
NINLARO kapseli, kova 2,3 mg, 3 mg, 4 mg
Vaikuttavat aineet ja niiden määrät
NINLARO 2,3 mg kovat kapselit
Yksi kapseli sisältää 3,3 milligrammaa iksatsomibisitraattia, joka vastaa 2,3 mg:aa iksatsomibia.
NINLARO 3 mg kovat kapselit
Yksi kapseli sisältää 4,3 milligrammaa iksatsomibisitraattia, joka vastaa 3 mg:aa iksatsomibia.
NINLARO 4 mg kovat kapselit
Yksi kapseli sisältää 5,7 milligrammaa iksatsomibisitraattia, joka vastaa 4 mg:aa iksatsomibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kova kapseli.
Kliiniset tiedot
Käyttöaiheet
NINLARO on tarkoitettu käytettäväksi yhdessä lenalidomidin ja deksametasonin kanssa sellaisten multippelia myeloomaa sairastavien aikuispotilaiden hoitoon, jotka ovat saaneet vähintään yhtä aiempaa hoitoa.
Ehto
Hoito tulee aloittaa ja sitä tulee seurata multippelin myelooman (MM) hoitoon perehtyneiden lääkäreiden valvonnassa.
Annostus ja antotapa
Hoidon aloitus ja seuranta tapahtuvat multippelin myelooman hoitoon perehtyneen lääkärin valvonnassa.
Annostus
Iksatsomibi-valmisteen suositeltu aloitusannos on 4 mg suun kautta kerran viikossa 28-päiväisen hoitojakson päivinä 1, 8 ja 15.
Lenalidomidin suositeltu aloitusannos on 25 mg vuorokaudessa 28-päiväisen hoitojakson päivinä 1–21.
Deksametasonin suositeltu aloitusannos on 40 mg 28-päiväisen hoitojakson päivinä 1, 8, 15 ja 22.
Antoaikataulu: iksatsomibi yhdessä lenalidomidin ja deksametasonin kanssa
28-päiväinen hoitojakso (4 viikon hoitojakso) | ||||||||
| Viikko 1 | Viikko 2 | Viikko 3 | Viikko 4 | ||||
| Päivä 1 | Päivät 2–7 | Päivä 8 | Päivät 9–14 | Päivä 15 | Päivät 16–21 | Päivä 22 | Päivät 23–28 |
Iksatsomibi | ✔ |
| ✔ |
| ✔ |
|
|
|
Lenalidomidi | ✔ | ✔Päivittäin | ✔ | ✔Päivittäin | ✔ | ✔Päivittäin |
|
|
Deksametasoni | ✔ |
| ✔ |
| ✔ |
| ✔ |
|
✔= lääkevalmisteen otto | ||||||||
Lisätietoa lenalidomidista ja deksametasonista, ks. kyseisten lääkevalmisteiden valmisteyhteenvedot.
Ennen uuden hoitojakson aloittamista:
-
Absoluuttisen neutrofiiliarvon on oltava ≥ 1 x 109/l
-
Trombosyyttiarvon on oltava ≥ 75 x 109/l
-
Ei-hematologisen toksisuuden on yleensä korjauduttava lääkärin harkinnan mukaan potilaan lähtötasolle tai asteen ≤ 1 tasolle.
Hoitoa jatketaan, kunnes tauti etenee tai potilas ei enää siedä hoitoa. iksatsomibi-valmisteen, lenalidomidin ja deksametasonin yhdistelmän käyttö yli 24 hoitojakson ajan perustuu aina potilaskohtaiseen hyöty-riskiarvioon, sillä yli 24 hoitojaksoa kestävästä käytöstä on niukasti siedettävyys- ja toksisuustietoja (ks. kohta Farmakodynamiikka).
Myöhästyneet tai ottamatta jääneet annokset
Jos iksatsomibiannoksen ottaminen myöhästyy tai annos jää ottamatta, kyseinen annos otetaan vain, jos seuraavan aikataulun mukaisen annoksen ottamiseen on ≥ 72 tuntia aikaa. Ottamatta jäänyttä annosta ei saa enää ottaa, jos seuraavan aikataulun mukaisen annoksen ottamiseen on alle 72 tuntia. Kaksinkertaista annosta ei saa ottaa ottamatta jääneen annoksen korvaamiseksi.
Jos potilas oksentaa annoksen ottamisen jälkeen, hänen ei pidä ottaa uutta annosta. Lääkkeen ottamista jatketaan ottamalla seuraava annos aikataulun mukaisesti.
Annosmuutokset
Iksatsomibiannoksen pienentäminen kuvataan Taulukko 1Taulukossa 1, ja annosmuutosohjeet esitetään Taulukossa 2.
Taulukko 1: iksatsomibi -annoksen pienentäminen
Suositeltu aloitusannos* | Ensimmäinen pienennyskerta | Toinen pienennyskerta | Lopetetaan |
4 mg | 3 mg | 2,3 mg |
*Pienennettyä 3 mg:n annosta suositellaan potilaille, joilla on keskivaikea tai vaikea maksan vajaatoiminta, vaikea munuaisten vajaatoiminta tai dialyysihoitoa vaativa loppuvaiheen munuaissairaus (ESRD).
Trombosytopenia, neutropenia ja ihottuma voivat johtua joko iksatsomibi-valmisteesta tai lenalidomidista. Niiden ilmetessä on suositeltavaa muuttaa iksatsomibi- ja lenalidomidiannoksia vuorotellen. Kyseisten toksisuuksien kohdalla ensimmäinen annosmuutos on lenalidomidihoidon tauottaminen tai sen annoksen pienentäminen. Annoksen pienentäminen näiden toksisuustyyppien vuoksi, ks. lenalidomidin valmisteyhteenvedon kohta Annostus ja antotapa.
Taulukko 2: iksatsomibi‑annosmuutosohjeet, kun valmistetta käytetään yhdessä lenalidomidin ja deksametasonin kanssa
Hematologinen toksisuus | Suositeltavat toimet |
Trombosytopenia (trombosyyttiarvo) | |
Trombosyyttiarvo < 30 x 109/l |
|
Neutropenia (absoluuttinen neutrofiiliarvo) | |
Absoluuttinen neutrofiiliarvo < 0,5 x 109/l |
|
Ei-hematologinen toksisuus | Suositeltavat toimet |
Ihottuma | |
Aste† 2 tai 3 |
|
Aste 4 | Hoito lopetetaan. |
Perifeerinen neuropatia | |
Asteen 1 perifeerinen neuropatia ja kipuja tai Asteen 2 perifeerinen neuropatia |
|
Asteen 2 perifeerinen neuropatia ja kipuja tai Asteen 3 perifeerinen neuropatia |
|
Asteen 4 perifeerinen neuropatia | Hoito lopetetaan. |
Muut ei-hematologiset toksisuudet | |
Muut Asteen 3 tai 4 ei-hematologiset toksisuudet |
|
*Jos toksisuus uusiutuu, lenalidomidiannosta ja iksatsomibi-annosta muutetaan vuorotellen.
†Vaikeusasteet perustuvat Yhdysvaltain National Cancer Institute ‑instituutin Common Terminology Criteria ‑kriteerien (CTCAE) versioon 4.03.
Muiden lääkevalmisteiden samanaikainen käyttö
Iksatsomibi-hoitoa saavien potilaiden kohdalla on harkittava profylaktisen viruslääkityksen käyttöä vyöruusun uudelleenaktivoitumisriskin pienentämiseksi. Iksatsomibi-tutkimuksissa vyöruusun ilmaantuvuus oli pienempi potilailla, jotka saivat profylaktista viruslääkitystä, kuin niillä, joille profylaktista lääkitystä ei annettu.
Iksatsomibi-valmisteen, lenalidomidin ja deksametasonin yhdistelmähoitoa saaville potilaille suositellaan potilaan taustariskien ja kliinisen tilan mukaista tromboosiprofylaksia.
Muut mahdollisesti tarvittavat samanaikaiset lääkevalmisteet, ks. lenalidomidin ja deksametasonin voimassa olevat valmisteyhteenvedot.
Erityisryhmät
Iäkkäät
Yli 65-vuotiaiden potilaiden iksatsomibiannosta ei tarvitse muuttaa.
