
GEFITINIB STADA tabletti, kalvopäällysteinen 250 mg
Vaikuttavat aineet ja niiden määrät
Yksi tabletti sisältää 250 mg gefitinibiä.
Apuaine, jonka vaikutus tunnetaan
Yksi tabletti sisältää 163,5 mg laktoosia (monohydraattina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen
Kliiniset tiedot
Käyttöaiheet
Gefitinib Stada on tarkoitettu monoterapiana paikallisesti edenneen tai metastaattisen ei-pienisoluisen keuhkosyövän (NSCLC) hoitoon aikuisille potilaille, joiden kasvaimessa on aktivoivia EGFR-tyrosiinikinaasin mutaatioita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Gefitinib Stada -hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus
Gefitinib Stada -valmisteen suositusannos on yksi 250 mg:n tabletti kerran vuorokaudessa. Jos annos unohtuu, se tulee ottaa heti muistettaessa. Jos seuraavan annoksen ottamiseen on alle 12 tuntia, annos jätetään väliin. Potilaiden ei tule ottaa kaksinkertaista annosta (kahta annosta samalla kertaa) unohtuneen annoksen korvaamiseksi.
Pediatriset potilaat
Gefitinib Stada -valmisteen turvallisuutta ja tehoa lasten ja alle 18-vuotiaiden nuorten hoidossa ei ole varmistettu. Gefitinibin käyttö ei-pienisoluisen keuhkosyövän hoitoon lapsille on merkityksetöntä.
Maksan vajaatoiminta
Gefitinibin pitoisuudet plasmassa suurenevat potilailla, joilla on maksakirroosista johtuva keskivaikea tai vaikea maksan vajaatoiminta (Child-Pugh B tai C). Näitä potilaita tulee seurata tarkoin haittavaikutusten riskin vuoksi. Plasmapitoisuudet eivät kohonneet potilailla, joilla aspartaattitransaminaasi- (ASAT), alkalinen fosfataasi- (AFOS) tai bilirubiiniarvo oli suurentunut maksametastaasien vuoksi (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen potilailla, joilla on munuaisten vajaatoiminta mikäli kreatiniinipuhdistuma on > 20 ml/min. Potilaista, joilla kreatiniinipuhdistuma on ≤ 20 ml/min, on saatavilla vain rajallisesti tietoa ja näiden potilaiden kohdalla tulee noudattaa varovaisuutta (ks. kohta Farmakokinetiikka).
Iäkkäät
Annoksen muuttaminen ei ole tarpeen potilaan iän perusteella (ks. kohta Farmakokinetiikka).
Heikot CYP2D6-metaboloijat
Annoksen muuttamista ei suositella heikon CYP2D6-metaboliagenotyypin potilailla, mutta näitä potilaita tulee tarkoin seurata haittavaikutusten riskin vuoksi (ks. kohta Farmakokinetiikka).
Annoksen muuttaminen toksisuuden vuoksi
Hoito voidaan keskeyttää lyhyeksi ajaksi (enintään 14 vrk) ja aloittaa uudelleen 250 mg:n annoksella potilailla, jotka sietävät huonosti haittavaikutuksina esiintyvää ripulia tai ihoreaktioita (ks. kohta Haittavaikutukset). Potilailla, jotka eivät siedä hoitoa keskeytyksen jälkeen, gefitinibihoito tulee lopettaa ja heille tulee harkita vaihtoehtoisia hoitoja.
Antotapa
Tabletti voidaan ottaa suun kautta ruoan kanssa tai tyhjään mahaan suurin piirtein samaan aikaan päivästä. Tabletti voidaan niellä kokonaisena veden kanssa tai jos se ei ole mahdollista, tabletti voidaan liuottaa puoleen lasilliseen hiilihapotonta vettä. Tablettia ei saa liuottaa muuhun nesteeseen. Tablettia ei saa murskata, vaan se liuotetaan hitaasti puoleen lasilliseen juomavettä. Nestettä sekoitetaan ajoittain, kunnes tabletti on liuennut veteen (saattaa kestää jopa 20 minuuttia). Liuos juodaan välittömästi, kun tabletti on liuennut (ts. tunnin kuluessa liuottamisesta). Lasiin lisätään uudelleen saman verran vettä ja neste juodaan. Liuos voidaan antaa myös nenä-mahaletkun tai gastrostomialetkun kautta.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Harkittaessa Gefitinib Stada -valmisteen käyttöä paikallisesti edenneen tai metastaattisen NSCLC:n hoitoon on tärkeää, että kasvainkudoksen EGFR-mutaatiostatus yritetään määrittää kaikilla potilailla. Jos kasvainnäytettä ei ole arvioitavissa, silloin verestä (plasmasta) eristettyä kiertävää kasvain DNA:ta (ctDNA) voidaan käyttää.
Vain vakaata, luotettavaa ja herkkää testiä (testejä), jonka on osoitettu soveltuvan EGFR mutaatiostatuksen määritykseen kasvaimesta tai ctDNA:sta, on käytettävä, jotta vältetään virheelliset negatiiviset tai virheelliset positiiviset määritykset (ks. kohta Farmakodynamiikka).
Interstitiaalinen keuhkosairaus (ILD)
ILD:ta, joka saattaa alkaa äkillisesti ja on joissain tapauksissa johtanut kuolemaan, on havaittu 1,3 %:lla gefitinibia saaneista potilaista (ks. kohta Haittavaikutukset). Jos potilaalla on pahenevia hengitystieoireita, kuten hengenahdistusta, yskää ja kuumetta, Gefitinib Stada -hoito on keskeytettävä ja potilas tutkittava viipymättä. Jos ILD todetaan, Gefitinib Stada -hoito on lopetettava ja potilasta hoidettava asianmukaisesti.
Japanilaisessa farmakoepidemiologisessa tapaus-verrokkitutkimuksessa, johon osallistui 3 159 gefitinibia tai solunsalpaajahoitoa käyttävää NSCLC-potilasta, joita seurattiin 12 viikon ajan, tunnistettiin seuraavat ILD:n riskitekijät (riippumatta siitä, saiko potilas gefitinibia vai solunsalpaajaa):
tupakointi, heikko suorituskyky (PS ≥ 2), tietokonetomografiassa osoitus normaalin keuhkokudoksen vähentymisestä (≤ 50 %), vastikään (< 6 kk) diagnosoitu NSCLC, aiemmin todettu ILD, vähintään 55 vuoden ikä ja samanaikainen sydänsairaus. Suurentunut ILD:n riski gefitinibiryhmässä verrattuna solunsalpaajaryhmään havaittiin lähinnä neljän ensimmäisen hoitoviikon aikana (vakioitu riskisuhde [OR] 3,8; 95 % luottamusväli [CI] 1,9–7,7). Sen jälkeen suhteellinen riski oli pienempi (vakioitu riskisuhde [OR] 2,5; 95 % luottamusväli [CI] 1,1–5,8). Kuoleman riski oli suurempi gefitinibi- tai solunsalpaajaryhmän potilailla, joille kehittyi ILD, mikäli heillä oli seuraavat riskitekijät: tupakointi, tietokonetomografiassa osoitus normaalin keuhkokudoksen vähenemisestä (≤ 50 %), aiemmin todettu ILD, vähintään 65 vuoden ikä ja laajat pleuraan kiinnikkeiset alueet (≥ 50 %).
Maksatoksisuus ja maksan vajaatoiminta
Maksan toimintakokeiden tuloksissa on havaittu poikkeavuuksia (mukaan lukien alaniiniaminotransferaasi-, aspartaattiaminotransferaasi- ja bilirubiiniarvojen nousu), melko harvoin hepatiittia (ks. kohta Haittavaikutukset). Yksittäisiä maksan vajaatoimintatapauksia on raportoitu, ja joissakin tapauksissa ne ovat olleet kuolemaan johtavia. Tämän vuoksi suositellaan maksan toimintakokeiden kontrolloimista ajoittain. Gefitinibia tulee käyttää varoen, jos potilaalla todetaan lieviä tai kohtalaisia maksan toiminnan muutoksia. Jos muutokset ovat vakavia, hoidon keskeyttämistä tulee harkita.
Maksakirroosista johtuvan maksan vajaatoiminnan on osoitettu johtavan gefitinibin plasmapitoisuuksien suurenemiseen (ks. kohta Farmakokinetiikka).
