiden munuaistoiminta oli normaali. Annossuositukset, ks. kohta Annostus ja antotapa.
Maksan vajaatoiminta
Lievä tai keskivaikea maksan vajaatoiminta ei vaikuttanut kliinisesti merkittävästi barisitinibin farmakokinetiikkaan. Barisitinibin käyttöä ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla.
Iäkkäät
Vähintään 65 vuoden tai vähintään 75 vuoden ikä ei vaikuta barisitinibialtistukseen (Cmax- ja AUC-arvoihin).
Pediatriset potilaat
Farmakokinetiikka lastenreumaa sairastavilla pediatrisilla potilailla
Puoliintumisaika vähintään 2- ja alle 18-vuotiailla pediatrisilla potilailla oli 8–9 tuntia.
Altistuminen pediatrisilla potilailla, jotka painoivat alle 30 kg ja vähintään 30 kg: Potilailla, jotka painoivat alle 30 kg ja joiden ikäkeskiarvo oli 8,1 vuotta (vaihteluväli 2,0-16,0 vuotta), AUC-arvon keskiarvo oli 381 h*ng/ml (CV = 76 %) ja Cmax-arvon keskiarvo 62,1 ng/ml (CV = 39 %). Potilailla, jotka painoivat vähintään 30 kg ja joiden ikäkeskiarvo oli 14,1 vuotta (vaihteluväli 9,0-17,0 vuotta), AUC-arvon keskiarvo oli 438 h*ng/ml (CV = 68 %) ja Cmax-arvon keskiarvo 60,7 ng/ml (CV = 30 %).
Altistuminen pediatrisilla potilailla, jotka painoivat vähintään 10 kg ja alle 20 kg ja vähintään 20 kg ja alle 30 kg: Potilailla, jotka painoivat vähintään 10 kg ja alle 20 kg ja joiden ikäkeskiarvo oli 5,1 vuotta (vaihteluväli 2,0–8,0 vuotta), AUC-arvon keskiarvo oli 458 h*ng/ml (CV = 81 %) ja Cmax-arvon keskiarvo 77,6 ng/ml (CV = 38 %). Potilailla, jotka painoivat vähintään 20 kg ja alle 30 kg ja joiden ikäkeskiarvo oli 10,3 vuotta (vaihteluväli 6,0–16,0 vuotta), AUC-arvon keskiarvo oli 327 h*ng/ml (CV = 66 %) ja Cmax-arvon keskiarvo 51,2 ng/ml (CV = 22 %).
Farmakokinetiikka atooppista ihottumaa sairastavilla pediatrisilla potilailla
Puoliintumisajan keskiarvo 2-vuotiailla ja alle 18‑vuotiailla pediatrisilla potilailla oli 13–18 tuntia.
Altistus pediatrisilla potilailla, jotka painoivat alle 30 kg tai vähintään 30 kg: Potilailla, jotka painoivat alle 30 kg ja joiden ikäkeskiarvo oli 6,4 vuotta (vaihteluväli 2,0–11,1 vuotta), AUC-arvon keskiarvo oli 404 h*ng/ml (CV = 78 %) ja Cmax-arvon keskiarvo 60,4 ng/ml (CV = 28 %). Potilailla, jotka painoivat vähintään 30 kg ja joiden ikäkeskiarvo oli 13,5 vuotta (vaihteluväli 6,2–17,9 vuotta), AUC-arvon keskiarvo oli 529 h*ng/ml (CV = 102 %) ja Cmax-arvon keskiarvo 57,0 ng/ml (CV = 42 %).
Altistus pediatrisilla potilailla, jotka painoivat vähintään 10 kg mutta alle 20 kg tai vähintään 20 kg mutta alle 30 kg: Potilailla, jotka painoivat vähintään 10 kg mutta alle 20 kg ja joiden ikäkeskiarvo oli 4,8 vuotta (vaihteluväli 2,0–6,9 vuotta), AUC-arvon keskiarvo oli 467 h*ng/ml (CV = 80 %) ja Cmax-arvon keskiarvo 73,4 ng/ml (CV = 21 %). Potilailla, jotka painoivat vähintään 20 kg mutta alle 30 kg ja joiden ikäkeskiarvo oli 7,5 vuotta (vaihteluväli 4,8–11,1 vuotta), AUC-arvon keskiarvo oli 363 h*ng/ml (CV = 72 %) ja Cmax-arvon keskiarvo 52,0 ng/ml (CV = 21 %).
