
ALPROLIX injektiokuiva-aine ja liuotin, liuosta varten 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU
Vaikuttavat aineet ja niiden määrät
ALPROLIX 250 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 250 IU hyytymistekijä IX:ää (rDNA), eftrenonakogi alfaa (eftrenonacogum alfa). Käyttökuntoon saattamisen jälkeen ALPROLIX sisältää noin 250 IU (50 IU/ml) ihmisen hyytymistekijä IX:ää (rDNA), eftrenonakogi alfaa.
ALPROLIX 500 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 500 IU hyytymistekijä IX:ää (rDNA), eftrenonakogi alfaa (eftrenonacogum alfa). Käyttökuntoon saattamisen jälkeen ALPROLIX sisältää noin 500 IU (100 IU/ml) ihmisen hyytymistekijä IX:ää (rDNA), eftrenonakogi alfaa.
ALPROLIX 1000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 1000 IU hyytymistekijä IX:ää (rDNA), eftrenonakogi alfaa (eftrenonacogum alfa). Käyttökuntoon saattamisen jälkeen ALPROLIX sisältää noin 1000 IU (200 IU/ml) ihmisen hyytymistekijä IX:ää (rDNA), eftrenonakogi alfaa.
ALPROLIX 2000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 2000 IU hyytymistekijä IX:ää (rDNA), eftrenonakogi alfaa (eftrenonacogum alfa). Käyttökuntoon saattamisen jälkeen ALPROLIX sisältää noin 2000 IU (400 IU/ml) ihmisen hyytymistekijä IX:ää (rDNA), eftrenonakogi alfaa.
ALPROLIX 3000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 3000 IU hyytymistekijä IX:ää (rDNA), eftrenonakogi alfaa (eftrenonacogum alfa). Käyttökuntoon saattamisen jälkeen ALPROLIX sisältää noin 3000 IU (600 IU/ml) ihmisen hyytymistekijä IX:ää (rDNA), eftrenonakogi alfaa.
Teho (IU) on määritetty käyttämällä Euroopan farmakopean yksivaiheista hyytymistestiä. ALPROLIX-valmisteen spesifinen aktiivisuus on 55‑84 IU/mg proteiinia.
Eftrenonakogi alfa (rekombinantti hyytymistekijä IX Fc-fuusioproteiini (rFIXFc)) sisältää 867 aminohappoa. Se on korkean puhtausasteen valmiste, jota valmistetaan yhdistelmä-DNA-tekniikalla ihmisalkion munuaisten (HEK) solulinjassa siten, että solunviljelyssä, puhdistamisessa tai lopullisessa valmisteessa ei ole käytetty lainkaan eksogeenista, ihmisestä tai eläimestä peräisin olevaa proteiinia.
Apuaine, jonka vaikutus tunnetaan
0,3 mmol (6,4 mg) natriumia / injektiopullo.
0,5 mg polysorbaattia 20 / injektiopullo.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Hemofilia B:tä (synnynnäinen hyytymistekijä IX:n puute) sairastavien potilaiden verenvuodon hoito ja ennaltaehkäisy.
ALPROLIX-valmistetta voidaan käyttää kaikille ikäryhmille.
Ehto
Hoito tulee aloittaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito pitää toteuttaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Hoidon valvonta
Hyytymistekijä IX:n pitoisuudet suositellaan määrittämään hoidon aikana tarkoituksenmukaisin väliajoin, jotta annettava annos ja toistettujen injektioiden antotiheys voidaan määrittää. Potilaiden tekijä IX -vasteet saattavat vaihdella yksilöllisesti siten, että puoliintumisajat ja saanto ovat erilaisia eri potilailla. Potilaan painoon perustuvaa annosta on ehkä muutettava ali- tai ylipainoisilla potilailla. Korvaushoidon tarkka seuranta hyytymistekijämääritysten (plasman FIX-aktiivisuus) avulla on välttämätöntä erityisesti suurten leikkausten yhteydessä.
Käytettäessä in vitro tromboplastiiniaikaan (aPTT) perustuvaa yksivaiheista hyytymismääritystä tekijän IX aktiivisuuden määrittämiseksi potilaiden verikokeista, plasman tekijä IX:n aktiivisuuden tuloksiin saattavat vaikuttaa huomattavasti sekä aPTT-reagenssin tyyppi että määrityksessä käytettävä viitestandardi. Tällä on merkitystä etenkin vaihdettaessa laboratoriota ja/tai määrityksessä käytettävää reagenssia.
Yksivaiheisella hyytymismäärityksellä tehdyt mittaukset, joissa käytetään kaoliinipohjaista aPTT-reagenssia, johtavat todennäköisesti aktiivisuustason aliarviointiin.
Annostus
Korvaushoidon annos ja kesto riippuvat hyytymistekijä IX:n puutoksen vaikeusasteesta, verenvuodon laajuudesta ja vuotokohdasta sekä potilaan kliinisestä tilasta.
Annettavien tekijä IX -yksiköiden määrä ilmaistaan kansainvälisinä yksikköinä (International Units, IU), mikä perustuu FIX-valmisteita koskevaan voimassaolevaan WHO-standardiin. FIX-aktiivisuus plasmassa ilmaistaan joko prosentteina (suhteessa normaaliin ihmisen plasmaan) tai IU-yksikköinä (plasman FIX-pitoisuuden kansainvälisen standardin mukaan).
Yksi kansainvälinen yksikkö (IU) rekombinanttitekijä IX Fc -aktiivisuutta vastaa FIX-määrää yhdessä millilitrassa normaalia ihmisen plasmaa.
Tarvittaessa toteutettava hoito
Tarvittavan rekombinanttitekijä IX Fc -annoksen laskeminen perustuu empiiriseen havaintoon, että 1 kansainvälinen yksikkö (IU) FIX-valmistetta painokiloa kohden suurentaa plasman FIX-aktiivisuutta 1 %:lla normaalista aktiivisuudesta (IU/dl). Tarvittava annos lasketaan seuraavalla laskukaavalla:
Tarvittava yksikkömäärä = potilaan paino (kg) × haluttu tekijä IX:n pitoisuuden lisäys (%) (IU/dl) × {käänteisarvo havaitusta saannosta (IU/kg per IU/dl)}
Annettavalla annoksella ja antotiheydellä on aina pyrittävä yksilölliseen kliiniseen tehokkuuteen. Jos vuodon hallitsemiseen tarvitaan toistoannosta, ALPROLIX-valmisteen pidentynyt puoliintumisaika on otettava huomioon (ks. kohta Farmakokinetiikka). Huippuaktiivisuuden saavuttamiseen kuluvan ajan ei odoteta viivästyvän.
