
CABOMETYX tabletti, kalvopäällysteinen 20 mg, 40 mg, 60 mg
Vaikuttavat aineet ja niiden määrät
CABOMETYX 20 mg:n kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää kabotsantinibi (S)-malaattia määrän, joka vastaa 20 mg:aa kabotsantinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 15,54 mg laktoosia.
CABOMETYX 40 mg:n kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää kabotsantinibi (S)-malaattia määrän, joka vastaa 40 mg:aa kabotsantinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 31,07 mg laktoosia.
CABOMETYX 60 mg:n kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää kabotsantinibi (S)-malaattia määrän, joka vastaa 60 mg:aa kabotsantinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 46,61 mg laktoosia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti.
Kliiniset tiedot
Käyttöaiheet
Munuaissyöpä (RCC)
CABOMETYX on tarkoitettu monoterapiana edenneen munuaissyövän
- ensilinjan hoitoon aikuispotilaille, joiden ennuste on kohtalainen tai huono (ks. kohta Farmakodynamiikka)
- aikuispotilaille aiemman endoteelikasvutekijään (VEGF) kohdistetun hoidon jälkeen (ks. kohta Farmakodynamiikka).
CABOMETYX on tarkoitettu yhdistelmänä nivolumabin kanssa edenneen munuaissyövän ensilinjan hoitoon aikuispotilaille (ks. kohta Farmakodynamiikka).
Maksasyöpä (HCC)
CABOMETYX on tarkoitettu monoterapiana maksasyövän (HCC:n) hoitoon aikuisille, jotka ovat aiemmin saaneet sorafenibihoitoa.
Erilaistunut kilpirauhassyöpä (DTC)
CABOMETYX on tarkoitettu monoterapiana aikuispotilaille, joilla on paikallisesti edennyt tai metastasoitunut, erilaistunut kilpirauhassyöpä ja joiden sairaus on radiojodihoitoon vastaamaton tai joita ei voida hoitaa radiojodilla, kun sairaus on edennyt aiemman systeemisen hoidon aikana tai sen jälkeen.
Neuroendokriiniset kasvaimet (NET)
CABOMETYX on tarkoitettu leikkaukseen soveltumattomien tai metastasoituneiden, hyvin erilaistuneiden haimanulkoisten (epNET) ja haimassa sijaitsevien (pNET) neuroendokriinisten kasvainten hoitoon aikuisille, joiden sairaus on edennyt vähintään yhden muun systeemisen hoidon kuin somatostatiinianalogihoidon jälkeen.
Ehto
Hoito tulee aloittaa syövän hoitoon tarkoitettujen lääkevalmisteiden antamiseen perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
CABOMETYX-hoito on aloitettava syövän hoitoon tarkoitettujen lääkevalmisteiden antamiseen perehtyneen lääkärin valvonnassa.
Annostus
CABOMETYX-tabletit ja kabotsantinibikapselit eivät ole keskenään biologisesti samanarvoisia eivätkä siten keskenään vaihtokelpoisia (ks. kohta Farmakokinetiikka).
CABOMETYX monoterapiana
Munuaissyövän, maksasyövän, erilaistuneen kilpirauhassyövän ja neuroendokriinisten kasvainten hoidossa suositeltu CABOMETYX-annos on 60 mg kerran vuorokaudessa.
Hoitoa on jatkettava, kunnes potilas ei enää kliinisesti hyödy hoidosta tai kunnes potilaalla ilmenee liiallista toksisuutta.
CABOMETYX yhdistelmänä nivolumabin kanssa edenneen munuaissyövän ensilinjan hoitoon
Suositeltu CABOMETYX-annos on 40 mg kerran vuorokaudessa yhdistelmänä nivolumabi-infuusionesteen, liuoksen, kanssa, joka annetaan laskimoon joko 240 mg:n annoksina 2 viikon välein tai 480 mg:n annoksina 4 viikon välein, tai nivolumabi-injektionesteen, liuoksen, kanssa, joka annetaan ihon alle joko 600 mg:n annoksina 2 viikon välein tai 1200 mg:n annoksina 4 viikon välein. Hoitoa pitää jatkaa, kunnes sairaus etenee tai ilmenee toksisuutta, joka ei ole hyväksyttävissä. Nivolumabihoitoa pitää jatkaa, kunnes sairaus etenee tai ilmenee toksisuutta, joka ei ole hyväksyttävissä, tai enintään 24 kuukauden ajan, jos potilaan sairaus ei etene (ks. nivolumabin annostus sen valmisteyhteenvedosta).
Hoitoon tehtävät muutokset
Lääkkeen epäiltyjen haittavaikutusten hoito saattaa edellyttää CABOMETYX-hoidon tilapäistä keskeyttämistä ja/tai annoksen pienentämistä (ks. taulukko 1). Kun annoksen pienentäminen on monoterapiassa tarpeen, suositellaan sen pienentämistä ensin 40 mg:aan vuorokaudessa ja sen jälkeen 20 mg:aan vuorokaudessa.
Annettaessa CABOMETYX yhdistelmänä nivolumabin kanssa CABOMETYX-annos suositellaan pienentämään 20 mg:aan kerran vuorokaudessa ja sen jälkeen 20 mg:aan joka toinen päivä (ks. nivolumabiannoksen muutoksia koskevat suositukset nivolumabin valmisteyhteenvedosta).
Hoidon keskeyttämistä suositellaan CTCAE-kriteerien mukaisen 3. asteen tai sitä pahemman toksisuuden hoitoon sekä sietämättömän 2. asteen toksisuuden hoitoon. Annoksen pienentämistä suositellaan, jos potilaalla esiintyy haittavaikutuksia, jotka jatkuessaan voivat kehittyä vakaviksi tai sietämättömiksi.
Jos potilaalta jää väliin yksi annos, väliin jäänyttä annosta ei saa ottaa, jos seuraavan annoksen ottamiseen on alle 12 tuntia.
Taulukko 1: Suositellut CABOMETYX-annosmuutokset haittavaikutusten yhteydessä
| Haittavaikutus ja vakavuus | Hoitoon tehtävä muutos |
| 1. ja 2. asteiden haittavaikutukset, jotka ovat siedettäviä ja helposti hallittavissa | Annoksen muutos ei yleensä ole tarpeen. Lisää tukihoitoja tarpeen mukaan. |
| 2. asteen haittavaikutukset, jotka ovat sietämättömiä ja joita ei voida hallita pienentämällä annosta tai tukihoidoilla | Keskeytä hoito, kunnes haittavaikutus lievenee asteeseen ≤ 1. Lisää tukihoitoja tarpeen mukaan. Harkitse hoidon aloittamista uudelleen pienemmällä annoksella. |
| 3. asteen haittavaikutukset (paitsi poikkeavat laboratoriolöydökset, jotka eivät ole kliinisesti oleellisia) | Keskeytä hoito, kunnes haittavaikutus lievenee asteeseen ≤ 1. Lisää tukihoitoja tarpeen mukaan. Aloita hoito uudelleen pienemmällä annoksella. |
| 4. asteen haittavaikutukset (paitsi poikkeavat laboratoriolöydökset, jotka eivät ole kliinisesti oleellisia) | Keskeytä hoito. Aloita asianmukaiset hoitotoimenpiteet. Jos haittavaikutus lievenee asteeseen ≤ 1, aloita hoito uudelleen pienemmällä annoksella. Jos haittavaikutus ei häviä, hoito on lopetettava pysyvästi. |
| CABOMETYX-hoitoa yhdistelmänä nivolumabin kanssa saavien munuaissyöpäpotilaiden kohonneet maksaentsyymipitoisuudet | |
| ALAT tai ASAT > 3 kertaa viitearvojen ylärajan (ULN), mutta ≤ 10 kertaa ULN eikä kokonaisbilirubiinipitoisuus samaan aikaan ≥ 2 kertaa ULN | Keskeytä CABOMETYX- ja nivolumabihoito, kunnes nämä haittavaikutukset lievenevät asteeseen ≤ 1 Jos epäillään immuunivälitteistä reaktiota, voidaan harkita kortikosteroidihoitoa (ks. nivolumabin valmisteyhteenveto). Toipumisen jälkeen voidaan harkita hoidon jatkamista vain yhdellä lääkevalmisteella tai hoidon aloittamista uudelleen kummallakin valmisteella peräkkäin. Jos nivolumabihoito aloitetaan uudelleen, ks. nivolumabin valmisteyhteenveto. |
| ALAT tai ASAT > 10 kertaa ULN tai > 3 kertaa ULN ja samaan aikaan kokonaisbilirubiinipitoisuus ≥ 2 kertaa ULN | Lopeta CABOMETYX- ja nivolumabihoito kokonaan. Jos epäillään immuunivälitteistä reaktiota, voidaan harkita kortikosteroidihoitoa (ks. nivolumabin valmisteyhteenveto). |
Huomautus: Toksisuusasteet perustuvat NCI-CTCAE (National Cancer Institute Common Terminology Criteria for Adverse Events) -luokituksen versioon 4.0.
Samanaikaiset lääkevalmisteet
Varovaisuutta on noudatettava, jos potilas saa samanaikaisesti lääkevalmisteita, jotka ovat voimakkaita CYP3A4:n estäjiä. Lisäksi on vältettävä sellaisten lääkevalmisteiden pitkään jatkuvaa samanaikaista käyttöä, jotka ovat voimakkaita CYP3A4:n induktoreja (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Samanaikaiseen hoitoon on harkittava vaihtoehtoista lääkevalmistetta, jolla ei ole CYP3A4:ää estävää tai indusoivaa vaikutusta tai jonka tällainen mahdollinen vaikutus on minimaalinen.
Erityisryhmät
Iäkkäät
Erityistä annoksen muuttamista ei suositella, kun kabotsantinibia annetaan iäkkäille potilaille (≥ 65-vuotiaille).
Etninen tausta
Annoksen muuttaminen etnisen taustan perusteella ei ole tarpeen (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Kabotsantinibia on käytettävä varoen potilaille, joilla on lievä tai kohtalainen munuaisten vajaatoiminta.
Kabotsantinibin käyttö ei ole suositeltavaa, jos potilaalla on vaikea munuaisten vajaatoiminta, koska sen turvallisuutta ja tehoa tämän potilasryhmän hoidossa ei ole varmistettu.
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa potilaille, joilla on lievä maksan vajaatoiminta. Kohtalaista maksan vajaatoimintaa (Child–Pugh-luokka B) sairastavista on vain vähän tietoa, joten annostussuosituksia ei voida antaa. Näiden potilaiden hoidossa suositellaan kokonaisturvallisuuden tarkkaa seurantaa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka). Käyttökokemusta lääkkeestä vaikeaa maksan vajaatoimintaa (Child–Pugh-luokka C) sairastaville ei ole, joten kabotsantinibin käyttö näille potilaille ei ole suositeltavaa (ks. kohta Farmakokinetiikka),
Sydämen vajaatoiminta
Sydämen vajaatoimintaa sairastavista potilaista on vain vähän tietoa, eikä erityisiä annostussuosituksia voida antaa.
Pediatriset potilaat
Kabotsantinibin turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole vielä varmistettu. Tällä hetkellä saatavilla olevat tiedot on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, mutta suosituksia annostuksesta ei voida antaa.
Antotapa
CABOMETYX otetaan suun kautta. Tabletit niellään kokonaisina eikä niitä saa murskata. Potilaita on neuvottava olemaan syömättä vähintään 2 tuntia ennen CABOMETYX-valmisteen ottamista ja 1 tunti sen jälkeen.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Koska useimmat haittavaikutukset ilmenevät hoidon alkuvaiheessa, lääkärin on seurattava potilaan tilaa huolellisesti kahdeksan ensimmäisen hoitoviikon aikana mahdollisten annosmuutosten tarpeen arvioimiseksi. Seuraavia haittavaikutuksia esiintyy yleensä hoidon alkuvaiheessa: hypokalsemia, hypokalemia, trombosytopenia, hypertensio, palmoplantaarinen erytrodysestesia (PPES) ‑oireyhtymä, proteinuria ja ruoansulatuselimistön haittavaikutukset (vatsakipu, limakalvojen tulehdus, ummetus, ripuli, oksentelu).
Epäiltyjen haittavaikutusten hoito voi edellyttää kabotsantinibihoidon keskeyttämistä tilapäisesti tai annoksen pienentämistä (ks. kohta Annostus ja antotapa):
Kliinisissä pivotaalitutkimuksissa, jotka koskivat monoterapiaa munuaissyövän hoidossa (METEOR, CABOSUN), maksasyövän hoidossa (CELESTIAL), kilpirauhassyövän hoidossa (COSMIC‑311) ja neuroendokriinisten kasvainten hoidossa (CABINET), annosta pienennettiin 46–67 %:lla ja hoito keskeytettiin 70–84 %:lla kabotsantinibihoitoa saaneista potilaista haittavaikutuksen vuoksi. Annosta jouduttiin pienentämään kahdesti 9,4–33 %:lla potilaista. Mediaaniaika ensimmäiseen annoksen pienentämiseen oli 38–106 päivää ja ensimmäiseen hoidon keskeytykseen 28–68 päivää.
Kliinisessä tutkimuksessa (CA2099ER), jossa kabotsantinibia annetaan yhdistelmänä nivolumabin kanssa edenneen munuaissyövän ensilinjan hoitoon,kabotsantinibiannosta pienennettiin haittavaikutuksen vuoksi 54,1 %:lla potilaista ja kabotsantinibihoito keskeytettiin haittavaikutuksen vuoksi 73,4 %:lla potilaista. Annosta jouduttiin pienentämään kahdesti 9,4 %:lla potilaista. Mediaaniaika ensimmäiseen annoksen pienentämiseen oli 106 päivää ja ensimmäiseen hoidon keskeytykseen 68 päivää.
Maksatoksisuus
Kabotsantinibihoitoa saaneilla potilailla on usein havaittu poikkeamia maksan toimintakokeissa (kohonnut alaniiniaminotransferaasi [ALAT], aspartaattiaminotransferaasi [ASAT] ja bilirubiini). On suositeltavaa ottaa maksan toimintakokeet (ALAT, ASAT ja bilirubiini) ennen kabotsantinibihoidon aloittamista ja seurata maksan toimintaa tarkkaan hoidon aikana. Jos maksan toimintakoetulokset huononevat ja sen katsotaan johtuvan kabotsantinibihoidosta (ts. jos muuta ilmeistä syytä ei ole), annosta on muutettava taulukossa 1 esitettyjen suositusten mukaisesti (ks. kohta Annostus ja antotapa).
Annettaessa kabotsantinibi yhdistelmänä nivolumabin kanssa asteiden 3 ja 4 kohonneita ALAT- ja ASAT-arvoja on raportoitu yleisemmin kuin edennyttä munuaissyöpää sairastavien potilaiden kabotsantinibimonoterapian yhteydessä (ks. kohta Haittavaikutukset). Maksaentsyymipitoisuuksia pitää seurata ennen hoidon aloittamista ja säännöllisesti koko hoidon ajan. Kumpaakin lääkettä koskevia lääketieteellisiä hoito-ohjeita pitää noudattaa (ks. kohta Annostus ja antotapa ja ks. nivolumabin valmisteyhteenveto). Harvinaisissa tapauksissa on raportoitu sappitiekatoa. Kaikki tapaukset ovat ilmenneet potilailla, jotka ovat saaneet immuuniaktivaation vapauttajia joko ennen kabotsantinibihoitoa tai samanaikaisesti sen kanssa.
Kabotsantinibi poistuu pääasiassa maksan kautta. Kokonaisturvallisuuden tarkkaa seurantaa suositellaan hoidettaessa potilaita, joilla on lievä tai kohtalainen maksan vajaatoiminta (ks. myös kohdat Annostus ja antotapa ja Farmakokinetiikka). Kabotsantinibihoidon yhteydessä maksaenkefalopatiaa ilmeni suhteellisesti enemmän kohtalaista (Child–Pugh-luokka B) maksan vajaatoimintaa sairastavilla potilailla. Kabotsantinibin käyttö ei ole suositeltavaa, jos potilaalla on vaikea maksan vajaatoiminta (Child–Pugh-luokka C, ks. kohta Annostus ja antotapa).
Maksaenkefalopatia
Maksasyöpää koskeneessa tutkimuksessa (CELESTIAL) maksaenkefalopatiaa ilmoitettiin kabotsantinibiryhmässä yleisemmin kuin lumeryhmässä. Kabotsantinibin käytön yhteydessä on ilmennyt ripulia, oksentelua, heikentynyttä ruokahalua ja elektrolyyttiarvojen poikkeavuuksia. Maksasyöpää sairastavilla potilailla, joiden maksan toiminta on heikentynyt, nämä muut kuin maksaan kohdistuvat vaikutukset saattavat olla maksaenkefalopatian laukaisevia tekijöitä. Potilaita on seurattava maksaenkefalopatian oireiden ja löydösten varalta.
Perforaatiot ja fistelit
Kabotsantinibia käytettäessä on havaittu vakavia ruoansulatuskanavan perforaatioita ja fisteleitä, jotka ovat joskus johtaneet kuolemaan. Ennen kabotsantinibihoidon aloittamista on arvioitava huolellisesti potilaat, joilla on tulehduksellinen suolistosairaus (esim. Crohnin tauti, ulseratiivinen koliitti, peritoniitti, divertikuliitti tai appendisiitti), kasvaimen infiltraatio ruoansulatuskanavaan tai komplikaatioita aiemmista vatsaelinkirurgisista toimenpiteistä (varsinkin, jos näihin liittyy viivästynyt tai epätäydellinen paraneminen). Hoidon aloittamisen jälkeen näitä potilaita on seurattava huolellisesti perforaation, fistelin sekä absessin ja sepsiksen oireiden varalta. Pitkään jatkuva tai toistuva ripuli hoidon aikana voi olla riskitekijä anaalifistelin kehittymiselle. Kabotsantinibi on lopetettava, jos potilaalla ilmenee ruoansulatuskanavan perforaatio tai fisteli, jota ei voida hoitaa riittävän tehokkaasti.
Ruoansulatuskanavan häiriöt
Yleisimpiä ruoansulatuskanavan tapahtumia olivat ripuli, pahoinvointi/oksentelu, ruokahalun heikkeneminen ja suutulehdus/suukipu (ks. kohta Haittavaikutukset). Kuivumisen, elektrolyyttihäiriöiden ja painon laskun välttämiseksi on välittömästi aloitettava lääkinnällinen hoito, mukaan lukien tukihoito pahoinvointilääkkeillä, ripulilääkkeillä tai antasideilla. Kabotsantinibihoidon keskeyttämistä, annoksen pienentämistä tai hoidon lopettamista pysyvästi on harkittava, jos merkittävät ruoansulatuskanavan häiriöt jatkuvat tai toistuvat (ks. taulukko 1).
Tromboemboliset tapahtumat
Kabotsantinibia käytettäessä on havaittu laskimoiden tromboembolisia tapahtumia, mukaan lukien keuhkoemboliaa, sekä valtimotromboemboliaa, joka on joskus johtanut kuolemaan. Kabotsantinibia on annettava varoen potilaille, joilla on ollut tällaisia tapahtumia aiemmin tai joilla on niiden riski. Maksasyöpää koskeneessa tutkimuksessa (CELESTIAL) kabotsantinibia saaneilla potilailla ilmoitettiin porttilaskimotromboosia, joka johti yhdessä tapauksessa kuolemaan. Porttilaskimon tromboosin vaara näyttäisi olevan tavanomaista suurempi, jos kasvain on aiemmin levinnyt porttilaskimoon. Kabotsantinibi on lopetettava, jos potilaalle kehittyy akuutti sydäninfarkti tai jokin muu kliinisesti merkittävä tromboembolinen komplikaatio.
CABINET-tutkimuksessa laskimotromboembolian yleisyys kabotsatinibihoitoa saaneilla potilailla oli pNET-kohortissa suurempi (19 %) kuin epNET-kohortissa (3,8 %).
Verenvuoto
Kabotsantinibia käytettäessä on havaittu vaikeaa verenvuotoa, joka on joskus johtanut kuolemaan. Potilaat, joilla on ennen hoidon aloittamista esiintynyt vaikeaa verenvuotoa, on arvioitava huolellisesti ennen kabotsantinibihoidon aloittamista. Kabotsantinibia ei saa antaa potilaille, joilla on vaikeaa verenvuotoa tai joilla on vaikean verenvuodon riski.
