
ILARIS injektioneste, liuos 150 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Potilaskortti: Jaksottaisten kuumeoireyhtymien hoitoon
Potilaskortti: Kihtiartriittikohtausten hoitoon
Potilaskortti: Stillin taudin hoitoon, mukaan lukien aikuisiän Stillin taudin ja yleisoireisen lastenreuman hoito
Vaikuttavat aineet ja niiden määrät
Ilaris 150 mg/ml injektioneste, liuos, injektiopullo
Yksi injektiopullo sisältää 150 mg kanakinumabia 1 ml:ssa.
Ilaris 150 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kynä sisältää 150 mg kanakinumabia 1 ml:ssa.
Kanakinumabi on ihmisperäinen monoklonaalinen vasta-aine, joka on tuotettu rekombinantti DNA-tekniikalla hiiren myeloomasoluissa (Sp2/0).
Apuaine, jonka vaikutus tunnetaan
Injektioneste, liuos sisältää 0,4 mg/ml polysorbaatti 80:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste).
Injektioneste, liuos (injektioneste), esitäytetty kynä (SensoReady-kynä).
Kliiniset tiedot
Käyttöaiheet
Jaksoittaiset kuumeoireyhtymät
Ilaris on tarkoitettu seuraavien autoinflammatoristen jaksoittaisten kuumeoireyhtymien hoitoon aikuisille, nuorille sekä 2-vuotiaille ja sitä vanhemmille lapsille:
Kryopyriiniin liittyvät oireyhtymät
Ilaris on tarkoitettu kryopyriiniin liittyvien oireyhtymien (CAPS) hoitoon, kuten:
- Muckle-Wellsin oireyhtymän (MWS) hoitoon
- imeväisiässä alkavan inflammatorisen monielintaudin (NOMID) / kroonisen infantiilin hermo-, iho- ja niveltulehdusoireyhtymän (CINCA) hoitoon
- periytyvän autoinflammatorisen kylmäurtikarian (FCAS) / familiaalisen kylmäurtikarian (FCU) vaikeiden muotojen hoitoon, kun potilaalla on kylmäurtikariaihottuman lisäksi muita oireita ja merkkejä sairaudestaan.
Tuumorinekroositekijään liittyvä jaksoittainen oireyhtymä (TRAPS)
Ilaris on tarkoitettu tuumorinekroositekijään (TNF) liittyvän jaksoittaisen oireyhtymän (TRAPS) hoitoon.
Hyperimmunoglobulinemia D -oireyhtymä (HIDS) / mevalonaattikinaasin vajaus (MKD)
Ilaris on tarkoitettu hyperimmunoglobulinemia D -oireyhtymän (HIDS) / mevalonaattikinaasin vajauksen (MKD) hoitoon.
Perinnöllinen Välimeren kuume (FMF)
Ilaris on tarkoitettu perinnöllisen Välimeren kuumeen (FMF) hoitoon. On suositeltavaa käyttää Ilarista yhdessä kolkisiinin kanssa silloin kun se on asianmukaista.
Ilaris on tarkoitettu myös seuraavien sairauksien hoitoon:
Stillin tauti
Ilaris on tarkoitettu aktiivisen Stillin taudin (mukaan lukien aikuisiän Stillin tauti ja yleisoireinen lastenreuma) hoitoon vähintään 2-vuotiaille potilaille, kun aiemmalla hoidolla (NSAID-lääkkeillä ja systeemisillä kortikosteroideilla) ei ole saavutettu riittävää vastetta. Ilaris-valmistetta voidaan käyttää monoterapiana tai yhdessä metotreksaatin kanssa.
Kihtiartriitti
Ilaris on tarkoitettu usein toistuvista kihtiartriittikohtauksista (vähintään 3 kohtausta viimeksi kuluneiden 12 kuukauden aikana) kärsivien aikuispotilaiden oireenmukaiseen hoitoon tilanteissa, joissa ei-steroidirakenteiset tulehduskipulääkkeet (NSAID-lääkkeet) ja kolkisiini ovat vasta-aiheisia, kun potilas ei siedä näitä lääkkeitä tai kun em. lääkkeillä ei saada riittävää vastetta, sekä potilaille, joille toistuvat kortikosteroidihoitojaksot eivät sovi (ks. kohta Farmakodynamiikka).
Ehto
Kryopyriiniin liittyvissä oireyhtymissä, tuumorinekroositekijään liittyvässä jaksoittaisessa oireyhtymässä, hyperimmunoglobulinemia D -oireyhtymässä / mevalonaattikinaasin vajauksessa, perinnöllisessä Välimeren kuumeessa ja Stillin taudissa hoito on aloitettava ja toteutettava kyseenomaisen käyttöaiheen diagnosoinnista ja hoidosta kokemusta saaneen erikoislääkärin seurannassa. Kihtiartriitin hoidossa lääkärillä tulee olla kokemusta biologisten lääkkeiden käytöstä ja terveydenhuollon ammattilaisen on annosteltava valmiste potilaalle.
Annostus ja antotapa
Kryopyriiniin liittyvissä oireyhtymissä, tuumorinekroositekijään liittyvässä jaksoittaisessa oireyhtymässä, hyperimmunoglobulinemia D -oireyhtymässä / mevalonaattikinaasin vajauksessa, perinnöllisessä Välimeren kuumeessa ja Stillin taudissa hoito on aloitettava ja toteutettava kyseenomaisen käyttöaiheen diagnosoinnista ja hoidosta kokemusta saaneen erikoislääkärin seurannassa.
Kihtiartriitin hoidossa lääkärillä on oltava kokemusta biologisten lääkkeiden käytöstä ja terveydenhuollon ammattilaisen on annosteltava Ilaris potilaalle.
Annostus
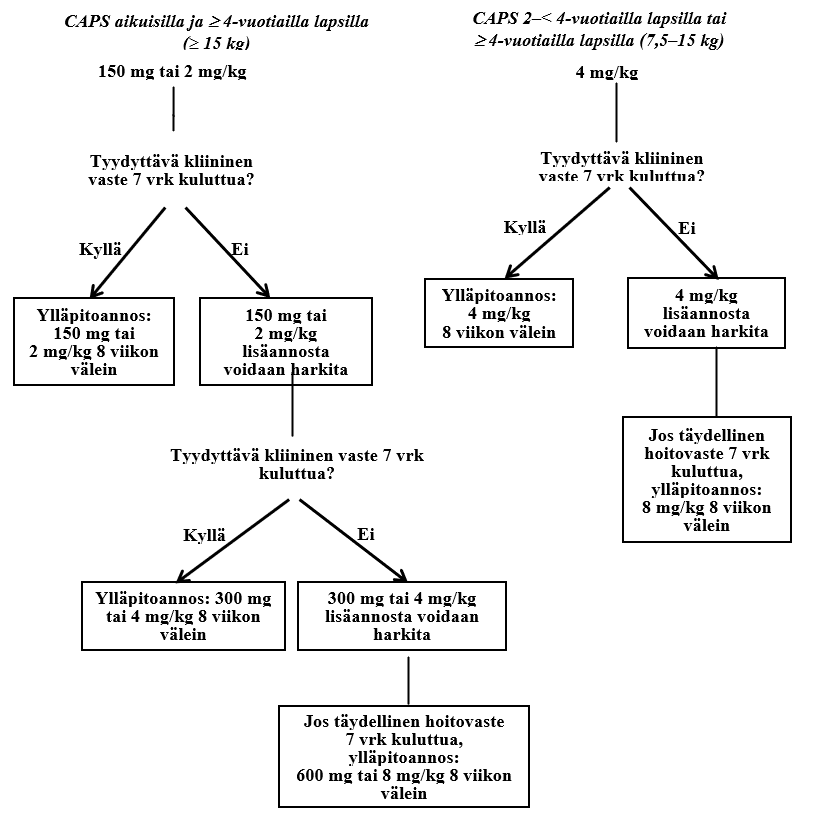
CAPS: Aikuiset, nuoret ja vähintään 2-vuotiaat lapset
Suositeltava kanakinumabin aloitusannos CAPS-potilaille on:
Aikuiset, nuoret ja ≥ 4-vuotiaat lapset:
- 150 mg potilaille, jotka painavat yli 40 kg
- 2 mg/kg potilaille, jotka painavat 15–40 kg
- 4 mg/kg potilaille, jotka painavat 7,5–15 kg
2–< 4-vuotiaat lapset:
- 4 mg/kg potilaille, jotka painavat vähintään 7,5 kg
Tämä annos annetaan injektiona ihon alle kerta-annoksena kahdeksan viikon välein.
Jos 150 mg tai 2 mg/kg aloitusannosta käyttävillä potilailla ei ole saatu tyydyttävää kliinistä vastetta (ihottuman tai muiden, yleistyneiden tulehdusoireiden häviäminen) 7 vuorokauden kuluttua hoidon alkamisesta, toisen 150 mg:n tai 2 mg:n/kg kanakinumabiannoksen antamista voidaan harkita. Jos tämän jälkeen saavutetaan täydellinen hoitovaste, on hoitoa jatkettava noudattaen tätä tehostettua hoito-ohjelmaa annoksella 300 mg tai 4 mg/kg 8 viikon välein. Jos tyydyttävää kliinistä vastetta ei ole saavutettu 7 vuorokauden kuluttua annoksen suurentamisesta, kolmatta 300 mg tai 4 mg/kg kanakinumabiannosta voidaan harkita. Jos täydellinen hoitovaste saavutetaan tämän jälkeen, on harkittava tehostettua hoitoa (600 mg tai 8 mg/kg 8 viikon välein) perustuen yksilölliseen kliiniseen arviointiin.
Jos 4 mg/kg aloitusannosta käyttävillä potilailla ei ole saavutettu tyydyttävää kliinistä vastetta 7 vuorokauden kuluttua hoidon aloittamisesta, toista 4 mg/kg kanakinumabiannosta voidaan harkita. Jos täydellinen hoitovaste saavutetaan tämän jälkeen, on harkittava tehostetun hoidon jatkamista (8 mg/kg 8 viikon välein) perustuen yksilölliseen kliiniseen arviointiin.
Kliininen kokemus alle 4 viikon antovälistä tai yli 600 mg tai 8 mg/kg annoksista on vähäistä.
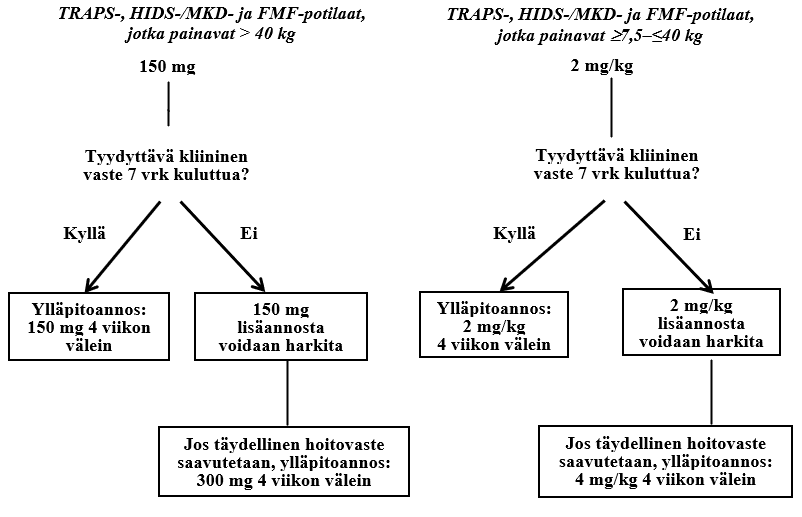
TRAPS, HIDS/MKD ja FMF: Aikuiset, nuoret ja vähintään 2-vuotiaat lapset
Suositeltava kanakinumabin aloitusannos TRAPS-, HIDS-/MKD- ja FMF-potilaille on:
- 150 mg potilaille, jotka painavat yli 40 kg
- 2 mg/kg potilaille, jotka painavat 7,5–40 kg
Lääke annetaan injektiona ihon alle kerta-annoksena neljän viikon välein.
Jos tyydyttävää kliinistä vastetta ei ole saavutettu 7 vuorokauden kuluttua hoidon alkamisesta, toista 150 mg tai 2 mg/kg kanakinumabiannosta voidaan harkita. Jos täydellinen hoitovaste saavutetaan tämän jälkeen, tehostettua hoitoa annostuksella 300 mg (tai 4 mg/kg ≤ 40 kg painavilla potilailla) 4 viikon välein jatketaan.
Jos potilaan kliininen tila ei parane, suositellaan, että hoitava lääkäri harkitsee uudelleen kanakinumabihoidon jatkamista.
Stillin tauti (yleisoireinen lastenreuma ja aikuisiän Stillin tauti)
Suositeltu kanakinumabiannos Stillin taudin hoitoon ≥ 7,5 kg painaville potilaille on 4 mg/kg (enintään 300 mg) injektiona ihon alle neljän viikon välein. Jos potilaan kliininen tila ei parane, suositellaan, että hoitava lääkäri harkitsee uudelleen kanakinumabihoidon jatkamista.