Jos potilaalla on jokin seuraavista verenvuototapahtumista, FIX-aktiivisuus ei saa laskea taulukossa mainitun pitoisuuden alapuolelle (% normaalista tai IU/dl) vastaavan hoitojakson aikana. Taulukkoa 1 voidaan käyttää annostusohjeena verenvuotojen ja kirurgisten toimenpiteiden yhteydessä:
Taulukko 1: ALPROLIX-annostusohje verenvuototapausten hoitoa ja leikkauksia varten
Verenvuodon määrä / kirurgisen toimenpiteen tyyppi | Tarvittava FIX-pitoisuus (%) (IU/dl) | Antotiheys (tuntia) / hoidon kesto (vrk) |
Verenvuoto | ||
Alkava hemartroosi, lihasverenvuoto tai suun limakalvovuoto | 20‑40 | Injektio toistetaan 48 tunnin välein, kunnes kipuna ilmenevä vuotoepisodi on mennyt ohi tai paraneminen on tapahtunut. |
Laajempi hemartroosi, lihasverenvuoto tai hematooma | 30‑60 | Injektio toistetaan 24‑48 tunnin välein, kunnes kipu lakkaa ja akuutti toimintakyvyn vajaus korjautuu. |
Hengenvaaralliset verenvuodot | 60‑100 | Injektio toistetaan 8‑24 tunnin välein, kunnes vaara on ohi. |
Leikkaus | ||
Pieni leikkaus kuten hampaanpoisto | 30‑60 | Injektio toistetaan 24 tunnin kuluttua tarvittaessa, kunnes paraneminen on tapahtunut 1. |
Suuri leikkaus | 80‑100 (pre- ja postoperatiivisesti) | Injektio toistetaan 8‑24 tunnin välein tarpeen mukaan, kunnes haava on parantunut riittävästi. Hoitoa jatketaan sen jälkeen vielä vähintään 7 vuorokauden ajan, jotta tekijä IX-aktiivisuus pysyy 30‑60 %:n (IU/dl) tasolla. |
1 Joillakin potilailla ja joissakin tilanteissa annosväliä voidaan pidentää korkeintaan 48 tuntiin (ks. farmakokineettiset tiedot kohdasta Farmakokinetiikka).
Estohoito eli profylaksi
Pitkäkestoisessa vuotojen estohoidossa suositeltavat aloitusohjelmat ovat joko:
-
50 IU/kg kerran viikossa, säädä annosta yksilöllisen vasteen perusteella, tai
-
100 IU/kg joka 10. päivä, säädä antoväliä yksilöllisen vasteen perusteella. Joillekin potilaille, joilla vuodot ovat hyvin hallinnassa heidän saadessaan hoitoa joka 10. päivä, voidaan mahdollisesti antaa hoitoa joka 14. päivä tai pitemmin väliajoin.
Suurin suositeltu estohoitoannos on 100 IU/kg.
Iäkkäät potilaat
Valmisteen käytöstä ≥ 65-vuotiaille on vain vähän kokemusta.
Pediatriset potilaat
Alle 12‑vuotiaille lapsille tiheämmät antovälit tai suuremmat annokset voivat olla tarpeen ja suositeltu aloitusannos on 50‑60 IU/kg joka 7. päivä. 12 vuotta täyttäneille nuorille annossuositukset ovat samat kuin aikuisille. Ks. kohdat Farmakodynamiikka ja Farmakokinetiikka.
Suurin suositeltu estohoitoannos on 100 IU/kg.
Antotapa
Laskimoon.
Jos potilas itse tai häntä hoitava omainen antaa valmisteen, heille on annettava asianmukainen opastus.
ALPROLIX injisoidaan laskimoon usean minuutin aikana. Antonopeus on määritettävä potilaan sietämän kiputason mukaan ja se saa olla enintään 10 ml/min.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yliherkkyys
Allergistyyppisiä yliherkkyysreaktioita on raportoitu ALPROLIX-valmistetta käytettäessä. Potilaita on neuvottava lopettamaan lääkevalmisteen käyttö välittömästi ja ottamaan yhteyttä lääkäriin, jos yliherkkyysoireita ilmaantuu.
Potilaille on kerrottava varhaisista yliherkkyysreaktioiden oireista, joita ovat esim. paukamat, yleistynyt nokkosihottuma, puristuksen tunne rinnassa, hengityksen vinkuminen, verenpaineen lasku ja anafylaksia.
Anafylaktisen sokin saanutta potilasta on hoidettava yleisten sokinhoito-ohjeiden mukaisesti.
Vasta-aineet eli inhibiittorit
Toistuvien ihmisen hyytymistekijä IX-valmisteilla tehtyjen hoitojen jälkeen on seurattava, kehittyykö potilaille neutraloivia vasta-aineita (inhibiittoreita), ja mahdollisten vasta-aineiden pitoisuus Bethesda-yksikköinä (BU) on määritettävä soveltuvan biologisen testin avulla.
Kirjallisuudessa on raportteja, joissa osoitetaan korrelaatio tekijä IX:n vasta-aineiden ja allergisten reaktioiden esiintymisen välillä. Siksi potilailta, jotka saavat allergisia reaktioita, pitäisi tutkia inhibiittorin esiintyminen. On huomattava, että potilailla, joilla esiintyy tekijä IX:n inhibiittoreita, voi olla myöhemmin lisääntynyt tekijä IX:n aiheuttaman anafylaksian vaara.
Tekijä IX-valmisteiden aiheuttamien allergisten reaktioiden vaaran vuoksi tekijä IX:n aloitusannokset pitäisi antaa hoitavan lääkärin arvioista riippuen lääketieteellisen valvonnan alaisena paikassa, missä allergisia reaktioita voidaan hoitaa lääketieteellisesti asianmukaisesti.
Tromboembolia
Tekijä IX-valmisteiden käyttöön liittyvien tromboembolisten komplikaatioiden mahdollisen riskin takia olisi aloitettava tromboottisen ja konsumptiokoagulopatian ensioireiden kliininen valvonta ja asianmukainen biologinen testaus, kun tätä tuotetta annetaan potilaille, joilla on jokin maksasairaus, leikkauksen jälkeen, vastasyntyneille/imeväisille tai potilaille, joilla on tromboosin tai disseminoituneen intravaskulaarisen koagulaation (DIC) riski. Näissä tapauksissa on punnittava ALPROLIX-hoidon mahdolliset edut ja edellä esitettyjen komplikaatioiden riskit.
Kardiovaskulaariset tapahtumat
Potilailla, joilla on ennestään kardiovaskulaarisia riskitekijöitä, korvaushoito tekijä IX -valmisteilla voi suurentaa kardiovaskulaarista riskiä.
Katetreihin liittyvät komplikaatiot
Jos toimenpide edellyttää keskuslaskimokatetria, on huomioitava keskuslaskimokatetriin liittyvät komplikaatiot, mukaan lukien paikalliset infektiot, bakteremia ja katetrointikohdan tromboosi.
Pediatriset potilaat
Luetellut varoitukset ja varotoimet koskevat sekä aikuisia että lapsia.
Apuaineeseen liittyvät huomioitavat seikat
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per injektiopullo eli sen voidaan sanoa olevan ”natriumiton”. Jos hoidossa käytetään useampaa injektiopulloa, natriumin kokonaismäärä on otettava huomioon.
Polysorbaatti
Tämä lääkevalmiste sisältää 0,5 mg polysorbaattia 20 per injektiopullo. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutuksia ALPROLIX-valmisteen ja muiden lääkevalmisteiden välillä ei ole raportoitu. Yhteisvaikutustutkimuksia ei ole tehty.
Raskaus ja imetys
Raskaus ja imetys
ALPROLIX-valmisteella ei ole tehty lisääntymistä koskevia eläinkokeita. Hiirillä on tehty koe, jossa tutkittiin valmisteen siirtymistä istukan läpi (ks. kohta Prekliiniset tiedot turvallisuudesta). Koska hemofilia B on naisilla harvinainen, raskauden ja imetyksen aikaisesta tekijä IX-hoidosta ei ole kokemuksia. Tämän vuoksi tekijä IX-valmistetta saa käyttää raskauden ja imetyksen aikana vain, jos se on selvästi välttämätöntä.