Maksasyöpää koskeneessa tutkimuksessa (CELESTIAL) kuolemaan johtaneita verenvuototapahtumia ilmoitettiin kabotsantinibiryhmässä yleisemmin kuin lumeryhmässä. Edennyttä maksasyöpää sairastavassa potilasjoukossa vaikealle verenvuodolle altistavia tekijöitä saattavat olla kasvaimen tunkeutuminen suuriin verisuoniin ja taustalla oleva maksakirroosi, joka on aiheuttanut ruokatorven suonikohjuja, porttilaskimon hypertensiota sekä trombosytopeniaa. CELESTIAL-tutkimuksesta suljettiin pois potilaat, jotka saivat samanaikaisesti hyytymisenestohoitoa tai verihiutaleiden estäjiä. Tutkimuksesta suljettiin pois myös potilaat, joilla oli hoitamattomia tai puutteellisesti hoidettuja suonikohjuja tai suonikohjuja, joihin liittyi verenvuotoa tai suuri verenvuodon riski.
Kabotsantinibin ja nivolumabin yhdistelmän käyttöä edenneen munuaissyövän ensilinjan hoitoon koskeneeseen tutkimukseen (CA2099ER) ei otettu mukaan potilaita, jotka saivat hyytymisenestohoitoa hoitoannoksina.
Aneurysmat ja valtimon dissekaatiot
VEGF-reitin estäjien käyttö potilailla, joilla on kohonnut verenpaine tai joilla ei ole kohonnutta verenpainetta, saattaa edistää aneurysmien ja/tai valtimon dissekaatioiden muodostumista. Tämä riski on arvioitava tarkoin ennen kabotsantinibihoidon aloittamista potilaille, joilla on riskitekijöitä, kuten kohonnut verenpaine tai aikaisempi aneurysma.
Trombosytopenia
Maksasyöpää koskeneessa tutkimuksessa (CELESTIAL), erilaistunutta kilpirauhassyöpää koskeneessa tutkimuksessa (COSMIC‑311) ja neuroendokriinisiä kasvaimia koskeneessa tutkimuksessa (CABINET) ilmoitettiin trombosytopeniaa ja verihiutaleiden määrän pienenemistä. Kabotsantinibihoidon aikana on seurattava verihiutaleiden määrää, ja annosta on muutettava trombosytopenian vaikeusasteen mukaan (ks. taulukko 1).
Haavakomplikaatiot
Kabotsantinibia käytettäessä on havaittu haavakomplikaatioita. Kabotsantinibihoito pitää lopettaa vähintään 28 vuorokautta ennen suunniteltua leikkausta, mukaan lukien hammaskirurgiset tai invasiiviset hammastoimenpiteet. Päätettäessä kabotsantinibihoidon jatkamisesta leikkauksen jälkeen on perusteena käytettävä kliinistä arviota haavan riittävästä paranemisesta. Kabotsantinibi on lopetettava, jos potilaan haavan paranemiseen liittyy lääketieteellisiä toimenpiteitä edellyttäviä komplikaatioita.
Hypertensio
Kabotsantinibia käytettäessä on havaittu verenpaineen kohoamista, mukaan lukien hypertensiivisiä kriisejä. Verenpaine on saatava hyvään hoitotasapainoon ennen kabotsantinibihoidon aloittamista. Kabotsantinibihoidon aloittamisen jälkeen verenpainetta on seurattava varhaisessa vaiheessa ja säännöllisesti, ja sitä on hoidettava tarpeen mukaan asianmukaisella verenpainelääkityksellä. Jos verenpaine on jatkuvasti koholla verenpainelääkityksestä huolimatta, kabotsantinibihoito on keskeytettävä, kunnes verenpaine on hallinnassa, minkä jälkeen kabotsantinibihoitoa voidaan jatkaa pienemmällä annoksella. Kabotsantinibi on lopetettava, jos hypertensio on vaikea ja jatkuu verenpainelääkityksestä ja kabotsantinibiannoksen pienentämisestä huolimatta. Hypertensiivisen kriisin ilmetessä kabotsantinibihoito on lopetettava.
Sydämen vajaatoiminta
Kabotsantinibin käyttöön on liittynyt suurentunut sydämen vajaatoiminnan riski. Kabotsantinibin yleiset haittavaikutukset (esim. kohonnut verenpaine, kilpirauhasen vajaatoiminta ja valtimoveritulppatapahtumat) voivat lisätä tätä riskiä ja siten johtaa sydämen vajaatoimintaan. Potilaita pitää seurata hoidon aikana sydämen vajaatoiminnan oireiden ja löydösten varalta. Nämä haittavaikutukset on hoidettava viipymättä. Tarvittaessa on harkittava hoidon tauottamista ja/tai annoksen muuttamista (ks. kohta Annostus ja antotapa) ja hoito tyrosiinikinaasi-inhibiittorilla (TKI) on lopetettava potilailla, joille ilmaantuu vaikea-asteinen sydämen vajaatoiminta.
Osteonekroosi
Kabotsantinibia käytettäessä on havaittu leuan osteonekroositapahtumia. Suu pitää tarkastaa ennen kabotsantinibin aloittamista ja säännöllisesti kabotsantinibihoidon aikana. Potilaita tulee neuvoa suun hygienian suhteen. Kabotsantinibihoidosta tulee pidättäytyä mahdollisuuksien mukaan vähintään 28 vuorokautta ennen suunniteltua hammaskirurgista tai invasiivista hammastoimenpidettä.
Varovaisuutta pitää noudattaa potilailla, jotka saavat leuan osteonekroosiin liittyviä lääkeaineita, kuten bisfosfonaatteja. Kabotsantinibi keskeytetään potilailla, joilla esiintyy leuan osteonekroosia.
Palmoplantaarinen erytrodysestesia
Kabotsantinibia käytettäessä joillakin potilailla on havaittu palmoplantaarinen erytrodysestesia ‑oireyhtymää (kämmeniin ja jalkapohjiin liittyvä oireyhtymä, PPES). Jos PPES on vaikea, kabotsantinibihoidon keskeyttämistä on harkittava. Kun PPES lievenee 1. asteeseen, kabotsantinibi on aloitettava uudelleen pienemmällä annoksella.
Proteinuria
Kabotsantinibia käytettäessä on havaittu proteinuriaa. Virtsan valkuaispitoisuutta on seurattava säännöllisesti kabotsantinibihoidon aikana. Kabotsantinibi on lopetettava, jos potilaalle kehittyy nefroottinen oireyhtymä.
Posteriorinen reversiibeli enkefalopatiaoireyhtymä
Kabotsantinibia käytettäessä on havaittu posteriorista reversiibeliä enkefalopatiaoireyhtymää (PRES). Oireyhtymän mahdollisuus on otettava huomioon, jos potilaalla on useita oireita, kuten kouristuskohtauksia, päänsärkyä, näköhäiriöitä, sekavuutta tai henkisen vireystilan muutoksia. Kabotsantinibihoito on lopetettava, jos potilaalla on PRES.
QT-ajan pidentyminen
Kabotsantinibia on käytettävä varoen potilaille, joilla on aiemmin esiintynyt QT-ajan pidentymistä, jotka käyttävät rytmihäiriölääkkeitä tai joilla on merkityksellinen sydänsairaus, bradykardia tai elektrolyyttihäiriö. Kabotsantinibia käytettäessä on harkittava ajoittaista hoidonaikaista EKG:n ja elektrolyyttipitoisuuksien (seerumin kalsium, kalium ja magnesium) seurantaa.
Kilpirauhasen toimintahäiriö
Kaikille potilaille suositellaan tekemään ennen hoidon aloittamista kilpirauhasen toimintaa selvittävät laboratoriomääritykset. Potilaat, joilla on ennestään hypotyreoosi tai hypertyreoosi, pitää hoitaa tavanomaisen hoitokäytännön mukaisesti ennen kabotsantinibihoidon aloittamista. Kaikkia potilaita pitää tarkkailla kabotsantinibihoidon aikana kilpirauhasen toimintahäiriöiden oireiden ja löydösten havaitsemiseksi. Kilpirauhasen toimintaa pitää seurata säännöllisin väliajoin koko kabotsantinibihoidon ajan. Potilaat, joille kehittyy kilpirauhasen toimintahäiriö, pitää hoitaa tavanomaisen hoitokäytännön mukaisesti.
Biokemiallisten laboratoriokokeiden tulosten poikkeavuudet
Kabotsantinibin käyttöön on liittynyt elektrolyyttiarvojen poikkeavuuksien (mukaan lukien hypo- ja hyperkalemian, hypomagnesemian, hypokalsemian, hyponatremian) tavanomaista suurempi ilmaantuvuus. Kabotsantinibin käytön yhteydessä esiintynyt hypokalsemia on ollut yleisempää ja vaikeampiasteista (mukana 3. ja 4. asteen tapauksia) kilpirauhassyöpää sairastavilla potilailla kuin muilla syöpäpotilailla. Kabotsantinibihoidon aikana on suositeltavaa seurata biokemiallisia parametreja ja tarvittaessa aloittaa asianmukainen korvaushoito tavanomaisen hoitokäytännön mukaisesti. Maksasyöpää sairastavilla potilailla on ilmennyt maksaenkefalopatiatapauksia, joiden voidaan katsoa johtuneen elektrolyyttitasapainon häiriöistä. Kabotsantinibihoidon keskeyttämistä, annoksen pienentämistä tai hoidon lopettamista pysyvästi on harkittava, jos merkittävät poikkeavuudet jatkuvat tai toistuvat (ks. taulukko 1).
CYP3A4-induktorit ja -estäjät
Kabotsantinibi on CYP3A4:n substraatti. Ketokonatsoli on voimakas CYP3A4:n estäjä, ja kabotsantinibin samanaikainen anto ketokonatsolin kanssa suurensi kabotsantinibin plasma-altistusta. Varovaisuutta on noudatettava, kun kabotsantinibia annetaan sellaisten lääkeaineiden kanssa, jotka ovat voimakkaita CYP3A4:n estäjiä. Rifampisiini on voimakas CYP3A4:n induktori, ja kabotsantinibin anto samanaikaisesti rifampisiinin kanssa pienensi kabotsantinibin plasma-altistusta. Siksi kabotsantinibia käytettäessä on vältettävä sellaisten lääkeaineiden pitkään jatkuvaa käyttöä, jotka ovat voimakkaita CYP3A4:n induktoreja (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
P-glykoproteiinin substraatit
Kabotsantinibi oli P-glykoproteiinin (P‑gp:n) kuljetustoiminnan estäjä (IC50 = 7,0 μM), mutta ei substraatti, kaksisuuntaisessa analyysijärjestelmässä, jossa käytettiin MDCK-MDR1-soluja. Siksi kabotsantinibi saattaa mahdollisesti suurentaa samanaikaisesti annettujen P‑gp:n substraattien pitoisuuksia plasmassa. Potilaita on varoitettava P‑gp:n substraattien (esim. feksofenadiinin, aliskireenin, ambrisentaanin, dabigatraanieteksilaatin, digoksiinin, kolkisiinin, maravirokin, posakonatsolin, ranolatsiinin, saksagliptiinin, sitagliptiinin, talinololin, tolvaptaanin) käytöstä kabotsantinibihoidon aikana (ks. kohta Yhteisvaikutukset).
MRP2-estäjät
MRP2-estäjien käyttö saattaa suurentaa kabotsantinibin pitoisuuksia plasmassa. Siksi MRP2-estäjien (esim. siklosporiinin, efavirentsin, emtrisitabiinin) samanaikaiseen käyttöön on suhtauduttava varoen (ks. kohta Yhteisvaikutukset).
Apuaineet
Laktoosi
Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkevalmistetta.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutus kabotsantinibiin
CYP3A4-estäjät ja -induktorit
Kun terveille vapaaehtoisille annettiin ketokonatsolia, joka on voimakas CYP3A4:n estäjä (400 mg vuorokaudessa 27 päivän ajan), kabotsantinibin puhdistuma väheni (29 %) ja kabotsantinibin kerta-annoksen plasma-altistus (AUC) suureni 38 %. Siksi voimakkaiden CYP3A4:n estäjien (esim. ritonaviirin, itrakonatsolin, erytromysiinin, klaritromysiinin, greippimehun) samanaikaiseen käyttöön kabotsantinibin kanssa on suhtauduttava varoen.
Kun terveille vapaaehtoisille annettiin rifampisiinia, joka on voimakas CYP3A4:n induktori (600 mg vuorokaudessa 31 päivän ajan), kabotsantinibin puhdistuma suureni (4,3-kertaisesti) ja kabotsantinibin kerta-annoksen plasma-altistus (AUC) pieneni 77 %. Siksi voimakkaiden CYP3A4:n induktorien (esim. fenytoiinin, karbamatsepiinin, rifampisiinin, fenobarbitaalin tai mäkikuismaa [Hypericum perforatum] sisältävien rohdosvalmisteiden) pitkään jatkuvaa käyttöä kabotsantinibin kanssa on vältettävä.
Mahan pH-arvoa muuttavat aineet
Protonipumpun estäjän (PPI:n) esomepratsolin (40 mg vuorokaudessa 6 päivän ajan) annolla terveille vapaaehtoisille samanaikaisesti yhden 100 mg:n kabotsantinibiannoksen kanssa ei ollut kliinisesti merkitsevää vaikutusta kabotsantinibin plasma-altistukseen (AUC-arvoon). Annoksen muuttaminen ei ole aiheellista, kun potilaille annetaan mahan pH-arvoa muuttavia aineita (esim. protonipumpun estäjiä, H2-reseptorisalpaajia tai antasideja) samanaikaisesti kabotsantinibin kanssa.
MRP2-estäjät
In vitro ‑tiedot osoittavat, että kabotsantinibi on MRP2:n substraatti. Siksi MRP2-estäjien antaminen saattaa suurentaa kabotsantinibin pitoisuutta plasmassa.
Sappisuoloja sitovat aineet
Sappisuoloja sitovilla aineilla, kuten kolestyramiinilla ja kolesevelaamilla (Cholestagel), voi olla yhteisvaikutuksia kabotsantinibin kanssa, ja ne voivat vaikuttaa sen imeytymiseen (tai takaisinimeytymiseen), mikä voi johtaa vähäisempään altistukseen (ks. kohta Farmakokinetiikka). Näiden mahdollisten yhteisvaikutusten kliininen merkitys ei ole tiedossa.
Kabotsantinibin vaikutus muihin lääkevalmisteisiin
Kabotsantinibin vaikutusta steroidiryhmän ehkäisyvalmisteiden farmakokinetiikkaan ei ole tutkittu. Koska ehkäisevän vaikutuksen pysyvyyttä ei voida taata, suositellaan lisäehkäisyn, kuten estemenetelmän, käyttöä.
Kabotsantinibin vaikutusta varfariinin farmakokinetiikkaan ei ole tutkittu. Yhteisvaikutus varfariinin kanssa voi olla mahdollinen. Tällaista yhdistelmää käytettäessä on seurattava INR-arvoja.
P-glykoproteiinin substraatit
Kabotsantinibi oli P‑gp:n kuljetustoiminnan estäjä (IC50 = 7,0 μM) mutta ei substraatti kaksisuuntaisessa analyysijärjestelmässä, jossa käytettiin MDCK-MDR1-soluja. Siksi kabotsantinibi saattaa mahdollisesti suurentaa samanaikaisesti annettujen P‑gp:n substraattien pitoisuuksia plasmassa. Potilaita on varoitettava P‑gp:n substraattien (esim. feksofenadiinin, aliskireenin, ambrisentaanin, dabigatraanieteksilaatin, digoksiinin, kolkisiinin, maravirokin, posakonatsolin, ranolatsiinin, saksagliptiinin, sitagliptiinin, talinololin, tolvaptaanin) käytöstä kabotsantinibihoidon aikana.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy miehille ja naisille
Naisia, jotka voivat tulla raskaaksi, on neuvottava välttämään raskaaksi tuloa kabotsantinibihoidon aikana. Kabotsantinibihoitoa saavien miespotilaiden naispuolisia kumppaneita on neuvottava välttämään raskaaksi tuloa. Mies- ja naispotilaiden sekä heidän kumppaneidensa on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään neljä kuukautta hoidon päättymisen jälkeen. Koska ehkäisypillereitä ei ehkä voida pitää ”tehokkaana ehkäisymenetelmänä”, niitä on käytettävä jonkun toisen menetelmän, kuten estemenetelmän, kanssa (ks. kohta Yhteisvaikutukset).
Raskaus
Kabotsantinibia käyttävistä raskaana olevista naisista ei ole tutkimuksia. Eläinkokeissa on havaittu alkio- ja sikiövaikutuksia sekä teratogeenisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmisille ei tunneta. Kabotsantinibia ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä kabotsantinibihoitoa.
Imetys
Ei tiedetä, erittyvätkö kabotsantinibi ja/tai sen metaboliitit ihmisen rintamaitoon. Lapseen kohdistuvan mahdollisen vahingollisen vaikutuksen takia äidin on lopetettava imetys kabotsantinibihoidon ajaksi ja vähintään neljän kuukauden ajaksi hoidon päättymisen jälkeen.
Hedelmällisyys
Kabotsantinibin vaikutuksesta ihmisen hedelmällisyyteen ei ole olemassa tietoja. Ei-kliinisten turvallisuutta koskevien löydösten perusteella kabotsantinibihoito saattaa vaarantaa miesten ja naisten hedelmällisyyden (ks. kohta Prekliiniset tiedot turvallisuudesta). Sekä miehiä että naisia on kehotettava hakeutumaan hedelmällisyysneuvontaan ja harkitsemaan mahdollisuuksia hedelmällisyyden säilyttämiseen ennen hoidon aloittamista.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Kabotsantinibilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Kabotsantinibiin on yhdistetty haittavaikutuksia, kuten väsymystä ja heikkoutta, ja siksi suositellaan varovaisuutta ajettaessa tai koneita käytettäessä.
Haittavaikutukset
Kabotsantinibi monoterapiana
Turvallisuusprofiilin yhteenveto
Munuaissyöpää sairastavilla yleisimmät vakavat haittavaikutukset (esiintyvyys ≥ 1 %) ovat keuhkokuume, vatsakipu, ripuli, pahoinvointi, hypertensio, embolia, hyponatremia, keuhkoembolia, oksentelu, kuivuminen, väsymys, voimattomuus, ruokahalun heikkeneminen, syvä laskimotromboosi, huimaus, hypomagnesemia ja palmoplantaarinen erytrodysestesia ˗oireyhtymä (PPES).
Maksasyöpää sairastavilla yleisimmät vakavat haittavaikutukset (esiintyvyys ≥ 1 %) ovat maksaenkefalopatia, voimattomuus, väsymys, palmoplantaarinen erytrodysestesia ‑oireyhtymä, ripuli, hyponatremia, oksentelu, vatsakipu ja trombosytopenia.
Erilaistunutta kilpirauhassyöpää sairastavilla yleisimmät vakavat haittavaikutukset (esiintyvyys ≥ 1 %) ovat ripuli, pleuraeffuusio, pneumoniitti, keuhkoembolia, hypertensio, anemia, syvä laskimotukos, hypokalsemia, leuan osteonekroosi, kipu, palmoplantaarinen erytrodysestesia ‑oireyhtymä, oksentelu ja munuaisten vajaatoiminta.
Potilailla, joilla on neuroendokriinisiä kasvaimia, yleisimmät vakavat haittavaikutukset (esiintyvyys ≥ 1 %) ovat hypertensio, väsymys, keuhkoembolia, oksentelu, ripuli, pahoinvointi ja embolia.
Munuaissyöpää, maksasyöpää ja erilaistunutta kilpirauhassyöpää sairastavilla potilailla sekä potilailla, joilla oli neuroendokriinisiä kasvaimia, yleisimmin esiintyneitä kaikenasteisia (esiintyivät vähintään 25 %:lla potilaista) haittavaikutuksia olivat ripuli, väsymys, pahoinvointi, ruokahalun heikkeneminen, palmoplantaarinen erytrodysestesia ‑oireyhtymä ja hypertensio.