Farmakokinetiikka pälvikaljua sairastavilla nuorilla potilailla
Puoliintumisajan keskiarvo vähintään 12-vuotiailla ja alle 18-vuotiailla nuorilla potilailla oli noin 10 tuntia.
Altistus nuorilla vähintään 12-vuotiailla ja alle 18-vuotiailla potilailla, jotka painoivat vähintään 30 kg: Potilailla, joiden ikäkeskiarvo oli 14,7 vuotta (vaihteluväli 12,0–18,0 vuotta), AUC-arvon keskiarvo oli 334 h*ng/ml (CV = 45 %) ja Cmax-arvon keskiarvo 52,9 ng/ml (CV = 23 %).
Potilaiden muut ominaisuudet
Paino, sukupuoli, ikä, rotu ja etninen tausta eivät vaikuttaneet kliinisesti merkittävästi barisitinibin farmakokinetiikkaan aikuispotilailla. Potilaiden ominaisuuksien keskimääräinen vaikutus farmakokinetiikan parametreihin (AUC- ja Cmax-arvoihin) oli yleensä barisitinibin farmakokinetiikan potilaskohtaisen vaihtelun puitteissa. Annosta ei siis tarvitse muuttaa näiden ominaisuuksien perusteella.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, geenitoksisuutta ja karsinogeenisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Hiirellä, rotalla ja koiralla todettiin lymfosyytti-, eosinofiili- ja basofiilimäärien laskua ja immuunijärjestelmän elinten/kudosten imukudoskatoa. Koiralla todettiin demodikoosiin liittyviä opportunistisia infektioita altistuksilla, jotka olivat ihmisen altistukseen nähden noin 7-kertaisia. Hiirellä, rotalla ja koiralla todettiin veren punasoluarvojen laskua, kun altistus oli noin 6–36 kertaa ihmisen altistuksen suuruinen. Rintalastan kasvulevyn degeneraatiota todettiin joillakin koirilla. Sen ilmaantuvuus oli pieni ja sitä esiintyi myös verrokkieläimillä, mutta sen vaikeusasteessa todettiin annosvastesuhde. Toistaiseksi ei tiedetä, onko ilmiöllä kliinistä merkitystä.
Rotalla ja kaniinilla tehdyissä lisääntymistoksikologian tutkimuksissa barisitinibin todettiin pienentävän sikiöiden kasvua/painoa (ihmisen altistukseen nähden noin 10-kertaisella altistuksella) ja aiheuttavan luuston epämuodostumia (ihmisen altistukseen nähden noin 39-kertaisella altistuksella). Sikiöhaittoja ei todettu, kun altistus oli AUC-arvon perusteella 2-kertainen verrattuna ihmisen altistukseen.
Uros- ja naarasrotilla toteutetussa yhdistetyssä hedelmällisyystutkimuksessa barisitinibi huononsi yleisiä parittelutuloksia (heikentyneet hedelmällisyys- ja hedelmöittymisindeksit). Naarasrotilla todettiin keltarauhasten ja implantaatiokohtien määrän vähenemistä, implantaatiota edeltävien alkiokuolemien lisääntymistä ja/tai haittoja, jotka kohdistuivat alkioiden eloonjäämiseen kohdussa. Siittiötuotantoon kohdistuneita vaikutuksia ei todettu (histopatologisessa arviossa), eivätkä urosrottien siemenneste/siittiöpäätetapahtumat muuttuneet. Yleisten parittelutulosten huononeminen johtui siis todennäköisesti aineen vaikutuksista naaraisiin.
Barisitinibia erittyi imettävien rottien maitoon. Pre- ja postnataalista kehitystä koskeneessa tutkimuksessa todettiin pentujen painon laskua 4 kertaa ihmisen altistuksen suuruisella altistuksella ja postnataalisen kuolleisuuden lisääntymistä 21 kertaa ihmisen altistuksen suuruisilla altistuksilla.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
mikrokiteinen selluloosa
kroskarmelloosinatrium
magnesiumstearaatti
mannitoli
Kalvopäällyste
punainen rautaoksidi (E172)
lesitiini (soija) (E322)
makrogoli
poly(vinyylialkoholi)
talkki
titaanidioksidi (E171)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
OLUMIANT tabletti, kalvopäällysteinen
2 mg (L:ei) 28 fol (838,77 €)
4 mg (L:ei) 28 fol (838,77 €)
PF-selosteen tieto
Olumiant 1 mg kalvopäällysteinen tabletti
Polyvinyylikloridi/polyeteeni/polyklooritrifluorieteeni–alumiini-läpipainopakkaukset pahvipakkauksissa, joissa on 14 tai 28 kalvopäällysteistä tablettia.
