
LIBTAYO infuusiokonsentraatti, liuosta varten 350 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Huomioitavaa

Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta
Haittavaikutukset
, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi millilitra konsentraattia sisältää 50 mg semiplimabia.
Yksi injektiopullo sisältää 350 mg semiplimabia 7 ml:ssa.
Semiplimabi tuotetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasolususpensioviljelmässä.
Apuaineet, joiden vaikutus tunnetaan
Yksi 7 ml:n injektiopullo sisältää 105 mg L-proliinia ja 14 mg polysorbaatti 80:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Ihon okasolusyöpä
LIBTAYO on tarkoitettu monoterapiana ihon metastasoituneen tai paikallisesti edenneen okasolusyövän hoitoon aikuisille potilaille, kun potilas ei sovellu saamaan parantavaa leikkaushoitoa tai parantavaa sädehoitoa.
LIBTAYO on tarkoitettu monoterapiana adjuvanttihoitoon aikuisille potilaille, joilla on ihon okasolusyöpä (CSCC) ja suuri uusiutumisriski leikkauksen ja sädehoidon jälkeen (ks. kohdasta Farmakodynamiikka valintakriteerit).
Tyvisolusyöpä
LIBTAYO on tarkoitettu monoterapiana paikallisesti edenneen tai metastasoituneen tyvisolusyövän (laBCC tai mBCC) hoitoon aikuisille potilaille, joilla tauti on edennyt hedgehog-reitin estäjän (HHI) käytön aikana tai jotka eivät siedä sitä.
Ei-pienisoluinen keuhkosyöpä
LIBTAYO on tarkoitettu monoterapiana aikuispotilaille ei-pienisoluisen keuhkosyövän (NSCLC) ensilinjan hoitoon, kun keuhkosyöpä ilmentää PD-L1:tä (≥ 50 %:ssa kasvainsoluista) ilman EGFR-, ALK- tai ROS1-poikkeavuuksia ja potilailla on:
- paikallisesti edennyt NSCLC, kun potilas ei sovellu saamaan definitiivistä kemosädehoitoa, tai
- metastasoitunut NSCLC.
LIBTAYO on tarkoitettu yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa aikuispotilaille ei‑pienisoluisen keuhkosyövän (NSCLC) ensilinjan hoitoon, kun keuhkosyöpä ilmentää PD-L1:tä (≥ 1 %:ssa kasvainsoluista) ilman EGFR-, ALK- tai ROS1‑poikkeavuuksia ja potilailla on:
- paikallisesti edennyt NSCLC, kun potilas ei sovellu saamaan definitiivistä kemosädehoitoa, tai
- metastasoitunut NSCLC.
Kohdunkaulan syöpä
LIBTAYO on tarkoitettu monoterapiana kohdunkaulan uusiutuneen tai metastasoituneen syövän hoitoon aikuisille potilaille, joilla tauti on edennyt platinapohjaisen solunsalpaajahoidon aikana tai sen jälkeen.
Ehto
Hoito tulee aloittaa syövän hoitoon tarkoitettujen lääkevalmisteiden antamiseen perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Syövän hoitoon perehtyneen lääkärin on aloitettava hoito ja valvottava sen toteuttamista.
PD-L1-testaus NSCLC-potilailla
NSCLC-potilaiden soveltuvuus hoitoon on arvioitava kasvaimen PD-L1:n ilmentymisen perusteella, ja PD-L1:n ilmentyminen on vahvistettava validoidulla testillä (ks. kohta Farmakodynamiikka).
Annostus
Suositeltu annos
Paikallisesti edennyt tai metastasoitunut ihon okasolusyöpä, ei-pienisoluinen keuhkosyöpä., tyvisolusyöpä ja uusiutunut tai metastasoitunut kohdunkaulan syöpä
Suositeltu semiplimabiannos on 350 mg kolmen viikon välein ja se annetaan 30 minuuttia kestävänä infuusiona laskimoon.
Hoitoa voidaan jatkaa, kunnes sairaus etenee tai kunnes ilmenee toksisia vaikutuksia, joita ei voida hyväksyä.
Korkean riskin ihon okasolusyövän adjuvanttihoito
Suositeltu semiplimabiannos 30 minuuttia kestävänä infuusiona laskimoon:
- 350 mg 3 viikon välein 12 viikon ajan, jonka jälkeen 700 mg 6 viikon välein tai
- 350 mg 3 viikon välein.
Hoitoa voidaan jatkaa, kunnes tauti uusiutuu, ilmenee ei-hyväksyttävää toksisuutta, tai hoitoa on annettu enintään yhteensä 48 viikkoa.
Annoksen muuttaminen
Annoksen pienentämistä ei suositella. Yksilöllinen turvallisuus ja siedettävyys saattaa edellyttää hoidon lykkäämistä tai lopettamista. Suositukset annoksen muuttamisesta haittavaikutusten vuoksi on esitetty taulukossa 1.
Taulukossa 1 on esitetty yksityiskohtaiset ohjeet immuunivälitteisten haittavaikutusten hoitoon (ks. myös kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Taulukko 1: Suositellut hoitoon tehtävät muutokset
Haittavaikutusa | Vaikeusasteb | Annoksen muuttaminen | Muut toimenpiteet |
Immuunivälitteiset haittavaikutukset | |||
Pneumoniitti | Aste 2 | LIBTAYO-hoidosta pidättäytyminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain |
LIBTAYO-hoitoa jatketaan, jos pneumoniitti lievittyy asteeseen 0–1 ja pysyy tällä tasolla, kun kortikosteroidiannos on pienennetty asteittain korkeintaan 10 mg:aan prednisonia vuorokaudessa tai vastaavalle tasolle muuta hoitoa | |||
Aste 3 tai 4 tai uusiutunut aste 2 | Pysyvä lopettaminen | Aloitusannos 2–4 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain | |
Paksusuolitulehdus | Aste 2 tai 3 | LIBTAYO-hoidosta pidättäytyminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain |
LIBTAYO-hoitoa jatketaan, jos paksusuolitulehdus tai ripuli lievittyy asteeseen 0–1 ja pysyy tällä tasolla, kun kortikosteroidiannos on pienennetty asteittain korkeintaan 10 mg:aan prednisonia vuorokaudessa tai vastaavalle tasolle muuta hoitoa | |||
Aste 4 tai uusiutunut aste 3 | Pysyvä lopettaminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain | |
Maksatulehdus | Aste 2 ja ASAT tai ALAT >3 – 5 × ULN tai kokonaisbilirubiini >1,5 – 3 × ULN | LIBTAYO-hoidosta pidättäytyminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain |
LIBTAYO-hoitoa jatketaan, jos maksatulehdus lievittyy asteeseen 0–1 ja pysyy tällä tasolla, kun kortikosteroidiannos on pienennetty asteittain korkeintaan 10 mg:aan prednisonia vuorokaudessa tai vastaavalle tasolle muuta hoitoa, tai ASAT- tai ALAT-arvo palautuu lähtötasolle kortikosteroidihoidon asteittaisen lopettamisen jälkeen | |||
Aste ≥3 ja ASAT tai ALAT >5 × ULN tai kokonaisbilirubiini >3 × ULN | Pysyvä lopettaminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain | |
Hypotyreoosi | Aste 3 tai 4 | LIBTAYO-hoidosta pidättäytyminen | Aloitetaan kilpirauhashormonikorvaushoito kliinisen tarpeen mukaan |
LIBTAYO-hoitoa jatketaan, kun hypotyreoosi lievittyy asteeseen 0 tai 1 tai on muuten kliinisesti vakaa | |||
Hypertyreoosi | Aste 3 tai 4 | LIBTAYO-hoidosta pidättäytyminen | Aloitetaan oireenmukainen hoito |
LIBTAYO-hoitoa jatketaan, kun hypertyreoosi lievittyy asteeseen 0 tai 1 tai on muuten kliinisesti vakaa | |||
Kilpirauhastulehdus | Aste 3 tai 4 | LIBTAYO-hoidosta pidättäytyminen | Aloitetaan oireenmukainen hoito |
LIBTAYO-hoitoa jatketaan, kun kilpirauhastulehdus lievittyy asteeseen 0 tai 1 tai on muuten kliinisesti vakaa | |||
Hypofysiitti | Aste 2–4 | LIBTAYO-hoidosta pidättäytyminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain, lisäksi hormonikorvaushoito kliinisen tarpeen mukaan |
LIBTAYO-hoitoa jatketaan, jos hypofysiitti lievittyy asteeseen 0–1 ja pysyy tällä tasolla, kun kortikosteroidiannos on pienennetty asteittain korkeintaan 10 mg:aan prednisonia vuorokaudessa tai vastaavalle tasolle muuta hoitoa, tai on muuten kliinisesti vakaa | |||
Lisämunuaisten vajaatoiminta | Aste 2–4 | LIBTAYO-hoidosta pidättäytyminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain, lisäksi hormonikorvaushoito kliinisen tarpeen mukaan |
LIBTAYO-hoitoa jatketaan, jos lisämunuaisten vajaatoiminta lievittyy asteeseen 0–1 ja pysyy tällä tasolla, kun kortikosteroidiannos on pienennetty asteittain korkeintaan 10 mg:aan prednisonia vuorokaudessa tai vastaavalle tasolle muuta hoitoa, tai on muuten kliinisesti vakaa | |||
Tyypin 1 diabetes | Aste 3 tai 4 (hyperglykemia) | LIBTAYO-hoidosta pidättäytyminen | Aloitetaan hoito veren glukoosipitoisuutta alentavilla lääkkeillä kliinisen tarpeen mukaan |
LIBTAYO-hoitoa jatketaan, kun diabetes lievittyy asteeseen 0 tai 1 tai on muuten kliinisesti vakaa | |||
Haittavaikutukset iholla | Aste 2 ja kesto yli 1 viikko, aste 3 tai epäilty Stevens–Johnsonin oireyhtymä (SJS) tai toksinen epidermaalinen nekrolyysi (TEN) | LIBTAYO-hoidosta pidättäytyminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain |
LIBTAYO-hoitoa jatketaan, jos ihoreaktio lievittyy asteeseen 0–1 ja pysyy tällä tasolla, kun kortikosteroidiannos on pienennetty asteittain korkeintaan 10 mg:aan prednisonia vuorokaudessa tai vastaavalle tasolle muuta hoitoa | |||
Aste 4 tai varmistettu SJS tai TEN | Pysyvä lopettaminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain | |
Immuunivälitteinen ihoreaktio tai muut immuunivälitteiset haittavaikutukset potilailla, jotka ovat aiemmin saaneet idelalisibia | Aste 2 | LIBTAYO-hoidosta pidättäytyminen | Aloitetaan välittömästi hoito, johon kuuluu prednisoni aloitusannoksena 1–2 mg/kg/vrk tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain |
LIBTAYO-hoitoa jatketaan, jos ihoreaktio tai muu immuunivälitteinen haittavaikutus lievittyy asteeseen 0–1 ja pysyy tällä tasolla, kun kortikosteroidiannos on pienennetty asteittain korkeintaan 10 mg:aan prednisonia vuorokaudessa tai vastaavalle tasolle muuta hoitoa | |||
Aste 3 tai 4 (umpierityssairauksia lukuun ottamatta) tai uusiutunut aste 2 | Pysyvä lopettaminen | Aloitetaan välittömästi hoito, johon kuuluu prednisoni aloitusannoksena 1–2 mg/kg/vrk tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain | |
Munuaistulehdus, johon liittyy munuaisen toimintahäiriö | Asteen 2 kreatiniiniarvon suureneminen | LIBTAYO-hoidosta pidättäytyminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain |
LIBTAYO-hoitoa jatketaan, jos munuaistulehdus lievittyy asteeseen 0–1 ja pysyy tällä tasolla, kun kortikosteroidiannos on pienennetty asteittain korkeintaan 10 mg:aan prednisonia vuorokaudessa tai vastaavalle tasolle muuta hoitoa | |||
Asteen 3 tai 4 kreatiniiniarvon suureneminen | Pysyvä lopettaminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito, minkä jälkeen annosta pienennetään asteittain | |
Muut immuunivälitteiset haittavaikutukset (kuten mm. paraneoplastinen enkefalomyeliitti, aivokalvotulehdus, lihastulehdus, kiinteän elinsiirteen hyljintä, käänteishyljintä, Guillain–Barrén oireyhtymä, keskushermoston tulehdus, krooninen tulehduksellinen demyelinoiva polyradikuloneuropatia, enkefaliitti, myasthenia gravis, perifeerinen neuropatia, sydänlihastulehdus, sydänpussitulehdus, immuunitrombosytopenia, vaskuliitti, nivelsärky, niveltulehdus, lihasheikkous, myalgia, polymyalgia rheumatica, Sjögrenin oireyhtymä, kutina, sarveiskalvotulehdus, immuunivälitteinen mahatulehdus, suutulehdus ja hemofagosyyttinen lymfohistiosytoosi) | Aste 2 tai 3 reaktion tyypin mukaan | LIBTAYO-hoidosta pidättäytyminen | Aloitetaan oireenmukainen hoito, johon kuuluu prednisoni aloitusannoksena 1–2 mg/kg/vrk tai vastaava hoito kliinisen tarpeen mukaan, minkä jälkeen annosta pienennetään asteittain |
LIBTAYO-hoitoa jatketaan, jos muu immuunivälitteinen haittavaikutus lievittyy asteeseen 0–1 ja pysyy tällä tasolla, kun kortikosteroidiannos on pienennetty asteittain korkeintaan 10 mg:aan prednisonia vuorokaudessa tai vastaavalle tasolle muuta hoitoa | |||
| Pysyvä lopettaminen | Aloitusannos 1–2 mg/kg/vrk prednisonia tai vastaava hoito kliinisen tarpeen mukaan, minkä jälkeen annosta pienennetään asteittain | |
Infuusioon liittyvät reaktiota | |||
Infuusioon liittyvä reaktio | Aste 1 tai 2 Aste 3 tai 4 | Infuusio keskeytettävä tai infuusionopeutta pienennettävä Pysyvä lopettaminen | Aloitetaan oireenmukainen hoito |
ALAT: alaniiniaminotransferaasi; ASAT: aspartaattiaminotransferaasi; ULN: viitealueen yläraja.
a. Ks. myös kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset.
b. Haittavaikutusten vaikeusasteluokat on arvioitava voimassa olevien NCI-CTCAE-kriteerien (National Cancer Institute Common Terminology Criteria for Adverse Events) mukaisesti.
Potilaskortti
Kaikkien LIBTAYO-valmistetta määräävien henkilöiden on tutustuttava koulutusmateriaaliin ja kerrottava potilaille potilaskortista, jossa selitetään, miten potilaan on toimittava, jos hänellä ilmenee oireita, jotka viittaavat immuunivälitteisiin haittavaikutuksiin tai infuusioon liittyviin reaktioihin. Lääkäri antaa jokaiselle potilaalle potilaskortin.
Erityisryhmät
Pediatriset potilaat
LIBTAYO-valmisteen turvallisuutta ja tehoa lasten ja alle 18‑vuotiaiden nuorten hoidossa ei ole varmistettu.
Saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta.
Iäkkäät potilaat
Annoksen muuttamista iäkkäillä potilailla ei suositella. Semiplimabialtistus on vastaavanlainen kaikissa ikäryhmissä (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka). Semiplimabimonoterapiasta ≥75-vuotiailla potilailla on vain vähän tietoa.
Munuaisten vajaatoiminta
LIBTAYO-annoksen muuttamista ei suositella munuaisten vajaatoimintaa sairastavilla potilailla. LIBTAYO-valmisteen käytöstä vaikeaa munuaisten vajaatoimintaa sairastaville potilaille, joilla kreatiniinipuhdistuma on 15–29 ml/min, on vain vähän tietoa (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttamista ei suositella lievää tai kohtalaista maksan vajaatoimintaa sairastavilla potilailla. LIBTAYO-valmistetta ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla. Vaikeaa maksan vajaatoimintaa sairastavista potilaista ei ole riittävästi tietoa annossuositusten antamiseen (ks. kohta Farmakokinetiikka).
Antotapa
LIBTAYO annetaan laskimoon. LIBTAYO annetaan infuusiona laskimoon 30 minuutin aikana käyttämällä laskimoyhteyttä, jossa on steriili, pyrogeeniton, vähän proteiineja sitova in-line- tai add-on-suodatin (huokoskoko 0,2–5 mikronia).
Samalla infuusiolinjalla ei saa antaa samanaikaisesti muita lääkevalmisteita.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet listatuille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Immuunivälitteiset haittavaikutukset
Semiplimabin käytön yhteydessä on havaittu vaikeita ja kuolemaan johtaneita immuunivälitteisiä haittavaikutuksia (ks. kohta Annostus ja antotapa ja kohta Haittavaikutukset). Tällaiset immuunivälitteiset reaktiot saattavat liittyä mihin tahansa elimiin. Immuunivälitteiset reaktiot voivat ilmetä milloin tahansa semiplimabihoidon aikana, mutta immuunivälitteisiä haittavaikutuksia voi ilmaantua myös semiplimabihoidon lopettamisen jälkeen.
Ohjeet immuunivälitteisistä haittavaikutuksista koskevat semiplimabin käyttöä sekä monoterapiana että yhdistelmähoitona solunsalpaajahoidon kanssa.
Semiplimabia tai muita PD-1:n/PD-L1:n estäjiä saaneilla potilailla voi ilmetä samanaikaisesti useampaan kuin yhteen elinjärjestelmään kohdistuvia immuunivälitteisiä haittavaikutuksia, kuten lihastulehdus ja sydänlihastulehdus tai myasthenia gravis.
Potilaita on tarkkailtava immuunivälitteisiin haittavaikutuksiin viittaavien oireiden ja löydösten varalta. Immuunivälitteisiä haittavaikutuksia on hoidettava muuttamalla semiplimabihoitoa ja antamalla hormonikorvaushoitoa (jos se on kliinisesti tarpeen) ja kortikosteroideja. Jos epäillään immuunivälitteistä haittavaikutusta, potilas on tutkittava immuunivälitteisen haittavaikutuksen varmistamiseksi ja muiden mahdollisten syiden, kuten infektion, poissulkemiseksi. Haittavaikutuksen vaikeusasteen mukaan semiplimabihoidosta on pidättäydyttävä toistaiseksi tai semiplimabihoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Havainnoivista tutkimuksista saadut tiedot viittaavat siihen, että potilailla, joilla on jo ennestään autoimmuunisairaus, immuunivälitteisten haittavaikutusten riski saattaa olla suurempi immuunijärjestelmän tarkistuspisteen estäjähoidon jälkeen verrattuna haittavaikutusten riskiin potilailla, joilla ei ole ennestään autoimmuunisairautta. Lisäksi taustalla olevan autoimmuunisairauden pahenemisvaiheet olivat yleisiä, mutta suurin osa niistä oli lieviä ja hoidettavissa olevia.
Immuunivälitteinen pneumoniitti
Semiplimabia saaneilla potilailla on havaittu immuunivälitteistä pneumoniittia, joka määritellään siten, että sen hoito edellyttää kortikosteroidien käyttöä eikä sille ole selviä vaihtoehtoisia syitä, ja jotkin tapauksista ovat johtaneet kuolemaan (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava pneumoniittiin viittaavien oireiden ja löydösten varalta, ja muut kuin immuunivälitteiseen pneumoniittiin viittaavat syyt on suljettava pois. Jos epäillään pneumoniittia, potilaalle on tehtävä kuvantamistutkimus kliiniseen arvioon perustuvan tarpeen mukaan ja potilasta on hoidettava muuttamalla semiplimabihoitoa ja antamalla kortikosteroideja (ks. kohta Annostus ja antotapa).