Kihtiartriitti
Hyperurikemian hoito on aloitettava tai säädettävä optimaaliseksi tilanteeseen sopivalla uraattipitoisuutta alentavalla lääkityksellä (ULT). Kanakinumabia on käytettävä tarpeen mukaan kihtiartriittikohtausten hoitoon annettavana lääkkeenä.
Kihtiartriitista kärsivien aikuisten hoidossa suositeltu kanakinumabiannos on 150 mg ihonalaisena kerta-annoksena kohtauksen aikana. Parhaan tehon saavuttamiseksi suositellaan kanakinumabin antamista mahdollisimman pian kihtiartriittikohtauksen alettua.
Potilaille, jotka eivät vastaa ensimmäiseen hoitoon, ei suositella uutta kanakinumabi-hoitokertaa. Sellaisten potilaiden kohdalla, jotka vastaavat hoitoon ja jotka myöhemmin tarvitsevat hoitoa uudestaan, on pidettävä vähintään 12 viikon tauko ennen uuden kanakinumabiannoksen antoa (ks. kohta Farmakokinetiikka).
Unohtuneet annokset
Jos CAPS-, TRAPS-, HIDS/MKD-, FMF- tai Stillin tauti (aikuisiän Stillin tauti tai yleisoireinen lastenreuma) -potilaan injektio jää väliin, on unohtunut annos annettava mahdollisimman pian odottamatta seuraavaan aikataulun mukaiseen annokseen. Seuraavat annokset on annettava suositelluin väliajoin.
Erityisryhmät
Pediatriset potilaat
CAPS, TRAPS, HIDS/MKD ja FMF
Kanakinumabin turvallisuutta ja tehoa alle 2 vuoden ikäisten lasten kryopyriiniin liittyvien oireyhtymien, tuumorinekroositekijään liittyvän jaksoittaisen oireyhtymän, hyperimmunoglobulinemia D ‑oireyhtymän / mevalonaattikinaasin vajauksen ja perinnöllisen Välimeren kuumeen hoidossa ei ole varmistettu. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta.
Yleisoireinen lastenreuma
Kanakinumabin turvallisuutta ja tehoa alle 2-vuotiaiden lasten yleisoireisen reuman hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Kihtiartriitti
Ei ole asianmukaista käyttää kanakinumabia pediatrisille potilaille kihtiartriitin hoitoon.
Iäkkäät potilaat
Erityistä annoksen säätöä ei tarvita.
Maksan vajaatoiminta
Kanakinumabia ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla. Suosituksia annostuksesta ei voida antaa.
Munuaisten vajaatoiminta
Lievää munuaisten vajaatoimintaa sairastavien potilaiden annostusta ei tarvitse muuttaa. Kliininen kokemus tästä potilasryhmästä on kuitenkin vähäistä.
Potilaskortti
Kaikkien Ilarista määräävien lääkäreiden on tunnettava valmisteyhteenveto ja ilmoitettava potilaalle/potilasta hoitavalle omaiselle potilaskortista ja kerrottava, mitä tehdä, jos heillä ilmenee infektion tai makrofagiaktivaatio-oireyhtymän oireita tai jos heidät rokotetaan ennen hoitoa. Lääkäri antaa potilaskortin jokaiselle potilaalle/potilasta hoitavalle omaiselle.
Antotapa
Ihon alle.
Sopivia pistoskohtia ovat: reiden yläosa, vatsa, olkavarsi tai pakara. Joka pistoskerralla suositellaan valittavan eri pistoskohta aristamisen välttämiseksi. Alueita, joiden iho on vaurioitunut, jotka ovat mustelmilla tai joissa on ihottumaa, on vältettävä. Arpikudokseen pistämistä on vältettävä, koska kanakinumabialtistus ei tällöin ole välttämättä riittävä.
Injektiopullo
Injektiopullo on tarkoitettu yhtä käyttökertaa varten kerta-annokseksi yhdelle potilaalle.
CAPS, TRAPS, HIDS/MKD, FMF ja Stillin tauti (aikuisiän Stillin tauti ja yleisoireinen lastenreuma)
Kun potilas tai potilasta hoitava omainen on saanut riittävän opastuksen oikeaan injektiotekniikkaan, hän voi pistää kanakinumabi-pistoksen, jos lääkäri arvioi sen olevan asianmukaista ja seuraa hoitoa tarvittaessa (ks. kohta Käyttö- ja käsittelyohjeet).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen annostelua varten.
Esitäytetty kynä
Injektiokynää ei saa ravistaa.
Esitäytetty kynä on tarkoitettu yhtä käyttökertaa varten kerta-annokseksi yhdelle potilaalle.
CAPS, TRAPS, HIDS/MKD, FMF ja Stillin tauti (aikuisiän Stillin tauti ja yleisoireinen lastenreuma)
Saatuaan riittävän opastuksen oikeaan injektiotekniikkaan, aikuiset ja vähintään 12-vuotiaat nuoret potilaat, jotka painavat yli 40 kg, tai heitä hoitava omainen, voivat pistää kanakinumabi-pistoksen, jos lääkäri arvioi sen olevan asianmukaista ja seuraa hoitoa tarvittaessa. Nuoret potilaat saattavat tarvita pistoksen suorittamisessa aikuisen omaisen valvontaa (ks. kohta Käyttö- ja käsittelyohjeet).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiivisessa vaiheessa olevat, vakavat infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Kanakinumabiin liittyy vakavien infektioiden esiintyvyyden lisääntymistä. Potilaita on seurattava huolellisesti infektio-oireiden ja -merkkien varalta kanakinumabihoidon aikana ja sen jälkeen (ks. kohta Haittavaikutukset). Lääkärin on noudatettava varovaisuutta antaessaan kanakinumabia potilaalle, jolla on infektio, jolla on aiemmin ollut toistuvia infektioita tai jolla on perussairaus, joka saattaa altistaa hänet infektioille.
CAPSin, TRAPSin, HIDS/MKD:n, FMF:n ja Stillin taudin (yleisoireinen lastenreuma ja AOSD) hoito
Kanakinumabihoitoa ei saa aloittaa eikä jatkaa, jos potilaalla on jokin lääketieteellistä hoitoa vaativa, aktiivinen infektio.
Kihtiartriitin hoito
Kanakinumabia ei saa antaa aktiivisen infektion aikana.
Kanakinumabin samanaikaista käyttöä tuumorinekroositekijän (TNF) estäjien kanssa ei suositella, koska se saattaa suurentaa vakavien infektioiden riskiä (ks. kohta Yhteisvaikutukset).
Yksittäisiä epätavallisia tai opportunistisia infektiotapauksia (mukaan lukien aspergilloosi, atyyppiset mykobakteeri-infektiot, vyöruusu) on ilmoitettu kanakinumabihoidon aikana. Kanakinumabin ja näiden tapahtumien syy-seuraussuhdetta ei voida sulkea pois.
Tuberkuloosin tartunnanjäljitys
Noin 12 %:lla CAPS-potilaista, joille tehtiin kliinisissä lääketutkimuksissa PPD (puhdistettu proteiinijohdannainen) -tuberkuliinikoe, kanakinumabihoidon aikana tehdyn seurantakokeen tulos oli positiivinen, vaikka potilaalla ei ollut piilevän tai aktiivisen tuberkuloosin kliinisiä löydöksiä.
Ei tiedetä, suurentaako kanakinumabin kaltaisten interleukiini-1:n (IL-1) estäjien käyttö tuberkuloosin uudelleenaktivoitumisriskiä. Ennen hoidon aloittamista kaikki potilaat on tutkittava sekä aktiivisen että piilevän tuberkuloosi-infektion poissulkemiseksi. Etenkin aikuispotilaiden kohdalla suositellaan, että tähän arviointiin kuuluu yksityiskohtainen sairaushistorian selvitys. Suositellaan, että asianmukaiset seulontakokeet (kuten tuberkuliinikoe, gammainterferonitutkimus tai keuhkojen röntgenkuvaus) tehdään kaikille potilaille (ottaen huomioon paikalliset suositukset). Potilaita on seurattava huolellisesti tuberkuloosi-oireiden ja -merkkien varalta kanakinumabihoidon aikana ja sen jälkeen. Kaikkia potilaita on neuvottava ottamaan yhteys lääkäriin, jos kanakinumabihoidon aikana ilmaantuu tuberkuloosin oireita tai löydöksiä (esimerkiksi pitkittynyttä yskää, painon laskua tai lievää lämpöilyä). Jos PPD-tuberkuliinikokeen negatiivinen tulos muuttuu positiiviseksi, etenkin riskiryhmään kuuluvilla potilailla, voidaan harkita tuberkuloosi-infektion seulontaa muilla menetelmillä.
Neutropenia ja leukopenia
Neutropeniaa (absoluuttinen neutrofiilimäärä [ANC] < 1,5 x 109/l) ja leukopeniaa on havaittu IL-1:n toimintaa estävien lääkkeiden, mukaan lukien kanakinumabin, käytön yhteydessä. Kanakinumabihoitoa ei saa aloittaa potilaille, joilla on neutropenia tai leukopenia. Valkosolujen, mukaan lukien neutrofiilien, määrittämistä suositellaan ennen hoidon aloittamista ja uudelleen 1‑2 kuukauden kuluttua. Jatkuvaa tai toistuvaa hoitoa saavien potilaiden osalta suositellaan lisäksi valkosolujen määrittämistä määräajoin hoidon aikana. Jos potilaalle kehittyy neutropenia tai leukopenia, on valkosolujen määrää seurattava huolellisesti ja harkittava hoidon keskeyttämistä.
Pahanlaatuiset kasvaimet
Maligniteetteja on ilmoitettu kanakinumabihoitoa saavilla potilailla. Pahanlaatuisten kasvainten kehittymisriskiä interleukiini-1:n (IL-1) estäjillä annetun hoidon yhteydessä ei tunneta.
Yliherkkyysreaktiot
Kanakinumabihoidon yhteydessä on raportoitu yliherkkyysreaktioita. Suurin osa näistä tapahtumista oli vakavuusasteeltaan lieviä. Kanakinumabi-hoitoon liittyviä anafylaktoidisia tai anafylaktisia reaktioita ei raportoitu kliinisen kanakinumabi-tutkimusohjelman aikana yli 2 600 potilaalla. Vakavien yliherkkyysreaktioiden riskiä, joka on yleinen injisoitavien proteiinien yhteydessä, ei kuitenkaan voida sulkea pois (ks. kohta Vasta-aiheet).
Maksan toiminta
Kliinisissä lääketutkimuksissa on raportoitu ohimenevää ja oireetonta seerumin transaminaasi- tai bilirubiinipitoisuuden suurenemista (ks. kohta Haittavaikutukset).
Rokotukset
Elävien (heikennettyjen) rokotteiden käyttöön mahdollisesti liittyvästä, infektion sekundaarisesta tartuntariskistä kanakinumabihoitoa saavilla potilailla ei ole tutkimustietoa. Siksi eläviä taudinaiheuttajia sisältäviä rokotteita ei saa antaa samanaikaisesti kanakinumabihoidon kanssa, ellei rokottamisesta saatava hyöty selvästi ylitä siihen liittyviä riskejä (ks. kohta Yhteisvaikutukset).
Lapsi- ja aikuispotilaiden kaikki rokotukset (mukaan lukien pneumokokkirokotus ja inaktivoitu influenssarokote) suositellaan antamaan asianmukaisesti ennen kanakinumabihoidon aloittamista (ks. kohta Yhteisvaikutukset).
CAPS-potilaiden NLRP3-geenimutaatio
Kliininen kokemus CAPS-potilaista, joilla ei ole varmistettua NLRP3-geenimutaatiota, on vähäistä.
Makrofagiaktivaatio-oireyhtymä Stillin tautia sairastavilla potilailla (yleisoireinen lastenreuma ja aikuisiän Stillin tauti)
Makrofagiaktivaatio-oireyhtymä (MAS) on tunnettu, henkeä uhkaava häiriö, joka saattaa kehittyä reumatautipotilaille, etenkin Stillin tautia sairastaville. Jos makrofagiaktivaatio-oireyhtymä todetaan tai sitä epäillään, arviointi ja hoito on aloitettava mahdollisimman varhain. Lääkärin on seurattava infektio-oireita ja Stillin taudin pahenemisen oireita, sillä näiden tiedetään aiheuttavan makrofagiaktivaatio-oireyhtymää. Kliinisistä tutkimuksista saatujen kokemusten perusteella kanakinumabi ei näytä lisäävän makrofagiaktivaatio-oireyhtymän ilmaantuvuutta Stillin tautia sairastavilla, mutta varmoja johtopäätöksiä ei kuitenkaan voida tehdä.
