
APREPITANT STADA kapsel, hård 125 mg+80 mg, 80 mg, 125 mg
Kvalitativ och kvantitativ sammansättning
Varje 125 mg kapsel innehåller 125 mg aprepitant.
Varje 80 mg kapsel innehåller 80 mg aprepitant.
Hjälpämne med känd effekt
Varje 125 mg kapsel innehåller 125 mg sackaros.
Varje 80 kapsel innehåller 80 mg sackaros.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Hård kapsel.
Kliniska uppgifter
Terapeutiska indikationer
Profylax mot illamående och kräkningar vid högemetogen och måttligt emetogen cytostatikabehandling vid cancer hos vuxna och ungdomar från 12 års ålder.
Aprepitant Stada 125 mg/80 mg ges som del av kombinationsbehandling (se avsnitt Dosering och administreringssätt).
Dosering och administreringssätt
Dosering
Vuxna
Aprepitant Stada ges i 3 dagar som en del av en behandling inkluderande en kortikosteroid och en 5-HT3-antagonist.
Den rekommenderade dosen är 125 mg oralt en gång dagligen en timme före påbörjande av cytostatikabehandling Dag 1 och 80 mg oralt en gång dagligen på morgonen Dag 2 och 3.
Följande dosregimer rekommenderas hos vuxna vid profylax mot illamående och kräkningar i samband med emetogen cytostatikabehandling vid cancer:
Högemetogen cytostatikabehandling
| Dag 1 | Dag 2 | Dag 3 | Dag 4 | |
| Aprepitant Stada | 125 mg oralt | 80 mg oralt | 80 mg oralt | ingen |
| Dexametason | 12 mg oralt | 8 mg oralt | 8 mg oralt | 8 mg oralt |
| 5-HT3-antagonister | Standarddos av 5-HT3-antagonister. För lämplig doseringsinformation hänvisas till produktinformationen för den valda 5-HT3-antagonisten | ingen | ingen | ingen |
Dexametason ska ges 30 minuter före cytostatikabehandling Dag 1 och på morgonen Dag 2 till 4. Dosen dexametason är vald med hänsyn till interaktioner med aktiva substanser.
Måttligt emetogen cytostatikabehandling
| Dag 1 | Dag 2 | Dag 3 | |
| Aprepitant Stada | 125 mg oralt | 80 mg oralt | 80 mg oralt |
| Dexametason | 12 mg oralt | ingen | ingen |
| 5-HT3-antagonister | Standarddos av 5-HT3-antagonister. För lämplig doseringsinformation hänvisas till produktinformationen för den valda 5-HT3-antagonisten | ingen | ingen |
Dexametason ska ges 30 minuter före cytostatikabehandling Dag 1. Dosen dexametason är vald med hänsyn till interaktioner med aktiva substanser.
Pediatrisk population
Ungdomar (i åldern 12 till och med 17 år)
Aprepitant Stada ges i 3 dagar som del av en behandling inkluderande en 5-HT3-antagonist. Den rekommenderade dosen av Aprepitant Stada kapslar är 125 mg oralt Dag 1 och 80 mg oralt Dag 2 och 3. Aprepitant Stada ges oralt en timme före cytostatika Dag 1, 2 och 3. Om cytostatika inte ges Dag 2 och 3 ska Aprepitant Stada ges på morgonen. Se produktresumén (SmPC) för den valda 5-HT3-antagonisten avseende lämplig doseringsinformation. Om en kortikosteroid, såsom dexametason, ges samtidigt med Aprepitant Stada ska dosen av kortikosteroiden ges i 50% av den vanliga dosen (se avsnitt Interaktioner och Farmakodynamiska egenskaper).
Säkerhet och effekt för 80 mg och 125 mg kapslar har inte fastställts hos barn yngre än 12 år. Inga data finns tillgängliga.
Allmänt
Effektdata för kombination med andra kortikosteroider och 5-HT3-antagonister är begränsade. För ytterligare information om administrering tillsammans med kortikosteroider, se avsnitt Interaktioner. För 5-HT3-antagonister som administreras samtidigt hänvisas till SmPC för dessa läkemedel.
Särskilda patientgrupper
Äldre (≥65 år)
Dosjustering är inte nödvändig för äldre (se avsnitt Farmakokinetiska egenskaper).
Kön
Dosjustering är inte nödvändig baserat på kön (se avsnitt Farmakokinetiska egenskaper).
Nedsatt njurfunktion
Dosjustering är inte nödvändig för patienter med nedsatt njurfunktion eller för patienter med njursjukdom i slutskedet och som genomgår hemodialys (se avsnitt Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Dosjustering är inte nödvändig för patienter med lätt nedsatt leverfunktion. Kliniska data för patienter med måttligt nedsatt leverfunktion är begränsade och data för patienter med gravt nedsatt leverfunktion saknas. Aprepitant ska användas med försiktighet hos denna patientgrupp (se avsnitt Varningar och försiktighet och Farmakokinetiska egenskaper).
Administreringssätt
Oral användning.
Den hårda kapseln ska sväljas hel.
Aprepitant Stada kan intas med eller utan mat.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Samtidig administrering med pimozid, terfenadin, astemizol eller cisaprid (se avsnitt Interaktioner).
Varningar och försiktighet
Patienter med måttlig till kraftig leverfunktionsnedsättning
Data för patienter med måttligt nedsatt leverfunktion är begränsade och data för patienter med gravt nedsatt leverfunktion saknas. Aprepitant Stada ska användas med försiktighet till dessa patienter (se avsnitt Farmakokinetiska egenskaper).
CYP3A4-interaktioner
Aprepitant Stada ska användas med försiktighet till patienter som samtidigt använder oralt administrerade aktiva substanser som huvudsakligen metaboliseras via CYP3A4 och som har ett smalt terapeutiskt fönster, t.ex. ciklosporin, takrolimus, sirolimus, everolimus, alfentanil, ergotalkaloidderivat, fentanyl och kinidin (se avsnitt Interaktioner). Dessutom bör samtidig administrering av irinotekan inledas med särskild försiktighet då kombinationen kan resultera i en ökad toxicitet.
Samtidig användning av warfarin (ett CYP2C9-substrat)
För patienter som står på kronisk warfarinbehandling ska International Normalised Ratio (INR) övervakas noggrant under behandling med Aprepitant Stada och i 14 dagar efter varje 3-dagarsbehandling med Aprepitant Stada (se avsnitt Interaktioner).
Samtidig användning av hormonella antikonceptionsmedel
Effekten av hormonella antikonceptionsmedel kan minska under och 28 dagar efter administrering av Aprepitant Stada. Alternativa icke-hormonella kompletterande preventivmetoder bör användas under behandling med Aprepitant Stada och ytterligare 2 månader efter den sista dosen av Aprepitant Stada (se avsnitt Interaktioner).
Hjälpämnen
Aprepitant Stada kapslar innehåller sackaros. Patienter med något av följande sällsynta ärftliga tillstånd bör inte använda detta läkemedel: fruktosöverkänslighet, glukos-galaktosmalabsorption eller sukras-isomaltas insufficiens.
Aprepitant Stada kapslar innehåller natrium. Detta läkemedel innehåller mindre än 1 mmol natrium (23 mg) per kapsel, dvs. är näst intill ”natriumfritt”.