Immuunivälitteinen paksusuolitulehdus
Semiplimabia saaneilla potilailla on havaittu immuunivälitteistä ripulia tai paksusuolitulehdusta, jotka määritellään siten, että niiden hoito edellyttää kortikosteroidien käyttöä eikä niille ole selviä vaihtoehtoisia syitä (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava ripuliin tai paksusuolitulehdukseen viittaavien oireiden ja löydösten varalta, ja potilasta on hoidettava muuttamalla semiplimabihoitoa ja antamalla ripulin hoitoon tarkoitettuja lääkkeitä ja kortikosteroideja (ks. kohta Annostus ja antotapa).
Immuunivälitteinen maksatulehdus
Semiplimabia saaneilla potilailla on havaittu immuunivälitteistä maksatulehdusta, joka määritellään siten, että sen hoito edellyttää kortikosteroidien käyttöä eikä sille ole selviä vaihtoehtoisia syitä, ja jotkin tapauksista ovat johtaneet kuolemaan (ks. kohta Haittavaikutukset). Potilaalle on tehtävä maksan toimintakokeet poikkeavuuksien varalta ennen hoitoa ja säännöllisesti hoidon aikana kliiniseen arvioon perustuvan tarpeen mukaan, ja potilasta on hoidettava muuttamalla semiplimabihoitoa ja antamalla kortikosteroideja (ks. kohta Annostus ja antotapa).
Immuunivälitteiset umpierityssairaudet
Semiplimabia saaneilla potilailla on havaittu immuunivälitteisiä umpierityssairauksia, joiden määritelmä on, että ne ovat hoidon aikana alkaneita tai pahentuneita umpierityssairauksia, joille ei ole selviä vaihtoehtoisia syitä (ks. kohta Haittavaikutukset).
Kilpirauhasen toimintahäiriöt (hypotyreoosi/hypertyreoosi/kilpirauhastulehdus)
Semiplimabia saaneilla potilailla on havaittu immuunivälitteisiä kilpirauhasen toimintahäiriöitä, ja niitä voi ilmetä milloin tahansa hoidon aikana. Kilpirauhastulehdus ei välttämättä näy kilpirauhasen toimintakokeissa. Hypotyreoosi voi seurata hypertyreoosin jälkeen. Potilaiden kilpirauhasen toiminnan muutoksia on seurattava hoidon alkaessa ja säännöllisesti hoidon aikana kliiniseen arvioon perustuvan tarpeen mukaan (ks. kohta Haittavaikutukset). Potilasta on hoidettava antamalla hormonikorvaushoitoa (tarvittaessa) ja muuttamalla semiplimabihoitoa. Hypertyreoosi hoidetaan normaalin hoitokäytännön mukaisesti (ks. kohta Annostus ja antotapa).
Hypofysiitti
Semiplimabia saaneilla potilailla on havaittu immuunivälitteistä hypofysiittiä (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava hypofysiittiin viittaavien oireiden ja löydösten varalta, ja potilasta on hoidettava muuttamalla semiplimabihoitoa sekä antamalla kortikosteroideja ja hormonikorvaushoitoa kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa).
Lisämunuaisten vajaatoiminta
Semiplimabia saaneilla potilailla on havaittu lisämunuaisten vajaatoimintaa (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava lisämunaisten vajaatoimintaan viittaavien oireiden ja löydösten varalta hoidon aikana ja sen jälkeen, ja potilasta on hoidettava muuttamalla semiplimabihoitoa sekä antamalla kortikosteroideja ja hormonikorvaushoitoa kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa).
Tyypin 1 diabetes
Semiplimabia saaneilla potilailla on havaittu immuunivälitteistä tyypin 1 diabetesta, myös diabeettista ketoasidoosia (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava hyperglykemian ja diabetekseen viittaavien oireiden ja löydösten varalta kliiniseen arvioon perustuvan tarpeen mukaan, ja potilasta on hoidettava veren glukoosipitoisuutta alentavilla lääkkeillä tai insuliinilla ja muuttamalla semiplimabihoitoa (ks. kohta Annostus ja antotapa).
Immuunivälitteiset haittavaikutukset iholla
Immuunivälitteisiä iholla ilmeneviä haittavaikutuksia, jotka määritellään siten, että niiden hoito edellyttää systeemisten kortikosteroidien käyttöä eikä niille ole selviä vaihtoehtoisia syitä, on raportoitu semiplimabihoidon yhteydessä, mukaan lukien vaikeita ihoreaktioita, kuten Stevens–Johnsonin oireyhtymää (SJS) ja toksista epidermaalista nekrolyysiä (TEN) (osa tapauksista kuolemaan johtaneita), ja muita reaktioita iholla, kuten ihottumaa, erythema multiformea ja pemfigoidia (ks. kohta Haittavaikutukset).
Potilasta on tarkkailtava epäiltyihin vaikeisiin ihoreaktioihin viittaavien oireiden varalta ja muut syyt on suljettava pois. Potilasta on hoidettava muuttamalla semiplimabihoitoa ja antamalla kortikosteroideja (ks. kohta Annostus ja antotapa). Jos potilaalla on Stevens–Johnsonin oireyhtymän tai toksisen epidermaalisen nekrolyysin oireita, hänet on ohjattava erikoissairaanhoidon piiriin arviointia ja hoitoa varten, ja hoitoon on tehtävä muutoksia (ks. kohta Annostus ja antotapa).
Stevens–Johnsonin oireyhtymää, kuolemaan johtanutta toksista epidermaalista nekrolyysiä ja suutulehduksia on ilmennyt yhden semiplimabiannoksen saamisen jälkeen aiemmin idelalisibia saaneilla potilailla, jotka osallistuivat kliiniseen tutkimukseen, jossa arvioitiin semiplimabia non-Hodgkin-lymfooman (NHL) hoidossa, ja jotka olivat saaneet sulfa-antibiootteja lyhyen ajan sisällä ennen tutkimukseen osallistumista (ks. kohta Haittavaikutukset). Potilasta on hoidettava muuttamalla semiplimabihoitoa ja antamalla kortikosteroideja, kuten edellä on kuvattu (ks. kohta Annostus ja antotapa).
Immuunivälitteinen munuaistulehdus
Semiplimabia saaneilla potilailla on havaittu immuunivälitteistä munuaistulehdusta, myös yksi kuolemaan johtanut tapaus. Immuunivälitteinen munuaistulehdus määritellään siten, että sen hoito edellyttää kortikosteroidien käyttöä eikä sille ole selviä vaihtoehtoisia syitä (ks. kohta Haittavaikutukset). Potilasta on tarkkailtava munuaisten toiminnan muutosten varalta. Potilasta on hoidettava muuttamalla semiplimabihoitoa ja antamalla kortikosteroideja (ks. kohta Annostus ja antotapa).
Muut immuunivälitteiset haittavaikutukset
Semiplimabia saaneilla potilailla on havaittu muita kuolemaan johtaneita ja henkeä uhanneita immuunivälitteisiä haittavaikutuksia, kuten paraneoplastista enkefalomyeliittiä, aivokalvotulehdusta, lihastulehdusta, sydänlihastulehdusta ja haimatulehdusta (ks. kohta Haittavaikutukset, muut immuunivälitteiset haittavaikutukset).
Muiden PD-1:n/PD-L1:n estäjien käytön yhteydessä on ilmoitettu ei-infektiivistä virtsarakkotulehdusta.
Epäillyt immuunivälitteiset haittavaikutukset on arvioitava muiden syiden poissulkemiseksi. Potilasta on seurattava immuunivälitteisiin haittavaikutuksiin viittaavien oireiden ja löydösten varalta ja hoidettava muuttamalla semiplimabihoitoa ja antamalla kortikosteroideja kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa ja kohta Haittavaikutukset).
PD-1:n estäjiä saaneilla potilailla on myyntiluvan myöntämisen jälkeisessä seurannassa ilmoitettu kiinteiden elinsiirteiden hyljintää. Semiplimabihoito saattaa suurentaa hyljinnän riskiä potilailla, joille on tehty kiinteän elimen siirto. Näiden potilaiden kohdalla on harkittava semiplimabihoidon hyötyjä verrattuna mahdollisen elinsiirteen hyljinnän riskiin. Allogeenisen hematopoieettisten kantasolujen siirron yhteydessä muita PD-1:n/PD-L1:n estäjiä saaneilla potilailla on myyntiluvan myöntämisen jälkeen ilmoitettu käänteishyljintätapauksia.
Semiplimabia saaneilla potilailla on ilmoitettu hemofagosyyttista lymfohistiosytoosia (HLH) (ks. kohta Haittavaikutukset). Potilasta on seurattava hemofagosyyttiseen lymfohistiosytoosiin viittaavien kliinisten oireiden ja löydösten varalta. Jos kyseinen tila varmistuu, semiplimabin anto on lopetettava ja hemofagosyyttisen lymfohistiosytoosin hoito on aloitettava (ks. kohta Annostus ja antotapa).
Infuusioon liittyvät reaktiot
Semiplimabi voi aiheuttaa vaikeita tai hengenvaarallisia infuusioon liittyviä reaktioita (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava infuusioon liittyviin reaktioihin viittaavien oireiden ja löydösten varalta, ja potilaita on hoidettava muuttamalla semiplimabihoitoa ja antamalla kortikosteroideja. Semiplimabi-infuusio on keskeytettävä tai infuusionopeutta on pienennettävä, jos potilaalla ilmenee lievä tai kohtalainen infuusioon liittyvä reaktio. Infuusion anto on lopetettava ja semiplimabihoito on lopetettava pysyvästi, jos potilaalla ilmenee vaikea (vaikeusasteen 3) tai hengenvaarallinen (vaikeusasteen 4) infuusioon liittyvä reaktio (ks. kohta Annostus ja antotapa).
Kliinisistä tutkimuksista poissuljetut potilaat
Potilaat, joilla oli aktiivinen infektio, immuunipuutos, joilla oli ollut autoimmuunisairauksia, ECOG-toimintakykyluokka ≥2 tai aikaisempi interstitiaalinen keuhkosairaus, suljettiin pois. Täydellinen luettelo tutkimuksista poissuljetuista potilaista, ks. kohta Farmakodynamiikka.
Koska semiplimabin käytöstä näille potilasryhmille ei ole tietoja, sen käytössä on noudatettava varovaisuutta, ja käytön on perustuttava hyötyjen ja riskien potilaskohtaiseen huolelliseen arviointiin.
Apuaineet
Yksi 7 ml:n injektiopullo sisältää 105 mg L-proliinia ja 14 mg polysorbaatti 80:tä.
L-proliini voi olla haitallinen tyypin I tai tyypin II hyperprolinemiaa sairastavilla potilailla.
Tämä lääkevalmiste sisältää polysorbaatti 80:tä, joka saattaa aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Systeemisten kortikosteroidien tai immuunisalpaajien käyttöä, lukuun ottamatta systeemisiä kortikosteroideja fysiologisilla annoksilla (≤ 10 mg/vrk prednisonia tai vastaava hoito), on vältettävä ennen semiplimabihoidon aloittamista, koska ne saattavat heikentää semiplimabin farmakodynaamista vaikutusta ja tehoa. Systeemisiä kortikosteroideja tai muita immuunisalpaajia voidaan kuitenkin käyttää semiplimabihoidon aloittamisen jälkeen immuunivälitteisten haittavaikutusten hoitoon (ks. kohta Annostus ja antotapa).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä semiplimabihoidon aikana ja vähintään neljän kuukauden ajan viimeisen semiplimabiannoksen saamisen jälkeen.
Raskaus
Semiplimabilla ei ole tehty eläinten lisääntymistutkimuksia. Tietoja semiplimabin käytöstä raskaana oleville naisille ei ole saatavilla. Eläinkokeissa on osoitettu, että PD-1-/PD-L1-reitin inhibointi voi suurentaa kehittyvän sikiön kuolemaan johtavan immuunivälitteisen hyljintäreaktion riskiä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Ihmisen IgG4:n tiedetään läpäisevän veri-istukkaesteen. Semiplimabi on IgG4, joten se saattaa siirtyä äidistä kehittyvään sikiöön. Semiplimabin käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä tehokasta ehkäisyä, ellei kliinisen hyödyn katsota olevan mahdollista riskiä suurempi.
Imetys
Ei tiedetä, erittyykö semiplimabi ihmisen rintamaitoon. Vasta-aineiden (IgG4 mukaan lukien) tiedetään erittyvän ihmisen rintamaitoon, joten rintaruokittavaan vastasyntyneeseen/vauvaan kohdistuvia riskejä ei voida poissulkea.
Jos imettävä nainen valitsee semiplimabihoidon, hänelle on kerrottava, ettei hän saa imettää semiplimabihoidon aikana eikä vähintään 4 kuukauteen viimeisen annoksen saamisen jälkeen.
Hedelmällisyys
Semiplimabin mahdollisista vaikutuksista hedelmällisyyteen ei ole saatavilla kliinisiä tietoja. Kolmen kuukauden mittaisessa toistuvilla annoksilla tehdyssä hedelmällisyyttä arvioineessa tutkimuksessa ei havaittu vaikutuksia hedelmällisyyden arviointiin liittyvissä muuttujissa tai urosten ja naaraiden sukupuolielimissä sukukypsyyden saavuttaneilla cynomolgus-apinoilla.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Semiplimabilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Semiplimabihoidon jälkeen on raportoitu ilmenneen väsymystä (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Semiplimabin käytön yhteydessä voi ilmetä immuunivälitteisiä haittavaikutuksia. Useimmat niistä, vaikeat reaktiot mukaan lukien, ovat hävinneet asianmukaisen hoidon aloittamisen tai semiplimabihoidon lopettamisen jälkeen (ks. Valikoitujen haittavaikutusten kuvaus jäljempänä).
Semiplimabi monoterapiana
Semiplimabimonoterapian turvallisuutta on arvioitu viidessä kliinisessä tutkimuksessa, joissa semiplimabia annettiin monoterapiana 1 281 potilaalle, joilla oli edennyt kiinteä pahanlaatuinen kasvain. Semiplimabi-altistuksen mediaanikesto oli 28 viikkoa (vaihteluväli: 2 päivää – 144 viikkoa).
Kliinisissä tutkimuksissa 21 %:lla semiplimabia saaneista potilaista ilmeni immuunivälitteisiä haittavaikutuksia, joihin sisältyi vaikeusasteen 5 (0,3 %), 4 (0,6 %), 3 (5,7 %) ja 2 (11,2 %) haittavaikutuksia. Immuunivälitteiset haittavaikutukset johtivat semiplimabihoidon pysyvään lopettamiseen 4,6 %:lla potilaista. Yleisimmät immuunivälitteiset haittavaikutukset olivat hypotyreoosi (6,8 %), hypertyreoosi (3,0 %), immuunivälitteinen pneumoniitti (2,6 %), immuunivälitteinen hepatiitti (2,4 %), immuunivälitteinen paksusuolitulehdus (2,0 %) ja immuunivälitteiset ihoreaktiot (1,9 %) (ks. Valikoitujen haittavaikutusten kuvaus jäljempänä, varoitukset ja käyttöön liittyvät varotoimet kohdassa Varoitukset ja käyttöön liittyvät varotoimet ja suositellut hoitoon tehtävät muutokset kohdassa Annostus ja antotapa).
Haittavaikutukset olivat vakavia 32,4 %:lla potilaista.
Haittavaikutukset johtivat semiplimabihoidon pysyvään lopettamiseen 9,4 %:lla potilaista.
Semiplimabihoidon yhteydessä on ilmoitettu vakavia ihoreaktioita, kuten Stevens–Johnsonin oireyhtymää (SJS) ja toksista epidermaalista nekrolyysiä (TEN) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Semiplimabi ihon okasolusyövän adjuvanttihoitona
Semiplimabin turvallisuutta monoterapiana adjuvanttihoitona ihon okasolusyöpää sairastavilla potilailla, joilla oli suuri uusiutumisriski, arvioitiin C-POST-tutkimuksessa 205 potilaalla. Altistuksen keston mediaani oli 47,9 viikkoa (vaihteluväli: 3–52 viikkoa) semiplimabiryhmässä.
Semiplimabin turvallisuusprofiili C-POST-tutkimuksen adjuvanttihoitoasetelmassa vastaa edenneitä syöpiä sairastavien semiplimabihoidon tunnettua turvallisuusprofiilia. Semiplimabin immuunivälitteisten haittavaikutusten esiintyvyys monoterapiana C-POST-tutkimuksessa oli 22,9 %. Niiden esiintyvyys oli 20,8 % monoterapiapopulaatiossa, jossa oli edenneitä kiinteitä maligniteetteja.
Haittavaikutukset olivat vakavia 17,6 %:lla potilaista.
Haittavaikutukset johtivat semiplimabihoidon pysyvään lopettamiseen 9,8 %:lla potilaista.
Semiplimabi yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa
Semiplimabin ja platinapohjaisen solunsalpaajahoidon yhdistelmän turvallisuutta on arvioitu kliinisessä tutkimuksessa, johon osallistui 465 potilasta, joilla oli paikallisesti edennyt tai metastasoitunut NSCLC. Altistuksen mediaanikesto oli 38,5 viikkoa (10 päivää – 102,6 viikkoa) semiplimabia ja solunsalpaajahoitoa saaneiden potilaiden ryhmässä ja 21,3 viikkoa (4 päivää – 95 viikkoa) solunsalpaajahoitoa saaneiden ryhmässä.
18,9 %:lla potilaista ilmeni immuunivälitteisiä haittavaikutuksia, joihin sisältyi vaikeusasteen 5 (0,3 %), 3 (2,6 %) ja 2 (7,4 %) haittavaikutuksia. Immuunivälitteiset haittavaikutukset johtivat semiplimabihoidon pysyvään lopettamiseen 1,0 %:lla potilaista. Yleisimmät immuunivälitteiset haittavaikutukset olivat hypotyreoosi (7,7 %), hypertyreoosi (5,1 %), kohonnut veren TSH-arvo (4,2 %), immuunivälitteiset ihoreaktiot (1,9 %), immuunivälitteinen pneumoniitti (1,9 %) ja alentunut veren TSH-arvo (1,6 %) (ks. Valikoitujen haittavaikutusten kuvaus jäljempänä, varoitukset ja käyttöön liittyvät varotoimet kohdassa Varoitukset ja käyttöön liittyvät varotoimet ja suositellut hoitoon tehtävät muutokset kohdassa Annostus ja antotapa).
Haittavaikutukset olivat vakavia 25,3 %:lla potilaista.
Haittavaikutukset johtivat semiplimabihoidon pysyvään lopettamiseen 5,1 %:lla potilaista.
Haittavaikutustaulukko
Taulukossa 2 on lueteltu haittavaikutusten ilmaantuvuus monoterapian turvallisuutta koskevassa aineistossa ja potilailla, jotka saivat semiplimabia yhdistelmänä solunsalpaajahoidon kanssa. Haittavaikutukset on esitetty elinjärjestelmän ja esiintymistiheyden mukaan. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥1/10); yleinen (≥1/100, <1/10); melko harvinainen (≥1/1 000, <1/100); harvinainen (≥1/10 000, <1/1 000); hyvin harvinainen (<1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutuksia, joiden tiedetään ilmenevän semiplimabin tai yhdistelmähoidon yksittäisten komponenttien käytön yhteydessä, saattaa ilmetä näiden lääkevalmisteiden yhdistelmällä annetun hoidon yhteydessä.