Yleisoireinen eosinofiilinen oireyhtymä (DRESS)
Ilaris-valmisteella hoidettavilla potilailla on raportoitu harvoin yleisoireista eosinofiilista oireyhtymää (DRESS). Ilmoitukset ovat koskeneet pääasiassa yleisoireista lastenreumaa (sJIA) sairastavia potilaita. DRESS-oireyhtymää sairastavat potilaat saattavat tarvita sairaalahoitoa, koska kyseinen tila voi johtaa kuolemaan. Jos DRESS-oireyhtymän merkkejä ja oireita ilmaantuu ja jollei oireille voida vahvistaa muuta syytä, Ilarista ei saa antaa uudelleen ja muuta hoitoa on harkittava.
Polysorbaatti 80
Tämä lääkevalmiste sisältää 0,4 mg polysorbaatti 80:tä per 1 ml injektionestettä. Polysorbaatit saattavat aiheuttaa allergisia reaktioita. Neuvo potilasta/potilasta hoitavaa omaista ottamaan yhteyttä lääkäriin, jos hänellä tai hänen lapsellansa on tiedossa olevia allergioita.
Yhteisvaikutukset
Systemaattisia tutkimuksia kanakinumabin ja muiden lääkevalmisteiden välisten yhteisvaikutusten selvittämiseksi ei ole tehty.
Vakavien infektioiden esiintyvyys suureni, kun toista IL-1:n estäjää annettiin yhdistelmänä TNF:n estäjien kanssa. Kanakinumabin samanaikaista käyttöä TNF:n estäjien kanssa ei suositella, koska se saattaa lisätä vakavien infektioiden riskiä.
Kroonista tulehdusta stimuloivat sytokiinit, kuten interleukiini-1-beeta (IL-1-beeta), saattavat hillitä maksan CYP450-entsyymien ekspressiota. CYP450:n ekspressio saattaa siten lisääntyä, kun aloitetaan voimakas sytokiinejä estävä hoito, kuten kanakinumabihoito. Tämä on kliinisesti merkitsevää kapean terapeuttisen indeksin omaavilla CYP450-substraateilla, joiden annosta säädetään yksilöllisesti. Kun kanakinumabihoito aloitetaan tämäntyyppisiä lääkkeitä käyttäville potilaille, on suositeltavaa seurata lääkkeen tehoa tai vaikuttavan aineen pitoisuutta ja säätää lääkevalmisteen annosta tarpeen mukaan.
Kanakinumabihoitoa saaneiden potilaiden vasteesta eläviä taudinaiheuttajia sisältäville rokotteille tai infektion sekundaarisesta tarttumisesta elävästä rokotteesta ei ole tietoa. Eläviä taudinaiheuttajia sisältäviä rokotteita ei siksi saa antaa samanaikaisesti kanakinumabihoidon kanssa, ellei rokotteista saatava hyöty selvästi ylitä siihen liittyviä riskejä. Jos eläviä taudinaiheuttajia sisältävien rokotteiden anto on aiheellista kanakinumabihoidon aloittamisen jälkeen, suositellaan pitämään ainakin kolmen (3) kuukauden tauko sekä edellisen kanakinumabi-injektion jälkeen että ennen seuraavan kanakinumabi-injektion antoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Terveillä aikuispotilailla tehdyn tutkimuksen tulokset osoittivat, että kanakinumabin 300 mg kerta-annos ei vaikuttanut influenssa- tai glykosyloidun proteiinipohjaisen meningokokkirokotteen vasta-ainevasteen alkamiseen tai kestoon.
56 viikkoa kestäneen 4-vuotiailla ja sitä nuoremmilla CAPS-potilailla tehdyn avoimen tutkimuksen tulokset osoittivat, että kaikille ei-eläviä lapsuusajan rokotusohjelman rokotteita saaneille potilaille kehittyi suojaava vasta-ainetaso.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Miesten ja naisten ehkäisy
Naisten on suositeltavaa käyttää tehokasta ehkäisyä kanakinumabihoidon aikana ja kolmen kuukauden ajan viimeisen annoksen jälkeen.
Raskaus
Kanakinumabin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja. Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria eikä epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Riskiä sikiölle/äidille ei tiedetä. Tämän vuoksi suositellaan antamaan valmistetta raskaana oleville tai raskautta suunnitteleville naisille vain hyötyjen ja riskien tarkan arvioinnin jälkeen.
Eläinkokeet viittaavat kanakinumabin läpäisevän istukan ja sitä on havaittu sikiössä. Ihmisiä koskevaa tietoa ei ole saatavilla, mutta kanakinumabi on G-luokan immunoglobuliini (IgG1), joten sen kulkeutuminen istukan läpi on odotettavissa. Kulkeutumisen kliinistä merkitystä ei tunneta. Elävien rokotteiden antamista ei kuitenkaan suositella 16 viikkoon äidin viimeisimmästä kanakinumabiannoksesta vastasyntyneille vauvoille, jotka ovat altistuneet kanakinumabille kohdussa. On suositeltavaa ohjeistaa kanakinumabia raskauden aikana saaneita naisia kertomaan asiasta vauvaa hoitaville terveydenhuollon ammattilaisille ennen kuin vastasyntyneelle annetaan mitään rokotteita.
Imetys
Ei tiedetä, erittyykö kanakinumabi ihmisillä äidinmaitoon. Tämän vuoksi suositellaan, että päätös imettämisestä kanakinumabihoidon aikana tehdään vasta hoidon hyötyjen ja riskien tarkan arvioinnin jälkeen.
Eläinkokeet ovat osoittaneet, että hiiren anti-hiiri-IL-1-beetavasta-aineella ei ollut haitallisia vaikutuksia imetettävien hiirenpoikasten kehitykseen ja että vasta-aine siirtyi niihin (ks. kohta Prekliiniset tiedot turvallisuudesta).
Hedelmällisyys
Kanakinumabin mahdollista vaikutusta ihmisen hedelmällisyyteen ei ole tutkittu järjestelmällisesti.
Kanakinumabilla ei ollut vaikutusta valkotupsu-urosmarmosettien (C. jacchus) hedelmällisyysparametreihin. Hiiren anti-hiiri-IL-1-beetavasta-aineella ei ollut haitallisia vaikutuksia uros- eikä naarashiirten hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ilaris-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Ilaris-hoito saattaa aiheuttaa huimausta/pyörrytystä tai voimattomuutta (ks. kohta Haittavaikutukset). Jos potilaalla ilmenee tällaisia oireita Ilaris-hoidon aikana, on hänen odotettava näiden oireiden poistumista kokonaan, ennen harkintakykyä tai motorisia taitoja vaativien tehtävien suorittamista.
Haittavaikutukset
Turvallisuustietojen yhteenveto
Yleisimpiä haittavaikutuksia olivat infektiot (pääasiassa ylähengitystieinfektiot). Vaikutusta haittavaikutusten tyyppiin tai esiintymistiheyteen ei havaittu pitkäaikaishoidossa.
Kanakinumabihoitoa saaneilla potilailla on ilmoitettu yliherkkyysreaktioita (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Kanakinumabihoitoa saaneilla potilailla on ilmoitettu opportunisti-infektioita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Haittavaikutukset on luokiteltu MedDRA-järjestelmän mukaan elinryhmittäin. Haittavaikutukset on luokiteltu kussakin elinjärjestelmäluokassa yleisyyden mukaan yleisimmistä alkaen. Esiintyvyysluokat on määritelty seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1Taulukko haittavaikutuksista
| MedDRA-elinjärjestelmäluokitus | Käyttöaiheet: Kryopyriiniin liittyvät oireyhtymät, tuumorinekroositekijään liittyvä jaksoittainen oireyhtymä, hyperimmunoglobulinemia D -oireyhtymä / mevalonaattikinaasin vajaus, perinnöllinen Välimeren kuume, yleisoireinen lastenreuma ja kihtiartriitti |
| Infektiot | |
| Hyvin yleiset | Hengitystieinfektiot (mukaan lukien keuhkokuume, keuhkoputkitulehdus, influenssa, virusinfektio, sivuontelotulehdus, nuha, nielutulehdus, tonsilliitti, nenänielutulehdus, ylähengitystieinfektio) Korvatulehdus Selluliitti Maha-suolitulehdus Virtsatieinfektio |
| Yleiset | Vulvovaginaalinen kandidiaasi |
| Hermosto | |
| Yleiset | Huimaus/kiertohuimaus |
| Ruoansulatuselimistö | |
| Hyvin yleiset | Ylävatsakipu1 |
| Melko harvinaiset | Ruokatorven refluksitauti2 |
| Iho ja ihonalainen kudos | |
| Hyvin yleiset | Pistoskohdan reaktio |
| Luusto, lihakset ja sidekudos | |
| Hyvin yleiset | Nivelkipu1 |
| Yleiset | Lihasten ja luiden kipu1 Selkäkipu2 |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Yleiset | Väsymys/voimattomuus2 |
| Tutkimukset | |
| Hyvin yleiset | Munuaisten kreatiniinipuhdistuman pieneneminen1,3 Proteinuria1,4 Leukopenia1,5 |
| Yleiset | Neutropenia5 |
| Melko harvinaiset | Trombosyyttiarvojen pieneneminen5 |
1 Yleisoireisessa lastenreumassa 2 Kihtiartriitissa 3 Arvioidun kreatiniinipuhdistuman perusteella; useimmiten ohimenevä 4 Useimmilla merkkejä lyhytaikaisesta 1+ -positiivisesta virtsan proteiinista testiliuskalla mitattuna 5 Lisätiedot, ks. jäljempänä | |
Stillin tauti (yleisoireinen lastenreuma ja aikuisiän Stillin tauti)
Yleisoireisen lastenreuman yhdistetty analyysi ja aikuisiän Stillin tauti
Yhteensä 445 yleisoireista lastenreumaa sairastavaa potilasta, jotka olivat iältään 2–<20-vuotiaita, sai kanakinumabia kliinisissä tutkimuksissa. Tämä ryhmä sisälsi 321 iältään 2–<12-vuotiasta potilasta, 88 iältään 12–<16-vuotiasta potilasta ja 36 iältään 16–<20-vuotiasta potilasta. Kaikkien yleisoireista lastenreumaa sairastavien potilaiden yhdistetty turvallisuusanalyysi osoitti, että nuorten, iältään 16–<20-vuotiaiden, yleisoireista lastenreumaa sairastavien potilaiden osajoukossa kanakinumabin turvallisuusprofiili oli yhdenmukainen alle 16-vuotiaiden, yleisoireista lastenreumaa sairastavien potilaiden kanssa. Kanakinumabin turvallisuusprofiili aikuisiän Stillin tautia sairastavilla potilailla satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa tutkimuksessa (GDE01T) 36 potilaalla (iältään 22–70-vuotiaita) oli samanlainen, kuin mitä on havaittu yleisoireista lastenreumaa sairastavilla potilailla.
Valittujen haittavaikutuksien kuvaus
Pitkäaikaistiedot ja laboratorioarvojen poikkeavuudet CAPS-potilailla
Kliinisten kanakinumabitutkimusten aikana CAPS-potilaiden keskimääräiset hemoglobiiniarvot nousivat ja valkosolujen, neutrofiilien ja verihiutaleiden keskimääräiset määrät laskivat.
CAPS-potilailla on harvoin havaittu transaminaasiarvojen kohoamista.
Kanakinumabihoitoa saaneilla CAPS-potilailla on havaittu oireetonta ja lievää seerumin bilirubiiniarvojen kohoamista ilman samanaikaista transaminaasiarvojen kohoamista.
Avoimissa, suurenevilla annoksilla toteutetuissa pitkäaikaistutkimuksissa 600 mg ja 8 mg/kg annosryhmissä ilmoitettiin useammin infektiotapahtumia (maha-suolitulehdus, hengitystieinfektio, ylähengitystieinfektio), oksentelua ja huimausta kuin muissa annosryhmissä.
Laboratorioarvojen poikkeavuudet TRAPS-, HIDS-/MKD- ja FMF-potilailla
Neutrofiilit
Vähintään asteen 2 neutrofiiliarvojen laskua esiintyi 6,5 %:lla potilaista (yleistä) ja asteen 1 laskua 9,5 %:lla potilaista, mutta lasku oli yleensä ohimenevää eikä neutropeniaan liittyviä infektioita ole havaittu haittavaikutuksena.
Trombosyytit
Trombosyyttiarvojen laskua (≥ aste 2) esiintyi 0,6 %:lla potilaista, mutta verenvuotoa ei ole havaittu haittavaikutuksena. Lievää ja ohimenevää asteen 1 trombosyyttiarvojen laskua esiintyi 15,9 %:lla potilaista ilman siihen liittyviä verenvuototapahtumia.
Laboratorioarvojen poikkeavuudet yleisoireista lastenreumaa sairastavilla
Hematologia
Koko yleisoireisen lastenreuman ohjelmassa ohimenevää valkosoluarvojen pienenemistä tasolle ≤ 0,8 x LLN ilmoitettiin 33 potilaalla (16,5 %).