Hedelmällisyys
Hedelmällisyyttä koskevia tietoja ei ole saatavilla. Eläimille tehtyjä hedelmällisyystutkimuksia ei ole suoritettu ALPROLIX-valmisteella.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
ALPROLIX-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Harvinaisissa tapauksissa on havaittu yliherkkyyttä tai allergisia reaktioita (näitä saattavat olla mm. angioedeema, infuusiokohdan polte ja kirvely, vilunväristykset, punoitus, yleistynyt nokkosihottuma, päänsärky, paukamat, alhainen verenpaine, letargia, pahoinvointi, levottomuus, takykardia, puristuksen tunne rinnassa, pistely, oksentelu, hengityksen vinkuminen), jotka voivat joissakin tapauksissa kehittyä vaikeaksi anafylaksiaksi (mukaan lukien sokki). Joissakin tapauksissa nämä oireet ovat pahentuneet vaikeaksi anafylaksiaksi ja ne ovat ilmenneet lähes samanaikaisesti tekijä IX:n inhibiittorien muodostumisen kanssa (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet). Siedätyshoidon jälkeen on raportoitu nefroottista oireyhtymää hemofilia B-potilailla, joilla on tekijä IX:n inhibiittoreita ja joilla on ollut allergisia reaktioita.
Hemofilia B-potilailla saattaa kehittyä neutraloivia tekijä IX:n vasta-aineita (inhibiittoreita). Jos näitä vasta-aineita muodostuu, potilaan kliininen hoitovaste on riittämätön. Tällaisissa tapauksissa suositellaan yhteydenottoa hemofiliaan erikoistuneeseen hoitoyksikköön.
Tekijä IX-valmisteiden käyttöön liittyy mahdollinen tromboembolisten komplikaatioiden riski, joka on tavallista suurempi matalan puhtausasteen valmisteita käytettäessä. Matalan puhtausasteen tekijä IX-valmisteiden käyttöön on liittynyt sydäninfarktia, disseminoitunutta intravaskulaarista koagulaatiota, laskimotromboosia ja keuhkoemboliaa. Korkean puhtausasteen tekijä IX-valmisteiden käyttöön on harvoin liittynyt tromboembolisia komplikaatioita.
Taulukoitu yhteenveto haittavaikutuksista
Aiemmin hoitoa saaneet potilaat: Faasin III kliinisissä tutkimuksissa ja yhdessä jatkotutkimuksessa tutkittiin yhteensä 153 potilasta, joilla oli vaikea hemofilia B. Haittatapahtumia seurattiin yhteensä 561 potilasvuoden ajan. Altistuspäivien yhteismäärä oli 26 106 mediaanin ollessa 165 (vaihteluväli 1‑528) altistuspäivää tutkimushenkilöä kohti.
Aiemmin hoitamattomat potilaat: Yhdessä kliinisessä tutkimuksessa tutkittiin yhteensä 33 potilasta, joilla oli vaikea hemofilia B. Haittatapahtumia seurattiin yhteensä 57,51 potilasvuoden ajan. Altistuspäivien yhteismäärä oli 2 233 mediaanin ollessa 76 (vaihteluväli 1‑137) altistuspäivää tutkimushenkilöä kohti.
Alla oleva taulukko 2 on laadittu MedDRA-elinjärjestelmäluokituksen mukaisesti.
Esiintymistiheydet on arvioitu seuraavan esitystavan mukaisesti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Taulukossa luetellaan kliinisissä tutkimuksissa ja markkinoille tulon jälkeisessä käytössä ilmoitetut haittavaikutukset.
Taulukko 2: ALPROLIX-valmisteen ilmoitetut haittavaikutukset
MedDRA-järjestelmän elinluokka | Haittavaikutukset | Esiintymistiheys |
Veri ja imukudos | Tekijä IX:n esto | Yleinen1 |
Immuunijärjestelmä | Yliherkkyys Anafylaktinen reaktio Anafylaktinen sokki | Yleinen1 Tuntematon Tuntematon |
Aineenvaihdunta ja ravitsemus | Alentunut ruokahalu | Melko harvinainen |
Hermosto | Päänsärky Huimaus Makuhäiriöt | Yleinen Melko harvinainen Melko harvinainen |
Sydän | Palpitaatio | Melko harvinainen |
Verisuonisto | Hypotensio | Melko harvinainen |
Ruoansulatuselimistö | Suun parestesia Pahanhajuinen hengitys | Yleinen Melko harvinainen |
Munuaiset ja virtsatiet | Obstruktiivinen uropatia Hematuria Munuaiskoliikki | Yleinen Melko harvinainen Melko harvinainen |
Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan eryteema Väsymys Infuusiokohdan kipu | Yleinen Melko harvinainen Melko harvinainen |
1 Esiintymistiheydet perustuvat aiemmin hoitamattomien potilaiden tutkimukseen. Sekä IX:n esto- että yliherkkyystapahtuma esiintyivät vain yhdellä aiemmin hoitamattomalla potilaalla tutkimuksessa IV. Katso valikoitujen haittavaikutusten kuvaus.
Valikoitujen haittavaikutusten kuvaus
Koko kliinisen tutkimusohjelman aikana yhdellä tutkimukseen IV osallistuneella (aiemmin hoitamattomalla) potilaalla kehittyi alhaisen titterin tekijä IX:n inhibiittori, johon liittyi yliherkkyyttä (ks. kohta Farmakodynamiikka). Markkinoille tulon jälkeen on havaittu inhibiittoreiden kehittymistä tekijä IX:lle ja yliherkkyysreaktioita (mukaan lukien anafylaksi).
Pediatriset potilaat
Haittavaikutusten yleisyyden, tyypin ja vaikeusasteen odotetaan olevan lapsilla samanlaiset kuin aikuisilla. Turvallisuustietokannan laajuus ja ikäluonnehdinta lapsilla, ks. kohta Farmakodynamiikka
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta.
Suomi/Finland
[Finnish]
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
[Swedish]
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Yliannostus
Suurempien kuin suositeltujen ALPROLIX-annosten vaikutuksia ei ole luonnehdittu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antihemorraginen veren hyytymistekijä IX, ATC-koodi: B02BD04
Vaikutusmekanismi
Tekijä IX on yksiketjuinen glykoproteiini, jonka molekyylipaino on noin 55 000 daltonia. Se on K‑vitamiinista riippuva hyytymistekijä. Tekijä IX:n aktivoi sisäisellä hyytymistiellä tekijä XIa ja ulkoisella hyytymistiellä tekijä VII/kudostekijäkompleksi. Aktivoitunut tekijä IX yhdessä aktivoituneen tekijä VIII:n kanssa aktivoi tekijä X:n. Aktivoitunut tekijä X muuttaa protrombiinin trombiiniksi. Trombiini muuttaa tämän jälkeen fibrinogeenin fibriiniksi, jolloin hyytymä muodostuu.
Hemofilia B on sukupuolisidonnainen perinnöllinen veren hyytymissairaus, joka johtuu tekijä IX:n pienentyneistä pitoisuuksista ja johtaa verenvuotoihin nivelissä, lihaksissa tai sisäelimissä joko spontaanisti tai tapaturman tai leikkausvamman seurauksena. Korvaushoidon ansiosta tekijä IX:n pitoisuus suurenee, jolloin hyytymistekijän puutos ja vuototaipumus hetkellisesti korjaantuvat.