Yhteisvaikutukset muiden lääkeaineiden kanssa
CYP3A4-induktorit saattavat voimistaa gefitinibin metaboliaa ja pienentää gefitinibin pitoisuuksia plasmassa. Sen vuoksi samanaikainen CYP3A4-induktorien (esim. fenytoiini, karbamatsepiini, rifampisiini, barbituraatit tai luontaistuotteet, jotka sisältävät mäkikuismaa/ Hypericum perforatum) käyttö saattaa heikentää hoidon tehoa ja niitä tulisi välttää (ks. kohta Yhteisvaikutukset).
Yksittäisillä potilailla, jotka ovat genotyypiltään heikkoja CYP2D6-metaboloijia, hoito tehokkaasti CYP3A4:ää estävällä lääkeaineella saattaa johtaa suurentuneisiin gefitinibipitoisuuksiin plasmassa. CYP3A4-inhibiittorihoitoa aloitettaessa potilaita tulee seurata tarkoin gefitinibin haittavaikutusten varalta (ks. kohta Yhteisvaikutukset).
International Normalised Ratio (INR) -arvojen suurenemista ja/tai verenvuototapahtumia on raportoitu joillakin varfariinia käyttävillä potilailla gefitinibin käytön yhteydessä (ks. kohta Yhteisvaikutukset). Varfariinia ja gefitinibiä samanaikaisesti käyttäviä potilaita tulee seurata säännöllisesti protrombiiniajan (PT) tai INR-arvojen muutosten varalta.
Lääkkeet, jotka nostavat merkitsevästi mahalaukun pH:ta, kuten protonipumpun estäjät ja H2-antagonistit, saattavat pienentää gefitinibin biologista hyötyosuutta ja pitoisuutta plasmassa ja siten heikentää sen tehoa. Jos antasideja otetaan säännöllisesti samaan aikaan kuin gefitinibia, vaikutus voi olla samankaltainen (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
Tiedot faasin II kliinisistä tutkimuksista, joissa gefitinibiä ja vinorelbiinia käytettiin samanaikaisesti, viittasivat siihen, että gefitinibi saattaa pahentaa vinorelbiinin neutropeenistä vaikutusta.
Laktoosi
Gefitinib Stada sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasin puutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol (23 mg) natriumia per kalvopäällysteinen tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Muut varoitukset
Potilaita tulee neuvoa hakeutumaan lääkäriin välittömästi, jos heille kehittyy vaikea tai jatkuva ripuli, pahoinvointi, oksentelu tai anoreksia, koska ne voivat johtaa elimistön kuivumiseen. Näitä oireita tulee hoitaa kliinisen tarpeen mukaan (ks. kohta Haittavaikutukset).
Potilaat, joiden oireet ja merkit viittaavat sarveiskalvotulehdukseen, kuten akuutti tai paheneva silmätulehdus, kyynelehtiminen, valoherkkyys, näön hämärtyminen, silmäkipu ja/tai punasilmäisyys, tulisi ohjata nopeasti silmätautien erikoislääkärille.
Jos haavaisen sarveiskalvotulehduksen diagnoosi varmistuu, gefitinibihoito on keskeytettävä, ja jos oireet eivät häviä tai ne palaavat gefitinibihoidon uudelleen aloittamisen jälkeen, on harkittava hoidon lopettamista.
Vaiheen I/II gefitinibi- ja sädehoitotutkimuksessa, johon osallistui 45 lasta, joilla oli vastatodettu aivorungon gliooma tai osittain poistettu supratentoriaalinen pahanlaatuinen gliooma, neljällä potilaalla raportoitiin keskushermoston verenvuotoa (joista yksi tapaus johti kuolemaan). Keskushermoston verenvuotoa raportoitiin lisäksi yhdellä ependymoomaa sairastavalla lapsella tutkimuksessa, jossa käytettiin ainoastaan gefitinibiä. Ei-pienisoluista keuhkosyöpää sairastavilla, gefitinibiä käyttävillä aikuispotilailla ei ole osoitettu suurentunutta aivoverenvuotoriskiä.
Gefitinibihoitoa saavilla potilailla on raportoitu ruoansulatuskanavan perforaatioita. Useimmiten näihin tapauksiin on liittynyt muita tunnettuja riskitekijöitä kuten muiden lääkkeiden, esim. steroidien tai NSAID-lääkkeiden samanaikainen käyttö, aiempia ruoansulatuskanavan haavaumia, ikä, tupakointi tai suolen metastaasit perforaatiokohdassa.
Yhteisvaikutukset
Gefitinibi metaboloituu sytokromi P450 isoentsyymi CYP3A4:n (ensisijaisesti) ja CYP2D6:n kautta.
Vaikuttavat aineet, jotka saattavat suurentaa gefitinibin pitoisuutta plasmassa
In vitro -tutkimukset ovat osoittaneet, että gefitinibi on P-glykoproteiinin (Pgp) substraatti. Käytettävissä olevat tiedot eivät viittaa siihen, että tällä in vitro -havainnolla olisi kliinisiä seuraamuksia.
CYP3A4:ää inhiboivat aineet voivat pienentää gefitinibin puhdistumaa. Samanaikainen tehokkaiden CYP3A4-inhibiittoreiden (esim. ketokonatsoli, posakonatsoli, vorikonatsoli, proteaasi-inhibiittorit, klaritromysiini, telitromysiini) annostelu saattaa suurentaa gefitinibin pitoisuutta plasmassa. Tämä suureneminen voi olla kliinisesti merkitsevää, koska annos ja altistus vaikuttavat haittavaikutusten ilmenemiseen. Suureneminen voi olla voimakkaampaa yksittäisillä potilailla, jotka ovat heikkoja CYP2D6-metaboloijia. Edeltävä itrakonatsolihoito (voimakas CYP3A4-inhibiittori) aiheutti 80 %:n suurenemisen gefitinibin keskimääräisessä AUC:ssa terveillä vapaaehtoisilla. Tilanteissa, joissa annetaan samanaikaisesti tehokkaita CYP3A4-inhibiittoreita, potilasta tulee tarkoin seurata gefitinibin haittavaikutusten varalta.
Tietoja samanaikaisesta hoidosta CYP2D6-inhibiittoreilla ei ole, mutta tämän entsyymin tehokkaat inhibiittorit voivat aiheuttaa gefitinibin plasmapitoisuuksien noin kaksinkertaista suurenemista voimakkailla CYP2D6-metaboloijilla (ks. kohta Farmakokinetiikka). Jos hoito tehokkaalla CYP2D6-inhibiittorilla aloitetaan, potilasta tulee tarkoin seurata haittavaikutusten varalta.
Vaikuttavat aineet, jotka saattavat pienentää gefitinibin pitoisuutta plasmassa
CYP3A4-induktorit voivat tehostaa aineenvaihduntaa ja pienentää gefitinibin pitoisuuksia plasmassa ja siten heikentää gefitinibin tehoa. CYP3A4-induktorien (esim. fenytoiini, karbamatsepiini, rifampisiini, barbituraatit tai mäkikuisma ( Hypericum perforatum)) samanaikaista käyttöä tulee välttää. Edeltävä rifampisiinihoito (tehokas CYP3A4-induktori) pienensi terveillä vapaaehtoisilla gefitinibin keskimääräistä AUC:ta 83 % (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Aineet, jotka aiheuttavat merkitsevää ja pitkäkestoista mahalaukun pH:n kohoamista, voivat pienentää gefitinibin pitoisuutta plasmassa ja siten heikentää gefitinibin tehoa. Suurilla lyhytvaikutteisten antasidien annoksilla voi olla samanlainen vaikutus, jos niitä otetaan säännöllisesti samaan aikaan gefitinibin kanssa. Gefitinibin käyttö samanaikaisesti ranitidiinin kanssa annoksella, joka aiheutti pitkäkestoisen mahalaukun pH:n kohoamisen (≥ 5), pienensi terveillä vapaaehtoisilla gefitinibin keskimääräistä AUC:ta 47 % (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Vaikuttavat aineet, joiden plasmapitoisuuteen gefitinibi voi vaikuttaa
In vitro -tutkimukset ovat osoittaneet, että gefitinibillä on rajallinen inhiboiva vaikutus CYP2D6:een. Kliinisessä tutkimuksessa potilaille annosteltiin metoprololia (CYP2D6-substraatti) samanaikaisesti gefitinibin kanssa. Tämä aiheutti 35 % suurenemisen altistumisessa metoprololille. Tämän kaltainen suureneminen saattaa olla merkitsevää CYP2D6-substraateille, joilla on kapea terapeuttinen indeksi. Kun CYP2D6-substraatteja harkitaan käytettäväksi samanaikaisesti gefitinibin kanssa, CYP2D6-substraatin annoksen muuttamista tulisi harkita erityisesti, jos on kyse valmisteista, joilla on kapea terapeuttinen ikkuna.