Yli 75-vuotiaiden potilaiden joukosta 13 iksatsomibiryhmän potilasta (28 %) ja 10 lumeryhmän potilasta (16 %) keskeytti hoidon. Yli 75-vuotiaiden potilaiden joukossa 10:llä iksatsomibiryhmän potilaalla (21 %) ja 9:llä lumeryhmän potilaalla (15 %) todettiin sydämen rytmihäiriöitä.
Maksan vajaatoiminta
Iksatsomibiannosta ei tarvitse muuttaa, jos potilaalla on lievä maksan vajaatoiminta (kokonaisbilirubiini ≤ viitearvojen yläraja [ULN] ja aspartaattiaminotransferaasi [ASAT] > ULN tai kokonaisbilirubiini > 1–1,5 x ULN ja ASAT mikä tahansa). Pienennettyä annosta, 3 mg, suositellaan, jos potilaalla on keskivaikea (kokonaisbilirubiini > 1,5–3 x ULN) tai vaikea (kokonaisbilirubiini > 3 x ULN) maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Iksatsomibiannosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma ≥ 30 ml/min). Pienennettyä annosta, 3 mg, suositellaan, jos potilaalla on vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma < 30 ml/min) tai dialyysihoitoa vaativa loppuvaiheen munuaissairaus (ESRD). Iksatsomibi ei dialysoidu, joten se voidaan antaa dialyysiajankohdasta riippumatta (ks. kohta Farmakokinetiikka).
Annostelusuositukset munuaisten vajaatoimintapotilaille, ks. lenalidomidin valmisteyhteenveto.
Pediatriset potilaat
Iksatsomibi-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Iksatsomibi otetaan suun kautta.
Iksatsomibi otetaan suunnilleen samaan kellonaikaan kunkin hoitojakson päivinä 1, 8 ja 15, viimeistään 1 tuntia ennen ruokaa tai aikaisintaan 2 tuntia ruoan jälkeen (ks. kohta Farmakokinetiikka). Kapseli nielaistaan kokonaisena veden kera. Sitä ei saa murskata, pureskella eikä avata (ks. kohta Käyttö- ja käsittelyohjeet).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Iksatsomibi-valmistetta käytetään yhdessä lenalidomidin ja deksametasonin kanssa; muut vasta-aiheet, ks. näiden lääkevalmisteiden valmisteyhteenvedot.
Varoitukset ja käyttöön liittyvät varotoimet
Iksatsomibi-valmistetta käytetään yhdessä lenalidomidin ja deksametasonin kanssa; muut varoitukset ja käyttöön liittyvät varotoimet, ks. näiden lääkevalmisteiden valmisteyhteenvedot.
Trombosytopenia
Iksatsomibihoidon yhteydessä on raportoitu trombosytopeniaa (ks. kohta Haittavaikutukset). Trombosyyttiarvojen nadiiri saavutetaan tyypillisesti kunkin 28-päiväisen hoitojakson päivien 14–21 välillä, ja arvot korjautuvat lähtötasolle seuraavan hoitojakson alkuun mennessä (ks. kohta Haittavaikutukset).
Trombosyyttiarvoja on seurattava iksatsomibihoidon aikana vähintään kerran kuukaudessa. Tiiviimpää seurantaa on harkittava ensimmäisten kolmen hoitojakson aikana lenalidomidin valmisteyhteenvedon mukaisella tavalla. Trombosytopeniaa voidaan hoitaa annosmuutoksin (ks. kohta Annostus ja antotapa) ja trombosyyttisiirroin tavanomaisten hoitosuositusten mukaisesti.
Ruoansulatuskanavaan kohdistuva toksisuus
Iksatsomibi-hoidon yhteydessä on raportoitu ripulia, ummetusta, pahoinvointia ja oksentelua, jotka ovat joskus vaatineet pahoinvointi- ja ripulilääkkeitä ja tukihoitoja (ks. kohta Haittavaikutukset). Annosta on muutettava, jos potilaalla on vaikeita (aste 3–4) oireita (ks. kohta Annostus ja antotapa). Jos potilaalla on vaikeita ruoansulatuskanavan oireita, on suositeltavaa seurata seerumin kaliumpitoisuutta.
Perifeerinen neuropatia
Iksatsomibi-valmisteen käytön yhteydessä on raportoitu perifeeristä neuropatiaa (ks. kohta Haittavaikutukset).Potilaita on seurattava perifeerisen neuropatian oireiden varalta. Perifeerisen neuropatian ilmaantuminen tai paheneminen voi vaatia annoksen muuttamista (ks. kohta Annostus ja antotapa).
Ääreisosien turvotus
Iksatsomibi-valmisteen käytön yhteydessä on raportoitu ääreisosien turvotusta (ks. kohta Haittavaikutukset). Mahdolliset perussyyt on arvioitava ja potilaalle on annettava tukihoitoa tarpeen mukaan. Jos potilaalla on asteen 3 tai 4 oireita, deksametasonin annosta on muutettava kyseisen valmisteen valmisteyhteenvedon mukaisesti, tai iksatsomibi-valmisteen annosta on muutettava (ks. kohta Annostus ja antotapa).
Ihoreaktiot
Iksatsomibin käytön yhteydessä on raportoitu ihottumaa (ks. kohta Haittavaikutukset). Ihottuman tukihoito on tarpeen, tai jos potilaalla on vähintään asteen 2 oireita, annosta muutetaan (ks. kohta Annostus ja antotapa). Iksatsomibihoidon yhteydessä on harvoin raportoitu myös vaikeita ihoon liittyviä haittavaikutuksia (severe cutaneous adverse reacion; SCAR), mukaan lukien toksinen epidermaalinen nekrolyysi (TEN) ja Stevens–Johnsonin oireyhtymä (SJS), jotka voivat olla henkeä uhkaavia tai kuolemaan johtavia (ks. kohta Haittavaikutukset).
Iksatsomibia määrättäessä potilaille pitää kertoa ihoreaktioiden merkeistä ja oireista ja heidän tilaansa on seurattava tarkasti niiden varalta. Jos näihin reaktioihin viittaavia merkkejä ja oireita ilmenee, iksatsomibin käyttö on lopetettava välittömästi ja harkittava vaihtoehtoista hoitoa (mikäli se on mahdollista).
Jos potilaalle on kehittynyt vaikea reaktio, kuten SJS tai TEN, iksatsomibin käytön yhteydessä, iksatsomibihoitoa ei saa enää aloittaa uudelleen kyseiselle potilaalle.
Tromboottinen mikroangiopatia
Tromboottisen mikroangiopatian (TMA) tapauksia, mukaan lukien tromboottista trombosytopeenistä purppuraa / hemolyyttis-ureemista oireyhtymää (TTP/HUS), on raportoitu iksatsomibia saaneilla potilailla. Osa tapahtumista on johtanut kuolemaan. Tromboottisen mikroangiopatian merkkien ja oireiden ilmaantumista pitää seurata. Jos tätä diagnoosia epäillään, lopeta iksatsomibin anto ja arvioi, onko potilaalla mahdollisesti tromboottinen mikroangiopatia. Jos tromboottinen mikroangiopatia suljetaan pois, iksatsomibin käyttö voidaan aloittaa uudelleen. Ei ole tiedossa, onko iksatsomibin käytön uudelleen aloittaminen turvallista, jos potilaalla on aiemmin ollut tromboottinen mikroangiopatia.
Maksatoksisuus
Iksatsomibi-valmisteen käytön yhteydessä on melko harvinaisena raportoitu lääkkeen aiheuttamia maksavaurioita, maksasoluvaurioita, rasvamaksaa, kolestaattista hepatiittia ja maksatoksisuutta (ks. kohta Haittavaikutukset). Maksaentsyymejä on seurattava säännöllisesti, ja jos potilaalla on asteen 3 tai 4 oireita, annosta on muutettava (ks. kohta Annostus ja antotapa).
Raskaus
Naisten on vältettävä raskautta iksatsomibi-hoidon aikana. Jos iksatsomibi-valmistetta käytetään raskauden aikana tai raskaus alkaa iksatsomibihoidon aikana, potilaalle on kerrottava sikiöön mahdollisesti kohdistuvista riskeistä.