Luettelo haittavaikutuksista taulukkomuodossa
Kabotsantinibimonoterapiaa saaneiden munuaissyöpää, maksasyöpää tai erilaistunutta kilpirauhassyöpää sairastavien potilaiden sekä potilaiden, joilla oli neuroendokriinisiä kasvaimia, (n = 1 355) yhdistetyissä tiedoissa raportoidut haittavaikutukset tai kabotsantinibin markkinoille tulon jälkeen raportoidut haittavaikutukset luetellaan taulukossa 2 MedDRA-elinjärjestelmä- ja yleisyysluokituksen mukaan. Esiintymistiheydet perustuvat kaikkiin asteisiin ja ne on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutusten vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2: Kliinisissä tutkimuksissa tai valmisteen markkinoille tulon jälkeen kabotsantinibia monoterapiana saaneilla potilailla raportoidut haittavaikutukset
| Infektiot | |
| Yleinen | absessi, keuhkokuume |
| Veri ja imukudos | |
| Hyvin yleinen | anemia, trombosytopenia |
| Yleinen | neutropenia, lymfopenia |
| Umpieritys | |
| Hyvin yleinen | hypotyreoosi* |
| Aineenvaihdunta ja ravitsemus | |
| Hyvin yleinen | ruokahalun heikkeneminen, hypomagnesemia, hypokalemia, hypoalbuminemia, hypokalsemia |
| Yleinen | kuivuminen, hypofosfatemia, hyponatremia, hyperkalemia, hyperbilirubinemia, hyperglykemia, hypoglykemia |
| Hermosto | |
| Hyvin yleinen | makuhäiriö, päänsärky, huimaus |
| Yleinen | perifeerinen neuropatiaa |
| Melko harvinainen | kouristukset, aivoverisuonitapahtuma, posteriorinen reversiibeli enkefalopatiaoireyhtymä |
| Kuulo ja tasapainoelin | |
| Yleinen | tinnitus |
| Sydän | |
| Melko harvinainen | akuutti sydäninfarkti, sydämen vajaatoiminta |
| Verisuonisto | |
| Hyvin yleinen | hypertensio, verenvuotob* |
| Yleinen | laskimotromboosic, hypotensio, embolia |
| Melko harvinainen | hypertensiivinen kriisi, valtimotromboosi, valtimoveritulppa |
| Tuntematon | aneurysmat ja valtimon dissekaatiot |
| Hengityselimet, rintakehä ja välikarsina | |
| Hyvin yleinen | äänen käheys, hengenahdistus, yskä |
| Yleinen | keuhkoembolia, allerginen nuha |
| Melko harvinainen | ilmarinta |
| Ruoansulatuselimistö | |
| Hyvin yleinen | ripuli*, pahoinvointi, oksentelu, suutulehdus, ummetus, vatsakipu, dyspepsia |
| Yleinen | ruoansulatuskanavan perforaatio*g, haimatulehdus, fisteli*, gastroesofageaalinen refluksitauti, peräpukamat, suukipu, suun kuivuminen, nielemisvaikeus, ilmavaivat |
| Melko harvinainen | kielikipu |
| Maksa ja sappi | |
| Yleinen | maksaenkefalopatia* |
| Melko harvinainen | kolestaattinen maksatulehdus |
| Iho ja ihonalainen kudos | |
| Hyvin yleinen | palmoplantaarinen erytrodysestesia ˗oireyhtymä, ihottumaf |
| Yleinen | kutina, hiustenlähtö, kuiva iho, hiusten värimuutos, hyperkeratoosi, eryteema |
| Tuntematon | ihovaskuliitti |
| Luusto, lihakset ja sidekudos | |
| Hyvin yleinen | raajakipu, nivelkipu |
| Yleinen | lihaskouristukset |
| Melko harvinainen | leuan osteonekroosi |
| Munuaiset ja virtsatiet | |
| Yleinen | valkuaisvirtsaisuus |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Hyvin yleinen | uupumus, limakalvojen tulehdus, voimattomuus, perifeerinen edeema |
| Tutkimuksetd | |
| Hyvin yleinen | painon lasku, seerumin ALAT- ja ASAT-arvojen nousu, veren AFOS-arvon nousu |
| Yleinen | GGT-arvojen nousu, veren kreatiniiniarvon nousu, amylaasiarvojen nousu, lipaasiarvojen nousu, veren kolesteroliarvojen nousu, veren triglyseridiarvojen nousu, veren valkosolujen määrän pieneneminen |
| Vammat, myrkytykset ja hoitokomplikaatiot | |
| Melko harvinainen | haavakomplikaatiote |
* Ks. tarkemmat tiedot kohdasta Haittavaikutukset Valikoitujen haittavaikutusten kuvaus.
a Mukaan lukien polyneuropatia; perifeerinen neuropatia on ollut pääasiassa sensorista
b Mukaan lukien nenäverenvuoto yleisimmin ilmoitettuna haittavaikutuksena
c Kaikentyyppiset laskimotromboosit, mukaan lukien syvä laskimotromboosi
d Perustuu raportoituihin haittavaikutuksiin
e Heikentynyt paraneminen, leikkausalueen komplikaatiot ja haavan aukeaminen
f Ihottuma on yhdistelmätermi, joka käsittää dermatiitin, aknen kaltaisen dermatiitin, rakkulaisen dermatiitin, kesivän ihottuman, punoittavan ihottuman, follikulaarisen ihottuman, makulaarisen ihottuman, makulopapulaarisen ihottuman, papulaarisen ihottuman, kutisevan ihottuman ja lääkeihottuman
g Kuolemaan johtaneita tapauksia on raportoitu
Kabotsantinibi yhdistelmänä nivolumabin kanssa edenneen munuaissyövän ensilinjan hoitoon
Turvallisuusprofiilin yhteenveto
Annettaessa kabotsantinibia yhdistelmänä nivolumabin kanssa, ks. nivolumabin valmisteyhteenveto ennen hoidon aloittamista. Lisätietoja nivolumabimonoterapian turvallisuusprofiilista, ks. nivolumabin valmisteyhteenveto.
Tietoaineistossa, joka koskee munuaissyövän hoitoa kabotsantinibiannosten 40 mg kerran päivässä ja nivolumabiannosten 240 mg:n joka toinen viikko yhdistelmällä (n = 320) ja jossa seuranta-aika on vähintään 16 kuukautta, yleisimmät vakavat haittavaikutukset (ilmaantuvuus ≥ 1 %) ovat ripuli, pneumoniitti, keuhkoembolia, keuhkokuume, hyponatremia, kuume, lisämunuaisten vajaatoiminta, oksentelu, kuivuminen.
Yleisimmät haittavaikutukset (≥ 25 %) olivat ripuli, uupumus, palmoplantaarinen erytrodysestesia ˗oireyhtymä (PPES), suutulehdus, luuston ja lihasten kipu, hypertensio, ihottuma, hypotyreoosi, heikentynyt ruokahalu, pahoinvointi, vatsakipu. Valtaosa haittavaikutuksista oli lieviä tai kohtalaisia (aste 1 tai 2).
Luettelo haittavaikutuksista taulukkomuodossa
Kabotsantinibin ja nivolumabin yhdistelmällä tehdyissä kliinisissä tutkimuksissa tunnistetut haittavaikutukset luetellaan taulukossa 3 MedDRA-elinjärjestelmä- ja yleisyysluokituksen mukaan. Esiintymistiheydet perustuvat kaikkiin asteisiin ja ne on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutusten vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3: Kabotsantinibin ja nivolumabin yhdistelmän käytössä raportoidut haittavaikutukset
| Infektiot | |
| Hyvin yleinen | ylähengitysteiden infektio |
| Yleinen | keuhkokuume |
| Veri ja imukudos | |
| Yleinen | eosinofilia |
| Immuunijärjestelmä | |
| Yleinen | yliherkkyys(mukaan lukien anafylaktinen reaktio) |
| Melko harvinainen | infuusioon liittyvä yliherkkyysreaktio |
| Umpieritys | |
| Hyvin yleinen | hypotyreoosi, hypertyreoosi |
| Yleinen | lisämunuaisten vajaatoiminta |
| Melko harvinainen | aivolisäkkeen tulehdus, kilpirauhastulehdus |
| Aineenvaihdunta ja ravitsemus | |
| Hyvin yleinen | heikentynyt ruokahalu |
| Yleinen | kuivuminen |
| Hermosto | |
| Hyvin yleinen | makuhäiriö, huimaus, päänsärky |
| Yleinen | perifeerinen neuropatia |
| Melko harvinainen | autoimmuunienkefaliitti, Guillain-Barrén oireyhtymä, myasteeninen oireyhtymä |
| Kuulo ja tasapainoelin | |
| Yleinen | tinnitus |
| Silmät | |
| Yleinen | kuivat silmät, näön sumeneminen |
| Melko harvinainen | uveiitti |
| Sydän | |
| Yleinen | eteisvärinä, takykardia |
| Melko harvinainen | sydänlihastulehdus |
| Verisuonisto | |
| Hyvin yleinen | hypertensio |
| Yleinen | tromboosia |
| Melko harvinainen | valtimoveritulppa |
| Hengityselimet, rintakehä ja välikarsina | |
| Hyvin yleinen | äänen käheys, hengenahdistus, yskä |
| Yleinen | pneumoniitti, keuhkoembolia, nenäverenvuoto, pleuraeffuusio |
| Melko harvinainen | ilmarinta |
| Ruoansulatuselimistö | |
| Hyvin yleinen | ripuli, oksentelu, pahoinvointi, ummetus, suutulehdus, vatsakipu, dyspepsia |
| Yleinen | koliitti, gastriitti, suukipu, suun kuivuminen, peräpukamat |
| Melko harvinainen | haimatulehdus, ohutsuolen perforaatiob, kielikipu |
| Maksa ja sappi | |
| Yleinen | hepatiitti |
| Tuntematon | sappitiekatoc |
| Iho ja ihonalainen kudos | |
| Hyvin yleinen | palmoplantaarinen erytrodysestesia ‑oireyhtymä, ihottumad, kutina |
| Yleinen | hiustenlähtö, kuiva iho, eryteema, hiusten värimuutos |
| Melko harvinainen | psoriaasi, urtikaria |
| Tuntematon | ihovaskuliitti |
| Luusto, lihakset ja sidekudos | |
| Hyvin yleinen | luuston ja lihasten kipue, nivelkipu, lihasspasmi |
| Yleinen | niveltulehdus |
| Melko harvinainen | myopatia, leuan osteonekroosi, fisteli |
| Munuaiset ja virtsatiet | |
| Hyvin yleinen | valkuaisvirtsaisuus |
| Yleinen | munuaisten vajaatoiminta, akuutti munuaisvaurio |
| Melko harvinainen | munuaistulehdus |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Hyvin yleinen | uupumus, kuume, turvotus |
| Yleinen | kipu, kipu rintakehässä |
| Tutkimuksetf | |
| Hyvin yleinen | ALAT-arvon nousu, ASAT-arvon nousu, hypofosfatemia, hypokalsemia, hypomagnesemia, hyponatremia, hyperglykemia, lymfopenia, veren AFOS-arvojen nousu, lipaasiarvojen nousu, amylaasiarvojen nousu, trombosytopenia, veren kreatiniiniarvon nousu, anemia, leukopenia, hyperkalemia, neutropenia, hyperkalsemia, hypoglykemia, hypokalemia, kokonaisbilirubiiniarvon nousu, hypermagnesemia, hypernatremia, painon lasku |
| Yleinen | veren kolesteroliarvojen nousu, hypertriglyseridemia |
Taulukossa 3 esitetyt haittavaikutusten esiintyvyydet eivät liity täysin yksinomaan kabotsantinibiin, vaan niihin saattavat osaltaan vaikuttaa perussairaus tai käyttö yhdistelmänä nivolumabin kanssa.
a Tromboosi yhdistelmätermi, joka käsittää porttilaskimotromboosin, keuhkolaskimotromboosin, keuhkotromboosin, aorttatromboosin, valtimotromboosin, syvän laskimotromboosin, lantion laskimotromboosin, alaonttolaskimon tromboosin, laskimotromboosin, raajan laskimotromboosin
b Kuolemaan johtaneita tapauksia on raportoitu
c Potilailla, jotka olivat saaneet aiempaa hoitoa immuuniaktivaation vapauttajilla tai saivat sitä samanaikaisesti
d Ihottuma on yhdistelmätermi, joka käsittää dermatiitin, aknen kaltaisen dermatiitin, rakkulaisen dermatiitin, kesivän ihottuman, punoittavan ihottuman, follikulaarisen ihottuman, makulaarisen ihottuman, makulopapulaarisen ihottuman, papulaarisen ihottuman, kutisevan ihottuman ja lääkeihottuman
e Luuston ja lihasten kipu on yhdistelmätermi, joka käsittää selkäkivun, luukivun, luusto- ja lihaskivun rintakehässä, luuston ja lihasten epämukavat tuntemukset, lihassäryn, niskakivun, raajakivun, selkärankakivun
f Laboratoriotermien yleisyydet kuvastavat niiden potilaiden osuutta, joilla laboratoriomääritysten arvot huononivat lähtötilanteesta, lukuun ottamatta painon laskua, veren kolesteroliarvon nousua ja hypertriglyseridemiaa
Valittujen haittavaikutusten kuvaus
Seuraavia haittavaikutuksia koskevat tiedot perustuvat potilaisiin, jotka saivat pivotaalitutkimuksissa monoterapiana CABOMETYX-lääkevalmistetta 60 mg kerran vuorokaudessa suun kautta joko munuaissyöpään aiemmin annetun endoteelikasvutekijään (VEGF) kohdistetun hoidon jälkeen tai aiemmin hoitamattoman munuaissyövän hoitoon, maksasyövän hoitoon aikaisemman systeemisen hoidon jälkeen, erilaistuneen kilpirauhassyövän hoitoon, kun potilaan sairaus oli radiojodihoitoon vastaamaton tai sitä ei voitu hoitaa radiojodilla ja erilaistunut kilpirauhassyöpä oli edennyt aiemman systeemisen hoidon aikana tai sen jälkeen, etenevään neuroendokriiniseen kasvaimeen annetun aiemman systeemisen hoidon jälkeen tai potilaisiin, jotka saivat CABOMETYX-lääkevalmistetta 40 mg kerran vuorokaudessa suun kautta yhdistelmänä nivolumabin kanssa edenneen munuaissyövän ensilinjan hoitoon (ks. kohta Farmakodynamiikka).
Ruoansulatuskanavan perforaatio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Munuaissyöpää koskeneessa tutkimuksessa (METEOR) ruoansulatuskanavan perforaatioita raportoitiin 0,9 prosentilla (3/331) kabotsantinibia saaneista munuaissyöpää sairastavista potilaista. Näiden tapahtumien vaikeusaste oli 2. tai 3. aste ja mediaaniaika tapahtuman alkamiseen 10,0 viikkoa.
Aiemmin hoitamatonta munuaissyöpää koskeneessa tutkimuksessa (CABOSUN) ruoansulatuskanavan perforaatioita raportoitiin 2,6 prosentilla (2/78) kabotsantinibia saaneista potilaista. Tapahtumien vaikeusaste oli 4. tai 5. aste.
Maksasyöpää koskeneessa tutkimuksessa (CELESTIAL) ruoansulatuskanavan perforaatioita raportoitiin 0,9 prosentilla (4/467) kabotsantinibia saaneista potilaista. Kaikkien tapahtumien vaikeusaste oli 3. tai 4. aste, ja mediaaniaika tapahtuman alkamiseen oli 5,9 viikkoa. Erilaistunutta kilpirauhassyöpää koskeneessa tutkimuksessa (COSMIC‑311) asteen 4 ruoansulatuskanavan perforaatio raportoitiin yhdellä (0,6 %) kabotsantinibia saaneista potilaista, ja se tapahtui 14 viikkoa hoidon jälkeen.
Neuroendokriinisiä kasvaimia koskeneessa tutkimuksessa (CABINET) ruoansulatuskanavan perforaatioita raportoitiin 1,3 prosentilla (3/227) kabotsantinibia saaneista potilaista. Näiden tapahtumien vaikeusaste oli 3., 4. tai 5. aste, ja mediaaniaika tapahtuman alkamiseen oli 21,6 viikkoa.
Nivolumabin kanssa yhdistelmänä edenneen munuaissyövän ensilinjan hoitoon käytettäessä (CA2099ER) ruoansulatuskanavan perforaatioita ilmaantui 1,3 %:lle (4/320) hoitoa saaneista potilaista. Yksi tapahtuma oli astetta 3, kaksi tapahtumaa oli astetta 4 ja yksi tapahtuma oli astetta 5 (johti kuolemaan).
Kabotsantinibin kliinisessä ohjelmassa on esiintynyt kuolemaan johtaneita perforaatioita.
Maksaenkefalopatia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Maksasyöpää koskeneessa tutkimuksessa (CELESTIAL) maksaenkefalopatiaa (maksaenkefalopatia, enkefalopatia, hyperammoneeminen enkefalopatia) raportoitiin 5,6 prosentilla (26/467) kabotsantinibia saaneista potilaista. Tapahtumista 2,8 prosentissa vaikeusaste oli 3. tai 4. aste, ja yhden tapahtuman (0,2 %) vaikeusaste oli 5. Mediaaniaika tapahtuman alkamiseen oli 5,9 viikkoa. Neuroendokriinisiä kasvaimia koskeneessa tutkimuksessa (CABINET) maksaenkefalopatiaa raportoitiin 0,9 prosentilla (2/227) kabotsantinibia saaneista potilaista. Tapahtumia oli yksi (0,4 %), ja sen vaikeusaste oli 3. aste. Mediaaniaika tapahtuman alkamiseen oli 14,3 viikkoa.
Munuaissyöpää sairastaville tehdyissä tutkimuksissa (METEOR, CABOSUN ja CA2099ER) ja erilaistunutta kilpirauhassyöpää sairastaville tehdyssä tutkimuksessa (COSMIC‑311) maksaenkefalopatiaa ei ilmoitettu.
Ripuli (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Munuaissyöpää koskeneessa tutkimuksessa (METEOR) ripulia ilmoitettiin esiintyneen 74 prosentilla (245/331) kabotsantinibia saaneista munuaissyöpäpotilaista. Tapahtumista 11 prosentissa vaikeusaste oli 3. Mediaaniaika tapahtuman alkamiseen oli 4,9 viikkoa.
Kliinisessä tutkimuksessa (CABOSUN), jossa oli mukana aiemmin hoitamattomia munuaissyöpäpotilaita, ripulia ilmoitettiin esiintyneen 73 prosentilla (57/78) kabotsantinibia saaneista munuaissyöpäpotilaista. Tapahtumista 10 prosentissa vaikeusaste oli 3. tai 4. aste.
Maksasyöpää sairastavilla potilailla tehdyssä tutkimuksessa (CELESTIAL) ripulia ilmoitettiin esiintyneen 54 prosentilla (251/467) kabotsantinibia saaneista potilaista. Tapahtumista 9,9 prosentissa vaikeusaste oli 3. tai 4. aste. Kaikkien tapahtumien alkamiseen kulunut mediaaniaika oli 4,1 viikkoa. Tutkittavia, joiden annosta muutettiin ripulin vuoksi, oli 84/467 (18 %); tutkittavia, joiden hoito keskeytettiin ripulin vuoksi, oli 69/467 (15 %) ja tutkittavia, joiden hoito lopetettiin ripulin vuoksi, oli 5/467 (1 %).
Erilaistunutta kilpirauhassyöpää koskeneessa tutkimuksessa (COSMIC‑311) ripulia ilmoitettiin esiintyneen 62 %:lla kabotsantinibihoitoa saaneista potilaista (105/170). Tapahtumista 7,6 prosentissa vaikeusaste oli 3–4. Tutkittavia, joiden annosta pienennettiin ripulin vuoksi, oli 24/170 (14 %), ja tutkittavia, joiden hoito keskeytettiin ripulin vuoksi, oli 36/170 (21 %).
Neuroendokriinisiä kasvaimia koskeneessa tutkimuksessa (CABINET) ripulia ilmoitettiin esiintyneen 63 prosentilla (144/227) kabotsantinibia saaneista potilaista. Tapahtumista 8,4 prosentissa vaikeusaste oli 3; vaikeusasteen 4 tapahtumia ei ollut. Mediaaniaika vaikeusasteen 3 tapahtuman alkamiseen oli 5,1 viikkoa.
Nivolumabin kanssa yhdistelmänä edenneen munuaissyövän ensilinjan hoitoon käytettäessä (CA2099ER) ripulia raportoitiin ilmaantuneen 64,7 %:lle (207/320) hoitoa saaneista potilaista; asteen 3-4 tapahtumien ilmaantuvuus oli 8,4 % (27/320). Kaikkien tapahtumien alkamiseen kuluneen ajan mediaani oli 12,9 viikkoa. Annosten antamista myöhennettiin tai annosta pienennettiin 26,3 %:lla (84/320) potilaista ja hoito lopetettiin 2,2 %:lla (7/320) potilaista, joilla oli ripulia.
Fistelit (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Munuaissyöpää koskeneessa tutkimuksessa (METEOR) fisteleitä ilmoitettiin esiintyneen 1,2 prosentilla (4/331) kabotsantinibia saaneista potilaista. Tämä luku sisältää anaalifistelit, joita esiintyi 0,6 prosentilla (2/331) kabotsantinibia saaneista potilaista. Yhden tapahtuman vaikeusaste oli 3. aste ja muiden 2. aste. Mediaaniaika tapahtuman alkamiseen oli 30,3 viikkoa.
Aiemmin hoitamatonta munuaissyöpää koskeneessa tutkimuksessa (CABOSUN) ei raportoitu fisteleitä.