Gefitinibi inhiboi BCRP-kuljettajaproteiinia in vitro, mutta havainnon kliininen merkitys ei ole tiedossa.
Muut mahdolliset yhteisvaikutukset
INR-arvon kohoamista ja/tai verenvuototapahtumia on raportoitu joillakin samanaikaisesti varfariinia käyttäneillä potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus ja imetys
Hedelmällisyys
Naisia, jotka voivat tulla raskaaksi, on neuvottava välttämään raskaaksi tuloa hoidon aikana.
Raskaus
Ei ole olemassa tietoja gefitinibin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmiselle ei tunneta. Gefitinib Stada -valmistetta ei pidä käyttää raskauden aikana, ellei se ole selvästi välttämätöntä.
Imetys
Ei tiedetä, erittyykö gefitinibi ihmisen rintamaitoon. Gefitinibi ja sen metaboliitit kertyivät imettävillä rotilla maitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Gefitinibin käyttö on vasta-aiheista rintaruokinnan aikana ja siksi imettäminen on keskeytettävä gefitinibihoidon ajaksi (ks. kohta Vasta-aiheet).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Gefitinibihoidon aikana on raportoitu heikotusta. Siksi potilaiden, joilla tätä oiretta esiintyy, tulee noudattaa varovaisuutta autolla ajon tai koneiden käytön aikana.
Haittavaikutukset
Yhteenveto haittavaikutuksista
Faasin III kliinisten tutkimusten, ISEL, INTEREST ja IPASS, yhdistetyt haittavaikutustiedot (2 462 gefitinibillä hoidettua potilasta) osoittivat, että useimmin raportoituja haittavaikutuksia, joita onollut yli 20 %:lla potilaista, ovat ripuli ja ihoreaktiot (sisältäen ihottuman, aknen, ihon kuivumisen ja kutinan). Haittavaikutukset ilmaantuvat yleensä ensimmäisen hoitokuukauden aikana, ja ne ovat yleensä korjautuvia. Noin 8 %:lle potilaista kehittyi vaikea haittavaikutus (common toxicity criteria (CTC) vaikeusaste 3 tai 4). Noin 3 % potilaista lopetti hoidon haittavaikutuksen takia.
Interstitiaalista keuhkosairautta (ILD) on esiintynyt 1,3 %:lla potilaista, usein vaikeana (vaikeusaste 3 tai 4). Kuolemaan johtaneita tapauksia on raportoitu.
Taulukoitu luettelo haittavaikutuksista
Taulukossa 1 esitetty turvallisuusprofiili perustuu gefitinibin kliinisestä kehitysohjelmasta saatuun sekä markkinoille tulon jälkeiseen kokemukseen. Taulukossa 1 haittavaikutukset on luokiteltu esiintymistiheyden mukaan, jos mahdollista, perustuen vertailukelpoisten haittatapahtumien esiintyvyyteen yhdistetyssä kliinisen faasin III ISEL-, INTEREST- ja IPASS-tutkimusten aineistossa (geitinibiä käytti 2 462 potilasta).
Haittavaikutusten esiintymistiheys on määritelty seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000 – < 1/100); harvinainen (≥ 1/10 000 – < 1/1 000); hyvin harvinainen (< 1/10 000), tuntematon (saatavissa oleva tieto ei riitä arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1. Haittavaikutukset
Haittavaikutukset elinjärjestelmän ja esiintymistiheyden mukaan | ||
Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Anoreksia, lievä tai keskivaikea (CTC vaikeusaste 1 tai 2). |
Silmät | Yleinen | Konjunktiviitti, blefariitti ja silmien kuivuminen*, yleensä lievä (CTC vaikeusaste 1). |
Melko harvinainen | Sarveiskalvon eroosio, korjautuva ja liittyy joskus poikkeavaan silmäripsien kasvuun. | |
Sarveiskalvotulehdus (0,12 %). | ||
Verisuonisto | Yleinen | Verenvuototapahtumat, kuten nenäverenvuoto ja verivirtsaisuus. |
Hengityselimet, rintakehä ja välikarsina | Yleinen | Interstitiaalinen keuhkosairaus (1,3 %), usein vaikea (CTC vaikeusaste 3-4). Kuolemaan johtaneita tapauksia on raportoitu. |
Ruoansulatuselimistö | Hyvin yleinen | Ripuli, yleensä lievä tai keskivaikea (CTC-vaikeusaste 1 tai 2). |
Oksentelu, yleensä lievä tai keskivaikea (CTC vaikeusaste 1 tai 2). | ||
Pahoinvointi, yleensä lievä (CTC vaikeusaste 1). | ||
Suutulehdus, yleensä lievä (CTC vaikeusaste 1). | ||
Yleinen | Ripulista, pahoinvoinnista, oksentelusta tai anoreksiasta johtuva kuivuminen. | |
Suun kuivuminen*, yleensä lievä (CTC vaikeusaste 1). | ||
Melko harvinainen | Haimatulehdus Ruuansulatuskanavan perforaatio. | |
Maksa ja sappi | Hyvin yleinen | Alaniiniaminotransferaasin nousu, pääasiassa lievä tai kohtalainen. |
Yleinen | Aspartaattiaminotransferaasin nousu, pääasiassa lievä tai kohtalainen. | |
Kokonaisbilirubiinin nousu, pääasiassa lievä tai kohtalainen. | ||
Melko havinainen | Hepatiitti**. | |
Iho ja ihonalainen kudos | Hyvin yleinen | Ihoreaktiot, yleensä lievä tai keskivaikea (CTC vaikeusaste 1 tai 2) märkärakkulainen ihottuma, toisinaan kutiseva ja kuiva, mukaan lukien ihon fissuurat, punoittavan ihon päällä. |
Yleinen | Kynsiongelmat. | |
Alopesia. | ||
Allergiset reaktiot (1,1 %) mukaan lukien angioödeema ja nokkosihottuma. | ||
Melko harvinainen | Palmoplantaarinen erytrodysestesia -oireyhtymä. | |
Harvinainen | Rakkulasairaudet, myös toksinen epidermaalinen nekrolyysi, Stevens-Johnsonin oireyhtymä ja erythema multiforme. | |
Kutaaninen vasuliitti. | ||
Munuaiset ja virtsatiet | Yleinen | Oireeton veren kreatiniinipitoisuuden suureneminen. |
Valkuaisvirtsaisuus. | ||
Kystiitti. | ||
Harvinainen | Hemorraginen kystiitti. | |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Voimattomuus, yleensä lievä (CTC vaikeusaste 1). |
Yleinen | Kuume. | |
Poikkeaviin laboratorioarvoihin liittyvien haittavaikutusten esiintymistiheys perustuu potilaisiin, joilla muutos oli vähintään 2 CTC-vaikeusastetta lähtötasosta kyseisissä olennaisissa laboratorioparametreissä.
* Tämä haittavaikutus voi liittyä gefitinibin käytön yhteydessä havaittuihin muihin kuivumisongelmiin (yleensä ihoreaktioihin).
** Tämä sisältää yksittäisiä maksan vajaatoimintatapauksia, jotka joissakin tapauksissa ovat olleet kuolemaan johtavia.
Interstitiaalinen keuhkosairaus (ILD)
INTEREST-tutkimuksessa ILD-tyyppisiä tapahtumia esiintyi 1,4 %:lla potilaista (10 potilasta) gefitinibiryhmässä ja 1,1 %:lla potilaista (8 potilasta) doketakseliryhmässä. Yksi ILD-tyyppinen tapaus johti kuolemaan, ja se ilmaantui gefitinibiä saaneelle potilaalle.