Hedelmällisessä iässä olevien naisten on käytettävä erittäin tehokasta ehkäisyä iksatsomibi-hoidon aikana ja 90 päivää hoidon päättymisen jälkeen (ks. kohdat Yhteisvaikutukset ja Raskaus ja imetys). Hormonaalista ehkäisyä käyttävien naisten on käytettävä lisäehkäisynä jotakin estemenetelmää.
Posteriorinen reversiibeli enkefalopatiaoireyhtymä
Posteriorista reversiibeliä enkefalopatiaoireyhtymää (pres) on ilmennyt iksatsomibi-valmistetta käyttävillä potilailla. PRES on harvinainen, palautuva neurologinen häiriö, jonka oireita voivat olla kouristuskohtaukset, verenpaineen kohoaminen, päänsärky, tajunnantilan muutokset ja näköhäiriöt. Diagnoosi vahvistetaan aivojen kuvauksella, mieluiten magneettikuvauksella. Jos potilaalle kehittyy PRES, iksatsomibi-valmisteen käyttö lopetetaan.
Vahvat CYP3A4:n induktorit
Vahvat induktorit saattavat heikentää iksatsomibi-valmisteen tehoa, joten voimakkaiden CYP3A:n induktorien, kuten karbamatsepiinin, fenytoiinin, rifampisiinin ja mäkikuisman (Hypericum perforatum), samanaikaista käyttöä on vältettävä (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka). Potilaiden tilaa pitää seurata huolellisesti sairauden hallinnassa pysymisen selvittämiseksi, jos samanaikaista vahvan CYP3A4:n induktorin käyttöä ei voida välttää.
Yhteisvaikutukset
Farmakokineettiset yhteisvaikutukset
CYP-entsyymien estäjät
Iksatsomibin käyttö samanaikaisesti klaritromysiinin (vahva CYP3A:n estäjä) kanssa ei muuttanut systeemistä iksatsomibialtistusta kliinisesti merkittävästi. Iksatsomibin Cmax pieneni 4 % ja AUC-arvo suureni 11 %. Iksatsomibiannosta ei siis tarvitse muuttaa, jos samanaikaisesti käytetään vahvoja CYP3A:n estäjiä.
Populaatiofarmakokinetiikan analyysin perusteella iksatsomibin käyttö samanaikaisesti vahvojen CYP1A2:n estäjien kanssa ei muuttanut systeemistä iksatsomibialtistusta kliinisesti merkittävästi. Iksatsomibiannosta ei siis tarvitse muuttaa, jos samanaikaisesti käytetään vahvoja CYP1A2:n estäjiä.
CYP-entsyymien induktorit
Iksatsomibin käyttö samanaikaisesti rifampisiinin kanssa pienensi iksatsomibin Cmax-arvoa 54 % ja AUC-arvoa 74 %. Vahvojen CYP3A:n induktorien ja iksatsomibin samanaikainen käyttö ei siis ole suositeltavaa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Iksatsomibin vaikutus muihin lääkevalmisteisiin
Iksatsomibi ei estä CYP-isoentsyymien 1A2, 2B6, 2C8, 2C9, 2C19, 2D6 eikä 3A4/5 toimintaa palautuvasti eikä aikariippuvaisesti. Iksatsomibi ei indusoinut CYP1A2-, CYP2B6- eikä CYP3A4/5-toimintaa eikä vastaavia immunoreaktiivisten proteiinien pitoisuuksia. Iksatsomibi ei oletettavasti aiheuta CYP-toiminnan estosta eikä indusoitumisesta johtuvia lääkeaineinteraktioita.
Kuljettajaproteiineista johtuvat yhteisvaikutukset
Iksatsomibi on heikon affiniteetin omaava P-gp:n substraatti. Iksatsomibi ei ole BCRP:n, MRP2:n eikä maksan OATP-kuljettajien substraatti. Iksatsomibi ei estä P-gp-, BCRP-, MRP2-, OATP1B1-, OATP1B3-, OCT2-, OAT1-, OAT3-, MATE1- eikä MATE2-K-toimintaa. Iksatsomibi ei oletettavasti aiheuta kuljettajaproteiinivälitteisiä lääkeaineinteraktioita.
Ehkäisytabletit
Deksametasonin tiedetään olevan heikko tai kohtalainen CYP3A4:n ja muiden entsyymien ja kuljettajaproteiinien induktori. Koska iksatsomibi-valmistetta käytetään yhdessä deksametasonin kanssa, on otettava huomioon, että ehkäisytablettien teho voi heikentyä. Hormonaalista ehkäisyä käyttävien naisten on käytettävä lisäehkäisynä jotakin estemenetelmää.
Raskaus ja imetys
Iksatsomibi-valmistetta käytetään yhdessä lenalidomidin ja deksametasonin kanssa; muut hedelmällisyyttä, raskautta ja imetystä koskevat seikat, ks. näiden lääkevalmisteiden valmisteyhteenvedot.
Hedelmällisessä iässä olevat naiset / Ehkäisy miehille ja naisille
Mies- ja naispotilaiden, jotka voivat saada lapsia, on käytettävä tehokasta ehkäisyä hoidon aikana ja 90 päivän ajan hoidon päättymisen jälkeen. iksatsomibi-valmisteen käyttöä ei suositella sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Deksametasonin tiedetään olevan heikko tai kohtalainen CYP3A4:n ja muiden entsyymien ja kuljettajaproteiinien induktori. Koska iksatsomibi-valmistetta käytetään yhdessä deksametasonin kanssa, on otettava huomioon, että ehkäisytablettien teho voi heikentyä. Tämän vuoksi ehkäisytabletteja käyttävien naisten on käytettävä lisäehkäisynä jotakin estemenetelmää.
Raskaus
Iksatsomibin käyttöä ei suositella raskauden aikana, sillä käyttö raskauden aikana voi aiheuttaa haittaa sikiölle. Naisten on siis vältettävä raskautta iksatsomibi-hoidon aikana.
Iksatsomibin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Iksatsomibi-valmistetta käytetään yhdessä lenalidomidin kanssa. Lenalidomidi on rakenteellisesti sukua talidomidille. Talidomidi on tunnettu ihmiselle teratogeeninen vaikuttava aine, joka aiheuttaa vaikeita, henkeä uhkaavia kehityshäiriöitä. Lenalidomidin käytön raskauden aikana odotetaan johtavan ihmisellä teratogeenisiin vaikutuksiin. Lenalidomidin raskaudenehkäisyohjelman ehtojen on täytyttävä kaikkien potilaiden kohdalla, ellei ole luotettavaa näyttöä siitä, ettei potilas voi saada lapsia. Ks. lenalidomidin ajantasainen valmisteyhteenveto.
Imetys
Ei tiedetä, erittyvätkö iksatsomibi tai sen metaboliitit ihmisen rintamaitoon. Tietoja eläimillä tehdyistä tutkimuksista ei ole saatavilla. Imettävään vauvaan kohdistuvia riskejä ei voida sulkea pois, joten imetys on lopetettava.
Iksatsomibi-valmistetta annetaan yhdistelmänä lenalidomidin kanssa, joten imetys on lopetettava lenalidomidin käytön vuoksi.
Hedelmällisyys
Iksatsomibi-valmisteen vaikutusta hedelmällisyyteen ei ole tutkittu (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Iksatsomibi-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Kliinisissä tutkimuksissa on todettu uupumusta ja huimausta. Potilaille on kerrottava, että heidän ei pidä ajaa eikä käyttää koneita, jos heillä esiintyy näitä oireita.
Haittavaikutukset
Iksatsomibi-valmistetta käytetään yhdessä lenalidomidin ja deksametasonin kanssa. Muut haittavaikutukset, ks. näiden lääkevalmisteiden valmisteyhteenvedot.
Turvallisuusprofiilin yhteenveto
NINLAROn turvallisuusprofiili perustuu saatavilla oleviin kliinisistä tutkimuksista saatuihin tietoihin sekä markkinoille tulon jälkeen saatuun kokemukseen. Alla sekä Taulukossa 3 kuvatut haittavaikutusten esiintymistiheydet on määritetty kliinisistä tutkimuksista saatujen tietojen perusteella.