Koko yleisoireisen lastenreuman ohjelmassa absoluuttisten neutrofiiliarvojen ohimenevää pienenemistä alle tason 1 x 109/l ilmoitettiin 12 potilaalla (6,0 %).
Koko yleisoireisen lastenreuman ohjelmassa verihiutalearvojen ohimenevää pienentymistä (< LLN) havaittiin 19 potilaalla (9,5 %).
ALAT/ASAT
Koko yleisoireisen lastenreuman ohjelmassa ALAT- ja/tai ASAT-arvojen suurenemista tasolle > 3 x ULN (normaalin viitealueen yläraja) ilmoitettiin 19 potilaalla (9,5 %).
Laboratorioarvojen poikkeavuudet kihtiartriittia sairastavilla potilailla
Hematologia
Veren valkosolujen määrän laskua ≤ 0,8 x normaaliarvojen alarajan raportoitiin 6,7 %:lla kanakinumabihoitoa saaneista potilaista verrattuna 1,4 %:iin triamsinoloniasetonidihoitoa saaneista. Vertailevissa tutkimuksissa absoluuttisen neutrofiilimäärän (ANC-arvon) vähenemistä alle arvon 1 x 109/l raportoitiin 2 %:lla potilaista. Yksittäistapauksissa todettiin myös ANC-arvoja < 0,5 x 109/l (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kihtiartriittia sairastavilla potilailla suoritetuissa aktiivikontrolloiduissa kliinisissä tutkimuksissa lievät (alle normaaliarvojen alarajan, mutta > 75 x 109/l) ja ohimenevät verihiutaleiden määrien laskut olivat yleisempiä kanakinumabihoitoa saaneilla (12,7 %) kuin vertailuvalmistetta saaneilla (7,7 %) potilailla.
Virtsahappo
Kihtiartriittia koskeneissa vertailututkimuksissa havaittiin virtsahappopitoisuuksien nousuja (0,7 mg/dl viikolla 12 ja 0,5 mg/dl viikolla 24) kanakinumabihoidon jälkeen. Eräässä toisessa tutkimuksessa virtsahappopitoisuuksien nousua ei havaittu ULT-hoitoa aloittavilla potilailla. Virtsahappopitoisuuden nousua ei havaittu kliinisissä tutkimuksissa ei-kihtiperäistä artriittia sairastavilla potilailla (ks. kohta Farmakodynamiikka).
ALAT/ASAT
Kanakinumabihoitoa saaneiden ryhmissä nähtiin keskimäärin 3,0 U/l:n ja mediaani 2,0 U/l:n alaniinitransaminaasiarvojen (ALAT) sekä keskimäärin 2,7 U/l:n ja mediaani 2,0 U/l:n aspartaattitransaminaasiarvojen (ASAT) nousuja lähtötasosta tutkimuksen päättymisajankohtaan saakka verrattuna triamsinoloniasetonidia saaneiden ryhmiin. Kliinisesti merkitsevien muutosten (≥ 3 x normaaliarvojen ylärajan) insidenssi oli kuitenkin suurempi triamsinoloniasetonidia saaneilla potilailla (sekä ALAT että ASAT: 2,5 %) kuin kanakinumabihoitoa saaneilla (ALAT: 1,6 %; ASAT: 0,8 %).
Triglyseridit
Aktiivikontrolloiduissa kihtiartriittitutkimuksissa kanakinumabihoitoa saaneilla potilailla havaittiin keskimäärin +33,5 mg/dl triglyseridipitoisuuden nousu verrattuna lievään, ‑3,1 mg/dl laskuun triamsinoloniasetonidia saaneilla potilailla. Triglyseridiarvojen nousua yli 5 x normaaliarvojen ylärajan ilmeni 2,4 %:lla kanakinumabihoitoa saaneista potilaista ja 0,7 %:lla triamsinoloniasetonidia saaneista potilaista. Tämän havainnon kliinistä merkitystä ei tunneta.
Pitkäaikaistiedot havainnoivasta tutkimuksesta
Yhteensä 243 CAPS-potilasta (85 pediatrista potilasta, iältään ≥ 2 – ≤ 17 vuotta, ja 158 aikuispotilasta, iältään ≥ 18 vuotta) sai kanakinumabihoitoa tavanomaisen hoitokäytönnön mukaisesti pitkäaikaisrekisteritutkimuksessa (kanakinumabialtistuksen keskiarvo 3,8 vuotta). Kanakinumabin pitkäaikaishoidon turvallisuusprofiili vastasi näissä olosuhteissa CAPS-potilailla toteutettujen interventiotutkimusten havaintoja.
Pediatriset potilaat
Interventiotutkimuksissa 80 CAPS-oireyhtymää sairastavaa lapsipotilasta (ikä 2‑17 vuotta) sai kanakinumabia. Yleisesti ottaen kanakinumabin turvallisuus- ja siedettävyysprofiilissa pediatrisilla potilailla ei ollut kliinisesti merkittäviä eroja verrattuna koko CAPS-populaatioon (aikuis- ja lapsipotilaat, N=211). Tämä koskee myös infektiojaksojen kokonaisesiintymistiheyttä ja ‑vaikeusastetta. Ylempien hengitysteiden infektiot olivat yleisimmin raportoituja infektiotapahtumia.
Lisäksi, kuusi alle 2-vuotiasta lapsipotilasta arvioitiin pienessä avoimessa kliinisessä tutkimuksessa. Kanakinumabin turvallisuusprofiili osoittautui samankaltaiseksi kuin 2-vuotiailla ja sitä vanhemmilla potilailla.
16 viikon tutkimuksessa kanakinumabia sai 102 TRAPS-, HIDS-/MKD- ja FMF-potilasta (ikä 2–17 vuotta). Yleisesti ottaen kanakinumabin turvallisuus- ja siedettävyysprofiilissa pediatrisilla potilailla ei ollut kliinisesti merkittäviä eroja verrattuna koko populaatioon.
Iäkkäät potilaat
Mitään merkittäviä eroja ≥ 65-vuotiaiden potilaiden turvallisuusprofiilissa ei ole todettu.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta.
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA.
Yliannostus
Ilmoitetut kokemukset yliannostustapauksista ovat rajallisia. Varhaisissa kliinisissä tutkimuksissa potilaat ja terveet vapaaehtoiset saivat jopa 10 mg/kg annoksia laskimonsisäisesti tai subkutaanisesti, ilman merkkejä akuutista toksisuudesta.
Yliannostustapauksessa suositellaan potilaan tilan seurantaa mahdollisten haittavaikutusten merkkien tai oireiden varalta. Lisäksi asianmukainen, oireenmukainen hoito olisi aloitettava välittömästi.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, interleukiinin estäjät, ATC-koodi: L04AC08
Vaikutusmekanismi
Kanakinumabi on ihmisperäinen IgG1/κ‑isotyypin monoklonaalinen anti-humaani interleukiini-1-beetavasta-aine (IL-1-beeta). Kanakinumabi sitoutuu spesifisesti ja suurella affiniteetilla ihmisperäiseen IL-1-beetaan ja neutraloi ihmisen IL-1-beetan biologisen aktiivisuuden salpaamalla sen vaikutuksen IL-1-reseptoreihin, ja näin ollen estäen IL-1-beetan indusoiman geeniaktivaation ja tulehduksenvälittäjäaineiden muodostumisen.
Farmakodynaamiset vaikutukset
CAPS, TRAPS, HIDS/MKD ja FMF
Kliinisissä tutkimuksissa CAPS-, TRAPS-, HIDS-/MKD- ja FMF-potilailla, joilla oli hallitsematon IL-1-beetan liikatuotanto, todettiin nopea ja pitkäkestoinen vaste kanakinumabihoitoon; eli laboratorioarvot, kuten kohonnut C-reaktiivinen proteiini (CRP) ja seerumin amyloidi A (SAA), kohonneet neutrofiili- ja verihiutalepitoisuudet ja leukosytoosi palautuivat nopeasti normaaleiksi.
Stillin tauti (yleisoireinen lastenreuma ja aikuisiän Stillin tauti)
Aikuisiän Stillin tauti ja yleisoireinen lastenreuma ovat vaikeita tulehduksellisia autoimmuunitauteja, joiden taustalla ovat synnynnäinen immuniteetti ja proinflammatoriset sytokiinit, joista tärkeimpiä on IL-1-beeta.
Yleisoireisen lastenreuman ja aikuisiän Stillin taudin yleisiä oireita ovat mm. kuume, ihottuma, hepatosplenomegalia, lymfadenopatia, polyserosiitti ja artriitti. Kanakinumabihoito lievitti yleisoireisen lastenreuman nivel- ja systeemisiä oireita nopeasti ja pitkäkestoisesti, vähensi tulehtuneiden nivelten määrää merkitsevästi, alensi kuumetta nopeasti ja vähensi akuutin vaiheen reaktantteja valtaosalla potilaista (ks. Kliininen teho ja turvallisuus).
Kihtiartriitti
Kihtiartriittikohtaus aiheutuu nivelissä ja niitä ympäröivissä kudoksissa olevista uraattikiteistä (mononatriumuraattimonohydraatti), jotka käynnistävät makrofagien IL-1-beeta-tuotannon NALP3-inflammasomisen kompleksin kautta. Makrofagien aktivoituminen ja samanaikainen IL-1-beetan ylituotanto johtavat akuuttiin ja kivuliaaseen tulehdusvasteeseen. Muut synnynnäisen immuunijärjestelmän aktivaattorit, kuten Tollin kaltaisten reseptorien endogeeniset agonistit, saattavat myötävaikuttaa IL-1-beeta-geenin transkriptionaaliseen aktivaatioon, mikä käynnistää kihtiartriittikohtauksen. Kanakinumabiannoksen jälkeen tulehdusmerkkiaineet CRP tai SAA sekä akuutin tulehduksen merkit sairaassa nivelessä (kuten kipu, turvotus, punoitus) vähenevät nopeasti.
Kliininen teho ja turvallisuus
CAPS
Kanakinumabin teho ja turvallisuus on osoitettu yhteensä 211 aikuisella ja pediatrisella potilaalla, joiden tauti oli vaikeusasteeltaan erilainen ja joilla oli erilaisia CAPS-fenotyyppejä (kuten FCAS/FCU, MWS ja NOMID/CINCA). Pivotaalitutkimukseen osallistui ainoastaan potilaita, joilla oli varmistettu NLRP3-geenimutaatio.
Vaiheen I/II tutkimuksissa kanakinumabihoidon vaikutus alkoi nopeasti, jolloin oireet hävisivät tai niissä oli nähtävissä kliinisesti merkittävää lievittymistä päivän kuluessa lääkkeen annon jälkeen. Laboratorioarvot, kuten korkea CRP- ja SAA-arvo, suuret neutrofiili- ja trombosyyttimäärät, palautuivat nopeasti normaaleiksi muutaman päivän kuluessa kanakinumabi-injektiosta.
Pivotaalitutkimus koostui 48 viikon pituisesta, kolmiosaisesta monikeskustutkimuksesta, eli 8 viikon pituisesta avoimesta jaksosta (osa I), 24 viikon pituisesta satunnaistetusta, kaksoissokkoutetusta, lumelääkekontrolloidusta lääkityksen lopettamista selvittävästä jaksosta (osa II) sekä 16 viikon pituisesta avoimesta jaksosta (osa III). Tutkimuksen tarkoituksena oli arvioida kanakinumabin (150 mg tai 2 mg/kg 8 viikon välein) tehoa, turvallisuutta ja siedettävyyttä CAPS-potilailla.
- Osa I: 97 %:lla potilaista havaittiin täydellinen kliininen ja biomerkkiaine-vaste kanakinumabille (määriteltiin tilaksi, jolloin sekä lääkärin yleisarvio autoinflammatorisesta ja ihosairaudesta oli ≤ minimaalinen että CRP- tai SAA-arvo < 10 mg/litra) ja vaste ilmaantui 7 vuorokauden kuluessa hoidon aloittamisesta. Lääkärin kliinisessä arviossa autoinflammatorisen sairauden aktiivisuudesta todettiin merkittävää paranemista: yleisarvio autoinflammatorisen sairauden aktiivisuudesta, ihosairauden (nokkosihottumatyyppinen ihottuma) arvio, nivelkipu, lihaskipu, päänsärky/migreeni, konjunktiviitti, uupumus/huonovointisuus, arvio muista tilaan liittyvistä oireista ja potilaan arvio oireista.
- Osa II: Pivotaalitutkimuksen lääkityksen lopettamista selvittävän jakson aikana ensisijaiseksi päätetapahtumaksi määriteltiin niiden potilaiden osuus, joilla sairaus uusiutui/aktivoitui: yhdenkään (0 %) kanakinumabihoitoa saamaan satunnaistetun potilaan sairaus ei aktivoitunut uudestaan, verrattuna 81 %:iin lumelääkehoitoa saamaan satunnaistetuista potilaista.