ISEL-tutkimuksessa interstitiaalisen keuhkosairauden kaltaisten tapahtumien ilmaantuvuus kokonaispopulaatiossa oli noin 1 % kummassakin hoitohaarassa. Valtaosa raportoiduista ILD-tapahtumista koski aasialaista alkuperää olevia potilaita ja interstitiaalisen keuhkosairauden ilmaantuvuus aasialaista alkuperää olevilla, gefitinibiä ja plaseboa käyttävillä potilailla oli vastaavasti noin 3 % ja 4 %. Yksi ILD-tyyppinen tapaus johti kuolemaan, kyseessä oli plaseboa saanut potilas.
Japanissa tehdyssä, valmisteen markkinoille tulon jälkeisessä tutkimuksessa (3 350 potilasta) interstitiaalisen keuhkosairauden kaltaisia tapahtumia raportoitiin gefitinibiä käyttävillä potilailla 5,8 %. Kuolemaan johtaneiden ILD-tyyppisten tapausten osuus näistä oli 38,6 %.
Avoimessa, faasin III kliinisessä tutkimuksessa (IPASS), johon osallistui 1 217 potilasta, verrattiin gefitinibiä karboplatiini-paklitakseli -yhdistelmäkemoterapiaan ensilinjan hoitona valikoiduille, edennyttä ei-pienisoluista keuhkosyöpää sairastaville potilaille Aasiassa. Interstitiaalisen keuhkosairauden kaltaisten tapahtumien ilmaantuvuus oli 2,6 % gefitinibi-hoitoryhmässä ja 1,4 % karboplatiini-paklitakseli -hoitoryhmässä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‐haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Gefitinibin yliannostukseen ei ole spesifistä hoitoa. Vaiheen I kliinisissä tutkimuksissa muutamat potilaat saivat kuitenkin jopa 1 000 mg:n vrk-annoksia. Joidenkin haittavaikutusten, pääasiassa ripulin ja ihottuman, esiintyvyyden ja vaikeusasteen lisääntymistä havaittiin. Yliannostukseen liittyviä haittavaikutuksia on hoidettava oireenmukaisesti, ja erityisesti vaikeaa ripulia on hoidettava kuten kliinisesti aiheellista. Yhdessä tutkimuksessa pienelle määrälle potilaita annettiin 1 500–3 500 mg:n annoksia. Tässä tutkimuksessa gefitinibialtistus ei lisääntynyt annoksen lisääntyessä ja haittavaikutukset olivat vakavuudeltaan yleensä lieviä tai keskivaikeita ja yhdenmukaisia tunnetun gefitinibi-turvallisuusprofiilin kanssa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Syöpälääkkeet, proteiinikinaasin estäjä, ATC-koodi: L01EB01.
Vaikutusmekanismi ja farmakodynaamiset vaikutukset
Epidermaalinen kasvutekijä (EGF) ja sen reseptori (EGFR [HER1; ErbB1]) on tunnistettu merkittäviksi tekijöiksi normaalien sekä syöpäsolujen kasvu- ja jakautumisprosessissa. EGFR:ää aktivoiva mutaatio syöpäsolussa on tärkeä tekijä tuumorisolujen kasvun edistämisessä, apoptoosin salpaamisessa, angiogeenisten tekijöiden tuotannon lisäämisessä ja metastaasiprosessien edesauttamisessa.
Gefitinibi on selektiivinen, pienimolekyylinen epidermaalisen kasvutekijäreseptorin tyrosiinikinaasin estäjä ja se on hoitolinjasta riippumatta tehokas hoito potilaille, joiden tuumoreissa on aktivoivia EGFR-tyrosiinikinaasin mutaatioita. Kliinisesti merkittävää tehoa ei ole osoitettu potilailla, joiden kasvaimien tiedetään olevan EGFR-mutaationegatiivisia.
Yleisten EGFR:ää aktivoivien mutaatioiden (eksoni 19 deletiot; L858R) hoitovastetulokset ovat voimakkaat tukien herkkyyttä gefitinibille; esimerkiksi taudin etenemisvapaa elinajan HR (95 % CI) on gefitinibille 0,489 (0,336; 0,710) verrattuna kaksoissolunsalpaajahoitoon (WJTOG3405).
Hoitovastetulokset gefitinibillä ovat niukemmat potilailla, joiden kasvaimissa on vähemmän yleisiä mutaatioita; saatavilla olevat tiedot viittaavat siihen, että G719X, L861Q ja S7681 ovat herkistäviä ja että T790M-mutaatio tai eksoni 20 -insertiot yksistään ovat resistenssimekanismeja.
Resistenssi
Useimmissa ei-pienisoluisen keuhkosyövän kasvaimissa, joissa on herkistäviä EGFR:n kinaasin mutaatioita, kehittyy lopulta resistenssi gefitinibihoidolle. Mediaaniaika taudin etenemiseen on 1 vuosi. Noin 60 %:ssa tapauksista resistenssiin liittyy sekundaarinen T790M-mutaatio, jolloin seuraavan linjan hoitovaihtoehdoksi voidaan harkita T790M-mutaatioon kohdistuvia EGFR:n tyrosiinikinaasin estäjiä. Muita mahdollisia resistenssimekanismeja, joita on ilmoitettu EGFR:n signaalivälitystä salpaavan lääkehoidon jälkeen, ovat mm. eston ohittava signaalivälitys (bypass signalling), kuten HER2- ja MET-geeniamplifikaatio ja PIK3CA-mutaatiot. 5–10 %:ssa tapauksista on myös ilmoitettu fenotyypin vaihtumista pienisoluiseksi keuhkosyöväksi.
Kiertävä kasvain-DNA (ctDNA)
IFUM-tutkimuksessa mutaatiostatus määritettiin kasvaimesta ja plasmasta peräisin olevasta ctDNA-näytteestä käyttämällä Therascreen EGFR RGQ PCR kit:iä (Qiagen). Sekä ctDNA- että kasvainnäyte olivat arvioitavissa 652 potilaalla 1 060 seulotusta. Objektiivinen hoitovaste oli 77 % (95 % CI: 66 % – 86 %) potilailla joiden kasvain ja ctDNA olivat mutaatiopositiivisia. Objektiivinen hoitovaste oli 60 % (95 % CI: 44 % – 74 %) potilailla, joilla vain kasvain oli mutaatiopositiivinen.
Taulukko 2. Yhteenveto alkuvaiheen mutaatiostatuksesta kaikkien niiden seulottujen potilaiden kasvain- ja ctDNA-näytteissä, joiden molemmat näytteet olivat arvioitavissa
| Mitta | Määritelmä | IFUM ilmaantuvuus % (CI) | IFUM N |
| Herkkyys | Niiden M+ kasvainten osuus, jotka ovat M+ ctDNA-näytteissä | 65,7 (55,8, 74,7) | 105 |
| Spesifisyys | Niiden M- kasvainten osuus, jotka ovat M- ctDNA-näytteissä | 99,8 (99,0, 100,0) | 547 |
Tiedot ovat yhtäpitäviä ennakkoon suunnitellun, eksploratiivisen japanilaisen IPASS-tutkimuksen alaryhmäanalyysin kanssa (Goto 2012). Kyseisessä tutkimuksessa käytettiin seerumista, ei plasmasta, peräisin olevaa ctDNA:ta EGFR-mutaatioanalyysiin käyttämällä EGFR Mutation Test Kit:iä (DxS) (N = 86). Kyseisessä tutkimuksessa herkkyys oli 43,1 % ja spesifisyys oli 100 %.
Kliininen teho ja turvallisuus
Ensilinjan hoito
Randomoituun faasin III ensilinjan IPASS-tutkimukseen osallistui Aasiasta1 potilaita, joilla oli edennyt (IIIB tai IV-vaihe), histologialtaan adenokarsinoomaksi luokiteltu, ei-pienisoluinen keuhkosyöpä ja jotka olivat joko aiemmin kevyesti tupakoineita (lopettaneet ≥ 15 vuotta aiemmin ja tupakoineet ≤ 10 askivuotta) tai tupakoimattomia (ks. taulukko 3).
