
TECENTRIQ infuusiokonsentraatti, liuosta varten 840 mg, 1200 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Tämä potilaskortti sisältää tärkeitä turvallisuutta koskevia tietoja, joista potilaan on oltava tietoinen ennen Tecentriq-hoidon aloittamista ja hoidon aikana.
Vaikuttavat aineet ja niiden määrät
Tecentriq 840 mg infuusiokonsentraatti, liuosta varten
Yksi 14 ml:n injektiopullo konsentraattia sisältää 840 mg atetsolitsumabia*.
Tecentriq 1200 mg infuusiokonsentraatti, liuosta varten
Yksi 20 ml:n injektiopullo konsentraattia sisältää 1200 mg atetsolitsumabia*.
Laimentamisen jälkeen (ks. kohta Käyttö- ja käsittelyohjeet) liuoksen lopullisen pitoisuuden pitää olla 3,2–16,8 mg/ml.
*Atetsolitsumabi on Fc-muunnettu, humanisoitu IgG1:n monoklonaalinen PD-L1-vasta-aine (anti-programmed death-ligand 1), joka tuotetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa.
Apuaine, jonka vaikutus tunnetaan
Yksi 840 mg:n Tecentriq-injektiopullo sisältää 5,6 mg polysorbaatti 20:tä.
Yksi 1 200 mg:n Tecentriq-injektiopullo sisältää 8 mg polysorbaatti 20:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Uroteelikarsinooma
Tecentriq on tarkoitettu monoterapiana paikallisesti edennyttä tai metastasoitunutta uroteelikarsinoomaa sairastavien aikuispotilaiden hoitoon
- aiemman platinaa sisältäneen solunsalpaajahoidon jälkeen tai
- jos sisplatiinin ei katsota sopivan potilaalle, ja potilaan kasvainten PDL1-ilmentymä on ≥ 5 % (ks. kohta Farmakodynamiikka).
Varhaisvaiheen ei-pienisoluinen keuhkosyöpä
Tecentriq on tarkoitettu monoterapiana täydellisen resektion ja platinaa sisältäneen solunsalpaajahoidon jälkeen ei-pienisoluisen keuhkosyövän adjuvanttihoitoon aikuispotilaille, joilla on suuri syövän uusiutumisriski, joiden kasvainten PD-L1-ilmentymä kasvainsoluissa on ≥ 50 % ja joiden ei-pienisoluisessa keuhkosyövässä ei ole EGFR-mutaatiota eikä se ole ALK-positiivinen (ks. valintakriteerit kohdasta Farmakodynamiikka).
Pitkälle edennyt ei-pienisoluinen keuhkosyöpä
Tecentriq on tarkoitettu yhdessä bevasitsumabin, paklitakselin ja karboplatiinin kanssa metastasoitunutta ei-levyepiteeliperäistä ei-pienisoluista keuhkosyöpää (NSCLC) sairastavien aikuispotilaiden ensilinjan hoitoon. Jos ei-pienisoluisessa keuhkosyövässä on EGFR-mutaatio tai jos syöpä on ALK-positiivinen ei-pienisoluinen keuhkosyöpä, Tecentriq-hoito yhdessä bevasitsumabin, paklitakselin ja karboplatiinin kanssa on aiheellinen vain sen jälkeen, kun asianmukainen kohdennettu hoito on epäonnistunut (ks. kohta Farmakodynamiikka).
Tecentriq on tarkoitettu yhdessä nab-paklitakselin ja karboplatiinin kanssa metastasoitunutta ei-levyepiteeliperäistä ei-pienisoluista keuhkosyöpää (NSCLC) sairastavien aikuispotilaiden ensilinjan hoitoon, kun ei-pienisoluisessa keuhkosyövässä ei ole EGFR-mutaatiota eikä se ole ALK-positiivinen ei-pienisoluinen keuhkosyöpä (ks. kohta Farmakodynamiikka).
Tecentriq on tarkoitettu monoterapiana metastasoitunutta ei-pienisoluista keuhkosyöpää (NSCLC) sairastavien aikuispotilaiden ensilinjan hoitoon, kun kasvaimen PD-L1-ilmentymä on kasvainsoluissa ≥ 50 % tai kasvaimeen infiltroivissa immuunisoluissa ≥ 10 % ja kun ei-pienisoluisessa keuhkosyövässä ei ole EGFR-mutaatiota eikä se ole ALK-positiivinen ei-pienisoluinen keuhkosyöpä (ks. kohta Farmakodynamiikka).
Tecentriq on tarkoitettu monoterapiana pitkälle edennyttä ei-pienisoluista keuhkosyöpää (NSCLC) sairastavien aikuispotilaiden ensilinjan hoitoon, kun platinapohjainen hoito ei sovellu potilaalle (ks. valintakriteerit kohdasta Farmakodynamiikka).
Tecentriq on tarkoitettu monoterapiana paikallisesti edennyttä tai metastasoitunutta ei-pienisoluista keuhkosyöpää (NSCLC) sairastavien aikuispotilaiden hoitoon aiemman solunsalpaajahoidon jälkeen. Jos ei-pienisoluisessa keuhkosyövässä on EGFR-mutaatio tai jos syöpä on ALK-positiivinen ei-pienisoluinen keuhkosyöpä (NSCLC), potilaalle on pitänyt antaa myös kohdennettuja hoitoja ennen Tecentriq-hoidon antamista (ks. kohta Farmakodynamiikka).
Pienisoluinen keuhkosyöpä
Tecentriq on tarkoitettu yhdessä karboplatiinin ja etoposidin kanssa levinnyttä pienisoluista keuhkosyöpää (SCLC) sairastavien aikuispotilaiden ensilinjan hoitoon (ks. kohta Farmakodynamiikka).
Kolmoisnegatiivinen rintasyöpä
Tecentriq on tarkoitettu yhdessä nab-paklitakselin kanssa aikuispotilaiden leikkaushoitoon soveltumattoman paikallisesti edenneen tai metastasoituneen kolmoisnegatiivisen rintasyövän hoitoon, kun potilaan kasvainten PD-L1-ilmentymä on ≥ 1 %, eikä potilas ole aiemmin saanut solunsalpaajahoitoa metastasoituneeseen tautiin.
Hepatosellulaarinen karsinooma
Tecentriq on tarkoitettu yhdessä bevasitsumabin kanssa edennyttä tai leikkauksella poistettavaksi soveltumatonta hepatosellulaarista karsinoomaa sairastavien aikuispotilaiden hoitoon, kun potilas ei ole aiemmin saanut systeemistä hoitoa (ks. kohta Farmakodynamiikka).
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Syöpälääkkeiden käyttöön perehtyneen lääkärin on aloitettava Tecentriq-hoito ja valvottava sen toteuttamista.
Uroteelikarsinoomaa tai kolmoisnegatiivista rintasyöpää tai ei-pienisoluista keuhkosyöpää sairastavien potilaiden PD-L1-testaus
Tecentriq-monoterapia
Jos käyttöaiheessa on niin määritelty, Tecentriq-hoidon valitsemisen potilaalle pitää tapahtua validoidulla testillä varmistetun kasvaimen PD-L1-ilmentymän perusteella (ks. kohdat Käyttöaiheet ja Farmakodynamiikka).
Tecentriq-yhdistelmähoito
Aiemmin hoitamatonta kolmoisnegatiivista rintasyöpää sairastavien potilaiden hoito pitää valita validoidulla testillä varmistetun kasvaimen PD-L1-ilmentymän perusteella (ks. kohta Farmakodynamiikka).
Annostus
Suositeltu Tecentriq-annos on joko 840 mg laskimoon kahden viikon välein tai 1200 mg laskimoon kolmen viikon välein tai 1680 mg laskimoon neljän viikon välein taulukon 1 mukaisesti.
Tutustu Tecentriq-yhdistelmähoidossa myös yhdistelmähoidossa käytettävien muiden valmisteiden täydellisiin tuotetietoihin (ks. kohta Farmakodynamiikka).
Taulukko 1. Suositellut Tecentriq-annokset laskimoon
| Käyttöaihe | Suositeltu annos ja hoitoaikataulu | Hoidon kesto |
| Tecentriq-monoterapia | ||
| Uroteelikarsinooman ensilinjan hoito |
| Kunnes tauti etenee tai ilmaantuu toksisuutta, joka ei ole hallittavissa. |
| Metastasoituneen ei-pienisoluisen keuhkosyövän ensilinjan hoito | ||
| Platinahoitoon soveltumattoman ei-pienisoluisen keuhkosyövän ensilinjan hoito | ||
| Varhaisvaiheen ei-pienisoluisen keuhkosyövän hoito |
| 1 vuoden ajan, paitsi jos tauti uusiutuu tai ilmaantuu toksisuutta, joka ei ole hyväksyttävissä. Yli 1 vuoden kestävää hoitoa ei ole tutkittu. |
| Uroteelikarsinooman toisen linjan hoito |
| Kunnes kliinistä hyötyä ei enää todeta tai ilmaantuu toksisuutta, joka ei ole hallittavissa. |
| Ei-pienisoluisen keuhkosyövän toisen linjan hoito | ||
| Tecentriq-yhdistelmähoito | ||
| Ei-levyepiteeliperäisen ei-pienisoluisen keuhkosyövän ensilinjan hoito yhdistelmänä bevasitsumabin, paklitakselin ja karboplatiinin kanssa | Induktio- ja ylläpitovaiheet:
Tecentriq pitää antaa ennen muita samana päivänä annettavia valmisteita. Samanaikaisesti annettavien valmisteiden induktiovaihe (neljä tai kuusi hoitosykliä): Bevasitsumabia, paklitakselia ja sen jälkeen karboplatiinia annetaan kolmen viikon välein. Ylläpitovaihe (ilman solunsalpaajia): bevasitsumabi 3 viikon välein. | Kunnes tauti etenee tai ilmaantuu toksisuutta, joka ei ole hallittavissa. Epätyypillisiä vasteita (eli kasvain pienenee taudin edettyä alkuvaiheessa) on havaittu, kun Tecentriq-hoitoa on jatkettu taudin edettyä. Hoidon jatkamista taudin edettyä voidaan harkita lääkärin arvion perusteella. |
| Ei-levyepiteeliperäisen ei-pienisoluisen keuhkosyövän ensilinjan hoito yhdistelmänä nab-paklitakselin ja karboplatiinin kanssa | Induktio- ja ylläpitovaiheet:
Tecentriq pitää antaa ennen muita samana päivänä annettavia valmisteita. Samanaikaisesti annettavien valmisteiden induktiovaihe (neljä tai kuusi hoitosykliä): Nab-paklitakseli ja karboplatiini annetaan päivänä 1; lisäksi nab-paklitakselia annetaan kunkin kolmiviikkoisen hoitosyklin päivinä 8 ja 15. | Kunnes tauti etenee tai ilmaantuu toksisuutta, joka ei ole hallittavissa. Epätyypillisiä vasteita (eli kasvain pienenee taudin edettyä alkuvaiheessa) on havaittu, kun Tecentriq-hoitoa on jatkettu taudin edettyä. Hoidon jatkamista taudin edettyä voidaan harkita lääkärin arvion perusteella. |
| Levinneen pienisoluisen keuhkosyövän ensilinjan hoito yhdistelmänä karboplatiinin ja etoposidin kanssa | Induktio- ja ylläpitovaiheet:
Tecentriq pitää antaa ennen muita samana päivänä annettavia valmisteita. Samanaikaisesti annettavien valmisteiden induktiovaihe (neljä hoitosykliä): Karboplatiini ja sen jälkeen etoposidi annetaan päivänä 1; etoposidia annetaan myös kunkin kolmiviikkoisen hoitosyklin päivinä 2 ja 3. | Kunnes tauti etenee tai ilmaantuu toksisuutta, joka ei ole hallittavissa. Epätyypillisiä vasteita (eli kasvain pienenee taudin edettyä alkuvaiheessa) on havaittu, kun Tecentriq-hoitoa on jatkettu taudin edettyä. Hoidon jatkamista taudin edettyä voidaan harkita lääkärin arvion perusteella. |
| Leikkaushoitoon soveltumattoman paikallisesti edenneen tai metastasoituneen kolmoisnegatiivisen rintasyövän ensilinjan hoito yhdistelmänä nab-paklitakselin kanssa |
Tecentriq pitää antaa ensin, jos samana päivänä annetaan nab-paklitakselia. Nab-paklitakselia pitää antaa 100 mg/m2 kunkin 28-päiväisen hoitosyklin päivinä 1, 8 ja 15. | Kunnes tauti etenee tai ilmaantuu toksisuutta, joka ei ole hallittavissa. |
| Edenneen tai leikkauksella poistettavaksi soveltumattoman hepatosellulaarisen karsinooman hoito yhdistelmänä bevasitsumabin kanssa |
Tecentriq pitää antaa ensin, jos samana päivänä annetaan bevasitsumabia. Bevasitsumabia annetaan annoksena 15 mg/kg 3 viikon välein. | Kunnes kliinistä hyötyä ei enää todeta tai ilmaantuu toksisuutta, joka ei ole hallittavissa. |
Annosten viivästyminen tai antamatta jääminen
Jos suunniteltu Tecentriq-annos jää antamatta, annos on annettava mahdollisimman pian. Antoaikataulua on muutettava niin, että asianmukainen antoväli säilyy.
Annoksen muuttaminen hoidon aikana
Tecentriq-annoksen pienentämistä ei suositella.
Annostelun siirtäminen tai hoidon lopettaminen (ks. myös kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset)
Taulukko 2. Ohjeet Tecentriq-annoksen muutoksiin
| Immuunivälitteinen haittavaikutus | Vaikeusaste | Hoidon muutos |
| Pneumoniitti | Aste 2 | Keskeytä Tecentriq-hoito. Hoitoa voidaan jatkaa, kun tapahtuma lievenee 12 viikon kuluessa asteeseen 0 tai asteeseen 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. |
| Aste 3 tai 4 | Lopeta Tecentriq-hoito pysyvästi. | |
| Hepatiitti potilailla, joilla ei ole hepatosellulaarista karsinoomaa | Aste 2: (ALAT tai ASAT > 3–5x viitevälin yläraja [upper limit of normal, ULN] tai veren bilirubiinipitoisuus > 1,5−3x ULN) | Keskeytä Tecentriq-hoito. Hoitoa voidaan jatkaa, kun tapahtuma lievenee 12 viikon kuluessa asteeseen 0 tai asteeseen 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. |
Aste 3 tai 4: (ALAT tai ASAT > 5x ULN tai veren bilirubiinipitoisuus > 3x ULN) | Lopeta Tecentriq-hoito pysyvästi. | |
| Hepatiitti potilailla, joilla on hepatosellulaarinen karsinooma | Jos ASAT/ALAT on lähtötilanteessa viitevälin rajoissa ja suurenee tasolle > 3x – ≤ 10x ULN tai Jos ASAT/ALAT on lähtötilanteessa > 1 – ≤ 3x ULN ja suurenee tasolle > 5x – ≤ 10x ULN tai Jos ASAT/ALAT on lähtötilanteessa > 3x – ≤ 5x ULN ja suurenee tasolle > 8x – ≤ 10x ULN | Keskeytä Tecentriq-hoito Hoitoa voidaan jatkaa, kun tapahtuma lievenee 12 viikon kuluessa asteeseen 0 tai asteeseen 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. |
Jos ASAT/ALAT suurenee tasolle > 10x ULN tai kokonaisbilirubiinipitoisuus suurenee tasolle > 3x ULN | Lopeta Tecentriq-hoito pysyvästi | |
| Koliitti | Asteen 2 tai 3 ripuli (pahentunut lähtötilanteesta ≥ 4 ulostuskertaa/vrk) tai oireinen koliitti | Keskeytä Tecentriq-hoito. Hoitoa voidaan jatkaa, kun tapahtuma lievenee 12 viikon kuluessa asteeseen 0 tai asteeseen 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. |
| Asteen 4 ripuli tai koliitti (hengenvaarallinen, kiireellinen hoito aiheellista) | Lopeta Tecentriq-hoito pysyvästi. | |
| Hypotyreoosi tai hypertyreoosi | Oireinen | Keskeytä Tecentriq-hoito Hypotyreoosi: Hoitoa voidaan jatkaa, kun oireet saadaan hallintaan kilpirauhasen korvaushoidolla ja TSH-pitoisuudet pienenevät. Hypertyreoosi: Hoitoa voidaan jatkaa, kun oireet saadaan hallintaan tyreostaattisella lääkevalmisteella ja kilpirauhasen toiminta paranee. |
| Lisämunuaisen vajaatoiminta | Oireinen | Keskeytä Tecentriq-hoito. Hoitoa voidaan jatkaa, kun oireet lievenevät 12 viikon kuluessa asteeseen 0 tai asteeseen 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon ja potilaan tila on korvaushoidon avulla vakaa. |
| Hypofysiitti | Aste 2 tai 3 | Keskeytä Tecentriq-hoito. Hoitoa voidaan jatkaa, kun oireet lievenevät 12 viikon kuluessa asteeseen 0 tai asteeseen 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon ja potilaan tila on korvaushoidon avulla vakaa. |
| Aste 4 | Lopeta Tecentriq-hoito pysyvästi. | |
| Tyypin 1 diabetes mellitus | Asteen 3 tai 4 hyperglykemia (paastotilan glukoosipitoisuus > 250 mg/dl eli 13,9 mmol/l) | Keskeytä Tecentriq-hoito. Hoitoa voidaan jatkaa, kun diabetes on saatu insuliinikorvaushoidolla hyvään hoitotasapainoon. |
| Ihottuma/vaikeat ihon lääkereaktiot (SCAR) | Aste 3 tai epäilty Stevens–Johnsonin oireyhtymä tai toksinen epidermaalinen nekrolyysi1 | Keskeytä Tecentriq-hoito. Hoitoa voidaan jatkaa, kun oireet lievenevät 12 viikon kuluessa asteeseen 0 tai asteeseen 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. |
Aste 4 tai varmistunut Stevens–Johnsonin oireyhtymä tai toksinen epidermaalinen nekrolyysi1 | Lopeta Tecentriq-hoito pysyvästi. | |
| Myasteeninen oireyhtymä/myasthenia gravis, Guillain–Barrén oireyhtymä, meningoenkefaliitti ja kasvohalvaus | Asteen 1 tai 2 kasvohalvaus | Keskeytä Tecentriq-hoito. Hoitoa voidaan jatkaa, jos tapahtuma häviää täysin. Jos tapahtuma ei häviä täysin Tecentriq-hoidon ollessa keskeytettynä, lopeta Tecentriq-hoito pysyvästi. |
Kaikkien vaikeusasteiden myasteeninen oireyhtymä/myasthenia gravis, Guillain–Barrén oireyhtymä ja meningoenkefaliitti tai asteen 3 tai 4 kasvohalvaus | Lopeta Tecentriq-hoito pysyvästi. | |
| Myeliitti | Aste 2, 3 tai 4 | Lopeta Tecentriq-hoito pysyvästi. |
| Haimatulehdus | Asteen 3 tai 4 seerumin amylaasi- tai lipaasipitoisuuden suureneminen (> 2,0x ULN) tai asteen 2 tai 3 haimatulehdus | Keskeytä Tecentriq-hoito. Hoitoa voidaan jatkaa, kun seerumin amylaasi- ja lipaasipitoisuus korjautuu 12 viikon kuluessa asteeseen 0 tai asteeseen 1 tai haimatulehduksen oireet ovat hävinneet ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. |
| Asteen 4 tai minkä tahansa vaikeusasteen toistuva haimatulehdus | Lopeta Tecentriq-hoito pysyvästi. | |
| Sydänlihastulehdus | ||
| Aste 2 tai vaikeampiasteinen | Lopeta Tecentriq-hoito pysyvästi. | |
| Munuaistulehdus | Aste 2: (kreatiniinipitoisuus > 1,5–3,0x lähtötilanteen arvo tai > 1,5–3,0x ULN) | Keskeytä Tecentriq-hoito. Hoitoa voidaan jatkaa, kun oireet lievenevät 12 viikon kuluessa asteeseen 0 tai asteeseen 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. |
Aste 3 tai 4: (kreatiniinipitoisuus > 3,0x lähtötilanteen arvo tai > 3,0 x ULN) | Lopeta Tecentriq-hoito pysyvästi. | |
| Myosiitti | Aste 2 tai 3 | Keskeytä Tecentriq-hoito. |
| Asteen 4 tai asteen 3 toistuva myosiitti | Lopeta Tecentriq-hoito pysyvästi. | |
| Sydänpussin sairaudet | Asteen 1 sydänpussitulehdus | Keskeytä Tecentriq-hoito.2 |
| Aste 2 tai vaikeampiasteinen | Lopeta Tecentriq-hoito pysyvästi. | |
| Hemofagosyyttinen lymfohistiosytoosi | Epäilty hemofagosyyttinen lymfohistiosytoosi1 | Lopeta Tecentriq-hoito pysyvästi. |
| Muut immuunivälitteiset haittavaikutukset | Aste 2 tai aste 3 | Keskeytä hoito, kunnes haittavaikutukset vähenevät 12 viikon kuluessa asteeseen 0–1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. |
| Aste 4 tai toistuva aste 3 | Lopeta Tecentriq-hoito pysyvästi (paitsi jos kyseessä on hormonikorvaushoidolla hoitotasapainossa pysyvä umpierityssairaus). | |
| Muut haittavaikutukset | Vaikeusaste | Hoidon muutos |
| Infuusioon liittyvät reaktiot | Aste 1 tai 2 | Hidasta infuusionopeutta tai keskeytä infuusion anto. Hoitoa voidaan jatkaa, kun tapahtuma on korjautunut. |
| Aste 3 tai 4 | Lopeta Tecentriq-hoito pysyvästi. |
ALAT = alaniiniaminotransferaasi; ASAT = aspartaattiaminotransferaasi; ULN = viitevälin yläraja (upper limit of normal).
Huom.: Toksisuuden vaikeusasteen pitää perustua voimassa olevaan National Cancer Institute Common Terminology Criteria for Adverse Event (NCI-CTCAE) ‑luokitukseen.
1 vaikeusasteesta riippumatta
2 tee tarkka sydäntutkimus syyn selvittämiseksi, ja hoida asianmukaisesti
Erityiset potilasryhmät
Pediatriset potilaat
Tecentriq-valmisteen turvallisuutta ja tehoa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole varmistettu. Tällä hetkellä saatavissa olevat tiedot kuvataan kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, mutta annostuksesta ei voida antaa suosituksia.
Iäkkäät potilaat
≥ 65-vuotiaiden potilaiden Tecentriq-annosta ei populaatiofarmakokineettisen analyysin perusteella tarvitse muuttaa (ks. kohdat Haittavaikutukset ja Farmakodynamiikka).
Aasialaista syntyperää olevat potilaat
Aasialaista syntyperää olevilla potilailla havaittiin IMpower150-tutkimuksessa lisääntynyttä hematologista toksisuutta, ja sen vuoksi paklitakselin aloitusannokseksi suositellaan 175 mg/m2 kolmen viikon välein.
Munuaisten vajaatoiminta
Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei populaatiofarmakokineettisen analyysin perusteella tarvitse muuttaa (ks. kohta Farmakokinetiikka). Tiedot vaikeaa munuaisten vajaatoimintaa sairastavista potilaista ovat liian suppeita, jotta tästä potilasryhmästä voitaisiin tehdä päätelmiä.
Maksan vajaatoiminta
Lievää tai keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden annosta ei populaatiofarmakokineettisen analyysin perusteella tarvitse muuttaa. Tecentriq-valmistetta ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla (ks. kohta Farmakokinetiikka).
ECOG (Eastern Cooperative Oncology Group) suorituskykyluokka ≥ 2
ECOG-suorituskykyluokan ≥ 2 potilaita ei otettu mukaan uroteelikarsinooman toisen linjan hoitoa, kolmoisnegatiivista rintasyöpää, levinnyttä pienisoluista keuhkosyöpää eikä hepatosellulaarista karsinoomaa koskeneisiin kliinisiin tutkimuksiin (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Antotapa
On tärkeää tarkistaa valmisteen etiketit ja siten varmistaa, että annetaan oikeaa potilaalle määrättyä lääkemuotoa (laskimoon tai ihon alle).
Tecentriq-valmisteen laskimoon annettava lääkemuoto ei ole tarkoitettu annettavaksi ihon alle, ja sitä saa antaa vain infuusiona laskimoon. Infuusioita ei saa antaa laskimoon paineella eikä boluksena.
Tecentriq-valmisteen laskimoon annettavaa lääkemuotoa parhaillaan saavien potilaiden hoitoon voidaan vaihtaa atetsolitsumabi-injektioneste, tai päinvastoin.
Ensimmäinen laskimoon annettava Tecentriq-annos on annettava 60 minuutin kestoisena. Jos potilas sietää hyvin ensimmäisen infuusion, seuraavat infuusiot voidaan antaa 30 minuutin kestoisina.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ja käsittelystä ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys atetsolitsumabille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Immuunivälitteiset haittavaikutukset
Useimmat atetsolitsumabihoidon aikana esiintyneet immuunivälitteiset haittavaikutukset korjautuivat, kun atetsolitsumabihoito keskeytettiin ja aloitettiin kortikosteroidi- ja/tai tukihoito. Useampaan kuin yhteen elinjärjestelmään vaikuttavia immuunivälitteisiä haittavaikutuksia on havaittu. Atetsolitsumabin immuunivälitteisiä haittavaikutuksia voi ilmaantua viimeisen atetsolitsumabiannoksen jälkeen.
Epäiltäessä immuunivälitteisiä haittavaikutuksia on tehtävä perusteelliset tutkimukset syyn varmistamiseksi tai muiden syiden poissulkemiseksi. Haittavaikutuksen vaikeusasteen perusteella atetsolitsumabihoito on keskeytettävä, ja potilaalle on annettava kortikosteroideja. Kun haittavaikutus lievenee asteeseen ≤ 1, kortikosteroidihoito on lopetettava vähitellen ≥ 1 kuukauden aikana. Kliinisistä tutkimuksista saadut suppeat tiedot potilaista, joiden immuunivälitteisiä haittavaikutuksia ei saatu hallintaan systeemisesti käytetyillä kortikosteroideilla, osoittavat, että muiden systeemisten immunosuppressiivisten lääkkeiden käyttöä voidaan harkita.
Atetsolitsumabin käyttö on lopetettava pysyvästi, jos jokin asteen 3 immuunivälitteinen haittavaikutus uusiutuu tai jos ilmaantuu jokin asteen 4 immuunivälitteinen haittavaikutus, lukuun ottamatta umpierityssairauksia, jotka ovat hormonikorvaushoidolla hoitotasapainossa (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Havainnoivista tutkimuksista saadut tiedot viittaavat siihen, että immuunivälitteisten haittavaikutusten riski voi autoimmuunitautia ennestään sairastavilla potilailla olla lisääntynyt immuuniaktivaation vapauttajahoidon jälkeen verrattuna riskiin potilailla, joilla ei ole ennestään autoimmuunitautia. Lisäksi perussairautena sairastetun autoimmuunitaudin äkillinen paheneminen oli yleistä, mutta valtaosin lievää ja hallittavissa.
Immuunivälitteinen pneumoniitti
Atetsolitsumabilla tehdyissä kliinisissä tutkimuksissa on havaittu pneumoniittitapauksia, myös kuolemaan johtaneita tapauksia (ks. kohta Haittavaikutukset). Potilaita pitää tarkkailla pneumoniitin oireiden ja löydösten havaitsemiseksi, ja muut kuin immuunivälitteiseen pneumoniittiin viittaavat syyt on suljettava pois.
Asteen 2 pneumoniitin yhteydessä atetsolitsumabihoito pitää keskeyttää ja aloittaa prednisonihoito annoksilla 1−2 mg/kg/vrk tai vastaava hoito. Jos oireet lievenevät asteeseen ≤ 1, kortikosteroidihoito pitää lopettaa vähitellen ≥ 1 kuukauden aikana. Atetsolitsumabihoitoa voidaan jatkaa, jos tapahtuma lievenee 12 viikon kuluessa asteeseen ≤ 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. Asteen 3 tai 4 pneumoniitin yhteydessä atetsolitsumabihoito pitää lopettaa pysyvästi.
Immuunivälitteinen hepatiitti
Atetsolitsumabilla tehdyissä kliinisissä tutkimuksissa on havaittu hepatiittitapauksia, joista osa on johtanut potilaan kuolemaan (ks. kohta Haittavaikutukset). Potilaita pitää tarkkailla hepatiitin oireiden ja löydösten havaitsemiseksi.
Aspartaattiaminotransferaasi- (ASAT), alaniiniaminotransferaasi- (ALAT) ja bilirubiinipitoisuuksia pitää seurata ennen atetsolitsumabihoidon aloittamista ja säännöllisesti hoidon aikana sekä kliinisen arvion perusteella tarpeen mukaan.
Jos potilaalla, jolla ei ole hepatosellulaarista karsinoomaa, asteen 2 tapahtuma (ALAT tai ASAT > 3–5x ULN tai veren bilirubiinipitoisuus > 1,5−3x ULN) jatkuu pidempään kuin 5–7 päivää, atetsolitsumabihoito pitää keskeyttää ja aloittaa prednisonihoito annoksilla 1−2 mg/kg/vrk tai vastaava hoito. Jos tapahtuma lievenee asteeseen ≤ 1, kortikosteroidihoito pitää lopettaa vähitellen ≥ 1 kuukauden aikana.
Atetsolitsumabihoitoa voidaan jatkaa, jos tapahtuma lievenee 12 viikon kuluessa asteeseen ≤ 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. Asteen 3 tai asteen 4 tapahtumien (ALAT tai ASAT > 5,0x ULN tai veren bilirubiinipitoisuus > 3x ULN) ilmaantuessa atetsolitsumabihoito on lopetettava pysyvästi.
Jos potilaalla, jolla on hepatosellulaarinen karsinooma, ALAT tai ASAT on lähtötilanteessa viitearvojen rajoissa ja suurenee tasolle > 3 – ≤ 10 x ULN tai on lähtötilanteessa > 1 ULN – ≤ 3 x ULN ja suurenee tasolle > 5 – ≤ 10 x ULN tai on lähtötilanteessa > 3 ULN – ≤ 5 x ULN ja suurenee tasolle > 8 – ≤ 10 x ULN yli 5–7 päivän ajaksi, atetsolitsumabihoito pitää keskeyttää ja aloittaa prednisonihoito annoksilla 1–2 mg/kg/vrk tai vastaava hoito. Jos tapahtuma lievenee asteeseen ≤ 1, kortikosteroidihoito pitää lopettaa vähitellen ≥ 1 kuukauden aikana.
Atetsolitsumabihoitoa voidaan jatkaa, jos tapahtuma lievenee 12 viikon kuluessa asteeseen ≤ 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. Jos ALAT tai ASAT suurenee tasolle > 10 x ULN tai kokonaisbilirubiinipitoisuus suurenee tasolle > 3 x ULN, atetsolitsumabihoito on lopetettava pysyvästi.
Immuunivälitteinen koliitti
Atetsolitsumabilla tehdyissä kliinisissä tutkimuksissa on havaittu ripulia ja koliittia (ks. kohta Haittavaikutukset). Potilaita pitää seurata koliitin oireiden ja löydösten havaitsemiseksi.
Atetsolitsumabihoito pitää keskeyttää, jos potilaalla on asteen 2 tai 3 ripulia (pahentunut lähtötilanteesta ≥ 4 ulostuskertaa/vrk) tai koliitti (oireinen). Jos oireet jatkuvat asteen 2 ripulin tai koliitin yhteydessä > 5 päivää tai uusiutuvat, potilaalle on aloitettava hoito prednisoniannoksilla 1−2 mg/kg/vrk tai vastaava hoito. Jos potilaalla on asteen 3 ripuli tai koliitti, hänelle on aloitettava laskimoon annettava kortikosteroidihoito (1−2 mg/kg/vrk metyyliprednisolonia tai vastaava hoito). Kun oireet lievenevät, aloitetaan hoito prednisoniannoksilla 1–2 mg/kg/vrk tai vastaava hoito. Jos oireet lievenevät asteeseen ≤ 1, kortikosteroidihoito pitää lopettaa vähitellen ≥ 1 kuukauden aikana. Atetsolitsumabihoitoa voidaan jatkaa, jos tapahtuma lievenee 12 viikon kuluessa asteeseen ≤ 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. Asteen 4 (hengenvaarallisen, kiireellistä hoitoa vaativan) ripulin tai koliitin ilmetessä atetsolitsumabihoito on lopetettava pysyvästi. Koliittiin liittyvä mahdollinen komplikaatio maha-suolikanavan perforaatio pitää ottaa huomioon.
Immuunivälitteiset umpierityssairaudet
Atetsolitsumabilla tehdyissä kliinisissä tutkimuksissa on havaittu hypotyreoosia, hypertyreoosia, lisämunuaisten vajaatoimintaa, hypofysiittiä ja tyypin 1 diabetes mellitusta, myös diabeettista ketoasidoosia (ks. kohta Haittavaikutukset).
Potilaita pitää seurata umpierityssairauksien kliinisten oireiden ja löydösten havaitsemiseksi. Kilpirauhasen toimintaa pitää seurata ennen atetsolitsumabihoitoa sekä säännöllisesti hoidon aikana. Jos potilaalla on kilpirauhasen toimintakokeiden poikkeavuuksia ennen hoitoa, potilaalle on harkittava tarkoituksenmukaista hoitoa.
Jos kilpirauhasen toimintakokeiden poikkeavuudet ovat oireettomia, potilaalle voidaan antaa atetsolitsumabihoitoa. Jos potilaalla on oireinen hypotyreoosi, atetsolitsumabihoito pitää keskeyttää ja aloittaa tarpeen mukaan kilpirauhashormonin korvaushoito. Pelkkä hypotyreoosi voidaan hoitaa korvaushoidolla ilman kortikosteroideja. Jos potilaalla on oireinen hypertyreoosi, atetsolitsumabihoito pitää keskeyttää ja aloittaa tarpeen mukaan hoito tyreostaattisella lääkevalmisteella. Kun oireet ovat hallinnassa ja kilpirauhasen toiminta paranee, atetsolitsumabihoitoa voidaan jatkaa.
Jos potilaalla on oireinen lisämunuaisten vajaatoiminta, atetsolitsumabihoito pitää keskeyttää ja aloittaa hoito laskimoon annettavilla kortikosteroideilla (1−2 mg/kg/vrk metyyliprednisolonia tai vastaava hoito). Kun oireet lievenevät, hoitoa pitää jatkaa prednisoniannoksilla 1−2 mg/kg/vrk tai vastaavalla hoidolla. Jos oireet lievenevät asteeseen ≤ 1, kortikosteroidihoito pitää lopettaa vähitellen ≥ 1 kuukauden aikana. Hoitoa voidaan jatkaa, jos oireet lievenevät 12 viikon kuluessa asteeseen ≤ 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia tai vastaavaa vuorokaudessa ja potilaan tila on korvaushoidolla (tarvittaessa) vakaa.
Jos potilaalle ilmaantuu asteen 2 tai asteen 3 hypofysiitti, atetsolitsumabihoito pitää keskeyttää ja aloittaa hoito laskimoon annettavilla kortikosteroideilla (1–2 mg/kg/vrk metyyliprednisolonia tai vastaavaa), ja tarvittaessa pitää aloittaa hormonikorvaushoito. Kun oireet lievenevät, hoitoa jatketaan prednisoniannoksilla 1–2 mg/kg/vrk tai vastaavalla hoidolla. Jos oireet lievenevät asteeseen ≤ 1, kortikosteroidihoito pitää lopettaa vähitellen ≥ 1 kuukauden aikana. Hoitoa voidaan jatkaa, jos oireet lievenevät 12 viikon kuluessa asteeseen ≤ 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia tai vastaavaa vuorokaudessa ja potilaan tila on korvaushoidolla (tarvittaessa) vakaa. Jos ilmaantuu asteen 4 hypofysiitti, atetsolitsumabihoito pitää lopettaa pysyvästi.
Tyypin 1 diabetekseen pitää aloittaa insuliinihoito. Jos potilaalla on asteen ≥ 3 hyperglykemia (paastotilan glukoosipitoisuus > 250 mg/dl eli 13,9 mmol/l), atetsolitsumabihoito pitää keskeyttää. Atetsolitsumabihoitoa voidaan jatkaa, jos diabetes on saatu insuliinikorvaushoidolla hyvään hoitotasapainoon.