- Osa III: Osassa II lumelääkehoitoa saaneet potilaat, joiden sairaus aktivoitui, saivat kliinisen ja serologisen vasteen uudelleen ja se säilyi, kun he aloittivat kanakinumabitutkimuksen avoimen jatkotutkimuksen.
Taulukko 2 Yhteenvetotaulukko valmisteen tehosta vaiheen III kliinisen tutkimuksen lääkityksen lopettamista selvittävän lumelääkekontrolloidun pivotaalijakson aikana (osa II)
Vaiheen III kliinisen tutkimuksen lääkityksen lopettamista selvittävä lumelääkekontrolloitu pivotaalijakso (osa II) | |||
Kanakinumabi N=15 n (%) | Lumelääke N=16 n (%) | p-arvo | |
Ensisijainen päätetapahtuma (taudin aktivoituminen) | |||
Niiden potilaiden osuus, joiden tauti aktivoitui osassa II | 0 (0 %) | 13 (81 %) | < 0,001 |
Tulehdusmerkkiaineet* | |||
C-reaktiivinen proteiini, mg/l | 1,10 (0,40) | 19,93 (10,50) | < 0,001 |
Seerumin amyloidi A, mg/l | 2,27 (‑0,20) | 71,09 (14,35) | 0,002 |
* keskimääräinen (mediaani) muutos osan II alusta | |||
Kahdesta avoimesta, kontrolloimattomasta, vaiheen III pitkäaikaistutkimuksesta toinen oli kanakinumabin turvallisuus-, siedettävyys- ja tehotutkimus CAPS-potilailla. Hoidon kokonaiskesto vaihteli 6 kuukaudesta 2 vuoteen. Toinen oli avoin kanakinumabitutkimus, jossa arvioitiin tehoa ja turvallisuutta japanilaisilla CAPS-potilailla 24 viikon ajan ja jossa oli jatkovaihe aina 48 viikkoon asti. Ensisijaisena tavoitteena oli arvioida niiden potilaiden osuutta, joilla ei ollut esiintynyt relapseja viikkoon 24 mennessä. Tähän lukeutuivat myös potilaat, joiden annosta suurennettiin.
Näiden kahden tutkimuksen yhdistetyssä tehoanalyysissä täydellisen vasteen 150 mg tai 2 mg/kg annoksilla saavutti 65,6 % potilaista, jotka eivät olleet aiemmin saaneet kanakinumabihoitoa. 85,2 % potilaista taas saavutti täydellisen vasteen kun kaikki annokset huomioitiin. Täydellisen vasteen saavutti 43,8 % niistä potilaista, jotka saivat 600 mg tai 8 mg/kg (tai jopa suurempia) annoksia. 2–< 4-vuotiaista potilaista harvempi saavutti täydellisen vasteen (57,1 %) kuin vanhemmista lapsi- ja aikuispotilaista. Täydellisen vasteen saavuttaneista potilaista vaste säilyi 89,3 prosentilla ilman relapseja.
Kokemukset annoksen suurentamisen jälkeen (tasolle 600 mg [8 mg/kg] 8 viikon välein) täydellisen vasteen saavuttaneilla yksittäisillä potilailla viittaavat siihen, että suuremmasta annoksesta saattavat hyötyä potilaat, jotka eivät saavuta täydellistä vastetta tai joilla täydellinen vaste ei säily suositusannoksia käytettäessä (150 mg tai 2 mg/kg potilaille, jotka painavat 15–40 kg). Suurennettuja annoksia annettiin useammin 2–< 4-vuotiaille potilaille ja potilaille, joilla oli NOMID-/CINCA-oireita verrattuna potilaisiin, joilla oli FCAS tai MWS.
Kuuden vuoden pituisen, havainnoivan rekisteritutkimuksen tarkoituksena oli saada tietoa kanakinumabihoidon pitkäaikaisturvallisuudesta ja -tehosta pediatrisilla ja aikuisilla CAPS-potilailla tavanomaisessa hoitokäytännössä. Tutkimukseen osallistui 243 CAPS-potilasta (mukaan lukien 85 alle 18-vuotiasta potilasta). Tautiaktiivisuuden arvioitiin olevan olematon tai lievä/keskivaikea yli 90 %:lla potilaista kaikkina tutkimuksen lähtötilanteen jälkeisinä ajankohtina, ja serologisten tulehdusmerkkiaineiden (CRP ja SAA) mediaani oli normaali (< 10 mg/l) kaikkina lähtötilanteen jälkeisinä ajankohtina. Vaikka annoksen muuttaminen oli tarpeen noin 22 %:lla kanakinumabihoitoa saaneista potilaista, vain pieni osa potilaista (1,2 %) lopetti kanakinumabin käytön terapeuttisen tehon puutteen vuoksi.
Pediatriset potilaat
CAPS-potilailla tehdyissä kanakinumabi-interventiotutkimuksissa oli mukana yhteensä 80 lapsipotilasta, jotka olivat iältään 2‑17-vuotiaita (heistä noin puolelle annostus määräytyi painon perusteella). Yleisesti ottaen kanakinumabin teho-, turvallisuus- ja siedettävyysprofiilissa pediatrisilla potilailla ei ollut kliinisesti merkityksellisiä eroja verrattuna koko CAPS-populaatioon. Kliiniset oireet ja objektiiviset tulehdusmerkkiaineet (esimerkiksi SAA ja CRP) vähenivät valtaosalla pediatrisista potilaista.
56 viikkoa kestänyt avoin tutkimus toteutettiin kanakinumabin tehon, turvallisuuden ja siedettävyyden arvioimiseksi 4-vuotiailla ja sitä nuoremmilla CAPS-potilailla. Tutkimuksessa arvioitiin 17 potilasta (mukaan lukien 6 alle 2‑vuotiasta potilasta), jotka käyttivät painoon perustuvia 2-8 mg/kg aloitusannoksia. Tutkimus arvioi myös kanakinumabin vaikutusta lapsuusajan rokotusohjelman rokotteiden vasta-aineiden muodostumiseen. Turvallisuuden tai tehon osalta ei havaittu eroa alle 2-vuotiaiden ja 2-vuotiaiden sekä sitä vanhempien potilaiden välillä. Kaikille potilaille, jotka saivat ei-eläviä lapsuusajan rokotusohjelman rokotteita (N=7), kehittyi suojaava vasta-ainetaso.
TRAPS, HIDS/MKD ja FMF
Kanakinumabihoidon teho ja turvallisuus TRAPS-, HIDS-/MKD- ja FMF-potilailla osoitettiin yhdessä keskeisessä neliosaisessa vaiheen III tutkimuksessa (N2301), johon kuului kolme erillistä tautikohorttia.
- Osa I: Kunkin tautikohortin vähintään 2-vuotiaat potilaat aloittivat 12 viikon seulontavaiheen, jonka aikana potilaat arvioitiin taudin aktivoitumisen suhteen.
- Osa II: Potilaat, joiden tauti oli aktivoitunut, satunnaistettiin 16 viikon kaksoissokkoutettuun, lumekontrolloituun hoitojaksoon, jonka aikana he saivat joko 150 mg kanakinumabia (2 mg/kg potilailla, joiden paino oli ≤ 40 kg) ihon alle tai lumelääkettä 4 viikon välein. Potilaat, jotka olivat iältään > 28 vrk–< 2 v, oli sallittua ottaa suoraan mukaan tutkimukseen osan II avoimeen ryhmään satunnaistamattomina potilaina (kyseiset potilaat suljettiin pois ensisijaisesta tehon analyysistä).
- Osa III: 16 viikkoa hoitoa saaneet ja vasteen saaneiksi luokitellut potilaat satunnaistettiin uudelleen 24 viikon kaksoissokkoutettuun lääkityksen lopettamista selvittävään jaksoon, jonka aikana he saivat kanakinumabia 150 mg (2 mg/kg potilaille, joiden paino oli ≤ 40 kg) ihon alle tai lumelääkettä 8 viikon välein.
- Osa IV: Kaikki kanakinumabihoitoa saaneet osaan III osallistuneet potilaat soveltuivat aloittamaan 72 viikon, avoimen hoidon jatkovaiheen.
Tutkimuksen osaan II otettiin yhteensä 185 potilasta (ikä vähintään 28 vrk), joista yhteensä 181 potilasta (ikä vähintään 2 v) satunnaistettiin.
Satunnaistetun hoitojakson (osa II) ensisijainen tehon päätetapahtuma oli kussakin kohortissa niiden vasteen saaneiden määrä, joiden taudin indeksiaktivoitumisvaihe oli päättynyt päivän 15 kohdalla ja joilla tauti ei aktivoitunut enää uudelleen 16 viikon hoitojakson aikana (määriteltiin täydelliseksi vasteeksi). Taudin indeksiaktivoitumisvaihe katsottiin päättyneeksi, jos PGA-pistemäärä (lääkärin yleisarvio [Physician’s Global Assessment] tautiaktiivisuudesta) oli < 2 (minimaalinen tai ei tautiaktiivisuutta) ja CRP-arvo oli viitealueella (≤ 10 mg/l) tai pienentynyt ≥ 70 % lähtötilanteesta. Uusi aktivoituminen määriteltiin PGA-pistemääräksi ≥ 2 (lievä, keskivaikea tai vaikea tauti) ja CRP-arvoksi ≥ 30 mg/l. Toissijaisia päätetapahtumia, jotka kaikki perustuivat viikon 16 tuloksiin (osan II päättyminen), olivat PGA-pistemäärän < 2 saavuttaneiden potilaiden määrä, serologisen remission (eli CRP ≤ 10 mg/l) saavuttaneiden potilaiden määrä ja niiden potilaiden määrä, joiden SAA-arvo normalisoitui (eli SAA ≤ 10 mg/l).
Kanakinumabi oli lumelääkettä parempi ensisijaisen tehon päätetapahtuman suhteen kaikissa kolmessa tautikohortissa. Kanakinumabi oli lumetta tehokkaampi kaikissa kolmessa kohortissa myös toissijaisten päätetapahtumien suhteen (PGA < 2 ja CRP ≤ 10 mg/l). Viikon 16 kohdalla SAA-arvo oli normalisoitunut (≤ 10 mg/l) suuremmalla potilasmäärällä kanakinumabihoitoryhmässä kuin lumeryhmässä kaikissa kolmessa kohortissa, ja TRAPS-potilailla ero oli tilastollisesti merkitsevä (tutkimustulokset, ks. taulukko 3 jäljempänä).
Taulukko 3 Yhteenvetotaulukko valmisteen tehosta vaiheen III tutkimuksen satunnaistetun, lumekontrolloidun pivotaalihoitojakson aikana (osa II)
Vaiheen III tutkimuksen satunnaistettu, lumekontrolloitu pivotaalihoitojakso (osa II) | |||
Kanakinumabi n/N (%) | Lumelääke n/N (%) | p-arvo | |
Ensisijainen päätetapahtuma (taudin aktivoituminen) – Niiden potilaiden määrä, joiden taudin indeksiaktivoitumisvaihe oli päättynyt päivän 15 kohdalla ja joilla tauti ei aktivoitunut enää uudelleen 16 viikon hoitojakson aikana | |||
FMF | 19/31 (61,29) | 2/32 (6,25) | < 0,0001* |
HIDS/MKD | 13/37 (35,14) | 2/35 (5,71) | 0,0020* |
TRAPS | 10/22 (45,45) | 2/24 (8,33) | 0,0050* |
Toissijaiset päätetapahtumat (taudin ja tulehduksen markkerit) | |||
PGA < 2 | |||
FMF | 20/31 (64,52) | 3/32 (9,38) | < 0,0001** |
HIDS/MKD | 17/37 (45,95) | 2/35 (5,71) | 0,0006** |
TRAPS | 10/22 (45,45) | 1/24 (4,17) | 0,0028** |
CRP ≤ 10 mg/l | |||
FMF | 21/31 (67,74) | 2/32 (6,25) | < 0,0001** |
HIDS/MKD | 15/37 (40,54) | 2/35 (5,71) | 0,0010** |
TRAPS | 8/22 (36,36) | 2/24 (8,33) | 0,0149** |
SAA ≤ 10 mg/l | |||
FMF | 8/31 (25,81) | 0/32 (0,00) | 0,0286 |
HIDS/MKD | 5/37 (13,51) | 1/35 (2,86) | 0,0778 |
TRAPS | 6/22 (27,27) | 0/24 (0,00) | 0,0235** |
n = vasteen saaneiden määrä; N = arviointikelpoisten potilaiden määrä * Osoittaa tilastollisen merkitsevyyden (yksitahoinen) tasolla 0,025 Fisherin eksaktin testin perusteella ** Osoittaa tilastollisen merkitsevyyden (yksitahoinen) tasolla 0,025 logistisen regressiomallin perusteella, kun hoitoryhmä ja PGA-, CRP- tai SAA-lähtöarvot olivat selittävinä muuttujina kussakin kohortissa | |||
Annoksen suurentaminen
Tutkimuksen osassa II kanakinumabihoitoa saaneet potilaat, joilla tautiaktiivisuus oli pitkittynyt, saivat 150 mg lisäannoksen (tai 2 mg/kg potilaille, joiden paino oli ≤ 40 kg) ensimmäisen kuukauden kuluessa. Lisäannos voitiin antaa aikaisintaan 7 päivän kuluttua ensimmäisestä hoitoannoksesta. Kaikilla potilailla, joiden annosta oli suurennettu, hoitoa jatkettiin suuremmalla annoksella eli annoksella 300 mg (tai 4 mg/kg potilaille, joiden paino oli ≤ 40 kg) 4 viikon välein.
