
DUPIXENT injektioneste, liuos, esitäytetty kynä 200 mg, injektioneste, liuos, esitäytetty ruisku 200 mg
Vaikuttavat aineet ja niiden määrät
Dupilumabi 200 mg injektioneste, liuos, esitäytetty ruisku
Yksi kertakäyttöinen esitäytetty ruisku sisältää 200 mg dupilumabia 1,14 ml:ssa liuosta (175 mg/ml).
Dupilumabi 200 mg injektioneste, liuos, esitäytetty kynä
Yksi kertakäyttöinen esitäytetty kynä sisältää 200 mg dupilumabia 1,14 ml:ssa liuosta (175 mg/ml).
Dupilumabi on täysin humaani monoklonaalinen vasta-aine, joka tuotetaan kiinanhamsterin munasarjasoluissa (CHO) yhdistelmä-DNA-tekniikalla.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää 2,28 mg polysorbaatti 80:tä per 200 mg:n (1,14 ml:n) annos. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Atooppinen ihottuma
Aikuiset ja nuoret
Dupixent on tarkoitettu keskivaikean tai vaikean atooppisen ihottuman hoitoon aikuisille ja vähintään 12-vuotiaille nuorille, joille harkitaan systeemistä hoitoa.
6 kuukauden – 11 vuoden ikäiset lapset
Dupixent on tarkoitettu vaikean atooppisen ihottuman hoitoon 6 kuukauden –11 vuoden ikäisille lapsille, joille harkitaan systeemistä hoitoa.
Astma
Aikuiset ja nuoret
Dupixent on tarkoitettu lisälääkkeeksi vaikean astman ylläpitohoitoon aikuisille ja vähintään 12-vuotiaille nuorille, joiden sairauteen liittyy veren kohonneen eosinofiilitason ja/tai kohonneen uloshengitysilman typpioksidiarvon (FeNO) perusteella määriteltävä tyypin 2 tulehdus, ks. kohta Farmakodynamiikka, ja joiden astmaa ei saada riittävän hyvään hallintaan suuriannoksisen inhaloitavan kortikosteroidin (ICS) ja toisen lääkevalmisteen yhdistelmän ylläpitohoidolla.
6–11-vuotiaat lapset
Dupixent on tarkoitettu lisälääkkeeksi vaikean astman ylläpitohoitoon 6–11‑vuotiaille lapsille, joiden sairauteen liittyy veren kohonneen eosinofiilitason ja/tai kohonneen uloshengitysilman typpioksidiarvon (FeNO) perusteella määriteltävä tyypin 2 tulehdus, ks. kohta Farmakodynamiikka, ja joiden astmaa ei saada riittävän hyvään hallintaan keskisuuren tai suuren inhaloitavan kortikosteroidin (ICS) annoksen ja toisen lääkevalmisteen ylläpitohoidolla.
Eosinofiilinen ruokatorvitulehdus
Dupixent on tarkoitettu eosinofiilisen ruokatorvitulehduksen hoitoon aikuisille, nuorille ja vähintään 1‑vuotiaille ja vähintään 15 kg painaville lapsille, joiden tautia ei saada riittävän hyvään hallintaan tavanomaisella lääkehoidolla, jotka eivät siedä tällaista hoitoa tai joille ei voida harkita tällaista hoitoa (ks. kohta Farmakodynamiikka).
Krooninen spontaani urtikaria
Dupixent on tarkoitettu keskivaikean tai vaikean kroonisen spontaanin urtikarian hoitoon aikuisille, nuorille ja lapsille (vähintään 2-vuotiaille), joiden vaste H1-antihistamiinihoitoon on riittämätön ja joiden kroonista spontaania urtikariaa ei ole hoidettu IgE:n vasta-aineilla.
Ehto
Valmistetta saavat määrätä vain käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneet lääkärit.
Annostus ja antotapa
Hoidon saa aloittaa dupilumabin käyttöaiheiden mukaisten sairauksien diagnosointiin ja hoitoon perehtynyt lääkäri (ks. kohta Käyttöaiheet).
Annostus
Atooppinen ihottuma
Aikuiset
Dupilumabin suositeltu annos aikuisille potilaille on aluksi 600 mg (kaksi 300 mg:n injektiota), minkä jälkeen annetaan 300 mg kahden viikon välein injektiona ihon alle.
Nuoret (12–17-vuotiaat)
Dupilumabin suositeltu annos nuorille (12–17-vuotiaille) potilaille on esitetty taulukossa 1.
Taulukko 1: Nuorille (12–17-vuotiaille) atooppista ihottumaa sairastaville potilaille ihon alle annettava dupilumabiannos
| Potilaan paino | Aloitusannos | Seuraavat annokset (kahden viikon välein) |
| alle 60 kg | 400 mg (kaksi 200 mg:n injektiota) | 200 mg |
| vähintään 60 kg | 600 mg (kaksi 300 mg:n injektiota) | 300 mg |
6–11-vuotiaat lapset
Dupilumabin suositeltu annos 6–11-vuotiaille lapsille on esitetty taulukossa 2.
Taulukko 2: 6–11-vuotiaille atooppista ihottumaa sairastaville lapsille ihon alle annettava dupilumabiannos
| Potilaan paino | Aloitusannos | Seuraavat annokset |
| vähintään 15 kg mutta alle 60 kg | 300 mg (yksi 300 mg:n injektio) päivänä 1, ja sitten 300 mg päivänä 15 | 300 mg neljän viikon välein* alkaen 4 viikon kuluttua päivänä 15 annetusta annoksesta |
| vähintään 60 kg | 600 mg (kaksi 300 mg:n injektiota) | 300 mg kahden viikon välein |
* Vähintään 15 kg mutta alle 60 kg painavilla potilailla annos voidaan lääkärin arvion perusteella suurentaa kahden viikon välein annettavaan 200 mg:n annokseen.
6 kuukauden – 5 vuoden ikäiset lapset
Dupilumabin suositeltu annos 6 kuukauden – 5 vuoden ikäisille lapsille on esitetty taulukossa 3.
| Taulukko 3: 6 kuukauden – 5 vuoden ikäisille atooppista ihottumaa sairastaville lapsille ihon alle annettava dupilumabiannos | ||
| Potilaan paino | Aloitusannos | Seuraavat annokset |
| vähintään 5 kg mutta alle 15 kg | 200 mg (yksi 200 mg:n injektio) | 200 mg neljän viikon välein |
| vähintään 15 kg mutta alle 30 kg | 300 mg (yksi 300 mg:n injektio) | 300 mg neljän viikon välein |
Dupilumabia voidaan käyttää paikallisten kortikosteroidien kanssa tai ilman niitä. Paikallishoitoon tarkoitettuja kalsineuriinin estäjiä voidaan käyttää, mutta niiden käyttö tulisi rajoittaa ainoastaan ongelmallisille alueille, kuten kasvoille, kaulalle, taive- ja genitaalialueille.
Hoidon lopettamista on harkittava atooppista ihottumaa sairastavilla potilailla, joilla ei ole todettu hoitovastetta 16 viikon hoidon jälkeen. Joidenkin aluksi osittaisen hoitovasteen saaneiden potilaiden tila saattaa parantua, kun hoitoa jatketaan yli 16 viikon ajan. Jos dupilumabihoito joudutaan keskeyttämään, potilaat voivat silti hyötyä hoidon aloittamisesta uudelleen.
Astma
Aikuiset ja nuoret
Dupilumabin suositeltu annos aikuisille ja nuorille (vähintään 12-vuotiaat) on:
- Aloitusannos 400 mg (kaksi 200 mg:n injektiota), minkä jälkeen annetaan 200 mg kahden viikon välein injektiona ihon alle.
- Potilaille, joilla on vaikea astma ja joita hoidetaan oraalisilla kortikosteroideilla, tai potilaille, joilla on vaikea astma ja lisäksi keskivaikea tai vaikea atooppinen ihottuma, tai aikuisille, joilla on lisäksi vaikea krooninen polypoottinen rinosinuiitti, aloitusannos 600 mg (kaksi 300 mg:n injektiota), minkä jälkeen annetaan 300 mg kahden viikon välein injektiona ihon alle.
6–11-vuotiaat lapset
Dupilumabin suositeltu annos 6–11-vuotiaille pediatrisille potilaille on esitetty taulukossa 4.
Taulukko 4: 6–11-vuotiaille astmaa sairastaville lapsille ihon alle annettava dupilumabiannos
| Paino | Aloitusannos ja seuraavat annokset |
| vähintään 15 kg mutta alle 30 kg | 300 mg neljän viikon välein |
vähintään 30 kg mutta alle 60 kg
| 200 mg kahden viikon välein tai 300 mg neljän viikon välein |
| vähintään 60 kg | 200 mg kahden viikon välein |
Suositeltu annos lapsipotilaille (6–11‑vuotiaat), joilla on hyväksytyn käyttöaiheen mukainen astma ja samanaikainen hyväksytyn käyttöaiheen mukainen vaikea atooppinen ihottuma, on esitetty taulukossa 2.
Samanaikaisesti oraalisilla kortikosteroideilla hoidettavien potilaiden steroidiannosta voidaan pienentää, kun dupilumabihoidolla on saavutettu kliininen vaste (ks. kohta Farmakodynamiikka). Steroidiannoksen pienentäminen on suositeltavaa tehdä asteittain (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Dupilumabi on tarkoitettu pitkäaikaiseen hoitoon. Lääkärin tulee arvioida jatkuvan hoidon tarvetta potilaan astman hallinnan perusteella vähintään kerran vuodessa.
Eosinofiilinen ruokatorvitulehdus
Dupilumabin suositeltu annos aikuisille, nuorille ja vähintään 1‑vuotiaille lapsille, jotka painavat vähintään 15 kg, on esitetty taulukossa 5.
Taulukko 5: Eosinofiilistä ruokatorvitulehdusta sairastaville aikuisille, nuorille ja vähintään 1‑vuotiaille lapsille ihon alle annettava dupilumabiannos
Paino | Annos |
vähintään 15 kg mutta alle 30 kg | 200 mg kahden viikon välein |
vähintään 30 kg mutta alle 40 kg | 300 mg kahden viikon välein |
vähintään 40 kg | 300 mg kerran viikossa |
Dupilumabi on tarkoitettu pitkäaikaiseen hoitoon.
Krooninen spontaani urtikaria
Aikuiset
Dupilumabin suositeltu annos aikuisille potilaille on aluksi 600 mg (kaksi 300 mg:n injektiota), minkä jälkeen annetaan 300 mg kahden viikon välein.
Lapset ja nuoret (6–17-vuotiaat)
Dupilumabin suositeltu annos lapsille ja nuorille (6–17-vuotiaille) potilaille on esitetty taulukossa 6.
Taulukko 6: Lapsille ja nuorille (6–17-vuotiaille) kroonista spontaania urtikariaa sairastaville potilaille ihon alle annettava dupilumabiannos*
| Paino | Aloitusannos | Seuraavat annokset |
vähintään 15 kg mutta alle 30 kg | 300 mg (yksi 300 mg:n injektio) päivänä 1, ja sitten 300 mg päivänä 15 | 300 mg neljän viikon välein alkaen 4 viikon kuluttua päivänä 15 annetusta annoksesta |
| vähintään 30 kg mutta alle 60 kg | 400 mg (kaksi 200 mg:n injektiota) | 200 mg kahden viikon välein |
| vähintään 60 kg | 600 mg (kaksi 300 mg:n injektiota) | 300 mg kahden viikon välein |
*Vähintään 5 kg mutta alle 15 kg painaville potilaille suositeltu annos on 200 mg neljän viikon välein.
2–5-vuotiaat lapset
Dupilumabin suositeltu annos 2–5-vuotiaille lapsille on esitetty taulukossa 7.
Taulukko 7: 2–5-vuotiaille kroonista spontaania urtikariaa sairastaville lapsille ihon alle annettava dupilumabiannos
Paino | Aloitusannos ja seuraavat annokset |
vähintään 5 kg mutta alle 15 kg | 200 mg neljän viikon välein |
vähintään 15 kg mutta alle 30 kg | 300 mg neljän viikon välein |
Yli 24 viikon pituista dupilumabin käyttöä ei ole tutkittu kroonisen spontaanin urtikarian hoidossa. 24 viikon jälkeen hoidon jatkamisen tarvetta tulee arvioida säännöllisesti. Hoidon lopettamista on harkittava kroonista spontaania urtikariaa sairastavilla potilailla, joilla ei ole todettu hoitovastetta 24 viikon hoidon jälkeen.
Unohtunut annos
Jos kerran viikossa otettava annos jää väliin, se on annettava mahdollisimman pian, ja tästä antopäivästä alkaen aloitetaan uusi annosaikataulu.
Jos kahden viikon välein otettava annos jää väliin, injektio on annettava 7 vuorokauden kuluessa siitä, kun väliin jäänyt annos olisi pitänyt antaa. Tämän jälkeen jatketaan potilaan alkuperäisen annosaikataulun mukaan. Jos väliin jäänyttä annosta ei anneta 7 vuorokauden kuluessa, on odotettava, kunnes on alkuperäisen aikataulun mukaan aika antaa seuraava annos.
Jos neljän viikon välein otettava annos jää väliin, injektio on annettava 7 vuorokauden kuluessa siitä, kun väliin jäänyt annos olisi pitänyt antaa. Tämän jälkeen jatketaan potilaan alkuperäisen annosaikataulun mukaan. Jos väliin jäänyttä annosta ei anneta 7 vuorokauden kuluessa, se on annettava mahdollisimman pian, ja tästä antopäivästä alkaen aloitetaan uusi annosaikataulu.
Erityisryhmät
Iäkkäät
Annoksen muuttaminen ei ole tarpeen iäkkäillä (≥65‑vuotiaat) potilailla (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta. Vaikeaa munuaisten vajaatoimintaa sairastavista potilaista on hyvin vähän tietoa (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Maksan vajaatoimintaa sairastavista potilaista ei ole saatavilla tietoja (ks. kohta Farmakokinetiikka).
Paino
Annoksen muuttamista potilaan painon perusteella ei suositella vähintään 12-vuotiaille astmaa sairastaville potilaille eikä atooppista ihottumaa tai kroonista spontaania urtikariaa sairastaville aikuisille (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Dupilumabin turvallisuutta ja tehoa alle 6 kuukauden ikäisten atooppista ihottumaa sairastavien lasten hoidossa ei ole varmistettu. Dupilumabin turvallisuutta ja tehoa alle 5 kg painavien lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Dupilumabin turvallisuutta ja tehoa alle 6-vuotiaiden vaikeaa astmaa sairastavien lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Dupilumabin turvallisuutta ja tehoa alle 1‑vuotiaiden tai alle 15 kg painavien eosinofiilistä ruokatorvitulehdusta sairastavien lasten hoidossa ei ole varmistettu.
Dupilumabin turvallisuutta ja tehoa alle 2-vuotiaiden kroonista spontaania urtikariaa sairastavien lasten hoidossa ei ole varmistettu.
Antotapa
Ihon alle
Esitäytetty dupilumabikynä on tarkoitettu käytettäväksi aikuisille sekä 2-vuotiaille ja sitä vanhemmille pediatrisille potilaille. Esitäytetty dupilumabiruisku on tarkoitettu käytettäväksi aikuisille sekä 6 kuukauden ikäisille ja sitä vanhemmille pediatrisille potilaille. Esitäytettyä dupilumabikynää ei ole tarkoitettu käytettäväksi alle 2-vuotiaille lapsille.
Dupilumabi annetaan injektiona ihon alle reiteen tai vatsaan, mutta ei alle 5 cm:n etäisyydelle navasta. Jos toinen henkilö antaa injektion, voidaan käyttää myös olkavartta.
Jokainen esitäytetty ruisku tai esitäytetty kynä on tarkoitettu vain kerta-antoon.
Jos käyttöaihe edellyttää 400 mg:n aloitusannosta (ks. Annostus kohdassa Annostus ja antotapa), annetaan kaksi 200 mg:n injektiota peräkkäin eri pistoskohtiin.
Pistoskohdan vaihtamista suositellaan jokaisella pistoskerralla. Vältä dupilumabin pistämistä aristavaan tai vaurioituneeseen ihoon tai ihoalueelle, jossa on mustelmia tai arpia.
Potilas voi pistää dupilumabi-injektion itse tai potilaan huoltaja voi antaa injektion, jos hoidosta vastaava terveydenhuollon ammattilainen katsoo tämän tarkoituksenmukaiseksi. Tätä ennen anna potilaalle ja/tai hänen huoltajalleen perusteellinen opastus dupilumabin käyttökuntoon saattamisesta ja antamisesta pakkausselosteen lopussa olevan käyttöohjeen mukaan. Vähintään 12-vuotiaille nuorille suositellaan, että dupilumabipistoksen antaa aikuinen tai pistos annetaan aikuisen valvonnassa. 6 kuukauden – alle 12 vuoden ikäisille lapsille suositellaan, että huoltaja antaa dupilumabipistoksen.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Astman tai keuhkoahtaumataudin akuutit pahenemisvaiheet
Dupilumabia ei saa käyttää astman tai keuhkoahtaumataudin akuuttien oireiden tai akuuttien pahenemisvaiheiden hoitoon. Dupilumabia ei saa käyttää akuutin bronkospasmin tai status asthmaticus ‑tilan hoitoon.
Kortikosteroidit
Systeemisten, paikallisten tai inhaloitavien kortikosteroidien käytön äkillistä keskeyttämistä dupilumabihoidon aloittamisen yhteydessä ei suositella. Jos kortikosteroidiannoksen pienentäminen on tarpeen, se on tehtävä asteittain lääkärin suorassa valvonnassa. Kortikosteroidiannoksen pienentämiseen saattaa liittyä systeemisiä vieroitusoireita, ja/tai annoksen pienentäminen saattaa tuoda esiin sairauksia, joita systeeminen kortikosteroidihoito on aiemmin peittänyt.
Systeemisten kortikosteroidien käyttö voi vähentää tyypin 2 tulehdukseen liittyvien biomerkkiaineiden pitoisuuksia. Tämä on otettava huomioon, kun arvioidaan, liittyykö oraalisia kortikosteroideja käyttävän potilaan sairauteen tyypin 2 tulehdusta (ks. kohta Farmakodynamiikka).
Yliherkkyys
Jos potilaalla ilmenee välitön tai viivästynyt systeeminen yliherkkyysreaktio, keskeytä dupilumabin antaminen välittömästi ja on aloitettava asianmukainen hoito. Joitakin tapauksia anafylaktisia reaktioita, angioedeemaa ja seerumitautia tai seerumitaudin kaltaista reaktiota on ilmoitettu. Anafylaktisten reaktioiden ja angioedeeman ilmaantumiseen kulunut viive dupilumabi-injektion jälkeen on ollut muutamasta minuutista seitsemään päivään asti (ks. kohta Haittavaikutukset).
Eosinofiiliset sairaudet
Astman tutkimusohjelmaan osallistuneilla, dupilumabia saaneilla aikuispotilailla on ilmoitettu eosinofiilisen keuhkokuumeen tapauksia ja eosinofiilisen granulomatoottisen polyangiitin (EGPA) tunnusmerkit täyttäneitä vaskuliittitapauksia. Kroonisen polypoottisen rinosinuiitin tutkimusohjelmaan osallistuneilla, dupilumabia tai lumelääkettä saaneilla ja samanaikaista astmaa sairastavilla aikuispotilailla on ilmoitettu EGPA:n tunnusmerkit täyttäneitä vaskuliittitapauksia. Lääkärin on tarkkailtava vaskuliittiin liittyvää ihottumaa, keuhko-oireiden pahenemista, sydänkomplikaatioita ja/tai neuropatiaa niillä potilailla, joilla esiintyy eosinofiliaa. Astmaan hoitoa saavilla potilailla saattaa ilmetä vakavaa systeemistä eosinofiliaa, johon liittyy joskus eosinofiilisen keuhkokuumeen kliinisiä piirteitä tai eosinofiilisen granulomatoottisen polyangiitin tunnusmerkit täyttävän vaskuliitin kliinisiä piirteitä. Näitä sairauksia hoidetaan usein systeemisillä kortikosteroideilla. Tällaisiin tapahtumiin saattaa yleensä, mutta ei aina, liittyä suun kautta annettavan kortikosteroidihoidon vähentäminen.
Loismatoinfektio
Potilaat, joilla oli todettu loismatoinfektio, suljettiin pois kliinisistä tutkimuksista. Koska dupilumabi estää IL-4/IL-13-signaalivälitystä, se saattaa vaikuttaa immuunivasteeseen matoinfektioita kohtaan. Potilaiden mahdollinen matoinfektio on hoidettava ennen dupilumabihoidon aloittamista. Jos potilas saa infektion dupilumabihoidon aikana eikä matoinfektioiden hoitoon tarkoitetuilla lääkkeillä saada hoitovastetta, dupilumabihoito on keskeytettävä, kunnes infektio on hävinnyt. Astmaa koskeneeseen pediatriseen kehitysohjelmaan osallistuneilla 6–11‑vuotiailla lapsilla ilmoitettiin kihomatotautitapauksia (ks. kohta Haittavaikutukset).
Silmän sidekalvo- ja sarveiskalvotulehdukseen liittyvät tapahtumat
Dupilumabin käytön yhteydessä on ilmoitettu sidekalvo- ja sarveiskalvotulehdukseen liittyviä tapahtumia erityisesti potilailla, joilla on atooppinen ihottuma. Osa potilaista ilmoitti sidekalvo- tai sarveiskalvotulehdukseen liittyneen näköhäiriöitä (esim. näön hämärtymistä) (ks. kohta Haittavaikutukset).
Kehota potilaita ilmoittamaan uusista tai pahenevista silmäoireista terveydenhuollon ammattilaiselle. Dupilumabia saaneille potilaille, joille kehittyy sidekalvotulehdus, joka ei parane tavanomaisella hoidolla, tai sarveiskalvotulehdukseen viittaavia oireita, on tehtävä tarvittaessa silmätutkimus (ks. kohta Haittavaikutukset).
Samanaikaisesti astmaa sairastavat potilaat
Neuvo dupilumabia käyttäviä, samanaikaisesti astmaa sairastavia potilaita, että heidän ei pidä itse säätää tai lopettaa astmalääkkeiden käyttöä keskustelematta lääkärin kanssa. Seuraa samanaikaista astmaa sairastavien potilaiden tilaa on seurattava huolellisesti dupilumabihoidon lopettamisen jälkeen.
Rokotukset
Elävien rokotteiden ja elävien heikennettyjen rokotteiden samanaikaista käyttöä dupilumabin kanssa on vältettävä, sillä samanaikaisen käytön kliinistä turvallisuutta ja tehoa ei ole tutkittu. Suositellaan, että potilaille annetaan kaikki ajantasaiset elävät ja elävät heikennetyt rokotteet voimassaolevien rokotussuositusten mukaisesti ennen dupilumabihoidon aloittamista. Ei ole saatavilla kliinisiä tietoja, joiden pohjalta voitaisiin antaa tarkempia ohjeita elävien rokotteiden tai elävien heikennettyjen rokotteiden antamisesta dupilumabia käyttäville potilaille. Immuunivasteita DTaP- ja meningokokkipolysakkaridirokotteelle on tutkittu (ks. kohta Yhteisvaikutukset).
Natriumpitoisuus
Tämä lääkevalmiste sisältää natriumia alle 1 mmol (23 mg) per 200 mg:n annos eli sen voidaan sanoa olevan "natriumiton”.