Ellei toisin ole ilmoitettu, alla esitetyt tiedot ovat yhdistettyjä turvallisuustietoja faasin 3 globaalista C16010‑avaintutkimuksesta (n = 720) ja kaksoissokkoutetusta, lumekontrolloidusta, Kiinassa toteutetusta C16010‑jatkotutkimuksesta (n = 115). 418 potilaan iksatsomibihoitoryhmässä ja 417 potilaan lumehoitoryhmässä yleisimmin raportoituja haittavaikutuksia (≥ 20 %) olivat ripuli (47 % iksatsomibi‑ ja 38 % lumeryhmässä), trombosytopenia (41 % ja 24 %), neutropenia (37 % ja 36 %), ummetus (31 % ja 24 %), ylähengitystieinfektio (28 % ja 24 %), perifeerinen neuropatia (28 % ja 22 %), pahoinvointi (28 % ja 20 %), selkäkipu (25 % ja 21 %), ihottuma (25 % ja 15 %), ääreisosien turvotus (24 % ja 19 %), oksentelu (23 % ja 12 %) ja keuhkoputkitulehdus (20 % ja 15 %). Vakavia haittavaikutuksia, joita ilmoitettiin ≥ 2 %:lla potilaista, olivat ripuli (3 %), trombosytopenia (2 %) ja keuhkoputkitulehdus (2 %).
Haittavaikutustaulukko
Haittavaikutusten esiintymistiheydet on luokiteltu seuraavan määritelmän mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Kussakin elinjärjestelmäluokassa haittavaikutukset on luokiteltu esiintymistiheyden mukaan, yleisimmät ensin mainittuina. Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3: Haittavaikutukset potilailla, jotka saivat iksatsomibia yhdessä lenalidomidin ja deksametasonin kanssa (kaikki asteet, aste 3 ja aste 4)
| Elinjärjestelmä/haittavaikutus | Haittavaikutukset (kaikki asteet) | Asteen 3 haittavaikutukset | Asteen 4 haittavaikutukset |
| Infektiot | |||
| Ylähengitystieinfektio | Hyvin yleinen | Yleinen | |
| Keuhkoputkitulehdus | Hyvin yleinen | Yleinen | |
| Vyöruusu | Yleinen | Yleinen | |
| Veri ja imukudos | |||
| Trombosytopenia* | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Neutropenia* | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Tromboottinen mikroangiopatia | Harvinainen | Harvinainen | |
| Tromboottinen trombosytopeeninen purppura† | Harvinainen | Harvinainen | Harvinainen |
| Immuunijärjestelmä | |||
| Anafylaktinen reaktio† | Harvinainen | Hyvin harvinainen | Hyvin harvinainen |
| Angioedeema† | Harvinainen | Harvinainen | |
| Aineenvaihdunta ja ravitsemus | |||
| Tuumorilyysioireyhtymä† | Harvinainen | Harvinainen | Harvinainen |
| Hermosto | |||
| Perifeerinen neuropatia* | Hyvin yleinen | Yleinen | |
| Posteriorinen reversiibeli enkefalopatiaoireyhtymä*† | Harvinainen | Harvinainen | Harvinainen |
| Transversaalimyeliitti† | Harvinainen | Harvinainen | |
| Ruoansulatuselimistö | |||
| Ripuli | Hyvin yleinen | Yleinen | |
| Ummetus | Hyvin yleinen | Melko harvinainen | |
| Pahoinvointi | Hyvin yleinen | Yleinen | |
| Oksentelu | Hyvin yleinen | Melko harvinainen | |
| Iho ja ihonalainen kudos | |||
| Ihottuma* | Hyvin yleinen | Yleinen | |
| Stevens-Johnsonin oireyhtymä† | Harvinainen | Harvinainen | |
| Akuutti kuumeinen neutrofiilinen dermatoosi | Harvinainen | Harvinainen | |
| Toksinen epidermaalinen nekrolyysi† | Harvinainen | Harvinainen | |
| Luusto, lihakset ja sidekudos | |||
| Selkäkipu | Hyvin yleinen | Melko harvinainen | |
| Artralgia | Hyvin yleinen | Yleinen | |
| Yleisoireet ja antopaikassa todettavat haitat | |||
| Ääreisosien turvotus | Hyvin yleinen | Yleinen | |
| Pyreksia | Hyvin yleinen | Melko harvinainen | |
*Yhdistetty useista haittavaikutustermeistä † Raportoitu faasin 3 tutkimusten ulkopuolella | |||
Tiettyjen haittavaikutusten kuvaus
Hoidon keskeyttäminen
Niiden iksatsomibi-ryhmän potilaiden osuus, joilla yksi tai useampia käytetyistä kolmesta lääkevalmisteesta lopetettiin, oli kaikkien haittavaikutusten kohdalla ≤ 3 %.
Trombosytopenia
Trombosyyttiarvon laskua tasolle ≤ 10 x 109/l todettiin hoidon aikana 2 %:lla sekä iksatsomibiryhmän että lumeryhmän potilaista. Molemmissa hoitoryhmissä trombosyyttiarvo laski hoidon aikana tasolle ≤ 5 x 109/l alle 1 %:lla potilaista. Iksatsomibiryhmässä 2 % ja lumeryhmässä 3 % potilaista lopetti yhden tai useampia käytetyistä kolmesta lääkevalmisteesta trombosytopenian vuoksi. Trombosytopenia ei johtanut verenvuototapahtumien eikä trombosyyttisiirtojen lisääntymiseen.
Ruoansulatuskanavaan kohdistuva toksisuus
Iksatsomibiryhmässä 2 % ja lumeryhmässä 1 % potilaista lopetti yhden tai useampia käytetyistä kolmesta lääkevalmisteesta ripulin vuoksi.
Ihottuma
Ihottumaa esiintyi 25 %:lla iksatsomibiryhmän potilaista ja 15 %:lla lumeryhmän potilaista. Molemmissa ryhmissä yleisin ilmoitettu ihottumatyyppi oli makulopapulaarinen tai makulaarinen ihottuma. Asteen 3 ihottumaa ilmoitettiin 3 %:lla iksatsomibiryhmän potilaista ja 2 %:lla lumeryhmän potilaista. Molemmissa hoitoryhmissä < 1 % potilaista lopetti yhden tai useampia käytetyistä kolmesta lääkevalmisteesta ihottuman vuoksi.
Perifeerinen neuropatia
Perifeeristä neuropatiaa esiintyi 28 %:lla iksatsomibiryhmän potilaista ja 22 %:lla lumeryhmän potilaista. Asteen 3 perifeeristä neuropatiaa ilmoitettiin haittavaikutuksena 2 %:lla iksatsomibiryhmän potilaista ja 1 %:lla lumeryhmän potilaista. Yleisimmin ilmoitettu haitta oli perifeerinen sensorinen neuropatia (21 % iksatsomibi‑ ja 15 % lumeryhmässä). Perifeeristä motorista neuropatiaa ei ilmoitettu yleisesti kummassakaan ryhmässä (< 1 %). Iksatsomibiryhmässä 3 % potilaista ja lumeryhmässä < 1 % potilaista lopetti yhden tai useampia käytetyistä kolmesta lääkevalmisteesta perifeerisen neuropatian vuoksi.
Silmät
Silmävaivoja ilmoitettiin monilla eri termeillä, mutta yhdessä tarkasteltuna esiintymistiheys oli iksatsomibiryhmän potilailla 34 % ja lumeryhmän potilailla 28 %. Yleisimmät haittavaikutukset olivat näön hämärtyminen (iksatsomibi‑ryhmässä 6 % ja lumeryhmässä 5 %), kuivasilmäisyys (iksatsomibiryhmässä 6 % ja lumeryhmässä 1 %), sidekalvotulehdus (iksatsomibiryhmässä 8 % ja lumeryhmässä 2 %) ja kaihi (iksatsomibiryhmässä 13 % ja lumeryhmässä 17 %). Asteen 3 haittavaikutuksia ilmoitettiin 6 %:lla iksatsomibiryhmän potilaista ja 8 %:lla lumeryhmän potilaista.
Muut haittavaikutukset
Faasin 3 globaalin C16010‑avaintutkimuksen (n = 720) ja kaksoissokkoutetun, lumekontrolloidun, Kiinassa toteutetun C16010‑jatkotutkimuksen (n = 115) yhdistetyissä tiedoissa seuraavia haittavaikutuksia esiintyi yhtä usein iksatsomibi‑ ja lumeryhmissä: uupumus (28 % iksatsomibi‑ ja 26 % lumeryhmässä), ruokahalun heikkeneminen (13 % ja 11 %), hypotensio (5 % ja 4 %), sydämen vajaatoiminta† (5 % molemmissa ryhmissä), rytmihäiriöt† (17 % ja 16 % ) ja maksan toimintahäiriö† (mukaan lukien maksaentsyymiarvojen muutokset; 11 % ja 9 %).
