
XOSPATA tabletti, kalvopäällysteinen 40 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Yleinen
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää 40 mg gilteritinibiä (fumaraattina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Xospata on tarkoitettu monoterapiana aikuisille potilaille, joilla on uusiutunut tai refraktaarinen akuutti myelooinen leukemia, jossa on FLT3-mutaatio (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Xospata-hoito tulee aloittaa ja hoitoa tulee jatkaa vain syöpähoitoihin perehtyneen lääkärin valvonnassa.
Ennen gilteritinibin ottamista potilaiden, joilla on uusiutunut tai refraktaarinen AML, on saatava validoidulla testillä vahvistus FMS:n kaltaisesta tyrosiinikinaasi 3 (FLT3) -mutaatiosta (sisäinen tandem-duplikaatio [ITD] tai tyrosiinikinaasidomeeni [TKD]).
Xospata-hoito voidaan aloittaa uudelleen hematopoieettisen kantasolusiirron (HSCT) jälkeen (ks. taulukko 1).
Annostus
Suositeltava aloitusannos on 120 mg gilteritinibiä (kolme 40 mg:n tablettia) kerran vuorokaudessa.
Veren koostumus, mukaan lukien kreatiinifosfokinaasi, tulee tutkia ennen hoidon aloittamista, hoidon 15. päivänä ja kuukausittain hoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilaalta tulee ottaa sydänsähkökäyrä (EKG) ennen gilteritinibihoidon aloittamista, ensimmäisen hoitojakson 8. ja 15. päivänä ja ennen seuraavien kolmen peräkkäisen hoitokuukauden aloittamista (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Naisia, jotka voivat tulla raskaaksi, on kehotettava tekemään raskaustesti seitsemän päivän sisällä ennen Xospata-hoidon aloittamista (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Raskaus ja imetys).
Hoitoa tulee jatkaa, kunnes Xospata-valmisteesta ei enää ole potilaalle kliinistä hyötyä, tai liiallisen toksisuuden ilmenemiseen saakka. Vaste voi ilmaantua viiveellä, ja siksi hoidon jatkamista määrätyllä annoksella jopa 6 kuukauden ajan pitää harkita, jotta kliininen vaste ehtii kehittyä.
Jos vastetta ei ole saatu [potilas ei ole saavuttanut täydellistä yhdistelmäremissiota (CRc)] 4 viikon hoidon jälkeen, annos voidaan suurentaa 200 mg:aan (viisi 40 mg:n tablettia) kerran vuorokaudessa, jos annos on siedettävä ja sen käyttäminen on kliinisesti perusteltua.
Annoksen muuttaminen
Taulukko 1: Suosituksia Xospata-annosten pienentämisestä, hoidon keskeyttämisestä tai lopettamisesta potilailla, joilla on uusiutunut tai refraktaarinen AML
| Kriteerit | Xospata-valmisteen annostus |
| Erilaistumisoireyhtymä |
|
| Posteriorinen reversiibeli enkefalopatiaoireyhtymä |
|
| QTcF-aika > 500 ms |
|
| QTcF-ajan piteneminen > 30 millisekunnilla EKG:ssä 1. hoitojakson päivänä 8 |
|
| Pankreatiitii |
|
| Muu vaikeusasteen 3a tai suuremman vaikeusasteen toksisuus, jota pidetään hoitoon liittyvänä. |
|
| Suunniteltu HSCT |
|
| a. Vaikeusaste 1 on lievä, vaikeusaste 2 on kohtalainen, vaikeusaste 3 on vaikea, vaikeusaste 4 on henkeä uhkaava. b. Vuorokausiannos voidaan pienentää 120 mg:sta 80 mg:aan tai 200 mg:sta 120 mg:aan. c. CRc:n määritelmä on remissio, joka pitää sisällään täydellisen remission (CR, ks. määritelmä kohdasta Farmakodynamiikka), CRp:n [saavutettu CR muuten, paitsi verihiutaleiden palautuminen on epätäydellistä (< 100 x 109/l)] ja CRi:n (saavutettu kaikki CR:n kriteerit muuten, paitsi hematologinen palautuminen on epätäydellistä; potilaalla edelleen neutropenia < 1 x 109/l, ja sen yhteydessä joko täydellinen verihiutaleiden palautuminen tai ei täydellistä verihiutaleiden palautumista). | |
Iäkkäät potilaat
Annosta ei tarvitse muuttaa ≥ 65-vuotiaille potilaille (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen potilailla, joilla on lievä (Child‑Pugh-luokka A) tai kohtalainen (Child‑Pugh-luokka B) maksan vajaatoiminta. Xospata-valmistetta ei suositella käytettäväksi potilaille, joilla on vaikea (Child-Pugh-luokka C) maksan vajaatoiminta, sillä turvallisuutta ja tehoa ei ole arvioitu tässä potilasryhmässä (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastaville potilaille (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Xospata-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla. 5HT2B:hen kohdistuvan in vitro ‑sitoutumisen vuoksi (ks. kohta Yhteisvaikutukset) vaikutus alle 6 kuukauden ikäisten potilaiden sydämen kehitykseen on mahdollista.
Antotapa
Xospata otetaan suun kautta.
Tabletit voidaan ottaa joko ruoan kanssa tai tyhjään mahaan. Ne pitää niellä kokonaisina veden kera, eikä niitä saa rikkoa tai murskata.
Xospata on annettava suunnilleen samaan aikaan joka päivä. Jos annos on jäänyt väliin tai sitä ei ole otettu tavalliseen aikaan, annos on annettava mahdollisimman pian samana päivänä ja seuraavana päivänä on palattava normaaliin aikatauluun. Jos potilas oksentaa annoksen ottamisen jälkeen, annosta ei tule ottaa uudelleen, vaan seuraavana päivänä tulee palata normaaliin aikatauluun.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle (vaikuttaville aineille) tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Erilaistumisoireyhtymä
Gilteritinibi on liitetty erilaistumisoireyhtymään (ks. kohta Haittavaikutukset). Erilaistumisoireyhtymään liittyy nopea proliferaatio ja myelooisten solujen erilaistuminen, ja se saattaa hoitamattomana olla henkeä uhkaava tai fataali. Erilaistumisoireyhtymän oireita ja kliinisiä löydöksiä ovat kuume, hengenahdistus, pleuraeffuusio, perikardiumeffuusio, keuhkopöhö, hypotensio, nopea painonnousu, perifeerinen turvotus, ihottuma ja munuaisten vajaatoiminta.
Jos epäillään erilaistumisoireyhtymää, on aloitettava kortikosteroidihoito ja hemodynaaminen valvonta, kunnes oireet häviävät. Jos vaikeat oireet jatkuvat yli 48 tuntia kortikosteroidien aloittamisen jälkeen, gilteritinibin käyttö pitää keskeyttää siihen asti, että oireet eivät ole enää vaikeita (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Kortikosteroideja voidaan vähentää, kun oireet ovat hävinneet, ja niitä pitää antaa vähintään 3 päivää. Erilaistumisoireyhtymän oireet voivat uusiutua, jos kortikosteroidihoito lopetetaan liian aikaisin.