Yksittäispakatut polyvinyylikloridi/alumiini/orientoitu polyamidi–alumiini-läpipainopakkaukset pahvipakkauksissa, joissa on 28 x 1 kalvopäällysteistä tablettia.
Olumiant 2 mg ja 4 mg kalvopäällysteinen tabletti
Polyvinyylikloridi/polyeteeni/polyklooritrifluorieteeni–alumiini-läpipainopakkaukset pahvipakkauksissa, joissa on 14, 28, 35, 56, 84 tai 98 kalvopäällysteistä tablettia.
Yksittäispakatut polyvinyylikloridi/alumiini/orientoitu polyamidi–alumiini-läpipainopakkaukset pahvipakkauksissa, joissa on 28 x 1 tai 84 x 1 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Olumiant 1 mg kalvopäällysteinen tabletti
Hyvin hennon vaaleanpunainen, pyöreä 6,75 mm tabletti, jonka toisella puolella on kaiverrus ”Lilly” ja toisella puolella kaiverrus ”1”.
Olumiant 2 mg kalvopäällysteinen tabletti
Hennon vaaleanpunainen, pitkänomainen tabletti, jonka koko on 9 x 7,5 mm ja jonka toisella puolella on kaiverrus ”Lilly” ja toisella puolella kaiverrus ”2”.
Olumiant 4 mg kalvopäällysteinen tabletti
Vaaleanpunainen, pyöreä 8,5 mm tabletti, jonka toisella puolella on kaiverrus ”Lilly” ja toisella puolella kaiverrus ”4”.
Tablettien molemmilla puolilla on painauma.
Käyttö- ja käsittelyohjeet
Pediatrisille potilaille, jotka eivät pysty nielemään kokonaista tablettia, voidaan harkita tabletin dispergoimista veteen. Vain vettä voidaan käyttää tabletin dispergoimiseen. Vain annokseen tarvittava määrä tabletteja dispergoidaan.
- Kokonainen tabletti asetetaan astiaan, jossa on 5–10 ml huoneenlämpöistä vettä, ja pyöritellään astiaa kevyesti niin, että tabletti dispergoituu. Tabletin dispergoituminen sameaksi, hennon vaaleanpunaiseksi suspensioksi voi kestää korkeintaan 10 minuuttia. Sakkautumista voi esiintyä.
- Kun tabletti on dispergoitunut, astiaa pyöritellään varovasti uudelleen, ja koko suspensio annostellaan välittömästi.
-
Lisää astiaan 5-10 millilitraa huoneenlämpöistä vettä, pyöräytä astiaa mahdollisen jäljelle jääneen lääkeaineen talteen saamiseksi, ja annostele välittömästi.
Veteen dispergoitu tabletti on stabiili korkeintaan 4 tunnin ajan huoneenlämmössä. Jos jostain syystä kaikkea näin valmistettua suspensiota ei saada annosteltua potilaalle, uutta tablettia ei pidä dispergoida ja annostella, vaan on odotettava seuraavaan aikataulun mukaiseen annoksenottoaikaan.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
OLUMIANT tabletti, kalvopäällysteinen
2 mg 28 fol
4 mg 28 fol
- Alempi erityiskorvaus (65 %). Barisitinibi: Aikuisten nivelreuman sekä vähintään 2-vuotiaiden juveniilin polyartriitin (mukaan lukien laajeneva oligoartriitti), entesiitteihin liittyvän artriitin tai nivelpsoriaasin hoito erityisin edellytyksin (293).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Barisitinibi: Aikuisten nivelreuman ja vähintään 2-vuotiaiden vaikean atooppisen ihottuman sekä juveniilin polyartriitin (mukaan lukien laajeneva oligoartriitti), entesiitteihin liittyvän artriitin tai nivelpsoriaasin hoito erityisin edellytyksin (3006).
ATC-koodi
L04AF02
Valmisteyhteenvedon muuttamispäivämäärä
27.04.2026
Yhteystiedot
 OY ELI LILLY FINLAND AB
OY ELI LILLY FINLAND AB Mannerheimintie 117
00280 Helsinki
09 854 5250
www.lilly.com/fi
medinfo_finland@lilly.com
Lääketietopalvelu puh. 0800-140 240