Vaikeiden (asteen 3–4) hypokalemiatapahtumien esiintymistiheys oli suurempi iksatsomibiryhmässä (7 %) kuin lumeryhmässä (2 %).
Kuolemaan johtaneita sieni- tai virusperäisiä keuhkokuumeita ilmoitettiin harvoin iksatsomibi-valmisteen, lenalidomidin ja deksametasonin yhdistelmähoitoa saaneilla potilailla.
†Vakioidut MedDRA-hakulausekkeet (SMQ)
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostusta on ilmoitettu NINLARO-valmistetta käyttävillä potilailla. Yliannostuksen oireet ovat yleisesti ottaen vastaavia kuin NINLARO-valmisteen tunnetut riskit (ks. kohta Haittavaikutukset). 12 mg:n yliannoksesta (otettu kerralla) on seurannut vakavia haittavaikutuksia, kuten vaikeaa pahoinvointia, aspiraatiokeuhkokuumetta, monielinhäiriötä ja kuolema.
Iksatsomibin yliannokselle ei tunneta spesifistä vastalääkettä. Yliannostustapauksessa potilaan tilaa seurataan tarkasti haittavaikutusten varalta (ks. kohta Haittavaikutukset) ja hänelle annetaan asianmukaista elintoimintoja tukevaa hoitoa. Iksatsomibi ei dialysoidu (ks. kohta Farmakokinetiikka).
Yliannostus oli yleisintä NINLARO-valmisteen käyttöä aloittavilla potilailla. Hoitoa aloittavien potilaiden kanssa pitää keskustella annostusohjeiden huolellisen noudattamisen tärkeydestä. Neuvo potilaita ottamaan suositeltu annos ohjeiden mukaan, sillä yliannostus on johtanut kuolemaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Solunsalpaajat, muut syöpälääkkeet, ATC-koodi: L01XG03
Vaikutusmekanismi
Iksatsomibisitraatti on aihiolääke, joka hydrolysoituu fysiologisissa oloissa nopeasti biologisesti aktiivisen iksatsomibin muotoon.
Iksatsomibi on suun kautta otettava, erittäin selektiivinen, reversiibeli proteasomin estäjä. Iksatsomibi sitoutuu ensisijaisesti 20S-proteasomin beeta-5-alayksikköön ja estää sen kymotrypsiinin kaltaista toimintaa.
Iksatsomibi indusoi useiden kasvainsolutyyppien apoptoosia in vitro. Iksatsomibilla todettiin olevan in vitro sytotoksinen vaikutus myeloomasoluihin, joita oli kerätty potilailta, joiden tauti oli uusiutunut useiden aiempien hoitojen (mm. bortetsomibin, lenalidomidin ja deksametasonin) jälkeen. Iksatsomibin ja lenalidomidin yhdistelmähoidolla todettiin olevan synergistinen sytotoksinen vaikutus multippelin myelooman solulinjoihin. Iksatsomibi esti kasvainten kasvua in vivo erilaisissa ksenograftikasvainmalleissa, mm. multippelin myelooman malleissa. In vitro iksatsomibi vaikutti luuytimen mikroympäristössä esiintyviin solutyyppeihin, mm. verisuonten endoteelisoluihin, osteoklasteihin ja osteoblasteihin.
Sydämen elektrofysiologia
Farmakokinetiikan ja farmakodynamiikan analyysissä, jossa oli mukana 245 potilasta, iksatsomibi ei pidentänyt QTc-aikaa kliinisesti merkittäviä altistuksia käytettäessä. Mallinnukseen perustuvan analyysin mukaan QTcF-ajan keskimuutos lähtötilanteesta oli 4 mg:n annoksilla arviolta 0,07 ms (90 % lv -0,22, 0,36). Iksatsomibipitoisuuksien ja RR-välin pituuden välillä ei ollut havaittavaa yhteyttä, mikä viittaa siihen, että iksatsomibilla ei ole kliinisesti merkittävää vaikutusta syketiheyteen.
Kliininen teho ja turvallisuus
Iksatsomibi-valmisteen tehoa ja turvallisuutta yhdessä lenalidomidin ja deksametasonin kanssa arvioitiin kansainvälisessä, satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa, superioriteettia arvioineessa faasin 3 monikeskustutkimuksessa (C16010) potilailla, joilla oli uusiutunut ja/tai refraktaarinen multippeli myelooma ja jotka olivat saaneet vähintään yhtä aiempaa hoitoa. Yhteensä 722 potilasta (lähtöryhmien mukainen populaatio [ITT]) satunnaistettiin suhteessa 1:1 saamaan joko iksatsomibi-valmisteen, lenalidomidin ja deksametasonin yhdistelmää (N = 360; iksatsomibihoito) tai lumelääkkeen, lenalidomidin ja deksametasonin yhdistelmää (N = 362; lumehoito), kunnes tauti eteni tai potilas ei enää sietänyt hoitoa. Tutkimukseen otettujen potilaiden multippeli myelooma oli refraktaarinen (mahdollisesti primaaristi refraktaarinen), uusiutunut aiemman hoidon jälkeen tai uusiutunut ja resistentti jollekin aiemmalle hoidolle. Tutkimukseen hyväksyttiin potilaita, jotka vaihtoivat hoitoa ennen taudin etenemistä, ja potilaita, joilla oli hallinnassa oleva sydän- tai verisuonisairaus. Faasin 3 tutkimuksesta suljettiin pois potilaat, jotka olivat resistenttejä lenalidomidille tai proteasomin estäjille tai jotka olivat saaneet yli kolmea aiempaa hoitoa. Tässä tutkimuksessa resistentin taudin määritelmänä oli taudin eteneminen hoidon aikana tai 60 vuorokauden kuluessa viimeisen lenalidomidiannoksen tai proteasomin estäjän annoksen jälkeen. Näistä potilasryhmistä on niukasti tietoa, joten on suositeltavaa tehdä huolellinen riski-hyötyarvio ennen iksatsomibihoidon aloittamista.
Tromboosiprofylaksia suositeltiin molemmissa hoitoryhmissä kaikille potilaille lenalidomidin valmisteyhteenvedon mukaisesti. Potilaat saivat samanaikaisesti muita lääkevalmisteita, kuten pahoinvointi- ja viruslääkkeitä ja antihistamiineja, lääkärin harkinnan mukaan ennaltaehkäisevästi ja/tai oireiden hoitoon.
Potilaat saivat 4 mg iksatsomibi-valmistetta tai lumelääkettä 28-päiväisten hoitojaksojen päivinä 1, 8 ja 15 sekä lenalidomidia (25 mg) päivinä 1–21 ja deksametasonia (40 mg) päivinä 1, 8, 15 ja 22. Munuaisten vajaatoimintapotilaille annettiin lenalidomidin valmisteyhteenvedon mukainen aloitusannos lenalidomidia. Hoitoa jatkettiin, kunnes tauti eteni tai potilas ei enää sietänyt hoitoa.
Hoitoryhmien demografiset tiedot ja taudin piirteet olivat lähtötilanteessa tasapainossa ja vertailukelpoiset. Mediaani-ikä oli 66 v (vaihteluväli 38–91 v), ja 58 % potilaista oli yli 65-vuotiaita. Potilaista 57 % oli miehiä. Potilaista 85 % oli valkoihoisia, 9 % aasialaisia ja 2 % mustia. ECOG-toimintakykyluokka oli 93 %:lla potilaista 0–1, ja 12 %:lla tauti oli lähtötilanteessa ISS-luokkaa III (N = 90). Kreatiniinipuhdistuma oli 25 %:lla potilaista < 60 ml/min. Potilaista 23 %:lla oli kevytketjutauti, ja 12 %:lla potilaista tauti oli mitattavissa vain vapaiden kevytketjujen määrityksellä. 19 %:lla oli suuren riskin sytogeneettisiä poikkeavuuksia (del[17], t[4;14], t[14;16], N = 137), 10 %:lla oli del(17) (N = 69) ja 34 %:lla oli 1q-amplifikaatio (1q21) (N = 247). Potilaat olivat saaneet aiemmin 1–3 hoitoa (mediaani 1); näihin kuuluivat aiempi bortetsomibihoito (69 %), karfiltsomibihoito (< 1 %), talidomidihoito (45 %), lenalidomidihoito (12 %) ja melfalaanihoito (81 %). Potilaista 57 %:lle oli tehty aiemmin kantasolusiirto. Potilaista 77 %:lla tauti oli uusiutunut aiempien hoitojen jälkeen, ja 11 % oli resistenttejä aiemmille hoidoille. Kaikkiaan 6 %:lla potilaista oli dokumentoitu primaaristi refraktaarinen tauti (määritelmä: kaikkien aiempien hoitojen parhaana vasteena stabiili tauti tai taudin eteneminen).