ALPROLIX (eftrenonakogi alfa) on pitkävaikutteinen, täysin rekombinantti fuusioproteiini, johon sisältyy ihmisen hyytymistekijä IX. Tämä on kovalenttisesti kiinnittynyt ihmisen immunoglobuliini G1:n Fc-domeeniin ja se on valmistettu yhdistelmä-DNA-tekniikalla.
Ihmisen immunoglobuliini G1:n Fc-osa sitoutuu vastasyntyneen Fc-reseptoriin. Tämä reseptori ilmentyy koko elämän ajan osana luonnollisesti esiintyvää reittiä, joka suojaa immunoglobuliineja lysosomaaliselta hajoamiselta kuljettamalla näitä proteiineja takaisin verenkiertoon, mikä johtaa niiden pitempään puoliintumisaikaan plasmassa.
Kliininen teho ja turvallisuus
ALPROLIX-valmisteen turvallisuutta, tehoa ja farmakokinetiikkaa on arvioitu kahdessa monikansallisessa avoimessa avaintutkimuksessa aiemmin hoitoa saaneilla potilailla: aikuisilla ja nuorilla potilailla tehty faasin 3 tutkimus, jota tässä kutsutaan nimellä tutkimus I, ja pediatrisilla potilailla tehty faasin 3 tutkimus, jota tässä kutsutaan nimellä tutkimus II (ks. Pediatriset potilaat). ALPROLIX-valmisteen turvallisuutta ja tehoa on arvioitu myös aiemmin hoitamattomilla potilailla, joilla oli vaikea hemofilia B (tutkimus IV), katso kohta Pediatriset potilaat.
Tutkimuksessa I verrattiin molempien estohoito-ohjelmien (kiinteästi viikon välein annoksella 50 IU/kg ja yksilöllisin välein annoksella 100 IU/kg toteutettava, 10 päivän välein alkava hoito) tehoa tarvittaessa toteutettavan hoidon tehoon. Tutkimukseen otettiin yhteensä 123 aiemmin hoitoa saanutta miespotilasta (12‑71-vuotiaita), joilla oli vaikea hemofilia B (≤ 2 % endogeeninen FIX:n aktiivisuus). Kaikki potilaat saivat ALPROLIX-hoitoa ja heidän tilannettaan seurattiin enintään viikolle 77 asti.
Tutkimuksen I loppuun asti suorittaneista 123 potilaasta 93 potilasta kirjautui tutkimukseen III (jatkotutkimus), jolloin kokonaisseuranta-ajan mediaani oli 6,5 vuotta.
On huomioitava, että vuositasolla lasketut mediaanivuotomäärät eivät ole vertailukelpoisia eri hyytymistekijäkonsentraattien välillä eivätkä eri kliinisten tutkimusten välillä.
Kiinteästi viikon välein ja yksilöllisin välein annettu estohoito
Mediaani viikottainen annos kiinteästi viikon välein annetun estohoidon haarassa oleville potilaille oli 45,17 IU/kg (neljännespisteiden välinen alue 38,1‑53,7) tutkimuksessa I. Vastaavat vuositasolla lasketut mediaanivuotomäärät tutkimushenkilöillä, joilla teho oli arvioitavissa, olivat seuraavat: 2,95 (neljännespisteiden välinen alue: 1,01‑4,35) ja ne pysyivät samankaltaisina koko tutkimuksen III ajan (1,85 (neljännespisteiden välinen alue: 0,76-4,0)). Tutkittavilla esiintyneiden nivelten verenvuototapahtumien mediaani oli 0,38 (neljännespisteiden välinen alue: 0,00-1,43) tutkimuksessa III.
Yksilöllisin välein annetun estohoidon haaran potilailla mediaani annosteluväli oli 12,53 päivää (neljännespisteiden välinen alue 10,4‑13,4) tutkimuksessa I. Vastaavat vuositasolla lasketut mediaanivuotomäärät olivat 1,38 (neljännespisteiden välinen alue: 0,00-3,43) ja ne pysyivät samankaltaisina koko tutkimuksen III ajan (1,85 (neljännespisteiden välinen alue: 0,76-4,0)).
Annosteluvälit ja hyytymistekijöiden kulutus pysyivät tutkimuksessa III (jatkotutkimus) samankaltaisina verrattuna tutkimukseen I molemmissa estohoidoissa.
Vuotoa ei ilmennyt lainkaan yksilöllistä estohoitoa saaneista tutkimushenkilöistä 42 %:lla ja viikoittaista estohoitoa saaneista 23,0 %:lla. Yksilöllisen annosteluvälin estohoitohaarassa potilaita, joilla oli ≥ 1 kohdenivel lähtötilanteessa, oli pienempi osuus kuin kerran viikossa annetun estohoidon haarassa (27,6 % edellisessä ja 57,1 % jälkimmäisessä ryhmässä).
Verenvuodon hoito
Tutkimuksen I aikana havaituista 636 vuototapahtumasta 90,4 % saatiin hallintaan yhdellä injektiolla ja yhteensä 97,3 % enintään kahdella injektiolla. Yhden vuototapahtuman hoidossa käytetty mediaani keskimääräinen annos injektiota kohti oli 46,07 (neljännespisteiden välinen alue 32,86‑57,03) IU/kg. Yhden vuototapahtuman hoidossa käytetty kokonaismediaaniannos oli 51,47 IU/kg (neljännespisteiden välinen alue 35,21‑61,73) kerran viikossa annetun estohoidon haarassa, 49,62 IU/kg (neljännespisteiden välinen alue:35,71‑94,82) yksilöllisin välein annetun estohoidon haarassa ja 46,58 IU/kg (neljännespisteiden välinen alue: 33,33‑59,41) tarvittaessa annetun hoidon haarassa.
Perioperatiivinen hoito (leikkaukseen liittyvä estohoito)
Tutkimuksessa I ja tutkimuksessa III suoritettiin ja arvioitiin yhteensä 35 suurta leikkausta 22 tutkimushenkilöllä (21 aikuista ja nuorta, 1 pediatrinen potilas, joka oli < 12‑vuotias). 35 suuresta leikkauksesta 28 leikkauksessa (80,0 %) tarvittiin yksi preoperatiivinen annos hemostaasin ylläpitämiseksi leikkauksen aikana. Leikkauksenaikaisen hemostaasin ylläpitämiseksi annettu mediaani keskimääräinen annos injektiota kohti oli 94,7 IU/kg (vaihteluväli 49‑152 IU/kg). Leikkauspäivänä annettu kokonaisannos vaihteli välillä 49‑341 IU/kg ja kokonaisannos 14 päivän perioperatiivisella jaksolla vaihteli välillä 60‑1 947 IU/kg.
Hemostaattinen vaste arvioitiin erinomaiseksi tai hyväksi 100 %:ssa suurista leikkauksista.
Pediatriset potilaat
Tutkimukseen II otettiin yhteensä 30 aiemmin hoitoa saanutta miespuolista pediatrista potilasta, joilla oli vaikea hemofilia B (≤ 2 % endogeeninen FIX:n aktiivisuus). Potilaat olivat alle 12‑vuotiaita (15 henkilöä oli < 6‑vuotiaita ja 15 henkilöä oli 6 - < 12‑vuotiaita). Kaikki potilaat saivat ALPROLIX-hoitoa ja heidän tilannettaan seurattiin enintään viikolle 52 asti.