Maksasyöpää koskeneessa tutkimuksessa (CELESTIAL) fisteleitä ilmoitettiin esiintyneen 1,5 prosentilla (7/467) maksasyöpää sairastaneista potilaista. Mediaaniaika tapahtuman alkamiseen oli 14 viikkoa.
Erilaistunutta kilpirauhassyöpää koskeneessa tutkimuksessa (COSMIC‑311) fisteleitä (kaksi anaalifisteliä ja yksi faryngeaalinen fisteli) ilmoitettiin 1,8 prosentilla kabotsantinibia saaneista potilaista (3/170).
Neuroendokriinisiä kasvaimia koskeneessa tutkimuksessa (CABINET) fisteleitä (kaksi anaalifisteliä ja yksi sappifisteli) ilmoitettiin 1,3 prosentilla (3/227) kabotsantinibia saaneista potilaista. Anaalifistelien vaikeusasteet olivat 1. aste ja 3. aste, sappifistelin vaikeusaste oli 2. aste. Mediaaniaika tapahtuman alkamiseen oli 19,3 viikkoa.
Nivolumabin kanssa yhdistelmänä edenneen munuaissyövän ensilinjan hoitoon käytettäessä (CA2099ER) fisteleitä raportoitiin ilmenneen 0,9 %:lle (3/320) hoitoa saaneista potilaista, ja fisteleiden vaikeusaste oli aste 1.
Kabotsantinibin kliinisessä ohjelmassa on esiintynyt kuolemaan johtaneita fisteleitä.
Verenvuoto (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Munuaissyöpää koskeneessa tutkimuksessa (METEOR) vaikea-asteisten verenvuototapahtumien ilmaantuvuus (aste ≥ 3) oli 2,1 % (7/331) kabotsantinibia saaneiden munuaissyöpää sairastavien potilaiden ryhmässä. Mediaaniaika tapahtuman alkamiseen oli 20,9 viikkoa.
Aiemmin hoitamatonta munuaissyöpää koskeneessa tutkimuksessa (CABOSUN) vaikea-asteisten verenvuototapahtumien (aste ≥ 3) ilmaantuvuus oli 5,1 % (4/78) kabotsantinibia saaneiden munuaissyöpää sairastavien potilaiden ryhmässä.
Maksasyöpää koskeneessa tutkimuksessa (CELESTIAL) vaikea-asteisten verenvuototapahtumien (aste ≥ 3) ilmaantuvuus oli 7,3 % (34/467) kabotsantinibia saaneiden potilaiden ryhmässä. Mediaaniaika tapahtuman alkamiseen oli 9,1 viikkoa.
Erilaistunutta kilpirauhassyöpää koskeneessa tutkimuksessa (COSMIC‑311) vaikea-asteisten verenvuototapahtumien (aste ≥ 3) ilmaantuvuus oli 2,4 % (4/170) kabotsantinibia saaneiden potilaiden ryhmässä. Mediaaniaika tapahtuman alkamiseen oli 11,5 viikkoa.
Neuroendokriinisiä kasvaimia koskeneessa tutkimuksessa (CABINET) vaikea-asteisten verenvuototapahtumien (aste ≥ 3) ilmaantuvuus oli 1,8 % (4/227) kabotsantinibia saaneiden potilaiden ryhmässä. Mediaaniaika tapahtuman alkamiseen oli 14,1 viikkoa.
Nivolumabin kanssa yhdistelmänä edenneen munuaissyövän ensilinjan hoitoon käytettäessä (CA2099ER) asteen ≥ 3 verenvuotoja ilmaantui 1,9 %:lle (6/320) hoitoa saaneista potilaista.
Kabotsantinibin kliinisessä ohjelmassa on esiintynyt kuolemaan johtaneita verenvuotoja.
Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
METEOR-, CABOSUN-, CA2099ER- ja CELESTIAL-tutkimuksissa ei raportoitu yhtään PRES-tapausta, mutta erilaistunutta kilpirauhassyöpää koskeneessa tutkimuksessa (COSMIC‑311) ja neuroendokriinisiä kasvaimia koskeneessa tutkimuksessa (CABINET) raportoitiin kummassakin PRES-tapaus yhdellä potilaalla. Muissa kliinisissä tutkimuksissa PRES-tapauksia on raportoitu harvoin (2/4872 potilasta; 0,04 %).
Maksaentsyymiarvon nousu yhdistettäessä kabotsantinibi nivolumabiin munuaissyövän hoidossa
Kliinisessä tutkimuksessa aiemmin hoitamatonta munuaissyöpää sairastavat potilaat saivat kabotsantinibia yhdistelmänä nivolumabin kanssa. Asteiden 3 ja 4 ALAT-arvon nousun (10,1 %) sekä ASAT-arvon nousun (8,2 %) ilmaantuvuuden havaittiin olevan tällöin suurempi verrattuna edennyttä munuaissyöpää sairastaviin kabotsantinibimonoterapiaa saaviin potilaisiin (METEOR-tutkimuksessa ALAT-arvon nousu 3,6 % ja ASAT-arvon nousu 3,3 %). Asteen ≥ 2 ALAT- tai ASAT-arvon nousuun kuluneen ajan mediaani oli 10,1 viikkoa (vaihteluväli: 2–106,6 viikkoa; n = 85). 91 %:lla potilaista, joilla todettiin asteen ≥ 2 ALAT- tai ASAT-arvon nousu, kohonnut arvo korjautui asteeseen 0–1, mihin kuluneen ajan mediaani oli 2,3 viikkoa (vaihteluväli: 0,4–108,1 viikkoa).
Niistä 45 potilaasta, joilla todettiin asteen ≥ 2 ALAT- tai ASAT-arvon nousu ja jotka altistettiin uudelleen joko pelkästään kabotsantinibille (n = 10) tai nivolumabille (n = 10) tai näille kummallekin (n = 25), asteen ≥ 2 ALAT- tai ASAT-arvon nousun havaittiin uusiutuneen neljällä kabotsantinibia saaneella potilaalla, kolmella nivolumabia saaneella potilaalla ja kahdeksalla sekä kabotsantinibia että nivolumabia saaneella potilaalla.
Hypotyreoosi
Munuaissyöpää koskeneessa tutkimuksessa (METEOR) hypotyreoosin ilmaantuvuus oli 21 % (68/331).
Tutkimuksessa, jossa oli mukana aiemmin hoitamattomia munuaissyöpäpotilaita (CABOSUN), hypotyreoosin ilmaantuvuus kabotsantinibihoitoa saaneilla munuaissyöpäpotilailla oli 23 % (18/78).
Maksasyöpää koskeneessa tutkimuksessa (CELESTIAL) hypotyreoosin ilmaantuvuus kabotsantinibihoitoa saaneilla potilailla oli 8,1 % (38/467) ja asteen 3 tapahtumien ilmaantuvuus oli 0,4 % (2/467).
Erilaistunutta kilpirauhassyöpää koskeneessa tutkimuksessa (COSMIC‑311) hypotyreoosin ilmaantuvuus oli 2,4 % (4/170). Kaikkien tapahtumien vaikeusaste oli 1–2, eivätkä ne vaatineet hoidon muuttamista tai keskeyttämistä.
Neuroendokriinisiä kasvaimia koskeneessa tutkimuksessa (CABINET) hypotyreoosin ilmaantuvuus kabotsantinibihoitoa saaneilla potilailla oli 26 % (59/227). Kaikkien tapahtumien vaikeusaste oli 1–2.
Nivolumabin kanssa yhdistelmänä edenneen munuaissyövän ensilinjan hoitoon käytettäessä (CA2099ER) hypotyreoosi ilmaantui 35,6 %:lle (114/320) hoitoa saaneista potilaista.
Pediatriset potilaat (ks. kohta Farmakodynamiikka)
ADVL1211-tutkimus oli kabotsantinibia koskenut suppea annoseskalaatiotutkimus pediatrisilla ja nuorilla potilailla, joilla oli toistuvia tai hoitoon reagoimattomia kiinteitä kasvaimia, mukaan lukien keskushermoston kasvaimia. Tutkimuksen turvallisuutta koskeneeseen potilasjoukkoon (N = 39) kuuluneiden kaikkien annosryhmien kaikilla tutkittavilla havaittiin seuraavia tapahtumia yleisemmin kuin aikuisilla: suurentunut aspartaattiaminotransferaasipitoisuus (ASAT) (hyvin yleinen, 76,9 %), suurentunut alaniiniaminotransferaasipitoisuus (ALAT) (hyvin yleinen, 71,8 %), pienentynyt lymfosyyttimäärä (hyvin yleinen, 48,7 %), pienentynyt neutrofiilimäärä (hyvin yleinen, 35,9 %) ja suurentunut lipaasipitoisuus (hyvin yleinen, 33,3 %). Näiden preferred term ‑termien lisääntynyt yleisyys koskee kaikkia vaikeusasteita sekä näiden haittavaikutusten 3./4. astetta. Raportoidut haittatapahtumat ovat laadultaan aikuispotilailla tunnetun kabotsantinibin turvallisuusprofiilin mukaisia. Tutkittavien pieni lukumäärä ei kuitenkaan mahdollista lopullisia arvioita kehityssuunnista ja esiintyvyyksistä eikä jatkovertailua kabotsantinibin tunnetun turvallisuusprofiilin kanssa.
Kabotsantinibia koskenut ADVL1622-tutkimus tehtiin lapsilla ja nuorilla aikuisilla seuraavien kiinteiden kasvainten mukaisesti ositettuna: Ewingin sarkooma, rabdomyosarkooma, muu pehmytkudossarkooma kuin rabdomyosarkooma (NRSTS), osteosarkooma, Wilmsin kasvain tai muita harvinaisia kiinteitä kasvaimia (ei-tilastollinen kohortti). Tutkimuksen kaikissa ositteissa kabotsantinibihoitoa saaneiden lasten ja nuorten aikuisten turvallisuusprofiili oli verrannollinen kabotsantinibihoitoa saaneiden aikuisten turvallisuusprofiilin kanssa.
Lapsilla, joiden kasvulevyt olivat kabotsantinibihoidon aikana avoinna, on havaittu kasvulevyjen laajenemista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kabotsantinibin yliannostukseen ei ole spesifistä hoitoa, eikä yliannostuksen mahdollisia oireita ole määritetty.
Jos yliannostusta epäillään, kabotsantinibin anto on heti lopetettava ja aloitettava tukihoito. Aineenvaihduntaan liittyviä kliinisiä laboratorioparametreja on seurattava vähintään viikoittain tai sopivaksi katsotulla aikavälillä mahdollisten muutostrendien arvioimiseksi. Yliannostukseen liittyviä haittavaikutuksia on hoidettava oireenmukaisesti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: solunsalpaajat, proteiinikinaasin estäjät, ATC-koodi: L01EX07.
Vaikutusmekanismi
Kabotsantinibi on pieni molekyyli ja se estää useita reseptorityrosiinikinaaseja (RTK), jotka liittyvät tuumorin kasvuun ja angiogeneesiin, patologiseen luun uudelleenmuotoutumiseen, lääkeresistenssiin ja syövän metastaattiseen etenemiseen. Kabotsantinibin kykyä estää useita kinaaseja arvioitiin, ja sen todettiin olevan MET-reseptorien (hepatosyyttikasvutekijän reseptoriproteiinin) ja endoteelikasvutekijän (VEGF) reseptorien estäjä. Kabotsantinibi estää myös muita tyrosiinikinaaseja, mukaan lukien GAS6-reseptori (AXL), RET, ROS1, TYRO3, MER, kantasolutekijän reseptori (KIT), TRKB, Fms:n kaltainen tyrosiinikinaasi 3 (FLT3) ja TIE-2.
Farmakodynaamiset vaikutukset
Kabotsantinibi esti annoksesta riippuvaisesti tuumorin kasvua, aiheutti tuumorin regressiota ja/tai esti etäpesäkkeiden muodostumista useissa prekliinisissä tuumorimalleissa.
Kardiologinen elektrofysiologia
Medullaarista kilpirauhassyöpää sairastaville potilaille tehdyssä kontrolloidussa kliinisessä tutkimuksessa havaittiin Friderician menetelmällä korjatun QT-ajan (QTcF) 10–15 ms:n piteneminen lähtötasosta 29. päivänä (mutta ei 1. päivänä) kabotsantinibihoidon (140 mg kerran vuorokaudessa) aloittamisen jälkeen. Tähän vaikutukseen ei liittynyt sydämen aaltomuodon morfologian muutoksia tai uusia rytmejä. Tässä tutkimuksessa tai munuaissyöpää tai maksasyöpää sairastaville tehdyissä tutkimuksissa tai neuroendokriinisiä kasvaimia koskeneessa tutkimuksessa (annos 60 mg) ei yhdenkään kabotsantinibihoitoa saaneen potilaan vahvistettu QTcF-aika ollut > 500 ms.
Kliininen teho ja turvallisuus
Munuaissyöpä
Satunnaistettu tutkimus munuaissyöpää sairastavilla potilailla, jotka ovat aiemmin saaneet endoteelikasvutekijään (VEGF) kohdistettua hoitoa (METEOR)
CABOMETYX-valmisteen turvallisuutta ja tehoa munuaissyövän hoitoon aiemmin annetun endoteelikasvutekijään (VEGF) kohdistetun hoidon jälkeen arvioitiin satunnaistetussa avoimessa 3. vaiheen monikeskustutkimuksessa (METEOR). Edennyttä munuaissyöpää (jossa oli kirkassolukomponentti) sairastavat potilaat (N = 658), jotka olivat aiemmin saaneet vähintään yhtä VEGF-reseptorin tyrosiinikinaasiestäjää (VEGFR TKI), satunnaistettiin (1:1) kabotsantinibiryhmään (N = 330) ja everolimuusiryhmään (N = 328). Potilaat olivat saattaneet saada aiempaa hoitoa sytokiineilla tai vasta-aineilla, joiden kohteena on VEGF, ohjelmoituun solukuolemaan vaikuttava PD-1-reseptori tai sen ligandit. Tutkimukseen otettiin potilaita, jotka olivat saaneet hoitoa aivometastaaseihin. Sokkoutettu riippumaton radiologinen arviointiryhmä arvioi etenemisvapaan elinajan (PFS), ja ensisijaisessa analyysissa oli mukana 375 ensimmäistä satunnaistettua tutkittavaa. Tehon toissijaisia päätetapahtumia olivat objektiivinen vasteosuus (ORR) ja kokonaiselossaoloaika (OS). Tuumorit arvioitiin 8 viikon välein ensimmäisen 12 kuukauden ajan ja sen jälkeen 12 viikon välein.
Lähtötason demografiset ja tautitiedot olivat samankaltaiset kabotsantinibi- ja everolimuusihaaroissa. Suurin osa potilaista oli miehiä (75 %), ja iän mediaani oli 62 vuotta. Potilaista 71 % sai aiemmin vain yhtä VEGFR TKI ‑hoitoa ja 41 % sai sunitinibia ainoana aiempana VEGFR TKI ‑hoitona. Memorial Sloan Kettering Cancer Center (MSKCC) ‑keskuksen ennustepisteiden mukaan 46 % potilaista kuului hyvän ennusteen ryhmään (0 riskitekijää), 42 % kohtalaisen ennusteen ryhmään (1 riskitekijä) ja 13 % huonon ennusteen ryhmään (2 tai 3 riskitekijää). Potilaista 54 prosentilla tauti oli metastasoinut kolmeen tai useampaan elimeen, kuten keuhkoihin (63 %), imusolmukkeisiin (62 %), maksaan (29 %) ja luustoon (22 %). Hoidon mediaanikesto oli 7,6 kuukautta (0,3–20,5) kabotsantinibiryhmän potilailla ja 4,4 kuukautta (0,21–18,9) everolimuusiryhmässä.
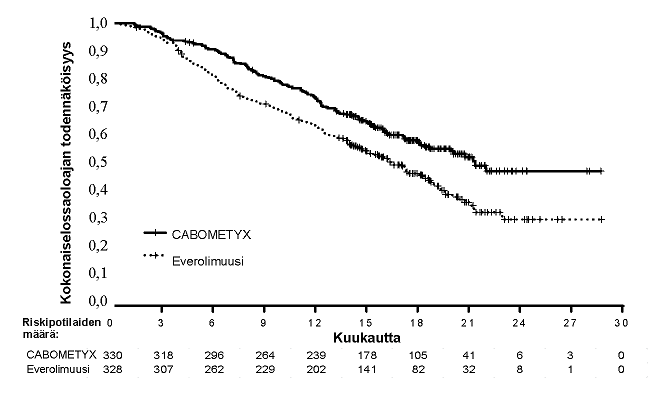
Kabotsantinibiryhmässä osoitettiin tilastollisesti merkitsevä etenemisvapaan elinajan paraneminen verrattuna everolimuusiryhmään (kuva 1, taulukko 4). Etenemisvapaan elinajan (PFS) analyysin kanssa samanaikaisesti toteutetussa suunnitellussa kokonaiselossaoloajan (OS) välianalyysissa ei saavutettu tilastollisen merkitsevyyden väliraja-arvoa (202 tapahtumaa, riskisuhde= 0,68 [0,51; 0,90], p = 0,006). Tämän jälkeisissä suunnittelemattomissa OS-välianalyyseissa osoitettiin tilastollisesti merkitsevä paraneminen kabotsantinibiryhmään satunnaistetuilla potilailla verrattuna everolimuusiryhmän potilaisiin (320 tapahtumaa, mediaani 21,4 kk vs. 16,5 kk; riskisuhde = 0,66 [0,53; 0,83], p = 0,0003; kuva 2). Seuranta-analyysin (deskriptiivinen) OS-tulokset 430 tapahtumasta olivat vertailukelpoiset.
Myös hoitoaikeen mukaisen potilasryhmän eksploratiivisten PFS- ja OS-analyysien tulokset ovat yhdenmukaisesti paremmat kabotsantinibilla kuin everolimuusilla erilaisissa alaryhmissä, jotka perustuvat ikään (< 65 vs. ≥ 65), sukupuoleen, MSKCC-riskiryhmään (hyvä, kohtalainen, huono), ECOG-luokitukseen (0 vs. 1), aikaan diagnoosista satunnaistamiseen (< 1 vuosi vs. ≥ 1 vuosi), tuumorin MET-luokitukseen (suuri vs. pieni vs. tuntematon), luuston metastaaseihin (ei ole vs. on), sisäelimistön metastaaseihin (ei ole vs. on), luuston ja sisäelimistön metastaaseihin (ei ole vs. on), aiempien VEGFR-TKI-hoitojen määrään (1 vs. ≥ 2), ensimmäisen VEGFR-TKI-hoidon kestoon (≤ 6 kk vs. > 6 kk).
Objektiivisten vasteosuuksien yhteenveto on taulukossa 5.