Posteriorinen reversiibeli enkefalopatiaoireyhtymä
Gilteritinibiä saaneilla potilailla on raportoitu posteriorista reversiibeliä enkefalopatiaoireyhtymää (PRES) (ks. kohta Haittavaikutukset). PRES on harvinainen, korjaantuva neurologinen sairaus, joka voi ilmetä nopeasti kehittyvinä oireina, kuten kouristeluna, päänsärkynä, sekavuutena, näköhäiriöinä ja neurologisina häiriöinä, joissa voi ilmetä tai olla ilmenemättä hypertensiota ja psyykkisen tilan vaihtelua. Kun epäillään PRES:ää, se pitää varmistaa aivokuvantamisella, mieluiten magneettikuvauksella (MRI). Gilteritinibin lopetus on suositeltavaa, jos potilaalle kehittyy PRES (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Pidentynyt QT-aika
Gilteritinibi on liitetty pidentyneeseen sydämen kammion repolarisaatioon (QT-aika) (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). QT-ajan pidentyminen voi ilmetä gilteritinibihoidon kolmen ensimmäisen kuukauden aikana. Siksi potilaalta tulee ottaa sydänsähkökäyrä (EKG) ennen hoidon aloittamista, ensimmäisen hoitojakson 8. ja 15. päivänä ja ennen seuraavien kolmen peräkkäisen hoitokuukauden aloittamista. Varovaisuutta on noudatettava, jos potilaalla on ollut aiempia merkityksellisiä sydäntapahtumia. Hypokalemia tai hypomagnesemia saattaa lisätä QT-ajan pidentymisriskiä. Hypokalemia tai hypomagnesemia pitää siksi hoitaa ennen gilteritinibihoitoa ja sen aikana.
Gilteritinibin käyttö pitää keskeyttää, jos potilaan QTcF on >500 ms (ks. kohta Annostus ja antotapa).
Päätöksen gilteritinibihoidon aloittamisesta uudelleen QT-ajan pidentymisen jälkeen pitää perustua huolelliseen hyötyjen ja riskien arviointiin. Jos gilteritinibin käyttö aloitetaan uudelleen pienennetyllä annoksella, sydänsähkökäyrä (EKG) tulee ottaa hoidon 15. päivänä ja ennen seuraavien kolmen peräkkäisen hoitokuukauden aloittamista. Kliinisissä tutkimuksissa 12 potilaalla todettiin QTcF >500 ms. Kolme potilasta keskeytti hoidon ja aloitti sen uudelleen ilman, että QT-ajan pidentyminen uusiutui.
Pankreatiitti
Pankreatiittitapauksia on raportoitu. Potilaat, joilla ilmenee pankreatiittiin viittaavia merkkejä ja oireita, on arvioitava ja heitä on valvottava. Gilteritinibi pitää keskeyttää, ja sitä voidaan jatkaa pienennetyllä annoksella, kun pankreatiitin merkit ja oireet ovat hävinneet (ks. kohta Annostus ja antotapa).
Vaikea munuaisten vajaatoiminta
Gilteritinibialtistus voi lisääntyä potilailla, joilla on vaikea munuaisten vajaatoiminta tai loppuvaiheen munuaissairaus. Potilaita on seurattava tarkoin toksisuuden varalta gilteritinibihoidon aikana (ks. kohta Farmakokinetiikka).
Yhteisvaikutukset
CYP3A:n/P-glykoproteiinin indusoijien samanaikainen antaminen saattaa johtaa vähentyneeseen gilteritinibialtistukseen, jolloin riskinä on gilteritinibin tehon häviäminen. Siksi gilteritinibin samanaikaista käyttöä vahvojen CYP3A4:n/P-glykoproteiinin indusoijien kanssa pitää välttää (ks. kohta Yhteisvaikutukset).
Gilteritinibin käytössä samanaikaisesti sellaisten lääkevalmisteiden kanssa, jotka ovat vahvoja CYP3A:n, P-glykoproteiinin ja/tai rintasyöpäresistenttien proteiinien (BCRP) estäjiä, on noudatettava varovaisuutta, sillä ne voivat lisätä gilteritinibialtistusta. On harkittava vaihtoehtoisia lääkevalmisteita, jotka eivät voimakkaasti estä CYP3A:n, P-glykoproteiinin ja/tai BCRP:n toimintaa. Tilanteissa, joissa hyväksyttäviä vaihtoehtoisia hoitoja ei ole, potilaita on seurattava tarkasti toksisuuden varalta gilteritinibin annon aikana (ks. kohta Yhteisvaikutukset).
Gilteritinibi saattaa heikentää sellaisten lääkevalmisteiden tehoa, jotka vaikuttavat 5HT2B-reseptoreihin tai epäspesifisiin sigma-reseptoreihin. Siksi gilteritinibin samanaikaista käyttöä näiden valmisteiden kanssa pitää välttää, paitsi jos käytön katsotaan olevan välttämätöntä potilaan hoidon kannalta (ks. kohta Yhteisvaikutukset).
Alkio- ja sikiötoksisuus ja ehkäisy
Raskaana oleville naisille on kerrottava sikiöön kohdistuvasta mahdollisesta riskistä (ks. kohdat Raskaus ja imetys ja Prekliiniset tiedot turvallisuudesta). Naisia, jotka voivat tulla raskaaksi, on kehotettava tekemään raskaustesti seitsemän päivän sisällä ennen gilteritinibihoidon aloittamista ja käyttämään tehokasta ehkäisyä gilteritinibihoidon aikana sekä ainakin 6 kuukautta hoidon päättymisen jälkeen. Hormonaalista ehkäisyä käyttävien naisten on käytettävä lisäksi jotakin estemenetelmää. Miehiä, joiden kumppani voi tulla raskaaksi, on kehotettava käyttämään tehokasta ehkäisyä gilteritinibihoidon aikana sekä ainakin 4 kuukautta viimeisen gilteritinibiannoksen jälkeen.
Yhteisvaikutukset
Gilteritinibiä metaboloivat pääasiassa CYP3A-entsyymit, joita jotkin samanaikaisesti käytettävät lääkevalmisteet voivat indusoida tai estää.