Kaikki 30 potilasta saivat ALPROLIX-hoitoa estohoito-ohjelmassa, joka aloitettiin antamalla 50‑60 IU/kg kerran viikossa. Annosta muutettiin enintään annokseen 100 IU/kg ja valmistetta annettiin vähintään kerran viikossa ja enintään kahdesti viikossa. Tutkimuksen II loppuun asti suorittaneista yhteensä 30:sta potilaasta 27 potilasta kirjautui tutkimukseen III (jatkotutkimus). Tutkimuksiin II+III osallistumisen mediaaniaika oli 2,88 vuotta ja altistuspäivien mediaani oli 166.
Tutkimukseen IV otettiin 33 aiemmin hoitamatonta pediatrista potilasta, joilla oli vaikea hemofilia B (≤ 2 % endogeeninen FIX:n aktiivisuus). Potilaiden mediaani-ikä tutkimukseen ottamisen hetkellä oli 0,6 vuotta (vaihteluväli 0,08–2 vuotta); 78,8 % tutkimushenkilöistä oli alle 1‑vuotiaita. ALPROLIX-valmisteen antamisen kokonaismediaaniaika oli 83,01 viikkoa (vaihteluväli 6,7–226,7 viikkoa) ja altistuspäivien mediaani oli kaiken kaikkiaan 76 päivää (vaihteluväli 1–137 päivää).
Yksilöllinen estohoito
Tutkimuksessa II mediaani keskimääräinen viikoittainen ALPROLIX-annos oli 59,40 IU/kg (neljännespisteiden välinen alue, 52,95‑64,78 IU/kg) < 6-vuotiailla potilailla ja 57,78 IU/kg (neljännespisteiden välinen alue, 51,67‑65,01 IU/kg) 6 - <12‑vuotiailla potilailla. Mediaani annosteluväli oli kaiken kaikkiaan 6,99 päivää (neljännespisteiden välinen alue, 6,94‑7,03) eikä mediaanissa annosteluvälissä ollut eroja ikäkohorttien välillä. Lukuun ottamatta yhtä potilasta, jonka viimeinen lääkärin määräämä annos oli 100 IU/kg viiden päivän välein, muiden 29 potilaan viimeiset määrätyt annokset olivat enintään 70 IU/kg kerran viikossa. 33 %:lla pediatrisista potilaista ei ilmennyt lainkaan vuotoja. Annosteluvälit ja hyytymistekijöiden kulutus pysyivät samankaltaisina tutkimuksessa III (jatkotutkimus) verrattuna tutkimukseen II.
Vuositasolla laskettu mediaanivuotomäärä < 12-vuotiailla potilailla, joilla teho oli arvioitavissa, oli 1,97 (neljännespisteiden välinen alue 0,00‑3,13) tutkimuksessa II ja se pysyi samankaltaisena koko tutkimuksen III (jatkotutkimus) ajan.
Aiemmin hoitamattomilla potilailla (tutkimus IV) mediaani keskimääräinen viikoittainen ALPROLIX-annos oli 57,96 IU/kg (neljännespisteiden välinen alue, 52,45–65,06 IU/kg) ja medaani annosteluväli oli 7 päivää (neljännespisteiden välinen alue, 6,95–7,12 päivää). Annosteluvälit ja hyytymistekijöiden kulutus pysyivät samankaltaisina tutkimuksessa IV verrattuna tutkimuksiin II ja III. Estohoitoa saavista aiemmin hoitamattomista potilaista 8 tutkimushenkilöllä (28,6 %) ei esiintynyt verenvuototapahtumia. Estohoito-ohjelmassa olevien tutkimushenkilöiden vuositasolla lasketut mediaanivuotomäärä oli 1,24 (neljännespisteiden välinen alue, 0,0–2,49).
Verenvuototapahtumien hoito
Tutkimuksen II aikana havaituista 60 vuototapahtumasta 75 % saatiin hallintaan yhdellä injektiolla ja yhteensä 91,7 % saatiin hallintaan enintään kahdella injektiolla. Verenvuototapahtuman hoitamiseksi annettu mediaani annos injektiota kohti oli 63,51 IU/kg (neljännespisteiden välinen alue, 48,92‑99,44). Verenvuototapahtuman hoitamiseksi annettu mediaani kokonaisannos oli 68,22 IU/kg (neljännespisteiden välinen alue, 50,89‑126,19).
Tutkimuksessa IV estohoitoa saavilla aiemmin hoitamattomilla potilailla havaituista 58 verenvuototapahtumasta 87,9 % saatiin hallintaan 1 injektiolla ja kaikista verenvuototapahtumista 96,6 % saatiin hallintaan enintään 2 injektiolla. Verenvuototapahtuman hoitamiseksi annettu mediaani annos injektiota kohti oli 71,92 IU/kg (neljännespisteiden välinen alue, 52,45–100,81 IU/kg). Verenvuototapahtuman hoitamiseksi annettu mediaani kokonaisannos oli 78,74 IU/kg (neljännespisteiden välinen alue, 53,57–104,90 IU/kg).
Farmakokinetiikka
Kaikki ALPROLIX-valmisteella tehdyt farmakokineettiset tutkimukset suoritettiin aiemmin hoitoa saaneilla potilailla, joilla oli vaikea hemofilia B. Tässä kohdassa esitettävät tiedot on saatu yksivaiheisen hyytymismäärityksen avulla käyttäen piioksidipohjaista aPTT-reagenssia, joka on kalibroitu tekijä IX: n plasmastandardeilla.
Farmakokineettisiä ominaisuuksia arvioitiin ALPROLIX-valmistetta (rFIXFc) saavilla 22 tutkimushenkilöllä (≥ 19‑vuotiaat). Vähintään 120 tuntia (5 päivää) kestäneen lääkkeettömän jakson jälkeen tutkimushenkilöille annettiin yksi 50 IU/kg:n annos. Farmakokineettiset näytteet kerättiin ennen annosta ja 11 eri ajankohtana enintään 240 tunnin (10 päivän) kuluttua annoksen jälkeen. Ei-kompartmentaalisesta analyysista saadut, 50 IU/kg:n ALPROLIX-annoksen jälkeiset farmakokineettiset muuttujat esitetään taulukossa 3.
Taulukko 3: ALPROLIX-valmisteen (50 IU/kg:n annos) farmakokineettiset muuttujat
Farmakokineettiset muuttujat1 | ALPROLIX (95 %:n CI) |
N=22 | |
Inkrementaalinen saanto (IU/dl per IU/kg) | 0,92 (0,77‑1,10) |
AUC/annos (IU*h/dl per IU/kg) | 31,58 (28,46‑35,05) |
Cmax (IU/dl) | 46,10 (38,56‑55,11) |
CL (ml/h/kg) | 3,17 (2,85‑3,51) |
t½ (h) | 77,60 (70,05‑85,95) |
t½α (h)2 | 5,03 (3,20‑7,89) |
t½β (h)2 | 82,12 (71,39‑94,46) |
MRT (h) | 95,82 (88,44‑106,21) |
Vss (ml/kg) | 303,4 (275,1‑334,6) |
Aika 1 %:iin (päivää)2 | 11,22 (10,20‑12,35) |
1 Farmakokineettiset parametrit esitetään geometrisena keskiarvona (95 %:n CI)
2 Nämä farmakokineettiset paramatrit on saatu kompartmentaalisesta analyysista
Lyhenteet: CI = luottamusväli; Cmax= maksimiaktiivisuus; AUC = FIX:n aktiivisuuden aikakäyrän alle jäävä alue; t½= terminaalinen puoliintumisaika; t½α = jakautumisen puoliintumisaika; t½β = eliminaation puoliintumisaika, CL = puhdistuma; Vss = jakaantumistilavuus vakaan tason vaiheessa; MRT = keskimääräinen elimistössä oloaika.