Immuunivälitteinen meningoenkefaliitti
Atetsolitsumabilla tehdyissä kliinisissä tutkimuksissa on havaittu meningoenkefaliittia (ks. kohta Haittavaikutukset). Potilaita pitää seurata meningiitin tai enkefaliitin kliinisten oireiden ja löydösten havaitsemiseksi.
Atetsolitsumabihoito on minkä tahansa asteen meningiitin tai enkefaliitin yhteydessä lopetettava pysyvästi. Potilaalle pitää aloittaa laskimoon annettava kortikosteroidihoito (1−2 mg/kg/vrk metyyliprednisolonia tai vastaava hoito). Kun oireet ovat hävinneet, hoitoa jatketaan prednisoniannoksilla 1−2 mg/kg/vrk tai vastaavalla hoidolla.
Immuunivälitteiset neuropatiat
Atetsolitsumabihoitoa saavilla potilailla on havaittu myasteenista oireyhtymää/myasthenia gravista tai Guillain–Barrén oireyhtymää, joka voi olla hengenvaarallinen, sekä kasvohalvauksia. Potilasta pitää seurata motorisen ja sensorisen neuropatian oireiden havaitsemiseksi (ks. tässä kohdassa myös ”Immuunivälitteinen sydänlihastulehdus-myosiitti-myasthenia gravis ‑limittymisoireyhtymä”).
Kliinisissä atetsolitsumabitutkimuksissa on havaittu myeliittiä (ks. kohta Haittavaikutukset). Potilaita pitää seurata tarkoin myeliittiin viittaavien oireiden ja löydösten havaitsemiseksi.
Jos potilaalle ilmaantuu minkä tahansa asteen myasteeninen oireyhtymä/myasthenia gravis tai Guillain–Barrén oireyhtymä, atetsolitsumabihoito on lopetettava pysyvästi. Systeemisen kortikosteroidihoidon aloittamista (prednisoniannoksina 1–2 mg/kg/vrk tai vastaavaa hoitoa) pitää harkita.
Jos potilaalle ilmaantuu asteen 1 tai 2 kasvohalvaus, atetsolitsumabihoito pitää keskeyttää ja on harkittava systeemistä kortikosteroidihoitoa (1–2 mg/kg/vrk prednisonia tai vastaava hoito). Hoitoa voidaan jatkaa vain, jos tapahtuma häviää täysin. Atetsolitsumabihoito pitää lopettaa pysyvästi, jos ilmaantuu asteen 3 tai asteen 4 kasvohalvaus tai muuta neuropatiaa, joka ei häviä täysin atetsolitsumabihoidon ollessa keskeytettynä.
Jos ilmaantuu asteen 2, 3 tai 4 myeliittiä, atetsolitsumabihoito pitää lopettaa pysyvästi.
Immuunivälitteinen haimatulehdus
Atetsolitsumabilla tehdyissä kliinisissä tutkimuksissa on havaittu haimatulehduksia, mukaan lukien suurentuneita seerumin amylaasi- ja lipaasipitoisuuksia (ks. kohta Haittavaikutukset). Potilaita pitää seurata tarkoin akuuttiin haimatulehdukseen viittaavien oireiden ja löydösten havaitsemiseksi.
Jos potilaalle ilmaantuu asteen ≥ 3 suurentuneita seerumin amylaasi- tai lipaasipitoisuuksia (> 2x ULN) tai asteen 2 tai 3 haimatulehdus, atetsolitsumabihoito pitää keskeyttää ja aloittaa hoito laskimoon annettavilla kortikosteroideilla (1–2 mg/kg/vrk metyyliprednisolonia tai vastaava hoito). Kun oireet lievenevät, hoitoa pitää jatkaa prednisoniannoksilla 1–2 mg/kg/vrk tai vastaavalla hoidolla. Atetsolitsumabihoitoa voidaan jatkaa, kun seerumin amylaasi- ja lipaasipitoisuus korjautuu 12 viikon kuluessa asteeseen ≤ 1 tai haimatulehduksen oireet ovat hävinneet ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. Asteen 4 tai minkä tahansa asteen toistuvan haimatulehduksen yhteydessä atetsolitsumabihoito pitää lopettaa pysyvästi.
Immuunivälitteinen sydänlihastulehdus
Atetsolitsumabihoidossa on havaittu sydänlihastulehdusta, mukaan lukien kuolemaan johtaneita tapauksia (ks. kohta Haittavaikutukset). Potilaita pitää seurata sydänlihastulehduksen oireiden ja löydösten havaitsemiseksi (ks. tässä kohdassa myös ”Immuunivälitteinen sydänlihastulehdus-myosiitti-myasthenia gravis ‑limittymisoireyhtymä”).
Potilailta, joilla on sydänoireita tai sydän- ja keuhko-oireita, pitää tutkia mahdollinen sydänlihastulehdus, jotta varmistetaan asianmukaisten toimenpiteiden aloittaminen varhaisvaiheessa. Jos sydänlihastulehdusta epäillään, atetsolitsumabihoito pitää keskeyttää ja aloittaa systeeminen kortikosteroidihoito viipymättä prednisoniannoksella 1–2 mg/kg/vrk tai vastaava hoito sekä konsultoida viipymättä kardiologia diagnoosin selvittämiseksi voimassa olevien kliinisten ohjeistojen mukaisesti. Kun sydänlihastulehdusdiagnoosi on varmistunut, atetsolitsumabihoito on lopetettava pysyvästi asteen ≥ 2 sydänlihastulehduksen vuoksi (ks. kohta Annostus ja antotapa).
Immuunivälitteinen munuaistulehdus
Atetsolitsumabilla tehdyissä kliinisissä tutkimuksissa on havaittu munuaistulehdusta (ks. kohta Haittavaikutukset). Potilaita pitää seurata munuaisten toiminnan muutosten havaitsemiseksi.
Jos potilaalla on asteen 2 munuaistulehdus, atetsolitsumabihoito pitää keskeyttää ja aloittaa systeeminen kortikosteroidihoito annoksina 1–2 mg/kg/vrk prednisonia tai vastaava hoito. Atetsolitsumabihoitoa voi jatkaa, jos tapahtuma lievenee 12 viikon kuluessa asteeseen ≤ 1 ja kortikosteroidihoidon annos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon. Jos potilaalla on asteen 3 tai 4 munuaistulehdus, atetsolitsumabihoito pitää lopettaa pysyvästi.
Immuunivälitteinen myosiitti
Atetsolitsumabihoidossa on havaittu myosiittia, myös kuolemaan johtaneita tapauksia (ks. kohta Haittavaikutukset). Potilaita pitää seurata myosiitin oireiden ja löydösten havaitsemiseksi (ks. tässä kohdassa myös ”Immuunivälitteinen sydänlihastulehdus-myosiitti-myasthenia gravis ‑limittymisoireyhtymä”).
Jos potilaalle kehittyy myosiitin oireita ja löydöksiä, potilasta on seurattava tarkoin ja hänelle on annettava viipymättä lähete erikoislääkärin tutkimuksiin ja hoitoon. Jos potilaalla on asteen 2 tai 3 myosiitti, atetsolitsumabihoito pitää keskeyttää ja aloittaa kortikosteroidihoito (1–2 mg/kg/vrk prednisonia tai vastaava hoito). Jos oireet lievenevät asteeseen ≤ 1, pienennä kortikosteroidiannosta kliinisen tilanteen mukaan. Atetsolitsumabihoitoa voi jatkaa, jos tapahtuma lievenee 12 viikon kuluessa asteeseen ≤ 1 ja kortikosteroidihoidon annos on pienennetty ≤ 10 mg:aan suun kautta otettavaa prednisonia vuorokaudessa tai vastaavaan hoitoon. Jos potilaalla on asteen 4 tai asteen 3 toistuva myosiitti tai jos kortikosteroidiannosta ei voida pienentää ≤ 10 mg prednisonia vuorokaudessa vastaavaan hoitoon 12 viikon kuluessa tapahtuman ilmaantumisesta, atetsolitsumabihoito pitää lopettaa pysyvästi.
Immuunivälitteiset vaikeat ihon lääkereaktiot
Atetsolitsumabia saavilla potilailla on raportoitu immuunivälitteisiä vaikeita ihon lääkereaktioita (severe cutaneous adverse reactions, SCARs), mukaan lukien Stevens–Johnsonin oireyhtymää ja toksista epidermaalista nekrolyysiä. Potilaita pitää seurata epäiltyjen vaikeiden ihon lääkereaktioiden havaitsemiseksi ja muut syyt pitää sulkea pois. Jos potilaalla epäillään vaikeaa ihon lääkereaktiota, hänelle on annettava lähete erikoislääkärille diagnoosia ja jatkohoitoa varten.
Jos potilaalle ilmaantuu asteen 3 ihoreaktio, atetsolitsumabihoito pitää haittavaikutuksen vaikeusasteen perusteella keskeyttää ja aloittaa systeeminen kortikosteroidihoito annoksella 1–2 mg/kg/vrk prednisonia tai vastaava hoito. Jos tapahtuma lievenee 12 viikon kuluessa asteeseen ≤ 1 ja kortikosteroidiannos on pienennetty ≤ 10 mg:aan prednisonia vuorokaudessa tai vastaavaan hoitoon, atetsolitsumabihoitoa voidaan jatkaa. Jos ilmenee asteen 4 ihoreaktioita, atetsolitsumabihoito pitää lopettaa pysyvästi ja potilaalle pitää antaa kortikosteroideja.
Jos potilaalla epäillään Stevens–Johnsonin oireyhtymää tai toksista epidermaalista nekrolyysiä, atetsolitsumabihoito pitää lopettaa. Stevens–Johnsonin oireyhtymän tai toksisen epidermaalisen nekrolyysin varmistuessa atetsolitsumabihoito pitää lopettaa pysyvästi.
Varovaisuutta on noudatettava harkittaessa atetsolitsumabihoitoa potilaalle, jolla on aiemmin ollut vaikea-asteinen tai henkeä uhkaava ihon haittavaikutus muiden immuniteettia stimuloivien syöpälääkkeiden aiemman käytön yhteydessä.
Immuunivälitteiset sydänpussin sairaudet
Atetsolitsumabin käytössä on havaittu sydänpussin sairauksia, mukaan lukien sydänpussitulehdusta, sydänpussin effuusiota ja sydämen tamponaatiota, jotka ovat toisinaan johtaneet kuolemaan (ks. kohta Haittavaikutukset). Potilaita pitää seurata sydänpussin sairauksien kliinisten oireiden ja löydösten havaitsemiseksi.
Jos asteen 1 sydänpussitulehdusta epäillään, atetsolitsumabihoito pitää keskeyttää sekä konsultoida viipymättä kardiologia diagnoosin selvittämiseksi voimassa olevien kliinisten ohjeistojen mukaisesti. Jos asteen ≥ 2 sydänpussin sairautta epäillään, atetsolitsumabihoito pitää keskeyttää ja aloittaa systeeminen kortikosteroidihoito viipymättä prednisoniannoksella 1–2 mg/kg/vrk tai vastaava hoito sekä konsultoida viipymättä kardiologia diagnoosin selvittämiseksi voimassa olevien kliinisten ohjeistojen mukaisesti. Kun sydänpussin sairauteen liittyvän tapahtuman diagnoosi on varmistunut, atetsolitsumabihoito on lopetettava pysyvästi asteen ≥ 2 sydänpussin sairauden vuoksi (ks. kohta Annostus ja antotapa).
Hemofagosyyttinen lymfohistiosytoosi
Atetsolitsumabia saavilla potilailla on raportoitu hemofagosyyttistä lymfohistiosytoosia (HLH), mukaan lukien kuolemaan johtaneita tapauksia (ks. kohta Haittavaikutukset). Hemofagosyyttinen lymfohistiosytoosi on otettava huomioon, jos sytokiinioireyhtymän ilmenemismuoto on epätyypillinen tai se pitkittyy. Potilaita on seurattava hemofagosyyttisen lymfohistiosytoosin oireiden ja löydösten havaitsemiseksi. Jos hemofagosyyttistä lymfohistiosytoosia epäillään, atetsolitsumabihoito on lopetettava pysyvästi, ja potilaalle pitää kirjoittaa lähete erikoislääkärin vastaanotolle jatkodiagnosointia ja -hoitoa varten.
Immuunivälitteinen sydänlihastulehdus-myosiitti-myasthenia gravis ‑limittymisoireyhtymä
Atetsolitsumabin markkinoille tulon jälkeen ja sillä tehdyissä kliinisissä tutkimuksissa on havaittu sydänlihastulehdus-myosiitti-myasthenia gravis ‑limittymisoireyhtymää (joka ilmenee joko kahden tai kaikkien kolmen sairauden limittymisenä), joka on joissakin tapauksissa johtanut kuolemaan. Oireyhtymän varhainen tunnistaminen ja tehokas hoito ovat välttämättömiä oireyhtymään liittyvän sairastuvuuden ja kuolleisuusriskin hallitsemiseksi.
On tärkeää tunnistaa limittyvät oireet, jotka voivat vaikeuttaa diagnoosia ja hoitoa ja joihin voi liittyä tavanomaista suurempi kuolleisuusriski. Potilaita, joilla on yhden sairauden oireita ja löydöksiä, on seurattava myös kahden muun mainitun sairauden varalta, ja heitä on hoidettava kliinisen tilanteen mukaan. Katso kohdan Annostus ja antotapa taulukosta 2 ohjeet suositelluista annosmuutoksista kunkin sairauden (sydänlihastulehdus, myosiitti, myasthenia gravis) yhteydessä ja noudata tiukimpia suosituksia.
Muut immuunivälitteiset haittavaikutukset
Atetsolitsumabin vaikutusmekanismin vuoksi voi esiintyä myös muita immuunivälitteisiä haittavaikutuksia, esimerkiksi ei-infektiivistä virtsarakkotulehdusta, uveiittia ja autoimmuunihemolyyttistä anemiaa.
Arvioi kaikki epäillyt immuunivälitteiset haittavaikutukset muiden syiden poissulkemiseksi. Potilaita on seurattava immuunivälitteisten haittavaikutusten merkkien ja oireiden varalta ja hoidettava reaktion vaikeuden perusteella muuttamalla hoitoa ja käyttämällä kortikosteroideja kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa ja kohta Haittavaikutukset).
Infuusioon liittyvät reaktiot
Atetsolitsumabin käytön yhteydessä on havaittu infuusioon liittyneitä reaktioita, mukaan lukien anafylaksia (ks. kohta Haittavaikutukset). Jos potilaalla on asteen 1 tai 2 infuusioon liittyvä reaktio, infuusionopeutta pitää hidastaa tai hoito pitää keskeyttää. Jos potilaalla on asteen 3 tai 4 infuusioon liittyvä reaktio, atetsolitsumabihoito pitää lopettaa pysyvästi. Jos potilaalla on asteen 1 tai 2 infuusioon liittyvä reaktio, atetsolitsumabihoitoa voidaan jatkaa tarkassa seurannassa. Esilääkitystä antipyreeteillä ja antihistamiinilla voidaan harkita.
Sairauskohtaiset varotoimet
Atetsolitsumabin käyttö yhdessä bevasitsumabin, paklitakselin ja karboplatiinin kanssa metastasoituneen ei-levyepiteeliperäisen ei-pienisoluisen keuhkosyövän hoitoon
Lääkärin pitää arvioida tarkoin neljästä lääkkeestä (atetsolitsumabi, bevasitsumabi, paklitakseli ja karboplatiini) koostuvan yhdistelmähoidon riskit ennen kuin tällainen hoito aloitetaan (ks. kohta Haittavaikutukset).
Atetsolitsumabin ja nab-paklitakselin yhdistelmän käyttö metastasoituneen kolmoisnegatiivisen rintasyövän hoitoon
Atetsolitsumabi- ja nab-paklitakselihoidon aikana ilmenevä neutropenia ja perifeeriset neuropatiat saattavat korjautua keskeyttämällä nab-paklitakselihoito. Lääkärin on luettava nab-paklitakselin käyttöön liittyvät erityiset varotoimet ja vasta-aiheet sen valmisteyhteenvedosta.
Atetsolitsumabin käyttö aiemmin hoitamatonta uroteelikarsinoomaa sairastaville potilaille, joille sisplatiinihoidon ei katsota sopivan
IMvigor210-tutkimuksen kohortin 1 tutkimuspotilasjoukon sairauden ominaisuudet lähtötilanteessa ja ennuste olivat yleisesti ottaen verrannolliset klinikan niihin potilaisiin, joille sisplatiinin ei katsottu sopivan, mutta jotka soveltuivat karboplatiinipohjaiseen yhdistelmäsolunsalpaajahoitoon. Siitä potilaiden osajoukosta, jolle ei sovi mikään solunsalpaajahoito, ei ole riittävästi tietoa, joten näiden potilasryhmien atetsolitsumabihoidossa pitää olla varovainen ja sen pitää perustua mahdollisen riski-hyötytasapainon tarkkaan yksilölliseen arviointiin.
Atetsolitsumabin käyttö yhdessä bevasitsumabin, paklitakselin ja karboplatiinin kanssa
Kliinisestä IMpower150-pivotaalitutkimuksesta poissuljettiin sellaiset ei-pienisoluista keuhkosyöpää (NSCLC) sairastaneet potilaat, joilla näkyi kuvantamisessa selvä kasvaimen infiltraatio rintakehän suuriin verisuoniin tai selvä keuhkoleesioiden kavitaatio. Poissulkukriteeri asetettiin sen jälkeen, kun useissa tapauksissa oli havaittu kuolemaan johtanut keuhkoverenvuoto, joka on bevasitsumabihoidon tunnettu riskitekijä.
Koska atetsolitsumabin käytöstä näille potilasjoukoille ei ole tietoja, sen käytössä pitää olla varovainen, ja käytön pitää perustua potilaan hyöty-riskitasapainon tarkkaan arviointiin.
Atetsolitsumabin käyttö yhdessä bevasitsumabin, paklitakselin ja karboplatiinin kanssa potilaille, joilla on EGFR-mutaatio ja joiden ei-pienisoluinen keuhkosyöpä on edennyt käytettäessä erlotinibin ja bevasitsumabin yhdistelmää
IMpower150-tutkimuksesta ei ole tietoja hoidon tehosta käytettäessä atetsolitsumabia yhdessä bevasitsumabin, paklitakselin ja karboplatiinin kanssa potilaille, joilla on EGFR-mutaatio ja joiden sairaus on aiemmin edennyt käytettäessä erlotinibin ja bevasitsumabin yhdistelmää.
Atetsolitsumabin käyttö yhdessä bevasitsumabin kanssa potilaille, joilla on hepatosellulaarinen karsinooma
Tiedot hepatosellulaarista karsinoomaa sairastavista potilaista, joilla on Child-Pugh-luokan B maksasairaus ja jotka saavat atetsolitsumabia yhdessä bevasitsumabin kanssa, ovat hyvin suppeita, eikä tällä hetkellä ole tietoja saatavissa hepatosellulaarista karsinoomaa sairastavista potilaista, joilla on Child-Pugh-luokan C maksasairaus.
Bevasitsumabihoitoa saavilla potilailla on tavanomaista suurempi verenvuotojen riski, ja vaikeaa maha-suolikanavan verenvuotoa, mukaan lukien kuolemaan johtaneita tapahtumia, on raportoitu hepatosellulaarista karsinoomaa sairastavilla potilailla, jotka saivat atetsolitsumabia yhdessä bevasitsumabin kanssa. Hepatosellulaarista karsinoomaa sairastavilta potilailta on seulottava ja sen jälkeen hoidettava ruokatorven suonikohjut kliinisen käytännön mukaan ennen kuin hoito atetsolitsumabin ja bevasitsumabin yhdistelmällä aloitetaan. Jos potilaalla on yhdistelmähoidon yhteydessä graduksen 3 tai 4 verenvuotoja, bevasitsumabin käyttö pitää lopettaa pysyvästi. Ks. bevasitsumabin valmisteyhteenveto.
Atetsolitsumabin ja bevasitsumabin yhdistelmähoidossa voi esiintyä diabetes mellitusta. Lääkärin pitää seurata veren glukoosipitoisuutta ennen atetsolitsumabin käyttöä yhdistelmänä bevasitsumabin kanssa sekä sen aikana kliinisen tarpeen mukaan.
Atetsolitsumabi monoterapiana metastasoituneen ei-pienisoluisen keuhkosyövän ensilinjan hoitoon
Ennen monoterapian aloittamista ei-pienisoluisen keuhkosyövän hoitoon lääkärin on huomioitava, että atetsolitsumabin vaikutus alkaa viiveellä. Atetsolitsumabihoidossa havaittiin 2,5 kuukauden kuluessa satunnaistamisesta enemmän kuolemia kuin solunsalpaajahoidossa. Varhaisvaiheen kuolemiin ei pystytty tunnistamaan mitään spesifistä tekijää (ks. kohta Farmakodynamiikka).
Kliinisistä tutkimuksista pois suljetut potilaat
Kliinisistä tutkimuksista suljettiin pois potilaat, joilla oli seuraavia sairauksia: aiemmin sairastettu autoimmuunisairaus, aiemmin sairastettu pneumoniitti, aktiivisia etäpesäkkeitä aivoissa, ECOG-suorituskyky ≥ 2 (paitsi potilailla, joilla oli platinapohjaiseen solunsalpaajahoitoon soveltumaton pitkälle edennyt ei-pienisoluinen keuhkosyöpä), HIV, hepatiitti B- tai hepatiitti C infektio (potilailla ei hepatosellulaarista karsinoomaa), merkittävä sydän- ja verisuonitauti tai jos potilaan hematologinen ja pääte-elimen toiminta olivat riittämättömät. Tutkimuksista suljettiin pois potilaat, jotka olivat saaneet 28 päivän kuluessa ennen tutkimukseen tuloa rokotuksen elävää, heikennettyä taudinaiheuttajaa sisältävällä rokotteella, 4 viikon kuluessa systeemisiä immuniteettia stimuloivia aineita tai 2 viikon kuluessa systeemisiä immunosuppressiivisia lääkevalmisteita; suun kautta otettavia tai laskimoon annettavia antibiootteja tutkimushoidon aloittamista edeltäneiden 2 viikon kuluessa saaneet potilaat suljettiin pois kliinisistä tutkimuksista.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää polysorbaatti 20:tä. Yksi injektiopullo Tecentriq 840 mg infuusiokonsentraattia liuosta varten, sisältää 5,6 mg polysorbaatti 20:tä, joka vastaa 0,4 mg/ml. Yksi injektiopullo Tecentriq 1 200 mg infuusiokonsentraattia liuosta varten, sisältää 8 mg polysorbaatti 20:tä, joka vastaa 0,4 mg/ml. Polysorbaatti 20 saattaa aiheuttaa allergisia reaktioita.
Potilaskortti
Lääkettä määräävän lääkärin on kerrottava potilaalle Tecentriq-hoidon riskeistä. Potilaalle annetaan potilaskortti, ja potilasta kehotetaan pitämään sitä aina mukana.
Yhteisvaikutukset
Atetsolitsumabilla ei ole tehty varsinaisia farmakokineettisiä yhteisvaikutustutkimuksia. Atetsolitsumabi poistuu verenkierrosta kataboloitumalla, joten metabolisia lääkkeiden yhteisvaikutuksia ei oletettavasti esiinny.
Systeemisten kortikosteroidien tai immunosuppressiivisten lääkkeiden käyttöä pitää välttää ennen atetsolitsumabihoidon aloittamista, koska ne saattavat häiritä atetsolitsumabin farmakodynaamista aktiivisuutta ja tehoa. Systeemisiä kortikosteroideja tai muita immunosuppressiivisia lääkkeitä voidaan kuitenkin käyttää atetsolitsumabihoidon aloittamisen jälkeen immuunivälitteisten haittavaikutusten hoitoon (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä atetsolitsumabihoidon aikana ja viisi kuukautta hoidon lopettamisen jälkeen.
Raskaus
Ei ole olemassa tietoja atetsolitsumabin käytöstä raskaana oleville naisille. Atetsolitsumabilla ei ole tehty kehitystä ja lisääntymistä koskevia tutkimuksia. Eläinkokeet ovat osoittaneet, että PDL1/PD-1-reitin estyminen hiiren tiineysmallissa voi aiheuttaa kehittyvän sikiön immuunivälitteisen hylkimisreaktion, mikä johtaa sikiön kuolemaan (ks. kohta Prekliiniset tiedot turvallisuudesta). Nämä tulokset osoittavat vaikutusmekanismiin perustuvan mahdollisen riskin, että atetsolitsumabin raskaudenaikaisesta käytöstä voi aiheutua sikiölle haittaa, mukaan lukien keskenmenojen ja kuolleena syntyneisyyden lisääntymistä.
Ihmisen G1-immunoglobuliinien (IgG1) tiedetään läpäisevän istukkaesteen, ja koska atetsolitsumabi on IgG1, atetsolitsumabi voi näin ollen kulkeutua äidistä kehittyvään sikiöön.
Atetsolitsumabia ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa atetsolitsumabilla.
Imetys
Ei tiedetä, erittyykö atetsolitsumabi ihmisen rintamaitoon. Atetsolitsumabi on monoklonaalinen vasta-aine, ja sitä oletetaan olevan ensimaidossa sekä maidossa myöhemminkin pieninä pitoisuuksina. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko Tecentriq-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Atetsolitsumabin mahdollisista vaikutuksista hedelmällisyyteen ei ole kliinisiä tietoja. Atetsolitsumabilla ei ole tehty lisääntymis- ja kehitystoksisuustutkimuksia, mutta 26 viikkoa kestäneessä toistuvaisannosten toksisuutta koskeneessa tutkimuksessa atetsolitsumabi vaikutti kuukautiskiertoon altistuksella (AUC), jonka arvioitiin olevan noin kuusinkertainen verrattuna altistukseen (AUC) suositusannoksia saaneilla potilailla. Tällainen vaikutus oli korjautuva (ks. kohta Prekliiniset tiedot turvallisuudesta). Vaikutuksia miesten lisääntymiselimiin ei todettu.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tecentriq-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Jos potilaalla on uupumusta, hän ei saa ajaa autoa eikä käyttää koneita ennen kuin oireet häviävät (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Atetsolitsumabimonoterapian turvallisuus perustuu yhdistettyihin tietoihin 5 039 potilaasta, joilla oli monia eri kasvaintyyppejä. Yleisimmät haittavaikutukset (> 10 %) olivat uupumus (29,3 %), heikentynyt ruokahalu (20,1 %), ihottuma (19,7 %), pahoinvointi (18,8 %), yskä (18,2 %), ripuli (18,1 %), kuume (17,9 %), hengenahdistus (16,6 %), nivelkipu (16,2 %), kutina (13,3 %), voimattomuus (13 %), selkäkipu (12,2 %), oksentelu (11,7 %), virtsatieinfektio (11 %) ja päänsärky (10,2 %).
Atetsolitsumabin ja muiden lääkkeiden yhdistelmähoidon turvallisuutta on arvioitu 4535 potilaalla useissa eri kasvaintyypeissä. Yleisimmät haittavaikutukset (≥ 20 %) olivat anemia (36,8 %), neutropenia (36,6 %), pahoinvointi (35,5 %), uupumus (33,1 %), alopesia (28,1 %), ihottuma (27,8 %), ripuli (27,6 %), trombosytopenia (27,1 %), ummetus (25,8 %), heikentynyt ruokahalu (24,7 %) ja perifeerinen neuropatia (24,4 %).
Atetsolitsumabin käyttö ei-pienisoluisen keuhkosyövän adjuvanttihoidossa
Atetsolitsumabin turvallisuusprofiili ei-pienisoluista keuhkosyöpää sairastavan potilasjoukon adjuvanttihoidossa (IMpower010) oli yleisesti yhdenmukainen monoterapian yhdistetyn kokonaisturvallisuusprofiilin kanssa edennyttä sairautta hoidettaessa. Atetsolitsumabin immuunivälitteisten haittavaikutusten ilmaantuvuus IMpower010-tutkimuksessa oli 51,7 % verrattuna 38,4 %:iin edennyttä sairautta sairastavassa monoterapiaa saaneessa yhdistetyssä potilasjoukossa. Adjuvanttihoidossa ei tunnistettu uusia immuunivälitteisiä haittavaikutuksia.
Atetsolitsumabin käyttö yhdessä bevasitsumabin, paklitakselin ja karboplatiinin kanssa
Ei-pienisoluisen keuhkosyövän ensilinjan hoitoa koskevassa tutkimuksessa (IMpower150) haittavaikutuksia havaittiin kaikkiaan yleisemmin neljästä lääkkeestä eli atetsolitsumabista, bevasitsumabista, paklitakselista ja karboplatiinista koostuvassa hoidossa verrattuna atetsolitsumabin, paklitakselin ja karboplatiinin yhdistelmän käyttöön, mukaan lukien asteen 3 ja 4 tapahtumia (63,6 % versus 57,5 %), asteen 5 tapahtumia (6,1 % versus 2,5 %), atetsolitsumabin osalta erityisesti seurattavia haittavaikutuksia (52,4 % versus 48,0 %) sekä minkä tahansa tutkimushoidon lopettamiseen johtaneita haittavaikutuksia (33,8 % versus 13,3 %). Pahoinvointia, ripulia, suutulehdusta, uupumusta, kuumetta, limakalvotulehdusta, ruokahalun heikkenemistä, painon laskua, hypertensiota ja proteinuriaa raportoitiin yleisemmin (≥ 5 %:n ero) potilailla, jotka saivat atetsolitsumabia yhdessä bevasitsumabin, paklitakselin ja karboplatiinin kanssa. Muita kliinisesti merkityksellisiä haittavaikutuksia, joita havaittiin yleisemmin atetsolitsumabia, bevasitsumabia, paklitakselia ja karboplatiinia saaneessa haarassa, olivat nenäverenvuoto, veriyskä, aivoverisuonitapahtuma, mukaan lukien kuolemaan johtaneet tapahtumat.
Kohdassa Varoitukset ja käyttöön liittyvät varotoimet on lisätietoa vakavista haittavaikutuksista.
Haittavaikutustaulukko
Haittavaikutukset luetellaan MedDRA-elinjärjestelmän ja esiintymistiheysluokan mukaisesti taulukossa 3 atetsolitsumabimonoterapian osalta ja yhdistelmähoidon osalta. Haittavaikutuksia, joita tiedetään esiintyvän käytettäessä pelkästään atetsolitsumabia tai solunsalpaajahoitoja, voi esiintyä käytettäessä näitä lääkevalmisteita yhdistelmänä, vaikka haittavaikutuksia ei olisikaan raportoitu yhdistelmähoitoja koskeneissa kliinisissä tutkimuksissa. Esiintymistiheydet on luokiteltu seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3. Yhteenveto haittavaikutuksista atetsolitsumabia saaneilla potilailla
| Atetsolitsumabimonoterapia | Atetsolitsumabiyhdistelmähoito | |
| Infektiot | ||
| Hyvin yleinen | virtsatieinfektioa | keuhkoinfektiob |
| Yleinen | sepsisaj | |
| Harvinainen | sytomegalovirusinfektio | sytomegalovirusinfektio |
| Veri ja imukudos | ||
| Hyvin yleinen | anemia, trombosytopeniad, neutropeniae, leukopeniaf | |
| Yleinen | trombosytopeniad, neutropeniae | lymfopeniag |
| Harvinainen | hemofagosyyttinen lymfohistiosytoosi, autoimmuunihemolyyttinen anemiaav | hemofagosyyttinen lymfohistiosytoosi, autoimmuunihemolyyttinen anemiaav |
| Immuunijärjestelmä | ||
| Yleinen | infuusioon liittyvä reaktioh | infuusioon liittyvä reaktioh |
| Harvinainen | sarkoidoosiar | |
| Umpieritys | ||
| Hyvin yleinen | hypotyreoosii | |
| Yleinen | hypotyreoosii, hypertyreoosij | hypertyreoosij |
| Melko harvinainen | diabetes mellitusk, lisämunuaisten vajaatoimintal, hypofysiittim | hypofysiittim |
| Aineenvaihdunta ja ravitsemus | ||
| Hyvin yleinen | heikentynyt ruokahalu | heikentynyt ruokahalu |
| Yleinen | hypokalemiaae, hyponatremiaaf, hyperglykemia, hypoalbuminemia, hypofosfatemia, hypokalsemia | hypokalemiaae, hyponatremiaaf, hypomagnesemian, hypoalbuminemia, hypofosfatemia, hypokalsemia |
| Hermosto | ||
| Hyvin yleinen | päänsärky | perifeerinen neuropatiao, päänsärky |
| Yleinen | perifeerinen neuropatiao | pyörtyminen, heitehuimaus |
| Melko harvinainen | Guillain–Barrén oireyhtymäp, meningoenkefaliittiq | |
| Harvinainen | myasteeninen oireyhtymär, kasvohalvaus, myeliitti | kasvohalvaus |
| Silmät | ||
| Melko harvinainen | uveiittias | |
| Harvinainen | uveiittias | |
| Sydän | ||
| Yleinen | sydänpussin sairaudetao | |
| Melko harvinainen | sydänpussin sairaudetao | |
| Harvinainen | sydänlihastulehduss | |
| Verisuonisto | ||
| Hyvin yleinen | hypertensioai | |
| Yleinen | hypotensio | |
| Hengityselimet, rintakehä ja välikarsina | ||
| Hyvin yleinen | hengenahdistus, yskä | hengenahdistus, yskä, nasofaryngiittiam |
| Yleinen | pneumoniittit, hypoksiaag, nasofaryngiittiam | dysfonia |
| Ruoansulatuselimistö | ||
| Hyvin yleinen | pahoinvointi, oksentelu, ripuliu | pahoinvointi, oksentelu, ripuliu, ummetus |
| Yleinen | koliittiv, vatsakipu, nielemishäiriöt, suunielun kipuw, suun kuivuminen | suutulehdus, makuaistin häiriö, koliittiv |
| Melko harvinainen | haimatulehdusx | |
| Harvinainen | keliakia | keliakia |
| Maksa ja sappi | ||
| Yleinen | suurentunut ASAT-pitoisuus, suurentunut ALAT-pitoisuus, hepatiittiy | suurentunut ASAT-pitoisuus, suurentunut ALAT-pitoisuus |
| Iho ja ihonalainen kudos | ||
| Hyvin yleinen | ihottumaz, kutina | ihottumaz, kutina, alopesiaah |
| Yleinen | kuiva ihoap | |
| Melko harvinainen | vaikeat ihon lääkereaktiotak, psoriaasian, jäkälätauditaq | vaikeat ihon lääkereaktiotak, psoriaasian |
| Harvinainen | pemfigoidi | pemfigoidi, jäkälätauditaq |
| Luusto, lihakset ja sidekudos | ||
| Hyvin yleinen | nivelkipu, selkäkipu | nivelkipu, luusto- ja lihaskipuaa, selkäkipu |
| Yleinen | luusto- ja lihaskipuaa, niveltulehdusat | niveltulehdusat |
| Melko harvinainen | myosiittiab, jännetuppitulehdusau | jännetuppitulehdusau |
| Munuaiset ja virtsatiet | ||
| Yleinen | suurentunut veren kreatiniinipitoisuusc | proteinuriaac, suurentunut veren kreatiniinipitoisuusc |
| Melko harvinainen | munuaistulehdusad | |
| Tuntematon | ei-infektiivinen virtsarakkotulehdusal | |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Hyvin yleinen | kuume, uupumus, voimattomuus | kuume, uupumus, voimattomuus, raajojen turvotus |
| Yleinen | influenssan kaltainen sairaus, vilunväreet | |
| Tutkimukset | ||
| Yleinen | suurentunut gammaglutamyylitransferaasipitoisuus | suurentunut veren alkalisen fosfataasin pitoisuus, suurentunut gammaglutamyylitransferaasipitoisuus |
| Melko harvinainen | suurentunut veren kreatiinikinaasipitoisuus | |
a Sisältää virtsatieinfektiota, kystiittiä, pyelonefriittiä, Escherichia-bakteerin aiheuttamaa virtsatieinfektiota, bakteeriperäistä virtsatieinfektiota, munuaisinfektiota, akuuttia pyelonefriittiä, kroonista pyelonefriittiä, pyeliittiä, munuaisabsessia, streptokokin aiheuttamaa virtsatieinfektiota, virtsaputkitulehdusta, sienen aiheuttamaa virtsatieinfektiota, Pseudomonas-bakteerin aiheuttamaa virtsatieinfektiota koskevat raportit.