Muiden lääkevalmisteiden vaikutus Xospata-valmisteeseen
CYP3A:n/P‑glykoproteiinin indusoijat
Xospata-valmisteen samanaikaista käyttöä vahvojen CYP3A:n/P-glykoproteiiniin indusoijien (kuten fenytoiinin, rifampisiinin ja mäkikuisman) kanssa pitää välttää, sillä ne voivat pienentää gilteritinibin pitoisuuksia plasmassa. Terveillä tutkittavilla samanaikainen rifampisiinin (600 mg), vahvan CYP3A:n/P‑glykoproteiinin indusoijan, antaminen vakaassa tilassa 20 mg:n gilteritinibiannoksen kanssa pienensi gilteritinibin keskimääräistä Cmax-arvoa 27 % ja keskimääräistä AUCinf-arvoa 70 % verrattuna tutkittaviin, joille annettiin ainoastaan yksi annos gilteritinibiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
CYP3A:n, P‑glykoproteiinin ja/tai BCRP:n estäjät
Vahvat CYP3A:n, P-glykoproteiinin ja/tai BCRP:n estäjät (muun muassa vorikonatsoli, itrakonatsoli, posakonatsoli, klaritromysiini, erytromysiini, kaptopriili, karvediloli, ritonaviiri ja atsitromysiini) voivat suurentaa gilteritinibipitoisuuksia plasmassa. Yhden 10 mg:n gilteritinibiannoksen antaminen samanaikaisesti itrakonatsolin (200 mg kerran vuorokaudessa 28 päivän ajan), vahvan CYP3A:n, P‑glykoproteiinin ja BCRP:n estäjän kanssa terveille tutkittaville suurensi keskimääräistä Cmax‑arvoa 20 %:lla ja suurensi AUCinf-arvon 2,2-kertaiseksi verrattuna tutkittaviin, jotka saivat ainoastaan yhden gilteritinibiannoksen. Gilteritinibialtistus suureni noin 1,5-kertaiseksi potilailla, joilla oli uusiutunut tai refraktaarinen AML, kun gilteritinibiä annettiin samanaikaisesti vahvan CYP3A:n, P-glykoproteiinin ja/tai BCRP:n estäjän kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Xospata-valmisteen vaikutus muihin lääkevalmisteisiin
Gilteritinibi estäjänä tai indusoijana
Gilteritinibi ei ole CYP3A4:n estäjä tai indusoija eikä MATE1:n estäjä in vivo. Vaikutukset midatsolaamin (sensitiivinen CYP3A4:n substraatti) farmakokinetiikkaan eivät olleet merkittäviä (Cmax ja AUC suurenivat noin 10 %) 15 päivän ajan kerran vuorokaudessa annetulla gilteritinibillä (300 mg) potilaille, joilla oli FLT3‑mutatoitunut uusiutunut tai refraktaarinen AML. Lisäksi vaikutukset kefaleksiinin (sensitiivinen MATE1:n substraatti) farmakokinetiikkaan eivät olleet merkittäviä (Cmax ja AUC pienenivät noin 10 %) 15 päivän ajan kerran vuorokaudessa annetulla gilteritinibillä (200 mg) potilaille, joilla oli FLT3‑mutatoitunut uusiutunut tai refraktaarinen AML.
Gilteritinibi on P-glykoproteiinin, BCRP:n ja OCT1:n estäjä in vitro. Koska kliinistä tietoa ei ole saatavilla, ei voida sulkea pois mahdollisuutta, että gilteritinibi voisi estää näitä kuljettajaproteiineja terapeuttisella annoksella. Varovaisuutta suositellaan jos gilteritinibiä annostellaan samanaikaisesti P-glykoproteiinin substraattien (esim. digoksiinin tai dabigatraanieteksilaatin), BCRP substraattien (esim. mitoksantroni, metotreksaatti, rosuvastatiini), tai OCT1 substraattien (esim. metformiini) kanssa.
5HT2B-reseptori tai epäspesifinen sigma-reseptori
In vitro-tietojen perusteella gilteritinibi saattaa heikentää sellaisten lääkevalmisteiden tehoa, jotka vaikuttavat 5HT2B-reseptoriin tai epäspesifiseen sigma-reseptoriin (serotoniinin takaisinoton estäjät, esimerkiksi essitalopraami, fluoksetiini, sertraliini). Siksi gilteritinibin samanaikaista käyttöä näiden lääkevalmisteiden kanssa pitää välttää, paitsi jos käytön katsotaan olevan välttämätöntä potilaan hoidon kannalta.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / miesten ja naisten ehkäisy
Naisille, jotka voivat tulla raskaaksi, suositellaan raskaustestin tekemistä seitsemän päivää ennen gilteritinibihoidon aloittamista. Naisten, jotka voivat tulla raskaaksi, on suositeltavaa käyttää tehokasta ehkäisyä (keinoja, joilla raskauden todennäköisyys on alle 1 %) hoidon aikana ja 6 kuukautta sen jälkeen. Ei tiedetä, heikentääkö gilteritinibi hormonaalisten ehkäisyvalmisteiden tehoa, ja siksi hormonaalista ehkäisyä käyttävien naisten on käytettävä lisäksi jotakin estemenetelmää. Lisääntymiskykyisiä miehiä on kehotettava käyttämään tehokasta ehkäisyä hoidon aikana ja ainakin 4 kuukautta viimeisen gilteritinibiannoksen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus
Gilteritinibi saattaa vahingoittaa sikiötä, jos sitä annetaan raskaana olevalle naiselle. Ei ole olemassa tietoja tai on vain vähän tietoja gilteritinibin käytöstä raskaana oleville naisille. Rotilla tehdyt lisääntymistutkimukset ovat osoittaneet, että gilteritinibi ehkäisi sikiön kasvua ja aiheutti alkioiden ja sikiöiden kuolemia ja teratogeenisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Gilteritinibin käyttöä ei suositella raskauden aikana eikä naisille, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyykö gilteritinibiä tai sen metaboliitteja ihmisen rintamaitoon. Saatavilla olevat tiedot eläinkokeista ovat osoittaneet gilteritinibin ja sen metaboliittien erittyvän eläimen maitoon imettävillä rotilla ja leviävän imeväisten rottien kudoksiin maidon kautta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Imeväiseen kohdistuvia riskejä ei voida poissulkea. Rintaruokinta on lopetettava gilteritinibihoidon ajaksi ja ainakin kahdeksi kuukaudeksi viimeisen annoksen jälkeen.
Hedelmällisyys
Tietoja gilteritinibin vaikutuksesta ihmisen hedelmällisyyteen ei ole saatavilla.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Gilteritinibillä on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Gilteritinibiä käyttävillä potilailla on raportoitu heitehuimausta, ja tämä on otettava huomioon potilaan ajokyvyn tai koneidenkäyttökyvyn arvioinnissa (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Xospata-valmisteen turvallisuutta arvioitiin 319 uusiutunutta tai refraktaarista AML:aa sairastavalla potilaalla, jotka olivat saaneet vähintään yhden 120 mg:n annoksen gilteritinibiä.