Polysorbaatti 80 (E433)
Tämä lääkevalmiste sisältää 2,28 mg polysorbaatti 80:tä per 200 mg:n (1,14 ml:n) annos. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Immuunivasteita rokotukselle arvioitiin tutkimuksessa, jossa atooppista ihottumaa sairastavat potilaat saivat dupilumabia 300 mg kerran viikossa 16 viikon ajan. 12 viikon kuluttua dupilumabin antamisesta potilaat rokotettiin T-soluista riippuvaisella DTaP-rokotteella tai T-soluista riippumattomalla meningokokkipolysakkaridirokotteella, ja immuunivasteet arvioitiin 4 viikon kuluttua. Vasta-ainevasteet sekä tetanus- että meningokokkipolysakkaridirokotteelle olivat samanlaiset dupilumabia ja lumelääkettä saaneilla tutkittavilla. Tutkimuksessa ei todettu haitallisia yhteisvaikutuksia kummankaan ei-eläviä taudinaiheuttajia sisältävän rokotteen ja dupilumabin välillä.
Näin ollen dupilumabia saaville potilaille voidaan samanaikaisesti antaa tällä hetkellä käytössä olevia inaktivoituja tai ei-eläviä taudinaiheuttajia sisältäviä rokotteita. Lisätietoa elävistä rokotteista, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Atooppista ihottumaa sairastavilla potilailla tehdyssä kliinisessä tutkimuksessa arvioitiin CYP-substraattien farmakokinetiikkaa. Tästä tutkimuksesta kerätyt tiedot eivät viitanneet siihen, että dupilumabilla olisi kliinisesti merkittäviä vaikutuksia CYP1A2‑, CYP3A4‑, CYP2C19‑, CYP2D6- tai CYP2C9-entsyymien toimintaan.
Dupilumabilla ei odoteta olevan vaikutusta samanaikaisesti annettujen muiden lääkevalmisteiden
farmakokinetiikkaan. Populaatioanalyysin perusteella yleisesti samanaikaisesti annetuilla lääkevalmisteilla ei ollut vaikutusta dupilumabin farmakokinetiikkaan keskivaikeaa tai vaikeaa astmaa sairastavilla potilailla.
Raskaus ja imetys
Raskaus
On vain vähän tietoja dupilumabin käytöstä raskaana olevilla naisilla. Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Dupilumabia saa käyttää raskauden aikana vain, jos hoidosta potilaalle mahdollisesti koituvat hyödyt ovat suuremmat kuin mahdolliset sikiölle aiheutuvat riskit.
Imetys
Ei tiedetä, erittyykö dupilumabi ihmisen rintamaitoon tai imeytyykö se nieltynä systeemisesti. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko dupilumabihoito, ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Eläinkokeissa ei todettu hedelmällisyyden heikkenemistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Dupilumabilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimpiä haittavaikutuksia atooppisen ihottuman, astman ja kroonisen polypoottisen rinosinuiitin hoidon yhteydessä ovat injektiokohdan reaktiot (mukaan lukien punoitus, edeema, kutina, kipu ja turpoaminen), silmän sidekalvotulehdus, allerginen sidekalvotulehdus, artralgia, huuliherpes ja eosinofilia. Lisäksi eosinofiilisen ruokatorvitulehduksen, keuhkoahtaumataudin ja kroonisen spontaanin urtikarian hoidon yhteydessä ilmoitettiin haittavaikutuksena injektiokohdan mustelma tai hematooma. Keuhkoahtaumataudin ja kroonisen spontaanin urtikarian hoidon yhteydessä ilmoitettiin haittavaikutuksina myös injektiokohdan kovettuma ja injektiokohdan dermatiitti. Keuhkoahtaumataudin hoidon yhteydessä ilmoitettiin haittavaikutuksena myös injektiokohdan ihottuma. Joitakin harvinaisia tapauksia, joissa potilaalla on ilmennyt seerumitauti, seerumitaudin kaltainen reaktio, anafylaktinen reaktio tai haavainen sarveiskalvotulehdus, on ilmoitettu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Taulukossa 8 esitetyt dupilumabin turvallisuustiedot on saatu ensisijaisesti 12 satunnaistetusta, lumekontrolloidusta tutkimuksesta, joihin osallistui atooppista ihottumaa, astmaa ja kroonista polypoottista rinosinuiittia sairastavia potilaita. Näihin tutkimuksiin osallistuneista potilaista 4 206 potilasta sai dupilumabia ja 2 326 potilasta sai lumelääkettä kontrolloidun vaiheen aikana, ja ne edustavat dupilumabin yleistä turvallisuusprofiilia.
Taulukossa 8 on lueteltu kliinisissä tutkimuksissa ja/tai myyntiluvan myöntämisen jälkeisessä seurannassa havaitut haittavaikutukset elinjärjestelmän ja esiintymistiheyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (saatavilla oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 8: Luettelo haittavaikutuksista
| MedDRA-elinjärjestelmä | Esiintymistiheys | Haittavaikutus |
| Infektiot | Yleinen | Sidekalvotulehdus* Huuliherpes* |
| Veri ja imukudos | Yleinen | Eosinofilia |
| Immuunijärjestelmä | Melko harvinainen | Angioedeema# |
| Harvinainen | Anafylaktinen reaktio Seerumitauti Seerumitaudin kaltainen reaktio | |
| Silmät | Yleinen | Allerginen sidekalvotulehdus* |
| Melko harvinainen | Sarveiskalvotulehdus*# Luomitulehdus*† Silmien kutina*† Kuivat silmät*† | |
| Harvinainen | Haavainen sarveiskalvotulehdus*† | |
| Iho ja ihonalainen kudos | Melko harvinainen | Kasvoihottuma# |
| Luusto, lihakset ja sidekudos | Yleinen | Artralgia# |
| Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Pistoskohdan reaktiot (mukaan lukien punoitus, edeema, kutina, kipu, turvotus ja mustelma) |
*Silmissä havaitut haittavaikutukset ja huuliherpes ilmenivät pääasiassa atooppista ihottumaa koskevissa tutkimuksissa.
†Silmien kutinaa, luomitulehdusta ja silmien kuivuutta todettiin esiintymistiheydellä yleinen ja haavaista sarveiskalvotulehdusta esiintymistiheydellä melko harvinainen atooppista ihottumaa koskevissa tutkimuksissa.
# Myyntiluvan myöntämisen jälkeisessä seurannassa.
Valikoitujen haittavaikutusten kuvaus
Yliherkkyys
Dupilumabin antamisen jälkeen on ilmoitettu anafylaktisia reaktioita, angioedeemaa ja seerumitautia tai seerumitaudin kaltaisia reaktioita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Silmän sidekalvo- ja sarveiskalvotulehdukseen liittyvät tapahtumat
Atooppista ihottumaa arvioineissa tutkimuksissa silmän sidekalvo- ja sarveiskalvotulehdusta ilmeni enemmän dupilumabia saaneilla atooppista ihottumaa sairastavilla potilailla verrattuna lumevalmistetta saaneisiin potilaisiin. Useimmilla potilailla sidekalvo- tai sarveiskalvotulehdus parani tai alkoi lievittyä hoitojakson aikana. Atooppista ihottumaa arvioineessa pitkäkestoisessa avoimessa jatkotutkimuksessa (AD-1225) silmän sidekalvo- ja sarveiskalvotulehduksen esiintyvyydet olivat 5 vuoden kohdalla edelleen vastaavat kuin lumekontrolloitujen atooppista ihottumaa arvioineiden tutkimusten dupilumabihaarassa. Astma- ja keuhkoahtaumatautipotilailla sidekalvo- tai sarveiskalvotulehduksen esiintyvyys oli pieni ja oiretta esiintyi yhtä paljon dupilumabi- ja lumeryhmissä. Kroonista polypoottista rinosinuiittia tai kyhmykutinaa (prurigo nodularis) sairastavilla potilailla sidekalvotulehdusta ilmeni useammin dupilumabi- kuin lumeryhmissä, mutta harvemmin kuin atooppista ihottumaa sairastavilla potilailla. Eosinofiilistä ruokatorvitulehdusta tai kroonista spontaania urtikariaa sairastavilla potilailla sidekalvotulehduksen esiintyvyys oli pieni ja oiretta esiintyi yhtä paljon dupilumabi- ja lumeryhmissä. Kroonisen polypoottisen rinosinuiitin, kyhmykutinan, eosinofiilisen ruokatorvitulehduksen ja kroonisen spontaanin urtikarian tutkimusohjelmissa ei ilmoitettu yhtäkään sarveiskalvotulehdustapausta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Herpeettinen ekseema
Herpeettistä ekseemaa ilmoitettiin alle 1 %:lla tutkittavista dupilumabiryhmissä ja alle 1 %:lla lumelääkeryhmässä 16 viikon pituisissa atooppista ihottumaa koskeneissa aikuisilla tehdyissä monoterapiatutkimuksissa. Dupilumabin ja paikallisen kortikosteroidin yhdistelmää aikuisilla arvioineessa 52 viikkoa kestäneessä atooppista ihottumaa koskeneessa tutkimuksessa herpeettistä ekseemaa ilmoitettiin 0,2 %:lla tutkittavista dupilumabin ja paikallisen kortikosteroidin yhdistelmää saaneiden ryhmässä ja 1,9 %:lla lumelääkkeen ja paikallisen kortikosteroidin yhdistelmää saaneiden ryhmässä. Pitkäkestoisessa avoimessa jatkotutkimuksessa (AD-1225) nämä esiintyvyydet olivat 5 vuoden kohdalla pysyneet vakaina.
Eosinofilia
Atooppisen ihottuman, astman, kroonisen polypoottisen rinosinuiitin, keuhkoahtaumataudin ja kroonisen spontaanin urtikarian käyttöaiheissa dupilumabia saaneilla potilailla eosinofiilien määrä suureni aluksi keskimäärin enemmän kuin lumelääkettä saaneilla potilailla lähtötilanteeseen verrattuna. Tutkimushoidon aikana eosinofiiliarvot pienenivät lähelle lähtötilanteen arvoja ja palautuivat lähtötasolle astman hoidon turvallisuutta arvioivan avoimen jatkotutkimuksen aikana (TRAVERSE). Pitkäkestoisessa avoimessa jatkotutkimuksessa (AD-1225) keskimääräiset veren eosinofiilipitoisuudet pienenivät alle lähtöarvon viikkoon 20 mennessä ja arvot pysyivät tällaisina jopa 5 vuoden ajan. Kyhmykutinan yhteydessä keskimääräisten veren eosinofiilipitoisuuksien ei todettu suurentuneen verrattuna lumelääkettä saaneisiin (PRIME ja PRIME2). Eosinofiilisen ruokatorvitulehduksen käyttöaiheessa ja keuhkoahtaumataudin käyttöaiheessa (BOREAS ja NOTUS) veren eosinofiilimäärien keskiarvot ja mediaanit pienenivät lähelle lähtötilanteen arvoja tai pysyivät lähtötilanteen arvoja pienempinä tutkimushoidon aikana.
Hoidon aikana ilmaantunutta eosinofiliaa (vähintään 5 000 solua/µl) havaittiin alle 3 %:lla dupilumabia saaneista ja alle 0,5 %:lla lumelääkettä saaneista potilaista (SOLO1-, SOLO2-, AD‑1021‑, DRI12544-, QUEST- ja VOYAGE-tutkimuksissa; SINUS-24- ja SINUS-52-tutkimuksissa; PRIME- ja PRIME2-tutkimuksissa; TREET-tutkimuksen osissa A ja B; BOREAS- ja NOTUS-tutkimuksissa; ja CUPID-tutkimuksissa A, B ja C).
AD‑1539-tutkimuksessa havaittiin hoidon aikana ilmaantunutta eosinofiliaa (vähintään 5 000 solua/µl) 8,4 %:lla dupilumabia saaneista ja 0 %:lla lumelääkettä saaneista potilaista. Hoitojakson päättyessä eosinofiilimäärien mediaani oli pienentynyt lähtötilanteen arvoja pienemmäksi.
Infektiot
Atooppista ihottumaa koskeneissa 16 viikon pituisissa aikuisilla tehdyissä kliinisissä monoterapiatutkimuksissa ilmoitettiin vakavia infektioita 1,0 %:lla lumelääkettä saaneista potilaista ja 0,5 %:lla dupilumabia saaneista potilaista. 52 viikon mittaisessa atooppista ihottumaa aikuisilla koskeneessa CHRONOS-tutkimuksessa ilmoitettiin vakavia infektioita 0,6 %:lla lumelääkettä saaneista potilaista ja 0,2 %:lla dupilumabia saaneista potilaista. Pitkäkestoisessa avoimessa jatkotutkimuksessa (AD-1225) vakavien infektioiden esiintyvyydet olivat 5 vuoden kohdalla pysyneet vakaina.
Infektioiden kokonaisilmaantuvuus dupilumabia saaneilla potilailla ei ollut suurempi kuin lumelääkettä saaneilla astmaa koskeneiden kliinisten tutkimusten yhdistetyissä turvallisuustiedoissa. 24 viikon mittaisella ajanjaksolla tarkastelluissa yhdistetyissä turvallisuustiedoissa ilmoitettiin vakavia infektioita 1,0 %:lla dupilumabia saaneista potilaista ja 1,1 %:lla lumelääkettä saaneista potilaista. 52 viikon mittaisessa QUEST-tutkimuksessa ilmoitettiin vakavia infektioita 1,3 %:lla dupilumabia saaneista potilaista ja 1,4 %:lla lumelääkettä saaneista potilaista.
Infektioiden kokonaisilmaantuvuus dupilumabia saaneilla potilailla ei ollut suurempi kuin lumelääkettä saaneilla kroonista polypoottista rinosinuiittia koskeneiden kliinisten tutkimusten yhdistetyissä turvallisuustiedoissa. 52 viikon mittaisessa SINUS‑52-tutkimuksessa ilmoitettiin vakavia infektioita 1,3 %:lla dupilumabia saaneista potilaista ja 1,3 %:lla lumelääkettä saaneista potilaista.
Infektioiden kokonaisilmaantuvuus dupilumabia saaneilla potilailla ei ollut suurempi kuin lumelääkettä saaneilla kyhmykutinaa koskeneiden kliinisten tutkimusten yhdistetyissä turvallisuustiedoissa. Yhdistetyissä turvallisuustiedoissa ilmoitettiin vakavia infektioita 1,3 %:lla dupilumabia saaneista potilaista ja 1,3 %:lla lumelääkettä saaneista potilaista.
Infektioiden kokonaisilmaantuvuus oli numeerisesti suurempi dupilumabia saaneilla potilailla (32,0 %) kuin lumelääkettä saaneilla (24,8 %) eosinofiilistä ruokatorvitulehdusta koskeneiden TREET-tutkimusten (osat A ja B) yhdistetyissä turvallisuustiedoissa 24 viikon ajalta. Infektioiden kokonaisilmaantuvuus oli numeerisesti suurempi lumelääkettä saaneilla potilailla (41,2 %) kuin dupilumabia saaneilla (35,8 %) eosinofiilistä ruokatorvitulehdusta koskeneessa EoE KIDS ‑tutkimuksessa (osa A). Eosinofiilistä ruokatorvitulehdusta koskeneiden TREET-tutkimusten (osat A ja B) 24 viikon mittaisella ajanjaksolla tarkastelluissa yhdistetyissä turvallisuustiedoissa vakavia infektioita ilmoitettiin 0,5 %:lla dupilumabia saaneista potilaista ja 0 %:lla lumelääkettä saaneista potilaista. Eosinofiilistä ruokatorvitulehdusta koskeneessa EoE KIDS ‑tutkimuksessa (osa A) ei ilmoitettu vakavia infektioita. Ylähengitystieinfektioiden (joihin kuului useita haittavaikutustermejä, kuten COVID‑19‑infektio, sinuiitti ja ylähengitystieinfektio) ilmaantuvuus oli numeerisesti suurempi dupilumabia saaneilla potilailla (17,2 %) kuin lumelääkettä saaneilla (10,3 %) eosinofiilistä ruokatorvitulehdusta koskeneessa TREET ‑tutkimuksessa (osat A ja B) ja numeerisesti suurempi dupilumabia saaneilla (26,9 %) kuin lumelääkettä saaneilla (20,6 %) eosinofiilistä ruokatorvitulehdusta koskeneessa EoE KIDS ‑tutkimuksessa (osa A).
Infektioiden kokonaisilmaantuvuus dupilumabia saaneilla potilailla ei ollut suurempi kuin lumelääkettä saaneilla keuhkoahtaumatautia koskeneiden kliinisten tutkimusten yhdistetyissä turvallisuustiedoissa. Vakavia infektioita ilmoitettiin 4,9 %:lla dupilumabia saaneista potilaista ja 4,8 %:lla lumelääkettä saaneista potilaista.
CUPID-tutkimusten (A, B ja C) yhdistetyissä turvallisuustiedoissa infektioiden kokonaisilmaantuvuus dupilumabia saaneilla potilailla ei ollut suurempi kuin lumelääkettä saaneilla. Yhdistetyissä turvallisuustiedoissa havaittiin vakavia infektioita 0,5 %:lla dupilumabia saaneista potilaista ja 0,5 %:lla lumelääkettä saaneista potilaista. PKM16982-tutkimuksessa ei havaittu vakavia infektioita dupilumabia saaneilla lapsilla.
Immunogeenisuus
Kaikkien terapeuttisten proteiinien tavoin myös dupilumabiin liittyy immunogeenisuuden mahdollisuus.
Lääkevasta-ainemäärityksissä (anti-drug-antibody, ADA) todetuilla vasteilla ei yleensä ollut vaikutusta dupilumabialtistukseen tai dupilumabin turvallisuuteen tai tehoon.
Noin 5 %:lla atooppista ihottumaa, astmaa tai kroonista polypoottista rinosinuiittia sairastavista potilaista, jotka saivat dupilumabia 300 mg kahden viikon välein 52 viikon ajan, kehittyi vasta-aineita dupilumabille; noin 2 %:lla todettiin pitkäkestoisia ADA-vasteita ja noin 2 %:lla todettiin neutraloivia vasta-aineita.
Samankaltaisia tuloksia havaittiin kyhmykutinaa sairastavilla aikuisilla, jotka saivat dupilumabia 300 mg kahden viikon välein 24 viikon ajan, atooppista ihottumaa sairastavilla lapsilla (6 kuukauden – 11 vuoden ikäisillä), jotka saivat dupilumabia joko 200 mg kahden viikon välein, 200 mg neljän viikon välein tai 300 mg neljän viikon välein 16 viikon ajan, sekä astmaa sairastavilla lapsilla (6–11‑vuotiailla), jotka saivat dupilumabia 100 mg kahden viikon välein tai 200 mg kahden viikon välein 52 viikon ajan. Samankaltaisia ADA-vasteita atooppista ihottumaa sairastavilla aikuisilla, jotka saivat dupilumabia enintään 5 vuoden ajan pitkäkestoisessa avoimessa jatkotutkimuksessa (AD-1225).
Noin 16 %:lle nuorista atooppista ihottumaa sairastavista potilaista, jotka saivat 300 mg tai 200 mg dupilumabia kahden viikon välein 16 viikon ajan, kehittyi vasta-aineita dupilumabille; noin 3 %:lla todettiin pitkäkestoisia ADA-vasteita ja noin 5 %:lla todettiin neutraloivia vasta-aineita.
Noin 9 %:lle astmapotilaista, jotka saivat dupilumabia 200 mg kahden viikon välein 52 viikon ajan, kehittyi vasta-aineita dupilumabille; noin 4 %:lla todettiin pitkäkestoisia ADA-vasteita ja noin 4 %:lla todettiin neutraloivia vasta-aineita.
Noin 1 %:lle eosinofiilistä ruokatorvitulehdusta sairastavista vähintään 1‑vuotiaista potilaista, jotka saivat dupilumabia 300 mg kerran viikossa (≥ 40 kg), 300 mg kahden viikon välein (≥ 30 –< 60 kg), 200 mg kahden viikon välein (≥ 15 –< 30 kg) tai 100 mg kahden viikon välein (≥ 5 –< 15 kg) 52 viikon ajan, kehittyi vasta-aineita dupilumabille; ADA‑vasteet eivät olleet pitkäkestoisia eivätkä neutraloivia.
Noin 8 %:lle keuhkoahtaumatautia sairastavista potilaista, jotka saivat dupilumabia 300 mg kahden viikon välein 52 viikon ajan, kehittyi vasta‑aineita dupilumabille; noin 3 %:lla todettiin pitkäkestoisia ADA‑vasteita ja noin 3 %:lla todettiin neutraloivia vasta‑aineita.
Noin 4,7 %:lle kroonista spontaania urtikariaa sairastavista aikuisista potilaista, jotka saivat dupilumabia 300 mg kahden viikon välein 24 viikon ajan, ja kroonista spontaania urtikariaa sairastavista nuorista potilaista, jotka saivat dupilumabia 300 mg tai 200 mg kahden viikon välein 24 viikon ajan, kehittyi vasta-aineita dupilumabille; noin 0,5 %:lla todettiin pitkäkestoisia ADA-vasteita ja noin 1 %:lla todettiin neutraloivia vasta-aineita. Kenellekään 2–11-vuotiaista kroonista spontaania urtikariaa sairastavista pediatrisista potilaista, jotka saivat dupilumabia 200 mg neljän viikon välein, 300 mg neljän viikon välein tai 200 mg kahden viikon välein 24 viikon ajan, ei kehittynyt positiivista ADA-vastetta dupilumabille.
Iästä ja populaatiosta riippumatta enintään 7 %:lla lumelääkeryhmien potilaista saatiin positiivinen tulos dupilumabivasta-aineille; enintään 3 %:lla todettiin pitkäkestoisia ADA-vasteita ja enintään 2 %:lla todettiin neutraloivia vasta-aineita.
Alle 1 %:lla hyväksyttyä dupilumabiannosta saaneista potilaista todettiin suuren titterin ADA-vasteita, joihin liittyi pienentynyt altistus ja heikentynyt teho. Lisäksi yhdellä potilaalla todettiin seerumitauti ja yhdellä seerumitaudin kaltainen reaktio (alle 0,1 %), joihin liittyi suuria ADA-tittereitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Atooppinen ihottuma
Nuoret (12–17‑vuotiaat)
Dupilumabin turvallisuutta arvioitiin tutkimuksessa, johon osallistui 250 iältään 12–17-vuotiasta potilasta, joilla oli keskivaikea tai vaikea atooppinen ihottuma (AD‑1526). Dupilumabin turvallisuusprofiili näillä potilailla, joita seurattiin viikolle 16 asti, oli samankaltainen kuin atooppista ihottumaa sairastavilla aikuisilla tehdyissä tutkimuksissa todettu turvallisuusprofiili.
6–11‑vuotiaat lapset
Dupilumabin turvallisuutta arvioitiin tutkimuksessa, johon osallistui 367 iältään 6–11‑vuotiasta potilasta, joilla oli vaikea atooppinen ihottuma (AD‑1652). Dupilumabin ja samanaikaisen paikallisen kortikosteroidihoidon (TCS) turvallisuusprofiili näillä potilailla oli viikkoon 16 asti samankaltainen kuin atooppista ihottumaa sairastavilla aikuisilla ja nuorilla tehdyissä tutkimuksissa todettu turvallisuusprofiili.
6 kuukauden – 5 vuoden ikäiset lapset
Dupilumabin ja samanaikaisen paikallisen kortikosteroidihoidon (TCS) turvallisuutta arvioitiin tutkimuksessa, johon osallistui 161 potilasta, jotka olivat 6 kuukauden – 5vuoden ikäisiä ja joilla oli keskivaikea tai vaikea atooppinen ihottuma (AD‑1539). Tutkimuksen alaryhmässä oli 124 potilasta, joilla oli vaikea atooppinen ihottuma. Dupilumabin ja samanaikaisen paikallisen kortikosteroidihoidon (TCS) turvallisuusprofiili tässä ikäryhmässä oli viikkoon 16 asti samankaltainen kuin atooppista ihottumaa sairastavilla aikuisilla ja 6–17‑vuotiailla pediatrisilla potilailla tehdyissä tutkimuksissa todettu turvallisuusprofiili.