Kuva 1. Riippumattoman radiologisen arviointiryhmän arvioimaa etenemisvapaata elinaikaa munuaissyöpää sairastavilla tutkittavilla aiemman endoteelikasvutekijään (VEGF) kohdistetun hoidon jälkeen kuvaava Kaplan-Meier-käyrä (ensimmäiset 375 satunnaistettua tutkittavaa) (METEOR)
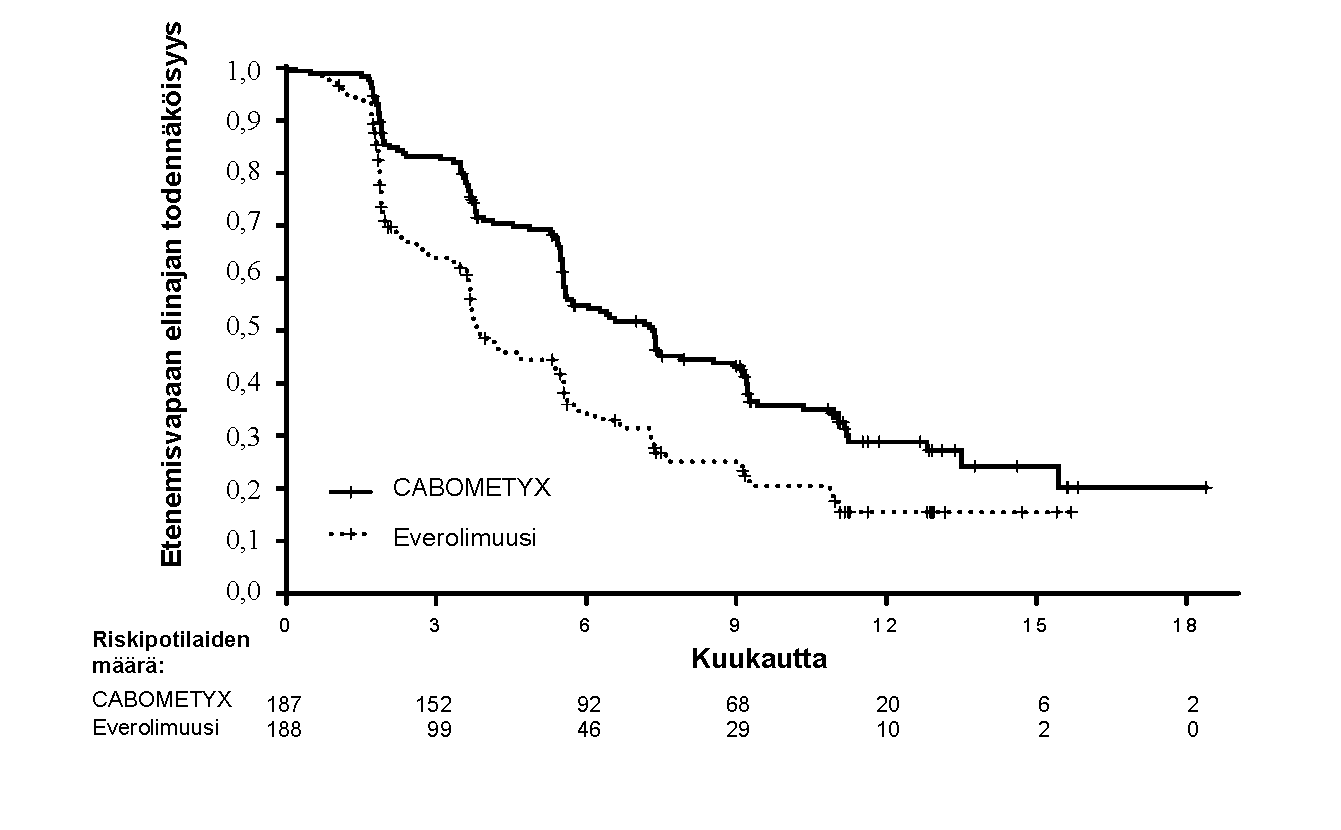
Taulukko 4. Yhteenveto riippumattoman radiologisen arviointiryhmän arvioimista etenemisvapaista elinajoista munuaissyöpää sairastavilla tutkittavilla aiemman endoteelikasvutekijään (VEGF) kohdistetun hoidon jälkeen (METEOR)
| Ensisijaisen PFS-analyysin populaatio | Lähtöryhmien muk. populaatio (ITT) | |||
| Päätetapahtuma | CABOMETYX | Everolimuusi | CABOMETYX | Everolimuusi |
| N = 187 | N = 188 | N = 330 | N = 328 | |
| Mediaani PFS (95 %:n luottamusväli), kk | 7,4 (5,6; 9,1) | 3,8 (3,7; 5,4) | 7,4 (6,6; 9,1) | 3,9 (3,7; 5,1) |
| Riskisuhde (95 %:n luottamusväli), p‑arvo1 | 0,58 (0,45; 0,74), p < 0,0001 | 0,51 (0,41; 0,62), p < 0,0001 | ||
1 stratifioidun log-rank-testin arvo
Kuva 2. Kokonaiselossaoloaikaa munuaissyöpää sairastavien tutkittavien aiemman endoteelikasvutekijään (VEGF) kohdistetun hoidon jälkeen kuvaava Kaplan-Meier-käyrä (METEOR)
Taulukko 5. Yhteenveto ORR-löydöksistä munuaissyöpää sairastavilla tutkittavilla aiemman endoteelikasvutekijään (VEGF) kohdistetun hoidon jälkeen, riippumattoman radiologisen arviointiryhmän (IRC) ja tutkijan arviot
| Ensisijaisen analyysin ORR, ITT-populaatio, IRC | ORR, tutkijan analyysi, ITT-populaatio | |||
| Päätetapahtuma | CABOMETYX | Everolimuusi | CABOMETYX | Everolimuusi |
| N = 330 | N = 328 | N = 330 | N = 328 | |
| ORR (vain osittaiset vasteet) (95 %:n luottamusväli) | 17 % (13 %, 22 %) | 3 % (2 %, 6 %) | 24 % (19 %, 29 %) | 4 % (2 %, 7 %) |
| p‑arvo1 | p < 0,0001 | p < 0,0001 | ||
| Osittainen vaste | 17 % | 3 % | 24 % | 4 % |
| Mediaaniaika, kk (95 %:n luottamusväli) | 1,91 (1,6; 11,0) | 2,14 (1,9; 9,2) | 1,91 (1,3; 9,8) | 3,50 (1,8; 5,6) |
| Stabiili tauti (SD) parhaana vasteena | 65 % | 62 % | 63 % | 63 % |
| Progressiivinen tauti (PD) parhaana vasteena | 12 % | 27 % | 9 % | 27 % |
1 khiin neliö -testi
Satunnaistettu tutkimus aiemmin hoitamattomilla munuaissyöpää sairastavilla potilailla (CABOSUN)
CABOMETYX-valmisteen turvallisuutta ja tehoa aiemmin hoitamattoman munuaissyövän hoitoon arvioitiin satunnaistetussa avoimessa monikeskustutkimuksessa (CABOSUN). Aiemmin hoitamatonta, paikallisesti edennyttä tai metastasoitunutta munuaissyöpää (jossa oli kirkassolukomponentti) sairastavat potilaat (N = 157) satunnaistettiin (1:1) kabotsantinibiryhmään (N = 79) ja sunitinibiryhmään (N = 78). Potilailla piti olla kohtalaisen tai huonon ennusteen tauti, joka oli määritelty International Metastatic RCC Database Consortiumin (IMDC) riskiryhmäluokituksen mukaan. Potilaat ositettiin IMDC-riskiryhmän ja luustometastaasien (kyllä/ei) mukaan. Noin 75 %:lle potilaista oli tehty munuaisenpoisto ennen hoidon aloittamista.
Kohtalaisen ennusteen taudissa oli yksi tai kaksi seuraavista riskitekijöistä, kun taas huonon ennusteen taudissa oli kolme tai useampia riskitekijöitä: aika diagnoosista munuaissyövän systeemiseen hoitoon < 1 vuosi, hemoglobiinipitoisuus < normaaliarvon alarajan (lower limit of normal, LLN), korjattu kalsiumpitoisuus > normaaliarvon ylärajan (upper limit of normal, ULN), Karnofskyn suorituspisteet < 80 %, neutrofiilimäärä > normaaliarvon ylärajan ja trombosyyttimäärä > normaaliarvon ylärajan.
Ensisijainen päätetapahtuma oli etenemisvapaa elinaika (PFS). Toissijaisia päätetapahtumia olivat objektiivinen vasteosuus (ORR) ja kokonaiselossaoloaika (OS). Tuumorit arvioitiin 12 viikon välein.
Lähtötason demografiset ja tautitiedot olivat samankaltaiset kabotsantinibi- ja sunitinibihaaroissa. Suurin osa potilaista oli miehiä (78 %), ja iän mediaani oli 62 vuotta. Potilaat jakautuivat IMDC-riskiryhmiin siten, että 81 prosentilla oli kohtalainen ennuste (1–2 riskitekijää) ja 19 prosentilla oli huono ennuste (≥ 3 riskitekijää). Suurimmalla osalla potilaista (87 %) ECOG-luokitus oli 0 tai 1; 13 prosentilla ECOG-luokitus oli 2. Kolmellakymmenelläkuudella prosentilla (36 %) potilaista oli luustometastaaseja.
Kabotsantinibiryhmässä osoitettiin sokkoutetun riippumattoman radiologisen arviointiryhmän arvion perusteella tilastollisesti merkitsevä etenemisvapaan elinajan paraneminen verrattuna sunitinibiryhmään (kuva 3 ja taulukko 6). Tutkijoiden arvioista tehdyn analyysin ja riippumattoman radiologisen arviointiryhmän arvioiden analyysin etenemisvapaata elinaikaa koskevat tulokset olivat yhdenmukaiset.
Kabotsantinibihoidon vaikutus potilaiden sekä MET-positiivisiin että MET-negatiivisiin tuumoreihin oli suotuisa verrattuna sunitinibiin, ja hoidon aktiivisuus oli MET-positiivisiin tuumoreihin suurempi kuin MET-negatiivisiin tuumoreihin (riskisuhde = 0,32 [0,16; 0,63] [MET-positiiviset] vs. 0,67 [0,37; 1,23] [MET-negatiiviset]).
Kabotsantinibihoidossa potilaiden elinaika oli yleensä pidempi verrattuna sunitinibihoitoon (taulukko 6). Tutkimuksella ei ollut osoitusvoimaa OS-analyysin suhteen, ja tiedot ovat keskeneräisiä.
Yhteenveto objektiivista vasteosuutta (ORR) koskevista löydöksistä on taulukossa 6.
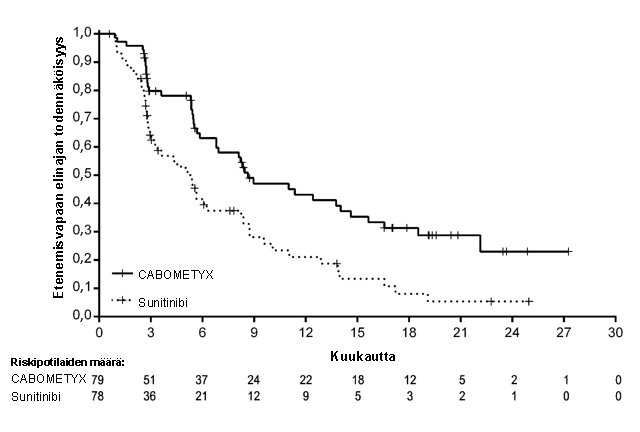
Kuva 3. Kaplan-Meierin käyrä etenemisvapaista elinajoista aiemmin hoitamatonta munuaissyöpää sairastavilla tutkittavilla, riippumattoman radiologisen arviointiryhmän arvio
Taulukko 6. Tehoa koskevat tulokset aiemmin hoitamatonta munuaissyöpää sairastavilla tutkittavilla (hoitoaikeen mukainen potilasjoukko [ITT], CABOSUN)
CABOMETYX (N = 79) | Sunitinibi (N = 78) | |
| Etenemisvapaa elinaika (PFS), riippumattoman radiologisen arviointiryhmän arvio a | ||
| Mediaani PFS (95 %:n luottamusväli), kk | 8,6 (6,2; 14,0) | 5,3 (3,0; 8,2) |
| Riskisuhde (95 %:n luottamusväli); ositettub,c | 0,48 (0,32, 0,73) | |
| Kaksitahoisen log-rank-testin p-arvo: ositettub | p = 0,0005 | |
| Etenemisvapaa elinaika (PFS), tutkijan arvio | ||
| Mediaani PFS (95 %:n luottamusväli), kk | 8,3 (6,5; 12,4) | 5,4 (3,4; 8,2) |
| Riskisuhde (95 %:n luottamusväli); ositettub,c | 0,56 (0,37; 0,83) | |
| Kaksitahoisen log-rank-testin p-arvo: ositettub | p = 0,0042 | |
| Kokonaiselossaoloaika (OS) | ||
| Mediaani OS (95 %:n luottamusväli), kk | 30,3 (14,6; ei arvioitavissa) | 21,0 (16,3; 27,0) |
| Riskisuhde (95 %:n luottamusväli); ositettu b,c | 0,74 (0,47, 1,14) | |
| Objektiivinen vasteosuus (ORR), riippumattoman radiologisen arviointiryhmän arvion (%) | ||
| Täydellinen vaste | 0 | 0 |
| Osittainen vaste | 16 (20) | 7 (9) |
| ORR (vain osittainen vaste) | 16 (20) | 7 (9) |
| Stabiili tauti | 43 (54) | 30 (38) |
| Progressiivinen tauti | 14 (18) | 23 (29) |
| Objektiivinen vasteosuus (ORR), tutkijan arvion (%) | ||
| Täydellinen vaste | 1 (1) | 0 |
| Osittainen vaste | 25 (32) | 9 (12) |
| ORR (vain osittainen vaste) | 26 (33) | 9 (12) |
| Stabiili tauti | 34 (43) | 29 (37) |
| Progressiivinen tauti | 14 (18) | 19 (24) |
aEU:n sensoroinnin mukaan
b IxRS:n mukaisia ositustekijöitä ovat IMDC-riskiluokat (kohtalainen ennuste, huono ennuste ja luustometastaasit [kyllä, ei])
c Arvioitu IxRS:n mukaisilla ositustekijöillä korjatulla Coxin suhteellisen vaaran regressiomallilla. Riskin suhde < 1 osoittaa etenemisvapaan elinajan olevan kabotsantinibin eduksi.
Satunnaistettu 3. vaiheen tutkimus kabotsantinibin ja nivolumabin yhdistelmän vertailusta sunitinibiin (CA2099ER)
Satunnaistetussa 3. vaiheen avoimessa tutkimuksessa (CA2099ER) arvioitiin kabotsantinibiannoksen 40 mg suun kautta kerran vuorokaudessa ja nivolumabiannoksen 240 mg laskimoon kahden viikon välein yhdistelmän turvallisuutta ja tehoa edenneen/metastasoituneen munuaissyövän ensilinjan hoitoon. Tutkimuksessa oli mukana (18-vuotiaita tai vanhempia) potilaita, jotka sairastivat edennyttä tai metastasoitunutta munuaissyöpää, johon liittyi kirkassolukomponentti. Tutkimuksessa mukana olleiden potilaiden KPS-suorituskyky (Karnofsky Performance Status, KPS) oli ≥ 70 % ja tauti oli mitattavissa RECIST v1.1 ‑kriteerien perusteella heidän PD-L1-statuksestaan tai IMDC-riskiryhmästään riippumatta. Tutkimukseen ei otettu mukaan potilaita, joilla oli autoimmuunisairaus tai muu systeemistä immunosuppressiota edellyttävä sairaus, jotka olivat aiemmin saaneet hoitoa PD-1-, PD-L1-, PD-L2-, CD137- tai CTLA-4-vasta-aineilla, joilla oli huonossa hoitotasapainossa oleva hypertensio verenpainelääkityksestä huolimatta, aktiivisia metastaaseja aivoissa tai huonossa hoitotasapainossa oleva lisämunuaisten vajaatoiminta. Potilaat ositettiin IMDC-ennustepisteiden, kasvaimen PD-L1:n ilmentämisen ja maantieteellisen alueen mukaan.
Yhteensä 651 potilasta satunnaistettiin saamaan joko 40 mg kabotsantinibia suun kautta kerran vuorokaudessa yhdistelmänä nivolumabiannosten 240 mg (n = 323) laskimoon kahden viikon välein kanssa tai 50 mg sunitinibia (n = 328) suun kautta vuorokaudessa neljän viikon ajan, jonka jälkeen seurasi kahden viikon hoitotauko. Hoitoa jatkettiin, kunnes sairaus eteni tai kunnes enintään 24 kuukauden pituisen nivolumabihoidon yhteydessä ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Hoitoa oli sallittua jatkaa RECIST 1.1 -version määritelmän perusteella tutkijan aluksi arvioimaa sairauden etenemistä pidempään, jos potilas tutkijan arvion mukaan oli hyötynyt hoidosta kliinisesti ja sieti tutkimuslääkkeen. Kasvain arvioitiin ensimmäisen kerran lähtötilanteen jälkeen 12 viikon (± 7 päivän) kuluttua satunnaistamisesta. Sen jälkeen kasvain arvioitiin 6 viikon (± 7 päivän) välein viikkoon 60 saakka, ja sen jälkeen 12 viikon (± 14 päivän) välein sairauden radiologiseen etenemiseen saakka, minkä sokkoutettu riippumaton keskitetty arvioijataho (Blinded Independent Central Review, BICR) varmisti. Tehoa koskevan ensisijaisen hoitotuloksen mittari oli sokkoutetun riippumattoman keskitetyn arvioijatahon arvioima etenemisvapaa elinaika (PFS). Muita tehon mittareita olivat keskeiset toissijaiset päätetapahtumat kokonaiselossaoloaika (OS) ja objektiivinen vasteosuus (ORR).
Ominaisuudet olivat lähtötilanteessa yleisesti tasapainossa näiden kahden ryhmän välillä. Iän mediaani oli 61 vuotta (vaihteluväli: 28–90), ja 38,4 % oli iältään ≥ 65-vuotiaita ja 9,5 % oli iältään ≥ 75-vuotiaita. Valtaosa potilaista oli miehiä (73,9 %) ja valkoihoisia (81,9 %). Kahdeksan prosenttia potilaista oli aasialaisia, 23,2 %:lla KPS-suorituskyky oli lähtötilanteessa 79–80 % ja 76,5 %:lla se oli 90–100 %. Potilaat jakautuivat IMDC-riskiluokituksen osalta siten, että 22,6 %:lla riski oli matala, 57,6 %:lla kohtalainen ja 19,7 %:lla suuri. Kasvaimen PD-L1:n ilmentymisen osalta 72,5 %:lla potilaista PD-L1:n ilmentymä oli < 1 % tai määrittämätön ja 24,9 %:lla potilaista PD-L1:n ilmentymä oli ≥ 1 %. Potilaista 11,5 %:lla oli kasvaimessa sarkomatoidisia piirteitä. Hoidon keston mediaani kabotsantinibin ja nivolumabin yhdistelmällä hoitoa saaneilla potilailla oli 14,26 kuukautta (vaihteluväli: 0,2–27,3 kuukautta) ja sunitinibihoitoa saaneilla potilailla 9,23 kuukautta (vaihteluväli: 0,8–27,6 kuukautta).
Tutkimuksessa kabotsantinibin ja nivolumabin yhdistelmähoitoon satunnaistetuilla potilailla osoitettiin tilastollisesti merkitsevä hyöty etenemisvapaan elinajan, kokonaiselossaoloajan ja objektiivisen vasteosuuden osalta sunitinibihoitoon verrattuna.
Ensisijaisen analyysin tehoa koskevat tulokset (vähimmäisseuranta-aika 10,6 kuukautta, seuranta-ajan mediaani 18,1 kuukautta) esitetään taulukossa 7.
Taulukko 7: Tehoa koskevat tulokset (CA2099ER)
| kabotsantinibi + nivolumabi (n = 323) | sunitinibi (n = 328) | |
| Sokkoutetun riippumattoman keskitetyn arvioijatahon arvioima etenemisvapaa elinaika (PFS) | ||
| Tapahtumia | 144 (44,6 %) | 191 (58,2 %) |
| Riskisuhdea | 0,51 | |
| 95 %:n luottamusväli | (0,41; 0,64) | |
| p‑arvob, c | < 0,0001 | |
| Mediaani (95 %:n luottamusväli)d | 16,59 (12,45; 24,94) | 8,31 (6,97; 9,69) |
| Kokonaiselossaoloaika (OS) | ||
| Tapahtumia | 67 (20,7 %) | 99 (30,2 %) |
| Riskisuhdea | 0,60 | |
| 98,89 %:n luottamusväli | (0,40; 0,89) | |
| p‑arvob,c,e | 0,0010 | |
| Mediaani (95 %:n luottamusväli) | N.E. | N.E. (22,6; N.E.) |
| Osuus (95 %:n luottamusväli) | ||
| 6 kuukauden aikapisteessä | 93,1 (89,7; 95,4) | 86,2 (81,9; 89,5) |
| Sokkoutetun riippumattoman keskitetyn arvioijatahon arvioima objektiivinen vasteosuus (ORR) (täydellinen vaste + osittainen vaste) | 180 (55,7 %) | 89 (27,1 %) |
| (95 %:n luottamusväli)f | (50,1; 61,2) | (22,4; 32,3) |
| Objektiivisten vasteosuuksien ero (95 %:n luottamusväli)g | 28,6 (21,7; 35,6) | |
| p‑rvoh | < 0,0001 | |
| Täydellinen vaste (CR) | 26 (8,0 %) | 15 (4,6 %) |
| Osittainen vaste (PR) | 154 (47,7 %) | 74 (22,6 %) |
| Stabiili tauti (SD) | 104 (32,2 %) | 138 (42,1 %) |
| Vasteen keston mediaanid | ||
| Kuukautta (vaihteluväli) | 20,17 (17,31; N.E.) | 11,47 (8,31; 18,43) |
| Vasteeseen kuluneen ajan mediaani | ||
| Kuukautta (vaihteluväli) | 2,83 (1,0–19,4) | 4,17 (1,7–12,3) |
a Ositettu Coxin suhteellinen riskimalli. Riskin suhde on kabotsantinibin ja nivolumabin osalta parempi kuin sunitinibin.
b 2-tahoiset p-arvot saatu ositetusta tavallisesta log-rank-testistä.
c Log-rank-testi ositettu IMDC-ennusteriskipisteiden (0, 1–2, 3–6), kasvaimen PD-L1:n ilmentymän (≥ 1 % versus < 1 % tai määrittämätön) ja maantieteellisen alueen (Yhdysvallat/Kanada/Länsi-Eurooppa/Pohjois-Eurooppa, muu maailma) mukaan siten kuin ne on syötetty IRT-järjestelmään (interaktiivinen vastausjärjestelmä).
d Perustuu Kaplan-Meierin estimaatteihin.
e Tilastollisen merkitsevyyden p-arvon raja < 0,0111.
f Luottamusväli perustuu Clopperin ja Pearsonin menetelmään.
g Ositteet korjattu DerSimonianin ja Lairdin perusteella objektiivisten vasteosuuksien erolla (kabotsantinibi + nivolumabi - sunitinibi).
h CMH-testin 2-tahoinen p-arvo.