Yleisimmät gilteritinibin haittavaikutukset olivat alaniiniaminotransferaasin (ALT) lisääntyminen (82,1 %), aspartaattiaminotransferaasin (AST) lisääntyminen (80,6 %), veren alkalisen fosfataasin lisääntyminen (68,7 %), veren kreatiinifosfokinaasin lisääntyminen (53,9 %), ripuli (35,1 %), uupumus (30,4 %), pahoinvointi (29,8 %), ummetus (28,2 %), yskä (28,2 %), perifeerinen turvotus (24,1 %), hengenahdistus (24,1 %), heitehuimaus (20,4 %), hypotensio (17,2 %), raajojen kipu (14,7 %), astenia (13,8 %), artralgia (12,5 %) ja myalgia (12,5 %).
Yleisimmät vakavat haittavaikutukset olivat akuutti munuaisvaurio (6,6 %), ripuli (4,7 %), ALT:n lisääntyminen (4,1 %), hengenahdistus (3,4 %), AST:n lisääntyminen (3,1 %) ja hypotensio (2,8 %). Muita kliinisesti merkittäviä vakavia haittavaikutuksia olivat muun muassa erilaistumisoireyhtymä (2,2 %), sydänsähkökäyrässä näkyvä pidentynyt QT-aika (0,9 %) ja posteriorinen reversiibeli enkefalopatiaoireyhtymä (0,6 %).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa havaitut haittavaikutukset on esitetty seuraavassa taulukossa MedDRA-elinjärjestelmäluokituksen mukaisesti ja yleisyysluokittain. Yleisyysluokat määritellään seuraavalla tavalla: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2: Haittavaikutukset
| MedDRA-elinjärjestelmä Suositeltu termi | Kaikki arvot % | Arvot ≥ 3 % | Yleisyysluokka |
| Immuunijärjestelmä | |||
| Anafylaksia | 1,3 | 1,3 | Yleinen |
| Hermosto | |||
| Heitehuimaus | 20,4 | 0,3 | Hyvin yleinen |
| Posteriorinen reversiibeli enkefalopatiaoireyhtymä | 0,6 | 0,6 | Melko harvinainen |
| Sydän | |||
| Sydänsähkökäyrän pidentynyt QT-aika | 8,8 | 2,5 | Yleinen |
| Perikardiumeffuusio | 4,1 | 0,9 | Yleinen |
| Sydänpussitulehdus | 1,6 | 0 | Yleinen |
| Sydämen vajaatoiminta | 1,3 | 1,3 | Yleinen |
| Verisuonisto | |||
| Hypotensio | 17,2 | 7,2 | Hyvin yleinen |
| Hengityselimet, rintakehä ja välikarsina | |||
| Yskä | 28,2 | 0,3 | Hyvin yleinen |
| Hengenahdistus | 24,1 | 4,4 | Hyvin yleinen |
| Erilaistumisoireyhtymä | 3,4 | 2,2 | Yleinen |
| Ruoansulatuselimistö | |||
| Ripuli | 35,1 | 4,1 | Hyvin yleinen |
| Pahoinvointi | 29,8 | 1,9 | Hyvin yleinen |
| Ummetus | 28,2 | 0,6 | Hyvin yleinen |
| Maksa ja sappi | |||
| Alaniiniaminotransferaasin lisääntyminen* | 82,1 | 12,9 | Hyvin yleinen |
| Aspartaattiaminotransferaasin lisääntyminen* | 80,6 | 10,3 | Hyvin yleinen |
| Luusto, lihakset ja sidekudos | |||
| Veren kreatiinikinaasin lisääntyminen* | 53,9 | 6,3 | Hyvin yleinen |
| Veren alkalisen fosfataasin lisääntyminen* | 68,7 | 1,6 | Hyvin yleinen |
| Raajojen kipu | 14,7 | 0,6 | Hyvin yleinen |
| Nivelkipu | 12,5 | 1,3 | Hyvin yleinen |
| Lihaskipu | 12,5 | 0,3 | Hyvin yleinen |
| Tuki- ja liikuntaelinkipu | 4,1 | 0,3 | Yleinen |
| Munuaiset ja virtsatiet | |||
| Akuutti munuaisvaurio | 6,6 | 2,2 | Yleinen |
| Yleisoireet ja antopaikassa todettavat haitat | |||
| Uupumus | 30,4 | 3,1 | Hyvin yleinen |
| Perifeerinen turvotus | 24,1 | 0,3 | Hyvin yleinen |
| Astenia | 13,8 | 2,5 | Hyvin yleinen |
| Huonovointisuus | 4,4 | 0 | Yleinen |
| * Yleisyys perustuu keskuslaboratorion arvoihin. | |||
Valikoitujen haittavaikutusten kuvaus
Erilaistumisoireyhtymä
Kliinisissä tutkimuksissa Xospata-hoitoa saaneesta 319 potilaasta 11:llä (3 %) ilmeni erilaistumisoireyhtymä. Erilaistumisoireyhtymään liittyy nopea proliferaatio ja myelooisten solujen erilaistuminen, ja se saattaa hoitamattomana olla henkeä uhkaava tai fataali. Xospata-hoitoa saaneilla potilailla erilaistumisoireyhtymän oireita ja kliinisiä löydöksiä olivat kuume, hengenahdistus, pleuraeffuusio, perikardiumeffuusio, keuhkopöhö, hypotensio, nopea painonnousu, perifeerinen turvotus, ihottuma ja munuaisten vajaatoiminta. Joissakin tapauksissa ilmeni samanaikaisesti Sweetin oireyhtymä. Erilaistumisoireyhtymä ilmeni aikaisimmillaan yksi päivä ja enimmillään 82 päivää Xospata-hoidon aloittamisen jälkeen ja osassa tapauksia sen kanssa havaittiin samanaikaisesti leukosytoosia. Erilaistumisoireyhtymän saaneista 11 potilaasta 9 (82 %) parani hoidon tai Xospata-annosten keskeyttämisen jälkeen. Erilaistumisoireyhtymäepäilyihin liittyvät suositukset ovat luettavissa kohdista Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.
PRES
Kliinisissä tutkimuksissa Xospata-hoitoa saaneesta 319 potilaasta 0,6 %:lla ilmeni posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES). PRES on harvinainen, korjaantuva neurologinen sairaus, joka voi ilmetä nopeasti kehittyvinä oireina, kuten kouristeluna, päänsärkynä, sekavuutena, näköhäiriöinä ja neurologisina häiriöinä ja jossa voi ilmetä tai olla ilmenemättä hypertensiota. Oireet ovat hävinneet hoidon lopettamisen jälkeen (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
QT-ajan pidentyminen
Kliinisissä tutkimuksissa 120 mg Xospata-valmistetta saaneista 317 potilaasta, joilta mitattiin lähtötilanteen jälkeinen QTc-arvo, 4 potilaalla (1 %) QTcF-arvo oli > 500 ms. Lisäksi kaikilla annosmäärillä 12 potilaalla (2,3 %), joilla oli uusiutunut tai refraktaarinen AML, suurin lähtötilanteen jälkeinen QTcF-aika oli > 500 ms (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Xospata-valmisteelle ei tiedetä olevan spesifistä vastalääkettä. Yliannostustapauksissa Xospata-hoito tulee lopettaa. Potilaita on tarkkailtava huolellisesti haittavaikutusten merkkien tai oireiden varalta ja aloitettava oireiden mukainen ja elintoimintoja tukeva hoito, ottaen huomioon pitkän puoliintumisajan, jonka arvioidaan olevan 113 tuntia.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, proteiinikinaasin estäjät, ATC-koodi: L01EX13
Vaikutusmekanismi
Gilteritinibifumaraatti on FLT3:n ja AXL:n estäjä.