Käsien ja jalkaterien atooppinen ihottuma
Dupilumabin turvallisuutta arvioitiin tutkimuksessa, johon osallistui 27 pediatrista potilasta, jotka olivat 12–17-vuotiaita ja joilla oli keskivaikea tai vaikea käsien ja jalkaterien atooppinen ihottuma (AD-1924). Dupilumabin turvallisuusprofiili näillä potilailla viikkoon 16 asti vastasi keskivaikeaa tai vaikeaa atooppista ihottumaa sairastavilla aikuisilla ja vähintään 6 kuukauden ikäisillä pediatrisilla potilailla tehdyissä tutkimuksissa todettua turvallisuusprofiilia.
Astma
Nuoret (12–17‑vuotiaat)
52 viikon mittaiseen QUEST-tutkimukseen otettiin mukaan yhteensä 107 astmaa sairastavaa 12–17-vuotiasta nuorta. Havaittu turvallisuusprofiili oli aikuisilla todetun kaltainen.
Dupilumabin pitkäaikaiskäytön turvallisuutta arvioitiin 89 nuorella potilaalla, jotka osallistuivat keskivaikeaa tai vaikeaa astmaa käsittelevään avoimeen jatkotutkimukseen (TRAVERSE). Tutkimuksessa potilaita seurattiin enintään 96 viikon ajan. Dupilumabin turvallisuusprofiili TRAVERSE-tutkimuksessa vastasi keskeisissä astmatutkimuksissa enintään 52 hoitoviikon aikana havaittua turvallisuusprofiilia.
6–11‑vuotiaat lapset
Keskivaikeaa tai vaikeaa astmaa sairastavilla 6–11‑vuotiailla lapsilla (VOYAGE) ilmoitettiin lisäksi haittavaikutuksena kihomatotautia 1,8 %:lla dupilumabiryhmien tutkittavista (5 tutkittavalla) mutta ei yhdelläkään lumelääkeryhmän tutkittavista. Kaikki kihomatotautitapaukset olivat lieviä tai keskivaikeita, ja tutkittavat toipuivat loismatoinfektioiden hoitoon tarkoitetuilla lääkkeillä ilman dupilumabihoidon keskeyttämistä.
Keskivaikeaa tai vaikeaa astmaa sairastavilla 6–11‑vuotiailla lapsilla ilmoitettiin eosinofiliaa (veren eosinofiilipitoisuus ≥ 3 000 solua/µl tai tutkijan haittatapahtumaksi arvioima tapahtuma) 6,6 %:lla dupilumabiryhmien tutkittavista ja 0,7 %:lla lumelääkeryhmän tutkittavista. Useimmat eosinofiliatapaukset olivat lieviä tai keskivaikeita, eikä niihin liittynyt kliinisiä oireita. Nämä tapaukset olivat ohimeneviä, vähenivät ajan myötä eivätkä johtaneet dupilumabihoidon keskeyttämiseen.
Dupilumabin pitkäaikaiskäytön turvallisuutta arvioitiin avoimessa jatkotutkimuksessa (EXCURSION) keskivaikeaa tai vaikeaa astmaa sairastavilla 6–11‑vuotiailla lapsilla, jotka olivat aiemmin osallistuneet VOYAGE-tutkimukseen. EXCURSION-tutkimukseen mukaan otetuista 365 potilaasta 350 oli mukana 52 viikon mittaisen hoidon loppuun asti ja 228 potilasta oli mukana 104 viikkoa kumulatiivisesti kestäneen hoidon loppuun asti (VOYAGE ja EXCURSION). Dupilumabin pitkän aikavälin turvallisuusprofiili EXCURSION-tutkimuksessa vastasi keskeisessä astmatutkimuksessa (VOYAGE) havaittua turvallisuusprofiilia 52 viikon mittaisen hoidon aikana.
Eosinofiilinen ruokatorvitulehdus
Nuoret (12–17‑vuotiaat)
TREET-tutkimuksiin (osat A ja B) otettiin mukaan yhteensä 99 eosinofiilistä ruokatorvitulehdusta sairastavaa 12–17‑vuotiasta nuorta. Havaittu turvallisuusprofiili oli aikuisilla todetun kaltainen.
1–11‑vuotiaat lapset
Dupilumabin turvallisuutta arvioitiin tutkimuksessa, johon osallistui 101 iältään 1–11‑vuotiasta lasta, joilla oli eosinofiilinen ruokatorvitulehdus (EoE KIDS, osa A). Dupilumabin turvallisuusprofiili näillä potilailla viikkoon 16 asti oli samankaltainen kuin eosinofiilistä ruokatorvitulehdusta sairastavilla aikuisilla ja 12–17‑vuotiailla nuorilla potilailla todettu turvallisuusprofiili.
Yhteensä 98 potilaalle, jotka olivat olleet mukana osan A loppuun asti, tarjottiin mahdollisuutta osallistua 36 viikon mittaiseen aktiivisella hoidolla toteutettuun jatkovaiheeseen (EoE KIDS, osa B). Dupilumabin turvallisuusprofiili oli viikkoon 52 asti samankaltainen kuin viikon 16 kohdalla havaittu turvallisuusprofiili.
Krooninen spontaani urtikaria
Nuoret (12–17-vuotiaat)
Dupilumabin turvallisuutta arvioitiin 12:lla 12–17-vuotiaalla nuorella, joilla oli krooninen spontaani urtikaria ja jotka osallistuivat CUPID-tutkimuksiin (tutkimukset A, B ja C). Yhdellä dupilumabihoitoa saaneella nuorella ilmoitettiin haittatapahtuma.
Lapset (2–11-vuotiaat)
Dupilumabin turvallisuutta arvioitiin 18:lla 2–11-vuotiaalla lapsella, joilla oli krooninen spontaani urtikaria ja jotka osallistuivat PKM16982-tutkimukseen ja CUPID-tutkimuksiin (tutkimukset A ja C). Nämä tiedot vastasivat yleisesti ottaen dupilumabia saaneilla kroonista spontaania urtikariaa sairastavilla potilailla todettua turvallisuusprofiilia.
Pitkäaikaiskäytön turvallisuus
Atooppinen ihottuma
Dupilumabin ja paikallisen kortikosteroidin yhdistelmän (CHRONOS-tutkimus) turvallisuusprofiili aikuisilla atooppista ihottumaa sairastavilla potilailla oli viikolle 52 asti vastaava kuin viikolla 16 havaittu turvallisuusprofiili.
Dupilumabin pitkäaikaiskäytön turvallisuutta arvioitiin avoimessa 6 kuukauden – 17 vuoden ikäisillä keskivaikeaa tai vaikeaa atooppista ihottumaa sairastavilla potilailla tehdyssä jatkotutkimuksessa (AD‑1434). Dupilumabin turvallisuusprofiili viikolle 52 asti seuratuilla potilailla oli samankaltainen kuin viikolla 16 AD‑1526-, AD‑1652- ja AD‑1539-tutkimuksissa havaittu turvallisuusprofiili. Lapsilla ja nuorilla havaittu dupilumabin pitkän aikavälin turvallisuusprofiili vastasi atooppista ihottumaa sairastavilla aikuisilla havaittua turvallisuusprofiilia.
Vaiheen 3 monikeskustutkimuksena toteutetussa avoimessa jatkotutkimuksessa (AD-1225) dupilumabin toistuvien annosten pitkäaikaisturvallisuutta arvioitiin 2 677 aikuisella, joilla oli keskivaikea tai vaikea atooppinen ihottuma. Potilaat saivat 300 mg annoksen viikoittain (99,7 %) ja näistä potilasta 179 osallistui tutkimukseen vähintään 260 viikon ajan. Tässä tutkimuksessa jopa 5 vuoden ajan havaittu dupilumabin pitkäaikaiskäytön turvallisuusprofiili oli yleisesti yhdenmukainen verrokkitutkimuksissa havaitun turvallisuusprofiilin kanssa.
Astma
Dupilumabin turvallisuusprofiili 96 viikon mittaisessa turvallisuustutkimuksessa (TRAVERSE) vastasi keskeisissä astmatutkimuksissa enintään 52 viikon ajan hoitoa saaneilla potilailla havaittua turvallisuusprofiilia.
Dupilumabin turvallisuusprofiili astmaa sairastavilla 6–11‑vuotiailla lapsilla, jotka osallistuivat 52 viikon mittaiseen pitkän aikavälin turvallisuustutkimukseen (EXCURSION), vastasi keskeisessä astmatutkimuksessa (VOYAGE) havaittua turvallisuusprofiilia 52 viikon mittaisen hoidon aikana.
Krooninen polypoottinen rinosinuiitti
Dupilumabin turvallisuusprofiili kroonista polypoottista rinosinuiittia sairastavilla aikuisilla oli viikolle 52 asti vastaava kuin viikon 24 kohdalla havaittu turvallisuusprofiili.
Eosinofiilinen ruokatorvitulehdus
Dupilumabin turvallisuusprofiili aikuisilla ja vähintään 12‑vuotiailla nuorilla (TREET, osa C) ja 1–11‑vuotiailla lapsilla (EoE KIDS, osa B) oli viikkoon 52 asti yleisesti yhdenmukainen TREET‑tutkimuksen osissa A ja B viikon 24 kohdalla ja EoE KIDS ‑tutkimuksen osassa A viikon 16 kohdalla havaitun turvallisuusprofiilin kanssa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Dupilumabin yliannostukseen ei ole spesifistä hoitoa. Yliannostustapauksissa tarkkaile potilasta haittavaikutusten oireiden tai merkkien varalta ja anna potilaalle välittömästi sopivaa oireenmukaista hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut ihotautien lääkkeet, ihottumalääkkeet, lukuun ottamatta kortikosteroideja, ATC-koodi: D11AH05
Vaikutusmekanismi
Dupilumabi on rekombinantti ihmisen monoklonaalinen IgG4-vasta-aine, joka estää interleukiini 4- ja interleukiini 13 ‑signaalivälitystä. Dupilumabi estää IL‑4-signaalivälitystä tyypin I reseptorin (IL4Rα/γc) kautta ja sekä IL‑4- että IL‑13-signaalivälitystä tyypin II reseptorin (IL‑4Rα/IL‑13Rα) kautta. IL‑4 ja IL‑13 ovat keskeisiä ihmisen tyypin 2 tulehdussairauksien, kuten atooppisen ihottuman, astman, eosinofiilisen ruokatorvitulehduksen ja kroonisen spontaanin urtikarian syntyyn vaikuttavia tekijöitä. Potilaiden IL-4/IL-13-signalointireitin toiminnan estäminen dupilumabilla vähentää monien tyypin 2 tulehduksen välittäjäaineiden pitoisuuksia.
Farmakodynaamiset vaikutukset
Kliinisissä atooppisen ihottuman tutkimuksissa dupilumabihoidon yhteydessä todettiin lähtötilanteen arvoista pienentyneitä tyypin 2 immuniteetin biomerkkiaineiden, kuten CCL17-kemokiinin (thymus and activation regulated chemokine, TARC), seerumin kokonais-IgE:n ja seerumin allergeenispesifisen IgE:n, pitoisuuksia. Dupilumabihoidon aikana atooppista ihottumaa sairastavilla aikuisilla ja nuorilla havaittiin atooppisen ekseeman aktiivisuuteen ja vaikeusasteeseen yhdistetyn biomarkkerin, laktaattidehydrogenaasin (LDH), pitoisuuksien pienenemistä.
Aikuisilla ja nuorilla astmapotilailla dupilumabihoito pienensi lumelääkkeeseen verrattuna huomattavasti kliinisissä tutkimuksissa arvioitujen tyypin 2 biomerkkiaineiden pitoisuuksia eli uloshengitysilman typpioksidin (FeNO) pitoisuutta sekä eotaksiini 3:n, kokonais-IgE:n, allergeenispesifisen IgE:n, TARC:n ja periostiinin pitoisuuksia veressä. Nämä tyypin 2 tulehdusreaktion biomerkkiaineiden pitoisuudet pienenivät vastaavanlaisesti sekä annostusohjelmalla 200 mg kahden viikon välein että annostusohjelmalla 300 mg kahden viikon välein. Pediatrisilla (6–11‑vuotiailla) astmapotilailla dupilumabihoito pienensi lumelääkkeeseen verrattuna huomattavasti kliinisissä tutkimuksissa arvioitujen tyypin 2 biomerkkiaineiden pitoisuuksia eli uloshengitysilman typpioksidin (FeNO) pitoisuutta sekä kokonais-IgE:n, allergeenispesifisen IgE:n ja TARC:n pitoisuuksia veressä. Näiden biomerkkiaineiden pitoisuudet laskivat 2 viikon hoidon aikana lähelle alinta mahdollista tasoa lukuun ottamatta IgE:tä, jonka pitoisuus pieneni hitaammin. Vaikutukset biomerkkiaineiden pitoisuuksiin säilyivät koko hoidon ajan.
Kliininen teho ja turvallisuus atooppisen ihottuman hoidossa
Atooppista ihottumaa sairastavat nuoret (12–17-vuotiaat)
Dupilumabin tehoa ja turvallisuutta monoterapiana nuorilla potilailla arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (AD-1526), johon osallistui 251 nuorta (12–17-vuotiasta) potilasta, joilla oli keskivaikea tai vaikea atooppinen ihottuma, mikä määriteltiin niin, että IGA-mittarin (tutkijalääkärin tekemä yleinen arviointi, Investigator’s Global Assessment) pistemäärä oli ≥ 3 kokonaisarviossa atooppisen ihottuman vaikeusastetta kuvaavalla asteikolla 0–4, ihottuman pinta-alaa ja vaikeusastetta kuvaavan EASI-mittarin (Eczema Area and Severity Index) pistemäärä oli ≥ 16 asteikolla 0–72 ja ihottuma-alueen osuus kehon pinta-alasta (BSA) oli vähintään 10 %. Tutkimuksen sisäänottokriteerit täyttävien potilaiden hoitovaste paikallisesti käytettyyn lääkitykseen oli ollut aiemmin riittämätön.
Potilaat saivat dupilumabia ihon alle injektioina, jotka annettiin seuraavasti: joko 1) 400 mg:n aloitusannos dupilumabia (kaksi 200 mg:n injektiota) päivänä 1, minkä jälkeen 200 mg dupilumabia kahden viikon välein, jos potilaiden paino oli lähtötilanteessa < 60 kg, tai 600 mg:n aloitusannos dupilumabia (kaksi 300 mg:n injektiota) päivänä 1, minkä jälkeen 300 mg dupilumabia kahden viikon välein, jos potilaiden paino oli lähtötilanteessa ≥ 60 kg, tai 2) 600 mg:n aloitusannos dupilumabia (kaksi 300 mg:n injektiota) päivänä 1, minkä jälkeen 300 mg dupilumabia 4 viikon välein riippumatta lähtötilanteen painosta, tai 3) vastaavanlaista lumelääkettä. Jos atooppisen ihottuman oireet olivat sietämättömiä, potilaat saivat tarvittaessa käyttää oirelääkitystä tutkijalääkärin harkinnan mukaan. Oirelääkitystä käyttäneiden katsottiin olevan potilaita, jotka eivät saaneet hoitovastetta.
Tässä tutkimuksessa tutkittavien keskimääräinen ikä oli 14,5 vuotta ja mediaanipaino 59,4 kg; tutkittavista 41,0 % oli naisia, 62,5 % valkoihoisia, 15,1 % aasialaisia ja 12,0 % mustaihoisia. 46,2 %:lla potilaista lähtötilanteen IGA-pistemäärä oli 3 (keskivaikea atooppinen ihottuma), 53,8 %:lla potilaista lähtötilanteen IGA oli 4 (vaikea atooppinen ihottuma), ihottuma-alueen osuus kehon pinta-alasta oli keskimäärin 56,5 % ja 42,4 % potilaista oli saanut aiemmin systeemisiä immuunisalpaajia. Lisäksi lähtötilanteen keskimääräinen EASI-pistemäärä oli 35,5, lähtötilanteen viikoittaisen kutinan voimakkuus oli NRS-asteikolla (Numerical Rating Scale) mitattuna keskimäärin 7,6, lähtötilanteen keskimääräinen POEM-pistemäärä (Patient Oriented Eczema Measure) oli 21,0 ja lähtötilanteen keskimääräinen ihoon liittyvää elämänlaatua kuvaava CDLQI-pistemäärä (Children Dermatology Life Quality Index) oli 13,6. Yhteensä 92,0 %:lla potilaista oli vähintään yksi muu samanaikainen allergiasairaus: 65,6 %:lla oli allerginen nuha, 53,6 %:lla oli astma ja 60,8 %:lla oli ruoka-allergioita.
Samanaikaiset ensisijaiset päätemuuttujat olivat niiden potilaiden osuus, joiden IGA-pistemäärä oli 0 tai 1 ("ei merkkejä atooppisesta ihottumasta" tai "ei juurikaan merkkejä atooppisesta ihottumasta") ja joilla pistemäärä oli pienentynyt vähintään 2 pistettä, ja niiden potilaiden osuus, joilla todettiin EASI-75 (vähintään 75 %:n paraneminen EASI-mittarilla arvioituna) lähtötilanteesta viikolla 16.
Kliininen vaste
Nuorten atooppista ihottumaa koskevan tutkimuksen tehoa koskevat tulokset viikolta 16 on esitetty taulukossa 9.
Taulukko 9: Dupilumabin tehoa koskevat tulokset viikolla 16 nuorten atooppista ihottumaa koskevasta tutkimuksesta (FAS)
| AD-1526 (FAS)a | ||
| Lumelääke | Dupilumabi 200 mg (< 60 kg) ja 300 mg (≥ 60 kg) Kahden viikon välein | |
| Satunnaistettuja potilaita | 85a | 82a |
| IGA 0 tai 1b, % vasteen saaneitac | 2,4 % | 24,4 %d |
| EASI-50, % vasteen saaneitac | 12,9 % | 61,0 %d |
| EASI-75, % vasteen saaneitac | 8,2 % | 41,5 %d |
| EASI-90, % vasteen saaneitac | 2,4 % | 23,2 %d |
| EASI, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta, % (+/- SE) | -23,6 % (5,49) | -65,9 %d (3,99) |
| Kutinan NRS, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta, % (+/- SE) | -19,0 % (4,09) | -47,9 %d (3,43) |
| Kutinan NRS (> 4 pisteen paraneminen), % vasteen saaneitac | 4,8 % | 36,6 %d |
CDLQI, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta (+/- SE) | -5,1 (0,62) | -8,5d (0,50) |
| CDLQI (≥ 6 pisteen paraneminen), % vasteen saaneita | 19,7 % | 60,6 %e |
POEM, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta (+/- SE) | -3,8 (0,96) | -10,1d (0,76) |
| POEM (≥ 6 pisteen paraneminen), % vasteen saaneita | 9,5 % | 63,4 %e |
a Täydellinen analyysisarja (Full Analysis Set, FAS) sisältää kaikki satunnaistetut potilaat.
b Vasteen saaneeksi määriteltiin tutkittava, jonka IGA-pistemäärä oli 0 tai 1 ("ei merkkejä atooppisesta ihottumasta" tai "ei juurikaan merkkejä atooppisesta ihottumasta") ja jolla pistemäärä IGA-asteikolla 0–4 oli pienentynyt vähintään 2 pistettä.
c Potilaat, jotka saivat oirelääkettä tai joiden tiedot olivat puutteelliset, luokiteltiin potilaiksi, jotka eivät saaneet hoitovastetta (58,8 % lumelääkehaarassa ja 20,7 % dupilumabihaarassa).
d p‑arvo < 0,0001 (tilastollisesti merkitsevä verrattuna lumelääkkeeseen p‑arvojen monivertailukorjauksen jälkeen)
e nimellinen p‑arvo < 0,0001
Suurempi osuus lumelääkeryhmään satunnaistetuista potilaista (58,8 %) kuin dupilumabiryhmään satunnaistetuista potilaista (20,7 %) tarvitsi oirelääkitystä (paikallisesti käytettäviä kortikosteroideja, systeemisiä kortikosteroideja tai systeemisiä ei-steroidaalisia immuunisalpaajia).
Lumeryhmään verrattuna merkittävästi suuremmalla osalla dupilumabiryhmään satunnaistetuista potilaista kutina lievittyi NRS-mittarilla arvioituna nopeasti (määriteltiin vähintään 4 pisteen parannukseksi jo viikolla 4; nimellinen p-arvo < 0,001), ja kutinaan hoitovasteen saaneiden potilaiden osuus suureni edelleen hoitojakson aikana NRS-mittarilla arvioituna.
Potilaiden ilmoittamat oireet sekä atooppisen ihottuman vaikutus uneen ja terveyteen liittyvään elämänlaatuun viikon 16 kohdalla POEM- ja CDLQI-pistemäärillä mitattuina paranivat merkittävästi dupilumabiryhmässä lumelääkeryhmään verrattuna.
Avoimessa jatkotutkimuksessa (AD-1434) arvioitiin dupilumabin pitkän aikavälin tehoa keskivaikeaa tai vaikeaa atooppista ihottumaa sairastavilla nuorilla potilailla, jotka olivat osallistuneet aiempiin dupilumabia koskeneisiin kliinisiin tutkimuksiin. Tutkimuksen tehoa koskevien tietojen perusteella viikolla 16 saatu kliininen hyöty säilyi viikolle 52.
Pediatriset potilaat (6–11-vuotiaat)
Samanaikaisesti paikallisen kortikosteroidin kanssa käytetyn dupilumabin tehoa ja turvallisuutta lapsilla arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (AD-1652) 367:llä iältään 6–11-vuotiaalla tutkittavalla, joilla oli vaikea atooppinen ihottuma, joka määriteltiin niin, että IGA-pistemäärä oli 4 (asteikolla 0–4), EASI-pistemäärä vähintään 21 (asteikolla 0–72) ja ihottuma-alueen osuus kehon pinta-alasta (BSA) vähintään 15 %. Tutkimuksen sisäänottokriteerit täyttävien potilaiden hoitovaste aiempaan paikallishoitoon oli ollut riittämätön. Potilaat jaettiin ryhmiin lähtötilanteessa mitatun painon mukaan (< 30 kg; ≥ 30 kg).
Ryhmässä, jossa potilaat saivat dupilumabia kahden viikon välein yhdessä paikallisen kortikosteroidin kanssa, potilaat, joiden paino oli lähtötilanteessa < 30 kg, saivat 200 mg:n aloitusannoksen päivänä 1 ja sen jälkeen 100 mg kahden viikon välein viikolta 2 viikolle 14, ja potilaat, joiden paino oli lähtötilanteessa ≥ 30 kg, saivat 400 mg:n aloitusannoksen päivänä 1 ja sen jälkeen 200 mg kahden viikon välein viikolta 2 viikolle 14. Ryhmässä, jossa potilaat saivat dupilumabia neljän viikon välein yhdessä paikallisen kortikosteroidin kanssa, potilaat saivat painosta riippumatta 600 mg:n aloitusannoksen päivänä 1 ja sen jälkeen 300 mg neljän viikon välein viikolta 4 viikolle 12.
Tässä tutkimuksessa tutkittavien keskimääräinen ikä oli 8,5 vuotta ja painon mediaani 29,8 kg; potilaista 50,1 % oli tyttöjä, 69,2 % valkoihoisia, 16,9 % mustaihoisia ja 7,6 % aasialaisia. Lähtötilanteessa ihottuma-alueen osuus kehon pinta-alasta oli keskimäärin 57,6 %, ja 16,9 % tutkittavista oli saanut aiemmin systeemisiä ei-steroidaalisia immuunisalpaajia. Lisäksi lähtötilanteen keskimääräinen EASI-pistemäärä oli 37,9, päivittäisen pahimman kutinan pistemäärän viikkokeskiarvo oli 7,8 asteikolla 0–10, lähtötilanteen keskimääräinen SCORAD-pistemäärä oli 73,6, lähtötilanteen POEM-pistemäärä oli 20,9 ja lähtötilanteen keskimääräinen CDLQI-pistemäärä oli 15,1. Yhteensä 91,7 %:lla tutkittavista oli vähintään yksi muu samanaikainen allergiasairaus: 64,4 %:lla oli ruoka-allergioita, 62,7 %:lla oli muita allergioita, 60,2 %:lla oli allerginen nuha ja 46,7 %:lla oli astma.