NE = ei arvioitavissa (non‑estimable)
Etenemisvapaan elinajan ensisijaiseen analyysiin sisältyi uuden syöpähoidon sensurointi (taulukko 7). Etenemisvapaata elinaikaa koskevat uuden syöpähoidon osalta sensuroidut ja sensuroimattomat tulokset olivat yhdenmukaiset.
Kabotsantinibin ja nivolumabin yhdistelmähoitoa saaneen ryhmän etenemisvapaassa elinajassa havaittiin hyötyä sunitinibihoitoa saaneeseen ryhmään verrattuna riippumatta PD L1:n ilmentymisestä kasvaimessa. Kasvaimen PD L1:n ilmentymän ollessa ≥ 1 % etenemisvapaan elinajan mediaani oli kabotsantinibin ja nivolumabin yhdistelmähoidossa 13,08 kuukautta ja sunitinibiryhmässä 4,67 kuukautta (riskisuhde = 0,45; 95 %:n luottamusväli: 0,29; 0,68). Kasvaimen PD L1:n ilmentymän ollessa < 1 % etenemisvapaan elinajan mediaani oli kabotsantinibin ja nivolumabin yhdistelmähoidossa 19,84 kuukautta ja sunitinibiryhmässä 9,26 kuukautta (riskisuhde = 0,50; 95 %:n luottamusväli: 0,38; 0,65).
Kabotsantinibin ja nivolumabin yhdistelmähoitoa saaneen ryhmän etenemisvapaassa elinajassa havaittiin hyötyä sunitinibiryhmään verrattuna riskiluokasta (IMDC) riippumatta. Matalan riskin ryhmässä kabotsantinibin ja nivolumabin yhdistelmän käytössä ei saavutettu etenemisvapaan elinajan mediaania ja sunitinibiryhmässä se oli 12,81 kuukautta (riskisuhde = 0,60; 95 %:n luottamusväli: 0,37; 0,98). Kohtalaisen riskin ryhmässä etenemisvapaan elinajan mediaani oli kabotsantinibin ja nivolumabin yhdistelmän käytössä 17,71 kuukautta ja sunitinibiryhmässä 8,38 kuukautta (riskisuhde = 0,54; 95 %:n luottamusväli: 0,41; 0,73). Suuren riskin ryhmässä etenemisvapaan elinajan mediaani oli kabotsantinibin ja nivolumabin yhdistelmän käytössä 12,29 kuukautta ja sunitinibiryhmässä 4,21 kuukautta (riskisuhde = 0,36; 95 %:n luottamusväli: 0,23; 0,58).
Etenemisvapaan elinajan ja kokonaiselossaoloajan päivitetty analyysi tehtiin, kun kaikkia potilaita oli seurattu vähintään 16 kuukauden ajan ja seuranta-ajan mediaani oli 23,5 kuukautta (ks. kuvat 4 ja 5). Etenemisvapaan elinajan riskisuhde oli 0,52 (95 %:n luottamusväli: 0,43; 0,64). Kokonaiselossaoloajan riskisuhde oli 0,66 (95 %:n luottamusväli: 0,50; 0,87). IMDC-riskiluokitusten alaryhmien tehoa koskevat päivitetyt tiedot (etenemisvapaa elinaika ja kokonaiselossaoloaika) ja PD-L1:n ilmentymätasot vahvistivat alkuperäiset tulokset. Matalan riskin ryhmä saavutti päivitetyssä analyysissä etenemisvapaan ajan mediaanin.
Kuva 4: Etenemisvapaan elinajan Kaplan‑Meier-käyrät (CA2099ER)
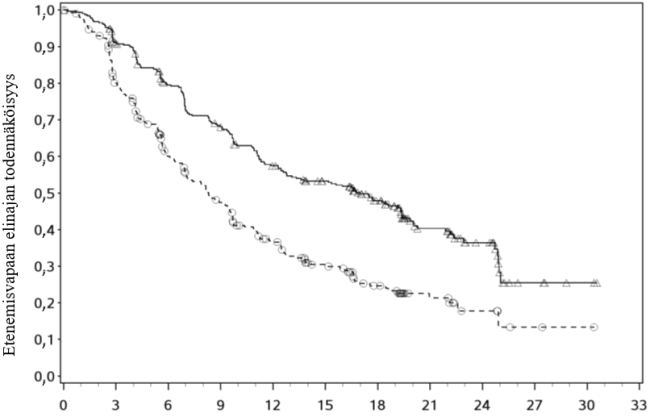
Sokkoutetun riippumattoman keskitetyn arvioijatahon arvioima etenemisvapaa elinaika (kuukautta)
Riskiryhmän tutkittavien lukumäärä
| Kabotsantinibi + nivolumabi | ||||||||||||
| 323 | 280 | 236 | 201 | 166 | 145 | 102 | 56 | 26 | 5 | 2 | 0 | |
| Sunitinibi | ||||||||||||
| 328 | 230 | 160 | 122 | 87 | 61 | 37 | 17 | 7 | 2 | 1 | 0 | |

Kabotsantinibi + nivolumabi (tapahtumia: 175/323), mediaani ja 95,0 %:n luottamusväli: 16,95 (12,58; 19,38)
Sunitinibi (tapahtumia: 206/328), mediaani ja 95,0 %:n luottamusväli: 8,31 (6,93; 9,69)
Kuva 5: Kokonaiselossaoloajan Kaplan-Meier-käyrät (CA2099ER)
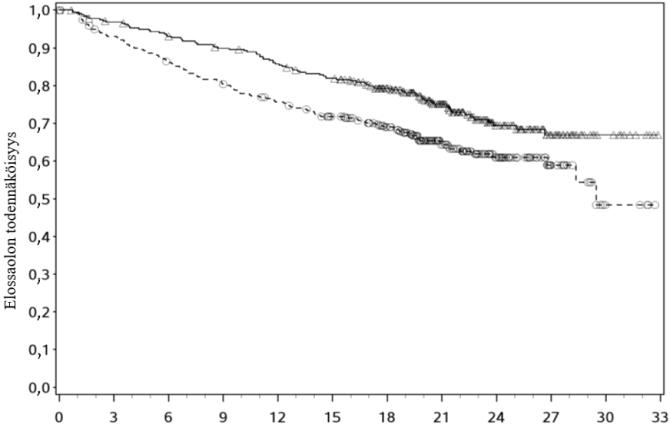
Kokonaiselossaoloaika (kuukautta)
Riskiryhmän tutkittavien lukumäärä
Kabotsantinibi + nivolumabi | |||||||||||
323 | 308 | 295 | 283 | 269 | 255 | 220 | 147 | 84 | 40 | 10 | 0 |
Sunitinibi | |||||||||||
328 | 295 | 272 | 254 | 236 | 217 | 189 | 118 | 62 | 22 | 4 | 0 |

Kabotsantinibi + nivolumabi (tapahtumia: 86/323), mediaani ja 95 %:n luottamusväli: NE
Sunitinibi (tapahtumia: 116/328), mediaani ja 95 %:n luottamusväli: 29,47 (28,35; NE)
Maksasyöpä
Kontrolloitu tutkimus sorafenibia saaneilla potilailla (CELESTIAL)
CABOMETYX-valmisteen turvallisuutta ja tehoa arvioitiin satunnaistetussa kaksoissokkoutetussa lumekontrolloidussa 3. vaiheen tutkimuksessa (CELESTIAL). Potilaat (N = 707), joiden maksasyöpä ei ollut hoidettavissa kuratiivisella hoidolla ja jotka olivat aiemmin saaneet sorafenibia edenneen taudin hoitoon, satunnaistettiin (2:1) kabotsantinibiryhmään (N = 470) tai lumeryhmään (N = 237). Potilaat olivat saattaneet saada sorafenibin lisäksi yhtä muuta aiempaa systeemistä hoitoa edenneeseen tautiin. Satunnaistaminen ositettiin sairauden etiologian (hepatiitti B ‑virus [HBV], johon saattoi liittyä hepatiitti C ‑virus [HBC], hepatiitti C ‑virus [ilman HBV:tä] tai muu) ja maantieteellisen alueen (Aasia, muut alueet) perusteella sekä sen mukaan, oliko sairaus levinnyt maksan ulkopuolelle ja/tai suuriin verisuoniin (kyllä, ei) perusteella.
Ensisijainen tehon päätetapahtuma oli kokonaiselossaoloaika (OS). Toissijaisia päätetapahtumia olivat tutkijan RECIST 1.1 ‑ kriteerien (Response Evaluation Criteria in Solid Tumours 1.1) perusteella arvioima etenemisvapaa elinaika (PFS) ja objektiivinen vasteosuus (ORR). Kasvaimet arvioitiin 8 viikon välein. Sairauden radiologisen etenemisen jälkeen tutkittavat jatkoivat sokkoutettua tutkimushoitoa niin kauan kuin hoidosta oli kliinistä hyötyä tai kunnes he tarvitsivat myöhempää systeemistä tai maksaan kohdistuvaa paikallista syöpähoitoa. Vaihtaminen lumelääkkeestä kabotsantinibiin ei ollut sallittua sokkoutetun tutkimusvaiheen aikana.
Lähtötason demografiset ja tautitiedot olivat samankaltaiset kabotsantinibi- ja lumehaaroissa. Seuraavassa esitetään kaikkia 707 satunnaistettua potilasta koskevat tiedot:
Valtaosa (82 %) potilaista oli miehiä: iän mediaani oli 64 vuotta. Valtaosa (56 %) potilaista oli valkoihoisia, ja 34 % oli aasialaisia. Viidelläkymmenelläkolmella prosentilla (53 %) potilaista ECOG-luokitus (PS) oli 0 ja 47 %:lla ECOG PS oli 1. Lähes kaikkien potilaiden (99 %) Child–Pugh-luokka oli A, ja 1 %:lla Child–Pugh-luokka oli B. Maksasyövän syynä oli 38 %:lla tutkittavista hepatiitti B ‑virus, 21 %:lla tutkittavista hepatiitti C ‑virus, 40 %:lla tutkittavista jokin muu (ei hepatiitti B- eikä hepatiitti C-virus). Seitsemälläkymmenelläkahdeksalla prosentilla (78 %) oli makroskooppinen verisuoni-invaasio ja/tai taudin leviäminen maksan ulkopuolelle, 41 %:lla alfafetoproteiinin (AFP) pitoisuus oli ≥ 400 mikrog/l, 44 %:lle oli tehty lokoregionaalinen transarteriaalinen embolisaatio tai kemoinfuusiotoimenpide, 37 % oli saanut sädehoitoa ennen kabotsantinibihoitoa. Sorafenibihoidon keston mediaani oli 5,32 kuukautta. Seitsemänkymmentäkaksi prosenttia (72 %) potilaista oli saanut yhtä ja 28 % potilaista oli saanut kahta aiempaa systeemistä hoitoa edenneeseen tautiin.
Kabotsantinibiryhmässä osoitettiin tilastollisesti merkitsevä kokonaiselossaoloajan paraneminen verrattuna lumeryhmään (taulukko 8 ja kuva 6).
Etenemisvapaan elinajan ja objektiivisten vasteosuuksien yhteenveto on taulukossa 8.
Taulukko 8: Tehoa koskevat tulokset maksasyöpää sairastavilla tutkittavilla (hoitoaikeen mukainen potilasjoukko [ITT], CELESTIAL)
CABOMETYX (N = 470) | Lume (N = 237) | |||
| Kokonaiselossaoloaika (OS) | ||||
| Mediaani OS (95 %:n luottamusväli), kk | 10,2 (9,1, 12,0) | 8,0 (6,8, 9,4) | ||
| Riskisuhde (95 %:n luottamusväli)1,2 | 0,76 (0,63, 0,92) | |||
| p-arvo1 | p = 0,0049 | |||
| Etenemisvapaa elinaika (PFS) 3 | ||||
| Mediaani PFS (95 %:n luottamusväli), kk | 5,2 (4,0, 5,5) | 1,9 (1,9, 1,9) | ||
| Riskisuhde (95 %:n luottamusväli)1 | 0,44 (0,36, 0,52) | |||
| p-arvo1 | p < 0,0001 | |||
| Kaplan–Meierin estimaatit (landmark estimates) niiden tutkittavien osuuksista (%), joilla ei ollut tapahtumia 3 kuukauden kohdalla | ||||
| % (95 %:n luottamusväli) | 67,0 % (62,2 %, 71,3 %) | 33,3 % (27,1 %, 39,7 %) | ||
| Objektiivinen vasteosuus (ORR), n (%)3 | ||||
| Täydellinen vaste | 0 | 0 | ||
| Osittainen vaste | 18 (4) | 1 (0,4) | ||
| ORR (täydellinen ja osittainen vaste) | 18 (4) | 1 (0,4) | ||
| p-arvo1,4 | p = 0,0086 | |||
| Stabiili tauti | 282 (60) | 78 (33) | ||
| Progressiivinen tauti | 98 (21) | 131 (55) | ||
1 Kaksitahoinen log-rank-testi, ositustekijät sairauden etiologia (HBV [johon saattoi liittyä HCV], HCV [ilman HBV:tä] tai muu), maantieteellinen alue (Aasia, muut alueet), sairauden leviäminen maksan ulkopuolelle ja/tai suuriin verisuoniin (kyllä, ei) (IVRS-tietojen mukaisesti)
2 Arvioitu Coxin suhteellisen vaaran regressiomallilla
3 Tutkijan RECIST 1.1‑kriteerien perusteella tekemän arvion mukaan
4 Ositettu Cochran–Mantel–Haenszelin (CMH) testi
Kuva 6: Kokonaiselossaoloaikaa kuvaava Kaplan–Meier-käyrä (CELESTIAL)
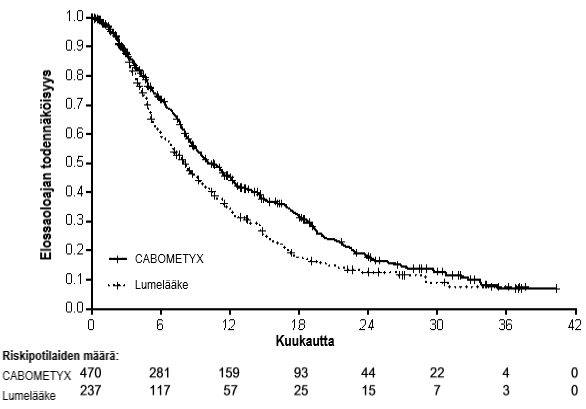
Kuva 7: Etenemisvapaata elinaikaa kuvaava Kaplan–Meier-käyrä (CELESTIAL)
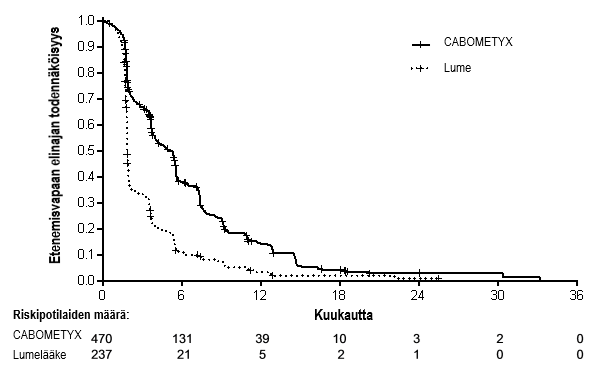
Systeemistä muuta kuin sädehoitoa ja paikallista maksaan kohdistuvaa systeemistä, tutkimussuunnitelmasta poikkeavaa syöpähoitoa (NPACT) sai kabotsantinibihaarassa 26 % ja lumehaarassa 33 % potilaista. Näitä hoitoja saavien tutkittavien täytyi lopettaa tutkimushoito. Myös NPACT-hoidon suhteen sensuroitu eksploratiivinen kokonaiselossaoloajan analyysi tuki ensisijaista analyysiä: ositustekijöillä (IxRS:n mukaisilla) korjattu riskisuhde oli 0,66 (95 %:n luottamusväli 0,52, 0,84; ositettu log-rank p-arvo = 0,0005). Kaplan–Meierin estimaatit kokonaiselossaoloajan mediaanista olivat kabotsantinibihaarassa 11,1 kuukautta ja lumehaarassa 6,9 kuukautta. Arvioitu mediaanien välinen ero oli 4,2 kuukautta.
Yleistä elämänlaatua (QoL) arvioitiin EuroQoL EQ-5D-5L ‑mittarilla. Ensimmäisten hoitoviikkojen aikana kabotsantinibihoidolla oli lumelääkkeeseen verrattuna negatiivinen vaikutus EQ-5D-utiliteetti-indeksiluvun pistemäärään. Tämän jakson jälkeiseltä ajalta on vain vähän yleistä elämänlaatua koskevia tietoja.
Erilaistunut kilpirauhassyöpä (DTC)
Aikuispotilaille tehty lumekontrolloitu tutkimus, johon osallistuneet potilaat olivat saaneet aiemmin systeemistä hoitoa ja joiden sairaus oli radiojodihoitoon vastaamaton tai joita ei voitu hoitaa radiojodilla (COSMIC‑311)
CABOMETYX-valmisteen turvallisuutta ja tehoa arvioitiin satunnaistetussa (2:1) kaksoissokkoutetussa lumekontrolloidussa monikeskustutkimuksessa (COSMIC‑311) aikuispotilailla, joilla oli sellainen paikallisesti edennyt tai metastasoitunut sairaus, johon liittyi erilaistunut kilpirauhassyöpä, joka oli edennyt jopa kahden aiemmin annetun endoteelikasvutekijään (VEGF) kohdistetun hoidon (esim. lenvatinibi tai sorafenibi) jälkeen ja joka oli radiojodihoitoon vastaamaton tai joka ei ollut hoidettavissa radiojodilla. Potilaat (N = 258), joilla oli mitattavissa oleva sairaus ja joiden sairauden radiologinen eteneminen oli todettu RECIST 1.1 ‑kriteereihin perustuvan tutkijan arvion mukaisesti joko VEGF-reseptorin tyrosiinikinaasiestäjään (VEGFR TKI) kohdistetun hoidon aikana tai sen jälkeen, satunnaistettiin saamaan joko kabotsantinibia 60 mg kerran vuorokaudessa suun kautta (N = 170) tai lumelääkettä (N = 88).
Satunnaistetut potilaat ositettiin aiemman lenvatinibihoidon (kyllä/ei) ja iän (≤ 65 vuotta vs. > 65 vuotta) mukaan. Tutkimukseen mukaan soveltuvat potilaat, jotka oli satunnaistettu saamaan lumelääkettä, saivat siirtyä kabotsantinibihoitoon sen jälkeen, kun sokkoutettu riippumaton radiologinen arviointitoimikunta oli todennut heidän sairautensa etenevän. Tutkittavat jatkoivat sokkoutuksen mukaista hoitoa niin kauan, kun he kokivat hyötyvänsä siitä tai kunnes heille ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Ensisijaisia tehon osoituksen mittareita olivat etenemisvapaa elinaika (progression-free survival, PFS) ITT-populaatiossa ja objektiivinen vasteosuus (ORR) 100 ensimmäisen satunnaistetun potilaan joukossa perustuen riippumattoman radiologisen arviointitoimikunnan antamaan RECIST 1.1 ‑kriteerien mukaiseen arvioon. Satunnaistamisen jälkeen kasvaimia arvioitiin tutkimuksen 12 ensimmäisen kuukauden aikana 8 viikon välein ja sitten 12 viikon välein. Muuna päätetapahtumana tutkimuksessa oli kokonaiselossaoloaika (overall survival, OS).