Gilteritinibi estää FLT3-reseptorin signaalivälityksen ja proliferaation soluissa, joissa ilmenee eksogeenisesti FLT3-mutaatioita, kuten FLT3‑ITD:tä, FLT3‑D835Y:tä ja FLT3‑ITD‑D835Y:tä, ja se indusoi apoptoosia leukemiasoluissa, joissa ilmenee FLT3‑ITD:tä.
Farmakodynaamiset vaikutukset
Potilailla, joilla oli uusiutunut tai refraktaarinen AML ja jotka saivat 120 mg gilteritinibiä, huomattava (> 90 %) FLT3:n fosforylaation estäminen oli nopeaa (24 tunnin sisällä ensimmäisestä annoksesta) ja pysyi käynnissä, kuten ex vivo plasman inhibitorisen toiminnan (PIA) analyysi osoitti.
Pidentynyt QT-aika
Pitoisuuteen liittyvä suureneminen QTcF:n lähtötilanteesta havaittiin 20–450 mg:n gilteritinibipitoisuuksilla. Ennustettu keskiarvomuutos QTcF:n lähtötilanteesta vakaalla keskiarvotasolla Cmax (282,0 ng/ml) 120 mg:n vuorokausiannoksella oli 4,96 ms ja ylempi yksipuolinen 95 %:n CI = 6,20 ms.
Kliininen teho ja turvallisuus
Uusiutunut tai refraktaarinen AML
Turvallisuutta ja tehoa arvioitiin aktiivisesti kontrolloidussa 3. vaiheen tutkimuksessa (2215-CL-0301).
ADMIRAL-tutkimus (2215‑CL‑0301)
ADMIRAL-tutkimus on vaiheen 3 satunnaistettu, avoin kliininen monikeskustutkimus aikuisilla potilailla, joilla on uusiutunut tai refraktaarinen AML ja FLT3-mutaatio, joka on määritetty LeukoStrat® CDx FLT3 -mutaatioanalyysillä. Tässä tutkimuksessa 371 potilasta satunnaistettiin 2:1 saamaan gilteritinibiä tai yhtä seuraavista salvage-kemoterapiahoidoista (247 sai gilteritinibiä ja 124 sai salvage-kemoterapiaa):
- sytarabiini 20 mg kahdesti vuorokaudessa injektiona ihon alle (s.c.) tai laskimoon infuusiona (i.v.) 10 päivän ajan (päivät 1–10) (LoDAC)
- atsasitidiini 75 mg/m2 kerran vuorokaudessa ihon alle tai laskimoon 7 päivän ajan (päivät 1–7)
- mitoksantroni 8 mg/m2, etoposidi 100 mg/m2 ja sytarabiini 1000 mg/m2 kerran vuorokaudessa laskimoon 5 päivän ajan (päivät 1–5) (MEC)
- granulosyyttiryhmiä stimuloiva kasvutekijä 300 µg/m2 kerran vuorokaudessa ihon alle 5 päivän ajan (päivät 1–5), fludarabiini 30 mg/m2 kerran vuorokaudessa laskimoon 5 päivän ajan (päivät 2–6), sytarabiini 2000 mg/m2 kerran vuorokaudessa laskimoon 5 päivän ajan (päivät 2–6), idarubisiini 10 mg/m2 kerran vuorokaudessa laskimoon 3 päivän ajan (päivät 2–4) (FLAG‑Ida).
Tutkimukseen otettujen potilaiden tauti oli uusiutunut tai refraktaarinen ensi linjan AML-hoidon jälkeen ja heidät stratifioitiin aiempaan AML-hoitoon saadun vasteen ja ennakolta valitun kemoterapian (joko korkea tai matala intensiteetti) mukaan. Tutkimuksessa oli mukana potilaita, joilla oli erilaisia AML:ään liittyviä sytogeneettisiä poikkeavuuksia, mutta mukaan ei otettu potilaita, joilla oli akuutti promyelosyyttileukemia (APL) tai hoitoon liittyvä AML.
Tutkimukseen satunnaistettiin 16 potilasta, jotka eivät saaneet hoitoa (1 potilas gilteritinibihaarassa ja 15 potilasta kemoterapiahaarassa). Gilteritinibiä annettiin suun kautta 120 mg:n vuorokausiannoksella, kunnes toksisuus oli liian suuri tai kliinistä hyötyä ei ollut. Annosten pienentäminen oli sallittua, jotta haittavaikutuksia voitiin hallita, ja annosten suurentaminen oli sallittua sellaisille potilaille, joilla ei saatu hoitovastetta 120 mg:n aloitusannoksella.
Niistä potilaista, jotka valittiin etukäteen saamaan Salvage-kemoterapiaa, 60,5 % satunnaistettiin korkean intensiteetin ja 39,5 % matalan intensiteetin hoitoon. MEC:tä ja FLAG-Idaa annettiin enintään kahdessa jaksossa, ensimmäisen jakson hoitovasteen mukaan. LoDACia ja atsasitidiinia annettiin jatkuvissa 4 viikon jaksoissa, kunnes toksisuus oli liian suuri tai kliinistä hyötyä ei ollut.
Kahden hoitohaaran demografiset piirteet ja lähtötilanteen piirteet olivat hyvässä tasapainossa. Satunnaistamisessa mediaani-ikä oli gilteritinibihaarassa 62 vuotta (vaihteluväli 20–84 vuotta) ja Salvage-kemoterapiahaarassa 62 vuotta (vaihteluväli 19–85 vuotta). Tutkimuksessa 42 % potilaista oli vähintään 65‑vuotiaita ja 12 % potilaista oli vähintään 75‑vuotiaita. 54 prosenttia potilaista oli naisia. Suurin osa potilaista oli valkoihoisia (59,3 %), 27,5 % aasialaisia, 5,7 % mustia, 4 % muita ja 3,5 % ei tiedossa. Suurimmalla osalla potilaista (83,8 %) ECOG-suorituskyky oli 0 tai 1. Potilailla oli seuraavat vahvistetut mutaatiot: vain FLT3‑ITD (88,4 %), vain FLT3‑TKD (8,4 %) tai sekä FLT3‑ITD että FLT3‑TKD (1,9 %). Kaksitoista prosenttia potilaista oli saanut aiempaa FLT3-estäjähoitoa. Suurimmalla osalla potilaista AML:n sytogeneettinen riski oli kohtalainen (73 %), 10 %:lla korkea, 1,3 %:lla pieni, ja 15,6 %:lla sytogeneettinen riski oli luokittelematon.