Ensisijaisen päätetapahtuman eksploratiivisessa analyysissä havaittiin, että jos potilaan vaste ensimmäiseen annokseen oli riittämätön, annoksen suurentaminen ensimmäisen kuukauden aikana tasolle 300 mg (tai 4 mg/kg) 4 viikon välein paransi tautiaktiivisuuden hallintaa, vähensi tautiaktiivisuutta ja normalisoi CRP- ja SAA-arvot.
Pediatriset potilaat:
Tutkimukseen otettiin kaksi satunnaistamatonta HIDS-/MKD-potilasta, joiden ikä oli > 28 vrk–< 2 v, ja heille annettiin kanakinumabia. Toisella potilaalla indeksiaktivoitumisvaihe päättyi päivään 15 mennessä yhden 2 mg/kg kanakinumabikerta-annoksen jälkeen, mutta potilas lopetti hoidon ensimmäisen annoksen jälkeen vakavien haittatapahtumien takia (pansytopenia ja maksan vajaatoiminta). Potilaalla oli tutkimukseenottovaiheessa anamneesissa immunologinen trombosytopeeninen purppura ja aktiivinen maksatoiminnan poikkeavuus. Toiselle potilaalle annettiin aloitusannoksena 2 mg/kg kanakinumabia ja lisäannoksena 2 mg/kg viikon 3 kohdalla. Annostusta suurennettiin viikon 5 kohdalla tasolle 4 mg/kg 4 viikon välein, ja potilas käytti kyseistä annostusta tutkimuksen osan II loppuun asti. Taudin aktivoitumisvaiheen päättyminen saavutettiin viikkoon 5 mennessä, eikä potilaan tauti aktivoitunut uudestaan tutkimuksen osan II loppuun mennessä (viikko 16).
Stillin tauti (yleisoireinen lastenreuma ja aikuisiän Stillin tauti)
Yleisoireinen lastenreuma
Kanakinumabin tehoa aktiivisen yleisoireisen lastenreuman hoidossa arvioitiin kahdessa faasin III pivotaalitutkimuksessa (G2305 ja G2301). Tutkimukseen otetut potilaat olivat iältään 2–< 20 vuotta (ikäkeskiarvo 8,5 vuotta ja taudin keston keskiarvo 3,5 vuotta lähtötilanteessa), ja heillä oli aktiivinen tauti (määritelmä: ≥ 2 tulehtunutta niveltä, kuume ja kohonnut CRP-arvo).
G2305-tutkimus
G2305-tutkimus oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu, 4 viikkoa kestävä tutkimus, jossa arvioitiin kanakinumabin lyhytaikaista tehoa 84 potilaalla. Potilaat satunnaistettiin kanakinumabiryhmään (4 mg/kg kerta-annos, enintään 300 mg) tai lumeryhmään. Ensisijainen tavoite oli niiden potilaiden osuus, joiden pediatriset American College of Rheumatology (ACR) ‑vastepisteet olivat kohentuneet vähintään 30 % päivänä 15. ACR-vastekriteerejä oli muokattu lisäämällä niihin kuumeen puuttuminen. Kanakinumabihoito paransi kaikkia pediatrisia ACR-vastepisteitä verrattuna lumeeseen päivinä 15 ja 29 (taulukko 4).
Taulukko 4Pediatrinen ACR-vaste ja tautitilanne päivinä 15 ja 29
Päivä 15 | Päivä 29 | |||
Kanakinumabi N = 43 | Lume N = 41 | Kanakinumabi N = 43 | Lume N = 41 | |
ACR30 | 84 % | 10 % | 81 % | 10 % |
ACR50 | 67 % | 5 % | 79 % | 5 % |
ACR70 | 61 % | 2 % | 67 % | 2 % |
ACR90 | 42 % | 0 % | 47 % | 2 % |
ACR100 | 33 % | 0 % | 33 % | 2 % |
Inaktiivinen tauti | 33 % | 0 % | 30 % | 0 % |
Hoitojen välinen ero oli kaikkien ACR-pistemäärien kohdalla merkitsevä (p ≤ 0,0001) | ||||
Muokattuihin pediatrisiin ACR-pisteytyksiin kuului systeemisiä ja artriittia koskevia komponentteja. Pisteytysten komponenttien tulokset olivat yhdenmukaiset ACR-kokonaisvastetulosten kanssa. Päivänä 15 aktiivisen artriitin affisioimien nivelten määrän mediaanimuutos lähtötilanteesta oli kanakinumabiryhmässä ‑67 % ja liikelaajuudeltaan rajoittuneiden nivelten määrän mediaanimuutos lähtötilanteesta ‑73 % (N = 43). Vastaavat arvot olivat lumeryhmässä 0 % ja 0 % (N = 41). Potilaiden kipupisteiden (0‑100 mm VAS-asteikko) keskimuutos päivänä 15 oli kanakinumabiryhmässä ‑50,0 mm (N = 43) ja lumeryhmässä +4,5 mm (N = 25). Kipupisteiden keskimuutos kanakinumabiryhmässä oli samaa luokkaa päivänä 29.
G2301-tutkimus
G2301-tutkimus oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu hoidon lopettamistutkimus, jossa arvioitiin kanakinumabia taudin aktivoitumisen ehkäisyssä. Tutkimus koostui kahdesta osasta, joissa oli kaksi erillistä ensisijaista päätetapahtumaa (steroidilääkityksen onnistunut vähentäminen ja aktivoitumiseen kulunut aika). Osaan I (avoin) otettiin mukaan 177 potilasta, jotka saivat 4 mg/kg (enintään 300 mg) kanakinumabia 4 viikon välein enintään 32 viikon ajan. Osan II (kaksoissokkoutettu) potilaat saivat joko 4 mg/kg kanakinumabia tai lumetta 4 viikon välein, kunnes 37 aktivoitumistapahtumaa oli todettu.
Kortikosteroidiannoksen pienentäminen:
Kortikosteroidiannosta pyrki pienentämään 92 potilasta yhteensä 128 potilaasta, jotka osallistuivat osaan I ja käyttivät kortikosteroideja. 57 potilasta (62 %) onnistui pienentämään kortikosteroidiannosta ja 42 potilasta (46 %) lopetti kortikosteroidihoidon 92 potilaasta, jotka pyrkivät pienentämään annosta.
Aktivoitumiseen kulunut aika:
Osassa II kanakinumabia käyttäneillä potilailla aktivoitumistapahtuman riski pieneni 64 % verrattuna lumeeseen (riskisuhde 0,36; 95 % lv 0,17–0,75; p = 0,0032). Tauti ei aktivoitunut havainnointivaiheen aikana (enintään 80 viikkoa) 63 potilaalla 100 potilaasta, jotka osallistuivat osaan II. Tämä koski sekä lume- että kanakinumabihoitoa.
Terveyttä ja elämänlaatua koskevat tulokset G2305- ja G2301-tutkimuksissa
Kanakinumabihoito paransi potilaiden fyysistä toimintakykyä ja elämänlaatua kliinisesti merkittävästi. G2305-tutkimuksessa Childhood Health Assessment Questionnaire ‑kyselylomakkeen pienimmän neliösumman menetelmällä laskettu keskiarvo parani kanakinumabiryhmässä 0,69 verrattuna lumeryhmään (3,6 kertaa pienin kliinisesti merkitsevä muutos eli 0,19) (p = 0,0002). Arvon kohentumisen mediaani lähtötilanteesta G2301-tutkimuksen osan I päättymiseen oli 0,88 (79 %). G2305-tutkimuksessa Child Health Questionnaire-PF50 ‑pisteiden ilmoitettiin kohentuneen kanakinumabiryhmässä tilastollisesti merkitsevästi verrattuna lumeeseen (fyysinen p = 0,0012; psykososiaalinen hyvinvointi p = 0,0017).
Yhdistetty tehoanalyysi
G2305- ja G2301-tutkimusten ja jatkotutkimuksen ensimmäisten 12 kanakinumabihoitoviikon tiedot yhdistettiin tehon säilymisen arviointia varten. Tietojen perusteella muokattujen pediatristen ACR-vasteiden ja komponenttien kohentuminen lähtötilanteen ja viikon 12 välisenä aikana oli samaa luokkaa kuin lumekontrolloidussa tutkimuksessa (G2305). Viikolla 12 muokattu pediatrinen ACR30-vaste oli 70 %, ACR50-vaste 69 %, ACR70-vaste 61 %, ACR90-vaste 49 % ja ACR100-vaste 30 %. Tauti oli inaktiivinen 28 %:lla potilaista (N = 178).
Kliinisten tutkimusten rajallinen näyttö viittaa siihen, että jos tosilitsumabilla tai anakinralla ei saavuteta vastetta, vaste saatetaan saavuttaa kanakinumabilla.
G2301E1-tutkimus
G2305- ja G2301-tutkimuksissa havaittu teho säilyi avoimessa, pitkäaikaisessa jatkotutkimuksessa G2301E1. Tutkimukseen osallistuneista 270 yleisoireista lastenreumaa sairastavista potilaista 147 oli saanut kanakinumabi-hoitoa G2305- ja G2301-tutkimuksissa (kohortti I), ja 123 potilasta ei ollut aikaisemmin saanut kanakinumabia (kohortti II). Kohortti I:ssä potilaiden hoidon mediaanikesto oli 3,2 vuotta (korkeintaan 5,2 vuotta), ja kohortti II:ssa potilaiden hoidon mediaanikesto oli 1,8 vuotta (korkeintaan 2,8 vuotta). Jatkotutkimuksessa kaikki potilaat saivat kanakinumabia 4 mg/kg (korkeintaan 300 mg) joka neljäs viikko. Molemmissa kohorteissa potilailla, jotka saivat hyvän hoitovasteen (jälkikäteen määritetty muokattu pediatrinen ACR-vaste ≥ 90) ja jotka eivät tarvinneet samanaikaista kortikosteroidihoitoa, kanakinumabi-annos voitiin laskea tasolle 2 mg/kg joka neljäs viikko (62/270; 23 %).
G2306-tutkimus
G2306-tutkimus oli avoin, hoitovasteen säilymistä arvioinut tutkimus, jossa kanakinumabia annettiin pienennetyllä annoksella (2 mg/kg 4 viikon välein) tai pidennetyllä annosvälillä (4 mg/kg 8 viikon välein) yleisoireista lastenreumaa sairastaville potilaille, jotka olivat saaneet kanakinumabia 4 mg/kg 4 viikon välein. 75 iältään 2–22-vuotiasta potilasta, joiden tauti oli ollut inaktiivinen vähintään 6 peräkkäistä kuukautta (kliininen remissio) potilaiden saadessa kanakinumabia ainoana hoitona (joillakin potilaista tauti oli pysynyt inaktiivisena vähintään 4 viikon ajan samanaikaisen kortikosteroidihoidon ja/tai metotreksaattihoidon lopettamisen jälkeen), satunnaistettiin saamaan kanakinumabia 2 mg/kg 4 viikon välein (N = 38) tai 4 mg/kg 8 viikon välein (N = 37). 24 viikon kuluttua satunnaistamisesta tauti oli pysynyt inaktiivisena 6 kuukauden ajan 71 prosentilla (27/38) potilaista, jotka saivat valmistetta pienennetyllä annoksella (2 mg/kg 4 viikon välein), ja 84 prosentilla (31/37) potilaista, jotka saivat valmistetta pidennetyllä annosvälillä (4 mg/kg 8 viikon välein). Tauti pysyi inaktiivisena 6 kuukauden ajan myös 93 prosentilla (26/28) kliinisen remission saavuttaneista potilaista, joiden annosta pienennettiin entisestään (annokseen 1 mg/kg 4 viikon välein), ja 91 prosentilla (30/33) potilaista, joiden annosväliä pidennettiin entisestään (annokseen 4 mg/kg 12 viikon välein). Potilaat, joiden tauti pysyi inaktiivisena toiset 6 kuukautta tällä pienimmällä annoksella, saivat lopettaa kanakinumabihoidon. Kaiken kaikkiaan 33 % (25/75) potilaista, jotka satunnaistettiin saamaan valmistetta pienennetyllä annoksella tai pidemmällä annosvälillä, pystyi lopettamaan kanakinumabihoidon ja säilyttämään inaktiivisen tautistatuksen 6 kuukauden ajan. Haittatapahtumien määrä kummassakin hoitoryhmässä oli samankaltainen kuin kanakinumabia 4 mg/kg 4 viikon välein saaneilla potilailla.