Valmisteen eliminaation puoliintumisaikaan (82 tuntia) vaikuttaa Fc-osa, jonka eläinmalleissa osoitettiin välittyvän neonataalisten Fc-reseptorin toistuvien reittien kautta.
Populaatiofarmakokineettinen malli kehitettiin perustuen FIX:n aktiivisuuden tietoihin, jotka saatiin kolmessa kliinisessä tutkimuksessa yhteensä 161 eri-ikäiseltä (2‑76‑vuotiaita) tutkimushenkilöltä, joiden paino oli 12,5‑186,7 kg (12 henkilöä faasin 1/2a tutkimuksessa, 123 henkilöä tutkimuksessa I ja 26 henkilöä tutkimuksessa II). Tyypillisellä 70 kg painavalla aikuisella valmisteen puhdistuman estimaatti on 2,30 dl/h ja valmisteen vakaan tilan jakautumistilavuus vastaavasti 194,8 dl. Tutkimuksissa havaittu keskimääräinen (SD) vaikutusaikaprofiili ALPROLIX-valmisteen kerta-annoksen jälkeen vaikeaa hemofilia B:tä sairastavilla potilailla on esitetty alla (ks. taulukko 4).
Taulukko 4: Havaittu keskimääräinen (SD) FIX:n aktiivisuus [IU/dl] yhden ALPROLIX-1kerta-annoksen (rFIXFc) jälkeen ≥ 12‑vuotiailla potilailla
Annos (IU/kg) | 10 min | 1 h | 3 h | 6 h | 24 h | 48 h | 96 h | 144 h | 168 h | 192 h | 240 h | 288 h |
50 | 52,9 (30,6) | 34,5 (7,3) | 28,7 (6,7) | 25,1 (5,1) | 15,1 (3,9) | 9,7 (3,0) | 5,0 (1,6) | 3,4 (1,1) | 3,2 (1,9) | 2,6 (1,0) | 2,1 (0,9) | NA |
100 | 112 (24) | NA | 77,1 (12,8) | NA | 36,7 (8,0) | 21,8 (4,8) | 10,1 (2,6) | NA | 4,81 (1,67) | NA | 2,86 (0,98) | 2,30 (0,94) |
1 Ks. kohta Annostus ja antotapa; NA: ei käytettävissä
Pediatriset potilaat
ALPROLIX-valmisteen farmakokineettiset muuttujat määritettiin nuorilla tutkimuksessa I (farmakokineettisiä näytteitä otettiin ennen annoksen antamista, minkä jälkeen arvioita tehtiin useina ajankohtina kunnes annoksen antamisesta oli kulunut 336 tuntia (14 päivää)) ja lapsilla tutkimuksessa II (farmakokineettisiä näytteitä otettiin ennen annoksen antamista, minkä jälkeen arvioita tehtiin 7 ajankohtana kunnes annoksen antamisesta oli kulunut 168 tuntia (7 päivää)). Taulukossa 5 esitetään 35:n alle 18-vuotiaan tutkimushenkilön pediatrisesta datasta lasketut farmakokineettiset muuttujat.
Taulukko 5: ALPROLIX-valmisteen (rFIXFc) farmakokineettisten muuttujien vertailu ikäluokittain
Farmakokineettiset parametrit1 | Tutkimus II | Tutkimus I | |
<6‑vuotiaat (2, 4) | 6-<12‑vuotiaat (6, 10) | 12-<18‑vuotiaat (12, 17) | |
N = 11 | N = 13 | N = 11 | |
IR (IU/dl per IU/kg) | 0,5989 (0,5152; 0,6752) | 0,7170 (0,6115; 0,8407) | 0,8470 (0,6767; 1,0600) |
AUC/annos (IU*h/dl per IU/kg) | 22,71 (20,32; 25,38) | 28,53 (24,47; 33,27) | 29,50 (25,13; 34,63) |
t½ (h) | 66,49 (55,86; 79,14) | 70,34 (60,95; 81,17) | 82,22 (72,30; 93,50) |
MRT (h) | 83,65 (71,76; 97,51) | 82,46 (72,65; 93,60) | 93,46 (81,77; 106,81) |
CL (ml/h/kg) | 4,365 (3,901; 4,885) | 3,505 (3,006; 4,087) | 3,390 (2,888; 3,979) |
Vss (ml/kg) | 365,1 (316,2; 421,6) | 289,0 (236,7; 352,9) | 316,8 (267,4; 375,5) |
1 Ei-kompartmentaalisesta analyysista saadut farmakokineettiset parametrit esitetään geometrisena keskiarvona (95 %:n CI)
Lyhenteet: CI = luottamusväli; IR= inkrementaalinen saanto, AUC = FIX:n aktiivisuuden aikakäyrän alle jäävä alue; t½ = terminaalinen puoliintumisaika; MRT = keskimääräinen elimistössä oloaika; CL = puhdistuma; Vss = jakaantumistilavuus vakaan tason vaiheessa
Prekliiniset tiedot turvallisuudesta
Kaneilla tehtyä trombogeenisyystestiä (Wesslerin staasimalli) sekä rotilla ja apinoilla tehtyjen toistuvan altistuksen aiheuttamaa toksisuutta koskevien tutkimusten (joihin sisältyi myös paikallisen toksisuuden, urosten lisääntymiselinten ja elektrokardiografisten parametrien arviointi) tulokset eivät viittaa erityiseen vaaraan ihmisille. Genotoksisuutta, karsinogeenisuutta, lisääntymistoksisuutta tai alkion-/sikiönkehitystä koskevia tutkimuksia ei ole tehty. Hiirillä tehdyssä kokeessa, jossa tutkittiin valmisteen siirtymistä istukan läpi, eftrenonakogi alfan (rFIXFc) on osoitettu läpäisevän istukan pieninä määrinä.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine
Sakkaroosi
Histidiini
Mannitoli
Polysorbaatti 20
Natriumhydroksidi (pH:n säätämiseen)
Suolahappo (pH:n säätämiseen)
Liuotin
Natriumkloridi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Vain mukana toimitettua infuusiovälineistöä saa käyttää, koska hyytymistekijä IX:n tarttuminen joidenkin injektiovälineiden sisäpintoihin voi aiheuttaa hoidon epäonnistumisen.
Kestoaika
Avaamaton injektiopullo
4 vuotta
Kestoajan puitteissa valmistetta voidaan säilyttää huoneenlämmössä (enintään 30 °C:ssa) yhtäjaksoisesti enintään 6 kuukautta. Päivämäärä, jolloin valmiste otetaan pois jääkaapista, on syytä merkitä pakkauksen päälle. Valmistetta ei saa siirtää enää takaisin jääkaappiin huoneenlämmössä säilyttämisen jälkeen. Valmistetta ei saa käyttää injektiopullon päälle painetun viimeisen käyttöpäivämäärän jälkeen tai 6 kuukauden kuluttua siitä, kun pakkaus on otettu jääkaapista, riippuen siitä, kumpi päivä on aikaisempi.
Käyttökuntoon saattamisen jälkeen
Kemiallisen ja fysikaalisen säilyvyyden on osoitettu olevan 6 tuntia, kun valmistetta säilytetään huoneenlämmössä (enintään 30 °C:ssa). Jos valmistetta ei käytetä 6 tunnin kuluessa, se on hävitettävä. Mikrobiologiselta kannalta valmiste on käytettävä heti käyttökuntoon saattamisen jälkeen. Ellei sitä käytetä heti, käytönaikainen varastointi ja käyttöä edeltävät olosuhteet ovat käyttäjään vastuulla. Suojaa valmiste suoralta auringonvalolta.