Ennen gilteritinibihoitoa 39,4 %:lla potilaista oli ensisijainen refraktaarinen AML, ja suurin osa näistä potilaista luokiteltiin refraktaarisiksi yhden kemoterapian induktiohoitojakson jälkeen, 19,7 %:lla oli uusiutunut AML allogeenisen hematopoieettisen kantasolusiirron (HSCT) jälkeen ja 41 %:lla oli uusiutunut AML ilman allogeenista hematopoieettista kantasolusiirtoa.
Ensisijainen hoidon tehon päätemuuttuja lopullisessa analyysissa oli hoitoaikomuspopulaation (ITT) kokonaiselinaika mitattuna satunnaistamispäivästä mistä tahansa syystä johtuvaan kuolemaan (analysoitujen tapahtumien määrä oli 261). Gilteritinibihaaraan satunnaistettujen potilaiden elinaika oli merkittävästi pidempi kemoterapiahaaraan verrattuna (HR 0,637; 95 % CI 0,490 – 0,830; 1-puolinen p‑arvo: 0,0004). Kokonaiselinajan mediaani oli gilteritinibiä saaneilla potilailla 9,3 kuukautta ja kemoterapiaa saaneilla 5,6 kuukautta. Tehoa tukivat myös täydellisen remission (CR) määrä/ täydellinen remissio, jossa osittainen hematologinen palautuminen (CRh) (taulukko 3, kuva 1).
Taulukko 3: ADMIRAL-tutkimuksen kokonaiselinaika ja täydellinen remissio potilailla, joilla oli uusiutunut tai refraktaarinen AML
Gilteritinibi (N = 247) | Kemoterapia (N = 124) | ||
| Selviytyminen | |||
| Kuolemat, n (%) | 171 (69,2) | 90 (72,6) | |
| Mediaani kuukausina (95 %:n CI) | 9,3 (7,7; 10,7) | 5,6 (4,7; 7,3) | |
| Riskisuhde (95 %:n CI) | 0,637 (0,490; 0,830) | ||
| p‑arvo (1‑puolinen) | 0,0004 | ||
| 1 vuoden eloonjäänti, % (95 %:n CI) | 37,1 (30,7; 43,6) | 16,7 (9,9; 25) | |
| Täydellinen remissio | |||
| CRa (95 %:n CIb) | 21,1 % (16,1; 26,7) | 10,5 % (5,7; 17,3) | |
| CRhc (95 %:n CIb) | 13 % (9; 17,8) | 4,8 % (1,8; 10,2) | |
| CR/CRh (95 %:n CIb) | 34 % (28,1; 40,3) | 15,3 % (9,5; 22,9) | |
CI: luottamusväli
| |||
Kuva 1: Kaplan-Meierin kaavio kokonaiselossaoloajasta ADMIRAL-tutkimuksessa
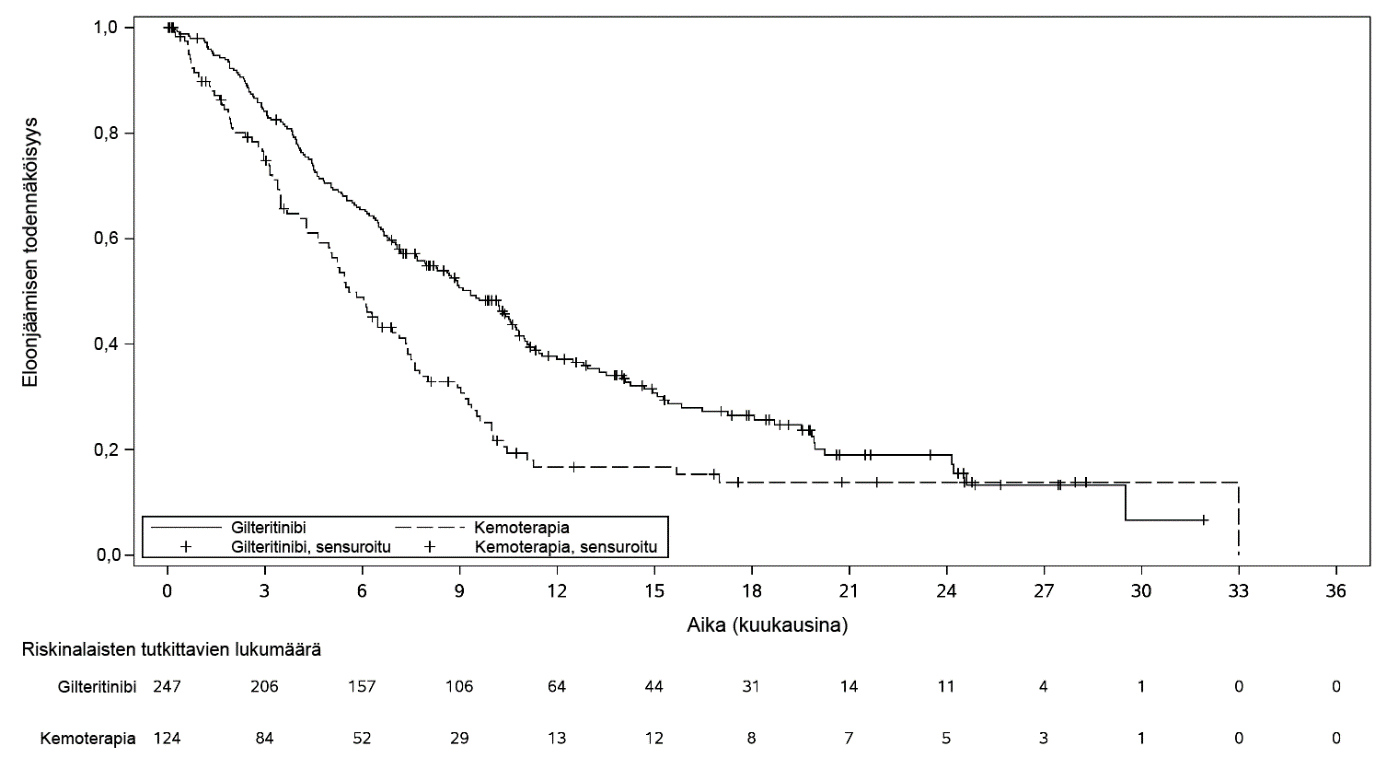
Potilaille, jotka saavuttivat CR:n/CRh:n, mediaaniaika primaarivasteeseen oli 3,7 kuukautta (vaihteluväli 0,9–10,6 kuukautta) gilteritinibiryhmässä ja 1,2 kuukautta (vaihteluväli 1–2,6 kuukautta) salvage-kemoterapiaryhmässä. Mediaaniaika CR:n/CRh:n parhaaseen vasteeseen oli 3,8 kuukautta (vaihteluväli 0,9–16 kuukautta) gilteritinibiryhmässä ja 1,2 kuukautta (vaihteluväli 1–2,6 kuukautta) salvage-kemoterapiaryhmässä.