Samanaikaiset ensisijaiset päätemuuttujat olivat niiden potilaiden osuus, joiden IGA-pistemäärä oli vähintään kahden pisteen parannuksen jälkeen 0 tai 1 ("ei merkkejä atooppisesta ihottumasta" tai "ei juurikaan merkkejä atooppisesta ihottumasta"), ja EASI‑75-vasteen saavuttaneiden potilaiden osuus (vähintään 75 %:n paraneminen EASI-mittarilla arvioituna) lähtötilanteesta viikolle 16.
Kliininen vaste
Taulukossa 10 on esitetty tulokset hyväksyttyjen annostusten ja lähtötilanteen painon mukaan jaoteltuina.
Taulukko 10: Dupilumabin ja samanaikaisen paikallisen kortikosteroidihoidon (TCS) tehoa koskevat tulokset viikon 16 kohdalla AD-1652-tutkimuksessa (FAS)a
Dupilumabi 300 mg neljän viikon väleind + TCS | Lumelääke + TCS | Dupilumabi 200 mg kahden viikon väleine + TCS | Lumelääke + TCS | |
| (N = 122) | (N = 123) | (N = 59) | (N = 62) | |
| ≥ 15 kg | ≥ 15 kg | ≥ 30 kg | ≥ 30 kg | |
| IGA 0 tai 1b, % vasteen saaneitac | 32,8 %f | 11,4 % | 39,0 %h | 9,7 % |
| EASI-50, % vasteen saaneitac | 91,0 %f | 43,1 % | 86,4 %g | 43,5 % |
| EASI-75, % vasteen saaneitac | 69,7 %f | 26,8 % | 74,6 %g | 25,8 % |
| EASI-90, % vasteen saaneitac | 41,8 %f | 7,3 % | 35,6 %h | 8,1 % |
| EASI, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta, % (+/- SE) | -82,1 %f (2,37) | -48,6 % (2,46) | -80,4 %g (3,61) | -48,3 % (3,63) |
| Kutinan NRS, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta, % (+/- SE) | -54,6 %f (2,89) | -25,9 % (2,90) | -58,2 %g (4,01) | -25,0 % (3,95) |
| Kutinan NRS (≥ 4 pisteen paraneminen), % vasteen saaneitac | 50,8 %f | 12,3 % | 61,4 %g | 12,9 % |
| CDLQI, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta (+/- SE) | -10,6f (0,47) | -6,4 (0,51) | -9,8g (0,63) | -5,6 (0,66) |
| CDLQI (≥ 6 pisteen paraneminen), % vasteen saaneita | 77,3 %g | 38,8 % | 80,8 %g | 35,8 % |
| POEM, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta (+/- SE) | -13,6f (0,65) | -5,3 (0,69) | -13,6g (0,90) | -4,7 (0,91) |
| POEM (≥ 6 pisteen paraneminen), % vasteen saaneita | 81,7 %g | 32,0 % | 79,3 %g | 31,1 % |
a Täydellinen analyysisarja (Full Analysis Set, FAS) sisältää kaikki satunnaistetut potilaat.
b Vasteen saaneeksi määriteltiin potilas, jonka IGA-pistemäärä oli 0 tai 1 ("ei merkkejä atooppisesta ihottumasta" tai "ei juurikaan merkkejä atooppisesta ihottumasta").
c Potilaat, jotka saivat oirelääkettä tai joiden tiedot olivat puutteelliset, luokiteltiin potilaiksi, jotka eivät saaneet hoitovastetta.
d Päivänä 1 potilaat saivat 600 mg dupilumabia (ks. kohta Farmakokinetiikka).
e Päivänä 1 potilaat saivat 400 mg dupilumabia (paino lähtötilanteessa ≥ 30 kg).
f p‑arvo < 0,0001 (tilastollisesti merkitsevä verrattuna lumelääkkeeseen p‑arvojen monivertailukorjauksen jälkeen)
g nimelliset p‑arvot < 0,0001
h nimellinen p‑arvo = 0,0002
NRS-asteikolla arvioitu pahin kutina lievittyi suuremmalla osalla dupilumabia yhdessä paikallisen kortikosteroidin kanssa saaneiden ryhmään satunnaistetuista potilaista verrattuna lumelääkettä ja paikallista kortikosteroidia saaneiden ryhmään (määriteltiin vähintään 4 pisteen parannukseksi viikolla 4).
Potilaiden ilmoittamat oireet sekä atooppisen ihottuman vaikutus uneen ja terveyteen liittyvään elämänlaatuun viikon 16 kohdalla POEM- ja CDLQI-pistemäärillä mitattuina paranivat merkittävästi dupilumabiryhmissä lumelääkettä saaneisiin verrattuna.
Avoimessa jatkotutkimuksessa (AD-1434) arvioitiin dupilumabin ja paikallisen kortikosteroidin yhdistelmän pitkän aikavälin tehoa ja turvallisuutta keskivaikeaa tai vaikeaa atooppista ihottumaa sairastavilla lapsipotilailla, jotka olivat osallistuneet aiempiin dupilumabin ja paikallisen kortikosteroidin yhdistelmää koskeneisiin kliinisiin tutkimuksiin. Tutkimuksen tehoa koskevien tietojen perusteella viikolla 16 saatu kliininen hyöty säilyi viikolle 52. Joillakin potilailla, jotka saivat 300 mg dupilumabia 4 viikon välein yhdessä paikallisen kortikosteroidin kanssa, todettiin kliinistä lisähyötyä, kun annosta suurennettiin niin, että potilaat saivat 200 mg dupilumabia kahden viikon välein yhdessä paikallisen kortikosteroidin kanssa. Dupilumabin turvallisuusprofiili potilailla, joiden seuranta jatkui viikolle 52, vastasi AD-1526- ja AD-1652-tutkimuksissa viikon 16 kohdalla havaittua turvallisuusprofiilia.
Pediatriset potilaat (6 kuukauden – 5 vuoden ikäiset)
Dupilumabin ja paikallisen kortikosteroidin yhdistelmän tehoa ja turvallisuutta lapsilla arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (AD‑1539) 162 potilaalla, jotka olivat 6 kuukauden – 5 vuoden ikäisiä ja joilla oli keskivaikea tai vaikea atooppinen ihottuma (lähtöryhmien mukainen populaatio). Keskivaikea tai vaikea atooppinen ihottuma määriteltiin niin, että IGA-pistemäärä oli vähintään 3 (asteikolla 0–4), EASI-pistemäärä vähintään 16 (asteikolla 0–72) ja ihottuma-alueen osuus kehon pinta-alasta (BSA) vähintään 10 %. 162 potilaasta 125:llä oli vaikea atooppinen ihottuma, joka määriteltiin niin, että IGA-pistemäärä oli 4. Tutkimukseen otettujen sisäänottokriteerit täyttävien potilaiden hoitovaste aiempaan paikallishoitoon oli ollut riittämätön. Potilaat jaettiin ryhmiin lähtötilanteessa mitatun painon mukaan (≥ 5 – < 15 kg ja ≥ 15 – < 30 kg).
Ryhmässä, jossa potilaat saivat dupilumabia neljän viikon välein yhdessä paikallisen kortikosteroidin kanssa, potilaat, joiden paino oli lähtötilanteessa ≥ 5 – < 15 kg, saivat 200 mg:n aloitusannoksen päivänä 1 ja sen jälkeen 200 mg neljän viikon välein viikolta 4 viikolle 12, ja potilaat, joiden paino oli lähtötilanteessa ≥ 15 – < 30 kg, saivat 300 mg:n aloitusannoksen päivänä 1 ja sen jälkeen 300 mg neljän viikon välein viikolta 4 viikolle 12. Potilaat saivat käyttää oirelääkitystä tutkijalääkärin harkinnan mukaan. Oirelääkitystä käyttäneiden katsottiin olevan potilaita, jotka eivät saaneet hoitovastetta.
AD‑1539-tutkimuksessa potilaiden keskimääräinen ikä oli 3,8 vuotta ja painon mediaani 16,5 kg; potilaista 38,9 % oli tyttöjä, 68,5 % valkoihoisia, 18,5 % mustaihoisia ja 6,2 % aasialaisia. Lähtötilanteessa ihottuma-alueen osuus kehon pinta-alasta oli keskimäärin 58,4 %, ja 15,5 % tutkittavista oli saanut aiemmin systeemisiä ei-steroidaalisia immuunisalpaajia. Lisäksi lähtötilanteen keskimääräinen EASI-pistemäärä oli 34,1, ja päivittäisen pahimman kutinan pistemäärän viikkokeskiarvo oli 7,6 asteikolla 0–10. Yhteensä 81,4 %:lla potilaista oli vähintään yksi muu samanaikainen allergiasairaus: 68,3 %:lla oli ruoka-allergioita, 52,8 %:lla oli muita allergioita, 44,1 %:lla oli allerginen nuha ja 25,5 %:lla oli astma.
Nämä sairauden ominaispiirteet lähtötilanteessa olivat vastaavanlaiset keskivaikeaa tai vaikeaa atooppista ihottumaa ja vaikeaa atooppista ihottumaa sairastavien tutkittavien populaatioissa.
Samanaikaiset ensisijaiset päätemuuttujat olivat niiden potilaiden osuus, joiden IGA-pistemäärä oli 0 tai 1 (”ei merkkejä atooppisesta ihottumasta” tai ”ei juurikaan merkkejä atooppisesta ihottumasta” ja pistemäärä pienentynyt vähintään 2 pistettä), ja niiden potilaiden osuus, joilla todettiin EASI‑75 (vähintään 75 %:n paraneminen EASI-mittarilla arvioituna) lähtötilanteesta viikolle 16. Ensisijainen päätemuuttuja oli niiden potilaiden osuus, joiden IGA-pistemäärä oli 0 (ei merkkejä atooppisesta ihottumasta) tai 1 (ei juurikaan merkkejä atooppisesta ihottumasta) viikolla 16.
Kliininen vaste
AD‑1539-tutkimuksen tehoa koskevat tulokset viikolta 16 on esitetty taulukossa 11.
Taulukko 11: Dupilumabin ja samanaikaisen paikallisen kortikosteroidihoidon (TCS) tehoa koskevat tulokset viikon 16 kohdalla AD‑1539-tutkimuksessa (FAS)a
Dupilumabi 200 mg (5 – < 15 kg) tai 300 mg (15 – < 30 kg) neljän viikon väleind + TCS (lähtöryhmien mukainen populaatio) (N = 83)a | Lumelääke (lähtöryhmien mukainen populaatio) (N = 79) | Dupilumabi 200 mg (5 – < 15 kg) tai 300 mg (15 – < 30 kg) neljän viikon väleind + TCS (vaikeaa atooppista ihottumaa sairastavien populaatio) (N = 63) | Lumelääke (vaikeaa atooppista ihottumaa sairastavien populaatio) (N = 62) | |
| IGA 0 tai 1b,c | 27,7 %e | 3,9 % | 14,3 %f | 1,7 % |
| EASI‑50, % vasteen saaneitac | 68,7 %e | 20,2 % | 60,3 %g | 19,2 % |
| EASI‑75c | 53,0 %e | 10,7 % | 46,0 %g | 7,2 % |
| EASI‑90c | 25,3 %e | 2,8 % | 15,9 %h | 0 % |
| EASI, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta, % (+/‑ SE) | ‑70,0 %e (4,85) | ‑19,6 % (5,13) | ‑55,4 %g (5,01) | ‑10,3 % (5,16) |
| Pahimman raapimisen/kutinan NRS, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta, % (+/‑ SE)* | ‑49,4 %e (5,03) | ‑2.2 % (5,22) | ‑41,8g (5,35) | 0,5 (5,40) |
| Pahimman raapimisen/kutinan NRS (≥ 4 pisteen paraneminen)c * | 48,1 %e | 8,9 % | 42,3 %i | 8,8 % |
| Potilaan unenlaadun NRS, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta (+/‑ SE)* | 2,0e (0,25) | 0,3 (0,26) | 1,7g (0,25) | 0,2 (0,25) |
| Potilaan ihokivun NRS, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta (+/‑ SE)* | ‑3,9e (0,30) | ‑0,6 (0,30) | ‑3,4g (0,29) | ‑0,3 (0,29) |
| POEM, pienimmän neliösumman keskimääräinen muutos lähtötilanteesta (+/‑ SE)* | ‑12,9e (0,89) | ‑3,8 (0,92) | ‑10,6g (0,93) | ‑2,5 (0,95) |
aTäydellinen analyysisarja (Full Analysis Set, FAS) sisältää kaikki satunnaistetut potilaat.
bVasteen saaneeksi määriteltiin potilas, jonka IGA-pistemäärä oli 0 tai 1 (”ei merkkejä atooppisesta ihottumasta” tai ”ei juurikaan merkkejä atooppisesta ihottumasta”).
cPotilaat, jotka saivat oirelääkettä (62 % lumelääkehaarassa ja 19 % dupilumabihaarassa) tai joiden tiedot olivat puutteelliset, luokiteltiin potilaiksi, jotka eivät saaneet hoitovastetta.
dPäivänä 1 potilaat saivat 200 mg (5 – < 15 kg) tai 300 mg (15 – < 30 kg) dupilumabia.
ep‑arvot < 0,0001, fnimellinen p‑arvo < 0,05, gnimellinen p‑arvo < 0,0001, hnimellinen p‑arvo < 0,005, inimellinen p‑arvo < 0,001
*Huoltajan ilmoittama tulos
Lumelääkettä ja paikallista kortikosteroidia saaneiden ryhmään verrattuna merkittävästi suuremmalla osalla dupilumabia yhdessä paikallisen kortikosteroidin kanssa saaneiden ryhmään satunnaistetuista potilaista pahin raapiminen/kutina lievittyi NRS-mittarilla arvioituna nopeasti (määriteltiin vähintään 4 pisteen parannukseksi jo viikolla 3, nimellinen p‑arvo < 0,005), ja pahimpaan raapimiseen/kutinaan hoitovasteen saaneiden potilaiden osuus suureni edelleen hoitojakson aikana NRS-mittarilla arvioituna.
Tässä tutkimuksessa dupilumabi paransi merkittävästi terveyteen liittyvää elämänlaatua, kun mittareina käytettiin CDLQI-pistemäärää (85 potilaalla, jotka olivat 4–5‑vuotiaita) ja IDQOL-pistemäärää (Infant’s Dermatology Quality of Life Index) (77 potilaalla, jotka olivat 6 kuukauden – 3 vuoden ikäisiä). Lähtöryhmien mukaisessa populaatiossa havaittiin CDLQI- ja IDQOL-pistemäärien pienimmän neliösumman suurempia keskimääräisiä muutoksia lähtötilanteesta viikkoon 16 mennessä dupilumabia yhdessä paikallisen kortikosteroidin kanssa saaneiden ryhmässä (CDLQI: ‑10,0, IDQOL: ‑10,9) verrattuna lumelääkettä ja paikallista kortikosteroidia saaneiden ryhmään (CDLQI: ‑2,5, IDQOL: ‑2,0) (p < 0,0001). Sekä CDLQI- että IDQOL-pistemäärien havaittiin parantuneen vastaavanlaisesti vaikeaa atooppista ihottumaa sairastavien tutkittavien populaatiossa.
Avoimessa jatkotutkimuksessa (AD‑1434) arvioitiin dupilumabin ja paikallisen kortikosteroidin yhdistelmän pitkän aikavälin tehoa ja turvallisuutta keskivaikeaa tai vaikeaa atooppista ihottumaa sairastavilla lapsipotilailla, jotka olivat osallistuneet aiempiin dupilumabin ja paikallisen kortikosteroidin yhdistelmää koskeneisiin kliinisiin tutkimuksiin. Tutkimuksen tehoa koskevien tietojen perusteella viikolla 16 saatu kliininen hyöty säilyi viikolle 52. Dupilumabin turvallisuusprofiili potilailla, joiden seuranta jatkui viikolle 52, vastasi AD‑1539-tutkimuksessa viikon 16 kohdalla havaittua turvallisuusprofiilia.
Käsien ja jalkaterien atooppinen ihottuma (aikuiset ja nuoret)
Dupilumabin tehoa ja turvallisuutta arvioitiin 16 viikkoa kestäneessä satunnaistetussa, kaksoissokkoutetussa, rinnakkaisryhmillä toteutetussa lumekontrolloidussa monikeskustutkimuksessa (AD-1924) 133 aikuisella ja 12–17-vuotiaalla pediatrisella potilaalla. Potilailla oli keskivaikea tai vaikea käsien ja jalkaterien atooppinen ihottuma, joka määriteltiin niin, että (käsien ja jalkaterien) IGA-pistemäärä oli vähintään 3 (asteikolla 0–4) ja käsien ja jalkaterien pahinta kutinaa kuvaava NRS-pistemäärä vähintään 4 (asteikolla 0–10). Sisäänottokriteerit täyttävien potilaiden hoitovaste aiempaan käsien ja jalkaterien atooppisen ihottuman paikallishoitoon oli ollut riittämätön tai potilaat eivät sietäneet tällaista hoitoa.
AD-1924-tutkimuksessa 38 % potilaista oli miespuolisia ja 80 % oli valkoihoisia, 72 %:lla tutkittavista lähtötilanteen IGA-pistemäärä (kädet ja jalkaterät) oli 3 (keskivaikea käsien ja jalkaterien atooppinen ihottuma) ja 28 %:lla tutkittavista lähtötilanteen IGA-pistemäärä (kädet ja jalat) oli 4 (vaikea käsien ja jalkaterien atooppinen ihottuma). Lähtötilanteen viikoittainen käsien ja jalkaterien pahin kutina oli NRS-asteikolla mitattuna keskimäärin 7,1.
Ensisijainen päätemuuttuja oli niiden potilaiden osuus, joiden käsien ja jalkaterien IGA-pistemäärä oli 0 (ei merkkejä atooppisesta ihottumasta) tai 1 (ei juurikaan merkkejä atooppisesta ihottumasta) viikolla 16. Keskeinen toissijainen päätemuuttuja oli kutinan lievittyminen mitattuna käsien ja jalkaterien pahimman kutinan NRS-pistemäärällä (≥ 4 pisteen paraneminen). Muita potilaiden ilmoittamia tuloksia olivat käsien ja jalkaterien ihokivun NRS-pistemäärä (0–10), unenlaadun NRS-pistemäärä (0–10), käsien ihottumaan liittyvä elämänlaatu mitattuna QoLHEQ-kyselyllä (quality of life in Hand Eczema Questionnaire) (0–117) sekä työhön liittyvän tuottavuuden ja toimintakyvyn heikkeneminen mitattuna WPAI-kyselyllä (0–100 %).
Niiden potilaiden osuus, joilla käsien ja jalkaterien IGA-pistemäärä oli viikon 16 kohdalla 0–1, oli dupilumabia saaneilla 40,3 % ja lumelääkettä saaneilla 16,7 % (hoitojen välinen ero 23,6, 95 %:n luottamusväli 8,84; 38,42). Niiden potilaiden osuus, joilla keskimääräinen viikoittainen käsien ja jalkaterien pahimman kutinan NRS-pistemäärä oli parantunut (pienentynyt) ≥ 4 pistettä viikon 16 kohdalla, oli dupilumabia saaneilla 52,2 % ja lumelääkettä saaneilla 13,6 % (hoitojen välinen ero 38,6, 95 %:n luottamusväli 24,06; 53,15).
Kun verrattiin dupilumabiryhmää lumeryhmään, havaittiin, että käsien ja jalkaterien ihokivun NRS-pistemäärä, unenlaadun NRS-pistemäärä ja QoLHEQ-pistemäärä olivat parantuneet ja WPAI-kyselyllä mitattu yleinen työkyvyn heikkeneminen ja rutiininomaisten toimintojen heikkeneminen olivat lievittyneet lähtötilanteesta viikkoon 16 mennessä enemmän dupilumabiryhmässä (pienimmän neliösumman keskimääräiset muutokset: käsien ja jalkaterien ihokivun NRS: dupilumabi ‑4,66 vs. lumelääke ‑1,93 [p < 0,0001]; unenlaadun NRS: dupilumabi 0,88 vs. lumelääke ‑0,00 [p < 0,05]; QoLHEQ: dupilumabi ‑40,28 vs. lumelääke ‑16,18 [p < 0,0001]; yleinen työkyvyn heikkeneminen: dupilumabi ‑38,57 % vs. lumelääke ‑22,83 % [nimellinen p < 0,001]; rutiininomaisten toimintojen heikkeneminen: dupilumabi ‑36,39 % vs. lumelääke ‑21,26 % [nimellinen p < 0,001]).
Atooppista ihottumaa sairastavat aikuiset
Katso atooppista ihottumaa sairastavia aikuisia koskevat kliiniset tiedot dupilumabin 300 mg:n vahvuuden valmisteyhteenvedosta.
Kliininen teho ja turvallisuus astmassa
Astmaa koskeva tutkimusohjelma sisälsi kolme satunnaistettua, kaksoissokkoutettua, lumekontrolloitua, rinnakkaisryhmillä toteutettua monikeskustutkimusta (DRI12544, QUEST ja VENTURE), joiden hoitojakso kesti 24–52 viikkoa ja joihin osallistui yhteensä 2 888 vähintään 12-vuotiasta potilasta. Tutkimukseen valituilta potilailta ei edellytetty veren eosinofiilipitoisuuden tai muiden tyypin 2 tulehdukseen liittyvien biomerkkiaineiden (kuten FeNO tai IgE) lähtötilanteen vähimmäisarvoja. Astman hoitosuosituksissa tyypin 2 tulehdus on määritelty tilaksi, jossa veren eosinofiilipitoisuus ≥ 150 solua/µl ja/tai FeNO ≥ 20 ppb. DRI12544- ja QUEST-tutkimusten ennalta määriteltyihin alaryhmäanalyyseihin sisältyivät veren eosinofiilipitoisuudet ≥ 150 ja ≥ 300 solua/µl sekä FeNO-pitoisuudet ≥ 25 ja ≥ 50 ppb.
DRI12544 oli 24 viikon pituinen annosmääritystutkimus, johon osallistui 776 vähintään 18-vuotiasta potilasta. Tutkimuksessa dupilumabia verrattiin lumelääkkeeseen keskivaikeaa tai vaikeaa astmaa sairastavilla aikuispotilailla, joiden astmaa hoidettiin keskisuurella tai suurella annoksella inhaloitavaa kortikosteroidia ja lisäksi pitkävaikutteisella beeta-agonistilla. Tutkimuksen ensisijainen päätemuuttuja oli FEV1 (l) -arvon muutos lähtötilanteesta 12 viikon kohdalla. Lisäksi potilailta määritettiin astman vaikeiden pahenemisvaiheiden vuotuinen ilmaantuvuus 24 viikon lumekontrolloidun hoitojakson aikana. Tuloksia arvioitiin koko tutkimuspopulaatiossa, jossa eosinofiilien tai muiden tyypin 2 tulehduksen biomerkkiaineiden lähtötilanteen vähimmäispitoisuuksia ei ollut määritelty, sekä lisäksi veren eosinofiilipitoisuuksien lähtötasoihin perustuvissa alaryhmissä.