Taulukko 2: Taulukko haittavaikutuksista potilailla, jotka saivat semiplimabia monoterapiana tai semiplimabia yhdistelmänä solunsalpaajahoidon kanssa | ||||||
Semiplimabi monoterapiana | Semiplimabi yhdistelmänä solunsalpaajahoidon kanssa | |||||
Elinjärjestelmä Suositeltu termi | Kaikki vaikeusasteet % | Vaikeusasteet 3–5 (%) | Kaikki vaikeusasteet % | Vaikeusasteet 3–5 (%) | ||
Infektiot | ||||||
Ylempien hengitysteiden infektioa | Hyvin yleinen | 10,9 | 0,4 | |||
Virtsateiden infektiob | Yleinen | 8,4 | 2,3 | |||
Veri ja imukudos | ||||||
Anemia | Hyvin yleinen | 15,0 | 5,2 | Hyvin yleinen | 43,6 | 9,9 |
Neutropenia | Hyvin yleinen | 15,4 | 5,8 | |||
Trombosytopenia | Hyvin yleinen | 13,1 | 2,6 | |||
Hemofagosyyttinen lymfohistiosytoosid | Tuntematon | -- | -- | |||
Immuunijärjestelmä | ||||||
Infuusioon liittyvä reaktio | Yleinen | 3,3 | <0,1 | Melko harvinainen | 0,3 | 0 |
Trombosytopeniac | Melko harvinainen | 0,9 | 0 | |||
Sjögrenin oireyhtymä | Melko harvinainen | 0,2 | 0 | |||
Kiinteän elinsiirteen hyljintäd | Tuntematon | -- | -- | |||
Umpieritys | ||||||
Hypotyreoosie | Yleinen | 6,8 | <0,1 | Yleinen | 7,7 | 0,3 |
Hypertyreoosi | Yleinen | 3,0 | <0,1 | Yleinen | 5,1 | 0 |
Kilpirauhastulehdusf | Melko harvinainen | 0,6 | 0 | Melko harvinainen | 0,6 | 0 |
Hypofysiittig | Melko harvinainen | 0,5 | 0,2 | |||
Lisämunuaisten vajaatoiminta | Melko harvinainen | 0,5 | 0,5 | |||
Tyypin 1 diabetesh | Harvinainen | <0,1 | <0,1 | Melko harvinainen | 0,3 | 0 |
Hermosto | ||||||
Päänsärky | Yleinen | 8,0 | 0,3 | |||
Perifeerinen neuropatiai | Yleinen | 1,3 | <0,1 | Hyvin yleinen | 21,2 | 0 |
Aivokalvotulehdusj | Harvinainen | <0,1 | <0,1 | |||
Aivotulehdus | Harvinainen | <0,1 | <0,1 | |||
Myasthenia gravis | Harvinainen | <0,1 | 0 | |||
Paraneoplastinen keskushermoston tulehdus | Harvinainen | <0,1 | <0,1 | |||
Krooninen tulehduksellinen demyelinoiva polyneuropatia | Harvinainen | <0,1 | 0 | |||
Silmät | ||||||
Sarveiskalvotulehdus | Harvinainen | <0,1 | 0 | |||
Suonikalvoston tulehdus | Harvinainen | < 0,1 | < 0,1 | |||
Sydän | ||||||
Sydänlihastulehdusk | Melko harvinainen | 0,5 | 0,3 | |||
Sydänpussitulehdusl | Melko harvinainen | 0,3 | 0,2 | |||
Verisuonisto | ||||||
Hypertensiom | Yleinen | 5,7 | 2,6 | |||
Aineenvaihdunta ja ravitsemus | ||||||
Heikentynyt ruokahalu | Hyvin yleinen | 13,0 | 0,6 | Hyvin yleinen | 17,0 | 1,0 |
Hyperglykemia | Hyvin yleinen | 17,6 | 1,9 | |||
Hypoalbuminemia | Hyvin yleinen | 10,3 | 0,6 | |||
Hengityselimet, rintakehä ja välikarsina | ||||||
Yskän | Hyvin yleinen | 10,8 | 0,2 | |||
Hengenahdistuso | Yleinen | 9,7 | 1,2 | Hyvin yleinen | 12,8 | 2,2 |
Pneumoniittip | Yleinen | 3,3 | 1,1 | Yleinen | 4,2 | 0,6 |
Ruoansulatuselimistö | ||||||
Pahoinvointi | Hyvin yleinen | 14,7 | 0,2 | Hyvin yleinen | 25,0 | 0 |
Ripuli | Hyvin yleinen | 16,3 | 0,7 | Hyvin yleinen | 10,6 | 1,3 |
Ummetus | Hyvin yleinen | 12,3 | 0,2 | Hyvin yleinen | 13,8 | 0,3 |
Vatsakipuq | Hyvin yleinen | 11,5 | 0,7 | |||
Oksentelu | Yleinen | 9,9 | 0,2 | Hyvin yleinen | 12,2 | 0 |
Paksusuolitulehdusr | Yleinen | 2,0 | 0,8 | Yleinen | 1,0 | 0,3 |
Suutulehdus | Yleinen | 1,8 | <0,1 | |||
Mahatulehduss | Melko harvinainen | 0,2 | 0 | |||
Haimatulehdust | Melko harvinainen | 0,2 | 0,2 | |||
Maksa ja sappi | ||||||
Maksatulehdusu | Yleinen | 2,7 | 1,8 | |||
Psyykkiset häiriöt | ||||||
Unettomuus | Hyvin yleinen | 10,9 | 0 | |||
Iho ja ihonalainen kudos | ||||||
Ihottumav | Hyvin yleinen | 21,4 | 1,6 | Hyvin yleinen | 12,5 | 1,3 |
Kutinaw | Hyvin yleinen | 12,7 | 0,2 | Yleinen | 3,5 | 0 |
Aktiininen keratoosi | Yleinen | 3,7 | 0 | |||
Hiusten lähtö | Hyvin yleinen | 36,9 | 0 | |||
Luusto, lihakset ja sidekudos | ||||||
Lihas- ja luukipux | Hyvin yleinen | 28,3 | 1,8 | Hyvin yleinen | 26,9 | 1,3 |
Niveltulehdusy | Melko harvinainen | 0,9 | 0,2 | Yleinen | 1,0 | 0 |
Lihastulehdusz | Melko harvinainen | 0,3 | <0,1 | |||
Lihasheikkous | Melko harvinainen | 0,2 | 0 | |||
Polymyalgia rheumatica | Melko harvinainen | 0,2 | 0 | |||
Munuaiset ja virtsatiet | ||||||
Munuaistulehdusaa | Yleinen | 1,2 | 0,2 | Yleinen | 2,6 | 0 |
Ei-infektiivinen virtsarakkotulehdus | Tuntematon | -- | -- | |||
Yleisoireet ja antopaikassa todettavat haitat | ||||||
Väsymysbb | Hyvin yleinen | 29,9 | 2,6 | Hyvin yleinen | 23,4 | 3,8 |
Kuumecc | Yleinen | 8,7 | 0,2 | |||
Edeemadd | Yleinen | 7,9 | 0,4 | |||
Tutkimukset | ||||||
Kohonnut alaniiniaminotransferaasiarvo | Yleinen | 4,6 | 0,5 | Hyvin yleinen | 16,3 | 2,2 |
Kohonnut aspartaattiaminotransferaasi-arvo | Yleinen | 4,4 | 0,7 | Hyvin yleinen | 14,7 | 0,3 |
Kohonnut veren alkalisen fosfataasin arvo | Yleinen | 1,9 | 0,2 | Yleinen | 4,5 | 0 |
Kohonnut veren kreatiniiniarvo | Yleinen | 1,6 | 0 | Yleinen | 8,7 | 0 |
Kohonnut veren TSH-arvo | Melko harvinainen | 0,8 | 0 | Yleinen | 4,2 | 0 |
Kohonneet transaminaasiarvot | Melko harvinainen | 0,4 | <0,1 | |||
Kohonnut veren bilirubiini | Melko harvinainen | 0,4 | <0,1 | Yleinen | 1,6 | 0,3 |
Alentunut veren TSH-arvo | Harvinainen | <0,1 | 0 | Yleinen | 1,6 | 0 |
Painonlasku | Hyvin yleinen | 11,2 | 1,3 | |||
Kohonnut gammaglutamyylitransferaasi-arvo | Melko harvinainen | 0,6 | 0,3 | |||
Haittavaikutusten vaikeusasteluokat arvioitiin NCI CTCAE:n version 4.03 kriteerien mukaisesti.
- Ylempien hengitysteiden infektio sisältää ylähengitystieinfektion, nasofaryngiitin, sinuiitin, hengitystieinfektion, riniitin, ylähengitysteiden virusinfektion, hengitysteiden virusinfektion, faryngiitin, laryngiitin, virusten aiheuttaman riniitin, akuutin sinuiitin, tonsilliitin ja trakeiitin.
- Virtsateiden infektio sisältää virtsatieinfektion, virtsarakkotulehduksen, pyelonefriitin, munuaisinfektion, akuutin pyelonefriitin, urosepsiksen, bakteerien aiheuttaman virtsarakkotulehduksen, Escherichia‑bakteerien aiheuttaman virtsatieinfektion, pyelokystiitin, bakteerien aiheuttaman virtsatieinfektion ja Pseudomonas-bakteerien aiheuttaman virtsatieinfektion.
- Trombosytopenia sisältää trombosytopenian ja immuunitrombosytopenian.
- Myyntiluvan myöntämisen jälkeinen tapahtuma.
- Hypotyreoosi sisältää hypotyreoosin ja immuunivälitteisen hypotyreoosin.
- Kilpirauhastulehdus sisältää tyreoidiitin, autoimmuunityreoidiitin ja immuunivälitteisen tyreoidiitin.
- Hypofysiitti sisältää hypofysiitin ja lymfosyyttisen hypofysiitin.
- Tyypin 1 diabetes sisältää diabeettisen ketoasidoosin ja tyypin 1 diabeteksen.
- Perifeerinen neuropatia sisältää perifeerisen sensorisen neuropatian, perifeerisen neuropatian, parestesian, polyneuropatian, neuriitin ja perifeerisen motorisen neuropatian.
- Aivokalvotulehdus sisältää aseptisen aivokalvotulehduksen.
- Sydänlihastulehdus sisältää sydänlihastulehduksen, autoimmuunisydänlihastulehduksen ja immuunivälitteisen sydänlihastulehduksen.
- Sydänpussitulehdus sisältää autoimmuunisydänpussitulehduksen ja sydänpussitulehduksen.
- Hypertensio sisältää hypertension ja hypertensiivisen kriisin.
- Yskä sisältää yskän, limaisen yskän ja ylähengitysteiden yskäoireyhtymän.
- Hengenahdistus sisältää hengenahdistuksen ja rasitukseen liittyvän hengenahdistuksen.
- Pneumoniitti sisältää pneumoniitin, immuunivälitteisen keuhkosairauden, interstitiaalisen keuhkosairauden ja keuhkofibroosin.
- Vatsakipu sisältää vatsakivun, ylävatsakivun, vatsan pingottuneisuuden, alavatsakivun, epämukavan tunteen vatsassa ja gastrointestinaalisen kivun.
- Koliitti sisältää koliitin, autoimmuunikoliitin, enterokoliitin ja immuunivälitteisen enterokoliitin.
- Mahatulehdus sisältää mahatulehduksen ja immuunivälitteisen mahatulehduksen.
- Haimatulehdusta (äkillistä haimatulehdusta ja immuunivälitteistä haimatulehdusta) ei havaittu monoterapia-aineistoon sisältyvissä tutkimuksissa (n = 1 281), ja esiintyvyys perustuu niiden potilaiden altistumiseen, jotka saivat semiplimabia monoterapiana asiaan kuuluvissa tutkimuksissa.
- Hepatiitti sisältää autoimmuunihepatiitin, immuunivälitteisen hepatiitin, hepatiitin, maksatoksisuuden, hyperbilirubinemian, maksasolujen vaurion, maksan vajaatoiminnan ja maksan poikkeavan toiminnan.
- Ihottuma sisältää ihottuman, makulopapulaarisen ihottuman, dermatiitin, eryteeman, kutiavan ihottuman, urtikarian, erytematoottisen ihottuman, rakkulaisen ihotulehduksen, aknen kaltaisen ihottuman, täpläisen ihottuman, psoriaasin, papulaarisen ihottuman, dyshidroottisen ekseeman, pemfigoidin, autoimmuunidermatiitin, allergisen ihottuman, atooppisen ihottuman, lääkeaineihottuman, kyhmyruusun, ihoreaktion, ihotoksisuuden, eksfoliatiivisen dermatiitin, yleistyneen eksfoliatiivisen dermatiitin, psoriasiformisen dermatiitin, erythema multiformen, eksfoliatiivisen ihottuman, immuunivälitteisen dermatiitin, lichen planuksen ja parapsoriaasin.
- Kutina sisältää kutinan ja allergisen kutinan.
- Lihas- ja luukipu sisältää nivelkivun, selkäkivun, raajakivun, myalgian, niskakivun, rintakehän lihas- ja luukivun, luukivun, lihas- ja luukivun, selkärangan kivun, muskuloskeletaalisen jäykkyyden ja muskuloskeletaalisen epämukavan tunteen.
- Niveltulehdus sisältää niveltulehduksen, moniniveltulehduksen, autoimmuuniniveltulehduksen ja immuunivälitteisen niveltulehduksen.
- Lihastulehdus sisältää myosiitin ja dermatomyosiitin.
Munuaistulehdus sisältää akuutin munuaisvaurion, heikentyneen munuaisten toiminnan, immuunivälitteisen munuaistulehduksen, munuaistulehduksen, munuaisten vajaatoiminnan, tubulointerstitiaalisen munuaistulehduksen ja toksisen nefropatian.
- Väsymys sisältää väsymyksen, voimattomuuden ja huonovointisuuden.
- Kuume sisältää pyreksian, hypertermian ja hyperpyreksian.
- Edeema sisältää perifeerisen edeeman, kasvojen edeeman, perifeerisen turvotuksen, kasvojen turvotuksen, paikallisen edeeman, yleistyneen edeeman ja turvotuksen.
Valikoitujen haittavaikutusten kuvaus
Alla kuvatut valikoidut haittavaikutukset perustuvat monoterapiana annetun semiplimabin turvallisuuteen 1 281 potilaalla, jotka osallistuivat kontrolloimattomiin kliinisiin tutkimuksiin.
Näiden valikoitujen haittavaikutusten esiintyvyys oli samankaltainen semiplimabimonoterapian yhteydessä potilailla, joilla oli edennyt kiinteä maligniteetti, monoterapiana adjuvanttihoitoasetelmassa ja käytettäessä semiplimabia yhdistelmänä solunsalpaajahoidon kanssa.
Immuunivälitteiset haittavaikutukset (ks. kohta Annostus ja antotapa ja kohta Varoitukset ja käyttöön liittyvät varotoimet)
Immuunivälitteinen pneumoniitti
Semiplimabia saaneista 1 281 potilaasta 33:lla (2,6 %) todettiin immuunivälitteinen pneumoniitti, ja heistä neljällä (0,3 %) oli vaikeusasteen 4 ja kahdeksalla (0,6 %) vaikeusasteen 3 immuunivälitteinen pneumoniitti. Immuunivälitteinen pneumoniitti johti semiplimabihoidon pysyvään lopettamiseen seitsemällätoista (1,3 %) 1 281 potilaasta. 33 potilaalla, joilla todettiin immuunivälitteinen pneumoniitti, mediaaniaika sen ilmaantumiseen oli 2,7 kuukautta (vaihteluväli: 7 vuorokautta – 22,2 kuukautta), ja pneumoniitin keston mediaani oli 1,1 kuukautta (vaihteluväli: 5 vuorokautta – 16,9 kuukautta). 33 potilaasta 27 (81,8 %) sai suurella annoksella kortikosteroidihoitoa, jonka keston mediaani oli 15 vuorokautta (vaihteluväli: 1 vuorokausi – 5,9 kuukautta). Viimeisen tietojenkeräyspäivän kohdalla pneumoniitti oli parantunut 20:llä (60,6 %) 33 potilaasta.
Immuunivälitteinen paksusuolitulehdus
Semiplimabia saaneista 1 281 potilaasta 25:llä (2,0 %) todettiin immuunivälitteinen ripuli tai paksusuolitulehdus, ja heistä kymmenellä (0,8 %) oli immuunivälitteinen vaikeusasteen 3 ripuli tai paksusuolitulehdus. Immuunivälitteinen ripuli tai paksusuolitulehdus johti semiplimabihoidon pysyvään lopettamiseen viidellä potilaalla (0,4 %) 1 281 potilaasta. 25 potilaalla, joilla todettiin immuunivälitteinen ripuli tai paksusuolitulehdus, mediaaniaika sen ilmaantumiseen oli 3,8 kuukautta (vaihteluväli: 1 vuorokausi – 16,6 kuukautta), ja immuunivälitteisen ripulin tai paksusuolitulehduksen keston mediaani oli 2,1 kuukautta (vaihteluväli: 4 vuorokautta – 26,8 kuukautta). 25 potilaasta 19 potilasta (76,0 %), joilla oli todettu immuunivälitteinen ripuli tai paksusuolitulehdus, sai suurella annoksella kortikosteroidihoitoa, jonka keston mediaani oli 22 vuorokautta (vaihteluväli: 2 vuorokautta – 5,2 kuukautta). Viimeisen tietojenkeräyspäivän kohdalla immuunivälitteinen ripuli tai paksusuolitulehdus oli parantunut neljällätoista (56,0 %) 25 potilaasta.
Immuunivälitteinen maksatulehdus
Semiplimabia saaneista 1 281 potilaasta 31:llä (2,4 %) todettiin immuunivälitteinen maksatulehdus, ja heistä yhdellä (<0,1 %) oli vaikeusasteen 5, neljällä (0,3 %) vaikeusasteen 4 ja 21:llä (1,6 %) vaikeusasteen 3 immuunivälitteinen maksatulehdus. Immuunivälitteinen maksatulehdus johti semiplimabihoidon pysyvään lopettamiseen kahdeksallatoista (1,4 %) 1 281 potilaasta. 31 potilaalla, joilla todettiin immuunivälitteinen maksatulehdus, mediaaniaika sen ilmaantumiseen oli 2,8 kuukautta (vaihteluväli: 7 vuorokautta – 22,5 kuukautta), ja maksatulehduksen keston mediaani oli 2,3 kuukautta (vaihteluväli: 5 vuorokautta – 8,7 kuukautta). Kaksikymmentäseitsemän 31 potilaasta (87,1 %), joilla oli todettu immuunivälitteinen maksatulehdus, sai suurella annoksella kortikosteroidihoitoa, jonka keston mediaani oli 24 vuorokautta (vaihteluväli: 2 vuorokautta – 3,8 kuukautta). Viimeisen tietojenkeräyspäivän kohdalla maksatulehdus oli parantunut 12:lla (38,7 %) 31 potilaasta.