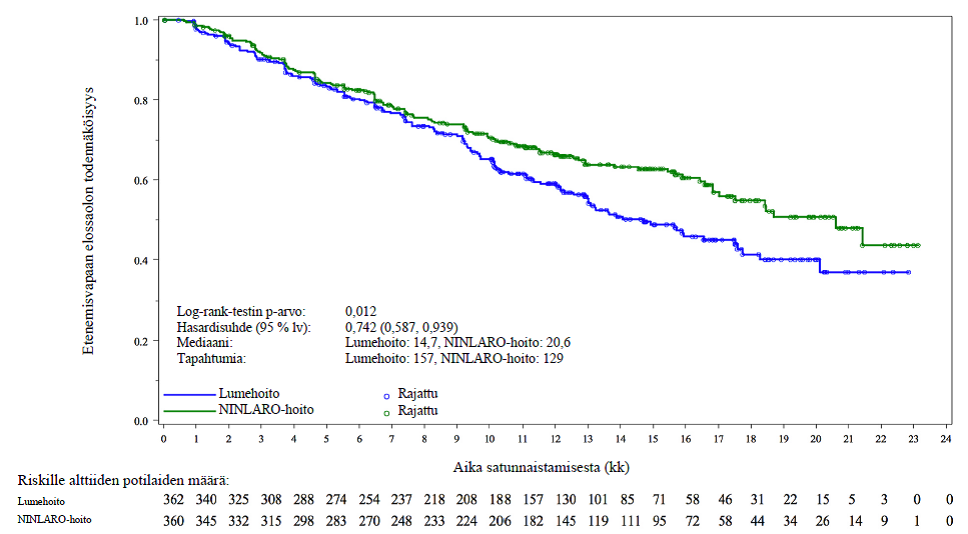
Ensisijainen päätetapahtuma oli etenemisvapaa elossaoloaika (PFS) International Myeloma Working Group ‑työryhmän (IMWG) vuoden 2011 Consensus Uniform Response Criteria ‑vastekriteereillä arvioituna. Riippumaton arviointilautakunta arvioi vastekriteerien täyttymisen sokkoutetusti keskuslaboratoriosta saatujen tulosten perusteella. Vaste arvioitiin 4 viikon välein taudin etenemiseen asti. Ensisijaisessa analyysissa (seurannan mediaanikesto 14,7 kk ja hoitojaksojen mediaani 13) etenemisvapaassa elossaoloajassa havaittiin tilastollisesti merkitsevä ero hoitoryhmien välillä. Etenemisvapaan elinajan tulokset esitetään yhteenvetona taulukossa 4 ja kuvassa 1. Iksatsomibi-ryhmässä kokonaisvasteprosentti oli parempi, mikä tuki etenemisvapaan elinajan pitenemistä.
Taulukko 4: Etenemisvapaa elinaika ja vastetulokset multippelia myeloomaa sairastavilla potilailla, jotka saivat iksatsomibi- tai lumelääkettä yhdessä lenalidomidin ja deksametasonin kanssa (lähtöryhmien mukainen populaatio, intent-to-treat, ensisijainen analyysi)
| Iksatsomibi + lenalidomidi ja deksametasoni (n = 360) | Lume + lenalidomidi ja deksametasoni (n = 362) | ||
Etenemisvapaa elossaoloaika | ||||
Tapahtumia, n (%) | 129 (36) | 157 (43) | ||
Mediaani (kk) | 20,6 | 14,7 | ||
p-arvo* | 0,012 | |||
Riskisuhde† (95 % lv) | 0,74 (0,59; 0,94) | |||
Kokonaisvaste‡, n (%) | 282 (78,3) | 259 (71,5) | ||
Vasteluokka, n (%) | ||||
Täydellinen vaste | 42 (11,7) | 24 (6,6) | ||
Erittäin hyvä osittainen vaste | 131 (36,4) | 117 (32,3) | ||
Osittainen vaste | 109 (30,3) | 118 (32,6) | ||
Vasteen saavuttamiseen kulunut aika (kk) | ||||
Mediaani | 1,1 | 1,9 | ||
Vasteen kesto§, kk | ||||
Mediaani | 20,5 | 15,0 | ||
*P-arvo perustuu stratifioituun log-rank-testiin.
†Hasardisuhde perustuu stratifioituun Coxin suhteellisen riskin regressiomalliin. Riskisuhde alle 1 viittaa siihen, että iksatsomibi-hoito oli parempi.
‡Kokonaisvasteprosentti (ORR) = täydellinen vaste (CR) + erittäin hyvä osittainen vaste (VGPR) + osittainen vaste (PR)
§Perustuu vasteen saavuttaneisiin potilaisiin vasteen suhteen arviointikelpoisten potilaiden populaatiossa
Kuva 1: Etenemisvapaan elinajan Kaplan–Meier-kuvio, lähtöryhmien mukainen populaatio (ITT) (ensisijainen analyysi)
Toisessa, ei‑inferentiaalisessa etenemisvapaan elossaoloajan analyysissä seuranta‑ajan mediaani oli 23 kuukautta. Tässä analyysissa etenemisvapaan elossaoloajan mediaani oli iksatsomibihoitoryhmässä arviolta 20 kk ja lumehoitoryhmässä arviolta 15,9 kk (hr = 0,82 [95 % lv (0,67; 1,0)]) lähtöryhmien mukaisessa populaatiossa. Yhtä aiempaa hoitoa saaneilla potilailla etenemisvapaan elossaolojan mediaani oli 18,7 kuukautta iksatsomibiryhmässä ja 17,6 kuukautta lumeryhmässä (HR = 0,99). Kahta tai kolmea aiempaa hoitoa saaneilla potilailla etenemisvapaan elossaoloajan mediaani oli 22,0 kuukautta iksatsomibiryhmässä ja 13,0 kuukautta lumeryhmässä (HR = 0,62).
Kokonaiselinajan loppuanalyysissä, kun seurannan keston mediaani oli noin 85 kuukautta, kokonaiselinajan mediaani ITT‑populaatiossa oli 53,6 kuukautta iksatsomibiryhmän potilailla ja 51,6 kuukautta lumeryhmän potilailla (HR = 0,94 [95 % lv: 0,78, 1,13; p = 0,495]). Yhtä aiempaa hoitoa saaneilla potilailla kokonaiselinajan mediaani oli 54,3 kuukautta iksatsomibiryhmässä ja 58,3 kuukautta lumeryhmässä (HR = 1,02 [95 % lv: 0,80, 1,29]). Kahta tai kolmea aiempaa hoitoa saaneilla potilailla kokonaiselinajan mediaani oli 53,0 kuukautta iksatsomibiryhmässä ja 43,0 kuukautta lumeryhmässä (HR = 0,85 [95 % lv: 0,64, 1,11]).
Kiinassa toteutettiin samankaltaisella tutkimusasetelmalla ja samankaltaisin soveltuvuuskriteerein satunnaistettu, kaksoissokkoutettu, lumekontrolloitu, faasin 3 tutkimus (n = 115). Monella tutkimukseen otetuista potilaista oli taudin toteamishetkellä pitkälle edennyt, Durie–Salmonin asteen III tauti (69 %), anamneesissa vähintään 2 aikaisempaa hoitoa (60 %) ja talidomidille refraktaarinen tauti (63 %). Ensisijaisessa analyysissa (seurannan mediaani 8 kuukautta ja hoitojaksojen mediaani 6) etenemisvapaan elinajan mediaani oli iksatsomibi‑hoitoryhmässä 6,7 kuukautta ja lumeryhmässä 4 kuukautta (p‑arvo = 0,035; hr = 0,60). Lopullisessa kokonaiselinajan analyysissa (seurannan mediaani 19,8 kuukautta) kokonaiselinaika oli pidempi iksatsomibihoitoryhmässä kuin lumeryhmässä (p‑arvo = 0,0014; HR = 0,42 [95 % lv: 0,242–0,726]).