b Sisältää keuhkokuumetta, keuhkoputkitulehdusta, alahengitystieinfektiota, infektiivistä pleuraeffuusiota, trakeobronkiittia, epätyypillistä keuhkokuumetta, keuhkopaisetta, keuhkoahtaumataudin infektiivistä pahenemista, syöpään liittyvää keuhkokuumetta, pyopneumothoraxia, keuhkopussin infektiota, toimenpiteen jälkeistä keuhkokuumetta koskevat raportit.
c Sisältää suurentunutta veren kreatiniinipitoisuutta, kohonnutta veren kreatiniinipitoisuutta koskevat raportit.
d Sisältää immunologista trombosytopeniaa (raportoitu yhdistetyn tietoaineiston ulkopuolisissa tutkimuksissa), trombosytopeniaa, verihiutalemäärän pienenemistä koskevat raportit.
e Sisältää neutropeniaa, pienentynyttä neutrofiilimäärää, kuumeista neutropeniaa, neutropeenistä sepsistä, granulosytopeniaa koskevat raportit.
f Sisältää vähentynyttä veren valkosolumäärää ja leukopeniaa koskevat raportit.
g Sisältää lymfopeniaa, vähentynyttä lymfosyyttimäärää koskevat raportit.
h Sisältää infuusioon liittyviä reaktioita, sytokiinioireyhtymää, yliherkkyyttä ja anafylaksiaa koskevat raportit.
i Sisältää kilpirauhasvasta-ainepositiivisuutta, autoimmuunihypotyreoosia, autoimmuunia kilpirauhastulehdusta, pienentynyttä veren tyreotropiinipitoisuutta, suurentunutta veren tyreotropiinipitoisuutta, matalan T3-arvon oireyhtymää, struumaa, hypotyreoosia, immuniteettivälitteistä hypotyreoosia, immuunivälitteistä kilpirauhastulehdusta, myksedeemaa, primaaria hypotyreoosia, kilpirauhassairautta, pienentynyttä kilpirauhashormonipitoisuutta, poikkeavuuksia kilpirauhasen toimintakokeissa, kilpirauhastulehdusta, akuuttia kilpirauhastulehdusta, pienentynyttä tyroksiinipitoisuutta, pienentynyttä vapaan tyroksiinin pitoisuutta, suurentunutta vapaan tyroksiinin pitoisuutta, suurentunutta tyroksiinipitoisuutta, pienentynyttä trijodityroniinipitoisuutta, suurentunutta trijodityroniinipitoisuutta, poikkeavaa vapaan trijodityroniinin pitoisuutta, pienentynyttä vapaan trijodityroniinin pitoisuutta, suurentunutta vapaan trijodityroniinin pitoisuutta, oireetonta kilpirauhastulehdusta koskevat raportit.
j Sisältää hypertyreoosia, Basedowin tautia, endokriinistä silmäoireyhtymää, eksoftalmusta koskevat raportit.
k Sisältää diabetes mellitusta, tyypin 1 diabetes mellitusta, diabeettista ketoasidoosia, ketoasidoosia koskevat raportit.
l Sisältää lisämunuaisten vajaatoimintaa, pienentynyttä veren kortikotropiinipitoisuutta, glukokortikoidin puutosta, primaaria lisämunuaisten vajaatoimintaa, sekundaarista lisämunuaiskuoren vajaatoimintaa koskevat raportit.
m Sisältää hypofysiittiä, hypopituitarismia, sekundaarista lisämunuaiskuoren vajaatoimintaa lämmönsäätelyhäiriötä koskevat raportit.
n Sisältää hypomagnesemiaa, pienentynyttä veren magnesiumpitoisuutta koskevat raportit.
o Sisältää perifeeristä neuropatiaa, autoimmuunineuropatiaa, perifeeristä sensorista neuropatiaa, polyneuropatiaa, vyöruusua, perifeeristä motorista neuropatiaa, neuralgista amyotrofiaa, perifeeristä sensomotorista neuropatiaa, toksista neuropatiaa, aksonaalista neuropatiaa, lanne-ristipunoksen neuropatiaa, neuropaattista nivelsairautta, ääreishermon infektiota, hermotulehdusta, immuunivälitteistä neuropatiaa koskevat raportit.
p Sisältää Guillain–Barrén oireyhtymää, nousevaa velttohalvausta ja demyelinoivaa polyneuropatiaa koskevat raportit.
q Sisältää enkefaliittia, autoimmuunienkefaliittia, meningiittiä, aseptista meningiittiä, valonarkuutta koskevat raportit.
r Sisältää myasthenia gravista koskevat raportit.
s Sisältää sydänlihastulehdusta, autoimmuuniperäistä sydänlihastulehdusta ja immuunivälitteistä sydänlihastulehdusta koskevat raportit.
t Sisältää pneumoniittia, keuhkoinfiltraatiota, bronkioliittia, immuunivälitteistä keuhkosairautta, immuunivälitteistä pneumoniittia, interstitiaalista keuhkosairautta, alveoliittia, keuhkon samentumaa, keuhkofibroosia, keuhkotoksisuutta, sädepneumoniittia koskevat raportit.
u Sisältää ripulia, ulosteen pakkokarkailua, suurta ulostustiheyttä, maha-suolikanavan hypermotiliteettia koskevat raportit.
v Sisältää koliittia, autoimmuunikoliittia, iskeemistä koliittia, mikroskooppista koliittia, haavaista koliittia, diversiokoliittia, eosinofiilistä koliittia, immuunivälitteistä enterokoliittia koskevat raportit.
w Sisältää suunielun kipua, suunielun epämukavia tuntemuksia, kurkun ärsytystä koskevat raportit.
x Sisältää autoimmuunia haimatulehdusta, haimatulehdusta, akuuttia haimatulehdusta, suurentunutta lipaasipitoisuutta, suurentunutta amylaasipitoisuutta koskevat raportit.
y Sisältää askitesta, autoimmuunihepatiittia, maksasolujen hajoamista, hepatiittia, akuuttia hepatiittia, toksista hepatiittia, maksatoksisuutta, immuunivälitteistä hepatiittia, maksan toimintahäiriöitä, lääkkeen aiheuttamaa maksavauriota, maksan vajaatoimintaa, rasvamaksaa, maksaleesioita, maksavauriota, ruokatorven suonikohjujen verenvuotoa, ruokatorven suonikohjuja, spontaania bakteeriperitoniittia koskevat raportit.
z Sisältää aknea, rakkuloita, dermatiittia, aknetyyppistä dermatiittia, allergista dermatiittia, lääkeaineihottumaa, ekseemaa, infektioekseemaa, eryteemaa, silmäluomien punoitusta, silmäluomien ihottumaa, toistopunoittumaa, follikuliittia, furunkkelia, käsien dermatiittia, immuunivälitteistä dermatiittia, huulten rakkuloita, suun verirakkuloita, käsi-jalkaoireyhtymää, pemfigoidia, ihottumaa, erytematoottista ihottumaa, makulaarista ihottumaa, makulopapulaarista ihottumaa, tuhkarokkomaista ihottumaa, papulaarista ihottumaa, papuloskvamoottista ihottumaa, kutisevaa ihottumaa, pustulaarista ihottumaa, vesikulaarista ihottumaa, kivespussien dermatiittia, seborrooista dermatiittia, ihon hilseilyä, ihotoksisuutta, ihon haavaumia, suoniyhteyteen liittyvää ihottumaa koskevat raportit.
aa Sisältää luusto- ja lihaskipua, lihassärkyä, luukipua koskevat raportit.
ab Sisältää myosiittia, rabdomyolyysiä, polymyalgia rheumaticaa, dermatomyosiittia, lihaksen märkäpesäkettä, myoglobinuriaa, myopatiaa, polymyosiittia koskevat raportit.
ac Sisältää proteinuriaa, proteiinin esiintymistä virtsassa, hemoglobinuriaa, virtsan poikkeavuuksia, nefroottista oireyhtymää ja albuminuriaa koskevat raportit.
ad Sisältää nefriittiä, autoimmuuniperäistä nefriittiä, Henoch-Schönleinin purppuranefriittiä, paraneoplastista glomerulonefriittiä, tubulointerstitiaalista nefriittiä koskevat raportit.
ae Sisältää hypokalemiaa, pienentynyttä veren kaliumpitoisuutta koskevat raportit.
af Sisältää hyponatremiaa, pienentynyttä veren natriumpitoisuutta koskevat raportit.
ag Sisältää hypoksiaa, pienentynyttä happisaturaatiota, pienentynyttä pO2-arvoa koskevat raportit.
ah Sisältää alopesiaa, madaroosia, pälvikaljuutta, täyskaljuutta, hypotrikoosia koskevat raportit.
ai Sisältää hypertensiota, verenpaineen nousua, hypertensiivistä kriisiä, systolisen verenpaineen nousua, diastolista hypertensiota, riittämättömässä hoitotasapainossa olevaa verenpainetta, hypertensiivistä retinopatiaa, hypertensiivistä nefropatiaa, essentiaalista hypertensiota, ortostaattista hypertensiota koskevat raportit.
aj Sisältää sepsistä, septistä sokkia, urosepsistä, neutropeenistä sepsistä, keuhkosepsistä, bakteerisepsistä, klebsiellasepsistä, vatsan alueen sepsistä, kandidasepsistä, eschericiasepsistä, pseudomonassepsistä, stafylokokkisepsistä koskevat raportit.
ak Sisältää rakkulaista dermatiittia, kesivää ihottumaa, erythema multiformea, eksfoliatiivista dermatiittia, yleistynyttä eksfoliatiivista dermatiittia, toksista ihottumaa, Stevens–Johnsonin oireyhtymää, lääkkeeseen liittyvää yleisoireista eosinofiilista reaktiota (DRESS), toksista epidermaalista nekrolyysiä, ihon vaskuliittia koskevat raportit.
al Sisältää ei-infektiivistä virtsarakkotulehdusta ja immuunivälitteistä virtsarakkotulehdusta koskevat raportit.
am Sisältää nasofaryngiittia, nenän tukkoisuutta ja rinorreaa koskevat raportit.
an Sisältää psoriaasia, psoriaasin kaltaista dermatiittia koskevat raportit.
ao Sisältää sydänpussitulehdusta, sydänpussin effuusiota, sydämen tamponaatiota ja kurovaa sydänpussitulehdusta koskevat raportit.
ap Sisältää kuivaa ihoa ja kseroosia koskevat raportit.
aq Sisältää likenoidia keratoosia, valkojäkälää ja punajäkälää koskevat raportit.
ar Sisältää sarkoidoosia, keuhkosarkoidoosia ja imusolmukkeen sarkoidoosia koskevat raportit.
as Sisältää uveiittia, värikalvon ja sädekehän tulehdusta ja värikalvotulehdusta koskevat raportit.
at Sisältää niveltulehdusta, nivelten turvotusta, nivelrikkoa, nivelreumaa, moniniveltulehdusta, selkärangan nivelrikkoa, autoimmuunia artriittia, immuunivälitteistä niveltulehdusta, spondyliittia, nivelen nestekertymää, nivelsairautta, oligoartriittia ja reumasairautta koskevat raportit.
au Sisältää jännetulehdusta, jännekipua, jännetuppitulehdusta ja synoviittia koskevat raportit.
av Sisältää autoimmuunihemolyyttistä anemiaa ja hemolyyttistä anemiaa koskevat raportit.
Valikoitujen haittavaikutusten kuvaus
Seuraavat tiedot kuvaavat atetsolitsumabimonoterapian merkitseviä haittavaikutuksia kliinisissä tutkimuksissa (ks. kohta Farmakodynamiikka). Atetsolitsumabiyhdistelmähoidon merkitsevien haittavaikutusten yksityiskohdat esitetään, jos havaittiin kliinisesti oleellisia eroja atetsolitsumabimonoterapiaan verrattuna. Näiden haittavaikutusten hoito-ohjeet ovat kohdissa Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.
Immuunivälitteinen pneumoniitti
Pneumoniittia esiintyi 3,0 %:lla (151/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Näistä potilaista kolmella oli kuolemaan johtanut tapahtuma. Ajan mediaani pneumoniitin ilmaantumiseen oli 3,7 kuukautta (vaihteluväli 3 päivästä 29,8 kuukauteen). Pneumoniitin keston mediaani oli 1,7 kuukautta (vaihteluväli 0 päivästä 27,8+ kuukauteen, jossa + tarkoittaa sensuroitua arvoa). Pneumoniitti johti atetsolitsumabihoidon lopettamiseen 41 (0,8 %) potilaalla. Kortikosteroidihoitoa vaatinut pneumoniitti todettiin 1,8 %:lla (92/5 039) atetsolitsumabimonoterapiaa saaneista potilaista.
Immuunivälitteinen hepatiitti
Hepatiittia esiintyi 1,7 %:lla (88/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Kolmella potilaalla 88:sta hepatiitti johti kuolemaan. Ajan mediaani hepatiitin ilmaantumiseen oli 1,4 kuukautta (vaihteluväli 0 päivästä 26,3 kuukauteen). Hepatiitin keston mediaani oli 1 kuukausi (vaihteluväli 0 päivästä 52,1+ kuukauteen; + tarkoittaa sensuroitua arvoa). Hepatiitti johti atetsolitsumabihoidon lopettamiseen 46 (0,9 %) potilaalla. Kortikosteroidihoitoa vaatinut hepatiitti todettiin 2,6 %:lla (130/5 039) atetsolitsumabimonoterapiaa saaneista potilaista.
Immuunivälitteinen koliitti
Koliittia esiintyi 1,2 %:lla (62/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Ajan mediaani koliitin ilmaantumiseen oli 4,5 kuukautta (vaihteluväli 15 päivästä 36,4 kuukauteen). Koliitin keston mediaani oli 1,4 kuukautta (vaihteluväli 3 päivästä 50,2+ kuukauteen, jossa + tarkoittaa sensuroitua arvoa). Koliitti johti atetsolitsumabihoidon lopettamiseen 24 (0,5 %) potilaalla. Kortikosteroidihoitoa vaatinut koliitti todettiin 0,6 %:lla (30/5 039) atetsolitsumabimonoterapiaa saaneista potilaista.
Immuunivälitteiset umpierityssairaudet
Kilpirauhashäiriöt
Hypotyreoosia esiintyi 8,5 %:lla (427/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Ajan mediaani hypotyreoosin ilmaantumiseen oli 4,2 kuukautta (vaihteluväli 0 päivästä 38,5 kuukauteen). Hypotyreoosia esiintyi 17,4 %:lla (86/495) potilaista, jotka saivat atetsolitsumabimonoterapiaa adjuvanttihoitona ei-pienisoluiseen keuhkosyöpään. Ajan mediaani hypotyreoosin ilmaantumiseen oli 4,0 kuukautta (vaihteluväli 22 päivästä 11,8 kuukauteen).
Hypertyreoosia esiintyi 2,4 %:lla (121/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Ajan mediaani hypertyreoosin ilmaantumiseen oli 2,7 kuukautta (vaihteluväli 0 päivästä 24,3 kuukauteen). Hypertyreoosia esiintyi 6,5 %:lla (32/495) potilaista, jotka saivat atetsolitsumabimonoterapiaa adjuvanttihoitona ei-pienisoluiseen keuhkosyöpään. Ajan mediaani hypertyreoosin ilmaantumiseen oli 2,8 kuukautta (vaihteluväli 1 päivästä 9,9 kuukauteen).
Lisämunuaisten vajaatoiminta
Lisämunuaisten vajaatoimintaa esiintyi 0,5 %:lla (25/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Ajan mediaani lisämunuaisten vajaatoiminnan ilmaantumiseen oli 6,2 kuukautta (vaihteluväli 3 päivästä 21,4 kuukauteen). Lisämunuaisten vajaatoiminta johti atetsolitsumabihoidon lopettamiseen viidellä potilaalla (0,1 %). Kortikosteroidihoitoa vaatinut lisämunuaisten vajaatoiminta todettiin 0,4 %:lla (20/5 039) atetsolitsumabimonoterapiaa saaneista potilaista.
Hypofysiitti
Hypofysiittiä esiintyi 0,2 %:lla (9/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Ajan mediaani hypofysiitin ilmaantumiseen oli 5,3 kuukautta (vaihteluväli 21 päivästä 13,7 kuukauteen). Kuusi (0,1 %) potilasta tarvitsi kortikosteroideja, ja atetsolitsumabihoito lopetettiin yhdellä (< 0,1 %) potilaalla.
Hypofysiittiä esiintyi 1,4 %:lla (15/1 093) atetsolitsumabia yhdistelmänä paklitakselin kanssa ja sen jälkeen atetsolitsumabia, tiheästi annettuja doksorubisiini- tai epirubisiiniannoksia ja syklofosfamidia saaneista potilaista. Ajan mediaani hypofysiitin ilmaantumiseen oli 3,8 kuukautta (vaihteluväli 2,4–10,7 kuukautta). Yksitoista (1,0 %) potilasta tarvitsi kortikosteroideja. Atetsolitsumabihoito lopetettiin seitsemällä (0,6 %) potilaalla.
Hypofysiittiä esiintyi 0,8 %:lla (3/393) potilaista, jotka saivat atetsolitsumabia yhdessä bevasitsumabin, paklitakselin ja karboplatiinin kanssa. Ajan mediaani hypofysiitin ilmaantumiseen oli 7,7 kuukautta (vaihteluväli 5,0–8,8 kuukautta). Kaksi potilasta tarvitsi kortikosteroideja.
Hypofysiittiä esiintyi 0,4 %:lla (2/473) potilaista, jotka saivat atetsolitsumabia yhdessä nab-paklitakselin ja karboplatiinin kanssa. Ajan mediaani hypofysiitin ilmaantumiseen oli 5,2 kuukautta (vaihteluväli: 5,1–5,3 kuukautta). Kumpikin potilas tarvitsi kortikosteroideja.
Diabetes mellitus
Diabetes mellitus kehittyi 0,6 %:lle (30/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Ajan mediaani diabeteksen kehittymiseen oli 5,5 kuukautta (vaihteluväli 3 päivästä 29,0 kuukauteen). Diabetes mellitus johti atetsolitsumabihoidon lopettamiseen < 0,1 %:lla (3/5 039) potilaista. Neljä (< 0,1 %) potilasta tarvitsi kortikosteroidihoitoa.
Diabetes mellitus kehittyi 2,0 %:lle (10/493) hepatosellulaarista karsinoomaa sairastavista potilaista, jotka saivat atetsolitsumabia yhdessä bevasitsumabin kanssa. Ajan mediaani diabetes mellituksen kehittymiseen oli 4,4 kuukautta (vaihteluväli: 1,2 kuukautta – 8,3 kuukautta). Yksikään diabetes mellitus tapahtuma ei johtanut atetsolitsumabihoidon lopettamiseen.
Immuunivälitteinen meningoenkefaliitti
Meningoenkefaliitti ilmaantui 0,4 %:lle (22/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Ajan mediaani meningoenkefaliitin ilmaantumiseen oli 15 päivää (vaihteluväli 0 päivästä 12,5 kuukauteen). Meningoenkefaliitin keston mediaani oli 24 päivää (vaihteluväli 6 päivästä 14,5+ kuukauteen; + tarkoittaa sensuroitua arvoa).
Kortikosteroidihoitoa vaatinut meningoenkefaliitti todettiin 0,2 %:lla (12/5 039) atetsolitsumabia saaneista potilaista, ja kahdeksan potilasta (0,2 %) lopetti atetsolitsumabihoidon.
Immuunivälitteiset neuropatiat
Guillain–Barrén oireyhtymä ja demyelinoiva polyneuropatia
Guillain–Barrén oireyhtymää ja demyelinoivaa polyneuropatiaa kehittyi 0,1 %:lle (6/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Ajan mediaani näiden tapahtumien kehittymiseen oli 4,1 kuukautta (vaihteluväli 18 päivästä 8,1 kuukauteen). Keston mediaani oli 8,0 kuukautta (vaihteluväli 18 päivästä 24,5+ kuukauteen; + tarkoittaa sensuroitua arvoa). Guillain–Barrén oireyhtymä johti atetsolitsumabihoidon lopettamiseen yhdellä potilaalla (< 0,1 %). Kortikosteroidihoitoa vaatinut Guillain–Barrén oireyhtymä todettiin < 0,1 %:lla (3/5 039) atetsolitsumabimonoterapiaa saaneista potilaista.
Immuunivälitteinen kasvohalvaus
Kasvohalvaus ilmaantui < 0,1 %:lle (1/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Aika kasvohalvauksen ilmaantumiseen oli 29 päivää. Kesto oli 1,1 kuukautta. Tapahtuma ei vaatinut kortikosteroidihoitoa eikä se johtanut atetsolitsumabihoidon lopettamiseen.
Immuunivälitteinen myeliitti
Myeliitti ilmaantui < 0,1 %:lle (1/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Aika myeliitin ilmaantumiseen oli 3 päivää. Tapahtuma vaati kortikosteroidihoitoa, mutta ei johtanut atetsolitsumabihoidon lopettamiseen.
Myasteeninen oireyhtymä
Myasthenia gravis ilmaantui < 0,1 %:lle (2/5 039) atetsolitsumabimonoterapiaa saaneista potilaista (mukaan lukien yksi kuolemaan johtanut tapaus). Aika (mediaani) myasthenia gravisin ilmaantumiseen oli 2,6 kuukautta (vaihteluväli: 1,2 kuukautta – 4 kuukautta).
Immuunivälitteinen haimatulehdus
Haimatulehdus, mukaan lukien suurentuneita amylaasi- ja lipaasipitoisuuksia, ilmaantui 0,8 %:lle (40/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Ajan mediaani näiden ilmaantumiseen oli 5 kuukautta (vaihteluväli 0 päivästä 24,8 kuukauteen). Niiden keston mediaani oli 24 päivää (vaihteluväli 3 päivästä 40,4+ kuukauteen; + tarkoittaa sensuroitua arvoa). Haimatulehdus johti atetsolitsumabihoidon lopettamiseen kolmella potilaalla (< 0,1 %). Kortikosteroidihoitoa vaatinut haimatulehdus todettiin 0,2 %:lla (8/5 039) atetsolitsumabimonoterapiaa saaneista potilaista.
Immuunivälitteinen sydänlihastulehdus
Sydänlihastulehdusta esiintyi < 0,1 %:lla (5/5 039) potilaista, jotka saivat atetsolitsumabimonoterapiaa. Näistä viidestä potilaasta yhdellä potilaalla ilmeni ei-pienisoluisen keuhkosyövän adjuvanttihoidossa kuolemaan johtanut tapahtuma. Ajan mediaani sydänlihastulehduksen ilmaantumiseen oli 3,7 kuukautta (vaihteluväli 1,5 kuukaudesta 4,9 kuukauteen). Sen keston mediaani oli 14 päivää (vaihteluväli 12 päivästä 2,8 kuukauteen). Sydänlihastulehdus johti atetsolitsumabihoidon lopettamiseen kolmella (< 0,1 %) potilaalla. Kolme (< 0,1 %) potilasta tarvitsi kortikosteroidihoitoa.
Immuunivälitteinen munuaistulehdus
Munuaistulehdusta esiintyi 0,2 %:lla (11/5 039) atetsolitsumabia saaneista potilaista. Ajan mediaani munuaistulehduksen ilmenemiseen oli 5,1 kuukautta (vaihteluväli 3 päivästä 17,5 kuukauteen). Munuaistulehdus johti atetsolitsumabihoidon lopettamiseen viidellä potilaalla (≤ 0,1 %). Viisi (0,1 %) potilasta tarvitsi kortikosteroidihoitoa.
Immuunivälitteinen myosiitti
Myosiittia esiintyi 0,6 %:lla (32/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Ajan mediaani myosiitin ilmenemiseen oli 3,5 kuukautta (vaihteluväli 12 päivästä 11,5 kuukauteen). Keston mediaani oli 3,2 kuukautta (vaihteluväli 9 päivästä 51,1+ kuukauteen; + tarkoittaa sensuroitua arvoa). Myosiitti johti atetsolitsumabihoidon lopettamiseen kuudella (0,1 %) potilaalla. Kymmenen (0,2 %) potilasta tarvitsi kortikosteroidihoitoa.
Immuunivälitteiset vaikeat ihon lääkereaktiot
Vaikeita ihon lääkereaktioita (SCAR) ilmaantui 0,6 %:lle (30/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. 30 potilaasta yhdellä oli kuolemaan johtanut tapahtuma. Ajan mediaani niiden ilmaantumiseen oli 4,8 kuukautta (vaihteluväli 3 päivästä 15,5 kuukauteen). Keston mediaani oli 2,4 kuukautta (vaihteluväli 1 päivästä 37,5+ kuukauteen; + tarkoittaa sensuroitua arvoa). Vaikeat ihon lääkereaktiot johtivat atetsolitsumabihoidon lopettamiseen kolmella (< 0,1 %) potilaalla. Systeemistä kortikosteroidihoitoa vaativia vaikeita ihon lääkereaktioita ilmaantui 0,2 %:lle (9/5 039) atetsolitsumabimonoterapiaa saaneista potilaista.
Immuunivälitteiset sydänpussin sairaudet
Sydänpussin sairauksia ilmaantui 1 %:lle (49/5 039) atetsolitsumabimonoterapiaa saaneista potilaista. Ajan mediaani niiden ilmaantumiseen oli 1,4 kuukautta (vaihteluväli 6 päivästä 17,5 kuukauteen). Keston mediaani oli 2,5 kuukautta (vaihteluväli 0 päivästä 51,5+ kuukauteen; + tarkoittaa sensuroitua arvoa). Sydänpussin sairaudet johtivat Tecentriq-hoidon lopettamiseen kolmella (< 0,1 %) potilaalla. Kortikosteroidihoitoa vaativia sydänpussin sairauksia ilmaantui 0,2 %:lle (7/5 039) potilaista.
Immuunijärjestelmän tarkistuspisteen estäjien luokkavaikutukset
Muilla immuunijärjestelmän tarkistuspisteen estäjillä annetun hoidon aikana on havaittu seuraavia haittavaikutuksia, joita voi ilmaantua myös atetsolitsumabihoidon aikana:
haiman eksokriininen vajaatoiminta.
Immunogeenisuus
Useissa vaiheen II ja III tutkimuksissa 13,1–54,1 %:lle potilaista kehittyi hoidonaikaisia atetsolitsumabin vasta-aineita. Potilailla, joille hoidonaikaisia atetsolitsumabin vasta-aineita kehittyi, oli yleensä lähtötilanteessa heikompi terveys ja sairauteen liittyi ennustetta heikentäviä ominaisuuksia. Tällainen terveyteen ja sairauden ominaisuuksiin liittyvä epätasapaino lähtötilanteessa voi sekoittaa farmakokineettisten, tehon ja turvallisuuden analyysien tulkintaa. Terveyttä ja sairauden ominaisuuksia lähtötilanteessa koskevan epätasapainon korjaamiseksi tehtiin eksploratiivisia analyyseja, jotta voitiin arvioida atetsolitsumabin vasta-aineiden vaikutusta tehoon. Nämä analyysit eivät sulkeneet pois tehoon liittyvien hyötyjen mahdollista vähenemistä potilailla, joille atetsolitsumabin vasta-aineita kehittyi, verrattuna potilaisiin, joille atetsolitsumabin vasta-aineita ei kehittynyt. Atetsolitsumabin vasta-aineiden ilmaantumiseen kuluneen ajan mediaani oli 3–5 viikkoa.
Atetsolitsumabimonoterapiahoitoa (N = 3 460) ja yhdistelmähoitoja (N = 2 285) saaneiden potilaiden yhdistetyistä tietoaineistoista havaittiin vertailtaessa potilaita, joilla oli todettu atetsolitsumabin vasta-aineita (vasta-ainestatus positiivinen) ja joilla ei ollut todettu atetsolitsumabin vasta-aineita (vasta-ainestatus negatiivinen), että haittatapahtumien esiintyvyys oli seuraava: monoterapiassa asteen 3–4 haittatapahtumia 46,2 % vs. 39,4 %, vakavia haittatapahtumia 39,6 % vs. 33,3 %, hoidon lopettamiseen johtaneita haittatapahtumia 8,5 % vs. 7,8 %, ja yhdistelmähoidossa asteen 3–4 haittatapahtumia 63,9 % vs. 60,9 %, vakavia haittatapahtumia 43,9 % vs. 35,6 %, hoidon lopettamiseen johtaneita haittatapahtumia 22,8 % vs. 18,4 %. Saatavissa olevien tietojen perusteella ei kuitenkaan voida tehdä varmoja päätelmiä haittavaikutusten mahdollisesta esiintyvyydestä.
Pediatriset potilaat
Atetsolitsumabin turvallisuutta lapsille ja nuorille ei ole varmistettu. Kliinisessä tutkimuksessa, jossa oli mukana 69 pediatrista (< 18-vuotiasta) potilasta, ei havaittu uusia turvallisuussignaaleja ja turvallisuusprofiili oli verrannollinen aikuisiin nähden.
Iäkkäät
Atetsolitsumabimonoterapiaa saavien < 65-vuotiaiden, 65–74-vuotiaiden ja 75–84-vuotiaiden potilaiden välillä ei yleisesti havaittu turvallisuutta koskevia eroja. Tiedot ≥ 85-vuotiaista potilaista ovat liian suppeita merkityksellisten päätelmien tekemiseksi tästä potilasjoukosta.
IMpower150-tutkimuksessa ≥ 65 vuoden ikään liittyi suurentunut haittavaikutusten kehittymisen riski niillä potilailla, jotka saivat atetsolitsumabia yhdessä bevasitsumabin, karboplatiinin ja paklitakselin kanssa.
Tutkimuksista IMpower150, IMpower133 ja IMpower110 saadut tiedot ≥ 75-vuotiaista potilaista olivat liian suppeita päätelmien tekemiseksi.
IPSOS-tutkimuksessa platinaa sisältävään ensilinjan hoitoon soveltumattomilla ei-pienisoluista keuhkosyöpää sairastavilla potilailla ensilinjan atetsolitsumabimonoterapian turvallisuusprofiilissa ei yleisesti ollut eroja eri ikäisten potilasalaryhmien välillä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Atetsolitsumabiyliannoksesta ei ole tietoa.
Yliannoksen saanutta potilasta on tarkkailtava huolellisesti haittavaikutusten oireiden ja löydösten havaitsemiseksi, ja oireiden tarkoituksenmukainen hoito on aloitettava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Solunsalpaajat, monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, PD‑1/PDL‑1 (ohjelmoitu solukuolemaproteiini 1/kuolemaligandi 1) estäjät. ATC-koodi: L01FF05.
Vaikutusmekanismi
PD-L1 (programmed death-ligand 1) saattaa ilmentyä kasvainsolujen ja/tai kasvaimeen infiltroivien immuunisolujen pinnalla. Kasvaimen mikroympäristössä se voi osaltaan estää immuunivastetta kasvainta vastaan. PD-L1:n sitoutuminen T-solujen ja antigeeniä esittelevien solujen pinnalla oleviin PD-1- ja B7.1-reseptoreihin hillitsee sytotoksisten T-solujen aktiivisuutta, T-solujen proliferaatiota ja sytokiinituotantoa.
Atetsolitsumabi on Fc-muunnettu, humanisoitu G1-immunoglobuliinin (IgG1) monoklonaalinen vasta-aine, joka sitoutuu suoraan PD-L1:een ja aiheuttaa PD-1- ja B7.1-reseptorien kaksoiseston, jolloin PD-L1/PD-1-välitteinen immuunivasteen esto vapautuu. Tällöin myös kasvaimen kasvua estävä immuunivaste aktivoituu uudelleen indusoimatta solujen vasta-aineriippuvaista sytotoksisuutta. Atetsolitsumabi säästää PD-L2:n ja PD-1:n välisen vuorovaikutuksen eikä PD-L2/PD-1-välitteinen estosignalointi siten esty.
Kliininen teho ja turvallisuus
Uroteelikarsinooma
IMvigor211 (GO29294): Satunnaistettu tutkimus paikallisesti edennyttä tai metastasoitunutta uroteelikarsinoomaa sairastavilla potilailla, jotka olivat aiemmin saaneet solunsalpaajahoitoa
Vaiheen III avoimessa, kansainvälisessä, satunnaistetussa monikeskustutkimuksessa (IMvigor211) arvioitiin atetsolitsumabin tehoa ja turvallisuutta solunsalpaajahoitoon verrattuna (tutkijan valinnan mukaan vinfluniini, dosetakseli tai paklitakseli) potilailla, joilla oli paikallisesti edennyt tai metastasoitunut uroteelikarsinooma, joka eteni platinaa sisältäneen hoidon aikana tai jälkeen. Tähän tutkimukseen ei otettu mukaan potilaita, joilla oli aiemmin ollut jokin autoimmuunisairaus, aktiivisia tai kortikosteroidiriippuvaisia aivometastaaseja, jotka olivat saaneet 28 päivän kuluessa ennen tutkimukseen tuloa elävää, heikennettyä taudinaiheuttajaa sisältävää rokotetta, 4 viikon kuluessa systeemisiä immuniteettia stimuloivia aineita tai 2 viikon kuluessa ennen tutkimukseen tuloa systeemisiä immunosuppressiivisia lääkevalmisteita. Kasvain arvioitiin ensimmäisten 54 viikon aikana 9 viikon välein ja tämän jälkeen 12 viikon välein. Kasvainnäytteistä tutkittiin prospektiivisesti PD-L1:n ilmentyminen kasvainta infiltroivissa immuunisoluissa, ja tulosten perusteella määriteltiin PD-L1:n ilmentymistä koskevat osajoukot jäljempänä kuvattuihin analyyseihin.
Tutkimukseen otettiin mukaan yhteensä 931 potilasta. Potilaat satunnaistettiin (1:1) saamaan joko atetsolitsumabia tai solunsalpaajahoitoa. Satunnaistaminen ositettiin solunsalpaajahoidon (vinfluniini vs. taksaani), PD-L1:n ilmentymisen immuunisoluissa (< 5 % vs. ≥ 5 %), ennustetta koskevien riskitekijöiden lukumäärän (0 vs. 1–3) ja maksametastaasien (kyllä vs. ei) perusteella. Ennustetta koskevia riskitekijöitä olivat aika aiemmasta solunsalpaajahoidosta < 3 kuukautta, ECOG-suorituskykyluokka > 0 ja hemoglobiinipitoisuus < 100 g/l (< 10 g/dl).
Atetsolitsumabia annettiin vakioannoksina 1200 mg infuusiona laskimoon 3 viikon välein. Atetsolitsumabiannosta ei saanut pienentää. Potilaat saivat hoitoa, kunnes kliinistä hyötyä ei tutkijan arvion mukaan enää todettu tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Vinfluniinia annettiin 320 mg/m2 infuusiona laskimoon kunkin 3 viikon pituisen hoitosyklin päivänä 1, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Paklitakselia annettiin 175 mg/m2 3 tunnin kestoisena infuusiona laskimoon kunkin 3 viikon pituisen hoitosyklin päivänä 1, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Dosetakselia annettiin 75 mg/m2 infuusiona laskimoon kunkin 3 viikon pituisen hoitosyklin päivänä 1, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Kaikkien hoitoa saaneiden potilaiden hoidon keston mediaani oli atetsolitsumabihaarassa 2,8 kuukautta, vinfluniini- ja paklitakselihaaroissa 2,1 kuukautta ja dosetakselihaarassa 1,6 kuukautta.
Primaarianalyysin potilasjoukon demografiset ominaisuudet ja sairauden ominaisuudet lähtötilanteessa olivat hyvin tasapainossa hoitohaarojen välillä. Iän mediaani oli 67 vuotta (vaihteluväli: 31–88), ja 77,1 % potilaista oli miehiä. Valtaosa potilaista oli valkoihoisia (72,1 %). Solunsalpaajahaarassa 53,9 % potilaista sai vinfluniinia, 71,4 %:lla potilaista oli vähintään yksi huonon ennusteen riskitekijä ja 28,8 %:lla oli lähtötilanteessa maksametastaaseja. Lähtötilanteen ECOG-suorituskykyluokka oli 0 (45,6 %) tai 1 (54,4 %). Potilaista 71,1 %:lla kasvaimen ensisijainen sijaintikohta oli virtsarakko, ja 25,4 %:lla potilaista oli ylempien virtsateiden uroteelikarsinooma. Potilaista 24,2 % oli saanut aiemmin ainoastaan platinaa sisältänyttä adjuvantti- tai neoadjuvanttihoitoa, ja sairaus oli edennyt 12 kuukauden kuluessa.