QUEST oli 52 viikon pituinen konfirmatorinen tutkimus, johon osallistui 1 902 vähintään 12-vuotiasta potilasta. Dupilumabia verrattiin tutkimuksessa lumelääkkeeseen 107 nuorella ja 1 795 aikuisella jatkuvaa astmaa sairastavalla potilaalla, jotka käyttivät tautinsa hoitoon keskisuurta tai suurta annosta inhaloitavaa kortikosteroidia sekä toista astman hoitotasapainoa ylläpitävää lääkettä. Myös kolmatta ylläpitolääkettä tarvinneet potilaat saivat osallistua tutkimukseen. Ensisijaiset päätemuuttujat olivat 52 viikon mittaisen lumekontrolloidun vaiheen aikana ilmenneiden vaikeiden pahenemisvaiheiden vuotuinen määrä ja ennen bronkodilataatiota mitattujen FEV1-arvojen muutos lähtötilanteesta 12 viikon kohdalla koko tutkimuspopulaatiossa (ilman eosinofiilien tai muiden tyypin 2 tulehdukseen liittyvien biomerkkiaineiden lähtötilanteen vähimmäispitoisuuksiin liittyviä vaatimuksia) sekä veren eosinofiilipitoisuuden ja FeNO-arvon lähtötasojen perusteella määritellyissä alaryhmissä.
VENTURE oli 24 viikon pituinen oraalisen kortikosteroidiannoksen pienentämistä koskeva tutkimus, johon osallistui 210 astmapotilasta tyypin 2 tulehdusmerkkiaineiden pitoisuuksista riippumatta. Tutkimukseen osallistujat tarvitsivat säännöllisesti käytettävän suuriannoksisen inhaloitavan kortikosteroidin ja toisen ylläpitolääkkeen lisäksi päivittäistä oraalista kortikosteroidihoitoa. Oraalisen kortikosteroidin annos optimoitiin seulontavaiheen aikana. Potilaat jatkoivat tutkimuksen aikana aiempaa astmalääkitystään, mutta oraalisen kortikosteroidin annosta pienennettiin 4 viikon välein annoksen pienentämisvaiheen aikana (viikot 4–20) niin kauan kuin astma pysyi hallinnassa. Ensisijainen päätemuuttuja oli oraalisen kortikosteroidiannoksen prosentuaalinen pieneneminen, jota arvioitiin koko tutkimuspopulaatiossa. Arviointi perustui viikkojen 20–24 oraalisiin kortikosteroidiannoksiin, joilla astma pysyi hallinnassa, ja näitä annoksia verrattiin aiemmin (lähtötilanteessa) optimoituihin oraalisiin kortikosteroidiannoksiin.
Näiden kolmen tutkimuksen demografiset tiedot ja potilaiden ominaisuudet lähtötilanteessa on esitetty alla olevassa taulukossa 12.
Taulukko 12: Astmatutkimusten demografiset tiedot ja potilaiden ominaisuudet lähtötilanteessa
| Muuttuja | DRI12544 (n = 776) | QUEST (n = 1 902) | VENTURE (n = 210) |
| Ikä, keskiarvo (vuotta) (keskihajonta) | 48,6 (13,0) | 47,9 (15,3) | 51,3 (12,6) |
| % naisia | 63,1 | 62,9 | 60,5 |
| % valkoihoisia | 78,2 | 82,9 | 93,8 |
| Astman kesto (vuotta), keskiarvo ± SD | 22,03 (15,42) | 20,94 (15,36) | 19,95 (13,90) |
| Ei koskaan tupakoinut (%) | 77,4 | 80,7 | 80,5 |
Pahenemisvaiheita edellisenä vuonna keskiarvo ± keskihajonta | 2,17 (2,14) | 2,09 (2,15) | 2,09 (2,16) |
| Suuriannoksisen inhaloitavan kortikosteroidin käyttö (%)a | 49,5 | 51,5 | 88,6 |
| Ennen annoksen antamista mitattu FEV1 (l) lähtötilanteessa ± keskihajonta | 1,84 (0,54) | 1,78 (0,60) | 1,58 (0,57) |
| Keskimääräinen prosentuaalinen FEV1 (% viitearvosta) lähtötilanteessa (± keskihajonta) | 60,77 (10,72) | 58,43 (13,52) | 52,18 (15,18) |
| % palautuvuus (± keskihajonta) | 26,85 (15,43) | 26,29 (21,73) | 19,47 (23,25) |
| ACQ-5-pistemäärän keskiarvo (± keskihajonta) | 2,74 (0,81) | 2,76 (0,77) | 2,50 (1,16) |
| AQLQ-pistemäärän keskiarvo (± keskihajonta) | 4,02 (1.09) | 4,29 (1,05) | 4,35 (1,17) |
Yleinen atooppinen sairaushistoria, % (AD %, NP %, AR %) | 72,9 (8,0, 10,6, 61,7) | 77,7 (10,3, 12,7, 68,6) | 72,4 (7,6, 21,0, 55,7) |
| FeNO, ppb, keskiarvo (± keskihajonta) | 39,10 (35,09) | 34,97 (32,85) | 37,61 (31,38) |
Osuus (%) potilaista, joilla FeNO ≥ 25 ppb ≥ 50 ppb | 49,9 21,6 | 49,6 20,5 | 54,3 25,2 |
| Kokonais-IgE, IU/ml, keskiarvo (± keskihajonta) | 435,05 (753,88) | 432,40 (746,66) | 430,58 (775,96) |
| Veren eosinofiilipitoisuus lähtötilanteessa, keskiarvo, solua/µl (± keskihajonta) | 350 (430) | 360 (370) | 350 (310) |
Osuus (%) potilaista, joilla EOS ≥ 150 solua/µl ≥ 300 solua/µl | 77,8 41,9 | 71,4 43,7 | 71,4 42,4 |
FEV1 = uloshengityksen sekuntikapasiteetti; ACQ-5 = astman hallintaa arvioiva ACQ-5-mittari (Asthma Control Questionnaire-5); AQLQ = astmapotilaan elämänlaatua arvioiva AQLQ-mittari (Asthma Quality of Life Questionnaire); AD = atooppinen ihottuma; NP = nenäpolyyppeja; AR = allerginen nuha; FeNO = uloshengitysilman typpioksidi (fraction of exhaled nitric oxide)
aDupilumabi-astmatutkimuksiin osallistui potilaita, jotka käyttivät joko keskisuurta tai suurta annosta inhaloitavaa kortikosteroidia. Keskisuureksi inhaloitavan kortikosteroidin annokseksi määriteltiin 500 µg flutikasonia tai sitä vastaava annos vuorokaudessa.
Pahenemisvaiheet
DRI12544- ja QUEST-tutkimusten kokonaispopulaatioissa astman vaikeiden pahenemisvaiheiden määrä väheni merkitsevästi enemmän tutkittavilla, jotka saivat dupilumabia 200 mg tai 300 mg kahden viikon välein, verrattuna lumelääkettä saaneisiin tutkittaviin. Pahenemisvaiheet vähenivät enemmän niillä tutkittavilla, joilla tyypin 2 tulehdusta kuvaavien biomerkkiaineiden, kuten veren eosinofiilien tai FeNO:n lähtötilanteen arvot olivat koholla (ks. taulukot 13 ja 14).
Taulukko 13: Vaikeiden pahenemisvaiheiden ilmaantuvuus DRI12544- ja QUEST-tutkimuksissa (veren eosinofiilipitoisuus lähtötilanteessa ≥ 150 ja ≥ 300 solua/µl)
| Hoito | Veren eosinofiilien pitoisuuksien lähtötaso | ||||||||||
| ≥ 150 solua/µl | ≥ 300 solua/µl | ||||||||||
| Pahenemisvaiheet vuodessa | % vähenemä | Pahenemisvaiheet vuodessa | % vähenemä | ||||||||
| N | Ilmaantuvuus (95 %:n luottamusväli) | Ilmaantuvuuksien suhde (95 %:n luottamusväli) | N | Ilmaantuvuus (95 %:n luottamusväli) | Ilmaantuvuuksien suhde (95 %:n luottamusväli) | ||||||
| Kaikki vaikeat pahenemisvaiheet | |||||||||||
| DRI12544-tutkimus | |||||||||||
| Dupilumabi 200 mg 2 vko:n välein | 120 | 0,29 (0,16; 0,53) | 0,28a (0,14; 0,55) | 72 % | 65 | 0,30 (0,13; 0,68) | 0,29c (0,11; 0,76) | 71 % | |||
| Dupilumabi 300 mg 2 vko:n välein | 129 | 0,28 (0,16; 0,50) | 0,27b (0,14; 0,52) | 73 % | 64 | 0,20 (0,08; 0,52) | 0,19d (0,07; 0,56) | 81 % | |||
| Lumelääke | 127 | 1,05 (0,69; 1,60) | 68 | 1,04 (0,57; 1,90) | |||||||
| QUEST-tutkimus | |||||||||||
| Dupilumabi 200 mg 2 vko:n välein | 437 | 0,45 (0,37; 0,54) | 0,44f (0,34; 0,58) | 56 % | 264 | 0,37 (0,29; 0,48) | 0,34f (0,24; 0,48) | 66 % | |||
| Lumelääke | 232 | 1,01 (0,81; 1,25) | 148 | 1,08 (0,85; 1,38) | |||||||
| Dupilumabi 300 mg 2 vko:n välein | 452 | 0,43 (0,36; 0,53) | 0,40 e (0,31; 0,53) | 60 % | 277 | 0,40 (0,32; 0,51) | 0,33e (0,23; 0,45) | 67 % | |||
| Lumelääke | 237 | 1,08 (0,88; 1,33) | 142 | 1,24 (0,97; 1,57) | |||||||
ap-arvo = 0,0003, bp-arvo = 0,0001, cp-arvo = 0,0116, dp-arvo = 0,0024, ep-arvo < 0,0001 (kaikki tilastollisesti merkitseviä verrattuna lumelääkkeeseen p‑arvojen monivertailukorjauksen jälkeen); fnimellinen p‑arvo < 0,0001
Taulukko 14: Vaikeiden pahenemisvaiheiden ilmaantuvuus QUEST-tutkimuksessa lähtötilanteen FeNO-alaryhmien mukaan määritettynä
| Hoito | Pahenemisvaiheet vuodessa | % vähenemä | ||
| N | Ilmaantuvuus (95 %:n luottamusväli) | Ilmaantuvuuksien suhde (95 %:n luottamusväli) | ||
| FeNO ≥ 25 ppb | ||||
| Dupilumabi 200 mg 2 viikon välein | 299 | 0,35 (0,27; 0,45) | 0,35 (0,25; 0,50)a | 65 % |
| Lumelääke | 162 | 1,00 (0,78; 1,30) | ||
| Dupilumabi 300 mg 2 viikon välein | 310 | 0,43 (0,35; 0,54) | 0,39 (0,28; 0,54) a | 61 % |
| Lumelääke | 172 | 1,12 (0,88; 1,43) | ||
| FeNO ≥ 50 ppb | ||||
| Dupilumabi 200 mg 2 viikon välein | 119 | 0,33 (0,22; 0,48) | 0,31 (0,18; 0,52) a | 69 % |
| Lumelääke | 71 | 1,057 (0,72; 1,55) | ||
| Dupilumabi 300 mg 2 viikon välein | 124 | 0,39 (0,27; 0,558) | 0,31 (0,19; 0,49) a | 69 % |
| Lumelääke | 75 | 1,27 (0,90; 1,80) | ||
animellinen p-arvo < 0,0001
DRI12544- ja QUEST-tutkimusten yhdistetyssä analyysissä sairaalahoitoa ja/tai päivystyskäyntiä vaatineet vaikeat pahenemisvaiheet vähenivät 25,5 %:a annettaessa 200 mg dupilumabia 2 viikon välein ja 46,9 %:a annettaessa 300 mg dupilumabia 2 viikon välein.
Keuhkofunktio
Ennen bronkodilataatiota mitattujen FEV1-arvojen havaittiin suurentuneen kliinisesti merkittävästi viikolla 12 DRI12544- ja QUEST-tutkimuksissa. Tutkittavilla, joilla tyypin 2 tulehdukseen liittyvien biomerkkiaineiden, kuten veren eosinofiilien ja FeNO:n, pitoisuudet olivat lähtötilanteessa suurempia, FEV1-arvot paranivat enemmän (taulukot 15 ja 16).
FEV1-arvojen havaittiin parantuneen merkittävästi jo viikolla 2 ensimmäisen dupilumabiannoksen antamisen jälkeen sekä 200 mg:n että 300 mg:n annosvahvuuksilla, ja saavutetut FEV1-arvot säilyivät viikkoon 24 asti DRI12544-tutkimuksessa ja viikkoon 52 asti QUEST-tutkimuksessa (ks. kuva 3).
Kuva 1: Ennen bronkodilataatiota mitattujen FEV1-arvojen (l) keskimääräinen muutos lähtötilanteesta ajan myötä (lähtötilanteen eosinofiilimäärä, EOS ≥ 150 ja ≥ 300 solua/µl ja uloshengitysilman typpioksidi, FeNO > 25 ppb) QUEST-tutkimuksessa
QUEST: Veren EOS ≥ 150 solua/µl QUEST: Veren EOS ≥ 300 solua/µl QUEST: FeNO ≥ 25 ppb
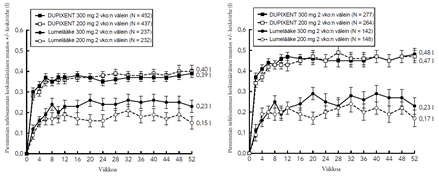
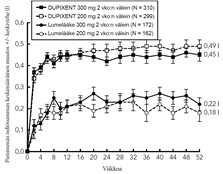
Taulukko 15: Ennen bronkodilataattorin antamista mitattujen FEV1-arvojen keskimääräinen muutos lähtötilanteesta viikolla 12 DRI12544- ja QUEST-tutkimuksissa (lähtötilanteessa todettu veren eosinofiilimäärä ≥ 150 ja ≥ 300 solua/µl)
| Hoito | Veren EOS lähtötilanteessa | ||||||
| ≥ 150 solua/µl | ≥ 300 solua/µl | ||||||
| N | Pienimmän neliösumman muutos lähtö-tilanteesta, keskiarvo l (%) | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | N | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo l (%) | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | ||
| DRI12544-tutkimus | |||||||
| Dupilumabi 200 mg 2 vko:n välein | 120 | 0,32 (18,25) | 0,23a (0,13; 0,33) | 65 | 0,43 (25,9) | 0,26c (0,11; 0,40) | |
| Dupilumabi 300 mg 2 vko:n välein | 129 | 0,26 (17,1) | 0,18b (0,08; 0,27) | 64 | 0,39 (25,8) | 0,21d (0,06; 0,36) | |
| Lumelääke | 127 | 0,09 (4,36) | 68 | 0,18 (10,2) | |||
| QUEST-tutkimus | |||||||
| Dupilumabi200 mg 2 vko:n välein | 437 | 0,36 (23,6) | 0,17f (0,11; 0,23) | 264 | 0,43 (29,0) | 0,21f (0,13; 0,29) | |
| Lumelääke | 232 | 0,18 (12,4) | 148 | 0,21 (15,6) | |||
| Dupilumabi300 mg 2 vko:n välein | 452 | 0,37 (25,3) | 0,15e (0,09; 0,21) | 277 | 0,47 (32,5) | 0,24e (0,16; 0,32) | |
| Lumelääke | 237 | 0,22 (14,2) | 142 | 0,22 (14,4) | |||
ap‑arvo < 0,0001, bp-arvo = 0,0004, cp-arvo = 0,0008, dp-arvo = 0,0063, ep-arvo < 0,0001 (kaikki tilastollisesti merkitseviä verrattuna lumelääkkeeseen p‑arvojen monivertailukorjauksen jälkeen); fnimellinen p‑arvo < 0,0001
Taulukko 16: Ennen bronkodilataatiota mitattujen FEV1-arvojen keskimääräinen muutos lähtötilanteesta viikolla 12 ja viikolla 52 QUEST-tutkimuksessa lähtötilanteen FeNO-alaryhmien mukaan
| Hoito | Viikko 12 | Viikko 52 | |||
| N | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo l (%) | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo l (%) | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | |
| FeNO ≥ 25 ppb | |||||
| Dupilumabi 200 mg 2 vko:n välein | 288 | 0,44 (29,0 %) | 0,23 (0,15; 0,31)a | 0,49 (31,6 %) | 0,30 (0,22; 0,39)a |
| Lumelääke | 157 | 0,21 (14,1 %) | 0,18 (13,2 %) | ||
| Dupilumabi 300 mg 2 vko:n välein | 295 | 0,45 (29,8 %) | 0,24 (0,16; 0,31)a | 0,45 (30,5 %) | 0,23 (0,15; 0,31)a |
| Lumelääke | 167 | 0,21 (13,7 %) | 0,22 (13,6 %) | ||
| FeNO ≥ 50 ppb | |||||
| Dupilumabi 200 mg 2 vko:n välein | 114 | 0,53 (33,5 %) | 0,30 (0,17; 0,44)a | 0,59 (36,4 %) | 0,38 (0,24; 0,53)a |
| Lumelääke | 69 | 0,23 (14,9 %) | 0,21 (14,6 %) | ||
| Dupilumabi 300 mg 2 vko:n välein | 113 | 0,59 (37,6 %) | 0,39 (0,26; 0,52)a | 0,55 (35,8 %) | 0,30 (0,16; 0,44)a |
| Lumelääke | 73 | 0,19 (13,0 %) | 0,25 (13,6 %) | ||
animellinen p-arvo < 0,0001
Elämänlaatu ja muut potilaan itse ilmoittamat tulokset astmassa
Tutkimusten ennalta määriteltyinä toissijaisina päätemuuttujina arvioitiin ACQ-5- ja AQLQ(S)-tuloksen perusteella hoitovasteen saaneiden potilaiden osuudet 24 viikon kohdalla (DRI12544 ja VENTURE) sekä 52 viikon kohdalla (QUEST, taulukko 17). Hoitovaste määriteltiin vähintään 0,5 pisteen paranemaksi pistemäärässä (ACQ-5:n arviointiasteikko 0–6 ja AQLQ(S):n 1–7). ACQ-5- ja AQLQ(S)-pistemäärien havaittiin parantuneen jo viikolla 2, ja tämä vaikutus säilyi 24 viikon ajan DRI12544-tutkimuksessa ja 52 viikon ajan QUEST-tutkimuksessa. Samankaltaisia tuloksia havaittiin VENTURE-tutkimuksessa.
Taulukko 17: ACQ-5‑ ja AQLQ(S)-tulosten perusteella määritetyt hoitovasteet viikolla 52 QUEST-tutkimuksessa
| Potilaiden ilmoittamat tulokset | Hoito | EOS ≥ 150 solua/µl | EOS ≥ 300 solua/µl | FeNO ≥ 25 ppb | |||
| N | Hoitovasteen saaneiden osuus (%) | N | Hoitovasteen saaneiden osuus (%) | N | Hoitovasteen saaneiden osuus (%) | ||
| ACQ-5 | Dupilumabi 200 mg 2 vko:n välein | 395 | 72,9 | 239 | 74,5 | 262 | 74,4 |
| Lumelääke | 201 | 64,2 | 124 | 66,9 | 141 | 65,2 | |
Dupilumabi 300 mg 2 vko:n välein | 408 | 70,1 | 248 | 71,0 | 277 | 75,8 | |
| Lumelääke | 217 | 64,5 | 129 | 64,3 | 159 | 64,2 | |
| AQLQ(S) | Dupilumabi 200 mg 2 vko:n välein | 395 | 66,6 | 239 | 71,1 | 262 | 67,6 |
| Lumelääke | 201 | 53,2 | 124 | 54,8 | 141 | 54,6 | |
Dupilumabi 300 mg 2 vko:n välein | 408 | 62,0 | 248 | 64,5 | 277 | 65,3 | |
| Lumelääke | 217 | 53,9 | 129 | 55,0 | 159 | 58,5 | |
Oraalisten kortikosteroidien vähentämistä koskeva tutkimus (VENTURE)
VENTURE-tutkimuksessa arvioitiin dupilumabin vaikutusta ylläpitohoitona annettavien oraalisten kortikosteroidien käytön vähentämiseen. Lähtötilanteen ominaisuudet on esitetty taulukossa 12. Kaikki tutkimuspotilaat olivat saaneet oraalista kortikosteroidihoitoa vähintään 6 kuukautta ennen tutkimuksen alkua. Oraalista kortikosteroidia käytettiin lähtötilanteessa keskimäärin 11,75 mg lumelääkeryhmässä ja 10,75 mg dupilumabiryhmässä.
Tässä 24 viikon mittaisessa tutkimuksessa astman pahenemisvaiheet (määritelty oraalisen kortikosteroidiannoksen vähintään 3 vuorokautta kestäneenä tilapäisenä suurentamisena) vähenivät dupilumabia saaneilla tutkittavilla 59 % suhteessa lumelääkettä saaneisiin (vuotuinen ilmaantuvuus dupilumabiryhmässä 0,65 ja lumelääkeryhmässä 1,60; ilmaantuvuuksien suhde 0,41 [95 %:n luottamusväli 0,26–0,63]). Lisäksi ennen bronkodilataatiota mitatut FEV1-arvot paranivat lähtötilanteesta viikkoon 24 mennessä enemmän dupilumabia kuin lumelääkettä saaneilla tutkittavilla (pienimmän neliösumman keskimääräinen ero dupilumabille verrattuna lumelääkkeeseen 0,22 l [95 %:n luottamusväli 0,09–0,34 l]). Vaikutukset keuhkojen toimintaan, oraalisten steroidien käyttöön ja pahenemisvaiheiden vähenemiseen olivat samankaltaisia riippumatta tyypin 2 tulehdukseen liittyvien biomerkkiaineiden (kuten veren eosinofiilien tai FeNO:n) lähtötasoista. VENTURE-tutkimuksessa arvioitiin myös ACQ-5- ja AQLQ(S)-mittareiden tuloksia, joiden osoitettiin parantuneen samankaltaisesti kuin QUEST-tutkimuksessa.
VENTURE-tutkimuksen tulokset lähtötilanteen biomerkkiainetasojen mukaan jaoteltuna on esitetty taulukossa 18.