Koska multippeli myelooma on heterogeeninen sairaus, hoidosta saatu hyöty voi vaihdella eri alaryhmien välillä faasin 3 tutkimuksessa (C16010) (ks. Kuva 2).
Kuva 2. Etenemisvapaan elossaoloajan Forest plot -kaavio alaryhmissä
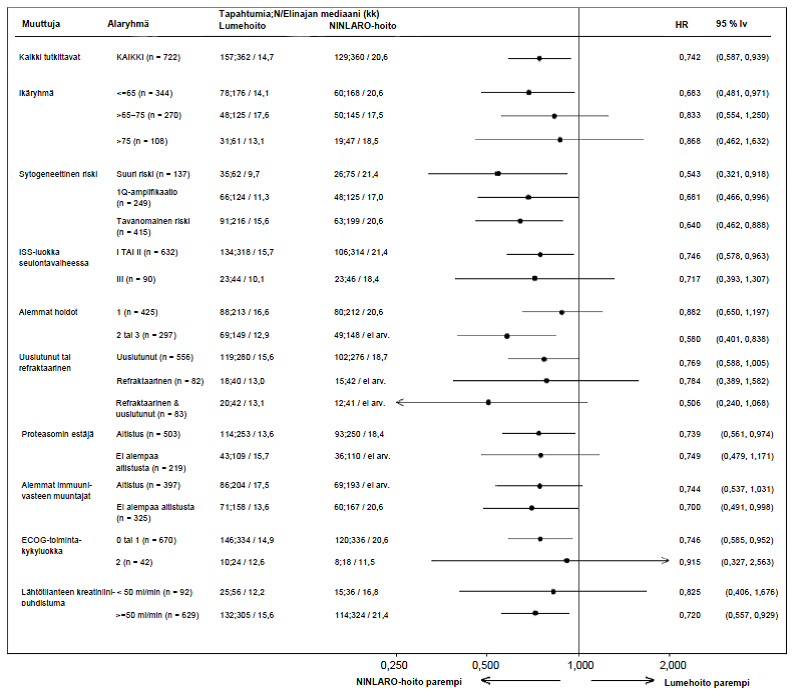
Faasin 3 tutkimuksessa (C16010) 10 potilaalla (5 kummassakin hoitoryhmässä) oli vaikea munuaisten vajaatoiminta lähtötilanteessa. iksatsomibiryhmän viidestä potilaasta yhdellä potilaalla oli vahvistettu osittainen vaste ja kolmella potilaalla oli vahvistettu stabiili tauti (kuitenkin kahdella oli vahvistamaton osittainen vaste ja yhdellä vahvistamaton erittäin hyvä osittainen vaste). Lumeryhmän viidestä potilaasta kahdella oli vahvistettu erittäin hyvä osittainen vaste.
Elämänlaatu pysyi hoidon aikana ennallaan ja oli molemmissa hoitoryhmissä samankaltainen, kun sitä tarkasteltiin EORTC QLQ‑C30- ja MY‑20-elämänlaatumittareilla faasin 3 tutkimuksessa (C16010).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset iksatsomibi-valmisteen käytöstä multippelin myelooman hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Suun kautta otetun iksatsomibin huippupitoisuudet plasmassa saavutettiin noin 1 tunnin kuluttua valmisteen ottamisesta. Suun kautta otetun valmisteen absoluuttinen biologinen hyötyosuus on keskimäärin 58 %. Iksatsomibin AUC-arvo suurenee suhteessa annokseen 0,2–10,6 mg:n alueella.
Valmisteen ottaminen runsasrasvaisen aterian kanssa pienensi iksatsomibin AUC-arvoa 28 % verrattuna valmisteen ottoon yön yli jatkuneen paaston jälkeen (ks. kohta Annostus ja antotapa).
Jakautuminen
Iksatsomibi sitoutuu 99-prosenttisesti plasman proteiineihin ja jakautuu veren punasoluihin. Veren ja plasman AUC-arvojen suhde on 10. Vakaan tilan jakautumistilavuus on 543 l.
Biotransformaatio
Kun radioaktiivisesti leimattu annos otettiin suun kautta, 70 % kaikesta plasman lääkemateriaalista oli iksatsomibin muodossa. Iksatsomibin tärkein puhdistumamekanismi on todennäköisesti useiden eri CYP-entsyymien ja muiden proteiinien kuin CYP-entsyymien välittämä metabolia. Kliinisesti relevanteilla iksatsomibipitoisuuksilla toteutetuissa in vitro -tutkimuksessa, joissa käytettiin cDNA-ekspressoituja ihmisen sytokromi P450 ‑isoentsyymejä, saatiin viitteitä siitä, että mikään tietty CYP-isoentsyymi ei ole vallitsevassa asemassa iksatsomibin metaboliassa ja että muut proteiinit kuin CYP-entsyymit osallistuvat kokonaismetaboliaan. Kliinisesti havaittavat pitoisuudet ylittäviä pitoisuuksia käytettäessä iksatsomibi metaboloitui useiden eri CYP-isoformien kautta, ja eri entsyymien arvioidut suhteelliset osuudet olivat seuraavat: 3A4 (42,3 %), 1A2 (26,1 %), 2B6 (16,0 %), 2C8 (6,0 %), 2D6 (4,8 %), 2C19 (4,8 %) ja 2C9 (< 1 %).
Eliminaatio
Iksatsomibin käyttäytymisprofiili elimistössä on multieksponentiaalinen. Populaatiofarmakokinetiikan analyysin mukaan systeeminen puhdistuma (CL) oli noin 1,86 l/h ja yksilöiden välinen vaihtelu 44 %. Iksatsomibin terminaalinen puoliintumisaika (t1/2) oli 9,5 vuorokautta. Kun valmistetta otettiin suun kautta kerran viikossa, päivänä 15 todettiin, että AUC-arvo oli kumuloitunut noin kaksinkertaiseksi.
Erittyminen
Kun viidelle pitkälle edennyttä syöpää sairastaneelle potilaalle annettiin 14C-iksatsomibikerta-annos suun kautta, 62 % annetusta radioaktiivisuudesta erittyi virtsaan ja 22 % ulosteeseen. Alle 3,5 % annetusta annoksesta erittyi muuttumattomana iksatsomibina virtsaan.
Erityisryhmät
Maksan vajaatoiminta
Populaatiofarmakokinetiikan analyysin mukaan iksatsomibin farmakokinetiikka on samankaltainen maksan toiminnaltaan normaaleilla potilailla ja potilailla, joilla on lievä maksan vajaatoiminta (kokonaisbilirubiini ≤ ULN ja ASAT > ULN tai kokonaisbilirubiini > 1–1,5 x ULN ja ASAT mikä tahansa).
Iksatsomibin farmakokinetiikkaa tutkittiin maksan toiminnaltaan normaaleilla potilailla 4 mg:n annoksilla (n = 12), keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla 2,3 mg:n annoksilla (kokonaisbilirubiini > 1,5–3 x ULN, n = 13) ja vaikeaa maksan vajaatoimintaa sairastavilla potilailla 1,5 mg:n annoksilla (kokonaisbilirubiini > 3 x ULN, n = 18). Sitoutumattoman lääkeaineen annosvakioitu AUC-arvo oli keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastavilla 27 % suurempi kuin maksan toiminnaltaan normaaleilla potilailla (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Populaatiofarmakokinetiikan analyysin mukaan iksatsomibin farmakokinetiikka on samankaltainen munuaistoiminnaltaan normaaleilla potilailla ja potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma ≥ 30 ml/min).