CHRYSALIS-tutkimus (2215-CL-0101)
Vaiheen 1/2 annoseskalaatiotutkimukseen 2215-CL-0101 (tukitutkimus) osallistui 157 potilasta, joilla oli FLT3-mutatoitunut AML ja jotka olivat aiemmin saaneet 1 tai > 1 hoitoa eri annosryhmissä (eli 80 mg, 120 mg tai 200 mg); 31,2 % oli saanut yhtä aiempaa hoitoa ja 68,8 % yli yhtä aiempaa hoitoa.
Tutkimuksessa 2215-CL-0101 yli yhtä aiempaa hoitoa saaneilla potilailla havaittu vasteosuus (CR/CRh) oli 21,4 % niillä potilailla, jotka olivat saaneet 120 mg:n annoksia ja 15,7 % niillä, jotka olivat saaneet eri annoksia. Kokonaiselossaoloajan mediaani oli 7,2 kuukautta niillä, joka saivat 120 mg ja 7,1 kuukautta niillä, jotka saivat eri annoksia.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Xospata-valmisteen käytöstä akuutin myelooisen leukemian hoidossa kaikissa pediatrisissa potilasryhmissä. Ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Farmakokinetiikka
Imeytyminen
Gilteritinibin suun kautta antamisen jälkeen huippupitoisuuksia plasmassa on havaittu mediaani tmax:n kohdalla: noin 4–6 tunnin kohdalla terveillä vapaaehtoisilla ja potilailla, joilla on uusiutunut tai refraktaarinen AML. Gilteritinibin ensimmäisen imeytymisen arvioitu imeytymisnopeus (ka) on 0,43 h–1 0,34 tunnin viiveellä perustuen populaation farmakokineettiseen mallinnukseen. Jatkuvan maksimipitoisuuden (Cmax) mediaani on 282,0 ng/ml (CV% = 50,8), ja plasman pitoisuuskäyrän alla oleva alue 24 tunnin annosväleillä (AUC0-24) on 6180 ng·h/ml (CV% = 46,4) päivittäisen 120 mg:n gilteritinibiannoksen jälkeen. Vakaat plasmatasot saavutetaan 15 päivän sisällä kerran vuorokaudessa otetuilla annoksilla noin 10-kertaisella kertymisellä.
Ruoan vaikutus
Terveillä aikuisilla gilteritinibin Cmax ja AUC pienenivät noin 26 % ja alle 10 % tässä järjestyksessä, kun yksi 40 mg:n annos otettiin yhdessä erittäin rasvapitoisen aterian kanssa, verrattuna gilteritinibialtistukseen paaston jälkeen. Mediaani tmax viivästyi 2 tuntia, kun gilteritinibi otettiin erittäin rasvapitoisen aterian kanssa.
Jakautuminen
Populaatioarvio jakautumisen keskus- ja ääreistilavuudesta olivat 1 092 l ja 1 100 l tässä järjestyksessä. Nämä tiedot viittaavat siihen, että gilteritinibi jakautuu laajasti plasman ulkopuolelle, mikä saattaa viitata laajaan kudosjakautumiseen. In vivo plasman proteiineihin sitoutuminen on ihmisillä noin 90 % ja gilteritinibi sitoutuu ensisijaisesti albumiiniin.
Biotransformaatio
In vitro-tietoihin perustuen gilteritinibi metaboloituu pääasiassa CYP3A4:n kautta. Ihmisen ensisijaisia metaboliitteja ovat muun muassa M17 (muodostuu N-dealkylaation ja hapettumisen kautta), M16 sekä M10 (molemmat muodostuvat N-dealkylaation kautta), ja niitä on havaittu eläimillä. Mikään näistä kolmesta metaboliitista ei ylittänyt 10 %:n parenteraalista altistusta. Metaboliittien farmakologisesta toiminnasta FLT3- ja AXL-reseptoreja kohtaan ei ole saatavilla tietoja.
Kuljettajaproteiinin lääkkeidenvälinen yhteisvaikutus
In vitro-kokeet osoittivat, että gilteritinibi on P‑glykoproteiinin ja BCRP:n substraatti. Gilteritinibi saattaa mahdollisesti estää BCRP:tä, P‑glykoproteiinia ja OCT1:tä kliinisesti merkittävillä pitoisuuksilla (ks. kohta Yhteisvaikutukset).
Eliminaatio
Yhden [14C] ‑gilteritinibiannoksen jälkeen gilteritinibiä erittyy pääasiassa ulosteeseen; ulosteessa on havaittu 64,5 % annetusta kokonaisannoksesta. Noin 16,4 % kokonaisannoksesta erittyi virtsaan muuttumattomana lääkkeenä ja metaboliitteina. Gilteritinibipitoisuudet plasmassa vähenivät kaksoiseksponentiaalisesti ja puoliintumisajan odotusarvo oli 113 tuntia. Arvioitu havaittava puhdistuma (CL/F) perustuen populaatiofarmakokineettiseen malliin on 14,85 l/h.
Lineaarisuus/ei-lineaarisuus
Yleisesti gilteritinibi osoitti lineaarista annokseen verrannollista farmakokinetiikkaa yhden ja usean annoksen antamisen jälkeen, kun annokset olivat 20–450 mg potilailla, joilla oli uusiutunut tai refraktaarinen AML.
Erityisryhmät
Populaatiofarmakokineettinen analyysi tehtiin sen arvioimiseksi, miten luontaiset ja ulkoiset kovariaatit vaikuttavat ennakoituun gilteritinibialtistukseen uusiutunutta tai refraktaarista AML:aa sairastavilla potilailla. Kovariaattianalyysin mukaan ikä (20–90 vuotta) ja paino (36–157 kg) olivat tilastollisesti merkitseviä; gilteritinibialtistuksen ennakoitu muutos oli kuitenkin alle 2-kertainen.