Taulukko 18. Dupilumabin vaikutus oraalisten kortikosteroidien annoksen vähenemiseen VENTURE-tutkimuksessa (lähtötilanteessa todettu veren eosinofiilipitoisuus ≥ 150 ja ≥ 300 solua/µl sekä FeNO ≥ 25 ppb)
Veren EOS lähtötilanteessa ≥ 150 solua/µl | Veren EOS lähtötilanteessa ≥ 300 solua/µl | FeNO ≥ 25 ppb | ||||
Dupilumabi 300 mg 2 vko:n välein N = 81 | Lume-lääke N = 69 | Dupilumabi 300 mg 2 vko:n välein N = 48 | Lume-lääke N = 41 | Dupilumabi 300 mg 2 vko:n välein N = 57 | Lume-lääke N = 57 | |
| Ensisijainen päätemuuttuja (viikko 24) | ||||||
| Oraalisten kortikosteroidien käytön prosentuaalinen väheneminen lähtötilanteesta | ||||||
Prosentuaalinen kokonaisvähenemä lähtötilanteesta, keskiarvo (%) Ero (% [95 % :n luottamusväli]) (dupilumabi vs. lumelääke) | 75,91 29,39b (15,67; 43,12) | 46,51 | 79,54 36,83b (18,94; 54,71) | 42,71 | 77,46 34,53b (19,08; 49,97) | 42,93 |
| Päivittäin käytettävän, oraalisen kortikosteroidiannoksen prosentuaalisen vähenemän mediaani lähtötilanteeseen verrattuna | 100 | 50 | 100 | 50 | 100 | 50 |
Prosentuaalinen vähenemä lähtötilanteesta 100 % ≥ 90 % ≥ 75 % ≥ 50 % > 0 % Oraalisen kortikosteroidin annos ei pienentynyt eikä suurentunut, tai tutkittava keskeytti tutkimukseen osallistumisen | 54,3 58,0 72,8 82,7 87,7 12,3 | 33,3 34,8 44,9 55,1 66,7 33,3 | 60,4 66,7 77,1 85,4 85,4 14,6 | 31,7 34,1 41,5 53,7 63,4 36,6 | 52,6 54,4 73,7 86,0 89,5 10,5 | 28,1 29,8 36,8 50,9 66,7 33,3 |
| Toissijainen päätemuuttuja (viikko 24)a | ||||||
| Niiden potilaiden osuus, joilla oraalisen kortikosteroidin annos pieneni < 5 mg:aan vuorokaudessa | 77 | 44 | 84 | 40 | 79 | 34 |
Vetosuhde (Odds ratio) (95 %:n luottamusväli) | 4,29c (2,04; 9,04) | 8,04d (2,71; 23,82) | 7,21b (2,69; 19,28) | |||
aLogistisen regressiomallin estimaatit, bnimellinenp-arvo < 0,0001, cnimellinen p-arvo = 0,0001, dnimellinen p-arvo = 0,0002
Pitkän aikavälin jatkotutkimus (TRAVERSE)
Avoimessa jatkotutkimuksessa (TRAVERSE) arvioitiin dupilumabin pitkäaikaiskäytön turvallisuutta keskivaikeaa tai vaikeaa astmaa sairastavilla 2 193 aikuisella ja 89 nuorella, jotka olivat osallistuneet aiempiin dupilumabilla tehtyihin kliinisiin tutkimuksiin (DRI12544, QUEST ja VENTURE) (ks. kohta Haittavaikutukset). 185 aikuisella tutkittavalla astman hallinta edellytti oraalisia kortikosteroideja. Toissijaisena päätemuuttujana mitattiin tehoa, joka vastasi keskeisten tutkimusten tuloksia ja säilyi jopa 96 viikon ajan. Aikuisilla, joiden astman hallinta edellytti oraalisia kortikosteroideja, pahenemisvaiheiden vähenemä ja keuhkofunktion paranema säilyivät viikolle 96 asti siitä huolimatta, että oraalisten kortikosteroidien annosta pienennettiin tai niiden käyttö lopetettiin.
Lapsitutkimus (6–11‑vuotiaat; VOYAGE)
Dupilumabin tehoa ja turvallisuutta pediatrisilla potilailla arvioitiin 52 viikkoa kestäneessä satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (VOYAGE), johon osallistui 408 tutkittavaa. Tutkittavat olivat 6–11‑vuotiaita, heillä oli keskivaikea tai vaikea astma, ja he käyttivät joko keskisuurta tai suurta annosta inhaloitavaa kortikosteroidia sekä yhtä astman hoitotasapainoa ylläpitävää lääkettä tai pelkästään suuriannoksista inhaloitavaa kortikosteroidia. Tutkittavat satunnaistettiin saamaan dupilumabia (N = 273) tai vastaavanlaista lumelääkettä (N = 135) kahden viikon välein painon (≤ 30 kg tai > 30 kg) perusteella. Tehoa arvioitiin potilailla, joilla oli tyypin 2 tulehdus joko veren eosinofiilipitoisuus ≥ 150 solua/µl tai FeNO:n (≥ 20 ppb) perusteella.
Ensisijainen päätemuuttuja oli vaikeiden pahenemisvaiheiden vuotuinen ilmaantuvuus 52 viikon lumekontrolloidun hoitojakson aikana, ja keskeinen toissijainen päätemuuttuja oli ennen bronkodilataatiota mitatun FEV1-arvon (% viitearvosta) muutos lähtötilanteesta viikon 12 kohdalla. Muita toissijaisia päätemuuttujia olivat ACQ-7-IA- ja PAQLQ(S)-IA-pistemäärien keskimääräiset muutokset lähtötilanteesta sekä pistemäärien perusteella hoitovasteen saaneiden osuus.
VOYAGE-tutkimuksen demografiset tiedot ja potilaiden ominaisuudet lähtötilanteessa on esitetty taulukossa 19.
Taulukko 19: VOYAGE-tutkimuksen demografiset tiedot ja potilaiden ominaisuudet lähtötilanteessa
| Parametri | EOS (N = 350) | EOS ≥ 300 solua/µl (N = 259)
|
| Ikä, keskiarvo (vuotta) (keskihajonta) | 8,9 (1,6) | 9,0 (1,6) |
| % tyttöjä | 34,3 | 32,8 |
| % valkoihoisia | 88,6 | 87,3 |
| Keskimääräinen paino (kg) | 36,09 | 35,94 |
| Pahenemisvaiheita edellisenä vuonna, keskiarvo (± keskihajonta) | 2,47 (2,30) | 2,64 (2,58) |
Inhaloitavan kortikosteroidin annos (%) Keskisuuri Suuri |
55,7 43,4 |
54,4 44,4 |
| Ennen annoksen antamista mitattu FEV1 (l) lähtötilanteessa (± keskihajonta) | 1,49 (0,41) | 1,47 (0,42) |
| Keskimääräinen prosentuaalinen FEV1 (% viitearvosta) (± keskihajonta) | 77,89 (14,40) | 76,85 (14,78) |
| % palautuvuus, keskiarvo (± keskihajonta) | 27,79 (19,34) | 22,59 (20,78) |
| ACQ-7-IA-pistemäärän keskiarvo (± keskihajonta) | 2,14 (0,72) | 2,16 (0,75) |
| PAQLQ(S)-IA-pistemäärän keskiarvo (± keskihajonta) | 4,94 (1,10) | 4,93 (1,12) |
Yleinen atooppinen sairaushistoria, % (AD %, AR %) | 94 (38,9; 82,6) | 96,5 (44,4; 85,7) |
| Kokonais‑IgE, IU/ml, mediaani (± keskihajonta) | 905,52 (1140,41) | 1077,00 (1230,83) |
| FeNO, ppb, keskiarvo (± keskihajonta) | 30,71 (24,42) | 33,50 (25,11) |
Osuus (%) potilaista, joilla FeNO ≥ 20 ppb | 58
| 64,1 |
| Veren eosinofiilipitoisuus lähtötilanteessa, keskiarvo, solua/µl (± keskihajonta) | 570 (380) | 710 (360) |
| Osuus (%) potilaista, joilla EOS ≥ 150 solua/µl ≥ 300 solua/µl |
94,6 74 |
0 100 |
FEV1 = uloshengityksen sekuntikapasiteetti; ACQ-7-IA = astman hallintaa arvioiva ACQ-7-IA-mittari (Asthma Control Questionnaire-7 Interviewer Administered); PAQLQ(S)-IA = pediatrisen astmapotilaan elämänlaatua arvioiva PAQLQ(S)-IA-mittari (Paediatric Asthma Quality of Life Questionnaire with Standardised Activities – Interviewer Administered); AD = atooppinen ihottuma; AR = allerginen nuha; EOS = veren eosinofiilipitoisuus; FeNO = uloshengitysilman typpioksidi (fraction of exhaled nitric oxide)
Dupilumabi vähensi lumelääkkeeseen verrattuna merkittävästi vaikeiden pahenemisvaiheiden vuotuista ilmaantuvuutta 52 viikon hoitojakson aikana potilailla, joilla oli tyypin 2 tulehdus sekä potilailla, joilla veren eosinofiilipitoisuus oli lähtötilanteessa ≥ 300 solua/µl tai joilla FeNO-arvo oli lähtötilanteessa ≥ 20 ppb. Kliinisesti merkittävä paranema ennen bronkodilataatiota mitatuissa FEV1-arvoissa (% viitearvosta) havaittiin viikolla 12. ACQ-7-IA- ja PAQLQ(S)-IA-pistemäärien todettiin myös parantuneen viikon 24 kohdalla, ja muutos säilyi viikolle 52. Sekä ACQ-7-IA- että PAQLQ(S)-IA-mittarilla arvioituna hoitovaste oli viikon 24 kohdalla saavutettu lumeryhmiä todennäköisemmin. VOYAGE-tutkimuksen tehoa koskevat tulokset on esitetty taulukossa 20.
Tyypin 2 tulehduksellisella potilaspopulaatiolla ennen bronkodilataatiota mitattujen FEV1-arvojen pienimmän neliösumman keskimääräinen muutos lähtötilanteesta viikon 12 kohdalla oli 0,22 l dupilumabiryhmässä ja 0,12 l lumelääkeryhmässä. Pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna oli 0,10 l (95 %:n luottamusväli 0,04; 0,16). Hoidon vaikutus säilyi koko 52 viikon hoitojakson ajan. Viikolla 52 pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna oli 0,17 l (95 %:n luottamusväli 0,09; 0,24).
Potilaspopulaatiolla, joilla veren eosinofiilipitoisuus oli lähtötilanteessa ≥ 300 solua/µl, ennen bronkodilataatiota mitattujen FEV1-arvojen pienimmän neliösumman keskimääräinen muutos lähtötilanteesta viikon 12 kohdalla oli 0,22 l dupilumabiryhmässä ja 0,12 l lumelääkeryhmässä. Pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna oli 0,10 l (95 %:n luottamusväli 0,03; 0,17). Hoidon vaikutus säilyi koko 52 viikon mittaisen hoitojakson ajan. Viikolla 52 pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna oli 0,17 l (95 %:n luottamusväli 0,09; 0,26).
Molemmissa ensisijaisissa tehopopulaatioissa FEF25–75 %-arvo ja FEV1/FVC-suhde paranivat nopeasti (ero alkoi näkyä jo viikolla 2) ja vaikutus säilyi koko 52 viikon mittaisen hoitojakson ajan, ks. taulukko 20.
Taulukko 20: Vaikeiden pahenemisvaiheiden ilmaantuvuus, FEV1-arvon keskimääräinen muutos lähtötilanteesta ja hoitovasteen saaneiden tutkittavien osuus ACQ-7-IA- ja PAQLQ(S)-IA-pistemäärien perusteella VOYAGE-tutkimuksessa
| Hoito | EOS ≥ 150 solua/µl tai FeNO ≥ 20 ppb | EOS ≥ 300 solua/µl | FeNO ≥ 20 ppb | |||||||
| Vaikeiden pahenemisvaiheiden vuotuinen ilmaantuvuus 52 viikon aikana | ||||||||||
| N | Ilmaantuvuus (95 %:n luottamusväli) | Ilmaantuvuuksien suhde (95 %:n luottamusväli) | N | Ilmaantuvuus (95 %:n luottamusväli) | Ilmaantuvuuksien suhde (95 %:n luottamusväli) | N | Ilmaantuvuus (95 %:n luottamusväli) | Ilmaantuvuuksien suhde (95 %:n luottamusväli) | ||
Dupilumabi 100 mg 2 viikon välein (< 30 kg) / 200 mg 2 viikon välein (≥ 30 kg) | 236 | 0,305 (0,223; 0,416) | 0,407b (0,274; 0,605) | 175 | 0,235 (0,160; 0,345) | 0,353b (0,222; 0,562) | 141 | 0,271 (0,170; 0,432) | 0,384c (0,227; 0,649) | |
| Lumelääke | 114 | 0,748 (0,542; 1,034) | 84 | 0,665 (0,467; 0,949) | 62 | 0,705 (0,421; 1,180) | ||||
| FEV1-arvon (% viitearvosta) keskimääräinen muutos lähtötilanteesta viikon 12 kohdalla | ||||||||||
| N | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | N | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | N | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | ||
Dupilumabi 100 mg 2 viikon välein (< 30 kg) / 200 mg 2 viikon välein (≥ 30 kg) | 229 | 10,53 | 5,21c (2,14; 8,27) | 168 | 10,15 | 5,32d (1,76; 8,88) | 141 | 11,36 | 6,74d (2,54; 10,93) | |
| Lumelääke | 110 | 5,32 | 80 | 4,83 | 62 | 4,62 | ||||
| FEF25–75 % ‑arvon (% viitearvosta) keskimääräinen muutos lähtötilanteesta viikon 12 kohdalla | ||||||||||
| N | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | N | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | N | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | ||
Dupilumabi 100 mg 2 viikon välein (< 30 kg) / 200 mg 2 viikon välein (≥ 30 kg) | 229 | 16,70 | 11,93e (7,44; 16,43) | 168 | 16,91 | 13,92e (8,89; 18,95) | 141 | 17,96 | 13,97e (8,30; 19,65) | |
| Lumelääke | 110 | 4,76 | 80 | 2,99 | 62 | 3,98 | ||||
| Prosentuaalisen FEV1/FVC-suhteen keskimääräinen muutos lähtötilanteesta viikon 12 kohdalla | ||||||||||
| N | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | N | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | N | Pienimmän neliösumman muutos lähtötilanteesta, keskiarvo | Pienimmän neliösumman keskimääräinen ero vs. lumelääke (95 %:n luottamusväli) | ||
Dupilumabi 100 mg 2 viikon välein (< 30 kg) / 200 mg 2 viikon välein (≥ 30 kg) | 229 | 5,67 | 3,73e (2,25; 5,21) | 168 | 6,10 | 4,63e (2,97; 6,29) | 141 | 6,84 | 4,95e (3,08; 6,81) | |
| Lumelääke | 110 | 1,94 | 80 | 1,47 | 62 | 1,89 | ||||
| ACQ-7-IA viikolla 24a | ||||||||||
| N | Hoitovasteen saaneiden osuus (%) | Kerroinsuhde vs. lumelääke (95 %:n luottamusväli) | N | Hoitovasteen saaneiden osuus (%) | Kerroinsuhde vs. lumelääke (95 %:n luottamusväli) | N | Hoitovasteen saaneiden osuus (%) | Kerroinsuhde vs. lumelääke (95 %:n luottamusväli) | ||
Dupilumabi 100 mg 2 viikon välein (< 30 kg) / 200 mg 2 viikon välein (≥ 30 kg) | 236 | 79,2 | 1,82g (1,02; 3,24) | 175 | 80,6 | 2,79f (1,43; 5,44) | 141 | 80,9 | 2,60g (1,21; 5,59) | |
| Lumelääke | 114 | 69,3 | 84 | 64,3 | 62 | 66,1 | ||||
| PAQLQ(S)-IA viikolla 24a | ||||||||||
| N | Hoitovasteen saaneiden osuus (%)
| Kerroinsuhde vs. lumelääke (95 %:n luottamusväli) | N | Hoitovasteen saaneiden osuus (%) | Kerroinsuhde vs. lumelääke (95 %:n luottamusväli) | N | Hoitovasteen saaneiden osuus (%) | Kerroinsuhde vs. lumelääke (95 %:n luottamusväli) | ||
Dupilumabi 100 mg 2 viikon välein (< 30 kg) / 200 mg 2 viikon välein (≥ 30 kg) | 211 | 73,0 | 1,57 (0,87; 2,84) | 158 | 72,8 | 1,84 (0,92; 3,65) | 131 | 75,6 | 2,09 (0,95; 4,61) | |
| Lumelääke | 107 | 65,4 | 81 | 63,0 | 61 | 67,2 | ||||
| aHoitovasteen saaneiden osuuden määritelmänä oli vähintään 0,5 pisteen paranema pistemäärässä (ACQ-7-IA:n arviointiasteikko 0–6 ja PAQLQ(S):n 1–7). | ||||||||||
bp‑arvo < 0,0001; cp‑arvo < 0,001, dp‑arvo < 0,01 (kaikki tilastollisesti merkitseviä verrattuna lumelääkkeeseen p‑arvojen monivertailukorjauksen jälkeen); enimellinen p‑arvo < 0,0001, fnimellinen p‑arvo < 0,01, gnimellinen p‑arvo < 0,05
VOYAGE-tutkimuksessa FEV1‑arvojen (% viitearvosta) havaittiin parantuneen merkitsevästi jo viikolla 2, ja saavutetut arvot säilyivät viikkoon 52 asti.
FEV1‑arvojen (% viitearvosta) paraneminen ajan kuluessa VOYAGE-tutkimuksessa on esitetty kuvassa 2.
Kuva 2: Ennen bronkodilataatiota mitatun FEV1‑arvon (l, % viitearvosta, ppFEV1) keskimääräinen muutos lähtötilanteesta ajan kuluessa VOYAGE-tutkimuksessa (veren eosinofiilipitoisuus ≥ 150 solua/µl tai FeNO ≥ 20 ppb lähtötilanteessa, eosinofiilipitoisuus ≥ 300 solua/µl lähtötilanteessa ja FeNO ≥ 20 ppb lähtötilanteessa)
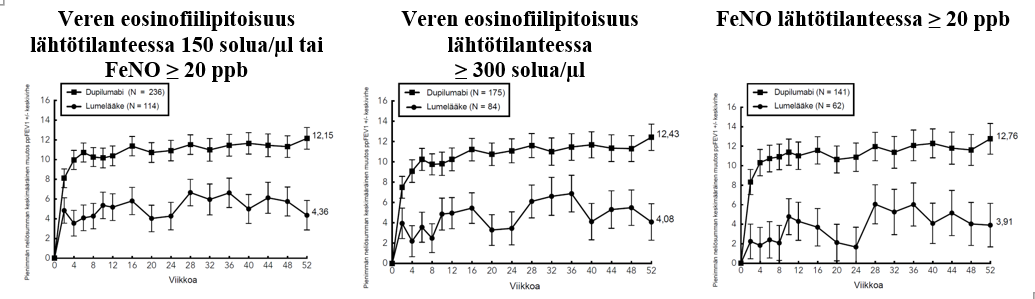
Tyypin 2 tulehduksellisella potilaspopulaatiolla vuoden aikana astman hoitoon käytettyjen systeemisten kortikosteroidikuurien lukumäärä pieneni VOYAGE-tutkimuksessa 59,3 % verrattuna lumelääkkeeseen (0,350 [95 %:n luottamusväli 0,256; 0,477] vs. 0,860 [95 %:n luottamusväli 0,616; 1,200]). Populaatiossa, jossa veren eosinofiilipitoisuus lähtötilanteessa oli ≥ 300 solua/µl, vuoden aikana astman hoitoon käytettyjen systeemisten kortikosteroidikuurien lukumäärä pieneni 66,0 % verrattuna lumelääkkeeseen (0,274 [95 %:n luottamusväli 0,188; 0,399] vs. 0,806 [95 %:n luottamusväli 0,563; 1,154]).
Dupilumabi paransi EQ-VAS-asteikolla (European Quality of Life 5-Dimension Youth Visual Analog Scale) mitattua kokonaisterveydentilaa viikkoon 52 mennessä sekä tyypin 2 tulehduksellisessa potilaspopulaatiossa että veren eosinofiiliarvon ( ≥ 300 solua/µl) perusteella määritellyssä potilaspopulaatiossa. Pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna oli 4,73 (95 %:n luottamusväli 1,18; 8,28) potilailla, joilla oli tyypin 2 tulehdus, ja 3,38 (95 %:n luottamusväli -0,66; 7,43) potilailla, joilla veren eosinofiilipitoisuus lähtötilanteessa oli ≥ 300 solua/µl.
Dupilumabi pienensi pediatristen potilaiden astman vaikutusta huoltajan elämänlaatuun PACQLQ-kyselyllä (Paediatric Asthma Quality of Life Questionnaire) viikolla 52 mitattuna sekä potilailla, joilla oli tyypin 2 tulehdus, että potilailla, joilla veren eosinofiilipitoisuus lähtötilanteessa oli ≥ 300 solua/µl. Pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna oli 0,47 (95 %:n luottamusväli 0,22; 0,72) potilailla, joilla oli tyypin 2 tulehdus, ja 0,50 (95 %:n luottamusväli 0,21; 0,79) potilailla, joilla veren eosinofiilipitoisuus lähtötilanteessa oli ≥ 300 solua/µl.
Pitkän aikavälin jatkotutkimus (EXCURSION)
Dupilumabin tehoa arvioitiin toissijaisena päätemuuttujana 365 pediatrisella (6-11-vuotiaalla) astmapotilaalla pitkän aikavälin jatkotutkimuksessa (EXCURSION). Sairaalahoitoa ja/tai päivystyskäyntiä vaatineet pahenemisvaiheet vähenivät pitkäkestoisesti ja altistus systeemisille kortikosteroideille pieneni. Keuhkofunktion havaittiin parantuneen pitkäkestoisesti useilla parametreillä, joita olivat esimerkiksi FEV1 (% viitearvosta), FVC (% viitearvosta), FEV1/FVC-suhde ja FEF25–75 % (% viitearvosta). Tämän lisäksi EXCURSION-tutkimuksen loppuun mennessä 75 %:lla potilaista saavutettiin ja/tai pystyttiin ylläpitämään normaali keuhkofunktio (ennen bronkodilataatiota mitattu FEV1 > 80 % viitearvosta). Teho säilyi kumulatiivisesti enimmillään 104 viikon mittaisen hoidon ajan (VOYAGE ja EXCURSION).
Kliininen teho ja turvallisuus eosinofiilisen ruokatorvitulehduksen hoidossa
1–11‑vuotiaat pediatriset potilaat, joilla on eosinofiilinen ruokatorvitulehdus
Dupilumabin tehoa ja turvallisuutta arvioitiin 1–11‑vuotiailla eosinofiilistä ruokatorvitulehdusta sairastavilla pediatrisilla potilailla kaksiosaisessa tutkimuksessa enintään 52 viikon ajalta (EoE KIDS, osa A ja osa B). Kaikilta tutkimukseen otetuilta potilailta edellytettiin, että tavanomainen lääkehoito (protonipumpun estäjillä) ei ollut tehonnut. Potilaista 77,5 % oli saanut aiemmin jotakin muuta tavanomaista lääkehoitoa (nieltäviä paikallisia kortikosteroideja) ennen tutkimukseen osallistumista, ja 53,5 %:lla potilaista tautia ei saatu riittävän hyvään hallintaan nieltävillä paikallisilla kortikosteroideilla tai he eivät sietäneet tällaista hoitoa tai tällainen hoito oli heille vasta‑aiheinen. Sisäänottokriteerit täyttävillä potilailla oli ≥ 15 intraepiteliaalista eosinofiiliä suurtehokenttää kohti (eos/hpf), vaikka he olivat saaneet protonipumpun estäjällä toteutetun hoitokuurin ennen seulontavaihetta tai seulontavaiheen aikana ja anamneesissa oli eosinofiilisen ruokatorvitulehduksen oireita ja löydöksiä. Osa A oli 16 viikon mittainen satunnaistettu, kaksoissokkoutettu, rinnakkaisryhmillä toteutettu, lumekontrolloitu monikeskustutkimus. Osa B oli aktiivisella hoidolla toteutettu jatkovaihe, jossa arvioitiin dupilumabihoito‑ohjelmia vielä 36 viikon ajan.
Osassa A dupilumabia arvioitiin vastaavanlaiseen lumelääkkeeseen verrattuna painoon perustuvissa hoito‑ohjelmissa (≥ 5 – < 15 kg [100 mg kahden viikon välein], ≥ 15 – < 30 kg [200 mg kahden viikon välein] ja ≥ 30 – < 60 kg [300 mg kahden viikon välein]). Dupilumabin suositeltu annostusohjelma 1–11‑vuotiaille, ≥ 40 kg painaville pediatrisille potilaille (300 mg kerran viikossa) valittiin käyttäen populaatiofarmakokineettistä mallia hyödyntäviä simulaatioita niin, että altistus vastasi altistusta sellaisilla eosinofiilistä ruokatorvitulehdusta sairastavilla, 300 mg kerran viikossa saaneilla aikuisilla ja 12–17‑vuotiailla pediatrisilla potilailla, joilla todettiin histologinen ja oireita koskeva teho (ks. kohta Farmakodynamiikka ja kohta Farmakokinetiikka).
Osaan A otettiin yhteensä 71 potilasta. Potilaiden keskimääräinen ikä oli 7 vuotta (1–11 vuotta) ja painon mediaani 24,8 kg. Potilaista 74,6 % oli miespuolisia, 87,3 % valkoihoisia, 9,9 % mustaihoisia ja 1,4 % aasialaisia. Yhteensä 55 potilasta, jotka olivat mukana osassa A, jatkoi osaan B.