Tutkimuksen IMvigor211 ensisijainen tehon päätetapahtuma on kokonaiselossaolo (overall survival, OS). Tutkijan RECIST v1.1 (Response Evaluation Criteria in Solid Tumours) kriteerien perusteella arvioimat toissijaiset tehon päätetapahtumat ovat kokonaisvasteosuus (objective response rate, ORR), taudin etenemättömyysaika (progression-free survival, PFS) ja vasteen kesto (duration of response, DOR). Kokonaiselossaolon vertailu hoitohaaran ja kontrollihaaran välillä IC2/3-, IC1/2/3- ja hoitoaikeen mukaisessa (Intention-to-treat eli PD-L1-statuksen suhteen valikoimattomat potilaat) potilasjoukossa testattiin hierarkkisesti määritellyllä sekvenssimenetelmällä, joka perustuu ositettuun log-rank-testiin kaksitahoisen testauksen 5 %:n merkitsevyystasolla seuraavasti: 1. vaihe IC2/3-potilasjoukko, 2. vaihe IC1/2/3-potilasjoukko, 3. vaihe PD-L1-statuksen suhteen valikoimattomat potilaat. Kunkin 2. ja 3. vaiheen kokonaiselossaolon tilastollinen merkitsevyys testattiin varsinaisesti vain, jos edellisen vaiheen tulos oli tilastollisesti merkitsevä.
Seurantavaiheen elossaolon seuranta-ajan mediaani oli 17 kuukautta. Tutkimuksen IMvigor211 ensisijainen analyysi ei saavuttanut kokonaiselossaoloa koskevaa ensisijaista päätetapahtumaa. Atetsolitsumabista ei osoitettu tilastollisesti merkitsevää hyötyä elossaolon suhteen verrattuna solunsalpaajahoitoa aiemmin saaneisiin paikallisesti edennyttä tai metastasoitunutta uroteelikarsinoomaa sairastaviin potilaisiin. Ennalta määritellyn hierarkkisen testausjärjestyksen mukaisesti IC2/3-potilasjoukko testattiin ensimmäisenä, ja kokonaiselossaolon riskisuhde oli 0,87 (95 %:n luottamusväli: 0,63, 1,21; elossaolon mediaani atetsolitsumabihoidossa 11,1 kuukautta vs. solunsalpaajahoidossa 10,6 kuukautta). Ositetun log-rank-testin p-arvo oli 0,41 eikä tämän potilasjoukon tuloksia siten katsottu tilastollisesti merkitseviksi. Näin ollen IC1/2/3-potilasjoukon tai PD-L1-statuksen suhteen valikoimattomien potilaiden kokonaiselossaolon tilastollista merkitsevyyttä ei voitu testata, ja näiden analyysien tulokset katsottiin eksploratiivisiksi. Yhteenveto PD-L1-statuksen suhteen valikoimattomien potilaiden keskeisistä tuloksista esitetään taulukossa 4. PD-L1-statuksen suhteen valikoimattomien potilaiden kokonaiselossaolon Kaplan–Meierin käyrä esitetään kuvassa 1.
ITT-potilasjoukon elossaolon analyysi päivitettiin eksploratiivisesti, kun elossaolon seuranta-ajan keston mediaani oli 34 kuukautta. Kokonaiselossaolon mediaani oli atetsolitsumabihaarassa 8,6 kuukautta (95 %:n luottamusväli: 7,8; 9,6) ja solunsalpaajahoitohaarassa 8,0 kuukautta (95 %:n luottamusväli: 7,2, 8,6), ja riskisuhde oli 0,82 (95 %:n luottamusväli: 0,71; 0,94). ITT-potilasjoukossa todettiin yhdenmukaisesti 12 kuukauden kokonaiselossaololukujen primaarianalyysissa havaitun kehityssuunnan kanssa, että 24 kuukauden ja 30 kuukauden kokonaiselossaololuvut olivat atetsolitsumabihaarassa numeerisesti suuremmat kuin solunsalpaajahoitohaarassa. Elossa olevien potilaiden prosenttiosuus 24 kuukauden hoidon jälkeen (KM-estimaatti) oli solunsalpaajahoitohaarassa 12,7 % ja atetsolitsumabihoitohaarassa 22,5 % ja 30 kuukauden hoidon jälkeen (KM-estimaatti) solunsalpaajahoitohaarassa 9,8 % ja atetsolitsumabihoitohaarassa 18,1 %.
Taulukko 4. Yhteenveto hoidon tehosta PD-L1-statuksen suhteen valikoimattomilla potilailla tutkimuksessa IMvigor211
| Tehon päätetapahtuma | Atetsolitsumabi (n = 467) | Solunsalpaajahoito (n = 464) |
| Ensisijainen tehon päätetapahtuma | ||
| Kokonaiselossaolo* | ||
| Kuolemien lkm (%) | 324 (69,4 %) | 350 (75,4 %) |
| Ajan mediaani tapahtumiin (kuukautta) | 8,6 | 8,0 |
| 95 %:n luottamusväli | 7,8, 9,6 | 7,2, 8,6 |
| Ositettuǂ riskisuhde (95 %:n luottamusväli) | 0,85 (0,73, 0,99) | |
| 12 kuukauden kokonaiselossaolo (%)** | 39,2 % | 32,4 % |
| Toissijaiset ja eksploratiiviset päätetapahtumat | ||
| Tutkijan arvioima taudin etenemättömyysaika (RECIST v1.1) | ||
| Tapahtumien lkm (%) | 407 (87,2 %) | 410 (88,4 %) |
| Taudin etenemättömyysajan mediaani (kuukautta) | 2,1 | 4,0 |
| 95 %:n luottamusväli | 2,1, 2,2 | 3,4, 4,2 |
| Ositettu riskisuhde (95 %:n luottamusväli) | 1,10 (0,95, 1,26) | |
| Tutkijan arvioima kokonaisvasteosuus (RECIST v1.1) | n = 462 | n = 461 |
| Varmistetun vasteen saaneiden lkm (%) | 62 (13,4 %) | 62 (13,4 %) |
| 95 %:n luottamusväli | 10,45, 16,87 | 10,47, 16,91 |
| Täydellisen vasteen saaneiden lkm (%) | 16 (3,5 %) | 16 (3,5 %) |
| Osittaisen vasteen saaneiden lkm (%) | 46 (10,0 %) | 46 (10,0 %) |
| Stabiilin taudin saavuttaneiden lkm (%) | 92 (19,9 %) | 162 (35,1 %) |
| Tutkijan arvioima vasteen kesto (RECIST v1.1) | n = 62 | n = 62 |
| Mediaani kuukausina*** | 21,7 | 7,4 |
| 95 %:n luottamusväli | 13,0, 21,7 | 6,1, 10,3 |
* PD-L1-statuksen suhteen valikoimattomien potilaiden kokonaiselossaolon analyysi tehtiin ositetun log-rank-testin perusteella, ja tulos esitetään vain deskriptiivisessä tarkoituksessa (p = 0,0378); ennalta määritellyn analyysihierarkian mukaan, PD-L1-statuksen suhteen valikoimattomien potilaiden kokonaiselossaololuvun analyysin p-arvoa ei voida katsoa tilastollisesti merkitseväksi.
ǂ Ositettu solunsalpaajahoidon (vinfluniini vs. taksaani), PD-L1:n ilmentymisen immuunisoluissa (< 5 % vs. ≥ 5 %), ennustetta koskevien riskitekijöiden lukumäärän (0 vs. 1–3) ja maksametastaasien (kyllä vs. ei) perusteella.
** Perustuu Kaplan–Meierin estimaattiin
*** Vaste säilyi edelleen 63 %:lla atetsolitsumabihaarassa vasteen saaneista ja 21 %:lla solunsalpaajahoitohaarassa vasteen saaneista.
Kuva 1. Kokonaiselossaolon Kaplan–Meierin käyrä (IMvigor211)
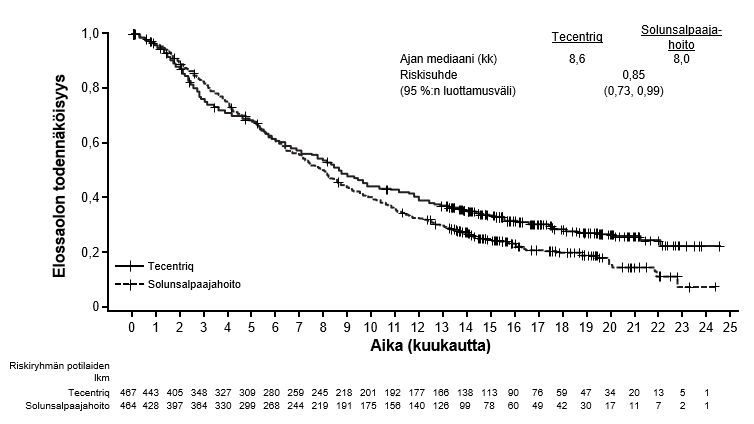
IMvigor210 (GO29293): Yhden hoitohaaran tutkimus aiemmin hoitamatonta uroteelikarsinoomaa sairastavilla potilailla, joille sisplatiinihoito ei sovi, sekä solunsalpaajahoitoa aiemmin saaneilla uroteelikarsinoomaa sairastavilla potilailla
Vaiheen II kansainvälisessä, kahden kohortin, yhden hoitoryhmän kliinisessä monikeskustutkimuksessa IMvigor210 oli mukana potilaita, jotka sairastivat paikallisesti edennyttä tai metastasoitunutta uroteelikarsinoomaa (jota kutsutaan myös virtsarakon välimuotoisen epiteelin [uroteelin] karsinoomaksi).
Tutkimukseen otettiin mukaan yhteensä 438 potilasta, ja heistä muodostettiin kaksi potilaskohorttia. Kohortissa 1 oli aiemmin hoitamattomia potilaita, jotka sairastivat paikallisesti edennyttä tai metastasoitunutta uroteelikarsinoomaa ja joille sisplatiinipohjainen solunsalpaajahoito ei sopinut tai he olivat liian huonokuntoisia tällaiseen hoitoon tai joiden sairaus oli edennyt aikaisintaan 12 kuukauden jälkeen siitä, kun platinaa sisältänyt neoadjuvantti- tai adjuvanttisolunsalpaajahoito oli päättynyt. Kohortissa 2 oli potilaita, jotka olivat saaneet paikallisesti edenneen tai metastasoituneen uroteelikarsinooman hoitoon vähintään yhden platinapohjaisen solunsalpaajahoito-ohjelman tai joiden sairaus oli edennyt 12 kuukauden kuluessa platinaa sisältäneestä neoadjuvantti- tai adjuvanttisolunsalpaajahoidosta.
Kohortissa 1 oli 119 potilasta, ja he saivat 1 200 mg atetsolitsumabia infuusiona laskimoon 3 viikon välein, kunnes tauti eteni. Iän mediaani oli 73 vuotta. Useimmat potilaat olivat miehiä (81 %), ja suurin osa potilaista oli valkoihoisia (91 %).
Kohortissa 1 oli 45 potilasta (38 %), joiden ECOG-suorituskykyluokka oli 0, 50 potilasta (42 %), joiden ECOG-suorituskykyluokka oli 1, ja 24 potilasta (20 %), joiden ECOG-suorituskykyluokka oli 2, 35 potilasta (29 %), joilla ei ollut Bajorin-riskitekijöitä (ECOG-suorituskykyluokka ≥ 2 ja viskeraalinen metastaasi), 66 potilasta (56 %), joilla oli yksi Bajorin-riskitekijä, ja 18 potilasta (15 %), joilla oli kaksi Bajorin-riskitekijää, 84 potilasta (71 %), joiden munuaisten toiminta oli heikentynyt (glomerulusten suodatusnopeus [GFR] < 60 ml/min), ja 25 potilasta (21 %), joilla oli etäpesäke maksassa.
Kohortin 1 ensisijainen tehon päätetapahtuma oli varmistettu kokonaisvasteosuus (objective response rate, ORR), jonka riippumaton arviointilautakunta (independent review facility, IRF) oli arvioinut RECIST v1.1 -kriteerien perusteella.
Primaarianalyysi tehtiin, kun kaikki potilaat olivat olleet seurannassa vähintään 24 viikkoa. Hoidon kestoajan mediaani oli 15,0 viikkoa, ja PD-L1-statuksen suhteen valikoimattomien potilaiden elossaolon seurannan mediaani oli 8,5 kuukautta. Riippumattoman arviointilautakunnan RECIST v1.1 kriteerien mukaisesti arvioima kokonaisvasteosuus oli kliinisesti oleellinen, mutta kun sitä verrataan ennalta määritetyn historiallisen verrokkiryhmän kokonaisvasteosuuteen 10 %, ei ensisijaisen päätetapahtuman osalta saavutettu tilastollista merkitsevyyttä. Riippumattoman arviointilautakunnan RECIST v1.1 kriteerien mukaisesti varmistamat kokonaisvasteosuudet olivat 21,9 % (95 %:n luottamusväli: 9,3, 40,0), jos kasvaimen PD-L1:n ilmentymisen taso oli ≥ 5 %, 18,8 % (95 %:n luottamusväli: 10,9, 29,0), jos kasvaimen PD-L1:n ilmentymisen taso oli ≥ 1 %, ja 19,3 % (95 %:n luottamusväli: 12,7, 27,6) PD-L1-statuksen suhteen valikoimattomien potilaiden ryhmässä. Objektiivisen vasteen keston mediaania ei saavutettu missään PD-L1:n ilmentymisen alaryhmässä eikä PD-L1-statuksen suhteen valikoimattomien potilaiden ryhmässä. Kokonaiselossaoloaikaa koskevat tiedot eivät olleet valmiit, ja niiden potilaiden osuus, joilla oli esiintynyt tapahtumia, oli noin 40 %. Potilaiden kaikkien alaryhmien (kasvaimen PD-L1:n ilmentymisen taso ≥ 5 % ja ≥ 1 %) ja PD-L1-statuksen suhteen valikoimattomien potilaiden ryhmässä kokonaiselossaoloajan mediaani oli 10,6 kuukautta.
Päivitetty analyysi tehtiin, kun kohortin 1 elossaolon seuranta-ajan mediaani oli 17,2 kuukautta. Yhteenveto esitetään taulukossa 5. Objektiivisen vasteen keston mediaania ei ollut saavutettu missään kasvaimen PD-L1:n ilmentymisen alaryhmässä eikä PD-L1-statuksen suhteen valikoimattomien potilaiden ryhmässä.
Taulukko 5. Päivitetty yhteenveto hoidon tehosta (IMvigor210, kohortti 1)
| Hoidon tehoa koskeva päätetapahtuma | PD-L1:n ilmentymisen taso ≥ 5 % kasvaimeen infiltroivissa immuunisoluissa | PD-L1:n ilmentymisen taso ≥ 1 % kasvaimeen infiltroivissa immuunisoluissa | PD-L1-statuksen suhteen valikoimattomat potilaat |
| Kokonaisvasteosuus (riippumattoman arviointilautakunnan arvio; RECIST v1.1) | n = 32 | n = 80 | n = 119 |
| Vasteen saaneiden lkm (%) | 9 (28,1 %) | 19 (23,8 %) | 27 (22,7 %) |
| 95 %:n luottamusväli | 13,8, 46,8 | 15,0, 34,6 | 15,5, 31,3 |
Täydellisen vasteen saaneiden lkm (%) 95 %:n luottamusväli | 4 (12,5 %) (3,5, 29,0) | 8 (10,0 %) (4,4, 18,8) | 11 (9,2 %) (4,7, 15,9) |
Osittaisen vasteen saaneiden lkm (%) 95 %:n luottamusväli | 5 (15,6 %) (5,3, 32,8) | 11 (13,8 %) (7,1, 23,3) | 16 (13,4 %) (7,9, 20,9) |
| Objektiivisen vasteen kesto (riippumattoman arviointilautakunnan arvio; RECIST v1.1) | n = 9 | n = 19 | n = 27 |
| Potilaat, joilla esiintyi tapahtuma (%) | 3 (33,3 %) | 5 (26,3 %) | 8 (29,6 %) |
| Mediaani (kk) (95 %:n luottamusväli) | NE (11,1, NE) | NE (NE) | NE (14,1, NE) |
| Taudin etenemättömyysaika (riippumattoman arviointilautakunnan arvio; RECIST v1.1) | n = 32 | n = 80 | n = 119 |
| Potilaat, joilla esiintyi tapahtuma (%) | 24 (75,0 %) | 59 (73,8 %) | 88 (73,9 %) |
| Mediaani (kk) (95 %:n luottamusväli) | 4,1 (2,3, 11,8) | 2,9 (2,1, 5,4) | 2,7 (2,1, 4,2) |
| Kokonaiselossaolo | n = 32 | n = 80 | n = 119 |
| Potilaat, joilla esiintyi tapahtuma (%) | 18 (56,3 %) | 42 (52,5 %) | 59 (49,6 %) |
| Mediaani (kk) (95 %:n luottamusväli) | 12,3 (6,0, NE) | 14,1 (9,2, NE) | 15,9 (10,4, NE) |
| 1 vuoden kokonaiselossaoloaika (%) | 52,4 % | 54,8 % | 57,2 % |
Luottamusväli = confidence interval (CI); Vasteen kesto = duration of response (DOR); Kasvaimeen infiltroivat immuunisolut = tumour-infiltrating immune cells (IC); Ei arvioitavissa = not estimable (NE); Kokonaisvasteosuus = objective response rate (ORR); Kokonaiselossaoloaika = overall survival (OS); Etenemättömyysaika = progression-free survival (PFS); RECIST = Response Evaluation Criteria in Solid Tumours v1.1.
Kohortin 1 loppuanalyysin ajankohtana potilaiden elossaolon seuranta-ajan mediaani oli 96,4 kuukautta. Kokonaiselossaolon mediaani potilailla, joilla PD-L1:n ilmentymisen taso kasvaimessa oli ≥ 5 % (käyttöaiheen mukaiset potilaat), oli 12,3 kuukautta (95 %:n luottamusväli: 6,0, 49,8).
Kohortissa 2 tehon muut ensisijaiset päätetapahtumat olivat riippumattoman arviointilautakunnan RECIST v1.1 kriteerien mukaisesti tekemän arvion perusteella varmistettu kokonaisvasteosuus ja tutkijan muokattujen RECIST-kriteerien (mRECIST) mukaisesti arvioima kokonaisvasteosuus. 310 potilasta sai 1200 mg atetsolitsumabia infuusiona laskimoon 3 viikon välein, kunnes kliinistä hyötyä ei enää todettu. Kohortin 2 primaarianalyysi tehtiin, kun kaikki potilaat olivat olleet seurannassa vähintään 24 viikkoa. Tutkimuksessa saavutettiin ensisijaiset päätetapahtumat kaikissa kohortin 2 alaryhmissä, mikä osoitti tilastollisesti merkitsevän kokonaisvasteosuuden riippumattoman arviointilautakunnan RECIST v1.1 kriteerien mukaisesti tekemän arvion perusteella ja tutkijan muokattujen RECIST-kriteerien mukaisesti tekemän arvion perusteella verrattuna ennalta määriteltyyn historiallisen vertailuryhmän 10 %:n kokonaisvasteosuuteen.
Analyysi tehtiin myös, kun kohortin 2 elossaolon seuranta-ajan mediaani oli 21,1 kuukautta. Riippumattoman arviointilautakunnan RECIST v1.1 kriteerien mukaisesti varmistamat kokonaisvasteosuudet olivat 28,0 % (95 %:n luottamusväli: 19,5, 37,9), jos PD-L1:n ilmentymisen taso kasvaimessa oli ≥ 5 %, 19,3 % (95 %:n luottamusväli: 14,2, 25,4), jos PD-L1:n ilmentymisen taso kasvaimessa oli ≥ 1 %, ja 15,8 % (95 %:n luottamusväli: 11,9, 20,4) PD-L1-statuksen suhteen valikoimattomien potilaiden ryhmässä. Tutkijan mRECIST-kriteerien mukaisesti tekemän arvion perusteella varmistettu kokonaisvasteosuus oli 29,0 % (95 %:n luottamusväli: 20,4, 38,9), jos PD-L1:n ilmentymisen taso kasvaimessa oli ≥ 5 %, 23,7 % (95 %:n luottamusväli: 18,1, 30,1), jos PD-L1:n ilmentymisen taso kasvaimessa oli ≥ 1 %, ja 19,7 % (95 %:n luottamusväli: 15,4, 24,6) PD-L1-statuksen suhteen valikoimattomien potilaiden ryhmässä. Riippumaton arviointilautakunta arvioi RECIST v1.1 kriteerien mukaisesti, että täydellisen vasteen saaneiden osuus PD-L1-statuksen suhteen valikoimattomien potilaiden joukossa oli 6,1 % (95 %:n luottamusväli: 3,7, 9,4). Kohortissa 2 ei saavutettu vasteen keston mediaania missään kasvaimen PD-L1:n ilmentymistason alaryhmässä eikä PD-L1-statuksen suhteen valikoimattomien potilaiden ryhmässä, mutta se saavutettiin potilailla, joiden PD-L1:n ilmentymisen taso oli < 1 % (13,3 kuukautta; 95 %:n luottamusväli: 4,2, NE). PD-L1-statuksen suhteen valikoimattomien potilaiden ryhmässä 12 kuukauden kokonaiselossaolo-osuus oli 37 %.
Kohortin 2 loppuanalyysin ajankohtana potilaiden elossaolon seuranta-ajan mediaani oli 46,2 kuukautta. Kokonaiselossaolon mediaani potilailla, joilla PD-L1:n ilmentymisen taso kasvaimessa oli ≥ 5 %, oli 11,9 kuukautta (95 %:n luottamusväli: 9,0, 22,8), potilailla, joilla PD-L1:n ilmentymisen taso kasvaimessa oli ≥ 1 %, se oli 9,0 kuukautta (95 %:n luottamusväli: 7,1, 11,1) ja PD‑L1-statuksen suhteen valikoimattomien potilaiden ryhmässä se oli 7,9 kuukautta (95 %:n luottamusväli: 6,7, 9,3).
IMvigor130 (WO30070): Vaiheen III tutkimus atetsolitsumabin käytöstä monoterapiana ja yhdistelmänä platinapohjaisten solunsalpaajien kanssa hoitamatonta paikallisesti edennyttä tai metastasoitunutta uroteelikarsinoomaa sairastavien potilaiden hoitoon
Satunnaistettu, lumekontrolloitu, osittain sokkoutettu (vain hoitohaarat A ja C) vaiheen III monikeskustutkimus IMvigor130 tehtiin potilailla, joilla oli paikallisesti edennyt tai metastasoitunut uroteelikarsinooma ja jotka eivät olleet aiemmin saaneet metastasoituneeseen sairauteen systeemistä hoitoa. Tässä tutkimuksessa verrattiin atetsolitsumabin ja platinapohjaisen solunsalpaajayhdistelmähoidon (joko sisplatiini tai karboplatiini yhdessä gemsitabiinin kanssa) yhdistelmän (hoitohaara A) tai atetsolitsumabimonoterapian (hoitohaara B, avoin haara) turvallisuutta ja tehoa lumelääkkeen ja platinapohjaisen solunsalpaajayhdistelmähoidon yhdistelmään (hoitohaara C). Tehon rinnakkaiset ensisijaiset päätetapahtumat olivat tutkijan arvioima taudin etenemättömyysaika (progression-free survival, PFS) hoitohaarassa A verrattuna hoitohaaraan C ja kokonaiselossaoloaika (overall survival, OS) hoitohaarassa A verrattuna haaraan C ja sitten hoitohaarassa B verrattuna haaraan C hierarkkisessa järjestyksessä analysoituna. Ero kokonaiselossaoloajassa ei ollut hoitohaaran A ja hoitohaaran C vertailun osalta tilastollisesti merkitsevä, joten ennalta määritellyn hierarkkisen analyysijärjestyksen mukaisia muita muodollisia testauksia ei voitu tehdä.
Niitä potilaita, joiden kasvaimet ilmensivät PD-L1:tä niukasti (immunohistokemiallinen PD-L1-värjäystulos VENTANA PD-L1 [SP142] ‑menetelmällä oli positiivinen alle 5 %:ssa immuunisoluista), ei otettu riippumattoman tietojenseurantakomitean (independent Data Monitoring Committee, iDMC) suosituksen perusteella mukaan atetsolitsumabin monoterapiahaaraan enää sen jälkeen, kun komitea oli tarkastellut varhaisvaiheen elossaolotietoja ja havainnut suunnittelemattomassa varhaisvaiheen analyysissa potilaiden tämän alaryhmän kokonaiselossaoloajan lyhentyneen. Tämä tapahtui kuitenkin vasta, kun valtaosa potilaista oli jo otettu tutkimukseen mukaan.
Atetsolitsumabimonoterapiaa saaneeseen hoitohaaraan ja pelkkää solunsalpaajahoitoa saaneeseen hoitohaaraan otetuista 719 potilaasta 50 potilasta atetsolitsumabihaarassa (n = 360) ja 43 potilasta solunsalpaajahaarassa (n = 359) ei Galskyn kriteerien mukaan soveltunut sisplatiinihoitoon, ja näillä potilailla PD-L1:n ilmentymä kasvaimissa oli suuri (immunohistokemiallinen PD‑L1-värjäystulos VENTANA PD‑L1 [SP142] ‑menetelmällä oli positiivinen vähintään 5 %:ssa immuunisoluista). Tässä potilaiden alaryhmässä tehdyssä eksploratiivisessa analyysissa kokonaiselossaoloajan osittamaton riskisuhde oli 0,56 (95 %:n luottamusväli: 0,34; 0,91). Kokonaiselossaoloajan mediaani oli atetsolitsumabimonoterapiaa saaneessa hoitohaarassa 18,6 kuukautta (95 %:n luottamusväli: 14,0; 49,4) verrattuna 10,0 kuukauteen (95 %:n luottamusväli: 7,4; 18,1) pelkkää solunsalpaajahoitoa saaneessa hoitohaarassa (ks. kuva 2).
Kuva 2. Kokonaiselossaolon Kaplan–Meierin kuvaaja sisplatiinihoitoon soveltumattomista potilaista, joiden kasvaimissa PD-L1:n ilmentymä on suuri (hoitohaara B vs. hoitohaara C)
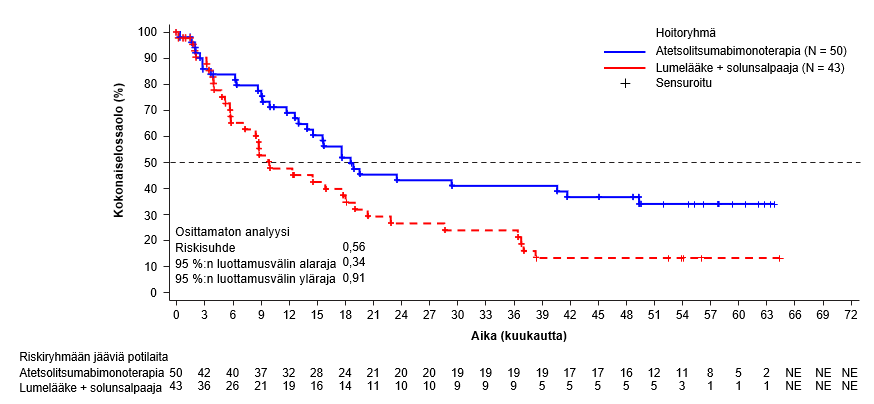
Ei-pienisoluinen keuhkosyöpä
Varhaisvaiheen ei-pienisoluisen keuhkosyövän adjuvanttihoito
IMpower010 (GO29527): Satunnaistettu vaiheen III tutkimus sisplatiinipohjaisen solunsalpaajahoidon jälkeen potilailla, joille oli tehty ei-pienisoluisen keuhkosyövän resektio
Avoin, satunnaistettu vaiheen III monikeskustutkimus GO29527 (IMpower010) tehtiin atetsolitsumabin tehon ja turvallisuuden arvioimiseksi levinneisyysasteen IB (kasvaimet ≥ 4 cm) – IIIA ei-pienisoluisen keuhkosyövän (Union for International Cancer Control/American Joint Committee on Cancer -levinneisyysastejärjestelmän, 7. painos, perusteella) adjuvanttihoidossa.
Suuren uusiutumisriskin potilaat, jotka soveltuvat hoidon käyttöaiheeseen ja jotka kuvastavat levinneisyysastejärjestelmän 7. painoksen mukaista levinneisyysasteen II–IIIA potilasjoukkoa, määriteltiin seuraavien valintakriteerien mukaan:
kasvaimen koko ≥ 5 cm tai minkä tahansa kokoinen kasvain, johon liittyy status N1 tai N2, tai rintakehän rakenteisiin tunkeutuvia kasvaimia (invaasio suoraan parietaaliseen pleuraan, rintakehän seinämään, palleaan, palleahermoon, mediastinaaliseen pleuraan, parietaaliseen sydänpussiin, välikarsinaan, sydämeen, suuriin verisuoniin, henkitorveen, recurrens-hermoon, ruokatorveen, nikamansolmuun, henkitorven harjuun) tai kasvaimiin liittyy invaasio pääkeuhkoputkeen < 2 cm distaalisesti henkitorven harjuun nähden, mutta ilman invaasiota henkitorven harjuun, tai kasvaimiin liittyy atelektaasi tai koko keuhkon obstruktiivinen pneumoniitti tai kasvaimissa on erillisiä pyörövarjoja samassa lohkossa tai primaarina toisessa samanpuoleisessa lohkossa.
Tutkimuksessa ei ollut mukana statuksen N2 potilaita, joilla kasvain olisi tunkeutunut välikarsinaan, sydämeen, suuriin verisuoniin, henkitorveen, recurrens-hermoon, ruokatorveen, nikamansolmuun tai henkitorven harjuun tai joilla olisi erillisiä pyörövarjoja toisessa samanpuoleisessa lohkossa.
Yhteensä 1 280:lle tutkimukseen mukaan otetulle potilaalle oli tehty kasvaimen täydellinen resektio ja he soveltuivat saamaan enimmillään 4 hoitosykliä sisplatiinipohjaista solunsalpaajahoitoa. Sisplatiinipohjaiset solunsalpaajahoito-ohjelmat kuvataan taulukossa 6.
Taulukko 6: Solunsalpaajia sisältävät adjuvanttihoito-ohjelmat (IMpower010)
Sisplatiinipohjaisia solunsalpaajia sisältävä adjuvanttihoito: 75 mg/m2 sisplatiinia laskimoon kunkin 21 päivän pituisen hoitosyklin päivänä 1 yhdessä jonkin seuraavien hoito-ohjelmien kanssa | 30 mg/m2 vinorelbiiniä laskimoon päivinä 1 ja 8 |
| 75 mg/m2 dosetakselia laskimoon päivänä 1 | |
| 1250 mg/m2 gemsitabiinia laskimoon päivinä 1 ja 8 | |
| 500 mg/m2 pemetreksediä laskimoon päivänä 1 (ei-levyepiteeliperäinen) |
Sisplatiinipohjaisen solunsalpaajahoidon (enintään neljä hoitosykliä) päätyttyä yhteensä 1 005 potilasta satunnaistettiin suhteessa 1:1 saamaan atetsolitsumabia (haara A) tai parasta tukihoitoa (haara B). Atetsolitsumabia annettiin vakioannoksena 1200 mg infuusiona laskimoon 3 viikon välein 16 hoitosykliä, ellei tauti uusiutunut tai ilmaantunut toksisuutta, joka ei ollut hyväksyttävissä. Satunnaistaminen ositettiin sukupuolen, taudin levinneisyysasteen, histologian ja PD-L1-ilmentymän mukaan.
Potilaita ei otettu tutkimukseen mukaan seuraavissa tilanteissa: anamneesissa oli autoimmuunisairaus, satunnaistamista edeltäneiden 28 päivän aikana oli annettu heikennettyjä eläviä taudinaiheuttajia sisältävää rokotetta, satunnaistamista edeltäneiden 4 viikon aikana oli annettu systeemisiä immuniteettia stimuloivia lääkeaineita tai satunnaistamista edeltäneiden 2 viikon aikana oli annettu systeemisiä immunosuppressiivisia lääkityksiä. Kasvain arvioitiin satunnaistamisvaiheen lähtötilanteessa sekä 4 kuukauden välein vuoden ajan ensimmäisen hoitosyklin 1 päivän jälkeen, sen jälkeen viiden vuoden ajan 6 kuukauden välein, minkä jälkeen vuosittain.
Hoitoaikeen mukaisen (ITT) potilasjoukon demografiset ominaisuudet ja taudin ominaisuudet olivat lähtötilanteessa hoitohaarojen välillä hyvin tasapainossa. Iän mediaani oli 62 vuotta (vaihteluväli: 26–84), ja 67 % potilaista oli miehiä. Valtaosa potilaista oli valkoihoisia (73 %), ja 24 % oli aasialaisia. Useimmat potilaat olivat joko nykyisiä tai entisiä tupakoitsijoita (78 %); potilaiden lähtötilanteen ECOG-suorituskykyluokka oli 0 (55 %) tai 1 (44 %). Kaikkiaan 12 %:lla potilaista oli levinneisyysasteen IB, 47 %:llä oli levinneisyysasteen II ja 41 %:llä oli levinneisyysasteen IIIA sairaus. Niiden potilaiden prosenttiosuus, joilla kasvainten VENTANA PD-L1 (SP263) -menetelmällä määritetty PD-L1-ilmentymä kasvainsoluissa oli ≥ 1 %, oli 55 % ja niiden potilaiden prosenttiosuus, joilla kyseinen ilmentymä kasvainsoluissa oli ≥ 50 %, oli 26 %.
Ensisijainen tehoa koskeva hoitotuloksen mittari oli tutkijan arvioima tautivapaa elossaoloaika (disease-free survival, DFS). Tautivapaaksi elossaoloajaksi määriteltiin satunnaistamispäivämäärästä jonkin seuraavista ilmaantumisen päivämäärään kulunut aika: sairauden ensimmäinen dokumentoitu uusiutuminen, uusi primaari ei-pienisoluinen keuhkosyöpä tai mistä tahansa syystä aiheutunut kuolema sen mukaan, mikä näistä tapahtui ensimmäisenä. Ensisijainen tehoa koskeva tavoite oli arvioida tautivapaata elossaoloaikaa leviämisasteen II–IIIA sairautta sairastavassa potilasjoukossa, jossa PD-L1-ilmentymä kasvainsoluissa oli ≥ 1 %. Keskeisiä toissijaisia tehoa koskevia tavoitteita oli arvioida tautivapaata elossaoloaikaa levinneisyysasteen II–IIIA potilasjoukossa, jossa PD-L1:n ilmentymätaso kasvainsoluissa oli ≥ 50 %, sekä kokonaiselossaoloa ITT-potilasjoukossa.
Tutkimuksessa saavutettiin tautivapaan elossaoloajan välianalyysin ajankohtana ensisijainen päätetapahtuma. Analyysissa levinneisyysasteen II–IIIA sairautta sairastavista potilaista, joilla PD-L1-ilmentymä kasvainsoluissa oli ≥ 50 % ja joilla ei ollut EGFR-mutaatioita eikä ALK:n uudelleenjärjestäytymistä (n = 209), havaittiin tautivapaan elossaoloajan pidentyneen atetsolitsumabihaarassa verrattuna parasta tukihoitoa saaneeseen haaraan. Tulokset olivat yhdenmukaisia tautivapaan elossaoloajan loppuanalyysin ajankohtana, jolloin seuranta-ajan mediaani oli 65 kuukautta.
Yhteenveto tautivapaan elossaoloajan ja kokonaiselossaoloajan keskeisistä tehoa koskevista tuloksista levinneisyysasteen II–IIIA sairautta sairastavassa potilasjoukossa, jossa PD-L1-ilmentymä kasvainsoluissa oli ≥ 50 % ja jossa ei ollut EGFR-mutaatioita eikä ALK:n uudelleenjärjestäytymistä, esitetään taulukossa 7. Tautivapaan elossaoloajan Kaplan–Meierin käyrä esitetään kuvassa 3.