Etenemisvapaan elinajan ensisijaiseen analyysiin sisältyi 187 satunnaistettua potilasta, joista 125 sai kabotsantinibia ja 62 lumelääkettä. Lähtötason demografiset ja tautitiedot olivat yleisesti tasapainossa molemmissa hoitoryhmissä. Iän mediaani oli 66 vuotta (vaihteluväli 32–85 vuotta), 51 % osallistujista oli ≥ 65‑vuotiaita ja 13 % ≥ 75‑vuotiaita. Suurin osa potilaista oli valkoihoisia (70 %), 18 % potilaista oli aasialaisia ja 55 % naisia. Potilaista 55 %:lla oli aiemmin histologisesti todettu papillaarinen kilpirauhassyöpä ja 48 %:lla follikulaarinen kilpirauhassyöpä, mukaan lukien 17 %, joilla oli todettu Hürthlen solusyöpä. Metastaaseja oli 95 %:lla potilaista: keuhkoissa 68 %:lla, imusolmukkeissa 67 %:lla, luustossa 29 %:lla, keuhkopussissa 18 %:lla ja maksassa 15 %:lla. Viittä potilaista ei ollut aiemmin hoidettu radioaktiivisella jodilla, koska se ei sopinut kyseisille potilaille, ja 63 % potilaista oli saanut aiemmin lenvatinibia, 60 % sorafenibia ja 23 % sekä sorafenibia että lenvatinibia. Potilaiden ECOG (Eastern Cooperative Oncology Group) ‑suorituskykyluokka oli lähtötilanteessa 0 (48 %) tai 1 (52 %).
Hoidon kestoajan mediaani oli kabotsantinibiryhmässä 4,4 kuukautta ja lumelääkeryhmässä 2,3 kuukautta.
Ensisijaisen analyysin (katkaisuajankohta 19.8.2020 ja etenemisvapaan elinajan seuranta-ajan mediaani 6,2 kuukautta) ja päivitetyn analyysin (katkaisuajankohta 8.2.2021 ja etenemisvapaan elinajan seuranta-ajan mediaani 10,1 kuukautta) tulokset on esitetty taulukossa 9. Tutkimuksessa ei pystytty osoittamaan tilastollisesti merkitsevää objektiivisen vasteosuuden parannusta potilailla, jotka oli satunnaistettu saamaan kabotsantinibia (n = 67), kun heitä verrattiin lumelääkettä saaneisiin potilaisiin (n = 33): 15 % vs. 0 %. Tutkimuksessa osoitettiin tilastollisesti merkitsevä etenemisvapaan elinajan parannus (seuranta-ajan mediaani 6,2 kuukautta) potilailla, jotka oli satunnaistettu saamaan kabotsantinibia (n = 125), kun heitä verrattiin lumelääkettä saaneisiin potilaisiin (n = 62).
Päivitettyyn etenemisvapaan elinajan ja kokonaiselossaoloajan analyysiin (seuranta-ajan mediaani 10,1 kuukautta) sisältyi 258 satunnaistettua potilasta, joista 170 oli saanut kabotsantinibia ja 88 lumelääkettä.
Kokonaiselossaoloajan analyysissa arvioidut tiedot sulautettiin, koska lumelääkkeellä hoidetut tutkittavat, joiden sairauden todettiin etenevän, saivat siirtyä kabotsantinibihoitoon.
Taulukko 9: Tehoa koskevat tulokset COSMIC‑311-tutkimuksessa
Ensisijainen analyysi1 (hoitoaikeen mukainen potilasjoukko, ITT) | Päivitetty analyysi2 (koko hoitoaikeen mukainen potilasjoukko, ITT) | ||||
| CABOMETYX (n = 125) | Lumelääke (n = 62) | CABOMETYX (n = 170) | Lumelääke (n = 88) | ||
| Etenemisvapaa elinaika (PFS)* | |||||
| Tapahtumien lukumäärä, (%) | 31 (25) | 43 (69) | 62 (36) | 69 (78) | |
| Progressiivinen tauti | 25 (20) | 41 (66) | 50 (29) | 65 (74) | |
| Kuolema | 6 (4,8) | 2 (3,2) | 12 (7,1) | 4 (4,5) | |
| Mediaani PFS (96 %:n luottamusväli), kk | NE (5,7; NE) | 1,9 (1,8; 3,6) | 11,0 (7,4; 13,8) | 1,9 (1,9; 3,7) | |
| Riskisuhde (96 %:n luottamusväli)3 | 0,22 (0,13; 0,36) | 0,22 (0,15; 0,32) | |||
| p‑arvo4 | < 0,0001 | ||||
| Kokonaiselossaoloaika (OS) | |||||
| Tapahtumat, n (%) | 17 (14) | 14 (23) | 37 (22) | 21 (24) | |
| Riskisuhde (95 %:n luottamusväli3 | 0,54 (0,27; 1,11) | 0,76 (0,45; 1,31) | |||
| Ensisijainen analyysi1 | |||||
| Objektiivinen vasteosuus (ORR)5 | |||||
| CABOMETYX (n = 67) | Lumelääke (n = 33) | ||||
| Kokonaisvaste, (%) | 10 (15) | 0 (0) | |||
| Täydellinen vaste | 0 | 0 | |||
| Osittainen vaste | 10 (15) | 0 | |||
| Stabiili tauti | 46 (69) | 14 (42) | |||
| Progressiivinen tauti | 4 (6) | 18 (55) | |||
* Etenemisvapaan elinajan ensisijaiseen analyysiin sisältyi uuden syöpähoidon sensurointi. Etenemisvapaata elinaikaa koskevat uuden syöpähoidon osalta sensuroidut ja sensuroimattomat tulokset olivat yhdenmukaiset.
NE, ei arvioitavissa (not evaluable).
1 Ensisijaisen analyysin katkaisuajankohta on 19.8.2020.
2 Toissijaisen analyysin katkaisuajankohta on 8.2.2021.
3 Arvioinnissa käytettiin Coxin verrannollisten riskisuhteiden mallia.
4 Log-rank-testi, jossa (IxRS:n mukaisina) ositustekijöinä olivat aiemmin saatu lenvatinibihoito (kyllä/ei) ja ikä (≤ 65 vuotta vs. > 65 vuotta).
5 Tutkimuksen 100 ensimmäisen potilaan perusteella seuranta-ajan mediaani on 8,9 kuukautta, CABOMETYX-ryhmässä n = 67 ja lumelääkeryhmässä n = 33. ORR:n parannus ei ollut tilastollisesti merkitsevä.
Kuva 8: Etenemisvapaata elinaikaa kuvaava Kaplan–Meier-käyrä COSMIC‑311-tutkimuksessa (päivitetty analyysi [katkaisuajankohta 8.2.2021], N = 258)
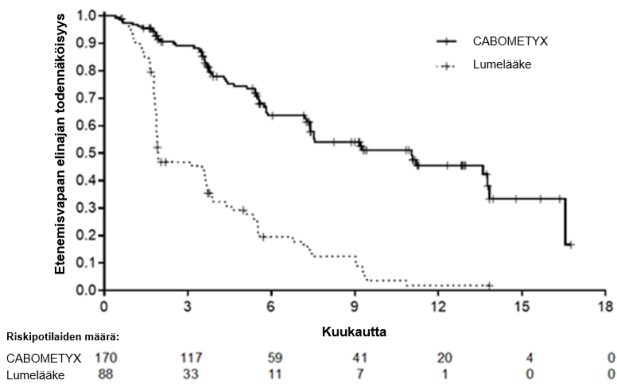
Neuroendokriiniset kasvaimet (NET)
Aikuispotilaille tehty lumekontrolloitu tutkimus, johon osallistuneilla potilailla oli paikallisesti edenneitä tai metastasoituneita epNET- tai pNET-kasvaimia, jotka olivat edenneet aiemman hoidon jälkeen (CABINET)
CABOMETYX-valmisteen turvallisuutta ja tehoa arvioitiin satunnaistetussa (2:1) kaksoissokkoutetussa lumekontrolloidussa 3. vaiheen monikeskustutkimuksessa (CABINET) aikuispotilailla, joilla oli paikallisesti edenneitä tai metastasoituneita hyvin erilaistuneita pNET-kasvaimia (kabotsantinibi: N = 64, lumelääke: N = 31) ja epNET-kasvaimia (kabotsantinibi: N = 134, lumelääke: N = 69) , jotka olivat edenneet aiemman hyväksytyn hoidon jälkeen.
Potilaat, joilla oli epNET- ja pNET-kasvaimia, osoitettiin kahteen eri kohorttiin, jotka satunnaistettiin ja analysoitiin erikseen.
Potilaat jatkoivat sokkoutettua tutkimushoitoa, kunnes sairaus eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä, tai tutkittava perui suostumuksensa. Tutkimukseen mukaan soveltuvat potilaat, jotka oli satunnaistettu saamaan lumelääkettä, saivat siirtyä avoimeen kabotsantinibihoitoon sen jälkeen, kun keskitetyn tosiaikaisen tarkastelun perusteella oli todettu heidän sairautensa etenevän. Ensisijainen tehon osoituksen mittari oli etenemisvapaa elinaika (progression-free survival, PFS) ITT-populaatiossa perustuen sokkoutetun riippumattoman arviointitoimikunnan antamaan RECIST 1.1 ‑kriteerien mukaiseen arvioon; ositustekijät satunnaistamisen yhteydessä olivat seuraavat:
- epNET: samanaikainen somatostatiinianalogihoito ja primaarikasvaimen sijaintikohta (keskisuoli/tuntematon vs. muu kuin keskisuoli / keuhkot / muu
- pNET: samanaikainen somatostatiinianalogihoito ja aiempi sunitinibihoito.
Kasvaimet arvioitiin 12 viikon välein tutkimuksen alusta taudin etenemiseen saakka. Kokonaiselossaoloaika oli toissijainen päätetapahtuma.
epNET-kohortti:
Valtaosa potilaista (51,7 %) oli naisia. Iän mediaani oli 66 vuotta. Valtaosa potilaista (83,7 %) oli valkoihoisia. Lisäksi potilaista 39,9 %:lla ECOG-toimintakykyluokka oli 0, ja 59,1 %:lla se oli 1. Primaarikasvaimen lähtökohta oli yleisimmin ohutsuolessa (32,5 %) ja seuraavaksi yleisimmin keuhkoissa (19,2 %), muualla (17,2 %), ja tuntemattomassa paikassa (11,8 %). Useimmilla potilailla oli toimimaton kasvain, joita oli 53,7 % tapauksista, ja 32,5 %:lla oli toiminnallinen kasvain. 13,8 %:lla potilaista kasvaimen toimintastatus oli tuntematon. Vaikeusasteeltaan kasvaimet olivat yleisimmin 2. asteen kasvaimia, joita todettiin 66 %:lla potilaista, ja 1. asteen kasvaimia, joita todettiin 25,6 %:lla potilaista. Valtaosa potilaista (69 %) käytti samanaikaisesti somatostatiinianalogeja, ja 92,6 % oli saanut aiemmin somatostatiinianalogihoitoa. 45,3 % potilaista oli saanut vain yhtä aiempaa muuta kuin somatostatiinianalogihoitoa. Useimmat kasvaimet olivat hyvin erilaistuneita (93,6 % tapauksista); 6,4 %:a ei ollut spesifioitu. Metastaaseja esiintyi yleisimmin maksassa (89,7 %:ssa tapauksista), imusolmukkeissa (70 %:ssa tapauksista), luustossa (49,3 %:ssa tapauksista), muissa paikoissa (35 %:ssa tapauksista) ja keuhkoissa (21,2 %:ssa tapauksista).
Taulukko 10: Tehoa koskevat tulokset CABINET-tutkimuksen epNET-kohorteissa
Päätetapahtuma | Kabotsantinibi | Lumelääke |
|---|---|---|
Etenemisvapaa elinaika | ||
Tapahtumien lukumäärä, n (%) | 71 (53) | 40 (58) |
Todettu eteneminen, n (%) | 53 (40) | 35 (51) |
Kuolema, n (%) | 18 (13) | 5 (7,2) |
Etenemisvapaan elinajan mediaani kuukausina1 (95 %:n luottamusväli) | 8,5 (7,5; 12,5) | 4,0 (3,0; 5,7) |
Riskisuhde2 (95 %:n luottamusväli) | 0,38 (0,25; 0,58) | |
Seuranta-ajan mediaani oli 23 kuukautta molemmissa haaroissa. Riippumattoman radiologisen arviointiryhmän sairauden etenemistä ja vastetta koskevien arviointien mukaisesti; tiedonkeruun katkaisuajankohta 24. elokuuta 2023
1 Perustuu Kaplan-Meierin estimaatteihin
2 Arvioinnissa käytetty Coxin verrannollisten riskitiheyksien mallia. CABINET-tutkimus lopetettiin tehoon liittyvistä syistä välianalyysin ajankohtana; välianalyysi oli suunniteltu ainoastaan tuloksettomuuden selvittämiseksi. Tyypin I virhettä ei kontrolloitu muodollisesti, eikä p‑arvoja esitetty. Esitetty 95 %:n luottamusväli on kuvaileva eikä tarkoita sitä, että tilastollinen merkitsevyys olisi saavutettu.
Kuva 9: epNET: Etenemisvapaan elinajan Kaplan-Meier-käyrät (tiedonkeruun katkaisuajankohta: 24. elokuuta 2023, N = 203)
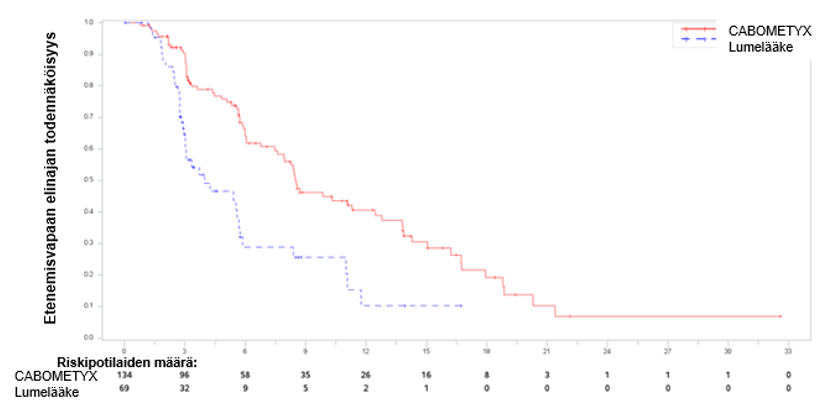
Päivitetyssä eksploratiivisessa OS-analyysissa (tietojen katkaisuajankohta: syyskuu 2024) 126 OS-tapahtumasta kokonaiselossaoloajan mediaani oli 21,95 kuukautta kabotsantinibihaarassa ja 22,47 kuukautta lumehaarassa. Riskisuhde oli 1,04 (95 %:n luottamusväli: 0,71, 1,52). Analyysiajankohtaan mennessä 28 potilasta (41 %) vaihtoi lumevalmisteesta kabotsantinibiin.
pNET-kohortti:
Valtaosa potilaista (57,9 %) oli miehiä. Iän mediaani oli kabotsantinibihaarassa 59,5 vuotta ja lumehaarassa 64 vuotta. Valtaosa potilaista (83,2 %) oli valkoihoisia. Potilaista 52,6 %:lla ECOG-toimintakykyluokka oli 0, ja 46,3 %:lla se oli 1. Useimmilla potilailla oli toimimaton kasvain, joita oli 73,7 % tapauksista, ja 16,8 %:lla oli toiminnallinen kasvain. 9,5 %:lla potilaista kasvaimen toimintastatus oli tuntematon. Vaikeusasteeltaan kasvaimet olivat yleisimmin 2. asteen kasvaimia, joita todettiin 61,1 %:lla potilaista; 1. asteen kasvaimia todettiin 22,1 %:lla potilaista, 3. asteen kasvaimia 11,6 %:lla potilaista, ja 5,3 %:lla potilaista kasvaimen vaikeusaste ei ollut tiedossa. Valtaosa potilaista (54,7 %) käytti samanaikaisesti somatostatiinianalogeja, ja 97,9 % oli saanut aiemmin somatostatiinianalogihoitoa. 28,4 % potilaista oli saanut vain yhtä aiempaa muuta kuin somatostatiinianalogihoitoa. Useimmat kasvaimet olivat hyvin erilaistuneita (97,9 % tapauksista); 2,1 %:a ei ollut spesifioitu. Metastaaseja esiintyi yleisimmin maksassa (96,8 %:ssa tapauksista), imusolmukkeissa (48,4 %:ssa tapauksista), luustossa (27,4 %:ssa tapauksista) ja muissa paikoissa (13,7 %:ssa tapauksista).
Taulukko 11: Tehoa koskevat tulokset CABINET-tutkimuksen pNET-kohorteissa
Kabotsantinibi | Lumelääke | |
|---|---|---|
Etenemisvapaa elinaika | ||
Tapahtumien lukumäärä, n (%) | 32 (50) | 25 (81) |
Todettu eteneminen, n (%) | 25 (39) | 21 (68) |
Kuolema, n (%) | 7 (11) | 4 (13) |
Etenemisvapaan ajan mediaani kuukausina1 (95 %:n luottamusväli) | 13,8 (8,9; 17,0) | 4,5 (3,0; 5,8) |
Riskisuhde2 (95 %:n luottamusväli) | 0,23 (0,12; 0,42) | |
Seuranta-ajan mediaani oli 23 kuukautta (kabotsantibini) ja 25 kuukautta (lumelääke). Riippumattoman radiologisen arviointiryhmän sairauden etenemistä ja vastetta koskevien arviointien mukaisesti; tiedonkeruun katkaisuajankohta 24. elokuuta 2023
1 Perustuu Kaplan-Meierin estimaatteihin
2 Arvioinnissa käytetty Coxin verrannollisten riskitiheyksien mallia. CABINET-tutkimus lopetettiin tehoon liittyvistä syistä välianalyysin ajankohtana; välianalyysi oli suunniteltu ainoastaan tuloksettomuuden selvittämiseksi. Tyypin I virhettä ei kontrolloitu muodollisesti, eikä p‑arvoja esitetty. Esitetty 95 %:n luottamusväli on kuvaileva eikä tarkoita sitä, että tilastollinen merkitsevyys olisi saavutettu.
Kuva 10: pNET: Etenemisvapaan elinajan Kaplan-Meier-käyrät, CABINET-tutkimus (tiedonkeruun katkaisuajankohta: 24. elokuuta 2023, N = 95)
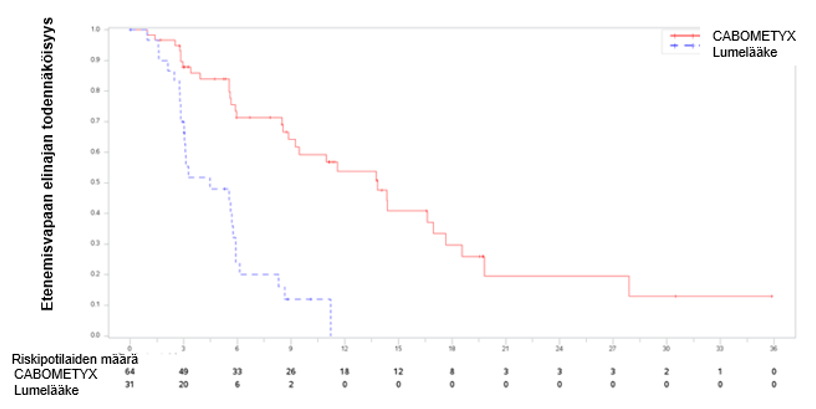
Päivitetyssä eksploratiivisessa OS-analyysissa (tietojen katkaisupiste: syyskuu 2024) 46 OS-tapahtumasta kokonaiselossaoloaikaa koskevan Kaplan-Meierin estimaatin mediaani oli 40,08 kuukautta kabotsantinibihaarassa ja 31,11 kuukautta lumehaarassa; riskisuhde oli 1,11 (0,59; 2,09). Analyysiajankohtaan mennessä 28 potilasta (41 %) vaihtoi lumevalmisteesta kabotsantinibiin.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa osa tutkimustuloksista CABOMETYX-valmisteen käytöstä kiinteiden syöpäkasvainten hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
ADVL 1211
COG-ryhmä (Children Oncology Group) on tehnyt kabotsantinibia koskevan vaiheen 1 tutkimuksen (ADVL1211) pediatrisilla potilailla, joilla on kiinteitä kasvaimia. Mukaan soveltuneet potilaat olivat iältään ≥ 2-vuotiaista ≤ 18-vuotiaisiin. Tähän tutkimukseen otettiin mukaan potilaita kolmeen annostasoon: 30 mg/m2, 40 mg/m2 ja 55 mg/m2 kerran päivässä jatkuvana hoito-ohjelmana (viikoittainen kehon pinta-alan mukainen annostus, joka pyöristetään lähimpään 20 mg:aan). Kabotsantinibin annostus perustui kehon pinta-alaan annostustaulukon mukaisesti.