Aikuisiän Stillin tauti
Kanakinumabin teho annoksella 4 mg/kg (korkeintaan 300 mg) annosteltuna joka neljäs viikko aikuisiän Stillin tautia sairastavilla potilailla satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa, jossa oli 36 potilasta (22–70-vuotiaita), oli vastaava teho kuin yleisoireista lastenreumaa sairastavilla potilailla. GDE01T-tutkimuksessa suuremmalla osuudella potilaista kanakinumabi-ryhmässä (12/18, 66,7 %) kuin potilailla lumelääkeryhmässä (7/17, 41,2 %) tulokset paranivat viikolla 12 lähtötasosta >1,2 pistettä mitattuna arviointiasteikolla DAS28-ERS (Disease Activity Score 28 Erythrocyte Sedimentation Rate). Tämä tulos ei kuitenkaan ollut tilastollisesti merkitsevä (vetosuhde 2,86; hoitojen erot [%] 25,49 [95 % lv: 9,43; 55,80]). Neljänteen tutkimusviikkoon mennessä seitsemän 18:sta kanakinumabilla hoidetusta potilaasta (38,9 %) oli jo saavuttanut DAS28-ESR-remission verrattuna kahteen potilaaseen 17:sta (11,8 %) lumelääkeryhmässä. Nämä tiedot ovat yhteneväisiä 418 yleisoireista lastenreumaa sairastavasta potilaasta tehdyn yhdistetyn tehoanalyysin kanssa, joka osoitti, että kanakinumabin teho 16–<20-vuotiaiden yleisoireista lastenreumaa sairastavien potilaiden alaryhmässä (n = 34) oli vastaava kuin kuin alle 16-vuotiailla potilailla (n = 384) havaittu teho.
Kihtiartriitti
Kanakinumabin teho akuuttien kihtiartriittikohtausten hoidossa on osoitettu kahdessa satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa monikeskustutkimuksessa, joihin osallistui usein toistuvista kihtiartriittikohtauksista kärsiviä potilaita (≥ 3 kohtausta tutkimusta edeltävien 12 kuukauden aikana), jotka eivät voineet käyttää NSAID-lääkkeitä tai kolkisiinia (johtuen joko vasta-aiheesta, yliherkkyydestä tai tehon puutteesta). Tutkimukset kestivät 12 viikkoa, ja niitä seurasi 12 viikon kaksoissokkoutettu jatkotutkimus. Yhteensä 225 potilasta sai 150 mg kanakinumabia ihonalaisesti ja 229 potilasta 40 mg triamsinoloniasetonidia lihakseen tutkimuksen alussa ja sen jälkeen uusien kohtausten yhteydessä. Kihtiartriittikohtausten keskimääräinen lukumäärä tutkimusta edeltävien 12 kuukauden aikana oli 6,5. Yli 85 %:lla potilaista oli jokin muu samanaikainen sairaus, kuten hypertensio (60 %), diabetes (15 %), iskeeminen sydänsairaus (12 %) ja ≥ 3. asteen krooninen munuaissairaus (25 %). Noin kolmanneksella [76 (33,8 %)] kanakinumabiryhmään osallistuneista potilaista ja 84:llä (36,7 %) triamsinoloniryhmään kuuluneista potilaista oli dokumentoidusti todettu este (yliherkkyys, vasta-aihe tai tehon puute) sekä NSAID-lääkkeiden että kolkisiinin käytölle. Tutkimukseenottovaiheessa 42 % potilaista ilmoitti samanaikaisesta uraattipitoisuutta alentavasta (ULT) hoidosta.
Ensisijaiset päätetapahtumat olivat (i) kihtiartriittikivun vaikeusaste (VAS-asteikolla, eli Visual Analogue Scale -mittarilla) 72 tunnin kuluttua annoksesta ja (ii) aika ensimmäiseen uuteen kihtiartriittikohtaukseen.
Koko tutkimuspopulaatiossa kivun vaikeusaste 72 tunnin kuluttua lääkkeen annosta oli tilastollisesti merkitsevästi alhaisempi 150 mg kanakinumabia saaneilla potilailla kuin triamsinoloniasetonidia saaneilla potilailla. Lisäksi kanakinumabi vähensi uusien kohtausten riskiä (ks. taulukko 5).
Tehoa koskevat tulokset sellaisten potilaiden osalta, jotka eivät voineet käyttää NSAID-lääkkeitä eivätkä kolkisiinia ja jotka saivat ULT-hoitoa, joiden ULT-hoito ei ollut tuottanut toivottua tulosta tai joille ULT-hoito oli vasta-aiheinen (N=101), olivat yhdenmukaiset koko tutkimuspopulaation tulosten kanssa siten, että tuloksissa todettiin tilastollisesti merkitsevä ero suhteessa triamsinoloniasetonidiin kivun vaikeusasteessa 72 tunnin kohdalla (‑10,2 mm; p = 0,0208) ja uusien kohtausten riskissä [riskisuhde (HR) 0,39; p = 0,0047 viikolla 24].
Taulukossa 5 esitetään tehoa koskevat tulokset sellaisen rajallisemman alaryhmän osalta, joiden potilaat saivat samanaikaista ULT-hoitoa (n = 62). Kanakinumabihoito sekä lievensi kipua että vähensi uusien kohtausten riskiä potilailla, jotka saivat ULT-hoitoa ja jotka eivät voineet käyttää NSAID-lääkkeitä eivätkä kolkisiinia. Havaittu hoidosta saatava ero verrattuna triamsinoloniasetonidiin oli kuitenkin vähemmän selkeä kuin tutkimuksen kokonaispopulaation osalta.
Taulukko 5 Koko tutkimuspopulaation ja ULT-hoitoa samanaikaisesti saavan alapopulaation (joka ei voinut käyttää NSAID-lääkitystä eikä kolkisiinia) tehoa koskevat tulokset
Tehoa koskeva päätetapahtuma | Koko tutkimuspopulaatio; n = 454 | Potilaat, jotka eivät voineet käyttää NSAID-lääkitystä eivätkä kolkisiinia, ja jotka saivat ULT-hoitoa n = 62 | |
Kihtiartriittikohtausten hoito mitattuna kivun intensiteettinä (VAS) 72 tunnin kohdalla | |||
Pienimmän neliösumman keskiarvon arvioitu ero triamsinoloniasetonidiin Luottamusväli Yksisuuntainen p-arvo | ‑10,7 (‑15,4 ‑ ‑6,0) p < 0,0001* | ‑3,8 (‑16,7 ‑ 9,1) p = 0,2798 | |
Uusien kihtiartriittikohtausten riskin väheneminen mitattuna aikana seuraavaan uuteen kohtaukseen (24 viikkoa) | |||
Riskisuhde (HR) triamsinoloniasetonidiin nähden Luottamusväli Yksisuuntainen p-arvo | 0,44 (0,32 ‑ 0,60) p < 0,0001* | 0,71 (0,29 ‑ 1,77) p = 0,2337 | |
*Ilmaisee merkittävän p-arvon ≤ 0,025 | |||
Turvallisuutta koskevat tulokset osoittivat lisääntynyttä haittavaikutusten ilmaantuvuutta kanakinumabilla verrattuna triamsinoloniasetonidiin (66 % vs 53 %), ja 20 %:lla kanakinumabia saaneista potilaista ilmoitettiin 24 viikon kuluessa haittavaikutuksena infektio, kun vastaava luku triamsinoloniasetonidia saaneiden ryhmässä oli 10 %.
Iäkkäät potilaat
Kanakinumabin teho, turvallisuus ja siedettävyys ≥ 65-vuotiailla potilailla olivat kokonaisuudessaan verrattavissa vastaaviin ominaisuuksiin < 65-vuotiailla.
Uraattipitoisuutta alentavaa hoitoa (ULT) saavat potilaat
Kliinisissä tutkimuksissa kanakinumabin ja ULT-hoidon samanaikainen käyttö oli turvallista. Tutkimuksen kokonaispopulaatiota tarkasteltaessa ULT-hoitoa saaneilla potilailla todettiin vähemmän huomattava hoitoero sekä kivun lievityksen että uusien kohtausten riskin vähenemisen osalta verrattuna potilaisiin, jotka eivät saaneet samanaikaista ULT-hoitoa.
Immunogeenisuus
Vasta-aineita kanakinumabille havaittiin noin 1,5 prosentilla kanakinumabi-hoitoa saaneista CAPS-potilaista, noin 3 prosentilla yleisoireista lastenreumaa sairastavista potilaista ja noin 2 prosentilla kihtiartriittipotilaista. Neutraloivia vasta-aineita ei havaittu. Vasta-aineiden kehittymisen ei todettu korreloivan kliiniseen vasteeseen eikä haittatapahtumiin.
Kanakinumabille ei havaittu kehittyvän vasta-aineita TRAPS-, HIDS-/MKD- eikä FMF-potilailla, jotka saivat 150 mg ja 300 mg annoksia 16 hoitoviikon ajan. Aikuisiän Stillin tautia sairastavilla potilailla ei myöskään havaittu vasta-aineita kanakinumabille.
Immuunivasteen havaitseminen on hyvin paljon riippuvaista käytetyn määrityksen herkkyydestä ja spesifisyydestä sekä testausolosuhteista. Siksi voi olla harhaanjohtavaa vertailla kanakinumabille ja muille valmisteille kehittyneiden vasta-aineiden esiintyvyyksiä.
Pediatriset potilaat
Myyntiluvan haltija on tehnyt neljä kanakinumabi-hoitoa koskevaa pediatrista tutkimussuunnitelmaa (kryopyriiniin liittyvät oireyhtymät, yleisoireinen lastenreuma, perinnöllinen Välimeren kuume – hyperimmunoglobulinemia D -oireyhtymä / mevalonaattikinaasin vajaus ja tuumorinekroositekijään liittyvä jaksoittainen oireyhtymä). Tähän valmisteyhteenvetoon on päivitetty pediatrisia potilaita koskevat kanakinumabi-tutkimusten tulokset.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset kanakinumabin käytöstä kihtiartriitin hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
CAPS
Imeytyminen
Kanakinumabin huippupitoisuus (Cmax) seerumissa havaittiin noin 7 vuorokauden kuluttua aikuisille CAPS-potilaille ihon alle annetun 150 mg:n kertainjektion jälkeen. Keskimääräinen terminaalinen puoliintumisaika oli 26 vuorokautta. Tyypillisen aikuisen CAPS-potilaan (70 kg) Cmax-keskiarvo 150 mg:n ihonalaisen kerta-annoksen jälkeen oli 15,9 mikrog/ml ja äärettömyyteen ekstrapoloidun käyrän alapuolisen alueen pinta-alan (AUC∞) keskiarvo 708 mikrog*vrk/ml. Ihon alle annetun kanakinumabin absoluuttisen hyötyosuuden arvioitiin olevan 66 %. Altistusta osoittavat muuttujat (kuten AUC ja Cmax) suurenivat suhteessa annokseen koko annosvälillä 0,30‑10,0 mg/kg, kun valmiste annettiin infuusiona laskimoon, tai annoksilla 150‑600 mg, kun valmiste annettiin injektiona ihon alle. Ennustetut vakaan tilan altistusarvot (Cmin,ss, Cmax,ss, AUC,ss,8w) 150 mg ihonalaisen annostelun jälkeen (tai vastaavasti 2 mg/kg) 8 viikon välein olivat lievästi suurempia 40‑70 kg painavilla (6,6 µg/ml, 24,3 µg/ml, 767 µg*d/ml) kuin alle 40 kg (4,0 µg/ml, 19,9 µg/ml, 566 µg*d/ml) ja yli 70 kg painavilla (4,6 µg/ml, 17,8 µg/ml, 545 µg*d/ml). Odotettu akkumulaatiosuhde oli 1,3 annettaessa 150 mg kanakinumabia ihon alle kuuden kuukauden ajan 8 viikon välein.
Jakautuminen
Kanakinumabi sitoutuu seerumin interleukiini-1-beetaan (IL-1-beeta). Kanakinumabin jakautumistilavuus (Vss) vaihteli painon mukaan. Vss oli arviolta 6,2 litraa 70 kg painavilla CAPS-potilailla.
Eliminaatio
Kanakinumabin näennäinen puhdistuma (CL/F) suurenee painon mukaan. CL/F oli arviolta 0,17 l/vrk 70 kg painavilla potilailla, joilla oli kryopyriiniin liittyvä oireyhtymä, ja 0,11 l/vrk 33 kg painavilla potilailla, joilla oli yleisoireinen lastenreuma. Kun painoerot otettiin huomioon, kanakinumabin farmakokinetiikassa ei havaittu kliinisesti merkitseviä eroja kryopyriiniin liittyvää oireyhtymää ja yleisoireista lastenreumaa sairastavien potilaiden välillä.