Taulukko 7. Yhteenveto tehosta levinneisyysasteen II–IIIA sairautta sairastavassa potilasjoukossa, jossa PD-L1-ilmentymä kasvainsoluissa on ≥ 50 % ja jossa ei ole EGFR-mutaatioita eikä ALK:n uudelleenjärjestäytymistä (IMpower010)
| Tehon päätetapahtuma | Haara A (atetsolitsumabi) | Haara B (paras tukihoito) |
| Tutkijan arvioima tautivapaa elossaoloaika* | n = 106 | n = 103 |
| Tapahtumien lkm (%) | 34 (32,1 %) | 55 (53,4 %) |
| Tautivapaan elossaoloajan keston mediaani (kuukautta) | NE | 42,9 |
| 95 %:n luottamusväli | (NE) | (32,0, NE) |
| Ositettuǂ riskisuhde (95 %:n luottamusväli) | 0,52 (0,33, 0,80) | |
| Kokonaiselossaoloaika* | n = 106 | n = 103 |
| Tapahtumien lkm (%) | 22 (20,8 %) | 41 (39,8 %) |
| Kokonaiselossaoloajan mediaani (kuukautta) | NE | 87,1 |
| 95 %:n luottamusväli | (NE) | (72,0, NE) |
| Ositettuǂ riskisuhde (95 %:n luottamusväli) | 0,47 (0,28, 0,80) | |
NE = ei arvioitavissa (not estimable)
* Päivitetty tautivapaan elossaoloajan ja kokonaiselossaoloajan analyysi kliinisten tietojen keruun katkaisuajankohtana 26. tammikuuta 2024
ǂ Ositettu levinneisyysasteen, sukupuolen ja histologian mukaan.
Kuva 3: Tautivapaan elossaoloajan Kaplan–Meierin käyrä levinneisyysasteen II–IIIA sairautta sairastavassa potilasjoukossa, jossa PD-L1-ilmentymä kasvainsoluissa on ≥ 50 % ja jossa ei ole EGFR-mutaatioita eikä ALK:n uudelleenjärjestäytymistä (IMpower010)
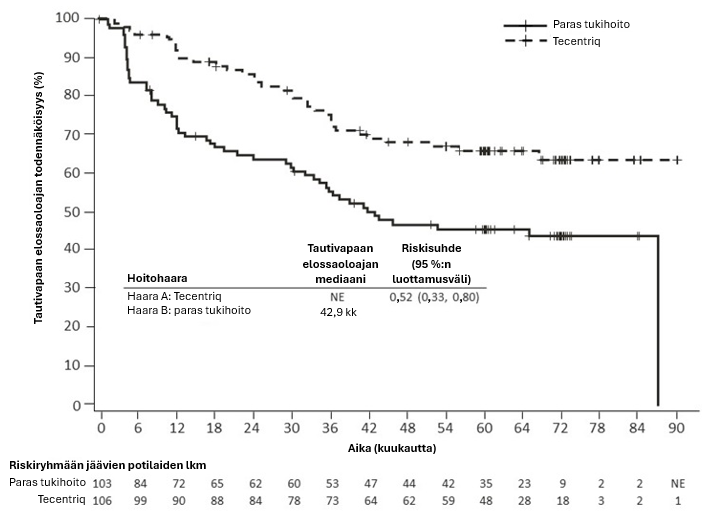
Tautivapaan elossaoloajan havaittu piteneminen atetsolitsumabihaarassa verrattuna parasta tukihoitoa saaneeseen haaraan osoitettiin yhdenmukaisesti suurimmassa osassa levinneisyysasteen II–IIIA sairautta sairastavassa potilasjoukossa, jossa PD-L1-ilmentymä kasvainsoluissa oli ≥ 50 % ja jossa ei ollut EGFR-mutaatioita eikä ALK:n uudelleenjärjestäytymistä, ennalta määritetyissä alaryhmissä, mukaan lukien sekä ei-levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastavat potilaat (osittamaton riskisuhde [HR] 0,40, 95 %:n luottamusväli: 0,23, 0,70; tautivapaan elossaoloajan mediaani NE vs. 36,8 kuukautta) ja levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastavat potilaat (osittamaton riskisuhde [HR] 0,67, 95 %:n luottamusväli: 0,34, 1,32; tautivapaan elossaoloajan ajan mediaania ei voitu arvioida).
Pitkälle edenneen ei-pienisoluisen keuhkosyövän ensilinjan hoito
IMpower150 (GO29436): Satunnaistettu vaiheen III tutkimus metastasoitunutta ei-levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastaneilla potilailla, jotka eivät olleet saaneet aiempaa solunsalpaajahoitoa; yhdistelmähoito paklitakselin ja karboplatiinin kanssa, joko bevasitsumabin kanssa tai ilman sitä
Kansainvälisessä, avoimessa, satunnaistetussa vaiheen III IMpower150-monikeskustutkimuksessa arvioitiin atetsolitsumabin, paklitakselin ja karboplatiinin yhdistelmähoidon tehoa ja turvallisuutta metastasoitunutta ei-levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastaneilla potilailla, jotka eivät olleet saaneet aiempaa solunsalpaajahoitoa ja joista osa sai bevasitsumabia, osa ei.
Tutkimukseen ei otettu mukaan potilaita, joilla oli aiemmin ollut jokin autoimmuunisairaus; potilaita, jotka olivat saaneet elävää, heikennettyä taudinaiheuttajaa sisältänyttä rokotetta satunnaistamista edeltäneiden 28 päivän aikana; potilaita, jotka olivat saaneet systeemisiä immuniteettia stimuloivia aineita satunnaistamista edeltäneiden 4 viikon aikana tai systeemisiä immunosuppressiivisia lääkevalmisteita satunnaistamista edeltäneiden 2 viikon aikana; potilaita, joilla oli aktiivisia tai hoitamattomia keskushermoston metastaaseja tai joilla näkyi kuvantamisessa selvä kasvaimen infiltraatio rintakehän suuriin verisuoniin tai selvä keuhkoleesioiden kavitaatio. Kasvain arvioitiin ensimmäisen hoitosyklin päivää 1 seuranneiden 48 viikon aikana 6 viikon välein ja tämän jälkeen 9 viikon välein. Kasvainnäytteistä tutkittiin PD-L1:n ilmentyminen kasvainsoluissa (TC) ja kasvainta infiltroivissa immuunisoluissa (IC), ja tulosten perusteella määriteltiin PD-L1:n ilmentymistä koskevat osajoukot jäljempänä kuvattuihin analyyseihin.
Tutkimukseen otettiin yhteensä 1 202 potilasta, jotka satunnaistettiin (1:1:1) saamaan yhtä taulukossa 8 kuvatuista hoito-ohjelmista. Satunnaistaminen ositettiin seuraavien mukaan: sukupuoli, maksametastaasien olemassaolo ja PDL1:n ilmentyminen kasvainsoluissa ja immuunisoluissa.
Taulukko 8. Laskimoon annettavat hoito-ohjelmat (IMpower150)
| Hoito-ohjelma | Induktio (Neljä tai kuusi 21 päivän sykliä) | Ylläpitohoito (21 päivän syklit) |
| A | Atetsolitsumabia (1200 mg) + paklitakseli (200 mg/m2)b,c + karboplatiinic (AUC 6) | Atetsolitsumabia (1200 mg) |
| B | Atetsolitsumabia (1200 mg) + bevasitsumabid (15 mg/kg) + paklitakseli (200 mg/m2)b,c + karboplatiinic (AUC 6) | Atetsolitsumabia (1200 mg) + bevasitsumabid (15 mg/kg) |
| C | Bevasitsumabid (15 mg/kg) + paklitakseli (200 mg/m2)b,c + karboplatiinic (AUC 6) | Bevasitsumabid (15 mg/kg) |
a Atetsolitsumabia annetaan, kunnes kliinistä hyötyä ei enää tutkijan arvion mukaan todettu.
b Paklitakselin aloitusannos aasialaista syntyperää oleville potilaille oli 175 mg/m2, koska hematologinen toksisuus on kaiken kaikkiaan yleisempää syntyperältään/etniseltä taustaltaan Aasian maista olevilla potilailla verrattuna muunlaista syntyperää oleviin potilaisiin.
c Paklitakselia ja karboplatiinia annettiin, kunnes jokin seuraavista ilmeni ensimmäisen kerran: potilas sai 4 tai 6 hoitosykliä, sairaus eteni tai potilaalla ilmeni toksisuutta, joka ei ollut hyväksyttävissä.
d. Bevasitsumabia annetaan, kunnes sairaus etenee tai potilaalla ilmenee toksisuutta, joka ei ole hyväksyttävissä.
Tutkimusjoukon demografiset ominaisuudet ja sairauden ominaisuudet lähtötilanteessa olivat hyvin tasapainossa hoitohaarojen välillä. Iän mediaani oli 63 vuotta (vaihteluväli: 31–90), ja 60 % potilaista oli miehiä. Valtaosa potilaista oli valkoihoisia (82 %). Noin 10 %:lla potilaista oli tunnettu EGFR-mutaatio, 4 %:lla oli tunnettua ALK-geenin uudelleenjärjestäytymistä, 14 %:lla oli maksametastaaseja lähtötilanteessa, ja useimmat potilaat olivat joko nykyisiä tai entisiä tupakoitsijoita (80 %). Lähtötilanteen ECOG-suorituskykyluokka oli 0 (43 %) tai 1 (57 %). 51 %:ssa potilaiden kasvaimista PD-L1:n ilmentymisen taso kasvainsoluissa ja immuunisoluissa oli ≥ 1 %, ja 49 %:ssa potilaiden kasvaimista PD-L1:n ilmentymisen taso kasvainsoluissa ja immuunisoluissa oli < 1 %.
Taudin etenemättömyysajan loppuanalyysin hetkellä potilaiden seuranta-ajan mediaani oli 15,3 kuukautta. ITT-potilasjoukossa, myös potilailla, joilla oli EGFR-mutaatioita tai ALK-geenin uudelleenjärjestäytymistä ja joiden olisi pitänyt saada aiempaa hoitoa tyrosiinikinaasin estäjillä, taudin etenemättömyysajan osoitettiin paranevan kliinisesti oleellisesti hoitohaarassa B verrattuna hoitohaaraan C (riskisuhde: 0,61, 95 %:n luottamusväli: 0,52, 0,72; taudin etenemättömyysajan mediaani 8,3 vs. 6,8 kuukautta).
Kokonaiselossaolon välianalyysin hetkellä potilaiden seuranta-ajan mediaani oli 19,7 kuukautta. Tämän analyysin päätulosten sekä ITT-potilasjoukon taudin etenemättömyysajan päivitetyn analyysin yhteenveto on esitetty taulukoissa 9 ja 10. ITT-potilasjoukon kokonaiselossaoloajan Kaplan–Meier-käyrä on esitetty kuvassa 4. Kuvassa 5 on yhteenveto ITT- ja PD-L1-alaryhmien kokonaiselossaoloajan tuloksista. Taudin etenemättömyysaikaa koskevat päivitetyt tulokset on esitetty myös kuvissa 6 ja 7.
Taulukko 9. Päivitetty yhteenveto hoidon tehosta ITT-potilasjoukossa (IMpower150)
| Hoidon tehoa koskeva päätetapahtuma | Hoitohaara A (atetsolitsumabi + paklitakseli + karboplatiini) | Hoitohaara B (atetsolitsumabi + bevasitsumabi + paklitakseli + karboplatiini) | Hoitohaara C (bevasitsumabi + paklitakseli + karboplatiini) |
| Toissijaiset päätetapahtumat# | |||
| Taudin etenemättömyysaika (tutkijan arvio; RECIST v1.1)* | n = 402 | n = 400 | n = 400 |
| Tapahtumien lkm (%) | 330 (82,1 %) | 291 (72,8 %) | 355 (88,8 %) |
| Etenemättömyysajan keston mediaani (kk) | 6,7 | 8,4 | 6,8 |
| 95 %:n luottamusväli | (5,7, 6,9) | (8,0, 9,9) | (6,0, 7,0) |
Ositettu riskisuhde‡ ^ (95 %:n luottamusväli) p-arvo1,2 | 0,91 (0,78, 1,06) 0,2194 | 0,59 (0,50, 0,69) < 0,0001 | --- |
| 12 kk:n etenemättömyysaika (%) | 24 | 38 | 20 |
| Kokonaiselossaolon välianalyysi* | n = 402 | n = 400 | n = 400 |
Kuolemien lkm (%) Ajan mediaani tapahtumiin (kk) 95 %:n luottamusväli | 206 (51,2 %) 19,5 (16,3, 21,3) | 192 (48,0 %) 19,8 (17,4, 24,2) | 230 (57,5 %) 14,9 (13,4, 17,1) |
Ositettu riskisuhde‡^ (95 %:n luottamusväli) p-arvo1,2 | 0,85 (0,71, 1,03) 0,0983 | 0,76 (0,63, 0,93) 0,006 | --- |
| 6 kk:n kokonaiselossaoloaika (%) | 84 | 85 | 81 |
| 12 kk:n kokonaiselossaoloaika (%) | 66 | 68 | 61 |
| Paras kokonaisvaste3* (tutkijan arvio; RECIST 1.1) | n = 401 | n = 397 | n = 393 |
| Vasteen saaneiden lkm (%) | 163 (40,6 %) | 224 (56,4 %) | 158 (40,2 %) |
| 95 %:n luottamusväli | (35,8, 45,6) | (51,4, 61,4) | (35,3, 45,2) |
| Täydellisen vasteen saaneiden lkm (%) | 8 (2,0 %) | 11 (2,8 %) | 3 (0,8 %) |
| Osittaisen vasteen saaneiden lkm (%) | 155 (38,7 %) | 213 (53,7 %) | 155 (39,4 %) |
| Vasteen kesto* (tutkijan arvio; RECIST v1.1) | n = 163 | n = 224 | n = 158 |
| Mediaani (kk) | 8,3 | 11,5 | 6,0 |
| 95 %:n luottamusväli | (7,1, 11,8) | (8,9, 15,7) | (5,5, 6,9) |
# Ensisijaiset tehon päätetapahtumat olivat taudin etenemättömyysaika ja kokonaiselossaolo. Nämä analysoitiin hoitoaikeen mukaisesta (ITT) potilasjoukosta, jossa potilailla oli villityypin kasvain, eli mukana ei ole potilaita, joilla oli EGFR-mutaatioita tai ALK-geenin uudelleenjärjestäytymistä.
1 Perustuu ositettuun log-rank-testiin
2 Tiedoksi: ITT-potilasjoukossa ei ole vielä tehty virallisia vertailuja hoitohaarojen B ja C välillä eikä hoitohaarojen A ja C välillä ennalta määritellyn analyysihierarkian mukaan.
3 Paras kokonaisvaste täydellisen vasteen ja osittaisen vasteen suhteen
‡ Ositettu seuraavien mukaan: sukupuoli, maksametastaasien olemassaolo ja PDL1:n ilmentyminen kasvainsoluissa ja immuunisoluissa.
^ Hoitohaara C on kaikkien riskisuhteiden vertailuryhmä.
* Etenemättömyysajan päivitetty analyysi ja kokonaiselossaolon välianalyysi kliinisten tietojen keruun katkaisuajankohtana 22. tammikuuta 2018.
RECIST = Response Evaluation Criteria in Solid Tumours v1.1.
Taulukko 10. Yhteenveto hoidon tehon päivitetystä vertailusta ITT-potilasjoukossa, hoitohaara A vs. hoitohaara B (IMpower150)
| Hoidon tehoa koskeva päätetapahtuma | Hoitohaara A (atetsolitsumabi + paklitakseli + karboplatiini) | Hoitohaara B (atetsolitsumabi + bevasitsumabi + paklitakseli + karboplatiini) |
| Taudin etenemättömyysaika (tutkijan arvio; RECIST v1.1)* | n = 402 | n = 400 |
| Tapahtumien lkm (%) | 330 (82,1 %) | 291 (72,8 %) |
| Etenemättömyysajan keston mediaani (kk) | 6,7 | 8,4 |
| 95 %:n luottamusväli | (5,7, 6,9) | (8,0, 9,9) |
Ositettu riskisuhde‡^ (95 %:n luottamusväli) p-arvo1,2 | 0,67 (0,57, 0,79) < 0,0001 | |
| Kokonaiselossaolon välianalyysi* | n = 402 | n = 400 |
Kuolemien lkm (%) Ajan mediaani tapahtumiin (kk) 95 %:n luottamusväli | 206 (51,2 %) 19,5 (16,3, 21,3) | 192 (48,0 %) 19,8 (17,4, 24,2) |
Ositettu riskisuhde‡^ (95 %:n luottamusväli) p-arvo1,2 | 0,90 (0,74, 1,10) 0,3000 | |
1Perustuu ositettuun log-rank-testiin.
2Tiedoksi: ITT-potilasjoukon osalta vertailut hoitohaaran A ja hoitohaaran B välillä eivät olleet mukana ennalta määritellyssä analyysihierarkiassa.
‡ Ositettu seuraavien mukaan: sukupuoli, maksametastaasien olemassaolo ja PD-L1:n ilmentyminen kasvainsoluissa ja immuunisoluissa.
* Etenemättömyysajan päivitetty analyysi ja kokonaiselossaolon välianalyysi kliinisten tietojen keruun katkaisuajankohtana 22. tammikuuta 2018.
^ Hoitohaara A on kaikkien riskisuhteiden vertailuryhmä.
Kuva 4. ITT-potilasjoukon kokonaiselossaolon Kaplan-Meier-käyrä (IMpower150)
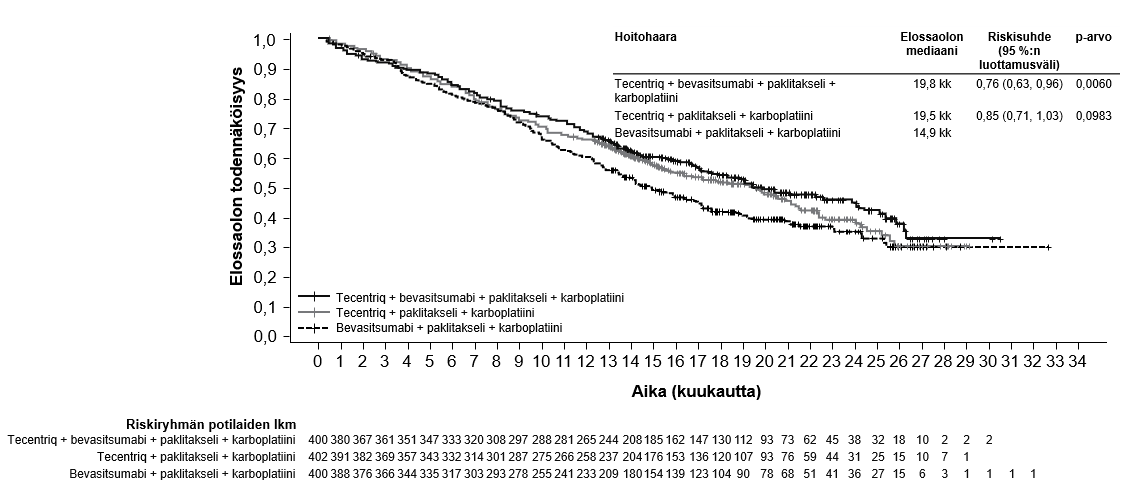
Kuva 5. ITT-potilasjoukon kokonaiselossaolon alaryhmäanalyysi (forest plot) PD-L1:n ilmentymistason mukaan, hoitohaara B vs. hoitohaara C (IMpower150)
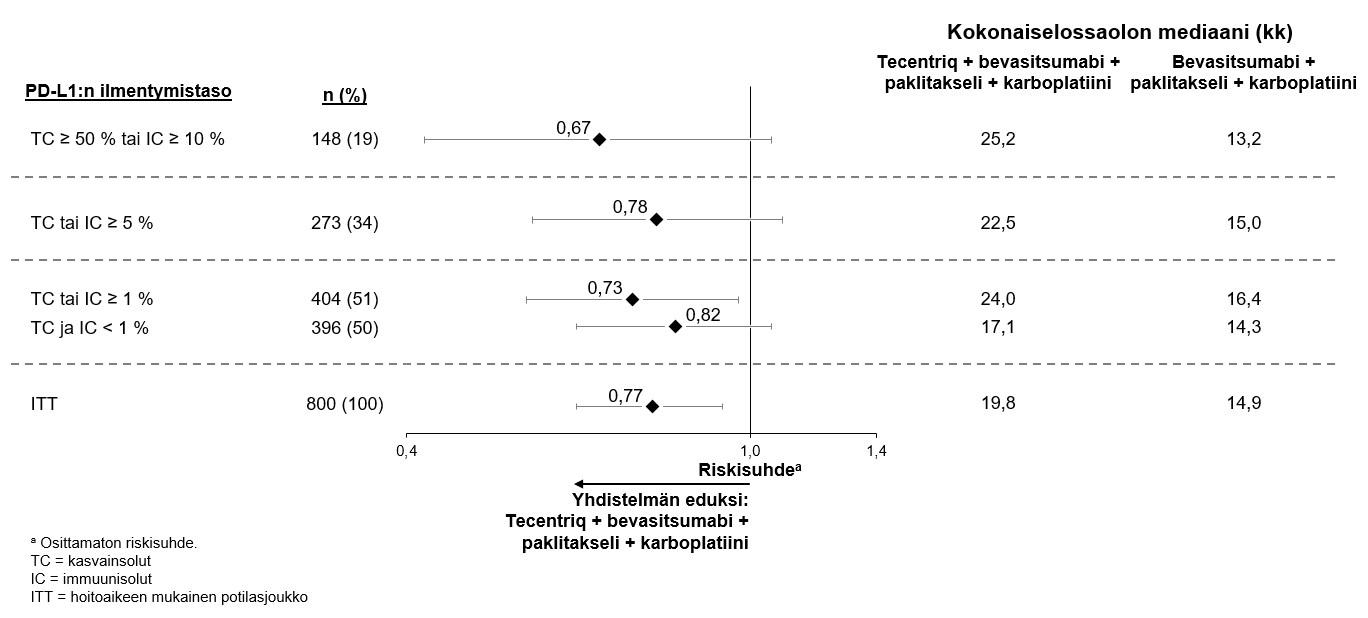
Kuva 6. Taudin etenemättömyysajan Kaplan-Meier-käyrä ITT-potilasjoukossa (IMpower150)
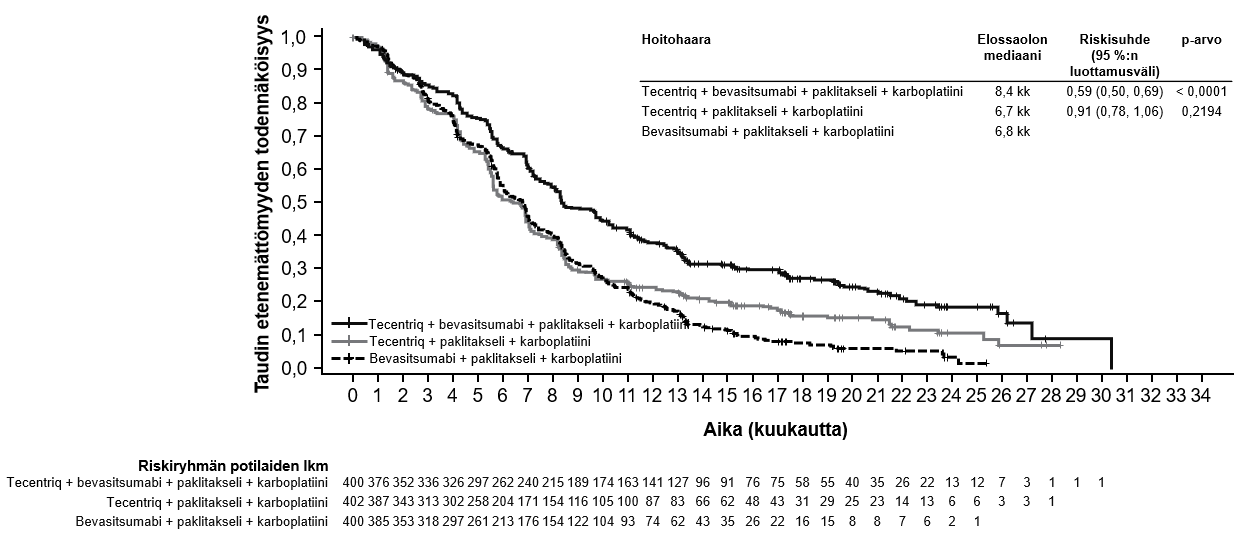
Kuva 7. Taudin etenemättömyysajan alaryhmäanalyysi (forest plot) PD-L1:n ilmentymistason mukaan ITT-potilasjoukossa, hoitohaara B vs. hoitohaara C (IMpower150)
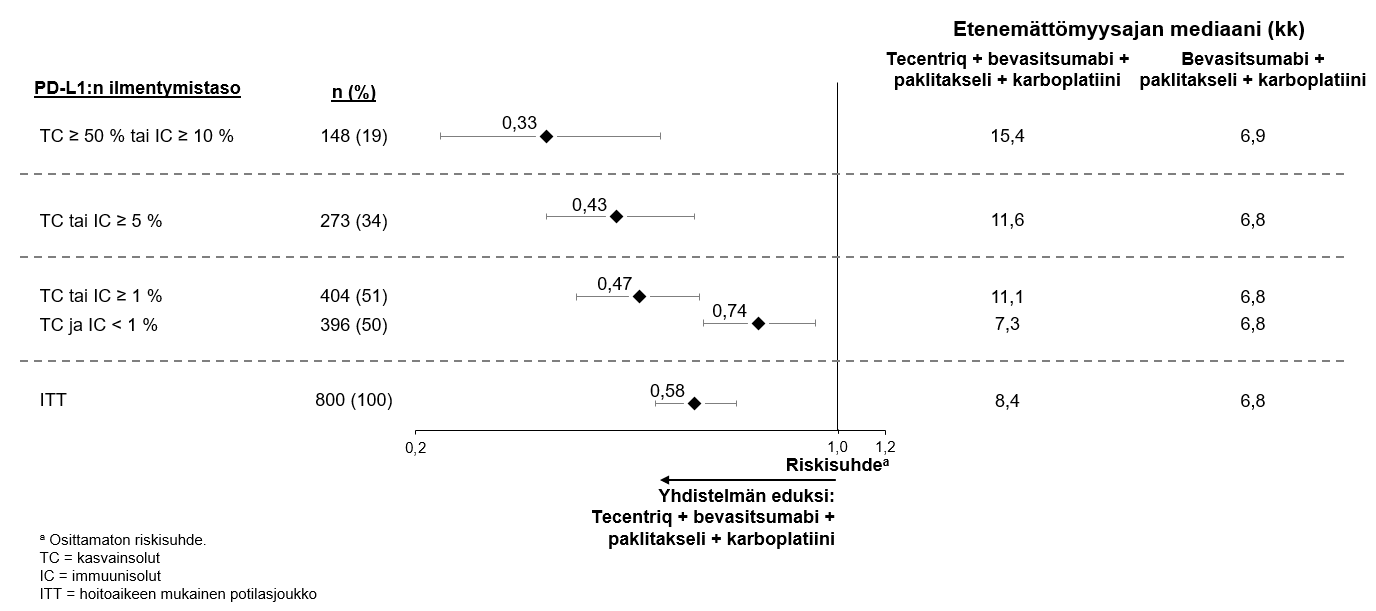
Kokonaiselossaolon välianalyysin ennalta määritellyt alaryhmäanalyysit osoittivat kokonaiselossaolon paranemista hoitohaarassa B hoitohaaraan C verrattuna potilailla, joilla oli EGFR-mutaatioita tai ALK-geenin uudelleenjärjestäytymistä (riskisuhde: 0,54, 95 %:n luottamusväli: 0,29, 1,03; kokonaiselossaolon mediaani NE vs. 17,5 kuukautta) ja maksametastaaseja (riskisuhde: 0,52 [95 %:n luottamusväli: 0,33, 0,82], kokonaiselossaolon mediaani 13,3 vs. 9,4 kuukautta). Myös taudin etenemättömyysajan paranemista osoitettiin potilailla, joilla oli EGFR-mutaatioita tai ALK-geenin uudelleenjärjestäytymistä (riskisuhde: 0,55 [95 %:n luottamusväli: 0,35, 0,87], taudin etenemättömyysajan mediaani 10,0 vs. 6,1 kuukautta) ja maksametastaaseja (riskisuhde: 0,41 [95 %:n luottamusväli: 0,26, 0,62], taudin etenemättömyysajan mediaani 8,2 vs. 5,4 kuukautta). Iältään < 65-vuotiaiden potilaiden alaryhmän kokonaiselossaolon tulokset olivat samankaltaiset iältään ≥ 65-vuotiaiden potilaiden alaryhmän kanssa. Iältään ≥ 75-vuotiaiden potilaiden tiedot ovat liian suppeita, jotta tästä potilasjoukosta voisi tehdä päätelmiä. Kaikkien alaryhmäanalyysien varsinaista tilastollista testausta ei suunniteltu.
IMpower130 (GO29537): Satunnaistettu vaiheen III tutkimus metastasoitunutta ei-levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastaneilla potilailla, jotka eivät olleet saaneet aiempaa solunsalpaajahoitoa; yhdistelmähoito nab-paklitakselin ja karboplatiinin kanssa
Vaiheen III avoimessa, satunnaistetussa tutkimuksessa GO29537 (IMpower130) arvioitiin atetsolitsumabin, nab-paklitakselin ja karboplatiinin yhdistelmähoidon tehoa ja turvallisuutta metastasoitunutta ei-levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastaneilla potilailla, jotka eivät olleet saaneet aiempaa solunsalpaajahoitoa. Potilaiden, joilla oli EGFR-mutaatioita tai ALK:n uudelleenjärjestäytymistä, tuli olla aiemmin tyrosiinikinaasin estäjillä hoidettuja.
Potilaiden syövän levinneisyys määriteltiin AJCC-luokituksen (American Joint Committee on Cancer, 7. painos) mukaan. Potilaita ei otettu tutkimukseen mukaan, jos heillä oli autoimmuunisairaustausta, he olivat saaneet heikennettyjä eläviä taudinaiheuttajia sisältävää rokotetta satunnaistamista edeltäneiden 28 päivän aikana, immuniteettia stimuloivia lääkeaineita edeltäneiden 4 viikon aikana tai systeemisiä immunosuppressiivisia lääkevalmisteita satunnaistamista edeltäneiden 2 viikon aikana, tai jos heillä oli aktiivisia tai hoitamattomia keskushermoston metastaaseja. Potilaat, jotka olivat saaneet aiempaa hoitoa CD137:n agonisteilla tai immuunivasteen tarkistuspisteen estäjillä (PD-1-vasta-aineet ja PD-L1-vasta-aineet), eivät soveltuneet tutkimukseen. Potilaat, jotka olivat aiemmin saaneet hoitoa CTLA-4-vasta-aineilla, voitiin kuitenkin ottaa tutkimukseen mukaan, kunhan potilas oli saanut viimeisen annoksen vähintään 6 viikkoa ennen satunnaistamista eikä potilaalle ollut aiemmin aiheutunut CTLA-4-vasta-aineista vaikea-asteisia immuunivälitteisiä haittatapahtumia (NCI CTCAE luokat 3 ja 4). Kasvain arvioitiin ensimmäisen hoitosyklin jälkeen 6 viikon välein ensimmäisten 48 viikon ajan, ja tämän jälkeen 9 viikon välein. Kasvaimista otetuista näytteistä tutkittiin PDL1:n ilmentyminen kasvainsoluissa ja kasvaimeen infiltroivissa immuunisoluissa. Tästä saatuja tuloksia käytettiin alaryhmien PDL1:n ilmentymistason määrittämiseen jäljempänä kuvattuja analyysejä varten.
Tutkimukseen mukaan otetut potilaat, mukaan lukien ne, joilla oli EGFR-mutaatioita tai ALK:n uudelleenjärjestäytymistä, satunnaistettiin suhteessa 2:1 saamaan toista taulukossa 11 kuvatuista hoitovaihtoehdoista. Satunnaistaminen ositettiin sukupuolen ja maksametastaasien esiintymisen mukaan sekä PD-L1:n ilmentymisen perusteella kasvainsoluissa ja immuunisoluissa. Verrokkiryhmän potilaiden (hoito-ohjelma B) oli mahdollista saada atetsolitsumabimonoterapiahoitoa taudin etenemisen jälkeen.
Taulukko 11. Laskimoon annettavat hoito-ohjelmat (IMpower130)
| Hoito-ohjelma | Induktiohoito (neljä tai kuusi 21 päivän pituista hoitosykliä) | Ylläpitohoito (21 päivän pituisia hoitosyklejä) |
| A | Atetsolitsumabi (1200 mg)a + nab-paklitakseli (100 mg/m2)b,c + karboplatiini (AUC 6)c | Atetsolitsumabi (1200 mg)a |
| B | Nab-paklitakseli (100 mg/m2)b,c + karboplatiini (AUC 6)c | Paras tukihoito tai pemetreksedi |
aAtetsolitsumabia annetaan, kunnes kliininen hyöty tutkijan arvion mukaan loppuu
b Nab-paklitakselia annetaan jokaisen hoitosyklin päivinä 1, 8 ja 15
c Nab-paklitakselia ja karboplatiinia annetaan siihen saakka, kunnes jokin seuraavista toteutuu: 4–6 hoitosykliä annettu kokonaan, tauti etenee tai ilmaantuu toksisuutta, joka ei ole hyväksyttävissä
Tutkimuspotilasjoukon (ITT-WT, n = 679) demografiset tiedot ja taudin ominaisuudet lähtötilanteessa olivat hyvin tasapainossa hoitohaarojen välillä. Iän mediaani oli 64 vuotta (vaihteluväli 18–86 vuotta). Valtaosa potilaista oli miehiä (59 %) ja valkoihoisia (90 %). 14,7 %:lla potilaista oli lähtötilanteessa maksametastaaseja, ja suurin osa potilaista oli parhaillaan tai oli aiemmin ollut tupakoijia (90 %). Valtaosalla potilaista lähtötilanteen ECOG-suorituskykyluokka oli 1 (59 %) ja PD-L1:n ilmentymistaso oli < 1 % (noin 52 %:lla). Niistä 107:stä B-haaran potilaasta, joilla induktiohoidon jälkeinen vaste oli stabiili tauti, osittainen vaste tai täydellinen vaste, 40 sai ylläpitohoitoon siirtymisen jälkeen pemetreksediä.
Kaikista potilaista, lukuun ottamatta niitä, joilla oli EGFR-mutaatioita tai ALK:n uudelleenjärjestäytymistä (määriteltiin hoitoaikeen mukaiseksi potilasjoukoksi, jossa potilailla oli villityypin kasvain [ITT-WT], n = 679), tehtiin primaarianalyysi. Potilaiden elossaoloajan seuranta-ajan mediaani oli 18,6 kuukautta, ja kokonaiselossaolon ja etenemättömyysajan todettiin parantuneen atetsolitsumabin, nab-paklitakselin ja karboplatiinin yhdistelmähoitoa saaneilla verrokkeihin verrattuna. Yhteenveto keskeisistä tuloksista on taulukossa 12. Kokonaiselossaolon Kaplan–Meier-käyrät ovat kuvassa 8, ja etenemättömyysajan Kaplan–Meier-käyrät ovat kuvassa 10. Yhteenvedot kokonaiselossaolon eksploratiivisista tuloksista PD-L1:n ilmentymätason mukaan ovat kuvassa 9 ja etenemättömyysajan eksploratiivisista tuloksista PD-L1:n ilmentymätason mukaan ovat kuvassa 11. Potilailla, joilla oli maksametastaaseja, etenemättömyysajan tai kokonaiselossaolon ei todettu parantuneen atetsolitsumabin, nab-paklitakselin ja karboplatiinin yhdistelmähoidossa verrattuna nab-paklitakselin ja karboplatiinin yhdistelmähoitoon (etenemättömyysajan riskisuhde 0,93, 95 %:n luottamusväli: 0,59, 1,47; kokonaiselossaolon riskisuhde 1,04, 95 %:n luottamusväli: 0,63, 1,72).
Nab-paklitakselia ja karboplatiinia saaneen verrokkihaaran potilaista 59 % sai taudin etenemisen jälkeen syövän hoitoon käytettävää immunoterapiaa, mukaan lukien atetsolitsumabia (41 % kaikista potilaista), verrattuna 7,3 %:iin potilaista atetsolitsumabin, nab-paklitakselin ja karboplatiinin yhdistelmää saaneessa haarassa.