Tavoitteena oli määrittää annosta rajoittava toksisuus, määrittää vaiheen 2 annossuositus, saada lapsista alustavia farmakokineettisiä tietoja ja selvittää teho kiinteisiin kasvaimiin. Mukaan otettiin 41 potilasta, joista 36 oli täysin arvioitavissa. Potilailla oli erilaisia kiinteitä kasvaimia: medullaarinen kilpirauhaskarsinooma (n = 5), osteosarkooma (n = 2), Ewingin sarkooma (n = 4), rabdomyosarkooma (n = 2), muu pehmytkudossarkooma (n = 4), Wilmsin kasvain (n = 2), hepatoblastooma (n = 2), hepatosellulaarinen karsinooma (n = 2), munuaiskarsinooma (n = 3), keskushermoston kasvaimia (n = 9) ja muita (n = 6).
Arvioitavissa olleen potilasjoukon 36 tutkittavasta neljä tutkittavaa (11,1 %) sai osittaisen vasteen parhaan kokonaisvasteen ja 8 tutkittavalla (22,2 %) oli stabiili tauti (vähintään 6 hoitosyklin ajan). Osittaisen vasteen tai stabiilin taudin vähintään 6 hoitosyklin ajaksi saavuttaneista 12 tutkittavasta 10 tutkittavaa oli ryhmissä, jotka saivat kabotsantinibiannoksia 40 mg/m2 (seitsemän tutkittavaa) tai 55 mg/m2 (kolme tutkittavaa).
Osittainen vaste todettiin keskitetyn tarkastelun perusteella kahdella potilaalla viidestä, joilla oli medullaarinen kilpirauhaskarsinooma, yhdellä potilaalla, jolla oli Wilmsin kasvain, ja yhdellä potilaalla, jolla oli kirkassolusarkooma.
ADVL1622
ADVL1622-tutkimuksessa arvioitiin kabotsantinibin aktiivisuutta valikoituihin pediatristen potilaiden kiinteisiin kasvaimiin. Tässä avoimessa kaksivaiheisessa vaiheen 2 monikeskustutkimuksessa oli mukana seuraavat kiinteiden kasvainten ositteet: muun kuin osteosarkooman osite (mukaan lukien Ewingin sarkooma, rabdomyosarkooma, muu pehmytkudossarkooma kuin rabdomyosarkooma ja Wilmsin kasvain), osteosarkoomaosite ja harvinaisten kiinteiden kasvainten osite (mukaan lukien medullaarinen kilpirauhaskarsinooma, munuaiskarsinooma, hepatosellulaarinen karsinooma, hepatoblastooma, lisämunuaisen kuorikerroksen karsinooma ja muut kiinteät kasvaimet). Kabotsantinibia annettiin suun kautta kerran päivässä jatkuvana hoito-ohjelmana, joka käsitti 28 päivän hoitosykleissä annoksen 40 mg/m2/vrk (kumulatiivinen viikoittainen annos 280 mg/m2 annostustaulukon mukaisesti). Tutkittavat olivat kaikissa ositteissa tutkimukseen mukaan tullessaan iältään ≥ 2-vuotiaista ≤ 30-vuotiaisiin, mutta medullaarisen kilpirauhaskarsinooman, munuaiskarsinooman ja hepatosellulaarisen karsinooman osalta yläikäraja oli ≤ 18 vuotta.
Muun kuin osteosarkooman ositteen ja harvinaisten kasvainten ositteen ensisijainen päätetapahtuma oli objektiivinen vasteosuus (ORR). Kaksivaiheiseen tutkimusasetelmaan sisältyi osteosarkoomaositteen osalta kaksoispäätetapahtumina objektiivinen vaste (täydellinen vaste + osittainen vaste), joka perustui RECIST-kriteerien (Response Evaluation Criteria in Solid Tumors) versioon 1.1, ja hoidon onnistuminen, joksi oli määritelty stabiili tauti ≥ 4 kuukauden ajan. Kabotsantinibin farmakokinetiikkaa arvioitiin pediatrisilla ja nuorilla tutkittavilla (ks. kohta Farmakokinetiikka).
Yhteenveto tehoa koskevista tuloksista
Tiedonkeruun katkaisupäivänä (30. kesäkuuta 2021) 108 tutkittavaa 109 tutkittavasta sai vähintään yhden kabotsantinibiannoksen. Muun kuin osteosarkooman ositteen kuhunkin tilastolliseen kohorttiin kuului 13 tutkittavaa. Näissä tilastollisissa kohorteissa ei havaittu vasteita. Osteosarkoomaositteeseen kuului yhteensä 29 tutkittavaa, joista 17 oli lapsia (iältään 9–17-vuotiaita) ja 12 oli aikuisia (iältään 18–22-vuotiaita).
Osteosarkoomaositteessa kaikki tutkittavat olivat saaneet aiempaa hoitoa. Yhdellä aikuisella ja yhdellä lapsella havaittiin osittainen vaste. Tauti pysyi hallinnassa 34,5 %:lla tutkittavista (95 %:n luottamusväli: 17,9; 54,3).
Farmakokinetiikka
Imeytyminen
Suun kautta otetun kabotsantinibin huippupitoisuus plasmassa saavutetaan 3–4 tuntia annoksen ottamisen jälkeen. Pitoisuus-aikaprofiilit plasmassa osoittavat toisen imeytymishuipun noin 24 tuntia annoksen ottamisen jälkeen, mikä viittaa siihen, että kabotsantinibi saattaa läpikäydä enterohepaattisen uudelleenkierron.
Toistuva kabotsantinibin anto 140 mg:n annoksella 19 vuorokauden ajan sai aikaan noin 4–5-kertaisen kabotsantinibin keskimääräisen kertymisen (AUC-käyrän perusteella) verrattuna kerta-annokseen. Vakaa tila saavutetaan suunnilleen 15. päivänä.
Runsaasti rasvaa sisältävä ateria nosti Cmax- ja AUC-arvoja (41 % ja 57 %) suhteessa paastoamiseen terveillä vapaaehtoisilla, joille annettiin yksi 140 mg:n kabotsantinibiannos suun kautta. Ruoan tarkasta vaikutuksesta ei ole tietoja, kun ruoka nautitaan 1 tunti kabotsantinibin ottamisen jälkeen.
Biologista samanarvoisuutta kabotsantinibikapselien ja -tablettien välillä ei voitu osoittaa terveille vapaaehtoisille annetun yhden 140 mg:n annoksen jälkeen. Kabotsantinibitabletin Cmax-arvossa todettiin 19 %:n nousu verrattuna kabotsantinibikapseliin. AUC-arvossa todettiin alle 10 %:n ero kabotsantinibitabletin ja kabotsantinibikapselin välillä.
Jakautuminen
Kabotsantinibi sitoutuu voimakkaasti ihmisen plasman proteiineihin in vitro (≥ 99,7 %). Populaatiofarmakokineettisen mallin (PK-mallin) perusteella keskustilan jakautumistilavuuden (Vc/F) on arvioitu olevan 212 l.
Biotransformaatio
Kabotsantinibi metaboloituu in vivo. Plasmassa todettiin neljä metaboliittia altistuksella (AUC), joka oli suurempi kuin 10 % emoyhdisteestä: XL184‑N‑oksidi, XL184-amidin jakautumistuote, XL184-monohydroksisulfaatti ja 6‑desmetyyliamidin jakautumistuote sulfaatti. Kumpikin kahdesta konjugoimattomasta metaboliitista (XL184-N‑oksidi ja XL184-amidin hajoamistuote), joilla on < 1 % emoyhdiste kabotsantinibin kohteena olevan kinaasin estopotentiaalista, vastaa < 10 %:a lääkkeeseen liittyvästä plasman kokonaisaltistuksesta.
Kabotsantinibi on CYP3A4:n substraatti in vitro, koska CYP3A4:ää neutraloiva vasta-aine estää metaboliitin XL184 N‑oksidin muodostumista > 80-prosenttisesti NADPH:n katalysoimassa ihmisen maksan mikrosomaalien inkubaatiossa (HLM), kun taas CYP1A2:ta, CYP2A6:ta, CYP2B6:ta, CYP2C8:aa, CYP2C19:ää, CYP2D6:ta ja CYP2E1:tä neutraloivilla vasta-aineilla ei ollut vaikutusta kabotsantinibin metaboliittien muodostumiseen. CYP2C9:n neutraloivalla vasta-aineella oli vähäinen vaikutus kabotsantinibin metaboliittien muodostumiseen (muodostuminen väheni < 20 %).
Eliminaatio
Populaatiofarmakokineettisessä analyysissa, jossa käytetyt tiedot kerättiin 1883 potilaalta ja 140 terveeltä vapaaehtoiselta, jotka saivat kabotsantinibia suun kautta 20–140 mg:n annoksina, kabotsantinibin terminaalinen puoliintumisaika oli noin 110 tuntia. Vakaan tilan keskimääräisen puhdistuman (CL/F) arvioitiin olevan 2,48 l/h. Terveille vapaaehtoisille annetun 14C-merkityn kabotsantinibin kerta-annoksen jälkeen noin 81 % annetusta radioaktiivisesta kokonaisannoksesta erittyi 48 päivän keräysjakson aikana ulosteeseen (54 %) ja virtsaan (27 %).
Farmakokinetiikka erityisryhmissä
Munuaisten vajaatoiminta
Tutkimuksessa, jossa tutkittavat saivat kerta-annoksena kabotsantinibia 60 mg, verrattuna normaaliin munuaisten toimintaan kokonaisplasman kabotsantinibin Cmax- ja AUC0-inf-arvojen pienimmän neliösumman geometristen keskiarvojen suhteet olivat 19 % ja 30 % suuremmat, kun munuaisten vajaatoiminta oli lievä (luottamusväli 90 %; Cmax 91,60–155,51 %; AUC0-inf 98,79–171,26 %), ja vastaavasti 2 % ja 6–7 % suuremmat (luottamusväli 90 %; Cmax 78,64–133,52 %; AUC0-inf 79,61–140,11 %), kun munuaisten vajaatoiminta oli kohtalainen. Plasman sitoutumattoman kabotsantinibin AUC0-inf-arvon pienimmän neliösumman geometrinen keskiarvo oli lievää munuaisten vajaatoimintaa sairastavilla tutkittavilla 0,2 % suurempi (90 %:n luottamusväli: 55,9–180 %) ja keskivaikeaa munuaisten vajaatoimintaa sairastavilla tutkittavilla 17 % suurempi (90 %:n luottamusväli: 65,1–209,7 %) kuin tutkittavilla, joiden munuaisten toiminta oli normaali. Tutkimuksia ei ole tehty vaikeaa munuaisten vajaatoimintaa sairastaville.
Maksan vajaatoiminta
Kabotsantinibin käytöstä terveille tutkittaville ja syöpää sairastaville potilaille (mukaan lukien maksasyöpää sairastavat potilaat) tehdyn integroidun populaatiofarmakokineettisen analyysin perusteella plasman keskimääräisessä kabotsantinibialtistuksessa ei ollut kliinisesti merkityksellisiä eroja niiden potilaiden välillä, joiden maksan toiminta oli normaali (n = 1425) tai joilla oli lievä maksan vajaatoiminta (n = 558). Kohtalaista maksan vajaatoimintaa sairastavista potilaista (n = 15) on vain vähän NCI-ODWG ((National Cancer Institute – Organ Dysfunction Working Group) -kriteerien mukaisia tietoja. Kabotsantinibin farmakokinetiikkaa ei ole arvioitu vaikeaa maksan vajaatoimintaa sairastavilla potilailla.
Etninen tausta
Populaatiofarmakokineettisessä analyysissa ei havaittu kliinisesti oleellisia, etniseen taustaan perustuvia eroja kabotsantinibin farmakokinetiikassa.
Pediatriset potilaat
Terveillä henkilöillä ja aikuispotilailla, joilla on erityyppisiä pahanlaatuisia sairauksia, kehitetyllä populaatiofarmakokineettisellä mallilla suoritetun simulaation tulokset osoittavat, että kun 12‑vuotiaille ja sitä vanhemmille erilaistunutta kilpirauhassyöpää sairastaville nuorille potilaille, joiden paino on < 40 kg, annetaan 40 mg:n kabotsantinibiannos kerran vuorokaudessa, tai kun potilaille, joiden paino on ≥ 40 kg, annetaan 60 mg:n annos kerran vuorokaudessa, saavutetaan vastaava altistus plasmassa kuin aikuisilla, jotka saavat 60 mg:n kabotsantinibiannoksen kerran vuorokaudessa (ks. kohta Annostus ja antotapa).
COG-ryhmän tekemissä kahdessa kliinisessä tutkimuksessa oli mukana pediatrisia potilaita, joilla oli kiinteitä kasvaimia (ADVL1211 ja ADVL1622). Niissä kabotsantinibin annostus perustui kehon pinta-alaan annostustaulukon mukaisesti ja hoidossa käytettiin aikuisille tarkoitettuja 20 mg:n ja 60 mg:n tabletteja. 55 potilaan iän mediaani oli 13 vuotta (vaihteluväli 4–18 vuotta). Populaatiofarmakokineettinen analyysi perustui kummassakin tutkimuksessa kerättyihin farmakokineettisiin tietoihin. Kabotsantinibin farmakokinetiikkaa kuvasti riittävästi kaksitilamalli, jossa oli ensimmäisen asteen eliminaatioprosessi ja ensimmäisen asteen imeytymisprosessi. Iän, sukupuolen, etnisen taustan ja kasvaimen tyypin ei havaittu vaikuttaneen kabotsantinibin farmakokinetiikkaan lapsipotilailla ja nuorilla potilailla. Vain kehon pinta-alan todettiin olevan kabotsantinibin farmakokinetiikkaa merkittävästi ennustava tekijä. Kehitetyssä mallissa ei havaittu annosriippuvuutta kolmen tutkitun annostason (30, 40 and 55 mg/m2) suhteen. Lapsilla ja nuorilla tutkittavilla kehon pinta-alaan perustuvan annoksen 40 mg/m2 jälkeen todetut altistukset ovat samankaltaiset kuin aikuisilla vakioannoksen 60 mg kerran päivässä yhteydessä.
Prekliiniset tiedot turvallisuudesta
Seuraavia haittavaikutuksia ei ole todettu kliinisissä tutkimuksissa, mutta niitä on havaittu koe-eläimillä, jotka olivat saaneet lääkevalmistetta hoitoannoksia vastaavina annoksina, ja siksi haitoilla voi olla kliinistä merkitystä:
Rotille ja koirille tehdyissä enintään 6 kuukautta kestävissä toistuvan annoksen toksisuutta koskevissa tutkimuksissa toksisuuden kohde-elimet olivat ruoansulatuskanava, luuydin, imukudokset, munuaiset, lisämunuaiset ja sukupuolielimet. Altistustaso, jolla ei havaittu haittavaikutuksia (NOAEL), oli näiden löydösten osalta aiotulla terapeuttisella annostuksella ihmisen kliinistä altistusta pienempi.
Kabotsantinibi ei ollut mutageeninen tai karsinogeeninen tavanomaisissa geenitoksisuutta koskevissa tutkimussarjoissa. Kabotsantinibin karsinogeenisuutta on arvioitu kahdella eläinlajilla: rasH2-siirtogeenisillä hiirillä ja Sprague-Dawley-rotilla. Rotilla tehdyssä 2‑vuotisessa karsinogeenisuustutkimuksessa kabotsantinibiin liittyviin kasvainlöydöksiin kuului hyvänlaatuisen feokromosytooman ilmaantuvuuden lisääntyminen: sitä ilmeni rotilla sukupuolesta riippumatta joko sellaisenaan tai yhdessä lisämunuaisytimen pahanlaatuisen feokromosytoooman / monimuotoisen pahanlaatuisen feokromosytoooman kanssa altistuksilla, jotka olivat huomattavasti ihmiselle tarkoitetun kliinisen altistuksen alapuolella. Rotilla havaittujen kasvainleesioiden kliininen merkitys on epäselvä, mutta todennäköisesti se on vähäinen.
Kabotsantinibi ei ollut karsinogeeninen rasH2-hiirimallissa altistuksella, joka oli hieman suurempi kuin aiotun hoitoannoksen ihmiselle aiheuttama altistus.
Rotilla tehdyissä hedelmällisyystutkimuksissa on havaittu uroksen ja naaraan hedelmällisyyden heikkenemistä. Lisäksi uroskoirien siittiöntuotannon havaittiin olevan epänormaalin niukkaa altistuksella, joka oli pienempi kuin aiotun hoitoannoksen ihmiselle aiheuttama kliininen altistus.
Alkion/sikiön kehitystä koskevia tutkimuksia on tehty rotilla ja kaneilla. Rotilla kabotsantinibi aiheutti sikiön menetyksen kiinnittymisen jälkeen, sikiön edeeman, huulihalkion/ristihuulen, ihon aplasian ja sykkyräisen tai alkeellisen hännän. Kaneilla kabotsantinibi aiheutti muutoksia sikiön pehmytkudoksissa (pienempi pernan koko, pieni tai puuttuva keuhkon keskilohko) ja lisäsi täydellisten epämuodostumien ilmaantuvuutta. Alkio-/sikiötoksisuuden ja teratogeenisten löydösten altistustaso, jolla ei havaittu haittavaikutuksia (NOAEL), oli pienempi kuin aiotun hoitoannoksen ihmiselle aiheuttama kliininen altistus.
Nuorilla rotilla (verrattavissa > 2-vuotiaisiin lapsipotilaisiin) esiintyi kabotsantinibin annon jälkeen valkosolujen määrän nousua, heikentynyttä hematopoieesia, puberteetti-ikäisiä/kypsymättömiä sukupuolielimiä naarailla (ilman hidastunutta emättimen aukeamista), hampaiden epämuodostumia, luuston mineraalipitoisuuden ja -tiheyden vähenemistä, maksan pigmentaatiota ja sappitiehyeen hyperplasiaa. Kohdussa/munasarjoissa havaitut löydökset ja heikentynyt hematopoieesi vaikuttivat olevan ohimeneviä, kun taas luuston parametrien ja maksan pigmentaation vaikutukset olivat pysyviä. Nuorilla rotilla (verrattavissa < 2-vuotiaisiin lapsipotilaisiin) ilmeni samansuuntaisia hoitoon liittyviä löydöksiä, minkä lisäksi uroksilla havaittiin myös muita löydöksiä lisääntymiselimissä (kivesten siementiehyiden degeneraatio ja/tai atrofia, lisäkiveksiin varastoituneen siemennesteen määrän pieneneminen), ja ne vaikuttivat olevan herkempiä kabotsantinibiin liittyvälle toksisuudelle vastaavilla annostasoilla.
Farmaseuttiset tiedot
Apuaineet
Tabletin koostumus
Mikrokiteinen selluloosa
Vedetön laktoosi
Hydroksipropyyliselluloosa
Kroskarmelloosinatrium
Vedetön kolloidinen piidioksidi
Magnesiumstearaatti
Kalvopäällyste
Hypromelloosi 2910
Titaniumdioksidi (E171)
Triasetiini
Keltainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
CABOMETYX tabletti, kalvopäällysteinen
20 mg (L:kyllä) 30 kpl (5121,14 €)
40 mg (L:kyllä) 30 kpl (5121,14 €)
60 mg (L:kyllä) 30 kpl (5121,14 €)
PF-selosteen tieto
HDPE-purkki, jossa on lapsiturvallinen polypropeenikorkki, kolme silikageeliä sisältävää kuivausainepakkausta ja polyesterikuituvanua. Kussakin purkissa on 30 kalvopäällysteistä tablettia.
Valmisteen kuvaus:
CABOMETYX 20 mg kalvopäällysteiset tabletit
Tabletit ovat keltaisia, pyöreitä, ilman jakouurretta ja niiden toisella puolella on teksti ”XL” ja toisella ”20”.
CABOMETYX 40 mg kalvopäällysteiset tabletit
Tabletit ovat keltaisia, kolmionmuotoisia, ilman jakouurretta ja niiden toisella puolella on teksti ”XL” ja toisella ”40”.
CABOMETYX 60 mg kalvopäällysteiset tabletit
Tabletit ovat keltaisia, soikeita, ilman jakouurretta ja niiden toisella puolella on teksti ”XL” ja toisella ”60”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
CABOMETYX tabletti, kalvopäällysteinen
20 mg 30 kpl
40 mg 30 kpl
60 mg 30 kpl
- Ylempi erityiskorvaus (100 %). Kabotsantinibi: Munuaissyövän hoito erityisin edellytyksin (1502).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Kabotsantinibi: Munuaissyövän ja kilpirauhassyövän hoito erityisin edellytyksin (3012).
ATC-koodi
L01EX07
Valmisteyhteenvedon muuttamispäivämäärä
15.01.2026
Yhteystiedot
Kista Science Tower, Färögatan 33
SE-164 51 Kista
Sweden
+46 8 451 60 00
www.ipsen.com
info.se@ipsen.com