1 Kiina, Hong Kong, Indonesia, Japani, Malesia, Filippiinit, Singapore, Taiwan, Thaimaa.
Taulukko 3. Tehokkuustulokset IPASS-tutkimuksesta, gefitinibi verrattuna karboplatiini-paklitakseli -yhdistelmään
| Potilasryhmä | N | Objektiivisten hoitovasteiden määrä ja 95 %:n luottamusväli hoitojen välisille eroillea | Ensisijainen päätetapahtuma Etenemisvapaa elinaikaab | Kokonaiselinaika ab |
| Kaikki | 1217 | 43,0 % vs. 32,2 % [5,3 % – 16,1 %] | HR 0,74 [0,65–0,85] 5,7 kk vs. 5,8 kk p < 0,0001 | HR 0,90 [0,79–1,02] 18,8 kk vs. 17, 4 kk p = 0,1087 |
| EGFR-mutaatiopositiiviset | 261 | 71,2 % vs. 47,3 % [12,0 % – 34,9 %] | HR 0,48 [0,36–0,64] 9,5 kk vs. 6,3 kk p < 0,0001 | HR 1,00 [0,76–1,33] 21,6 kk vs. 21,9 kk |
| EGFR-mutaationegatiiviset | 176 | 1,1 % vs. 23,5 % [-32,5 % – 13,3 %] | HR 2,85 [2,05–3,98] 1,5 kk vs. 5,5 kk p < 0,0001 | HR 1,18 [0,86–1,63] 11,2 kk vs. 12,7 kk |
| EGFR-mutaatiostatus ei tiedossa | 780 | 43,3 % vs. 29,2 % [7,3 % – 20,6 %] | HR 0,68 [0,58–0,81] 6,6 kk vs. 5,8 kk p < 0,0001 | HR 0,82 [0,70–0,96] 18,9 kk vs. 17,2 kk |
a Tulokset on esitetty gefitinibi vs. karboplatiini-paklitakseli -yhdistelmä.
b kk tarkoittaa mediaania kuukausissa. Numerot hakasuluissa ovat 95 %:n luottamusvälejä HR:lle.
N Randomoitujen potilaiden lukumäärä
HR Vaarasuhde (hazard ratio), < 1 vaarasuhteet suosivat gefitinibiä
Elämänlaatua mittaavat tulokset erosivat EGFR-mutaatiostatuksen mukaisesti. Potilaista, joilla oli positiivinen EGFR-mutaatiostatus, merkitsevästi useampi gefitinibiä käyttänyt potilas koki elämänlaadun paranemista ja keuhkosyöpäoireiden helpottumista verrattuna karboplatiini-paklitakselia saaneisiin potilaisiin (ks. taulukko 4).
Taulukko 4. Elämänlaatua mittaavat tulokset IPASS-tutkimuksesta, gefitinibi verrattuna
karboplatiini-paklitakseli -yhdistelmään
| Potilasryhmä | N | FACT-L QoL parantuminena % | LCS-oireiden väheneminena % |
| Kaikki | 1 151 | (48,0 % vs. 40,8 %) p = 0,0148 | (5,5 % vs. 48.5 %) p = 0,3037 |
| EGFR-mutaatiopositiiviset | 259 | (70,2 % vs. 44,5 %) p < 0,0001 | (75,6 % vs. 53,9 %) p = 0,0003 |
| EGFR-mutaationegatiiviset | 169 | (14,6 % vs. 36,3 %) p = 0,0021 | (20,2 % vs. 47,5 %) p = 0,0002 |
Tutkimuksen elämänlaatua mittaavan indeksin tulokset tukivat FACT-L ja LCS tuloksia.
a Tulokset on esitetty gefitinibi vs. karboplatiini-paklitakseli -yhdistelmä.
N Elämänlaadun arviointiin sopivien potilaiden määrä.
QoL Elämänlaatu (Quality of life)
FACT-L Functional assessment of cancer therapy-lung
LCS Lung cancer subscale
IPASS-tutkimuksessa gefitinibi-hoidolla saavutettiin paremmat tulokset etenemisvapaan elinajan, objektiivisten hoitovasteiden määrän, elämänlaadun ja oireiden lievittymisen suhteen eikä kokonaiselinajassa ollut merkitsevää eroa, kun hoitoa verrattiin karboplatiini-paklitakseli -yhdistelmähoitoon aiemmin hoitamattomilla potilailla, joilla oli paikallisesti edennyt tai etäpesäkkeinen ei-pienisoluinen keuhkosyöpä ja joiden syövässä oli aktivoivia EGFR-tyrosiinikinaasin mutaatioita.
Aikaisempaa hoitoa saaneet potilaat
Satunnaistettuun faasin III INTEREST-tutkimukseen osallistui potilaita, joilla oli paikallisesti edennyt tai metastasoitunut ei-pienisoluinen keuhkosyöpä ja jotka olivat aiemmin saaneet platinaa sisältävää solunsalpaajahoitoa. Kokonaispopulaatiossa ei havaittu tilastollisesti merkitsevää eroa gefitinibin ja doketakselin (75 mg/m2) välillä kokonaiselinajan, taudin etenemisvapaan elinajan sekä objektiivisten hoitovasteiden määrän suhteen (ks. taulukko 5).
Taulukko 5. Tehokkuustulokset INTEREST-tutkimuksesta, gefitinibi verrattuna doketakseliin
| Potilasryhmä | N | Objektiivisten hoitovasteiden määrä ja 95 %:n luottamusväli hoitojen välisille eroillea | Etenemisvapaa elinaikaab | Ensisijainen päätetapahtuma Kokonaiselinaikaab |
| Kaikki | 1466 | 9,1 % vs. 7,6 % [-1,5 % – 4,5 %] | HR 1,04 [0,93–1,18] 2,2 kk vs. 2,7 kk p = 0,4658 | HR 1,020 [0,905–1,150] c 7,6 kk vs. 8,0 kk p = 0,7332 |
| EGFR-mutaatiopositiiviset | 44 | 42,1 % vs. 21,1 % [-8,2 % – 46,0 %] | HR 0,16 [0,05–0,49] 7,0 kk vs. 4,1 kk p = 0,0012 | HR 0,83 [0,41–1,67] 14,2 kk vs. 16,6 kk p = 0,6043 |
| EGFR-mutaationegatiiviset | 253 | 6,6 % vs. 9,8 % [-10,5 % – 4,4 %] | HR 1,24 [0,94–1,64] 1,7 kk vs. 2,6 kk p = 0,1353 | HR 1,02 [0,78–1,33] 6,4 kk vs. 6,0 kk p = 0,9131 |
| Aasialaisetc | 323 | 19,7 % vs. 8,7 % [3,1 % – 19,2 %] | HR 0,83 [0,64–1,08] 2,9 kk vs. 2,8 kk p = 0,1746 | HR 1,04 [0,80–1,35] 10,4 kk vs. 12,2 kk p = 0,7711 |
| Ei-Aasialaiset | 1143 | 6,2 % vs. 7,3 % [-4,3 % – 2,0 %] | HR 1,12 [0,98–1,28] 2,0 kk vs. 2,7 kk p = 0,1041 | HR 1,01 [0,89–1,14] 6,9 kk vs. 6,9 kk p = 0,9259 |
a Tulokset on esitetty gefitinibi vs. doketakseli.
b kk tarkoittaa mediaania kuukausissa. Numerot hakasuluissa ovat 96 %:n luottamusvälejä kokonaiselinajalle HR:lle kokonaispopulaatiossa tai muuten HR:n 95 %:n luottamusvälejä.
c Luottamusväli kokonaan yhdenvertaisuusrajan ( non-inferiority) 1,154 alapuolella.