Eksploratiivisessa analyysissa, jossa seuranta-aika oli pidempi (mediaani: 24,1 kuukautta), kummankin hoitohaaran kokonaiselossaolon mediaani oli muuttumaton primaarianalyysiin nähden, jolloin riskisuhde = 0,82 (95 %:n luottamusväli: 0,67; 1,01).
Taulukko 12. Yhteenveto hoidon tehosta primaarianalyysin potilasjoukossa (ITT-WT) (IMpower130)
| Hoidon tehoa koskevat päätetapahtumat | Hoitohaara A Atetsolitsumabi + nab-paklitakseli + karboplatiini | Hoitohaara B Nab-paklitakseli + karboplatiini |
| Muut ensisijaiset päätetapahtumat | ||
| Kokonaiselossaolo | n = 451 | n = 228 |
| Kuolemien lkm (%) | 226 (50,1 %) | 131 (57,5 %) |
| Ajan mediaani tapahtumiin (kk) | 18,6 | 13,9 |
| 95 %:n luottamusväli | (16,0, 21,2) | (12,0, 18,7) |
| Ositettu riskisuhde‡ (95 %:n luottamusväli) | 0,79 (0,64, 0,98) | |
| p-arvo | 0,033 | |
| 12 kk:n kokonaiselossaoloaika (%) | 63 | 56 |
| Taudin etenemättömyysaika (tutkijan arvio; RECIST v1.1) | n = 451 | n = 228 |
| Tapahtumien lkm (%) | 347 (76,9 %) | 198 (86,8 %) |
| Etenemättömyysajan keston mediaani (kk) | 7,0 | 5,5 |
| 95 %:n luottamusväli | (6,2, 7,3) | (4,4, 5,9) |
| Ositettu riskisuhde‡ (95 %:n luottamusväli) | 0,64 (0,54, 0,77) | |
| p-arvo | < 0,0001 | |
| 12 kk:n etenemättömyysaika (%) | 29 % | 14 % |
| Muut päätetapahtumat | ||
| Kokonaisvasteosuus (tutkijan arvio; RECIST v1.1)^ | n = 447 | n = 226 |
| Varmistetun vasteen saaneiden lkm (%) | 220 (49,2 %) | 72 (31,9 %) |
| 95 %:n luottamusväli | (44,5, 54,0) | (25,8, 38,4) |
| Täydellisen vasteen saaneiden lkm (%) | 11 (2,5 %) | 3 (1,3 %) |
| Osittaisen vasteen saaneiden lkm (%) | 209 (46,8 %) | 69 (30,5 %) |
| Vahvistettu vasteen kesto (tutkijan arvio; RECIST 1.1)^ | n = 220 | n = 72 |
| Mediaani (kk) | 8,4 | 6,1 |
| 95 %:n luottamusväli | (6,9, 11,8) | (5,5, 7,9) |
‡ Ositus perustuu sukupuoleen sekä PDL1:n ilmentymiseen kasvainsoluissa ja immuunisoluissa
^ Vahvistettu kokonaisvasteosuus ja vasteen kesto ovat eksploratiivisia päätetapahtumia
RECIST = Response Evaluation Criteria in Solid Tumours v1.1
Kuva 8. Kokonaiselossaolon Kaplan–Meier-kuvaaja (IMpower130)
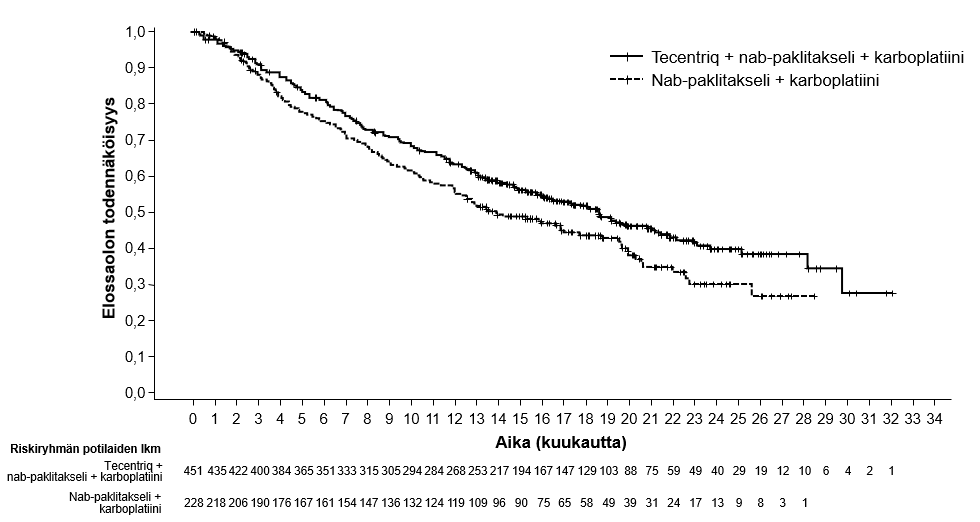
Kuva 9. Kokonaiselossaoloajan Forest plot alaryhmäanalyysi kasvaimen PDL1:n ilmentymistason mukaan (Impower130)
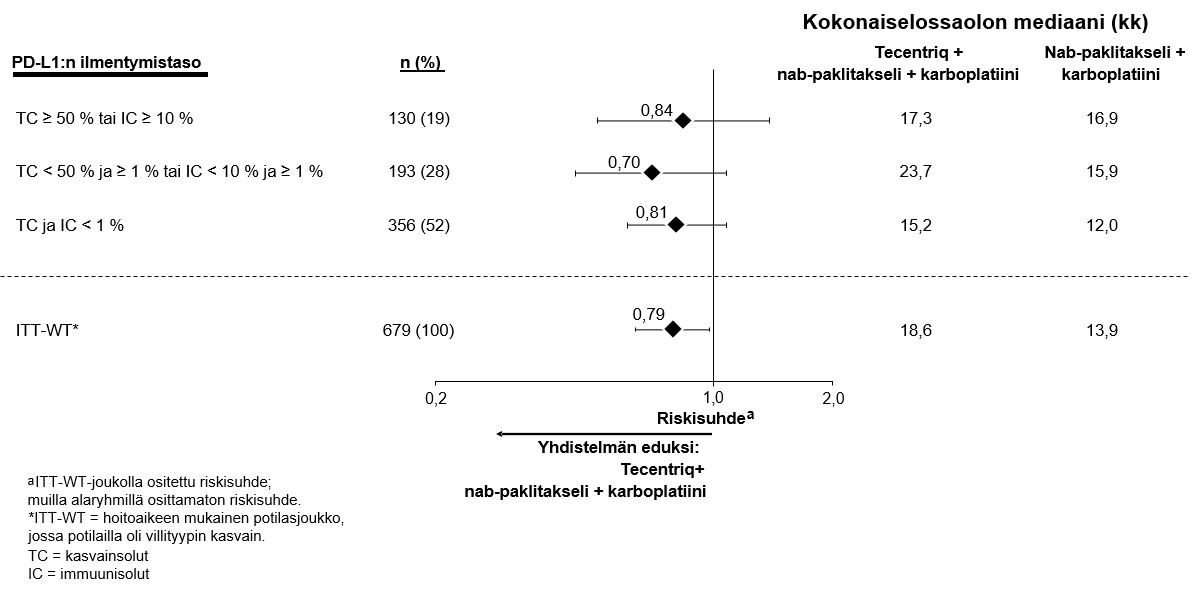
Kuva 10. Taudin etenemättömyysajan Kaplan–Meier-kuvaaja (Impower130)
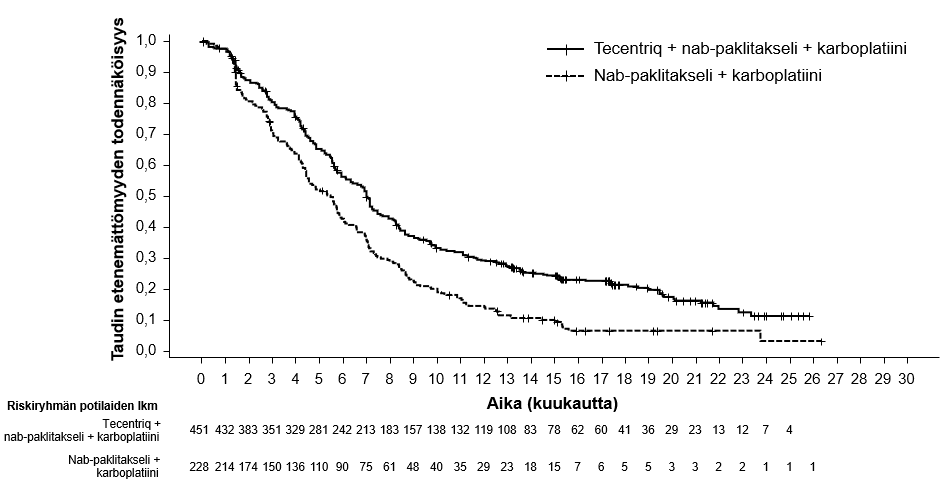
Kuva 11. Taudin etenemättömyysajan Forest plot alaryhmäanalyysi kasvaimen PDL1:n ilmentymistason mukaan (Impower130)
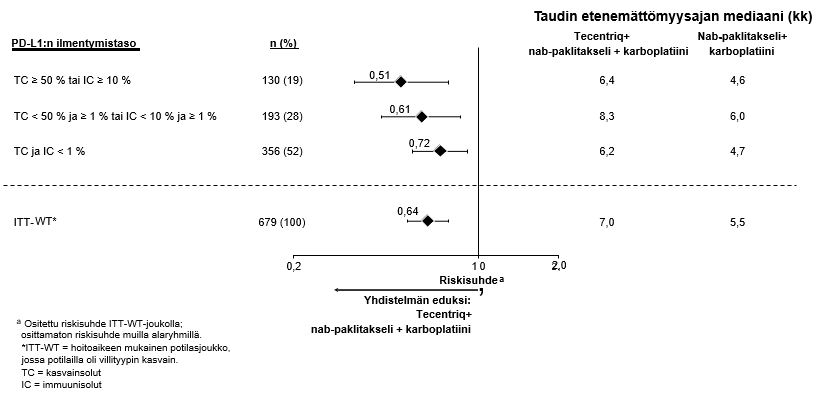
Impower110 (GO29431): Satunnaistettu vaiheen III tutkimus metastasoitunutta ei-pienisoluista keuhkosyöpää sairastaneilla potilailla, jotka eivät olleet saaneet aiempaa solunsalpaajahoitoa
Avoimessa, satunnaistetussa, vaiheen III Impower110-monikeskustutkimuksessa arvioitiin atetsolitsumabin tehoa ja turvallisuutta metastasoitunutta ei-pienisoluista keuhkosyöpää sairastavilla potilailla, jotka eivät olleet saaneet aiempaa solunsalpaajahoitoa. PD-L1:n ilmentymisen taso oli kasvainsoluissa ≥ 1 % (PD-L1-värjäytyviä ≥ 1 % kasvainsoluista) tai immuunisoluissa ≥ 1 % (PD-L1-värjäytyviä kasvaimeen infiltroivia immuunisoluja, jotka kattoivat ≥ 1 %:n kasvaimen pinta-alasta), mikä todettiin VENTANA PD-L1 (SP142) -määrityksellä.
Yhteensä 572 potilasta satunnaistettiin suhteessa 1:1 saamaan atetsolitsumabia (hoitohaara A) tai solunsalpaajahoitoa (hoitohaara B). Atetsolitsumabia annettiin vakioannoksina 1200 mg infuusiona laskimoon 3 viikon välein, kunnes kliinistä hyötyä ei tutkijan arvion mukaan enää todettu tai kunnes ilmeni toksisuutta, joka ei ollut hyväksyttävissä. Solunsalpaajahoito-ohjelmat kuvataan taulukossa 13. Satunnaistaminen ositettiin sukupuolen, ECOG-suorituskyvyn, histologian ja kasvaimen PD-L1:n ilmentymisen mukaan kasvainsoluissa ja immuunisoluissa.
Taulukko 13. Laskimoon annettavat solunsalpaajahoito-ohjelmat (Impower110)
| Hoito-ohjelma | Induktiohoito (Neljä tai kuusi 21 päivän sykliä) | Ylläpitohoito (21 päivän syklit) |
| B (ei-levyepiteeli-peräinen) | Sisplatiinia (75 mg/m²) + pemetreksedia (500 mg/m²) TAI karboplatiinia (AUC 6) + pemetreksedia (500 mg/m²) | Pemetreksedib,d (500 mg/m²) |
| B (levyepiteeli-peräinen) | Sisplatiinia (75 mg/m²) + gemsitabiinia,c (1250 mg/m2) TAI karboplatiinia (AUC 5) + gemsitabiinia,c (1000 mg/m2) | Paras tukihoitod |
a Sisplatiinia, karboplatiinia, pemetreksediä ja gemsitabiinia annetaan 4 tai 6 syklin loppuun saakka tai kunnes tauti etenee tai ilmaantuu toksisuutta, joka ei ole hyväksyttävissä.
b Pemetreksediä annetaan ylläpitohoitona 21 päivän välein, kunnes tauti etenee tai ilmaantuu toksisuutta, joka ei ole hyväksyttävissä.
c Gemsitabiinia annetaan kunkin syklin päivinä 1 ja 8.
d Vertailuryhmästä (platinapohjainen solunsalpaajahoito) ei ollut sallittua siirtyä atetsolitsumabihaaraan (hoitohaara A)
Tutkimukseen ei otettu mukaan potilaita, joilla oli aiemmin ollut jokin autoimmuunisairaus; potilaita, jotka olivat saaneet elävää heikennettyä taudinaiheuttajaa sisältänyttä rokotetta satunnaistamista edeltäneiden 28 päivän aikana; potilaita, jotka olivat saaneet systeemisiä immuniteettia stimuloivia lääkevalmisteita satunnaistamista edeltäneiden 4 viikon aikana tai systeemisiä immunosuppressiivisia lääkevalmisteita satunnaistamista edeltäneiden 2 viikon aikana; potilaita, joilla oli aktiivisia tai hoitamattomia metastaaseja keskushermostossa. Kasvain arvioitiin ensimmäisen hoitosyklin jälkeisten 48 viikon aikana 6 viikon välein ja tämän jälkeen 9 viikon välein.
Potilailla, joilla PD-L1:n ilmentymisen taso oli kasvainsoluissa ≥ 1 % tai immuunisoluissa ≥ 1 % ja joilla ei ollut EGFR-mutaatioita eikä ALK-geenin uudelleenjärjestäytymistä (n = 554), demografiset ja sairauden ominaisuudet lähtötilanteessa olivat hyvin tasapainossa hoitohaarojen välillä. Iän mediaani oli 64,5 vuotta (vaihteluväli: 30–87), ja 70 % potilaista oli miehiä. Suurin osa potilaista oli valkoihoisia (84 %) ja aasialaisia (14 %). Valtaosa potilaista oli parhaillaan tai oli aiemmin ollut tupakoijia (87 %) ja potilaiden lähtötilanteen ECOG-suorituskyky oli 0 (36 %) tai 1 (64 %). Kaikkiaan 69 %:lla potilaista oli ei-levyepiteeliperäinen sairaus ja 31 %:lla oli levyepiteeliperäinen sairaus. Potilailla, joilla PD-L1:n ilmentymä on suuri (PD-L1:n ilmentymisen taso kasvainsoluissa ≥ 50 % tai immuunisoluissa ≥ 10 %) ja joilla ei ole EGFR-mutaatioita eikä ALK-geenin uudelleenjärjestäytymistä (n = 205), demografiset ja sairauden lähtötilanteen ominaisuudet edustivat yleisesti hyvin laajempaa tutkimuspotilasjoukkoa ja olivat hyvin tasapainossa hoitohaarojen välillä.
Ensisijainen päätetapahtuma oli kokonaiselossaoloaika. Kokonaiselossaoloajan välianalyysin ajankohtana potilaista, joilla PD-L1:n ilmentymä oli suuri, pois lukien kuitenkin ne, joilla oli EGFR-mutaatioita tai ALK-geenin uudelleenjärjestäytymistä (n = 205), atetsolitsumabihoitoon satunnaistetuilla (hoitohaara A) osoitettiin tilastollisesti merkitsevä kokonaiselossaoloajan piteneminen solunsalpaajahoitoon (hoitohaara B) verrattuna (riskisuhde [HR] 0,59, 95 %:n luottamusväli: 0,40, 0,89; kokonaiselossaoloajan mediaani 20,2 kuukautta vs 13,1 kuukautta) ja kaksitahoinen p-arvo 0,0106. Potilailla, joilla PD-L1:n ilmentyminen oli suuri, elossaolon seuranta-ajan mediaani oli 15,7 kuukautta.
Kokonaiselossaoloajan eksploratiivisessa analyysissa, jossa näiden potilaiden seuranta-aika oli pidempi (mediaani: 31,3 kuukautta), kokonaiselossaoloajan mediaani oli atetsolitsumabihaarassa muuttumaton kokonaiselossaolon ensisijaiseen välianalyysiin nähden (20,2 kuukautta), ja solunsalpaajahoitohaarassa se oli 14,7 kuukautta (riskisuhde [HR] 0,76, 95 %:n luottamusväli: 0,54, 1,09). Yhteenveto eksploratiivisen analyysin keskeisistä tuloksista on taulukossa 14. Kaplan–Meier-käyrät kokonaiselossaoloajasta ja etenemättömyysajasta potilailla, joilla PD-L1:n ilmentymä on suuri, esitetään kuvissa 12 ja 13. Ensimmäisten 2,5 kuukauden aikana kuolleiden potilaiden osuus oli suurempi atetsolitsumabihoitohaarassa (16/107, 15,0 %) verrattuna solunsalpaajahoitohaaraan (10/98, 10,2 %). Varhaisiin kuolemiin liittyviä spesifisiä tekijöitä ei pystytty tunnistamaan.
Taulukko 14. Yhteenveto hoidon tehosta potilailla, joilla on suuri PD-L1 ilmentymä eli kasvainsoluissa ≥ 50 % tai immuunisoluissa ≥ 10 % (Impower110)
| Tehon päätetapahtumat | Hoitohaara A (Atetsolitsumabi) | Hoitohaara B (Solunsalpaajahoito) |
| Ensisijainen päätetapahtuma | ||
| Elossaoloaika | n = 107 | n = 98 |
| Kuolleiden lkm (%) | 64 (59,8 %) | 64 (65,3 %) |
| Ajan mediaani tapahtumaan (kk) | 20,2 | 14,7 |
| 95 %:n luottamusväli | (17,2, 27,9) | (7,4, 17,7) |
| Ositettu riskisuhde‡ (95 %:n luottamusväli) | 0,76 (0,54, 1,09) | |
| 12 kuukauden kokonaiselossaolo (%) | 66,1 | 52,3 |
| Toissijaiset päätetapahtumat | ||
| Tutkijan arvioima taudin etenemättömyysaika (RECIST v1.1) | n = 107 | n = 98 |
| Tapahtumien lkm (%) | 82 (76,6 %) | 87 (88,8 %) |
| Taudin etenemättömyysajan mediaani (kk) | 8,2 | 5,0 |
| 95 %:n luottamusväli | (6,8, 11,4) | (4,2, 5,7) |
| Ositettu riskisuhde‡ (95 %:n luottamusväli) | 0,59 (0,43, 0,81) | |
| 12 kuukauden taudin etenemättömyysaika (%) | 39,2 | 19,2 |
| Tutkijan arvioima kokonaisvasteosuus (RECIST 1.1) | n = 107 | n = 98 |
| Vasteen saaneiden lkm (%) | 43 (40,2 %) | 28 (28,6 %) |
| 95 %:n luottamusväli | (30,8, 50,1) | (19,9, 38,6) |
| Täydellisen vasteen saaneiden lkm (%) | 1 (0,9 %) | 2 (2,0 %) |
| Osittaisen vasteen saaneiden lkm (%) | 42 (39,3 %) | 26 (26,5 %) |
| Tutkijan arvioima objektiivisen vasteen kesto (RECIST 1.1) | n = 43 | n = 28 |
| Mediaani (kk) | 38,9 | 8,3 |
| 95 %:n luottamusväli | (16,1, NE) | (5,6, 11,0) |
‡ Ositettu sukupuolen ja ECOG-suorituskyvyn mukaan (0 vs. 1)
RECIST = Response Evaluation Criteria in Solid Tumours v1.1; NE = ei arvioitavissa (not estimable).
Kuva 12. Kaplan–Meier-käyrä kokonaiselossaoloajasta potilailla, joilla PD-L1:n ilmentymisen taso on suuri eli kasvainsoluissa ≥ 50 % tai immuunisoluissa ≥ 10 % (Impower110)
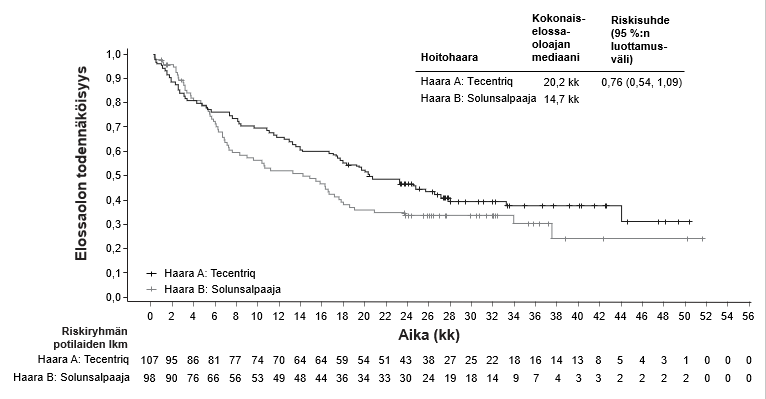
Kuva 13. Kaplan–Meier-käyrä etenemättömyysajasta potilailla, joilla PD-L1:n ilmentymisen taso on suuri eli kasvainsoluissa ≥ 50 % tai immuunisoluissa ≥ 10 % (Impower110)
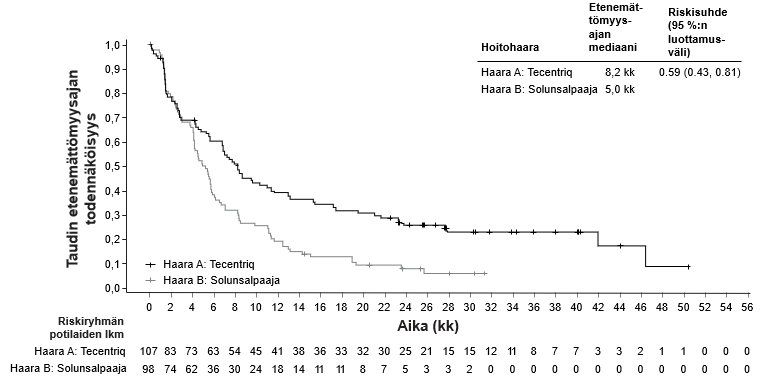
Kokonaiselossaoloajan havaitun pitenemisen atetsolitsumabihoitohaarassa solunsalpaajahoitohaaraan verrattuna osoitettiin olleen yhdenmukainen kaikissa potilaiden alaryhmissä, joissa PD-L1:n ilmentymä oli suuri, mukaan lukien sekä ei-levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastavat potilaat (riskisuhde [HR] 0,62, 95 %:n luottamusväli: 0,40, 0,96; kokonaiselossaoloajan mediaani 20,2 vs. 10,5 kuukautta) että levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastavat potilaat (riskisuhde [HR] 0,56, 95 %:n luottamusväli: 0,23, 1,37; kokonaiselossaoloajan mediaania ei saavutettu vs. 15,3 kuukautta). Tiedot ≥ 75-vuotiaista potilaista ja potilaista, jotka eivät olleet koskaan tupakoineet, olivat liian vähäisiä, jotta näistä alaryhmistä voitaisiin tehdä päätelmiä.
IPSOS-tutkimus (MO29872): Satunnaistettu vaiheen III tutkimus aiemmin hoitamatonta paikallisesti edennyttä leikkaukseen soveltumatonta tai metastasoitunutta ei-pienisoluista keuhkosyöpää sairastavilla potilailla, joille platinapohjainen solunsalpaajahoito ei sovellu
Avoin, satunnaistettu vaiheen III kontrolloitu tutkimus MO29872 (IPSOS) tehtiin atetsolitsumabin tehon ja turvallisuuden arvioimiseksi yhdestä lääkevalmisteesta (tutkijan valinnan mukaan vinorelbiini tai gemsitabiini) koostuvaan solunsalpaajahoitoon verrattuna aiemmin hoitamatonta pitkälle edennyttä tai uusiutunutta (levinneisyysaste IIIB [AJCC-luokituksen 7. painoksen mukaan], joka ei ole hoidettavissa useita hoitomuotoja käsittävällä hoidolla) tai metastasoitunutta (levinneisyysaste IV) ei-pienisoluista keuhkosyöpää sairastavilla potilailla, joille platinapohjaisen solunsalpaajahoidon ei katsottu soveltuvan.
Käyttöaiheen mukaiset potilaat, joille platinapohjainen solunsalpaajahoito ei sovellu, määriteltiin seuraavien valintakriteerien perusteella: potilaan ikä > 80 vuotta tai ECOG-suorituskykyluokka 3 tai potilaan ECOG-suorituskykyluokka 2 ja muita oleellisia samanaikaisia sairauksia tai iäkäs (≥ 70-vuotias) ja muita oleellisia samanaikaisia sairauksia. Oleelliset samanaikaiset sairaudet liittyvät hoitavan lääkärin arvion mukaan sydänsairauksiin, hermostosairauksiin, psyykkisiin sairauksiin, verisuonisairauksiin, munuaissairauksiin, aineenvaihduntaan ja ravitsemukseen liittyviin sairauksiin tai keuhkosairauksiin, jotka ovat platinapohjaisen hoidon vasta-aiheita.
Tutkimukseen ei otettu mukaan potilaita, jotka olivat alle 70-vuotiaita ja joilla ECOG-suorituskykyluokka 0 tai 1; potilaita joilla oli aktiivisia tai hoitamattomia etäpesäkkeitä keskushermostossa; potilaita joille annettu eläviä, heikennettyjä taudinaiheuttajia sisältävää rokotetta satunnaistamista edeltävien 4 viikon aikana; potilaita joille annettu systeemisiä immuniteettia stimuloivia tai systeemisiä immuniteettia lamaavia lääkevalmisteita satunnaistamista edeltävien 4 viikon aikana. Myös potilaat, joilla oli EGFR-mutaatioita tai ALK-geenin uudelleenjärjestäytymistä, suljettiin pois tutkimuksesta. Potilaat soveltuivat mukaan tutkimukseen kasvaimen PD-L1-statuksesta riippumatta.
Potilaat satunnaistettiin suhteessa 2:1 saamaan atetsolitsumabia (haara A) tai solunsalpaajahoitoa (haara B). Atetsolitsumabia annettiin 1200 mg:n vakioannos infuusiona laskimoon 3 viikon välein. Solunsalpaajahoito-ohjelmat kuvataan taulukossa 15. Hoitoa annettiin, kunnes sairaus RECIST v1.1 ‑kriteerien perusteella eteni tai kunnes ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Satunnaistaminen ositettiin histologian (levyepiteeli/ei levyepiteeli), PD-L1-ilmentymän (PD-L1:n immunohistokemiallinen status VENTANA PD-L1 (SP142) ‑määrityksellä mitattuna: TC3 tai IC3 vs. TC0/1/2 ja IC0/1/2 vs. tuntematon) ja aivoissa olevien etäpesäkkeiden (kyllä/ei) perusteella.
Taulukko 15: Hoito-ohjelmat (IPSOS)
| Hoito-ohjelma | |
| A | Atetsolitsumabi 1200 mg infuusiona laskimoon jokaisen 21 päivän pituisen syklin päivänä 1. |
| B | Vinorelbiini: infuusiona laskimoon annoksena 25–30 mg/m2 tai suun kautta annoksena 60–80 mg/m2 jokaisen 21 päivän pituisen hoitosyklin päivinä 1 ja 8 tai jokaisen 28 päivän pituisen syklin päivinä 1, 8 ja 15 tai viikoittain tai Gemsitabiini: infuusiona laskimoon annoksena 1000–1250 mg/m2 jokaisen 21 päivän pituisen syklin päivinä 1 ja 8 tai jokaisen 28 päivän pituisen syklin päivinä 1, 8 ja 15. |
Tutkimukseen otettiin mukaan yhteensä 453 potilasta (ITT-potilasjoukko). Potilasjoukkoon kuuluneet potilaat olivat pääasiassa valkoihoisia (65,8 %) ja miehiä (72,4 %). Potilaiden iän mediaani oli 75 vuotta, ja 72,8 % potilaista oli vähintään 70-vuotiaita. ECOG-suorituskykyluokka oli 1,5 %:lla potilaista 0, 15,0 %:lla potilaista 1, 75,9 %:lla potilaista 2 ja 7,5 %:lla potilaista 3. Kaikkiaan 13,7 %:lla potilaista oli levinneisyysasteen IIIB tauti, joka ei ollut hoidettavissa useita hoitomuotoja käsittävällä hoidolla, ja 86,3 %:lla oli levinneisyysasteen IV tauti. Potilaiden kasvainten VENTANA PD-L1 (SP263) ‑määrityksellä mitatut PD-L1-ilmentymän prosenttiosuudet olivat seuraavat: 46,8 %:lla potilaista TC < 1 %, 28,7 %:lla potilaista 1–49 % ja 16,6 %:lla potilaista ≥ 50 %, ja 7,9 %:lla potilaista PD-L1-ilmentymän status oli tuntematon.
Tutkimuksen ensisijainen päätetapahtuma oli kokonaiselossaoloaika. Kokonaiselossaoloajan lopullisen analyysin ajankohtana seurannan mediaani oli 41,0 kuukautta. Tehoa koskevat tulokset esitetään taulukossa 16 ja kuvassa 14.
Taulukko 16: Yhteenveto tehosta ei-pienisoluista keuhkosyöpää sairastavilla potilailla, joille platinapohjainen solunsalpaajahoito ei sovellu (IPSOS)
| Tehon päätetapahtuma | Atetsolitsumabi (n = 302) | Solunsalpaajahoito (n = 151) |
| Ensisijainen päätetapahtuma | ||
| Kokonaiselossaoloaika | ||
| Tapahtumien lkm (%) | 249 (82,5 %) | 130 (86,1 %) |
| Ajan mediaani tapahtumaan (kk) (95 %:n luottamusväli) | 10,3 (9,4, 11,9) | 9,2 (5,9, 11,2) |
| Ositettu riskisuhde (95 %:n luottamusväli) ǂ | 0,78 (0,63, 0,97) | |
| p-arvo (ositettu log-rank) | p = 0,028 | |
| Toissijaiset päätetapahtumat | ||
| Tutkijan arvioima taudin etenemättömyysaika (RECIST 1.1) | ||
| Tapahtumien lkm (%) | 276 (91,4 %) | 138 (91,4 %) |
| Taudin etenemättömyysajan mediaani (kk) (95 %:n luottamusväli) | 4,2 (3,7, 5,5) | 4,0 (2,9, 5,4) |
| Ositettu riskisuhde (95 %:n luottamusväli) ǂ | 0,87 (0,70, 1,07) | |
| Kokonaisvasteosuus (RECIST 1.1) | ||
| Varmistetun vasteen saaneiden lkm (%) | 51 (16,9 %) | 12 (7,9 %) |
| Objektiivisen vasteen kesto (RECIST 1.1) | ||
| Mediaani (kk) (95 %:n luottamusväli) | 14,0 (8,1, 20,3) | 7,8 (4,8, 9,7) |
RECIST = Response Evaluation Criteria in Solid Tumours v1.1. ǂ Arvioitu riskisuhde ja 95 %:n luottamusväli saatu Coxin mallilla, jossa kovariaattina on hoitoryhmä. Ositetussa analyysissä ositetekijöiksi lisättiin histologinen alatyyppi, PD-L1:n immunohistokemiallinen status ja etäpesäkkeet aivoissa (kyllä/ei). | ||
Kuva 14: Kaplan–Meier-käyrät kokonaiselossaoloajasta ei-pienisoluista keuhkosyöpää sairastavilla potilailla, joille platinapohjainen solunsalpaajahoito ei sovellu (IPSOS)
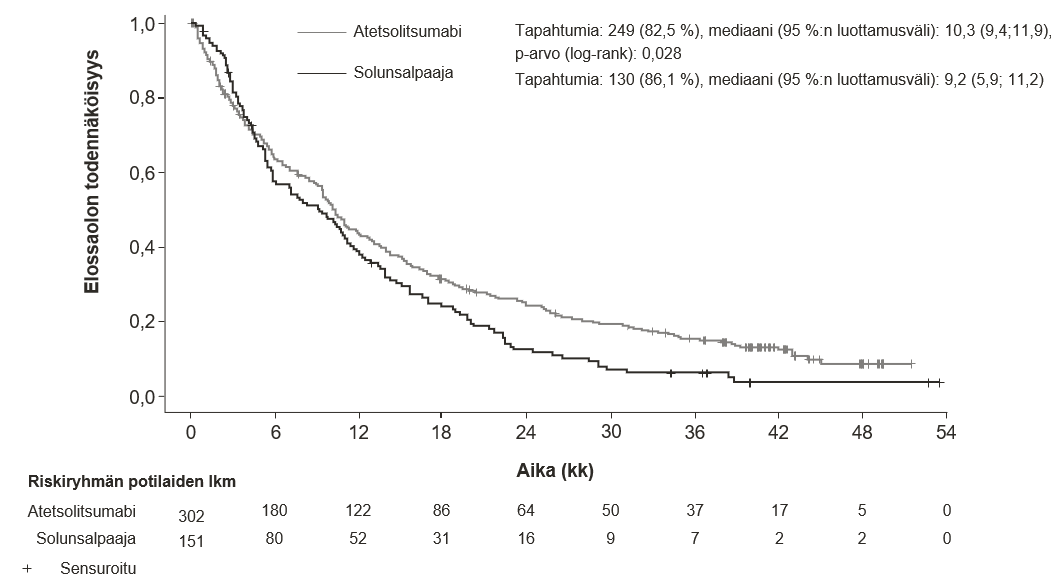
Ei-pienisoluisen keuhkosyövän toisen linjan hoito
OAK (GO28915):Satunnaistettu vaiheen III tutkimus paikallisesti edennyttä tai metastasoitunutta ei-pienisoluista keuhkosyöpää sairastavilla potilailla, jotka olivat aiemmin saaneet solunsalpaajahoitoa
Vaiheen III avoimessa, kansainvälisessä, satunnaistetussa, monikeskustutkimuksessa OAK verrattiin atetsolitsumabin tehoa ja turvallisuutta dosetakseliin paikallisesti edennyttä tai metastasoitunutta ei-pienisoluista keuhkosyöpää sairastavilla potilailla, joiden sairaus oli edennyt platinaa sisältävän hoidon aikana tai sen jälkeen. Potilaita ei otettu tutkimukseen mukaan, jos heillä oli aiemmin ollut autoimmuunisairaus, aktiivisia tai kortikosteroideista riippuvaisia etäpesäkkeitä aivoissa, potilas oli saanut 28 päivän kuluessa ennen tutkimukseen mukaan tuloa rokotuksen elävää, heikennettyä taudinaiheuttajaa sisältävällä rokotteella tai potilas oli saanut systeemisiä immuniteettia stimuloivia aineita 4 viikon aikana tai systeemisiä immunosuppressiivisia lääkevalmisteita 2 viikon aikana ennen tutkimukseen mukaan tuloa. Kasvaimet arvioitiin kuuden viikon välein ensimmäisten 36 viikon ajan ja sen jälkeen 9 viikon välein. Kasvainnäytteistä arvioitiin prospektiivisesti PDL1:n ilmentyminen kasvainsoluissa ja kasvaimeen infiltroivissa immuunisoluissa.
Tutkimukseen otettiin mukaan yhteensä 1 225 potilasta, ja analyysisuunnitelman mukaisesti 850 ensimmäisenä satunnaistettua potilasta otettiin mukaan ensisijaiseen tehon analyysiin. Satunnaistaminen ositettiin kasvaimeen infiltroivien immuunisolujen PDL1:n ilmentymistason mukaan, aiempien solunsalpaajahoitojen lukumäärän mukaan sekä histologian mukaan. Potilaat satunnaistettiin (1:1) saamaan joko atetsolitsumabia tai dosetakselia.