Interaktioner
Aprepitant (125 mg/80 mg) är ett substrat, en måttlig hämmare och en inducerare av CYP3A4. Aprepitant är även en CYP2C9-inducerare. Under behandling med Aprepitant Stada hämmas CYP3A4. Efteravslutad behandling ger Aprepitant Stada en övergående lätt induktion av CYP2C9, CYP3A4 och glukuronidering. Aprepitant interagerar troligtvis inte med transportproteinet P-glykoprotein, vilket visats genom att aprepitant inte interagerar med digoxin.
Effekten av aprepitant på farmakokinetiken hos andra läkemedel
CYP3A4-hämning
Som en måttlig CYP3A4-hämmare kan aprepitant (125 mg/80 mg) öka plasmakoncentrationerna av samtidigt administrerade aktiva substanser och som metaboliseras via CYP3A4. Den totala exponeringen för oralt administrerade CYP3A4-substrat kan öka upp till 3-faldigt under en 3-dagars behandling med Aprepitant Stada. Effekten av aprepitant på plasmakoncentrationen av intravenöst administrerade CYP3A4-substrat förväntas vara mindre. Aprepitant Stada ska inte användas samtidigt med pimozid, terfenadin, astemizol eller cisaprid (se avsnitt Kontraindikationer). Aprepitants hämning av CYP3A4 kan resultera i förhöjda plasmakoncentrationer av dessa aktiva substanser, vilket kan orsaka allvarliga eller livshotande reaktioner. Försiktighet bör iakttas under samtidig administrering av Aprepitant Stada och oralt administrerade aktiva substanser som huvudsakligen metaboliseras via CYP3A4 och som har ett smalt terapeutiskt fönster, t.ex. ciklosporin, takrolimus, sirolimus, everolimus, alfentanil, diergotamin, ergotamin, fentanyl och kinidin (se avsnitt Varningar och försiktighet).
Kortikosteroider
Dexametason: Vid samtidig administrering med Aprepitant Stada 125 mg/80 mg regim bör den vanliga orala dosen av dexametason minskas med ungefär 50%. I kliniska studier vid illamående och kräkningar inducerade av cytostatikabehandling valdes dosen dexametason med hänsyn till läkemedelsinteraktioner (se avsnitt Dosering och administreringssätt). När Aprepitant gavs i en behandling om 125 mg tillsammans med oralt administrerat dexametason 20 mg Dag 1, och när aprepitant, 80 mg/dag, gavs tillsammans med oralt administrerat dexametason 8 mg Dag 2 till 5, ökade AUC för dexametason (ett CYP3A4-substrat), 2,2 gånger Dag 1 och 5.
Metylprednisolon: Vid samtidig administrering med Aprepitant Stada 125 mg/80 mg regim bör den vanliga intravenösa dosen av metylprednisolon minskas med ca 25 %, och den vanliga orala dosen med ca 50 %. När aprepitant gavs i en behandling om 125 mg Dag 1 + 80/dag Dagarna 2 och 3, ökade AUC för metylprednisolon, ett CYP3A4-substrat, 1,3 gånger Dag 1 och 2,5 gånger Dag 3 när 125 mg metylprednisolon samtidigt administrerades intravenöst Dag 1 och 40 mg oralt Dag 2 och 3.
Under pågående behandling med metylprednisolon kan AUC för metylprednisolon, på grund av den inducerande effekten av aprepitant på CYP3A4, minska vid senare tidpunkter inom 2 veckor efter påbörjad dosering med aprepitant. Denna effekt kan förväntas vara mer uttalad för oralt administrerat metylprednisolon.
Cytostatika
I farmakokinetiska studier, där aprepitant gavs i en regim om 125 mg Dag 1 och 80 mg/dag Dag 2 och 3 påverkades inte farmakokinetiken för docetaxel som administrerades intravenöst Dag 1 och inte heller vinorelbin som gavs intravenöst Dag 1 eller Dag 8. Eftersom effekten av aprepitant i högre grad påverkar farmakokinetiken för oralt givna CYP3A4-substrat än för intravenöst administrerade CYP3A4-substrat kan interaktion med följande oralt administrerade cytostatika, som huvudsakligen eller delvis metaboliseras av CYP3A4, (såsom etoposid, vinorelbin) inte uteslutas. Försiktighet bör iakttas och ytterligare övervakning kan vara lämpligt för patienter som får läkemedel vilka huvudsakligen eller delvis metaboliseras av CYP3A4 (se avsnitt Varningar och försiktighet). Efter marknadsföring har fall av neurotoxicitet, en möjlig biverkning av ifosfamid, rapporterats efter samtidig administrering av aprepitant och ifosfamid.
Immunosuppressiva läkemedel
Vid 3-dagars behandling mot CINV förväntas en övergående måttligt ökad exponering av immunosuppressiva läkemedel som metaboliseras via CYP3A4 (t.ex. ciklosporin, takrolimus, everolimus och sirolimus), med en efterföljande lätt minskad exponering. Med anledning av den korta behandlingstiden och de tidsberoende begränsade förändringarna vad gäller exponering, rekommenderas inte dosreduktion av immunosuppressiva läkemedel under samtidig 3-dagars behandling med Aprepitant Stada.
Midazolam
Den potentiella effekten av ökade plasmakoncentrationer av midazolam eller andra bensodiazepiner metaboliserade via CYP3A4 (alprazolam, triazolam) bör övervägas vid samtidig administrering av dessa läkemedel och Aprepitant Stada (125 mg/80 mg).
Aprepitant ökade AUC för midazolam (ett känsligt CYP3A4-substrat), 2,3 gånger Dag 1 och 3,3 gånger Dag 5 när en oral engångsdos av 2 mg midazolam administrerades samtidigt Dag 1 och Dag 5 under en behandling med aprepitant 125 mg Dag 1 och 80 mg/dag under Dag 2 till 5.
I en annan studie med intravenös administrering av midazolam, gavs aprepitant 125 mg Dag 1 och 80 mg/dag under Dag 2 och 3. Midazolam 2 mg gavs intravenöst före administreringen av 3-dagarsbehandlingen med aprepitant och Dag 4, 8 och 15. Aprepitant ökade AUC för midazolam med 25 % Dag 4 och minskade AUC för midazolam med 19 % Dag 8, och 4 % Dag 15. Dessa effekter betraktades inte som kliniskt betydelsefulla.
I en tredje studie med intravenös och oral administrering av midazolam, gavs 125 mg aprepitant Dag 1 och 80 mg/dag Dag 2 och 3, samtidigt gavs 32 mg ondansetron Dag 1 samt 12 mg dexametason Dag 1 och 8 mg dagarna 2–4. Denna kombination (dvs aprepitant, ondansetron och dexametason) minskade AUC för oralt givet midazolam med 16% Dag 6,9 % Dag 8,7 % Dag 15 och 17 % Dag 22. Dessa effekter bedömdes inte vara kliniskt betydelsefulla.
Ytterligare en studie med intravenös administrering av midazolam och aprepitant har avslutats. 2 mg intravenöst midazolam gavs 1 timme efter oral administrering av en oral engångsdos av aprepitant 125 mg. Plasma AUC för midazolam ökade 1,5 gånger. Denna effekt bedömdes inte vara kliniskt betydelsefull.