Immuunivälitteiset umpierityssairaudet
Semiplimabia saaneista 1 281 potilaasta 87:llä (6,8 %) todettiin hypotyreoosi, ja heistä yhdellä (<0,1 %) oli vaikeusasteen 3 hypotyreoosi. Kolme (0,2 %) potilasta 1 281 potilaasta lopetti semiplimabihoidon hypotyreoosin vuoksi. 87 potilaalla, joilla todettiin hypotyreoosi, mediaaniaika sen ilmaantumiseen oli 4,0 kuukautta (vaihteluväli: 15 vuorokautta – 18,9 kuukautta), ja sen keston mediaani oli 9,2 kuukautta (vaihteluväli: 1 vuorokausi – 37,1 kuukautta). Viimeisen tietojenkeräyspäivän kohdalla hypotyreoosi oli parantunut viidellä (5,7 %) 87 potilaasta.
Semiplimabia saaneista 1 281 potilaasta 39:llä (3,0 %) todettiin hypertyreoosi, ja heistä yhdellä (<0,1 %) oli vaikeusasteen 3 ja yhdellätoista (0,9 %) vaikeusasteen 2 hypertyreoosi. Yksikään potilas ei lopettanut semiplimabihoitoa hypertyreoosin vuoksi. 39 potilaalla, joilla todettiin hypertyreoosi, mediaaniaika sen ilmaantumiseen oli 1,9 kuukautta (vaihteluväli: 20 vuorokautta – 23,8 kuukautta) ja keston mediaani oli 1,9 kuukautta (vaihteluväli: 9 vuorokautta – 32,7 kuukautta). Viimeisen tietojenkeräyspäivän kohdalla hypertyreoosi oli parantunut 22:lla (56,4 %) 39 potilaasta.
Semiplimabia saaneista 1 281 potilaasta kahdeksalla (0,6 %) todettiin kilpirauhastulehdus, ja heistä neljällä (0,3 %) oli vaikeusasteen 2 kilpirauhastulehdus. Yksikään potilas ei lopettanut semiplimabihoitoa kilpirauhastulehduksen vuoksi. Viimeisen tietojenkeräyspäivän kohdalla kilpirauhastulehdus oli parantunut yhdellä (12,5 %) kahdeksasta potilaasta.
Semiplimabia saaneista 1 281 potilaasta kuudella (0,5 %) todettiin lisämunuaisten vajaatoiminta, ja heistä kuudella (0,5 %) lisämunuaisten vajaatoiminta oli vaikeusastetta 3. Lisämunuaisten vajaatoiminta johti semiplimabihoidon pysyvään lopettamiseen yhdellä (<0,1 %) 1 281 potilaasta. Kuudella potilaalla, joilla todettiin lisämunuaisten vajaatoiminta, mediaaniaika sen ilmaantumiseen oli 7,5 kuukautta (vaihteluväli: 4,2 kuukautta – 18,3 kuukautta) ja keston mediaani oli 2,9 kuukautta (vaihteluväli: 22 vuorokautta – 6,1 kuukautta). Kaksi kuudesta (33,3 %) potilaasta sai systeemistä kortikosteroidihoitoa suurella annoksella. Viimeisen tietojenkeräyspäivän kohdalla lisämunuaisten vajaatoiminta oli parantunut yhdellä (16,7 %) kuudesta potilaasta.
Seitsemällä (0,5 %) 1 281:stä semiplimabia saaneesta potilaasta todettiin immuunivälitteinen hypofysiitti, ja heistä kolmella (0,2 %) immuunivälitteinen hypofysiitti oli vaikeusastetta 3. Yksi (<0,1 %) potilas 1 281:stä lopetti semiplimabihoidon hypofysiitin vuoksi. Seitsemällä potilaalla, joilla todettiin hypofysiitti, mediaaniaika sen ilmaantumiseen oli 7,4 kuukautta (vaihteluväli: 2,5 kuukautta – 10,4 kuukautta) ja keston mediaani 2,7 kuukautta (vaihteluväli 9 vuorokautta – 34,9 kuukautta). Kolme seitsemästä potilaasta (42,9 %) sai suurella annoksella kortikosteroidihoitoa. Viimeisen tietojenkeräyspäivän kohdalla hypofysiitti oli parantunut yhdellä (14,3 %) seitsemästä potilaasta.
1 281 potilaasta yhdellä (<0,1 %) todettiin tyypin 1 diabetes (vaikeusaste 4), jolle ei ollut vaihtoehtoisia syitä.
Immuunivälitteiset haittavaikutukset iholla
Semiplimabia saaneista 1 281 potilaasta 24:llä (1,9 %) todettiin immuunivälitteinen haittavaikutus iholla, ja heistä yhdellätoista (0,9 %) se oli vaikeusasteen 3 immuunivälitteinen haittavaikutus iholla. Immuunivälitteiset haittavaikutukset iholla johtivat semiplimabihoidon pysyvään lopettamiseen kolmella (0,2 %) 1 281 potilaasta. 24 potilaalla, joilla todettiin immuunivälitteinen haittavaikutus iholla, mediaaniaika sen ilmaantumiseen oli 2,0 kuukautta (vaihteluväli: 2 vuorokautta – 17,0 kuukautta), ja sen keston mediaani oli 2,9 kuukautta (vaihteluväli: 8 vuorokautta – 38,8 kuukautta). Seitsemäntoista (70,8 %) 24 potilaasta, joilla oli todettu immuunivälitteinen haittavaikutus iholla, sai suurella annoksella kortikosteroidihoitoa, jonka keston mediaani oli 10 vuorokautta (vaihteluväli: 1 vuorokausi – 2,9 kuukautta). Viimeisen tietojenkeräyspäivän kohdalla immuunivälitteinen ihoreaktio oli parantunut seitsemällätoista (70,8 %) 24 potilaasta.
Immuunivälitteinen munuaistulehdus
Semiplimabia saaneista 1 281 potilaasta yhdeksällä (0,7 %) todettiin immuunivälitteinen munuaistulehdus, ja heistä yhdellä (<0,1 %) oli vaikeusasteen 5 ja yhdellä (<0,1 %) vaikeusasteen 3 immuunivälitteinen munuaistulehdus. Immuunivälitteinen munuaistulehdus johti semiplimabihoidon pysyvään lopettamiseen kahdella potilaalla (0,2 %) 1 281 potilaasta. Yhdeksällä potilaalla, joilla todettiin immuunivälitteinen munuaistulehdus, mediaaniaika sen ilmaantumiseen oli 2,1 kuukautta (vaihteluväli: 14 vuorokautta – 12,5 kuukautta), ja munuaistulehduksen keston mediaani oli 1,5 kuukautta (vaihteluväli: 9 vuorokautta – 5,5 kuukautta). Kuusi yhdeksästä potilaasta (66,7 %), joilla oli todettu immuunivälitteinen munuaistulehdus, sai suurella annoksella kortikosteroidihoitoa, jonka keston mediaani oli 18 vuorokautta (vaihteluväli: 3 vuorokautta – 1,3 kuukautta). Viimeisen tietojenkeräyspäivän kohdalla munuaistulehdus oli parantunut seitsemällä (77,8 %) yhdeksästä potilaasta.
Muut immuunivälitteiset haittavaikutukset
Seuraavien kliinisesti merkittävien immuunivälitteisten haittavaikutusten ilmaantuvuus semiplimabia monoterapiana saaneilla 1 281 potilaalla oli alle 1 % (ellei toisin ole ilmoitettu). Tapahtumat olivat korkeintaan vaikeusastetta 3, ellei toisin ole mainittu:
Hermosto: aseptinen aivokalvotulehdus, paraneoplastinen enkefalomyeliitti (vaikeusaste 5), krooninen tulehduksellinen demyelinoiva polyradikuloneuropatia, enkefaliitti, myasthenia gravis, perifeerinen neuropatiaa
Sydän: sydänlihastulehdusb (vaikeusaste 5), sydänpussitulehdusc
Immuunijärjestelmä: immuunitrombosytopenia
Luusto, lihakset ja sidekudos: nivelsärky (1,2 %), niveltulehdusd, lihasheikkous, myalgia, myosiittie (vaikeusaste 4), polymyalgia rheumatica, Sjögrenin oireyhtymä
Iho ja ihonalainen kudos: kutina
Silmät: sarveiskalvotulehdus, suonikalvoston tulehdusf (vaikeusaste 4)
Ruoansulatuselimistö: suutulehdus, immuunivälitteinen mahatulehdus, haimatulehdus (vaikeusaste 4)
a. sisältää neuriitin, perifeerisen neuropatian, perifeerisen sensorisen neuropatian ja polyneuropatian
b. sisältää autoimmuunin sydänlihastulehduksen, immuunivälitteisen sydänlihastulehduksen ja sydänlihastulehduksen
c. sisältää autoimmuunin sydänpussitulehduksen ja sydänpussitulehduksen
d. sisältää niveltulehduksen, immuunivälitteisen niveltulehduksen ja moniniveltulehduksen
e. sisältää myosiitin ja dermatomyosiitin
f. raportoitu kliinisissä tutkimuksissa yhdistetyn aineiston ulkopuolelta
Lisäksi on havaittu seuraavia immuunivälitteisiä haittavaikutuksia kliinisissä tutkimuksissa yhdistelmähoitoa saaneilla potilailla: vaskuliitti, Guillain-Barrén oireyhtymä, keskushermoston tulehdus ja aivokalvotulehdus (vaikeusaste 4), joista jokaisen esiintymistiheys harvinainen.
Immuunijärjestelmän tarkistuspisteen estäjien luokkavaikutukset
Muilla immuunijärjestelmän tarkistuspisteen estäjillä annetun hoidon aikana on havaittu seuraavia haittavaikutuksia, joita voi ilmaantua myös semiplimabihoidon aikana: keliakia, haiman eksokriininen vajaatoiminta.
Infuusioon liittyvät reaktiot
Semiplimabimonoterapiaa saaneista 1 281 potilaasta 94:llä (7,3 %) todettiin infuusioon liittyviä reaktioita, ja heistä kahdella (0,2 %) potilaalla oli infuusioon liittyvä vaikeusasteen 3 tai 4 reaktio. Infuusioon liittyvä reaktio johti semiplimabihoidon pysyvään lopettamiseen yhdellä potilaalla (<0,1 %). Yleisiä infuusioon liittyvän reaktion oireita olivat pahoinvointi, kuume ja oksentelu. Viimeisen tietojenkeräyspäivän kohdalla infuusioon liittyvän reaktion oireet olivat hävinneet 93 potilaalla 94:stä (98,9 %).
Immunogeenisuus
Kaikkien terapeuttisten proteiinien tavoin myös semiplimabihoitoon liittyy immunogeenisuuden mahdollisuus. Kliinisissä tutkimuksissa semiplimabia saaneista 1 029 potilaista 2,1 %:lla kehittyi vasta-aineita semiplimabille hoidon vuoksi, ja noin 0,3 %:lla todettiin pitkäkestoinen vasta-ainevaste. Neutraloivia vasta-aineita ei havaittu. Farmakokineettisen profiilin tai turvallisuusprofiilin muutoksista semiplimabivasta-aineiden kehittymisen yhteydessä ei todettu näyttöä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksissa potilaan tilaa on seurattava tarkoin haittavaikutuksiin viittaavien oireiden tai löydösten havaitsemiseksi, ja sopiva oireenmukainen hoito on aloitettava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, PD‑1/PD‑L1 (ohjelmoituneen solukuoleman proteiinin 1 / ligandin 1) estäjät, ATC-koodi: L01FF06
Vaikutusmekanismi
Semiplimabi on täysin humaani immunoglobuliini G4 (IgG4) eli monoklonaalinen vasta-aine, joka sitoutuu ohjelmoidun solukuoleman reseptori 1:een (PD‑1) ja estää sen vuorovaikutuksen sen ligandien PD‑L1:n ja PD‑L2:n kanssa. PD‑1-reseptorin sitoutuminen sen ligandeihin PD‑L1:een ja PD‑L2:een, joita antigeenejä esittelevät solut ilmentävät ja joita kasvainsolut ja/tai muut solut kasvaimen mikroympäristössä saattavat ilmentää, johtaa T‑solujen toiminnan, kuten lisääntymisen, sytokiinien erityksen ja sytotoksisen vaikutuksen, estoon. Semiplimabi voimistaa T‑soluvasteita, kuten antituumorivastetta, estämällä PD‑1-reseptorin sitoutumisen PD‑L1- ja PD‑L2-ligandeihin.
Kliininen teho ja turvallisuus
Ihon okasolusyöpä
Edennyt ihon okasolusyöpä
Semiplimabin tehoa ja turvallisuutta on tutkittu yhdessä kliinisessä tutkimuksessa, R2810-ONC-1540 (tutkimus 1540), potilailla, joilla oli metastasoitunut (nodaalinen tai systeeminen) ihon okasolusyöpä tai paikallisesti edennyt ihon okasolusyöpä ja jotka eivät soveltuneet parantavaan leikkaushoitoon tai parantavaan sädehoitoon. Tutkimus 1540 oli vaiheen 2 avoin monikeskustutkimus, jossa ryhmiin 1–3 osallistui 193 potilasta, joilla oli metastasoitunut ihon okasolusyöpä tai paikallisesti edennyt ihon okasolusyöpä, ja jossa seuranta-ajan yhdistetty mediaani oli 15,7 kuukautta. Seuranta-ajan keston mediaani oli 18,5 kuukautta metastasoitunutta ihon okasolusyöpää sairastavien, annosta 3 mg/kg kahden viikon välein saaneiden potilaiden ryhmässä (ryhmä 1), 15,5 kuukautta paikallisesti edennyttä ihon okasolusyöpää sairastavien, annosta 3 mg/kg kahden viikon välein saaneiden potilaiden ryhmässä (ryhmä 2) ja 17,3 kuukautta metastasoitunutta ihon okasolusyöpää sairastavien, annosta 350 mg kolmen viikon välein saaneiden potilaiden ryhmässä (ryhmä 3). 165 potilaan lisäkohortissa, jossa edennyttä ihon okasolusyöpää (metastasoitunutta ihon okasolusyöpää tai paikallisesti edennyttä ihon okasolusyöpää) sairastavat potilaat saivat annosta 350 mg kolmen viikon välein (ryhmä 6), seuranta-ajan keston mediaani oli 8,7 kuukautta.
Mikä tahansa seuraavista aiheutti potilaan sulkemisen pois tutkimuksista: autoimmuunisairaus, joka oli 5 vuoden sisällä edellyttänyt systeemistä hoitoa immuunisalpaajilla; aiemmin tehty kiinteän elimen siirto; pneumoniitti viimeksi kuluneiden 5 vuoden aikana; aiempi hoito PD-1:n/PD-L1:n vasta-aineilla tai muilla immuunijärjestelmän tarkistuspisteen estäjillä; aktiivinen hoitoa vaativa infektio, mukaan lukien tiedossa oleva ihmisen immuunikatovirusinfektio (HIV) ja aktiivinen hepatiitti B- tai hepatiitti C -infektio; krooninen lymfaattinen leukemia; etäpesäkkeet aivoissa tai ECOG (Eastern Cooperative Oncology Group) -toimintakykyluokka ≥2.
Tutkimuksessa 1540 potilaat saivat semiplimabia laskimoon, kunnes tauti eteni, potilaalla ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä, tai suunniteltu hoito [3 mg/kg kahden viikon välein 96 viikon ajan (ryhmät 1 ja 2) tai 350 mg kolmen viikon välein 54 viikon ajan (ryhmä 3)] oli päättynyt. Parantava leikkaushoito sallittiin, jos paikallisesti edennyttä tautia sairastavilla potilailla todettiin riittävä vaste hoitoon. Kasvainvasteet arvioitiin 8 viikon välein (potilailta, joilla annos oli 3 mg/kg kahden viikon välein) tai 9 viikon välein (potilailta, joilla annos oli 350 mg kolmen viikon välein). Tutkimuksen 1540 ensisijainen tehon päätemuuttuja oli vahvistettu objektiivinen vasteluku (ORR), jonka määritti riippumaton keskitetty arvioijataho (Independent Central Review, ICR). Niiden potilaiden kohdalla, joilla oli metastasoitunut ihon okasolusyöpä ilman ulkoisia näkyviä kohdeleesioita, objektiivinen vasteluku määritettiin RECIST 1.1 ‑kriteerien (Response Evaluation Criteria in Solid Tumours) mukaisesti. Niiden potilaiden kohdalla, joilla oli ulkoisia näkyviä kohdeleesioita (paikallisesti edennyt ihon okasolusyöpä tai metastasoitunut ihon okasolusyöpä), ORR määritettiin käyttämällä yhdistelmäpäätemuuttujaa, joka sisälsi radiologisten tietojen (RECIST 1.1) ja digitaalisten lääketieteellisten valokuvien (WHO:n kriteerit) ICR-arviot. Keskeinen toissijainen päätemuuttuja oli vasteen kesto (DOR) ICR-arvion mukaan. Muita toissijaisia päätemuuttujia olivat ORR ja DOR tutkijan arvion mukaan, elossaoloaika ilman taudin etenemistä (PFS) ICR-arvion ja tutkijan arvion mukaan, kokonaiselossaoloaika (OS), täydellisen vasteen saaneiden osuus (CR) ICR-arvion mukaan ja EORTC QLQ-C30 -kyselyssä (European Organisation for Research and Treatment of Cancer, Quality of Life Questionnaire Core 30) potilaiden ilmoittamien hoitotulosten pistemäärien muutos.
Tehoa koskevassa analyysissä 193 potilaasta, joilla oli edennyt ihon okasolusyöpä ja jotka osallistuivat tutkimukseen 1540 ryhmissä 1–3, 115:llä oli metastasoitunut ihon okasolusyöpä ja 78:lla oli paikallisesti edennyt ihon okasolusyöpä. Mediaani-ikä oli 72 vuotta (vaihteluväli: 38–96): 78 potilasta (40,4 %) oli vähintään 75-vuotiaita, 66 potilasta (34,2 %) oli vähintään 65- ja alle 75-vuotiaita ja 49 potilasta (25,4 %) oli alle 65-vuotiaita. Yhteensä 161 potilasta (83,4 %) oli miehiä ja 187 potilasta (96,9 %) oli valkoihoisia. ECOG-toimintakykyluokka oli 0 (44,6 %) tai 1 (55,4 %). 33,7 % potilaista oli saanut aiemmin ainakin yhtä systeemistä syöpähoitoa, 81,3 %:lle potilaista oli tehty aiemmin syöpään liittyvä leikkaus ja 67,9 % potilaista oli saanut aiemmin sädehoitoa. Metastasoitunutta ihon okasolusyöpää sairastavista potilaista 76,5 %:lla oli systeemisiä etäpesäkkeitä ja 22,6 %:lla oli ainoastaan nodaalisia etäpesäkkeitä.
Tutkimuksen 1540 ryhmien 1–3 lopulliseen analyysiin perustuvat tehoa koskevat tulokset on esitetty taulukossa 3.