Atetsolitsumabia annettiin vakioannoksina 1200 mg laskimoon 3 viikon välein. Annoksen pienentäminen ei ollut sallittua. Potilaita hoidettiin, kunnes kliinistä hyötyä ei tutkijan arvion mukaan enää todettu. Dosetakselia annettiin 75 mg/m2 laskimoon kunkin 3 viikon pituisen hoitosyklin päivänä 1, kunnes sairaus eteni. Kaikkien hoidettujen potilaiden hoidon keston mediaani oli dosetakseliryhmässä 2,1 kuukautta ja atetsolitsumabiryhmässä 3,4 kuukautta.
Primaarianalyysin potilasjoukon demografiset ominaisuudet sekä sairauden lähtötilanteen ominaisuudet olivat hyvin tasapainossa hoitoryhmien välillä. Iän mediaani oli 64 vuotta (vaihteluväli: 33–85), ja 61 % potilaista oli miehiä. Valtaosa potilaista oli valkoihoisia (70 %). Noin kolmella potilaalla neljästä oli ei-levyepiteeliperäinen histologia (74 %), 10 %:lla tiedettiin olevan EGFR-mutaatio, 0,2 %:lla tiedettiin olevan ALK-geenin uudelleenjärjestäytymistä, 10 %:lla oli lähtötilanteessa etäpesäkkeitä keskushermostossa. Useimmat potilaat olivat parhaillaan tai olivat aiemmin olleet tupakoijia (82 %). Lähtötilanteen ECOG-suorituskykyluokka oli 0 (37 %) tai 1 (63 %). Seitsemänkymmentäviisi prosenttia potilaista oli saanut aiemmin vain yhtä platinapohjaista hoitoa.
Tehon ensisijainen päätetapahtuma oli kokonaiselossaoloaika. Yhteenveto tämän tutkimuksen keskeisistä tuloksista sekä elossaoloajan mediaani 21 kuukauden seurannassa esitetään taulukossa 17. ITT-potilasjoukon kokonaiselossaoloajan Kaplan–Meier-käyrät esitetään kuvassa 15. Kuvassa 16 esitetään yhteenveto ITT- ja PD-L1-alaryhmien kokonaiselossaoloajan tuloksista, jotka osoittavat atetsolitsumabin hyödyn kokonaiselossaoloajan suhteen kaikissa alaryhmissä, mukaan lukien potilaat, joiden kasvainsoluissa ja immuunisoluissa PD-L1:n ilmentymisen taso on < 1 %.
Taulukko 17. Yhteenveto hoidon tehosta primaarianalyysin potilasjoukossa (PD-L1-statuksen suhteen valikoimattomat potilaat)* (OAK)
| Tehon päätetapahtuma | Atetsolitsumabi (n = 425) | Dosetakseli (n = 425) |
| Ensisijainen tehon päätetapahtuma | ||
| Kokonaiselossaoloaika | ||
| Kuolleiden lkm (%) | 271 (64 %) | 298 (70 %) |
| Ajan mediaani tapahtumaan (kk) | 13,8 | 9,6 |
| 95 %:n luottamusväli | (11,8, 15,7) | (8,6, 11,2) |
| Ositettuǂ riskisuhde (95 %:n luottamusväli) | 0,73 (0,62, 0,87) | |
| p-arvo** | 0,0003 | |
| 12 kuukauden kokonaiselossaolo-osuus (%)*** | 218 (55 %) | 151 (41 %) |
| 18 kuukauden kokonaiselossaolo-osuus (%)*** | 157 (40 %) | 98 (27 %) |
| Toissijaiset päätetapahtumat | ||
| Tutkijan arvioima taudin etenemättömyysaika (RECIST v1.1) | ||
| Tapahtumien lkm (%) | 380 (89 %) | 375 (88 %) |
| Taudin etenemättömyysajan mediaani (kk) | 2,8 | 4,0 |
| 95 %:n luottamusväli | (2,6, 3,0) | (3,3, 4,2) |
| Ositettu riskisuhde (95 %:n luottamusväli) | 0,95 (0,82, 1,10) | |
| Tutkijan arvioima kokonaisvasteosuus (RECIST v1.1) | ||
| Vasteen saaneiden lkm (%) | 58 (14 %) | 57 (13 %) |
| 95 %:n luottamusväli | (10,5, 17,3) | (10,3, 17,0) |
| Tutkijan arvioima objektiivisen vasteen kesto (RECIST v1.1) | n = 58 | n = 57 |
| Mediaani (kk) | 16,3 | 6,2 |
| 95 %:n luottamusväli | (10,0, NE) | (4,9, 7,6) |
Luottamusväli = confidence interval (CI); Vasteen kesto = duration of response (DOR); Kasvaimeen infiltroivat immuunisolut = tumour-infiltrating immune cells (IC); Ei arvioitavissa = not estimable (NE); Kokonaisvasteosuus = objective response rate (ORR); Kokonaiselossaoloaika = overall survival (OS); Etenemättömyysaika = progression-free survival (PFS); RECIST = Response Evaluation Criteria in Solid Tumours v1.1
* Primaarianalyysin potilasjoukko, jossa on mukana ensimmäisinä satunnaistetut 850 potilasta. ǂ Ositettu PD-L1:n ilmentymistaso kasvaimeen infiltroivissa immuunisoluissa, aiempien solunsalpaajahoitojen lukumäärän ja histologian mukaan.
** Perustuu ositettuun log-rank-testiin.
*** Perustuu Kaplan–Meierin estimaatteihin.
Kuva 15. Primaarianalyysin potilasjoukon (PD-L1-statuksen suhteen valikoimattomat potilaat) kokonaiselossaoloajan Kaplan–Meier-käyrä (OAK)
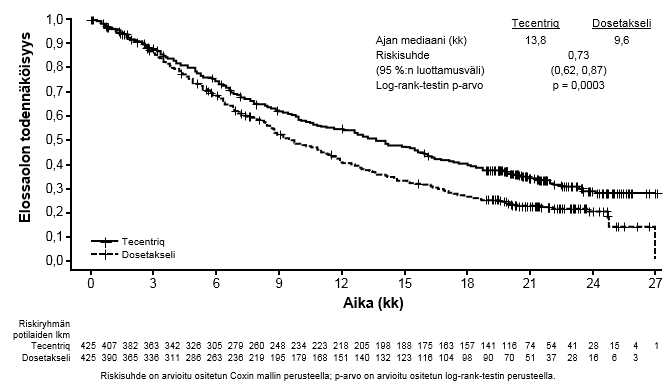
Kuva 16. Kokonaiselossaoloajan Forest plot alaryhmäanalyysi kasvaimen PD-L1:n ilmentymistason mukaan primaarianalyysin potilasjoukossa (OAK)
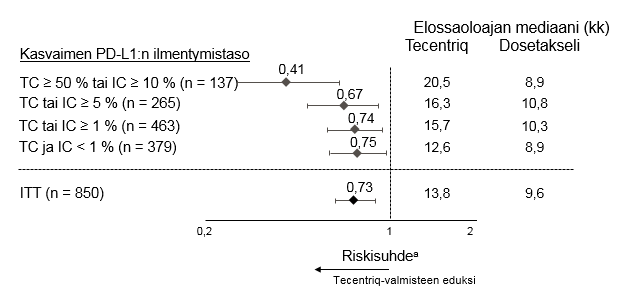
aITT-potilasjoukon ositettu riskisuhde, ja kasvainsolut (TC) tai immuunisolut (IC) ≥ 1 %. Osittamaton riskisuhde muiden eksploratiivisten osajoukkojen osalta.
Kokonaiselossaoloajan havaittiin parantuneen atetsolitsumabiryhmässä dosetakseliin verrattuna muuta kuin levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastavilla potilailla (riskisuhde [HR] 0,73, 95 %:n luottamusväli: 0,60, 0,89; kokonaiselossaoloajan mediaani 15,6 kuukautta [atetsolitsumabi] vs. 11,2 kuukautta [dosetakseli]) ja levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastavilla potilailla (HR 0,73, 95 %:n luottamusväli: 0,54, 0,98; kokonaiselossaoloajan mediaani 8,9 kuukautta [atetsolitsumabi] vs. 7,7 kuukautta [dosetakseli]). Kokonaiselossaoloajan havaittu piteneminen osoitettiin yhdenmukaisesti kaikissa potilaiden alaryhmissä, myös niillä potilailla, joilla oli lähtötilanteessa etäpesäkkeitä aivoissa (HR 0,54, 95 %:n luottamusväli: 0,31, 0,94; kokonaiselossaoloajan mediaani 20,1 kuukautta [atetsolitsumabi] vs. 11,9 kuukautta [dosetakseli]) sekä potilailla, jotka eivät olleet koskaan tupakoineet (HR 0,71, 95 %:n luottamusväli: 0,47, 1,08; kokonaiselossaoloajan mediaani 16,3 kuukautta [atetsolitsumabi] vs. 12,6 kuukautta [dosetakseli]). Potilailla, joilla oli EGFR-mutaatioita, atetsolitsumabin ei todettu parantavan kokonaiselossaoloaikaa dosetakseliin verrattuna (HR 1,24, 95 %:n luottamusväli: 0,71, 2,18; kokonaiselossaoloajan mediaani 10,5 kuukautta [atetsolitsumabi] vs. 16,2 kuukautta [dosetakseli]).
Atetsolitsumabihoidossa havaittiin dosetakseliin verrattuna pidempi aika potilaan raportoimaan rintakehässä tuntuneen kivun pahenemiseen, jota mitattiin EORTC QLQ-LC13 elämänlaatumittarilla (HR 0,71, 95 %:n luottamusväli: 0,49, 1,05; kummassakaan hoitohaarassa ei saavutettu mediaania). Aika keuhkosyövän muiden oireiden (eli yskä, hengenahdistus ja käsi-/hartiakipu) EORTC QLQ-LC13 elämänlaatumittarilla mitattuun pahenemiseen oli atetsolitsumabilla ja dosetakselilla samanpituinen. Koska tutkimuksen koeasetelma oli avoin, tulosten tulkinnassa pitää olla varovainen.
POPLAR (GO28753):Satunnaistettu vaiheen II tutkimus solunsalpaajahoitoa saaneilla paikallisesti edennyttä tai metastasoitunutta ei-pienisoluista keuhkosyöpää sairastavilla potilailla
GO28753 (POPLAR) oli vaiheen II kansainvälinen, satunnaistettu, avoin kontrolloitu monikeskustutkimus, jossa oli mukana paikallisesti edennyttä tai metastasoitunutta ei-pienisoluista keuhkosyöpää sairastavia potilaita, joiden syöpä oli edennyt platinaa sisältävän hoito-ohjelman aikana tai sen jälkeen PD-L1:n ilmentymisestä riippumatta. Ensisijainen tehon päätetapahtuma oli kokonaiselossaoloaika. Yhteensä 287 potilasta satunnaistettiin suhteessa 1:1 saamaan joko atetsolitsumabia (1200 mg infuusiona laskimoon 3 viikon välein, kunnes kliinistä hyötyä ei enää todettu) tai dosetakselia (75 mg/m2 infuusiona laskimoon kunkin 3 viikon pituisen hoitosyklin päivänä 1, kunnes sairaus eteni). Potilaat satunnaistettiin immuunisolujen PD-L1:n ilmentymistason, aiempien solunsalpaajahoitojen lukumäärän ja histologian perusteella. Päivitetty analyysi osoitti, kun oli havaittu yhteensä 200 kuolemaa ja kun elossaolon seurannan mediaani oli 22 kuukautta, että atetsolitsumabihoitoa saaneiden potilaiden kokonaiselossaoloajan mediaani oli 12,6 kuukautta verrattuna dosetakselihoitoa saaneiden 9,7 kuukauteen (HR 0,69, 95 %:n luottamusväli: 0,52, 0,92). Kokonaisvasteosuus oli 15,3 % [atetsolitsumabi] vs. 14,7 % [dosetakseli], ja objektiivisen vasteen keston mediaani oli atetsolitsumabihoidossa 18,6 kuukautta verrattuna dosetakselihoidon 7,2 kuukauteen.
Pienisoluinen keuhkosyöpä
Impower133 (GO30081): Satunnaistettu vaiheen I/III tutkimus levinnyttä pienisoluista keuhkosyöpää sairastavilla potilailla, jotka eivät olleet saaneet aiempaa solunsalpaajahoitoa; yhdistelmähoito karboplatiinin ja etoposidin kanssa
Vaiheen I/III satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (Impower133) arvioitiin atetsolitsumabin, karboplatiinin ja etoposidin yhdistelmähoidon tehoa ja turvallisuutta levinnyttä pienisoluista keuhkosyöpää sairastavilla potilailla, jotka eivät olleet saaneet aiempaa solunsalpaajahoitoa.
Potilaita ei otettu tutkimukseen mukaan, jos heillä oli aktiivisia tai hoitamattomia keskushermoston metastaaseja, aiemmin ollut autoimmuunisairaus, jos potilas oli saanut heikennettyjä eläviä taudinaiheuttajia sisältävää rokotetta satunnaistamista edeltäneiden 4 viikon aikana tai systeemisiä immunosuppressiivisia lääkevalmisteita satunnaistamista edeltäneen 1 viikon aikana. Kasvain arvioitiin ensimmäisen hoitosyklin ensimmäisen päivän jälkeen 6 viikon välein ensimmäisten 48 viikon ajan, ja tämän jälkeen 9 viikon välein. Jos potilas täytti sovitut kriteerit ja halusi hoitoa taudin etenemisen jälkeenkin, kasvain arvioitiin 6 viikon välein, kunnes hoito lopetettiin.
Tutkimukseen otettiin yhteensä 403 potilasta, jotka satunnaistettiin (1:1) saamaan toista taulukossa 18 kuvatuista hoitovaihtoehdoista. Satunnaistaminen ositettiin sukupuolen, ECOG-suorituskyvyn ja aivometastaasien esiintymisen mukaan.
Taulukko 18. Laskimoon annettavat hoito-ohjelmat (Impower133)
| Hoito-ohjelma | Induktiohoito (neljä 21 päivän pituista hoitosykliä) | Ylläpitohoito (21 päivän pituisia hoitosyklejä) |
| A | Atetsolitsumabi (1200 mg)a + karboplatiini (AUC 5)b + etoposidi (100 mg/m2)b,c | Atetsolitsumabi (1200 mg) a |
| B | Lumelääke + karboplatiini (AUC 5)b + etoposidi (100 mg/m2)b,c | Lumelääke |
aAtetsolitsumabia annettiin, kunnes kliininen hyöty tutkijan arvion mukaan loppui
bKarboplatiinia ja etoposidia annettiin siihen saakka, kunnes jokin seuraavista toteutui: 4 hoitosykliä annettu kokonaan, tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä
cEtoposidia annettiin jokaisen hoitosyklin päivinä 1, 2 ja 3
Tutkimuspotilasjoukon demografiset tiedot ja taudin ominaisuudet lähtötilanteessa olivat hyvin tasapainossa hoitohaarojen välillä. Iän mediaani oli 64 vuotta (vaihteluväli: 26–90 vuotta), ja 10 % potilaista oli ≥ 75-vuotiaita. Valtaosa potilaista oli miehiä (65 %) ja valkoihoisia (80 %). 9 %:lla potilaista oli aivometastaaseja, ja suurin osa potilaista oli parhaillaan tai oli aiemmin ollut tupakoijia (97 %). Lähtötilanteen ECOG-suorituskykyluokka oli 0 (35 %) tai 1 (65 %).
Primaarianalyysin ajankohtana potilaiden elossaolon seuranta-ajan mediaani oli 13,9 kuukautta. Kokonaiselossaolon havaittiin pidentyneen tilastollisesti merkitsevästi atetsolitsumabin, karboplatiinin ja etoposidin yhdistelmää käytettäessä vertailuhaaraan verrattuna (riskisuhde = 0,70, 95 %:n luottamusväli: 0,54, 0,91; kokonaiselossaolon mediaani 12,3 kuukautta vs. 10,3 kuukautta). Kokonaiselossaolon eksploratiivisessa loppuanalyysissa, jossa seuranta-aika oli pidempi (mediaani: 22,9 kuukautta), kummankin hoitohaaran kokonaiselossaolon mediaani oli muuttumaton kokonaiselossaolon primaariin välianalyysiin nähden. Taulukossa 19 esitetään yhteenveto taudin etenemättömyysajan, kokonaisvasteosuuden ja vasteen keston tulosten primaarianalyysista sekä kokonaiselossaolon eksploratiivisen loppuanalyysin tuloksista. Kuvissa 17 ja 18 esitetään kokonaiselossaolon ja taudin etenemättömyysajan Kaplan–Meier-käyrät. Tiedot potilaista, joilla oli aivometastaaseja, ovat liian suppeita, jotta niistä voisi tehdä päätelmiä tästä potilasjoukosta.
Taulukko 19. Yhteenveto hoidon tehosta (Impower133)
| Keskeiset hoidon tehoa koskevat päätetapahtumat | Hoitohaara A (atetsolitsumabi + karboplatiini + etoposidi) | Hoitohaara B (lumelääke + karboplatiini + etoposidi) |
| Muut ensisijaiset päätetapahtumat | ||
| Kokonaiselossaolon analyysi* | n = 201 | n = 202 |
| Kuolemien lkm (%) | 142 (70,6 %) | 160 (79,2 %) |
| Ajan mediaani tapahtumiin (kk) | 12,3 | 10,3 |
| 95 %:n luottamusväli | (10,8, 15,8) | (9,3, 11,3) |
| Ositettu riskisuhde‡ (95 %:n luottamusväli) | 0,76 (0,60, 0,95) | |
| p-arvo | 0,0154 | |
| 12 kk:n kokonaiselossaoloaika (%) | 51,9 | 39,0 |
| Taudin etenemättömyysaika (tutkijan arvio; RECIST v1.1) ** | n = 201 | n = 202 |
| Tapahtumien lkm (%) | 171 (85,1 %) | 189 (93,6 %) |
| Etenemättömyysajan keston mediaani (kk) | 5,2 | 4,3 |
| 95 %:n luottamusväli | (4,4, 5,6) | (4,2, 4,5) |
| Ositettu riskisuhde‡ (95 %:n luottamusväli) | 0,77 (0,62, 0,96) | |
| p-arvo | 0,0170 | |
6 kk:n etenemättömyysaika (%) 12 kk:n etenemättömyysaika (%) | 30,9 12,6 | 22,4 5,4 |
| Muut päätetapahtumat | ||
| Kokonaisvasteosuus (tutkijan arvio; RECIST 1.1)** ^ | n = 201 | n = 202 |
| Vasteen saaneiden lkm (%) | 121 (60,2 %) | 130 (64,4 %) |
| 95 %:n luottamusväli | (53,1, 67,0) | (57,3, 71,0) |
| Täydellisen vasteen saaneiden lkm (%) | 5 (2,5 %) | 2 (1,0 %) |
| Osittaisen vasteen saaneiden lkm (%) | 116 (57,7 %) | 128 (63,4 %) |
| Vasteen kesto (tutkijan arvio; RECIST 1.1)** ^ | n = 121 | n = 130 |
| Mediaani (kk) | 4,2 | 3,9 |
| 95 %:n luottamusväli | (4,1, 4,5) | (3,1, 4,2) |
RECIST=Response Evaluation Criteria in Solid Tumours v1.1
‡ Ositus perustuu sukupuoleen ja ECOG-suorituskykyyn
* Kokonaiselossaolon eksploratiivinen loppuanalyysi kliinisten tietojen keruun katkaisupäivämääränä 24. tammikuuta 2019
** Taudin etenemättömyysajan, kokonaisvasteosuuden ja vasteen keston analyysit kliinisten tietojen keruun katkaisupäivämääränä 24. huhtikuuta 2018
*** Tarkoitettu vain havainnollistamiseen
^ Vahvistettu kokonaisvasteosuus ja vasteen kesto ovat eksploratiivisia päätetapahtumia
Kuva 17. Kokonaiselossaolon Kaplan–Meier-käyrät (Impower133)
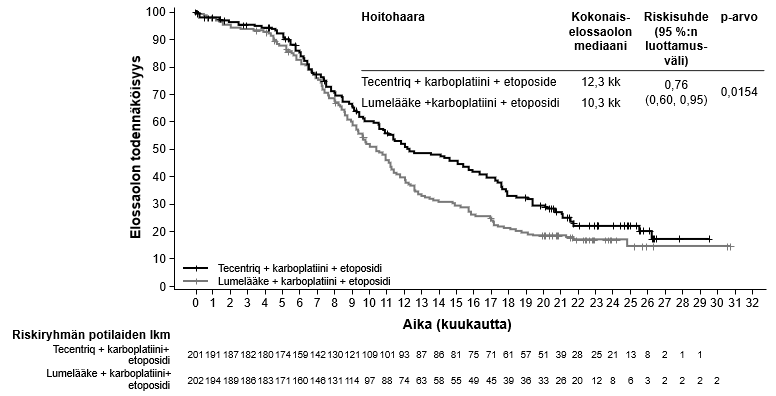
Kuva 18. Taudin etenemättömyysajan Kaplan–Meier-käyrät (Impower133)
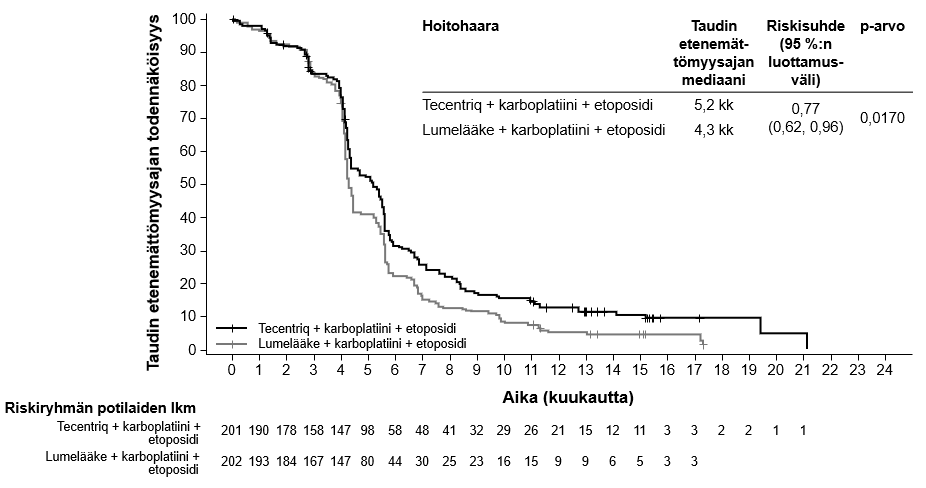
Kolmoisnegatiivinen rintasyöpä
Impassion130 (WO29522): Satunnaistettu vaiheen III tutkimus paikallisesti edennyttä tai metastasoitunutta kolmoisnegatiivista rintasyöpää sairastavilla potilailla, jotka eivät olleet aikaisemmin saaneet hoitoa metastasoituneeseen tautiin
Vaiheen III kaksoissokkoutettu, kahden hoitohaaran kansainvälinen, satunnaistettu, lumekontrolloitu monikeskustutkimus (Impassion130) tehtiin atetsolitsumabin ja nab-paklitakselin turvallisuuden ja tehon arvioimiseksi potilailla, joilla oli paikallisesti edennyt tai metastasoitunut kolmoisnegatiivinen rintasyöpä, joka ei ollut poistettavissa leikkauksella, ja jotka eivät olleet saaneet aiemmin solunsalpaajahoitoa metastasoituneeseen tautiin. Potilaiden piti soveltua taksaanimonoterapiaan (eli ei taudin nopeaa kliinistä etenemistä, henkeä uhkaavia viskeraalisia etäpesäkkeitä eikä tarvetta oireiden ja/tai taudin saamiseen nopeasti hallintaan). Tähän tutkimukseen ei otettu mukaan potilaita, jotka olivat viimeksi kuluneiden 12 kuukauden aikana saaneet aiemmin solunsalpaajia neoadjuvantti- tai adjuvanttihoitona, joilla oli aiemmin ollut autoimmuunisairaus, jotka olivat saaneet 4 viikon kuluessa ennen satunnaistamista elävää, heikennettyä taudinaiheuttajaa sisältävää rokotetta, jotka olivat saaneet 4 viikon kuluessa systeemisiä immuniteettia stimuloivia lääkeaineita tai 2 viikon kuluessa ennen satunnaistamista systeemisiä immunosuppressiivisia lääkevalmisteita, eikä potilaita, joilla oli hoitamaton, oireinen tai kortikosteroideista riippuvainen etäpesäke aivoissa. Kasvain arvioitiin hoitosyklin 1 päivän 1 jälkeen 8 viikon (± 1 viikon) välein ensimmäisten 12 kuukauden ajan ja tämän jälkeen 12 viikon (± 1 viikon) välein.
Tutkimukseen otettiin mukaan yhteensä 902 potilasta, ja ositus perustui maksametastaasien esiintymiseen, aiempaan taksaanihoitoon ja PD-L1:n ilmentymiseen kasvaimeen infiltroivien immuunisolujen pinnalla (PD-L1-värjäytyviä kasvaimeen infiltroivia immuunisoluja < 1 %:lla kasvaimen alueesta vs. ≥ 1 %:lla kasvaimen alueesta), joka selvitettiin VENTANA PD-L1 (SP142) määrityksellä.
Potilaat satunnaistettiin saamaan 840 mg atetsolitsumabia tai lumevalmistetta infuusiona laskimoon kunkin 28 päivän pituisen hoitosyklin päivinä 1 ja 15 yhdistelmänä nab-paklitakselin (100 mg/m2) kanssa, jota annettiin infuusiona laskimoon kunkin 28 päivän pituisen hoitosyklin päivinä 1, 8 ja 15. Potilaat saivat hoitoa, kunnes tauti eteni radiologisesti RECIST v1.1 kriteerien perusteella tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Atetsolitsumabihoitoa voitiin jatkaa sen jälkeen, kun nab-paklitakselihoito oli lopetettu sen vuoksi, että ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Jokaisessa hoitohaarassa hoitosyklien lukumäärän mediaani oli atetsolitsumabin osalta 7 ja nab-paklitakselin osalta 6.
Tutkimuspotilasjoukon demografiset ominaisuudet ja sairauden ominaisuudet lähtötilanteessa olivat hyvin tasapainossa hoitohaarojen kesken. Valtaosa potilaista oli naisia (99,6 %), ja 67,5 % oli valkoihoisia ja 17,8 % oli aasialaisia. Iän mediaani oli 55 vuotta (vaihteluväli: 20–86 vuotta). Lähtötilanteen ECOG-suorituskykyluokka oli 0 (58,4 %) tai 1 (41,3 %). Lähtötilanteessa kaikkiaan 41 %:lla tutkimukseen mukaan otetuista potilaista PD-L1:n ilmentymä oli ≥ 1 %, 27 %:lla oli etäpesäkkeitä maksassa ja 7 %:lla oli oireettomia etäpesäkkeitä aivoissa. Noin puolet potilaista oli saanut (neo)adjuvanttihoitona jotakin taksaania (51 %) tai antrasykliiniä (54 %). Potilaiden demografiset ominaisuudet ja potilaiden kasvaimen PD-L1:n ilmentymä ≥ 1 % lähtötilanteessa olivat yleisesti laajempaa tutkimuspotilasjoukkoa edustavia.
Muita ensisijaisia tehon päätetapahtumia olivat tutkijan RECIST v1.1 kriteerien perusteella arvioima taudin etenemättömyysaika (PFS) hoitoaikeen mukaisessa (intention-to-treat, ITT) potilasjoukossa ja potilailla, joilla PD-L1:n ilmentymä oli ≥ 1 %, sekä kokonaiselossaolo (overall survival, OS) hoitoaikeen mukaisessa potilasjoukossa ja potilailla, joilla PD-L1:n ilmentymä oli ≥ 1 %. Toissijaisia tehon päätetapahtumia olivat kokonaisvasteosuus (objective response rate, ORR) ja vasteen kesto (duration of response, DOR) RECIST v1.1 kriteerien perusteella.
Impassion130-tutkimuksen taudin etenemättömyysaikaa, kokonaisvasteosuutta ja vasteen kestoa koskevista tuloksista potilailla, joilla PD-L1:n ilmentymä oli ≥ 1 % taudin etenemättömyysajan loppuanalyysin ajankohtana ja joiden elossaoloa seurattiin enimmillään 13 kuukauden ajan (mediaani), esitetään yhteenveto taulukossa 20 sekä taudin etenemättömyysajan Kaplan-Meierin käyrät kuvassa 19. Potilailla, joilla PD-L1:n ilmentymä oli < 1 %, ei todettu taudin etenemättömyysajan pidentymistä, kun nab-paklitakseliin lisättiin atetsolitsumabi (riskisuhde 0,94, 95 %:n luottamusväli 0,78, 1,13).
Niiden potilaiden kokonaiselossaolosta, joilla PD-L1:n ilmentymä oli ≥ 1 %, tehtiin loppuanalyysi, kun seurannan kesto oli enimmillään 19,12 kuukautta (mediaani). Kokonaiselossaolon tulokset esitetään taulukossa 20, ja Kaplan-Meierin käyrät esitetään kuvassa 20. Potilailla, joilla PD-L1:n ilmentymä oli < 1 %, ei todettu kokonaiselossaolon pidentymistä, kun nab-paklitakseliin lisättiin atetsolitsumabi (riskisuhde 1,02, 95 %:n luottamusväli 0,84, 1,24).
Potilaista, joilla PD-L1:n ilmentymä oli ≥ 1 %, tehtiin eksploratiivinen alaryhmäanalyysi, jossa selvitettiin aiempaa (neo)adjuvanttihoitoa, BRCA1/2:n mutaatioita ja oireettomia etäpesäkkeitä aivoissa lähtötilanteessa.
Aiempaa (neo)adjuvanttihoitoa saaneiden potilaiden (n = 242) taudin ensisijaisen (lopullisen) etenemättömyysajan (PFS) riskisuhde oli 0,79 ja lopullisen kokonaiselossaolon riskisuhde oli 0,77, kun taas potilailla, jotka eivät olleet saaneet aiempaa (neo)adjuvanttihoitoa (n = 127), taudin ensisijaisen (lopullisen) etenemättömyysajan (PFS) riskisuhde oli 0,44 ja lopullisen kokonaiselossaolon riskisuhde oli 0,54.
Impassion130-tutkimuksessa testatuista 614 potilaasta 89 potilaalla (15 %) oli patogeenisia BRCA1/2:n mutaatioita. Alaryhmässä, jossa potilailla oli PD-L1+/BRCA1/2-mutaatio, 19 potilasta sai atetsolitsumabin ja nab-paklitakselin yhdistelmää ja 26 potilasta sai lumelääkkeen ja nab-paklitakselin yhdistelmää. Eksploratiivisen analyysin perusteella, kun pieni otoskoko otetaan huomioon, BRCA1/2-mutaatio ei näytä vaikuttavan atetsolitsumabin ja nab-paklitakselin yhdistelmästä taudin etenemättömyysajan (PFS) suhteen saatavaan kliiniseen hyötyyn.
Tehosta potilaille, joilla oli lähtötilanteessa oireettomia etäpesäkkeitä aivoissa, ei ole näyttöä, mutta hoidettujen potilaiden lukumäärä oli pieni. Taudin etenemättömyysajan mediaani oli atetsolitsumabin ja nab-paklitakselin yhdistelmää saaneessa haarassa (n = 15) 2,2 kuukautta verrattuna 5,6 kuukauteen lumelääkkeen ja nab-paklitakselin yhdistelmää saaneessa haarassa (n = 11) (riskisuhde 1,40; 95 %:n luottamusväli 0,57, 3,44).
Taulukko 20. Yhteenveto hoidon tehosta potilailla, joilla PD-L1:n ilmentymä oli ≥ 1 % (Impassion130)
| Keskeiset tehon päätetapahtumat | Atetsolitsumabi + nab‑paklitakseli | Lumelääke + nab‑paklitakseli |
| Ensisijaiset tehon päätetapahtumat | n = 185 | n = 184 |
| Tutkijan arvioima taudin etenemättömyysaika (RECIST v1.1) – primaarianalyysi3 | ||
| Tapahtumien lkm (%) | 138 (74,6 %) | 157 (85,3 %) |
| Taudin etenemättömyysajan mediaani (kuukautta) | 7,5 | 5,0 |
| 95 %:n luottamusväli | (6,7, 9,2) | (3,8, 5,6) |
| Ositettu riskisuhde‡ (95 %:n luottamusväli) | 0,62 (0,49, 0,78) | |
| p-arvo1 | < 0,0001 | |
| 12 kuukauden etenemättömyysaika (%) | 29,1 | 16,4 |
| Tutkijan arvioima taudin etenemättömyysaika (RECIST v1.1) – päivitetty eksploratiivinen analyysi4 | ||
| Tapahtumien lkm (%) | 149 (80,5 %) | 163 (88,6 %) |
| Taudin etenemättömyysajan mediaani (kuukautta) | 7,5 | 5,3 |
| 95 %:n luottamusväli | (6,7, 9,2) | (3,8, 5,6) |
| Ositettu riskisuhde‡ (95 %:n luottamusväli) | 0,63 (0,50–0,80) | |
| p-arvo1 | < 0,0001 | |
| 12 kuukauden etenemättömyysaika (%) | 30,3 | 17,3 |
| Kokonaiselossaolo1,2,5 | ||
| Kuolemien lkm (%) | 120 (64,9 %) | 139 (75,5 %) |
| Ajan mediaani tapahtumiin (kuukautta) | 25,4 | 17,9 |
| 95 %:n luottamusväli | (19,6, 30,7) | (13,6, 20,3) |
| Ositettu riskisuhde‡ (95 %:n luottamusväli) | 0,67 (0,53, 0,86) | |
| Toissijaiset ja eksploratiiviset päätetapahtumat | ||
| Tutkijan arvioima kokonaisvasteosuus (RECIST 1.1) 3 | n = 185 | n = 183 |
| Vasteen saaneiden lkm (%) | 109 (58,9 %) | 78 (42,6 %) |
| 95 %:n luottamusväli | (51,5, 66,1) | (35,4, 50,1) |
| Täydellisen vasten saaneiden lkm (%) | 19 (10,3 %) | 2 (1,1 %) |
| Osittaisen vasteen saaneiden lkm (%) | 90 (48,6 %) | 76 (41,5 %) |
| Stabiilin taudin saavuttaneiden lkm (%) | 38 (20,5 %) | 49 (26,8 %) |
| Tutkijan arvioima vasteen kesto 3 | n = 109 | n = 78 |
| Mediaani kuukausina | 8,5 | 5,5 |
| 95 %:n luottamusväli | (7,3, 9,7) | (3,7, 7,1) |
‡ Ositettu maksametastaasien ja aiemman taksaanihoidon perusteella. | ||
Kuva 19. Etenemättömyysajan Kaplan–Meierin käyrä (potilaat, joilla PD-L1:n ilmentymä ≥ 1 %) (IMpassion130)
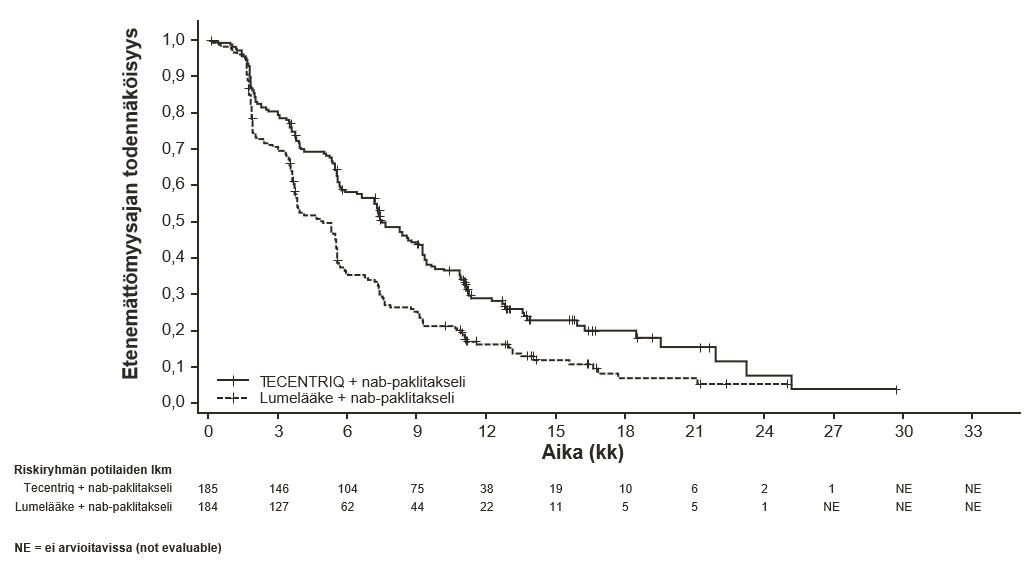
Kuva 20. Kokonaiselossaolon Kaplan–Meierin käyrä (potilaat, joilla PD-L1:n ilmentymä ≥ 1 %) (IMpassion130)
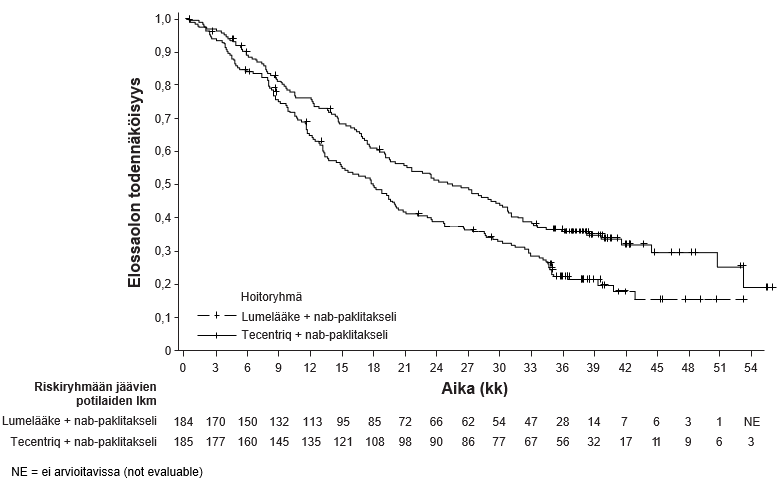
EORTC QLQ-C30 kyselyllä mitattu aika potilaan raportoiman yleisen terveydentilan / terveyteen liittyvän elämänlaadun huononemiseen (≥ 10 pisteen pitkäkestoinen vähenemä lähtötilanteen pisteistä) oli kummassakin hoitoryhmässä samankaltainen, mikä osoittaa sen, että lähtötilanteen terveyteen liittyvä elämänlaatu säilyi kaikilla potilailla ennallaan yhtä pitkään.