Induktion
Som en lätt inducerare av CYP2C9, CYP3A4 och glukuronidering kan aprepitant under två veckor efter inledning och behandling minska plasmakoncentrationerna av substrat som elimineras med hjälp av dessa system. Induktionen kan bli tydlig först efter att en 3-dagars behandling med aprepitant avslutats. För CYP2C9- och CYP3A4-substrat är induktionen övergående med en maximal effekt 3–5 dagar efter en avslutad 3-dagars behandling med aprepitant. Effekten kvarstår under några dagar, avtar därefter långsamt och saknar klinisk betydelse två veckor efter avslutad behandling med aprepitant. Lätt induktion av glukuronidering ses även då 80 mg aprepitant ges oralt i 7 dygn. Uppgifter beträffande effekter på CYP2C8 och CYP2C19 saknas. För warfarin, acenokumarol, tolbutamid, fenytoin och andra aktiva läkemedelssubstanser som man vet metaboliseras av CYP2C9 bör försiktighet iakttas vid administrering under denna tidsperiod.
Warfarin
För patienter som står på kronisk warfarin-behandling ska protrombintiden (INR) övervakas noggrant under behandling med Aprepitant Stada och i två veckor efter varje 3-dagarsbehandling med Aprepitant Stada vid illamående och kräkningar inducerade av cytostatikabehandling (se avsnitt Varningar och försiktighet). När en engångsdos om 125 mg aprepitant administrerades Dag 1 och 80 mg/dag Dag 2 och 3 till friska frivilliga som var stabilt inställda på kronisk warfarinbehandling, sågs ingen effekt av aprepitant på plasma-AUC av R(+) eller S(-) warfarin uppmätt Dag 3. Det förekom dock en minskning med 34 % av dalvärdeskoncentrationen S(-) warfarin (ett CYP2C9-substrat) vilken åtföljdes av en minskning med 14 % av INR 5 dagar efter avslutad behandling.
Tolbutamid
När aprepitant gavs som 125 mg Dag 1 och 80 mg/dag dag 2 och 3, minskade AUC för tolbutamid (ett CYP2C9-substrat) med 23 % Dag 4, 28% Dag 8 och 15 % Dag 15. Detta då en engångsdos av tolbutamid 500 mg administrerades oralt före 3-dagarsbehandlingen med aprepitant och Dag 4, 8 och 15.
Hormonella antikonceptionsmedel
Effekten av hormonella antikonceptionsmedel kan minska under och 28 dagar efter administrering av Aprepitant Stada. Alternativa icke-hormonella kompletterande preventivmetoder bör användas vid behandling med Aprepitant Stada och ytterligare 2 månader efter den sista dosen av aprepitant.
I en klinisk studie gavs en engångsdos oralt antikonceptionsmedel innehållande etinylestradiol och noretindron under dagarna 1 till 21 tillsammans med aprepitant 125 mg Dag 8 och 80 mg/dag Dag 9 och 10. Dessutom gavs ondansetron 32 mg intravenöst Dag 8 och oralt dexametason 12 mg Dag 8 och 8 mg/dag dagarna 9, 10 och 11. Under dagarna 9 till 21 i denna studie, minskade de dalvärdeskoncentrationerna av etinylestradiol med så mycket som 64 % och de dalvärdeskoncentrationerna av noretindron med så mycket som 60 %.
5-HT3-antagonister
I kliniska interaktionsstudier hade aprepitant ingen kliniskt signifikant effekt på farmakokinetiken för ondansetron, granisetron eller hydrodolasetron (den aktiva metaboliten av dolasetron).
Andra läkemedels effekt på farmakokinetiken för aprepitant
Samtidig administrering av Aprepitant Stada och aktiva substanser som hämmar CYP3A4-aktivitet (t.ex. ketokonazol, itrakonazol, vorikonazol, posakonazol, klaritromycin, telitromycin, nefazodon och proteashämmare) bör inledas försiktigt då kombinationen resulterar i flerfaldigt ökade plasmakoncentrationer av aprepitant (se avsnitt Varningar och försiktighet).
Samtidig administrering av Aprepitant Stada och aktiva substanser som kraftigt inducerar CYP3A4-aktivitet (t.ex. rifampicin, fenytoin, karbamazepin, fenobarbital), bör undvikas då kombinationen resulterar i minskade plasmakoncentrationer av aprepitant, vilket kan resultera i en minskad effekt av Aprepitant Stada.
Samtidig administrering av Aprepitant Stada och naturläkemedel innehållande johannesört ( Hypericum perforatum) rekommenderas inte.
Ketokonazol
När en 125 mg engångsdos av aprepitant administrerades Dag 5 under en 10-dagarsbehandling med 400 mg ketokonazol/dag, en stark hämmare av CYP3A4, ökade AUC för aprepitant ca 5 gånger och den genomsnittliga terminala halveringstiden för aprepitant ökade ca 3 gånger.
Rifampicin
När en 375 mg engångsdos av aprepitant administrerades Dag 9 under en 14-dagarsbehandling med 600 mg/dag rifampicin (en stark inducerare av CYP3A4), minskade AUC för aprepitant 91 % och den genomsnittliga terminala halveringstiden minskade 68 %.
Pediatrisk population
Interaktionsstudier har endast utförts på vuxna.
Fertilitet, graviditet och amning
Preventivmetoder för män och kvinnor
Effekten av hormonella antikonceptionsmedel kan minska under och 28 dagar efter administrering av Aprepitant Stada. Alternativa icke-hormonella kompletterande preventivmetoder bör användas vid behandling med Aprepitant Stada och ytterligare 2 månader efter den sista dosen av Aprepitant Stada (se avsnitt Varningar och försiktighet och Interaktioner).
Graviditet
Inga data från exponering under graviditet finns tillgängliga för aprepitant. Risken för reproduktionstoxikologiska effekter av aprepitant har inte fullt karakteriserats då exponeringsnivåer över de terapeutiska hos människa vid 125 mg/80 mg doser inte kunde uppnås i djurstudier. Dessa studier indikerade inte direkta eller indirekta skadliga effekter på graviditet, embryonal/fosterutveckling, förlossning eller utveckling efter födseln (se avsnitt Prekliniska säkerhetsuppgifter). De potentiella effekterna på reproduktion av förändringar av neurokininregleringen är okänd. Aprepitant Stada ska användas under graviditet endast då det är absolut nödvändigt.
Amning
Aprepitant utsöndras i mjölken hos digivande råttor. Det är inte känt om aprepitant utsöndras i modersmjölken hos människor. Amning rekommenderas därför inte under behandling med Aprepitant Stada.
Fertilitet
Sannolikheten för att aprepitant ska påverka fertiliteten har inte fastställts eftersom exponeringsnivåer över de terapeutiska exponeringsnivåerna hos människa inte kunnat uppnås i djurstudier.
Fertilitetsstudier indikerade inte direkta eller indirekta skadliga effekter vad gäller parningsförmåga, fertilitet, embryonal-/fosterutveckling eller spermieantal och spermierörlighet (se avsnitt Prekliniska säkerhetsuppgifter).
Effekter på förmågan att framföra fordon och använda maskiner
Aprepitant Stada kan ha mindre effekt på förmågan att framföra fordon och använda maskiner. Yrsel och trötthet kan förekomma efter administrering av Aprepitant Stada (se avsnitt Biverkningar).