N Randomoitujen potilaiden lukumäärä
HR Vaarasuhde (hazard ratio), < 1 vaarasuhteet suosivat gefitinibiä
Kuvat 1 ja 2 Tehokkuustulokset ei-aasialaisten potilaiden ryhmässä INTEREST tutkimuksessa
(N potilaat = randomoitujen potilaiden määrä)
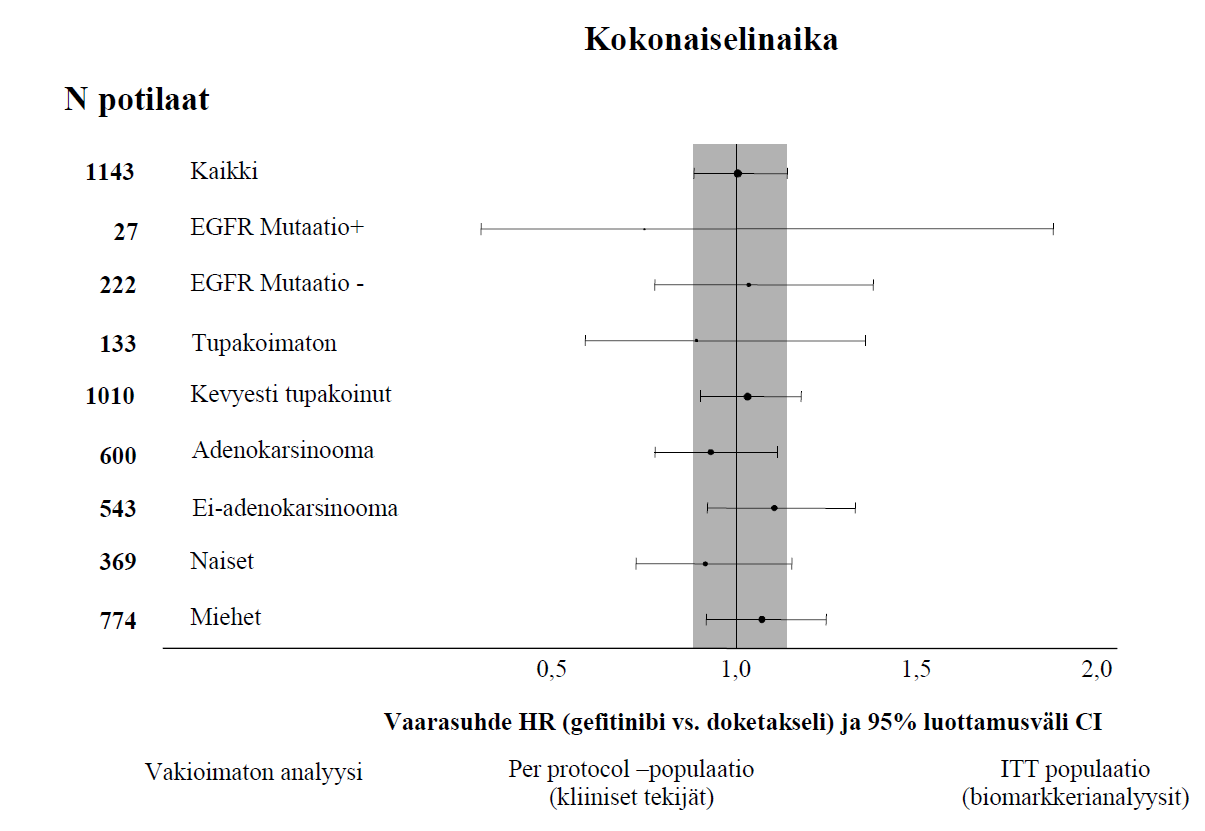
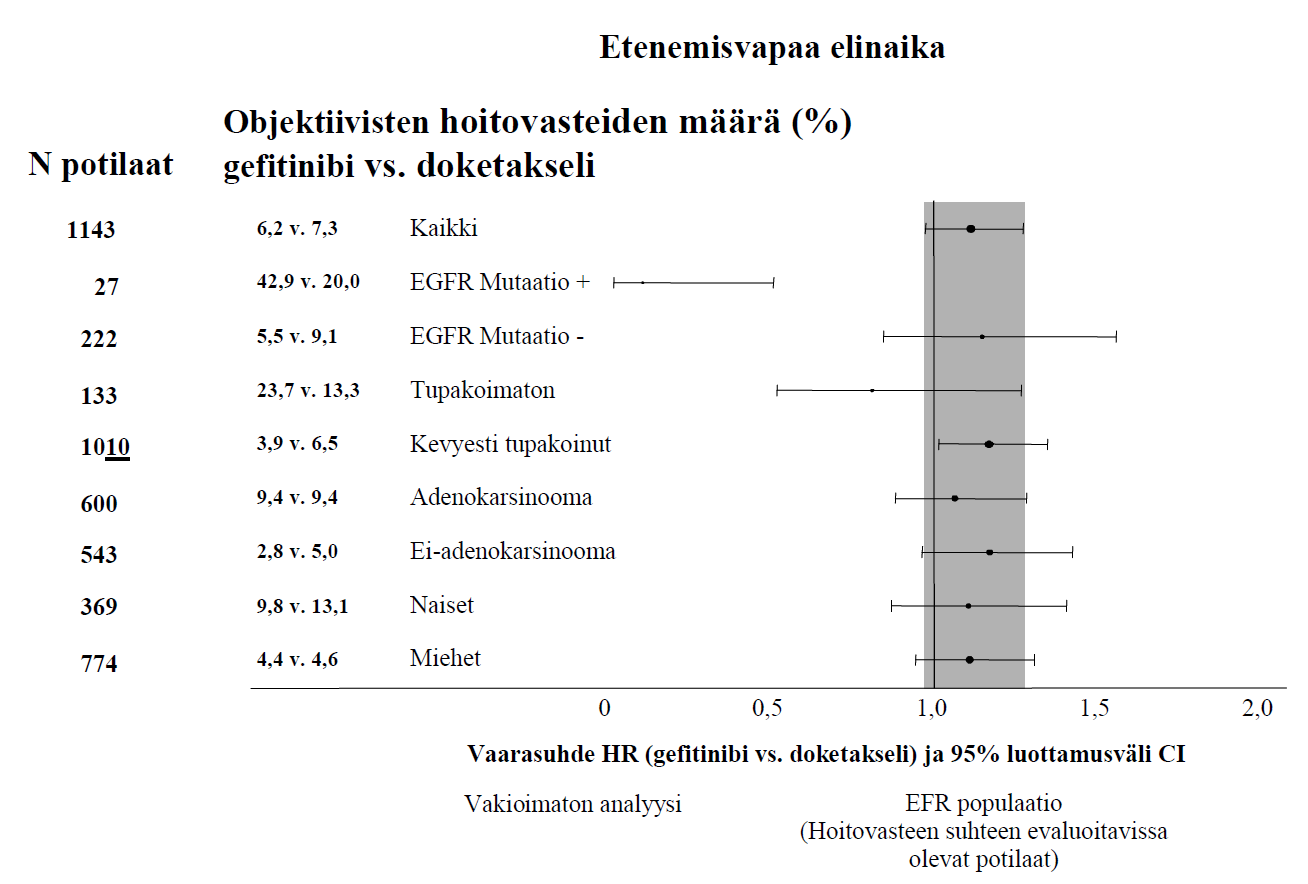
Randomoituun faasin III ISEL-tutkimukseen osallistui edennyttä ei-pienisoluista keuhkosyöpää sairastavia potilaita, joita oli aiemmin hoidettu yhdellä tai kahdella solunsalpaajahoidolla, joista viimeksi käytettyyn hoitoon potilaat joko eivät saaneet vastetta tai eivät sietäneet sitä. Gefitinibiä yhdistettynä parhaaseen mahdolliseen tukihoitoon verrattiin plaseboon yhdistettynä parhaaseen mahdolliseen tukihoitoon. Gefitinibi ei pidentänyt elinaikaa koko tutkimusjoukossa. Elinaikatulokset vaihtelivat tupakointihistorian ja etnisen ryhmän mukaan (ks. taulukko 6).