Osan A ensisijainen tehoa mittaava päätemuuttuja oli niiden potilaiden osuus, joilla saavutettiin histologinen remissio (ruokatorven intraepiteliaalisten eosinofiilien suurin määrä ≤ 6 eos/hpf) viikolla 16. Toissijaisiin päätemuuttujiin kuuluivat niiden potilaiden osuus, joilla saavutettu ruokatorven intraepiteliaalisten eosinofiilien suurin määrä oli < 15 eos/hpf, sekä seuraavien muuttujien muutokset lähtötilanteesta: ruokatorven intraepiteliaalisten eosinofiilien suurin määrä (eos/hpf), EoEHSS‑järjestelmän mukaan arvioidun taudin vaikeusastetta kuvaavan pistemäärän keskiarvon absoluuttinen muutos, EoEHSS‑järjestelmän mukaan arvioidun taudin laajuutta kuvaavan pistemäärän keskiarvon absoluuttinen muutos ja EoE‑EREFS‑pistemäärän absoluuttinen muutos. Vaikutusta eosinofiilisen ruokatorvitulehduksen merkkeihin mitattiin havainnoijan ilmoittamilla tuloksilla. PESQ‑C‑kyselylomakkeella (Paediatric EoE Sign/Symptom Questionnaire‑Caregiver) arvioitiin niiden päivien osuutta, joina potilaalla ilmeni vähintään yksi eosinofiilisen ruokatorvitulehduksen merkki, ja PEESS‑pisteytyksellä (Paediatric Eosinophilic Esophagitis Symptom Score) arvioitiin eosinofiilisen ruokatorvitulehduksen merkkien esiintymistiheyttä ja vaikeusastetta.
Osan A tehoa koskevat tulokset on esitetty taulukossa 21 ja jäljempänä.
Taulukko 21: Dupilumabin tehoa koskevat tulokset viikolla 16 1–11‑vuotiailla tutkittavilla, joilla on eosinofiilinen ruokatorvitulehdus (EoE KIDS, osa A)
| Dupilumabia N = 37 | Lumelääke N = 34 | Ero vs. lumelääke |
|---|---|---|---|
Ensisijainen päätemuuttuja | |||
Niiden tutkittavien osuus, joilla saavutettiin histologinen remissio (ruokatorven intraepiteliaalisten eosinofiilien suurin määrä ≤ 6 eos/hpf), n (%)b | 25 (67,6) | 1 (2,9) | 64,5 (48,19, 80,85) |
Toissijaiset päätemuuttujat | |||
Niiden tutkittavien osuus, joilla saavutettu ruokatorven intraepiteliaalisten eosinofiilien suurin määrä oli < 15 eos/hpf), n (%)b | 31 (83,8) | 1 (2,9) | 81 (68,07, 94,10) |
Ruokatorven intraepiteliaalisten eosinofiilien suurimman määrän prosentuaalinen muutos lähtötilanteesta (eos/hpf), pienimmän neliösumman keskiarvo (SE)c | ‑86,09 (11,84) | 20,98 (12,23) | ‑107,07 (‑139,25, ‑74,90) |
EoEHSS‑järjestelmän mukaan arvioidun taudin vaikeusastetta kuvaavan pistemäärän keskiarvon (0–3d) absoluuttinen muutos lähtötilanteesta (eos/hpf), pienimmän neliösumman keskiarvo (SE) | ‑0,879 (0,05) | 0,023 (0,05) | ‑0,902 (‑1,03, ‑0,77) |
EoEHSS‑järjestelmän mukaan arvioidun taudin laajuutta kuvaavan pistemäärän keskiarvon (0–3b) absoluuttinen muutos lähtötilanteesta, pienimmän neliösumman keskiarvo (SE) | ‑0,835 (0,05) | 0,048 (0,05) | ‑0,883 (‑1,01, ‑0,76) |
EoE‑EREFS‑pistemäärän (0–18e) absoluuttinen muutos lähtötilanteesta, pienimmän neliösumman keskiarvo (SE) | ‑3,5 (0,42) | 0,3 (0,45) | ‑3,8 (‑4,94, ‑2,63) |
a DUPIXENT‑valmistetta arvioitiin porrastetuissa, painoon perustuvissa hoito‑ohjelmissa (≥ 5 – < 15 kg [100 mg kahden viikon välein], ≥ 15 – < 30 kg [200 mg kahden viikon välein] ja ≥ 30 – < 60 kg [300 mg kahden viikon välein]).
b Histologisen remission osalta prosenttiosuuksien ero on arvioitu käyttämällä Mantel–Haenszelin menetelmää, jossa tiedot on korjattu lähtötilanteen painoluokan suhteen (≥ 5 – < 15 kg, ≥ 15 – < 30 kg ja ≥ 30 – < 60 kg).
c Absoluuttisen muutoksen tai prosentuaalisen muutoksen ero on arvioitu käyttämällä kovarianssianalyysiä, jossa lähtötilanteen mittaus on kovariaatti ja hoito ja lähtötilanteen painoluokka (≥ 5 – < 15 kg, ≥ 15 – < 30 kg ja ≥ 30 – < 60 kg) kiinteitä tekijöitä.
d EoEHSS‑pistemäärät mitattiin asteikolla 0–3; suuremmat pistemäärät viittaavat histologisten poikkeavuuksien suurempaan vaikeusasteeseen ja laajuuteen.
e EoE‑EREFS‑kokonaispistemäärät mitattiin asteikolla 0–18; suuremmat pistemäärät viittaavat huonompiin endoskooppisiin tulehdus‑ ja remodellaatiolöydöksiin.
Osassa A suuremmalla osuudella dupilumabiryhmään satunnaistetuista potilaista saavutettiin histologinen remissio (ruokatorven intraepiteliaalisten eosinofiilien suurin määrä ≤ 6 eos/hpf) lumeryhmään verrattuna. Niiden tutkittavien osuus, joilla havaittiin histologinen remissio 16 viikon hoidon jälkeen osassa A, säilyi viikkoon 52 asti osassa B.
Osassa A havaittiin numeerista paranemista niiden päivien osuudessa, joina havaittiin vähintään yksi eosinofiilisen ruokatorvitulehduksen merkki (PESQ‑C), 16 viikon hoidon jälkeen, ja tulokset säilyivät viikkoon 52 asti osassa B.
Osassa A havaittiin nimellisesti merkitsevää paranemista eosinofiilisen ruokatorvitulehduksen merkkien esiintymistiheydessä ja vaikeusasteessa (PEESS‑Caregiver) 16 viikon hoidon jälkeen. PEESS‑Caregiver‑mittaria ei käytetty osassa B.
Eosinofiilistä ruokatorvitulehdusta sairastavat aikuiset ja nuoret
Katso eosinofiilistä ruokatorvitulehdusta sairastavia aikuisia ja nuoria koskevat kliiniset tiedot dupilumabin 300 mg:n vahvuuden valmisteyhteenvedosta.
Kliininen teho kroonisen spontaanin urtikarian hoidossa
Kroonista spontaania urtikariaa koskeva tutkimusohjelma sisälsi kolme satunnaistettua, kaksoissokkoutettua, lumekontrolloitua, rinnakkaisryhmillä toteutettua 24 viikon mittaista hoitoon liittyvää monikeskustutkimusta (CUPID-tutkimus A, CUPID-tutkimus B ja CUPID-tutkimus C). Kaikissa kroonista spontaania urtikariaa koskevissa kliinisissä tutkimuksissa dupilumabia käytettiin H1-antihistamiinihoidon lisänä. Dupilumabin tehoa kroonista spontaania urtikariaa sairastavien potilaiden hoidossa tukevat CUPID-tutkimukset A ja C, joihin osallistui aikuisia ja pediatrisia (6–17 vuotiaita) potilaita, joilla oli oireita H1-antihistamiinihoidosta huolimatta ja joita ei ollut hoidettu IgE:n vasta-aineilla. Dupilumabin turvallisuutta kroonista spontaania urtikariaa sairastavien potilaiden hoidossa tukevat CUPID-tutkimukset A, B ja C. CUPID-tutkimuksissa dupilumabiryhmän potilaat saivat 600 mg dupilumabia injektiona ihon alle päivänä 1 ja sen jälkeen 300 mg joka toinen viikko. Alle 60 kg painavat nuoret potilaat saivat 400 mg dupilumabia päivänä 1 ja sen jälkeen 200 mg joka toinen viikko.
CUPID-tutkimukset A ja C
CUPID-tutkimuksissa A ja C arvioitiin dupilumabin tehoa kroonista spontaania urtikariaa sairastavilla potilailla, joilla oli oireita H1-antihistamiinihoidosta huolimatta ja joita ei ollut hoidettu IgE:n vasta-aineilla. Näihin tutkimuksiin osallistui 289 vähintään 6 vuotiasta potilasta, jotka satunnaistettiin saamaan dupilumabia kahden viikon välein (N = 144) tai lumelääkettä (N = 145) lisättynä tavanomaiseen antihistamiinilääkitykseen.
Ensisijainen tehoa mittaava päätemuuttuja oli urtikaria-aktiivisuutta 7 päivän ajalta mittaavan pistemäärän (UAS7) muutos lähtötilanteesta viikolla 24. Sairauden vaikeusasteen mittarina käytettiin viikoittaista urtikaria-aktiivisuutta mittaavaa pistemäärää (UAS7, vaihteluväli 0–42), johon kuuluvat viikoittaista kutinan vaikeusastetta mittaava pistemäärä (ISS7, vaihteluväli 0–21) ja viikoittaista nokkosihottumapaukamien määrää mittaava pistemäärä (HSS7, vaihteluväli 0–21).
Keskeinen toissijainen päätemuuttuja oli kutinan vaikeusastetta 7 päivän ajalta mittaavan pistemäärän (ISS7) muutos lähtötilanteesta viikolla 24. ISS7-pistemäärä (vaihteluväli 0–21) määriteltiin päiväkohtaista kutinan vaikeusastetta mittaavien pistemäärien summaksi 7 päivän ajalta. Päiväkohtainen pistemäärä kirjattiin samaan aikaan joka päivä. Muita toissijaisia päätemuuttujia olivat nokkosihottuman vaikeusastetta 7 päivän ajalta mittaavan pistemäärän (HSS7) muutos lähtötilanteesta viikolla 24 sekä niiden potilaiden osuudet viikolla 24, joilla UAS7 oli ≤ 6 ja joilla UAS7 oli 0.
CUPID-tutkimusten A ja C demografiset tiedot ja potilaiden ominaisuudet lähtötilanteessa on esitetty alla olevassa taulukossa 22.
Taulukko 22: Demografiset tiedot ja potilaiden ominaisuudet lähtötilanteessa CUPID-tutkimuksissa A ja C
| Muuttuja | CUPID-tutkimus A (N = 138) | CUPID-tutkimus C (N = 151) | Yhdistetyt tulokset (N = 289) |
| Ikä (vuotta), keskiarvo (keskihajonta) | 41,3 (15,5) | 44,7 (16,9) | 43,1 (16,3) |
| % miehiä | 34,1 | 29,8 | 31,8 |
| Painoindeksi (kg/m2), keskiarvo (keskihajonta) | 27,67 (6,47) | 26,81 (6,16) | 27,22 (6,31) |
| Sairauden kesto, keskiarvo (keskihajonta) | 5,7 (8,5) | 6,5 (9,8) | 6,1 (9,2) |
| UAS7-pistemäärä lähtötilanteessa, keskiarvo (keskihajonta) | 31,3 (7,7) | 28,3 (7,5) | 29,8 (7,7) |
| Vaikeasti aktiivinen krooninen spontaani urtikaria (UAS7 ≥ 28) | 70,3 | 59,6 | 64,7 |
| ISS7-pistemäärä lähtötilanteessa, keskiarvo (keskihajonta) | 15,9 (4,0) | 15,1 (3,8) | 15,5 (3,9) |
| HSS7-pistemäärä lähtötilanteessa, keskiarvo (keskihajonta) | 15,4 (4,3) | 13,2 (4,7) | 14,2 (4,7) |
| UCT-pistemäärä lähtötilanteessa, keskiarvo (keskihajonta) | 3,7 (2,3) | 5,2 (3,2) | 4,5 (2,9) |
| Kokonais-IgE:n pitoisuus lähtötilanteessa (IU/ml), mediaani | 101,0 | 107,3 | 103,0 |
CUPID-tutkimusten A ja C ensisijaisten ja toissijaisten päätemuuttujien tulokset on esitetty taulukossa 23.
Taulukko 23: CUPID-tutkimusten A ja C ensisijaisten ja toissijaisten päätemuuttujien tulokset
| CUPID-tutkimus A | CUPID-tutkimus C | |||||
|---|---|---|---|---|---|---|
| Dupilumabi (N = 70) | Lumelääke (N = 68) | Ero (95 %:n luottamusväli), dupilumabi vs. lumelääkeb | Dupilumabi (N = 74) | Lumelääke (N = 77) | Ero (95 %:n luottamusväli), dupilumabi vs. lumelääkeb | |
| Ensisijainen päätemuuttuja | ||||||
| UAS7-pistemäärän muutos lähtötilanteesta viikolla 24a | -20,53 (1,76) | -12,00 (1,81) | -8,53 (-13,16; -3,90) | -15,86 (2,66) | -11,21 (2,65) | -4,65 (-8,65; -0,65) |
| Toissijaiset päätemuuttujat | ||||||
| ISS7-pistemäärän muutos lähtötilanteesta viikolla 24a | -10,24 (0,91) | -6,01 (0,94) | -4,23 (-6,63; -1,84) | -8,64 (1,41) | -6,10 (1,40) | -2,54 (-4,65; -0,43) |
| HSS7-pistemäärän muutos lähtötilanteesta viikolla 24a | -10,28 (0,91) | -5,90 (0,93) | -4,38 (-6,78; -1,98) | -7,27 (1,32) | -5,11 (1,31) | -2,17 (-4,15; -0,19) |
| Dupilumabi (N = 70) | Lumelääke (N = 68) | Kerroinsuhde (95 %:n luottamusväli), dupilumabi vs. lumelääkeb | Dupilumabi (N = 74) | Lumelääke (N = 77) | Kerroinsuhde (95 %:n luottamusväli), dupilumabi vs. lumelääkeb | |
| Niiden potilaiden osuus, joilla UAS7-pistemäärä oli ≤ 6 viikon 24 kohdallaa | 32 (45,7) | 16 (23,5) | 2,848 (1,301; 6,234) | 30 (40,5) | 18 (23,4) | 3,137 (1,371; 7,176)
|
| Niiden potilaiden osuus, joilla UAS7-pistemäärä = 0 viikon 24 kohdallaa | 22 (31,4) | 9 (13,2) | 2,908 (1,173; 7,209) | 22 (29,7) | 14 (18,2) | 2,677 (1,127; 6,359) |
a Esitetyt arvot ovat pienimmän neliösumman keskimääräisiä muutoksia lähtötilanteesta (keskivirhe) jatkuvien muuttujien osalta ja vastaajien lukumääriä ja prosenttiosuuksia binääristen muuttujien osalta.
b Ero on pienimmän neliösumman keskimääräinen ero jatkuvien muuttujien osalta ja kerroinsuhde binääristen muuttujien osalta.
Dupilumabihoito johti UAS7-pistemäärän paranemiseen ajan mittaan 24 viikon hoitojakson aikana (kuva 3).
Kuva 3: UAS7-pistemäärän pienimmän neliösumman keskimääräinen muutos viikolla 24 CUPID-tutkimusten A ja C lähtöryhmien mukaisessa populaatiossa (ITT-populaatio)
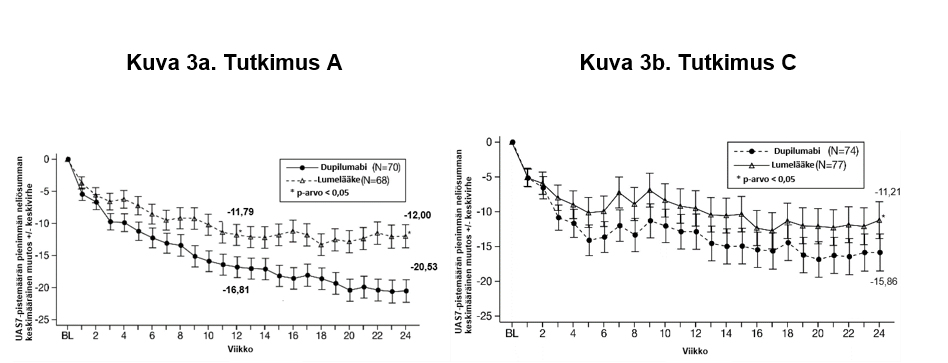
HSS7- ja ISS7-pistemäärien havaittiin parantuneen vastaavanlaisesti 24 viikon aikana.
Pediatriset potilaat
Atooppinen ihottuma
Dupilumabin teho ja turvallisuus on varmistettu vähintään 6 kuukauden ikäisillä atooppista ihottumaa sairastavilla lapsipotilailla. Dupilumabin käyttöä tässä ikäryhmässä tukevat AD‑1526-tutkimus, johon osallistui 251 nuorta, jotka olivat 12–17‑vuotiaita ja joilla oli keskivaikea tai vaikea atooppinen ihottuma, AD‑1652-tutkimus, johon osallistui 367 lapsipotilasta, jotka olivat 6–11‑vuotiaita ja joilla oli vaikea atooppinen ihottuma, sekä AD‑1539-tutkimus, johon osallistui 162 lasta, jotka olivat 6 kuukauden – 5 vuoden ikäisiä ja joilla oli keskivaikea tai vaikea atooppinen ihottuma (heistä 125:llä oli vaikea atooppinen ihottuma). Pitkäaikaiskäyttöä tukee AD‑1434-tutkimus, johon osallistui 823 lapsipotilasta, jotka olivat 6 kuukauden – 17 vuoden ikäisiä. Näistä potilaista 275 oli nuoria, 368 oli 6–11‑vuotiaita lapsia ja 180 oli 6 kuukauden – 5 vuoden ikäisiä lapsia. Turvallisuus ja teho olivat yleisesti yhdenmukaiset atooppista ihottumaa sairastavilla 6 kuukauden – 5 vuoden ikäisillä lapsipotilailla, 6–11-vuotiailla lapsipotilailla, nuorilla (12–17‑vuotiailla) potilailla ja aikuisilla potilailla (ks. kohta Haittavaikutukset). Turvallisuutta ja tehoa atooppista ihottumaa sairastavien alle 6 kuukauden ikäisten lasten hoidossa ei ole varmistettu.
Astma
QUEST-tutkimukseen osallistui yhteensä 107 keskivaikeaa tai vaikeaa astmaa sairastavaa 12–17-vuotiasta nuorta, jotka saivat joko 200 mg (N = 21) tai 300 mg (N = 18) dupilumabia (tai vastaavaa lumelääkettä joko 200 mg [N = 34] tai 300 mg [N = 34]) kahden viikon välein. Tehoa tarkasteltiin astman vaikeiden pahenemisvaiheiden ja keuhkofunktion suhteen sekä nuorilla että aikuisilla. Sekä 200 mg:n että 300 mg:n annoksella kahden viikon välein annosteltuna FEV1-arvon (pienimmän neliösumman keskimääräinen muutos lähtötilanteesta viikolla 12) havaittiin parantuneen merkittävästi (200 mg:n annoksella 0,36 l ja 300 mg:n annoksella 0,27 l). Annoksella 200 mg joka toinen viikko annosteltuna vaikeat pahenemisvaiheet vähenivät nuorilla potilailla vastaavasti kuin aikuispotilailla. Turvallisuusprofiili oli nuorilla yleisesti samankaltainen kuin aikuisilla.
Pitkäkestoiseen avoimeen tutkimukseen (TRAVERSE) otettiin mukaan yhteensä 89 iältään 12–17-vuotiasta nuorta, joilla oli keskivaikea tai vaikea astma. Tässä tutkimuksessa mitattiin toissijaisena päätemuuttujana tehoa, joka vastasi keskeisten tutkimusten tuloksia ja säilyi jopa 96 viikon ajan.
VOYAGE-tutkimukseen otettiin mukaan yhteensä 408 iältään 6–11‑vuotiasta lasta, joilla oli keskivaikea tai vaikea astma. Tutkimuksessa arvioitiin kahden viikon välein annettua 100 mg:n annosta ja kahden viikon välein annettua 200 mg:n annosta. Neljän viikon välein annetun 300 mg:n dupilumabiannoksen teho 6–11‑vuotiailla lapsilla on ekstrapoloitu kahden viikon välein annettujen 100 mg:n ja 200 mg:n annosten VOYAGE-tutkimuksessa todetun tehon perusteella, sekä QUEST-tutkimuksessa kahden viikon välein annettujen 200 mg:n ja 300 mg:n annosten aikuisilla ja nuorilla todetun tehon perusteella. Tutkittavilla, jotka olivat mukana VOYAGE-tutkimuksessa hoitojakson loppuun asti, oli mahdollisuus osallistua avoimeen jatkotutkimukseen (EXCURSION). Tässä tutkimuksessa 18 tutkittavaa (vähintään 15 kg mutta alle 30 kg) 365:stä sai 300 mg dupilumabia neljän viikon välein, ja turvallisuusprofiili oli samankaltainen kuin VOYAGE-tutkimuksessa. Turvallisuutta ja tehoa astmaa sairastavien alle 6‑vuotiaiden lasten hoidossa ei ole varmistettu.
Eosinofiilinen ruokatorvitulehdus
Dupilumabin teho ja turvallisuus eosinofiilisen ruokatorvitulehduksen hoidossa on varmistettu 1–17‑vuotiailla pediatrisilla potilailla. Dupilumabin käyttöä tässä potilasryhmässä tukevat riittävät ja hyvin kontrolloidut tutkimukset ja muut farmakokineettiset tiedot. Yhteensä 72 iältään 12–17‑vuotiasta pediatrista potilasta sai 300 mg dupilumabia kerran viikossa tai lumelääkettä 24 viikon ajan (TREET‑tutkimuksen osat A ja B). Näistä potilaista 37 sai dupilumabihoitoa osissa A ja B, ja 34 jatkoi 300 mg:n annoksen käyttöä kerran viikossa 28 lisäviikon ajan (TREET‑tutkimus, osa C). Yhteensä 71 iältään 1–11‑vuotiasta pediatrista potilasta sai dupilumabia 100 mg kahden viikon välein, 200 mg kahden viikon välein tai 300 mg kahden viikon välein tai lumelääkettä 16 viikon ajan (EoE KIDS ‑tutkimus, osa A). Näistä potilaista 37 sai dupilumabia osassa A, ja heistä kaikki jatkoivat dupilumabihoito‑ohjelmaansa 36 lisäviikon ajan (EoE KIDS ‑tutkimus, osa B). Myös populaatiofarmakokineettinen analyysi tukee dupilumabin käyttöä annostuksella 300 mg kerran viikossa eosinofiilistä ruokatorvitulehdusta sairastavilla 1–11‑vuotiailla potilailla, jotka painavat ≥ 40 kg (ks. kohta Farmakodynamiikka). Dupilumabin turvallisuus ja teho olivat samankaltaiset aikuisilla ja pediatrisilla potilailla (ks. kohta Haittavaikutukset ja kohta Farmakodynamiikka).
Krooninen spontaani urtikaria
Dupilumabin teho ja turvallisuus kroonisen spontaanin urtikarian hoidossa on varmistettu 2–17-vuotiailla pediatrisilla potilailla.
Turvallisuutta ja tehoa alle 2-vuotiaiden kroonista spontaania urtikariaa sairastavien lasten hoidossa ei ole varmistettu (ks. kohta Annostus ja antotapa).