Biverkningar
Sammanfattning av säkerhetsprofilen
Säkerhetsprofilen för aprepitant utvärderades hos ca 6 500 vuxna i fler än 50 studier och 184 barn och ungdomar i två pivotala pediatriska kliniska studier.
De vanligaste biverkningarna rapporterades med en högre frekvens hos vuxna behandlade med aprepitant jämfört med standardterapi hos patienter som fick högemetogen cytostatikabehandling (HEC) var: hicka (4,6% mot 2,9%), förhöjt alaninaminotransferas (ALAT) (2,8% mot 1, 1%), dyspepsi (2,6% mot 2,0%), förstoppning (2,4% mot 2,0%), huvudvärk (2, 0% mot 1,8%) och aptitnedsättning (2,0% mot 0,5%). Den vanligaste biverkning som rapporterades i högre frekvens hos patienter som behandlades med aprepitant jämfört med standardterapi, hos patienter som fick måttligt emetogen cytostatikabehandling MEC, var trötthet (1,4% mot 0,9%).
De vanligaste biverkningarna rapporterade med en högre frekvens hos pediatriska patienter som fick emetogen cytostatikabehandling och behandlades med aprepitant jämfört med kontrollbehandling var hicka (3,3% mot 0,0%) och flush (1,1% mot 0,0%).
Biverkningstabell
Följande biverkningar observerades i en sammantagen analys av HEC- och MEC-studier i en högre frekvens med aprepitant jämfört med standardterapi hos vuxna eller pediatriska patienter eller i studier efter godkännandet av läkemedlet. De angivna frekvenskategorierna i tabellen baseras på studier på vuxna. De observerade frekvenserna i den pediatriska populationen var desamma eller lägre såvida det inte anges i tabellen. Några mindre vanliga biverkningar hos den vuxna populationen sågs inte i de pediatriska studierna.
Frekvenserna definieras enligt följande: mycket vanlig (≥1/10); vanlig (≥1/100, <1/10); mindre vanlig (≥1/1 000, <1/100); sällsynt (≥1/10 000, <1/1 000) och mycket sällsynt (<1/10 000), ingen känd frekvens (kan inte beräknas från tillgängliga data).
| Systemorganklass | Biverkningar | Frekvens |
| Infektioner och infestationer | candidiasis, stafylokockinfektion | sällsynt |
| Blodet och lymfsystemet | febril neutropeni, anemi | mindre vanlig |
| Immunsystemet | överkänslighetsreaktioner inkluderande anafylaktiska reaktioner | ingen känd |
| Metabolism och nutrition | aptitnedsättning | vanlig |
| polydipsi | sällsynt | |
| Psykiska störningar | ångest | mindre vanlig |
| desorientering, euforisk sinnesstämning | sällsynt | |
| Centrala och perifera nervsystemet | huvudvärk | vanlig |
| yrsel, somnolens | mindre vanlig | |
| kognitiv störning, letargi, dysgeusi | sällsynt | |
| Ögon | konjunktivit | sällsynt |
| Öron och balansorgan | tinnitus | sällsynt |
| Hjärtat | palpitationer | mindre vanlig |
| bradykardi, hjärt-kärlsjukdom | sällsynt | |
| Blodkärl | värmevallningar/flush | mindre vanlig |
| Andningsvägar, bröstkorg och mediastinum | hicka | vanlig |
orofaryngeal smärta, nysningar, hosta, postnasalt dropp, halsirritation | sällsynt | |
| Magtarmkanalen | förstoppning, dyspepsi | vanlig |
| rapning, illamående†, kräkningar†, gastroesofagal refluxsjukdom, buksmärta, muntorrhet, flatulens | mindre vanlig | |
| perforerande duodenalsår, stomatit, bukspänning, hård avföring, neutropen kolit | sällsynt | |
| Hud och subkutan vävnad | utslag, akne | mindre vanlig |
| fotosensitivitetsreaktion, hyperhidros, seborré, hudförändring, kliande utslag, Stevens-Johnsons syndrom/toxisk epidermal nekrolys | sällsynt | |
| klåda, urtikaria | ingen känd | |
| Muskuloskeletala systemet och bindväv | muskelsvaghet, muskelspasmer | sällsynt |
| Njurar och urinvägar | dysuri | mindre vanlig |
| pollakisuri | sällsynt | |
| Allmänna symtom och/eller symtom vid administreringsstället | trötthet | vanlig |
| asteni, sjukdomskänsla | mindre vanlig | |
| ödem, obehagskänsla i bröstet, gångstörning | sällsynt | |
| Undersökningar | förhöjt ALAT | vanlig |
| förhöjt ASAT, förhöjt alkaliskt fosfatas | mindre vanlig | |
positivt test för röda blodkroppar i urinen, minskat natrium i blodet, viktminskning, minskat antal neutrofiler, glukosuri, ökad urinmängd | sällsynt |
†Illamående och kräkningar var effektparametrar under de 5 första dagarna efter cytostatikabehandling och rapporterades bara som biverkningar därefter.
Beskrivning av utvalda biverkningar
Biverkningsprofilen hos vuxna under upprepade behandlingscykler i HEC- och MEC-studier i upp till 6 ytterligare behandlingsomgångar med cytostatika motsvarade i allmänhet de som observerades i första behandlingscykeln. I ytterligare en kontrollerad klinisk studie på 1 169 vuxna patienter som fick aprepitant och HEC var biverkningsprofilen i allmänhet lik den som setts i de andra HEC studierna med aprepitant.
Ytterligare biverkningar har observerats hos vuxna patienter som behandlats med aprepitant mot postoperativt illamående och kräkningar (PONV), och med incidenser högre än för ondansetron: övre buksmärtor, onormala tarmljud, förstoppning*, dysartri, dyspné, hypestesi, insomnia, mios, illamående, sensorisk störning, magbesvär, sub-ileus*, nedsatt synskärpa, väsande andning.
*Rapporterades hos patienter som tog en högre dos aprepitant
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till (se detaljer nedan):
webbplats: www.fimea.fi
Säkerhets‐ och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Vid överdosering ska Aprepitant Stada sättas ut och symtomatisk terapi ges samt övervakning ske. Eftersom aprepitant verkar antiemetiskt, kan framkallning av kräkning med hjälp av läkemedel vara verkningslöst.
Aprepitant är inte dialyserbart.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Antiemetika, ATC-kod A04AD12.
Aprepitant är en selektiv antagonist med hög affinitet till humana substans P-neurokinin-1-(NK1)-receptorer.
3-dagars behandling med aprepitant hos vuxna
I två randomiserade, dubbelblinda studier innefattande totalt 1094 vuxna patienter som fick cytostatikabehandling inkluderande cisplatin >70 mg/m2, jämfördes aprepitant i kombination med ondansetron/dexametason behandling (se avsnitt Dosering och administreringssätt) med standardterapi (placebo plus intravenöst ondansetron 32 mg intravenöst under Dag 1 samt oralt dexametason 20 mg under Dag 1 och 8 mg två gånger dagligen under Dag 2 till 4). Trots att dosen ondansetron 32 mg intravenöst användes i kliniska studier, är detta inte längre den rekommenderade dosen. För lämplig doseringsinformation hänvisas till produktinformationen för vald 5-HT3-receptorantagonist.