Taulukko 3: Tehoa koskevat tulokset – Tutkimus 1540 – metastasoitunut ihon okasolusyöpä (mCSCC) eri annostuksilla ja paikallisesti edennyt ihon okasolusyöpä (laCSCC)
Tehon päätetapahtumat | mCSCC semiplimabi: 3 mg/kg 2 viikon välein (ryhmä 1) (n = 59) | laCSCC semiplimabi: 3 mg/kg 2 viikon välein (ryhmä 2) (n = 78) | mCSCC semiplimabi: 350 mg 3 viikon välein (ryhmä 3) (n = 56) | |
ICR | ICR | ICR | ||
Vahvistettu objektiivinen vasteluku (ORR)a | ||||
ORR | 50,8 % | 44,9 % | 46,4 % | |
95 %:n luottamusväli ORR:lle | (37,5; 64,1) | (33,6; 56,6) | (33,0; 60,3) | |
Täydellinen vaste (CR)b | 20,3 % | 12,8 % | 19,6 % | |
Osittainen vaste (PR) | 30,5 % | 32,1 % | 26,8 % | |
Vakaa tauti (SD) | 15,3 % | 34,6 % | 14,3 % | |
Etenevä tauti (PD) | 16,9 % | 12,8 % | 25,0 % | |
Vasteen kesto (DOR) | ||||
Mediaanic (kk) 95 %:n luottamusväli | NR (20,7; NE) | 41,9 (20,5; 54,6) | 41,3 (40,8; 46,3) | |
Vaihteluväli (kk) | 2,8–38,9 | 1,9–54,6 | 4,2–46,3 | |
Potilaat, joilla DOR ≥6 kk, % | 93,3 % | 88,6 % | 96,2 % | |
Aika vasteeseen (TTR) | ||||
Mediaani (kk) vaihteluväli (min–maks.) | 1,9 (1,7–21,8) | 2,1 (1,8–8,8) | 2,1 (2,0–22,8) | |
Elossaoloaika ilman taudin etenemistä (PFS)a, c | ||||
6 kk (95 %:n luottamusväli) | 66,4 % (52,5; 77,1) | 72,4 % (60,1; 81,5) | 60,7 % (46,7; 72,1) | |
12 kk (95 %:n luottamusväli) | 53,8 % (40,0; 65,8) | 60,8 % (47,8; 71,5) | 53,4 % (39,5; 65,4) | |
Kokonaiselossaoloaika (OS)a, c | ||||
12 kk (95 %:n luottamusväli) | 81,3 % (68,7; 89,2) | 91,8 % (82,6; 96,2) | 72,5 % (58,6; 82,5) | |
ICR: riippumaton, keskitetty arvioijataho; NR: Ei saavutettu (Not reached); NE: Ei arvioitavissa (Not evaluable)
c. Perustuu Kaplan-Meierin estimaatteihin. | ||||
Teho ja PD-L1-status
Kliinistä aktiivisuutta havaittiin kasvaimen PD‑L1-ilmentymisstatuksesta riippumatta.
Ihon okasolusyövän adjuvanttihoito korkean riskin potilailla
C-POST-tutkimuksessa semiplimabin tehoa adjuvanttihoitona arvioitiin ihon okasolusyöpää sairastavilla potilailla, joilla oli suuri taudin uusiutumisriski leikkauksen ja sitä seuranneen sädehoidon jälkeen. Tutkimus oli satunnaistettu, kaksoissokkoutettu, lumelääkekontrolloitu vaiheen 3 monikeskustutkimus. Tutkittavilla oli suuri uusiutumisriski imusolmukkeisiin liittyvien piirteiden (ekstrakapsulaarinen laajentuminen tai leviäminen vähintään kolmeen imusolmukkeeseen) ja/tai muiden kuin imusolmukkeisiin liittyvien piirteiden (in-transit -etäpesäkkeet, T4-leesio, perineuraalinen invaasio tai paikallisesti uusiutunut kasvain, jossa oli vähintään yksi uusi haitallinen piirre). Tutkittavat saivat adjuvanttisädehoidon 2–10 viikon sisällä satunnaistamisesta.
Tutkimuksesta suljettiin pois potilaat, joilla oli autoimmuunisairaus, joka oli 5 vuoden sisällä edellyttänyt systeemistä hoitoa immuunisalpaajilla; aiemmin tehty kiinteän elimen siirto; aiempi allogeeninen tai autologinen kantasolusiirto; huonossa hoitotasapainossa oleva HIV-infektio, hepatiitti B tai hepatiitti C -infektio; tai ECOG-toimintakykyluokka ≥ 2. Kroonista lymfaattista leukemiaa sairastavat potilaat soveltuivat tutkimukseen, jos he eivät olleet tarvinneet systeemistä hoitoa krooniseen lymfosyyttiseen leukemiaan kuuden kuukauden sisällä.
C-POST-tutkimuksessa 415 potilasta satunnaistettiin suhteessa 1:1 saamaan semiplimabia (N = 209) tai lumelääkettä (N = 206). Tutkimuksen ensimmäisessä osassa 1 334 potilasta sai 350 mg semiplimabia (N = 171) tai lumelääkettä (N = 163) laskimoon 3 viikon välein 12 viikon ajan, minkä jälkeen 700 mg semiplimabia tai lumelääkettä laskimoon 6 viikon välein 36 lisäviikon ajan, ja 81 potilasta sai 350 mg semiplimabia (N = 38) tai lumelääkettä (N = 43) laskimoon 3 viikon välein enintään 48 viikon ajan. Hoitoa jatkettiin, kunnes tauti uusiutui, ilmeni ei-hyväksyttävää toksisuutta, tai kun hoitoa oli annettu enintään 48 viikon ajan.
Tutkimuksen osassa 2, joka oli avoin ja valinnainen, potilailla, joiden sairaus uusiutui milloin tahansa tutkimuksen aikana ja jotka olivat lumelääkeryhmässä, oli mahdollisuus saada myöhemmin annettavaa semiplimabia 350 mg laskimoon 3 viikon välein. Potilailla, joiden tauti uusiutui ≥ 3 kuukautta suunnitellun 48 viikon pituisen semiplimabihoidon päättymisen jälkeen ja jotka olivat semiplimabia saavassa ryhmässä, oli mahdollisuus saada semiplimabia 350 mg laskimoon 3 viikon välein. Potilaita voitiin hoitaa enintään 96 viikon ajan osassa 2.
Ensisijainen päätetapahtuma oli elossaoloaika ilman tautia (DFS), joka määriteltiin aikana satunnaistamisesta ensimmäiseen dokumentoituun taudin uusiutumiseen tutkijan arvioinnin tai mistä tahansa syystä johtuvan kuoleman perusteella. Kuvantamisarvioinnit tehtiin kunkin 12 viikon syklin lopussa 48 viikon aikana. Seurantajakson aikana kuvantaminen suoritettiin 4 kuukauden välein suunnitellun seurannan ensimmäisten 2 vuoden aikana ja sen jälkeen 6 kuukauden välein taudin uusiutumiseen asti.
Tutkimuspopulaation ominaisuudet olivat seuraavat: mediaani-ikä 71 vuotta (vaihteluväli: 33–95); 83,9 % miehiä; 91,1 % valkoihoisia, 3,1 % aasialaisia; 63,6 %:lla oli ECOG-toimintakykyluokka 0 ja 36,4 %:lla ECOG-toimintakykyluokka 1. Kasvaimen sijaintipaikka oli pään ja kaulan alueella 82,7 %:lla ja muualla 17,3 %:lla potilaista. Korkean riskin piirre oli nodaalinen 58,3 %:lla potilaista ja yksinomaan ei-nodaalinen 41,7 %:lla potilaista.
C-POST-tutkimuksen tehoa koskevat tulokset on esitetty taulukossa 4 ja kuvassa 1.
Taulukko 4: Tehoa koskevat tulokset C-POST-tutkimuksessa korkean riskin ihon okasolusyövän hoidossa adjuvanttihoitoasetelmassa- ensisijainen analyysi
Tehon päätetapahtumata | Semiplimabi | Lumelääke |
N = 209 | N = 206 | |
Elossaoloaika ilman tautia (DFS) | ||
Tapahtumien määrä, n (%) | 24 (11,5 %) | 65 (31,6 %) |
Taudin uusiutuminen, n (%) | 18 (8,6 %) | 61 (29,6 %) |
Kuolemat, n (%)b | 6 (2,9 %) | 4 (1,9 %) |
Mediaani (95 %:n luottamusväli) kuukausinac | NR (NE; NE) | 49,4 (48,5; NE) |
Hasardisuhde (95 %:n luottamusväli)d | 0,32 (0,20; 0,51) | |
p-arvoe | < 0,0001 | |
CI: Luottamusväli; NE: Ei arvioitavissa (Not evaluable); NR: Ei saavutettu (Not reached, viimeinen tietojenkeräyspäivä 4.10.2024) | ||
- Seurannan keston mediaani: semiplimabi: 24,5 kuukautta; lumelääke: 23,8 kuukautta
- Kuolemat, jotka laskettiin tutkijan arvion mukaan elossaoloajan tapahtumiksi ilman tautia; eivät sisällä kuolemantapauksia potilailla, joilla oli aiemmin ollut taudin uusiutuminen
- Perustuu Kaplan-Meier-menetelmään
- Perustuu ositettuun suhteelliseen hasardimalliin
- Perustuu kaksisuuntaiseen p-arvoon
Päivitetyn analyysin ajankohtana (viimeinen tietojen keräyspäivä 7.4.2025), taudittoman elossaoloajan tapahtumia (DFS) raportoitiin 29 (13,9 %) semiplimabiryhmässä ja 68 (33,0 %) lumelääkeryhmässä seuranta-ajan ollessa 31 kuukautta semiplimabiryhmässä ja 30 kuukautta lumelääkeryhmässä (HR 0,35; 95 %:n luottamusväli 0,23; 0,55). Kokonaiselossaoloajan tulokset eivät olleet valmiita ennalta määritetyn ensisijaisen analyysin hetkellä.
Kuva 1: Elossaoloaika ilman tautia C-POST-tutkimuksessa korkean riskin ihon okasolusyövän hoidossa adjuvanttiasetelmassa - päivitetty analyysia
a. Perustuu päivitettyyn analyysiin (viimeinen tietojenkeräyspäivä 7.4.2025)
Tyvisolusyöpä
Semiplimabin tehoa ja turvallisuutta on arvioitu avoimessa ei-satunnaistetussa kliinisessä monikeskustutkimuksessa (tutkimus 1620), potilailla, joilla oli paikallisesti edennyt (laBCC) tai metastasoinut (mBCC) ihon tyvisolusyöpä ja joilla tauti oli edennyt HHI-hoidon aikana tai jotka eivät sietäneet aiempaa HHI-hoitoa tai hoitotulos ei ollut vakaata tautitilannetta parempi 9 kuukauden HHI-hoidon jälkeen (pois lukien hoidon tauot). Tutkimuksesta suljettiin pois potilaat, joilla oli autoimmuunisairaus, joka oli 5 vuoden sisällä edellyttänyt systeemistä hoitoa immuunisalpaajilla; aiemmin tehty kiinteän elimen siirto; aiempi hoito anti-PD-1/PD-L1-vasta-aineilla tai muilla immuunijärjestelmän tarkistuspisteen estäjillä; HIV-infektio, hepatiitti B tai hepatiitti C -infektio; tai ECOG-toimintakykyluokka ≥2.
Potilaat saivat semiplimabia 350 mg laskimoon joka 3. viikko viiden 9 viikon mittaisen syklin ajan ja sen jälkeen neljän 12 viikon syklin ajan, yhteensä 93 viikon ajan. Hoito jatkui, kunnes tauti eteni, toksiset vaikutukset olivat sietämättömiä tai suunniteltu hoito päättyi. Kasvaimet arvioitiin 9 viikon välein syklien 1–5 aikana ja 12 viikon välein syklien 6–9 aikana. Ensisijaiset tehon päätemuuttujat olivat vahvistettu objektiivinen vasteluku (ORR) ja vasteen kesto (DOR) ICR:n arvioimana. Toissijaisia tehon päätemuuttujia olivat ORR ja DOR tutkijan arvion mukaan, elossaoloaika ilman taudin etenemistä, kokonaiselossaoloaika, täydellisen vasteen saaneiden osuus ICR:n arvion mukaan ja aika vasteen saamiseen. Potilailla, joilla oli mBCC ilman ulkoisia näkyviä kohdeleesioita, ORR määritettiin RECIST 1.1 ‑kriteerien mukaisesti. Potilailla, joilla oli ulkoisia näkyviä kohdeleesioita (laBCC ja mBCC), ORR määritettiin käyttämällä yhdistelmäpäätemuuttujaa, joka sisälsi radiologisten tietojen (RECIST 1.1) ja digitaalisten lääketieteellisten valokuvien (WHO:n kriteerit) ICR-arviot.
Tutkimuksen 1620 tehoanalyysiin kuului yhteensä 138 potilasta, joilla oli edennyt tyvisolusyöpä; 84 potilaalla oli paikallisesti edennyt tyvisolusyöpä ja 54 potilaalla metastasoinut tyvisolusyöpä.
Paikallisesti edennyttä tyvisolusyöpää sairastavien ryhmässä mediaani-ikä oli 70,0 vuotta (vaihteluväli: 42–89): potilaista 31 (37 %) oli <65-vuotiaita ja 53 (63 %) oli vähintään 65-vuotiaita. Yhteensä 56 (67 %) oli miehiä ja 57 (68 %) oli valkoihoisia; ECOG-toimintakykyluokka oli 0 (61 %) ja 1 (39 %); 83 %:lle potilaista oli tehty vähintään yksi syöpäleikkaus ja 35 %:lla potilaista oli ollut >3 aikaisempaa syöpäleikkausta (mediaani: 3,0 leikkausta, vaihteluväli: 1–43); 50 % potilaista oli saanut vähintään kerran sädehoitoa (RT) syövän hoitoon (mediaani: 1,0 RT, vaihteluväli: 1–6).
Metastasoinutta tyvisolusyöpää sairastavien ryhmässä mediaani-ikä oli 63,5 vuotta (vaihteluväli: 38‑90): potilaista 27 (50 %) oli <65-vuotiaita ja 27 (50 %) oli vähintään 65-vuotiaita. Yhteensä 38 (70 %) oli miehiä ja 47 (87 %) oli valkoihoisia; ECOG-toimintakykyluokka oli 0 (67 %) ja 1 (33 %); 85 %:lle potilaista oli tehty vähintään yksi syöpäleikkaus ja 28 %:lla potilaista oli ollut >3 aikaisempaa syöpäleikkausta (mediaani: 2,0 leikkausta, vaihteluväli: 1–8); 59 % potilaista oli saanut vähintään kerran sädehoitoa (RT) syövän hoitoon (mediaani: 1,0 RT, vaihteluväli: 1–4).
Kaikki 138 potilasta olivat aiemmin saaneet HHI-hoitoa, ja 12 %:a (16/138) oli aiemmin hoidettu sekä vismodegibilla että sonidegibilla (erillisinä hoitolinjoina). Paikallisesti edennyttä tyvisolusyöpää sairastavista 84 potilaasta 71 % (60/84) lopetti HHI-hoidon taudin etenemisen takia, 38 % (32/84) potilaista lopetti HHI-hoidon haittavaikutusten takia ja 2 % (2/84) lopetti yksinomaan vasteen puuttumisen takia. Metastasoinutta tyvisolusyöpää sairastavista 54 potilaasta 76 % (41/54) lopetti HHI-hoidon taudin etenemisen takia, 33 % (18/54) potilaista lopetti HHI-hoidon haittavaikutusten takia ja 6 % (3/54) lopetti yksinomaan vasteen puuttumisen takia. Tutkijat saattoivat yksittäisen potilaan kohdalla valita useamman kuin yhden syyn HHI-hoidon lopettamiseen.
Tutkimuksen tehoa koskevat tulokset on esitetty taulukossa 5.
Taulukko 5: Tehoa koskevat tulokset tutkimuksesta 1620, paikallisesti edennyt ja metastasoitunut tyvisolusyöpä
Tehon päätetapahtumat | laBCC semiplimabi 350 mg 3 viikon välein | mBCC semiplimabi 350 mg 3 viikon välein |
N=84 | N=54 | |
ICR | ICR | |
Paras kokonaisvaste (BOR)a, b, c | ||
Objektiivinen vasteluku (ORR: CR+ PR) (95 % luottamusväli) | 27 (32,1 %) (22,4, 43,2) | 12 (22,2 %) (12,0; 35,6) |
Täydellinen vaste (CR)d (95 % luottamusväli) | 6 (7,1 %) (2,7, 14,9) | 1 (1,9 %) (0,0, 9,9) |
Osittainen vaste (PR) | 21 (25,0 %) | 11 (20,4 %) |
Etenevä tauti (PD) | 9 (10,7 %) | 16 (29,6 %) |
Vasteen kesto (DOR) | N=27, joilla vaste | N=12, joilla vaste |
Mediaanie (kk) (95 % luottamusväli) | NR (15,5, NE) | 16,7 (9,8, NE) |
Vaihteluväli (havaittu) (kk) | 2,1–36,8+ | 9,0–25,8+ |
Potilaat, joilla DOR ≥6 kk, %e (95 % luottamusväli) | 88,5 % (68,4, 96,1) | 100,0 % (100, 100) |
Aika vasteeseen (TTR) | N=27, joilla vaste | N=12, joilla vaste |
Mediaani (kuukausia) (Vaihteluväli) | 4,3 (2,1–21,4) | 3,1 (2,0–10,5) |
+: Merkitsee jatkumista viimeisen arvioinnin aikana; ICR: riippumaton, keskitetty arvioijataho (Independent Central Review); NR: Ei saavutettu (Not reached); NE: Ei arvioitavissa (Not evaluable)
- Seurannan keston mediaani: laBCC: 15.9 kuukautta, mBCC: 8.4 kuukautta
- Sisältää 2 laBCC-potilasta, jotka täyttivät inkluusiokriteerit ainoastaan määritelmän “Ei parempi kuin vakaa tautitila (SD) 9 kuukauden HHI-hoidon jälkeen” perusteella. BOR-tulokset ICR:n mukaan olivat SD yhden potilaan kohdalla ja NE yhden potilaan kohdalla.
- Sisältää 3 mBCC-potilasta, jotka täyttivät inkluusiokriteerit ainoastaan määritelmän “Ei parempi kuin vakaa tautitila (SD) 9 kuukauden HHI-hoidon jälkeen” perusteella. BOR-tulokset ICR:n mukaan olivat osittainen vaste (PR) yhden potilaan kohdalla ja SD kahden potilaan kohdalla.
- Paikallisesti edennyttä tyvisolusyöpää sairastavien potilaiden täydellinen vaste edellytti biopsiaa tuloksen varmistamiseksi tutkimuksessa 1620.
- Perustuu Kaplan Meierin estimaatteihin.
Teho ja PD-L1-status:
Kliininen aktiivisuus oli riippumaton kasvaimen PD-L1-ekspressiostatuksesta.
Ei-pienisoluinen keuhkosyöpä
Semiplimabimonoterapia ei-pienisoluisen keuhkosyövän ensilinjan hoidossa
Semiplimabin tehoa ja turvallisuutta arvioitiin satunnaistetussa, avoimessa monikeskustutkimuksessa 1624 verrattuna platinapohjaisen solunsalpaajan ja toisen solunsalpaajan yhdistelmään potilailla, joilla oli joko definitiiviseen kemosädehoitoon soveltumaton paikallisesti levinnyt ei-pienisoluinen keuhkosyöpä (NSCLC) tai metastaattinen ei-pienisoluinen keuhkosyöpä ja joilla PD-L1 ilmeni ≥50 % kasvainsoluista IHC 22C3 pharmDx -määrityksellä.