Hepatosellulaarinen karsinooma
IMbrave150 (YO40245): Satunnaistettu vaiheen III tutkimus yhdistelmähoidosta bevasitsumabin kanssa leikkauksessa poistettavaksi soveltumatonta hepatosellulaarista karsinoomaa sairastavilla potilailla, jotka eivät ole aiemmin saaneet systeemistä hoitoa
Vaiheen III satunnaistetussa, kansainvälisessä, avoimessa monikeskustutkimuksessa (IMbrave150) arvioitiin atetsolitsumabin ja bevasitsumabin yhdistelmän tehoa ja turvallisuutta potilailla, joilla on paikallisesti edennyt tai metastasoitunut ja/tai leikkauksessa poistettavaksi soveltumaton hepatosellulaarinen karsinooma ja jotka eivät olleet aiemmin saaneet systeemistä hoitoa. Yhteensä 501 potilasta satunnaistettiin (2:1) saamaan joko atetsolitsumabia (1200 mg) ja 15 mg/kg bevasitsumabia 3 viikon välein infuusiona laskimoon tai 400 mg sorafenibia suun kautta kaksi kertaa päivässä. Satunnaistaminen ositettiin maantieteellisen alueen, makrovaskulaarisen infiltraation ja/tai maksan ulkopuolelle leviämisen, lähtötilanteen α-fetoproteiinipitoisuuden (AFP) ja ECOG-suorituskykyluokan mukaan. Kummankin haaran potilaat saivat hoitoa, kunnes siitä ei enää saatu kliinistä hyötyä tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Potilaiden oli mahdollista lopettaa joko atetsolitsumabin tai bevasitsumabin käyttö (esim. haittatapahtumien vuoksi) ja jatkaa hoitoa yhdellä lääkeaineella, kunnes yhdestä lääkeaineesta ei enää saatu kliinistä hyötyä tai ilmaantui siihen liittyvää toksisuutta, joka ei ollut hyväksyttävissä.
Tutkimukseen otettiin mukaan aikuisia, joiden sairaus ei ollut parannettavissa tai eteni kirurgisten ja/tai lokoregionaalisten hoitojen jälkeen ja joiden Child-Pugh luokka oli A ja ECOG-suorituskykyluokka oli 0/1 ja jotka eivät olleet aiemmin saaneet systeemistä hoitoa. Verenvuoto (mukaan lukien kuolemaan johtavat tapahtumat) on bevasitsumabin tunnettu haittavaikutus, ja maha-suolikanavan alkuosan verenvuodot ovat hepatosellulaarista karsinoomaa sairastavilla potilailla yleinen ja hengenvaarallinen komplikaatio. Potilailta oli näin ollen tutkittava suonikohjut hoitoa edeltäneiden 6 kuukauden aikana, ja jos heillä oli verenvuotoa suonikohjuista hoitoa edeltävien 6 kuukauden aikana, hoitamattomia tai riittämättömästi hoidettuja suonikohjuja, joihin liittyi verenvuotoa tai suuri verenvuotoriski, heitä ei otettu tutkimukseen mukaan. Jos potilaalla oli aktiivinen B-hepatiitti, edellytyksenä olivat hepatiitti B viruksen DNA-pitoisuus < 500 IU/ml tutkimushoidon aloittamista edeltävien 28 päivän aikana sekä tavanomainen hoito hepatiitti B virusinfektioon vähintään 14 päivän ajan ennen tutkimukseen mukaan tuloa ja koko tutkimuksen ajan.
Potilaita ei otettu tutkimukseen mukaan myöskään, jos jokin seuraavista koski heitä: keskivaikea tai vaikea askites, anamneesissa maksaenkefalopatia, tiedossa oleva fibrolamellaarinen hepatosellulaarinen karsinooma, sarkooman kaltainen hepatosellulaarinen karsinooma, sekamuotoinen kolangiokarsinooma ja hepatosellulaarinen karsinooma, aktiivinen hepatiitti B- ja C yhteisinfektio, anamneesissa autoimmuunisairaus, rokotus eläviä, heikennettyjä taudinaiheuttajia sisältävällä rokotteella satunnaistamista edeltäneiden 4 viikon aikana, immuunijärjestelmää vahvistavien lääkeaineiden systeeminen käyttö satunnaistamista edeltävien 4 viikon aikana tai immuunijärjestelmää lamaavien lääkevalmisteiden systeeminen käyttö satunnaistamista edeltävien 2 viikon aikana, hoitamattomia tai kortikosteroidiriippuvaisia metastaaseja aivoissa. Kasvain tutkittiin 1. hoitosyklin 1 päivästä lähtien ensimmäisten 54 viikon ajan 6 viikon välein ja sen jälkeen 9 viikon välein.
Tutkimuspotilasjoukon demografiset ominaisuudet ja sairauden ominaisuudet lähtötilanteessa olivat hyvin tasapainossa hoitohaarojen välillä. Iän mediaani oli 65 vuotta (vaihteluväli: 26–88 vuotta), ja 83 % oli miehiä. Valtaosa potilaista oli aasialaisia (57 %) ja valkoihoisia (35 %). Potilaista 40 % oli Aasiasta (muualta kuin Japanista), ja 60 % oli muualta maailmasta. Noin 75 %:lla potilaista oli makrovaskulaarinen infiltraatio ja/tai tauti oli levinnyt maksan ulkopuolelle, ja 37 %:lla α‑fetoproteiinipitoisuus oli lähtötilanteessa ≥ 400 ng/ml. Lähtötilanteen ECOG-suorituskykyluokka oli 0 (62 %) tai 1 (38 %). Hepatosellulaarisen karsinooman ensisijainen riskitekijä oli 48 %:lla potilaista hepatiitti B virusinfektio, 22 %:lla potilaista hepatiitti C virusinfektio ja 31 %:lla potilaista muu kuin virustauti. Hepatosellulaarinen karsinooma luokiteltiin 82 %:lla potilaista BCLC (Barcelona Clinic Liver Cancer) luokaksi C, 16 %:lla potilaista BCLC-luokaksi B ja 3 %:lla potilaista BCLC-luokaksi A.
Muut ensisijaiset tehon päätetapahtumat olivat kokonaiselossaolo ja riippumattoman arviointilautakunnan RECIST v1.1 kriteerien mukaisesti arvioima etenemättömyysaika. Primaarianalyysin ajankohtana potilaiden elossaolon seuranta-aika oli 8,6 kuukautta (mediaani). Tiedot osoittivat, että kokonaiselossaolo ja etenemättömyysaika olivat atetsolitsumabin ja bevasitsumabin yhdistelmän käytössä riippumattoman arviointilautakunnan RECIST v1.1 kriteerien mukaisesti tekemän arvion perusteella pidentyneet tilastollisesti merkitsevästi sorafenibiin verrattuna. Myös varmistetun kokonaisvasteosuuden (ORR) havaittiin riippumattoman arviointilautakunnan RECIST v1.1 kriteerien mukaisesti tekemän arvion sekä hepatosellulaarista karsinoomaa koskevien muunnettujen RECIST-kriteerien (mRECIST) perusteella pidentyneen tilastollisesti merkitsevästi. Yhteenveto primaarianalyysin keskeisistä tehoa koskevista tuloksista esitetään taulukossa 21.
Päivitetty tehon deskriptiivinen analyysi tehtiin, kun elossaolon seuranta-ajan mediaani oli 15,6 kuukautta. Kokonaiselossaolon mediaaniaika oli atetsolitsumabin ja bevasitsumabin yhdistelmää saaneessa haarassa 19,2 kuukautta (95 %:n luottamusväli: 17,0, 23,7) verrattuna sorafenibihaaran 13,4 kuukauteen (95 %:n luottamusväli: 11,4, 16,9) ja riskisuhde (HR) oli 0,66 (95 %:n luottamusväli: 0,52, 0,85). Riippumattoman arviointilautakunnan RECIST v1.1 -kriteerien mukaisesti arvioima etenemättömyysajan mediaani oli atetsolitsumabin ja bevasitsumabin yhdistelmää saaneessa haarassa 6,9 kuukautta (95 %:n luottamusväli: 5,8, 8,6) verrattuna sorafenibihaaran 4,3 kuukauteen (95 %:n luottamusväli: 4,0, 5,6) ja riskisuhde (HR) oli 0,65 (95 %:n luottamusväli: 0,53, 0,81).
Riippumattoman arviointilautakunnan RECIST v1.1 -kriteerien mukaisesti arvioima kokonaisvasteosuus oli atetsolitsumabin ja bevasitsumabin yhdistelmää saaneessa haarassa 29,8 % (95 %:n luottamusväli: 24,8, 35,0) ja sorafenibihaarassa 11,3 % (95 %:n luottamusväli: 6,9, 17,3). Riippumattoman arviointilautakunnan RECIST v1.1 -kriteerien mukaisesti arvioima objektiivisen vasteen kesto varmistetun vasteen saaneilla oli atetsolitsumabin ja bevasitsumabin yhdistelmää saaneessa haarassa 18,1 kuukautta (95 %:n luottamusväli: 14,6, ei arvioitavissa) verrattuna sorafenibihaaran 14,9 kuukauteen (95 %:n luottamusväli: 4,9, 17,0).
Kokonaiselossaolon Kaplan–Meier-käyrät (päivitetty analyysi) esitetään kuvassa 21 ja etenemättömyysajan Kaplan–Meier-käyrät (primaarianalyysi) esitetään kuvassa 22.
Taulukko 21. Yhteenveto tehosta (IMbrave150-tutkimuksen primaarianalyysi)
| Keskeiset tehon päätetapahtumat | Atetsolitsumabi + bevasitsumabi | Sorafenibi |
| Kokonaiselossaolo | n = 336 | n = 165 |
| Kuolemien lkm (%) | 96 (28,6 %) | 65 (39,4 %) |
| Ajan mediaani tapahtumaan (kuukautta) | NE | 13,2 |
| 95 %:n luottamusväli | (NE, NE) | (10,4, NE) |
| Ositettu riskisuhde‡ (95 %:n luottamusväli) | 0,58 (0,42, 0,79) | |
| p-arvo1 | 0,0006 | |
| 6 kuukauden kokonaiselossaolo (%) | 84,8 % | 72,3 % |
| Riippumattoman arviointilautakunnan arvioima etenemättömyysaika (RECIST 1.1) | n = 336 | n = 165 |
| Tapahtumien lkm (%) | 197 (58,6 %) | 109 (66,1 %) |
| Taudin etenemättömyysajan mediaani (kuukautta) | 6,8 | 4,3 |
| 95 %:n luottamusväli | (5,8, 8,3) | (4,0, 5,6) |
| Ositettu riskisuhde‡ (95 %:n luottamusväli) | 0,59 (0,47, 0,76) | |
| p-arvo1 | < 0,0001 | |
| 6 kuukauden etenemättömyysaika | 54,5 % | 37,2 % |
| Riippumattoman arviointilautakunnan arvioima kokonaisvasteosuus (RECIST 1.1) | n = 326 | n = 159 |
| Varmistetun vasteen saaneiden lkm (%) | 89 (27,3 %) | 19 (11,9 %) |
| 95 %:n luottamusväli | (22,5, 32,5) | (7,4, 18,0) |
| p-arvo2 | < 0,0001 | |
| Täydellisen vasteen saaneiden lkm (%) | 18 (5,5 %) | 0 |
| Osittaisen vasteen saaneiden lkm (%) | 71 (21,8 %) | 19 (11,9 %) |
| Stabiilin taudin saavuttaneiden lkm (%) | 151 (46,3 %) | 69 (43,4 %) |
| Riippumattoman arviointilautakunnan arvioima objektiivisen vasteen kesto (RECIST 1.1) | n = 89 | n = 19 |
| Mediaani (kuukautta) | NE | 6,3 |
| 95 %:n luottamusväli | (NE, NE) | (4,7, NE) |
| Vaihteluväli (kuukautta) | (1,3+, 13,4+) | (1,4+, 9,1+) |
| Riippumattoman arviointilautakunnan arvioima kokonaisvasteosuus (HCC mRECIST) | n = 325 | n = 158 |
| Varmistetun vasteen saaneiden lkm (%) | 108 (33,2 %) | 21 (13,3 %) |
| 95 %:n luottamusväli | (28,1, 38,6) | (8,4, 19,6) |
| p-arvo2 | < 0,0001 | |
| Täydellisen vasteen saaneiden lkm (%) | 33 (10,2 %) | 3 (1,9 %) |
| Osittaisen vasteen saaneiden lkm (%) | 75 (23,1 %) | 18 (11,4 %) |
| Stabiilin taudin saavuttaneiden lkm (%) | 127 (39,1 %) | 66 (41,8 %) |
| Riippumattoman arviointilautakunnan arvioima objektiivisen vasteen kesto (HCC mRECIST) | n = 108 | n = 21 |
| Mediaani (kuukautta) | NE | 6,3 |
| 95 %:n luottamusväli | (NE, NE) | (4,9, NE) |
| Vaihteluväli (kuukautta) | (1,3+, 13,4+) | (1,4+, 9,1+) |
| ‡ Ositettu maantieteellisen alueen mukaan (Aasia, pois lukien Japani vs muu maailma), makrovaskulaarinen infiltraatio ja/tai leviäminen maksan ulkopuolelle (kyllä vs. ei) ja lähtötilanteen α-fetoproteiinipitoisuus (< 400 vs. ≥ 400 ng/ml) 1 Perustuu kaksitahoiseen ositettuun log-rank-testiin 2 Nimelliset p-arvot perustuvat kaksitahoiseen Cochran-Mantel-Haenszelin testiin + Tarkoittaa sensuroitua arvoa RECIST = Response Evaluation Criteria in Solid Tumors v1.1; HCC mRECIST = Modified RECIST Assessment for Hepatocellular Carcinoma; NE = ei arvioitavissa (not estimable) | ||
Kuva 21. Kokonaiselossaolon Kaplan-Meier-käyrä ITT-potilasjoukossa (IMbrave150-tutkimuksen päivitetty analyysi)
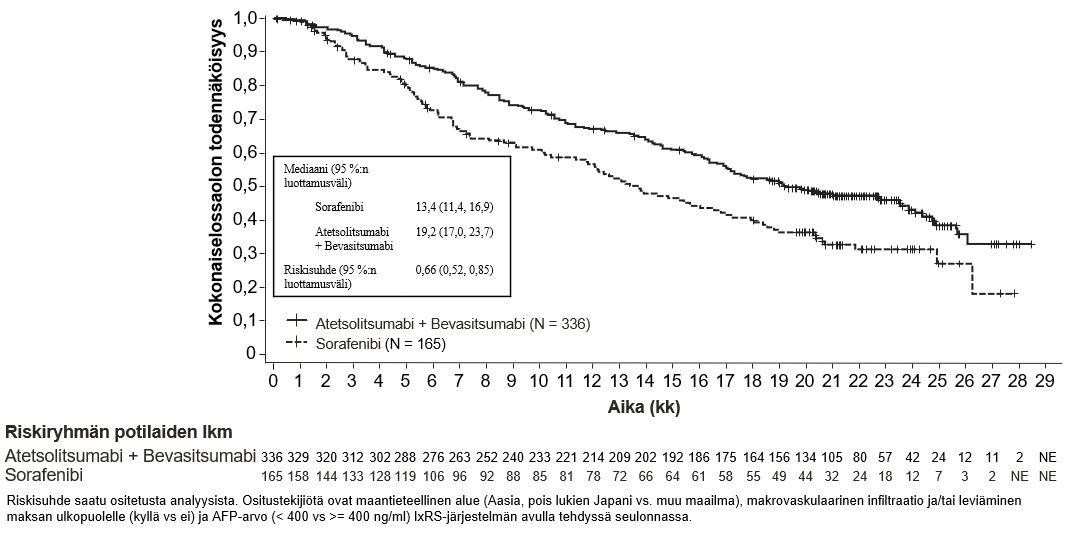
Kuva 22. Riippumattoman arviointilautakunnan RECIST v1.1 kriteerien mukaan arvioiman etenemättömyysajan Kaplan–Meier-käyrä ITT-potilasjoukossa (IMbrave150-tutkimuksen primaarianalyysi)
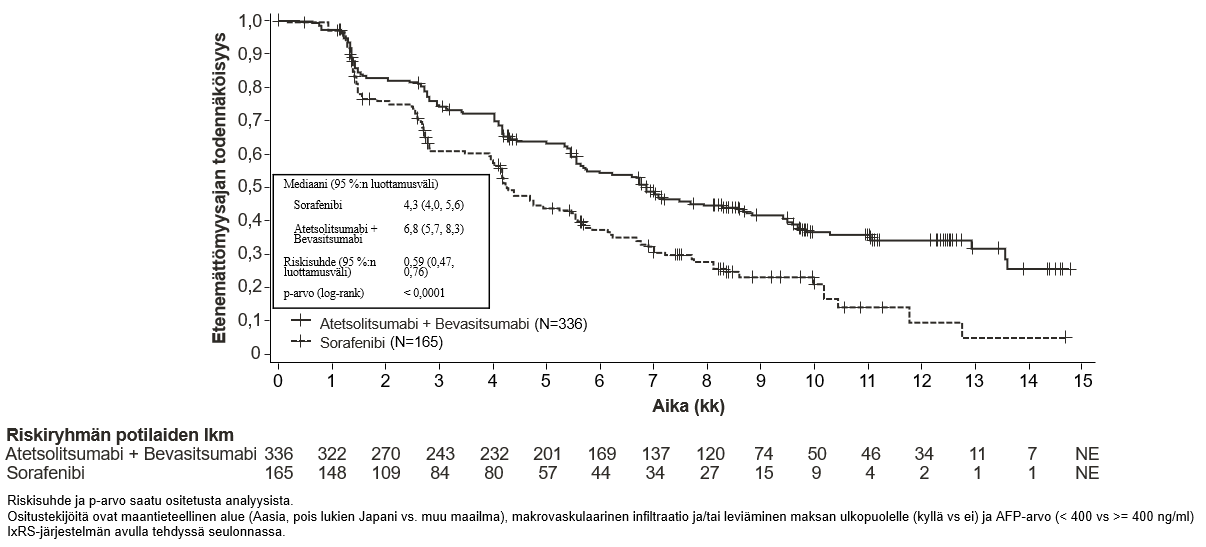
Hoidon teho iäkkäillä
Atetsolitsumabimonoterapiaa saavien ≥ 65-vuotiaiden ja nuorempien potilaiden välillä ei yleisesti havaittu hoidon tehoa koskevia eroja. IMpower150-tutkimuksessa ≥ 65 vuoden ikään liittyi atetsolitsumabin heikompi teho potilailla, jotka saivat atetsolitsumabia yhdessä karboplatiinin ja paklitakselin kanssa.
Tutkimuksista IMpower150, IMpower133 ja IMpower110 saadut tiedot ≥ 75-vuotiaista potilaita ovat liian suppeita, jotta tästä potilasjoukosta voisi tehdä päätelmiä.
Pediatriset potilaat
Varhaisvaiheen avoin monikeskustutkimus tehtiin atetsolitsumabihoidon turvallisuuden ja farmakokinetiikan selvittämiseksi; tutkimuksessa oli mukana pediatrisia (< 18 vuotta, n = 69) ja nuoria aikuisia potilaita (18–30 vuotta, n = 18), joilla oli uusiutuneita tai eteneviä kiinteitä kasvaimia sekä Hodgkinin lymfooma ja non-Hodgkin-lymfooma. Potilaat saivat hoitoa atetsolitsumabiannoksilla 15 mg/kg laskimoon 3 viikon välein (ks. kohta Farmakokinetiikka).
Farmakokinetiikka
Altistus atetsolitsumabille suureni suhteessa annokseen annosvälillä 1 mg/kg – 20 mg/kg, mukaan lukien vakioannos 1200 mg, jota annettiin 3 viikon välein. Potilasjoukon analyysissa, jossa oli mukana 472 potilasta, atetsolitsumabin annosvälin 1−20 mg/kg farmakokinetiikaksi kuvattiin lineaarisen kaksitila-altistusmalli ja ensimmäisen asteen eliminaatio. Laskimoon annettavien atetsolitsumabiannosten 840 mg 2 viikon välein, 1200 mg 3 viikon välein ja 1680 mg 4 viikon välein farmakokineettiset ominaisuudet ovat samat; kokonaisaltistukset näillä kolmella annostuksella oletetaan olevan verrannolliset. Populaatiofarmakokineettinen analyysi viittaa siihen, että vakaa tila saavutetaan useita annoksia annettaessa 6–9 viikossa. Systeeminen kertyminen oli pitoisuus-aikakäyrän alla olevan pinta-alan suhteen 1,91-kertainen, maksimipitoisuuden suhteen 1,46-kertainen ja pienimmän pitoisuuden suhteen 2,75-kertainen.
Imeytyminen
Atetsolitsumabi annetaan infuusiona laskimoon.
Jakautuminen
Populaatiofarmakokineettinen analyysi osoittaa, että tyypillisen potilaan keskustilan jakautumistilavuus on 3,28 l ja tilavuus vakaassa tilassa on 6,91 l.
Biotransformaatio
Atetsolitsumabin metaboliaa ei ole suoranaisesti tutkittu. Vasta-aineet poistuvat elimistöstä pääasiassa kataboloitumalla.
Eliminaatio
Populaatiofarmakokineettinen analyysi osoittaa, että atetsolitsumabin puhdistuma on 0,200 l/vrk ja tyypillinen terminaalisen eliminaation puoliintumisaika on 27 vuorokautta.
Erityispotilasryhmät
Seuraavilla tekijöillä ei populaatiofarmakokineettisen analyysin ja altistus–vasteanalyysin perusteella ole vaikutusta atetsolitsumabin farmakokinetiikkaan: ikä (21–89 vuotta), maantieteellinen alue, etninen tausta, munuaisten vajaatoiminta, lievä maksan vajaatoiminta, kasvaimen PD-L1:n ilmentymisen taso tai ECOG-suorituskykyluokka. Painolla, sukupuolella, vasta-ainepositiivisuudella, albumiinipitoisuuksilla ja kasvaintaakalla on tilastollisesti merkitsevä, mutta ei kliinisesti oleellista vaikutusta atetsolitsumabin farmakokinetiikkaan. Annosmuutoksia ei suositella.
Iäkkäät potilaat
Atetsolitsumabia ei ole tutkittu erityisesti iäkkäillä potilailla. Iän vaikutusta atetsolitsumabin farmakokinetiikkaan tutkittiin populaatiofarmakokineettisen analyysin avulla. Iän ei todettu olevan merkittävä atetsolitsumabin farmakokinetiikkaan vaikuttava yhteismuuttuja, mikä perustuu iältään 21−89-vuotiaaseen (n = 472) potilasjoukkoon, jonka iän mediaani oli 62 vuotta. Atetsolitsumabin farmakokinetiikassa ei havaittu kliinisesti merkittävää eroa, jos potilaan ikä oli < 65 vuotta (n = 274), 65−75 vuotta (n = 152) tai > 75 vuotta (n = 46) (ks. kohta Annostus ja antotapa).
Pediatriset potilaat
Farmakokineettiset tulokset pediatrisilla (< 18 vuotta, n = 69) ja nuorilla aikuisilla potilailla (18–30 vuotta, n = 18) tehdystä yhdestä varhaisvaiheen avoimesta monikeskustutkimuksesta osoittavat, että atetsolitsumabin puhdistuma ja jakautumistilavuus olivat painon mukaan normalisoituina verrannolliset annoksia 15 mg/kg saaneilla pediatrisilla potilailla ja 1 200 mg atetsolitsumabia 3 viikon välein saaneilla nuorilla aikuisilla potilailla, ja altistus oli pediatrisilla potilailla tyypillisesti pienempi, sillä paino laski. Näihin eroihin ei liittynyt atetsolitsumabipitoisuuksien laskua alle terapeuttisen tavoitealtistuksen. Alle 2vuotiaista lapsista on vähän tietoja, joten varmoja päätelmiä ei voida tehdä.
Munuaisten vajaatoiminta
Atetsolitsumabia ei ole tutkittu erityisesti munuaisten vajaatoimintaa sairastavilla potilailla. Atetsolitsumabin puhdistumassa ei havaittu populaatiofarmakokineettisessä analyysissä kliinisesti merkityksellisiä eroja, jos potilaalla oli lievä (laskennallinen glomerulusten suodatusnopeus [eGFR] 60–89 ml/min/1,73 m2; n = 208) tai keskivaikea (eGFR 30–59 ml/min/1,73 m2; n = 116) munuaisten vajaatoiminta verrattuna potilaisiin, joiden munuaisten toiminta oli normaali (eGFR vähintään 90 ml/min/1,73 m2; n = 140). Vain muutamalla potilaalla oli vaikea munuaisten vajaatoiminta (eGFR 15–29 ml/min/1,73 m2; n = 8) (ks. kohta Annostus ja antotapa). Vaikean munuaisten vajaatoiminnan vaikutusta atetsolitsumabin farmakokinetiikkaan ei tunneta.
Maksan vajaatoiminta
Atetsolitsumabia ei ole tutkittu erityisesti maksan vajaatoimintaa sairastavilla potilailla. Atetsolitsumabin puhdistumassa ei havaittu populaatiofarmakokineettisessä analyysissä kliinisesti tärkeitä eroja potilailla, joilla oli lievä maksan vajaatoiminta (bilirubiinipitoisuus ≤ ULN ja ASAT > ULN tai bilirubiinipitoisuus > 1,0–1,5 × ULN ja ASAT-arvo mikä tahansa) tai keskivaikea maksan vajaatoiminta (bilirubiinipitoisuus > 1,5–3 × ULN ja ASAT-arvo mikä tahansa), verrattuna potilaisiin, joiden maksan toiminta oli normaali (bilirubiinipitoisuus ≤ ULN ja ASAT ≤ ULN). Vaikeaa maksan vajaatoimintaa (bilirubiinipitoisuus > 3 × ULN ja ASAT-arvo mikä tahansa) sairastavista potilaista ei ole tietoja. Maksan vajaatoiminta määriteltiin maksan toimintahäiriöitä koskevien National Cancer Institute-Organ Dysfunction Working Group (NCI-ODWG) kriteerien mukaisesti (ks. kohta Annostus ja antotapa). Keskivaikean tai vaikean maksan vajaatoiminnan (bilirubiinipitoisuus > 1,5–3 × ULN ja ASAT-arvo mikä tahansa tai bilirubiinipitoisuus > 3 × ULN ja ASAT-arvo mikä tahansa) vaikutusta atetsolitsumabin farmakokinetiikkaan ei tunneta.
Prekliiniset tiedot turvallisuudesta
Karsinogeenisuus
Atetsolitsumabin karsinogeenisuuden selvittämiseksi ei ole tehty karsinogeenisuustutkimuksia.
Mutageenisuus
Atetsolitsumabin mutageenisuuden selvittämiseksi ei ole tehty mutageenisuustutkimuksia. Monoklonaaliset vasta-aineet eivät kuitenkaan oletettavasti muuta DNA:ta tai kromosomeja.
Hedelmällisyys
Atetsolitsumabilla ei ole tehty hedelmällisyystutkimuksia. Kroonisissa toksisuustutkimuksissa on kuitenkin tutkittu cynomolgus-apinaurosten ja -naaraiden lisääntymiselimiä. Atetsolitsumabin viikoittainen anto naarasapinoille potilaille suositeltujen annosten AUC-arvoon verrattuna 6-kertaisiksi arvioituina altistuksina (AUC) aiheutti epäsäännöllisen kiimakierron ja vastamuodostuneiden keltarauhasten puuttumisen munasarjoista, mikä oli korjautuvaa. Urosten lisääntymiselimissä ei todettu vaikutuksia.
Teratogeenisuus
Atetsolitsumabilla ei ole tehty lisääntymistä tai teratogeenisuutta koskevia eläinkokeita. Eläinkokeet ovat osoittaneet, että PDL1/PD-1-reitin estyminen voi aiheuttaa kehittyvään sikiöön kohdistuvaa immuunivälitteistä hylkimistä, mikä johtaa sikiön kuolemaan. Atetsolitsumabin antaminen voi vahingoittaa sikiötä ja aiheuttaa myös alkio- ja sikiökuolleisuutta.
Farmaseuttiset tiedot
Apuaineet
L-histidiini
Väkevä etikkahappo
Sakkaroosi
Polysorbaatti 20 (E 432)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta.
Laimennettu liuos
Käytönaikaiseksi kemialliseksi ja fysikaaliseksi säilyvyydeksi valmistamisen jälkeen on osoitettu enintään 24 tuntia ≤ 30 °C:ssa ja enintään 30 vuorokautta 2 °C – 8 °C:ssa.
Valmistettu infuusioliuos tulisi mikrobiologiselta kannalta käyttää heti. Jos valmistetta ei käytetä heti, käytönaikainen säilytysaika ja -olosuhteet ovat käyttäjän vastuulla eivätkä saa tavallisesti ylittää 24 tuntia 2 °C – 8 °C:ssa tai 8 tuntia vallitsevassa lämpötilassa (≤ 25 °C), paitsi jos valmiste on laimennettu kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä jääkaapissa (2 °C – 8 ºC).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TECENTRIQ infuusiokonsentraatti, liuosta varten
840 mg (L:ei) 14 ml (60 mg/ml) (2863,54 €)
1200 mg (L:ei) 20 ml (60 mg/ml) (3994,62 €)
PF-selosteen tieto
Tyypin I lasinen injektiopullo, jossa butyylikumitulppa ja alumiinisinetti, jossa vaaleansininen irti napsautettava muovilevy, ja joka sisältää 14 ml tai 20 ml infuusiokonsentraattia, liuosta varten.
Pakkauksessa yksi injektiopullo.
Valmisteen kuvaus:
Kirkas, väritön tai hieman kellertävä liuos. Liuoksen pH on 5,5–6,1 ja osmolaalisuus on 129–229 mosm/kg.
Käyttö- ja käsittelyohjeet
Tecentriq ei sisällä antimikrobiaalista säilytysainetta eikä bakteriostaattisia aineita, joten terveydenhuollon ammattilaisen pitää noudattaa sen valmistamisessa aseptista tekniikkaa käyttövalmiiksi sekoitetun liuoksen steriiliyden varmistamiseksi. Käytä Tecentriq-valmisteen valmisteluun steriiliä neulaa ja ruiskua.
Aseptinen valmistus, käsittely ja säilytys:
Infuusion valmistamisessa on varmistettava aseptinen käsittelytapa.
- Koulutuksen saanut henkilökunta valmistaa infuusion aseptisissa olosuhteissa hyviä toimintatapoja, etenkin parenteraalisten valmisteiden aseptista valmistamista koskevia toimintatapoja, noudattaen.
- Valmistaminen tehdään laminaarivirtauskaapissa tai biologisessa turvakaapissa laskimoon annettavien aineiden turvallista käsittelyä koskevia tavanomaisia varotoimia noudattaen.
- Valmistamisessa on noudatettava laskimoon annettavaksi valmistettuja infuusioliuoksia koskevia riittäviä säilytysolosuhteita, jotta varmistetaan aseptisten olosuhteiden säilyminen.
Ei saa ravistaa.
Laimennusohjeet
Suositusannos 840 mg: Injektiopullosta vedetään neljätoista millilitraa Tecentriq-konsentraattia, joka laimennetaan polyvinyylikloridista (PVC), polyolefiinista (PO), polyeteenistä (PE) tai polypropeenista (PP) valmistetussa infuusiopussissa 0,9-prosenttiseen natriumkloridi-injektioliuokseen (9 mg/ml).
Suositusannos 1200 mg: Injektiopullosta vedetään kaksikymmentä millilitraa Tecentriq-konsentraattia, joka laimennetaan polyvinyylikloridista (PVC), polyolefiinista (PO), polyeteenistä (PE) tai polypropeenista (PP) valmistetussa infuusiopussissa 0,9-prosenttiseen natriumkloridi-injektioliuokseen (9 mg/ml).
Suositusannos 1680 mg: Kahdesta Tecentriq 840 mg:n injektiopullosta vedetään kaksikymmentäkahdeksan millilitraa Tecentriq-konsentraattia, joka laimennetaan polyvinyylikloridista (PVC), polyolefiinista (PO), polyeteenistä (PE) tai polypropeenista (PP) valmistetussa infuusiopussissa 0,9-prosenttiseen natriumkloridi-injektioliuokseen (9 mg/ml).
Laimentamisen jälkeen liuoksen lopullisen pitoisuuden pitää olla 3,2–16,8 mg/ml.
Pussia on käänneltävä varovasti ylösalaisin liuoksen sekoittamiseksi, jotta vältetään vaahdonmuodostus. Infuusio on annettava heti sen käyttökuntoon saattamisen jälkeen (ks. kohta Kestoaika).
Parenteraaliset lääkevalmisteet on tarkistettava ennen antoa silmämääräisesti, ettei niissä ole hiukkasia ja värimuutoksia havaittavissa. Jos liuoksessa havaitaan hiukkasia tai värimuutoksia, sitä ei saa käyttää.
Tecentriq-valmisteen ja sellaisten infuusiopussien välillä, joiden valmisteeseen kosketuksissa oleva pinta on polyvinyylikloridia (PVC), polyolefiinia (PO), polyeteeniä (PE) tai polypropeenia (PP), ei ole havaittu yhteensopimattomuutta. Myöskään polyeetterisulfonista tai polysulfonista valmistettujen letkunsisäisten suodatinkalvojen tai PVC:stä, PE:stä, polybutadieenistä tai polyeetteriuretaanista valmistettujen infuusion annossa käytettyjen muiden laitteiden käytössä ei ole havaittu yhteensopimattomuutta. Letkunsisäisen suodatinkalvon käyttö on valinnaista.
Ei saa antaa samanaikaisesti muiden lääkevalmisteiden kanssa saman infuusioletkun kautta.
Hävittäminen
Tecentriq-valmisteen joutumista luontoon on vältettävä. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TECENTRIQ infuusiokonsentraatti, liuosta varten
840 mg 14 ml
1200 mg 20 ml
- Ei korvausta.
ATC-koodi
L01FF05
Valmisteyhteenvedon muuttamispäivämäärä
26.03.2026
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com