Effekten baserades på utvärderingen av följande sammansatta mått: fullständigt svar (definierat som ingen emesis och ingen användning av undsättande behandling) främst under behandlingscykel 1. Resultaten utvärderades för varje enskild studie och för de två studierna i kombination.
En sammanfattning av huvudresultaten från den kombinerade analysen visas i tabell 1.
Tabell 1
Procent av vuxna patienter, som fick högemetogen cytostatikabehandling, med behandlingssvar uppdelat i behandlingsgrupp och fas – Behandlingscykel 1.
Aprepitantbehandling (N= 521) † | Standardterapi (N= 524) † | Skillnader* | ||
| % | % | % | (95 % KI) | |
| SAMMANSATTA MÅTT | ||||
| Fullständigt svar (ingen emesis och ingen användning av undsättande behandling) | ||||
| Generellt (0-120 timmar) | 67,7 | 47,8 | 19,9 | (14,0, 25,8) |
| 0-24 timmar | 86,0 | 73,2 | 12,7 | (7,9, 17,6) |
| 25-120 timmar | 71,5 | 51,2 | 20,3 | (14,5, 26,1) |
| INDUVIDUELLA MÅTT | ||||
| Ingen emesis (inga episoder av emesis oavsett användning av undsättande behandling) | ||||
| Generellt (0-120 timmar) | 71,9 | 49,7 | 22,2 | (16,4, 28,0) |
| 0-24 timmar | 86,8 | 74,0 | 12,7 | (8,0, 17,5) |
| 25-120 timmar | 76,2 | 53,5 | 22,6 | (17,0, 28,2) |
| Inget signifikant illamående (maximalt VAS <25 mm på en skala mellan 0–100 mm) | ||||
| Generellt (0-120 timmar) | 72,1 | 64,9 | 7,2 | (1,6, 12,8) |
| 25-120 timmar | 74,0 | 66,9 | 7,1 | (1,5, 12,6) |
* Konfidensintervallen beräknades utan justering för kön och samtidig cytostatikabehandling, vilka inkluderades i den primära analysen av oddskvoten och logistiska modeller.
† En patient med aprepitantbehandling hade data enbart från den akuta fasen och exkluderades från den totala analysen och analys av sen fas. En patient med standardterapi hade data enbart från den sena fasen och exkluderades från den totala analysen och analys av akut fas.
Den uppskattade tiden till första emesis i den kombinerade analysen skildras av Kaplan-Meier plotten i Figur 1.
Figur 1
Procent av vuxna patienter som fick högemetogen cytostatikabehandling som var fria från emesis över tid – Behandlingscykel 1
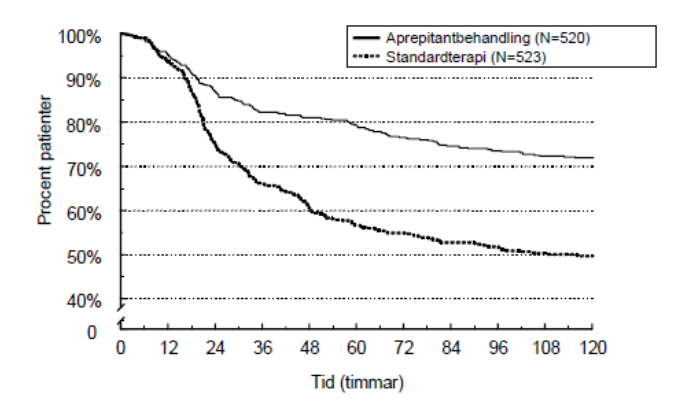
Statistiskt signifikanta effektskillnader sågs även i var och en av de 2 individuella studierna.
I samma 2 kliniska studier fortsatte 851 vuxna patienter in i den förlängda behandlingscykeln i upp till 5 behandlingscykler med cytostatikabehandling. Effekten av aprepitantbehandlingen upprätthölls uppenbarligen under alla behandlingscykler.
I en randomiserad, dubbel-blind studie med totalt 866 vuxna patienter (864 kvinnor, 2 män) som fick cytostatikabehandling inkluderande cyklofosfamid 750 – 1 500 mg/m2; eller cyklofosfamid 500–1500 mg/m2 och doxirubicin (≤60 mg/m2) eller epirubicin (≤100 mg/m2), jämfördes aprepitant i kombination med en ondansetron/dexametason behandling (se avsnitt Dosering och administreringssätt) med standardterapi (placebo plus ondansetron 8 mg oralt (två gånger Dag 1 och var 12:e timme Dag 2 och 3) plus dexametason 20 mg oralt Dag 1).
Effekten baserades på bedömning av det sammansatta måttet: fullständigt svar (definierat som ingen emesis och ingen användning av undsättande behandling) i första hand under behandlingscykel 1.
En sammanfattning av de huvudsakliga studieresultaten visas i tabell 2:
Tabell 2
Procentandel vuxna patienter med behandlingssvar uppdelat i behandlingsgrupp och fas
Behandlingscykel 1
Måttligt emetogen cytostatikabehandling
Aprepitant-behandling (N= 433) † | Standardterapi (N=424) | Skillander* | ||
| % | % | % | (95 % KI) | |
| SAMMANSATTA MÅTT | ||||
| Fullständigt svar (ingen emesis och ingen användning av undsättande behandling) | ||||
| Generellt (0-120 timmar) | 50,8 | 42,5 | 8,3 | (1,6, 15,0) |
| 0-24 timmar | 75,7 | 69,0 | 6,7 | (0,7, 12,7) |
| 25-120 timmar | 55,4 | 49,1 | 6,3 | (-0,4, 13,0) |
| INDIVUDUELLA MÅTT | ||||
| Ingen emesis (inga episoder av emesis oavsett användning av undsättande behandling) | ||||
| Generellt (0-120 timmar) | 75,7 | 58,7 | 17,0 | (10,8, 23,2) |
| 0-24 timmar | 87,5 | 77,3 | 10,2 | (5,1, 15,3) |
| 25-120 timmar | 80,8 | 69,1 | 11,7 | (5,9, 17,5) |
| Inget signifikant illamående (maximalt VAS <25 mm på en skala 0–100 mm) | ||||
| Generellt (0-120 timmar) | 60,9 | 55,7 | 5,3 | (-1,3, 11,9) |
| 0-24 timmar | 79,5 | 78,3 | 1,3 | (-4,2, 6,8) |
| 25-120 timmar | 65,3 | 61,5 | 3,9 | (-2,6, 10,3) |
* Konfidensintervallen beräknades utan justering för ålderskategori (<55 år, ≥55 år) och prövargrupp, vilka inkluderades i den primära analysen av oddskvoten och logistiska modeller.
† En patient med aprepitantbehandling hade data enbart från akut fas och exkluderades från den totala analysen och analys av sen fas.
I samma kliniska studie, fortsatte 744 vuxna patienter i en förlängningsdel med flera ytterligare behandlingscykler med cytostatikabehandling. Effekten av aprepitantbehandlingen upprätthölls under alla behandlingscykler.