Maksan vajaatoiminta
Maksan vajaatoiminnan vaikutusta gilteritinibin farmakokinetiikkaan tutkittiin tutkittavilla, joilla oli lievä (Child‑Pugh-luokka A) ja kohtalainen (Child‑Pugh-luokka B) maksan vajaatoiminta. Tulokset osoittivat, että altistus vapaalle gilteritinibille lievää tai kohtalaista maksan vajaatoimintaa sairastavilla on verrannainen altistukseen, joka havaittiin tutkittavilla, joiden maksan toiminta on normaalia. Lievän maksan vajaatoiminnan (NCI-ODWG:n määritelmä) vaikutus gilteritinibialtistukseen arvioitiin myös käyttämällä populaatiofarmakokineettistä mallia ja tulokset osoittavat jatkuvalla gilteritinibialtistuksella olevan hyvin pieniä eroja suhteessa tyypilliseen uusiutunutta tai refraktaarista AML:ää sairastavaan potilaaseen, jonka maksan toiminta on normaalia.
Gilteritinibiä ei ole tutkittu potilailla, joilla on vaikea maksan vajaatoiminta (Child-Pugh-luokka C).
Munuaisten vajaatoiminta
Gilteritinibin farmakokinetiikkaa arvioitiin viidellä henkilöllä, joilla oli vaikea (CrCL 15 ‑ < 30 ml/min) munuaisten vajaatoiminta ja neljällä potilaalla, joilla oli loppuvaiheen munuaissairaus (CrCL < 15 ml/min). Gilteritinibin keskimääräisen Cmax-arvon 1,4-kertaistuminen ja keskimääräisen AUCinf-arvon 1,5-kertaistuminen havaittiin potilailla, joilla oli vaikea munuaisten vajaatoiminta tai loppuvaiheen munuaissairaus verrattuna potilaisiin, joiden munuaisten toiminta oli normaali (n=8) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Prekliiniset tiedot turvallisuudesta
Seuraavia haittavaikutuksia ei ole todettu kliinisissä tutkimuksissa, mutta niitä on todettu koe-eläimillä (turvallisuusfarmakologia / toistuvan annoksen toksisuus), jotka ovat saaneet hoitoannoksia vastaavia määriä lääkeainetta. Siksi haitoilla voi olla kliinistä merkitystä.
Turvallisuusfarmakologia
Rotilla havaittiin virtsaamisen vähentymistä, kun annos oli 30 mg/kg tai suurempi ja ulostamisen vähentymistä, kun annos oli 100 mg/kg. Koirilla havaittiin 10 mg/kg ja sitä suuremmilla annoksilla piilevää verta ulosteessa, 30 mg/kg annoksella veren kalsiumpitoisuuden pienenemistä ja 100 mg/kg annoksella veren kalsiumpitoisuuden suurenemista, jota seurasi pieneneminen. Näitä muutoksia havaittiin, kun altistumistasot plasmassa olivat samat tai pienemmät kuin hoitoannoksilla. Näiden havaintojen mahdollinen kliininen merkitys on tuntematon.
Toistuvan annoksen toksisuus
Rotilla ja koirilla tehdyissä toistuvan annoksen toksisuustutkimuksissa toksisuuden kohde-elimet olivat maha-suolikanava (verenvuotoa koirilla), lymfohematopoieettinen järjestelmä (lymfosyyttinekroosia sekä luuytimen solumäärän pienenemistä ja muutoksia hematologisissa parametreissa), silmät (tulehduksia ja mykiönsamentumia rotilla, silmänpohjan värimuutoksia koirilla, verkkokalvon vakuolisaatiota), keuhkot (interstitiaalista pneumoniaa rotilla ja tulehdusta koirilla), munuaiset (munuaistiehyeiden muutoksia ja positiivinen virtsan piilevän veren koe) sekä maksa (hepatosyyttien vakuolisaatiota), virtsarakko (epiteelivakuolisaatio), epiteelikudos (haavoja ja tulehduksia) ja fosfolipidoosi (rottien keuhkot ja munuaiset). Näitä muutoksia havaittiin, kun altistumistasot plasmassa olivat samat tai pienemmät kuin hoitoannoksilla. Useimmat muutokset osoittivat palautumista 4 viikon toipumisvaiheen loppuun mennessä. Näiden havaintojen mahdollinen kliininen merkitys on tuntematon.
Genotoksisuus
Gilteritinibi ei indusoinut geenimutaatiota tai kromosomipoikkeamia in vitro. In vivo mikrotumatesti osoitti, että gilteritinibin on mahdollista indusoida mikrotumia hiirillä.
Lisääntymistoksisuus
Rotilla tehdyt alkioiden ja sikiöiden kehitystutkimukset, joissa rotat saivat hoitoannoksia vastaavia määriä lääkeainetta, ovat osoittaneet, että gilteritinibi ehkäisi sikiön kasvua ja aiheutti alkioiden ja sikiöiden kuolemia ja teratogeenisuutta. Gilteritinibi siirtyi istukan kautta rotilla, mikä aiheutti samanlaisen radioaktiivisuuden siirtymisen sikiöön kuin emon plasmassa havaittu määrä.
Gilteritinibiä erittyi imettävien rottien maitoon, ja sen pitoisuus maidossa oli suurempi kuin emon plasmassa. Gilteritinibi jakautui maidon välityksellä imeväisten eri kudoksiin, paitsi aivoihin.
Nuorten eläinten toksisuustutkimus
Nuorten rottien toksisuustutkimuksessa pienin kuolettava annostaso (2,5 mg/kg päivässä) oli paljon pienempi kuin aikuisilla rotilla (20 mg/kg). Maha-suolikanava tunnistettiin yhdeksi kohde-elimistä, samoin kuin aikuisillakin rotilla.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mannitoli (E421)
Hydroksipropyyliselluloosa
Hydroksipropyyliselluloosa, matalasubstituutioasteinen
Magnesiumstearaatti
Kalvopäällyste
Hypromelloosi
Talkki
Makrogoli
Titaanidioksidi
Keltainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita. Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
XOSPATA tabletti, kalvopäällysteinen
40 mg (L:ei) 84 fol (18034,15 €)
PF-selosteen tieto
OPA/alumiini/PVC/alumiiniläpipainopakkaukset, joissa on 21 kalvopäällysteistä tablettia.
Yksi pakkaus sisältää 84 kalvopäällysteistä tablettia.
Valmisteen kuvaus:
Pyöreä, vaaleankeltainen kalvopäällysteinen tabletti, jonka koko on n. 7,1 mm ja jonka toiselle puolelle on painettu yrityksen logo ja numero '235'.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
XOSPATA tabletti, kalvopäällysteinen
40 mg 84 fol
- Ylempi erityiskorvaus (100 %). Gilteritinibi: Aikuisten akuutin myelooisen leukemian hoito erityisin edellytyksin (1531).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Gilteritinibi: Monoterapiana akuutin myelooisen leukemian hoito erityisin edellytyksin (3033).
ATC-koodi
L01EX13
Valmisteyhteenvedon muuttamispäivämäärä
27.06.2024
Yhteystiedot
Hatsinanpuisto 8
02600 Espoo
09 8560 6000
www.astellas.fi