Taulukko 6. Tehokkuustulokset ISEL-tutkimuksesta, gefitinibi verrattuna plaseboon
| Potilasryhmä | N | Objektiivisten hoitovasteiden määrä ja 95 %:n luottamusväli hoitojen välisille eroille3 | Aika hoidon epäonnistumiseenab | Ensisijainen päätetapahtuma Kokonaiselinaikaabc |
| Kaikki | 1692 | 8,0 % vs. 1,3 % [4,7 % – 8,8 %] | HR 0,82 [0,73–0,92] 3,0 kk vs. 2,6 kk p = 0,0006 | HR 0,89 [0,77–1,02] 5,6 kk vs. 5,1 kk p = 0,0871 |
| EGFR-mutaatiopositiiviset | 26 | 37.5 % vs. 0 % [-15.1 % – 61,4 %] | HR 0,79 [0,20–3,12] 10,8 kk vs. 3,8 kk p = 0,7382 | HR NC NR vs. 4,3 kk |
| EGFR-mutaationegatiiviset | 189 | 2,6 % vs. 0 % [-5,6 % – 7,3 %] | HR 1,10 [0,78–1,56] 2,0 kk vs. 2,6 kk p = 0,5771 | HR 1,16 [0,79–1,72] 3,7 kk vs. 5,9 kk p = 0,4449 |
| Tupakoimattomat | 375 | 18,1 % vs. 0 % [12,3 % – 24,0 %] | HR 0,55 [0,42–0,72] 5,6 kk vs. 2,8 kk p < 0,0001 | HR 0,67 [0,49–0,92] 8,9 kk vs. 6,1 kk p = 0,0124 |
| Joskus tupakoineet | 1317 | 5,3 % vs. 1,6 % [1,4 % – 5,7 %] | HR 0,89 [0,78–1,01] 2,7 kk vs. 2,6 kk p = 0,0707 | HR 0,92 [0,79–1,06] 5,0 kk vs. 4,9 kk p = 0,2420 |
| Aasialaisetd | 342 | 12,4 % vs. 2,1 % [4,0 % – 15,8 %] | HR 0,69 [0,52–0,91] 4,4 kk vs. 2,2 kk p = 0,0084 | HR 0,66 [0,48–0,91] 9,5 kk vs. 5,5 kk p = 0,0100 |
| Ei-aasialaiset | 1350 | 6,8 % vs. 1,0 % [3,5 % – 7,9 %] | HR 0,86 [0,76–0,98] 2,9 kk vs. 2,7 kk p = 0,0197 | HR 0,92 [0,80–1,07] 5,2 kk vs. 5,1 kk p = 0,2942 |
a Tulokset on esitetty gefitinibi vs. plasebo.
b kk on mediaanit kuukausissa. Numerot hakasuluissa ovat HR:n 95 %:n luottamusvälejä
c Stratified log-rank -testi koko tutkimusjoukolle; muuten cox proportional hazards -malli
d Etninen ryhmä aasialaiset ei sisällä intialaista syntyperää olevia potilaita ja ryhmä viittaa potilaiden rotuun, ei välttämättä syntymäpaikkaan
N Randomoitujen potilaiden lukumäärä
NC Ei laskettu kokonaiselinajan vaarasuhteen osalta, koska tapahtumien lukumäärä on liian pieni
NR Ei saavutettu (not reached)
HR Vaarasuhde (hazard ratio), < 1 vaarasuhteet suosivat gefitinibiä
IFUM-tutkimus oli yhden hoitohaaran monikeskustutkimus ja siihen osallistui valkoihoisia ei-pienisoluista keuhkosyöpää sairastavia potilaita (n = 106), joiden kasvaimessa oli aktivoivia ja herkistäviä EGFR-tyrosiinikinaasin mutaatioita. Tutkimuksen tarkoituksena oli osoittaa gefitinibin aktiivisuuden samankaltaisuus valkoihoisessa ja aasialaisessa väestössä. Tutkijalääkärin arvioimaobjektiivinen hoitovaste oli 70 % ja etenemisvapaan elinajan mediaani oli 9,7 kuukautta.
Tulokset olivat verrannolliset IPASS-tutkimuksen tulosten kanssa.
EGFR-mutaatiostatus ja kliiniset tekijät
Kliinisten tekijöiden, joita ovat tupakoimattomuus, kasvaimen adenokarsinoomahistologia ja naissukupuoli, on todettu olevan riippumattomia positiivista EGFR-mutaatiostatusta ennustavia tekijöitä gefitinibitutkimuksiin* osallistuneista 786 valkoihoisesta potilaasta tehdyssä monimuuttuja-analyysissa (ks. taulukko 7). EGFR-mutaatiopositiivisten kasvainten esiintyvyys on lisäksi korkeampi aasialaisilla potilailla.
Taulukko 7. Yhteenveto monimuuttujaisesta logistisesta regressioanalyysistä, jolla pyrittiin tunnistamaan riippumattomia, EGFR-mutaatiostatusta ennustavia tekijöitä 786 valkoihoisella potilaalla*
EGFR-mutaatioiden esiintyvyyttä ennustavat tekijät | p-value | Mahdollisuus EGFR-mutaatioon | Positiivinen ennustava tekijä (9,5 % koko tutkimuspopulaatiosta on EGFR-mutaatiopositiivisia (M+)) |
| Tupakointi | < 0,0001 | 6,5 kertaa suurempi mahdollisuus tupakoimattomilla verrattuna joskus tupakoineisiin | 28/70 (40 %) tupakoimattomista on M+ 47/716 (7 %) joskus tupakoineista on M+ |
| Histologia | < 0,0001 | 4,4 kertaa suurempi mahdollisuus, jos kasvaimen histologia on adenokarsinooma verrattuna ei-adenokarsinoomaan | 63/396 (16 %) potilaista, joiden kasvaimen histologia on adenokarsinooma, on M+ 12/390 (3 %) potilaista, joiden kasvaimen histologia on ei-adenokarsinooma, on M+ |
| Sukupuoli | 0,0397 | 1,7 kertaa suurempi mahdollisuus naisilla verrattuna miehiin | 40/235 (17 %) naisista on M+ 35/551 (6 %) miehistä on M+ |
* Seuraavista tutkimuksista: INTEREST, ISEL, INTACT 1&2, IDEAL 1&2, INVITE
Farmakokinetiikka
Imeytyminen
Suun kautta otettuna gefitinibin imeytyminen on kohtalaisen hidasta ja huippupitoisuudet plasmassa saadaan yleensä 3–7 tunnin kuluttua annostuksesta. Keskimääräinen absoluuttinen biologinen hyötyosuus on syöpäpotilailla 59 %. Ruoka ei vaikuta merkitsevästi gefitinibin systeemiseen altistukseen. Gefitinibialtistus pieneni 47 % tutkimuksessa, johon osallistui terveitä vapaaehtoisia, ja jossa mahalaukun pH pidettiin yli arvon 5. Tämä johtui todennäköisesti gefitinibin heikentyneestä liukenemisesta mahalaukussa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Jakautuminen
Gefitinibin keskimääräinen jakaantumistilavuus vakiintuneessa (steady-state) tilanteessa on 1 400 l, mikä viittaa siihen, että gefitinibi jakautuu laajalti kudoksiin. Sitoutuminen plasman proteiineihin on noin 90-prosenttista. Gefitinibi sitoutuu seerumin albumiiniin ja happamaan alfa-1-glykoproteiiniin.
In vitro -tutkimustiedot viittaavat siihen, että gefitinibi on kalvoproteiini Pgp:n substraatti.
Biotransformaatio
In vitro -tiedot osoittavat, että gefitinibin oksidatiiviseen metaboliaan vaikuttavat pääasiassa CYP3A4- ja CYP2D6-isoentsyymit.
In vitro -tutkimukset viittaavat siihen, että gefitinibi estää vain vähän CYP2D6:ta. Gefitinibillä ei ole havaittu entsyymejä indusoivia vaikutuksia eläintutkimuksissa eikä minkään muun sytokromi-P450-entsyymin merkitsevää estoa ( in vitro).
Gefitinibin metabolia ihmisellä on laajaa. Viisi metaboliittia on tunnistettu eritteistä ja kahdeksan plasmasta. Päämetaboliitti on O-desmetyyligefitinibi, joka estää 14-kertaa heikommin EGFR:n stimuloimaa solukasvua kuin gefitinibi, eikä sillä ole inhiboivaa vaikutusta tuumorisolujen kasvuun hiiressä. Sen vuoksi on pidetty epätodennäköisenä, että se myötävaikuttaisi gefitinibin kliiniseen aktiviteettiin.
O-desmetyyligefitinibin muodostuminen on osoittautunut, in vitro, olevan CYP2D6-välitteistä.
CYP2D6:n roolia gefitinibin metabolisessa puhdistumassa on arvioitu kliinisessä tutkimuksessa terveillä vapaaehtoisilla, jotka genotyypitettiin CYP2D6 statuksen suhteen. Heikot metaboloijat eivät tuottaneet mitattavia O-desmetyyligefitinibin pitoisuuksia. Gefitinibialtistuksen erot olivat suuria sekä nopeilla että hitailla metaboloijilla ja altistustasot olivat osin päällekkäisiä, mutta keskimääräinen altistuminen gefitinibille oli kaksinkertaista hitaiden metaboloijien ryhmässä. Suurempi keskimääräinen altistus henkilöillä, joilla CYP2D6 ei ole aktiivinen, saattaa olla kliinisesti merkittävä, koska haittavaikutuksilla on yhteys annokseen ja altistukseen.