I en andra kliniska multicenter, randomiserad, dubbelblind, parallellgruppsstudie jämfördes aprepitantbehandlingen med standardterapi hos 848 vuxna patienter (652 kvinnor, 196 män) som fick en cytostatikabehandling med intravenös dos av oxaliplatin, karboplatin, epirubicin, idarubicin, ifosfamid, irinotekan, daunorubicin, doxorubicin; cyclofosfamid intravenöst (<1 500 mg/m2) eller cytarabin intravenöst (>1 g/m2). Patienter som fick aprepitantbehandlingen fick cytostatikabehandling mot ett antal olika tumörer, däribland 52% bröstcancer, 21% gastrointestinala cancerformer inklusive kolorektal cancer, 13% lungcancer och 6% gynekologiska cancerformer. Aprepitantbehandlingen i kombination med ondansetron/dexametasonbehandling (se avsnitt Dosering och administreringssätt) jämfördes med standardterapi (placebo i kombination med ondansetron 8 mg oralt (två gånger Dag 1, och var tolfte timme Dag 2 och 3) plus dexametason 20 mg oralt Dag 1).
Effekt baserades på utvärderingen av följande primära och viktiga sekundära effektmått: Ingen emesis under den generella perioden (0 till 120 timmar efter cytostatikabehandling), utvärdering av säkerhet och tolerabilitet vid aprepitantbehandlingen mot illamående och kräkningar inducerade av cytostatikabehandling (CINV) och fullständigt behandlingssvar (definierat som avsaknad av emesis och ingen användning av undsättande behandling) under den generella perioden (0 till 120 timmar efter cytostatikabehandling). Därutöver utvärderades inget signifikant illamående under den generella perioden (0 till 120 timmar efter cytostatikabehandling) som ett explorativt effektmått, och under akut respektive fördröjd fas som post-hoc-analys.
En sammanfattning av huvudresultaten visas i tabell 3.
Tabell 3
Procentandel vuxna patienter med behandlingssvar uppdelat i behandlingsgrupp och fas i studie 2 – Behandlingscykel 1
Måttlig emetogen cytostatikabehandling
Aprepitantbehandling (N= 425) | Standard terapi (N=406) | Skillnader* | ||
| % | % | % | (95 % KI) | |
Fullständigt svar (ingen emesis och ingen användning av undsättande behandling) | ||||
| Generellt (0-120 timmar) | 68,7 | 56,3 | 12,4 | (5,9, 18,9) |
| 0-24 timmar | 89,2 | 80,3 | 8,9 | (4,0, 13,8) |
| 25-120 timmar | 70,8 | 60,9 | 9,9 | (3,5, 16,3) |
| Ingen emesis (ingen emesis oavsett användning av undsättande behandling) | ||||
| Generellt (0-120 timmar) | 76,2 | 62,1 | 14,1 | (7,9, 20,3) |
| 0-24 timmar | 92,0 | 83,7 | 8,3 | (3,9, 12,7) |
| 25-120 timmar | 77,9 | 66,8 | 11,1 | (5,1, 17,1) |
| Inget signifikant illamående (maximalt VAS <25 mm på en skala 0–100 mm) | ||||
| Generellt (0-120 timmar) | 73,6 | 66,4 | 7,2 | (1,0, 13,4) |
| 0-24 timmar | 90,9 | 86,3 | 4,6 | (0,2, 9,0) |
| 25-120 timmar | 74,9 | 69,5 | 5,4 | (-0,7, 11,5) |
* Konfidensintervallen beräknades utan justering för kön och område, vilka inkluderades i den primära analysen med logistiska modeller.
Nyttan av kombinationsbehandling med aprepitant den totala studiepopulationen berodde huvudsakligen på resultat hos patienter med otillräcklig kontroll med standardbehandlingen såsom hos kvinnor, även om resultaten var numeriskt överlägsna standardterapi oberoende av ålder, tumörtyp eller kön. Fullständigt svar på aprepitantbehandlingen och standardbehandlingen nåddes hos 209 av 324 (65%) respektive 161 av 320 (50%) hos kvinnor och 83 av 101 (82%) respektive 68 av 87 (78%) hos män.
Pediatrisk population
I en randomiserad, dubbel-blind, kontrollerad klinisk studie med aktiv jämförelse som inkluderade 302 barn och ungdomar (i åldern 6 månader till 17 år) som fick måttlig eller högemetogen cytostatika, jämfördes aprepitantregimen mot en kontrollregim för profylax mot CINV. Effekten av aprepitantregimen utvärderades efter en enstaka behandlingscykel (behandlingscykel 1). Patienter hade möjlighet att erhålla öppen aprepitantbehandling i efterföljande cykler (frivillig behandlingscykel 2–6), emellertid utvärderades inte effekten i dessa frivilliga cykler.
Aprepitantregimen för ungdomar i åldern 12 till och med 17 års ålder (n=47) bestod av aprepitant kapslar 125 mg oralt på Dag 1 och 80 mg/dag på Dag 2 och 3 i kombination med ondansetron på Dag 1. Aprepitantregimen för barn i åldern 6 månader till yngre än 12 år (n=105) bestod av aprepitant pulver till oral suspension 3,0 mg/kg (upp till 125 mg) oralt på Dag 1 och 2,0 mg/kg (upp till 80 mg) oralt på Dag 2 och 3 i kombination med ondansetron på Dag 1. Kontrollregimen hos ungdomar i åldern 12 år till och med 17 år (n=48) och barn i åldern 6 månader till yngre än 12 år (n=102) bestod av placebo för aprepitant på Dag 1, 2 och 3 i kombination med ondansetron på Dag 1. aprepitant eller placebo samt ondansetron gavs 1 timme respektive 30 minuter före påbörjande av cytostatikabehandling. Dexametason givet intravenöst var efter beslut av läkaren tillåtet som del av den antiemetiska behandlingen hos pediatriska patienter i båda åldersgrupperna. En dosreduktion (50%) av dexametason krävdes för pediatriska patienter som fick aprepitant. Ingen dosreduktion krävdes för pediatriska patienter som fick kontrollregimen. Bland de pediatriska patienterna använde 29% i aprepitantregimen och 28% i kontrollregimen dexametason som del av behandlingen i behandlingscykel 1.
Den antiemetiska aktiviteten för aprepitant utvärderades under en 5-dagarsperiod (120 timmar) efter initiering av cytostatikabehandling på Dag 1. Det primära effektmåttet var fullständigt svar i den fördröjda fasen (25 till 120 timmar efter påbörjande av cytostatika) i cykel 1. En sammanfattning av huvudresultaten från studien visas i tabell 4.
Tabell 4
Antal (%) pediatriska patienter med fullständigt behandlingssvar och ingen emesis uppdelat efter behandlingsgrupp och fas – Behandlingscykel 1 (Intent to treat population)
Aprepitantregim n/m (%) | Kontrollregim n/m (%) | |
| PRIMÄRT EFFEKTMÅTT | ||
| Fullständigt svar* – fördröjd fas | 77/152 (50,7)† | 39/150 (26,0) |
| ANDRA FÖRUTBESTÄMDA EFFEKTMÅTT | ||
| Fullständigt svar * – akut fas | 101/152 (66,4)‡ | 78/150 (52,0) |
| Fullständigt svar * – generell fas | 61/152 (40,1)† | 30/150 (20,0) |
| Ingen emesis§ – generell fas | 71/152 (46,7)† | 32/150 (21,3) |
* Fullständigt svar = Ingen emesis eller kväljningar eller ulkningar och ingen användning av undsättande läkemedel. † p <0,01 jämfört med kontrollregim ‡ p <0,05 jämfört med kontrollregim § Ingen emesis = Ingen emesis eller kväljning eller ulkning n/m = Antal patienter med önskat svar/antal patienter inkluderade vid tidpunkt Akut fas: 0 till 24 timmar efter påbörjande av cytostatika Fördröjd fas: 25 till 120 timmar efter påbörjande av cytostatika Generell fas: 0 till 120 timmar efter påbörjande av cytostatika | ||
Den uppskattade tiden till första emesis efter påbörjande av cytostatikabehandling var längre vid aprepitantregimen (uppskattad mediantid till första emesis var 94,5 timmar) jämfört med kontrollregimen (uppskattad mediantid till första emesis var 26,0 timmar) vilket visas i Kaplan-Meierkurvorna i figur 2.