Säilytys
Säilytä jääkaapissa (2°C ‑ 8°C). Ei saa jäätyä. Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ALPROLIX injektiokuiva-aine ja liuotin, liuosta varten
250 IU (L:ei) 1 kpl (50 IU/ml) (290,96 €)
500 IU (L:ei) 1 kpl (100 IU/ml) (565,87 €)
1000 IU (L:ei) 1 kpl (200 IU/ml) (1095,42 €)
2000 IU (L:ei) 1 kpl (400 IU/ml) (2126,96 €)
3000 IU (L:ei) 1 kpl (600 IU/ml) (3100,13 €)
PF-selosteen tieto
Jokainen pakkaus sisältää:
- kuiva-ainetta lasisessa (tyyppi 1) injektiopullossa, jossa on klorobutyylikumitulppa
- 5 ml liuotinta lasisessa (tyyppi 1) esitäytetyssä ruiskussa, jossa on bromobutyylikumista valmistettu mäntätulppa
- männän varsi
- steriili injektiopullon adapteri käyttökuntoon saattamista varten
- steriili infuusiovälineistö
- alkoholipyyhe/-pyyhkeitä
- laastari/laastareita
- sideharsotaitos /-taitoksia.
Pakkauskoko 1.
Valmisteen kuvaus:
Kuiva-aine: kylmäkuivattu, valkoinen tai luonnonvalkoinen jauhe tai kakku.
Liuotin: kirkas tai väritön liuos.
pH: 6,5‑7,5
Osmolaarisuus: 255‑345 mOsm/kg
Käyttö- ja käsittelyohjeet
Kussakin injektiopullossa oleva injektiokuiva-aine liuosta varten valmistetaan käyttövalmiiksi lisäämällä siihen pakkauksessa olevan esitäytetyn ruiskun sisältämä liuotin (natriumkloridiliuos) käyttäen tähän tarkoitettua injektiopullon steriiliä adapteria.
Injektiopulloa on pyöriteltävä varovasti, kunnes injektiokuiva-aine on kokonaan liuennut.
Käyttökuntoon saatetun liuoksen on oltava kirkasta tai hiukan opaalinhohtoista sekä väritöntä. Käyttökuntoon saatetut lääkevalmisteet on tarkastettava silmämääräisesti ennen antoa mahdollisten hiukkasten ja värimuutosten varalta. Älä käytä liuosta, jos se on sameaa tai sisältää sakkaa.
Tämä valmiste on vain kertakäyttöön.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Valmistus- ja anto-ohjeet
Alla kuvataan menettelytavat ALPROLIX-valmisteen käyttökuntoon valmistamiseen ja antoon.
ALPROLIX annetaan injektiona laskimoon (i.v.) sen jälkeen, kun se on tehty käyttövalmiiksi liuottamalla injektiokuiva-aine pakkauksessa olevasta esitäytetystä ruiskusta saatavaan liuottimeen. ALPROLIX-pakkaus sisältää:
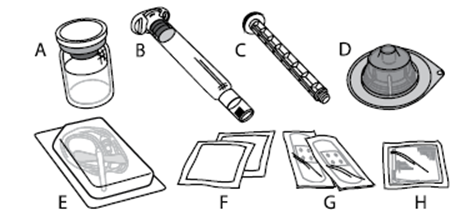
A) 1 injektiokuiva-ainepullo B) 5 ml liuotinta esitäytetyssä ruiskussa C) 1 männän varsi D) 1 injektiopullon adapteri E) 1 infuusiovälineistö F) 2 alkoholipyyhettä G) 2 laastaria H) 1 sideharsotaitos
ALPROLIX-valmistetta ei saa sekoittaa muiden injektio- tai infuusionesteiden kanssa.
Pese kädet ennen pakkauksen avaamista.
Valmistus:
1. Tarkista pakkauksesta valmisteen nimi ja vahvuus varmistaaksesi, että lääke on oikea. Tarkista viimeinen käyttöpäivämäärä ALPROLIX-pakkauksesta. Älä käytä, jos lääke on vanhentunut. | |
2. Jos ALPROLIX-valmistetta on säilytetty jääkaapissa, odota jonkin aikaa, jotta ALPROLIX-valmistetta sisältävä injektiopullo (A) ja liuotinta (B) sisältävä ruisku lämpiävät huoneenlämpöisiksi ennen käyttöä. Älä käytä ulkoisia lämmönlähteitä. | |
3. Aseta injektiopullo puhtaalle, tasaiselle alustalle. Poista muovinen irti napsautettava korkki injektiopullosta. | 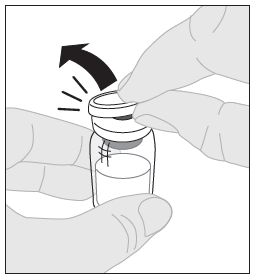 |
4. Pyyhi injektiopullon yläosa pakkauksessa olevalla alkoholipyyhkeellä (F) ja anna sen kuivua. Älä koske injektiopullon yläosaan äläkä anna sen koskettaa mihinkään pyyhkimisen jälkeen. | 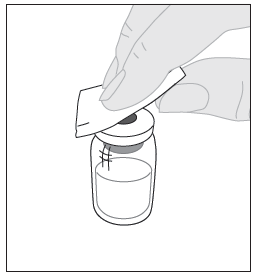 |
5. Irrota paperinen suojus injektiopullon kirkkaasta, muovisesta adapterista (D). Älä irrota adapteria sen suojakorkista. Älä koske injektiopullon adapteripakkauksen sisäpuolta. | |
6. Aseta injektiopullo tasaiselle alustalle. Pitele injektiopullon adapteria suojakorkista ja aseta se suoraan injektiopullon yläosan päälle. Paina tiukasti alaspäin, kunnes adapteri napsahtaa paikalleen injektiopullon yläosaan siten, että adapterin piikki läpäisee injektiopullon tulpan. | 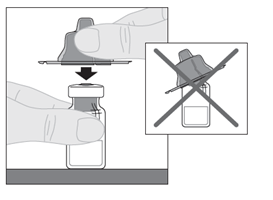 |
7. Kiinnitä männän varsi (C) liuotinruiskuun työntämällä männän varren kärki ruiskun männässä olevaan aukkoon. Käännä männän vartta tiukasti myötäpäivään, kunnes se on varmasti kiinni ruiskun männässä. | 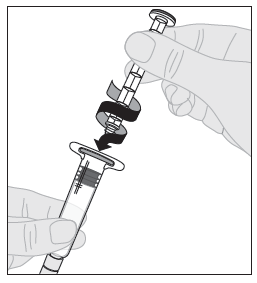 |