Toistuvan annostuksen yhteydessä ei havaittu viitteitä puhdistuman nopeutumisesta tai kanakinumabin farmakokineettisten ominaisuuksien aikariippuvaisista muutoksista. Farmakokinetiikassa ei havaittu sukupuoleen tai ikään liittyviä eroja, kun tulokset oli korjattu painon mukaan.
TRAPS, HIDS/MKD ja FMF
Biologista hyötyosuutta ei ole määritetty erikseen TRAPS-, HIDS-/MKD- ja FMF-potilailla. Näennäinen puhdistuma (CL/F) TRAPS-, HIDS-/MKD- ja FMF-populaatiossa painon ollessa 55 kg (0,14 l/vrk) oli verrattavissa CAPS-populaatioon painon ollessa 70 kg (0,17 l/vrk). Näennäinen jakautumistilavuus (V/F) oli 4,96 l painon ollessa 55 kg.
Kun 150 mg annoksia annettiin toistuvasti ihon alle 4 viikon välein, kanakinumabin minimipitoisuudet viikon 16 kohdalla (Cmin) olivat arviolta 15,4 ± 6,6 μg/ml. Arvioitu vakaan tilan AUCtau-arvo oli 636,7 ± 260,2 µg*d/ml.
Stillin tauti (yleisoireinen lastenreuma ja aikuisiän Stillin tauti)
Yleisoireista lastenreumaa sairastavien biologisesta hyötyosuudesta ei ole tehty erillistä määritystä. Näennäinen puhdistuma painokiloa kohti (CL/F/kg) oli samaa luokkaa yleisoireisen lastenreuman ja kryopyriiniin liittyvän oireyhtymän populaatioissa (0,004 l/vrk/kg). Näennäinen jakautumistilavuus painokiloa kohti (V/F/kg) oli 0,14 l/kg. Niukat farmakokineettiset tiedot aikuisiän Stillin tautia sairastavilla potilailla viittaavat vastaavaan kanakinumabin farmakokinetiikkaan kuin yleisoireista lastenreumaa sairastavilla potilailla ja muilla potilasryhmillä.
Kun yleisoireista lastenreumaa sairastaville potilaille annettiin toistuvat 4 mg/kg:n annokset 4 viikon välein, kanakinumabin akkumulaatiosuhde oli 1,6. Vakaa tila saavutettiin 110 vuorokaudessa. Ennustettu keskimääräinen (±SD) Cmin,ss oli 14,7 ± 8,8 µg/ml, Cmax,ss 36,5 ± 14,9 µg/ml ja AUCss,4w 696,1 ± 326,5 µg*d/ml.
AUCss oli 2–3-vuotiaiden ikäryhmässä 692 µg*d/ml, 4–5-vuotiailla 615 µg*d/ml, 6–11-vuotiailla 707 µg*d/ml ja 12–19-vuotiailla 742 µg*d/ml. Painon perusteella stratifioituna todettiin alhaisemmat (30–40 %) keskimääräiset altistusarvot Cmin,ss:n (11,4 vs 19 µg/ml) ja AUCss:n (594 vs 880 µg*d/ml) osalta pienempipainoisten (≤ 40 kg) potilaiden ryhmässä kuin painavampien (> 40 kg) lasten ryhmään.
Populaatiofarmakokineettisen mallinnusanalyysin perusteella kanakinumabin farmakokinetiikka nuorilla, 16–20-vuotiailla, yleisoireista lastenreumaa sairastavilla aikuisilla on samanlainen kuin alle 16-vuotiailla potilailla. Kanakinumabin ennustettu vakaan tilan pitoisuus annostasolla 4 mg/kg (enimmäisannos 300 mg) on yli 20-vuotiailla vastaava kuin alle 20-vuotiailla yleisoireista lastenreumaa sairastavilla potilailla.
Kihtiartriittipotilaat
Erillistä analyysiä kihtiartriittipotilaiden biologisen hyötyosuuden määrittämiseksi ei ole tehty. Näennäiset puhdistumat painokiloa kohden (CL/F/kg) olivat toisiinsa verrannollisia kihtiartriitti- ja CAPS-potilailla (0,004 l/vrk/kg). Tyypillisen kihtiartriittipotilaan (93 kg) keskimääräinen altistuminen ihonalaisen 150 mg:n kerta-annoksen jälkeen (Cmax: 10,8 mikrog/ml ja AUC∞: 495 mikrog*vrk/ml) jäi pienemmäksi kuin tyypillisen 70 kg painavan CAPS-potilaan vastaava altistuminen (15,9 mikrog/ml ja 708 mikrog*vrk/ml). Tämä tulos on linjassa sen havaitun tosiasian kanssa, että CL/F-arvo suurenee potilaan painon kasvaessa.
Odotettu akkumulaatiosuhde oli 1,1 annettaessa 150 mg kanakinumabia ihon alle 12 viikon välein.
Pediatriset potilaat
4-vuotiaille ja vanhemmille lapsipotilaille kanakinumabin huippupitoisuudet ilmaantuivat 2‑7 vuorokauden (Tmax) kuluttua ihon alle annetun kerta-injektion 150 mg tai 2 mg/ml jälkeen. Terminaalinen puoliintumisaika oli 22,9–25,7 vuorokautta, mikä on samankaltainen kuin aikuisilla havaitut farmakokineettiset ominaisuudet. Populaatiofarmakokineettisen mallinnusanalyysin perusteella kanakinumabin farmakokinetiikka 2–< 4-vuotiailla lapsilla oli samaa luokkaa kuin 4-vuotiailla ja sitä vanhemmilla potilailla. Ihonalaisen imeytymisnopeuden arvioitiin laskevan iän myötä, ja se vaikutti olevan nopeinta kaikkein nuorimmilla potilailla. Tämän mukaisesti Tmax oli lyhyempi (3,6 vuorokautta) nuoremmilla yleisoireista lastenreumaa sairastavilla potilailla (2‑3-vuotiaat) verrattuna vanhempiin yleisoireista lastenreumaa sairastaviin potilaisiin (12‑19-vuotiaat; Tmax 6 vuorokautta). Iällä ei ollut vaikutusta lääkkeen biologiseen hyötyosuuteen (AUCss).
Täydentävä farmakokineettinen analyysi osoitti, että kanakinumabin farmakokinetiikka kuudella alle 2-vuotiaalla pediatrisella CAPS-potilaalla oli vastaava kuin 2‑4 -vuotiailla pediatrisilla potilailla. Populaatiofarmakokineettisestä mallista tehdyn analyysin perusteella odotettu altistuminen on 2 mg/kg annoksen antamisen jälkeen vertailukelpoinen kaikilla pediatrisilla CAPS-ikäryhmillä. Altistuminen oli kuitenkin 40% vähäisempää pediatrisilla potilailla, joilla oli hyvin alhainen kehonpaino (esim. 10 kg) kuin aikuisilla potilailla (150 mg annos). Havainto on yhdenmukainen aikaisempien havaintojen kanssa, joissa altistuminen on ollut suurempaa painavammilla CAPS-potilasryhmillä.
TRAPS-, HIDS-/MKD- ja FMF-oireyhtymissä altistusparametrit (minimipitoisuudet) olivat samaa luokkaa 2–< 20-vuotiaiden ikäryhmissä ihon alle annetun kanakinumabin jälkeen (2 mg/kg 4 viikon välein).
Farmakokinetiikka on samaa luokkaa yleisoireisen lastenreuman, kryopyriiniin liittyvän oireyhtymän, tuumorinekroositekijään liittyvän jaksoittaisen oireyhtymän, hyperimmunoglobulinemia D ‑oireyhtymän / mevalonaattikinaasin vajauksen ja perinnöllisen Välimeren kuumeen pediatrisissa populaatioissa.
Iäkkäät potilaat
Eroja puhdistumaan tai jakaantumistilavuuteen liittyvien farmakokineettisten parametrien osalta ei havaittu iäkkäiden potilaiden ja < 65-vuotiaiden aikuisten potilaiden välillä.
Prekliiniset tiedot turvallisuudesta
Ristireaktiivisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, immunotoksisuutta, lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Kanakinumabilla ei ole tehty systemaattisia karsinogeenisuustutkimuksia.
Farmaseuttiset tiedot
Apuaineet
Mannitoli (E421)
Histidiini
Histidiinihydrokloridimonohydraatti
Polysorbaatti 80 (E433)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Injektiopullo
3 vuotta.
Mikrobiologiselta kannalta valmiste on käytettävä heti avaamisen jälkeen.
Esitäytetty kynä
3 vuotta.
Käytä 14 päivän kuluessa jääkaapista poistamisen jälkeen (ennen kotelossa ilmoitettua viimeistä käyttöpäivämäärää [EXP]). Säilytä alle 30 °C.
Säilytys
Injektiopullo
Säilytä jääkaapissa (2 °C ‑ 8 ºC).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle
Esitäytetty kynä
Säilytä jääkaapissa (2 °C ‑ 8 ºC).
Käytä 14 päivän kuluessa jääkaapista poistamisen jälkeen (ennen kotelossa ilmoitettua viimeistä käyttöpäivämäärää [EXP]). Säilytä alle 30 °C.
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ILARIS injektioneste, liuos
150 mg/ml (L:ei) 1 kpl (1 ml) (12842,88 €)
PF-selosteen tieto
Injektiopullo
Injektioneste (liuos) injektiopullossa (tyypin I lasia), jossa tulppa (laminoitua klooributyylikumia) ja irti napsautettava korkki (alumiinia).
Pakkaus sisältää 1 injektiopullon.
Esitäytetty kynä
Injektioneste, liuos, esitäytetty kynä toimitetaan kolmionmuotoisen injektiokynän sisällä olevassa kertakäyttöisessä esitäytetyssä ruiskussa. Injektiokynässä on läpinäkyvä tarkistusikkuna ja etiketti. Injektiokynän sisällä on esitäytetty 1 ml:n lasinen ruisku, jossa on kiinteä neula (27 G x 0,5 ”) ja jäykkä styreenibutadieenikumia sisältävä neulansuojus. Männän pysäytin on silikonipäällysteistä kumia ja se on laminoitu suojakalvolla.
Pakkaus sisältää yhden esitäytetyn kynän.
Valmisteen kuvaus:
Liuos on kirkasta tai opaalinhohtoista ja väritöntä tai hieman ruskehtavan keltaista ja sen pH on noin 6,5 ja osmolaliteetti 350–450 mOsm/kg.
Käyttö- ja käsittelyohjeet
Injektiopullo
Ilaris 150 mg/ml injektioneste, liuos toimitetaan yhtä käyttökertaa varten tarkoitetussa injektiopullossa.
Ohjeet annostelua varten
Anna injektiopullon lämmetä huoneenlämpöiseksi ennen pistämistä. Liuoksessa ei saa olla havaittavia hiukkasia, ja sen on oltava kirkasta tai opaalinhohtoista. Liuoksen on oltava väritöntä tai hieman ruskehtavan keltaisen sävyistä. Käytä 18 G tai 21 G x 2” neulaa (tai vastaavaa markkinasaatavuuden mukaan) ja 1 ml ruiskua ja vedä ruiskuun varovasti tarvittava tilavuus annettavan annoksen mukaisesti. Kun tarvittava tilavuus on vedetty ruiskuun, kiinnitä neulansuojus takaisin siirtoneulaan ja irrota neula ruiskusta. Kiinnitä ruiskuun 27 G x 0,5” neula (tai vastaava markkinasaatavuuden mukaan) ja pistä liuos välittömästi ihon alle. Pakkausselosteessa annetaan yksityiskohtaiset käyttöohjeet.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Esitäytetty kynä
Ilaris 150 mg injektioneste, liuos toimitetaan yhtä käyttökertaa varten tarkoitetussa esitäytetyssä kynässä.
Ohjeet annostelua varten
Kun pakkaus on poistettu jääkaapista, esitäytetyn kynän on annettava lämmetä huoneenlämpöiseksi (alle 30 °C) 30 minuutin ajan.
Ennen käyttöä suositellaan esitäytetyn kynän silmämääräistä tarkastusta. Liuoksen on oltava kirkasta tai opaalinhohtoista. Sen väri voi vaihdella värittömästä hieman ruskehtavan keltaiseen. Liuoksessa saattaa näkyä pieni ilmakupla, mikä on normaalia. Älä käytä, jos liuoksessa näkyy hiukkasia tai se on selkeästi ruskeaa. Pakkausselosteessa annetaan yksityiskohtaiset käyttöohjeet.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ILARIS injektioneste, liuos
150 mg/ml 1 kpl
- Ei korvausta.
ATC-koodi
L04AC08
Valmisteyhteenvedon muuttamispäivämäärä
30.04.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com