Figur 2
Tid till första episod av emesis från start av administrering av cytostatika - pediatriska patienter i den generella fasen – Behandlingscykel 1 (Intent to treat population)
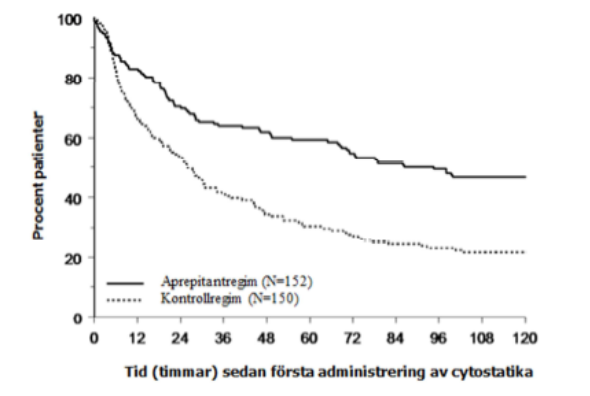
En effektanalys av subpopulationer i behandlingscykel 1 visade, oavsett åldersgrupp, kön, användning av dexametason som antiemetikaprofylax och cytostatikas emetogenicitet, att aprepitantregimen gav bättre kontroll avseende effektmåttet fullständigt svar än kontrollregimen.
Farmakokinetiska egenskaper
Aprepitant uppvisar icke-linjär farmakokinetik. Både clearance och absolut biotillgänglighet minskar med ökad dos.
Absorption
Den genomsnittliga absoluta orala biotillgängligheten av aprepitant är 67% för 80 mg kapseln och 59% för 125 mg kapseln. Den genomsnittliga maximala plasmakoncentrationen (Cmax) inträffade efter ca 4 timmar (tmax). Oral administrering av kapseln tillsammans med en ca 800 kcal standardfrukost resulterade i en ökning av AUC för aprepitant med upp till 40%. Denna ökning ansågs inte vara kliniskt betydelsefull.
Farmakokinetiken för aprepitant är icke-linjär i det terapeutiska intervallet. Hos friska, unga vuxna var ökningarna av AUC0-∞ 26% större än dosproportionellt mellan engångsdoser av 80 mg och 125 mg administrerade efter födointag.
Efter oral administrering av en 125 mg engångsdos av aprepitant Dag 1 och 80 mg dagligen Dag 2 och 3, var AUC0-24hr (medelvärde ± SD) 19,6 ± 2,5 μg∙h /ml respektive 21,2 ± 6,3 μg∙h/ml Dag 1 och 3. Cmax var 1,6 ± 0,36 μg/ml och 1,4 ± 0,22 μg/ml Dag 1 respektive Dag 3.
Distribution
Aprepitant binds i hög grad till plasmaproteiner, i genomsnitt 97%. Den genomsnittliga distributionsvolymen vid steady state (Vdss) är ca 66 liter hos människa.
Metabolism
Aprepitant genomgår omfattande metabolism. Hos friska unga vuxna står aprepitant för ca 19% av radioaktiviteten i plasma under 72 timmar efter intravenös administrering av en 100 mg engångsdos av [14C]-fosaprepitant, en prodrug av aprepitant. Detta indikerar en påtaglig närvaro av metaboliter i plasman. Tolv metaboliter av aprepitant har identifierats i plasma hos människa. Metabolismen av aprepitant sker till stor del via oxidation av morfolinringen och dess sidokedjor, och de resulterande metaboliterna är enbart svagt aktiva. In vitro studier på levermikrosomer från människa indikerar att aprepitant metaboliseras primärt av CYP3A4 och potentiellt med ett mindre bidrag av CYP1A2 och CYP2C19.
Eliminering
Aprepitant utsöndras inte oförändrad i urinen. Metaboliterna utsöndras i urinen och via gallvägarna i faeces. Efter en intravenöst administrerad 100 mg engångsdos av [14C]-fosaprepitant, en prodrug av aprepitant, till friska frivilliga återfanns 57% av radioaktiviteten i urinen och 45% i faeces.
Aprepitants plasmaclearance är dosberoende, minskar med ökad dos och varierar mellan ca 60 till 72 ml/min i det terapeutiska doseringsintervallet. Den terminala halveringstiden varierade mellan ca 9 och 13 timmar.
Farmakokinetiken hos särskilda patientgrupper
Äldre: Efter oral administrering av en 125 mg engångsdos aprepitant Dag 1 och 80 mg dagligen Dag 2 till 5 var AUC0-24hr för aprepitant 21% högre Dag 1 och 36% högre Dag 5 hos äldre (>65 år) jämfört med yngre vuxna. Cmax var 10% högre Dag 1 och 24% högre Dag 5 hos äldre jämfört med yngre vuxna. Dessa skillnader anses inte vara kliniskt betydelsefulla. Ingen dosjustering av Aprepitant Stada är nödvändig för äldre patienter.
Kön: Efter oral administrering av en 125 mg engångsdos aprepitant, är Cmax för aprepitant 16% högre för kvinnor jämfört med män. Aprepitants halveringstid är 25% lägre för kvinnor jämfört med män och tmax inträffar vid ungefär samma tidpunkt. Dessa skillnader anses inte vara kliniskt betydelsefulla.
Ingen dosjustering av Aprepitant Stada är nödvändig baserat på kön.
Nedsatt leverfunktion: Lätt nedsatt leverfunktion (Child-Pugh skala klass A) påverkar inte farmakokinetiken för aprepitant i en klinisk relevant grad. Dosjustering är inte nödvändig för patienter med lätt nedsatt leverfunktion. Baserat på tillgängliga data kan inte slutsatser dras rörande påverkan av måttligt nedsatt leverfunktion på farmakokinetiken för aprepitant (Child-Pugh klass B). Det finns inga kliniska eller farmakokinetiska data från patienter med gravt nedsatt leverfunktion (Child-Pugh klass C).
Nedsatt njurfunktion: En 240 mg engångsdos av aprepitant administrerades till patienter med gravt nedsatt njurfunktion (CrCl <30 ml/min) och till patienter med njursjukdom i slutskedet (ESRD) som behöver hemodialys.
Hos patienter med gravt nedsatt njurfunktion minskade AUC0-∞ för totalt aprepitant (obundet och proteinbundet) med 21% och Cmax minskade med 32% jämfört med friska frivilliga. Hos patienter med ESRD som genomgick hemodialys minskade AUC0-∞ för totalt aprepitant med 42% och Cmax minskade med 32%. På grund av små minskningar i proteinbindningsgraden av aprepitant hos patienter med njursjukdom, påverkades AUC av det farmakologiskt aktiva obundna läkemedlet inte signifikant jämfört med friska frivilliga. Hemodialys utförd 4 eller 48 timmar efter dosering hade ingen signifikant effekt på farmakokinetiken för aprepitant; mindre än 0,2% av dosen återfanns i dialysatet.