8. Katkaise valkoinen, avaamattomuuden osoittava muovinen korkki liuotinruiskusta taivuttamalla lävistettyä korkkia, kunnes se irtoaa naksahtaen. Siirrä korkki syrjään asettamalla se nurinpäin tasaiselle alustalle. Älä koske korkin sisäpuolta tai ruiskun kärkeä. | 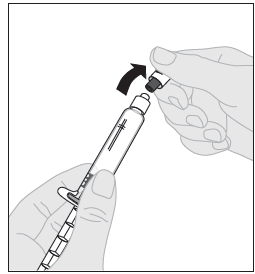 |
9. Ota suojakorkki pois adapterista ja hävitä se. | 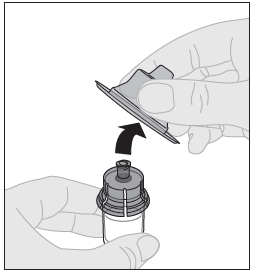 |
10. Liitä liuotinruisku injektiopullon adapteriin työntämällä ruiskun kärki adapterin aukkoon. Paina ja käännä ruiskua tiukasti myötäpäivään, kunnes se on varmasti kiinni. | 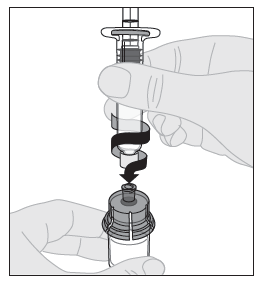 |
11. Paina männän vartta hitaasti alaspäin kunnes kaikki liuotin on ALPROLIX-injektiopullossa. | 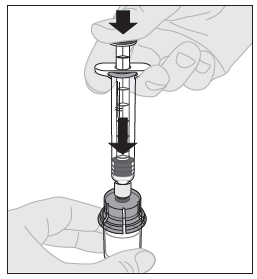 |
12. Ruiskun ollessa yhä kiinni adapterissa ja männän varren ollessa alhaalla pyörittele injektiopulloa varovasti, kunnes kuiva-aine on kokonaan liuennut. Ei saa ravistaa. | 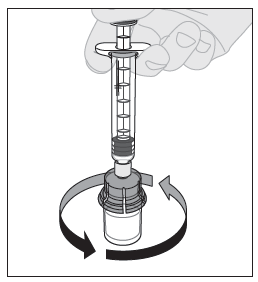 |
13. Lopullinen liuos pitää tarkastaa silmämääräisesti ennen antoa. Liuoksen on oltava kirkasta tai hieman opaalinhohtoista (helmiäismäistä) sekä väritöntä. Älä käytä liuosta, jos se on sameaa tai jos siinä näkyy hiukkasia. | |
14. Varmista, että ruiskun männän varsi on edelleen painettuna kokonaan alas ja käännä injektiopullo ylösalaisin. Vedä männän vartta hitaasti niin, että kaikki liuos virtaa injektiopullon adapterin läpi ruiskuun. Huom! Jos käytät yhdellä injektiokerralla enemmän kuin yhden injektiopullon ALPROLIX-valmistetta, kukin injektiopullo on saatettava käyttökuntoon erikseen edellä esitettyjen ohjeiden mukaisesti (vaiheet 1‑13) ja liuotinruisku on vedettävä pois, mutta injektiopullon adapteri jätettävä paikalleen. Käyttövalmiit liuokset voidaan vetää kustakin injektiopullosta käyttämällä yhtä isoa luer-lukollista ruiskua. | 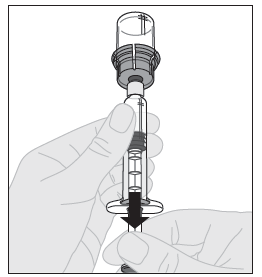 |
15. Irrota ruisku injektiopullon adapterista vetämällä sitä varovasti ja kääntämällä ruiskua vastapäivään. | 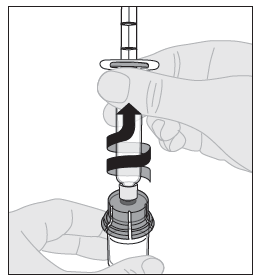 |
16. Hävitä injektiopullo ja adapteri. Huom! Jos liuosta ei oteta heti käyttöön, ruiskun korkki pitää laittaa varovasti takaisin ruiskun kärkeen. Älä koske ruiskun kärkeä tai korkin sisäpuolta. Käyttövalmista ALPROLIX-valmistetta voidaan säilyttää huoneenlämmössä enintään 6 tunnin ajan ennen antoa. Tämän ajan kuluttua käyttövalmis ALPROLIX on hävitettävä. Suojaa valmiste suoralta auringonvalolta. | |
Antotapa (injektio laskimoon):
ALPROLIX annetaan käyttäen pakkauksessa olevaa infuusiovälineistöä (E).
1. Avaa infuusiovälineistön pakkaus ja poista letkun päässä oleva korkki. Kiinnitä käyttövalmista ALPROLIX-liuosta sisältävä ruisku infuusiovälineistön letkun päähän kääntämällä ruiskua myötäpäivään. | 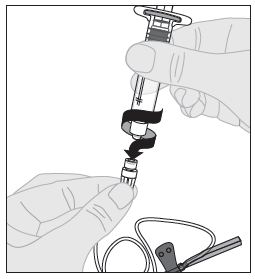 |
2. Käytä tarvittaessa kiristyssidettä ja valmistele injektiokohta pyyhkimällä iho hyvin pakkauksen toisella alkoholipyyhkeellä. 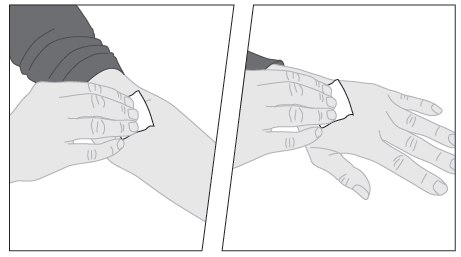 | |
3. Poista kaikki ilma infuusioletkustosta painamalla männän vartta hitaasti alaspäin kunnes neste on infuusiovälineistön neulan kohdalla. Älä työnnä liuosta neulan läpi. Poista neulasta sen kirkas, muovinen suojus. | |
4. Työnnä infuusiovälineistön neula laskimoon lääkärin tai sairaanhoitajan opettamalla tavalla ja poista kiristysside. Halutessasi voit kiinnittää toisella pakkauksen laastareista (G) neulan muovisiivekkeet paikalleen injektiokohtaan. Käyttövalmis valmiste on injisoitava laskimoon usean minuutin aikana. Lääkäri saattaa muuttaa suositeltua injektionopeutta, jotta olosi tuntuisi mukavammalta. | |
5. Injektion lopettamisen ja neulan poistamisen jälkeen neulan suojus on käännettävä takaisin ja napsautettava neulan päälle. | 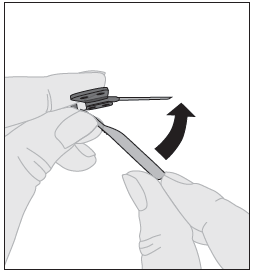 |
6. Hävitä käytetty neula, käyttämättä jäänyt liuos, ruisku ja tyhjä injektiopullo turvallisesti asianmukaiseen sairaalajätteen säiliöön, koska nämä materiaalit voivat vahingoittaa toisia, ellei niitä hävitetä oikeaoppisesti. Tarvikkeita ei saa käyttää uudelleen. | |
Korvattavuus
ALPROLIX injektiokuiva-aine ja liuotin, liuosta varten
250 IU 1 kpl
500 IU 1 kpl
1000 IU 1 kpl
2000 IU 1 kpl
3000 IU 1 kpl
- Ylempi erityiskorvaus (100 %). Krooniset hyytymishäiriöt (126).
- Peruskorvaus (40 %).
ATC-koodi
B02BD04
Valmisteyhteenvedon muuttamispäivämäärä
30.04.2025
Yhteystiedot
Äyritie 18
01510 Vantaa
0201 558 840
www.sobi.fi
mail.fi@sobi.com