Kaikkiaan 710 potilasta rekrytoitiin.
Tutkimuksesta suljettiin pois potilaat, joilla oli kasvaimessa EGFR-, ALK- tai ROS1-genomipoikkeamia, ECOG-toimintakykyluokka (PS) ≥2, sairauksia, jotka edellyttivät systeemistä immunosuppressiota, kontrolloimaton B-hepatiitti- tai C-hepatiitti- tai HIV-infektio, aikaisempi interstitiaalinen keuhkotauti, sekä potilaat, jotka eivät olleet koskaan tupakoineet, tai joilla oli autoimmuunitauti, joka edellytti systeemistä hoitoa 2 vuoden sisällä hoidosta. Aivojen metastaasien hoito oli sallittu, ja potilaat voitiin rekrytoida, jos heitä oli hoidettu asianmukaisesti ja he olivat neurologisesti palanneet lähtötasolle vähintään 2 viikkoa ennen satunnaistamista. Stabiiliuden tai vasteen radiologista vahvistamista ei vaadittu.
Satunnaistaminen ositettiin histologian mukaan (ei-levyepiteeli vs. levyepiteeli) ja maantieteellisen alueen mukaan (Eurooppa, Aasia, tai muu maailma). Potilaat satunnaistettiin (1:1) saamaan semiplimabia 350 mg laskimoon 3 viikon välein enintään 108 viikon ajan tai tutkijan valinnan mukaan seuraavia platina-solunsalpaajahoitoja 4–6 syklin ajan: paklitakseli + sisplatiini tai karboplatiini; gemsitabiini + sisplatiini tai karboplatiini; tai pemetreksedi + sisplatiini tai karboplatiini, jota seurasi valinnainen ylläpitohoito pemetreksedillä (tätä hoitoa ei suositeltu potilaille, joilla oli levyepiteeli-NSCLC).
Semiplimabihoito jatkui, kunnes tapahtui jokin seuraavista: RECIST 1.1:n mukaan määritelty etenevä tauti, ei-hyväksyttävä toksisuus tai hoito oli kestänyt enintään 108 viikon ajan. Potilaat, joilla ilmeni riippumattoman arviointilautakunnan (IRC) arvioima RECIST 1.1:n mukaan määritelty etenevä tauti semiplimabihoidon aikana, saivat jatkaa semiplimabihoitoa niin, että siihen lisättiin 4 sykliä histologiaspesifistä solunsalpaajahoitoa, kunnes havaittiin taudin eteneminen uudelleen. Potilaat, joilla ilmeni IRC:n arvioima RECIST 1.1:n mukaan määritelty etenevä tauti solunsalpaajahoidolla, saivat jatkaa semiplimabihoitoa enintään 108 viikon ajan tai kunnes tauti eteni edelleen tai ilmeni ei-hyväksyttävää toksisuutta. 203 potilaasta, jotka satunnaistettiin saamaan solunsalpaajahoitoa ja joilla oli IRC:n arvioima RECIST 1.1:n mukaan määritelty etenevä tauti, 150 (73,9 %) potilasta siirtyi saamaan semiplimabihoitoa. Kasvainstatus arvioitiin 9 viikon välein. Ensisijaiset tehon päätetapahtumat olivat kokonaiselossaoloaika (OS) ja elossaoloaika ilman taudin etenemistä (PFS), jonka arvioi sokkoutettu IRC käyttämällä RECIST 1.1-kriteereitä. Keskeinen toissijainen päätetapahtuma oli objektiivinen vasteluku (ORR).
710 potilaan lähtötason muuttujat olivat: mediaani-ikä 63 vuotta (45 % oli vähintään 65-vuotiaita), 85 % miespuolisia, 86 % valkoihoisia, ECOG-toimintakykyluokka 0 27 %:lla ja 1 73 %:lla, ja 12 %:lla oli ollut metastaasi aivoissa. Taudin ominaispiirteet olivat paikallisesti edennyt (16 %), metastaattinen (84 %), levyepiteeli (44 %) ja ei-levyepiteeli (56 %).
Tutkimus osoitti, että OS parani tilastollisesti merkitsevästi potilailla, jotka satunnaistettiin saamaan semiplimabia solunsalpaajahoitoon verrattuna.
Tehon tulokset esitetään taulukossa 6, kuvassa 2 ja kuvassa 3.
Taulukko 6: Tehotulokset tutkimuksesta 1624, ei-pienisoluinen keuhkosyöpä
Tehon päätetapahtumata | Semiplimabi 350 mg 3 viikon välein N=356 | Solunsalpaajahoito N=354 |
Kokonaiselossaoloaika (OS) | ||
Kuolemantapaukset, n (%) | 108 (30,3) | 141 (39,8) |
Mediaani kuukausina (95 % luottamusväli)b | 22,1 (17,7, NE) | 14,3 (11,7, 19,2) |
Hasardisuhde (95 % luottamusväli)c | 0,68 (0,53, 0,87) | |
p-arvod | 0,0022 | |
OS 12 kuukauden kohdalla (95 % luottamusväli)b | 70 % (64, 75) | 56 % (49, 62) |
Elossaoloaika ilman taudin etenemistä (PFS) | ||
Tapahtumat, n (%) | 201 (56,5) | 262 (74,0) |
Mediaani kuukausina (95 % luottamusväli)b | 6,2 (4,5, 8,3) | 5,6 (4,5, 6,1) |
Hasardisuhde (95 % luottamusväli)c | 0,59 (0,49, 0,72) | |
PFS 12 kuukauden kohdalla (95 % luottamusväli)b | 38 % (32, 44) | 7 % (4, 11) |
Objektiivinen vasteluku (%)e | ||
ORR (95 % luottamusväli) | 36,5 (31,5, 41,8) | 20,6 (16,5, 25,2) |
Täydellinen vaste (CR) -luku | 3,1 | 0,8 |
Osittainen vaste (PR) -luku | 33,4 | 19,8 |
Vasteen kesto | N=130 vasteen saanutta | N=73 vasteen saanutta |
Mediaani (kuukautta)b | 21,0 | 6,0 |
Vaihteluväli (kuukautta) | (1,9+, 23,3+) | (1,3+, 16,5+) |
Potilaat, joilla oli havaittu DOR ≥6 kuukautta, % | 69 % | 41 % |
NE: ei arvioitavissa (Not evaluable); +: meneillään oleva vaste
a. Seurannan mediaanikesto: semiplimabi: 13,1 kuukautta; solunsalpaajahoito: 13,1 kuukautta
b. Perustuu Kaplan-Meier-estimaatteihin
c. Perustuu ositettuun suhteelliseen hasardimalliin
d. Perustuu kaksisuuntaiseen p-arvoon
e. Perustuu Clopper-Pearsonin eksaktiin luottamusväliin
Kuva 2: OS tutkimuksessa 1624, ei-pienisoluinen keuhkosyöpä
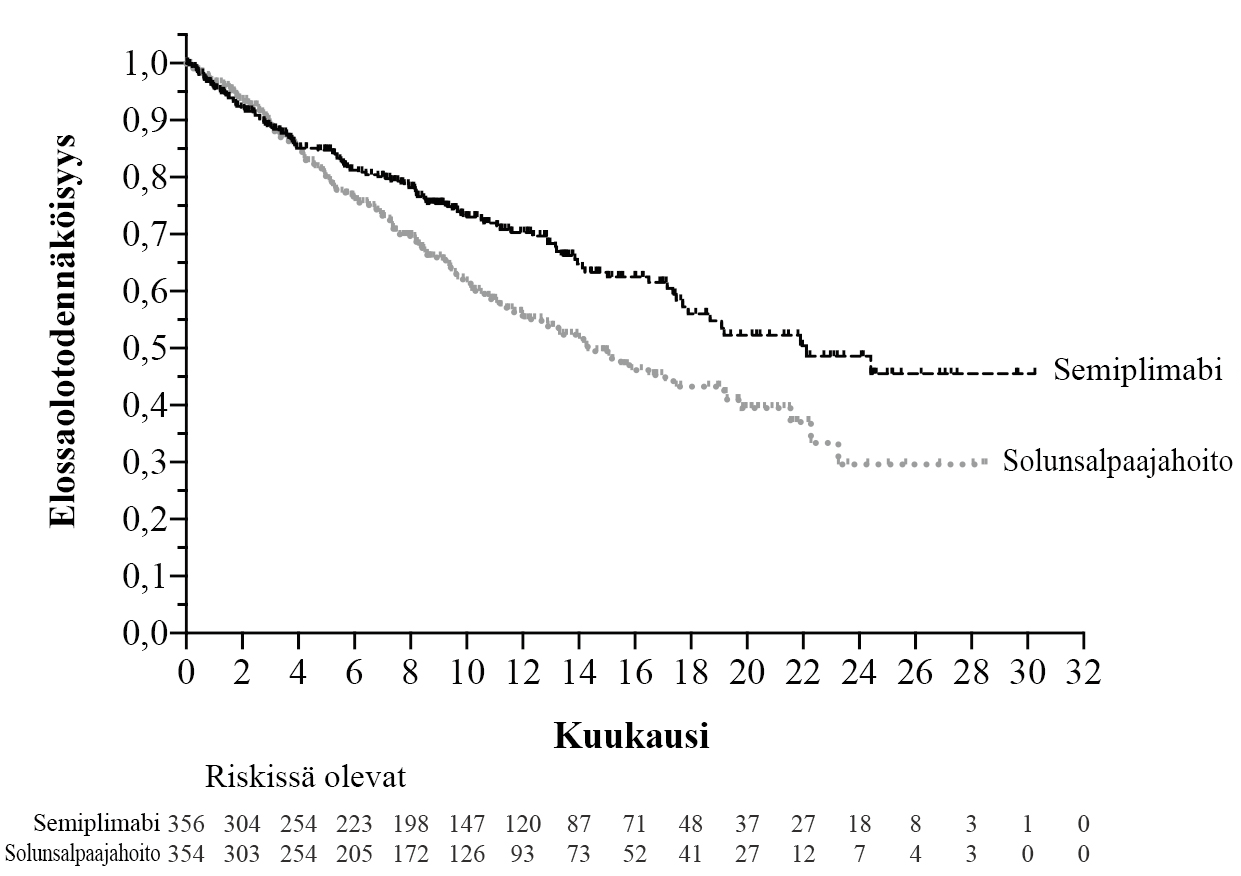
Kuva 3: PFS tutkimuksessa 1624, ei-pienisoluinen keuhkosyöpä
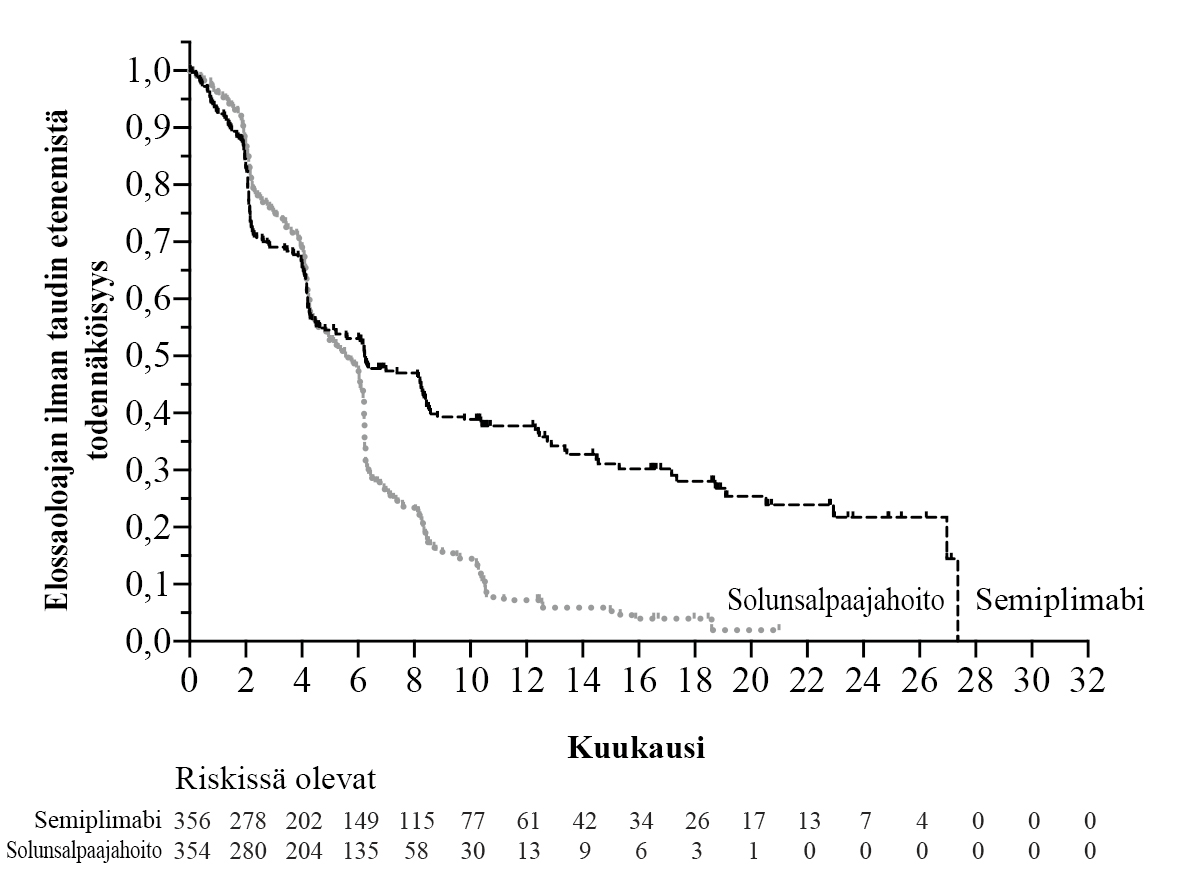
NSCLC:n ensilinjan hoito semiplimabilla yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa
Semiplimabin ja platinapohjaisen solunsalpaajahoidon yhdistelmän tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa monikeskustutkimuksessa 16113, johon osallistuneilla 466 potilaalla oli joko definitiiviseen kemosädehoitoon soveltumaton paikallisesti levinnyt NSCLC tai metastaattinen NSCLC (kasvaimen PD-L1-ilmentymisstatuksesta riippumatta). Potilaat eivät olleet aiemmin saaneet systeemistä hoitoa metastaattisen NSCLC:n vuoksi. Testaus kasvaimen genomipoikkeamien määrittämiseksi ei ollut edellytys tutkimukseen 16113 osallistumiselle EGFR-, ALK- ja ROS1-poikkeamien testausta lukuun ottamatta.
Potilaat eivät soveltuneet tutkimukseen, jos heillä oli kasvaimessa EGFR-, ALK- tai ROS1-genomipoikkeamia, jokin sairaus, joka edellytti systeemistä immunosuppressiota, aktiivinen B‑hepatiitti- tai C‑hepatiitti-infektio, kontrolloimaton HIV tai senhetkinen tai äskettäin sairastettu autoimmuunitauti, joka edellytti systeemistä hoitoa. Potilaat, joilla oli ollut metastaasi aivoissa, soveltuivat tutkimukseen, jos heitä oli hoidettu asianmukaisesti ja he olivat neurologisesti palanneet lähtötasolle vähintään 2 viikkoa ennen satunnaistamista. Stabiiliuden tai vasteen radiologista vahvistamista ei vaadittu.
Satunnaistaminen ositettiin histologian mukaan (ei-levyepiteeli vs. levyepiteeli) ja PD‑L1:n ilmentymisen mukaan (<1 % vs. 1–49 % vs. ≥50 %) hyödyntäen VENTANA PD-L1 (SP263) ‑määritystä. Potilaat satunnaistettiin (2:1) saamaan joko semiplimabia 350 mg laskimoon 3 viikon välein 108 viikon ajan ja platinapohjaista solunsalpaajahoitoa 3 viikon välein 4 syklin ajan tai lumelääkettä laskimoon 3 viikon välein 108 viikon ajan ja platinapohjaista solunsalpaajahoitoa 3 viikon välein 4 syklin ajan.
Semiplimabi- tai lumelääkehoito jatkui, kunnes potilaalla ilmeni RECIST 1.1:n mukaan määritelty etenevä tauti tai ei-hyväksyttävää toksisuutta tai enintään 108 viikon ajan. Solunsalpaajahoitoa annettiin 4 syklin ajan, ja sen jälkeen annettiin ylläpitohoito pemetreksedillä kliinisen tarpeen mukaan tai kunnes ilmeni RECIST 1.1:n mukaan määritelty etenevä tauti tai ei-hyväksyttävää toksisuutta. Tutkimuksessa 16113 käytettyyn solunsalpaajahoitoon kuului karboplatiini tai sisplatiini yhdistettynä paklitakseliin tai pemetreksediin, ja pemetreksedihoito-ohjelmiin kuului pakollinen ylläpitohoito. Kasvainstatus arvioitiin 9 viikon välein viikosta 9 alkaen vuonna 1 ja 12 viikon välein viikosta 55 alkaen vuonna 2. Ensisijainen tehon päätetapahtuma oli kokonaiselossaoloaika (OS). Keskeisiä toissijaisia päätetapahtumia, joita sokkoutettu riippumaton arviointilautakunta (IRC) arvioi RECIST 1.1:n perusteella, olivat elossaoloaika ilman taudin etenemistä (PFS) ja objektiivinen vasteluku (ORR).
466 potilaasta 327:llä (70 %) oli kasvaimia, jotka ilmensivät PD-L1:tä (≥1 %:ssa kasvainsoluista). Näistä potilaista 217 oli semiplimabia ja solunsalpaajahoitoa saaneessa ryhmässä ja 110 oli lumelääkettä ja solunsalpaajahoitoa saaneessa ryhmässä. Niillä 327 potilaalla, joiden kasvaimet ilmensivät PD-L1:tä ≥1 %:ssa kasvainsoluista, lähtötason muuttujat olivat: mediaani-ikä 62 vuotta (38 % oli vähintään 65-vuotiaita), 83 % miespuolisia, 87 % valkoihoisia, ECOG-toimintakykyluokka oli 16 %:lla 0 ja 83 %:lla 1, ja 6 %:lla oli ollut metastaasi aivoissa. Potilaista 51 % tupakoi edelleen, 34 % oli tupakoinut aiemmin ja 15 % ei ollut koskaan tupakoinut (alle 100 savuketta koko elämän aikana). Taudin ominaispiirteet olivat paikallisesti edennyt (14 %), metastaattinen (86 %), histologialtaan levyepiteeliperäinen (45 %) ja histologialtaan ei-levyepiteeliperäinen (55 %).
Koko tutkimuspopulaation primaarianalyysissä, jonka kohdalla seurannan keston mediaani oli 16,4 kuukautta, tutkimus osoitti, että OS parani tilastollisesti merkitsevästi potilailla, jotka satunnaistettiin saamaan semiplimabia yhdistelmänä solunsalpaajahoidon kanssa, verrattuna potilaisiin, jotka saivat lumelääkettä yhdistelmänä solunsalpaajahoidon kanssa.
Tehon tulokset potilailla, joiden kasvaimet ilmensivät PD-L1:tä ≥1 %:ssa kasvainsoluista, esitetään taulukossa 7, kuvassa 4 ja kuvassa 5.