Iksatsombin farmakokinetiikkaa tutkittiin 3 mg:n annoksilla munuaistoiminnaltaan normaaleilla potilailla (kreatiniinipuhdistuma ≥ 90 ml/min, n = 18), vaikeaa munuaisten vajaatoimintaa sairastavilla (kreatiniinipuhdistuma < 30 ml/min, n = 14) ja dialyysihoitoa vaativaa loppuvaiheen munuaissairautta (ESRD) sairastavilla (n = 6). Sitoutumattoman lääkeaineen AUC-arvo oli vaikeaa munuaisten vajaatoimintaa tai dialyysihoitoa vaativaa loppuvaiheen munuaissairautta (ESRD) sairastavilla 38 % suurempi kuin munuaistoiminnaltaan normaaleilla potilailla. Hemodialyysin aikana dialysaattoriin tulevasta ja siitä lähtevästä verestä mitattiin samankaltaiset iksatsomibipitoisuudet, mikä viittaa siihen, että iksatsomibi ei dialysoidu (ks. kohta Annostus ja antotapa).
Ikä, sukupuoli, etninen tausta
Populaatiofarmakokinetiikan analyysin mukaan ikä (23–91 v), sukupuoli, kehon pinta-ala (1,2–2,7 m2) ja etninen tausta eivät vaikuttaneet iksatsomibin puhdistumaan kliinisesti merkittävästi. Keskimääräinen AUC-arvo oli 35 % suurempi aasialaisilla potilailla. Iksatsomibin AUC-arvot valkoihoisilla potilailla olivat kuitenkin osittain päällekkäiset aasialaisten potilaiden AUC-arvojen kanssa.
Prekliiniset tiedot turvallisuudesta
Mutageenisuus
Iksatsomibi ei ollut mutageeninen bakteerien käänteismutaatiokokeessa (Amesin testi) eikä klastogeeninen luuytimen mikrotumakokeessa hiirillä. Iksatsomibi tuotti positiiviset tulokset in vitro ‑klastogeenisuuskokeessa ihmisen ääreisveren lymfosyyteillä. Toisaalta iksatsomibi tuotti negatiiviset tulokset hiiren in vivo Comet-tutkimuksessa, jossa arvioitiin DNA:n pyrstöalueen prosenttiosuutta mahalaukussa ja maksassa. Kokonaisuutena ajatellen näyttö viittaa siis siihen, että iksatsomibihoitoon ei liity geenitoksisuusriskiä.
Lisääntyminen ja alkion- ja sikiönkehitys
Iksatsomibi aiheutti alkio- ja sikiötoksisuutta tiineille rotille ja kaneille vain emolle toksisia annoksia käytettäessä ja tilanteissa, joissa altistukset olivat hieman suurempia kuin suositusannoksia saavilla potilailla todetaan. Iksatsomibilla ei tehty hedelmällisyyttä, varhaista alkionkehitystä eikä pre- tai postnataalista toksisuutta arvioineita tutkimuksia, mutta yleisissä toksisuustutkimuksissa arvioitiin lisääntymiskudoksia. Iksatsomibihoidon käyttö enintään 6 kk ajan (rotta) ja enintään 9 kk ajan (koira) ei vaikuttanut urosten eikä naaraiden lisääntymiselimiin.
Eläintoksikologia ja/tai -farmakologia
Useita hoitojaksoja kestäneissä toistuvien annosten toksisuustutkimuksissa rotilla ja koirilla tärkeimpiä toksisuuden kohde-elimiä olivat ruoansulatuskanava, imukudokset ja hermosto. Kun koirille annettiin valmistetta suun kautta 9 kuukauden tutkimuksessa (10 hoitojaksoa), jossa antoaikataulu muistutti kliinistä hoitoprotokollaa (28 päivän hoitojaksot), mikroskooppiset vaikutukset neuroneihin olivat yleensä erittäin vähäisiä ja niitä havaittiin vain annoksilla 0,2 mg/kg (4 mg/m2). Valtaosa kohde-elinlöydöksistä korjautui osittain tai kokonaan, kun hoito lopetettiin. Poikkeuksena olivat lannerangan takajuuriganglioiden ja selkäytimen takasarven neuronilöydökset.
Kun valmistetta annettiin suun kautta, kudoksiin jakautumista koskeneessa tutkimuksessa rotilla todettiin, että aivot ja selkäydin kuuluivat lääkepitoisuuksiltaan pienimpiin kudoksiin. Tämä viittaa siihen, että iksatsomibin pääsy veri-aivoesteen läpi on rajallista. Ilmiön merkitystä ihmisille ei kuitenkaan tunneta.
Ei-kliinisissä turvallisuusfarmakologian tutkimuksissa in vitro (hERG-kanavilla) ja in vivo (koirilla, joita seurattiin telemetrialla peroraalisen kerta-annoksen jälkeen) todettiin, että iksatsomibi ei vaikuttanut kardiovaskulaari- tai hengitystoimintaan, kun lääkkeen AUC-arvo oli yli 8-kertainen verrattuna kliinisiin pitoisuuksiin.
Farmaseuttiset tiedot
Apuaineet
NINLARO 2,3 mg,kovat kapselit
Kapselin sisältö
Mikrokiteinen selluloosa, magnesiumstearaatti, talkki.
Kapselin kuori
Liivate, titaanidioksidi (E171). punainen rautaoksidi (E172).
Painomuste
Shellakka, propyleeniglykoli, kaliumhydroksidi, musta rautaoksidi (E172).
NINLARO 3 mg kovat kapselit
Kapselin sisältö
Mikrokiteinen selluloosa, magnesiumstearaatti, talkki.
Kapselin kuori
Liivate, titaanidioksidi (E171), musta rautaoksidi (E172).
Painomuste
Shellakka, propyleeniglykoli, kaliumhydroksidi, musta rautaoksidi (E172).
NINLARO 4 mg kovat kapselit
Kapselin sisältö
Mikrokiteinen selluloosa, magnesiumstearaatti, talkki.
Kapselin kuori
Liivate, titaanidioksidi (E171), keltainen rautaoksidi (E172), punainen rautaoksidi (E172).
Painomuste
Shellakka, propyleeniglykoli, kaliumhydroksidi, musta rautaoksidi (E172).
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Säilytä alle 30 °C. Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
NINLARO kapseli, kova
2,3 mg (L:ei) 3 fol (4980,08 €)
3 mg (L:ei) 3 fol (4980,08 €)
4 mg (L:ei) 3 fol (4980,08 €)
PF-selosteen tieto
PVC-Al/Al-läpipainopakkaus, joka sisältää kolme kapselia ja on suljettu taskupakkauksen sisään.
Yksi taskupakkaus on pakattu yhteen ulkopakkaukseen.
Valmisteen kuvaus:
NINLARO 2,3 mg, kovat kapselit
Vaalean pinkki koon 4 kova liivatekapseli, jossa kannessa merkintä ”Takeda” ja rungossa merkintä ”2.3 mg” mustalla musteella
NINLARO 3 mg, kovat kapselit
Vaalean harmaa koon 4 kova liivatekapseli, jossa kannessa merkintä ”Takeda” ja rungossa merkintä ”3 mg” mustalla musteella
NINLARO 4 mg, kovat kapselit
Vaalean oranssi koon 3 kova liivatekapseli, jossa kannessa merkintä ”Takeda” ja rungossa merkintä ”4 mg” mustalla musteella
Käyttö- ja käsittelyohjeet
Iksatsomibi on sytotoksinen. Kapselin saa ottaa pakkauksesta vasta juuri ennen lääkkeen ottamista. Kapseleita ei saa avata eikä murskata. Suoraa kosketusta kapselin sisällön kanssa on vältettävä. Jos kapseli rikkoutuu, siivouksen aikana on vältettävä lääkepölyn nousemista ilmaan. Jos kosketusta lääkkeeseen tapahtuu, kosketusalue pestään perusteellisesti vedellä ja saippualla.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
NINLARO kapseli, kova
2,3 mg 3 fol
3 mg 3 fol
4 mg 3 fol
- Ylempi erityiskorvaus (100 %). Iksatsomibi: Multippelin myelooman hoito erityisin edellytyksin (1505).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Iksatsomibi: Multippelin myelooman hoito erityisin edellytyksin (398).
ATC-koodi
L01XG03
Valmisteyhteenvedon muuttamispäivämäärä
21.05.2026
Yhteystiedot
 TAKEDA OY
TAKEDA OY PL 1406, Ilmalankuja 3
00101 Helsinki
0800 774 051
www.takeda.fi
etunimi.sukunimi@takeda.com