CUPID-tutkimuksiin A, B ja C osallistui yhteensä 12 nuorta, jotka olivat 12–17-vuotiaita ja joilla oli krooninen spontaani urtikaria. He saivat 200 mg dupilumabia kahden viikon välein (30 kg – < 60 kg painavat nuoret), 300 mg dupilumabia kahden viikon välein (≥ 60 kg painavat nuoret) tai lumelääkettä. Dupilumabin vaikuttavuus kroonisen spontaanin urtikarian hoidossa nuorilla on ekstrapoloitu tiedoista, jotka koskevat hoidon tehoa tätä sairautta sairastavilla aikuisilla. Nuorille suositeltu annostus perustuu painoon.
CUPID-tutkimuksiin (A ja C) ja PKM16982-tutkimukseen osallistui yhteensä 20 pediatrista potilasta, jotka olivat 2–11-vuotiaita ja joilla oli krooninen spontaani urtikaria. 18 lasta sai dupilumabia (200 mg neljän viikon välein [5 kg – < 15 kg], 300 mg neljän viikon välein [15 kg – < 30 kg] tai 200 mg kahden viikon välein [30 kg – < 60 kg]) ja 2 sai lumelääkettä. Dupilumabin vaikuttavuus kroonisen spontaanin urtikarian hoidossa 2–11-vuotiailla pediatrisilla potilailla on ekstrapoloitu tiedoista, jotka koskevat hoidon tehoa tätä sairautta sairastavilla aikuisilla. Pediatrisille potilaille suositeltu annostus perustuu painoon.
Dupilumabin tehoa kroonisen spontaanin urtikarian hoidossa 2–11-vuotiailla lapsilla tukevat myös PKM16982-tutkimuksesta saadut tiedot. Tutkimukseen otettiin 15 potilasta, jotka saivat dupilumabia 200 mg neljän viikon välein (N = 1), 300 mg neljän viikon välein (N = 6) tai 200 mg kahden viikon välein (N = 8).
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Dupixent-valmisteen käytöstä astman hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa). Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset dupilumabin käytöstä nenäpolypoosin, kyhmykutinan, keuhkoahtaumataudin ja rakkulaisen pemfigoidin hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Dupilumabin farmakokinetiikka on samankaltainen atooppista ihottumaa, astmaa, eosinofiilista ruokatorvitulehdusta ja kroonista spontaania urtikariaa sairastavilla potilailla.
Imeytyminen
Aikuisille ihon alle annetun dupilumabin 75–600 mg:n kerta-annoksen jälkeen mediaaniajat huippupitoisuuden saavuttamiseen seerumissa (tmax) olivat 3–7 vuorokautta. Populaatiofarmakokineettisellä analyysillä määritetty dupilumabin absoluuttinen hyötyosuus ihon alle antamisen jälkeen on samansuuruinen atooppista ihottumaa, astmaa ja kroonista spontaania urtikariaa sairastavilla potilailla ja vaihtelee välillä 61–64 %.
Vakaan tilan pitoisuudet saavutettiin viikkoon 16 mennessä, kun oli annettu 600 mg:n aloitusannos ja 300 mg:n annos kahden viikon välein. Kaikissa kliinisissä tutkimuksissa vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli aikuisilla 69,2 ± 36,9 µg/ml – 80,2 ± 35,3 µg/ml kahden viikon välein annetulla 300 mg:n annoksella ja 29,2 ± 18,7 µg/ml – 36,5 ± 22,2 µg/ml kahden viikon välein annetulla 200 mg:n annoksella.
Jakautuminen
Populaatiofarmakokineettisellä analyysillä arvioitu dupilumabin noin 4,6 litran jakautumistilavuus viittaa siihen, että dupilumabi jakautuu pääasiassa verisuonistoon.
Biotransformaatio
Erityisiä metaboliatutkimuksia ei ole tehty, koska dupilumabi on proteiini. Dupilumabin odotetaan hajoavan pieniksi peptideiksi ja yksittäisiksi aminohapoiksi.
Eliminaatio
Dupilumabin eliminaatiota välittävät rinnakkaiset lineaariset ja ei-lineaariset reitit. Suurilla pitoisuuksilla dupilumabi eliminoituu pääasiassa saturoitumatonta proteolyyttistä reittiä, kun taas pienillä pitoisuuksilla on vallitsevana ei-lineaarinen saturoituva IL‑4Rα‑kohdevälitteinen eliminaatio.
Mediaaniaikaa vakaan tilan viimeisen dupilumabiannoksen antamisesta pitoisuuksien pienenemiseen toteamisrajan alapuolelle arvioitiin populaatiofarmakokineettisellä analyysillä. Käytetyt dupilumabiannostukset olivat 300 mg kerran viikossa, 300 mg kahden viikon välein, 200 mg kahden viikon välein, 300 mg neljän viikon välein tai 200 mg neljän viikon välein. Aikuisilla ja nuorilla mediaaniajat olivat 9–13 viikkoa, 6–11‑vuotiailla lapsipotilailla noin 1,5 kertaa pidemmät ja alle 6‑vuotiailla lapsipotilailla noin 2,5 kertaa pidemmät.
Lineaarisuus/ei-lineaarisuus
Ei-lineaarisen puhdistuman vuoksi pitoisuus-aikakäyrän alle jäävänä pinta-alana mitattu dupilumabialtistus suurenee enemmän kuin suhteessa annokseen 75–600 mg:n kerta-annosten ihon alle antamisen jälkeen.
Erityisryhmät
Sukupuoli
Populaatiofarmakokineettisessä analyysissä sukupuolella ei todettu olevan kliinisesti merkittäviä vaikutuksia systeemiseen dupilumabialtistukseen.
Iäkkäät
Vaiheen 2 annosmääritystutkimuksessa tai vaiheen 3 lumekontrolloiduissa tutkimuksissa dupilumabille altistetuista 1 539:stä atooppista ihottumaa sairastavasta potilaasta (myös potilaita, joilla oli käsien ja jalkaterien atooppinen ihottuma)yhteensä 71 oli iältään vähintään 65-vuotiaita. Vaikka valmisteen turvallisuudessa ja tehossa vanhempien ja nuorempien atooppista ihottumaa sairastavien aikuispotilaiden välillä ei havaittu eroja, vähintään 65-vuotiaiden potilaiden määrä ei ollut riittävä, jotta olisi voitu määrittää, eroaako iäkkäiden hoitovaste nuorempien potilaiden hoitovasteesta.
Populaatiofarmakokineettisessä analyysissä iällä ei todettu olevan kliinisesti merkittäviä vaikutuksia systeemiseen dupilumabialtistukseen. Tässä analyysissä oli kuitenkin mukana vain 61 yli 65-vuotiasta potilasta.
Dupilumabille altistetuista 1 977 astmapotilaasta yhteensä 240 potilasta oli vähintään 65-vuotiaita ja 39 potilasta oli vähintään 75-vuotiaita. Teho ja turvallisuus olivat tässä ikäryhmässä samanlaiset kuin koko tutkimuspopulaatiossa.
Dupilumabille altistetuista 198:stä kroonista spontaania urtikariaa sairastavasta potilaasta yhteensä 30 potilasta oli vähintään 65-vuotiaita, ja näistä 7 potilasta oli vähintään 75-vuotiaita. Teho ja turvallisuus olivat tässä ikäryhmässä samanlaiset kuin koko tutkimuspopulaatiossa.
Rotu
Populaatiofarmakokineettisessä analyysissä rodulla ei todettu olevan kliinisesti merkittäviä vaikutuksia systeemiseen dupilumabialtistukseen.
Maksan vajaatoiminta
Koska dupilumabi on monoklonaalinen vasta-aine, sen ei odoteta merkittävästi eliminoituvan maksan kautta. Kliinisiä tutkimuksia maksan vajaatoiminnan vaikutuksen arvioimiseksi dupilumabin farmakokinetiikkaan ei ole tehty.
Munuaisten vajaatoiminta
Koska dupilumabi on monoklonaalinen vasta-aine, sen ei odoteta merkittävästi eliminoituvan munuaisten kautta. Kliinisiä tutkimuksia munuaisten vajaatoiminnan vaikutuksen arvioimiseksi dupilumabin farmakokinetiikkaan ei ole tehty. Populaatiofarmakokineettisessä analyysissä lievällä tai keskivaikealla munuaisten vajaatoiminnalla ei todettu olevan kliinisesti merkittävää vaikutusta systeemiseen dupilumabialtistukseen. Vaikeaa munuaisten vajaatoimintaa sairastavista potilaista on hyvin vähän tietoa.
Paino
Dupilumabin minimipitoisuudet olivat pienempiä painavammilla tutkittavilla, mutta tällä ei ollut merkittävää vaikutusta tehoon.
Pediatriset potilaat
Atooppinen ihottuma
Populaatiofarmakokineettisen analyysin perusteella ikä ei vaikuttanut dupilumabin puhdistumaan aikuisilla eikä 6–17‑vuotiailla pediatrisilla potilailla. 6 kuukauden – 5 vuoden ikäisillä pediatrisilla potilailla puhdistuma suureni iän myötä, mutta tämä on otettu huomioon suositellussa annostusohjelmassa.
Dupilumabin farmakokinetiikkaa ei ole tutkittu atooppista ihottumaa sairastavilla (alle 6 kuukauden ikäisillä tai alle 5 kg painavilla) pediatrisilla potilailla.
Atooppista ihottumaa sairastavilla 12–17-vuotiailla nuorilla, jotka saivat joko 200 mg:n (< 60 kg) tai 300 mg:n (≥ 60 kg) annoksen kahden viikon välein, dupilumabin vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli 54,5 ± 27,0 µg/ml.
AD-1652-tutkimuksessa vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli 76,3 ± 37,2 µg/ml 6–11-vuotiailla atooppista ihottumaa sairastavilla lapsilla, jotka saivat 300 mg:n annoksen neljän viikon välein (≥ 15 kg). AD-1434-tutkimuksessa vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli viikon 16 kohdalla 108 ± 53,8 µg/ml 6–11-vuotiailla lapsilla, jotka saivat aluksi 300 mg neljän viikon välein (≥ 15 kg) ja joiden annosta suurennettiin niin, että tutkittavat saivat kahden viikon välein joko 200 mg (vähintään 15 kg ja alle 60 kg) tai 300 mg (≥ 60 kg). Farmakokineettisten simulaatioiden perusteella 6–11-vuotiailla lapsilla, jotka saavat 300 mg neljän viikon välein, 300 mg:n aloitusannokset päivinä 1 ja 15 saavat aikaan vastaavan vakaan tilan altistuksen kuin 600 mg:n aloitusannos päivänä 1.
6 kuukauden – 5 vuoden ikäisillä atooppista ihottumaa sairastavilla lapsilla vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli 110 ± 42,8 µg/ml, kun annostus oli 300 mg neljän viikon välein (≥ 15 – < 30 kg), ja 109 ± 50,8 µg/ml, kun annostus oli 200 mg neljän viikon välein (≥ 5 – < 15 kg).
Astma
Dupilumabin farmakokinetiikkaa astmaa sairastavien pediatristen potilaiden (alle 6‑vuotiaiden) hoidossa ei ole tutkittu.
QUEST-tutkimukseen osallistui yhteensä 107 astmaa sairastavaa 12–17-vuotiasta nuorta. Dupilumabin vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli 107 ± 51,6 µg/ml kahden viikon välein annetulla 300 mg:n annoksella ja 46,7 ± 26,9 µg/ml kahden viikon välein annetulla 200 mg:n annoksella. Painon suhteen korjatuissa tuloksissa nuorilla potilailla ei havaittu iästä riippuvaisia farmakokineettisiä eroja.
VOYAGE-tutkimuksessa dupilumabin farmakokinetiikkaa tutkittiin 270 potilaalla, joilla oli keskivaikea tai vaikea astma. Tutkittavat saivat ihon alle dupilumabia joko 100 mg kahden viikon välein (91 alle 30 kg painavaa lasta) tai 200 mg kahden viikon välein (179 vähintään 30 kg painavaa lasta). Dupilumabin noin 3,7 litran jakautumistilavuus arvioitiin populaatiofarmakokineettisellä analyysillä. Vakaan tilan pitoisuudet saavutettiin viikkoon 12 mennessä. Vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli 58,4 ± 28,0 µg/ml tutkittavilla, jotka saivat 100 mg dupilumabia, ja 85,1 ± 44.9 µg/ml tutkittavilla, jotka saivat 200 mg dupilumabia. Simulaatiossa, jossa mallinnettiin 300 mg:n annoksen antamista ihon alle neljän viikon välein vähintään 15 kg mutta alle 30 kg ja vähintään 30 kg mutta alle 60 kg painaville 6–11‑vuotiaille lapsille, ennustetut vakaan tilan minimipitoisuudet olivat vähintään 30 kg painavilla lapsilla vastaavia kuin kahden viikon välein annettujen 200 mg:n annosten saamisen jälkeen ja alle 30 kg painavilla lapsilla vastaavia kuin kahden viikon välein annettujen 100 mg:n annosten saamisen jälkeen. Lisäksi simulaatiossa, jossa mallinnettiin 300 mg:n annoksen antamista ihon alle neljän viikon välein vähintään 15 kg mutta alle 60 kg painaville 6–11‑vuotiaille lapsille, ennustetut vakaan tilan minimipitoisuudet vastasivat aikuisilla ja nuorilla tehokkaiksi osoitettuja pitoisuuksia. Populaatiofarmakokineettisellä analyysillä arvioitu vakaan tilan viimeisen annoksen antamisesta kulunut mediaaniaika dupilumabipitoisuuksien pienenemiseen toteamisrajan alapuolelle oli 14–18 viikkoa annostusohjelmalla 100 mg kahden viikon välein, 200 mg kahden viikon välein tai 300 mg neljän viikon välein.
Eosinofiilinen ruokatorvitulehdus
Kliinisessä tutkimuksessa (EoE KIDS, osa A) dupilumabin farmakokinetiikkaa tutkittiin 36:lla eosinofiilistä ruokatorvitulehdusta sairastavalla 1–11‑vuotiaalla lapsella, jotka saivat dupilumabia (≥ 5 – < 15 kg [100 mg kahden viikon välein], ≥ 15 – < 30 kg [200 mg kahden viikon välein] ja ≥ 30 – < 60 kg [300 mg kahden viikon välein]). Dupilumabin vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli 163 ± 60,8 µg/ml.
1–11-vuotiaita pediatrisia potilaita koskevissa simuloinneissa käytettiin populaatiofarmakokineettistä mallia, jolla ennustettiin dupilumabin vakaan tilan minimipitoisuudet seuraavasti: 170 ± 78 µg/ml ≥ 15 – < 30 kg painavilla potilailla, jotka saivat 200 mg dupilumabia kahden viikon välein, 158 ± 63 µg/ml ≥ 30 – < 40 kg painavilla potilailla, jotka saivat 300 mg dupilumabia kahden viikon välein, tai 276 ± 99 µg/ml ≥ 40 kg painavilla potilailla, jotka saivat 300 mg dupilumabia kerran viikossa. Vakaan tilan minimipitoisuudet simuloitiin myös aikuisille ja 12–17-vuotiaille pediatrisille potilaille ja potilaille, jotka painoivat ≥ 30 kg – < 40 kg ja jotka saivat 300 mg kahden viikon välein (159 ± 61 µg/ml).
Krooninen spontaani urtikaria
Populaatiofarmakokineettinen analyysi tukee dupilumabin käyttöä annostuksella 200 mg neljän viikon välein, 300 mg neljän viikon välein tai 200 mg kahden viikon välein kroonista spontaania urtikariaa sairastavilla 2–11-vuotiailla potilailla, jotka painavat < 60 kg.
Farmakokinetiikkaa ei ole selvitetty kroonista spontaania urtikariaa sairastavien alle 2‑vuotiaiden pediatristen potilaiden kohdalla.
CUPID-tutkimuksiin A, B ja C osallistui yhteensä kaksitoista 12–17 vuotiasta nuorta, jotka sairastivat kroonista spontaania urtikariaa. Havaittu vakaan tilan minimipitoisuus niillä 5 nuorella kroonista spontaania urtikariaa sairastavalla potilaalla, jotka saivat dupilumabia joko 300 mg kahden viikon välein tai 200 mg kahden viikon välein 24 viikon ajan, oli samalla alueella kuin potilaskohtaiset vakaan tilan minimipitoisuudet kroonista spontaania urtikariaa sairastavilla aikuisilla potilailla, jotka saivat 300 mg dupilumabia kahden viikon välein 24 viikon ajan.
PKM16982-tutkimukseen ja CUPID-tutkimuksiin (A ja C) osallistui yhteensä 20 lasta, jotka olivat 2–11-vuotiaita ja joilla oli krooninen spontaani urtikaria. 18:lla 2–11-vuotiaalla kroonista spontaania urtikariaa sairastavalla pediatrisella potilaalla viikon 24 kohdalla havaittu vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli 91,1 ± 39,4 µg/ml, kun dupilumabiannostus oli 200 mg neljän viikon välein, 300 mg neljän viikon välein tai 200 mg kahden viikon välein.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta (turvallisuutta koskevat farmakologiset päätemuuttujat mukaan lukien) ja lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Dupilumabin mahdollista mutageenisuutta ei ole arvioitu, mutta monoklonaalisten vasta-aineiden ei odoteta muuttavan DNA:ta tai kromosomeja.
Dupilumabilla ei ole tehty karsinogeenisuustutkimuksia. IL‑4Rα:n inhibitiota koskevan saatavilla olevan näytön ja surrogaattivasta-aineilla tehdyistä eläinkokeista saatujen toksikologisten tietojen arvioinnissa ei ole todettu viitteitä dupilumabin mahdollisesti lisääntyneestä karsinogeenisuudesta.
Apinoilla tehdyssä lisääntymistoksisuutta arvioineessa tutkimuksessa, jossa käytettiin apinan IL-4Rα:lle spesifistä surrogaattivasta-ainetta, ei havaittu sikiön poikkeavuuksia IL-4Rα:n saturoivilla annoksilla.
Tehostetussa pre- ja postnataalista kehitystä arvioineessa tutkimuksessa ei todettu haittavaikutuksia emoilla tai niiden jälkeläisillä 6 kuukauden kuluessa synnytyksen/syntymän jälkeen.
Uros- ja naarashiirillä tehdyissä hedelmällisyystutkimuksissa, joissa käytettiin surrogaattivasta-ainetta IL‑4Rα:aa vastaan, ei todettu hedelmällisyyden heikkenemistä (ks. kohta Raskaus ja imetys).
Farmaseuttiset tiedot
Apuaineet
L-arginiinimonohydrokloridi
L-histidiini
L-histidiinimonohydrokloridimonohydraatti
Polysorbaatti 80 (E433)
Natriumasetaattitrihydraatti
Väkevä etikkahappo (E260)
Sakkaroosi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta.
Esitäytettyä ruiskua tai esitäytettyä kynää voidaan tarvittaessa säilyttää jääkaapista ottamisen jälkeen pakkauksessaan enintään 14 vuorokauden ajan huoneenlämmössä (alle 25 °C) valolta suojattuna. Päivämäärä, jolloin pakkaus on otettu jääkaapista, on kirjoitettava muistiin ulkopakkauksessa sille varattuun tilaan. Pakkaus täytyy hävittää, jos se on ollut poissa jääkaapista pidempään kuin 14 vuorokautta tai jos viimeinen käyttöpäivämäärä on mennyt.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
DUPIXENT injektioneste, liuos, esitäytetty kynä
200 mg (L:ei) 2 x 1,14 ml (175 mg/ml, soikea ikkuna (nuoli puuttuu)) (1305,79 €)
DUPIXENT injektioneste, liuos, esitäytetty ruisku
200 mg (L:ei) 2 x 1,14 ml (175 mg/ml, neulansuojamekanismi) (1305,79 €)
PF-selosteen tieto
Dupixent 200 mg injektioneste, liuos, esitäytetty ruisku
1,14 ml liuosta silikonoidussa, tyypin 1 kirkkaasta lasista valmistetussa esitäytetyssä ruiskussa, jossa on neulan suojamekanismi ja 27 G:n, 12,7 mm:n (½ tuuman) ohutseinäinen, ruostumattomasta teräksestä valmistettu kiinteä neula, jossa viistetty kärki.
Pakkauskoko:
- 1 esitäytetty ruisku
- 2 esitäytettyä ruiskua
- Monipakkaus, joka sisältää 6 (3 kpl 2 ruiskun pakkauksia) esitäytettyä ruiskua.
Dupixent 200 mg injektioneste, liuos, esitäytetty kynä
1,14 ml liuosta silikonoidussa, tyypin 1 kirkkaasta lasista valmistetussa ruiskussa, joka on esitäytetyssä kynässä ja jossa on 27 G:n, 12,7 mm:n (½ tuuman) ohutseinäinen, ruostumattomasta teräksestä valmistettu kiinteä neula, jossa viistetty kärki.
Esitäytetyssä kynässä on joko pyöreä korkki ja soikea tarkistusikkuna, jonka ympärillä on nuoli, tai nelikulmainen korkki, jossa on uurteita ja soikea tarkistusikkuna ilman nuolta.
Pakkauskoko:
- 1 esitäytetty kynä
- 2 esitäytettyä kynää
- 6 esitäytettyä kynää
- Monipakkaus, joka sisältää 6 (2 kpl 3 kynän pakkauksia) esitäytettyä kynää.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai läpikuultava, väritön tai vaaleankeltainen steriili liuos, jossa ei ole näkyviä hiukkasia ja jonka pH-arvo on noin 5,9.
Käyttö- ja käsittelyohjeet
Pakkausselosteen lopussa on tarkat ohjeet Dupixent-valmisteen antamisesta esitäytetyllä ruiskulla tai esitäytetyllä kynällä.
Liuoksen on oltava kirkasta tai läpikuultavaa ja väritöntä tai vaaleankeltaista. Jos liuos on sameaa tai värjäytynyttä tai siinä näkyy hiukkasmaista ainetta, liuosta ei pidä käyttää.
Kun 200 mg:n esitäytetty ruisku tai esitäytetty kynä on otettu jääkaapista, sen on annettava lämmetä 30 minuuttia huoneenlämmössä, korkeintaan 25 °C:iseksi, ennen Dupixent-injektion antamista.
Esitäytettyä ruiskua tai esitäytettyä kynää ei saa altistaa kuumuudelle tai suoralle auringonvalolle eikä sitä saa ravistaa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti. Laita esitäytetty ruisku tai esitäytetty kynä käytön jälkeen terävälle jätteelle tarkoitettuun astiaan ja hävitä paikallisten määräysten mukaisesti. Älä toimita astiaa kierrätykseen.
Korvattavuus
DUPIXENT injektioneste, liuos, esitäytetty kynä
200 mg 2 x 1,14 ml
DUPIXENT injektioneste, liuos, esitäytetty ruisku
200 mg 2 x 1,14 ml
- Ylempi erityiskorvaus (100 %). Dupilumabi (vaikea atooppinen ihottuma): Vaikeaan atooppiseen ihottumaan liittyvän yleisen erytrodermian hoito aikuisille ja 6 kuukautta täyttäneille lapsille ja nuorille erityisin edellytyksin (1553).
- Alempi erityiskorvaus (65 %). Dupilumabi (vaikea astma): Vaikean astman hoito aikuisille ja 12 vuotta täyttäneille nuorille erityisin edellytyksin (260).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Dupilumabi: Vaikean astman, vaikean keuhkoahtaumataudin, vaikean atooppisen ihottuman ja vaikean kroonisen polypoottisen rinosinuiitin hoito erityisin edellytyksin (395).
ATC-koodi
D11AH05
Valmisteyhteenvedon muuttamispäivämäärä
08.04.2026
Yhteystiedot
 SANOFI OY
SANOFI OY Revontulenkuja 1
02100 Espoo
0201 200 300
www.sanofi.fi