Taulukko 7: Tehotulokset tutkimuksesta 16113, ei-pienisoluinen keuhkosyöpä (potilaat, joiden kasvaimet ilmensivät PD-L1:tä ≥1 %:ssa kasvainsoluista)a | ||
Päätetapahtumata | semiplimabi ja solunsalpaajahoito N = 217 | lumelääke ja solunsalpaajahoito N = 110 |
Kokonaiselossaoloaika (OS) | ||
Kuolemantapaukset, n (%) | 78 (35,9) | 55 (50,0) |
Mediaani kuukausina (95 % luottamusväli)b | 21,9 (17,3, NE) | 12,6 (10,3, 16,4) |
Hasardisuhde (95 % luottamusväli)c | 0,55 (0,39, 0,78) | |
Elossaoloaika ilman taudin etenemistä (PFS) | ||
Tapahtumat, n (%) | 134 (61,8) | 86 (78,2) |
Mediaani kuukausina (95 % luottamusväli)b | 8,5 (6,7, 10,7) | 5,5 (4,3, 6,2) |
Hasardisuhde (95 % luottamusväli)c | 0,48 (0,36, 0,63) | |
Objektiivinen vasteluku (ORR) (%) | ||
ORR (95 % luottamusväli)d | 47,9 (41,1, 54,8) | 22,7 (15,3, 31,7) |
Täydellinen vaste (CR) ‑luku | 2,8 | 0 |
Osittainen vaste (PR) ‑luku | 45,2 | 22,7 |
Vasteen kesto (DOR) | ||
Mediaani kuukausinab (vaihteluväli) | 15,6 (1,7, 18,7+) | 4,9 (1,9, 18,8+) |
NE: ei arvioitavissa (Not evaluable); +: meneillään oleva vaste (viimeinen tietojenkeräyspäivä 14.6.2021)
- Seurannan mediaanikesto: semiplimabi ja solunsalpaajahoito: 15,9 kuukautta, lumelääke ja solunsalpaajahoito: 16,1 kuukautta
- Perustuu Kaplan–Meier-menetelmään
- Perustuu ositettuun suhteelliseen hasardimalliin
- Clopper–Pearsonin eksakti luottamusväli
Ennalta määritellyn lopullisen analyysin ajankohtana potilailla, joiden kasvaimet ilmensivät PD-L1:tä ≥1 %:ssa kasvainsoluista ja jotka oli satunnaistettu saamaan semiplimabia yhdistelmänä solunsalpaajahoidon kanssa, oli havaittavissa kliinisesti merkittävä jatkuva hyöty elossaolon ja elossaoloajan ilman taudin etenemistä suhteen verrattuna pelkkää solunsalpaajahoitoa saaneisiin potilaisiin. Seurannan keston mediaani oli 27,9 kuukautta.
Kuva 4: OS tutkimuksessa 16113, ei-pienisoluinen keuhkosyöpä (potilaat, joiden kasvaimet ilmensivät PD-L1:tä ≥1 %:ssa kasvainsoluista) – (lopullinen analyysi)a
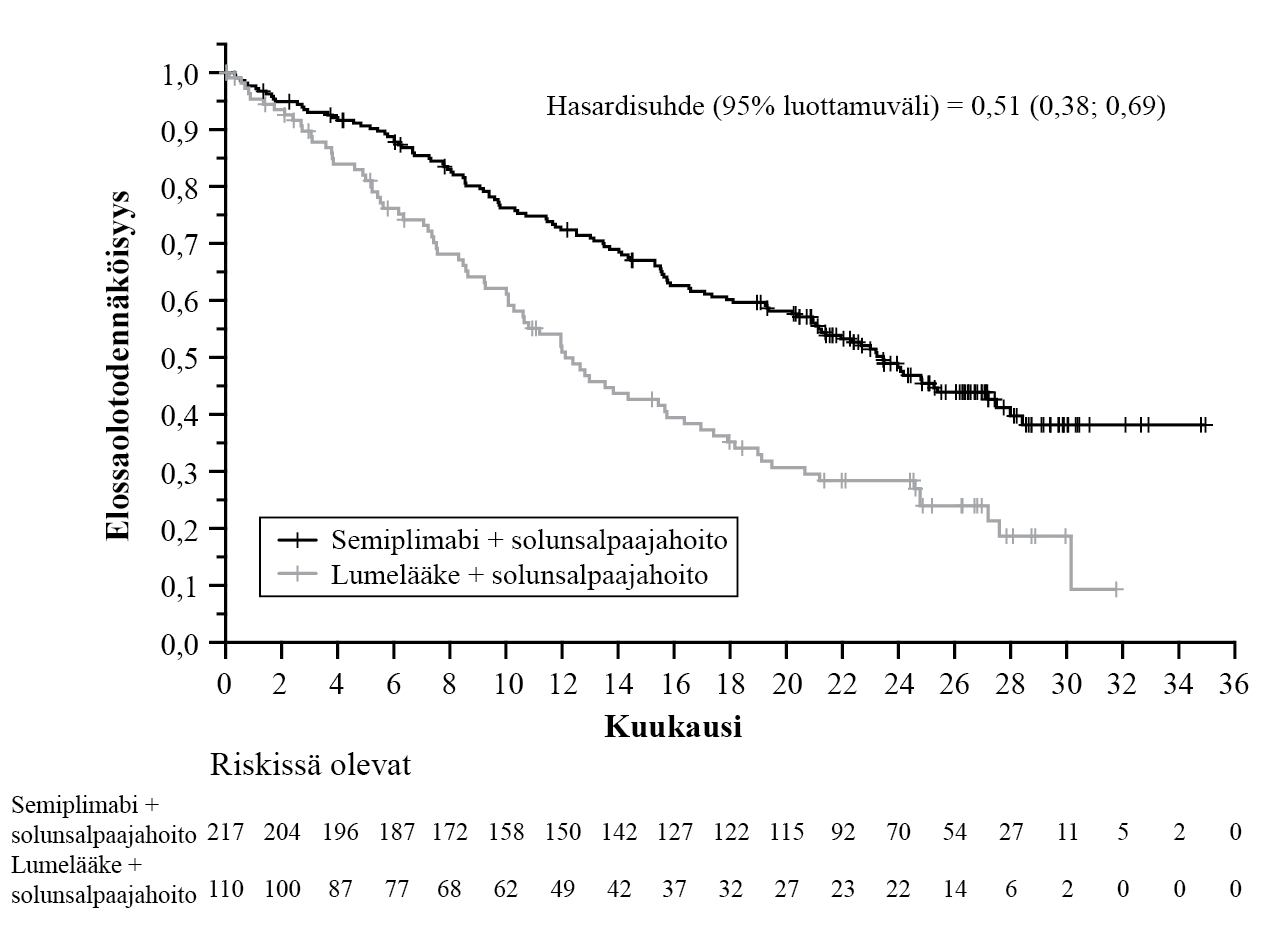
a. Perustuu OS:n lopulliseen analyysiin (viimeinen tietojenkeräyspäivä 14.6.2022)
Kuva 5 PFS tutkimuksessa 16113, ei-pienisoluinen keuhkosyöpä (potilaat, joiden kasvaimet ilmensivät PD-L1:tä ≥1 %:ssa kasvainsoluista) – (lopullinen analyysi)a
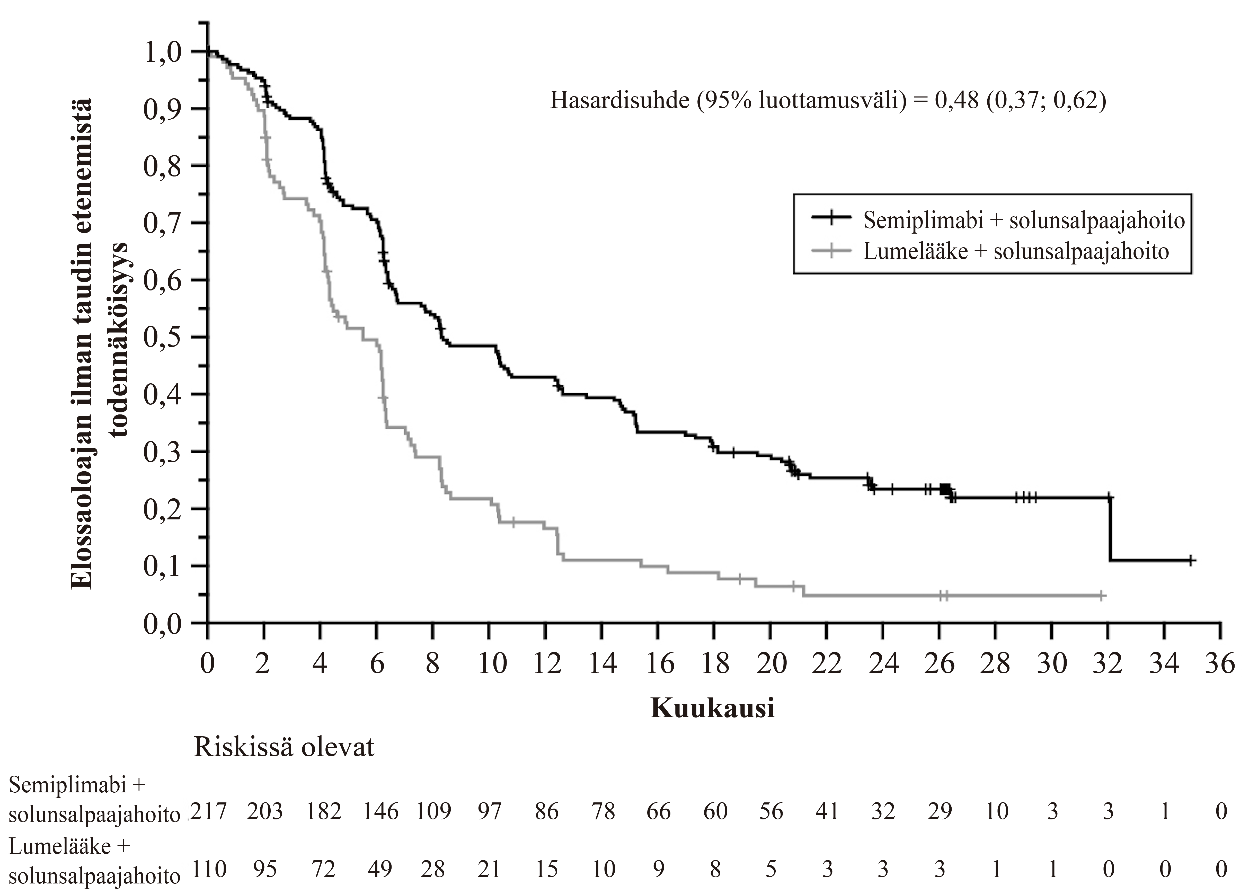
a. Perustuu PFS:n lopulliseen analyysiin (viimeinen tietojenkeräyspäivä 14.6.2022)
Kohdunkaulan syöpä
Semiplimabin tehoa ja turvallisuutta bevasitsumabin kanssa tai ilman bevasitsumabia on arvioitu satunnaistetussa avoimessa monikeskustutkimuksessa (tutkimus 1676) potilailla, joilla oli uusiutunut tai metastasoitunut kohdunkaulan syöpä ja joilla kasvaimet olivat edenneet platinapohjaisen solunsalpaajahoidon aikana tai sen jälkeen. Potilaita otettiin tutkimukseen kasvaimen PD‑L1-ilmentymisstatuksesta riippumatta. Tutkimuksesta suljettiin pois potilaat, joilla oli autoimmuunisairaus, joka oli 5 vuoden sisällä edellyttänyt systeemistä hoitoa immuunisalpaajilla, sekä potilaat, jotka olivat aiemmin saaneet hoitoa PD‑1:n/PD‑L1:n estäjillä.
Tehoa koskevan analyysin ositustekijöitä olivat maantieteellinen alue (Pohjois-Amerikka, Aasia, muu maailma) ja histologia [histologisesti levyepiteelisyöpä (SCC) tai histologisesti adenokarsinooma tai adenoskvamoosi syöpä (AC)]. Satunnaistaminen ositettiin myös sen mukaan, olivatko potilaat saaneet aiemmin bevasitsumabia, sekä potilaiden ECOG-toimintakykyluokan mukaan. Potilaat satunnaistettiin (1:1) saamaan semiplimabia 350 mg laskimoon 3 viikon välein tai laskimoon annettavaa solunsalpaajaa, joka oli tutkijan valinnan mukaan pemetreksedi, topotekaani, irinotekaani, gemsitabiini tai vinorelbiini, enintään 96 viikon ajan.
Hoitoa jatkettiin, kunnes tauti eteni, ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä, tai suunniteltu hoito päättyi. Kasvaimet arvioitiin ensimmäisten 24 viikon aikana 6 viikon välein ja sen jälkeen 12 viikon välein. Ensisijainen tehon päätetapahtuma oli kokonaiselossaoloaika (OS) levyepiteelisyöpäpotilailla ja sen jälkeen koko populaatiossa. Toissijaisia päätetapahtumia olivat elossaoloaika ilman taudin etenemistä (PFS), objektiivinen vasteluku (ORR) RECIST 1.1 ‑kriteerien mukaan ja vasteen kesto (DOR) tutkijan arvion mukaan.
Mediaani-ikä oli 51 vuotta (22–87 vuotta); 63 % oli valkoihoisia, 29 % aasialaisia ja 3,5 % mustaihoisia; 49 % oli saanut aiemmin bevasitsumabia, ECOG-toimintakykyluokka oli 47 %:lla 0 ja 53 %:lla 1, 78 %:lla oli SCC ja 22 %:lla AC, 94 %:lla oli metastasoitunut tauti; 57 % oli saanut yhtä aiemman linjan hoitoa uusiutuneen tai metastasoituneen taudin vuoksi ja 43 % oli saanut useampaa kuin yhtä aiemman linjan hoitoa uusiutuneen tai metastasoituneen taudin vuoksi. Seurannan keston mediaani primaarianalyysissä koko populaatiossa oli 18,2 kuukautta.
OS:n osoitettiin olevan tilastollisesti merkitsevästi parempi semiplimabin käytön yhteydessä sekä levyepiteelisyöpäpotilailla että koko populaatiossa solunsalpaajahoitoon verrattuna.
Tehoa koskevat tulokset on esitetty taulukossa 8, kuvassa 6 ja kuvassa 7.
Taulukko 8: Tehoa koskevat tulokset tutkimuksesta 1676, kohdunkaulan syöpä
Levyepiteelihistologia (SCC) (N = 477) | Koko populaatio (N = 608) | |||
Tehon päätetapahtumat | Semiplimabi 350 mg 3 viikon välein (n = 239) | Solunsalpaaja-hoito (n = 238) | Semiplimabi 350 mg 3 viikon välein (n = 304) | Solunsalpaaja-hoito (n = 304) |
Kokonaiselossaoloaika (OS)a | ||||
Kuolemantapaukset, n (%) | 143 (59,8 %) | 161 (67,6 %) | 184 (60,5 %) | 211 (69,4 %) |
Mediaani kuukausina (95 % luottamusväli)b | 11,1 (9,2, 13,4) | 8,8 (7,6, 9,8) | 12,0 (10,3, 13,5) | 8,5 (7,5, 9,6) |
Hasardisuhde (95 % luottamusväli)c | 0,73 (0,58, 0,91) | 0,69 (0,56, 0,84) | ||
p‑arvod | 0,00306 | 0,00011 | ||
Elossaoloaika ilman taudin etenemistä (PFS)a | ||||
Tapahtumat, n (%) | 197 (82,4 %) | 214 (89,9 %) | 253 (83,2 %) | 269 (88,5 %) |
Mediaani kuukausina (95 % luottamusväli)b | 2,8 (2,6, 4,0) | 2,9 (2,7, 3,9) | 2,8 (2,6, 3,9) | 2,9 (2,7, 3,4) |
Hasardisuhde (95 % luottamusväli)c | 0,71 (0,58, 0,86) | 0,75 (0,62, 0,89) | ||
p‑arvod | 0,00026 | 0,00048 | ||
Objektiivinen vasteluku (%)a | ||||
ORR (95 % luottamusväli)e | 17,6 (13,0, 23,0) | 6,7 (3,9, 10,7) | 16,4 (12,5, 21,1) | 6,3 (3,8, 9,6) |
Vasteen kesto (DOR)a | N = 42 | N = 16 | N = 50 | N = 19 |
Mediaani (kk)b (95 % luottamusväli) | 16,4 (12,4, NE) | 6,9 (4,2, 7,7) | 16,4 (12,4, NE) | 6,9 (5,1, 7,7) |
a. Seuranta-ajan mediaani: 18,2 kuukautta. (Viimeinen tietojenkeräyspäivä 4.1.2021.)
b. Perustuu Kaplan–Meier-estimaatteihin.
c. Perustuu ositettuun suhteelliseen hasardimalliin, jossa ositus on tehty histologian ja maantieteellisen alueen mukaan.
d. Yksitahoinen p‑arvo, joka perustuu ositettuun suhteelliseen hasardimalliin (semiplimabi vs. solunsalpaaja).
e. Perustuu Clopper–Pearsonin eksaktiin luottamusväliin.
Päivitetyssä kokonaiselossaoloajan analyysissä (viimeinen tietojenkeräyspäivä 4.1.2022) seurannan keston mediaanin ollessa 30,2 kuukautta semiplimabihoidolla osoitettiin olevan jatkuva hyöty elossaolon suhteen verrattuna solunsalpaajahoitoon (hasardisuhde [HR]: 0,66, 95 %:n luottamusväli [0,55; 0,79]) (ks. kuva 6).