Eliminaatio
Gefitinibi erittyy pääasiassa metaboliitteina ulosteen kautta. Gefitinibin ja metaboliittien munuaispuhdistuma muodostaa alle 4 % annetusta annoksesta.
Syöpäpotilailla gefitinibin kokonaisplasmapuhdistuma on noin 500 ml/min ja keskimääräinen terminaalinen puoliintumisaika on 41 tuntia. Gefitinibin kerran päivässä annostelu aiheuttaa 2–8-kertaisen kumuloitumisen siten, että steady-state-altistus saavutetaan 7–10 annoksen jälkeen.
Steady-state-tilassa verenkierrossa olevat plasmapitoisuudet säilyvät tyypillisesti 2–3-kertaluokan sisällä 24 tunnin välein annosteltuna.
Erityisryhmät
Syöpäpotilailla populaatiofarmakokineettisissä analyyseissä ei ole havaittu suhdetta ennustetun steady-state-tason pienimmän pitoisuuden ja potilaan iän, painon, sukupuolen, etnisen alkuperän tai kreatiniinipuhdistuman (yli 20 ml/min) välillä.
Maksan vajaatoiminta
Faasin I avoimessa tutkimuksessa potilaat, joilla oli maksakirroosista johtuva lievä, keskivaikea tai vaikea maksan vajaatoiminta (Child-Pugh-luokituksen mukaan) ja jotka saivat 250 mg gefitinibiä kerta-annoksena, systeemisen altistuksen havaittiin olevan kaikissa ryhmissä suurempi terveisiin verrokkeihin verrattuna. Potilailla, joilla oli keskivaikea tai vaikea maksan vajaatoiminta havaittiin keskimäärin 3,1-kertainen altistumisen suureneminen gefitinibille. Yhdelläkään potilaalla ei ollut syöpää, kaikilla oli maksakirroosi ja joillakin hepatiitti. Systeemisen altistumisen lisääntymisellä saattaa olla kliinistä merkitystä, koska haittavaikutuksilla on yhteys annokseen ja altistukseen.
Gefitinibiä on arvioitu kliinisessä tutkimuksessa, johon osallistui 41 potilasta, joilla oli kiinteä kasvain ja normaali maksan toiminta tai keskivaikea tai vaikea maksametastaaseista johtuva maksan vajaatoiminta (luokiteltu lähtötason ASAT:n, AFOS:n ja bilirubiinin Common Toxicity Criteria (CTC) -asteiden mukaisesti). Ilmeni, että päivittäisen gefitinibin annostelun (250 mg) jälkeen steady-state-tason saavuttamiseen kulunut aika, kokonaispuhdistuma plasmasta (CmaxSS) ja steady-state-altistus (AUC24SS) olivat samankaltaisia potilailla, joiden maksan toiminta oli normaali, kuin potilailla, joilla oli keskivaikea maksan vajaatoiminta. Tiedot neljästä potilaasta, joilla oli maksametastaasien aiheuttama vaikea maksan vajaatoiminta viittaavat siihen, että steady-state-tason altistus näillä potilailla on myös vastaavanlainen kuin potilailla, joilla maksan toiminta on normaali.
Prekliiniset tiedot turvallisuudesta
Haittavaikutukset, joita ei ole havaittu kliinisissä tutkimuksissa, mutta joita on todettu eläinkokeissa kliinistä käyttöä vastaavilla altistustasoilla ja jotka saattavat olla kliinisessä käytössä olennaisia, ovat seuraavat:
- sarveiskalvoepiteelin atrofia ja sarveiskalvon samentuma
- munuaisnystykuolio
- maksasolukuolio ja eosinofiilinen sinusoidien makrofagi-infiltraatio
Tiedot ei-kliinisistä ( in vitro) tutkimuksista viittaavat siihen, että gefitinibi pystyy estämään sydämen aktiopotentiaalin repolarisaatioprosessia (esim. QT-aika). Kliininen kokemus ei ole osoittanut kausaalista suhdetta QT-ajan pidentymisen ja gefitinibin välillä.
Naarasrottien hedelmällisyyden havaittiin heikkenevän 20 mg/kg vuorokausiannoksella.
Julkaistut tutkimukset ovat osoittaneet, että geneettisesti muunnelluille hiirille, joilta puuttuu EGFR-ekspressio, ilmaantui kehityshäiriöitä liittyen eri elimien, kuten iho, maha-suolikanava ja keuhkot, epiteliaaliseen kypsymättömyyteen. Kun gefitibiniä annosteltiin rotille organogeneesin aikana, sen ei havaittu vaikuttavan sikiönkehitykseen suurimmalla vuorokausiannoksella, 30 mg/kg, mutta kanilla sikiön paino pieneni 20 mg/kg ja sitä suuremmalla vuorokausiannoksella. Kummallakaan lajilla ei havaittu lääkeaineen aiheuttamia sikiön epämuodostumia. Kun lääkeainetta annosteltiin rotille koko raskauden ajan poikimiseen asti, poikasten eloonjääminen aleni 20 mg/kg vuorokausiannoksella.
Kun imettäville rotille annettiin oraalisesti C-14-gefitinibiä 14 vuorokauden ajan poikimisen jälkeen, maidon radioaktiivisuuspitoisuus oli 11–19 kertaa suurempi kuin veren.
Gefitinibillä ei ole havaittu genotoksisia vaikutuksia.
Rotilla tehdyssä kaksi vuotta kestäneessä karsinogeenisuustutkimuksessa todettiin sekä uros- että naarasrotilla pieni, mutta tilastollisesti merkitsevä hepatosellulaaristen adenoomien esiintyvyyden kasvu, sekä naarasrotilla mesenteeristen imusolmukkeiden hemangiosarkoomien esiintyvyyden suureneminen pelkästään suurimmalla annoksella (10 mg/kg/vrk). Hepatosellulaarisia adenoomia havaittiin myös kaksi vuotta kestäneissä karsinogeenisuustutkimuksissa hiirillä, ja niissä osoitettiin pieni löydöksen esiintyvyyden suureneminen uroshiirillä, jotka saivat keskisuuria annoksia, ja sekä uros- että naarashiirillä suurimmalla annoksella. Vaikutus oli tilastollisesti merkitsevä naarashiirillä, mutta ei uroshiirillä. Hiiriin ja rottiin vaikuttamattomat tasot eivät olleet rajattavissa kliinisen altistuksen yhteydessä mitattavista tasoista. Näiden löydösten kliinistä merkitystä ei tiedetä.
In vitro -fototoksisuustutkimuksen tulokset osoittivat, että gefitinibi saattaa olla fototoksinen.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Natriumlauryylisulfaatti
Laktoosimonohydraatti
Mikrokiteinen selluloosa
Povidoni
Kroskarmelloosinatrium
Magnesiumstearaatti
Tabletin päällys
Polyvinyylialkoholi
Makrogoli
Talkki
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
GEFITINIB STADA tabletti, kalvopäällysteinen
250 mg (L:kyllä) 30 fol (160,08 €)
PF-selosteen tieto
Perforoitu tai perforoimaton oPA/Al/PVC-Al-läpipainopakkaus.
Pakkaus sisältää 30 tablettia tai 30 x 1 tabletti. Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Ruskea, pyöreä, kaksoiskupera päällystetty tabletti (halkaisija noin 11 mm), jonka toisella puolella painatus ”G9FB 250”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
GEFITINIB STADA tabletti, kalvopäällysteinen
250 mg 30 fol
- Ylempi erityiskorvaus (100 %). Afatinibi ja gefitinibi: Paikallisesti edenneen tai etäpesäkkeitä lähettäneen ei-pienisoluisen keuhkosyövän hoito erityisin edellytyksin (155).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Afatinibi ja gefitinibi: Paikallisesti edenneen tai etäpesäkkeitä lähettäneen ei-pienisoluisen keuhkosyövän hoito erityisin edellytyksin (341).
ATC-koodi
L01EB01
Valmisteyhteenvedon muuttamispäivämäärä
20.10.2022
Yhteystiedot
PL 1310, Puolikkotie 8, 02230 Espoo (käyntiosoite)
00101 Helsinki
0207 416 888