Dosjustering av Aprepitant Stada är inte nödvändig för patienter med nedsatt njurfunktion eller för patienter med ESRD som genomgår hemodialys.
Pediatrisk population: Som del av en 3-dagarsregim gav dosering med aprepitant kapslar (125/80/80 mg) hos ungdomar (i åldern 12 till och med 17 år) AUC0-24hr över 17 μg∙h/ml på Dag 1 med koncentrationer (Cmin) vid slutet av Dag 2 och 3 över 0,4 μg/ml hos en majoritet av patienter. Medianvärdet för den maximala plasmakoncentrationen (Cmax) var ungefär 1,3 μg/ml på Dag 1 och inträffade efter ungefär 4 timmar. Som del av en 3-dagarsregim gav dosering med aprepitant pulver till oral suspension (3/2/2 mg/kg) hos patienter i åldern 6 månader till yngre än 12 år AUC0-24hr över 17 μg∙h /ml på Dag 1 med koncentrationer (Cmin) vid slutet av Dag 2 och 3 över 0,1 μg/ml hos en majoritet av patienter. Medianvärdet för den maximala plasmakoncentrationen (Cmax) var ungefär 1,2 μg/ml på Dag 1 och inträffade efter 5 till 7 timmar.
En populationsfarmakokinetisk analys av aprepitant hos pediatriska patienter (i åldern 6 månader till och med 17 år) tyder på att kön och etnisk tillhörighet inte har någon klinisk betydelse på farmakokinetiken för aprepitant.
Förhållande mellan koncentration och effekt
Med hjälp av användning av en högspecifik NK1-receptor markör, har positronemissionstomografi (PET)-studier med friska unga män visat att aprepitant penetrerar hjärnan och upptar NK1-receptorer på ett dos- och plasma-koncentrationsberoende sätt. De plasmakoncentrationer av aprepitant som uppnåddes med 3-dagarsbehandlingen med Aprepitant Stada hos vuxna förutspås uppta mer än 95% av hjärnans NK1-receptorer.
Prekliniska säkerhetsuppgifter
Gängse studier avseende allmäntoxicitet, gentoxicitet, karcinogenicitet, reproduktionseffekter och effekter på utveckling visade inte några särskilda risker för människa. Det bör dock noteras att systemexponeringen hos gnagare var densamma eller till och med lägre än terapeutisk exponering hos människa vid 125 mg/80 mg doser. Dessutom, även om inga biverkningar observerades i reproduktionsstudierna vid humana exponeringsnivåer, var exponeringarna i djur inte tillräckliga för att göra en adekvat riskbedömning hos människa.
I en toxicitetsstudie med unga råttor som behandlats från postnatal dag 10 till dag 63, ledde aprepitant från 250 mg/kg b.i.d till en tidigare vaginalöppning hos honor och från 10 mg/kg b.i.d. till en fördröjd preputial separation hos hanar. Det fanns inga marginaler till kliniskt relevant exponering. Det fanns inga behandlingsrelaterade effekter på parning, fertilitet eller embryonal/fetal överlevnad och inga patologiska förändringar i de reproduktiva organen. I en toxicitetsstudie med unga hundar som behandlats från postnatal dag 14 till dag 42, sågs från 4 mg/kg/dag en minskad testikelvikt och storlek på Leydig-celler hos hanar från 6 mg/kg/dag och ökad livmodervikt, hypertrofi av uterus och cervix samt ödem i vaginalvävnad hos hondjur från 4 mg/kg/dag. Det fanns inga marginaler till kliniskt relevant exponering för aprepitant. Vid korttidsbehandling i enlighet med rekommenderad dosregim anses det osannolikt att dessa fynd är kliniskt relevanta.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Kapselns innehåll
Hypromellose
Poloxamer
Sackaros
Mikrokristallin cellulosa
Kapsel hölje (125 mg)
Gelatin
Natriumlaurilsulfat (E487)
Titandioxid (E171)
Röd järnoxid (E172)
Kapsel hölje (80 mg)
Gelatin
Natriumlaurilsulfat (E487)
Titandioxid (E171)
Tryckbläck
Shellack
Svart järnoxid (E172)
Propylenglykol (E1520)
Inkompatibiliteter
Ej relevant.
Hållbarhet
30 månader.
Särskilda förvaringsanvisningar
Förvaras i originalförpackningen. Fuktkänsligt.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
APREPITANT STADA kapseli, kova
125 mg+80 mg (L:ei) 1x125 mg + 2x80 mg (42,57 €)
80 mg (L:ei) 2 fol (29,08 €), 5 fol (36,82 €)
125 mg (L:ei) 5 fol (126,35 €)
PF-selosteen tieto
Aprepitant Stada är förpackad i en kartong innehållande lämpligt antal OPA/ALU/PVC-aluminiumblister med en bipacksedel.
Aprepitant Stada 125 mg hårda kapslar tillhandahållas i följande förpackningsstorlekar:
- Aluminiumblister innehållande en 125 mg kapsel
- 5 aluminiumblister som innehåller en 125 mg kapsel.
Aprepitant Stada 80 mg hårda kapslar tillhandahållas i följande förpackningsstorlekar:
- Aluminiumblister innehållande en 80 mg kapsel
- 2-dagars behandlingsförpackning innehållande två 80 mg kapslar
- 5 aluminiumblister som innehåller en 80 mg kapsel.
Aprepitant Stada 125 mg + 80 mg hårda kapslar tillhandahållas i följande förpackningsstorlekar:
- 3-dagars behandlingsförpackning innehållande en 125 mg kapsel och två 80 mg kapslar.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Läkemedlets utseende:
125 mg kapslarna är ogenomskinliga hårda gelatinkapslar storlek 1, med en rosa överdel och vit underdel, med ”125 mg” tryckt i svart bläck på underdelen.
80 mg kapslarna är ogenomskinliga hårda gelatinkapslar storlek 2, med vit under- och överdel, och med”80 mg” tryckt i svart bläck på underdelen.
Särskilda anvisningar för destruktion och övrig hantering
Inga särskilda anvisningar för destruktion.
Ersättning
APREPITANT STADA kapseli, kova
125 mg+80 mg 1x125 mg + 2x80 mg
80 mg 2 fol
- Ylempi erityiskorvaus (100 %). Rintasyöpä (115), Eturauhassyöpä (116), Leukemiat, muut pahanlaatuiset veri- ja luuydintaudit sekä pahanlaatuiset imukudostaudit (117), Gynekologiset syövät (128), Pahanlaatuiset kasvaimet, joita ei ole edellä erikseen mainittu (130).
- Peruskorvaus (40 %).
APREPITANT STADA kapseli, kova
80 mg 5 fol
125 mg 5 fol
- Ei korvausta.
Atc-kod
A04AD12
Datum för översyn av produktresumén
18.02.2020
Yhteystiedot
PL 1310, Puolikkotie 8, 02230 Espoo (käyntiosoite)
00101 Helsinki
0207 416 888