Kuva 6: OS tutkimuksessa 1676, kohdunkaulan syöpä – koko populaatio (päivitetty analyysi)a
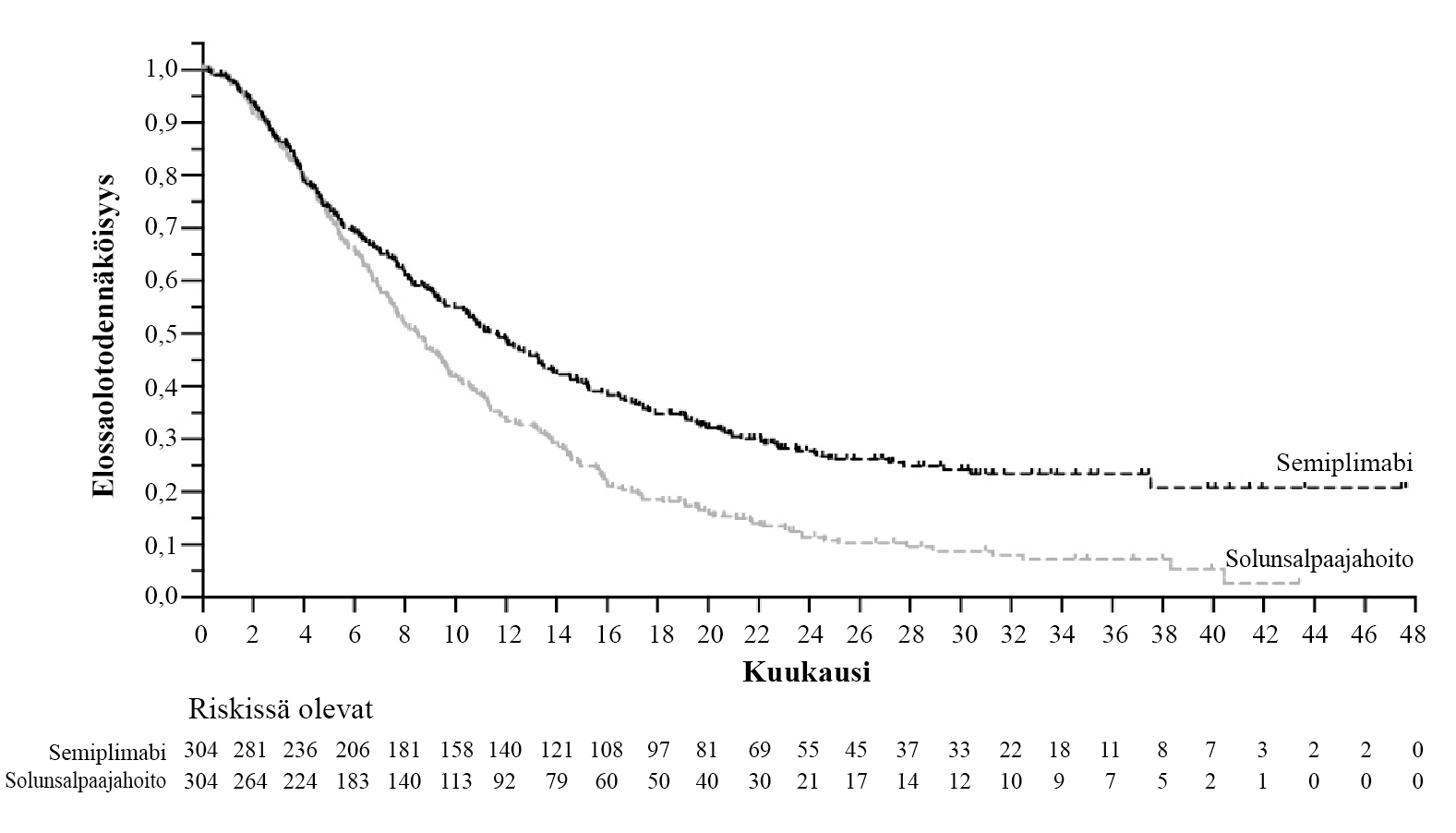
a. Perustuu tuloksiin päivitetystä kokonaiselossaolon analyysistä, joka toteutettiin vuoden kuluttua primaarianalyysistä.
Kuva 7: PFS tutkimuksessa 1676, kohdunkaulan syöpä – koko populaatio (primaarianalyysi)
Alaryhmäanalyysit:
Alaryhmäanalyysissä, jossa arvioitiin kokonaiselossaoloa eri histologioissa päivitetyn eksploratiivisen kokonaiselossaolon analyysin perusteella, HR oli SCC-ryhmässä 0,69 (95 %:n luottamusväli, 0,56; 0,85) ja AC-ryhmässä 0,55 (95 %:n luottamusväli, 0,36; 0,81).
Elossaolon yhteyttä kasvainsolujen PD‑L1-ilmentymisstatukseen arvioitiin eksploratiivisessa alaryhmäanalyysissä hyödyntäen kliinisissä tutkimuksissa käytettävää määritystä (VENTANA PD‑L1 SP263 ‑määritystä). Tutkimukseen otetuista 608 potilaasta 42 %:lta oli näytteitä, joista tutkittiin PD‑L1. Näistä näytteistä 64 %:ssa PD‑L1 oli ≥1 % ja 36 %:ssa PD‑L1 oli <1 %. Päivitetyssä eksploratiivisessa kokonaiselossaolon analyysissä seurannan keston mediaanin ollessa 30,2 kuukautta HR oli 0,70 (95 %:n luottamusväli 0,48; 1,01) ryhmässä, jossa PD‑L1 oli ≥1 %, ja 0,85 (95 %:n luottamusväli 0,53; 1,36) ryhmässä, jossa PD‑L1 oli <1 %.
Iäkkäät potilaat
Monoterapia
1 281 potilaasta, joita oli hoidettu semiplimabimonoterapialla kliinisissä tutkimuksissa, 52,2 % (669 / 1 281) oli alle 65-vuotiaita, 25,9 % (332 / 1 281) oli 65–74-vuotiaita ja 21,9 % (280 / 1 281) oli vähintään 75-vuotiaita.
Kaiken kaikkiaan tehon suhteen ei havaittu eroa iäkkäiden ja nuorempien potilaiden välillä. 65‑vuotiailla ja sitä vanhemmilla potilailla oli suuntaus vakavien haittavaikutusten ja hoidon keskeyttämisen suurempaan esiintymistiheyteen kuin alle 65-vuotiailla potilailla, jotka saivat semiplimabia monoterapiana.
Yhdistelmähoito
312 potilaasta, joita oli hoidettu semiplimabilla yhdistelmänä solunsalpaajahoidon kanssa, 59 % (184/312) oli alle 65-vuotiaita, 35,3 % (110/312) oli 65–74-vuotiaita ja 5,8 % (18/312) oli vähintään 75-vuotiaita.
Kaiken kaikkiaan turvallisuuden ja tehon suhteen ei havaittu eroa iäkkäiden ja nuorempien potilaiden välillä, kun potilaat saivat semiplimabia yhdistelmänä solunsalpaajahoidon kanssa.
Pediatriset potilaat
Tutkimuksessa 1690 arvioitiin semiplimabin tehoa, turvallisuutta ja farmakokinetiikkaa 57 pediatrisella potilaalla ja nuorella aikuispotilaalla, joilla oli uusiutuneita tai hoitoon reagoimattomia kiinteitä kasvaimia ja keskushermoston kasvaimia, vastikään diagnosoitu diffuusi sisäsyntyinen aivosillan gliooma (DIPG), vastikään diagnosoitu korkean asteen gliooma (HGG) tai uusiutunut korkean asteen gliooma. Tämä avoin monikeskustutkimus koostui kahdesta vaiheesta, vaiheesta 1 ja tehon arviointivaiheesta, jotka toteutettiin rinnakkain.
Vaiheessa 1 semiplimabimonoterapian turvallisuutta ja farmakokinetiikkaa arvioitiin 25 potilaalla (0 – < 18-vuotiaita): 8 potilaalla oli uusiutuneita tai hoitoon reagoimattomia kiinteitä kasvaimia ja 17 potilaalla oli uusiutuneita tai hoitoon reagoimattomia keskushermoston kasvaimia. 16 potilasta, joilla oli kiinteitä tai keskushermoston kasvaimia, sai semiplimabiannoksena 3 mg/kg kahden viikon välein, ja 9 potilasta, joilla oli keskushermoston kasvaimia, sai semiplimabiannoksena 4,5 mg/kg kahden viikon välein. Tehon arviointivaiheessa semiplimabin tehoa ja turvallisuutta yhdistelmänä sädehoidon kanssa arvioitiin 32 potilaalla (3–25-vuotiaita), joilla oli keskushermoston kasvaimia: 11 potilaalla oli vastikään diagnosoitu DIPG, 12 potilaalla oli vastikään diagnosoitu HGG ja 9 potilaalla oli uusiutunut HGG. Kaikki vähintään 12-vuotiaat potilaat saivat semiplimabiannoksena 3 mg/kg kahden viikon välein, ja 3 – < 12-vuotiaat potilaat saivat semiplimabiannoksena 4,5 mg/kg kahden viikon välein. Semiplimabi annettiin 30 minuutin pituisena laskimoinfuusiona.
Semiplimabilla ei todettu olevan tehoa yhdistelmänä sädehoidon kanssa tutkittavissa potilasjoukoissa, koska kokonaiselossaoloajan (OS) tai elossaoloajan ilman taudin etenemistä (PFS) paranemista ei ollut osoitettavissa historiatietoihin verrattuna.
Uusia riskejä tai turvallisuussignaaleja ei todettu.
Farmakokinetiikka
Pitoisuustiedot kerättiin 1 063 potilaasta, joilla oli erilaisia kiinteitä kasvaimia ja jotka saivat semiplimabia laskimoon, ja tiedot yhdistettiin populaatiofarmakokineettiseen analyysiin.
Annostuksella 350 mg kolmen viikon välein semiplimabin vakaan tilan pitoisuuksien keskiarvot vaihtelivat välillä Ctrough 59 mg/l – Cmax (infuusion päätyttyä) 171 mg/l. Vakaan tilan altistus saavutetaan, kun hoito on kestänyt noin 4 kuukautta.
Vakaan tilan semiplimabialtistus potilailla, joilla oli kiinteitä kasvaimia, oli samansuuruinen annoksilla 350 mg kolmen viikon välein ja 3 mg/kg kahden viikon välein.
Suuren riskin ihon okasolusyöpää sairastavilla potilailla simuloitujen pitoisuuksien keskiarvo luotiin päivitetyllä populaatiofarmakokineettisellä mallilla ja 1 000 virtuaalipotilaalla hoito-ohjelmaa kohti. Näillä potilailla annoksella 350 mg 3 viikon välein 12 viikon ajan, minkä jälkeen 700 mg 6 viikon välein 36 lisäviikon ajan, semiplimabin keskimääräiset pitoisuudet vakaassa tilassa vaihtelivat alimman pitoisuuden (Ctrough) 52,5 mg/l ja infuusion lopussa olevan enimmäispitoisuuden (Cmax) 233 mg/l välillä. Nämä pitoisuudet olivat vakaassa tilassa 66,3 mg/l ja 154 mg/l käytettäessä hoito-ohjelmana 350 mg 3 viikon välein 48 viikon ajan.
Imeytyminen
Semiplimabi annetaan laskimoon, joten sillä saavutetaan täysi hyötyosuus.
Jakautuminen
Semiplimabi jakautuu ensisijaisesti verisuonistoon, ja sen jakautumistilavuus vakaassa tilassa (Vss) on 5,9 litraa. Tmax:n mediaani on 30 minuutin infuusion lopussa.
Biotransformaatio
Erityisiä metaboliatutkimuksia ei ole tehty, koska semiplimabi on proteiini. Semiplimabin odotetaan hajoavan pieniksi peptideiksi ja yksittäisiksi aminohapoiksi.
Eliminaatio
Semiplimabin puhdistuma on lineaarinen kahden viikon välein annettavilla 1–10 mg/kg:n annoksilla. Semiplimabin puhdistuma on ensimmäisen annoksen saamisen jälkeen noin 0,25 l vuorokaudessa. Kokonaispuhdistuma vaikuttaa pienenevän noin 11 % ajan myötä, mikä johtaa 0,22 l:n puhdistumaan vuorokaudessa vakaassa tilassa (CLss). Puhdistuman pienenemisellä ei katsota olevan kliinistä merkitystä. Puoliintumisaika annosvälillä on vakaassa tilassa 22 vuorokautta.
Lineaarisuus/ei-lineaarisuus
Annostuksilla 1–10 mg/kg kahden viikon välein semiplimabin farmakokinetiikan havaittiin olevan lineaarinen ja suhteessa annokseen, mikä viittaa systeemisen kohdevälitteisen reitin saturoitumiseen.
Erityisryhmät
Populaatiofarmakokineettinen analyysi viittaa siihen, että seuraavilla tekijöillä ei ole kliinisesti merkittävää vaikutusta semiplimabialtistukseen: ikä, sukupuoli, paino, rotu, syöpätyyppi, albumiinipitoisuus, munuaisten vajaatoiminta ja lievä tai kohtalainen maksan vajaatoiminta.
Pediatriset potilaat
Farmakokinetiikkaa pediatrisilla potilailla arvioitiin päivitetyn populaatiofarmakokineettisen mallin perusteella, joka sisälsi yhdistetyt farmakokineettiset tiedot 1227 aikuiselta, joilla oli erilaisia kiinteitä kasvaimia ja jotka saivat semiplimabimonoterapiaa laskimoon, sekä 55 pediatriselta potilaalta tai nuorelta aikuispotilaalta (ikä 1–24 vuotta), jotka saivat semiplimabia laskimoon annoksella 3 mg/kg tai 4,5 mg/kg kahden viikon välein, sädehoidon kanssa tai ilman sitä. Pediatristen potilaiden altistus oli samanlaista kuin aikuisilla, jotka saivat semiplimabia laskimoon 350 mg kolmen viikon välein. Altistuksen havaittiin olevan hieman suurempaa 0 – < 12-vuotiailla pediatrisilla potilailla, jotka saivat 4,5 mg/kg kahden viikon välein. Kokonaisuutena pienin ennustettu mediaaniarvoinen Ctrough,ss -pitoisuus ja suurin Cmax,ss -pitoisuus kaikkien pediatristen potilaiden osalta oli vaihteluvälin sisällä, joka havaittiin 350 mg laskimoon kolmen viikon välein saaneilla aikuispotilailla.
Munuaisten vajaatoiminta
Munuaisten vajaatoiminnan vaikutusta semiplimabialtistukseen arvioitiin populaatiofarmakokineettisellä analyysillä potilailla, joilla oli lievä (kreatiniinipuhdistuma 60‑89 ml/min; n = 396), kohtalainen (kreatiniinipuhdistuma 30–59 ml/min; n = 166) tai vaikea (kreatiniinipuhdistuma 15–29 ml/min; n = 7) munuaisten vajaatoiminta. Semiplimabialtistuksessa munuaisten vajaatoimintaa sairastavilla potilailla ei todettu kliinisesti merkittäviä eroja verrattuna potilaisiin, joilla munuaiset toimivat normaalisti. Semiplimabia ei ole tutkittu potilailla, joiden kreatiniinipuhdistuma on <21 ml/min (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Maksan vajaatoiminnan vaikutusta semiplimabialtistukseen arvioitiin populaatiofarmakokineettisellä analyysillä. Potilailla, joilla oli lievä maksan vajaatoiminta (n = 22) (kokonaisbilirubiini [TB] yli 1,0‑1,5-kertainen viitealueen ylärajaan nähden ja mikä tahansa aspartaattiaminotransferaasi [ASAT] ‑arvo) ja kohtalainen maksan vajaatoiminta (n = 3), (kokonaisbilirubiini [TB] yli 1,5–3,0-kertainen viitealueen ylärajaan nähden ja mikä tahansa aspartaattiaminotransferaasi [ASAT] -arvo), ei todettu kliinisesti merkittäviä eroja semiplimabialtistuksessa verrattuna potilaisiin, joilla maksa toimi normaalisti. Semiplimabia ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla. Vaikeaa maksan vajaatoimintaa sairastavista potilaista ei ole riittävästi tietoa annossuositusten antamiseen (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Semiplimabin mahdollisen karsinogeenisuuden tai genotoksisuuden arvioimiseksi ei ole tehty tutkimuksia. Semiplimabilla ei ole tehty eläinten lisääntymistutkimuksia (ks. kohta Raskaus ja imetys). Kuten kirjallisuudessa on raportoitu, PD‑1/PD‑L1-signaalireitti edesauttaa raskauden jatkumista ylläpitämällä immunologista toleranssia, ja tutkimukset ovat osoittaneet, että PD‑1-reseptorin estäminen johtaa raskauden keskeytymiseen varhaisessa vaiheessa. Spontaanien keskenmenojen ja/tai resorptioiden on osoitettu lisääntyneen sekä hiirillä että apinoilla, joilla PD‑L1:n ilmentyminen on vähäistä (knockout- tai anti-PD1/PD‑L1:n monoklonaaliset vasta-aineet). Näillä eläinlajeilla emon ja sikiön välinen yhteys on samankaltainen kuin ihmisillä.
Farmaseuttiset tiedot
Apuaineet
L-histidiini
L-histidiinimonohydrokloridimonohydraatti
Sakkaroosi
L-proliini
Polysorbaatti 80
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
4 vuotta
Avaamisen jälkeen
Avaamisen jälkeen lääkevalmiste on laimennettava ja annettava infuusiona välittömästi (ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa).
Infuusioliuoksen valmistamisen jälkeen
Mikrobiologiselta kannalta valmistettu infuusioneste on annettava välittömästi. Jos laimennettua liuosta ei anneta välittömästi, käytön aikaiset säilytysajat ja säilytysolosuhteet ennen käyttöä ovat käyttäjän vastuulla.
Kemiallinen ja fysikaalinen käytön aikainen stabiilius on osoitettu seuraavasti:
- huoneenlämmössä korkeintaan 25°C:ssa enintään 8 tunnin ajan infuusioliuoksen valmistamisesta infuusion päättymiseen
tai
- jääkaapissa 2–8°C:ssa enintään 10 päivän ajan infuusioliuoksen valmistamisesta infuusion päättymiseen. Anna laimennetun liuoksen lämmetä huoneenlämpöön ennen antoa.
Ei saa jäätyä.
Säilytys
Avaamaton injektiopullo
Säilytä jääkaapissa (2°C - 8°C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Avatun tai laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LIBTAYO infuusiokonsentraatti, liuosta varten
350 mg (L:ei) 1 kpl (7 ml (50 mg/ml)) (6714,12 €)
PF-selosteen tieto
LIBTAYO on 10 ml:n kirkkaasta tyypin 1 lasista valmistetussa injektiopullossa, jossa on harmaa FluroTec‑materiaalilla pinnoitettu klooributyylitulppa ja repäisykorkillinen suljin.
Yksi pakkaus sisältää yhden injektiopullon.
Valmisteen kuvaus:
Kirkas tai hieman opaalinhohtoinen, väritön tai vaaleankeltainen liuos, jonka pH on 6,0 ja osmolaalisuus 300–360 mmol/kg. Kertakäyttöön tarkoitetussa injektiopullossa oleva liuos saattaa sisältää hyvin pieniä määriä hiukkasia, joiden väri vaihtelee läpikuultavasta valkoiseen.
Käyttö- ja käsittelyohjeet
Valmistelu ja anto
- Tarkista silmämääräisesti ennen antoa, näkyykö lääkevalmisteessa hiukkasia tai värimuutoksia. LIBTAYO on kirkas tai hieman opaalinhohtoinen, väritön tai vaaleankeltainen liuos, joka saattaa sisältää hyvin pieniä määriä läpikuultavia tai valkoisia hiukkasia.
- Hävitä injektiopullo, jos liuos on sameaa tai värjäytynyttä tai sisältää muita ylimääräisiä hiukkasia kuin hyvin pieniä määriä läpikuultavia tai valkoisia hiukkasia.
- Älä ravista injektiopulloa.
- Vedä LIBTAYO-injektiopullosta 7 ml (350 mg) liuosta ruiskuun ja siirrä se laskimoon antamista varten tarkoitettuun infuusiopussiin, jossa on natriumkloridi-injektioliuosta (9 mg/ml, 0,9 %) tai glukoosi-injektioliuosta (50 mg/ml, 5 %). Sekoita laimennettu liuos kääntelemällä varovasti. Älä ravista liuosta. Laimennetun liuoksen lopullisen pitoisuuden on oltava 1‑20 mg/ml. Käytä 2 injektiopulloa 700 mg:n annokseen.
- LIBTAYO annetaan infuusiona laskimoon 30 minuutin aikana käyttämällä laskimoyhteyttä, jossa on steriili, pyrogeeniton, vähän proteiineja sitova in-line- tai add-on-suodatin (huokoskoko 0,2–5 mikronia).
- Älä anna samanaikaisesti muita lääkevalmisteita samalla infuusiolinjalla.
LIBTAYO on tarkoitettu vain kerta-antoon. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
LIBTAYO infuusiokonsentraatti, liuosta varten
350 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FF06
Valmisteyhteenvedon muuttamispäivämäärä
17.11.2025
Yhteystiedot
Warrington Place
D02 HH27 Dublin 2
Ireland